 Rachel Darley1
Rachel Darley1 Patricia T. Illing2Patrick Duriez3
Patricia T. Illing2Patrick Duriez3 Alistair Bailey1
Alistair Bailey1 Anthony W. Purcell2
Anthony W. Purcell2 Andy van Hateren1†
Andy van Hateren1† Tim Elliott4*†
Tim Elliott4*†- 1Institute for Life Sciences and Centre for Cancer Immunology, Faculty of Medicine, University of Southampton, Southampton, United Kingdom
- 2Department of Biochemistry and Molecular Biology and Biomedicine Discovery Institute, Monash University, Melbourne, VIC, Australia
- 3Cancer Research UK Protein Core Facility, Faculty of Medicine, University of Southampton, Southampton, United Kingdom
- 4Centre for Immuno-Oncology and Chinese Academy of Medical Sciences (CAMS)-Oxford Institute, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom
Major Histocompatibility Complex class I (MHC-I) molecules bind and present peptides to cytotoxic T cells, protecting against pathogens and cancer. MHC-I is highly polymorphic and each allotype is promiscuous, and capable of binding a unique and diverse repertoire of peptide ligands. The peptide editing chaperone tapasin optimizes this allotype specific repertoire of peptides, resulting in the selection of high affinity peptides. MHC-I allotypes differ in the extent they engage tapasin. This suggests that tapasin-dependent MHC-I allotypes should present a less diverse repertoire that is enriched in higher-affinity peptides, and which are present in higher abundance, than tapasin independent MHC-I allotypes, which should present a broader repertoire containing peptides with a lower average affinity. Experimental verification of this hypothesis has been confounded by the different peptide binding specificities of MHC-I allotypes. Here, we independently investigated the peptide focusing function of tapasin by introducing a point mutation into a tapasin independent MHC-I allotype that dramatically increased its tapasin dependence without substantially altering its peptide binding specificity. This allowed us to demonstrate ligand focusing by tapasin at both the repertoire level in cellulo, and by using an in vitro system in which tapasin was artificially tethered to MHC-I, at the individual peptide level. We found that tapasin had a greater influence on tapasin dependent MHC-I molecules, and that tapasin modulated peptide selection according to peptide-MHC-I complex stability, disfavoring short-lived peptide-MHC-I complexes. Thus, tapasin dependent MHC-I molecules experience greater tapasin filtering, resulting in less diverse MHC-I immunopeptidomes that are enriched in high affinity peptide-MHC-I complexes.
1 Introduction
Major Histocompatibility Complex class I (MHC-I) molecules are an important component of the adaptive immune response and provide protection from pathogens and cancer by binding intracellular peptides and presenting them at the cell surface to specialized immune cells including cytotoxic T cells. The peptides presented by MHC-I molecules (the MHC-I immunopeptidome) are predominantly selected from a diverse pool of peptides transported into the endoplasmic reticulum following proteasome mediated degradation of intracellular proteins and defective nascent polypeptides, which can be further refined by aminopeptidases within the ER.
MHC-I peptide selection is orchestrated by peptide loading complexes (PLC), which are centered upon TAP peptide transporters and include a number of chaperones: tapasin; calreticulin; and ERp57, and which synergistically co-ordinate recruitment of nascent MHC-I molecules and their peptide loading (1). Of the PLC constituents, tapasin assists MHC-I to preferentially select high affinity peptides for presentation, forming stable, long-lived peptide-MHC-I complexes (2–5). Once loaded with peptide, MHC-I complexes are released from the PLC and exit the ER, where some MHC-I allotypes encounter further scrutiny from the tapasin homologue TAPBPR (6). Like tapasin, TAPBPR refines the peptides presented by MHC-I, preferentially favoring high affinity peptides (7, 8). Empty or sub-optimally loaded MHC-I (i.e. those MHC-I molecules containing low affinity binding peptides) may also be returned to the ER following retrieval from the endoplasmic-reticulum–Golgi intermediate compartment in a calreticulin dependent manner (9–11).
MHC-I molecules are highly polymorphic, with MHC-I allotypes binding different repertoires of peptides depending on the molecular composition of their peptide-binding grooves (12, 13). In addition, MHC-I allotypes differ in their dependence upon tapasin for the selection of peptide cargoes that permit stable cell surface expression (2, 14–16). While all MHC-I allotypes benefit from tapasin to some extent, this ranges from allotypes such as HLA-B*44:02 for which tapasin is essentially obligatory for peptide selection and cell surface expression, to allotypes such as HLA-B*44:05 which can efficiently select and present a repertoire of peptides at the cell surface in the absence of tapasin (2, 14, 15, 17). Additionally, TAPBPR has specificity for a select group of MHC-I allotypes, with a strong preference for some HLA-A gene products (6, 18, 19).
Tapasin and TAPBPR mediated peptide editing therefore underpins the diversity of the MHC-I immunopeptidome that is presented to T cells and is an important factor in determining the breadth of an immune response and protection against potentially lethal infections and cancer (14, 20, 21). Experimental and computational studies have suggested that the diversity of peptides presented by different human MHC-I allotypes varies (22–26). In humans, the cell surface expression levels of four MHC-I allotypes was shown to be inversely correlated with the breadth of their immunopeptidomes (24, 25, 27). Interestingly, Chappell et al. noted that the cell surface expression levels of these allotypes correlated with their tapasin dependence (15, 27). Indeed, the tapasin dependence of a wide variety of HLA-A and HLA-B allotypes was measured and shown to be inversely correlated with the number of peptides derived from HIV that elicited an immunogenic response (14). Collectively, these studies suggest that tapasin dependent MHC-I allotypes may present a less diverse range of peptides, which is enriched in high affinity peptides and present at higher surface expression levels, compared with tapasin independent MHC-I allotypes, and that these factors are important determinants for successful immune responses. Similarly, a more diverse repertoire of peptides was identified in TAPBPR depleted cells compared with TAPBPR expressing cells, suggesting that TAPBPR restricts the diversity of peptides presented by some MHC-I molecules (7). However, tapasin or TAPBPR mediated focusing of MHC-I peptide repertoires has not been formally demonstrated, mostly because of the difficulty of distinguishing between whether the differential peptide focusing experienced by MHC-I allotypes is a consequence of variation in their ability to bind tapasin, or TAPBPR, and exploit their peptide editing potential, or because of differences in peptide selectivity imposed by the composition of the peptide binding grooves.
We recently demonstrated the relationship between tapasin activity and repertoire focusing by tuning the level of tapasin expression and measuring changes in peptide editing intensity (28). Here, we build upon these findings by measuring the influence that tapasin dependence has on the composition of immunopeptidomes selected by highly similar human MHC-I molecules. We next characterized how tapasin modulates MHC-I peptide selection and the magnitude of tapasin optimization experienced by MHC-I allotypes using an in vitro system.
2 Materials and methods
2.1 Analysis of HLA-B*44:02, HLA-B*44:05 and HLA-B*44:05-W147A immunopeptidomes
Plasmids encoding full length HLA-B*44:02, HLA-B*44:05 and HLA-B*44:05-W147A proteins, and the generation of stable transfectants of 721.220 cells reconstituted with human tapasin have been described previously (29). Approximately 5x108 cells were grown for each cell line, and cell pellets were snap frozen. Cell lysis and MHC-I peptide isolation were performed as previously described in detail (30). Briefly, cell pellets were lysed by cryomilling and incubation in a non-denaturing lysis buffer (0.5% Igepal CA-630, 50 mM Tris pH 8.0, 150 mM NaCl, 1 x Roche Complete protease inhibitor cocktail). MHC-I complexes were captured from the lysate using 7.5 mg of anti-MHC-I antibody, W6/32, immobilized on protein A Sepharose and subsequently dissociated with 10% acetic acid. The peptides were fractionated and separated from β2m and heavy chain components by reversed-phase high performance liquid chromatography (RP-HPLC). The peptide containing fractions were concentrated and chromatographically distant fractions were combined to generate nine pools, reconstituted in 15 µL 0.1% formic acid containing 500 fmol iRT reference peptides (31). Pools were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using a SCIEX 5600+ mass spectrometer, equipped with a NanoUltra cHiPLC system (Eksigent) and a Nanospray III ion source. Peptides were trapped on a cHiPLC trap column (3 µm, ChromXP C18CL, 120 Å, 0.5 mm x 200 µm), by loading at 5 µL/min in 0.1% formic acid, 2% acetonitrile, prior to elution over a cHiPLC column (3 µm, ChromXP C18CL, 120 Å, 15 cm x 75 µm) at 300 nL/min with increasing acetonitrile over 75 minutes. The mass spectrometer was operated as follows: ion spray voltage 2400 V, curtain gas 25 l/min, ion source gas 20 l/min, interface heater temperature 150°C, MS1 range 200-1800, MS1 accumulation time 200 ms, MS2 range 60-1800, and MS2 accumulation time 150 ms. The top 20 ions meeting the following criteria were selected for MS/MS fragmentation with rolling collision energy: >200 Da, charge state +2 to +5, >40 cps, dynamic exclusion 30 seconds after two occurrences. Mass spectra were interpreted by database search against the Uniprot/SwissProt (32) reviewed human proteome accessed October 2018 using PEAKS Studio XPRO (10.6 build, Bioinformatics Solutions Inc.) and a contaminant database contain the iRT reference peptide sequences using the following parameters: Instrument Triple TOF, fragmentation CID, parent mass error tolerance 25ppm, fragment mass error tolerance 0.1Da, enzyme none, digest mode unspecific, variable modifications Oxidation (M) +15.99 and Deamidation (NQ) +0.98, max variable modifications per peptide 3, False discovery rate (FDR) estimation enabled. A 5% peptide FDR was applied. Peptides identified from similar isolations of MHC class II from closely related 721.221 derived cell lines were excluded from downstream analyses. Analyses were performed using peptides of 8–13 amino acids, non-redundant by sequence (i.e. modifications were not considered), consistent with MHC-I ligands.
Peptide motifs were assigned via MixMHCp (v2.1, ref (33)) with two motifs specified for HLA-B*44:02 as described in Ref (34). Peptide affinities were predicted using NetMHCpan 4.1 (35), and were represented in nM values, with HLA-B*44:05 specified as the reference HLA supertype for HLA-B*44:05-W147A. Gibbs clustering was performed using Gibbs Cluster 2.0 using pre-set parameters for MHC class I ligands of length 8-13 (36).
2.2 Data availability
Mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE (37) partner repository under the accession code PXD054743.
2.3 Synthetic peptides
The following peptides were used in in vitro peptide competition experiments:
HLA-B*44:05 and HLA-B*44:05-W147A: SEIKETNDTW, AETYVEGQRI, YEGQFKDNMF, AEDELAMRGF, EEVEQGVKF, SEIVGSIKM, EEGPSSVRF, SEDEGNLRF, SEMKVSSTW, SEMKVSSTWL, QEMPWNVRM, HESGASIKI, AENEELHQLW, TEVLSNVKF, YEHEKDLVW, DENPQQLKL, KEQNYSDDVL, LESDSFLKF, AELESVLSHL, LEAEADKIGL, AEFAKLVEEF, AELFMEQQHL, AETEGILQKL, EEFGKAFSF. Apart from EEFGKAFSF, the HLA-B*44:05 and HLA-B*44:05-W147A peptides used in the peptide competition experiments were selected from the analysis of the immunopeptidomes or from the Immune Epitope Database (38).
HLA-B*35:01 and HLA-B*35:03: YPLHEQHGM, APLHEQHGM, YALHEQHGM, YPAHEQHGM, YPLKEQHGM, YPLHAQHGM, YPLHEAHGM, YPLHEQAGM, YPLHEQHAM, YPLHEQHGA, LPSSADVEF, LPSKADVEF, FPSDSWCYF, FPSDSWAYF, QFADVIVLF, FPFKYAAAF, KPIVVLHGY, LPPLDITPY, LPPAWQPFL, TPERMAEAGF, CPTENEPDL, CPTENEPDY, EPDLAQCFF, EPDLAQCFY. The HLA-B*35:01 and HLA-B*35:03 peptides used in the peptide competition experiments were selected from references (39–46).
HLA-A*02:01: YLENGKETL, YLVAEKVTV, KLWEAESKL, KLVKEVIAV, GLDDIKDLKV, FLLAEDTKV, SLLENLEKI, FLFEPVVKA, YVVPFVAKV, FLPSDCFPSV, NLVPMVATV, NAVPMVATV, IYSYMDDLYV, IYQYMDDLYV, ICQYMDDLYV, YQYMDDLYV, VLIGPTPVNII, VLVGPTPVNI, VLVGPTPINI, VLIGPTPVNI. The HLA-A*02:01 peptides used in the peptide competition experiments were selected from references (7, 47).
The fluorescent tetramethylrhodamine (TAMRA) labelled peptides: EEFGKTAMRAAFSF and YPLKTAMRAEQHGM, where KTAMRA denotes TAMRA labelled lysine and the unlabeled competing peptides were synthesized by Syn Peptides (Shanghai, China). The following UV-labile conditional peptide ligands were utilized: SEIDTVAjY, KILGFVFjV, KPIVVLjGY and LPSSADjEF, where j represents 3-amino-3-(2-nitro)phenyl-propionic acid. The UV conditional peptides and the TAMRA labelled FLPSDCTAMRAFPSV peptide, where the side chain of cysteine was labelled with 5-TAMRA-maleimide, were synthesized by Peptide Synthetics (Fareham UK). All peptides were reconstituted in dimethyl sulfoxide (DMSO).
