 Jill A. Poole1*
Jill A. Poole1* Aaron Schwab1
Aaron Schwab1 Geoffrey M. Thiele1,2
Geoffrey M. Thiele1,2 Todd A. Wyatt1,2,3
Todd A. Wyatt1,2,3 Amy J. Nelson1
Amy J. Nelson1 Oliver W. Schanze1Angela Gleason1
Oliver W. Schanze1Angela Gleason1 Michael J. Duryee1,2Bryant R. England1,2
Michael J. Duryee1,2Bryant R. England1,2 Ted R. Mikuls1,2
Ted R. Mikuls1,2- 1Department of Internal Medicine, College of Medicine, Omaha, NE, United States
- 2Research Service, Veterans Affairs Nebraska-Western Iowa Health Care System, Omaha, NE, United States
- 3Department of Environmental, Agricultural and Occupational Health, College of Public Health, University of Nebraska Medical Center, Omaha, NE, United States
Background: Lung disease is the most overrepresented cause of death in rheumatoid arthritis (RA). Animal studies have demonstrated potentiated autoimmunity, arthritis, and profibrotic/inflammatory lung disease with a combination of airborne exposures and collagen-induced arthritis (CIA), a model that recapitulates features of RA-associated interstitial lung disease (RA-ILD). As patients with RA-ILD demonstrate unique circulating monocyte subpopulations, this study aims to characterize lung infiltrating monocytes/macrophages in a mouse model of RA-ILD and determine whether reducing these cells mitigates the development of lung disease.
Methods: Autoimmune-prone DBA/1J mice received intranasal inhalation of lipopolysaccharide (LPS) daily for up to 5 weeks and CIA induction. Experimental groups included Sham (saline injection/saline inhalation), CIA (CIA/saline), LPS (saline/LPS), and CIA+LPS (CIA/LPS). Lung disease was assessed by longitudinal imaging, lung function measurements, bronchoalveolar lavage fluid, lung tissues, and lung histopathology. Cell subpopulations were analyzed by single cell RNA-sequencing and flow cytometry. Intravenous clodronate liposome administration was employed to reduce circulating monocytes.
Results: Longitudinal imaging demonstrated increased lung volume and tissue density in CIA+LPS mice. Lung function assessment showed reduced compliance and increased airway resistance with dual exposure. Unsupervised clustering revealed 16 discrete clusters among the experimental groups with robust clusters of monocytes/macrophages of overlapping characteristics for both CIA+LPS and CIA. By flow cytometry, dual CIA+LPS exposure induced activated CD11c+CD206+CD11b+MHC Class IIhiCD80+ alveolar macrophages, CD11cmidCD206-CD11b+Ly6Chi(and Ly6Clo)MHC Class IIhiCD80+CD86+ interstitial macrophages, and CD11c-CD11b+Ly6ChiMHC Class IIhiCD80+CD86+ monocytic-like cells. MHC Class IIhi-expressing cells across monocyte/macrophage subpopulations of CIA+LPS treated mice were more aligned with CIA than LPS alone. Intravenous clodronate liposome administration reduced CIA+LPS-induced both CD11c+CD11b+ and CD11cmidCD11b+ lung macrophages, neutrophils, lymphocytes, inflammatory/pro-fibrotic mediators, and expression of vimentin and citrullinated and malondialdehyde acetaldehyde (MAA)-modified proteins/lung autoantigens.
Conclusion: The interaction of inhalation-induced airway inflammation and autoimmune arthritis results in lung disease associated with uniquely activated infiltrating inflammatory interstitial macrophages. Moreover, depletion of circulating monocytes attenuated lung disease. Whereas the induced interstitial macrophage immunophenotype is more aligned to CIA than endotoxin exposure, co-exposure modeling renders unique features that potentially inform the pathogenesis and treatment of RA-ILD.
Introduction
Rheumatoid arthritis (RA) is a debilitating autoimmune disease that is frequently associated with a number of extra-articular manifestations including interstitial lung disease (ILD) (1). Whereas subclinical lung disease affects up to 40% of individuals with RA, clinically relevant ILD impacts up to 10% of patients and respiratory-related diseases are among the leading causes of death in men and women with RA (2–4). The pathogenesis of RA-associated ILD (RA-ILD) is complex with roles for autoimmunity, dysregulation of inflammatory and fibrotic pathways, oxidative stress, and exposures to (5) various environmental and occupational factors in genetically susceptible persons (2, 6, 7). Several inhalant environmental and occupational factors are associated with RA and RA-related lung disease including tobacco smoke, air pollutants, military-related exposures, agriculture, textile, silica, construction, and more (7). As endotoxin is an important component of many of these exposures including agriculture, textile, tobacco smoke, silica, military-related exposures and air pollutants (8–13), it is often utilized to model environmental/occupational inhalant exposures. Interestingly, the link between occupational and other environmental exposures and RA risk is well established in men but less clear in women (14). Whereas cellular mediators of RA-ILD have been postulated to be neutrophils, macrophages, and lymphocytes (15), recent studies have identified increasing myeloid cell subpopulations (particularly monocytes/macrophages) as playing a central pathogenic role in disease development (16–18).
High peripheral blood monocyte counts have been associated with poor prognosis in RA-ILD (16), and we recently found increases in circulating intermediate and nonclassical monocytes (as opposed to classical monocytes) as well as increased monocytic myeloid derived suppressor cells in RA-ILD patients (17). Moreover, circulating monocytes in individuals with RA-ILD demonstrate unique innate immune and fibrotic gene expression profiles compared to patients with idiopathic pulmonary fibrosis, patients with RA without lung disease, and those without autoimmunity and lung disease (18). Although it is likely that these alternatively activated and/or suppressed circulating monocytes ultimately impact lung disease, their potential role in mediating RA-ILD remains unclear. Animal modeling of RA-ILD represents an important strategy to understand disease mechanisms and explore potential interventional strategies. We have previously combined repetitive inhalant airborne hazard exposures (e.g., endotoxin, agriculture organic dust extracts) with collagen-induced arthritis (CIA) in mice as a model of RA-ILD. This model demonstrates several features resembling human RA-ILD including potentiated autoimmunity, arthritis, expression of post-translationally modified lung autoantigens (i.e., citrullinated and malondialdehyde acetaldehyde (MAA)-modified proteins), and pro-fibrotic and pro-inflammatory lung disease characterized by increased lung infiltrates comprised of macrophages, lymphocytes, and neutrophils (19–22).
With the development of newer technologies, new dimensions of the richness and heterogeneity of lung mononuclear phagocytes, including resident and recruited monocytes and macrophages that can adapt to the lung microenvironment to contribute to lung homeostasis, inflammatory stimuli, scavenging, fibrosis processes and immune surveillance exist (23–28). Although tissue-resident macrophages such as alveolar macrophages (AM) may be seeded during embryonic development and maintained in the absence of monocyte recruitment (29), it has also been suggested that circulating monocytes participate in the response to stimuli and the renewal of both AM and interstitial macrophages (IM) as well as persist in the lung as monocytic-like cells (26, 27, 30). The immunophenotype of combined airborne hazard exposure with arthritis induction (CIA)-induced lung monocyte-macrophage subpopulations has not been detailed nor has their role been known, which is the aim of this current study.
The objectives of this study were to leverage the combination of inhalant endotoxin (lipopolysaccharide, LPS) and CIA (CIA+LPS) to further characterize the lung pathology and lung function consequences of this model relative to LPS alone, CIA alone, and control mice and apply single-cell RNA sequencing and unsupervised clustering to resultant lung immune cells. Next, lung monocyte-macrophage subpopulations were delineated across groups followed by studies examining the impact of selective depletion of circulating monocytes to better understand the contribution of these cells to RA-ILD development.
Methods
Animal exposure model
An established 5-week exposure model (19, 20) was utilized whereby mice were randomized to 1 of 4 experimental groups including Sham (saline injection/saline inhalation), LPS alone (saline injection/LPS inhalation), collagen-induced arthritis (CIA) alone (CIA induction/saline inhalation), and CIA+LPS co-exposure (CIA injection/LPS inhalation). DBA/1J mice (age 6-8 weeks) were purchased from Inovtiv (Indianapolis, IN) and allowed to acclimate for one week prior to initiation of experiments. Male mice were utilized for all studies because we have previously demonstrated that female mice were profoundly less susceptible to development of lung disease in the setting of arthritis induction (19). CIA was induced as per the Chondrex protocol (Chondrex, Inc, Redmond, WA) (22). Airway inflammation was induced using an established model of repetitive intranasal LPS inhalation (19). Under light sedation with isoflurane, mice received 100 ng of LPS from gram-negative Escherichia coli (O55:B5; Sigma, St. Louis, MO) in 50 μl of sterile saline or saline alone daily for up to 5 weeks (weekends excluded). Animals were euthanized 1 day after the final LPS exposure by isoflurane followed by exsanguination (right axillary blood collection). No respiratory distress, signs of stress, or significant weight loss (defined as >20%) were observed throughout the exposure period.
All animal studies are detailed in accordance with the Animal Research Reporting In Vivo Experiments (ARRIVE) guidelines (https://arriveguidelines.org/). Animal procedures were approved by the Institutional Animal Care and Use Committee and were in accordance with the NIH guidelines for the use of rodents.
Clodronate-induced systemic monocyte/macrophage depletion
In separate proof-of-principle studies, Sham and CIA+LPS co-exposed mice were administered encapsulated clodronate liposomes intravenously to deplete systemic monocytes and recruited monocyte-derived macrophages (31, 32). Prior to CIA injections and LPS inhalation exposures, control and clodronate liposomes (Liposoma Technology, Amsterdam, Netherlands; 200 μl x 5 mg/ml) were injected weekly x 2 doses into the tail vein. The timing used was based on the reported duration of effect with repletion of macrophages observed 1-2 weeks following clodronate administration (33). In these studies, co-exposure duration was limited to 2 weeks due to increased tail vein irritation/inflammation noted with weekly liposome injections plus CIA at ≥ 3 weeks.
Micro-CT imaging
Progression of lung pathology was longitudinally and quantitatively determined using a live animal Quantum GX-2 micro-CT scanner (Perkin Elmer, Waltham, MA) with measurements at baseline and weekly for 5 weeks of treatment conditions. High-speed, 4-minute scans were performed utilizing an X-ray tube voltage of 90 kV and a current of 88 μA as previously described (34). Model construction and quantification were completed using 3D Slicer (5.6.2) software. Registration between baseline and subsequent scans was performed using the Elastix extension (35). Fibrosis progression is associated with increased lung volumes (i.e., increase in non-aerated lung volume) (36–39) and increased Hounsfield Units (HU) density distribution (i.e., increased tissue density) (40, 41).
Invasive pulmonary function measurements
Lung function was assessed for dynamic compliance (Cdyn) and total lung resistance (RL) was invasively assessed via a tracheostomy tube at 5 weeks following treatment conditions using a computerized small-animal ventilator (FinePointe, Buxco Electronics, Wilmington, NC) as previously described (42).
Arthritis evaluation
A semi-quantitative arthritis scoring system (Chondrex; www.chondrex.com) was applied weekly and based on hind-foot examination with score ranges from 0 (no inflammation) to 4 (erythema and severe swelling encompassing ankle, foot, and digits).
Single-cell RNA sequencing
For these studies, 4-5 mice per treatment group were euthanized with whole lungs harvested after removal of blood from the pulmonary vasculature. Lungs were dissociated with gentleMACS Dissociator (Miltenyi Biotech, Gaithersburg, MD) in a digestion solution as previously described (43). Sample were then pooled by treatment group. Single cell suspensions processed for RNAseq generated from whole lungs were quantified and viability tested using a LUNA-FL™ Dual Fluorescence Cell Counter (Logos Biosystems, Annandale, VA). Single cells were isolated using a 10x Chromium controller per manufacturer’s suggested protocol (10x Genomics, Pleasanton, CA). Reverse transcription was performed on a thermocycler (C100 Touch™ Thermal cycler, Bio-Rad, Hercules, CA) per recommended protocol followed by cDNA amplification. Amplified products were solid phase reversible immobilization (SPIR) bead-purified and evaluated by Fragment Analyzer (Agilent, Santa Clara, CA). Twenty-five percent of the cDNA volume was subjected to fragmentation and double-sided SPRIselect (Beckman Coulter, Indianapolis, IN) was used for PCR purification. PCR products were purified, quantified, and library size distribution was determined by fragment Analyzer. Libraries were sequenced per the manufacturer’s suggested parameters on a NextSeq500 and sequenced to an average depth of 50,000 reads per cell.
Single-cell RNA sequencing data processing
Cell Ranger Count tool uses STAR to align the reads to the mouse reference transcriptome. Aligned reads were filtered for valid cell barcodes and unique molecular identifiers (UMIs) and observed cell barcodes were retained. The default estimated cell count was used for this experiment. The resulting analysis files for each sample were then aggregated using the Cell Ranger aggr pipeline, which performed a between-sample normalization step and merged all samples into one.
The aggregated cell-by-gene count matrix was then used as the input for downstream analysis. The aggregated matrix was analyzed using the Loupe Browser (v.6.4; 10X Genomics) that allows easy visualization and analysis of Chromium single-cell 5′ and 3′ gene expression data and visualize UMAP plots, as well as normalized and annotated cell clustering and projection. To find the Differential Expression or Marker genes, we used the “globally distinguished” from the Significant Feature Comparison option to distinguish a cluster from every other cluster by using the p-value threshold (0.05) and log2 fold-change (FC) > 1 (upregulated) or < -1 (downregulated) to filter for significant genes. The data sets are deposited to the Gene Expression Omnibus (GEO) database with access number GSE284234.
Flow cytometry
Total lung cells (bronchoalveolar lavage fluid remained intact) were quantified following tissue dissociation of right lung lobes (19). Single-cell suspensions were stained with LIVE/DEAD Fixable Dead Cell stain (Invitrogen, Carlsbad, CA) and incubated with CD16/32 (Fc Block, Biolegend, San Diego, CA) to minimize non-specific antibody staining. Cells were then stained with antibodies (BD Biosciences, Franklin Lakes, NJ unless otherwise stated) against mouse cell surface markers including rat anti-mouse CD45 (clone:30-F11), CD11b (M1/70), Ly6G (1A8), CD86 (GL-1), Ly6C (HK1.4), CD206 (C068C2), F4/80 (T45-2342), CD4 (RM4-5), C8a (53-6.7), CD19 (1D3, Invitrogen), MHC Class II (I-A/I-E) (M5/114.15.2, Cell Signaling Technology), hamster anti-mouse CD3e (145-2C11), Armenian Hamster anti-Mouse CD11c (N418, Invitrogen), or Armenian Hamster anti-Mouse CD80 (16-10A1). Cells were acquired on a BD LSRII Yellow/Green cytometer configured with 355-nm, 405-nm, 488-nm, 561-nm, 633-nm lasers. Post-acquisition, data were exported and stored using the flow cytometry standard (FCS) 3.1 format and analyzed using FlowJo software version 10.10 (FlowJo, Ashland, OR).
The gating strategy for identification of non-debris, singlets, live CD45+ cells is depicted in Supplementary Figure 1. Lung immune cells were gated and defined as neutrophils (CD11c-Ly6G+), CD3+CD4+ T cells, CD3+CD8+ T cells and CD19+ B cells, as previously reported (19, 31, 44). Monocytic and macrophage populations were first broadly categorized as alveolar macrophages (AM) (CD11c+CD206+CD11bvar), interstitial macrophages (IM) (CD11cmidCD206-CD11b+), and monocytic-like cells (CD11c-CD206-CD11b+), based upon reports by others (45–47). Note, Fluorescence Minus One (FMO) controls were used to set the upper boundary for background signal for monocyte/macrophage subpopulation expression of CD206, MHC Class II, Ly6C, CD80, CD86 (Supplementary Figure 2). To further delineate subpopulations utilizing reports by others (45–47), “resting” AMs were defined as CD11c+CD206+CD11b-CD80+MHC Class IIlo (47). As recent reports by others have defined lung CD11c+(hi)CD206+CD80+ macrophages that highly express CD11b and/or MHC Class II as either activated AM or interstitial macrophages (IM) (27, 31, 45–48), in this current study, CD11c+CD206+CD80+MHC Class IIhiCD11b- were referred to as alternatively activated AMs and CD11c+CD206+CD80+MHC Class IIhiCD11b+ as activated AMs. IMs (CD11cmidCD206-CD11b+) and monocytic-like cells (CD11cloCD206-CD11b+) were delineated into descriptive subpopulations based upon MHC Class II and Ly6C expression with corresponding co-stimulatory molecule (CD80, CD86) expression by subpopulation. Ly6C is a glycoprotein utilized to identify lung macrophage and monocyte subpopulations in a steady state vs. activated/disease state with high Ly6C expression associated with inflammatory and “recruited” monocyte and macrophage status with conversion to low expression with resolution of disease processes (27). Moreover, Ly6Chi-expressing lung monocytes/macrophages has been associated with pro-fibrotic features (49). Correspondingly, MHC Class II high expression is characteristically associated with a “M1”-activated/inflammatory-like phenotype (27, 45–48). Lung cell population numbers were determined by multiplying percent gated by total lung cells.
