 Shiho Kurosaka1,2‡
Shiho Kurosaka1,2‡ Hiromasa Tanno1*‡§Minako Hirose1Wakana Kamada1Rena Takayashiki1Ikue Sone1Yuki Sato1Takumi Watanabe2
Hiromasa Tanno1*‡§Minako Hirose1Wakana Kamada1Rena Takayashiki1Ikue Sone1Yuki Sato1Takumi Watanabe2 Shinyo Ishi3Miki Shoji3Yoshimichi Imai3
Shinyo Ishi3Miki Shoji3Yoshimichi Imai3 Ko Sato4†Keiko Ishii4
Ko Sato4†Keiko Ishii4 Hiromitsu Hara5
Hiromitsu Hara5 Sho Yamasaki6,7,8
Sho Yamasaki6,7,8 Shinobu Saijo8
Shinobu Saijo8 Yoichiro Iwakura9,10Kazuyoshi Kawakami4
Yoichiro Iwakura9,10Kazuyoshi Kawakami4 Emi Kanno1
Emi Kanno1- 1Department of Translational Science for Nursing, Tohoku University Graduate School of Medicine, Sendai, Japan
- 2Bio-Lab Co., Ltd., Hidaka, Japan
- 3Department of Plastic and Reconstructive Surgery, Tohoku University Graduate School of Medicine, Sendai, Japan
- 4Department of Medical Microbiology, Mycology and Immunology, Tohoku University Graduate School of Medicine, Sendai, Japan
- 5Department of Immunology, Graduate School of Medical and Dental Sciences, Kagoshima University, Kagoshima, Japan
- 6Department of Molecular Immunology, Research Institute for Microbial Diseases, Osaka University, Suita, Japan
- 7Laboratory of Molecular Immunology, Immunology Frontier Research Center, Osaka University, Suita, Japan
- 8Division of Molecular Immunology, Medical Mycology Research Center, Chiba University, Chiba, Japan
- 9Department of Veterinary Medical Sciences, Graduate School of Agricultural and Life Sciences, The University of Tokyo, Tokyo, Japan
- 10Laboratory for Human Disease Models, Louis Pasteur Center for Medical Research, Kyoto, Japan
Introduction: Lactic acid bacteria (LAB) are well known for their beneficial effects on the regulation of immune responses and host protection against microbial infections. We previously reported that heat-killed Enterococcus faecalis strain KH2 (heat-killed KH2), a species of LAB, enhances inflammatory responses at wound sites and accelerates the skin wound healing process. In this study, we aimed to clarify the pathway underlying the wound-healing effects of heat-killed KH2. We focused on CARD9, a common adaptor molecule for C-type lectin receptors and Dectin-2, the upstream receptor for this adaptor molecule.
Methods: Four full-thickness dermal wounds were created on the backs of wild-type (WT) mice, CARD9 KO mice, and Dectin-2 KO mice, and the effects of heat-killed KH2 administration were examined. We analyzed the percent wound closure, re-epithelialization, granulation tissue formation, and the production of inflammatory cytokines and chemokines.
Results: Heat-killed KH2 administration enhanced wound closure, granulation tissue formation, and re-epithelialization in WT mice. However, these effects were absent in heat-killed KH2-treated CARD9 KO mice. Similar results were observed in the migration of neutrophils and the production of TNF-α, IL-6, KC, and MIP-2 in heat-killed KH2-treated CARD9 KO mice. Furthermore, heat-killed KH-2 induced activation of reporter cells expressing Dectin-2. Finally, heat-killed KH-2 treatment in Dectin-2 KO mice did not promote skin wound healing.
Conclusion: These results suggest that recognition of heat-killed KH2 by Dectin-2 may activate CARD9-mediated signaling, which may contribute to the promotion of skin wound healing through KH2 treatment.
1 Introduction
Skin wound healing consists of three phases: inflammation, proliferation, and remodeling (1). In the inflammatory phase, keratinocytes and fibroblasts produce inflammatory cytokines and chemokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, keratinocyte-derived chemokine (KC), and macrophage inflammatory proteins (MIP)-1 and MIP-2. These molecules are important for leukocyte recruitment, and the migrated leukocytes, including neutrophils and macrophages, phagocytose pathogens and necrotic tissue, help to cleanse the wound and protect against infection. In the proliferative phase, numerous growth factors are essential for the formation of granulation tissue, angiogenesis, and epithelialization, which play critical roles in the healing process (1, 2). Lactic acid bacteria (LAB) are well known for their beneficial effects on intestinal regulation, anti-allergic activity, host protection against microbial infections, and anti-tumor effects (3, 4). LAB are generally classified into several groups, including Lactobacillus spp., Lactococcus spp., Streptococcus sp., and Enterococcus spp (5). Several studies have demonstrated the beneficial effects of LAB on wound healing. For example, genetically modified Lactobacillus enhances wound repair in a mouse and pig wound model (6, 7). Topical administration of heat-killed L. plantarum accelerates skin wound healing by increasing macrophage infiltration at the wound sites (8). We previously examined the effect of heat-killed E. faecalis strain KH2 (heat-killed KH2) on skin wound healing and reported that heat-killed KH2 induces inflammatory cytokines including TNF-α and IL-6 and promotes skin wound healing by accelerating re-epithelialization and granulation tissue formation (9). However, the mechanism by which heat-killed KH2 induces inflammation and promotes wound healing is not yet fully understood. In general, LABs are recognized by pattern recognition receptors, such as Toll-like receptors (TLRs) and C-type lection receptors (CLRs). TLRs are the most commonly reported pattern recognition receptors that recognize LAB (10, 11). However, in recent years, several studies have demonstrated the involvement of CLRs in the recognition of LAB (12, 13). There are various types of CLRs, including dendritic cell-associated C-type lectin (Dectin)-1, Dectin-2, Dectin-3, and macrophage-inducible C-type lectin (Mincle). Dectin-1, 2, 3, and Mincle are mainly expressed on dendritic cells (DCs) and macrophages. When ligands bind to these CLRs, these cells produce inflammatory cytokines via caspase recruitment domain-containing protein 9 (CARD9), a downstream signaling adaptor molecule of CLRs (14). We have previously shown that the CLRs-CARD9 pathway is involved in the skin wound healing process (15, 16).
Based on this background, in this study, we aimed to define the roles of CLRs and CARD9 in wound healing. We found that heat-killed KH2 is recognized by Dectin-2, not Dectin-1, and that CARD9-mediated signaling is essential for the promotion of skin wound healing by heat-killed KH2.
2 Materials and methods
2.1 Animals
CARD9, Dectin-1, and Dectin-2 KO mice were generated as previously described (17, 18). These KO mice were backcrossed to C57BL/6 mice for more than eight generations. Wild-type (WT) C57BL/6 mice, obtained from CLEA Japan (Tokyo, Japan), were used as controls. Male and female mice, aged 7 to 10 weeks, were housed under specific pathogen-free conditions at the Institute for Animal Experimentation, Tohoku University Graduate School of Medicine (Sendai, Japan) with ad libitum access to food and water. All animal experiments were approved by the Ethics Review Committee for Animal Experimentation of Tohoku University and were performed under anesthesia to minimize animal suffering.
2.2 Preparation of heat-killed E. faecalis KH2 and dextrin
E. faecalis KH2 (NITEP-14444; GenBank AB534553), isolated from a human fecal sample, was obtained from Bio-Lab Co., Ltd. (Saitama, Japan) (19). The strain was cultured aerobically overnight at 37°C in MRS broth (Difco, Detroit, MI, USA), centrifuged, and heat-killed at 105°C for 30 min using an autoclave (HV-2IILB; Hirayama Manufacturing Corp., Saitama, Japan). To improve water dispersibility, the heat-killed bacteria were homogenized at 15 MPa (ECONIZER LABO-01; Sanmaru Machinery Co., Ltd., Shizuoka, Japan) and mixed with an equal amount of dextrin (NSD300; San-ei Sucrochemical Co., Ltd., Aichi, Japan). The final suspension was diluted to 200 mg/mL with normal saline.
2.3 Wound creation and tissue collection
As previously described (9), mice were anesthetized with isoflurane (1.0–2.0%; Mairan Pharma, Osaka, Japan) and the dorsal skin was shaved and sterilized with 70% ethanol. Four 6-mm full-thickness wounds were created using a biopsy punch (Kai Industries Co., Ltd., Gifu, Japan). The wounds were covered with a polyurethane film (Tegaderm Transparent Dressing, 3M, Health Care, Saint Paul, MN, USA) for occlusive dressing, and an elastic bandage (Hilate, Iwatsuki, Tokyo, Japan) was applied to prevent the film from peeling off. At specified time points, mice were euthanized, and 1 cm² skin samples were excised for histopathological analysis and cytokine quantification.
2.4 Heat-killed KH2 treatment of wounds
Wounds were created following the method described above. Immediately after wounding, a 5-μL suspension of heat-killed KH2 (1000 μg), or an equal volume of dextrin as a vehicle control, was applied to the base of the wound in mice using a pipette.
2.5 Measurement of the wound area
Digital images of the wounds were acquired at baseline and at designated time points using a digital camera (CX4, Ricoh, Tokyo, Japan). The wound area was quantified using ImageJ software (NIH, Bethesda, MD, USA). Wound healing was evaluated as percent wound closure, calculated as follows: % wound closure = (1 − wound area at the indicated time point/wound area on day 0) × 100.
2.6 Histopathology and immunohistochemistry
The wounded tissues were fixed in 4% paraformaldehyde, paraffin-embedded, sectioned, and stained with hematoxylin-eosin (HE) (16, 20). Re-epithelialization was assessed by measuring the distance from the wound edge to the advancing epithelial cells in HE-stained sections. The re-epithelialization index was calculated as the percentage of the wound area covered by new epithelium. The granulation tissue area was also quantified from the HE-stained sections.
