 Eugenia Belcastro1†‡
Eugenia Belcastro1†‡ Annamaria Cudini1‡
Annamaria Cudini1‡ Irene Mezzani1
Irene Mezzani1 Stefania Petrini2
Stefania Petrini2 Valentina D’Oria2
Valentina D’Oria2 Riccardo Schiaffini1Marco Scarsella1
Riccardo Schiaffini1Marco Scarsella1 Anna Lo Russo1
Anna Lo Russo1 Alessandra Fierabracci1*
Alessandra Fierabracci1*- 1Bambino Gesù Children’s Hospital, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Rome, Italy
- 2Confocal Microscopy Core Facility, Bambino Gesù Children’s Hospital, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Rome, Italy
Introduction: Recent evidence highlights neutrophils’ role in initiating/sustaining aberrant immune responses in type 1 diabetes (T1D). The PTPN22 C1858T variant, a risk factor for several autoimmune conditions including T1D, affects T/B-cell receptor signaling. This study investigates its contribution to the altered neutrophil activation and function in T1D.
Materials and methods: Neutrophils were isolated from peripheral blood mononuclear cells (PBMCs) of wild-type (WT) C1858C, heterozygous (HET) C1858T T1D patients, and healthy controls (HD). Reactive oxygen species (ROS) levels were assessed using a fluorogenic probe by Fluorescence- Activated Cell Sorting (FACS) in tumor necrosis factor-alpha (TNF-α) primed, unprimed-N-formylmethionyl-leucyl-phenylalanine (fMLP) stimulated, and primed-stimulated neutrophils. Neutrophil adhesion/transmigration was evaluated via brightfield and epifluorescence microscopy on human umbilical vein endothelial cells (HUVECs).
Results: Neutrophil counts were increased in the female patients than in HD. ROS production was enhanced in the neutrophils of the HET patients versus WT controls under the different culture conditions. Furthermore, ROS levels were increased in the HET (n = 10) versus WT (n = 6) patients (p < 0.05). Neutrophil adhesion to HUVECs and transmigration through the monolayer were increased in the HET (n = 4) versus WT (n = 6) patients under both basal and TNF-α conditions (p < 0.0001).
Conclusion: Neutrophils from C1858T patients are more intrinsically active with increased ROS production and HUVEC adhesion/transmigration, suggesting enhanced contribution to the migration of other immunotypes from vessels into the pancreatic islets during T1D etiopathogenesis.
1 Introduction
Type 1 diabetes (T1D) is recognized as a multifactorial etiopathogenesis with several gene polymorphisms affecting the regulation of the innate and adaptive immune responses (1, 2). Half of the genetic risk is linked to the Human Leukocyte Antigen (HLA) region, i.e., class II and class I genes (3). Outside the HLA region, the insulin variable number of tandem repeats (INS-VNTR) polymorphism in the INS promoter displays a stronger T1D association (4). Other robustly linked genes are cytotoxic T lymphocyte-associated antigen 4 (CTLA4) (5), protein tyrosine phosphatase N22 (PTPN22) (6), and interleukin-2 receptor subunit alpha (IL2RA)https://www.genecards.org/cgi-bin/carddisp.pl?gene=IL2RA (7). In recent years, the idea that autoimmune diseases are mediated exclusively by autoreactive T lymphocytes, the predominant immunotype observed at histopathology of a few reported cases of early-stage human insulitis (8), has been revised (9). Macrophages and neutrophils, which physiologically reside in the pancreatic islet, participate in tissue homeostasis and organ damage, playing a central role in the initiation and perpetuation of aberrant immune responses (10). During the development of insulitis, a strong interaction occurs between cells of innate and adaptive immunity (10). Proteins released from beta (β) cells are taken up and processed by innate immune cells and presented to autoreactive T lymphocytes against the pancreatic islet after their migration into the draining lymph nodes (11). β-Cell death, in turn, triggers the recruitment and activation of B1a cells, neutrophils, and plasmacytoid dendritic cells (12).
In T1D, reduced circulating neutrophil numbers precede and accompany the disease, a phenomenon that parallels the active autoimmune destruction of β-cells (13). Furthermore, β-cell function declines more significantly in individuals with the lowest percentages of circulating neutrophils. Neutrophils infiltrate the pancreas and extrude neutrophil extracellular traps in coordination with other immune cells, even prior to the onset of clinical symptoms in presymptomatic at-risk subjects (13). Neutrophils direct and guide the innate immune response through complex interactions with macrophages, natural killer cells (14), dendritic cells, and effector cell mediators, i.e., soluble recognition molecules, which have the property of increasing phagocytosis, stimulating the complement system, and modulating inflammation, cytokine production, tissue damage molecules, and pathogens that contribute to the innate inflammatory environment (15, 16).
The reduction of circulating neutrophils in T1D subjects can be caused by their impaired reduced bone marrow egression compared with that in non-diabetic subjects (17), a feature that in mice was associated with a defective bone marrow microenvironment (17). Additionally, the heightened consumption and destruction of circulating neutrophils can be attributed to increased apoptosis or anti-neutrophil specific antibodies and increased homing and recruitment to the target organ (18, 19). The reduced number of circulating neutrophils can also be related to the possible triggering of viral infections that initiate and accelerate the process of islet autoimmunity (20).
Among the genetic polymorphisms that regulate the innate and adaptive immune responses in T1D etiopathogenesis, the C1858T variant of PTPN22 is a prominent risk factor for several autoimmune conditions, altering signaling through T- and B-cell receptors (21–23), thus supporting the survival of autoreactive T lymphocytes. In particular, in humans, this variant is significantly associated with T1D, augmenting the risk of developing the disorder by two- to fourfold (24). The Lyp protein encoded by PTPN22 controls the effect of tyrosine kinases and regulates signaling pathways by phosphorylating tyrosine residues in target proteins (22). The Lyp protein has an effect not only on lymphocytes but also on neutrophils, where it is highly expressed (25). The variant is responsible for a more precocious onset of T1D compared to WT patients and more severe clinical manifestations due to a rapid decline in the β-cell reservoir. The variant halts the pathogenetic mechanism, causing a “gain of function” in the encoded Lyp protein with a paradoxical reduced T-cell activation (21).
This study aimed to investigate the contribution of the C1858T PTPN22 variant encoding the Lyp variant protein (R620W) in the altered activation and function of neutrophils in T1D. Indeed, altered tyrosine phosphorylation levels were observed in neutrophils (26), and enhanced neutrophil activation and function have been reported in patients affected by rheumatoid arthritis harboring variant PTPN22 (27). In the present investigation, the impact of the variant on reactive oxygen species (ROS) production by neutrophils (28) and their adhesion and transmigration properties to endothelial cells was specifically evaluated.
2 Materials and methods
2.1 Subjects
Neutrophil counts were assessed in 74 T1D patients recruited at onset and 66 healthy blood donors (Supplementary Tables 1, 2). Among the T1D patients, 37 harbored the C1858T PTPN22 variant, while 37 were wild-type (WT). For in-depth experimental analyses of neutrophil activation and function, a cohort of 27 unrelated T1D patients (13 female and 14 male patients with an age range at referral of between 1.1 and 16.8 years) were recruited from the University Department of Pediatrics (DPUO) at Bambino Gesù Children’s Hospital (OPBG) in Rome (Table 1). Fifteen healthy blood donors representing the control group were recruited from the OPBG Blood Transfusion Centre (Supplementary Table 2). The overall experimental design is illustrated in Supplementary Figure 1.
