 Ceyda Bayraktar Eltutan1*
Ceyda Bayraktar Eltutan1* Simon Rinaldi2
Simon Rinaldi2 Atay Vural3Bagdagul Aksu4Hülya Maras Genc1
Atay Vural3Bagdagul Aksu4Hülya Maras Genc1 Edibe Pembegul Yildiz1
Edibe Pembegul Yildiz1- 1Division of Pediatric Neurology, Department of Pediatrics, Istanbul Faculty of Medicine, Istanbul University, Istanbul, Türkiye
- 2Nuffield Department of Clinical Neurosciences, John Radcliffe Hospital, University of Oxford, Oxford, United Kingdom
- 3Research Center for Translational Medicine, Koc University, Istanbul, Türkiye
- 4Division of Pediatric Nephrology, Department of Pediatrics, Istanbul Faculty of Medicine, Istanbul University, Istanbul, Türkiye
We present a 12-year-old boy with acute onset sensorimotor neuropathy and membranous glomerulonephritis associated with contactin-1 antibodies. This prompted us to explore the clinical characteristics of this condition and assess whether its presentation differs between pediatric and adult patients. A comprehensive search was conducted across multiple online databases, including PubMed and EMBASE, using MeSH terms such as “chronic inflammatory demyelinating polyradiculopathy”, “acute inflammatory demyelinating polyradiculopathy “, “CIDP”, “Guillain Barre syndrome”, “proteinuria”, “nephrotic syndrome”, “nephropathy”, “renal disease”, “glomerulonephritis”, “membranous nephropathy”, “autoimmune nodopathies”, and “membranous glomerulonephritis”. We reviewed publications up to October 2024 and identified 39 patients with anti-contactin associated CIDP (chronic inflammatory demyelinating polyradiculopathy) with membranous glomerulonephritis (MGN), including our case. This rare coexistence typically occurs at advanced ages, with only two pediatric cases. Clinical features were similar regardless of age at onset. We compared the onset, symptoms, progression, renal histopathology, and treatment responses between pediatric and adult patients.
Introduction
Antibodies against neurofascin-155 (Nf155), neurofascin 140/186 (Nf140/186), contactin-1 (CNTN1) and contactin associated protein-1 (Caspr1) are found in some patients with acquired, immune mediated neuropathies (1). In the latest guideline published in 2021 by the Joint Task Force of the European Academy of Neurology and the Peripheral Nerve Society, patients with these autoantibodies are classified as having autoimmune nodopathies, rather than chronic inflammatory demyelinating polyneuropathy (CIDP) (2).
Both acute inflammatory demyelinating polyneuropathies (AIDP) and CIDP are acquired, immune-mediated, peripheral neuropathies. AIDP, an electrophysiologically defined subtype of Guillain Barre Syndrome (GBS), has an acute onset and is usually self-limited, while CIDP has a persistent or relapsing course of disease. The association between acute polyneuropathy and nephropathy has been long reported (3–5). GBS-associated nephropathy usually does not require histological diagnosis, as it often resolves spontaneously. In cases associated with CIDP, the renal disease appears to persist (6). Several reports have described an association between CIDP and nephropathy (7–14). More specifically, the relationship between autoimmune nodopathy and membranous glomerulonephritis (MGN) has recently become a subject of further research and its physiopathology has been partially elucidated (10–12).
MGN is also an immune-mediated disease. Although it is one of the most common causes of nephrotic syndrome in adults, it is rare among children. When MGN develops secondary to an underlying condition -such as infection, malignancy, or autoimmune disease- it is classified as secondary MGN. Primary and secondary MGN differ in their pathological appearances. While both types can show lesions in the glomerular basement membrane, primary MGN typically involves only the capillary walls. In contrast, secondary MGN may present with additional findings such as mesangial hypercellularity, extraglomerular deposits, and full-house immunostaining. While IgG4-predominant staining is associated with primary MGN, IgG1 and IgG2 predominance is expected in secondary MGN (15). In 80% of cases of primary MGN, anti-phospholipase A2 receptor (PLA2R) is positive, while it is mostly negative in secondary MGN. In 15% of cases of primary MGN, the target antigen remains unknown. Previous case reports have suggested an association between antibody mediated neuropathies and nephropathy, indicating that an immune-mediated neuro-renal disease may be targeting both myelin and podocytes (12).
Case report
A 12-year-old Turkish boy presented with mild, gradually progressive muscle weakness over several weeks. He was unable to walk without support for the last three days. There was weakness of hip flexors, hip extensors, knee flexors, knee extensors, ankle flexors and extensors. Deep tendon reflexes were decreased in the upper extremities and absent in the lower extremities. Vibration sensation was reduced up to the knees. Romberg’s test was positive, suggesting sensory ataxia. Cranial nerve examination was normal. There was no respiratory compromise. The remainder of the examination was normal.
Cerebrospinal fluid (CSF) analysis revealed elevated protein levels (236 mg/dl, normal value <45 mg/dl) with no leukocytes. Contrast-enhanced spinal cord MRI showed thickening of the spinal roots of the cauda equine with contrast enhancement (Figure 1). Nerve conduction studies (NCS) showed significant reduced motor conduction velocity, decreased compound muscle action potentials (CMAPs), and prolonged motor distal latency in bilateral common peroneal and tibial nerves. Sensory nerve action potentials (SNAPs) were absent in the lower extremities and was relatively normal in the upper extremities. Initially he was diagnosed with AIDP and received intravenous immunoglobulin (0,4 gr/kg/d for five days). After the initial treatment there was minimal improvement in lower extremity numbness only. At this time, the modified Rankin Scale (mRS) score was 4.
