 Víctor Casanova1,2†
Víctor Casanova1,2† Andrea Rodríguez-Agustín1,2†
Andrea Rodríguez-Agustín1,2† Rubén Ayala-Suárez1
Rubén Ayala-Suárez1 Elisa Moraga1
Elisa Moraga1 María José Maleno1
María José Maleno1 Josep Mallolas1,3,4
Josep Mallolas1,3,4 Esteban Martínez1,3,4,5Sonsoles Sánchez-Palomino1,2,4
Esteban Martínez1,3,4,5Sonsoles Sánchez-Palomino1,2,4 José M. Miró1,3,4,5
José M. Miró1,3,4,5 José Alcamí1,2,4,6*‡
José Alcamí1,2,4,6*‡ Núria Climent1,2,4*‡
Núria Climent1,2,4*‡- 1AIDS and HIV Infection Group, Fundació de Recerca Clínic Barcelona-Institut d’Investigacions Biomèdiques August Pi i Sunyer (FRCB-IDIBAPS), Barcelona, Spain
- 2Department of Medicine, Universitat de Barcelona (UB), Barcelona, Spain
- 3Infectious Diseases Unit, Hospital Clínic de Barcelona, University of Barcelona, Barcelona, Spain
- 4Centro de Investigación Biomédica en Red sobre Enfermedades Infecciosas (CIBERINFEC), Instituto de Salud Carlos III (ISCIII), Madrid, Spain
- 5Reial Academia de Medicina de Catalunya (RAMC), Barcelona, Spain
- 6AIDS Immunopathology Unit, Centro Nacional de Microbiología, Instituto de Salud Carlos III (ISCIII), Madrid, Spain
Introduction: Current antiretroviral therapy (ART) for HIV infection reduces plasma viral loads to undetectable levels and has increased the life expectancy of people with HIV (PWH). However, this increased lifespan is accompanied by signs of accelerated aging and a higher prevalence of age-related comorbidities. Tat (Trans-Activator of Transcription) is a key protein for viral replication and pathogenesis. Tat is encoded by 2 exons, with the full-length Tat ranging from 86 to 101 aa (Tat101). Introducing a stop codon in position 73 generates a 1 exon, synthetic 72aa Tat (Tat72). Intracellular, full-length Tat activates the NF-κB pro-inflammatory pathway and increases antiapoptotic signals and ROS generation. These effects may initiate a cellular senescence program, characterized by cell cycle arrest, altered cell metabolism, and increased senescence-associated secretory phenotype (SASP) mediator release However, the precise role of HIV-Tat in inducing a cellular senescence program in CD4+ T-cells is currently unknown.
Methods: Jurkat Tetoff cell lines stably transfected with Tat72, Tat101, or an empty vector were used. Flow cytometry and RT-qPCR were used to address senescence biomarkers, and 105 mediators were assessed in cell supernatants with an antibody-based membrane array. Key results obtained in Jurkat-Tat cells were addressed in primary, resting CD4+ T-cells by transient electroporation of HIV-Tat-FLAG plasmid DNA.
Results: In the Jurkat cell model, expression of Tat101 increased the levels of the senescence biomarkers BCL-2, CD87, and p21, and increased the release of sCD30, PDGF-AA, and sCD31, among other factors. Tat101 upregulated CD30 and CD31 co-expression in the Jurkat cell surface, distinguishing these cells from Tat72 and Tetoff Jurkats. The percentage of p21+, p16+, and γ-H2AX+ cells were higher in Tat-expressing CD4+ T-cells, detected as a FLAG+ population compared to their FLAG- (Tat negative) counterparts. Increased levels of sCD31 and sCD26 were also detected in electroporated CD4+ T-cell supernatants.
Discussion: Intracellular, full-length HIV-Tat expression increases several senescence biomarkers in Jurkat and CD4+ T-cells, and SASP/Aging mediators in cell supernatants. Intracellular HIV-Tat may initiate a cellular senescence program, contributing to the premature aging phenotype observed in PWH.