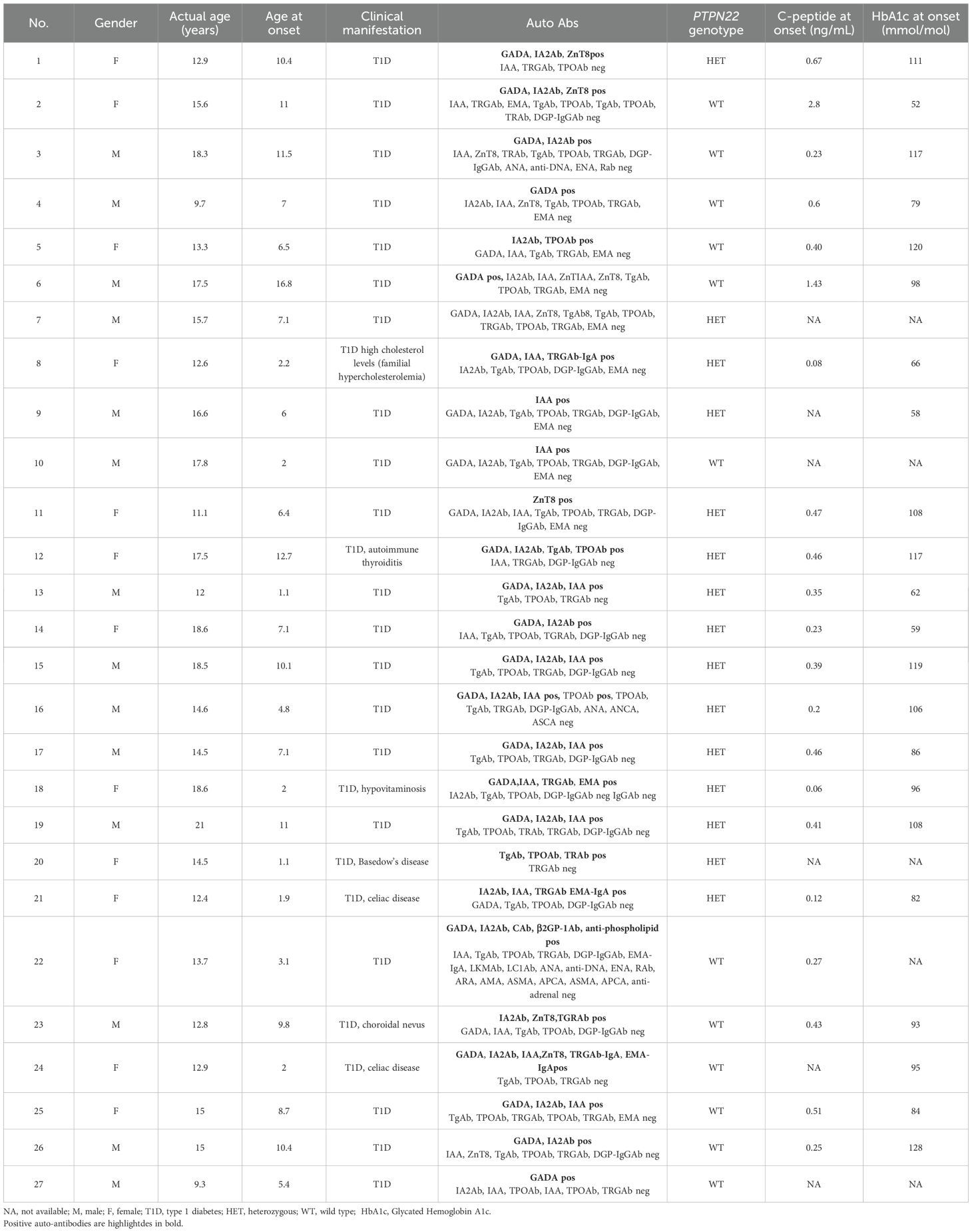
Table 1. Clinical characteristics of T1D patients (n = 27) included for in-depth experimental analyses.
The patients’ sera were assayed for insulin-dependent diabetes mellitus (T1D)-related Abs, i.e., anti-glutamic acid decarboxylase isoform 65 (GADAb) (First anti-GAD ELISA RSR, Cardiff, UK), anti-tyrosine phosphatase-related islet antigen 2 (IA2Ab) (First antiIA2 ELISA RSR), anti-insulin (IAA) (Medzyme Corporation, Montreal, QC, Canada), and anti-zinc transporter 8 Abs (ZnT8Ab) (anti-ZnT8 RSR) by enzyme-linked immunosorbent assay (ELISA); and for thyroid-related Abs, i.e., anti-thyrotropin (TSH) receptor (TRAb) immunoassay (Immulite TSI, Siemens Healthcare, Tarrytown, NY, USA), thyroglobulin (TgAb), and thyroperoxidase (TPOAb) via electrochemiluminescence immunoassay (ECLIA) (Siemens, Erlangen, Germany). Celiac-disease-related Abs were screened by chemiluminescence (CLIA; Quanta-Flash-Werfen, Monza, Milan, Italy), i.e., anti-transglutaminase (TRGAb) (CLIA; Quanta-Flash-Werfen, Monza, Milan, Italy), and by fluorimetric enzyme-linked immunoassay (FEIA), endomysial Ab (EMA) (Werfen, Barcelona, Spain) by immunofluorescence (IFL), and anti-deamidated gliadin (DPG-IgG Ab) by EliA. Specific hepatic Abs, i.e., anti-liver–kidney microsomal (LKMAb) and anti-liver cytosol type 1 (LC1Ab), anti-ribosomal (RAb), anti-adrenal (ADR Ab), and parietal cell antibodies (APCA), were measured by indirect IFL (KSL-Werfen). Non-organ-specific Abs anti-nuclear (ANA), anti-neutrophil cytoplasmic (ANCA) (Elettrochimica s.r.l., Malnate, Varese), anti-double-stranded DNA (anti-dsDNA) (bioMérieux Italia S.p.A, Bagno a Ripoli, Florence, Italy), anti-mitochondrial (AMA), anti-smooth muscle (ASMA), and anti-reticulin Ab (ARA) were tested by IFL (KSL-Werfen). Extractable nuclear antigen Abs (ENA, SSA/RoAb) were tested by ELiA (Thermo Fisher, Waltham, MA, USA). Anti-cardiolipin (ACA) and anti-beta 2 glycoprotein 1 Ab (anti-β2GP1) were measured by ELISA. Informed consent was obtained from all those who took part in the present study in accordance with the Declaration of Helsinki. All subjects entered the study after written informed consent was obtained. The investigation was approved by the local institutional review board (IRB) of Bambino Gesù Children’s Hospital, which regulates human sample usage for experimental studies (Study protocol no. 2436_OPBG_2020); all procedures followed were in accordance with institutional guidelines. Written informed consent was obtained from the next of kin in the case of children. Participant consent was recorded using a paper-based inventory system. The IRB approved the consent procedure.
2.2 Screening for the presence of C1858T PTPN22 variant
Peripheral blood from patients and healthy control donors was collected in Ethyleneidiaminetetraacetic acid (EDTA), and leukocyte genomic DNA was extracted from whole blood samples using QIAmp DNA blood mini kit (Qiagen, Hilden, Germany), according to the manufacturer’s guidelines. The detection of the C1858T variant in the PTPN22 gene (protein tyrosine phosphatase N22, GenBank ID: 26191) was performed via PCR using specific primers for the amplification of exon 14 of the PTPN22 gene: forward 5′-GATAATGTTGCTTCAACGGAATTT-3′ and reverse 5′-CCTCAAACTCAAGGCTCACAC-3′ (annealing temperature 58.5°C). The amplification lasted 35 cycles, generating PCR products of 318 bp that were purified using the NucleoSpin Gel and PCR Clean-up kit (Bioanalysis). Purified PCR product sequencing was carried out using the BigDye Terminator v.3.1 Cycle sequencing protocol (Life Technologies, Applied Biosystems, Paisley, Scotland, UK).