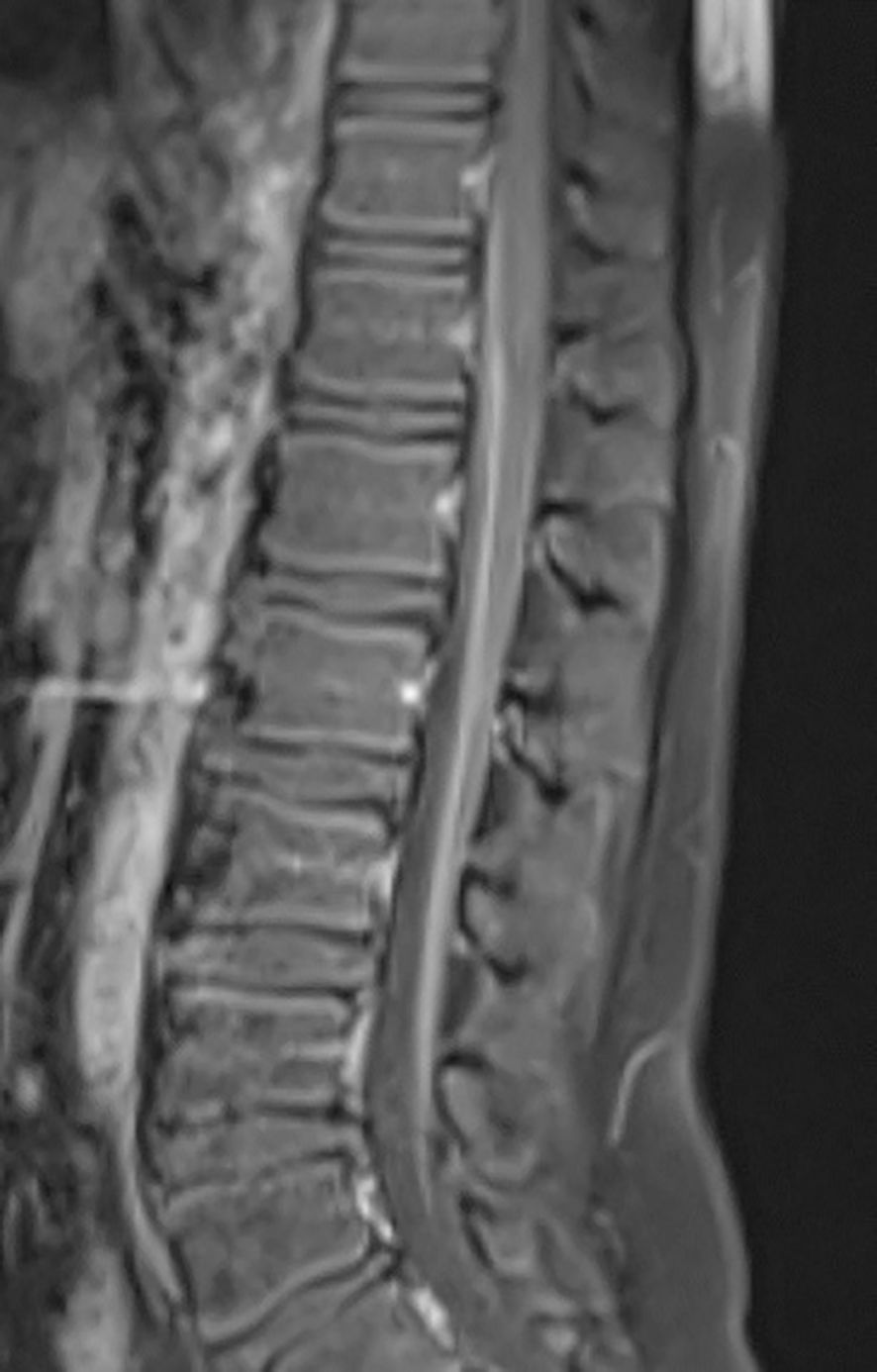
Figure 1. Sagittal T1-weighted post-contrast MRI of the spine demonstrates enhancement of the cauda equina nerve roots.
The patient also had concomitant new-onset nephrotic range proteinuria. Blood urea nitrogen and serum creatinine levels were normal, and there was no edema. A 24-hour urine collection demonstrated 4.2 g/L of protein. Renal ultrasonography was unremarkable. A renal biopsy was performed which was compatible with stage II membranous nephropathy. Light microscopy revealed global sclerosis in 1 out of 20 glomeruli. Direct immunofluorescence showed diffuse granular subepithelial IgG deposits, staining along capillary loops [(IgG (3+), IgM (-), C3 (2+), fibrinogen (-), C1q (-), C4d (+), kappa (2+), lambda (3+)]. PLA2R was negative by immunohistochemistry. There was no amyloid deposition. Electron microscopy showed uniform diffuse thickening of the glomerular basement membrane with the formation of segmental spikes. There was no increase in mesangial cellularity, and the tubules and interstisium showed no significant abnormalities. The absence of mesangial hypercellularity, extraglomerular deposits, and full-house immunostaining, along with subepithelial IgG deposits and spike formation on electron microscopy, were suggestive of primary MGN despite negative PLA2R staining.