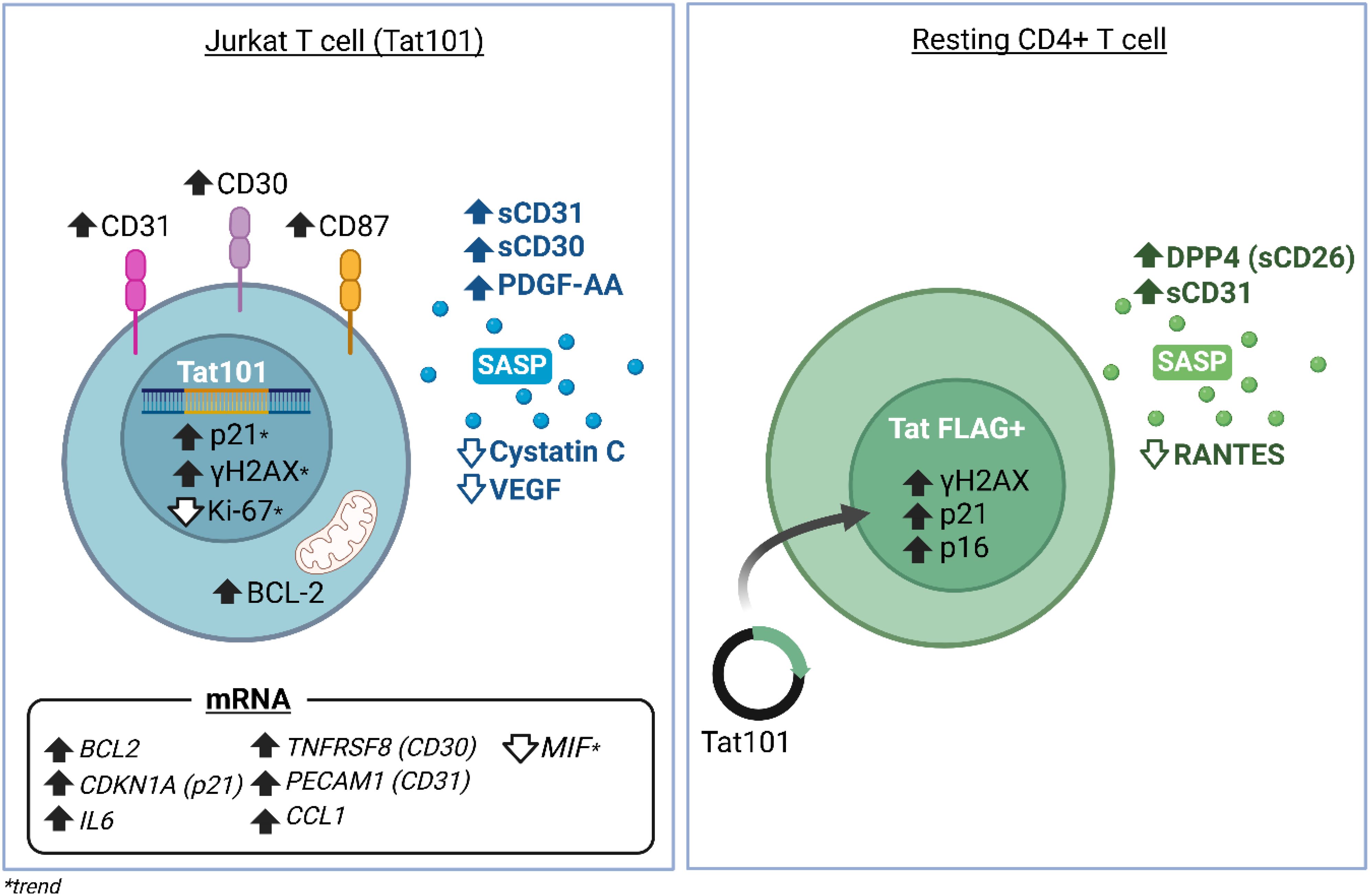
Graphical Abstract.
1 Introduction
Current antiretroviral therapy (ART) has transformed HIV from a progressive and fatal infection into a manageable chronic condition. ART has enabled people with HIV (PWH) to live longer, with estimations showing that more than 70% of PWH will be over 50 years of age by 2030 (1, 2). Despite ART success, PWH have a higher prevalence of age-associated comorbidities like cancer, cardiovascular diseases, frailty, and neurocognitive impairment than those without HIV (3, 4). Importantly, up to 64% of PWH of 50 years of age present 2 or more comorbidities, compared to 43% of HIV-negative, aged-matched controls (5). This phenotypic resemblance to the elderly led to the hypothesis that HIV infection may cause ‘premature aging’ (6, 7). Aging is defined as a functional decline in several organs and systems, leading to susceptibility to disease and death (8). The cumulative effects of HIV replication, chronic inflammation, and long exposures to ART treatment may contribute to the premature aging of PWH (9, 10).
