 Tian Zhang
Tian Zhang Liqun Li
Liqun Li Shengshi Huang
Shengshi Huang Maria N. Starodubtseva
Maria N. Starodubtseva Ju Liu
Ju Liu- 1Laboratory of Translational Medicine in Microvascular Regulation, Medical Research Center, The First Affiliated Hospital of Shandong First Medical University and Shandong Provincial Qianfoshan Hospital, Jinan, Shandong, China
- 2Shandong Provincial Key Medical and Health Laboratory of Translational Medicine in Microvascular Aging, The First Affiliated Hospital of Shandong First Medical University and Shandong Provincial Qianfoshan Hospital, Jinan, Shandong, China
- 3Laboraotry for Future Industry in Gene Editing in Vascular Endothelial Cells of Universities of Shandong Province, The First Affiliated Hospital of Shandong First Medical University and Shandong Provincial Qianfoshan Hospital, Jinan, Shandong, China
- 4Department of Clinical Laboratory, Qingdao Municipal Hospital, University of Health and Rehabilitation Sciences, Qingdao, Shandong, China
- 5Gomel State Medical University, Gomel, Belarus
- 6Institute of Radiobiology of NAS of Belarus, Gomel, Belarus
Vasculitis is a group of syndromes characterized by inflammation, presence of autoantibodies and endothelial cells (ECs) damage, which lead to stenosis or occlusion of the vascular lumen. Anti-endothelial cell antibodies (AECAs) are a heterogeneous group of autoantibodies in vasculitis. AECAs bind to antigens and membrane-bound proteins of ECs, inducing inflammation, coagulation, and apoptosis. In this review, we discuss the pathological role of AECAs in different types of vasculitis. In addition, AECAs potentially induce alterations of ECs mechanical properties, and subsequently promotes angiogenic phenotypes in the occurrence of vasculitis.
Introduction
Vasculitis encompasses a spectrum of immune-mediated diseases marked by the immune system’s aberrant attack on blood vessels in various body organs, such as the skin, lungs, and kidneys. This disorder is categorized based on the size of the affected blood vessel (1). In 2012, the Chapel Hill International Consensus Conferences established and standardized vasculitis nomenclature. The classification of vasculitis includes seven primary categories: large vessel vasculitis, medium vessel vasculitis, small vessel vasculitis, variable vessel vasculitis, single-organ vasculitis, vasculitis associated with systemic diseases, and vasculitis linked to probable etiology (2) (Table 1).
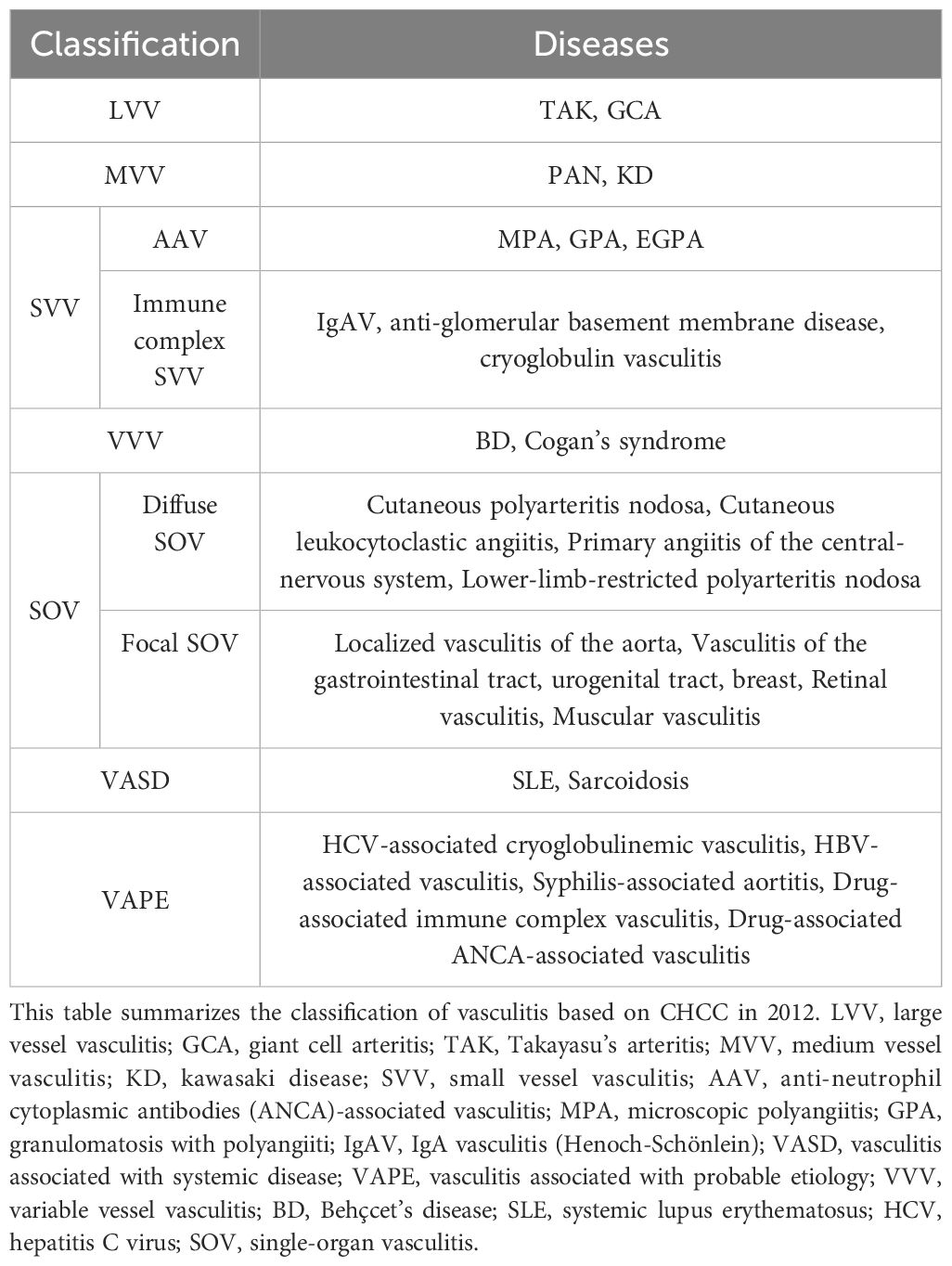
Table 1. The classification of vasculitis.
The occurrence of vasculitis typically precipitates vascular stenosis or occlusion, which in turn induces organ ischemia or contributes to the attenuation of blood vessel walls, thereby facilitating the formation of aneurysms or provoking hemorrhage. Central to this pathophysiological process is ECs injury, accompanied by the infiltration of leukocytes, which collectively underpin the common pathological mechanism inherent in such conditions (3). Contemporary research emphasizes the critical role of autoantibodies in the etiology of vasculitis. These autoantibodies in vasculitis predominantly target elements of the immune system or act directly against ECs (4–6). Anti-Endothelial Cell Antibodies (AECAs) are identified as one of the key autoantibodies in the context of vasculitis. These AECAs are often detected in patients diagnosed with vasculitis syndrome and are considered to be reliable indicators of both the activity and severity of the condition, as evidenced by various studies (7–10). AECAs have been identified in a spectrum of vasculitis disorders, encompassing Takayasu arteritis (TAK) (11), giant cell arteritis (GCA) (12), Kawasaki disease (KD) (13), granulomatosis with polyangiitis (GPA) (14), microscopic polyangiitis (MPA) (15), IgA vasculitis (IgAV) (16), Behçet’s disease (BD) (17), systemic lupus erythematosus (SLE) (18), and sarcoidosis (19). The presence of AECAs in vasculitis highlights their potential role in the pathogenesis and clinical progression of vasculitis. The pathological mechanisms include activation of ECs, induction of coagulation and apoptosis. The above conditions in turn lead to angiogenesis and changes in the mechanical properties of ECs. In this review, we summarized our current understanding of the diverse mechanisms of AECAs in ECs dysfunction, highlighting the angiogenesis and cellular mechanics properties of AECAs in vasculitis.
AECAs characteristics
Antigen types
AECAs are circulating antibodies that recognize various antigenic determinants on ECs (20). Most scholars believe that AECAs’ antigens are classified into three categories: “planted” antigens, constitutively expressed antigens, and cryptic antigens (21, 22).
“Planted” antigens adhere to the endothelium either directly, such as myeloperoxidase (MPO) and β2-glycoprotein I (β2-GPI), or indirectly adhere through DNA or DNA-histone complexes. Because of the presence of “planted” antigens, AECAs are capable cross-link with many autoantibodies (23). Constitutively expressed antigens include human leukocyte antigen (HLA) I antigens, cardiolipin, and other phospholipid components, as well as extracellular matrix components (24). HLA I antigens present on ECs represent some of the antigenic epitopes for AECAs. Cardiolipin and other phospholipid structures are some of the antigenic determinants for a subset of AECAs in vasculitis. In addition, different extracellular matrix components such as collagen type II, IV, VII, vimentin, tropomyosin, and laminin are also target antigens for AECAs (25). Cryptic antigens include HLA II antigen and proteinase 3 (PR3). HLA II and PR3 determinants are present only on activated ECs and represent target antigens for AECAs (26). Cytokine activation human umbilical vein endothelial cells (HUVECs) lead to PR3 translocation from the cytoplasm to the cell membrane. This is followed by the development of vasculitis in conjunction with AECAs.
Other antigens include α-enolase, heat shock protein (HSP) 60, HSP70, ribosomal P protein P0, calreticulin, and tubulin (27–29). Besides, adhesion molecules such as intercellular cell adhesion molecule-1 (ICAM-1) also serve as specific target antigens for AECAs (29) (Table 2). Numerous target antigens for AECAs in vasculitis have been studied, including HLA in Takayasu’s arteritis and KD (30, 31), β2-GPI in GCA (32) and IgAV (33), DNA, PR3, MPO and phospholipid in anti-neutrophil cytoplasmic antibodies-associated vasculitis (34), HSP60, HSP70 in GCA and KD (35). However, the specific pathogenesis has not been elucidated (Table 2).