2.7 Measurement of cytokine and chemokine concentrations
The wound tissues were homogenized with a saline solution using a stainless-steel mesh, and the supernatant was collected after centrifugation. Supernatants were analyzed for cytokine and chemokine levels using enzyme-linked immunosorbent assay (ELISA) kits (BioLegend, San Diego, CA, USA, for TNF-α and IL-6; R&D Systems, Minneapolis, MN, USA, for KC, MIP-2, MIP-1α, and MIP-β). The results were expressed as values per wound.
2.8 Preparation of leukocytes in the wound tissue
As previously described (8), mice were sacrificed on days 1, 3 and 7 post-wounding. Wound tissues were excised, minced, and digested with a mixture of enzymes (Roche, Mannheim, Germany, 0.2 mg/mL Liberase TL, 2.5 mg/mL collagenase D, 0.1 mg/mL DNase I, 2.0 mg/mL Dispase II) in RPMI 1640 medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10 mM HEPES (Sigma) and 10% fetal calf serum (FCS; Biowest, Nuaillé, France). Cell suspensions were filtered through a 70 μm cell strainer (BD Falcon, Bedford, MA, USA), washed, and used for flow cytometric analysis.
2.9 Flow cytometric analysis
The cells obtained from the wounds were incubated with anti-mouse CD16/CD32 (clone 2.4G2, BD Biosciences, Franklin Lakes, NJ, USA) on ice for 15 min in phosphate-buffered saline (PBS) that contained 1% FCS and 0.1% sodium azide. The cells were then stained with the following antibodies: APC/Cy7-anti-Ly6G mAb (clone 1A8, BioLegend), APC-anti-CD11b mAb (clone M1/70, BioLegend), PE-anti-F4/80 mAb (clone BM8, BioLegend), Pacific Blue-anti-CD45 mAb (clone 30-F11, BioLegend), FITC-anti-CD45R/B220 mAb (clone RA3-6B2, BioLegend), FITC-anti-NK1.1 mAb (clone PK136, BioLegend), FITC-anti-T-cell receptor γδ (TCRγδ) mAB (clone GL3, BioLegend), FITC-anti-CD3ϵ mAb (clone 145-2C11, BioLegend), 7-AAD Viability Staining Solution (BioLegend), APC-anti-Ly6C mAb (clone HK1.4, BioLegend), and FITC- I-A/I-E (MHC II) (clone M5/114.15.2, BioLegend). To analyze macrophage subtypes, the CD45+ cells were gated on Ly6G-F4/80+ cells and subsequently analyzed the expression of Ly6C and MHC II within the Ly6G-F4/80+ population. Isotype-matched irrelevant IgG was used as a control for staining. The gating strategy is shown in Supplementary Figures 1 and 2.
2.10 Generation and culture of bone marrow-derived dendritic cells
BM-DCs were prepared following the protocol of Luts et al. (21). Briefly, BM cells from WT, CARD9 KO, Dectin-1 KO, and Dectin-2 KO mice were cultured at 2 × 105 cells/mL in RPMI 1640 medium supplemented with 10% FCS, 100 U/mL penicillin G, 100 μg/mL streptomycin, and 50 μM 2-mercaptoethanol (Sigma-Aldrich) and 20 ng/mL murine granulocyte-macrophage colony-stimulating factor (GM-CSF; FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan). On days 3 and 6, the cultures were supplemented with fresh medium containing GM-CSF. On day 8, non-adherent cells were harvested and used as BM-DCs. The obtained cells were then cultured with heat-killed KH2, LPS (Sigma-Aldrich), CpG1826 ODN (CpG) (Hokkaido System Science, Hokkaido, Japan), and heat-killed Candida albicans (HKCA) for 24 h at 37°C in a 5% CO2 incubator. The culture supernatants and cell pellets were collected and stored at -30°C before use.
2.11 Reporter assay
As described in previous reports (22), 2B4 T-cell hybridomas were transfected with an NFAT-GFP reporter construct, which consisted of three tandem NFAT-binding sites fused to enhanced GFP cDNA (23). This cell line was further transfected with Dectin-1 or Dectin-2 and FcRγ genes, and a control cell line lacking Dectin-2 was used. The cells were stimulated for 20 h in 37°C at 1.0 × 105 cells/mL with heat-killed KH2 (1, 10, 100 μg/mL), zymosan-depleted (dZymosan; 60 μg/mL, InvivoGen, San Diego, USA), and furfurman (100 μg/mL, InvivoGen). The reporter cells were incubated with anti-mouse CD16/CD32 (clone 2.4G2, BD Biosciences) on ice for 15 min in PBS that contained 1% FCS and 0.1% sodium azide. The cells were then stained with APC-anti-CD3ϵ mAb (clone 145-2C11, BioLegend) and washed three times in the same buffer. Isotype-matched IgG was used for control staining. GFP expression in gated CD3-positive reporter cells was evaluated using a BD FACS Canto II flow cytometer (BD Bioscience).
2.12 Statistical analysis
Data were analyzed using JMP® Pro 17 0.0 software (SAS Institute Japan, Tokyo, Japan). Results are expressed as the mean ± SD. Differences between groups were assessed for statistical significance using Dunnett’s or Tukey–Kramer’s honestly significant difference (HSD) test for comparisons involving more than three experimental groups after confirming normality using the Shapiro–Wilk test. A p-value of less than 0.05 was considered statistically significant.
3 Results
3.1 Effects of CARD9 deficiency on the promotion of skin wound healing by heat-killed KH2 administration
To investigate the involvement of CLRs in the recognition of heat-killed KH2 and the induction of inflammatory cytokines, we examined the role of CARD9, a downstream adaptor molecule of CLRs, by stimulating BM-DCs from WT and CARD9 KO mice with heat-killed KH2 and comparing TNF-α production. TNF-α production by BM-DCs upon stimulation with heat-killed KH2 was significantly reduced in CARD9 KO mice compared to WT mice (Supplementary Figure 3). Based on this result, we hypothesized that CLRs-CARD9 pathway is involved in the recognition of heat-killed KH2. Consequently, we considered the possibility that the CLRs-CARD9 pathway contributes to the mechanism by which KH2 promotes skin wound healing. To investigate this further, we used CARD9 KO mice to analyze the impact of CARD9 deficiency on the promotion of wound healing by heat-killed KH2. As shown in Figures 1A, B, WT mice treated with heat-killed KH2 exhibited significantly accelerated wound closure by day 7 post-wounding compared with WT mice treated with vehicle control. However, this effect was not observed in heat-killed KH2-treated CARD9 KO mice. In heat-killed KH2-treated WT mice, the granulation area and re-epithelialization rate were significantly increased by day 7 after wounding, compared with vehicle-treated WT mice. However, this effect was not observed in heat-killed KH2-treated CARD9 KO mice (Figures 1C–E). Additionally, wound closure, granulation area, and re-epithelialization rate were significantly delayed in heat-killed KH2-treated CARD9 KO mice compared to heat-killed KH2-treated WT mice (Figures 1C–E). These results suggest that CARD9 may be involved in the heat-killed KH2-mediated promotion of wound healing.
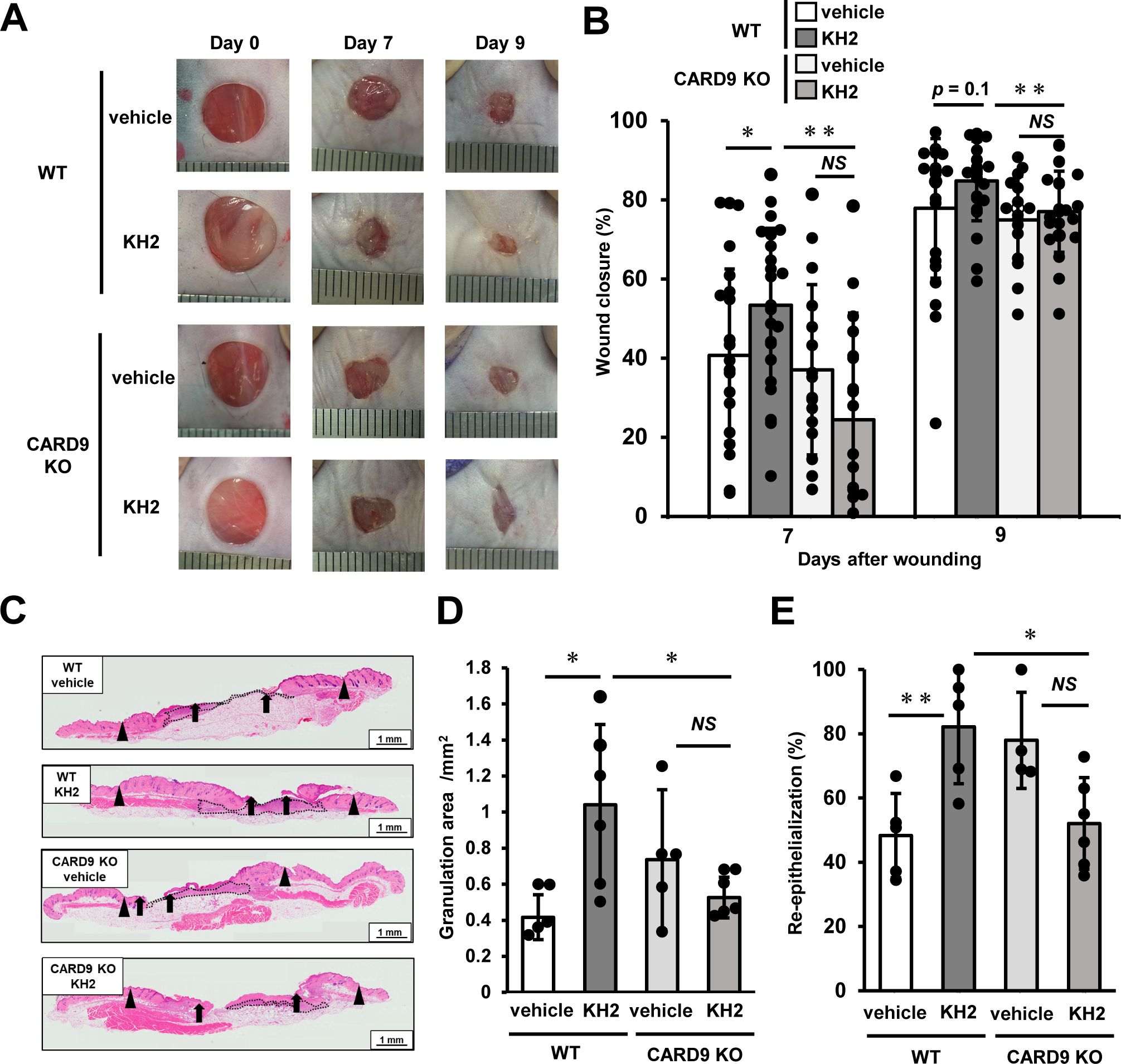
Figure 1. Effects of CARD9 deficiency on the promotion of skin wound healing by heat-killed KH2 administration. Wounds were created on the backs of WT mice or CARD9 KO mice treated with vehicle control or heat-killed KH2. Photographs (A) were taken, and the percentages of wound closure (B) were evaluated on days 7 and 9 after wound creation (n = 20–24 wounds). (C) Representative histological images of skin wounds on day 7. Arrowheads, arrows, and the dotted line indicate the original wound edges, granulation area, and re-epithelialized leading edges, respectively. The wound edges were defined as the border between normal epithelium and thick proliferative epithelium. (D) Granulation tissue area was quantified 7 days after wound creation (n = 4–6 wounds). (E) The re-epithelialization ratio was calculated 7 days post-wounding (n = 4–6 wounds). Each column represents the mean ± standard deviation. Results are representative of at least two independent experiments. *p < 0.05, **p < 0.01, NS, Not significant.