Serological tests for possible etiological factors, including hepatitis B virus DNA, antibodies to hepatitis C virus and cytomegalovirus, HIV, and Treponema pallidum, were all within normal limits. There was no significant family history of neurological or renal disease. Complement and immunoglobulin levels were within normal ranges. Serum anti-ganglioside antibodies were negative. Folate, vitamin B12 and HbA1c levels were also within normal ranges. Anti-nuclear antibody was positive with a titer of 1:100. Anti-SSa and anti-Mi antibodies were also positive. Anti-neutrophil cytoplasmic antibodies (ANCA), anti-PLA2R, anti-GBM, and anti-double-stranded DNA (anti-dsDNA) antibodies were all negative. Ophthalmological examination was unremarkabele, and the Schirmer test was negative. The salivary gland biopsy showed sparse periductal lymphocytic infiltration, with no significant inflammatory changes.
Due to the progression period exceeding 8 weeks, acute-onset CIDP was diagnosed according to the European Academy of Neurology and the Peripheral Nerve Society (EAN/PNS) (2). We administered intravenous methylprednisolone 1 gr/day for five days. He was able to take a few steps without help, by the fifth day. Neurological symptoms, including ataxia, limb numbness and gait disturbance were slightly alleviated, the mRS score was 3 (Table 1). There was only a slight decrease in proteinuria. Due to partial improvement in neurological findings, and incomplete response for nephrologic findings, rituximab therapy was initiated. After 4 weeks of rituximab treatment, neurological symptoms further improved, the mRS score decreased to 2, and the spot urine protein/creatinine ratio gradually declined from about 3–4 to near 1 (Figure 2). As autoimmune nodopathy panel testing was not available at our center, the serum sample was sent to the Oxford Autoimmune Neurology Diagnostic Laboratory. Serum samples were analyzed using a live cell-based assay, with initial screening dilutions of 1:100 for NF155 and NF186, and 1:40 for CNTN1/Caspr1. Our patient tested positive for anti-CNTN1 antibodies at screening level, and subsequent subclass analysis revealed IgG1 predominance. Monthly maintenance IVIG therapy was continued. An increase in proteinuria was observed 3–4 months after the loading rituximab doses. He receives regular physiotherapy. On his last examination, five months after rituximab, he exhibited no motor weakness; however, proteinuria persisted at a non-nephrotic level. We follow the patient in terms of relapse of both neurological and renal symptoms.
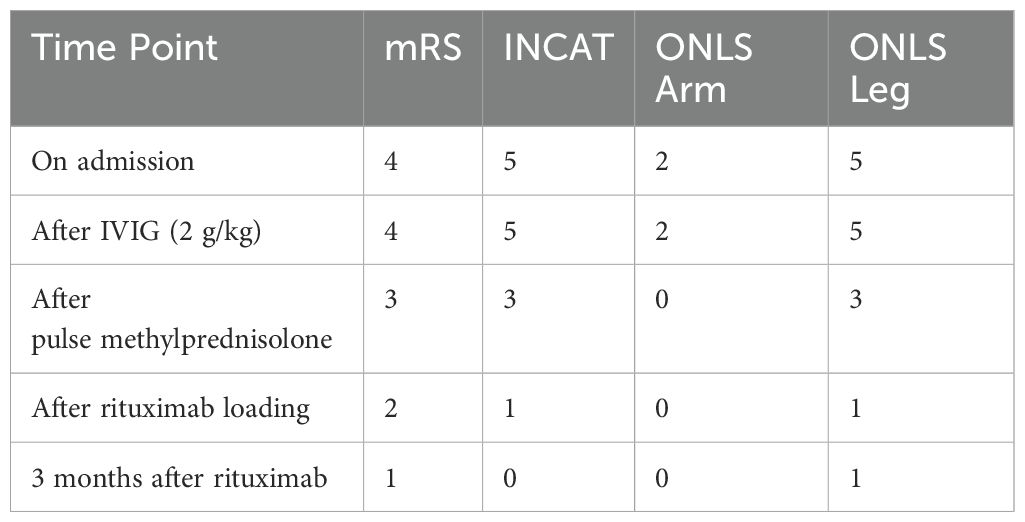
Table 1. Changes in disability scores over time.