2.4 Production of peptide-receptive MHC-I molecules
Plasmids encoding human β2-microglobulin and HLA-A*02:01-fos have been described previously (7). Nucleotides encoding amino acids 1 to 275 of the mature HLA-B*44:05, HLA-B*44:05-W147A, HLA-B*35:01 and HLA-B*35:03 allotypes were amplified with primers 5’-ATACATATGGGCTCCCACTCCATGA-3’ and 5’-GGAACCTCCCTCCCATCTCAGGGTGAG-3’ from DNA encoding HLA-B*44:05 (2), HLA-B*44:05 W147A (29), HLA-B*35:01 and HLA-B*35:03 (originally kindly supplied by Prof. Raghavan and described in Ref (15), and which had been subsequently sub-cloned into pCDNA3.1). Nucleotides encoding the fos leucine zipper were amplified with primers 5’-AGATGGGAGGGAGGTTCC-3’ and 5’-CGCAAGCTTTTAATGGGC-3’ from DNA encoding HLA-A*02:01-fos. The purified products from the MHC-I and fos PCR reactions were used in a third reaction using primers 5’-ATACATATGGGCTCCCACTCCATGA-3’ and 5’-CGCAAGCTTTTAATGGGC-3’ to create constructs encoding the MHC-I-fos allotypes. Following agarose gel electrophoresis and digestion of the purified products with restriction enzymes the MHC-I-fos sequences were cloned into pET22b (Invitrogen). The fos leucine zipper sequences were removed from HLA-B*44:05-fos and HLA-B*44:05-W147A-fos by amplifying with primers 5’-ATACATATGGGCTCCCACTCCATGA-3’ and 5’-GCCAAGCTTCTACTCCCATCTCAGG-3’ to create HLA-B*44:05 and HLA-B*44:05-W147A constructs without the fos leucine zipper. Following agarose gel electrophoresis and digestion of the purified products with restriction enzymes the HLA-B*44:05 and HLA-B*44:05-W147A sequences were cloned into pET22b (Invitrogen).
Peptide loaded MHC-I complexes were obtained by combining solubilized heavy chain inclusion bodies with solubilized human β2m inclusion bodies and the appropriate UV labile peptide: HLA-B*44:05, HLA-B*44:05-fos, HLA-B*44:05-W147A and HLA-B*44:05-W147A-fos: SEIDTVAjY; HLA-B*35:01-fos: KPIVVLjGY; HLA-B*35:03-fos: LPSSADjEF; HLA-A*02:01-fos: KILGFVFjV in 8 M urea, 50 mM MES pH 6.5, 0.1 mM EDTA. Refolding was initiated by 14-fold dilution with cold 100 mM Tris pH 8, 2 mM EDTA, 0.4 M l-arginine hydrochloride, 5 mM reduced glutathione and 0.5 mM oxidized glutathione added over three hours whilst stirring to achieve final concentrations of 1 µM heavy chain, 2 µM β2-microglobulin and either 10 µM (HLA-A*02:01), 30 µM (HLA-B*35:03) or 40 µM (HLA-B*35:01, HLA-B*44:05 and HLA-B*44:05-W147A) peptide. Two days later, the protein mixture was concentrated and purified by size exclusion chromatography using a Superdex 200 packed 26/600 gel filtration column (Cytiva) and phosphate buffered saline.
2.5 Production of conjugated tapasin-jun-ERp57 C60A proteins and TAPBPR proteins
Plasmids encoding human tapasin-jun with a twin strep affinity purification tag, and ERp57 containing the C60A mutation (48), and the purification of tapasin-jun-ERp57 C60A conjugates, have been described before (49).
Nucleotides encoding amino acids 22 to 406 of human TAPBPR and a His6 affinity purification tag were amplified by PCR using primers 5’-AGCGCGTCTCCAATGAAGCCCCACCCAGCAGAG–3’ and 5’-AGCGCGTCTCCTCCCTTAGTGATGGTGATGGTGGTG–3’ and a plasmid encoding human TAPBPR with a His6 tag kindly supplied by Prof. Louise Boyle (50), and subcloned after the BM40 signal peptide of pDSG102 vector (IBA Life Technologies). DNA encoding human TAPBPR with His6 tag (hTAPBPR-His6) was transfected into MEXI29E cells (IBA Life Technologies) and transfectants were grown in culture for seven days. The cell suspension was centrifuged at 4,000 rpm for 60 minutes and the supernatant passed through a 0.2 µm filter. hTAPBPR-His6 protein was then purified using a 5 ml Nickel Excel column (Cytiva) and equilibration (20 mM sodium phosphate, 0.5 M NaCl, pH 7.4) and elution buffers (20 mM sodium phosphate, 0.5 M NaCL, 500 mM imidazole, pH 7.4). The eluted material was concentrated using 10 k Da spin concentrator columns (Amicon) and further purified using a Superdex 200 packed 26/600 gel filtration column (Cytiva) and 20 mM sodium phosphate, 100 mM NaCl, pH 7.2. The protein was concentrated to around 4 mg/ml, aliquoted and frozen.
2.6 In vitro peptide competition experiments
Peptide competition experiments were prepared in PBS supplemented with 0.5 mg/ml bovine serum albumin (BSA) and a final concentration of 1.67% DMSO. The indicated concentrations of MHC-I molecules were supplemented with 20x excess β2-microglobulin and exposed to 366 nm light for 20 minutes at 4°C. Twenty µl of the UV exposed proteins were added to a 96 well microplate, with each well containing 40 µl of a titration (0 - 83.33 µM) of an unlabeled peptide competitor, 2–3 nM of the appropriate TAMRA labelled peptide, and 300 nM tapasin-jun-ERp57 (HLA-B*44 and HLA-B*35 experiments), or 300 nM TAPBPR (HLA-A*02:01 experiments) or neither tapasin-jun-ERp57 or TAPBPR. Samples were prepared in duplicate and incubated overnight at 25°C.
HLA-B*44:05-fos and HLA-B*44:05-W147A-fos proteins were used at 375 nM with 2 nM EEFGK*AFSF peptide. HLA-B*44:05 and HLA-B*44:05-W147A proteins were used at 160 nM with 2 nM EEFGK*AFSF peptide. HLA-B*35:01-fos and HLA-B*35:03-fos proteins were used at 225 nM with 3 nM YPLK*EQHGM peptide. HLA-A*02:01-fos protein was used at 50 nM with 2 nM FLPSDC*FPSV peptide.
Fluorescence polarization measurements were taken using an I3x (Molecular Devices) with rhodamine detection cartridge. Binding of TAMRA-labelled peptide is reported in milli polarization units (mP) and is obtained from the equation: mP = 1000 x (S – G x P)/(S + G x P), where S and P are background subtracted fluorescence count rates (S = polarization emission filter is parallel to the excitation filter; P = polarization emission filter is perpendicular to the excitation filter and G (grating) is an instrument and assay dependent factor. IC50 values were calculated by performing non-linear regression in GraphPad Prism using the one phase decay model, with plateaus constrained to 50. Apart from the experiments involving the HLA-B*44:05 and HLA-B*44:05-W147A proteins, each peptide was tested in at least two independent experiments, with the mean IC50 values being taken from the replicate experiments.
2.7 In vitro indirect measurements of peptide-MHC-I complex half-lives
Indirect peptide dissociation experiments were conducted essentially as described in ref (51). Experiments were performed in PBS supplemented with 0.5 mg/ml BSA and a final concentration of 1.67% DMSO. The indicated concentrations of MHC-I molecules were supplemented with 20x excess β2-microglobulin and exposed to 366 nm light for 20 minutes at 4°C, before being incubated with an equimolar concentration of each of the unlabeled peptides, or no peptide (no peptide control), overnight at 25°C in a volume of 105.6 µl. The next day 48 µl was added to each well of a 96 well microplate, before 12 µl of the appropriate TAMRA labelled peptide was added to each well, and fluorescence polarization measurements were periodically taken at 25°C for ~200 hours. Samples were prepared in duplicate. Each peptide was tested in at least two independent experiments.
HLA-B*44:05-fos and HLA-B*44:05-W147A-fos proteins were used at 375 nM with 4 nM EEFGK*AFSF. HLA-B*35:01-fos and HLA-B*35:03-fos proteins were used at 225 nM with 3 nM YPLK*EQHGM peptide. HLA-A*02:01-fos protein was used at 50 nM with 2 nM FLPSDC*FPSV peptide.
Peptide-MHC-I half-lives were calculated by performing non-linear regression in GraphPad Prism using the one phase association model, with plateaus constrained to the maximum polarization that was measured in the no peptide control.
3 Results
3.1 Tapasin skews the immunopeptidome selected by tapasin dependent MHC-I molecules in favor of high affinity peptides
The HLA-B*44:02 and HLA-B*44:05 MHC-I allotypes have been frequently used to investigate tapasin function as they differ by just one amino acid residue (HLA-B*44:02: Asp116, HLA-B*44:05: Tyr116). They share similar peptide binding preferences (17, 26) and are both similarly poor substrates for TAPBPR (18), but vary drastically in tapasin dependence (2, 14, 15, 17, 29). We have found that in vivo, the W147A mutation increases the tapasin dependence of the otherwise highly tapasin independent HLA-B*44:05 molecules (Supplementary Figures 1b, c, e and ref (29)). Thus, tapasin enhances peptide loading of W147A molecules to a level that is intermediate between highly tapasin dependent HLA-B*44:02 and highly tapasin independent HLA-B*44:05 molecules. Importantly, this mutation did not substantially change peptide binding specificity as assessed by peptide stabilization assays, where five known HLA-B*44 binding peptides increased the recovery of radiolabeled W147A molecules to an approximately similar extent as HLA-B*44:02 and HLA-B*44:05 molecules (Supplementary Figure 2). Therefore, the W147A mutant allowed us to investigate the effect of tapasin on the peptide presentation profile of the HLA-B*44:05 molecule in relative isolation of differences in peptide binding specificities. We therefore sought to compare the immunopeptidomes selected by HLA-B*44:02, HLA-B*44:05 and HLA-B*44:05-W147A molecules that experience differential benefit from tapasin, using monoallelic antigen presenting cells.
Most of the peptides identified from each cell line were unique to each immunopeptidome, with only 696 peptides (around 6.8% of all peptides) identified in all three data sets (Figure 1a). There were between ~3400-~5500 peptides identified in each immunopeptidome (Figure 1b). All three MHC-I allotypes had similar ligand length distributions, with peptides of nine or ten residues being most prevalent (Figure 1c). Whilst most peptides showed the P2 glutamic acid anchor residue anticipated for HLA-B*44 ligands, Gibbs cluster analysis of the pooled immunopeptidomes revealed 1228 peptides with a highly distinct motif characterized by enrichment of hydrophobic residues at P2, proline at P3 and leucine at the C-terminus (Figure 1d, non-B44). This motif is similar to that reported for HLA-C*01:02 (52) and 66% of these peptides were predicted to bind HLA-C*01:02 by NetMHCpan4.1 (35) (Supplementary Table 1). These peptides are likely to be derived from residual HLA-Cw1 expression reported for the 721.220 line (16) which high resolution typing of the related 721.221 cell line reveals to be HLA-C*01:02 (53). Although it should be noted that the majority of these peptides are also predicted to bind non-classical HLA-E (and HLA-G) (Supplementary Table 1). Indeed, this cluster contained, VMAPRTLIL which is a well-recognized HLA-E ligand from certain classical HLA leader sequences, including HLA-C*01:02 (54, 55). Thus, except for 51 peptides in this cluster that possessed glutamic acid at P2, we termed these peptides likely non-HLA-B*44 (Supplementary Table 1) due to potential contribution by endogenous HLA-C and non-classical HLA of the parental 721.220 cell line.