3.2 Effects of CARD9 deficiency on leukocyte infiltration by heat-killed KH2 administration
To elucidate the effects of CARD9 on heat-killed KH2-mediated leukocyte infiltration, we examined the kinetics of leukocyte infiltration in the wounded tissues. As shown in Figure 2A, the percentage of CD45+ leukocytes was significantly decreased in heat-killed KH2-treated CARD9 KO mice compared with heat-killed KH2-treated WT mice at 1 and 3 days post-wounding. The total number of leukocytes in the wounded tissues of heat-killed KH2-treated CARD9 KO mice was significantly reduced on day 1 post-wounding compared to heat-killed KH2-treated WT mice. There was no significant difference in the percentage of neutrophils between groups of heat-killed KH2-treated WT mice and CARD9 KO mice. On day 1, the number of neutrophils in heat-killed KH2-treated CARD9 KO mice showed a decreasing trend compared to heat-killed KH2-treated WT mice, with a significant reduction observed on day 3 (Figure 2B). The percentage of macrophages was significantly increased in heat-killed KH2-treated CARD9 KO mice compared with heat-killed KH2-treated WT mice at day 3. Additionally, the number of macrophages was significantly decreased in heat-killed KH2-treated CARD9 KO mice compared with heat-killed KH2-treated WT mice at day 1 post-wounding, with a trend toward a decrease at day 7 (Figure 2C). In contrast, there were no significant differences in the number of lymphocytes between heat-killed KH2-treated CARD9 KO and WT mice (Figure 2D). These results suggested that CARD9 may contribute to neutrophil accumulation in the early phase and macrophage accumulation in the late phase of wound healing after treatment with heat-killed KH2.
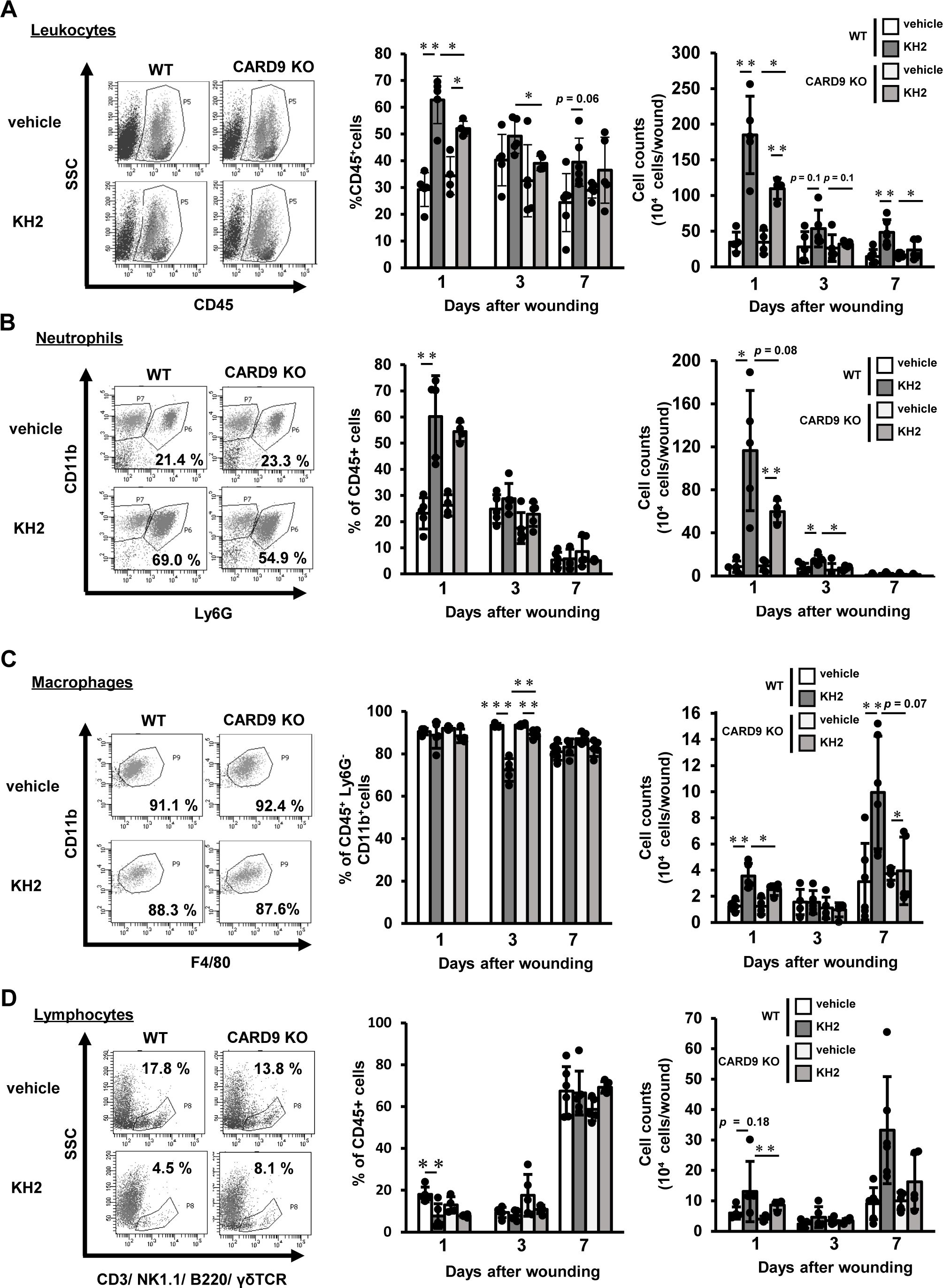
Figure 2. Effects of CARD9 deficiency on neutrophil infiltration by heat-killed KH2 administration. Flow cytometry was used to quantify the percentage and number of (A) leukocytes, (B) neutrophils, (C) macrophages, and (D) lymphocytes in wounded tissues on days 1, 3, and 7 post-wounding, as represented in the plots of wound-infiltrating cells. Leukocytes were prepared from four wounds at the indicated time points, and neutrophils were defined as CD45+Ly6G+CD11b+ cells. Macrophages were identified as CD45+F4/80+CD11b+Ly6G− cells. Lymphocytes were identified as CD45+ cells expressing CD3, NK1.1, TCRγδ, or B220 (n = 4–6 mice). The flow cytometry plots shown are representative of those from 1 day post-wounding. Each column represents the mean ± standard deviation. Results are representative of at least two independent experiments. *p < 0.05, **p < 0.01.
3.3 Effects of CARD9 deficiency on macrophage phenotype by heat-killed KH2 administration
Next, we analyzed macrophage subtypes at 7 days post-wounding. Following the classification by Rodero et al. (24), who reported Ly6C+MHCII− macrophages as the pro-inflammatory M1 phenotype and Ly6C−MHCII+ macrophages as the anti-inflammatory M2 phenotype, we also categorized macrophages accordingly. As shown in Figures 3A–C, the proportion of Ly6C+MHCII− macrophages was significantly lower in heat-killed KH2-treated WT mice compared with vehicle-treated WT mice, and also showed a trend toward a lower proportion compared with heat-killed KH2-treated KO mice. Furthermore, the numbers of Ly6C+MHCII− macrophages and Ly6C−MHCII+ macrophages significantly increased in heat-killed KH2-treated WT mice compared with vehicle-treated WT mice. However, this effect was not observed in heat-killed KH2-treated CARD9 KO mice. These results suggested that CARD9 may be crucial for heat-killed KH2 to modulate macrophage recruitment and polarization.