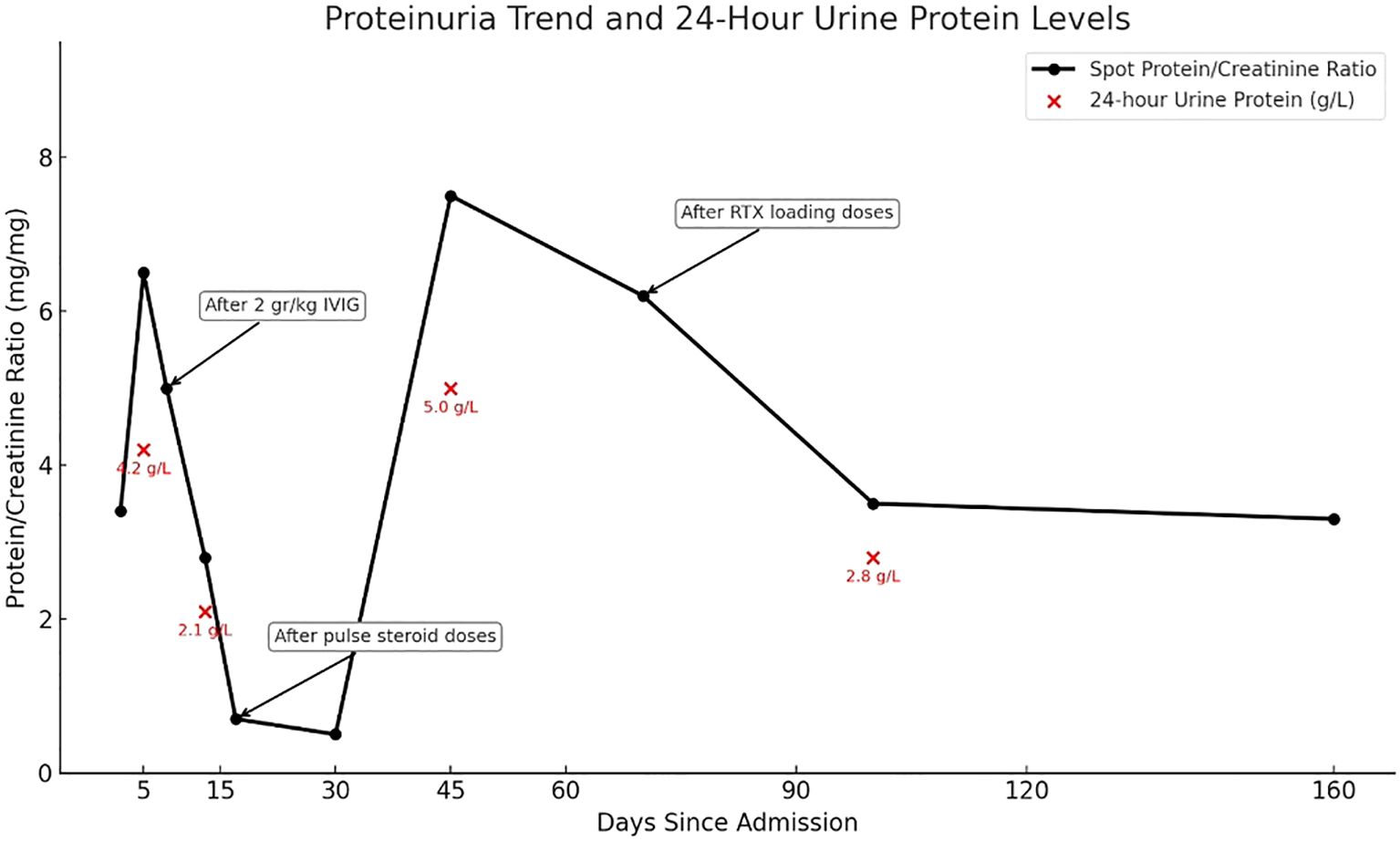
Figure 2. Spot urine protein/creatinine ratio and 24-hour urine protein levels during follow-up. The black line shows spot urine protein/creatinine ratio trends, and red dots indicate 24-hour urine protein levels. Arrows mark the timing og IVIG, pulse steroids, and rituximab treatments.
Discussion
We conducted a comprehensive literature review of cases related to our patient, carefully examining the references of relevant articles to avoid any reported patients. To our knowledge, the first pediatric patient of CIDP with MGN was reported in 1992 by Kohli et al. (9), followed by a second case reported by Kanemoto in 1999 (16). Tang et al. later identified the first adolescent-onset case presenting with a GBS-like illness and positive for anti-CNTN1 antibodies (7). As the first two pediatric cases were reported in the 1990s, the clinical information was limited, and anti-contactin antibodies were not studied at that time. Including our case, a total of 39 patients with anti-contactin antibody-associated CIDP and membranous glomerulonephritis (MGN) have been reported in the literature (Table 2), with only one previous pediatric case - a 14-year-old-boy- described by Tang et al. (7). Therefore, our patient represents the second pediatric case with this association. This condition predominantly occurs in older adults. Among the 37 adult patients, age at onset ranged from 39 to 80 years, with a mean of 59.7 years. Of these, 25 were male, 6 were female, and gender was not reported in 6 cases.