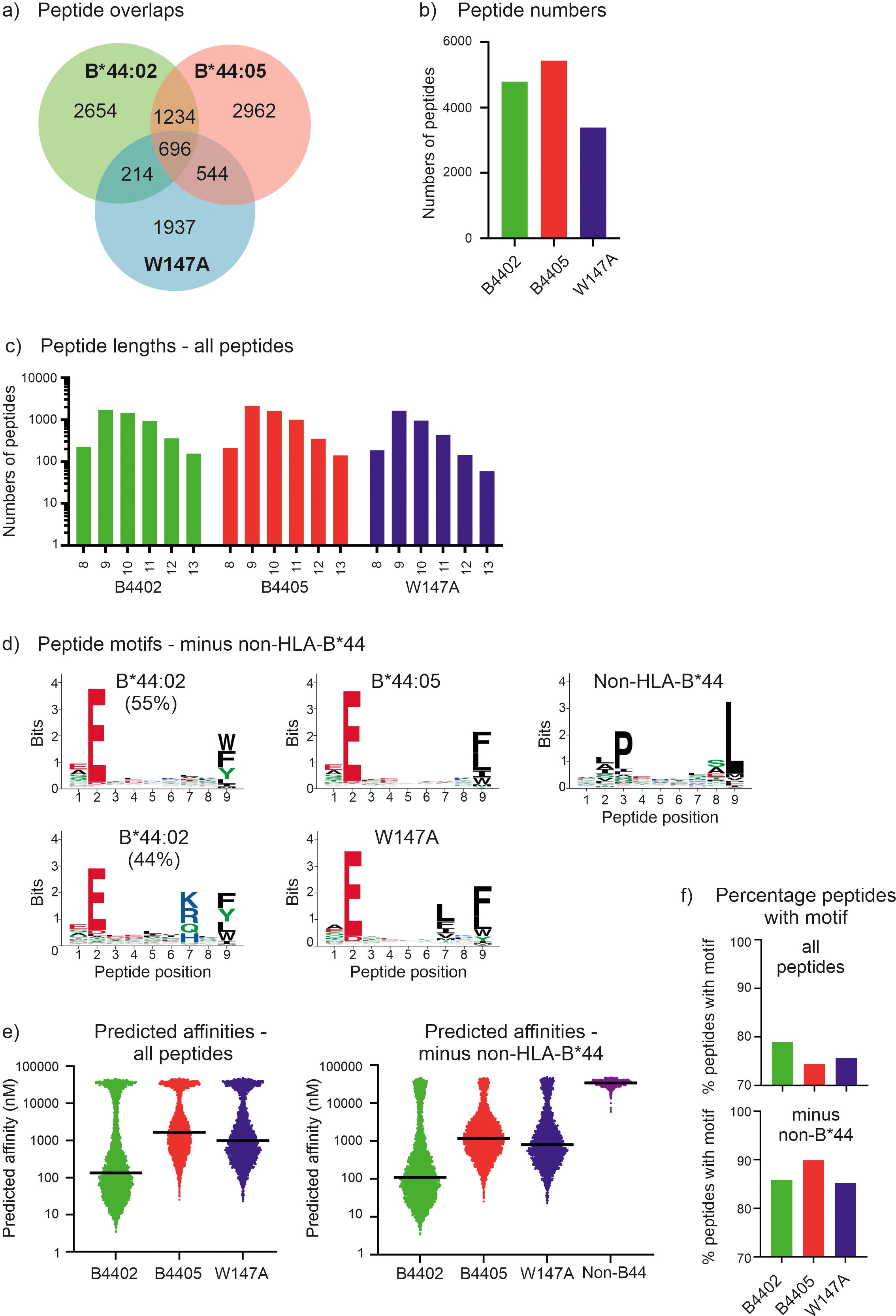
Figure 1. The immunopeptidomes of the tapasin dependent HLA-B*44:02 and HLA-B*44:05-W17A molecules contain greater proportions of high affinity peptides than tapasin independent HLA-B*44:05 molecules. (a) Venn diagram showing the number of peptides that were unique to each immunopeptidome, or which were shared. (b) Bar graph depicting the number of peptides identified within each immunopeptidome that were 8–13 amino acids long. (c) Bar graph depicting the frequency of peptides identified within the immunopeptidomes that were 8–13 amino acids long. (d) Motifs representing the immunopeptidomes of the HLA-B*44:02, HLA-B*44:05 and HLA-B*44:05-W17A molecules after Gibbs clustering and omission of potential non-HLA-B*44 binding peptides that did not have glutamic acid at position 2. At each position of a nonameric peptide there is a stack of amino acids with the height of the letter representing the frequency at which that residue was found. Only peptides that were 8–13 amino acids long were considered and were assigned to one (HLA-B*44:05, HLA-B*44:05-W147A, and non-HLA-B*44) or two motifs (HLA-B*44:02, as described previously ref (34)). For HLA-B*44:02, ~6% of peptides had no clear motif, and are not presented. (e) Graphs depicting the predicted affinities of the peptides identified within each immunopeptidome with or without omission of the potential non-HLA-B*44 derived peptides. The y axes represent the predicted affinity with nM units, calculated using NetMHCpan as detailed in the methods, with high affinity peptides having low nM values. Each dot represents a peptide, with the median indicated by a black horizontal bar. The left-hand graph shows the predicted affinities when all peptides are included, the right-hand graph shows the predicted affinities when the potential non-HLA-B*44 derived peptides were omitted. (f) Graphs depicting the proportion of peptides containing the preferred motif of the indicated MHC-I molecule. This was calculated as the percentage of peptides that contained the preferred motif within each immunopeptidome (“all”, upper plot), or after potential non-HLA-B*44 derived peptides were omitted (“minus non-HLA-B*44”, lower plot). The HLA-B*44:02 and HLA-B*44:05-W147A motifs were glutamic acid at position 2 and any of tryptophan, phenylalanine, tyrosine, leucine, isoleucine or methionine at the C-terminal position. The HLA-B*44:05 motif was glutamic acid at position 2 and any of tryptophan, phenylalanine, leucine, isoleucine or methionine at the C-terminal position.
When the potential non-HLA-B*44 peptides were omitted, we found that, as previously reported (34), the HLA-B*44:02 immunopeptidome was best represented by two peptide motifs, with ~55% of peptides having a motif with almost exclusively glutamic acid at position 2, while tryptophan, phenylalanine, tyrosine, or leucine dominate the C-terminal position (Figure 1d, Supplementary Figure 3 shows the motifs of the molecules without the omission of the potential non-HLA-B*44 peptides). In comparison ~44% of HLA-B*44:02 bound peptides had a motif with similar preferences at positions 2 and 9 but had an additional specificity for amino acids with basic or polar side chains at position 7.
The HLA-B*44:05 immunopeptidome was best represented by a single motif, which was like the most prevalent HLA-B*44:02 motif, with glutamic acid strongly preferred at position 2, and amino acids with hydrophobic side chains preferred at the C-terminal position. The HLA-B*44:05 W147A immunopeptidome closely resembled that of HLA-B*44:05, having a single motif with very similar specificities at positions 2 and 9. One slight difference was a preference for hydrophobic side chains at position 7, particularly leucine, phenylalanine, isoleucine, and valine, which would, presumably, be prevented from binding to wildtype HLA-B*44:05 by the bulky tryptophan side chain of position 147. This analysis therefore confirms that the specificity of the HLA-B*44:05-W147A peptide binding groove is highly similar to that of wild-type HLA-B*44:05.
We next compared the predicted affinities of the peptides recovered from these MHC-I molecules. As we could not definitively determine whether the potential non-HLA-B*44 derived peptides were eluted from HLA-C/E/G molecules co-immunoprecipitated with HLA-B*44 molecules, or if these peptides were low affinity peptides eluted from the HLA-B*44 molecules we compared the effect of omitting these potential non-HLA-B*44 derived peptides from our analysis. We found that regardless of whether the potential non-HLA-B*44 derived peptides were included (Figure 1e, left) or omitted (Figure 1e, right), the predicted affinity of the median peptide was highest for the peptides identified from HLA-B*44:02 expressing cells and lowest for the HLA-B*44:05 immunopeptidome, with HLA-B*44:05-W147A peptides being intermediate (Figure 1e where high affinity peptides have lower nM values than low affinity peptides, Table 1). As expected, the peptides likely to derive from non-HLA-B*44 molecules endogenously expressed in the 721.220 cells were predicted to bind HLA-B*44 with very low affinity (Figure 1e, non-HLA-B*44, where low affinity peptides have higher nM values than high affinity peptides), and their removal resulted in an increase in the proportion of peptides with the expected B44 anchor residues (Figure 1f).
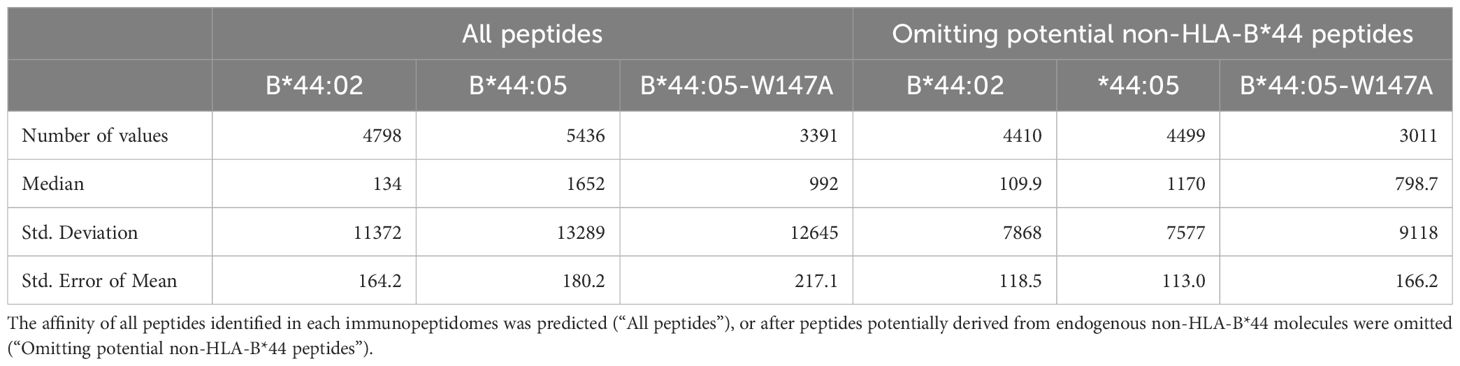
Table 1. Descriptive statistics for HLA-B*44:02, HLA-B*44:05 and HLA-B*44:05-W147A immunopeptidomes.
Taken together, these results are consistent with tapasin preferentially skewing the repertoires of peptides presented by tapasin-dependent HLA-B*44:02 and HLA-B*44:05-W147A molecules in favor of the most highly stable peptides, with an increased prevalence of preferred amino acid side chains at positions 2 and 9. This suggests tapasin-dependent MHC-I molecules have less diverse peptide repertoires than tapasin independent MHC-I molecules.
3.2 Enhanced tapasin mediated focusing of peptides competing for binding to HLA-B*44:05-W147A compared with HLA-B*44:05
To test whether HLA-B*44:05-W147A selects a higher affinity peptide cargo than HLA-B*44:05 as a result of its interaction with tapasin, we utilized an approach pioneered by Chen and Bouvier to observe tapasin function in vitro, in which MHC-I are placed in close proximity to monomeric tapasin molecules, or tapasin-ERp57 heterodimers, via a jun/fos leucine zipper (5). We conducted in vitro peptide competition experiments in which each of 24 peptides (twelve 10 mers and twelve 9 mers) that were predicted to cover a wide range of affinities individually competed against the high affinity tetramethyl rhodamine (TAMRA) labelled index peptide EEFGKTAMRAAFSF for binding to either HLA-B*44:05-fos or HLA-B*44:05-W147A-fos. EEFGKTAMRAAFSF binding and dissociation experiments are shown in Supplementary Figure 4. We found for both HLA-B*44:05 and HLA-B*44:05-W147A the affinities that were predicted were correlated with experimentally determined binding affinities as measured by IC50 values (Figure 2a, Table 2).