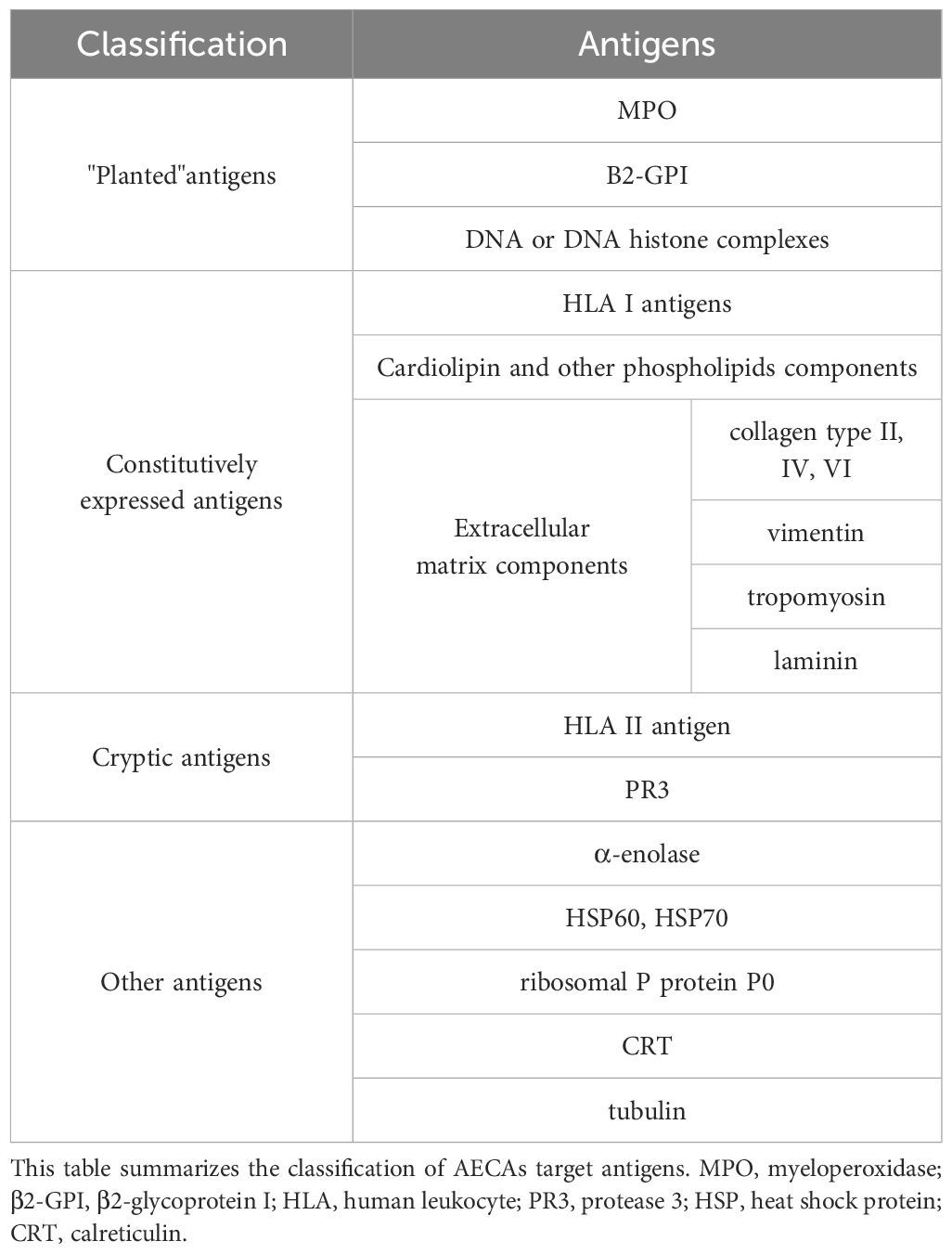
Table 2. The classification of AECAs target antigens.
However, it is crucial to emphasize that AECAs exhibit cross-reactivity with multiple cell types, as their epitopes are expressed not only on ECs but also on fibroblasts, leukocytes, and monocytes (36). Although ECs models are commonly used in AECA studies, their biological properties differ from pathological targets. For example, HUVECs derive from embryonic umbilical veins. These cells exhibit distinct phenotypic features, including short-chain von Willebrand factor (vWF) multimers, compared to adult arterial or capillary ECs. This model-target mismatch causes discrepancies between experimental and clinical findings. Vascular bed heterogeneity influences AECAs targeting specificity. In sarcoidosis, AECAs react more strongly with bone marrow ECs than HUVECs. In BD, they show higher reactivity with omental microvascular ECs versus HUVECs. Conversely, in TAK, AECAs activate HUVECs but not microvascular ECs (23, 37). Additionally, epigenetic modifications (e.g., DNA methylation) in ECs show significant heterogeneity, potentially influencing antigen accessibility and immunogenicity. Furthermore, AECAs detection has limited diagnostic value in clinical practice. These antibodies lack a specific target antigen to guarantee immune specificity. Current detection methods show suboptimal accuracy and face technical challenges in standardization (38).
Pro-inflammatory effects
Under normal conditions, leukocytes race in laminar blood and do not adhere to ECs (39). As inflammation progresses, ECs produce tumor necrosis factor-α (TNF-α), interleukin (IL)-1, IL-6, enhance the expression of ICAM-1, vascular cell adhesion molecule 1 (VCAM-1), and synthesize E-selectin, causing leukocytes to roll on ECs (40–43). E-selectin binds to leukocytes through their ligands and promotes leukocyte adhesion (44). There is evidence that AECAs bind to β2-GPI on the surface of ECs, up-regulating IL-1, TNF-α, ICAM-1, VCAM-1 and E-selectin, and increasing leukocyte adhesion to ECs (45–47). Leukocyte adhesion leads to endothelial dysfunction, decreased endothelium-dependent vasodilation, excessive capillary filtration, and increased protein leakage in venules (48) (Figure 1). The increased expression of AECAs mediates ECs injury and activates the nuclear factor-κB (NF-κB) signaling pathway (49). NF-κB activation occurs through the degradation of the inhibitor of NF-κB (IκB). The degradation of IκB requires the activation of IκB kinase (IKK) to phosphorylate IκB. IKK increases IκB degradation, leading to nuclear translocation of NF-κB subunit and NF-κB mediated expression of pro-inflammatory cytokines (50). Leukocyte adhesion is further promoted by up-regulating the expression of IL-1, TNF-α, ICAM-1, VCAM-1, and E-selectin (51–53) (Figures 1, 2).

Figure 1. AECAs pro-inflammatory effects in ECs. AECAs bind to antigens on the surface of ECs and release IL-1β, TNF-α, E-selectin, ICAM-1, and VCAM-1 to promote leukocyte rolling. ICAM-1, VCAM-1, and E-selectin bind to leukocytes via ligands to promote adhesion and eventual leukocyte crossing of the endothelial gap (AECAs, anti-endothelial cell antibodies; ICAM-1, intercellular cell adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1; IL-1β, Interleukin-1β; TNF-α, tumor necrosis factor-α).

Figure 2. AECAs pro-apoptotic and pro-inflammatory effects in ECs. On the left, AECAs regulate mitochondrial pathway-mediated apoptosis through NO and HSP60 production. When ECs are injured, HSP60 is located on the plasma membrane and separated from Bax. With the decrease of HSP60, Bax moves from the cytoplasm to the mitochondria and is accompanied by the release of Cytoc, which activates caspase-3 and induces apoptosis in ECs. FcγRШ mediates NK cells, binds to AECAs and directly kills antibody-linked target cells. AECAs causes ECs apoptosis through Fas/FasL interaction. Upon binding of Fas to its cognate ligand FasL, Fas is recruited in the cytoplasm via the death domain of the intracellular FADD and caspase-8-associated Fas, forming a death-inducing signaling complex consisting of Fas, FADD and caspase-8, which activates caspase-3 and ultimately leads to apoptosis. On the right, AECAs interact with ECs antigens and activate IKK. Activated IKK phosphorylates IκB protein and induces ubiquitination and degradation of IκB, which subsequently leads to NF-κB activation. After NF-κB translocation into the nucleus, the expression of IL-1, TNF-α, ICAM-1 and VCAM-1 is upregulated (AECAs, anti-endothelial cell antibodies; ECs, endothelial cells; NO, nitric oxide; HSP60, heat shock protein 60; NK, Natural Killer; IκB, inhibitor of NF-κB; IKK, IκB Kinase; Fas, factor associated suicide; FasL, Fas ligand; FADD, Fas-associating protein with a novel death domain; IL-1, Interleukin-1; TNF-α, tumor necrosis factor-α; ICAM-1, intercellular cell adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1).
Pro-coagulant effects
ECs have the function of maintaining blood flow and regulating blood coagulation (54). When ECs are damaged, coagulation becomes dysfunctional, leading to occlusive thrombosis, local tissue and organ necrosis (55). vWF and tissue factor (TF) are essential indicators of thrombosis caused by ECs injury (56–58). Evidence suggests that AECAs stimulate ECs to secrete vWF and its ultra-large polymers (ULvWF) (13, 56). ULvWF located on the surface of damaged ECs interacts with platelets (59). Platelets are activated by vWF and form thrombus with vWF and fibrin in damaged ECs. Activated platelets release P-selectin and promote the release of neutrophil extracellular trap (NET), comprising condensed chromatin and histone. Histone induce platelet aggregation and platelet accumulation (60, 61). In addition, the interaction between AECAs and ECs surface antigen leads to the production of tissue factor (TF), and TF activity is dose-dependent on the AECAs titer, contributing to thrombosis (13, 56, 62). In summary, AECAs binding to ECs promotes the release of vWF, ULvWF, and TF, causing inflammation and thrombosis (Figure 3).

Figure 3. AECAs pro-coagulant effects in ECs. AECAs act on ECs, resulting in damage to ECs and release of coagulation factors such as vWF and TF. Platelets are activated by vWF and TF. Thrombus formation with vWF, TF and fibrinogen in damaged ECs (AECAs, anti-endothelial cell antibodies; ECs, endothelial cells; vWF, Von Willebrand factor; TF, tissue factor).
Pro-apoptotic effects
At present, there are two ways to induce apoptosis: the mitochondrial pathway, also known as the endogenous pathway, activates caspase by regulating mitochondrial membrane permeability and releasing apoptosis-activating factors (63). The death receptor-mediated pathway activates caspase by combining extracellular death receptors and corresponding ligands (64, 65). Both pathways are characterized by caspase activation. The following evidence provides that how AECAs induce ECs apoptosis through two pathways (66) (Figure 2).
Mitochondrial pathway
Mitochondrial proteins such as heat shock protein 60 (HSP60) and nitric oxide (NO) mediate ECs apoptosis. HSP60 exists in mitochondria. 15% to 20% of HSP60 exists in the cytoplasm and forms a complex with Bax in the cytoplasm (67). When ECs are stressed or damaged, HSP60 is located on the plasma membrane and separated from Bax. With the decrease of HSP60, Bax moves from the cytoplasm to mitochondria and is accompanied by the release of cytochrome c, and apoptosis is triggered. As a result, apoptosis is initiated, HSP60 is released, and this, in turn, accelerates the activation of procaspase-3 (67). The abnormal expression of HSP60 on the cytoplasmic membrane makes ECs susceptible to apoptosis (68). As a result, HSP60 dissociates from the plasma membrane and interacts with AECAs. Bax moves to mitochondria, leading to the release of cytochrome c and further activation of caspases-3, triggering apoptosis (69). Furthermore, ECs undergo apoptosis via the mitochondria-dependent pathway regulated by NO production (70). There is evidence that AECAs induce ECs apoptosis through a NO-mediated mechanism in dengue virus infection (66). NO-regulated ECs injury thus plays a role in disrupting ECs integrity and contributing to the pathogenesis of vasculopathy induced by AECAs. In conclusion, AECAs exert apoptosis by acting on ECs through the mitochondrial pathway via mitochondrial-associated proteins such as HSP60 and NO.