Clustering T-distributed stochastic neighbor embedding
We utilized t-distributed stochastic neighbor embedding (tSNE) as an unsupervised nonlinear dimensionality reduction algorithm to visualize the high dimensional flow cytometry data generated (50). The FlowJo tSNE platform computes clustering of data from user defined selection of cytometric parameters (50). To reduce noise, non-debris singlet, live CD45+, Ly6G-, lymphocyte gate- events from a representative sample from each experimental group at 30,000 events/sample were concatenated into a single file and analyzed by tSNE with selection of cytometric parameters including FSC-A, SSC-A, and uncompensated CD11c, CD11b, Ly6C, MHC Class II, CD86, CD80, CD206, and F4/80 with the following technical options: iterations:1000, perplexity:20, learning rate:14,000, k-nearest neighbors (KNN) algorithm: Exact-vantage point tree, and gradient algorithm: Barnes-Hut. After computation, the embedded tSNE mapping of the combined 4 experimental groups is shown by contour plots of tSNE_1 vs. tSNE_2 with overlay labeling based upon the gating strategies. The 4 representative groups were also separated for visualization.
Phagocytosis
To assay functional consequences associated with experimental conditions, phagocytic ability was determined for lung monocyte/macrophage subpopulations. Following dissociation of the left lung, cells from 2-3 mice were pooled and single cell suspensions were incubated with opsonized, fluorescein-conjugated E. coli BioParticles (Invitrogen) to quantify phagocytic activity. Cells were then placed on ice and incubated as described above for markers indicative of monocytes and macrophages (i.e., live/dead, CD45, CD11b, CD11c). Cells were then cold washed, fixed with 4% paraformaldehyde, and analyzed on a BD LSRII YG (Green Profile). CD11c+CD11bvar, CD11cmidCD11b+, CD11c-CD11b+ cells were analyzed for BioParticle-associated fluorescence (44) with results presented as the percent of the cell population exhibiting Bioparticles (% Bioparticle +). To compare phagocytosis across experiments (2 independent experimental runs, n=10 mice/group total with 4 samples/group), the % Bioparticle + per mouse was divided by mean for same specific cell subpopulation in Sham mice to generate a phagocytic index.
Lavage fluid cells and lung homogenate analyses
Bronchoalveolar lavage fluid (BALF) was collected using 3 x 1 mL of phosphate-buffered saline (PBS; pH 7.2). Total BALF cell counts from pooled lavages were enumerated using a BioRad TC 20 cell counter. Differential cell counts were determined from cytospin-prepared slides (Wescor Cytopro 7620 Cytocentrifuge) with Diff-Quick (Siemens, Newark, DE). Cell-free BALF from the first lavage fraction was evaluated for cytokines and chemokines. After BALF isolation and removal of blood from pulmonary vasculature, right lobe homogenates were prepared (19). BALF and lung homogenates were analyzed for inflammatory/profibrotic mediators by ELISA (R&D Systems, Minneapolis, MN and Abcam, Waltham, MA). Analytes quantified (and thresholds for detection in pg/ml) included tumor necrosis factor (TNF)-⍺ (1.88), IL-6 (1.6), CCL2 (0.304), CCL7 (1.53), CXCL2 (1.5), neutrophil elastase (1.60), fibronectin (0.48 ng/ml), matrix metalloproteinase (MMP)-3 (125), MMP-8 (53), MMP-9 (78.1), and tissue inhibitor of metalloproteinase (TIMP)-1 (31.3).
Lung histopathology and autoantigens
Left lung lobes were excised and inflated to 15 cm H20 pressure with 10% formalin (Fisher Scientific, Fair Lawn, NJ) for 24 hours to preserve pulmonary architecture (19). Lungs were processed and cut (4-5 μM) at midpoint sections to include regions of both large and small airways as well as blood vessels. Tissues were stained for hematoxylin and eosin (H&E), modified Masson’s Trichrome, or preserved for subsequent immunohistochemistry (IHC) by the Tissue Science Core Facility at the Department of Pathology and Microbiology as previously described (19). H&E-stained slides of the entire lung section from each animal were reviewed and semi-quantitatively scored by an expert pathologist blinded to experimental conditions (19, 51). This scoring system (scored 0 to 4) evaluates the spectrum of inflammatory changes for alveolar and bronchiolar compartments with higher scores indicating greater inflammation (19, 51).
The CC motif chemokine receptor 2 (CCR2) is a critical facilitator of monocyte recruitment and activation that upon reaching inflamed tissue can differentiate into macrophages (52, 53). To quantify CCR2+ inflammatory monocytes/macrophages and neutrophils, lung sections were stained with anti-CCR2 (1:100, ab273050 Abcam) and anti-myeloperoxidase (MPO), 1:100 Cat# ab9535, Abcam) and cross absorbed with donkey anti-rabbit AlexaFluor488 (A21206, Lot#2156521, Invitrogen) and AlexFluor555 (Cat#A31572, Invitrogen), respectively, at 1:100 dilution as secondary antibodies. Slides were mounted with VECTASHIELD® Antifade Mounting Medium with 4’,6-diamidino-2-phenylindole; to identify nuclei) (Cat#H-1200, Vector Laboratories, Newark, CA) and visualized under a Zeiss fluorescent microscope (Zeiss Observer.Z1 Zeiss, White Plains, NY). Vimentin is an extracellular matrix protein that is also targeted by post-translation modification generated during the process of inflammation and previously demonstrated to be increased with CIA+LPS co-exposure (19, 20). Citrullinated (CIT) and malondialdehyde acetaldehyde (MAA)-modified proteins are post-translationally modified proteins and lung autoantigens that have also been increased with CIA+LPS co-exposure (19, 20). We have previously demonstrated increased expression and robust co-localization of MAA and CIT with vimentin in the lungs of mice and humans with inflammatory arthritis and lung disease including individuals with RA-ILD (21, 54). Lung sections were stained with Cy5 rabbit anti-vimentin (Bioss, Woburn, MA), Zenon AF594 label (Invitrogen), mouse monoclonal anti-peptidyl-citrulline antibody (clone F95 IgMκ, Millipore Sigma, Burlington, MA) or rabbit polyclonal IgG antibody to MAA (21). Detection of the F95 antibody was done using an AF488 AffiniPure donkey anti-mouse IgM, μ chain specific antibody (Jackson Immunoresearch, West Grove, PA). DAPI was added and samples sealed with Fluoromount-G (Southern Biotech, Birmingham, AL). Fluorochromes were detected using a Revolve fluorescent microscope (ECHO, Sand Diego, CA). For IHC, 10 images of the lung parenchyma per mouse were captured and antigen expression was quantified by integrated density using Image J FIJI plugin (20, 21).
Statistical analysis
Unless otherwise noted, sample sizes were extrapolated for modulating lung monocyte-macrophage subpopulations from previous work assessing combined CIA+LPS models (19) and LPS administration in the presence/absence of clodronate liposomes (31). The mean (± SD) number of CD11cintCD11bhi transition/recruited monocyte-Mϕ was 0.15x105 (0.04 x 105) with Sham and 2.5 x 105 (0.7 x 105) with CIA+LPS co-exposure at 5 weeks; thus, a sample size of n=2 in each group achieved 80% power at the 0.05 level of significance to detect differences in this cell subpopulation following an experimental exposure vs Sham. Likewise, a sample size of n=5 achieved 80% power at the 0.05 level of significance to detect a 60% reduction following targeted cell depletion with clodronate. For micro-CT and lung function studies, there were 5 mice/group. For immunophenotyping of cell infiltrates across 4 groups there were 10 mice in each of the 4 experimental groups. For the clodronate liposome studies using 3 groups (i.e., Sham, vehicle liposome in CIA+LPS, and clodronate liposome in CIA+LPS), there were 8 mice in each group. Sample numbers below the maximum number reflect limitations in available sample quantity.
All data are depicted as mean with standard deviation (SD). The Shapiro-Wilk test was utilized to test for normality among treatment groups. If the assumption of normality was satisfied, a one-way ANOVA was used. For non-parametric data, a Kruskal-Wallis test was applied. The two-stage Benjamini, Krieger, & Yekutieli procedure was applied to control for false discovery (55). All statistical analyses were performed using GraphPad Prism (version: 10.2.2) software, and statistical significance was accepted at a p value < 0.05.
Results
Combined CIA+LPS exposure results in increased non-aerated lung volume and tissue density
In the first set of experiments, mice randomized to Sham, LPS, CIA, and CIA+LPS (experimental schematic, Figure 1A) underwent live-animal micro-CT imaging to visualize and quantitate lung parenchymal changes including lung volume and density (quantified in Hounsfield Units or HU)(Figures 1B–D). Across all treatment groups, quantitative CT measurements demonstrated increases in lung volumes over time as compared to respective baseline measurements (Figure 1C). There were significant changes in lung volume demonstrated with CIA+LPS exposure, but not LPS and CIA alone, as compared to Sham at 5 weeks (Figure 1D). In contrast, lung density (HU) increased significantly in both CIA and CIA+LPS mice vs. their respective baseline measurements (Figures 1B, C) as well as compared to Sham and LPS (Figure 1D).
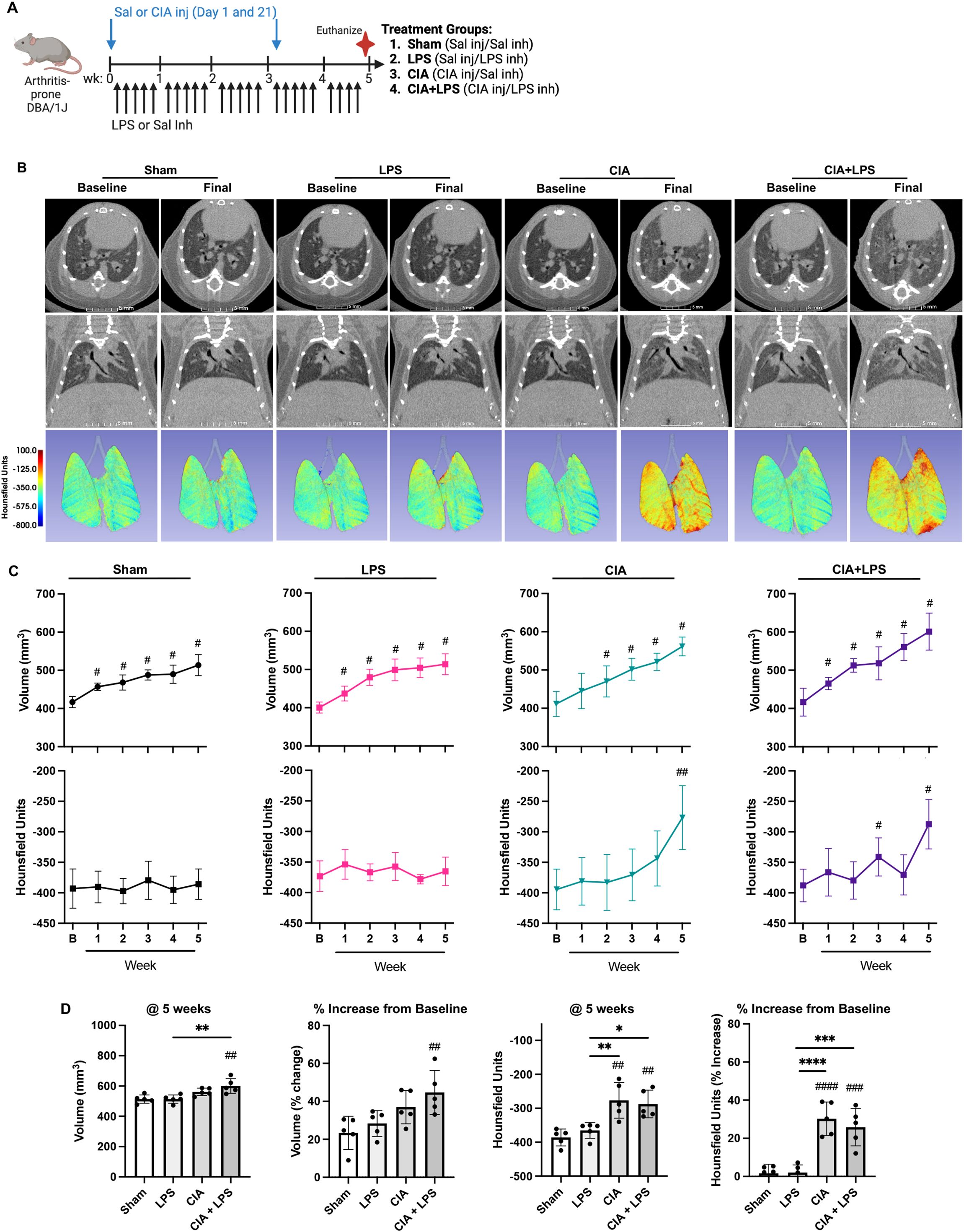
Figure 1. CIA+LPS co-exposure results in increased non-aerated lung volume and tissue density. (A) Experimental schematic with mice randomized to receive saline (Sal) or collagen-induced arthritis (CIA) injection (inj) and either daily lipopolysaccharide (LPS) or Sal intranasal inhalation (inh) daily for 5 weeks. (B) Representative micro-CT images (axial and coronal plane) at baseline and week 5 (final) from one mouse per treatment group. Density of lung tissues demonstrated by colored range of Hounsfield units (HU). (C) Line graphs depict mean volume and averaged HU by treatment group at baseline (B) and over time. (D) Scatter plots depict source measurement and % change from baseline of final (5 week) of volume and HU across treatment groups. All data expressed as mean with SD bars of n=5 mice/group. # represent statistical significance versus Sham or baseline and brackets with * between groups. ####,****<0.0001; ###,***0.0001 to 0.001; ##,**0.001 to 0.1, #,*0.01 to 0.05.
CIA+LPS co-exposure results in a decrease in lung compliance, increase in airway resistance and adverse lung pathology
Following final micro-CT imaging, the same mice were invasively assessed for lung function using a small animal ventilator. Dynamic compliance (Cdyn) was significantly reduced in CIA+LPS mice compared to all other treatment groups (Figure 2A). Correspondingly, total airway resistance (RL) was significantly increased in CIA+LPS mice compared to all other experimental groups (Figure 2A).
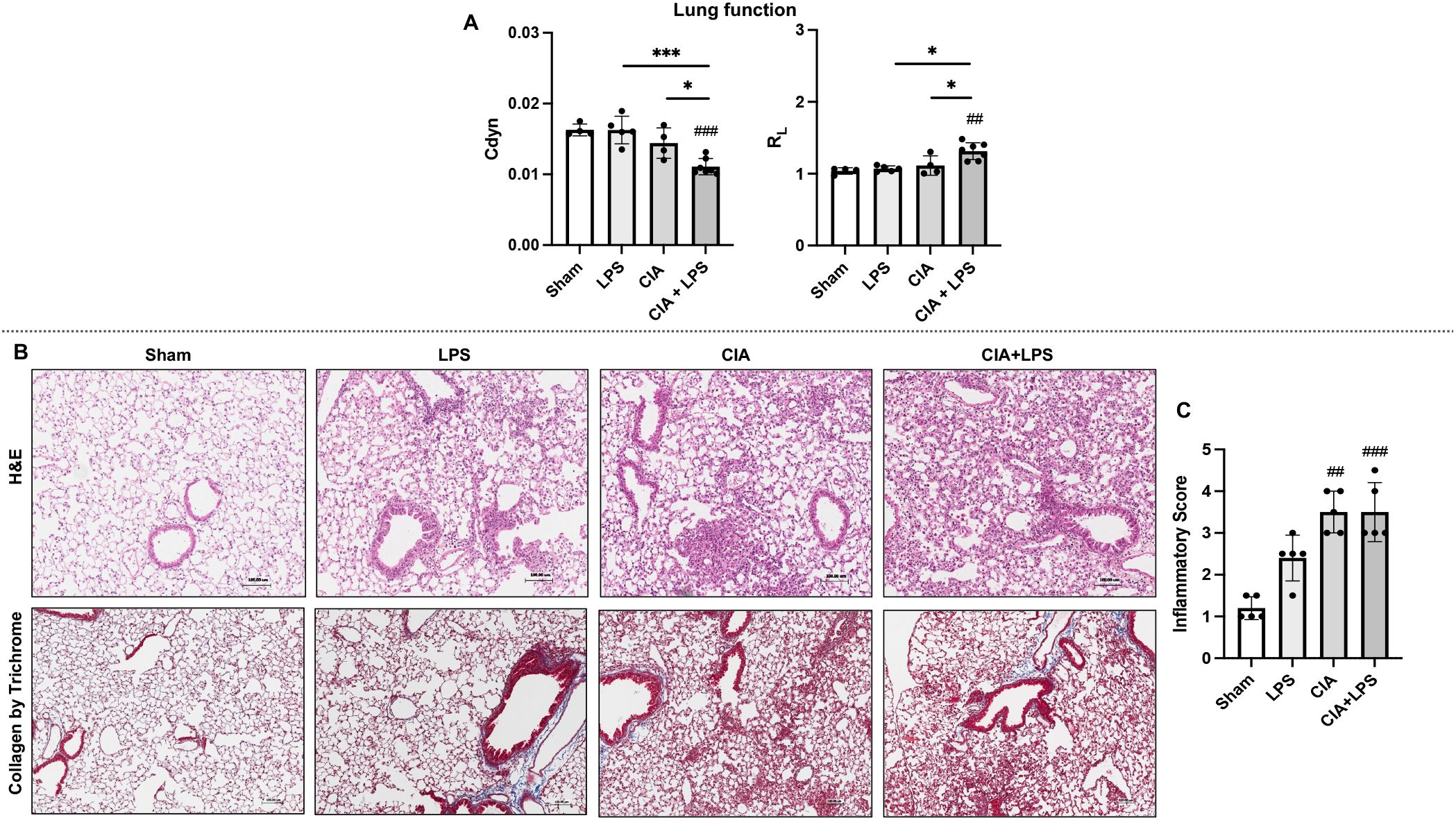
Figure 2. CIA+LPS co-exposure results in a decrease in lung compliance, increase in airway resistance and adverse lung pathology. (A) Lung function as determined by invasive pulmonary function testing represented by dynamic compliance (Cdyn) and total lung resistance (RL) at 5-week across treatment groups. (B) Representative H&E and trichrome stained lung images from one mouse per treatment group with corresponding scatter plot of (C) semiquantitative inflammatory scores. All data expressed as mean with SD bars of n=5 mice/group. # represent statistical significance versus Sham or baseline and brackets with * between groups. ###,***0.0001 to 0.001; ##0.001 to 0.1, *0.01 to 0.05.