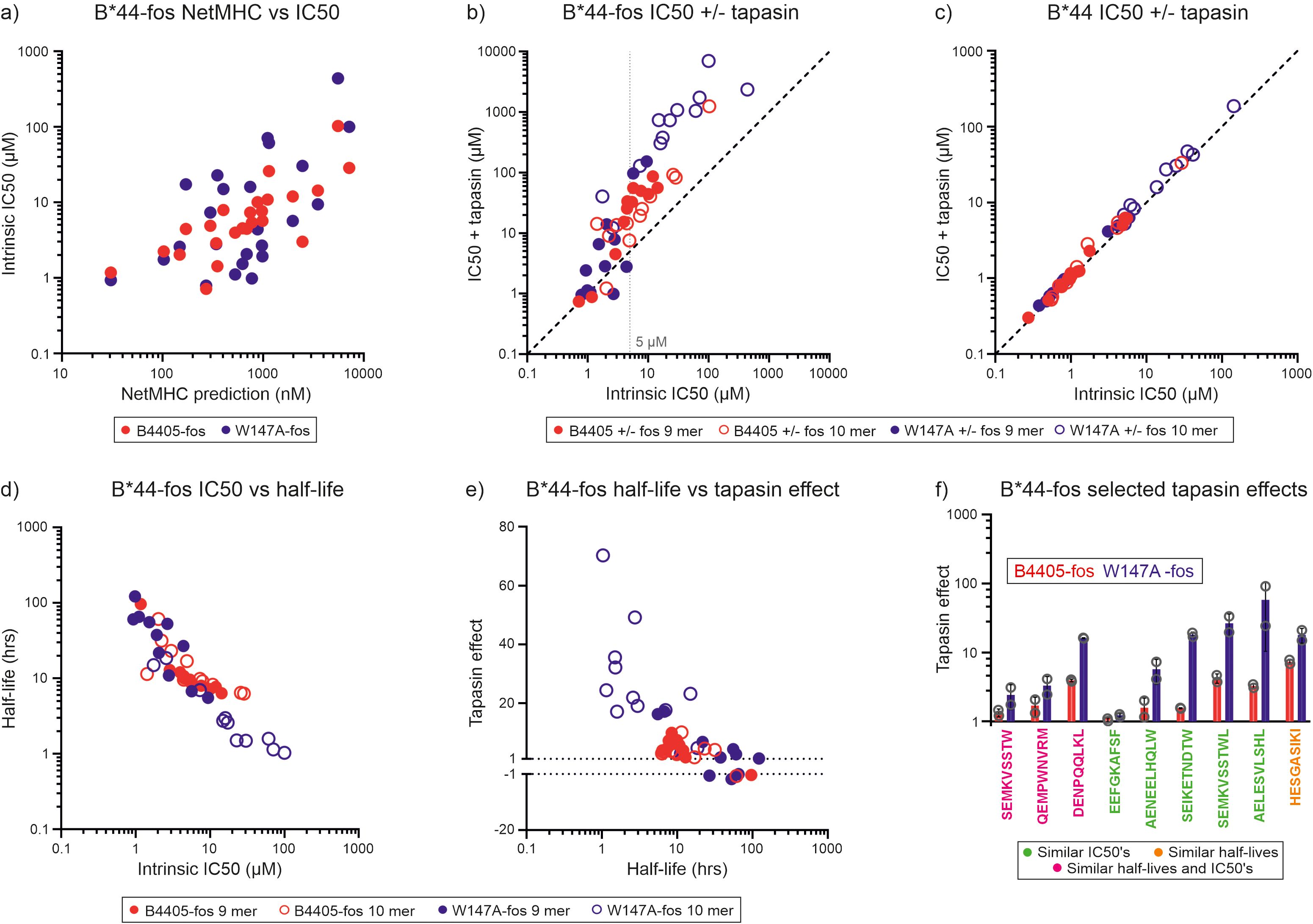
Figure 2. Tapasin-jun-ERp57 focuses the repertoire of peptides selected by HLA-B*44:05-fos and HLA-B*44:05-W147A-fos. (a) For HLA-B*44:05-fos and HLA-B*44:05-W147A-fos molecules, the affinities of the peptides used in the competition experiments were predicted and plotted against the measured mean intrinsic IC50 values. The predicted affinities of the competing peptides are represented in nM units, with high affinity peptides having low nM values. The intrinsic IC50 values are the IC50 values measured in the absence of tapasin-jun-Erp57 and are shown in µM units, with high affinity peptides having low µM values. (b) Peptide competition experiments were conducted in which unlabeled peptides individually competed against EEFGKTAMRAAFSF peptide for binding to either HLA-B*44:05-fos or HLA-B*44:05-W147A-fos in the presence or absence of tapasin-jun-ERp57. Binding of EEFGKTAMRAAFSF peptide was measured by fluorescence polarization and IC50 values calculated for each peptide (IC50 value is shown in µM units, with high affinity peptides having low µM values). Each peptide was tested at least twice, and the mean of the replicate experiments is reported. Peptides whose ability to compete for binding is not affected by tapasin-jun-ERp57 will fall along the diagonal dashed line. A faint dashed vertical line indicates a concentration discussed in the text. (c) Peptide competition experiments were conducted with HLA-B*44:05 or HLA-B*44:05-W147A molecules without fos leucine zipper sequences in which peptides competed for binding in the presence or absence of tapasin-jun-ERp57. (d) The half-lives of the complexes formed between the unlabeled peptides and HLA-B*44:05-fos or HLA-B*44:05-W147A-fos were indirectly measured and plotted against the mean intrinsic IC50 values. Each peptide was tested at least twice, and the mean half-life of the replicate experiments is reported in hours (high affinity peptide-MHC-I complexes have long half-lives). (e) The magnitude that tapasin-jun-ERp57 changed the ability of peptides to compete for binding to the HLA-B*44:05-fos or HLA-B*44:05-W147A-fos molecules was calculated and is reported as the “tapasin editing effect” (shortened to “tapasin effect” on the y axis of the graph). When tapasin-jun-ERp57 made a peptide a weaker competitor, this was calculated as the ratio of IC50 measured in the presence of tapasin divided by the intrinsic IC50 and was plotted as a positive number. When tapasin-jun-ERp57 made a peptide a stronger competitor, this was calculated as the ratio of the intrinsic IC50 divided by the IC50 measured in the presence of tapasin and was plotted as a negative number. For each peptide, the tapasin editing effect was plotted against the mean half-life measured for that peptide-MHC-I-fos complex. (f) Bar graph showing the “tapasin editing effect” observed for selected peptides competing for binding to HLA-B*44:05-fos or HLA-B*44:05-W147A-fos. Peptides which either competed for binding to HLA-B*44:05-fos or HLA-B*44:05-W147A-fos with similar intrinsic IC50s (peptide sequence in green), or formed complexes with HLA-B*44:05-fos or HLA-B*44:05-W147A-fos with similar half-lives (peptide sequence in orange), or had both similar intrinsic IC50s and similar half-lives (peptide sequence in pink) were selected and the magnitude by which tapasin-jun-ERp57 changed the ability to compete was compared. The tapasin editing effect (“tapasin effect” on y axis) measured in each experiment is shown as an open symbol, with the height of the bar indicating the mean, and the error bar indicating the standard deviation between replicates. To facilitate comparison of how tapasin changed the ability of different peptides to compete for binding, the data is plotted to show the magnitude by tapasin changed their ability to compete irrespective of whether a peptide became a poorer or stronger competitor.
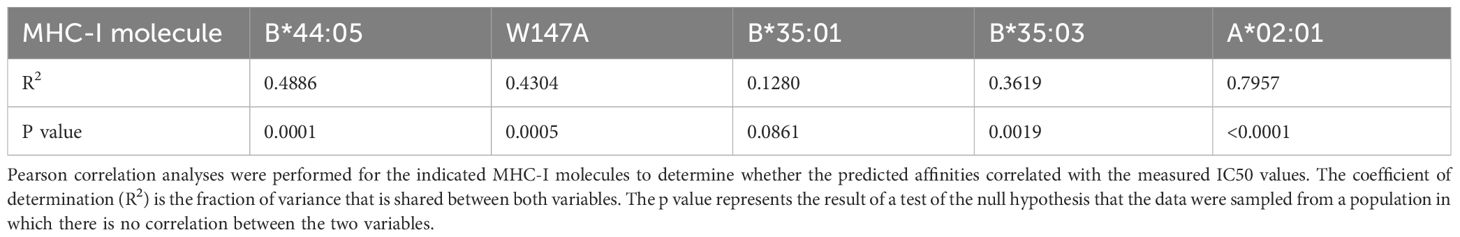
Table 2. Correlation analyses of the relationships between predicted affinity and measured IC50.
Using this approach, we could directly demonstrate tapasin-mediated peptide focusing as tapasin-jun-ERp57 diminished the ability of experimental peptides with lower affinity than the index peptide to compete for binding to both HLA-B*44:05-fos and HLA-B*44:05-W147A-fos (i.e. there was a higher IC50 in the presence of tapasin-jun-ERp57 in Figure 2b). This effect was most apparent for peptides with intrinsic IC50s of 5 µM or greater and was dependent on the leucine zipper to tether MHC-I and tapasin-jun-ERp57, as MHC-I molecules lacking the C-terminal fos sequence did not undergo substantial repertoire editing (Figure 2c).
Peptide binding half-lives measured for the peptide-MHC-I-fos complexes were inversely related to the intrinsic IC50 measurements (Figure 2d). We quantified the magnitude by which tapasin-jun-ERp57 changed the ability of peptides to compete for binding to HLA-B*44:05-fos or HLA-B*44:05-W147A-fos (the “tapasin editing effect”) and plotted this against the peptide-MHC-I complex half-lives (Figure 2e). For HLA-B*44:05-fos and HLA-B*44:05-W147A-fos proteins, the magnitude by which tapasin-jun-ERp57 modulated peptide competition increased as the half-life of the peptide-MHC-I-fos complex decreased, such that the least stable peptide-MHC-I-fos complexes experienced the greatest tapasin-jun-ERp57 function and became poorer competitors: i.e. these peptides would be more susceptible to being edited out of the repertoire (Figure 2e).
By comparing only those peptides that bound to HLA-B*44:05-fos and HLA-B*44:05-W147A-fos similarly (Supplementary Figures 5a, b) we found that, with the exception of EEFGKAFSF peptide, tapasin-jun-ERp57 had a greater influence on peptides competing for binding to HLA-B*44:05-W147A compared with HLA-B*44:05 (Figure 2f). This indicates that some low affinity peptides that are selected for presentation by HLA-B*44:05 are likely to be preferentially edited out of the repertoire by introducing the W147A mutation – as a direct result of higher tapasin dependence and more aggressive peptide filtering.
3.3 Tapasin modulates the ability of peptides to compete for binding to HLA-B*35:01-fos and HLA-B*35:03-fos
The HLA-B*44:05 and HLA-B*44:05-W147A allotype pair provides a convenient way of isolating the impact of tapasin on peptide repertoire editing, independent of substantial differences in MHC-I peptide binding specificity. We next compared the impact of tapasin on two naturally occurring, related alleles to seek further evidence that the intensity of tapasin-jun-ERp57 optimization increases with the tapasin dependency of an MHC-I allotype. We undertook the same analysis of HLA-B*35:01-fos and HLA-B*35:03-fos, which differ by a single residue at position 116 (HLA-B*35:01: Ser116, HLA-B*35:03: Phe116), and bind similar, although not identical peptide repertoires (56–58). Although both allotypes can efficiently assemble with peptides independently of tapasin, we and others have observed a modest difference in tapasin dependence of these MHC-I allotypes, with HLA-B*35:01 being slightly more independent than HLA-B*35:03 (Supplementary Figure 1 and refs (14, 15)).
We conducted in vitro peptide competition and indirect peptide dissociation experiments using YPLKTAMRAEQHGM TAMRA labelled index peptide (YPLKTAMRAEQHGM binding and dissociation experiments shown in Supplementary Figure 6) and a panel of 24 peptides that covered a wide range of predicted binding affinities for each allotype (Figure 3a, Table 2). We found that tapasin-jun-ERp57 decreased the ability of intermediate affinity peptides (those with intrinsic IC50 values between 0.4 µM and 50 µM in peptide competition experiments, and half-lives of 2–25 hours in indirect dissociation assays) to compete for binding to both HLA-B*35:01-fos and HLA-B*35:03-fos (Figures 3b–d). In contrast, tapasin-jun-ERp57 had comparatively much smaller effects on the competitive abilities of low affinity peptides (intrinsic IC50 values greater than 50 µM and half-lives shorter than 2 hours) which competed poorly against the fluorescent index peptide even in the absence of tapasin, and of high affinity peptides (intrinsic IC50 values smaller than 0.4 µM and half-lives longer than 25 hours), which competed potently against the fluorescent index peptide. Fourteen peptides bound to HLA-B*35:01-fos and HLA-B*35:03-fos with similar affinity (Supplementary Figures 5c, d), and we found that tapasin-jun-ERp57 had a greater influence on the ability of many of these 14 peptides to compete for binding to HLA-B*35:03-fos than was apparent for HLA-B*35:01-fos (Figure 3e). This was consistent with the slightly greater tapasin dependence of HLA-B*35:03 observed by us and others (Supplementary Figure 1 and ref (14)) and consistent with HLA-B*44:05-W147A receiving greater optimization from tapasin than HLA-B*44:05.