2.3 Neutrophil count
Neutrophil counts were analyzed in a cohort of 74 T1D patients and 66 age- and sex-matched healthy blood donors. Comparison was also conducted between 35 T1D female patients and 25 controls, and between 39 male T1D patients and 41 healthy donors. The mean age of healthy donors was less than 24 years. The mean age at referral for all T1D patients was 17.74 years, with a mean age at T1D onset of 8.03 years. Specifically, for the WT PTPN22 T1D patients, the mean age at referral was 16.88 years, and the age at T1D onset was 7.88 years. For the heterozygous (HET) C1858T PTPN22 patients, the mean age at referral was 18.49 years, with a mean onset age of 7.82 years. Neutrophil counts were also compared between 37 wild-type and 36 heterozygous PTPN22 T1D patients vs. the healthy control group. Additionally, neutrophil counts were examined in a subset of diabetes-related autoantibody-positive WT T1D patients (n = 37) and HET for the PTPN22 polymorphism (n = 37) vs. autoantibody-negative healthy controls (n = 66).
2.4 Isolation of peripheral blood neutrophils
Polymorphonuclear (PMN) leukocytes were isolated from whole blood samples collected in EDTA obtained from 27 patients and 15 controls using discontinuous gradient centrifugation according to English et al. (29). Briefly, each whole blood sample (approximately 6 mL) was carefully stratified over a double Pancoll human 1077 (PAN-Biotech GmbH, Aidenbach, Germany)/Histopaque 1119 (Sigma-Aldrich Co., St Louis, MO, USA) gradient and centrifuged at 3,500 rpm for 40 minutes, with low acceleration and without brake at room temperature. Granulocyte ring was recovered; cells were washed twice with sterile 0.9% NaCl solution. Contaminated red blood cells were lysed by adding 2 mL of cold sterile H2O and gently mixing the tubes by inverting them for 20 seconds at room temperature. The cells were then immediately washed with 2% NaCl solution and resuspended in fresh phosphate-buffered saline (PBS) before being counted in a Burker chamber.
2.5 Reactive oxygen species assessment
The intracellular ROS levels were assessed using a 5-(and 6-)-chloromethyl-2′,7′-dichilorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA; Life Technologies, Carlsbad, CA, USA) fluorogenic probe. Specifically, among the T1D cohort (Table 1), ROS analysis was performed in neutrophils isolated from 16 T1D patients. The mean age at referral for all T1D patients was 12.4 years, with a mean age at T1D onset of 5.9 years. Specifically, for the WT T1D patients, the mean age at referral was 11 years, and the age at T1D onset was 6.6 years. For the HET C1858T patients, the mean age at referral was 13.2 years, with a mean onset age of 5.5 years. For the healthy donor group, a subgroup of five individuals were included in the ROS production analysis. Neutrophils (1.0 × 106/mL in PBS) were primed with 5 ng/mL tumor necrosis factor-alpha (TNF-α) (PeproTech, Cranbury, NJ, USA) for 15 minutes at 37°C and then incubated with 2 μM CM-H2DCFDA for 20 minutes at 37°C in the dark. Samples were also stimulated with 1 μM N-formylmethionyl-leucyl-phenylalanine (fMLP) (Sigma-Aldrich Co., USA) for 30 minutes at 37°C, followed by washing twice in PBS before flow cytometry analysis. Negative and positive controls were also included. Data were acquired using the BD LSRFortessa X-20 flow cytometer (BD Biosciences, Sean Jose, CA, USA) and revealed by mean fluorescence intensity (MFI) values. Dead cells were excluded from analysis by side/forward scatter (SSC/FSC) gating prior to propidium iodide staining. To evaluate potential age-dependent effects on neutrophil function, a correlation analysis was conducted between age and ROS production across all samples, as well as within the T1D patient subgroup.
2.6 Adhesion and transmigration assays
Neutrophil adhesion and transmigration through human umbilical vein endothelial cell (HUVEC; Life Technologies) monolayers was conducted in 10 T1D patients, with (n = 4) or without (n = 6) the C1858T PTPN22 gene variant. The mean age at referral for all T1D patients was 14.4 years, with a mean age at T1D onset of 8.5 years. Specifically, for the WT T1D patients, the mean age at referral was 12.7 years, and the age at T1D onset was 9.1 years. For the heterozygous C1858T patients, the mean age at referral was 14.4 years, with a mean onset age of 6.4 years; within both groups (WT and HET), patients were age matched. HUVECs were cultured at recommended seeding density (2.5 × 105 cells/cm2) in Endothelial Growth Medium-2 (EGM™-2, Euroclone, Milan, Italy), according to the manufacturing datasheet. For adhesion and transmigration assays, HUVECs (1.5 × 105 cells in 300 μL) were seeded onto 24-well cellQART® Cell Culture Inserts (3-μm PET Translucent, SABEU GmbH & Co. KG, Northeim, Germany) and cultured overnight to generate confluent monolayers. As previously described (30, 31), TNF-α (5 ng/mL) was added to confluent monolayers for 4 hours before the adhesion. The concentration of 5 ng/mL was chosen since it was shown as the most effective with neutrophils from C1858T patients with rheumatoid arthritis (27). Isolated neutrophils were then added for 2 hours at 37°C to allow adhesion and migration. The number of neutrophils added ranged from 1 × 105 to 2 × 106/mL, depending on the blood volume collected from each patient. After the removal of non-adherent neutrophils and washing the inserts with PBS, samples were fixed with 4% paraformaldehyde for 15 minutes in the dark. After two additional PBS washes, the cells’ nuclei were counterstained with Hoechst (1:5,000 in PBS) for 15 minutes. Digitized images of the endothelial surface were made using brightfield and epifluorescence microscopy at different levels of depth, above and below the transwell surface, respectively. Adhesion and transmigration were determined by counting all adherent/transmigrated cells per image and then calculating the number of adherent/transmigrated cells per cellQART transwell (total area, 33.16 mm2). Thus, a ratio was expressed by dividing this number by the total number of neutrophils added in each of the eight independent experiments. The following formula was employed for calculation:
Analogously, the correlation between age and neutrophil transmigration was assessed within the T1D patient subgroup.
2.7 Statistical analysis
Neutrophil counts and ROS production levels between the T1D patient groups (wild-type and C1858T PTPN22 variant) and the control group, under different conditions, e.g., unstimulated, TNF-α, and TNF-α+fMLP, were assessed for normality using the Kolmogorov–Smirnov (KS) test. For data satisfying normality criteria (KS test: p > 0.10), statistical significance was evaluated using one-way ANOVA, followed by Tukey’s multiple comparison test. The analysis of multiple biological replicates was performed using the GraphPad Prism software (version 5, San Diego, CA). Additionally, differences in neutrophil counts and ROS production between the patient and control groups were analyzed using an unpaired t-test. Pearson’s correlation analysis was applied to assess potential age-dependent effects on neutrophil function and transmigration. Values are expressed as means ± SEM. A p-value of less than 0.05 was considered statistically significant. Neutrophil adhesion and transmigration through HUVECs were evaluated using proportion Z-tests. The total sum of neutrophils added, adhered, and transmigrated in each of eight independent experiments was used to compare the different experimental conditions, e.g., with and without TNF-α, by means of the one- or two-proportion Z-test. A result with p < 0.01 was considered statistically significant.
3 Results
We tested the hypothesis that the presence of C1858T PTPN22 variant may affect neutrophil function in a cohort of 27 T1D patients recruited and genotyped for this SNP (Table 1). Fifteen patients were heterozygous for variant C1858T, and 12 patients were wild-type. Patients were closely matched for age and sex. There were no significant differences in patient demographics, including disease duration from onset and treatment.