Fas/FasL pathway
AECAs contribute to the apoptosis of ECs through antibody-dependent cell-mediated cytotoxicity (ADCC), underscoring their role in the pathophysiological mechanisms affecting vascular integrity and function (71). Natural Killer (NK) cells are the mediators of ADCC. FcγRIII mediates on NK cells, binding to AECAs (72), and directly kills the target cells attached to the antibody. NK cells have two different cytotoxic mechanisms. The first is granule-mediated apoptosis, which depends on the synergistic effect of the perforin and serine protease granzymes (73). The other is factor-associated suicide (Fas)/Fas ligand (FasL) interaction mediated apoptosis (74). Existing research indicates that the granzyme A gene is expressed in systemic sclerosis, suggesting that granzyme mediates ECs injury (71), but whether AECAs are induced by granzyme A expression has not been functionally confirmed. A study has confirmed that AECAs induce apoptosis of human dermal microvascular ECs through Fas, while blocking anti-FasL antibodies inhibited ECs apoptosis (75). Therefore, AECAs may cause apoptosis of ECs through the Fas/FasL interaction. After Fas binds to its homologous ligand FasL, Fas recruits Fas-associated death domain (FADD) and caspase-8/10 in the cytoplasm through the death domain of the intracellular segment, forming a death-inducing signal complex composed of Fas, FADD and caspase-8/10, which activates caspases-3 and eventually leads to apoptosis (76, 77).
Pathological role of AECAs in vasculitis
In examining the pathological role of AECAs in vasculitis, it is imperative to acknowledge their pervasive presence across a multitude of vasculitic disorders. The function of AECAs extends beyond serving merely as biomarkers of disease; they play a pivotal role in the pathophysiology of disease development and progression (12, 78). We will delve into the specific roles of AECAs in various forms of vasculitis, as well as their impact on clinical manifestations of these diseases (as shown in Table 3).
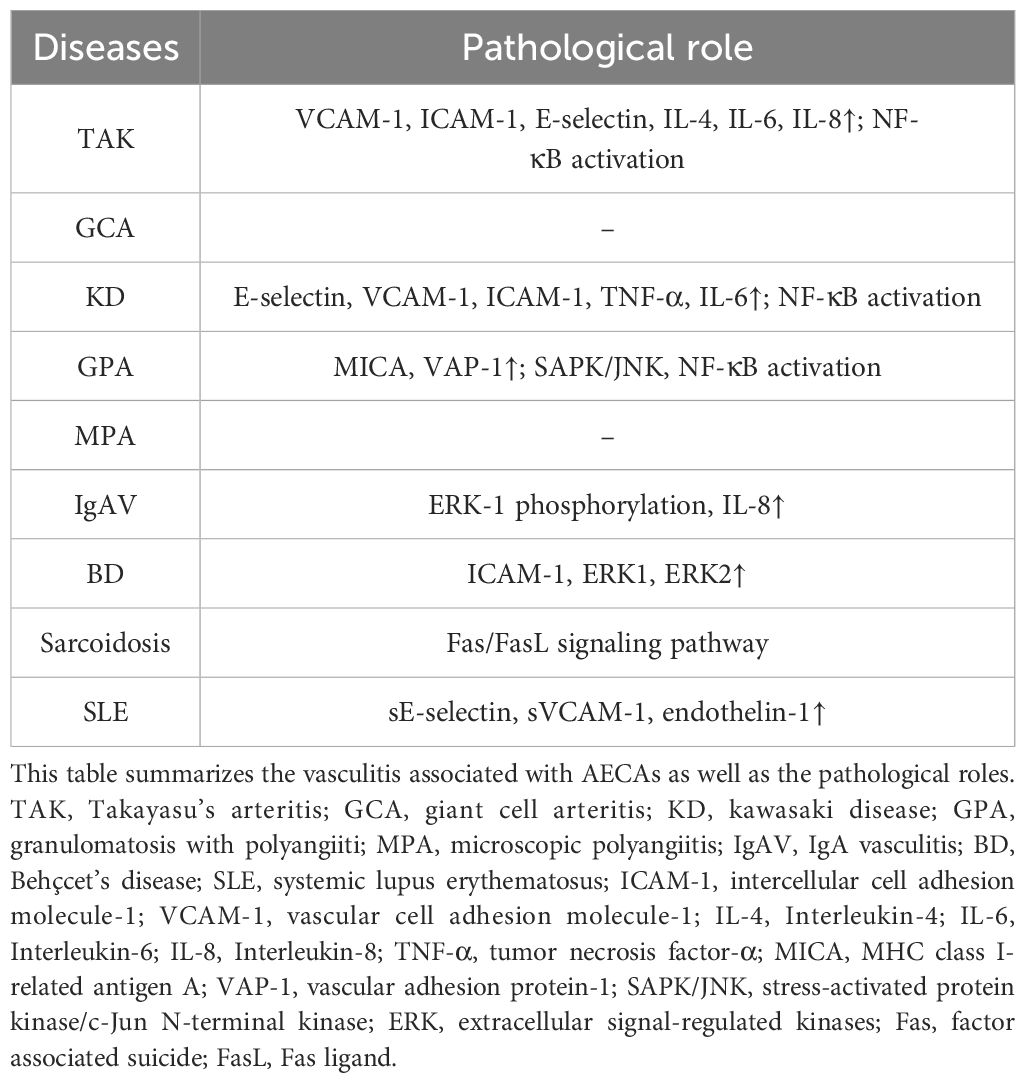
Table 3. The pathological role of AECAs in vasculitis.
Large vessel vasculitis
TAK is a chronic idiopathic granulomatous large-vessel vasculitis that affects the aorta, its main branches, and pulmonary arteries (79). Evidence has shown that AECAs are related to the pathogenesis of TAK (78). The positive rates of IgG and IgM AECAs in TAK patients were over 68%, and the titer of IgM AECA was higher (80). Study shows that most patients with TAK have circulating AECAs, which are directed predominantly to a triplet of aortic ECs antigens ranging in size from 60kD to 65kD. These AECAs induce the expression of VCAM-1 and E-selectin, as well as the production of inflammatory cytokines such as IL-4, IL-6, and IL-8 by aortic ECs, leading to the apoptosis of aortic ECs (81). Another study found that AECAs derived from a single patient with TAK were found to activate HUVEC, as shown by increased secretion of IL-6, vWF and increased expression of VCAM-1, ICAM-1, E-selectin, associated with NF-κB activation and increased adhesion of monocytes to these cells (82).
GCA, also known as temporal arteritis, is a primary systemic vasculitis involving large and medium vessels. However, with the advancement of vascular imaging technology, GCA is now considered to be a systemic disease beyond the superficial temporal artery, which causes large artery stenosis or aortic involvement (aortitis, aneurysm formation, or dissection) (83–86). A study has confirmed that IgG, IgM, and IgA AECA are positive in GCA patients, with IgA AECA is highly expressed, and IgG AECA plays a vital role in maintaining homeostasis (12).
Medium vessel vasculitis
KD is the most representative disease of medium vessel vasculitis. The research indicates that AECAs are related to KD pathogenesis and clinical diagnosis. E. Grunebaum (87) et al. confirmed that AECAs activated ECs, thereby increasing the secretion of IL-6, the expression of adhesion molecules such as E-selectin, VCAM-1, ICAM-1, and the adhesion of U937 cells to HUVECs. A study has found that the high expression of E-selectin in acute KD is related to the activation of NF-κB in vascular ECs and then increase the release of TNF-α (88). In conclusion, AECAs activate ECs through the NF-κB signaling pathway and increase the expression of cytokines such as IL-6, TNF-α and adhesion factors such as VCAM-1, ICAM-1, E-selectin, leading to impaired function of ECs, and participating in the pathogenesis of KD.
Small vessel vasculitis
GPA was formerly known as Wegener’s granuloma, and is characterized by necrotizing vasculitis of small vessels and granulomatous inflammation (89). Current research has confirmed that AECAs are a critical factor in the pathogenesis of GPA. Many patients with GPA have AECAs that react with human kidney microvascular ECs. Stimulation of human kidney microvascular ECs with IgG AECA upregulated MHC class I - related antigen A (MICA) and vascular adhesion protein-1 (VAP-1) expression, triggered rapid Ca2+ flux, induced stress-activated protein kinase (SAPK)/c-Jun N-terminal kinase (JNK), specific phosphorylation of transcription factor c-Jun and activating transcription factor-2, and activated NF-κB. However, specific SAPK/JNK inhibitors significantly reduced AECAs-induced chemokine production and phosphorylation of c-Jun and activating transcription factor-2 and eliminated MICA protein expression (14). Taken together, the elevated expression of AECAs mediates the pathogenesis of GPA and is associated with the SAPK/JNK pathway and the endothelial inflammatory protein VAP-1.
MPA is a systemic autoimmune necrotizing vasculitis. A reported study showed the presence of AECAs in patients with MPA and suggested a correlation between AECAs titer and disease activity (15). Régent et al. confirmed that purified serum IgG from patients with MPA induced extracellular regulated kinase (ERK) phosphorylation in ECs than IgG from healthy controls in vitro, which supports a possible pathogenic role of AECAs in MPA (90).
IgAV, also known as Henoch-Schönlein. IgAV is a type of vasculitis with IgA1-dominated immune deposits affecting small vessels (capillaries, veins or arterioles) (91). Studies found that AECAs are involved in the pathogenesis of IgAV. Yang YH (92)and Cynthia C (16)et al showed that AECAs were significantly elevated in patients with acute Henoch-Schönlein (16, 92). The mechanism of occurrence is related to the binding of AECAs to EC antigen, leading to ERK-1 phosphorylation, which activates protein-1 phosphorylation and promotes IL-8 expression (93).
Variable vessel vasculitis
BD is a variable-vessel vasculitis with a predominance of recurrent thrombophlebitis, thrombosis and cutaneous vasculitis (94). Direskeneli H (95) found a high rate of positive AECAs in patients with BD. AECAs are associated with disease activity in BD. Studies have found that AECAs positive sera from BD patients lead to changes in the expression of adhesion molecules on the cell surface of human dermal microvascular ECs (HDMECs) and promote the adherence of T lymphocytes to HDMECs and, thereby initiating or amplifying inflammatory vascular injury (96). IgM AECAs play a pathogenetic role in BD by activating ECs directly. In addition, extracellular signal-regulated kinases (ERK) 1 and 2 were involved in the expression of ICAM-1 on HDMECs stimulated by AECAs (97, 98). Although the exact pathogenesis of BD is still unknown, it seems that it involves at least three steps: activation of ECs, adhesion molecule expression, and lymphocyte adhesion. Activation of ECs facilitates leukocyte traffic and thus initiate an inflammatory injury.