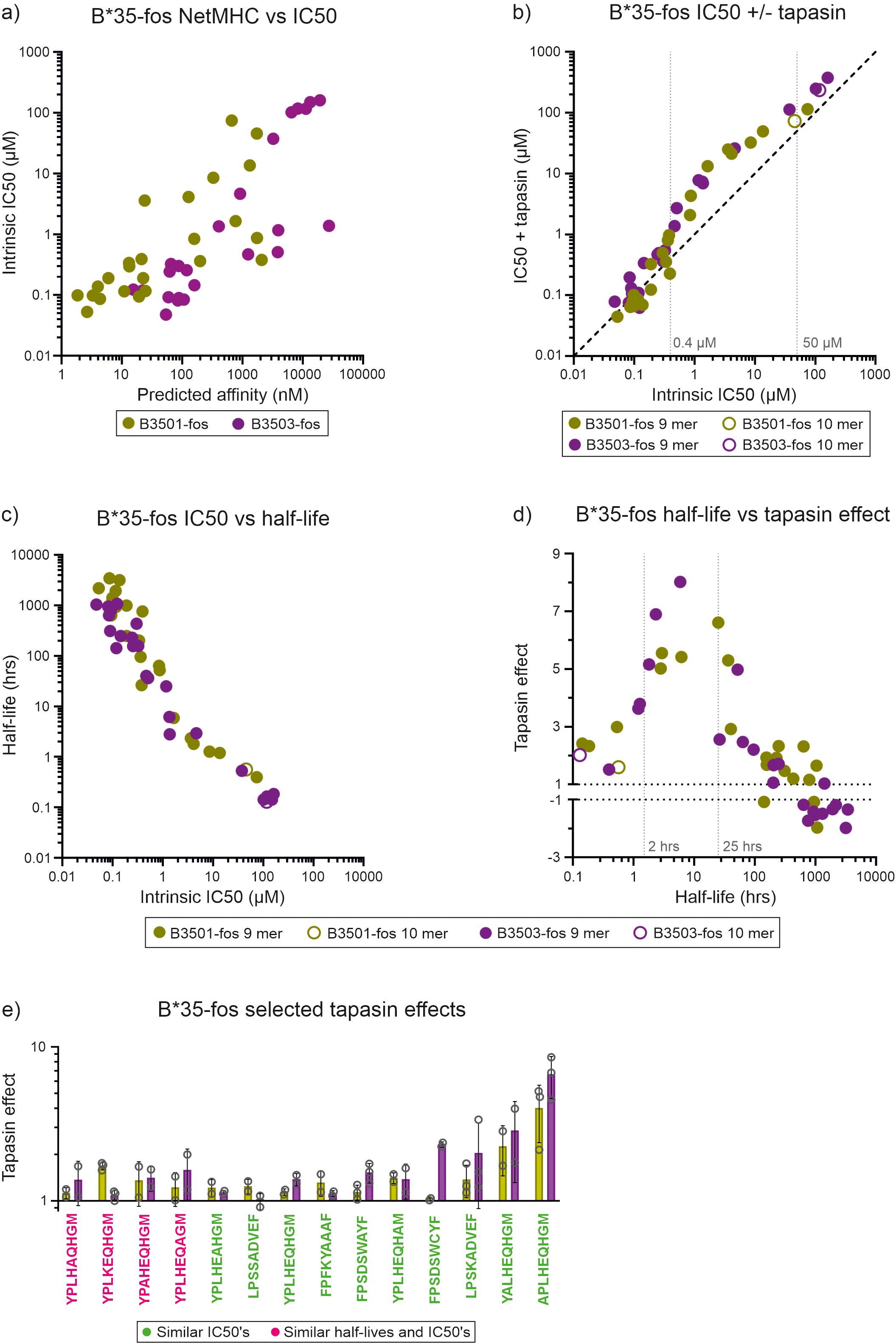
Figure 3. Tapasin-jun-ERp57 focuses the repertoire of peptides selected by HLA-B*35:01-fos and HLA-B*35:03-fos. (a) For HLA-B*35:01-fos and HLA-B*35:03-fos molecules, the affinities of the peptides used in the competition experiments were predicted and plotted against the measured mean intrinsic IC50 values as in Figure 2a. (b) Peptide competition experiments were conducted as described in Figure 2b with HLA-B*35:01-fos or HLA-B*35:03-fos molecules. Faint dashed vertical lines indicate concentrations discussed in the text. (c) The half-lives of the complexes formed between the unlabeled peptides and HLA-B*35:01-fos or HLA-B*35:03-fos were indirectly measured and plotted against the mean intrinsic IC50 values as in Figure 2d. (d) The magnitude that tapasin-jun-ERp57 changed the ability of peptides to compete for binding to the HLA-B*35:01-fos or HLA-B*35:03-fos molecules was calculated as before (“tapasin editing effect”) and plotted against the mean half-lives measured for that peptide-MHC-I-fos complex. Faint dashed vertical lines indicate half-lives discussed in the text. (e) Bar graph showing the “tapasin editing effect” observed for selected peptides competing for binding to HLA-B*35:01-fos or HLA-B*35:03-fos. Peptides which either competed for binding to HLA-B*35:01-fos or HLA-B*35:03-fos with similar intrinsic IC50s (peptide sequence in green) or had both similar intrinsic IC50s and formed complexes with HLA-B*44:05-fos or HLA-B*44:05-W147A-fos with similar half-lives (peptide sequence in pink) were selected and the magnitude by which tapasin-jun-ERp57 changed the ability to compete was compared and is presented as in Figure 2f.
3.4 TAPBPR modulates the ability of peptides to compete for binding to HLA-A*02:01-fos
We, and others, have previously shown that the tapasin orthologue, TAPBPR, which is not part of the peptide loading complex and most likely acts on peptide-MHC-I complexes released from the PLC as a further quality control checkpoint, also has a peptide editing function similar to tapasin (6–8). We therefore sought to determine whether TAPBPR might focus the peptide repertoire of HLA-A*02:01, a relatively tapasin independent allotype that, unlike HLA-B*44:02, HLA-B*44:05, HLA-B*35:01 and HLA-B*35:03 receives considerable benefit from TAPBPR mediated peptide editing (7, 8, 18). We carried out peptide competition experiments with a panel of 20 peptides that were predicted to cover a wide range of affinities (Figure 4a, Table 2) and the high affinity FLPSDCTAMRAFPSV index peptide (FLPSDCTAMRAFPSV binding and dissociation experiments shown in Supplementary Figure 7). TAPBPR modulated the ability of peptides to compete for binding to HLA-A*02:01 (Figures 4b–d). TAPBPR made low affinity peptides, with intrinsic IC50 values of 3 µM or greater, and half-lives of less than 50 hours, substantially poorer competitors for binding. Conversely, TAPBPR made most peptides with intrinsic IC50 values less than 3 µM, and half-lives of 50 hours or longer, stronger competitors for binding to HLA-A*02:01 (Figure 4b–d). The divergent effects that TAPBPR had on the ability of peptides to compete for binding to HLA-A*02:01 suggests that the index FLPSDCTAMRAFPSV peptide has an IC50 value of around 3 µM, (approximately 20-fold lower than was measured for unlabeled FLPSDCFPSV [0.14 µM]). Thus, we found that TAPBPR can also modulate the peptide repertoire in an analogous fashion to tapasin.
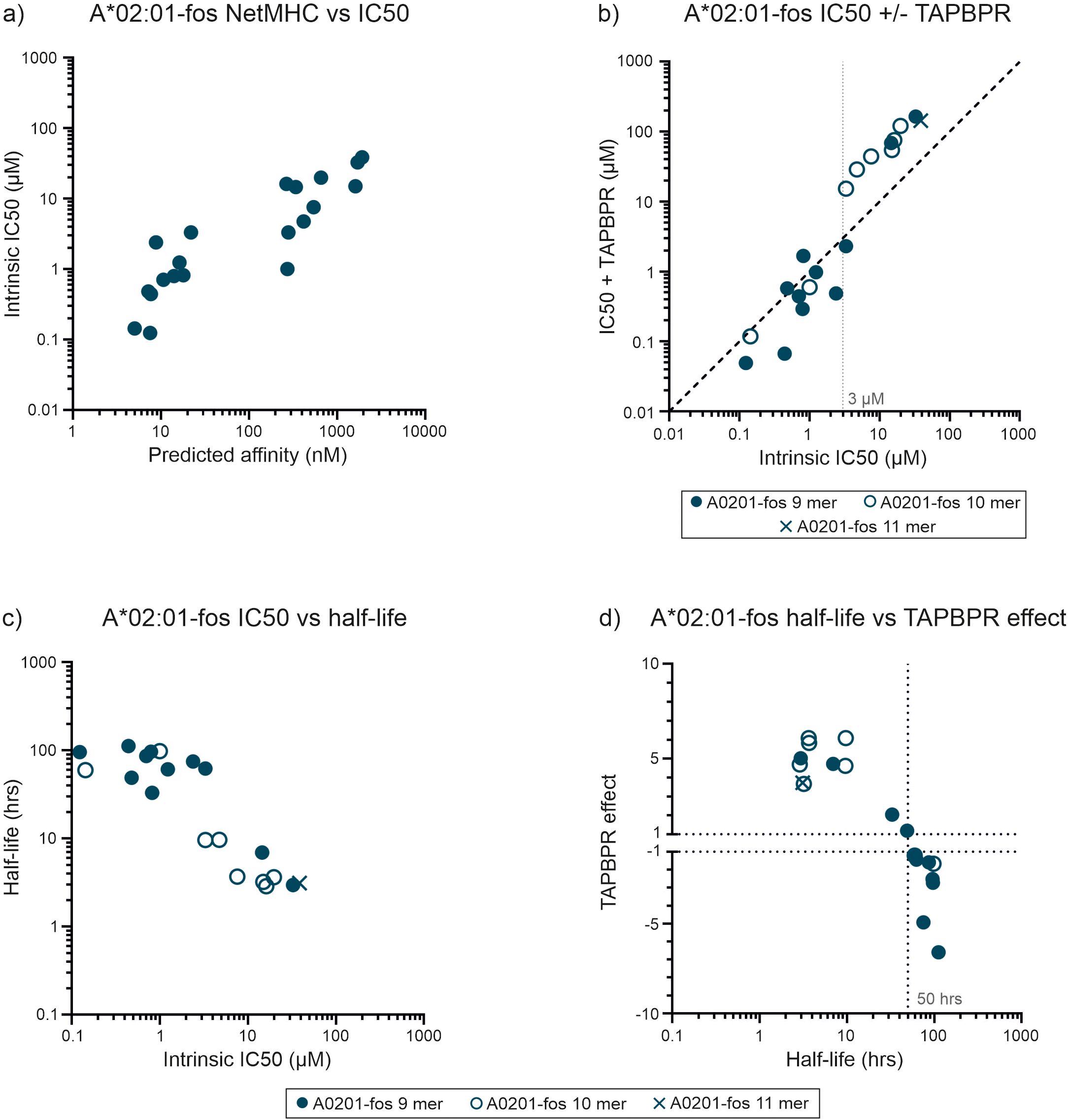
Figure 4. TAPBPR focuses the repertoire of peptides selected by HLA-A*02:01-fos. (a) For HLA-A*02:01-fos molecules, the affinities of the peptides used in the competition experiments were predicted and plotted against the measured mean intrinsic IC50 values as in Figure 2a. (b) Peptide competition experiments were conducted in the presence or absence of TAPBPR. Each peptide was tested at least twice, and the mean of the replicate experiments is reported. A faint dashed vertical line indicates a concentration discussed in the text. (c) The half-lives of the complexes formed between the unlabeled peptides and HLA-A*02:01-fos were indirectly measured and plotted against the mean intrinsic IC50 values as in Figure 2d. (d) The magnitude that TAPBPR changed the ability of peptides to compete for binding to the HLA-A*02:01-fos molecules was calculated (in the same manner as for tapasin) and plotted as the “TAPBPR editing effect” as in Figure 2e (shortened to “TAPBPR effect” on the y axis of the graph). This was plotted against the mean half-lives measured for the peptide-MHC-I-fos complexes on the x axis.
4 Discussion
The highly polymorphic nature of MHC-I molecules has long been known to define peptide binding specificity and, at least in mammals, dependence upon tapasin and TAPBPR for the acquisition of high affinity peptides. Recently, it has been suggested that the tapasin dependence of human MHC-I allotypes is inversely correlated with the diversity of their immunopeptidome (14, 27, 59). Our analysis of the HLA-B*44:02, HLA-B*44:05 and HLA-B*44:05-W147A immunopeptidomes provides direct evidence that tapasin dependent MHC-I allotypes generally bind peptide repertoires that are enriched in higher affinity peptides, closely complimenting the specificities of MHC-I anchor pockets. In comparison, tapasin independent MHC-I allotypes generally bind peptide repertoires that have lower average affinity and are less well suited to MHC-I anchor pocket specificities, and are likely to contain a greater diversity of peptide sequences. This suggests, that when tapasin is allowed to engage HLA-B*44:02 molecules or HLA-B*44:05 molecules in cells because of either aspartic acid at position 116 (HLA-B*44:02) or the W147A mutation (HLA-B*44:05-W147A), this results in more aggressive peptide focusing.
It is thought that MHC-I molecules transition between “closed” peptide bound states with low free energy and “open” peptide receptive states with higher free energy, with iterations of this process underpinning peptide exchange (22, 23, 29, 60–65). The mechanism by which tapasin independent MHC-I allotypes can self-edit their peptide repertoire remains to be determined. One possibility is that MHC-I allotypes like HLA-B*44:05 can independently self-edit their peptide repertoire because they have an intrinsic ability to adopt and transition between open and closed states. Indeed, as differences in MHC-I-fos-tapasin-jun-ERp57 binding affinities are unlikely in the context of an artificially tethered interaction, a potential mechanistic explanation for the greater tapasin optimization experienced by tapasin-dependent (HLA-B*44:05-W147A-fos or HLA-B*35:03-fos) molecules is that for tapasin dependent molecules there is a slower rate of transition from open to closed states, as has been suggested previously (29). Thus, tapasin dependent MHC-I allotypes have less intrinsic potential to transition between states and are consequently hard-wired to experience greater benefit from tapasin when it is available.
An alternate mechanism by which the W147A mutation increased the tapasin dependence of HLA-B*44:05 involves the hydrogen bond formed between the tryptophan at position 147, which is highly conserved in classical MHC-I, and the penultimate carbonyl group of the peptide. It is likely that in the absence of this hydrogen bond, there is increased conformational flexibility surrounding the C-terminal portion of the peptide and in the α2–1 sub-helix, which are key interaction surfaces for tapasin (1, 66–68) or TAPBPR (69, 70). The abrogation of this hydrogen bond tethering peptide to the MHC-I peptide binding groove is likely to have facilitated a higher affinity interaction with tapasin, allowing greater potential for tapasin assisted peptide editing to occur (65). A third possibility is that the ability to self-edit peptide repertoires involves the displacement of low affinity peptides from the peptide binding groove without substantial rearrangements of protein domains. In such a scenario, our experiments indicate that the combination of tryptophan at position 147 and tyrosine at position 116 are integral to the self-editing ability of HLA-B*44:05 while the combination of tryptophan and aspartic acid at these positions does not permit self-editing for HLA-B*44:02. However, the mechanism by which these residues are involved in self-editing remains to be determined. While molecular models of MHC-I self-editing have been proposed based on analysis of selected MHC-I allotypes (29, 71–76), these models do not provide a universal mechanism of self-editing that is applicable for all tapasin independent MHC-I allotypes, and it is possible that different mechanisms may operate in different MHC-I allotypes.