3.1 Neutrophil counts in T1D patients
No significant differences in neutrophil counts were observed when comparing total T1D patients to healthy donor controls or male T1D patients to male healthy donors (Figures 1A, B). However, neutrophil counts were significantly higher in female T1D patients compared to female healthy donors (Figure 1C). Neutrophil counts did not differ between the T1D subgroups (wild-type or heterozygous for the PTPN22 variant) and healthy donor controls (Figure 1D), between the autoantibody-positive T1D patients and healthy donor controls (Figure 1E), or between the heterozygous (C1858T) and wild-type (C1858C) T1D patients and healthy donors (Figure 1F).
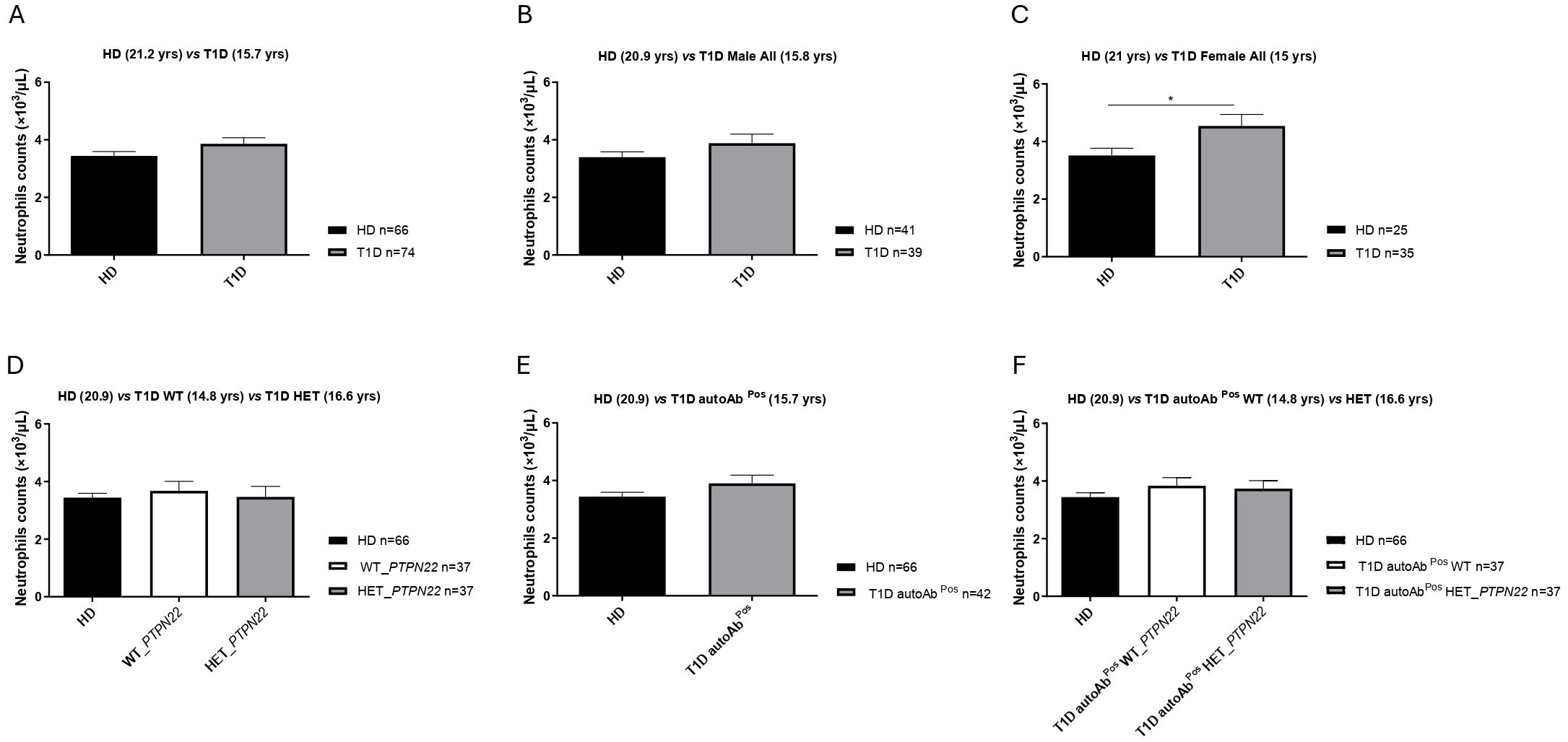
Figure 1. Neutrophil counts in the peripheral blood. Neutrophil counts in type 1 diabetes (T1D) patients (n = 74) versus healthy controls (HD) (n = 66) (A), male T1D patients (n = 39) vs. male HD (n = 41) (B), female T1D patients (n = 35) vs. female HD (n = 25) (C), heterozygous (HET) PTPN22 T1D patients (n = 37) compared to PTPN22 wild-type (WT) patients (n = 37) and HD (n = 66) (D), autoantibody-positive T1D patients (n = 42) vs. HD (n = 66) (E), and autoantibody-positive HET (C1858T) (n = 37) and WT (C1858C) T1D patients (n = 37) compared to HD (n = 66) (F). Data are expressed as mean ± SEM, with the sample size (n) indicated in the histogram of each respective panel, and mean age (years) is shown in brackets. *p < 0.05.
3.2 Enhanced ROS production in neutrophils from C1858T T1D patients
Neutrophils isolated from the healthy donors WT (n = 5, mean age 31.10 years), PTPN22 C1858C T1D patients (n = 6, mean age 11.58 years), and PTPN22 C1858T T1D patients (n = 10, mean age 13.57 years) exhibited a significantly greater ability to produce ROS in response to TNF-α priming, fMLP stimulation, or the combination of TNFα+fMLP (Figures 2A–C). Specifically, the combination treatment resulted in significantly higher ROS levels than either TNF-α or fMLP alone. Moreover, these findings were further confirmed by flow cytometric histograms of DCF fluorescence, which showed higher ROS levels in neutrophils from the T1D patient groups (wild-type and C1858T PTPN22 variant) (n = 16) compared to healthy donors (n = 5) (Supplementary Figure 2). As shown in Figure 3A, ROS production was increased following combined treatment (TNF-α+fMLP) in both the PTPN22 C1858C T1D patients (n = 6) and healthy individuals (n = 5) compared to the respective controls (unstimulated). Furthermore, enhanced ROS production in response to priming with TNF-α and then fMLP-treated neutrophils from the PTPN22 C1858C T1D patients than in those treated with TNF-α or fMLP alone was observed. When the T1D patients were categorized and compared based on their PTPN22 genotype (WT and HET), differences in ROS production were more evident. ROS levels were significantly higher in the heterozygous T1D patients (n = 10) compared to wild-type T1D patients (n = 6) following treatments with TNF-α and TNF-α+fMLP (p < 0.001). Enhanced ROS levels were observed under fMLP treatment, although this difference was not statistically significant (Figure 3B). In addition, fold change in ROS production was markedly increased in the heterozygous C1858T PTPN22 (n = 10) vs. wild-type C1858C PTPN22 (n = 6) T1D patients (p < 0.05) (Figure 3C); although a broader comparison among all genotypes (HD, WT, and HET) revealed a similar trend, the differences were not statistically significant (Supplementary Figure 3). A significant negative correlation between age and ROS production was observed across all subjects (HD, WT, and HET) under all tested conditions (Supplementary Table 3), likely reflecting the age gap between the adult HD and pediatric T1D patients. In contrast, within the pediatric T1D subgroup (WT + HET), a significant positive correlation with age emerged under TNF-α (r = 0.53, p = 0.033), fMLP (r = 0.49, p = 0.052), and TNF-α+fMLP (r = 0.64, p = 0.008) stimulation, with a similar upward trend under all stimulated conditions (Supplementary Table 4). Despite this trend, HET patients—who were on average younger than WT patients (11.58 vs. 13.57 years)—exhibited significantly higher ROS levels, indicating that enhanced ROS production is likely driven by the PTPN22 variant rather than age.