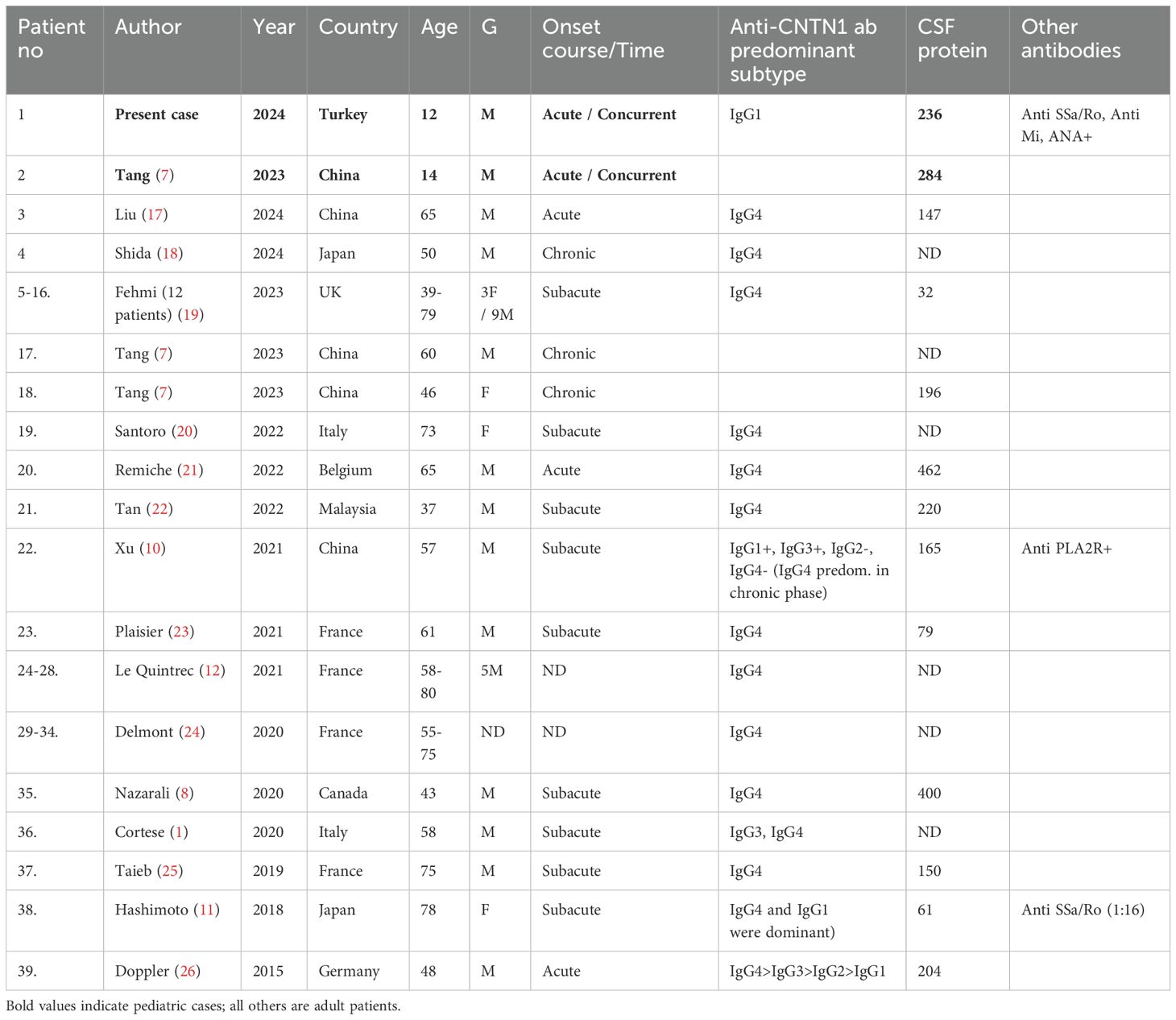
Table 2. Patients with neuropathy and membranous glomerulonephritis with positive anti-CNTN1 antibody.
Previous studies have indicated that anti-CNTN1 associated nodopathy exhibit distinct characteristics (27), which are even more pronounced in patients with concomitant membranous glomerulonephritis (MGN) (10, 11, 14). Xu et al. compared anti-CNTN1 positive patients with and without MGN, finding that those with MGN had an earlier age of onset (10). We observed that the disease onset was more often subacute or chronic in adults. In contrast, both pediatric patients were initially diagnosed with AIDP, which was later revised to CIDP as the disease course progressed. Following the detection of the anti-contactin antibodies, the diagnosis of autoimmune nodopathy was confirmed.
Clinical characteristics of anti-CNTN1 associated nodopathy include acute or subacute onset, rapid progression, worse disability, distal-dominant sensorimotor neuropathy, frequent proprioceptive sensory loss leading to sensory ataxia, elevated CSF protein levels, early axonal involvement, and poor response to intravenous immunoglobulin. The majority of patients including the pediatric cases exhibited these findings. All patients had nephrotic range proteinuria. Renal biopsy showed granular deposits of IgG along the glomerular basement membrane consistent with membranous glomerulonephritis for all patients. High intensity of IgG staining on glomeruli, paucity of C3 deposits and the absence of C1q staining were observed, consistent with primary MGN. Antibodies against phospholipase A2 receptor (PLA2R) were negative in all cases except one. Interestingly, this patient was also one of the three patients who had anti-contactin antibodies with IgG1 subclass predominance (10). Including the other reported pediatric patient, the majority of patients exhibited IgG4 subclass anti-CNTN1 antibodies. IgG1 subclass predominance in three patients was attributed to early sample collection during the acute phase of the disease (26, 28). In the case reported by Xu et al., when retested in the chronic phase, a subclass switch from IgG1 to IgG4 was revealed (10). Notably, all three patients with IgG1 predominance also exhibited positivity for other autoantibodies. In the case reported by Xu et al. anti PLA2R antibodies were positive, and in the case reported by Hashimoto et al. anti SSA/Ro positivity was observed (11). Our patient is the third case with IgG1-predominant anti-CNTN1 antibodies. Serum sample was also obtained during the acute phase, and the additional antibodies -including anti SSA/Ro, anti-Mi and ANA- were also detected.