Lung sections were next evaluated for histopathological changes by H&E and collagen deposition was evaluated by Masson’s trichrome staining (Figure 2B). There were increases in semi-quantitative inflammatory scores across treatment groups reaching statistical significance for CIA (p=0.0009) and CIA+LPS (p=0.0012) but not LPS (p=0.16) vs. Sham (Figure 2C). Arthritis inflammatory scores (mean ± SD) at 5 weeks were increased for LPS (0.4 ± 0.2, p=0.019), CIA (0.75 ± 0.5, p=0.0001), and CIA+LPS (1.1 ± 0.5, p<0.0001) vs. Sham (0 ± 0).
Collectively, these studies laid the foundation for subsequent studies aimed at delineating critical cells with corresponding gene expression data through high-throughput analyses.
Single-cell RNA-seq identifies 16 unique immune cell subsets with CIA and LPS driving unique distributions of immune cells within clusters
To cumulatively capture lung immune cells, the 10x genomics platform was utilized with a total of 16,748 cells analyzed (Sham: 4467, LPS: 3220, CIA: 3930, and CIA+LPS: 5131 cells). Unsupervised clustering of UMAP-projected CD45+ cells revealed 16 unique immune cell subsets differentially distributed by treatment group (Figure 3A) and graphically shown using unique colors and arbitrary numbers (Figure 3B). Major lung cell types were identified by signature genes (Figure 3C). Common gene markers of murine lung macrophages include Itgax (Cd11c), Cd68, Siglecf, and Cd206 (45–47, 56). Cd11b gene is predominately expressed by myeloid-derived cells including macrophages, monocytes, and neutrophils (57). Thus, macrophage cells were identified for clusters 2, 3, and 5 and partially 4 and 8, noting gene expression of Itgax/Cd11c (clusters 2, 3, 5, partially 4 and 8), Cd68 (clusters 2, 3, 5, partially 4 and 8), Siglecf (clusters 3,5), and Cd206 (clusters 2, 3, 5, partially 4). Cd11b was expressed in clusters 2, 4, 8 as well as 1, 7, and 10 (the neutrophil cluster). The F13a1 gene, which encodes for Factor XIIIA, has been linked to inflammatory-like monocytes with strong association with monocytes differentiating into interstitial macrophages as well as monocytes promoting and facilitating monocyte migration, adhesion, and phagocytosis within inflamed tissues (58, 59). CCR2 is a critical facilitator of monocyte recruitment and activation that upon reaching inflamed tissue can differentiate into macrophages (52, 53). Thus, inflammatory-like monocytes were identified with F13a1 and Ccr2 expression in clusters 2 and 4. (Note, Ccr2 expression was also demonstrated in lymphocyte clusters 9, 13, 15, recognizing that lymphocytes can also express Ccr2 (60, 61)). Clusters 1, 7 and 10 were identified as neutrophil subsets based on distribution of Cxcr2 and Retnlg expression (62). Dendritic cells (DCs) were sparse, with conventional DCs characterized by Irf4 and Xcr1 gene expression (45, 63) and plasmacytoid DCs characterized by Siglech expression (64) located within cluster 4. Clusters 9, 11, 13, and 15 were identified as T lymphocytes based on Trbc2 expression, and clusters 6, 12, and 16 were identified as B lymphocytes based on Cd19 expression. The UMAP-projected pattern of immune cell infiltrates of CIA+LPS co-exposure was similar to that of CIA alone as opposed to LPS alone or Sham. In addition, a heatmap was generated by experimental group to show that the top 20-25 genes for CIA and CIA+LPS showed similar patterns that differed from LPS and Sham (Figure 3D). Next, the pattern of expression of the top 3 to 7 genes that distinguished each unique cluster from every other cluster are shown (Figure 3E). All significant gene expressing data is summarized for unique sample-based gene features (Supplementary Data Files 1–3), treatment group gene features as compared to Sham (Supplementary Data File 2), and unique cluster-based gene features (Supplementary Data File 3).

Figure 3. Unsupervised single-cell transcriptional profiling of lung CD45+ cells demonstrate differences in distribution of aggregated gene clusters with identification of 16 unique clusters among experimental groups. (A) UMAP plot with all the treatment groups by color merged (combined) and experimental groups plotted individually to show cell distribution. (B) UMAP plot demonstrating the 16 unique unsupervised clusters identified with (C) major lung cell types identified by signature genes including Cd11c/Itgax, Cd68, Siglecf, Mrc1 (macrophages), F13a1, Ccr2 (monocytes), Cxcr2, Retnlg (neutrophils), Irf4, Xcr1, Siglech (dendritic cells), Cd19 (B cells), and Trbc2 (T cells). (D) Heatmap demonstrates relationships by treatment-based groups based upon the top 20-25 genes per group. (E) Heatmap demonstrates relationships between the top 3 to 7 genes that distinguish each unique cluster from every other cluster. N=4-5 mice/group pooled per sample.
Notably, immune cell cluster 2, which expresses Itgax (CD11c), Cd68, Mrc1 (Cd206), Itgam (Cd11b), F13a1, and Ccr2 was strikingly increased with both CIA+LPS and CIA (vs. Sham and LPS), suggesting an importance for this infiltrating inflammatory and/or activated macrophage-monocyte subpopulation. Top/unique genes expressed in cluster 2 included C1qc, C1qb, C1qa, Saa3, MS4a7, Apoe, Cxcl16, and Cadm1. Cluster 4 (monocytic-like with high Cd11b, F13a1 and Ccr2) was also increased, but to a lesser degree, with CIA+LPS and CIA. Top/unique genes expressed in cluster 4 included Plac8, Plbd1, Ifitm3, Ifitm6, Ly6c2, F1da1, Ccr2, Ms4a4c. Cluster 7, attributed to the neutrophil cluster expressing the Cxcr2 gene, was predominant in both CIA+LPS and LPS (vs. Sham and CIA). Top/unique genes expressed in cluster 7 included Rsad2, Ccl3, Ccl4, Pde1a, Ifit3b, Ifit1, Ifit3, Ltf, and Cxcl10. Finally, T lymphocytes were increased with both CIA and CIA+LPS with predominant increases particularly observed for clusters 9 and 13 of CIA+LPS that included the following high expressing unique genes: Cxcr6, IL17a, Ifng, Il1r1, Nebl, ctla4, Trdc, Cd5, Lat, IL19r1, Cd3g, Icos, and Sf6galnac3.
Thus, in the following set of studies, we focused on delineating the immunophenotype of the lung monocyte-macrophage subpopulations by cell surface protein expression (as opposed to gene expression) to further characterize their role in CIA+LPS co-exposure by flow cytometry analysis.
Co-exposure with CIA+LPS increases total cells and neutrophils and modulates CD11c+CD206+CD11bvar alveolar macrophages toward an activated status.
CIA+LPS co-exposure significantly increased total lung cell infiltrates vs. Sham, LPS, and CIA (Figure 4A). Neutrophil infiltrates (defined as CD11c-Ly6G+) were also significantly increased in CIA+LPS vs. Sham, LPS, and CIA with LPS exposure in isolation also increasing neutrophil infiltrates vs. Sham (Figure 4B). As outlined in the Methods section, monocyte-macrophage cell subpopulations were first broadly delineated based upon CD11c and CD11b expression into 3 groups: CD11c+CD11bvar, CD11cmidCD11b+, and CD11c-CD11b+ cells. Alveolar macrophages (AMs) were defined as CD11c+CD206+CD11bvar (45–47) (Figure 4C). Moreover, these cells lacked expression of Ly6C and had high expression of the monocyte and macrophage marker F4/80 (ADGRE1) (65) (Supplementary Figure 3) as well as high expression of CD80 (Figure 2C) as AMs constitutively express CD80 (66). Here, we define 3 AM subpopulations: resting AMs (MHC Class IIloCD11bloCD80+), alternatively activated AMs (MHC Class IIhiCD11bloCD80+), and activated AMs (MHC Class IIhiCD11bhiCD80+) (Figure 4C).
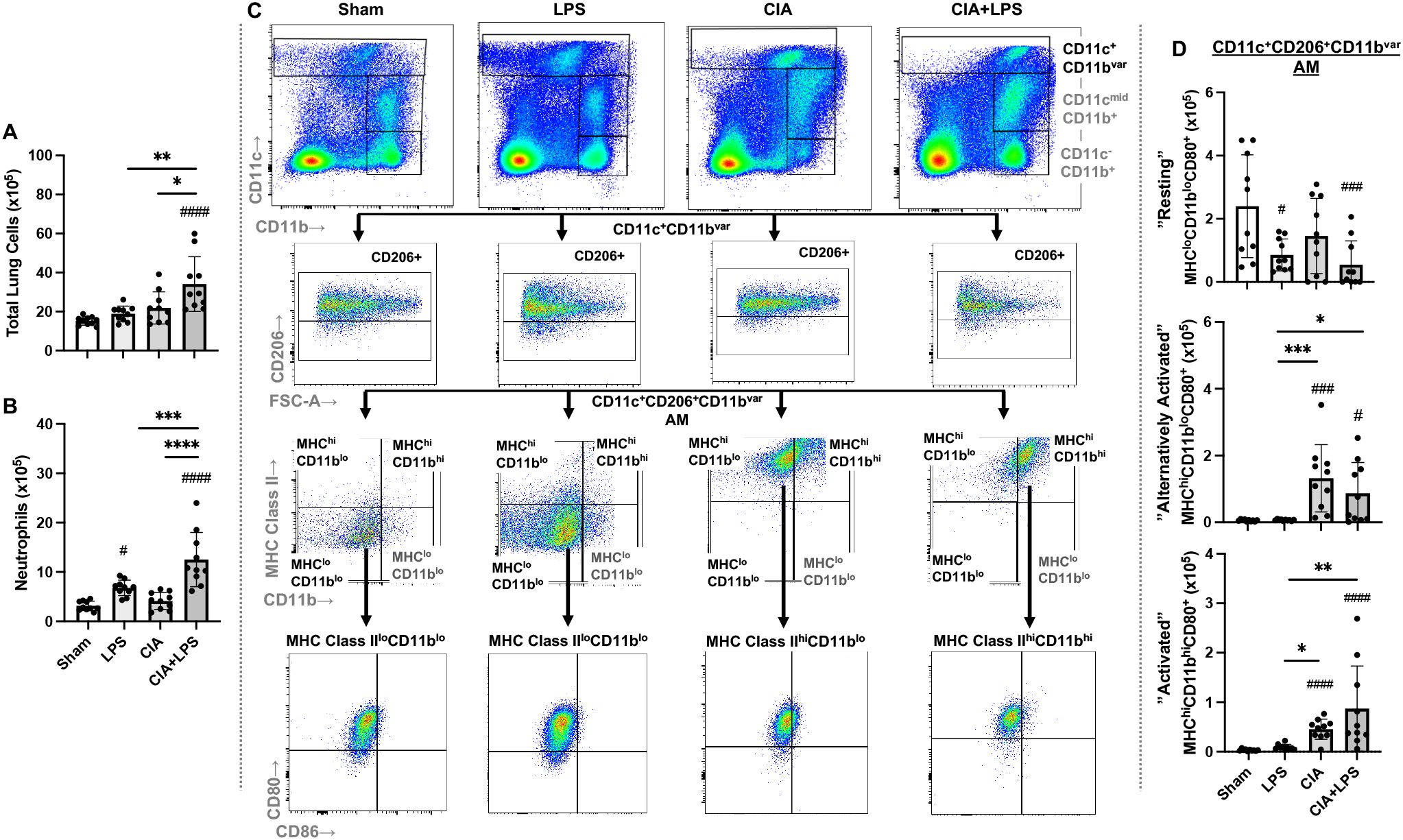
Figure 4. Lung total cell, neutrophil and CD11c+CD206+CD11bvar macrophage subpopulations vary by specific treatment group. (A) Total lung cells and (B) Ly6G+ neutrophils enumerated. (C) Representative contour plot highlighting the CD11c+CD11bvar macrophage subpopulations by one mouse per treatment group after removal of neutrophils gated from live CD45+ cells after excluding debris and doublets that are further delineated by CD206, MHC Class II and CD11b expression and costimulatory CD80 and CD86 expression. (D) CD11c+CD206+CD11bvar macrophage subpopulations defined as resting (MHC Class IIlo, CD11blo, CD80+), alternatively activated (MHC Class IIhi, CD11blo, CD80+), and activated (MHC Class IIhi, CD11bhi, CD80+), differ across treatment groups. Scatter dot plots depict mean with SD bars of n=10 mice/group. # represent statistical significance versus Sham and brackets with * between groups. ####,****<0.0001; ###,***0.0001 to 0.001; **0.001 to 0.1, #,*0.01 to 0.05.
Overall, there was a decrease in resting AMs with LPS, CIA, and CIA+LPS treatment as compared to Sham. Instead, there was an increase in alternatively activated AMs and activated AMs with CIA and CIA+LPS vs. Sham (and vs. LPS) (Figure 4D).
Co-exposure CIA+LPS induces high MHC Class II expression across both high and low Ly6C subpopulations of CD11cmidCD206-CD11b+ interstitial macrophages
The CD11cmidCD206-CD11b+ interstitial macrophage (IM) subpopulation was further delineated and enumerated by MHC Class II expression followed by Ly6C expression and CD80/CD86 expression across all treatment groups (Figures 5A–E). There was no difference across Sham, LPS, CIA, and CIA+LPS groups in MHC Class II low expressing IMs, regardless of Ly6C high vs. low expression status (Figures 5A, B). However, there were significant and robust increases in MHC Class II high-expressing IMs with CIA (5.8-fold increase) and CIA+LPS (7.9-fold increase) vs. Sham (Figure 5C). MHC Class IIhi IMs were then further characterized by Ly6C expression (Figure 5C). As compared to Sham, there were increases in both MHC Class IIhiLy6Chi IMs and MHC Class IIhiLy6Clo IMs (Figure 5D) with CIA (4.3-fold and 6.4-fold increase, respectively) and CIA+LPS (11.5-fold and 6.7-fold increase, respectively). These IM subpopulations were further delineated by co-stimulatory CD80 and CD86 expression (Figure 5C). There were significant increases in double CD80+/CD86+ expressing MHC Class IIhiLy6Chi IMs for CIA+LPS vs. Sham (14.8-fold increase), vs. LPS (4.1-fold increase), and vs. CIA (3.6-fold increase) (Figure 5E). As compared to shame, there were also significant increases, but to a lesser degree, for these MHC Class IIhiLy6Chi IMs for LPS (3.7-fold increase) and CIA (4.1-fold increase). For double CD80+/CD86+-expressing MHC Class IIhiLy6Clo cells, there were increases with both CIA and CIA+LPS treatment groups vs. Sham and vs. LPS (Figure 5E). Complete expression patterns by co-stimulatory molecules (CD80, CD86, double positive and double negative) of the MHC Class IIhi IMs subpopulations by Ly6C expression are shown in Table 1.