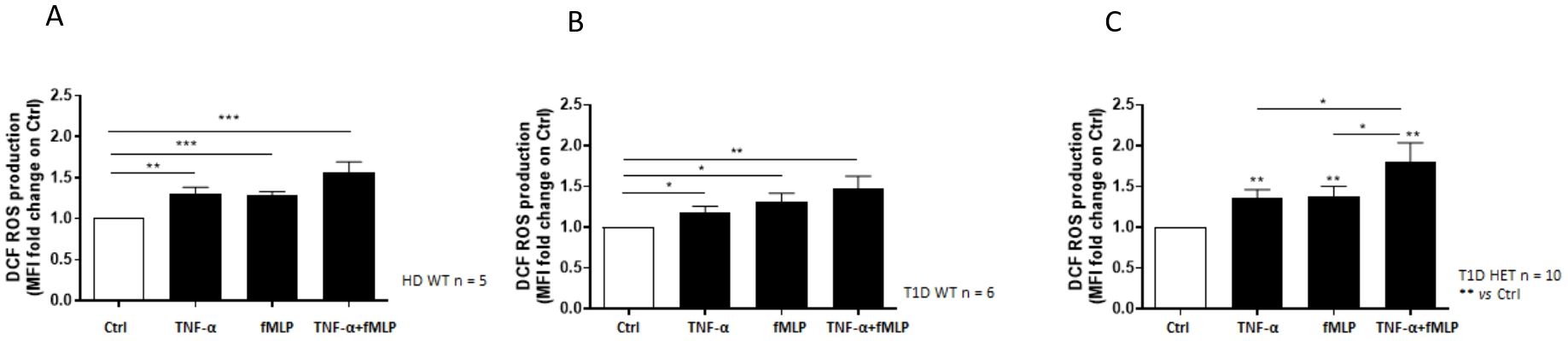
Figure 2. Neutrophil reactive oxygen species (ROS) production measured by DCF fluorescence. Effect of the different stimulatory conditions on ROS production, expressed as mean fluorescence intensity (MFI) fold change in neutrophils isolated from healthy blood donors wild-type (WT) (n = 5) (A), WT type 1 diabetes (T1D) patients (n = 6) (B), and heterozygous (HET) T1D patients (n = 10) (C). Data are expressed as mean ± SEM. *p<0.05, **p<0.01 and ***p<0.001.
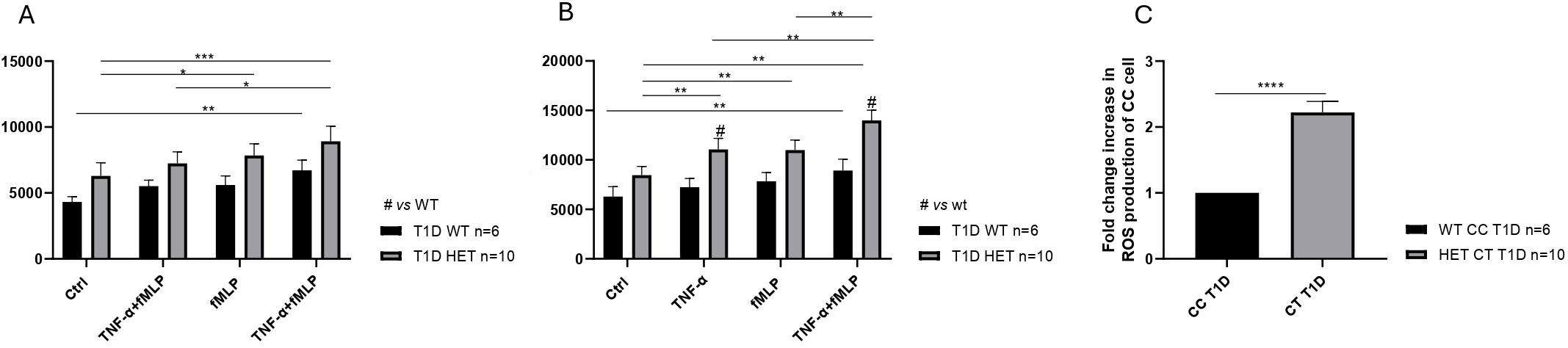
Figure 3. Increased reactive oxygen species (ROS) production in neutrophils of diabetic patients. ROS generation in neutrophils isolated from wild-type (WT) type 1 diabetes (T1D) (n = 6) and healthy controls (HD) (n = 5), compared to the respective controls (A). Enhanced mean fluorescence intensity of ROS production in neutrophils from heterozygous (n = 10) compared to wild-type T1D patients (n = 6) and respective controls in response to various stimulatory conditions (B). Increased fold change in ROS production in T1D patients categorized by genotype [heterozygous (HET) C1858T PTPN22, n = 10 vs. WT C1858C PTPN22, n = 6] (C). Data are expressed as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
3.3 Increased neutrophil adhesion and transmigration in C1858T PTPN22 T1D patients
To investigate whether the presence of the C1858T PTPN22 variant in T1D patients altered the ability of neutrophils to be recruited to the pancreas, potentially leading to insulitis, the behavior (adhesion and transmigration) of neutrophils isolated from the wild-type (C1858C) and heterozygous (C1858T) T1D patients on endothelial cell monolayers stimulated with TNF-α was examined. Neutrophil adhesion and transmigration were significantly enhanced in the heterozygous C1858T PTPN22 (n = 4) compared to WT PTPN22 (n = 6) T1D patients, both under basal conditions and following TNF-α stimulation (Figure 4). Specifically, neutrophils from the C1858T patients exhibited higher adhesion (Figures 4A, B) and greater transmigration across the monolayer after TNF-α stimulation (Figures 4C, D) compared to those from the wild-type T1D patients (Z-test, p < 0.0001). No significant correlation was observed between age and neutrophil transmigration in the pediatric T1D patients carrying (HET) or not carrying (WT) the PTPN22 C1858T variant (r = −0.21 and −0.05 for unstimulated and TNF-α-stimulated conditions, respectively) (Supplementary Table 5). This suggests that the C1858T PTPN22 variant may enhance neutrophil recruitment and migration independently of age, especially through inflamed endothelium, by contributing to the development of insulitis in T1D.