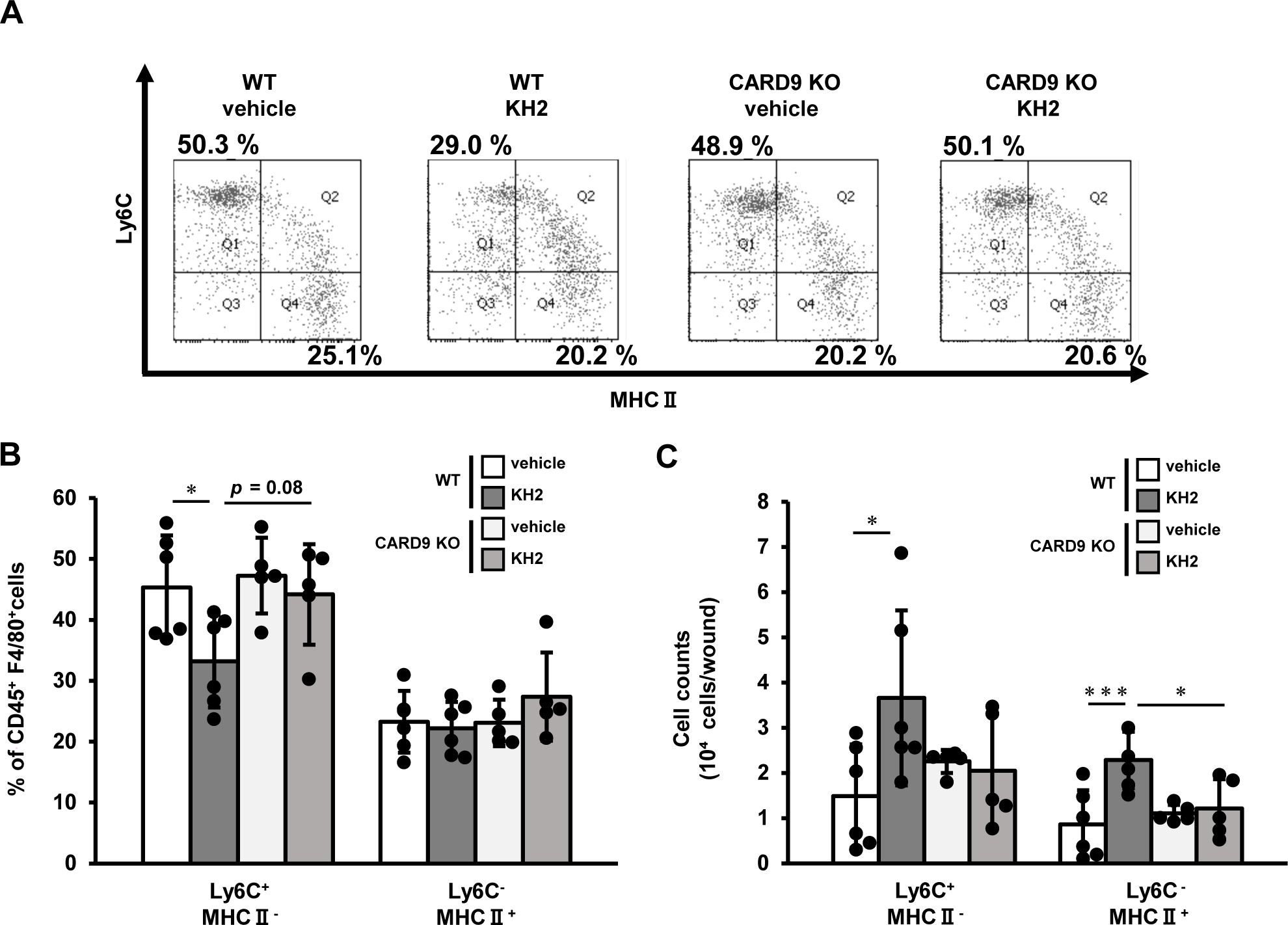
Figure 3. Effects of CARD9 deficiency on macrophage phenotype by heat-killed KH2 administration. Flow cytometry was used to quantify the percentage and number of macrophage subtypes in wounded tissues on day 7 after wound creation. (A) Representative plots of inflammatory and anti-inflammatory macrophages are shown. (B) The percentages of macrophages are shown, and (C) the number of macrophages is shown. Inflammatory macrophages were identified as CD45+F4/80+Ly6G−Ly6C+MHCII− cells. Anti-inflammatory macrophages were identified as CD45+F4/80+Ly6G−Ly6C−MHCII+ cells. (n = 5–6 mice). Each column represents the mean ± standard deviation. Results are representative of at least two independent experiments. *p < 0.05, ***p < 0.001.
3.4 Effects of CARD9 deficiency on cytokine and chemokine production by heat-killed KH2 administration
Next, we evaluated how heat-killed KH2 treatment affected the synthesis of inflammatory cytokines and chemokines in the wounded tissues. As shown in Figure 4, TNF-α levels were significantly lower in heat-killed KH2-treated CARD9 KO mice compared to heat-killed KH2-treated WT mice on day 1 post-wounding. As shown in Figure 4, IL-6 was also significantly decreased in heat-killed KH2-treated CARD9 KO mice compared to heat-killed KH2-treated WT mice on days 1 and 3 after wounding. The production of KC and MIP-2, chemokines involved in neutrophil migration (25), was significantly reduced in heat-killed KH2-treated CARD9 KO mice compared to heat-killed KH2-treated WT mice on days 1 and 3 after wounding (Figure 4). The production of MIP-1α and MIP-1β, chemokines involved in macrophage migration (26), was significantly decreased in heat-killed KH2-treated CARD9 KO mice compared to heat-killed KH2-treated WT mice on day 1 post-wounding (Figure 4). These results indicated that CARD9 plays a key role in the early inflammatory response to heat-killed KH2, facilitating immune cell recruitment for wound healing.
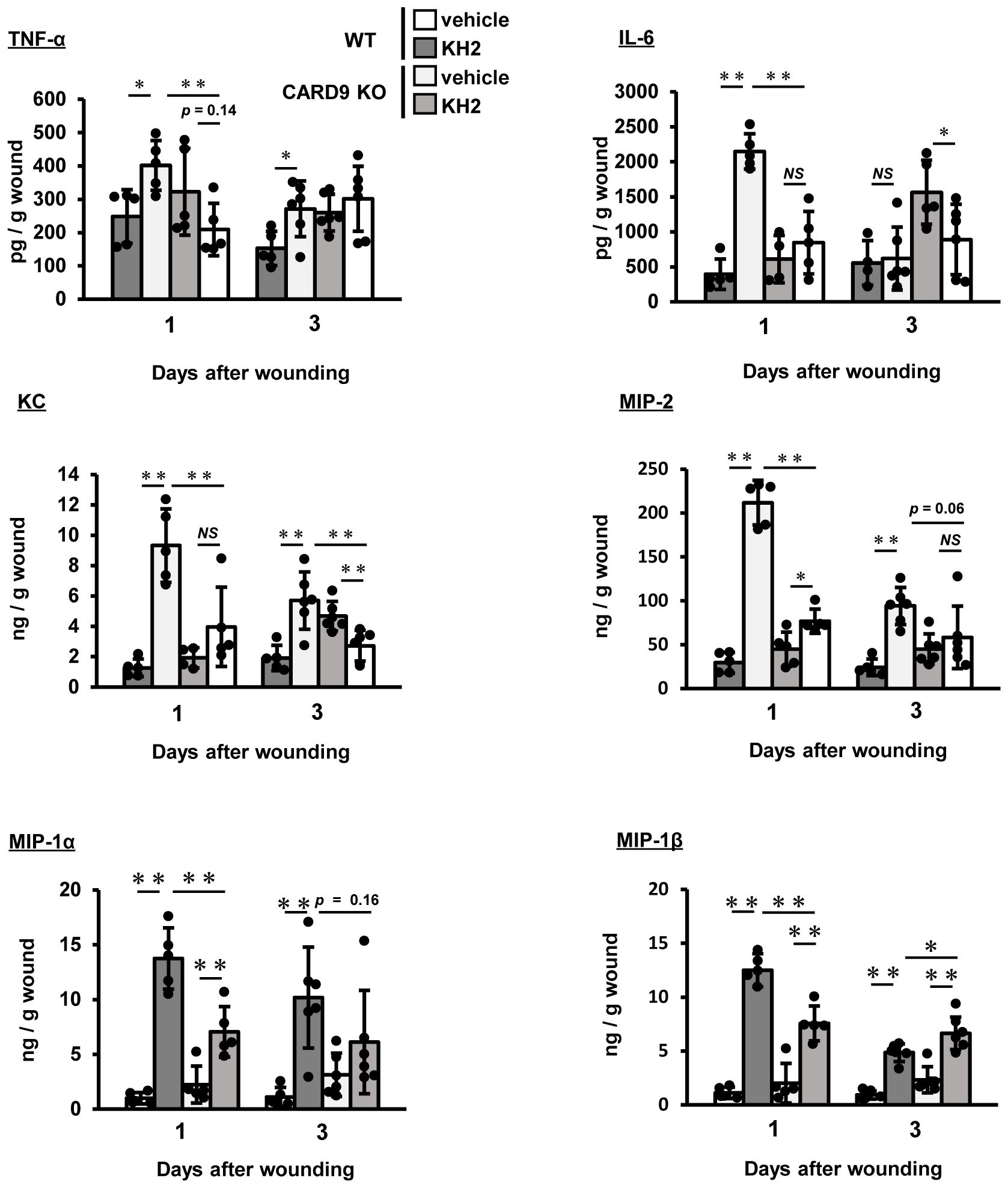
Figure 4. Effects of CARD9 deficiency on cytokine and chemokine production by heat-killed KH2 administration. Effects of CARD9 deficiency on the production of TNF-α, IL-6, KC, MIP-2, MIP-1α, and MIP-1β. Chemokine production in wound tissue homogenates was measured on days 1 and 3 post-wounding (n = 4–6 mice). Each column represents the mean ± standard deviation. Results are representative of at least two independent experiments. *p < 0.05, **p < 0.01, NS, Not significant.
3.5 Involvement of Dectin-1 and Dectin-2 in heat-killed KH2 recognition
To determine whether the loss of heat-killed KH2-mediated wound healing acceleration in CARD9 KO mice was due to the recognition of heat-killed KH2 by upstream CLRs, we examined the ability of Dectin-1 and Dectin-2 to recognize heat-killed KH2 using BM-DCs and an NFAT-GFP reporter assay.