Vasculitis associated with systemic disease
Sarcoidosis is an inflammatory disease characterized by granuloma (abnormal inflammatory cell mass) in almost all organs that usually affects the lungs, lymph nodes, skin, and eyes (99). Naoki Inui et al. showed that AECAs positive rate and AECAs level in sarcoidosis patients’ serum and bronchoalveolar lavage fluid were significantly increased (20). Initial AECAs levels were further elevated in patients with multiple organ involvement or requiring glucocorticoid therapy. AECAs leading to ECs apoptosis were found to be mediating the pathogenesis of sarcoidosis (100). The mechanism induced by ADCC via Fas/FasL interactions (75). Taken together, AECAs induces ECs apoptosis through the Fas/FasL pathway, which is one of the pathological mechanisms in the development of sarcoid vasculitis.
SLE is a chronic autoimmune disease characterized by a multisystem disorder caused by immune dysregulation (101). Study confirms that AECAs are useful diagnostic and prognostic tools for SLE patients (102, 103). The expression of AECAs is increased at the initial stage of vascular injury in SLE. In SLE, AECAs demonstrate significant correlations with elevated serum concentrations of sE-selectin and sVCAM-1, mechanistically contributing to ECs activation through enhanced leukocyte adhesion and pro-inflammatory signaling (46, 104, 105). Notably, ribosomal P0 protein has been identified as an autoantigen recognized by AECAs, with its immunoreactivity showing specific association with SLE disease manifestations (106). Experimental evidence indicates that IgM-class AECAs co-localizing with immune complexes induce endothelial endothelin-1 overexpression, mediating the initiation and progression of microvascular injury in SLE patients (107). Furthermore, anti-phospholipid antibodies and AECAs exhibit functional cross-reactivity. Specific anti-annexin V antibodies bind to membrane-associated epitopes, inducing phosphatidylserine exposure and activating apoptotic pathways in ECs. These mechanisms are implicated in SLE-associated vascular pathology (29, 108).
Pathogenic mechanisms of AECAs in vasculitis
Angiogenesis
Angiogenesis in vasculitis is related to inflammatory factors secreted by ECs. Research has shown that in GCA, when a large amount of carriers of free hemoglobin with angiogenic properties are produced, the levels of TNF-α and IL-6 were increased. In GCA vasculopathy, leukocyte constitutive (PECAM-1, ICAM-1, ICAM-2, and P-selectin) and inducible (E-selectin and VCAM-1) ECs adhesion molecules are overexpressed by ECs of adventitial micro-vessels and neo-vessels (109–111). The interactions of leukocytes with these ligands are responsible for forming inflammatory infiltrates in GCA lesions (112). GCA is characterized by the release of pro-inflammatory cytokines such as IL-1, TNF-α, and IL-6 during the acute systemic phase (113), which influence vascular responses such as vessel occlusion or regeneration and participate in the pathogenesis of GCA inflammatory lesions (114). In KD, ECs of newly formed vessels in coronary aneurysms express E-selectinandVCAM-1, which is involved in leukocyte adhesion to ECs (115). In contrast, luminal ECs of coronary arteries without aneurysms do not express E-selectin and VCAM-1, such as ECs of newly formed vessels in polyarteritis nodosa and GCA (112). In conclusion, angiogenesis is involved in the occurrence and development of vasculitis. Newly formed blood vessels express leukocyte adhesion molecules, such as VCAM-1, ICAM-1, and E-selectin, providing a new location for leukocytes to invade the blood vessel wall (112). In addition, new vessels expand the surface of ECs, providing an additional source of cytokines and chemokines that amplify the inflammatory process (116).
Studies have shown that AECAs induce the activation of ECs and secretion of inflammatory cytokines such as IL-1β and TNF-α, coagulation factors such as vWF, and adhesion molecules such as ICAM-1, VCAM-1, E-selectin, leading to vascular inflammation and occlusion, and participating in the occurrence of vasculitis (117, 118). After AECAs induce vascular damage, minimal proliferation, fibrosis, and thrombosis lead to narrowing or occlusion of the vascular lumen, causing tissue hypoxia and ischemia. The hypoxia-ischemic environment resulting from vascular lumen stenosis or occlusion is a powerful signal for new angiogenesis, ultimately leading to angiogenesis (119).
Mechanical properties
Mechanical properties are significant for the normal functioning of ECs. They determine the integrity and mechanical stress resistance of an ЕС monolayer and regulate the functions of the endothelium under constant mechanical load from the side of the blood flow. Cells are known to be viscoelastic materials, and the origin of their mechanical properties is determined by the cytoskeleton (120). The cytoskeleton is a multi-hierarchical network structure within the cell with protein fibers as the main component, consisting of three types of protein fibers: actin filaments, tubulin microtubules, and a group of polymers known collectively as intermediate filaments. The cytoskeleton has several broad functions: it spatially organizes cellular contents; it connects the cell physically and biochemically to the external environment; it is involved in intracellular and extracellular transport and signal transduction; it generates coordinated forces that enable cells to move and change shape (121).
Several works reported cytoskeletal remodeling in response to the ligation of AECAs to ECs (122–125). Stimulation of ECs with HLA class I was revealed to activate stress fiber formation via a mechanism that did not include any detectable change in intracellular Ca2+ concentration, but induced Myosin light-chain phosphorylation and stress fiber assembly involving myosin light-chain (MLC) kinase and Rho-kinase (ROCK) in an ERK1/2-dependent manner. Molecular aggregation of HLA class I molecules with antibodies leads to the recruitment of integrin ß4 and the subsequent activation of intracellular signals that increase Rho-GTP activity, induce phosphorylation of ROCK, and trigger the assembly and phosphorylation of focal adhesion kinase, Src and paxillin at the focal adhesions to stimulate actin reorganization (124). The ligation of HLA class II to ECs was shown to induce necrotic cell death via a mechanism of lysosomal membrane permeabilization involving the reorganization of the actin cytoskeleton and the formation of actin stress fibers. The effect was downregulated by the actin polymerization inhibitor cytochalasin D and inhibition of Rho GTPases (125). More early work revealed that autoantibodies from a subset of advanced type 2 diabetes activate ROCK, and induce stress fiber formation and apoptosis in ECs (122). Changes in the stiffness of ECs occur in vascular inflammation (126). The stress fiber formation can increase the EC stiffness by a factor of 2-10 (127).
The mechanical properties of ECs depend also on the mechanical properties of their environment. An increase in substrate rigidity correlates with an overall increase in EC stiffness and apparent viscosity that is associated with the reorganization of actin cytoskeleton. Conversely, the cells on the soft substrate were more deformable and less viscous that is related to actin disordering (128). Inflammation, as a pathological factor of vasculitis, leads to increased vascular stiffness, and vascular stiffness is correlated with the degree of inflammation (129, 130). Inflammatory mediators alter the mechanical properties and permeability of ECs (131, 132). ECs barrier protection mediators, such as prostaglandin (PG) E2, sphingosine 1-phosphate and PGI2, reduce EC stiffness and permeability (131, 133). AECAs bind to specific antigens on the ECs and support to the EC release of mediators such as ICAM-1 and TNF-α which increase EC stiffness and permeability, leading to ECs barrier impairment (126, 134).
Pro-inflammatory cytokines stimulate or inhibit the formation of actin cytoskeletal structures. EC stiffness increases when affected by TNF-α (135). The RhoA pathway regulates TNF-α-induced cytoskeleton rearrangement, leading to increased ECs permeability (134, 136). The RhoA/ROCK pathway can regulate vascular function because changes in the actin cytoskeleton are fundamental to vascular contraction and remodeling, inflammatory cell recruitment, and cell proliferation (137). RhoA has been extensively studied as a regulator of vascular leakage and leukocyte migration through the endothelium. RhoA signals through the activation of ROCK, which inhibits MLC phosphatase and induces the phosphorylation of MLC (p-MLC) (138, 139). This process enhances the formation of actin bundles, fibers, and stretched actin structures and promotes the loss of VE-cadherin mediated cell-cell contacts, leading to vascular leakage (140–142). However, whether AECAs activate the RhoA signaling pathway by binding to antigens on the ECs’ surface and producing inflammatory factors, which leads to cytoskeletal rearrangement and increased cell stiffness and permeability, ultimately resulting in the development of vasculitis, has not been investigated and requires follow-up experiments for verification (Figure 4).

Figure 4. AECAs alter ECs mechanics properties. AECAs bind to specific antigens on ECs, and upon activation of the RhoA signaling pathway, ROCK is activated, inhibiting MLCP and inducing p-MLC, leading to cytoskeletal reorganization, increased ECs stiffness, and increased permeability (ROCK, Rho-related kinase; MLCP, myosin light chain phosphatase; p-MLC, MLC phosphorylation).
Despite the types of AECAs that are already known today (143, 144), there is little information about their specific antigens in ECs. Only a few antigens targeted by AECAs were identified in vasculitis (38). Among these antigens, some antigens are directly related to the cytoskeleton, such as tropomyosin and vimentin. Literature data show that tropomyosin regulates cell stiffness in a complex way via the generation of specific populations of actin filaments containing tropomyosin isoforms (145). Vimentin maintains cell mechanical properties, motility, adhesion, and other signaling pathways that protect against compressive stress and preserve mechanical integrity by enhancing cell elastic behavior (146). Several antigens, like peroxiredoxins and MPO, affect cytoskeletal activity. MPO induces actin cytoskeleton reorganization and affects mechanical stiffness, as found in human platelets (147). Peroxiredoxins interact with collapsing response mediator protein 2 that regulates microtubule structure, for example, during lymphocyte migration and neuronal development (148). Other molecular targets, like HSP60 are associated with mechanisms for regulating the functions and pathways of cell death, such as apoptosis, resulting in decreased arterial elasticity (69, 149, 150). A recent AFM study highlighted the vital role of microtubules in shaping ECs mechanics. It showed that the disruption of microtubules by exposing the cells to colchicine caused the cell to stiffen, the relaxation times increased, and the adhesion between the tip and cell decreased (151). Actin and vimentin cytoskeleton reorganization and cell stiffening have been recently detected in ECs after blocking CD109 antigen, a regulator of many signaling pathways, using anti-CD109 antibodies (152).
Conclusion
AECAs target diverse antigenic epitopes, including planted antigens, constitutively expressed surface molecules, and cryptic antigens exposed during cellular stress. These interactions can induce ECs injury through ADCC mechanisms. Additionally, AECAs exhibit pro-angiogenic properties and may alter ECs mechanical properties, such as cytoskeletal integrity and intercellular junction stability, thereby promoting vascular hyperpermeability. Collectively, these pathophysiological processes contribute to the pathogenesis of vasculitis. However, AECAs exhibit considerable heterogeneity in their prevalence across vasculitis, and the correlation between antibody titers and clinical disease activity remains inconsistent across studies. Current limitations in their clinical utility stem from suboptimal specificity and the absence of standardized detection protocols. Therefore, methodologically rigorous studies are required to establish reproducible assay systems for AECAs quantification. Such standardization is critical to elucidate the precise mechanistic roles of AECAs in vasculitis progression and to evaluate their potential as therapeutic targets or disease-monitoring biomarkers.