Cellular senescence, one of the hallmarks of aging (8), is a complex cellular response to different stressors, such as oncogene activation (11, 12), irradiation, cytotoxic stimuli or oxidative stress, among others (13). Senescence was first defined in cultured lung fibroblasts that reached replicative exhaustion (14), where progressive telomere attrition is recognized by the DNA damage response (DDR) proteins γH2AX and 53BP1 (P53 pathway) (15), inducing the transcription of the cyclin-dependent kinase inhibitors CDKN1A (p21CIP1) and CDKN2A (p16INK4A) (16). This ultimately leads to cell cycle arrest and reduced expression of the proliferation marker MKI67 (Ki-67) (16). Additionally, senescence involves profound morphological changes, chromatin reorganization, apoptotic resistance (BCL2), metabolic reprogramming (SA-β-Gal) and the establishment of a complex secretory program, termed the senescence-associated secretory phenotype (SASP) (17). The SASP includes cytokines, chemokines, angiogenic factors, proteases, collagens and other factors with a paracrine and inflammatory effect, able to induce senescence on surrounding cells (18–20). The exact composition and intensity of the SASP varies according to the senescent stimuli and cellular type. Cytokine induction leads to inflammation and the recruitment of immune cells that survey and clear senescent cells (21). This is beneficial in limiting tumorigenesis and promoting wound healing, however, aberrant accumulation of senescent cells occurs during aging and in many pathologies, contributing to chronic inflammation and organ damage (13).
In this regard, different hallmarks of cellular senescence have been detected in PBMCs from PWH, such as telomere shortening (22, 23), and increased p16INK4A expression (24–26). BCL2, a senescence biomarker contributing to the apoptotic resistance seen in senescent cells, is also important for HIV infection, as it supports the survival of infected cells and HIV persistence (27). Preliminary results from our group (Climent et al, manuscript in preparation) indicate that cellular senescence biomarkers are upregulated in ex-vivo CD4+ T-cells from PWH during untreated HIV infection. Importantly, ART does not fully revert these changes after 1 year of treatment. HIV replication and viral products such as gp120, Nef or Tat have been proposed as potential inducers of cellular senescence in cells from HIV individuals (9). Only a few studies explore the role of HIV proteins in cellular senescence induction (reviewed in (28). Specifically, X4 and R5 HIV gp120 proteins increase SA-β-gal staining in endothelial cells (29, 30). Nef increases oxidative stress and mitochondrial dysfunction in human bone marrow mesenchymal stem cells (31) and SA-β-gal Levels, p16 and p53 activity in human adipose tissue (32), globally inducing early senescence in those cells. Finally, chronic exposure of human bone marrow mesenchymal stem cells to p55-gag results in reduced cell proliferation and increased senescence biomarker expression (33). Interestingly, most of these effects have also been observed in the presence of HIV-Tat, mostly due to increased NF-κB pathway signaling and ROS generation (29, 31, 32, 34, 35).
Tat is a regulatory protein expressed early in HIV transcription. It is encoded by two exons that translate into a 101-residue protein (Tat101 or full-length Tat). Tat is critical for viral replication by facilitating efficient elongation of viral transcripts through binding to the RNA polymerase II (RNAPII) complex and recruiting cellular elongation factors (36). HIV-Tat is also actively secreted by HIV‐1‐infected cells to the extracellular space where it mediates additional effects on surrounding cells (37). The first exon of Tat comprises amino acids from 1–72 (Tat72) and introducing a stop codon in position 73 produces an active protein (Tat72) that partially maintains the elongation ability of the full-length Tat (Tat101) (38). The second exon, consisting of amino acids from 73–101, enhances the protein’s transcriptional competence and adds several additional functions. Previous work from our group showed that the presence of the second exon of Tat was necessary to increase NF-κB pathway signaling, BCL2 expression, resistance to FASL-mediated apoptosis, ROS generation, and changes in cytoskeleton organization both in Jurkat T-cell models and primary CD4+ T-cells (38–42). Interestingly, some of these changes are now considered hallmarks of cellular senescence (43). Ultimately, full-length Tat has been shown to reprogram CD4+ T-cells by directly regulating transcription of over 400 genes (44).
In this regard, Tat has been shown to induce cellular senescence in endothelial cells (29), microglia (35), bone marrow mesenchymal stem cells (31) and human adipose tissue (32). Furthermore, Tat can be detected in the serum (45) and spinal fluid (46) of ART-treated, and virally suppressed PWH. Even low doses of a chronic Tat expression are sufficient to cause a neurodegenerative phenotype (47) and neuronal age-related diseases (48) in mouse models, underscoring the importance of Tat effects even in the absence of full viral replication. However, the specific impact of the different Tat forms in eliciting a CD4+ T-cell senescence program is currently unknown. Given the above, we wanted to explore the role of Tat in inducing the cellular senescence program in CD4+ T cells.