Stimulation of BM-DCs from Dectin-1 or Dectin-2 KO mice with heat-killed KH2 for 24 h revealed TNF-α production, as measured by ELISA. TNF-α production in heat-killed KH2 stimulated BM-DCs from Dectin-1 KO mice was comparable to that of BM-DCs from WT mice. While the positive control dZymosan (Dectin-1 ligand) stimulation resulted in decreased TNF-α production, stimulation with the non-Dectin-1 ligands furfurman (Dectin-2 ligand) and CpG (TLR9 ligand) resulted in TNF-α production in Dectin-1 KO mice comparable with that observed in WT mice. On the other hand, heat-killed KH2-induced TNF-α production was significantly reduced in BM-DCs from Dectin-2 KO mice. Furthermore, stimulation with the Dectin-2 ligand, furfurman, did not induce TNF-α production, while stimulation with the Dectin-1 and TLR9 ligands, dZymosan and CpG, did not result in a decrease in TNF-α production in Dectin-2 KO mice (Figure 5A). In addition, GFP expression was not observed in either control reporter cells or Dectin-1-expressing reporter cells when stimulated with heat-killed KH2 (Figures 5B, C). In contrast, GFP expression in Dectin-2-expressing reporter cells was increased in a dose-dependent manner when these cells were stimulated with heat-killed KH2 (Figure 5D). These results suggested that Dectin-2 recognizes heat-killed KH2.
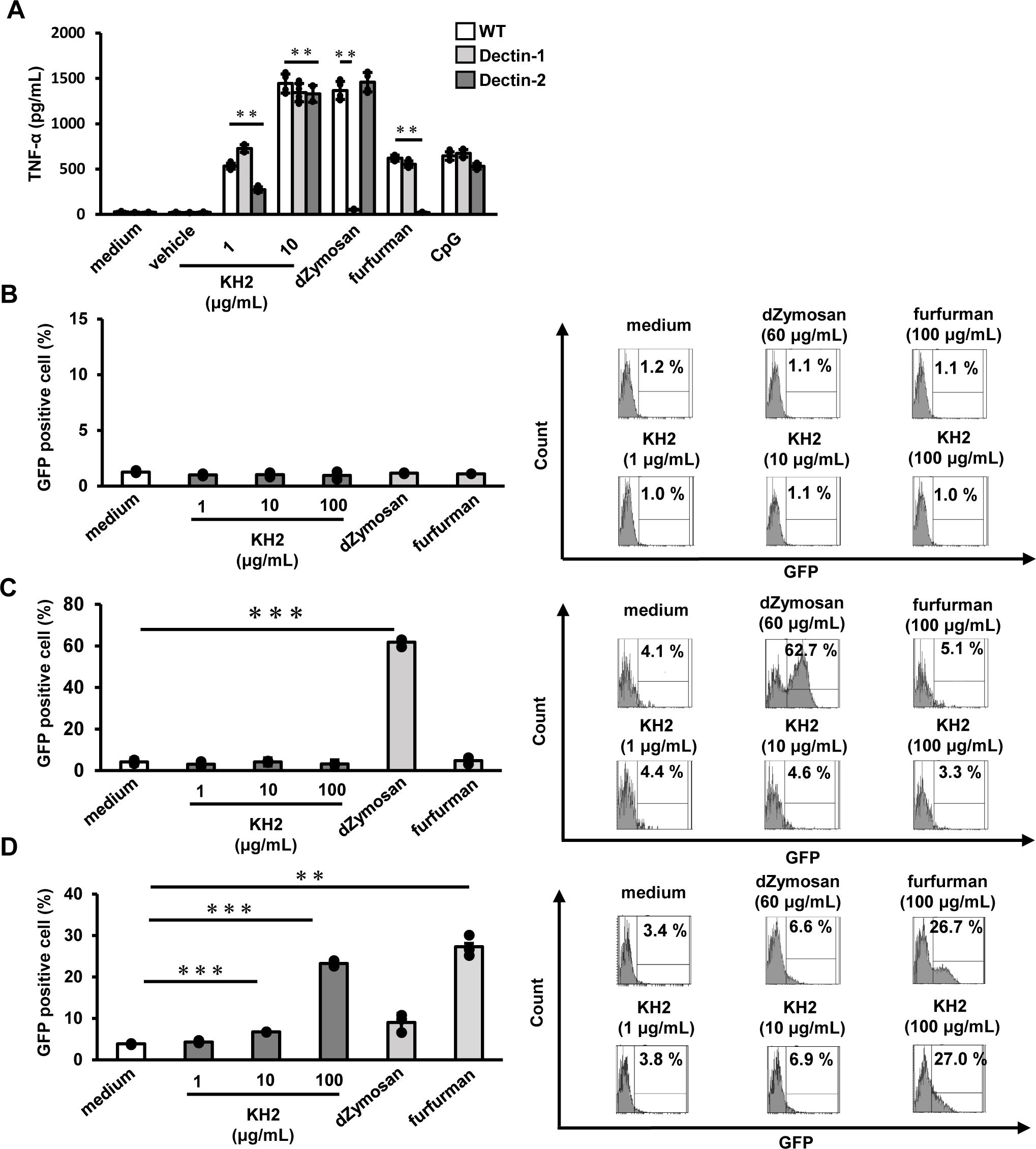
Figure 5. The effect of Dectin-1 and Dectin-2 deficiency on TNF-α production by heat-killed KH2 treatment by BM-DCs. Investigating the role of Dectin-1 and Dectin-2 reporter cells in response to heat-killed KH2. (A) BM-DCs were prepared from WT, Dectin-1 KO, and Dectin-2 KO mice and stimulated with heat-killed KH2 (1, 10 μg/mL), CpG (1 μg/mL), dZymosan (60 μg/mL) and furfurman (100 μg/mL) for 24 h. Production of TNF-α in the culture supernatants was analyzed. (B) Control, (C) Dectin-1, or (D) Dectin-2-NFAT-GFP reporter cells were cultured with heat-killed KH2, vehicle, dZymosan (60 μg/mL) and furfurman (100 μg/mL). GFP expression was measured by flow cytometry. dZymosan, which is a Dectin-1 ligand, furfurman, which is a Dectin-2 ligand, and CpG, which is a TLR9 ligand, were used as the positive controls. Representative histograms from three independent experiments are shown. Each column represents the mean ± standard deviation of triplicate cultures. Results are representative of at least two independent experiments. **p < 0.01, ***p < 0.001.
3.6 Effects of Dectin-1 and Dectin-2 deficiency on the promotion of skin wound healing by heat-killed KH2 administration
To further elucidate the role of Dectin-1 and Dectin-2 in promoting wound healing induced by heat-killed KH2, we investigated its involvement in wound healing using Dectin-1 KO mice and Dectin-2 KO mice. As shown in Figures 6A, B, WT mice treated with heat-killed KH2 showed significant acceleration of wound closure on day 7 post-wounding compared to mice treated with vehicle control. However, this effect was not observed in heat-killed KH2-treated Dectin-2 KO mice. Furthermore, similar to Dectin-2 KO mice, heat-killed KH2 did not promote wound closure in Dectin-1 KO mice (Supplementary Figure 4). In heat-killed KH2-treated WT mice, the granulation area was significantly increased compared to vehicle-treated mice on day 7 post-wounding, but this effect was not observed in heat-killed KH2-treated Dectin-2 KO mice (Figures 6C, D). The re-epithelialization rate was similar in both heat-killed KH2-treated Dectin-2 KO mice and WT mice (Figures 6C, E). These results suggested that Dectin-2 may be involved in promoting wound closure and granulation tissue formation, although not in heat-killed KH2-mediated re-epithelialization, and that Dectin-1, while not involved in heat-killed KH2 recognition, may contribute to promoting wound closure.
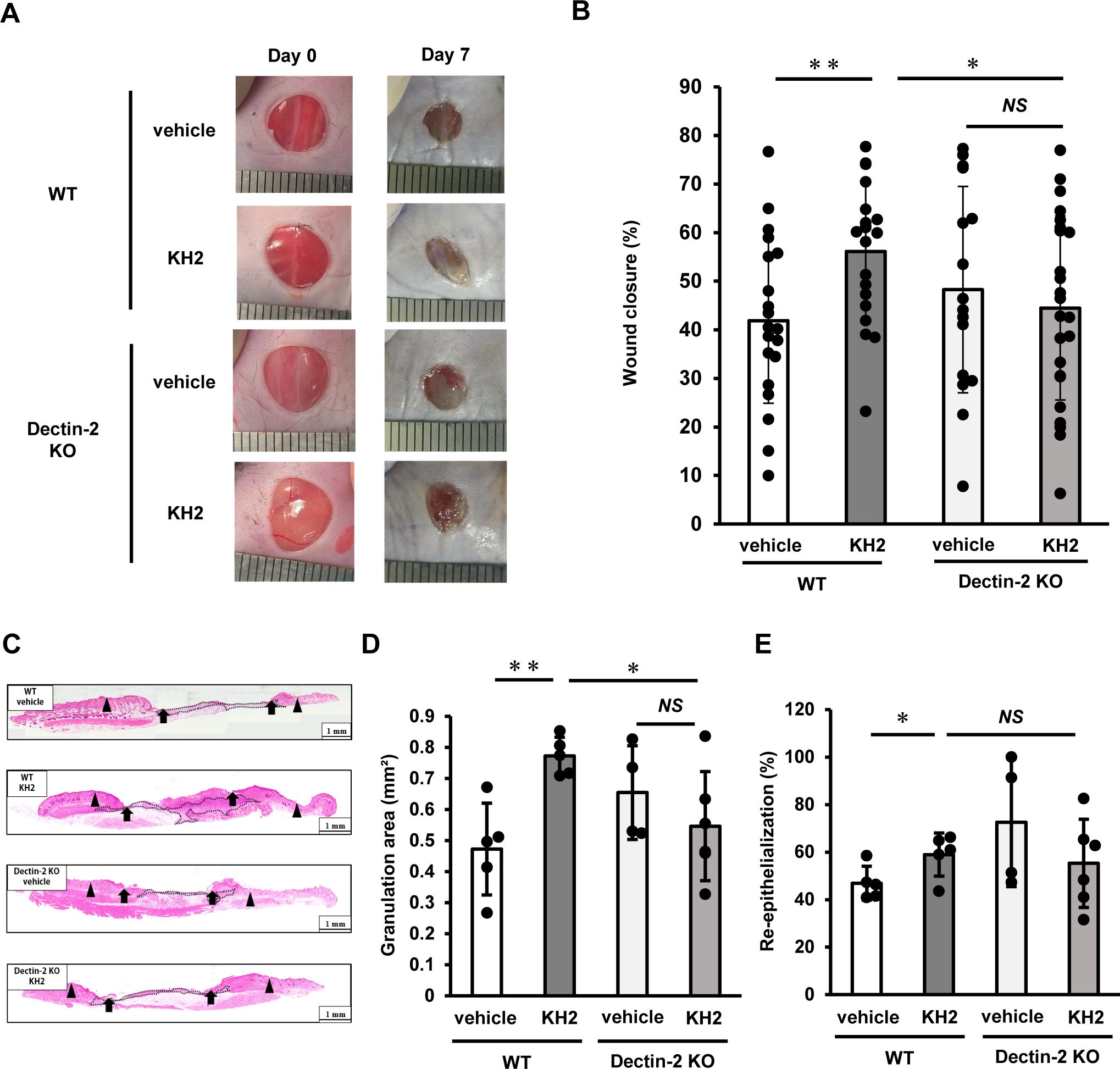
Figure 6. Effects of Dectin-1 and Dectin-2 deficiency on skin wound healing promoted by heat-killed KH2 administration. Wounds were created on the backs of WT mice or Dectin-2 KO mice treated with vehicle control or heat-killed KH2. Photographs (A) were taken, and the percentages of wound closure (B) were evaluated on day 7 post-wounding (n = 20–24 wounds). (C) Representative histological views of skin wounds on day 7. Arrowheads, arrows, and the dotted line indicate the original wound edges, granulation area, and re-epithelialized leading edges, respectively. The wound edges were defined as the border between normal epithelium and thick proliferative epithelium. (D) Granulation tissue area was quantified 7 days after wound creation (n = 4–6 wounds). (E) The re-epithelialization ratio was calculated 7 days post-wounding (n = 4–6 wounds). Each column represents the mean ± standard deviation. Results are representative of at least two independent experiments. *p < 0.05, **p < 0.01, NS, Not significant.