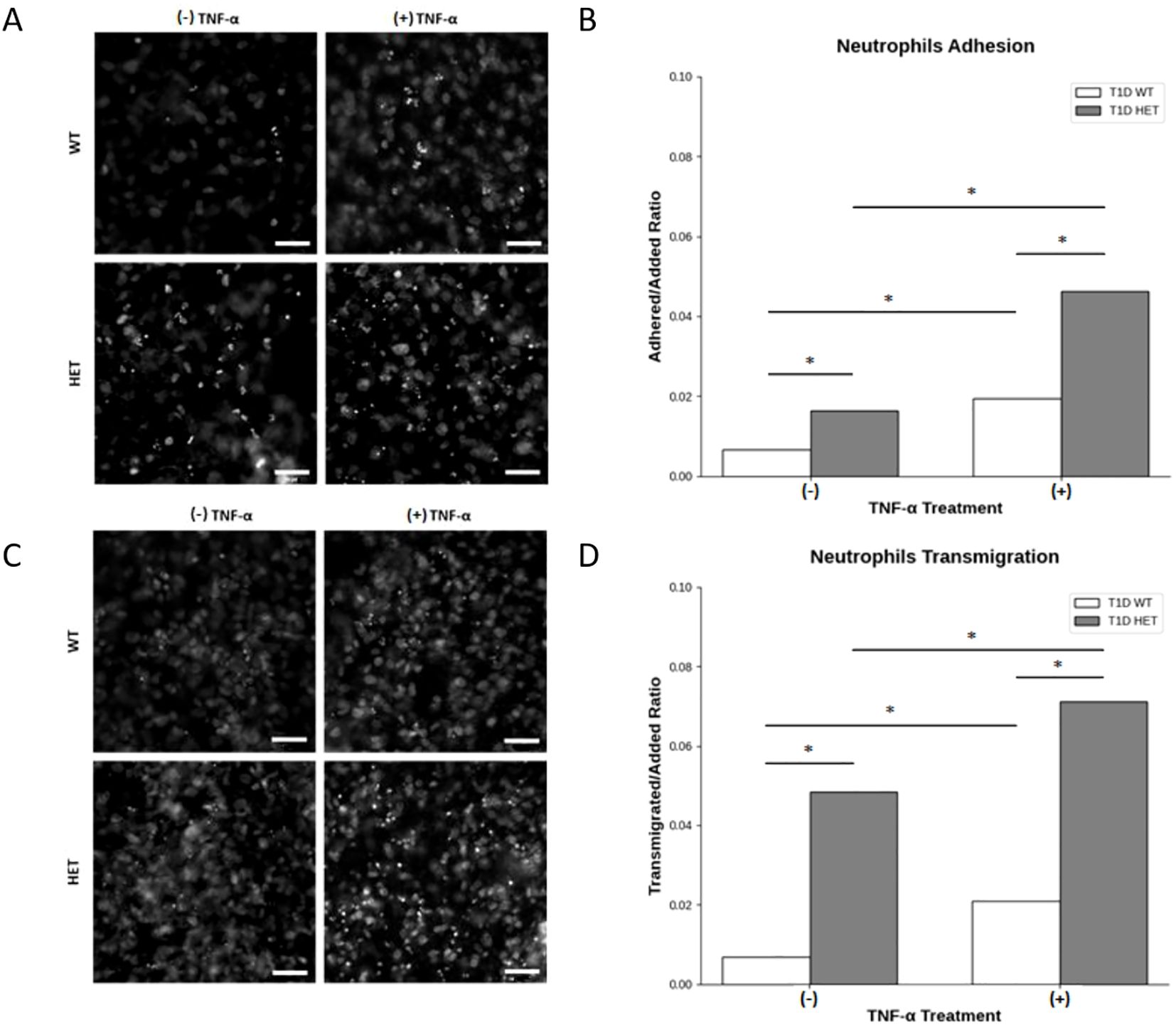
Figure 4. Neutrophil adhesion to human umbilical vein endothelial cells (HUVECs) and transmigration across the endothelial monolayer. Representative phase-contrast microscopy of neutrophils from PTPN22 wild-type and heterozygous C1858T PTPN22 type 1 diabetes (T1D) patients adhering to a HUVEC monolayer (A) and quantitative comparison of the adherent-to-added neutrophil ratio in heterozygous C1858T PTPN22 (n = 4) vs. wild-type T1D patients (n = 6) in basal and TNF-α-stimulated culture conditions (B). Representative phase-contrast microscopy of neutrophils from PTPN22 wild-type (WT) and heterozygous (HET) C1858T PTPN22 T1D patients transmigrating across the HUVEC monolayer (C) and comparison of the transmigrated-to-added ratio of neutrophils in heterozygous C1858T PTPN22 (n = 4) vs. wild-type T1D patients (n = 6) in basal and TNF-α-stimulated culture conditions (D). Images and data are representative of n = 8 independent experiments. Bars show the cumulative adhered-to-added and transmigrated-to-added neutrophil ratios across all the experiments. Proportion Z-test, *p < 0.001. Scale bars in A and C, 50 μm.
4 Discussion
The main finding from our study demonstrates that the C1858T PTPN22 variant contributes to enhanced neutrophil activation in T1D. Specifically, neutrophils from the HET C1858T PTPN22 variant patients exhibited significantly increased ROS production, as well as greater adhesion and transmigration across endothelial cells, compared to those from the WT C1858T PTPN22 patients. These observations suggest that the C1858T PTPN22 variant may play a role in amplifying immune cell recruitment during the inflammatory response, independently of age, underscoring the synergistic impact of genetic predisposition and immune dysregulation on neutrophil function in T1D.
Specifically, ROS production tends to increase with age, in contrast to the overall cohort analysis, where a negative correlation was observed, likely reflecting the inclusion of older healthy donors. Notably, this positive age-related trend in T1D was consistent across all stimulated conditions. This pattern reinforces that the observed differences in ROS production between the WT and HET T1D patients are not attributable to age. Crucially, the comparison underlying our primary conclusions involves two pediatric groups with similar mean ages, thereby minimizing age as a confounding factor. Despite being younger on average, HET patients exhibited significantly higher ROS levels, strongly suggesting that the enhanced neutrophil function is likely attributable to the PTPN22 variant rather than age. Thus, the direction and magnitude of the age effect further support a genotype-driven functional difference rather than an age-related artifact.
Furthermore, adhesion and transmigration of neutrophils through HUVEC monolayers were increased in the heterozygous C1858T PTPN22 vs. wild-type PTPN22 T1D patients, both under basal conditions and following TNF-α stimulation, with no significant correlation with age. These findings suggest that the presence of the C1858T PTPN22 variant in T1D patients alters the ability of neutrophils to be recruited to the target organ, the pancreas, migrating more rapidly, especially through inflamed endothelium, thus leading to insulitis (32).
Of note, contrasting results were reported on the neutrophil counts in T1D patients at disease onset, found either decreased (17, 33, 34) or increased (35, 36). In the present investigation, neutrophil percentages were similar in the T1D patients at onset and healthy controls. However, neutrophil counts were significantly higher in the female T1D patients than in female healthy controls. Other studies have reported that the size effect was dependent on both age and sex, with diabetic men exhibiting lower circulating neutrophil levels (37). Neutrophil functions were also reported to change at different steps in T1D. Indeed, the current literature is contradictory regarding the host defense functions of neutrophils, including oxidative burst activity (38–40) and migration (41, 42). Increased production of ROS by neutrophils in both T1D patients and rat models (40, 43) not only drives pancreatic β-cell destruction but also compromises neutrophil antioxidant defense, heightening susceptibility to infections in diabetic patients (44). This dysregulated immune response is further compounded by genetic predisposition, such as the C1858 variant of PTPN22.
The interplay between oxidative stress and immune dysfunction shows implications in all stages of autoimmune T1D, as described by the literature (45). Specifically, ROS production and impaired neutrophil function not only contribute to cellular injury but also can activate inflammatory, redox-dependent transcription factors, such as NF-κB and cytokine production (46), thereby sustaining inflammation. Therefore, the redox modulation may be more important than once thought, even though newer therapeutic approaches allow for precise targeting of specific immune cells. These results pave the way for future immunotherapeutic strategies targeting the PTPN22 variant using lipoplexes (47–50). These approaches could effectively mitigate the pathogenetic mechanisms driving diabetic insulitis by modulating not only T- and B-lymphocyte activation but also neutrophil function, including their heightened ROS production, adhesion, and transmigration. These interventions may offer new possibilities to improve outcomes in T1D, considering the multifaceted contribution of neutrophils to disease progression.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Institutional Review Board (IRB) of Bambino Gesù Children’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
EB: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Conceptualization. AC: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft. IM: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft. SP: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft. VD’O: Data curation, Methodology, Software, Writing – original draft. RS: Resources, Writing – original draft. MS: Methodology, Writing – original draft. AR: Methodology, Writing – original draft. AF: Conceptualization, Funding acquisition, Investigation, Methodology, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Italian Ministry of Health with funding from Ricerca Finalizzata Grant RF-2019–12369889 to Alessandra Fierabracci.