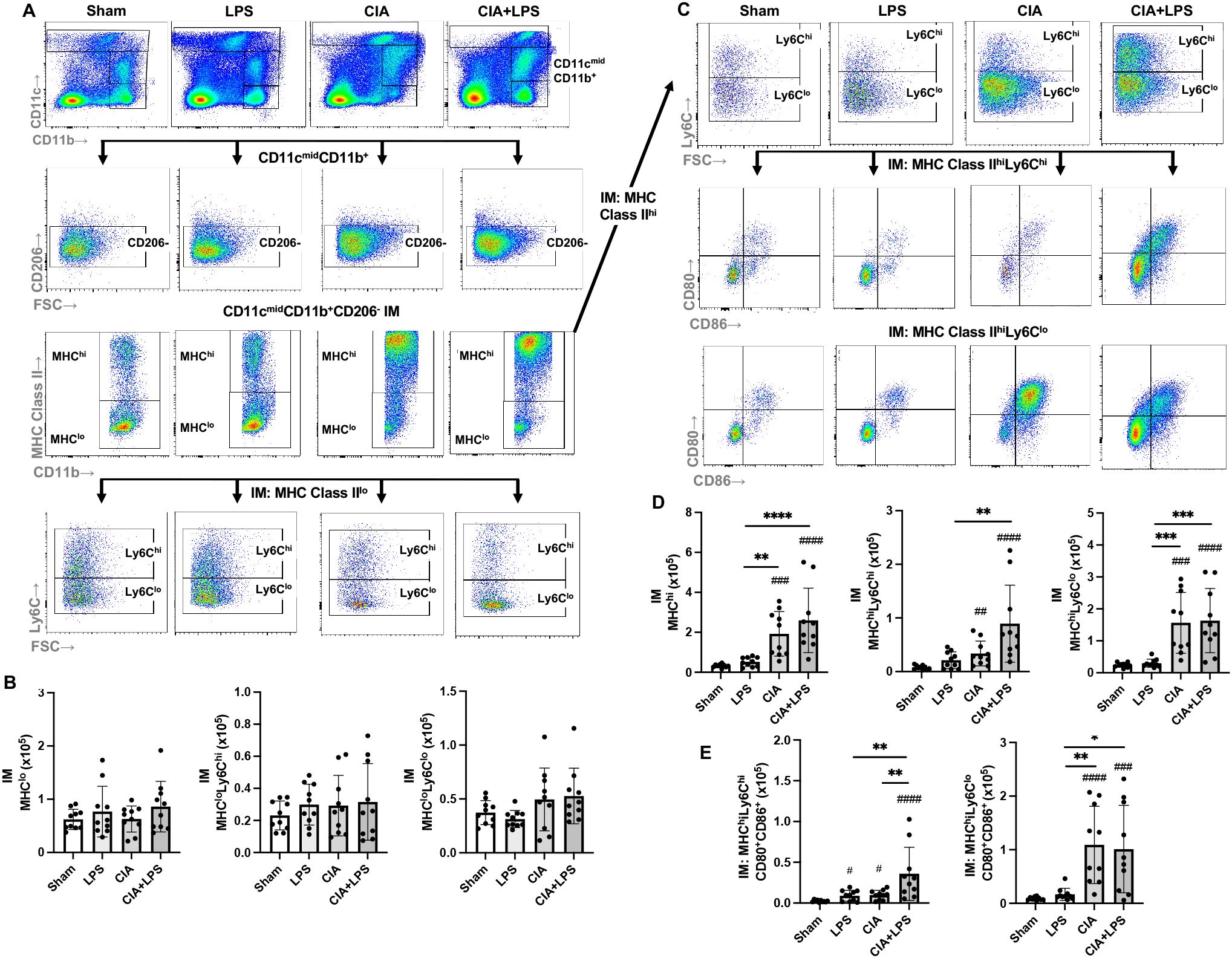
Figure 5. Lung CD11cmidCD11b+ interstitial macrophage subpopulations vary by specific treatment group. (A) Representative contour plot from one mouse per treatment group highlighting the CD11cmidCD11b+ interstitial macrophages (IM) after removal of neutrophils gated from live CD45+ cells debris and doublets that are further delineated by CD206 and MHC Class II with Ly6C expression of MHC Class IIlo-expressing cells. (B) There is no difference in number of CD11cmidCD11b+CD206-MHC Class IIlo interstitial macrophages across treatment groups and correspondingly, there is no difference in number of these cells when further delineated by high or low Ly6C expression. (C) CD11cmidCD11b+MHC Class II high expressing cells were further delineated by Ly6C expression followed by CD80 and CD86 expression. (D) CD11cmidCD11b+CD206-MHC Class IIhi expressing interstitial macrophages and those cells with/without Ly6C expression cells were enumerated across treatment groups. (E) Double positive (CD80+CD86+) of MHC Class IIhi expressing macrophages were enumerated based on high/low Ly6C expression across treatment groups. (Cell number distribution across treatment groups based upon CD80/CD86 expression is shown in Table 1). Scatter dot plots depict mean with SD bars of n=10 mice/group. # represent statistical significance versus Sham and brackets with * between groups. ####,****<0.0001; ###,***0.0001 to 0.001; ##,**0.001 to 0.1, #,*0.01 to 0.05.
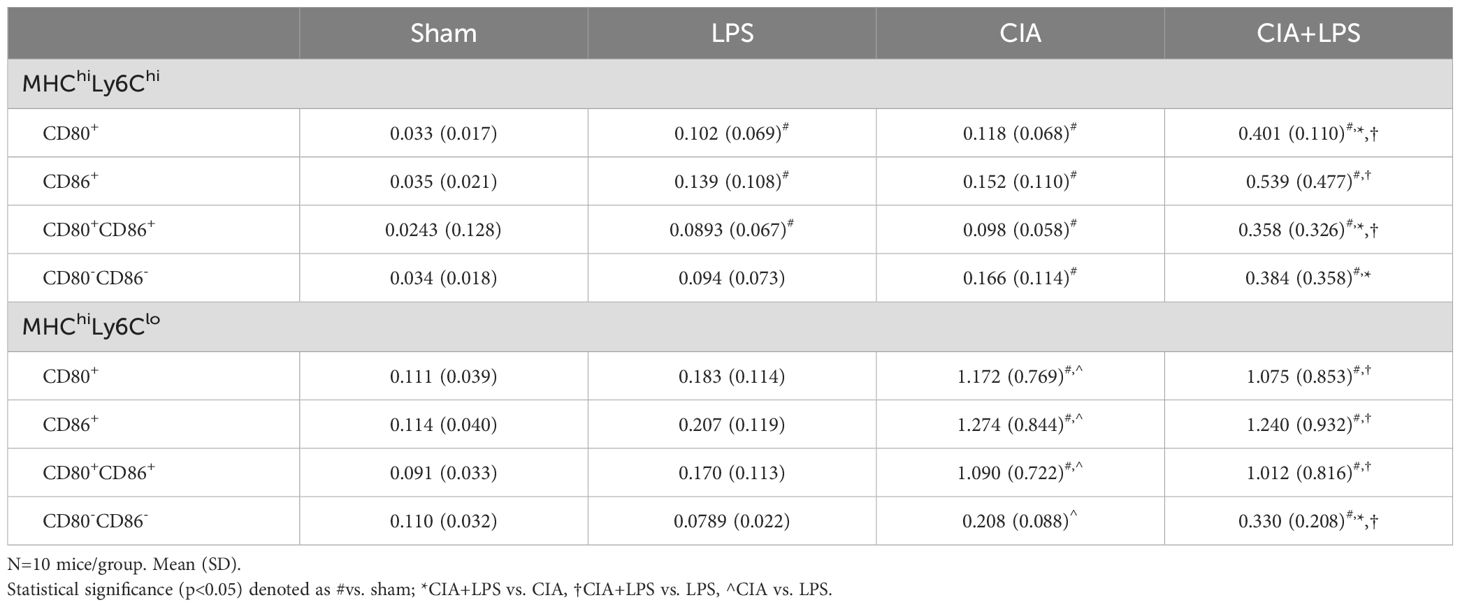
Table 1. Expression patterns of co-stimulatory molecules of CD11cmidCD206-CD11b+ interstitial macrophage (IM) subpopulations.
Co-exposure CIA+LPS increased lung CD11c-CD206-CD11b+ monocytic-like cell subpopulations defined by high MHC Class II and/or high Ly6C expression
The CD11c-CD206-CD11b+ monocytic-like cell subpopulation was further delineated by MHC Class II expression followed by delineation by Ly6C and CD80/CD86 expression across treatment groups (Figure 6A). Overall, there was a lower frequency of MHC Class IIhi vs. MHC Class IIlo monocytic-like cells (Figures 6B, C). CIA+LPS co-exposure increased the number of MHC Class IIloLy6Chi monocytic-like cells as compared to all other treatment groups (Figure 6B). Whereas there was no difference between CIA+LPS vs. Sham for MHC Class IIloLy6Clo monocytic-like cells, this cell subpopulation was increased with CIA alone.
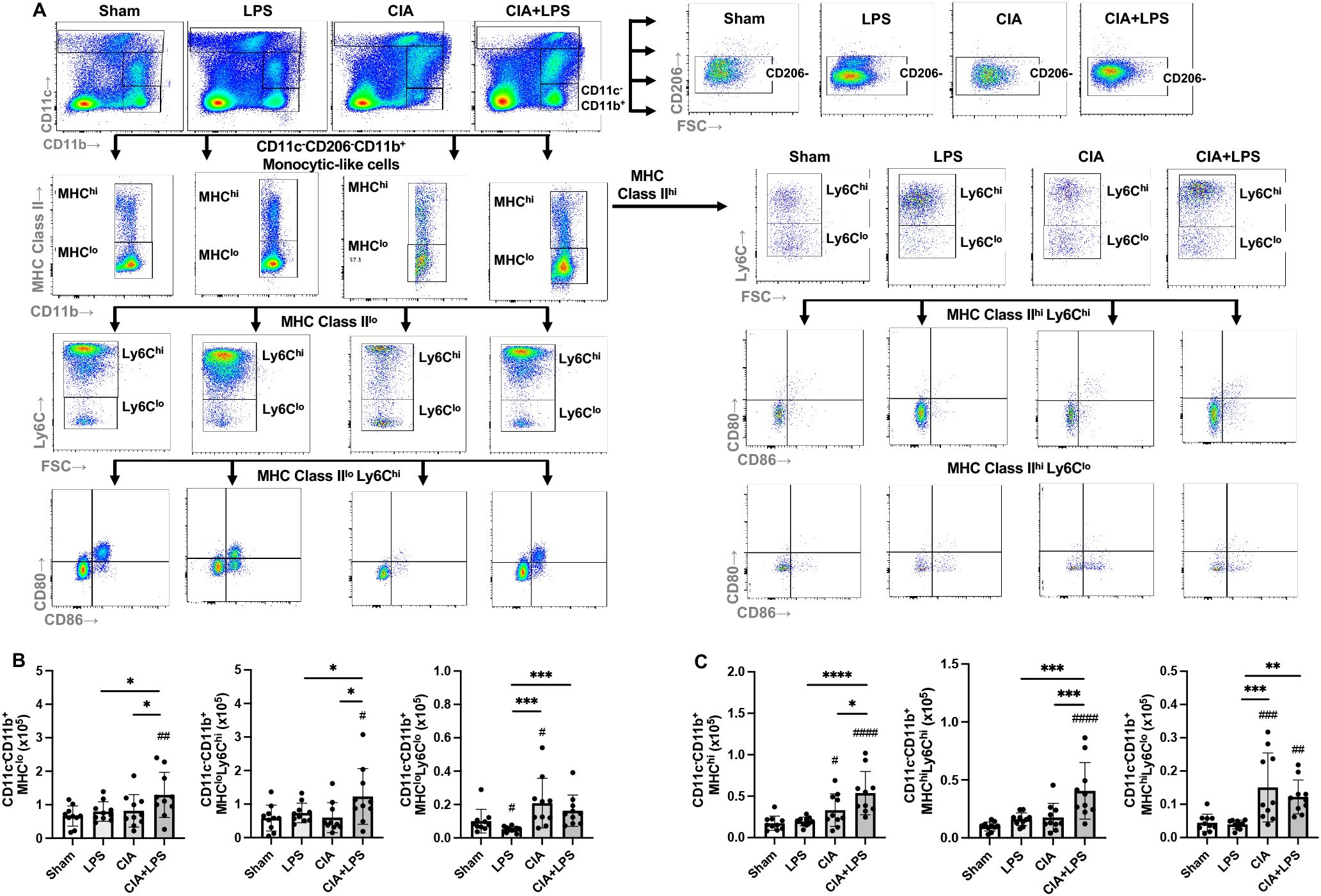
Figure 6. Lung CD11c-CD11b+ monocytic-like cell subpopulations vary by specific treatment group. (A) Representative contour plot from one mouse per treatment group highlighting the CD11c-CD11b+ monocytic-like cells after removal of neutrophils gated from live CD45+ cells debris and doublets that are further delineated in sequential manner by CD206, MHC Class II, Ly6C, and CD80/CD86 expression. Numbers of monocytic-like subpopulations based upon (B) MHC Class IIlo and Ly6C expression and (C) MHC Class IIhi and Ly6C expression enumerated across treatment groups. (Cell number distribution across treatment groups based upon CD80/CD86 expression is shown in Table 2). Scatter dot plots depict mean with SD bars of n=10 mice/group. # represent statistical significance versus Sham and brackets with * between groups. ####,****<0.0001; ###,***0.0001 to 0.001; ##,**0.001 to 0.1, #,*0.01 to 0.05.
For the MHC Class IIhi monocytic-like cells, these cells were increased for both CIA and CIA+LPS (vs. Sham), but there were only increases in MHC ClassIIhiLy6Chi for CIA+LPS as compared to all other groups (e.g., a 4-fold increase vs. Sharm) (Figure 6C). The MHC ClassIIhiLy6Clo cells were increased for both CIA and CIA+LPS. The expression patterns by co-stimulatory molecules (CD80, CD86, double positive and double negative) of these monocytic-like subpopulations are summarized in Table 2.
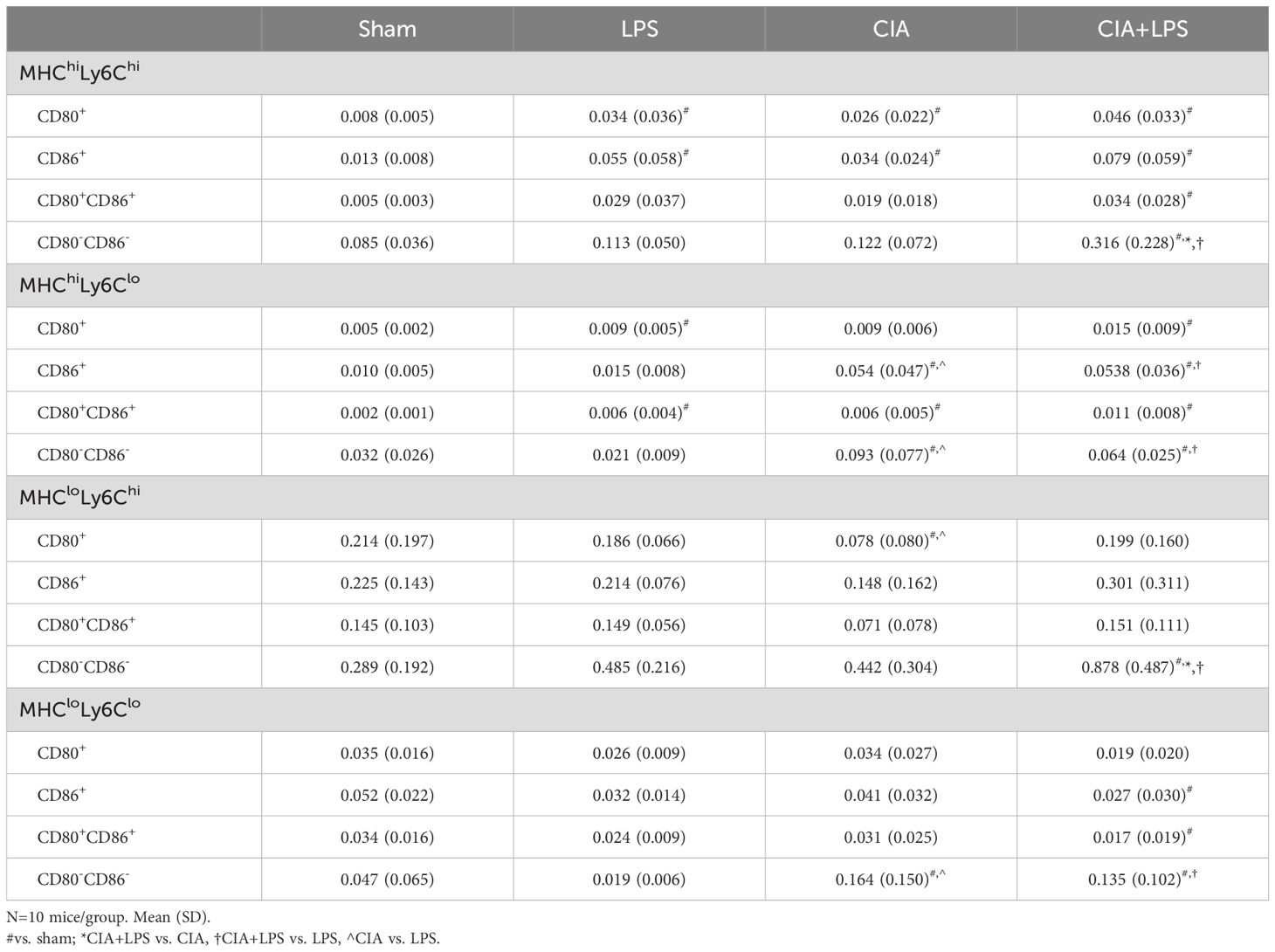
Table 2. Expression patterns of co-stimulatory molecules of CD11c-CD206-CD11b+ monocytic-like cell subpopulations.