4 Discussion
In this study, we found that heat-killed KH2 was recognized by Dectin-2, not Dectin-1, and that Dectin-2-CARD9 signaling contributed to the acceleration of wound healing.
CARD9 is well known for its involvement in neutrophilic inflammatory responses, particularly through the production of pro-inflammatory cytokines such as TNF-α and IL-6, as well as chemokines like KC and MIP-2, which are involved in neutrophil migration. Previous studies have demonstrated that fungal infection in CARD9 KO mice leads to a marked decrease in neutrophil infiltration, accompanied by reduced production of the chemokines KC and MIP-2 (27, 28). Furthermore, in Streptococcus pneumoniae infection, another Gram-positive bacterium, CARD9 KO mice exhibit decreased neutrophil numbers due to reduced chemokine production (29). Consistent with these earlier studies, we observed that CARD9 KO mice had reduced neutrophil numbers and diminished production of pro-inflammatory cytokines (TNF-α, IL-6) and chemokines (KC, MIP-2) in response to heat-killed KH2 treatment compared to WT mice. These findings suggest that CARD9 is required for heat-killed KH2-induced production of inflammatory cytokines and chemokines, as well as the recruitment of neutrophils.
Neutrophils are essential for clearing pathogens from wounds. However, prolonged neutrophil infiltration can impede the resolution of inflammation and delay wound healing (30, 31). In this study, we observed a significant increase in neutrophil infiltration in heat-killed KH2-treated WT mice on day 1 post-wounding, followed by a decline by day 3, suggesting that neutrophils did not persistently accumulate at the wound site. These findings indicate that the recruited neutrophils may have contributed to accelerated wound healing. In contrast, the number of neutrophils was decreased in heat-killed KH2-treated CARD9 KO mice compared to heat-killed KH2-treated WT mice, although the kinetics of neutrophil infiltration post-wounding were similar between the two groups. While the pattern of neutrophil infiltration was comparable between heat-killed KH2-treated WT and CARD9 KO mice, the impaired wound healing observed in heat-killed KH2-treated CARD9 KO mice may indicate a potential impairment of neutrophil activity due to CARD9 deficiency. As previously mentioned, CLRs upstream of CARD9 are primarily expressed in macrophages and DCs; however, these receptors are also found in neutrophils (16) (32),. In addition, Sun et al. reported that neutrophils from CARD9-deficient mice produced less TNF-α and IL-6 in response to fungal stimulation (33). Our study showed reduced TNF-α and IL-6 production from CARD9 KO in response to heat-killed KH2, suggesting that CARD9 may be important for neutrophil function in inflammation. Additionally, our previous study demonstrated that neutrophil-derived TNF-α promotes wound healing (34). Therefore, we speculate that in this study, neutrophils producing TNF-α via the CARD9 signaling pathway may have contributed to accelerated wound healing.
Macrophages are also known to be the most important cells in skin wound healing. Macrophages are broadly classified into pro-inflammatory and anti-inflammatory macrophages (35). Both types of macrophages are important for wound healing. Pro-inflammatory macrophages accumulate early after injury and contribute to healing by phagocytosing necrotic tissue and pathogenic microorganisms, and clear apoptotic neutrophils. Subsequently, anti-inflammatory macrophages accumulate or polarize at the wound site and function to complete wound healing, through the production of growth factors (36).
In this study, the number of macrophages in heat-killed KH2-treated CARD9 KO mice was lower compared with heat-killed KH2-treated WT mice on day 1 post-wounding, with a trend toward lower numbers on day 7. The number of macrophages on day 1 post-wounding also correlated with the production of the macrophage chemokines MIP-1α and MIP-1β. Conversely, the number of macrophages on day 3 post-wounding did not correlate with chemokine production levels. In CARD9 KO mice, the discrepancy between macrophage accumulation and chemokine levels has been reported in a cryptococcal infection model, but the reason for this inconsistency was unclear (37). Investigations into expression of CCR1, -4, and -5, which are chemokine receptors for MIP-1α and MIP-1β (26), on macrophages in CARD9 KO mice will be necessary in the future.
Furthermore, in this study, we analyzed both pro-inflammatory (Ly6C+MHCII−) and anti-inflammatory (Ly6C−MHCII+) macrophages on day 7 post-wounding. We found that the proportion of pro-inflammatory macrophages was significantly lower in WT mice treated with heat-killed KH2 compared with vehicle-treated WT mice. This effect that was not observed in CARD9 KO mice.
Rodero et al. reported that a decrease in the proportion of pro-inflammatory macrophages is important for promoting healing (24). Therefore, we speculated that the decrease in pro-inflammatory macrophages also contributed to promoting healing in this study. However, regarding absolute numbers, both pro-inflammatory and anti-inflammatory macrophages were increased in heat-killed KH2-treated WT mice.
As mentioned earlier, macrophages are crucial immune cells in skin wound healing. Lucas and colleagues showed the importance of macrophages at all stages of wound healing, demonstrated by macrophage depletion at different times, indicating that depletion of both pro-inflammatory and anti-inflammatory macrophages delayed wound healing (38). Lörchner H, et al. have also shown the potential for Ly6C+MHCII− macrophages to promote healing through angiogenesis after myocardial infarction (39), suggesting that the increased levels of Ly6C+MHCII− macrophages observed in our study may have contributed to skin wound repair. Further research is needed to elucidate the role of Ly6C+MHCII− pro-inflammatory macrophages.
In the current study, we observed that heat-killed KH2 was recognized by Dectin-2, not Dectin-1. This suggests that heat-killed KH2 contains a ligand that can bind to Dectin-2. In a previous study, Yoshikawa et al. demonstrated that L. paracasei KW3110 strain upregulated Dectin-2 expression in macrophages, suggesting that Dectin-2 is involved in the recognition and subsequent phagocytosis of L. paracasei KW3110 (12). Furthermore, Bene et al. demonstrated that Dectin-2 recognizes the mucosa-binding protein of L. reuteri ATCC 53608 strain, leading to the induction of TNF-α and IL-6 production (40). To the best of our knowledge, while there have been some reports of Dectin-2 involvement in recognizing Lactobacillus spp., no studies have yet demonstrated a role for Dectin-2 in recognizing Enterococcus spp. Currently, the involvement of CLRs in E. faecalis is understood to include the role of mannose receptors (MRs) in the phagocytosis of E. faecalis EC-12 strain (41) and the reported involvement of Mincle in E. faecalis recognition (42).
Dectin-2 is a pattern recognition receptor that binds to high-mannose structures, such as α-mannan and lipoarabinomannan (18, 43). The presence of mannose in E. faecalis has been reported (44), suggesting that Dectin-2 may interact with the mannose components of the exopolysaccharide in heat-killed KH2. Furthermore, earlier studies have shown that MR and Dectin-2 share common ligands (45, 46). Therefore, the ligand recognized by MR may also be detected by Dectin-2. Mincle is also known to be a CLR that recognizes α-mannose (47). To investigate its potential role, we stimulated BM-DCs from Mincle KO mice with heat-killed KH2, but did not observe a decrease in TNF-α production in Mincle KO mice (Supplementary Figure 5). Based on these results, we conclude that Mincle is not involved in the recognition of heat-killed KH2. This result differs from a previous study that reported that Mincle recognizes E. faecalis (42). The previous study used E. faecalis that had translocated from the intestines of mice to bone marrow, whereas this study used E. faecalis KH2 isolated from human feces. Additionally, previous studies used LB broth for culturing, while we used MRS broth. Given that research showed strain and culture methods affect the cell wall components of LAB (44), therefore the different results could be due to differences in strain or culture methods.