Acknowledgments
We acknowledge the technical support of Alessia Palma for her contribution to the PTPN22 polymorphism screening.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1554570/full#supplementary-material
References
1. Ilonen J, Lempainen J, and Veijola R. The heterogeneous pathogenesis of type 1 diabetes mellitus. Nat RevEndocrinol. (2019) 15:635–50. doi: 10.1038/s41574-019-0254-y
2. Redondo MJ, Oram RA, and Steck AK. Genetic risk scores for type 1 diabetes prediction and diagnosis. Curr Diabetes Rep. (2017) 17:129. doi: 10.1007/s11892-017-0961-5
3. Rich SS, Weitkamp LR, and Barbosa J. Genetic heterogeneity of insulin-dependent (type I) diabetes mellitus: evidence from a study of extended haplotypes. Am J Hum Genet. (1984) 36:1015–23.
4. Pugliese A, Zeller M, Fernandez A Jr, Zalcberg LJ, Bartlett RJ, Ricordi C, et al. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet. (1997) 15:293–7. doi: 10.1038/ng0397-293
5. Marron MP, Raffel LJ, Garchon HJ, Jacob CO, Serrano-Rios M, Martinez Larrad MT, et al. Insulin-dependent diabetes mellitus (IDDM) is associated with CTLA4 polymorphisms in multiple ethnic groups. Hum Mol Genet. (1997) 6:1275–82. doi: 10.1093/hmg/6.8.1275
6. Su Y, Li X, Wu PD, Zhang YL, Fang PF, Wu FF, et al. The association between PTPN22 SNPs and susceptibility to type 1 diabetes: An updated meta-analysis. PloS One. (2025) 20:e0321624. doi: 10.1371/journal.pone.0321624
7. Tang W, Cui D, Jiang L, Zhao L, Qian W, Long SA, et al. Association of common polymorphisms in the IL2RA gene with type 1 diabetes: evidence of 32,646 individuals from 10 independent studies. J Cell Mol Med. (2015) 19:2481–8. doi: 10.1111/jcmm.12642
8. Bottazzo GF, Dean BM, McNally JM, MacKay EH, Swift PG, and Gamble DR. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N Engl J Med. (1985) 313:353–60. doi: 10.1056/NEJM198508083130604
9. Sun L, Xi S, He G, Li Z, Gang X, Sun C, et al. Two to tango: dialogue between adaptive and innate immunity in type 1 diabetes. J Diabetes Res. (2020) 2020:4106518. doi: 10.1155/2020/4106518
10. Citro A, Campo F, Dugnani E, and Piemonti L. Innate immunity mediated inflammation and beta cell function: neighbors or enemies? Front Endocrinol (Lausanne). (2020) 11:606332. doi: 10.3389/fendo.2020.606332
11. Zinselmeyer BH, Vomund AN, Saunders BT, Johnson MW, Carrero JA, and Unanue ER. The resident macrophages in murine pancreatic islets are constantly probing their local environment, capturing beta cell granules and blood particles. Diabetologia. (2018) 61:1374–83. doi: 10.1007/s00125-018-4592-4
12. Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, et al. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med. (2013) 19:65–73. doi: 10.1038/nm.3042
13. Valle A, Giamporcaro GM, Scavini M, Stabilini A, Grogan P, Bianconi E, et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes. (2013) 62:2072–7. doi: 10.2337/db12-1345
14. Gianchecchi E, Delfino DV, and Fierabracci A. NK cells in autoimmune diseases: Linking innate and adaptive immune responses. Autoimmun Rev. (2018) 17:142–54. doi: 10.1016/j.autrev.2017.11.018
15. Battaglia M, Petrelli A, and Vecchio F. Neutrophils and type 1 diabetes: current knowledge and suggested future directions. Curr Opin Endocrinol Diabetes Obes. (2019) 26:201–6. doi: 10.1097/med.0000000000000485
16. Huang J, Xiao Y, Xu A, and Zhou Z. Neutrophils in type 1 diabetes. J Diabetes Investig. (2016) 7:652–63. doi: 10.1111/jdi.12469
17. Ferraro F, Lymperi S, Méndez-Ferrer S, Saez B, Spencer JA, Yeap BY, et al. Diabetes impairs hematopoietic stem cell mobilization by altering niche function. Sci Transl Med. (2011) 3:104ra1. doi: 10.1126/scitranslmed.3002191
18. Akgul C, Moulding DA, and Edwards SW. Molecular control of neutrophil apoptosis. FEBS Lett. (2001) 487:318–22. doi: 10.1016/s0014-5793(00)02453-2
19. Capsoni F, Sarzi-Puttini P, and Zanella A. Primary and secondary autoimmune neutropenia. Arthritis Res Ther. (2005) 7:1–7. doi: 10.1186/ar1727
20. Coppieters KT, Boettler T, and von Herrath M. Virus infections in type 1 diabetes. Cold Spring Harb Perspect Med. (2012) 2:a007682. doi: 10.1101/cshperspect.a007682
21. Vang T, Congia M, Macis MD, Musumeci L, Orrù V, Zavattari P, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. (2005) 37:1317–9. doi: 10.1038/ng1673
22. Gianchecchi E, Palombi M, and Fierabracci A. The putative role of the C1858T polymorphism of protein tyrosine phosphatase PTPN22 gene in autoimmunity. Autoimmun Rev. (2013) 12:717–25. doi: 10.1016/j.autrev.2012.12.003
23. Gianchecchi E, Crinò A, Giorda E, Luciano R, Perri V, Russo AL, et al. Altered B cell homeostasis and toll-like receptor 9-driven response in type 1 diabetes carriers of the C1858T PTPN22 allelic variant: implications in the disease pathogenesis. PloS One. (2014) 9:e110755. doi: 10.1371/journal.pone.0110755
24. Petrone A, Spoletini M, Zampetti S, Capizzi M, Zavarella S, Osborn J, et al. The PTPN22 1858T gene variant in type 1 diabetes is associated with reduced residual beta-cell function and worse metabolic control. Diabetes Care. (2008) 31:1214–8. doi: 10.2337/dc07-1158
25. Chien W, Tidow N, Williamson EA, Shih LY, Krug U, Kettenbach A, et al. Characterization of a myeloid tyrosine phosphatase, Lyp, and its role in the Bcr-Abl signal transduction pathway. J Biol Chem. (2003) 278:27413–20. doi: 10.1074/jbc.M304575200
26. Lloyds D, Davies EV, Williams BD, and Hallett MB. Tyrosine phosphorylation in neutrophils from synovial fluid of patients with rheumatoid arthritis. Br J Rheumatol. (1996) 35:846–52. doi: 10.1093/rheumatology/35.9.846
27. Bayley R, Kite KA, McGettrick HM, Smith JP, Kitas GD, Buckley CD, et al. The autoimmune-associated genetic variant PTPN22 R620W enhances neutrophil activation and function in patients with rheumatoid arthritis and healthy individuals. Ann Rheum Dis. (2015) 74:1588–95. doi: 10.1136/annrheumdis-2013-204796
28. Ellson CD, Riça IG, Kim JS, Huang YM, Lim D, Mitra T, et al. An integrated pharmacological, structural, and genetic analysis of extracellular versus intracellular ROS production in neutrophils. J Mol Biol. (2022) 434:167533. doi: 10.1016/j.jmb.2022.167533
29. English D and Andersen BR. Single-step separation of red blood cells, granulocytes and mononuclear leukocytes on discontinuous density gradients of Ficoll-Hypaque. J Immunol Methods. (1974) 5:249–52. doi: 10.1016/0022-1759(74)90109-4
30. McGettrick HM, Butler LM, and Nash GB. Analysis of leukocyte migration through monolayers of cultured endothelial cells. Methods Mol Biol. (2007) 370:37–54. doi: 10.1007/978-1-59745-353-0_4
31. McGettrick HM, Hunter K, Moss PA, Buckley CD, Ed Rainger G, and Nash GB. Direct observations of the kinetics of migrating T cells suggest active retention by endothelial cells with continual bidirectional migration. J Leukoc Biol. (2009) 85:98–107. doi: 10.1189/jlb.0508301
32. Eizirik DL, Colli ML, and Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. (2009) 5:219–26. doi: 10.1038/nrendo.2009.21
33. Harsunen MH, Puff R, D’Orlando O, Giannopoulou E, Lachmann L, Beyerlein A, et al. Reduced blood leukocyte and neutrophil numbers in the pathogenesis of type 1 diabetes. Horm Metab Res. (2013) 45:467–70. doi: 10.1055/s-0032-1331226
34. Wang Y, Xiao Y, Zhong L, Ye D, Zhang J, Tu Y, et al. Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with β-cell autoimmunity in patients with type 1 diabetes. Diabetes. (2014) 63:4239–48. doi: 10.2337/db14-0480
35. Collier A, Jackson M, Bell D, Patrick AW, Matthews DM, Young RJ, et al. Neutrophil activation detected by increased neutrophil elastase activity in type 1 (insulin-dependent) diabetes mellitus. Diabetes Res. (1989) 10:135–8.