There may be multiple physiologically relevant consequences of variable degrees of repertoire editing by tapasin and TAPBPR. For tapasin dependent MHC-I allotypes, and for those MHC-I molecules that bind TAPBPR strongly, the extent of peptide focusing is likely to change in line with the expression levels of tapasin and TAPBPR, along with other components of the MHC-I antigen processing and presentation pathway. While cytokines released in response to inflammation may enhance MHC-I mediated antigen presentation, viral immune evasion proteins may target key proteins, including tapasin, to avoid MHC-I antigen presentation (77, 78). Similarly, some cancers lose expression of proteins involved in MHC-I mediated antigen presentation, including tapasin, generally leading to tumor progression and poorer prognoses (20, 79, 80). Thus, those MHC-I molecules that can independently optimize their peptide repertoire may be less susceptible to down regulation of tapasin.
Our data supports recent suggestions that in humans, tapasin dependence and immunopeptidome diversity are inversely correlated (14, 27, 59). Thus, tapasin dependency is likely to result in a more focused profile of presented peptides in which fewer, higher affinity peptides are presented at relatively higher abundance. There are also substantial differences in the diversity of MHC-I immunopeptidomes presented by different chicken MHC-I allotypes, with MHC-I immunopeptidome diversity being correlated, not with tapasin dependence, but with the structure of the MHC-I peptide binding groove and the specificity of the co-evolving polymorphic TAP peptide transporters (27, 59, 81, 82). Importantly, in both chickens and humans, diverse MHC-I immunopeptidomes have been shown to correlate with resistance to certain infectious pathogens (14, 27, 59). Additionally, vaccination studies in rhesus macaques have shown that presentation of a diverse pool of peptides by non-classical Mamu-E MHC-I molecules, equivalent to HLA-E in humans, resulted in enhanced immune responses (83). Thus, the protective benefit of diverse MHC-I immunopeptidomes appears to be an evolutionarily conserved feature of MHC-I molecules. MHC-I molecules with promiscuous peptide binding specificities, or the ability to select and present a broad peptide repertoire in the absence of tapasin/TAPBPR might be considered survival “generalists” (59). By comparison, MHC-I molecules with fastidious peptide binding specificities, or high dependency on tapasin/TAPBPR for repertoire editing may be beneficial for responding to new, and perhaps especially virulent pathogens, and therefore may be classified as survival “specialists” (59). Similarly, in the context of tumor immunology, a generalist approach (e.g. low tapasin dependence, mild editing) may be more protective against tumors that express multiple tumor specific neoepitopes, whereas for those that express a paucity of neoepitopes a more specialist approach (e.g. high tapasin dependence, aggressive editing) may be preferable.
In conclusion, our data are consistent with a scenario in which the dependence of individual MHC-I allotypes upon tapasin for optimal peptide selection underpins the diversity of their immunopeptidomes (14, 27, 59). Our in vitro observations of tapasin-jun-ERp57 and TAPBPR mediated peptide focusing illustrate how tapasin or TAPBPR filters the immunopeptidome according to individual peptide-MHC-I complex stability.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/pride/archive/, PXD054743 10.6019/PXD054743.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. No potentially identifiable images or data are presented in this study.
Author contributions
RD: Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Validation, Visualization, Writing – review & editing. PI: Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Validation, Writing – review & editing. PD: Resources, Writing – review & editing. AB: Formal Analysis, Validation, Writing – review & editing. AP: Funding acquisition, Supervision, Writing – review & editing. AVH: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. TE: Conceptualization, Funding acquisition, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by a Cancer Research UK program grant A28279 (award to TE), by National Health and Medical Research Council Australia (NHMRC) project grants 1122099 and 1165490 (awarded to AP), while PI was supported by a NHMRC Early Career Fellowship (1072159) and a Monash University Faculty of Medicine, Nursing and Health Sciences Senior Postdoctoral Fellowship. AP acknowledges salary support from a NHMRC Principal Research Fellowship (1137739).
Acknowledgments
The authors acknowledge Charlotte Barton for her technical help during a project placement.
Conflict of interest
AP is a member of the Scientific advisory board of Bioinformatic Solutions Inc., Canada and is a shareholder and Scientific advisory board member of Evaxion Biotech Denmark. He is a co-founder of Resseptor Therapeutics Australia.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1563789/full#supplementary-material
Supplementary Methods 1 | DNA constructs encoding HLA-B35:01 or HLA-B35:03 in the pMSCVneo plasmid were kindly provided by Prof M. Raghavan (15) and sub-cloned into the pMCFRpuro plasmid. Stable transfectants of tapasin-deficient 721.220 cells, or tapasin-reconstituted 721.220-tapasin cells, expressing HLA-B*35:01, HLA-B*35:03 were generated as described (29). MHC-I pulse chase thermostability assays. MHC-I thermostability assays were performed essentially as described (2). In brief, tapasin-deficient.220 cells, or tapasin-reconstituted.220-tapasin cells, expressing the indicated MHC-I molecules were radiolabeled with 10 μ Ci/ml 35S Translabel for six minutes, chased for the indicated period, before the cells were lysed. The lysates were heated to 37°C for 12 minutes, before being cooled, and MHC-I-β2m complexes immunoprecipitated with W6/32 antibody. The eluted proteins were separated by SDS PAGE, and the gels were stained with Coomassie brilliant blue and an image of the gel taken, before the gels were dried and exposed to phosphor screens. The intensities of the radiolabeled heavy chain bands were measured using Personal Molecular Imager FX and quantified using Quantity One software and normalized according to the corresponding intensity of immunoprecipitated W6/32 antibody heavy chain bands in the Coomassie stained gel image. Flow cytometry measurements of MHC-I cell surface expression. Cells were stained with W6/32 antibody followed by anti-mouse FITC secondary antibody in the dark, washed and analyzed on a FACSCalibur flow cytometer (BD Biosciences). MHC-I pulse chase maturation assays. MHC-I pulse chase maturation assays were performed as described (2, 29). MHC-I peptide stabilization assays. Tapasin-deficient.220 cells expressing the indicated MHC-I molecules were incubated at 37°C for 40 minutes in 4 ml of methionine and cysteine free RPMI media that was supplemented with 10% dialyzed FCS and 2 mM L-glutathione. The cells were radiolabeled with 7.8 MBq of 35S Translabel for 30 minutes at 37°C before the radiolabeling was quenched by the addition of 20 ml ice cold PBS, and the cells lysed. The lysates were divided into five aliquots and incubated for 30 minutes on ice with either 0, 1, 2, 5 or 20 µM of the following known HLA-B*44 binding peptides (FEDLRVLSF, VETPIRNEW, TENGSFVAGY, EENLLDFVRF or SEIDTVAKY, refs (84–87)). MHC-I molecules were immunoprecipitated using W6/32 antibody as detailed above and separated by SDS PAGE gels.
Supplementary Figure 1 | The HLA-B*35:01, HLA-B*35:03, HLA-B*44:02, HLA-B*44:05 and HLA-B*44:05-W147A MHC-I molecules differ in their dependence upon tapasin. (a) Representative images of 35S pulse labelled MHC-I thermostability assays of the indicated MHC-I molecules expressed in tapasin deficient 721.220 cells (- tapasin) or tapasin reconstituted 721.220-tapasin cells (+ tapasin). Only the portion of the gel showing the heavy chain band is shown. (b) Bar graph depicting how much tapasin improved the thermostability of the indicated MHC-I molecules. This was calculated as the ratio of the percentage of thermostable MHC-I recovered in the presence of tapasin divided by the percentage of thermostable MHC-I recovered in the absence of tapasin after a chase of 120 minutes. Dots represent results from individual experiments, and vertical lines depict the standard deviation observed between experiments. The data for HLA-B*44:02, HLA-B*44:05 and HLA-B*44:05-W147A is published in ref (29). (c) Bar graph showing the cell surface expression level of the indicated MHC-I molecules transfected into tapasin deficient 721.220 cells (- tapasin) or tapasin reconstituted 721.220-tapasin cells (+ tapasin) following staining with W6/32 antibody and flow cytometry analysis. Dots represent the mean fluorescent intensity measured in an experiment, and vertical lines depict the standard deviation observed between experiments. The data for HLA-B*44:02, HLA-B*44:05 and HLA-B*44:05-W147A is published in ref (29). (d) Representative images of 35S pulse chase MHC-I maturation assays of the indicated MHC-I molecules expressed in tapasin deficient 721.220 cells (- tapasin) or tapasin reconstituted 721.220-tapasin cells (+ tapasin). W6/32 immunoprecipitated samples were digested (+) or mock digested (-) with endoglycosidase H (endo H). ER and ES denote immunoprecipitated MHC-I molecules that are resistant (ER) or sensitive (ES) to digestion with endoglycosidase H. (e) Graphs depicting the percentage of immunoprecipitated MHC-I molecules that were resistant to digestion with endoglycosidase H at each chase point in the absence (left) or presence of tapasin (right). Vertical error bars denote the standard deviation observed between experiments. The maturation of HLA-B*44:02, HLA-B*44:05 and HLA-B*44:05-W147A in 721.220 or 721.220-tapasin cells is published in ref (29).
Supplementary Figure 2 | Peptide stabilization assays with tapasin-deficient cells expressing HLA-B*44:02, HLA-B*44:05 or HLA-B*44:05-W147A. MHC-I peptide stabilization assays of the indicated MHC-I molecules expressed in tapasin deficient 721.220 cells. The cells were radiolabeled before the cells were lysed. Aliquots of lysates were incubated with the indicated peptide for 30 minutes before MHC-I molecules were immunoprecipitated and separated by SDS PAGE. Only the portion of the gel showing the heavy chain band is shown. Graphs show the density of the heavy chain bands for the indicated MHC-I molecules. The anomalous data point for HLA-B*44:05-W147A incubated with 2 µM FEDLRVLSF (denoted with an asterisk above) was omitted from the graph for clarity.
Supplementary Figure 3 | Analysis of the immunopeptidomes without peptides potentially derived from non-HLA-B*44 molecules. Motifs representing the immunopeptidomes of the HLA-B*44:02, HLA-B*44:05, HLA-B*44:05-W17A molecules without any peptides being omitted from the analysis. Peptides were assigned to one (HLA-B*44:05 and HLA-B*44:05-W147A) or two motifs (HLA-B*44:02, as described previously ref (34)). For HLA-B*44:02, ~11% of peptides had no clear motif, and are not presented.
Supplementary Figure 4 | Peptide binding and peptide dissociation experiments with HLA-B*44:05-fos and HLA-B*44:05-W147A-fos in the presence or absence of tapasin-jun-ERp57. (a) Peptide binding experiment in which 160 nM HLA-B*44:05-fos was supplemented with 3.2 µM β2-microglobulin and UV exposed, before being added to 2 nM EEFGK*AFSF peptide in the presence or absence of 300 nM tapasin-jun-ERp57. Fluorescence polarization was measured at 25°C. (b) Peptide binding experiment with HLA-B*44:05-W147A-fos as in Supplementary Figure 4a. (c) Peptide dissociation experiment in which 160 nM HLA-B*44:05-fos was supplemented with 3.2 µM β2-microglobulin, UV exposed and incubated overnight with 2 nM EEFGK*AFSF peptide. The next day, 66.7 µM EENLLDFVRF peptide competitor was added in the presence or absence of 300 nM tapasin-jun-ERp57. Fluorescence polarization was measured at 25°C. (d) Peptide dissociation experiment with HLA-B*44:05-W147A-fos as in Supplementary Figure 4c.
Supplementary Figure 5 | Identification of similar binding peptides. (a) The mean intrinsic IC50 value of each peptide competing for binding to HLA-B*44:05-fos is shown on the x-axis, while the mean intrinsic IC50 value measured for each peptide competing for binding to HLA-B*44:05-W147A-fos is shown on the y-axis. To identify peptides with similar abilities to compete for binding to both HLA-B*44:05-fos and HLA-B*44:05-W147A-fos, the ratio by which the intrinsic IC50s differed was calculated. Peptides with an intrinsic IC50 ratio of less than 2 are shown in green or shown in pink if the peptides also have a half-life ratio of less than 2, all other peptides are shown in grey. (b) The mean half-lives of the peptide-HLA-B*44:05-fos complexes are shown on the x-axis, while the mean half-lives of the peptide-HLA-B*44:05-W147A-fos complexes are shown on the y-axis. To identify peptides that formed complexes with HLA-B*44:05-fos and HLA-B*44:05-W147A-fos with similar half-lives, the ratio by which the half-lives differed was calculated. Peptides with a half-life ratio of less than 2 are shown in orange or shown in pink if the peptides also have an intrinsic IC50 ratio of less than 2, all other peptides are shown in grey. (c) The mean intrinsic IC50 value of each peptide competing for binding to HLA-B*35:01-fos is shown on the x-axis, while the mean intrinsic IC50 value measured for each peptide competing for binding to HLA-B*35:03-fos is shown on the y-axis. Peptides with similar intrinsic IC50 ratios were calculated as in Supplementary Figure 5a. (d) The mean half-lives of the peptide-HLA-B*35:01-fos complexes are shown on the x-axis, while the mean half-lives of the peptide-HLA-B*35:03-fos complexes are shown on the y-axis. Peptides with similar half-life ratios were calculated as in Supplementary Figure 5b.