Next, we assessed the potential contribution of Dectin-2 to wound healing in response to heat-killed KH2 treatment. We observed a decrease in both the percent wound closure and the granulation area in heat-killed KH2-treated Dectin-2 KO mice compared to heat-killed KH2-treated WT mice. These findings were consistent with those observed in CARD9 KO mice treated with heat-killed KH2. However, in contrast to the effects observed in CARD9 KO mice, KH2 administration did not significantly affect re-epithelialization in Dectin-2 KO mice. These results suggest that Dectin-2 recognition of KH2 is primarily involved in granulation tissue formation rather than re-epithelialization. To further elucidate the mechanisms underlying re-epithelialization, it is necessary to consider the potential involvement of other CLRs upstream of CARD9, such as Dectin-1, Dectin-3, and Mincle (46, 48). Among these, Dectin-3 has been reported to form heterodimers with Dectin-2 (49), suggesting a potential role in promoting re-epithelialization upon KH2 recognition. Since the role of Dectin-3 in KH2 recognition has not been investigated in this study, further studies are required to clarify its involvement.
Furthermore, as KH2 is a lactic acid bacterium, it likely possesses ligands for receptors other than Dectin-2, such as TLR2 and TLR9, which recognize peptidoglycan, lipoteichoic acid, and DNA (50). Both TLR2 and TLR9 have been reported to promote wound healing (51, 52) so it is possible that stimulation of these TLRs may have contributed to the observed healing effects. Therefore, further studies are needed to elucidate the roles of other receptors involved in KH2 recognition, including TLRs, in wound healing. While Dectin-1 did not seem to participate in KH2 recognition, our results indicated its potential involvement in heat-killed KH2-induced wound closure. Dectin-1 also synergizes with TLR2 to enhance cytokine production (53). Therefore, although further investigation is needed, Dectin-1 may be involved in cytokine production following heat-killed KH2 recognition by TLR2 and, consequently, in promoting wound closure.
In conclusion, this study demonstrated that topical administration of heat-killed KH2 strain induced inflammation and accelerated wound closure, granulation tissue formation, and re-epithelialization. The CARD9 pathway was shown to be involved in the wound healing-promoting effects of heat-killed KH2. In this study, we used heat-killed KH2, which has been reported to elicit an immune response equivalent to that of live bacteria, but with the advantages of reduced risk of antibiotic resistance and systemic infection (3, 54). Furthermore, as heat-killed bacteria do not proliferate, they do not cause persistent infection or inflammation. Indeed, the inflammation induced by heat-killed KH2 administration was transient in this study. These findings suggest that the topical application of heat-killed KH2 could be a promising therapeutic strategy for wound healing.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by the Ethics Review Committee for Animal Experimentation of Tohoku University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SK: Formal Analysis, Investigation, Writing – original draft. HT: Formal Analysis, Investigation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing. MH: Formal Analysis, Investigation, Writing – review & editing. WK: Formal Analysis, Investigation, Writing – review & editing. RT: Formal Analysis, Investigation, Writing – review & editing. IS: Investigation, Writing – review & editing. YS: Formal Analysis, Writing – review & editing. TW: Methodology, Resources, Writing – review & editing. SI: Formal Analysis, Writing – review & editing. MS: Formal Analysis, Writing – review & editing. YIm: Formal Analysis, Writing – review & editing. KS: Formal Analysis, Writing – review & editing. KI: Formal Analysis, Writing – review & editing. HH: Resources, Writing – review & editing. SY: Resources, Writing – review & editing. SS: Resources, Writing – review & editing. YIw: Resources, Writing – review & editing. KK: Methodology, Supervision, Writing – review & editing. EK: Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported in part by a Grant-in-Aid for Scientific Research (B) (23K24627), Grant-in-Aid for Challenging Research (Exploratory) (23K18384), and Japan Agency for Medical Research and Development (AMED) (24hma322031h001).
Acknowledgments
A part of this study was supported by the Support System for Young Researchers to use research equipment, instruments, and devices at Tohoku University.
Conflict of interest
Authors SK and TW were employed by the company Bio-Lab Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1550934/full#supplementary-material
References
1. Rodrigues M, Kosaric N, Bonham CA, and Gurtner GC. Wound healing: A cellular perspective. Physiol Rev. (2019) 99:665–706. doi: 10.1152/physrev.00067.2017
2. Chen L and DiPietro LA. Toll-like receptor function in acute wounds. Adv Wound Care. (2017) 6:344–55. doi: 10.1089/wound.2017.0734
3. Piqué N, Berlanga M, and Miñana-Galbis D. Health benefits of heat-killed (Tyndallized) probiotics: an overview. Int J Mol Sci. (2019) 20:2534. doi: 10.3390/ijms20102534
4. Tsukahara T, Nakamura S, Romero-Pèrez GA, Ohwaki M, Yanagisawa T, and Kan T. Stimulation of murine cell-mediated immunity by dietary administration of a cell preparation of Enterococcus faecalis strain KH-2 and its possible activity against tumour development in mice. Biosci Microbiota Food Health. (2018) 37:49–57. doi: 10.12938/bmfh.17-021
5. Mokoena MP. Lactic acid bacteria and their bacteriocins: classification, biosynthesis and applications against uropathogens: A mini-review. Molecules. (2017) 22:1255. doi: 10.3390/molecules22081255
6. Vågesjö E, Öhnstedt E, Mortier A, Lofton H, Huss F, Proost P, et al. Accelerated wound healing in mice by on-site production and delivery of CXCL12 by transformed lactic acid bacteria. Proc Natl Acad Sci U.S.A. (2018) 115:1895–900. doi: 10.1073/pnas.1716580115
7. Öhnstedt E, Lofton Tomenius H, Frank P, Roos S, Vågesjö E, and Phillipson M. Accelerated wound healing in minipigs by on-site production and delivery of CXCL12 by transformed lactic acid bacteria. Pharmaceutics. (2022) 14:229. doi: 10.3390/pharmaceutics14020229
8. Ishi S, Kanno E, Tanno H, Kurosaka S, Shoji M, Imai T, et al. Cutaneous wound healing promoted by topical administration of heat-killed Lactobacillus plantarum KB131 and possible contribution of CARD9-mediated signaling. Sci Rep. (2023) 13:15917. doi: 10.1038/s41598-023-42919-z
9. Tanno H, Kanno E, Kurosaka S, Oikawa Y, Watanabe T, Sato K, et al. Topical Administration of Heat-Killed Enterococcus faecalis Strain KH2 Promotes Re-Epithelialization and Granulation Tissue Formation during Skin Wound-Healing. Biomedicines. (2021) 9:1520. doi: 10.3390/biomedicines9111520
10. Inoue R, Nagino T, Hoshino G, and Ushida K. Nucleic acids of Enterococcus faecalis strain EC-12 are potent Toll-like receptor 7 and 9 ligands inducing interleukin-12 production from murine splenocytes and murine macrophage cell line J774.1. FEMS Immunol Med Microbiol. (2011) 61:94–102. doi: 10.1111/j.1574-695X.2010.00752.x
11. Rocha-Ramírez LM, Hernández-Ochoa B, Gómez-Manzo S, Marcial-Quino J, Cárdenas-Rodríguez N, Centeno-Leija S, et al. Evaluation of immunomodulatory activities of the heat-killed probiotic strain lactobacillus casei IMAU60214 on macrophages in vitro. Microorganisms. (2020) 8:79. doi: 10.3390/microorganisms8010079
12. Yoshikawa M, Yamada S, Sugamata M, Kanauchi O, and Morita Y. Dectin-2 mediates phagocytosis of Lactobacillus paracasei KW3110 and IL-10 production by macrophages. Sci Rep. (2021) 11:17737. doi: 10.1038/s41598-021-97087-9
13. Prado Acosta M, Goyette-Desjardins G, Scheffel J, Dudeck A, Ruland J, and Lepenies B. S-Layer From Lactobacillus brevis Modulates Antigen-Presenting Cell Functions via the Mincle-Syk-Card9 Axis. Front Immunol. (2021) 12:602067. doi: 10.3389/fimmu.2021.602067
14. Drummond RA, Saijo S, Iwakura Y, and Brown GD. The role of Syk/CARD9 coupled C-type lectins in antifungal immunity. Eur J Immunol. (2011) 41:276–81. doi: 10.1002/eji.201041252
15. Kanno E, Kawakami K, Tanno H, Suzuki A, Sato N, Masaki A, et al. Contribution of CARD9-mediated signalling to wound healing in skin. Exp Dermatol. (2017) 26:1097–104. doi: 10.1111/exd.13389
16. Yamaguchi K, Kanno E, Tanno H, Sasaki A, Kitai Y, Miura T, et al. Distinct roles for dectin-1 and dectin-2 in skin wound healing and neutrophilic inflammatory responses. J Invest Dermatol. (2021) 141:164–176.e8. doi: 10.1016/j.jid.2020.04.030
17. Hara H, Ishihara C, Takeuchi A, Imanishi T, Xue L, Morris SW, et al. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat Immunol. (2007) 8:619–29. doi: 10.1038/ni1466
18. Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, et al. Dectin-2 Recognition of α-Mannans and Induction of Th17 Cell Differentiation Is Essential for Host Defense against Candida albicans. Immunity. (2010) 32:681–91. doi: 10.1016/j.immuni.2010.05.001