Unique clustering by tSNE algorithm of monocytes-macrophages by treatment group highlight robust MHC Class II and Ly6C expression with co-exposure CIA+LPS
To provide visualization of the complex multi-dimensional data, the tSNE algorithm was applied to CD45+, neutrophil (Ly6G) negative, lymphocyte gate negative cells, combined and separated from a representative murine lung from each group (Figure 7A). The embedded tSNE mapping is demonstrated by contour plot with overlay colors by expression intensity, labeling cell marker expression from the combined map to provide visualization of marker expression differences (Figure 7B). CIA+LPS co-exposure demonstrates unique distribution with a striking cellular cluster shift towards the left that is delineated by high MHC Class II and CD11c expression as well as a predominant top and rightward cluster delineated by high Ly6C expression. Collectively, these flow cytometric studies highlight that co-exposure CIA+LPS uniquely and robustly upregulates MHC Class II and Ly6C expression across the heterogenous lung monocyte-macrophage cell spectrum, and this would imply a polarization state consistent of cells with activated, inflammatory, and pro-fibrotic features (27, 46, 49).
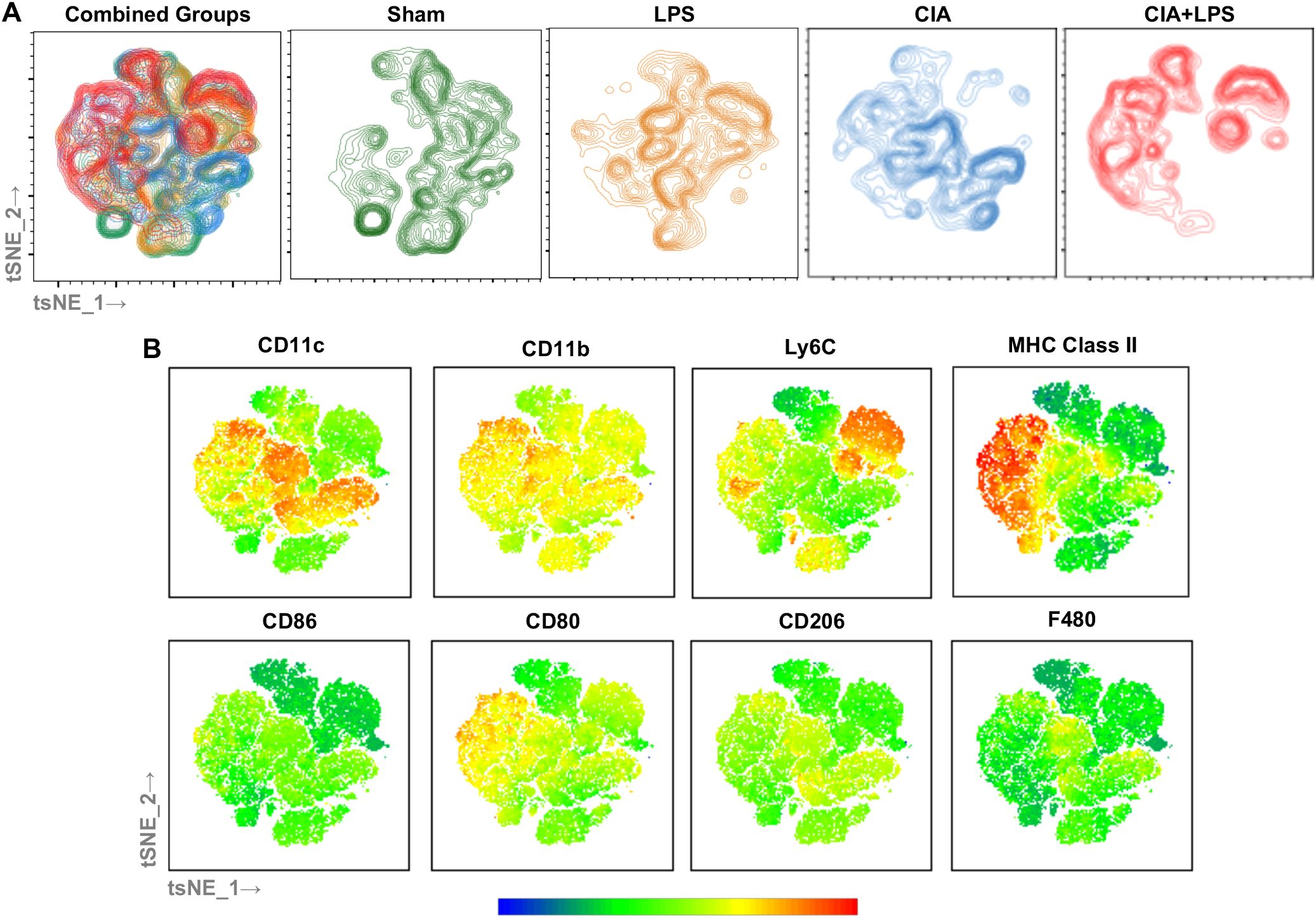
Figure 7. Clustering by tSNE algorithm of lung monocytes-macrophages from representative of each Sham, LPS, CIA, and CIA+LPS treatment group. (A) tSNE mapping of the data applied to non-debris, single, live CD45+Ly6G-lymphocyte gate- cells (30,000 cells/sample) of a representative mouse lung combined and from each group (sham, LPA, CIA, CIA+LPS). (B) Multigraph overlays of staining intensity (blue: low and red: high) of cell surface markers utilized for delineating lung monocyte-macrophage subpopulations demonstrated by contour plots.
Phagocytic activity of lung monocytes-macrophages differs across treatment condition and subpopulation
Phagocytic ability reflects an important innate immune function for host clearance of lung particulates and debris that was assessed in the 3 broadly defined monocyte-macrophage subpopulations: AM (CD11c+CD11bvar), IM (CD11cmidCD11b+), and monocytic-like cells (CD11c-CD11b+) (Figures 8A, B). Representative histograms are shown for all treatment groups for each cell subpopulation (Figure 8A) with the phagocytic index of 4 samples of pooled mice (n=10 total mice/condition) for all treatment groups across each of the 3 cell subpopulations (Figure 8B). There were significant increases in phagocytic ability of both AMs and monocytic-like cells with LPS and CIA+LPS but not CIA vs. Sham. There was no difference in phagocytic ability for IMs across treatment groups. Next, phagocytic ability differences within treatment groups across cell subpopulations were assessed (Figure 8C). Within Sham mice and as compared to AMs, IMs demonstrated increased phagocytic ability and monocytic-like cells demonstrated decreased phagocytic ability. As phagocytic ability was generally increased with treatment groups for AMs, there was no difference between the phagocytic ability of AMs to that of IMs for LPS, CIA, and CIA+LPS. For all experimental groups, the phagocytic ability of monocytic-like cells was reduced in comparison to AMs and/or IMs.
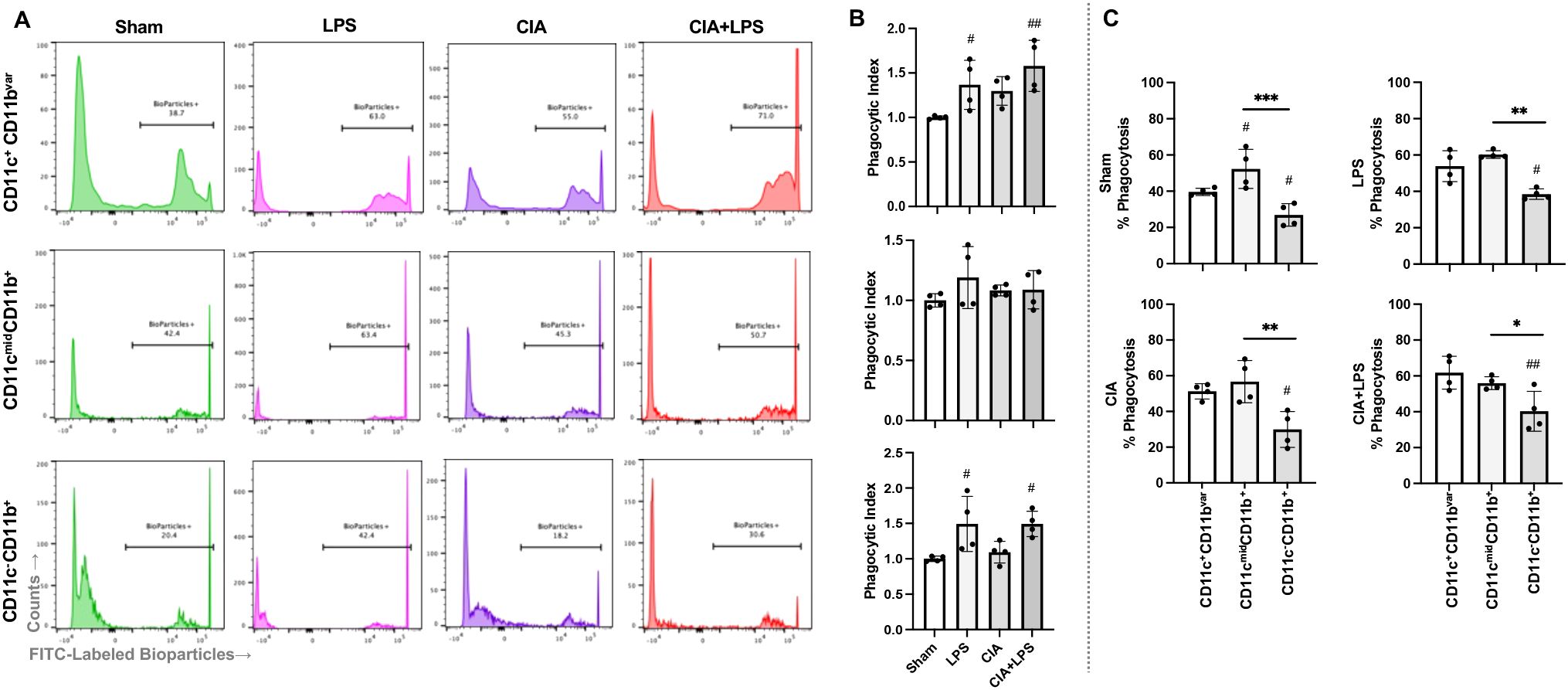
Figure 8. Phagocytic activity of lung monocyte-macrophage subpopulations across treatment groups. (A) Representative histograms from one mouse per treatment group depicting the phagocytic ability of the 3 generalized monocyte-macrophage subpopulations including CD11c+CD11bvar, CD11cmidCD11b+, and CD11c-CD11b+ cells upon incubation with fluorescently labeled E coli. (B) Scatter plots with bars depict mean with SD of phagocytic index normalized to respective Sham cell population (% bioparticle positive per mouse divided by averaged Sham per experimental run). (C) % Phagocytosis (Bioparticle +) by treatment group across these 3 cell subpopulations. N=2-3 mice pooled per sample for 4 samples/group (N=10 mice/condition). ***0.0001 to 0.001; ##,**0.001 to 0.01, #,*0.01 to 0.05.
As these studies collectively highlight the potential role for monocyte-derived lung macrophages in CIA+LPS, the following set of studies sought to determine the functional consequence of reducing the infiltration of lung macrophages by depleting the reservoir of circulating monocytes focused on CIA+LPS co-exposed mice.
Systemic clodronate liposome delivery reduces CIA+LPS co-exposure-induced airway and cellular influx
To reduce the circulating/systemic reservoir of available monocytes-macrophages, mice were treated with intravenous clodronate liposomes (vs. vehicle control liposomes) one day prior to initiation of CIA+LPS co-exposure modeling with a dose repeated 1-week later (Figure 9A). In these proof-of-concept studies, the exposure model was terminated at 2 weeks (as opposed to 5 weeks) because of unacceptable tail vein irritation and inflammation from repeated clodronate and CIA injections. In the CIA+LPS mice, there were significant reductions (% reduction) in airway influx (BALF) of total cells (64%), neutrophils (73%), and lymphocytes (80%) but not macrophages with clodronate liposomes vs. vehicle (Figure 9B). Likewise, there were significant reductions in lung tissue infiltrates of total lung cells (50%), activated AMs (CD11c+CD11b+, 68%), IMs (71%), neutrophils (42%), CD4+ T cells (55%), CD8+ T cells (48%), and CD19+ B cells (44%) but not monocytic-like cells in CIA+LPS mice treated with clodronate liposomes vs. vehicle liposomes (Figure 9C).
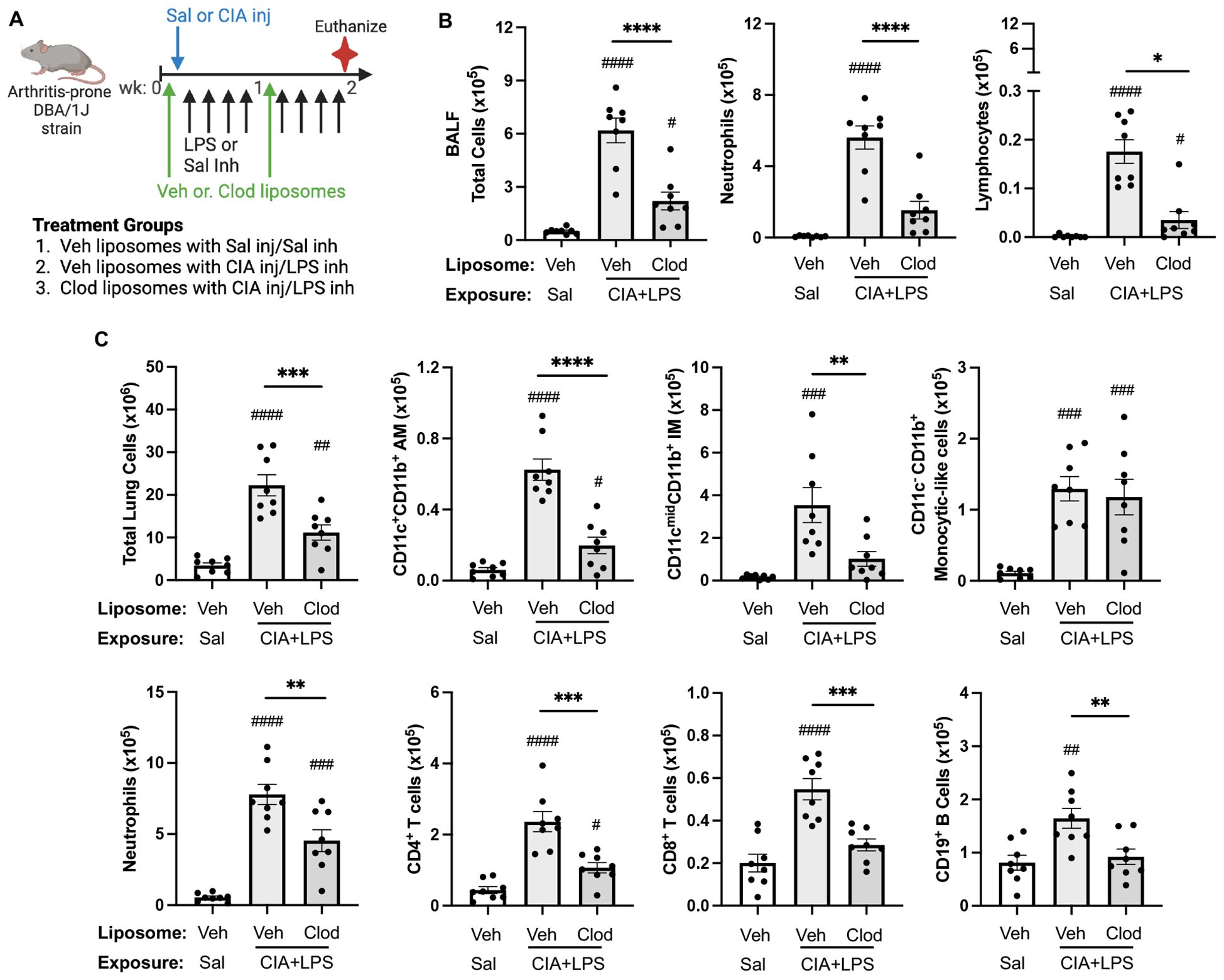
Figure 9. Systemic clodronate liposome delivery reduces 2-week CIA+LPS co-exposure-induced airway and lung tissue cellular influx. (A) Mice were treated with vehicle (Veh) control or clodronate (Clod) liposome twice (baseline and week 1), saline or CIA injection (inj) once at baseline, and then received intranasal inhalation with LPS (100 ng) or saline daily for 2 weeks (weekends excluded). (B) Bronchoalveolar lavage fluid (BALF) total cells, neutrophils, and lymphocytes and (C), lung tissue cellular infiltrates. Total lung cells enumerated with number of lung cells determined by multiplying lung cell % gated population by flow cytometry by total lung cells enumerate from lung sample. AM, alveolar macrophages; IM, interstitial macrophages. All data expressed as mean with SD bars of n=8 mice/group. Symbols represent statistical significance versus vehicle liposome+saline exposures (#) or between CIA+LPS mice treated with vehicle versus clodronate liposomes (*). ####,****<0.0001; ###,***0.0001 to 0.001; ##,**0.001 to 0.1, #,*0.01 to 0.05.