There are only four reported cases in the literature, in which patients diagnosed with both CIDP and MGN tested negative for anti-CNTN1 antibodies. Two cases were reported by Tang et al. (7), one by Zhang et al. (29), and one by Gupta et al. (30). In the case reported by Zhang et al., renal histopathology was consistent with secondary MGN. In the case reported by Gupta et al., the patient tested positive for PLA2R and exhibited features consistent with Sjogren's syndrome. These findings suggest that the MGN in both patients may be related to a condition other than anti-contactin associated nodopathy. However, in the study by Tang et al., despite the clinical and laboratory findings being highly consistent with autoimmune nodopathy, both patients were tested negative. This raises the question of whether serum antibody testing was performed after immunotherapy, or whether seropositivity might have detected during subsequent follow-up. Several cases were reported prior to 2015, with the coexistence of CIDP with MGN, when anti contactin antibodies were not tested. Given the high probability of antibody positivity in these patients, testing even during follow-up may offer valuable guidance for immunotherapy decisions.
Our literature review revealed that, among patients with contactin-positive nodopathy and nephropathy, membranous glomerulonephritis (MGN) was the only renal pathology reported. Similarly, in patients with anti-neurofascin155 associated nodopathy and nephropathy, the histopathological diagnosis has consistently been focal segmental glomerulosclerosis (FSGS) (31–33). This suggests a potential relationship between the type of autoantibody and the glomerular pathology. The absence of glomerulonephritis in seronegative patients across various cohort studies further supports the hypothesis of an immune-mediated neuro-renal disease (24).
Smyth et al. reported a patient who developed MGN two decades after a CIDP diagnosis, and had been followed for essential hypertension for 10 years prior (34). Therefore, in patients diagnosed with CIDP at a relatively young age, it would be beneficial to follow blood pressure and proteinuria.
Several reports have emphasized that patients with autoimmune nodopathies have poor response to first-line treatments like corticosteroids or IVIG, in contrast to CIDP patients, which show a good response. When we reviewed the overall treatment responses, we found that while IVIG initially improved sensorimotor functions, it was not consistently effective in follow-up, and had minimal impact on proteinuria. The limited response to IVIG treatment in nodopathy is often attributed to the predominance of IgG4 subclass antibodies. Recent studies suggested that IgG1 and IgG3 subclasses may contribute to acute inflammation through complement activation, which could explain the transient initial response to IVIG (11, 35). It is also stated that, a subclass switch to IgG4 during the chronic stage, may explain the subsequent decline in IVIG responsiveness (36, 37). Early identification of autoimmune nodopathies is crucial for guiding treatment and preventing long term axonal degeneration. Corticosteroids provided transient improvement in nephrotic symptoms. Rituximab was generally more effective than both IVIG and corticosteroids in managing both nodopathy and nephropathy (23). However, resistance to rituximab has also been reported (25). In both pediatric cases, the treatment responses were consistent with these overall findings. Furthermore, longitudinal studies are needed to fully understand the complex interplay between autoantibodies and complement activation in autoimmune nodopathies.
Conclusion
Our case provides further evidence for the association between autoimmune nodopathy and nephropathy in pediatric population. Due to the limited number of reported pediatric cases and their short follow-up durations, it remains unclear whether early-onset correlates with disease severity. The distinct phenotype of anti-contactin-1 mediated nodopathies suggests a shared immune-mediated pathogenesis affecting both the peripheral nervous system and the kidneys. Antibody testing should be considered in male patients with acute or subacute onset of severe sensorimotor neuropathy, especially when response to IVIG and corticosteroids is poor. Although the coexistence of nodopathy and nephropathy is rare, urine analysis for proteinuria and blood pressure measurements may help detect subclinical renal involvement at an earlier stage. Different types of nephrotic syndrome may occur depending on the type of antibody present. Rituximab appears to be an effective treatment option for both neurological and renal involvement in these cases. The main limitation was the inability to repeat antibody subclass testing during follow-up, which could have provided further insight.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