36. Jackson MH, Collier A, Nicoll JJ, Muir AL, Dawes J, Clarke BF, et al. Neutrophil count and activation in vascular disease. Scott Med J. (1992) 37:41–3. doi: 10.1177/003693309203700205
37. Vecchio F, Lo Buono N, Stabilini A, Nigi L, Dufort MJ, Geyer S, et al. Abnormal neutrophil signature in the blood and pancreas of presymptomatic and symptomatic type 1 diabetes. JCI Insight. (2018) 3. doi: 10.1172/jci.insight.122146
38. Marhoffer W, Stein M, Schleinkofer L, and Federlin K. Evidence of ex vivo and in vitro impaired neutrophil oxidative burst and phagocytic capacity in type 1 diabetes mellitus. Diabetes Res Clin Pract. (1993) 19:183–8. doi: 10.1016/0168-8227(93)90112-i
39. Thomson GA, Fisher BM, Gemmell CG, MacCuish AC, and Gallacher SJ. Attenuated neutrophil respiratory burst following acute hypoglycaemia in diabetic patients and normal subjects. Acta Diabetol. (1997) 34:253–6. doi: 10.1007/s005920050084
40. Wierusz-Wysocka B, Wysocki H, Sieklerka H, Wykretowicz A, Szczepanik A, and Klimas R. Evidence of polymorphonuclear neutrophils (PMN) activation in patients with insulin-dependent diabetes mellitus. J Leukoc Biol. (1987) 42:519–23. doi: 10.1002/jlb.42.5.519
41. Chanchamroen S, Kewcharoenwong C, Susaengrat W, Ato M, and Lertmemongkolchai G. Human polymorphonuclear neutrophil responses to Burkholderia pseudomallei in healthy and diabetic subjects. Infect Immun. (2009) 77:456–63. doi: 10.1128/IAI.00503-08
42. Debczyński W and Pietruska Z. Chemotaxis and spontaneous migration of neutrophil leukocytes from patients with diabetes. Pol Tyg Lek. (1994) 49:11–3.
43. Rocha RER, Coelho I, Pequito DCT, Yamagushi A, Borghetti G, Yamazaki RK, et al. Interval training attenuates the metabolic disturbances in type 1 diabetes rat model. Arq Bras Endocrinol Metabol. (2013) 57:594–602. doi: 10.1590/S0004-27302013000800003
44. Dinçer Y, Akçay T, İlkova H, Alademir Z, Özbay G, and damage DNA. and antioxidant defense in peripheral leukocytes of patients with Type I diabetes mellitus. Mutat Res. (2003) 527:49–55. doi: 10.1016/S0027-5107(03)00073-3
45. Novoselova EG, Glushkova OV, Khrenov MO, Lunin SM, Novoselova TV, and Parfenuyk SB. Role of innate immunity and oxidative stress in the development of type 1 diabetes mellitus. Peroxiredoxin 6 as a new anti-diabetic agent. Biochem (Mosc). (2021) 86:1579–89. doi: 10.1134/S0006297921120075
46. Dinić S, Arambašić Jovanović J, Uskoković A, Mihailović M, Grdović N, Tolić A, et al. Oxidative stress-mediated beta cell death and dysfunction as a target for diabetes management. Front Endocrinol. (2022) 13:1006376. doi: 10.3389/fendo.2022.1006376
47. Perri V, Pellegrino M, Ceccacci F, Scipioni A, Petrini S, Gianchecchi E, et al. Use of short interfering RNA delivered by cationic liposomes to enable efficient down-regulation of PTPN22 gene in human T lymphocytes. PloS One. (2017) 12:e0175784. doi: 10.1371/journal.pone.0175784
48. Pellegrino M, Ceccacci F, Petrini S, Scipioni A, De Santis S, Cappa M, et al. Exploiting novel tailored immunotherapies of type 1 diabetes: Short interfering RNA delivered by cationic liposomes enables efficient down-regulation of variant PTPN22 gene in T lymphocytes. Nanomedicine. (2019) 18:371–9. doi: 10.1016/j.nano.2018.11.001
49. Arena A, Belcastro E, Accardo A, Sandomenico A, Pagliarosi O, Rosa E, et al. Preparation and in vitro evaluation of RITUXfab-decorated lipoplexes to improve delivery of siRNA targeting C1858T PTPN22 variant in B lymphocytes. Int J Mol Sci. (2021) 23:408. doi: 10.3390/ijms23010408
Keywords: type 1 diabetes, variant PTPN22, reactive oxygen species, neutrophils activation, endothelial cells
Citation: Belcastro E, Cudini A, Mezzani I, Petrini S, D’Oria V, Schiaffini R, Scarsella M, Russo AL and Fierabracci A (2025) Effect of the autoimmune-associated genetic variant PTPN22 R620W on neutrophil activation and function in patients with insulin-dependent diabetes mellitus. Front. Immunol. 16:1554570. doi: 10.3389/fimmu.2025.1554570
Received: 02 January 2025; Accepted: 30 July 2025;
Published: 08 September 2025.
Edited by:
Ricardo Pujol Borrell, Autonomous University of Barcelona, SpainReviewed by:
Bergithe Eikeland Oftedal, University of Bergen, NorwayAhmad Mahfuz Gazali, University Malaysia Pahang, Malaysia
Copyright © 2025 Belcastro, Cudini, Mezzani, Petrini, D’Oria, Schiaffini, Scarsella, Russo and Fierabracci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandra Fierabracci, YWxlc3NhbmRyYS5maWVyYWJyYWNjaUBvcGJnLm5ldA==
†Present address: Eugenia Belcastro, Department of Translational Research and New Technologies in Medicine and Surgery, University of Pisa, Pisa, Italy
‡These authors have contributed equally to this work and share first authorship