CIA+LPS co-exposure induced mediators are reduced with systemic clodronate liposome delivery
Treatment with intravenous clodronate liposomes (vs. vehicle) also resulted in reductions (% reduction) in CIA+LPS-induced BALF levels of IL-6 (74%), CCL2 (85%, p=0.014), CCL7 (83%), neutrophil elastase (75%), and fibronectin (72%) (Figure 10A). There were also reductions in lung tissue homogenate levels of MMP-3 (18), MMP-9 (30%), and TIMP-1 (48%), in CIA+LPS mice treated with clodronate liposomes vs. vehicle liposomes (Figure 10B). However, there were no treatment differences between clodronate and vehicle in BALF levels of TNF-⍺ and CXCL2 or lung tissue homogenate levels of IL-6, TNF-⍺, CXCL2, CCL2, CCL7, neutrophil elastase, and fibronectin following CIA+LPS exposure (Table 3).
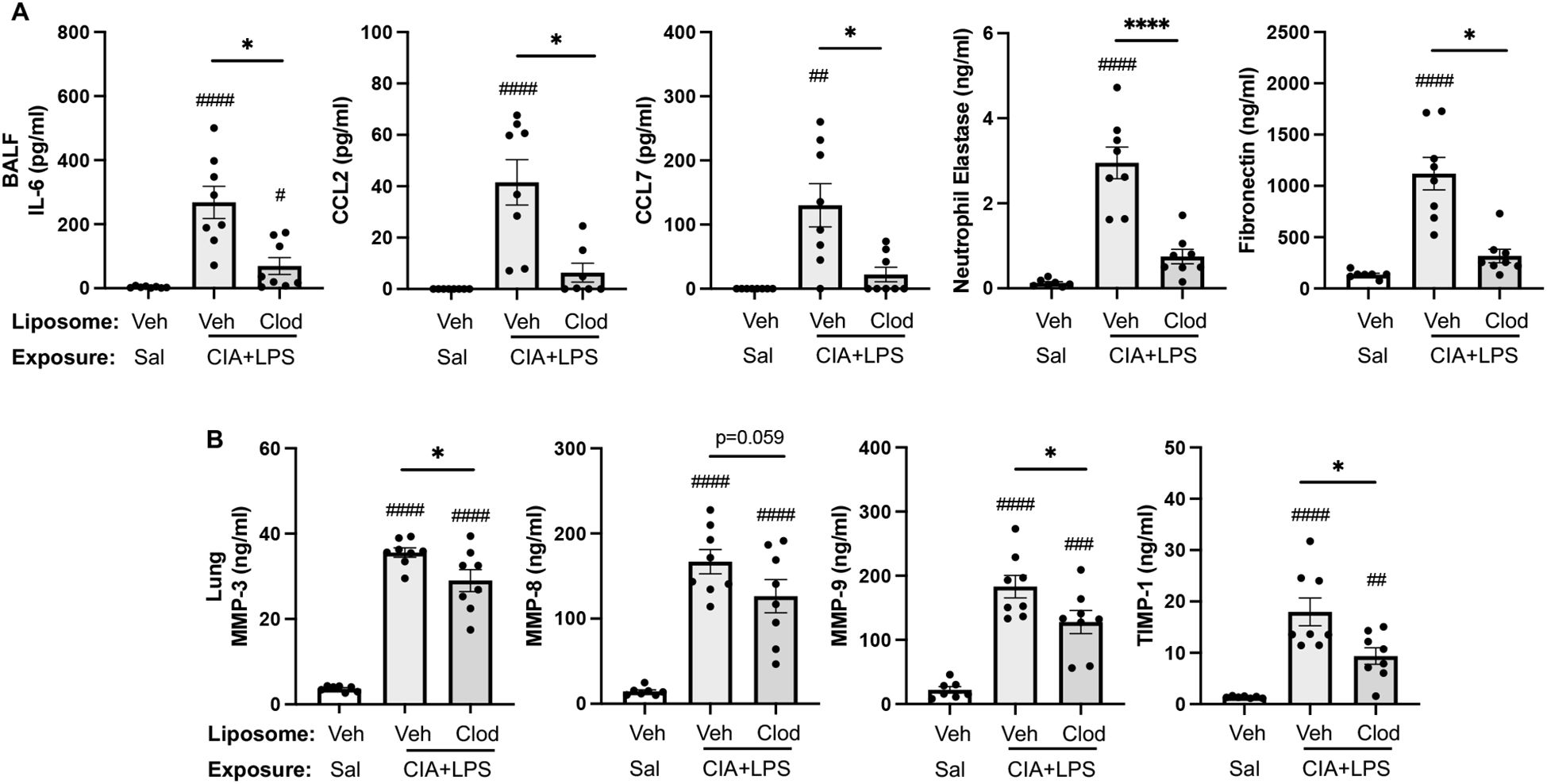
Figure 10. CIA+LPS co-exposure induced mediators reduced with systemic clodronate liposome delivery. (A) Bronchoalveolar lavage fluid (BALF) levels of IL-6, CCL2, CCL7, neutrophil elastase, and fibronectin. (B) Lung tissue homogenate levels of MMP-3, -8, -9, and TIMP-1. All data expressed as mean with SD bars of n=8 mice/group. Symbols represent statistical significance versus vehicle liposome+saline exposures (#) or between CIA+LPS mice treated with vehicle versus clodronate liposomes (*). ####,****<0.0001; ###0.0001 to 0.001; ##0.001 to 0.1, *0.01 to 0.05.
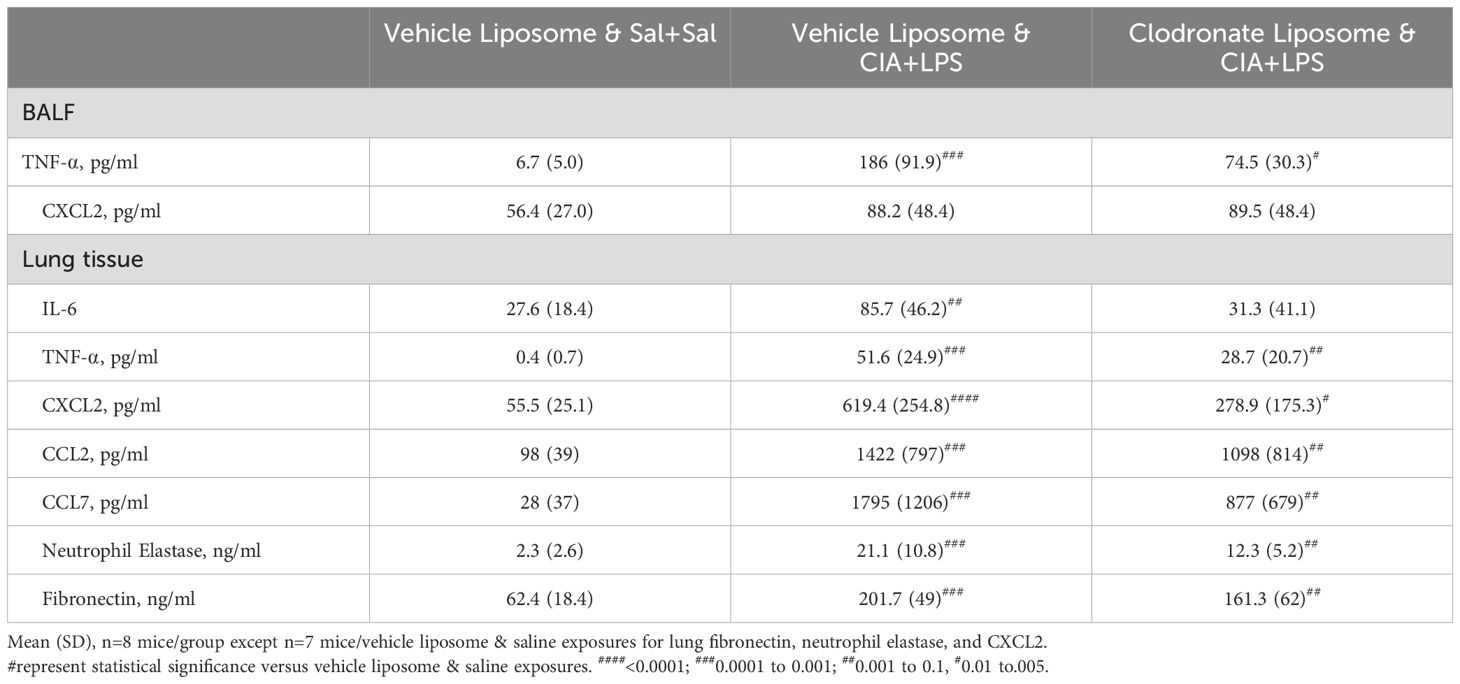
Table 3. CIA+LPS co-exposure induced mediators not significantly affected by systemic clodronate liposome delivery.
Systemic clodronate liposome delivery reduces CIA+LPS co-exposure induced lung tissue inflammation, infiltrating CCR2+ monocyte-macrophages and MPO+ neutrophils as well as vimentin and lung CIT and MAA autoantigen expression
Lung sections from mice treated with vehicle and clodronate liposomes with CIA+LPS co-exposure and control mice treated with vehicle liposomes and saline+saline were evaluated for histopathological changes by H&E, collagen by trichrome, CCR2+, and MPO+ cell infiltrates, vimentin expression, and expression of CIT and MAA autoantigens (Figure 11A). Semi-quantitative inflammatory scores following CIA+LPS exposure were significantly reduced with clodronate vs. vehicle control liposomes (Figure 11B). Correspondingly, CIA+LPS exposure-induced collagen deposition was reduced with clodronate vs. vehicle control liposome (Figure 11C). CIA+LPS-induced CCR2+ monocyte/macrophage cell infiltrates, quantified by flow cytometry, were reduced by 78% (Figure 11D) and MPO+ neutrophils were reduced by 53% (Figure 11E). Next, the expression of vimentin, an extracellular matrix protein that is also targeted by post-translational modifications generated during the process of inflammation and oxidative stress and expressed by monocyte/macrophages and lung mesenchymal cells, was reduced 98% in CIA+LPS mice treated with clodronate vs. vehicle (Figure 11F). Correspondingly, there were significant treatment-related reductions in the expression of lung autoantigens including reductions in the expression of CIT (76% reduction) and MAA (84% reduction) (Figures 11G, H).
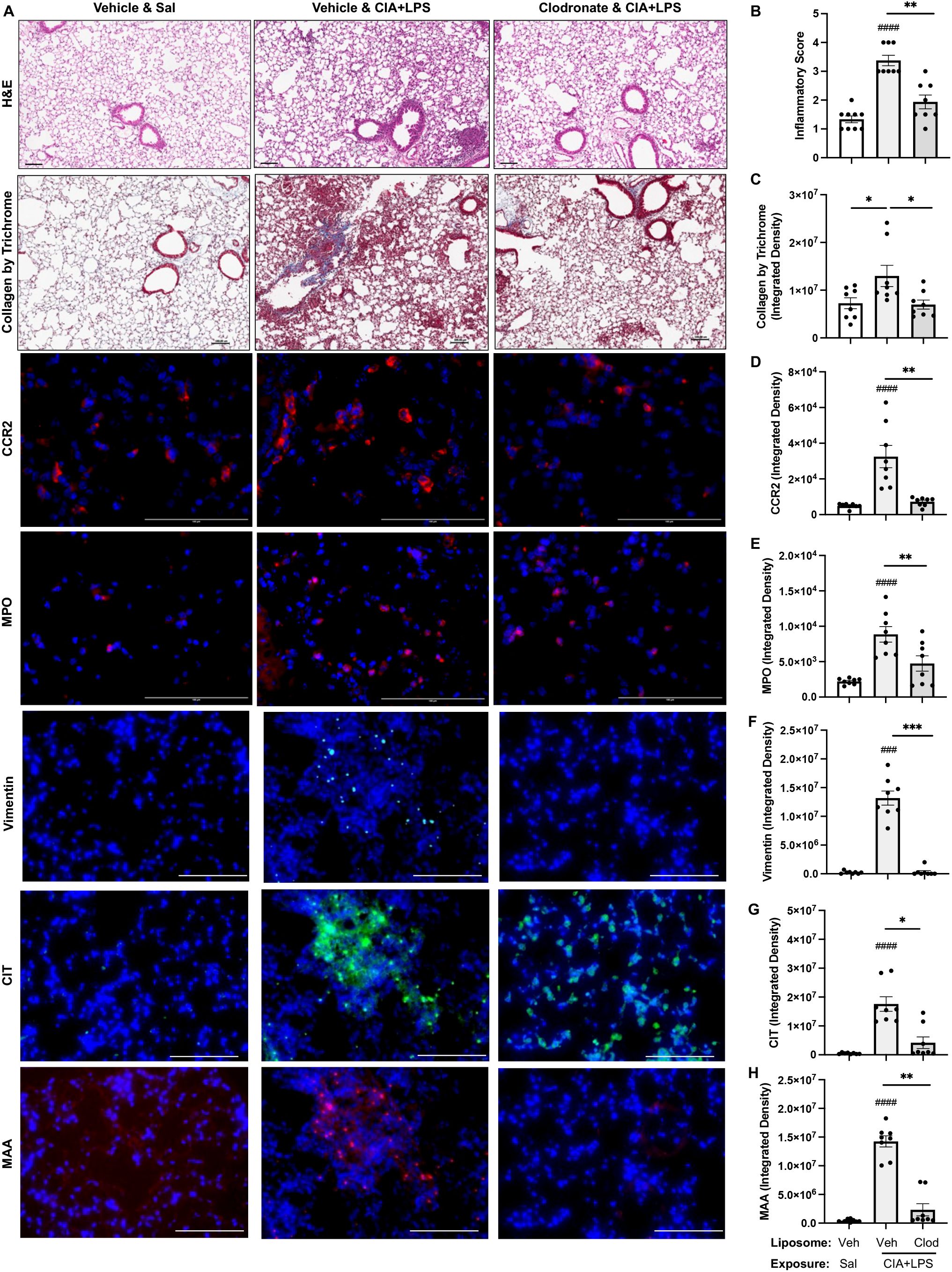
Figure 11. Systemic clodronate liposome delivery reduces CIA+LPS co-exposure induced lung tissue inflammation, collagen deposition, infiltrating CCR2+ monocyte-macrophage cells and myeloperoxidase (MPO)+ neutrophils, vimentin expression, and lung CIT and MAA autoantigen expression. (A) Representative images from treatment groups stained by H&E, collagen by trichrome, CCR2 (red), and MPO (red), vimentin (teal), citrulline (CIT, green), and malondialdehyde-acetaldehyde (MAA, red) modified proteins with DAPI nuclei staining (blue) by confocal microscopy. Scatter plots with bars depict mean with SD of (B) semi-quantitative lung inflammatory score and integrated density of (C) Collagen by trichrome, (D) CCR2 and (E) MPO, (F) vimentin, (G) CIT, and (H) MAA quantified per each mouse. Line scale denotes 100 μm. n=8 mice/group. Symbols represent statistical significance versus vehicle liposome+saline exposures (#) or between CIA+LPS mice treated with vehicle versus clodronate liposomes (*). ####<0.0001; ###,***0.0001 to 0.001; **0.001 to 0.1, *0.01 to 0.05.
Discussion
RA-ILD represents the most overrepresented cause of death in RA (4, 67, 68). Knowledge of the critical cellular players, biomarkers of disease, and efficacious treatment options are limited, and the factors driving the development and progression of lung disease in RA are unclear. Studies herein demonstrate that the interaction of inhalation-induced airway inflammation and autoimmune arthritis resulted in lung disease associated with robust and activated monocyte/macrophages, which directly mediate the adverse lung consequences observed in this informative disease model. Moreover, CIA+LPS induced several subpopulations of activated AMs, recruited, inflammatory, and activated IMs, and activated/inflammatory monocytic-like cells, all sharing high expression levels of MHC Class II antigen, co-stimulatory molecule(s), and inflammatory markers (Ly6C, CD11b). Strikingly, IMs generated with CIA+LPS was more aligned to autoimmune arthritis induction than inhalant LPS exposure. Finally, the targeted depletion of circulating monocytes was associated with marked reductions in CIA+LPS-induced IMs, neutrophils, lymphocytes, inflammatory/pro-fibrotic mediators, and expression of vimentin and citrullinated and malondialdehyde acetaldehyde (MAA)-modified proteins/lung autoantigens. Collectively, the results of this study highlight the central role that infiltrating/recruited IMs appear to play in the pathogenesis of RA-ILD.