19. Watanabe T, Hayashi K, Takahashi I, Ohwaki M, Kan T, and Kawahara T. Physical properties of lactic acid bacteria influence the level of protection against influenza infection in mice. PloS One. (2021) 16:e0251784. doi: 10.1371/journal.pone.0251784
20. Miura T, Kawakami K, Kanno E, Tanno H, Tada H, Sato N, et al. Dectin-2–mediated signaling leads to delayed skin wound healing through enhanced neutrophilic inflammatory response and neutrophil extracellular trap formation. J Invest Dermatol. (2019) 139:702–11. doi: 10.1016/j.jid.2018.10.015
21. Lutz MB, Kukutsch N, Ogilvie ALJ, Rößner S, Koch F, Romani N, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. (1999) 223:77–92. doi: 10.1016/S0022-1759(98)00204-X
22. Tanno D, Yokoyama R, Kawamura K, Kitai Y, Yuan X, Ishii K, et al. Dectin-2-mediated signaling triggered by the cell wall polysaccharides of Cryptococcus neoformans. Microbiol Immunol. (2019) 63:500–12. doi: 10.1111/1348-0421.12746
23. Ohtsuka M, Arase H, Takeuchi A, Yamasaki S, Shiina R, Suenaga T, et al. NFAM1, an immunoreceptor tyrosine-based activation motif-bearing molecule that regulates B cell development and signaling. Proc Natl Acad Sci. (2004) 101:8126–31. doi: 10.1073/pnas.0401119101
24. Rodero MP, Hodgson SS, Hollier B, Combadiere C, and Khosrotehrani K. Reduced il17a expression distinguishes a ly6cloMHCIIhi macrophage population promoting wound healing. J Invest Dermatol. (2013) 133:783–92. doi: 10.1038/jid.2012.368
25. Armstrong DA, Major JA, Chudyk A, and Hamilton TA. Neutrophil chemoattractant genes KC and MIP-2 are expressed in different cell populations at sites of surgical injury. J Leukoc Biol. (2004) 75:641–8. doi: 10.1189/jlb.0803370
26. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, and Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
27. Drummond RA, Swamydas M, Oikonomou V, Zhai B, Dambuza IM, Schaefer BC, et al. CARD9+ microglia promote antifungal immunity via IL-1β- and CXCL1-mediated neutrophil recruitment. Nat Immunol. (2019) 20:559–70. doi: 10.1038/s41590-019-0377-2
28. Drummond RA, Collar AL, Swamydas M, Rodriguez CA, Lim JK, Mendez LM, et al. CARD9-dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PloS Pathog. (2015) 11:e1005293. doi: 10.1371/journal.ppat.1005293
29. Ishizuka S, Yokoyama R, Sato K, Shiroma R, Nakahira A, Yamamoto H, et al. Effect of CARD9 Deficiency on Neutrophil-Mediated Host Defense against Pulmonary Infection with Streptococcus pneumoniae. Infect Immun. (2020) 89:e00305–20. doi: 10.1128/iai.00305-20
30. Tanno H, Kanno E, Sato S, Asao Y, Shimono M, Kurosaka S, et al. Contribution of invariant natural killer T cells to the clearance of pseudomonas aeruginosa from skin wounds. Int J Mol Sci. (2021) 22:3931. doi: 10.3390/ijms22083931
31. Tanno H, Kawakami K, Kanno E, Suzuki A, Takagi N, Yamamoto H, et al. Invariant NKT cells promote skin wound healing by preventing a prolonged neutrophilic inflammatory response. Wound Repair Regener. (2017) 25:805–15. doi: 10.1111/wrr.12588
32. Sheng R, Zhong X, Yang Z, and Wang X. The role of CARD9 deficiency in neutrophils. Mediators Inflammation. (2021) 2021:6643603. doi: 10.1155/2021/6643603
33. Sun L, Zhang S, Wan Z, Li R, and Yu J. In Vivo and In Vitro Impairments in T Helper Cell and Neutrophil Responses against Mucor irregularis in Card9 Knockout Mice. Infect Immun. (2021) 89:e00040. doi: 10.1128/IAI.00040-21
34. Kanno E, Kawakami K, Miyairi S, Tanno H, Otomaru H, Hatanaka A, et al. Neutrophil-derived tumor necrosis factor-α contributes to acute wound healing promoted by N-(3-oxododecanoyl)-l-homoserine lactone from Pseudomonas aeruginosa. J Dermatol Sci. (2013) 70:130–8. doi: 10.1016/j.jdermsci.2013.01.004
35. Krzyszczyk P, Schloss R, Palmer A, and Berthiaume F. The role of macrophages in acute and chronic wound healing and interventions to promote pro-wound healing phenotypes. Front Physiol. (2018) 9:419. doi: 10.3389/fphys.2018.00419
36. Aitcheson SM, Frentiu FD, Hurn SE, Edwards K, and Murray RZ. Skin wound healing: normal macrophage function and macrophage dysfunction in diabetic wounds. Molecules. (2021) 26:4917. doi: 10.3390/molecules26164917
37. Campuzano A, Castro-Lopez N, Martinez AJ, Olszewski MA, Ganguly A, Leopold Wager C, et al. CARD9 is required for classical macrophage activation and the induction of protective immunity against pulmonary cryptococcosis. mBio. (2020) 11:e03005–19. doi: 10.1128/mbio.03005-19
38. Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Müller W, et al. Differential roles of macrophages in diverse phases of skin repair. J Immunol. (2010) 184:3964–77. doi: 10.4049/jimmunol.0903356
39. Lörchner H, Hou Y, Adrian-Segarra JM, Kulhei J, Detzer J, Günther S, et al. Reg proteins direct accumulation of functionally distinct macrophage subsets after myocardial infarction. Cardiovasc Res. (2018) 114:1667–79. doi: 10.1093/cvr/cvy126
40. Bene KP, Kavanaugh DW, Leclaire C, Gunning AP, MacKenzie DA, Wittmann A, et al. Lactobacillus reuteri surface mucus adhesins upregulate inflammatory responses through interactions with innate C-type lectin receptors. Front Microbiol. (2017) 8:321. doi: 10.3389/fmicb.2017.00321
41. Tsuruta T, Inoue R, Nagino T, Nishibayashi R, Makioka Y, and Ushida K. Role of the mannose receptor in phagocytosis of Enterococcus faecalis strain EC-12 by antigen-presenting cells. MicrobiologyOpen. (2013) 2:610–7. doi: 10.1002/mbo3.99
42. Robles-Vera I, Jarit-Cabanillas A, Brandi P, Martínez-López M, Martínez-Cano S, Rodrigo-Tapias M, et al. Microbiota translocation following intestinal barrier disruption promotes Mincle-mediated training of myeloid progenitors in the bone marrow. Immunity. (2025) 58:381–396.e9. doi: 10.1016/j.immuni.2024.12.012
43. Yonekawa A, Saijo S, Hoshino Y, Miyake Y, Ishikawa E, Suzukawa M, et al. Dectin-2 is a direct receptor for mannose-capped lipoarabinomannan of mycobacteria. Immunity. (2014) 41:402–13. doi: 10.1016/j.immuni.2014.08.005
44. Kavitake D, Devi PB, Delattre C, Reddy GB, and Shetty PH. Exopolysaccharides produced by Enterococcus genus — An overview. Int J Biol Macromol. (2023) 226:111–20. doi: 10.1016/j.ijbiomac.2022.12.042
45. Kerscher B, Willment JA, and Brown GD. The Dectin-2 family of C-type lectin-like receptors: an update. Int Immunol. (2013) 25:271–7. doi: 10.1093/intimm/dxt006
46. Brown GD and Crocker PR. Lectin receptors expressed on myeloid cells. Microbiol Spectr. (2016). doi: 10.1128/microbiolspec.MCHD-0036-2016
47. Yamasaki S, Matsumoto M, Takeuchi O, Matsuzawa T, Ishikawa E, Sakuma M, et al. C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc Natl Acad Sci. (2009) 106:1897–902. doi: 10.1073/pnas.0805177106
48. Graham LM and Brown GD. The Dectin-2 family of C-type lectins in immunity and homeostasis. Cytokine. (2009) 48:148–55. doi: 10.1016/j.cyto.2009.07.010
49. Zhu L-L, Zhao X-Q, Jiang C, You Y, Chen X-P, Jiang Y-Y, et al. C-type lectin receptors dectin-3 and dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity. (2013) 39:324–34. doi: 10.1016/j.immuni.2013.05.017
50. Bermudez-Brito M, Plaza-Díaz J, Muñoz-Quezada S, Gómez-Llorente C, and Gil A. Probiotic mechanisms of action. Ann Nutr Metab. (2012) 61:160–74. doi: 10.1159/000342079
51. Suga H, Sugaya M, Fujita H, Asano Y, Tada Y, Kadono T, et al. TLR4, rather than TLR2, regulates wound healing through TGF-β and CCL5 expression. J Dermatol Sci. (2014) 73:117–24. doi: 10.1016/j.jdermsci.2013.10.009
52. Sato T, Yamamoto M, Shimosato T, and Klinman DM. Accelerated wound healing mediated by activation of toll-like receptor 9. Wound Repair Regener. (2010) 18:586–93. doi: 10.1111/j.1524-475X.2010.00632.x
53. Ferwerda G, Meyer-Wentrup F, Kullberg B-J, Netea MG, and Adema GJ. Dectin-1 synergizes with TLR2 and TLR4 for cytokine production in human primary monocytes and macrophages. Cell Microbiol. (2008) 10:2058–66. doi: 10.1111/j.1462-5822.2008.01188.x
Keywords: skin wound healing, Enterococcus faecalis KH2, C-type lectin receptors, CARD9, Dectin-2
Citation: Kurosaka S, Tanno H, Hirose M, Kamada W, Takayashiki R, Sone I, Sato Y, Watanabe T, Ishi S, Shoji M, Imai Y, Sato K, Ishii K, Hara H, Yamasaki S, Saijo S, Iwakura Y, Kawakami K and Kanno E (2025) Contribution of CARD9 signaling to wound healing in skin promoted by topical administration of heat-killed Enterococcus faecalis strain KH2 and the involvement of Dectin-2. Front. Immunol. 16:1550934. doi: 10.3389/fimmu.2025.1550934
Received: 24 December 2024; Accepted: 27 May 2025;
Published: 12 June 2025.
Edited by:
Toshikazu Kondo, Wakayama Medical University, JapanReviewed by:
Liliana Oliveira, Universidade do Porto, PortugalFukumi Furukawa, Takatsuki Red Cross Hospital, Japan
Copyright © 2025 Kurosaka, Tanno, Hirose, Kamada, Takayashiki, Sone, Sato, Watanabe, Ishi, Shoji, Imai, Sato, Ishii, Hara, Yamasaki, Saijo, Iwakura, Kawakami and Kanno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiromasa Tanno, aGlyb21hc2EudGFubm8uZDNAdG9ob2t1LmFjLmpw
†Present address: Ko Sato, Department of Clinical Microbiology and Infection, Tohoku University Graduate School of Medicine, Sendai, Japan
‡These authors have contributed equally to this work
§ORCID: Hiromasa Tanno, orcid.org/0000-0002-6031-6432