CE: Conceptualization, Data curation, Project administration, Writing – original draft. SR: Writing – review & editing. AV: Writing – review & editing. BA: Writing – review & editing. HG: Writing – review & editing. EY: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We would like to thank Oxford Autoimmune Neurology Diagnostic Laboratory for their assistance in processing and analyzing the blood sample of our patient.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cortese A, Lombardi R, Briani C, Callegari I, Benedetti L, Manganelli F, et al. Antibodies to neurofascin, contactin-1, and contactin-associated protein 1 in CIDP: Clinical relevance of IgG isotype. Neurol Neuroimmunol Neuroinflamm. (2020) 7(1):e639. doi: 10.1212/NXI.0000000000000639
2. Van den Bergh PYK, van Doorn PA, Hadden RDM, Avau B, Vankrunkelsven P, Allen JA, et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint Task Force-Second revision. Eur J Neurol. (2021) 28:3556–83. doi: 10.1111/ene.v28.11
3. Rodriguez-Iturbe B, Garcia R, Rubio L, Zabala J, Moros G, and Torres R. Acute glomerulonephritis in the Guillain-Barre-Strohl syndrome. Report of nine cases. Ann Intern Med. (1973) 78:391–5. doi: 10.7326/0003-4819-78-3-391
4. Behan PO, Lowenstein LM, Stilmant M, and Sax DS. Landry-Guillain-Barre-Strohl syndrome and immune-complex nephritis. Lancet. (1973) 1:850–4. doi: 10.1016/S0140-6736(73)91420-7
5. Peters DK, Sevitt LH, Direkze M, and Bayliss SG. Landry-Guillain-Barre-Strohl polyneuritis and the nephrotic syndrome. Lancet. (1973) 1:1183–4. doi: 10.1016/S0140-6736(73)91181-1
6. Filippone EJ, Kanzaria M, Bell R, Newman E, and J LF. Secondary membranous nephropathy associated with guillain-barre syndrome. Case Rep Nephrol Urol. (2013) 3:34–9. doi: 10.1159/000350903
7. Tang Y, Liu J, Gao F, Hao H, Jia Z, Zhang W, et al. CIDP/autoimmune nodopathies with nephropathy: a case series study. Ann Clin Transl Neurol. (2023) 10:706–18. doi: 10.1002/acn3.51754
8. Nazarali S, Mathey EK, Tang D, Margetts PJ, and Baker SK. Chronic inflammatory demyelinating polyneuropathy and concurrent membranous nephropathy. Can J Neurol Sci. (2020) 47:585–7. doi: 10.1017/cjn.2020.46
9. Kohli A, Tandon P, and Kher V. Chronic inflammatory demyelinating polyradiculoneuropathy with membranous glomerulonephritis: report of one case. Clin Neurol Neurosurg. (1992) 94:31–3. doi: 10.1016/0303-8467(92)90115-J
10. Xu Q, Liu S, Zhang P, Wang Z, Chang X, Liu Y, et al. Characteristics of anti-contactin1 antibody-associated autoimmune nodopathies with concomitant membranous nephropathy. Front Immunol. (2021) 12:759187. doi: 10.3389/fimmu.2021.759187
11. Hashimoto Y, Ogata H, Yamasaki R, Sasaguri T, Ko S, Yamashita K, et al. Chronic inflammatory demyelinating polyneuropathy with concurrent membranous nephropathy: an anti-paranode and podocyte protein antibody study and literature survey. Front Neurol. (2018) 9:997. doi: 10.3389/fneur.2018.00997
12. Le Quintrec M, Teisseyre M, Bec N, Delmont E, Szwarc I, Perrochia H, et al. Contactin-1 is a novel target antigen in membranous nephropathy associated with chronic inflammatory demyelinating polyneuropathy. Kidney Int. (2021) 100:1240–9. doi: 10.1016/j.kint.2021.08.014
13. Hu N, Niu J, and Liu M. Chronic inflammatory demyelinating polyradiculoneuropathy concomitant with nephropathy. Neurol Sci. (2022) 43:5885–98. doi: 10.1007/s10072-022-06215-4
14. Chen KH, Chang CT, and Hung CC. Glomerulonephritis associated with chronic inflammatory demyelinating polyneuropathy. Ren Fail. (2006) 28:255–9. doi: 10.1080/08860220600580415
15. Beck LH Jr., Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. (2009) 361:11–21. doi: 10.1056/NEJMoa0810457
16. Kanemoto K, Nakahara C, Saitoh H, Fukushima T, Kashiwagi R, Takahashi M, et al. Renal glucosuria and membranous glomerulonephritis in chronic inflammatory demyelinating polyradiculoneuropathy: CIDP. Nihon Jinzo Gakkai Shi. (1999) 41:511–6.
17. Liu Y, Yang CL, Zhao XL, Zhao YJ, Du T, Wang CC, et al. Characteristics of anti-contactin1 antibody positive autoimmune nodopathies combined with membranous nephropathy. J Neuroimmunol. (2024) 396:578460. doi: 10.1016/j.jneuroim.2024.578460
18. Shida R, Iwakura T, Ohashi N, Ema C, Aoki T, Tashiro T, et al. Anti-contactin 1 antibody-associated membranous nephropathy in chronic inflammatory demyelinating polyneuropathy with several autoantibodies. Intern Med. (2024) 63:699–705. doi: 10.2169/internalmedicine.2126-23
19. Fehmi J, Davies AJ, Antonelou M, Keddie S, Pikkupeura S, Querol L, et al. Contactin-1 links autoimmune neuropathy and membranous glomerulonephritis. PloS One. (2023) 18:e0281156. doi: 10.1371/journal.pone.0281156
20. Santoro D, Debiec H, Longhitano E, Torreggiani M, Barreca A, Vegezzi E, et al. Contactin 1, a potential new antigen target in membranous nephropathy: A case report. Am J Kidney Dis. (2022) 80:289–94. doi: 10.1053/j.ajkd.2021.08.025