Author contributions
TZ: Conceptualization, Writing – review & editing, Writing – original draft. LL: Writing – review & editing. SH: Writing – review & editing. MS: Writing – review & editing. JL: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Nature Science Foundation of China (82241030,82171318 82011530024), Medical and Health Science and Technology Department Project of Shandong (202403020702), State Scientific Research Program "Natural Resources and the Environment"(3.01,2021-2025).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mishra K, Ramcharitar RK, Sharma AM. Vasculitis: when to consider this diagnosis? Med Clinics North America. (2023) 107:845–59. doi: 10.1016/j.mcna.2023.05.005
2. Jennette JC. Overview of the 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Clin Exp Nephrol. (2013) 17:603–6. doi: 10.1007/s10157-013-0869-6
3. Morita T, Trés GFS, Criado RFJ, Sotto MN, Criado PR. Update on vasculitis: an overview and dermatological clues for clinical and histopathological diagnosis - part I. An Bras Dermatologia. (2020) 95:355–71. doi: 10.1016/j.abd.2020.01.003
4. Csernok E. The diagnostic and clinical utility of autoantibodies in systemic vasculitis. Antibodies (Basel Switzerland). (2019) 8(2):31. doi: 10.3390/antib8020031
5. Segelmark M, Björck L. Streptococcal enzymes as precision tools against pathogenic igg autoantibodies in small vessel vasculitis. Front Immunol. (2019) 10:2165. doi: 10.3389/fimmu.2019.02165
6. Li W, Huang H, Cai M, Yuan T, Sheng Y. Antineutrophil cytoplasmic antibody-associated vasculitis update: genetic pathogenesis. Front Immunol. (2021) 12:624848. doi: 10.3389/fimmu.2021.624848
7. Christophe J, Christophe D, Jean-Eric A, Sandrine J, Alain S, Lo?C G, et al. Induction of endothelial cell apoptosis by the binding of anti-endothelial cell antibodies to hsp60 in vasculitis-associated systemic autoimmune diseases. Arthritis Rheumatol. (2014) 52:4028–38. doi: 10.1002/art.21401
8. Sebastian JK, Mahr AD, Ahmed SS, Stone JH, Zurina RP, Davis JC, et al. Antiendothelial cell antibodies in patients with wegener’s granulomatosis: prevalence and correlation with disease activity and manifestations. J Rheumatol. (2007) 34:1027–31.
9. Karasawa R, Yudoh K, Ozaki S, Kato T. Anti-endothelial cell antibodies (Aeca) in patients with systemic vasculitis: our research using proteomics. Expert Opin Biol Ther. (2011) 11:77–87. doi: 10.1517/14712598.2011.540234
10. Guilpain P, Mouthon L. Antiendothelial cells autoantibodies in vasculitis-associated systemic diseases. Clin Rev Allergy Immunol. (2008) 35:59–65. doi: 10.1007/s12016-007-8069-3
11. Mutoh T, Shirai T, Ishii T, Shirota Y, Fujishima F, Takahashi F, et al. Identification of two major autoantigens negatively regulating endothelial activation in takayasu arteritis. Nat Commun. (2020) 11:1253. doi: 10.1038/s41467-020-15088-0
12. Régent A, Dib H, Ly KH, Agard C, Tamby MC, Tamas N, et al. Identification of target antigens of anti-endothelial cell and anti-vascular smooth muscle cell antibodies in patients with giant cell arteritis: A proteomic approach. Arthritis Res Ther. (2011) 13:R107. doi: 10.1186/ar3388
13. Sakurai Y. Autoimmune aspects of kawasaki disease. J Investig Allergol Clin Immunol. (2019) 29:251–61. doi: 10.18176/jiaci.0300
14. Holmén C, Elsheikh E, Christensson M, Liu J, Johansson AS, Qureshi AR, et al. Anti endothelial cell autoantibodies selectively activate sapk/jnk signalling in wegener’s granulomatosis. J Am Soc Nephrol: JASN. (2007) 18:2497–508. doi: 10.1681/asn.2006111286
15. Savage CO, Pottinger BE, Gaskin G, Lockwood CM, Pusey CD, Pearson JD. Vascular damage in wegener’s granulomatosis and microscopic polyarteritis: presence of anti-endothelial cell antibodies and their relation to anti-neutrophil cytoplasm antibodies. Clin Exp Immunol. (1991) 85:14–9. doi: 10.1111/j.1365-2249.1991.tb05675.x
16. Patterson CC, Ross P Jr., Pope-Harman AL, Knight DA, Magro CM. Alpha-1 anti-trypsin deficiency and henoch-schönlein purpura associated with anti-neutrophil cytoplasmic and anti-endothelial cell antibodies of immunoglobulin-a isotype. J Cutan Pathol. (2005) 32:300–6. doi: 10.1111/j.0303-6987.2005.00304.x
17. Cho SB, Ahn KJ, Kim DH, Zheng Z, Cho S, Kang SW, et al. Identification of hnrnp-A2/B1 as a target antigen of anti-endothelial cell iga antibody in behçet’s disease. J Invest Dermatol. (2012) 132:601–8. doi: 10.1038/jid.2011.397
18. van Paassen P, Duijvestijn A, Debrus-Palmans L, Damoiseaux J, Vroomen M, Tervaert JW. Induction of endothelial cell apoptosis by igg antibodies from sle patients with nephropathy: A potential role for anti-endothelial cell antibodies. Ann New York Acad Sci. (2007) 1108:147–56. doi: 10.1196/annals.1422.017
19. Domiciano DS, Carvalho JF, Shoenfeld Y. Pathogenic role of anti-endothelial cell antibodies in autoimmune rheumatic diseases. Lupus. (2009) 18:1233–8. doi: 10.1177/0961203309346654
20. Inui N, Matsui T, Suda T, Chida K. Anti-endothelial cell antibodies in patients with sarcoidosis. Chest. (2008) 133:955–60. doi: 10.1378/chest.07-0850
21. Del Papa N, Conforti G, Gambini D, La Rosa L, Tincani A, D’Cruz D, et al. Characterization of the endothelial surface proteins recognized by anti-endothelial antibodies in primary and secondary autoimmune vasculitis. Clin Immunol Immunopathol. (1994) 70:211–6. doi: 10.1006/clin.1994.1031
22. Belizna C, Tervaert JW. Specificity, pathogenecity, and clinical value of antiendothelial cell antibodies. Semin Arthritis Rheum. (1997) 27:98–109. doi: 10.1016/s0049-0172(97)80010-8
23. Shoenfeld Y. Classification of Anti-Endothelial Cell Antibodies into Antibodies against Microvascular and Macrovascular Endothelial Cells: The Pathogenic and Diagnostic Implications. Cleve Clin J Med. (2002) 69 Suppl 2:Sii65–7. doi: 10.3949/ccjm.69.suppl_2.sii65
24. Direskeneli H, D’Cruz D, Khamashta MA, Hughes GR. Autoantibodies against endothelial cells, extracellular matrix, and human collagen type iv in patients with systemic vasculitis. Clin Immunol Immunopathol. (1994) 70:206–10. doi: 10.1006/clin.1994.1030
25. Delunardo F, Scalzi V, Capozzi A, Camerini S, Misasi R, Pierdominici M, et al. Streptococcal-vimentin cross-reactive antibodies induce microvascular cardiac endothelial proinflammatory phenotype in rheumatic heart disease. Clin Exp Immunol. (2013) 173:419–29. doi: 10.1111/cei.12135
26. Sánchez-Zapardiel E, Mancebo E, Díaz-Ordoñez M, de Jorge-Huerta L, Ruiz-Martínez L, Serrano A, et al. Isolated de novo antiendothelial cell antibodies and kidney transplant rejection. Am J Kidney Dis. (2016) 68:933–43. doi: 10.1053/j.ajkd.2016.07.019
27. Hirohata S. Anti-ribosomal P antibodies and lupus nephritis. Clin Exp Nephrol. (2011) 15:471–7. doi: 10.1007/s10157-011-0462-9
28. Yazici ZA, Behrendt M, Cooper D, Goodfield M, Partridge L, Lindsey NJ. The identification of endothelial cell autoantigens. J Autoimmun. (2000) 15:41–9. doi: 10.1006/jaut.2000.0391
29. Dieudé M, Senécal JL, Raymond Y. Induction of endothelial cell apoptosis by heat-shock protein 60-reactive antibodies from anti-endothelial cell autoantibody-positive systemic lupus erythematosus patients. Arthritis Rheum. (2004) 50:3221–31. doi: 10.1002/art.20564
30. Wen X, Chen S, Li J, Li Y, Li L, Wu Z, et al. Association between genetic variants in the human leukocyte antigen-B/mica and takayasu arteritis in chinese han population. Int J Rheum Dis. (2018) 21:271–7. doi: 10.1111/1756-185x.13012
31. Bossi G, Mannarino S, Pietrogrande MC, Salice P, Dellepiane RM, Cremaschi AL, et al. Genetic epistasis between killer immunoglobulin-like receptors and human leukocyte antigens in kawasaki disease susceptibility. Genes Immun. (2015) 16:481–7. doi: 10.1038/gene.2015.34
32. Meyer O, Nicaise P, Moreau S, de Bandt M, Palazzo E, Hayem G, et al. Antibodies to cardiolipin and beta 2 glycoprotein I in patients with polymyalgia rheumatica and giant cell arteritis. Rev Du Rhumatisme (English ed). (1996) 63:241–7.
33. Yang YH, Chang CJ, Chuang YH, Hsu HY, Yu HH, Lee JH, et al. Identification and characterization of iga antibodies against B2-glycoprotein I in childhood henoch-schönlein purpura. Br J Dermatol. (2012) 167:874–81. doi: 10.1111/j.1365-2133.2012.11068.x
34. Richard Kitching A, Anders H-J, Basu N, Brouwer E, Gordon J, David R, et alAnca-associated vasculitis. Nat Rev Dis Primers. (2020) 6:72. doi: 10.1038/s41572-020-0212-y
35. López-Hoyos M, Alvarez L, Ruiz Soto M, Blanco R, José Bartolomé M, Martínez-Taboada VM. Serum levels of antibodies to chlamydia pneumoniae and human hsp60 in giant cell arteritis patients. Clin Exp Rheumatol. (2008) 26:1107–10.