The complexity of the pathogenesis of RA-associated lung disease involves the dynamic interplay between environmental triggers and genetic susceptibility ultimately leading to a specific loss of tolerance and autoimmunity with increasing evidence suggesting that initial immune responses may be initiated at the level of pulmonary mucosa (69), noting that subclinical lung disease occurs in upwards of 30-40% of RA patients. In addition to measures of inflammation and histopathologic assessments, additional metrics were deployed in this study to better characterize lung disease in the CIA+LPS co-exposure animal model. Live micro-CT imaging revealed a gradual increase in non-aerated lung volume, a feature of lung fibrosis in mice, with corresponding increases in lung tissue density that were most apparent after 5 weeks. Interestingly, arthritis induction alone also increased lung density but not non-aerated lung volume, consistent with our data demonstrating increases in lung inflammation observed with CIA alone. Moreover, declines in lung function including reduced tissue compliance and increased resistance, were only observed in the co-exposed animals. Collectively, these imaging and functional studies support the use of this model as they align with observations demonstrated with other experimental lung fibrosis models including those employing bleomycin and silica.
In patients with RA-ILD, circulating monocytes demonstrate high expression of genes involved in macrophage differentiation-activation and pro-fibrosis processes (18). In this current study, single-cell RNA-seq studies identified a striking immune cell cluster (i.e., cluster 2) that was unique to CIA and CIA+LPS as opposed to Sham or LPS with a predominance of features characteristic of activated/inflammatory monocyte-derived macrophages. This observation coupled with human data of altered circulating monocytes in patients with RA-ILD may suggest that future approaches targeting and/or reducing lung monocyte/macrophage burden may be advantageous in RA lung disease. Moreover, complement genes were highly expressed with co-exposure CIA+LPS. Although dysregulation of the complement system appears to be an important factor in autoimmune and pulmonary disease and current therapies such as glucocorticoids, pirfenidone, and disease modifying antirheumatic drugs and targeted biologics may act indirectly on the complement system (70, 71), there are currently no therapies approved or recommended in RA-ILD that target the complement system. Other prominent monocyte/macrophage cluster specific genes expressed included interferon-related genes and genes involved with inflammation (Apoe, Saa3), cell adhesion (Cadm1), lipid metabolism (Apoe, Pldbd1), as well as those with less known immune cell functions involved in differentiation and activation (Plac8, Ms4a4c, Ms4s7) (72–75). Future studies may be warranted to further explore targeting these molecules and/or related signaling pathways for the development of novel approaches.
Macrophages in RA-ILD are heterogeneous and not only initiate the inflammatory response but also contribute to repair and resolution as these cells possess high plasticity and various immune functions (69). Without the environmental stimulant (i.e., inhalant endotoxin), our animal modeling studies demonstrated that systemic arthritis induction alone (i.e., CIA) strikingly increases MHC Class II expression across all monocyte-macrophage subpopulations in the lung with less robust expression of “inflammatory” and/or pro-fibrosis markers such as Ly6C (27, 49). High MHC Class II expression implies an increased potential for antigen presentation to T cells to potentially facilitate the activation of autoreactive T cells that target self-antigens (76). Thus, this suggests that CIA (and perhaps RA) alone acts to prime the lung for an adverse inflammatory response in the wake of an environmental stimulus with this “priming” effect mediated by monocytes. The addition of inhalant endotoxin potentiated monocyte/macrophage numbers, inflammatory marker expression, and phagocytic ability, underscoring that clinical lung disease development in RA likely requires multiple factors including environmental triggers. On the converse, it is well-recognized that repetitive endotoxin exposures are associated with chronic adaptation or tolerance responses; yet the current studies may imply that arthritis induction/autoimmunity may break endotoxin tolerance. Thus, combined modeling (CIA+LPS) strikingly potentiated/activated lung macrophages as compared to one-hit alone.
Depleting the reservoir of circulating monocytes in the setting of co-exposure suggests that systemic monocytes are a critical to orchestrating disease severity and progression. Monocyte-macrophage secretory products including IL-6 that contributes to the proliferation of T cells and inflammation/fibrosis (69) as well as leukocyte chemoattractants (CCL2 and CCL7), fibronectin and vimentin, were all markedly reduced in CIA+LPS mice following depletion of the circulating monocyte pool. Furthermore, there were robust reductions in both CIT- and MAA-modified protein generation following co-exposure with CIA+LPS resulting from the depletion of circulating monocytes. This is highly relevant as CIT and MAA stimulate undifferentiated macrophages and soluble factor secretion that drive an aggressive fibroblast phenotype and extracellular matrix deposition that characterize lung fibrosis (77–79). Moreover, these modifications have been implicated in promoting tolerance loss leading to disease-related autoimmunity (54, 69, 80). Although reductions were observed herein with the expression of extracellular matrix proteins, these reductions were relatively modest, suggesting that circulating and recruited monocytes/macrophages are not the major cellular source of these wound repair factors.
There are limitations to this study. This study focused on LPS as the inhalant exposure as it is commercially available, standardized to promote reproducibility, and is frequently contained in environmental/occupational exposures that have been implicated in RA (8–14, 81–83). Other airborne hazards relevant to RA-associated lung disease such as cigarette smoke, silica, complex air pollution/particulates, and chemical toxins/gases as well as combination of these exposures will need to be explored with and without systemic arthritis induction in future work. We also did not delineate and evaluate DCs; DCs are critical antigen presenting cells that also express high MHC Class II and co-stimulatory molecules as well as can express CD11c, CD11b, and Ly6C in various states, but uniquely express CD103 (as well Irf4, Xcr1, Siglec-H) and do not express CD206, CD68, and Siglec-F (45, 63, 64, 84–86), and as such, future studies are needed to careful delineate the smaller subset of DCs. Other potential future studies could include isolating specific cell populations to investigate ex vivo cellular and functional responses and/or the adoptive transfer of cells from a diseased mouse to a naïve mouse to determine whether adverse biological responses could be reproduced. Furthermore, direct depletion of lung macrophages by lung-delivery of clodronate liposomes at various time points in this co-exposure model, as opposed to systemic depletion, could also be investigated to determine functional consequences.
In conclusion, the interaction of inhalation-induced airway inflammation and autoimmune arthritis results in lung disease associated with uniquely activated infiltrating inflammatory monocytes-macrophages that mediate adverse lung consequences in mice. Whereas the induced IM immunophenotype is more aligned to arthritis induction than endotoxin exposure, co-exposure modeling offers unique features that informs RA-ILD pathogenesis that could be leveraged in future pre-clinical work. It will also be important for future studies to translate these observations to humans to fully understand the mechanisms and impact of the unique lung macrophage subpopulations of RA-ILD.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The animal study was approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
JP: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AS: Data curation, Formal analysis, Investigation, Supervision, Visualization, Writing – original draft, Writing – review & editing, Software. GT: Conceptualization, Data curation, Formal analysis, Investigation, Resources, Software, Supervision, Visualization, Writing – review & editing. TW: Data curation, Formal analysis, Investigation, Methodology, Resources, Software, Supervision, Writing – review & editing. AN: Data curation, Formal analysis, Investigation, Methodology, Project administration, Supervision, Validation, Writing – review & editing, Software, Visualization. OS: Data curation, Formal analysis, Visualization, Writing – review & editing, Investigation, Methodology, Software. AG: Data curation, Formal analysis, Investigation, Supervision, Visualization, Writing – review & editing, Methodology, Software. MD: Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Visualization, Writing – review & editing. BE: Funding acquisition, Investigation, Methodology, Software, Supervision, Visualization, Writing – review & editing. TM: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The main funding support is from a grant by the Department of Defense, PR200793 (JAP, TRM). Other funding support includes National Institute of Occupational Safety and Health (R01OH012045; JAP and U54 OH010162; TAW), VA (BLR&D Merits I01 BX004660 and I01 BX003635, TRM, I01 BX005886 and IS1 BX004790, TAW and CSR&D CX002203 BRE), National Institute of Health (2U54GM115458, TRM), Rheumatology Research Foundation (TRM, BRE). TAW is the recipient of a Research Career Scientist Award (IK6 BX005962). ADS was supported by NIH grant 1F30ES036063-01 through the National Institute of Environmental Health Sciences (NIEHS).
Acknowledgments
The authors acknowledge Craig Semerad, PhD, Victoria B. Smith, BS, and Holly Britton, MS, in the Flow Cytometry Research Core Facility for aiding with flow cytometry studies. We also thank Babu Guda, PhD, and Sankarasubramanian Jagadesan, PhD, of the UNMC Bioinformatics and Systems Biology Core as well as James Eudy, PhD, Jennifer Bushing, MS, Alok Dhar, PhD, and Greg Kubik, BS, of the UNMC Genomics Core for assistance with the single cell RNA sequencing studies and analyses. The UNMC Flow Cytometry Research Facility is administrated through the Office of the Vice Chancellor for Research and supported by state funds from the Nebraska Research Initiative and The Fred and Pamela Buffett Cancer Center’s National Cancer Institute Cancer Support Grant (P30 CA036727). Major instrumentation has been provided by the Office of the Vice Chancellor for Research, The University of Nebraska Foundation, the Nebraska Banker’s Fund, and by the NIH-NCRR Shared Instrument Program. The University of Nebraska DNA Sequencing Core receives partial support from the National Institute for General Medical Science (NIGMS) INBRE - 5P20GM103427-21 grant as well as The Fred & Pamela Buffett Cancer Center Support Grant - 5P30CA036727-36. This publication’s contents are the sole responsibility of the authors and do not necessarily represent the official views of the NIH or NIGMS. We also thank Marie Nguyen for article preparation assistance and submission.
Conflict of interest
Author JP has received research reagents anti-IL-33/ST2 blocking antibody reagent, no monies from AstraZeneca. Author JP is a site recruiter no monies for clinical industry studies for asthma, sinus disease, and urticaria/angioedemaGlaxoSmithKline, AstraZeneca, Regeneron Pharmaceuticals, CellDex Therapeutics. Author BE has consulted with and received research support from Boehringer-Ingelheim. Author TM has consulted for Horizon Therapeutics, Otaltech Therapeutics, Pfizer, and UCB and has received research support from Horizon.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs, the Department of Defense, or the United States government.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1557583/full#supplementary-material
References
1. Erickson Ar CACMTR. Clinical features of rheumatoid arthritis. In: Budd RC GSBMIBODJR, editor. Kelley & Firestein’s textbook of rheumatology. Elsevier, Philadelphia (2017). p. 1167–86.
2. Shaw M, Collins BF, Ho LA, Raghu G. Rheumatoid arthritis-associated lung disease. Eur Respir review: an Off J Eur Respir Society. (2015) 24:1–16. doi: 10.1183/09059180.00008014
3. Olson AL, Swigris JJ, Sprunger DB, Fischer A, Fernandez-Perez ER, Solomon J, et al. Rheumatoid arthritis-interstitial lung disease-associated mortality. Am J Respir Crit Care Med. (2011) 183:372–8. doi: 10.1164/rccm.201004-0622OC
4. Kelly CA, Saravanan V, Nisar M, Arthanari S, Woodhead FA, Price-Forbes AN, et al. Rheumatoid arthritis-related interstitial lung disease: associations, prognostic factors and physiological and radiological characteristics—a large multicentre UK study. Rheumatology. (2014) 53:1676–82. doi: 10.1093/rheumatology/keu165
5. Sagawa T, Ichinose T, Honda A, Kuroda E, Ishikawa R, Miyasaka N, et al. Acceleration of acute lung inflammation by IL-1alpha released through cell death of alveolar macrophages upon phagocytosis of fine Asian sand dust particles. Environ Int. (2024) 194:109178. doi: 10.1016/j.envint.2024.109178
6. Van Kalsbeek D, Brooks R, Shaver D, Ebel A, Hershberger D, Schmidt C, et al. Peripheral blood biomarkers for rheumatoid arthritis-associated interstitial lung disease: A systematic review. ACR Open Rheumatol. (2023) 5:201–26. doi: 10.1002/acr2.11535
7. Luedders BA, Mikuls TR, Thiele GM, Poole JA, England BR. Inhalant and additional mucosal-related environmental risks for rheumatoid arthritis. Rheum Dis Clin North Am. (2022) 48:781–98. doi: 10.1016/j.rdc.2022.06.002
8. Davidson ME, Schaeffer J, Clark ML, Magzamen S, Brooks EJ, Keefe TJ, et al. Personal exposure of dairy workers to dust, endotoxin, muramic acid, ergosterol and ammonia on large-scale dairies in the high plains western United States. J Occup Environ hygiene. (2018) 15:182–93. doi: 10.1080/15459624.2017.1403610
9. Hasday JD, Bascom R, Costa JJ, Fitzgerald T, Dubin W. Bacterial endotoxin is an active component of cigarette smoke. Chest. (1999) 115:829–35. doi: 10.1378/chest.115.3.829
10. d’Errico A, Peraza S, Weiss I, Martinez W, Monge EA, Wouters IM, et al. Occupational exposure to respirable and inhalable dust and its components in a Nicaraguan sugarcane plantation. Occup Environ Med. (2025) 82:36–43. doi: 10.1136/oemed-2024-109604
11. Van De Graaff J, Poole JA. A clinician’s guide to occupational exposures in the military. Curr Allergy Asthma Rep. (2022) 22:259–64. doi: 10.1007/s11882-022-01051-0
12. Sun Y, Kang J, Zhang FY, Wang H, Lai PS, Washko GR, et al. Characterization of occupational endotoxin-related small airway disease with longitudinal paired inspiratory/expiratory CT scans. Chest. (2025). doi: 10.1016/j.chest.2024.12.037
13. Grant TL, Wood RA. The influence of urban exposures and residence on childhood asthma. Pediatr Allergy Immunol. (2022) 33:e13784. doi: 10.1111/pai.13784
14. Murphy D, Hutchinson D. Is male rheumatoid arthritis an occupational disease? A review. Open Rheumatol J. (2017) 11:88–105. doi: 10.2174/1874312901711010088
15. Gilligan D, O’Connor C, Ward K, Moloney D, Bresnihan B, FitzGerald M. Bronchoalveolar lavage in patients with mild and severe rheumatoid lung disease. Thorax. (1990) 45:591–6. doi: 10.1136/thx.45.8.591
16. Saku A, Fujisawa T, Nishimoto K, Yoshimura K, Hozumi H, Karayama M, et al. Prognostic significance of peripheral blood monocyte and neutrophil counts in rheumatoid arthritis-associated interstitial lung disease. Respir Med. (2021) 182:106420. doi: 10.1016/j.rmed.2021.106420
17. Poole JA, Cole KE, Thiele GM, Talmadge JE, England BR, Nelson AJ, et al. Expansion of distinct peripheral blood myeloid cell subpopulations in patients with rheumatoid arthritis-associated interstitial lung disease. Int Immunopharmacol. (2024) 127:111330. doi: 10.1016/j.intimp.2023.111330
18. Poole JA, Schwab A, Thiele GM, England BR, Nelson AJ, Gleason A, et al. Unique transcriptomic profile of peripheral blood monocytes in rheumatoid arthritis-associated interstitial lung disease. Rheumatol (Oxford). (2024). doi: 10.1093/rheumatology/keae572