Supplementary Figure 6 | Peptide binding and peptide dissociation experiments with HLA-B*35:01-fos and HLA-B*35:03-fos in the presence or absence of tapasin-jun-ERp57. (a) Peptide binding experiment in which 225 nM HLA-B*35:01-fos was supplemented with 4.5 µM β2-microglobulin and UV exposed, before being added to 3 nM YPLK*EQHGM peptide in the presence or absence of 300 nM tapasin-jun-ERp57. Fluorescence polarization was measured at 25°C. (b) Peptide binding experiment with HLA-B*35:03-fos as in Supplementary Figure 6a. (c) Peptide dissociation experiment in which 225 nM HLA-B*35:01-fos was supplemented with 4.5 µM β2- microglobulin and UV exposed and incubated overnight with 3 nM YPLK*EQHGM peptide. The next day, 66.7 µM YPLHEQHGM peptide competitor was added in the presence or absence of 300 nM tapasin-jun-ERp57. Fluorescence polarization was measured at 25°C. (d) Peptide dissociation experiment with HLA-B*35:03-fos as in Supplementary Figure 6c.
Supplementary Figure 7 | Peptide binding and peptide dissociation experiments with HLA-A*02:01-fos in the presence or absence of TAPBPR. (a) Peptide binding experiment in which 50 nM HLA-A*02:01-fos was supplemented with 1 µM β2-microglobulin and UV exposed, before being added to 2 nM FLPSDC*FPSV peptide in the presence or absence of 300 nM TAPBPR. Fluorescence polarization was measured at 25°C. (b) Peptide dissociation experiment in which 50 nM HLA-A*02:01-fos was supplemented with 1 µM β2-microglobulin and UV exposed and incubated overnight with 2 nM FLPSDC*FPSV peptide. The next day, 66.7 µM FLPSDCFPSV peptide competitor was added in the presence or absence of 300 nM TAPBPR and fluorescence polarization was measured at 25°C.
Supplementary Table 1 | Peptide lists and analysis of potential non-HLA-B44 peptides. The first three tabs of the spreadsheet provide lists of peptides that are 8–13 amino acids and non-redundant by sequence (i.e. modifications were not considered) that were identified within each immunopeptidome (B4402 peptides, B4405 peptides, W147A peptides). The potential non-HLA-B*44 peptides identified by Gibbs clustering are designated 1 for potential non-HLA-B*44 ligands or 0 for potential HLA-B*44 ligands. The fourth tab provides a list of 1228 potential non-HLA-B*44 binding peptides that were identified by pooling and clustering all peptides (NonB44 cluster analysis). This tab provides an analysis of the potential sources of these peptides and their predicted binding affinity to HLA-B*44, HLA-C*01:02, HLA-E or HLA-G molecules. The fifth tab (Non B44 peptides) provides a list of 1177 potential non-HLA-B*44 binding peptides, i.e. peptides identified by clustering that do not have glutamic acid at position 2. The sixth tab (Overlaps) details the extent to which peptides are shared between immunopeptidomes or were unique. The analysis was conducted including potential non-B*44 peptides and is replicated omitting these peptides.
References
1. Domnick A, Winter C, Susac L, Hennecke L, Hensen M, Zitzmann N, et al. Molecular basis of MHC I quality control in the peptide loading complex. Nat Commun. (2022) 13:4701. doi: 10.1038/s41467-022-32384-z
2. Williams AP, Peh CA, Purcell AW, McCluskey J, Elliott T. Optimization of the MHC class I peptide cargo is dependent on tapasin. Immunity. (2002) 16:509–20. doi: 10.1016/S1074-7613(02)00304-7
3. Howarth M, Williams A, Tolstrup AB, Elliott T. Tapasin enhances MHC class I peptide presentation according to peptide half-life. Proc Natl Acad Sci U S A. (2004) 101:11737–42. doi: 10.1073/pnas.0306294101
4. Wearsch PA, Cresswell P. Selective loading of high-affinity peptides onto major histocompatibility complex class I molecules by the tapasin-ERp57 heterodimer. Nat Immunol. (2007) 8:873–81. doi: 10.1038/ni1485
5. Chen M, Bouvier M. Analysis of interactions in a tapasin/class I complex provides a mechanism for peptide selection. EMBO J. (2007) 26:1681–90. doi: 10.1038/sj.emboj.7601624
6. Boyle LH, Hermann C, Boname JM, Porter KM, Patel PA, Burr ML, et al. Tapasin-related protein TAPBPR is an additional component of the MHC class I presentation pathway. Proc Natl Acad Sci U S A. (2013) 110:3465–70. doi: 10.1073/pnas.1222342110
7. Hermann C, van Hateren A, Trautwein N, Neerincx A, Duriez PJ, Stevanovic S, et al. TAPBPR alters MHC class I peptide presentation by functioning as a peptide exchange catalyst. Elife. (2015) 4. doi: 10.7554/eLife.09617
8. Morozov GI, Zhao H, Mage MG, Boyd LF, Jiang J, Dolan MA, et al. Interaction of TAPBPR, a tapasin homolog, with MHC-I molecules promotes peptide editing. Proc Natl Acad Sci U S A. (2016) 113:E1006–1015. doi: 10.1073/pnas.1519894113
9. Howe C, Garstka M, Al-Balushi M, Ghanem E, Antoniou AN, Fritzsche S, et al. Calreticulin-dependent recycling in the early secretory pathway mediates optimal peptide loading of MHC class I molecules. EMBO J. (2009) 28:3730–44. doi: 10.1038/emboj.2009.296
10. Wearsch PA, Peaper DR, Cresswell P. Essential glycan-dependent interactions optimize MHC class I peptide loading. Proc Natl Acad Sci U S A. (2011) 108:4950–5. doi: 10.1073/pnas.1102524108
11. Neerincx A, Hermann C, Antrobus R, van Hateren A, Cao H, Trautwein N, et al. TAPBPR bridges UDP-glucose:glycoprotein glucosyltransferase 1 onto MHC class I to provide quality control in the antigen presentation pathway. Elife. (2017) 6. doi: 10.7554/eLife.23049
12. Falk K, Rotzschke O, Rammensee HG. Cellular peptide composition governed by major histocompatibility complex class I molecules. Nature. (1990) 348:248–51. doi: 10.1038/348248a0
13. Rotzschke O, Falk K, Deres K, Schild H, Norda M, Metzger J, et al. Isolation and analysis of naturally processed viral peptides as recognized by cytotoxic T cells. Nature. (1990) 348:252–4. doi: 10.1038/348252a0
14. Bashirova AA, Viard M, Naranbhai V, Grifoni A, Garcia-Beltran W, Akdag M, et al. HLA tapasin independence: broader peptide repertoire and HIV control. Proc Natl Acad Sci U S A. (2020) 117:28232–8. doi: 10.1073/pnas.2013554117
15. Rizvi SM, Salam N, Geng J, Qi Y, Bream JH, Duggal P, et al. Distinct assembly profiles of HLA-B molecules. J Immunol. (2014) 192:4967–76. doi: 10.4049/jimmunol.1301670
16. Greenwood R, Shimizu Y, Sekhon GS, DeMars R. Novel allele-specific, post-translational reduction in HLA class I surface expression in a mutant human B cell line. J Immunol. (1994) 153:5525–36. doi: 10.4049/jimmunol.153.12.5525
17. Zernich D, Purcell AW, Macdonald WA, Kjer-Nielsen L, Ely LK, Laham N, et al. Natural HLA class I polymorphism controls the pathway of antigen presentation and susceptibility to viral evasion. J Exp Med. (2004) 200:13–24. doi: 10.1084/jem.20031680
18. Ilca FT, Drexhage LZ, Brewin G, Peacock S, Boyle LH. Distinct polymorphisms in HLA class I molecules govern their susceptibility to peptide editing by TAPBPR. Cell Rep. (2019) 29:1621–32.e1623. doi: 10.1016/j.celrep.2019.09.074
19. Sun Y, Papadaki GF, Devlin CA, Danon JN, Young MC, Winters TJ, et al. Xeno interactions between MHC-I proteins and molecular chaperones enable ligand exchange on a broad repertoire of HLA allotypes. Sci Adv. (2023) 9:eade7151. doi: 10.1126/sciadv.ade7151
20. DhatChinamoorthy K, Colbert JD, Rock KL. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol. (2021) 12:636568. doi: 10.3389/fimmu.2021.636568
21. Walker-Sperling V, Digitale JC, Viard M, Martin MP, Bashirova A, Yuki Y, et al. Genetic variation that determines TAPBP expression levels associates with the course of malaria in an HLA allotype-dependent manner. Proc Natl Acad Sci U S A. (2022) 119:e2205498119. doi: 10.1073/pnas.2205498119
22. Carignano A, Dalchau N. A theory for how the antigen presentation profile influences the timing of T-cell detection. bioRxiv. (2018). doi: 10.1101/480301
23. Dalchau N, Phillips A, Goldstein LD, Howarth M, Cardelli L, Emmott S, et al. A peptide filtering relation quantifies MHC class I peptide optimization. PLoS Comput Biol. (2011) 7:e1002144. doi: 10.1371/journal.pcbi.1002144
24. Paul S, Weiskopf D, Angelo MA, Sidney J, Peters B, Sette A. HLA class I alleles are associated with peptide-binding repertoires of different size, affinity, and immunogenicity. J Immunol. (2013) 191:5831–9. doi: 10.4049/jimmunol.1302101
25. Kosmrlj A, Read EL, Qi Y, Allen TM, Altfeld M, Deeks SG, et al. Effects of thymic selection of the T-cell repertoire on HLA class I-associated control of HIV infection. Nature. (2010) 465:350–4. doi: 10.1038/nature08997
26. Kaur A, Surnilla A, Zaitouna AJ, Mumphrey MB, Basrur V, Grigorova I, et al. Mass spectrometric profiling of HLA-B44 peptidomes provides evidence for tapasin-mediated tryptophan editing. J Immunol. (2023) 211:1298–307. doi: 10.4049/jimmunol.2300232
27. Chappell P, Meziane el K, Harrison M, Magiera L, Hermann C, Mears L, et al. Expression levels of MHC class I molecules are inversely correlated with promiscuity of peptide binding. Elife. (2015) 4:e05345. doi: 10.7554/eLife.05345
28. Boulanger DSM, Douglas LR, Duriez PJ, Kang Y, Dalchau N, James E, et al. Tapasin-mediated editing of the MHC I immunopeptidome is epitope specific and dependent on peptide off-rate, abundance, and level of tapasin expression. Front Immunol. (2022) 13:956603. doi: 10.3389/fimmu.2022.956603
29. Bailey A, Dalchau N, Carter R, Emmott S, Phillips A, Werner JM, et al. Selector function of MHC I molecules is determined by protein plasticity. Sci Rep. (2015) 5:14928. doi: 10.1038/srep14928
30. Purcell AW, Ramarathinam SH, Ternette N. Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat Protoc. (2019) 14:1687–707. doi: 10.1038/s41596-019-0133-y
31. Escher C, Reiter L, MacLean B, Ossola R, Herzog F, Chilton J, et al. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics. (2012) 12:1111–21. doi: 10.1002/pmic.201100463
32. UniProt C. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. (2023) 51:D523–31. doi: 10.1093/nar/gkac1052
33. Bassani-Sternberg M, Gfeller D. Unsupervised HLA peptidome deconvolution improves ligand prediction accuracy and predicts cooperative effects in peptide-HLA interactions. J Immunol. (2016) 197:2492–9. doi: 10.4049/jimmunol.1600808
34. Gfeller D, Guillaume P, Michaux J, Pak HS, Daniel RT, Racle J, et al. The length distribution and multiple specificity of naturally presented HLA-I ligands. J Immunol. (2018) 201:3705–16. doi: 10.4049/jimmunol.1800914
35. Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. (2020) 48:W449–54. doi: 10.1093/nar/gkaa379
36. Andreatta M, Alvarez B, Nielsen M. GibbsCluster: unsupervised clustering and alignment of peptide sequences. Nucleic Acids Res. (2017) 45:W458–63. doi: 10.1093/nar/gkx248
37. Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. (2019) 47:D442–50. doi: 10.1093/nar/gky1106
38. Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. (2019) 47:D339–43. doi: 10.1093/nar/gky1006
39. Khanna R, Silins SL, Weng Z, Gatchell D, Burrows SR, Cooper L. Cytotoxic T cell recognition of allelic variants of HLA B35 bound to an Epstein-Barr virus epitope: influence of peptide conformation and TCR-peptide interaction. Eur J Immunol. (1999) 29:1587–97. doi: 10.1002/(SICI)1521-4141(199905)29:05<1587::AID-IMMU1587>3.0.CO;2-W
40. Thammavongsa V, Schaefer M, Filzen T, Collins KL, Carrington M, Bangia N, et al. Assembly and intracellular trafficking of HLA-B*3501 and HLA-B*3503. Immunogenetics. (2009) 61:703–16. doi: 10.1007/s00251-009-0399-2