36. Renaudineau Y, Revelen R, Levy Y, Salojin K, Gilburg B, Shoenfeld Y, et al. Anti-endothelial cell antibodies in systemic sclerosis. Clin Diagn Lab Immunol. (1999) 6:156–60. doi: 10.1128/cdli.6.2.156-160.1999
37. Renaudineau Y, Grunebaum E, Krause I, Praprotnik S, Revelen R, Youinou P, et al. Anti-endothelial cell antibodies (Aeca) in systemic sclerosis–increased sensitivity using different endothelial cell substrates and association with other autoantibodies. Autoimmunity. (2001) 33:171–9. doi: 10.3109/08916930109008045
38. Legendre P, Régent A, Thiebault M, Mouthon L. Anti-endothelial cell antibodies in vasculitis: A systematic review. Autoimmun Rev. (2017) 16:146–53. doi: 10.1016/j.autrev.2016.12.012
39. Kreuger J, Phillipson M. Targeting vascular and leukocyte communication in angiogenesis, inflammation and fibrosis. Nat Rev Drug Discov. (2016) 15:125–42. doi: 10.1038/nrd.2015.2
40. Kang S, Kishimoto T. Interplay between interleukin-6 signaling and the vascular endothelium in cytokine storms. Exp Mol Med. (2021) 53:1116–23. doi: 10.1038/s12276-021-00649-0
41. Chen JX, Huang XY, Wang P, Lin WT, Xu WX, Zeng M. Effects and mechanism of arachidonic acid against tnf-A Induced apoptosis of endothelial cells. Clin Hemorheol Microcirc. (2021) 77:259–65. doi: 10.3233/ch-200946
42. Bui TM, Wiesolek HL, Sumagin R. Icam-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J Leukoc Biol. (2020) 108:787–99. doi: 10.1002/jlb.2mr0220-549r
43. Singh V, Kaur R, Kumari P, Pasricha C, Singh R. Icam-1 and vcam-1: gatekeepers in various inflammatory and cardiovascular disorders. Clin Chim Acta. (2023) 548:117487. doi: 10.1016/j.cca.2023.117487
44. Balta S. Endothelial dysfunction and inflammatory markers of vascular disease. Curr Vasc Pharmacol. (2021) 19:243–9. doi: 10.2174/1570161118666200421142542
45. Del Papa N, Raschi E, Moroni G, Panzeri P, Borghi MO, Ponticelli C, et al. Anti-endothelial cell igg fractions from systemic lupus erythematosus patients bind to human endothelial cells and induce a pro-adhesive and a pro-inflammatory phenotype in vitro. Lupus. (1999) 8:423–9. doi: 10.1177/096120339900800603
46. Carvalho D, Savage CO, Isenberg D, Pearson JD. Igg anti-endothelial cell autoantibodies from patients with systemic lupus erythematosus or systemic vasculitis stimulate the release of two endothelial cell-derived mediators, which enhance adhesion molecule expression and leukocyte adhesion in an autocrine manner. Arthritis Rheum. (1999) 42:631–40. doi: 10.1002/1529-0131(199904)42:4<631::Aid-anr5>3.0.Co;2-x
47. Meroni PL, D’Cruz D, Khamashta M, Youinou P, Hughes GR. Anti-endothelial cell antibodies: only for scientists or for clinicians too? Clin Exp Immunol. (1996) 104:199–202. doi: 10.1046/j.1365-2249.1996.20707.x
48. Krieglstein CF, Granger DN. Adhesion molecules and their role in vascular disease. Am J Hypertens. (2001) 14:44s–54s. doi: 10.1016/s0895-7061(01)02069-6
49. Lee KH, Cho HJ, Kim HS, Lee WJ, Lee S, Bang D. Activation of extracellular signal regulated kinase 1/2 in human dermal microvascular endothelial cells stimulated by anti-endothelial cell antibodies in sera of patients with behcet’s disease. J Dermatol Sci. (2002) 30:63–72. doi: 10.1016/S0923-1811(02)00062-2
50. Prescott JA, Mitchell JP, Cook SJ. Inhibitory feedback control of nf-Kb signalling in health and disease. Biochem J. (2021) 478:2619–64. doi: 10.1042/bcj20210139
51. Mussbacher M, Salzmann M, Brostjan C, Hoesel B, Schoergenhofer C, Datler H, et al. Cell type-specific roles of nf-Kb linking inflammation and thrombosis. Front Immunol. (2019) 10:85. doi: 10.3389/fimmu.2019.00085
52. Theofilis P, Sagris M, Oikonomou E, Antonopoulos AS, Siasos G, Tsioufis C, et al. Inflammatory mechanisms contributing to endothelial dysfunction. Biomedicines. (2021) 9(7):781. doi: 10.3390/biomedicines9070781
53. Zhong L, Simard MJ, Huot J. Endothelial micrornas regulating the nf-Kb pathway and cell adhesion molecules during inflammation. FASEB J. (2018) 32:4070–84. doi: 10.1096/fj.201701536R
54. Trimm E, Red-Horse K. Vascular endothelial cell development and diversity. Nat Rev Cardiol. (2023) 20:197–210. doi: 10.1038/s41569-022-00770-1
55. Schmaier AA, Anderson PF, Chen SM, El-Darzi E, Aivasovsky I, Kaushik MP, et al. Tmem16e regulates endothelial cell procoagulant activity and thrombosis. J Clin Invest. (2023) 133(11):e163808. doi: 10.1172/jci163808
56. Grover SP, Mackman N. Tissue factor: an essential mediator of hemostasis and trigger of thrombosis. Arterioscler Thromb Vasc Biol. (2018) 38:709–25. doi: 10.1161/atvbaha.117.309846
57. Shahidi M. Thrombosis and von willebrand factor. Adv Exp Med Biol. (2017) 906:285–306. doi: 10.1007/5584_2016_122
58. Manz XD, Bogaard HJ, Aman J. Regulation of vwf (Von willebrand factor) in inflammatory thrombosis. Arterioscler Thromb Vasc Biol. (2022) 42:1307–20. doi: 10.1161/atvbaha.122.318179
59. Chang JC. Disseminated intravascular coagulation: new identity as endotheliopathy-associated vascular microthrombotic disease based on in vivo hemostasis and endothelial molecular pathogenesis. Thromb J. (2020) 18:25. doi: 10.1186/s12959-020-00231-0
60. Martinod K, Wagner DD. Thrombosis: tangled up in nets. Blood. (2014) 123:2768–76. doi: 10.1182/blood-2013-10-463646
61. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. (2015) 126:242–6. doi: 10.1182/blood-2015-01-624023
62. Shenkman B, Budde U, Angerhaus D, Lubetsky A, Savion N, Seligsohn U, et al. Adamts-13 regulates platelet adhesion under flow. A new method for differentiation between inherited and acquired thrombotic thrombocytopenic purpura. Thromb Haemost. (2006) 96:160–6.
63. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. (2020) 21:85–100. doi: 10.1038/s41580-019-0173-8
64. Xie J, Li B, Yao B, Zhang P, Wang L, Lu H, et al. Transforming growth factor-B1-regulated fas/fasl pathway activation suppresses nucleus pulposus cell apoptosis in an inflammatory environment. Biosci Rep. (2020) 40(2):BSR20191726. doi: 10.1042/bsr20191726
65. Qian QZ, Cao XK, Liu HY, Zheng GY, Qian QQ, Shen FH. Tnfr/tnf-A Signaling pathway regulates apoptosis of alveolar macrophages in coal workers’ Pneumoconiosis. Oncotarget. (2018) 9:1302–10. doi: 10.18632/oncotarget.18921
66. Lin YS, Lin CF, Lei HY, Liu HS, Yeh TM, Chen SH, et al. Antibody-mediated endothelial cell damage via nitric oxide. Curr Pharm Des. (2004) 10:213–21. doi: 10.2174/1381612043453469
67. Gupta S, Knowlton AA. Hsp60, bax, apoptosis and the heart. J Cell Mol Med. (2005) 9:51–8. doi: 10.1111/j.1582-4934.2005.tb00336.x
68. Alard JE, Dueymes M, Youinou P, Jamin C. Modulation of endothelial cell damages by anti-hsp60 autoantibodies in systemic autoimmune diseases. Autoimmun Rev. (2007) 6:438–43. doi: 10.1016/j.autrev.2007.01.012
69. Jamin C, Dugué C, Alard JE, Jousse S, Saraux A, Guillevin L, et al. Induction of endothelial cell apoptosis by the binding of anti-endothelial cell antibodies to hsp60 in vasculitis-associated systemic autoimmune diseases. Arthritis Rheum. (2005) 52:4028–38. doi: 10.1002/art.21401
70. Morikawa Y, Shibata A, Okumura N, Ikari A, Sasajima Y, Suenami K, et al. Sibutramine provokes apoptosis of aortic endothelial cells through altered production of reactive oxygen and nitrogen species. Toxicol Appl Pharmacol. (2017) 314:1–11. doi: 10.1016/j.taap.2016.11.003
71. Kahaleh MB, Fan PS. Mechanism of serum-mediated endothelial injury in scleroderma: identification of a granular enzyme in scleroderma skin and sera. Clin Immunol Immunopathol. (1997) 83:32–40. doi: 10.1006/clin.1996.4322
72. Holt CM, Lindsey N, Moult J, Malia RG, Greaves M, Hume A, et al. Antibody-dependent cellular cytotoxicity of vascular endothelium: characterization and pathogenic associations in systemic sclerosis. Clin Exp Immunol. (1989) 78:359–65.
73. Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. (2015) 15:388–400. doi: 10.1038/nri3839
74. Cruz-Muñoz ME, Valenzuela-Vázquez L, Sánchez-Herrera J, Santa-Olalla Tapia J. From the “Missing self” Hypothesis to adaptive nk cells: insights of nk cell-mediated effector functions in immune surveillance. J Leukoc Biol. (2019) 105:955–71. doi: 10.1002/jlb.Mr0618-224rr
75. Sgonc R, Gruschwitz MS, Boeck G, Sepp N, Gruber J, Wick G. Endothelial cell apoptosis in systemic sclerosis is induced by antibody-dependent cell-mediated cytotoxicity via cd95. Arthritis Rheum. (2000) 43:2550–62. doi: 10.1002/1529-0131(200011)43:11<2550::Aid-anr24>3.0.Co;2-h
76. Jimbo H, Nagai H, Fujiwara S, Shimoura N, Nishigori C. Fas-fasl interaction in cytotoxic T cell-mediated vitiligo: the role of lesional expression of tumor necrosis factor-A and interferon-Γ in fas-mediated melanocyte apoptosis. Exp Dermatol. (2020) 29:61–70. doi: 10.1111/exd.14053
77. Wang M, Su P. The role of the fas/fasl signaling pathway in environmental toxicant-induced testicular cell apoptosis: an update. Syst Biol Reprod Med. (2018) 64:93–102. doi: 10.1080/19396368.2017.1422046
78. Wang H, Ma J, Wu Q, Luo X, Chen Z, Kou L. Circulating B lymphocytes producing autoantibodies to endothelial cells play a role in the pathogenesis of takayasu arteritis. J Vasc Surg. (2011) 53:174–80. doi: 10.1016/j.jvs.2010.06.173
79. Joseph G, Goel R, Thomson VS, Joseph E, Danda D. Takayasu arteritis: jacc focus seminar 3/4. J Am Coll Cardiol. (2022) S0735-1097(22)07305-3. doi: 10.1016/j.jacc.2022.09.051
80. Park MC, Park YB, Jung SY, Lee KH, Lee SK. Anti-endothelial cell antibodies and antiphospholipid antibodies in takayasu’s arteritis: correlations of their titers and isotype distributions with disease activity. Clin Exp Rheumatol. (2006) 24:S10–6.