19. Poole JA, Thiele GM, Ramler E, Nelson AJ, Duryee MJ, Schwab AD, et al. Combined repetitive inhalant endotoxin and collagen-induced arthritis drive inflammatory lung disease and arthritis severity in a testosterone-dependent manner. Am J Physiol Lung Cell Mol Physiol. (2024) 326:L239–L51. doi: 10.1152/ajplung.00221.2023
20. Mikuls TR, Gaurav R, Thiele GM, England BR, Wolfe MG, Shaw BP, et al. The impact of airborne endotoxin exposure on rheumatoid arthritis-related joint damage, autoantigen expression, autoimmunity, and lung disease. Int Immunopharmacol. (2021) 100:108069. doi: 10.1016/j.intimp.2021.108069
21. Poole JA, Mikuls TR, Thiele GM, Gaurav R, Nelson AJ, Duryee MJ, et al. Increased susceptibility to organic dust exposure-induced inflammatory lung disease with enhanced rheumatoid arthritis-associated autoantigen expression in HLA-DR4 transgenic mice. Respir Res. (2022) 23:160. doi: 10.1186/s12931-022-02085-8
22. Poole JA, Thiele GM, Janike K, Nelson AJ, Duryee MJ, Rentfro K, et al. Combined collagen-induced arthritis and organic dust-induced airway inflammation to model inflammatory lung disease in rheumatoid arthritis. J Bone Miner Res. (2019) 34:1733–43. doi: 10.1002/jbmr.3745
23. Laskin DL, Malaviya R, Laskin JD. Role of macrophages in acute lung injury and chronic fibrosis induced by pulmonary toxicants. Toxicological sciences: an Off J Soc Toxicology. (2019) 168:287–301. doi: 10.1093/toxsci/kfy309
24. Puttur F, Gregory LG, Lloyd CM. Airway macrophages as the guardians of tissue repair in the lung. Immunol Cell Biol. (2019) 97:246–57. doi: 10.1111/imcb.2019.97.issue-3
25. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. (2017) 214:2387–404. doi: 10.1084/jem.20162152
26. Li YH, Zhang Y, Pan G, Xiang LX, Luo DC, Shao JZ. Occurrences and functions of ly6C(hi) and ly6C(lo) macrophages in health and disease. Front Immunol. (2022) 13:901672. doi: 10.3389/fimmu.2022.901672
27. Draijer C, Florez-Sampedro L, Reker-Smit C, Post E, van Dijk F, Melgert BN. Explaining the polarized macrophage pool during murine allergic lung inflammation. Front Immunol. (2022) 13:1056477. doi: 10.3389/fimmu.2022.1056477
28. Rodero MP, Poupel L, Loyher PL, Hamon P, Licata F, Pessel C, et al. Immune surveillance of the lung by migrating tissue monocytes. Elife. (2015) 4:e07847. doi: 10.7554/eLife.07847
29. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
30. Jenkins SJ, Hume DA. Homeostasis in the mononuclear phagocyte system. Trends Immunol. (2014) 35:358–67. doi: 10.1016/j.it.2014.06.006
31. Schwab AD, Wyatt TA, Moravec G, Thiele GM, Nelson AJ, Gleason A, et al. Targeting transitioning lung monocytes/macrophages as treatment strategies in lung disease related to environmental exposures. Respir Res. (2024) 25:157. doi: 10.1186/s12931-024-02804-3
32. Tatham KC, O’Dea KP, Romano R, Donaldson HE, Wakabayashi K, Patel BV, et al. Intravascular donor monocytes play a central role in lung transplant ischaemia-reperfusion injury. Thorax. (2018) 73:350–60. doi: 10.1136/thoraxjnl-2016-208977
33. Nguyen T, Du J, Li YC. A protocol for macrophage depletion and reconstitution in a mouse model of sepsis. STAR Protoc. (2021) 2:101004. doi: 10.1016/j.xpro.2021.101004
34. Bauer CD, Mosley DD, Samuelson DR, Poole JA, Smith DR, Knoell DL, et al. Zinc protects against swine barn dust-induced cilia slowing. Biomolecules. (2024) 14:843. doi: 10.3390/biom14070843
35. Klein S, Staring M, Murphy K, Viergever MA, Pluim JP. elastix: a toolbox for intensity-based medical image registration. IEEE Trans Med Imaging. (2010) 29:196–205. doi: 10.1109/TMI.2009.2035616
36. De Langhe E, Vande Velde G, Hostens J, Himmelreich U, Nemery B, Luyten FP, et al. Quantification of lung fibrosis and emphysema in mice using automated micro-computed tomography. PloS One. (2012) 7:e43123. doi: 10.1371/journal.pone.0043123
37. Redente EF, Jacobsen KM, Solomon JJ, Lara AR, Faubel S, Keith RC, et al. Age and sex dimorphisms contribute to the severity of bleomycin-induced lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol. (2011) 301:L510–8. doi: 10.1152/ajplung.00122.2011
38. Dekoster K, Decaesteker T, Berghen N, Van den Broucke S, Jonckheere AC, Wouters J, et al. Longitudinal micro-computed tomography-derived biomarkers quantify non-resolving lung fibrosis in a silicosis mouse model. Sci Rep. (2020) 10:16181. doi: 10.1038/s41598-020-73056-6
39. Singh N, Arora N. Diesel exhaust exposure in mice induces pulmonary fibrosis by TGF-beta/Smad3 signaling pathway. Sci Total Environ. (2022) 807:150623. doi: 10.1016/j.scitotenv.2021.150623
40. Mecozzi L, Mambrini M, Ruscitti F, Ferrini E, Ciccimarra R, Ravanetti F, et al. In-vivo lung fibrosis staging in a bleomycin-mouse model: a new micro-CT guided densitometric approach. Sci Rep. (2020) 10:18735. doi: 10.1038/s41598-020-71293-3
41. Buccardi M, Ferrini E, Pennati F, Vincenzi E, Ledda RE, Grandi A, et al. A fully automated micro−CT deep learning approach for precision preclinical investigation of lung fibrosis progression and response to therapy. Respir Res. (2023) 24:126. doi: 10.1186/s12931-023-02432-3
42. Johnson AN, Harkema JR, Nelson AJ, Dickinson JD, Kalil J, Duryee MJ, et al. MyD88 regulates a prolonged adaptation response to environmental dust exposure-induced lung disease. Respir Res. (2020) 21:97. doi: 10.1186/s12931-020-01362-8
43. Gaurav R, Mikuls TR, Thiele GM, Nelson AJ, Niu M, Guda C, et al. High-throughput analysis of lung immune cells in a combined murine model of agriculture dust-triggered airway inflammation with rheumatoid arthritis. PloS One. (2021) 16:e0240707. doi: 10.1371/journal.pone.0240707
44. Schwab AD, Nelson AJ, Gleason AM, Schanze OW, Wyatt TA, Shinde DD, et al. Aconitate decarboxylase 1 mediates the acute airway inflammatory response to environmental exposures. Front Immunol. (2024) 15:1432334. doi: 10.3389/fimmu.2024.1432334
45. Li X, Mara AB, Musial SC, Kolling FW, Gibbings SL, Gerebtsov N, et al. Coordinated chemokine expression defines macrophage subsets across tissues. Nat Immunol. (2024) 25:1110–22. doi: 10.1038/s41590-024-01826-9
46. Starke N, Challa NVD, Yuan H, Chen S, Duncan MR, Cabrera Ranaldi E, et al. Extracellular vesicle ASC: A novel mediator for lung-brain axis in preterm brain injury. Am J Respir Cell Mol Biol. (2024) 71:464–80. doi: 10.1165/rcmb.2023-0402OC
47. Dovhyi R, Dvukhriadkina A, Ostrovska K, Rudyk M, Verhovcova I, Vaivode K, et al. Bacteriophage derived dsRNA induces polarized activation of alveolar macrophages from Balb/c and C57Bl/6 mice in vitro in sex- and age-dependent manner. Cell Immunol. (2025) 408:104916. doi: 10.1016/j.cellimm.2025.104916
48. Yao Y, Jeyanathan M, Haddadi S, Barra NG, Vaseghi-Shanjani M, Damjanovic D, et al. Induction of autonomous memory alveolar macrophages requires T cell help and is critical to trained immunity. Cell. (2018) 175:1634–50 e17. doi: 10.1016/j.cell.2018.09.042
49. Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med. (2011) 184:569–81. doi: 10.1164/rccm.201010-1719OC
51. Tantawy MA, Hatesuer B, Wilk E, Dengler L, Kasnitz N, Weiß S, et al. The interferon-induced gene Ifi27l2a is active in lung macrophages and lymphocytes after influenza A infection but deletion of Ifi27l2a in mice does not increase susceptibility to infection. PloS One. (2014) 9:e106392. doi: 10.1371/journal.pone.0106392
52. Okabe Y, Medzhitov R. Tissue biology perspective on macrophages. Nat Immunol. (2016) 17:9–17. doi: 10.1038/ni.3320
53. Oliveira VLS, Pollenus E, Berghmans N, Queiroz-Junior CM, Blanter M, Mattos MS, et al. Absence of CCR2 promotes proliferation of alveolar macrophages that control lung inflammation in acute respiratory distress syndrome in mice. Int J Mol Sci. (2022) 23:12920. doi: 10.3390/ijms232112920
54. England BR, Duryee MJ, Roul P, Mahajan TD, Singh N, Poole JA, et al. Malondialdehyde-acetaldehyde adducts and antibody responses in rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheumatol. (2019) 71:1483–93. doi: 10.1002/art.40900
55. Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. (2001) 125:279–84. doi: 10.1016/S0166-4328(01)00297-2
56. Cheminant JR, Deering-Rice CE, Massa CB, Adhikari U, Noll J, Reilly CA, et al. Parenchymal and inflammatory responses to ozone exposure in the aging healthy and surfactant protein C mutant lung. Am J Physiol Lung Cell Mol Physiol. (2025) 328:L334–L49. doi: 10.1152/ajplung.00261.2024
57. Gonzalez-Juarrero M, Shim TS, Kipnis A, Junqueira-Kipnis AP, Orme IM. Dynamics of macrophage cell populations during murine pulmonary tuberculosis. J Immunol. (2003) 171:3128–35. doi: 10.4049/jimmunol.171.6.3128
58. Porrello A, Leslie PL, Harrison EB, Gorentla BK, Kattula S, Ghosh SK, et al. Factor XIIIA-expressing inflammatory monocytes promote lung squamous cancer through fibrin cross-linking. Nat Commun. (2018) 9:1988. doi: 10.1038/s41467-018-04355-w
59. Ning DS, Zhou ZQ, Zhou SH, Chen JM. Identification of macrophage differentiation related genes and subtypes linking atherosclerosis plaque processing and metabolic syndrome via integrated bulk and single-cell sequence analysis. Heliyon. (2024) 10:e34295. doi: 10.1016/j.heliyon.2024.e34295
60. Milger K, Yu Y, Brudy E, Irmler M, Skapenko A, Mayinger M, et al. Pulmonary CCR2(+)CD4(+) T cells are immune regulatory and attenuate lung fibrosis development. Thorax. (2017) 72:1007–20. doi: 10.1136/thoraxjnl-2016-208423
61. Yang S, Yu J, Dong X, Zeng J, Tan L, Zhang H, et al. CCR2 signaling regulates anti-chlamydia T cell immune responses in the airway. PloS Pathog. (2025) 21:e1012912. doi: 10.1371/journal.ppat.1012912
62. Xie X, Shi Q, Wu P, Zhang X, Kambara H, Su J, et al. Single-cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat Immunol. (2020) 21:1119–33. doi: 10.1038/s41590-020-0736-z
63. Jirmo AC, Grychtol R, Gaedcke S, Liu B, DeStefano S, Happle C, et al. Single cell RNA sequencing reveals distinct clusters of Irf8-expressing pulmonary conventional dendritic cells. Front Immunol. (2023) 14:1127485. doi: 10.3389/fimmu.2023.1127485
64. Takagi H, Fukaya T, Eizumi K, Sato Y, Sato K, Shibazaki A, et al. Plasmacytoid dendritic cells are crucial for the initiation of inflammation and T cell immunity in vivo. Immunity. (2011) 35:958–71. doi: 10.1016/j.immuni.2011.10.014
65. Waddell LA, Lefevre L, Bush SJ, Raper A, Young R, Lisowski ZM, et al. ADGRE1 (EMR1, F4/80) is a rapidly-evolving gene expressed in mammalian monocyte-macrophages. Front Immunol. (2018) 9:2246. doi: 10.3389/fimmu.2018.02246
66. Fulton SA, Reba SM, Pai RK, Pennini M, Torres M, Harding CV, et al. Inhibition of major histocompatibility complex II expression and antigen processing in murine alveolar macrophages by Mycobacterium bovis BCG and the 19-kilodalton mycobacterial lipoprotein. Infect Immun. (2004) 72:2101–10. doi: 10.1128/IAI.72.4.2101-2110.2004
67. England BR, Sayles H, Michaud K, Thiele GM, Poole JA, Caplan L, et al. Chronic lung disease in U.S. Veterans with rheumatoid arthritis and the impact on survival. Clin Rheumatol. (2018) 37:2907–15. doi: 10.1007/s10067-018-4314-9
68. England BR, Sayles H, Michaud K, Caplan L, Davis LA, Cannon GW, et al. Cause-specific mortality in male US veterans with rheumatoid arthritis. Arthritis Care Res. (2016) 68:36–45. doi: 10.1002/acr.22642
69. Anton ML, Cardoneanu A, Burlui AM, Mihai IR, Richter P, Bratoiu I, et al. The lung in rheumatoid arthritis-friend or enemy? Int J Mol Sci. (2024) 25:6460. doi: 10.3390/ijms25126460
70. Triggianese P, Conigliaro P, De Martino E, Monosi B, Chimenti MS. Overview on the link between the complement system and auto-immune articular and pulmonary disease. Open Access Rheumatol. (2023) 15:65–79. doi: 10.2147/OARRR.S318826
71. Holers VM, Banda NK. Complement in the initiation and evolution of rheumatoid arthritis. Front Immunol. (2018) 9:1057. doi: 10.3389/fimmu.2018.01057
72. Thompson JC, Wilson PG, Shridas P, Ji A, de Beer M, de Beer FC, et al. Serum amyloid A3 is pro-atherogenic. Atherosclerosis. (2018) 268:32–5. doi: 10.1016/j.atherosclerosis.2017.11.011
73. Saito-Sasaki N, Sawada Y, Okada E, Nakamura M. Cell adhesion molecule 1 (CADM1) is an independent prognostic factor in patients with cutaneous squamous cell carcinoma. Diagnostics (Basel). (2021) 11:830. doi: 10.3390/diagnostics11050830
74. Jia Y, Ying X, Zhou J, Chen Y, Luo X, Xie S, et al. The novel KLF4/PLAC8 signaling pathway regulates lung cancer growth. Cell Death Dis. (2018) 9:603. doi: 10.1038/s41419-018-0580-3
75. Mattiola I, Tomay F, De Pizzol M, Silva-Gomes R, Savino B, Gulic T, et al. The macrophage tetraspan MS4A4A enhances dectin-1-dependent NK cell-mediated resistance to metastasis. Nat Immunol. (2019) 20:1012–22. doi: 10.1038/s41590-019-0417-y
76. Tsai S, Santamaria P. MHC class II polymorphisms, autoreactive T-cells, and autoimmunity. Front Immunol. (2013) 4:321. doi: 10.3389/fimmu.2013.00321
77. Aripova N, Duryee MJ, Zhou W, England BR, Hunter CD, Klingemann LE, et al. Citrullinated and malondialdehyde-acetaldehyde modified fibrinogen activates macrophages and promotes profibrotic responses in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. (2024). doi: 10.1152/ajplung.00153.2024
78. Aripova N, Duryee MJ, England BR, Hunter CD, Mordeson JE, Ryan EM, et al. Citrullinated and malondialdehyde-acetaldehyde modified fibrinogen activates macrophages and promotes an aggressive synovial fibroblast phenotype in patients with rheumatoid arthritis. Front Immunol. (2023) 14:1203548. doi: 10.3389/fimmu.2023.1203548
79. Aripova N, Duryee MJ, Hunter CD, Ryan EM, Daubach EC, Jones SQ, et al. Peptidyl arginine deiminase expression and macrophage polarization following stimulation with citrullinated and malondialdehyde-acetaldehyde modified fibrinogen. Int Immunopharmacol. (2022) 110:109010. doi: 10.1016/j.intimp.2022.109010
80. Matson S, Lee J, Eickelberg O. Two sides of the same coin? A review of the similarities and differences between idiopathic pulmonary fibrosis and rheumatoid arthritis-associated interstitial lung disease. Eur Respir J. (2021) 57:2002533. doi: 10.1183/13993003.02533-2020
81. Stolt P, Yahya A, Bengtsson C, Kallberg H, Ronnelid J, Lundberg I, et al. Silica exposure among male current smokers is associated with a high risk of developing ACPA-positive rheumatoid arthritis. Ann Rheumatic Diseases. (2010) 69:1072–6. doi: 10.1136/ard.2009.114694
82. Ebel AV, Lutt G, Poole JA, Thiele GM, Baker JF, Cannon GW, et al. Association of agricultural, occupational, and military inhalants with autoantibodies and disease features in US veterans with rheumatoid arthritis. Arthritis Rheumatol. (2021) 73:392–400. doi: 10.1002/art.41559
83. Too CL, Muhamad NA, Ilar A, Padyukov L, Alfredsson L, Klareskog L, et al. Occupational exposure to textile dust increases the risk of rheumatoid arthritis: results from a Malaysian population-based case-control study. Ann Rheumatic Diseases. (2016) 75:997–1002. doi: 10.1136/annrheumdis-2015-208278
84. Izumi G, Nakano H, Nakano K, Whitehead GS, Grimm SA, Fessler MB, et al. CD11b(+) lung dendritic cells at different stages of maturation induce Th17 or Th2 differentiation. Nat Commun. (2021) 12:5029. doi: 10.1038/s41467-021-25307-x
85. Carter H, Costa RM, Adams TS, Gilchrist TM, Emch CE, Bame M, et al. CD103+ dendritic cell - fibroblast crosstalk via TLR9, TDO2, and AHR signaling drives lung fibrogenesis. JCI Insight. (2025). doi: 10.1172/jci.insight.177072
Keywords: lung, autoimmunity, macrophage, monocyte, inflammation, fibrosis, transcriptome, imaging
Citation: Poole JA, Schwab A, Thiele GM, Wyatt TA, Nelson AJ, Schanze OW, Gleason A, Duryee MJ, England BR and Mikuls TR (2025) Lung disease in relation to unique monocyte-macrophage subpopulations induced by combined inhalant endotoxin and collagen-induced arthritis. Front. Immunol. 16:1557583. doi: 10.3389/fimmu.2025.1557583
Received: 08 January 2025; Accepted: 11 March 2025;
Published: 09 April 2025.
Edited by:
Thiago Almeida Pereira, Stanford University, United StatesReviewed by:
Junyi Wang, Chengdu Third People’s Hospital, ChinaQi Li, Capital Medical University, China
Copyright © 2025 Poole, Schwab, Thiele, Wyatt, Nelson, Schanze, Gleason, Duryee, England and Mikuls. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jill A. Poole, amFwb29sZUB1bm1jLmVkdQ==