41. Morel S, Ooms A, Van Pel A, Wolfel T, Brichard VG, van der Bruggen P, et al. A tyrosinase peptide presented by HLA-B35 is recognized on a human melanoma by autologous cytotoxic T lymphocytes. Int J Cancer. (1999) 83:755–9. doi: 10.1002/(SICI)1097-0215(19991210)83:6<755::AID-IJC10>3.0.CO;2-S
42. Mandruzzato S, Stroobant V, Demotte N, van der Bruggen P. A human CTL recognizes a caspase-8-derived peptide on autologous HLA-B*3503 molecules and two unrelated peptides on allogeneic HLA-B*3501 molecules. J Immunol. (2000) 164:4130–4. doi: 10.4049/jimmunol.164.8.4130
43. Sidney J, del Guercio MF, Southwood S, Engelhard VH, Appella E, Rammensee HG, et al. Several HLA alleles share overlapping peptide specificities. J Immunol. (1995) 154:247–59. doi: 10.4049/jimmunol.154.1.247
44. Reker S, Becker JC, Svane IM, Ralfkiaer E, Straten PT, Andersen MH. HLA-B35-restricted immune responses against survivin in cancer patients. Int J Cancer. (2004) 108:937–41. doi: 10.1002/ijc.v108:6
45. Hourigan CS, Harkiolaki M, Peterson NA, Bell JI, Jones EY, O'Callaghan CA. The structure of the human allo-ligand HLA-B*3501 in complex with a cytochrome p450 peptide: steric hindrance influences TCR allo-recognition. Eur J Immunol. (2006) 36:3288–93. doi: 10.1002/eji.200636234
46. Menssen R, Orth P, Ziegler A, Saenger W. Decamer-like conformation of a nona-peptide bound to HLA-B*3501 due to non-standard positioning of the C terminus. J Mol Biol. (1999) 285:645–53. doi: 10.1006/jmbi.1998.2363
47. Abdul-Jawad S, Ondondo B, van Hateren A, Gardner A, Elliott T, Korber B, et al. Increased valency of conserved-mosaic vaccines enhances the breadth and depth of epitope recognition. Mol Ther. (2016) 24:375–84. doi: 10.1038/mt.2015.210
48. Dick TP, Bangia N, Peaper DR, Cresswell P. Disulfide bond isomerization and the assembly of MHC class I-peptide complexes. Immunity. (2002) 16:87–98. doi: 10.1016/S1074-7613(02)00263-7
49. Illing PT, van Hateren A, Darley R, Croft NP, Mifsud NA, King S, et al. Kinetics of abacavir-induced remodelling of the major histocompatibility complex class I peptide repertoire. Front Immunol. (2021) 12:672737. doi: 10.3389/fimmu.2021.672737
50. Porter KM, Hermann C, Traherne JA, Boyle LH. TAPBPR isoforms exhibit altered association with MHC class I. Immunology. (2014) 142:289–99. doi: 10.1111/imm.2014.142.issue-2
51. Brown LV, Wagg J, Darley R, van Hateren A, Elliott T, Gaffney EA, et al. De-risking clinical trial failure through mechanistic simulation. Immunother Adv. (2022) 2:ltac017. doi: 10.1093/immadv/ltac017
52. Di Marco M, Schuster H, Backert L, Ghosh M, Rammensee HG, Stevanovic S. Unveiling the peptide motifs of HLA-C and HLA-G from naturally presented peptides and generation of binding prediction matrices. J Immunol. (2017) 199:2639–51. doi: 10.4049/jimmunol.1700938
53. Creary LE, Guerra SG, Chong W, Brown CJ, Turner TR, Robinson J, et al. Next-generation HLA typing of 382 International Histocompatibility Working Group reference B-lymphoblastoid cell lines: Report from the 17th International HLA and Immunogenetics Workshop. Hum Immunol. (2019) 80:449–60. doi: 10.1016/j.humimm.2019.03.001
54. Lin Z, Bashirova AA, Viard M, Garner L, Quastel M, Beiersdorfer M, et al. HLA class I signal peptide polymorphism determines the level of CD94/NKG2-HLA-E-mediated regulation of effector cell responses. Nat Immunol. (2023) 24:1087–97. doi: 10.1038/s41590-023-01523-z
55. Llano M, Lee N, Navarro F, Garcia P, Albar JP, Geraghty DE, et al. HLA-E-bound peptides influence recognition by inhibitory and triggering CD94/NKG2 receptors: preferential response to an HLA-G-derived nonamer. Eur J Immunol. (1998) 28:2854–63. doi: 10.1002/(SICI)1521-4141(199809)28:09<2854::AID-IMMU2854>3.0.CO;2-W
56. Hill AV, Elvin J, Willis AC, Aidoo M, Allsopp CE, Gotch FM, et al. Molecular analysis of the association of HLA-B53 and resistance to severe malaria. Nature. (1992) 360:434–9. doi: 10.1038/360434a0
57. Falk K, Rotzschke O, Grahovac B, Schendel D, Stevanovic S, Jung G, et al. Peptide motifs of HLA-B35 and -B37 molecules. Immunogenetics. (1993) 38:161–2. doi: 10.1007/BF00190906
58. Steinle A, Falk K, Rotzschke O, Gnau V, Stevanovic S, Jung G, et al. Motif of HLA-B*3503 peptide ligands. Immunogenetics. (1996) 43:105–7. doi: 10.1007/BF00186615
59. Kaufman J. Generalists and specialists: A new view of how MHC class I molecules fight infectious pathogens. Trends Immunol. (2018) 39:367–79. doi: 10.1016/j.it.2018.01.001
60. Bouvier M, Wiley DC. Structural characterization of a soluble and partially folded class I major histocompatibility heavy chain/beta 2m heterodimer. Nat Struct Biol. (1998) 5:377–84. doi: 10.1038/nsb0598-377
61. Kurimoto E, Kuroki K, Yamaguchi Y, Yagi-Utsumi M, Igaki T, Iguchi T, et al. Structural and functional mosaic nature of MHC class I molecules in their peptide-free form. Mol Immunol. (2013) 55:393–9. doi: 10.1016/j.molimm.2013.03.014
62. Yanaka S, Ueno T, Shi Y, Qi J, Gao GF, Tsumoto K, et al. Peptide-dependent conformational fluctuation determines the stability of the human leukocyte antigen class I complex. J Biol Chem. (2014) 289:24680–90. doi: 10.1074/jbc.M114.566174
63. Wieczorek M, Sticht J, Stolzenberg S, Gunther S, Wehmeyer C, El Habre Z, et al. MHC class II complexes sample intermediate states along the peptide exchange pathway. Nat Commun. (2016) 7:13224. doi: 10.1038/ncomms13224
64. Wieczorek M, Abualrous ET, Sticht J, Alvaro-Benito M, Stolzenberg S, Noe F, et al. Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front Immunol. (2017) 8:292. doi: 10.3389/fimmu.2017.00292
65. van Hateren A, Elliott T. The role of MHC I protein dynamics in tapasin and TAPBPR-assisted immunopeptidome editing. Curr Opin Immunol. (2021) 70:138–43. doi: 10.1016/j.coi.2021.06.016
66. Muller IK, Winter C, Thomas C, Spaapen RM, Trowitzsch S, Tampe R. Structure of an MHC I-tapasin-ERp57 editing complex defines chaperone promiscuity. Nat Commun. (2022) 13:5383. doi: 10.1038/s41467-022-32841-9
67. Jiang J, Taylor DK, Kim EJ, Boyd LF, Ahmad J, Mage MG, et al. Structural mechanism of tapasin-mediated MHC-I peptide loading in antigen presentation. Nat Commun. (2022) 13:5470. doi: 10.1038/s41467-022-33153-8
68. van Hateren A, Elliott T. Visualising tapasin- and TAPBPR-assisted editing of major histocompatibility complex class-I immunopeptidomes. Curr Opin Immunol. (2023) 83:102340. doi: 10.1016/j.coi.2023.102340
69. Thomas C, Tampe R. Structure of the TAPBPR-MHC I complex defines the mechanism of peptide loading and editing. Science. (2017) 358:1060–4. doi: 10.1126/science.aao6001
70. Jiang J, Natarajan K, Boyd LF, Morozov GI, Mage MG, Margulies DH. Crystal structure of a TAPBPR-MHC I complex reveals the mechanism of peptide editing in antigen presentation. Science. (2017) 358:1064–8. doi: 10.1126/science.aao5154
71. Bailey A, van Hateren A, Elliott T, Werner JM. Two polymorphisms facilitate differences in plasticity between two chicken major histocompatibility complex class I proteins. PLoS One. (2014) 9:e89657. doi: 10.1371/journal.pone.0089657
72. Zacharias M, Springer S. Conformational flexibility of the MHC class I alpha1-alpha2 domain in peptide bound and free states: a molecular dynamics simulation study. Biophys J. (2004) 87:2203–14. doi: 10.1529/biophysj.104.044743
73. Sieker F, Springer S, Zacharias M. Comparative molecular dynamics analysis of tapasin-dependent and -independent MHC class I alleles. Protein Sci. (2007) 16:299–308. doi: 10.1110/ps.062568407
74. Lan H, Abualrous ET, Sticht J, Fernandez LMA, Werk T, Weise C, et al. Exchange catalysis by tapasin exploits conserved and allele-specific features of MHC-I molecules. Nat Commun. (2021) 12:4236. doi: 10.1038/s41467-021-24401-4
75. Abualrous ET, Fritzsche S, Hein Z, Al-Balushi MS, Reinink P, Boyle LH, et al. F pocket flexibility influences the tapasin dependence of two differentially disease-associated MHC Class I proteins. Eur J Immunol. (2015) 45:1248–57. doi: 10.1002/eji.201445307
76. Sun Y, Pumroy RA, Mallik L, Chaudhuri A, Wang C, Hwang D, et al. CryoEM structure of an MHC-I/TAPBPR peptide-bound intermediate reveals the mechanism of antigen proofreading. Proc Natl Acad Sci U S A. (2025) 122:e2416992122. doi: 10.1073/pnas.2416992122
77. Bennett EM, Bennink JR, Yewdell JW, Brodsky FM. Cutting edge: adenovirus E19 has two mechanisms for affecting class I MHC expression. J Immunol. (1999) 162:5049–52. doi: 10.4049/jimmunol.162.9.5049
78. Park B, Kim Y, Shin J, Lee S, Cho K, Fruh K, et al. Human cytomegalovirus inhibits tapasin-dependent peptide loading and optimization of the MHC class I peptide cargo for immune evasion. Immunity. (2004) 20:71–85. doi: 10.1016/S1074-7613(03)00355-8
79. Sokol L, Koelzer VH, Rau TT, Karamitopoulou E, Zlobec I, Lugli A. Loss of tapasin correlates with diminished CD8(+) T-cell immunity and prognosis in colorectal cancer. J Transl Med. (2015) 13:279. doi: 10.1186/s12967-015-0647-1
80. Shionoya Y, Kanaseki T, Miyamoto S, Tokita S, Hongo A, Kikuchi Y, et al. Loss of tapasin in human lung and colon cancer cells and escape from tumor-associated antigen-specific CTL recognition. Oncoimmunology. (2017) 6:e1274476. doi: 10.1080/2162402X.2016.1274476
81. Walker BA, Hunt LG, Sowa AK, Skjodt K, Gobel TW, Lehner PJ, et al. The dominantly expressed class I molecule of the chicken MHC is explained by coevolution with the polymorphic peptide transporter (TAP) genes. Proc Natl Acad Sci U S A. (2011) 108:8396–401. doi: 10.1073/pnas.1019496108
82. Tregaskes CA, Harrison M, Sowa AK, van Hateren A, Hunt LG, Vainio O, et al. Surface expression, peptide repertoire, and thermostability of chicken class I molecules correlate with peptide transporter specificity. Proc Natl Acad Sci U S A. (2016) 113:692–7. doi: 10.1073/pnas.1511859113
83. Hansen SG, Wu HL, Burwitz BJ, Hughes CM, Hammond KB, Ventura AB, et al. Broadly targeted CD8(+) T cell responses restricted by major histocompatibility complex E. Science. (2016) 351:714–20. doi: 10.1126/science.aac9475
84. DiBrino M, Parker KC, Margulies DH, Shiloach J, Turner RV, Biddison WE, et al. Identification of the peptide binding motif for HLA-B44, one of the most common HLA-B alleles in the Caucasian population. Biochemistry. (1995) 34:10130–8. doi: 10.1021/bi00032a005
85. Archbold JK, Macdonald WA, Gras S, Ely LK, Miles JJ, Bell MJ, et al. Natural micropolymorphism in human leukocyte antigens provides a basis for genetic control of antigen recognition. J Exp Med. (2009) 206:209–19. doi: 10.1084/jem.20082136
86. Walker S, Fazou C, Crough T, Holdsworth R, Kiely P, Veale M, et al. Ex vivo monitoring of human cytomegalovirus-specific CD8+ T-cell responses using QuantiFERON-CMV. Transpl Infect Dis. (2007) 9:165–70. doi: 10.1111/j.1399-3062.2006.00199.x
Keywords: MHC class I, tapasin, TAPBPR, peptide editing, peptide selection, immunopeptidome
Citation: Darley R, Illing PT, Duriez P, Bailey A, Purcell AW, van Hateren A and Elliott T (2025) Evidence of focusing the MHC class I immunopeptidome by tapasin. Front. Immunol. 16:1563789. doi: 10.3389/fimmu.2025.1563789
Received: 20 January 2025; Accepted: 14 April 2025;
Published: 08 May 2025.
Edited by:
Efstratios Stratikos, National and Kapodistrian University of Athens, GreeceReviewed by:
David H. Margulies, National Institute of Allergy and Infectious Diseases (NIH), United StatesAnne Halenius, University of Freiburg Medical Center, Germany
Copyright © 2025 Darley, Illing, Duriez, Bailey, Purcell, van Hateren and Elliott. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tim Elliott, dGltLmVsbGlvdHRAaW1tb25jLm94LmFjLnVr
†These authors share senior authorship