81. Chauhan SK, Tripathy NK, Nityanand S. Antigenic targets and pathogenicity of anti-aortic endothelial cell antibodies in takayasu arteritis. Arthritis Rheum. (2006) 54:2326–33. doi: 10.1002/art.21921
82. Blank M, Krause I, Goldkorn T, Praprotnik S, Livneh A, Langevitz P, et al. Monoclonal anti-endothelial cell antibodies from a patient with takayasu arteritis activate endothelial cells from large vessels. Arthritis Rheum. (1999) 42:1421–32. doi: 10.1002/1529-0131(199907)42:7<1421::Aid-anr16>3.0.Co;2-o
83. Muratore F, Kermani TA, Crowson CS, Green AB, Salvarani C, Matteson EL, et al. Large-vessel giant cell arteritis: A cohort study. Rheumatol (Oxford England). (2015) 54:463–70. doi: 10.1093/rheumatology/keu329
84. de Boysson H, Daumas A, Vautier M, Parienti JJ, Liozon E, Lambert M, et al. Large-vessel involvement and aortic dilation in giant-cell arteritis. A multicenter study of 549 patients. Autoimmun Rev. (2018) 17:391–8. doi: 10.1016/j.autrev.2017.11.029
85. van der Geest KSM, Sandovici M, Brouwer E, Mackie SL. Diagnostic accuracy of symptoms, physical signs, and laboratory tests for giant cell arteritis: A systematic review and meta-analysis. JAMA Internal Med. (2020) 180:1295–304. doi: 10.1001/jamainternmed.2020.3050
86. Saadoun D, Vautier M, Cacoub P. Medium- and large-vessel vasculitis. Circulation. (2021) 143:267–82. doi: 10.1161/circulationaha.120.046657
87. Grunebaum E, Blank M, Cohen S, Afek A, Kopolovic J, Meroni PL, et al. The role of anti-endothelial cell antibodies in kawasaki disease - in vitro and in vivo studies. Clin Exp Immunol. (2002) 130:233–40. doi: 10.1046/j.1365-2249.2002.02000.x
88. Suzuki Y, Ichiyama T, Ohsaki A, Hasegawa S, Shiraishi M, Furukawa S. Anti-inflammatory effect of 1alpha,25-dihydroxyvitamin D(3) in human coronary arterial endothelial cells: implication for the treatment of kawasaki disease. J Steroid Biochem Mol Biol. (2009) 113:134–8. doi: 10.1016/j.jsbmb.2008.12.004
89. Puéchal X. Granulomatosis with polyangiitis (Wegener’s). Joint Bone Spine. (2020) 87:572–8. doi: 10.1016/j.jbspin.2020.06.005
90. Régent A, Lofek S, Dib H, Bussone G, Tamas N, Federici C, et al. Identification of target antigens of anti-endothelial cell antibodies in patients with anti-neutrophil cytoplasmic antibody-associated vasculitides: A proteomic approach. Clin Immunol (Orlando Fla). (2014) 153:123–35. doi: 10.1016/j.clim.2014.03.020
91. Pillebout E, Sunderkötter C. Iga vasculitis. Semin Immunopathol. (2021) 43:729–38. doi: 10.1007/s00281-021-00874-9
92. Yang YH, Wang SJ, Chuang YH, Lin YT, Chiang BL. The level of iga antibodies to human umbilical vein endothelial cells can be enhanced by tnf-alpha treatment in children with henoch-schönlein purpura. Clin Exp Immunol. (2002) 130:352–7. doi: 10.1046/j.1365-2249.2002.01964.x
93. Yang YH, Huang YH, Lin YL, Wang LC, Chuang YH, Yu HH, et al. Circulating iga from acute stage of childhood henoch-schönlein purpura can enhance endothelial interleukin (Il)-8 production through mek/erk signalling pathway. Clin Exp Immunol. (2006) 144:247–53. doi: 10.1111/j.1365-2249.2006.03076.x
94. Alibaz-Oner F, Direskeneli H. Update on the diagnosis of behçet’s disease. Diagn (Basel Switzerland). (2022) 13(1):41. doi: 10.3390/diagnostics13010041
95. Direskeneli H, Keser G, D’Cruz D, Khamashta MA, Akoğlu T, Yazici H, et al. Anti-endothelial cell antibodies, endothelial proliferation and von willebrand factor antigen in behçet’s disease. Clin Rheumatol. (1995) 14:55–61. doi: 10.1007/BF02208085
96. Lee KH, Chung HS, Bang D, Lee S. Behçet’s disease sera containing antiendothelial cell antibodies promote adhesion of T lymphocytes to cultured human dermal microvascular endothelial cells. Yonsei Med J. (1999) 40:152–8. doi: 10.3349/ymj.1999.40.2.152
97. Kwang Hoon L, Hae-Shin C, Hyoung Sup K, Sang-Ho O, Moon-Kyung H, Ja-Hyun B, et al. Human alpha-enolase from endothelial cells as a target antigen of anti-endothelial cell antibody in beh?Et’s disease. Arthritis Rheumatol. (2003) 48:2025–35. doi: 10.1002/art.11074
98. Lee JH, Cho SB, Bang D, Oh SH, Ahn KJ, Kim J, et al. Human anti-alpha-enolase antibody in sera from patients with behçEt’s disease and rheumatologic disorders. Clin Exp Rheumatol. (2009) 27:S63.
99. Rossides M, Darlington P, Kullberg S, Arkema EV. Sarcoidosis: epidemiology and clinical insights. J Internal Med. (2023) 293:668–80. doi: 10.1111/joim.13629
100. Sgonc R, Gruschwitz MS, Dietrich H, Recheis H, Gershwin ME, Wick G. Endothelial cell apoptosis is a primary pathogenetic event underlying skin lesions in avian and human scleroderma. J Clin Invest. (1996) 98:785–92. doi: 10.1172/jci118851
101. Lazar S, Kahlenberg JM. Systemic lupus erythematosus: new diagnostic and therapeutic approaches. Annu Rev Med. (2023) 74:339–52. doi: 10.1146/annurev-med-043021-032611
102. Horváthová M, Jahnová E, Nyulassy S. Detection of antiendothelial cell antibodies in patients with connective tissue diseases by flow cytometry and their relation to endothelial cell activation. Physiol Res. (2002) 51:613.
103. Perricone C, Pendolino M, Olivieri M, Conti F, Valesini G, Alessandri C. Neuropsychiatric manifestations associated with anti-endothelial cell antibodies in systemic lupus erythematosus. Israel Med Assoc J Imaj. (2015) 17:171.
104. Cieślik P, Semik-Grabarczyk E, Hrycek A, Holecki M. The impact of anti-endothelial cell antibodies (Aecas) on the development of blood vessel damage in patients with systemic lupus erythematosus: the preliminary study. Rheumatol Int. (2022) 42:791–801. doi: 10.1007/s00296-022-05104-5
105. Kondo A, Takahashi K, Mizuno T, Kato A, Hirano D, Yamamoto N, et al. The level of iga antibodies to endothelial cells correlates with histological evidence of disease activity in patients with lupus nephritis. PloS One. (2016) 11:e0163085. doi: 10.1371/journal.pone.0163085
106. Frampton G, Moriya S, Pearson JD, Isenberg DA, Ward FJ, Smith TA, et al. Identification of candidate endothelial cell autoantigens in systemic lupus erythematosus using a molecular cloning strategy: A role for ribosomal P protein P0 as an endothelial cell autoantigen. Rheumatol (Oxford). (2000) 39:1114–20. doi: 10.1093/rheumatology/39.10.1114
107. Yoshio T, Masuyama J, Mimori A, Takeda A, Minota S, Kano S. Endothelin-1 release from cultured endothelial cells induced by sera from patients with systemic lupus erythematosus. Ann Rheum Dis. (1995) 54:361–5. doi: 10.1136/ard.54.5.361
108. Matsuda J, Gotoh M, Gohchi K, Kawasugi K, Tsukamoto M, Saitoh N. Anti-endothelial cell antibodies to the endothelial hybridoma cell line (Eahy926) in systemic lupus erythematosus patients with antiphospholipid antibodies. Br J Haematol. (1997) 97:227–32. doi: 10.1046/j.1365-2141.1997.d01-2147.x
109. Cid MC, Hernández-Rodríguez J, Esteban MJ, Cebrián M, Gho YS, Font C, et al. Tissue and serum angiogenic activity is associated with low prevalence of ischemic complications in patients with giant-cell arteritis. Circulation. (2002) 106:1664–71. doi: 10.1161/01.cir.0000030185.67510.c0
110. Kotulska-Kucharz A, Kopeć-Mędrek M, Kucharz EJ. Serum angiostatin and endostatin levels in patients with granulomatosis with polyangiitis and immune complex small vessel vasculitis. Reumatologia. (2018) 56:285–8. doi: 10.5114/reum.2018.79498
111. Hernández-Rodríguez J, García-Martínez A, Casademont J, Filella X, Esteban MJ, López-Soto A, et al. A strong initial systemic inflammatory response is associated with higher corticosteroid requirements and longer duration of therapy in patients with giant-cell arteritis. Arthritis Rheum. (2002) 47:29–35. doi: 10.1002/art1.10161
112. Cid MC, Cebrián M, Font C, Coll-Vinent B, Hernández-Rodríguez J, Esparza J, et al. Cell adhesion molecules in the development of inflammatory infiltrates in giant cell arteritis: inflammation-induced angiogenesis as the preferential site of leukocyte-endothelial cell interactions. Arthritis Rheum. (2000) 43:184–94. doi: 10.1002/1529-0131(200001)43:1<184::Aid-anr23>3.0.Co;2-n
113. Baumann H, Gauldie J. The acute phase response. Immunol Today. (1994) 15:74–80. doi: 10.1016/0167-5699(94)90137-6
114. Hernández-Rodríguez J, Segarra M, Vilardell C, Sánchez M, García-Martínez A, Esteban MJ, et al. Tissue production of pro-inflammatory cytokines (Il-1beta, tnfalpha and il-6) correlates with the intensity of the systemic inflammatory response and with corticosteroid requirements in giant-cell arteritis. Rheumatol (Oxford England). (2004) 43:294–301. doi: 10.1093/rheumatology/keh058
115. Miura M, Garcia FL, Crawford SE, Rowley AH. Cell adhesion molecule expression in coronary artery aneurysms in acute kawasaki disease. Pediatr Infect Dis J. (2004) 23:931–6. doi: 10.1097/01.inf.0000142171.91235.fc
116. Cid MC, Segarra M, García-Martínez A, Hernández-Rodríguez J. Endothelial cells, antineutrophil cytoplasmic antibodies, and cytokines in the pathogenesis of systemic vasculitis. Curr Rheumatol Rep. (2004) 6:184–94. doi: 10.1007/s11926-004-0067-3
117. Cacciola R, Gentilini Cacciola E, Vecchio V, Cacciola E. Impact of anti-endothelial cell antibodies (Aecas) in patients with polycythemia vera and thrombosis. Diagn (Basel Switzerland). (2022) 12(5):1077. doi: 10.3390/diagnostics12051077
118. Jackson AM, Sigdel TK, Delville M, Hsieh SC, Dai H, Bagnasco S, et al. Endothelial cell antibodies associated with novel targets and increased rejection. J Am Soc Nephrol: JASN. (2015) 26:1161–71. doi: 10.1681/asn.2013121277
119. Maruotti N, Cantatore FP, Nico B, Vacca A, Ribatti D. Angiogenesis in vasculitides. Clin Exp Rheumatol. (2008) 26:476–83.