21. Remiche G, Monteiro MLS, Catalano C, Hougardy JM, Delmont E, Boucraut J, et al. Rituximab responsive relapsing-remitting igG4 anticontactin 1 chronic inflammatory demyelinating polyradiculoneuropathy associated with membranous nephropathy: A case description and brief review. J Clin Neuromuscul Dis. (2022) 23:219–26. doi: 10.1097/CND.0000000000000395
22. Tan CY, Goh KJ, Oh AW, Devaux J, and Shahrizaila N. Autoantibody profile in a Malaysian cohort of chronic inflammatory demyelinating polyneuropathy. Neuromuscul Disord. (2022) 32:255–62. doi: 10.1016/j.nmd.2022.01.006
23. Plaisier E, Not A, Buob D, Ronco P, and Debiec H. Contactin-1-associated membranous nephropathy: complete immunologic and clinical remission with rituximab. Kidney Int. (2021) 100:1342–4. doi: 10.1016/j.kint.2021.08.029
24. Delmont E, Brodovitch A, Kouton L, Allou T, Beltran S, Brisset M, et al. Antibodies against the node of Ranvier: a real-life evaluation of incidence, clinical features and response to treatment based on a prospective analysis of 1500 sera. J Neurol. (2020) 267:3664–72. doi: 10.1007/s00415-020-10041-z
25. Taieb G, Le Quintrec M, Pialot A, Szwarc I, Perrochia H, Labauge P, et al. Neuro-renal syndrome” related to anti-contactin-1 antibodies. Muscle Nerve. (2019) 59:E19–21. doi: 10.1002/mus.26392
26. Doppler K, Appeltshauser L, Wilhelmi K, Villmann C, Dib-Hajj SD, Waxman SG, et al. Destruction of paranodal architecture in inflammatory neuropathy with anti-contactin-1 autoantibodies. J Neurol Neurosurg Psychiatry. (2015) 86:720–8. doi: 10.1136/jnnp-2014-309916
27. Vural A, Doppler K, and Meinl E. Autoantibodies against the node of ranvier in seropositive chronic inflammatory demyelinating polyneuropathy: diagnostic, pathogenic, and therapeutic relevance. Front Immunol. (2018) 9:1029. doi: 10.3389/fimmu.2018.01029
28. Miura Y, Devaux JJ, Fukami Y, Manso C, Belghazi M, Wong AH, et al. Contactin 1 IgG4 associates to chronic inflammatory demyelinating polyneuropathy with sensory ataxia. Brain. (2015) 138:1484–91. doi: 10.1093/brain/awv054
29. Zhang Q, Tan Y, Meng L, Xu R, Zhou F, and Zhao M. A rare case of membranous nephropathy associated with chronic inflammatory demyelinating polyradiculoneuropathy. Ren Fail. (2023) 45:2209659. doi: 10.1080/0886022X.2023.2209659
30. Gupta N, Khullar D, Prasad P, Grover R, Chabbra G, Gandhi KR, et al. Rare occurrence of PLA2R-positive membranous nephropathy in a patient with sjogren’s syndrome and CIDP. Indian J Nephrol. (2020) 30:117–20. doi: 10.4103/ijn.IJN_152_19
31. Bukhari S, Bettin M, Cathro HP, Gwathmey K, Gautam J, and Bowman B. Anti-neurofascin-associated nephrotic-range proteinuria in chronic inflammatory demyelinating polyneuropathy. Kidney Med. (2020) 2:797–800. doi: 10.1016/j.xkme.2020.06.016
32. Delmont E, Manso C, Querol L, Cortese A, Berardinelli A, Lozza A, et al. Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain. (2017) 140:1851–8. doi: 10.1093/brain/awx124
33. Querol L, Nogales-Gadea G, Rojas-Garcia R, Diaz-Manera J, Pardo J, Ortega-Moreno A, et al. Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology. (2014) 82:879–86. doi: 10.1212/WNL.0000000000000205
34. Smyth S and Menkes DL. Coincident membranous glomerulonephritis and chronic inflammatory demyelinating polyradiculoneuropathy: questioning the autoimmunity hypothesis. Muscle Nerve. (2008) 37:130–5. doi: 10.1002/mus.20841
35. Appeltshauser L, Weishaupt A, Sommer C, and Doppler K. Complement deposition induced by binding of anti-contactin-1 auto-antibodies is modified by immunoglobulins. Exp Neurol. (2017) 287:84–90. doi: 10.1016/j.expneurol.2016.10.006
36. Huang CC, Lehman A, Albawardi A, Satoskar A, Brodsky S, Nadasdy G, et al. IgG subclass staining in renal biopsies with membranous glomerulonephritis indicates subclass switch during disease progression. Mod Pathol. (2013) 26:799–805. doi: 10.1038/modpathol.2012.237
Keywords: chronic inflammatory demyelinating polyradiculopathy, membranous glomerulonephritis, autoimmune nodopathy, anti-contactin antibody, child
Citation: Bayraktar Eltutan C, Rinaldi S, Vural A, Aksu B, Maras Genc H and Pembegul Yildiz E (2025) Case Report: Pediatric age onset CNTN1 antibody-associated neuropathy with nephropathy and literature review. Front. Immunol. 16:1549363. doi: 10.3389/fimmu.2025.1549363
Received: 21 December 2024; Accepted: 28 May 2025;
Published: 18 June 2025.
Edited by:
Chris Wincup, King’s College Hospital NHS Foundation Trust, United KingdomReviewed by:
Christian Moritz, Centre Hospitalier Universitaire (CHU) de Saint-Étienne, FranceLuca Leonardi, Sapienza University of Rome, Italy
Copyright © 2025 Bayraktar Eltutan, Rinaldi, Vural, Aksu, Maras Genc and Pembegul Yildiz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ceyda Bayraktar Eltutan, Y2V5ZGEuYmF5cmFrdGFyZWx0dXRhbkBpc3RhbmJ1bC5lZHUudHI=