120. Pegoraro AF, Janmey P, Weitz DA. Mechanical properties of the cytoskeleton and cells. Cold Spring Harbor Perspect Biol. (2017) 9(11):a022038. doi: 10.1101/cshperspect.a022038
121. Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature. (2010) 463:485–92. doi: 10.1038/nature08908
122. Zimering MB, Pan Z. Autoantibodies in type 2 diabetes induce stress fiber formation and apoptosis in endothelial cells. J Clin Endocrinol Metab. (2009) 94:2171–7. doi: 10.1210/jc.2008-2354
123. Zhang X, Valenzuela NM, Reed EF. Hla class I antibody-mediated endothelial and smooth muscle cell activation. Curr Opin Organ Transplant. (2012) 17:446–51. doi: 10.1097/MOT.0b013e328355f1c2
124. Ziegler ME, Souda P, Jin YP, Whitelegge JP, Reed EF. Characterization of the endothelial cell cytoskeleton following hla class I ligation. PloS One. (2012) 7:e29472. doi: 10.1371/journal.pone.0029472
125. Aljabri A, Vijayan V, Stankov M, Nikolin C, Figueiredo C, Blasczyk R, et al. Hla class ii antibodies induce necrotic cell death in human endothelial cells via a lysosomal membrane permeabilization-mediated pathway. Cell Death Dis. (2019) 10:235. doi: 10.1038/s41419-019-1319-5
126. Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. (2005) 25:932–43. doi: 10.1161/01.Atv.0000160548.78317.29
127. Satcher RL Jr., Dewey CF Jr. Theoretical estimates of mechanical properties of the endothelial cell cytoskeleton. Biophys J. (1996) 71:109–18. doi: 10.1016/s0006-3495(96)79206-8
128. Jannatbabaei A, Tafazzoli-Shadpour M, Seyedjafari E, Fatouraee N. Cytoskeletal remodeling induced by substrate rigidity regulates rheological behaviors in endothelial cells. J Biomed Mater Res Part A. (2019) 107:71–80. doi: 10.1002/jbm.a.36533
129. Ucar AK, Ozdede A, Kayadibi Y, Adaletli I, Melikoglu M, Fresko I, et al. Increased arterial stiffness and accelerated atherosclerosis in takayasu arteritis. Semin Arthritis Rheum. (2023) 60:152199. doi: 10.1016/j.semarthrit.2023.152199
130. Hussein MA, Ramadan MM, Moneam MAE, Halim HAE, Ghaffar NAE, Fawzy MW. Interleukin 37; a possible marker of arterial stiffness in behçet’s disease. Am J Med Sci. (2022) 364:425–32. doi: 10.1016/j.amjms.2022.04.013
131. Byfield FJ, Wen Q, Leszczynska K, Kulakowska A, Namiot Z, Janmey PA, et al. Cathelicidin ll-37 peptide regulates endothelial cell stiffness and endothelial barrier permeability. Am J Physiol Cell Physiol. (2011) 300:C105–12. doi: 10.1152/ajpcell.00158.2010
132. Okamoto T, Kawamoto E, Takagi Y, Akita N, Hayashi T, Park EJ, et al. Gap junction-mediated regulation of endothelial cellular stiffness. Sci Rep. (2017) 7:6134. doi: 10.1038/s41598-017-06463-x
133. Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann New York Acad Sci. (2008) 1123:134–45. doi: 10.1196/annals.1420.016
134. Thompson PW, Randi AM, Ridley AJ. Intercellular adhesion molecule (Icam)-1, but not icam-2, activates rhoa and stimulates C-fos and rhoa transcription in endothelial cells. J Immunol (Baltimore Md: 1950). (2002) 169:1007–13. doi: 10.4049/jimmunol.169.2.1007
135. Huveneers S, Daemen MJ, Hordijk PL. Between rho(K) and a hard place: the relation between vessel wall stiffness, endothelial contractility, and cardiovascular disease. Circ Res. (2015) 116:895–908. doi: 10.1161/circresaha.116.305720
136. McKenzie JA, Ridley AJ. Roles of rho/rock and mlck in tnf-alpha-induced changes in endothelial morphology and permeability. J Cell Physiol. (2007) 213:221–8. doi: 10.1002/jcp.21114
137. Sawma T, Shaito A, Najm N, Sidani M, Orekhov A, El-Yazbi AF, et al. Role of rhoa and rho-associated kinase in phenotypic switching of vascular smooth muscle cells: implications for vascular function. Atherosclerosis. (2022) 358:12–28. doi: 10.1016/j.atherosclerosis.2022.08.012
138. Chen S, Guo F, Liu X, Xi J, Xue M, Guo Y, et al. Roles of the rhoa-rock signaling pathway in the endothelial H(2)S production and vasodilation in rat cerebral arteries. ACS Omega. (2022) 7:18498–508. doi: 10.1021/acsomega.2c00996
139. You LJ, Li PW, Zhang WW, Feng MF, Zhao WP, Hou HM, et al. Schisandrin a ameliorates increased pulmonary capillary endothelial permeability accompanied with sepsis through inhibition of rhoa/rock1/mlc pathways. Int Immunopharmacol. (2023) 118:110124. doi: 10.1016/j.intimp.2023.110124
140. Mu X, Tseng C, Hambright WS, Matre P, Lin CY, Chanda P, et al. Cytoskeleton stiffness regulates cellular senescence and innate immune response in hutchinson-gilford progeria syndrome. Aging Cell. (2020) 19:e13152. doi: 10.1111/acel.13152
141. Szulcek R, Beckers CM, Hodzic J, de Wit J, Chen Z, Grob T, et al. Localized rhoa gtpase activity regulates dynamics of endothelial monolayer integrity. Cardiovasc Res. (2013) 99:471–82. doi: 10.1093/cvr/cvt075
142. Hsieh SL, Wang JJ, Su KH, Kuo YL, Hsieh S, Wu CC. Suppressive effects of the gynura bicolor ether extract on endothelial permeability and leukocyte transmigration in human endothelial cells induced by tnf-A. Evidence-Based Complement Altern Med: eCAM. (2020) 2020:9413724. doi: 10.1155/2020/9413724
143. Praprotnik S, Blank M, Meroni PL, Rozman B, Eldor A, Shoenfeld Y. Classification of Anti-Endothelial Cell Antibodies into Antibodies against Microvascular and Macrovascular Endothelial Cells: The Pathogenic and Diagnostic Implications. Arthritis Rheum. (2001) 44:1484–94. doi: 10.1002/1529-0131(200107)44:7<1484::Aid-art269>3.0.Co;2-q
144. Belizna C, Duijvestijn A, Hamidou M, Tervaert JW. Antiendothelial cell antibodies in vasculitis and connective tissue disease. Ann Rheum Dis. (2006) 65:1545–50. doi: 10.1136/ard.2005.035295
145. Prasad TK, Hack E, Hallberg RL. Function of the maize mitochondrial chaperonin hsp60: specific association between hsp60 and newly synthesized F1-atpase alpha subunits. Mol Cell Biol. (1990) 10:3979–86. doi: 10.1128/mcb.10.8.3979
146. Mendez MG, Restle D, Janmey PA. Vimentin enhances cell elastic behavior and protects against compressive stress. Biophys J. (2014) 107:314–23. doi: 10.1016/j.bpj.2014.04.050
147. Gorudko IV, Sokolov AV, Shamova EV, Grudinina NA, Drozd ES, Shishlo LM, et al. Myeloperoxidase modulates human platelet aggregation via actin cytoskeleton reorganization and store-operated calcium entry. Biol Open. (2013) 2:916–23. doi: 10.1242/bio.20135314
148. Pace PE, Peskin AV, Konigstorfer A, Jasoni CJ, Winterbourn CC, Hampton MB. Peroxiredoxin interaction with the cytoskeletal-regulatory protein crmp2: investigation of a putative redox relay. Free Radic Biol Med. (2018) 129:383–93. doi: 10.1016/j.freeradbiomed.2018.10.407
149. Alard JE, Hillion S, Guillevin L, Saraux A, Pers JO, Youinou P, et al. Autoantibodies to endothelial cell surface atp synthase, the endogenous receptor for hsp60, might play a pathogenic role in vasculatides. PloS One. (2011) 6:e14654. doi: 10.1371/journal.pone.0014654
150. Ellins E, Shamaei-Tousi A, Steptoe A, Donald A, O’Meagher S, Halcox J, et al. The relationship between carotid stiffness and circulating levels of heat shock protein 60 in middle-aged men and women. J Hypertens. (2008) 26:2389–92. doi: 10.1097/HJH.0b013e328313918b
151. Weber A, Iturri J, Benitez R, Zemljic-Jokhadar S, Toca-Herrera JL. Microtubule disruption changes endothelial cell mechanics and adhesion. Sci Rep. (2019) 9:14903. doi: 10.1038/s41598-019-51024-z
Keywords: vasculitis, endothelial cells, anti-endothelial cell antibodies (AECAs), angiogenesis, mechanical properties, stiffness
Citation: Zhang T, Li L, Huang S, Starodubtseva MN and Liu J (2025) Anti-endothelial cell antibodies in pathogenesis of vasculitis. Front. Immunol. 16:1567293. doi: 10.3389/fimmu.2025.1567293
Received: 27 January 2025; Accepted: 09 April 2025;
Published: 30 April 2025.
Edited by:
Alexandre Wagner Silva De Souza, Federal University of São Paulo, BrazilReviewed by:
Paweł Cieślik, Medical University of Silesia, PolandRenate Kain, Medical University of Vienna, Austria
Copyright © 2025 Zhang, Li, Huang, Starodubtseva and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ju Liu, anUubGl1QHNkdS5lZHUuY24=