We hypothesized that full-length Tat expression in CD4+ T-cells would lead to the onset of cellular senescence. To this end, we addressed the effect of Tat72 or Tat101 on the expression of several canonical senescence biomarkers encompassing different biological characteristics of senescent cells, according to the SenNet recommendations (43). We addressed BCL2, p21CIP1, p16INK4A, Ki-67, γH2AX, CD87/uPAR, SA-βGAL, and an array of SASP factors both at the mRNA and at the protein level, in an established and well-characterized Jurkat cell model stably expressing Tat (38, 39, 41, 42). We validated these important changes in primary CD4+ T-cells. We show that full-length Tat increases critical senescence biomarkers and SASP factors. We propose that full-length Tat expression may contribute to chronic inflammation and the HIV premature aging phenotype seen in PWH.
2 Material and methods
2.1 Cell lines
Jurkat-Tat72 and Jurkat-Tat101 stably express HIV-Tat first exon (1 exon, 1-72aa; Tat72) or full-length HIV-Tat (2 exons, 1-101aa; Tat101), respectively. These stable transfectants were created and obtained from Alcamí and Coiras’ lab (Instituto de Salud Carlos III, Madrid, Spain). Shortly, stable transfectants were generated by electroporation of pTRE2hyg-Tat72, pTRE2hyg-Tat101, or a pTRE2hyg empty vector (control; Tetoff) in the Jurkat-TEToff cell line (Clontech, BD Biosciences) and stabilized with hygromycin B. In this TEToff system, Tat expression can be repressed by adding 1 μg/ml of Doxycycline (DOX) (Takara Bio, Mountain View, CA, USA) for 48 h. Tat expression in these cells and DOX silencing have been extensively characterized before (38, 39, 41, 42, 49). Jurkat cells were grown in RPMI 1640 medium supplemented with 10% (v/v) fetal calf serum (FCS), 2 mM l-glutamine, 100 μg/ml streptomycin, and 100 U/ml penicillin (GIBCO, Thermo Fisher, Waltham, MA USA), termed R10 media. Culture media in Jurkat-Tat cells was supplemented with 300 μg/ml Geneticin (G418 Sulfate) and 300 μg/ml Hygromycin B (GIBCO). Cells were maintained in a humidified air 5% CO2 atmosphere at 37°C.
2.2 Primary CD4+ T-cells
Peripheral blood mononuclear cells (PBMCs) from buffy coats (Banc de Sang i Teixits, Barcelona, Spain) were isolated by density-gradient centrifugation (Lymphoprep, Stem Cell Technologies, Vancouver, BC, Canada), at 800g for 30 min at room temperature. The collected PBMCs were then washed twice with phosphate-buffered saline (PBS, Corning, Glendale, AZ, USA), counted, and viability addressed with an automatic counter Luna FL system (Logos Biosystems, Villeneuve d’Ascq, France) using an Acridine Orange/Propidium Iodide dual stain. CD4+ T-cells were negatively isolated using the EasySep™ Human CD4+ T Cell Enrichment Kit (Stem Cell Technology), following the manufacturer’s instructions. The purity and viability of isolated CD4+ T-cells were routinely checked by flow cytometry and found to be above 93% and 90% respectively.
2.3 Plasmids
Long terminal repeat (LTR)-LUC and LTR-GFP plasmids were obtained from Alcami’s lab and were previously described (38, 39, 50). pEGFP-N1 plasmid was a gift from Alcami’s lab and was originally from Clontech (BD Biosciences). For transient transfections of HIV-Tat, the same Tat72 and Tat101 cDNA sequences used to generate the stable Jurkat-Tat transfectants, were cloned into pcDNA3.1(+) backbones using Genescript Express Cloning services (Genscript, Oxford, UK). pcDNA3.1(+) -Tat72, a pcDNA3.1(+) -Tat101, and these two same constructs but with a C-Terminal DYKDDDDK (FLAG) tag fused to HIV-Tat were generated, yielding pcDNA3.1(+) -Tat72-DYK and pcDNA3.1(+) -Tat101-DYK plasmids. An identical, empty pcDNA3.1(+) vector was used as a control plasmid. Heat shock transformation of competent bacteria (Library Efficiency DH5α, Invitrogen, Waltham, MA, USA) was used to amplify all DNA plasmids, which were purified using a PureYield™ Plasmid Maxiprep System (Promega Corporation, WI, USA). Nucleic acid concentrations were determined based on 260nm absorbance with an EzDrop 1000 Spectrophotometer (Blue-Ray Biotech, New Taipei City, Taiwan).
2.4 Cell electroporation
Cells were electroporated using a NEON NXT device (Invitrogen). Jurkat cells were split and grown in fresh media the day before electroporation, then washed twice with PBS and seeded at 2×107 cells/ml in R-Buffer, following manufacturer recommendations. For Neon 10 μl Tips, a total of 2×105 cells and 0.75 μg of LTR-GFP DNA or pEGFP-N1 plasmid were used in 10 μl R-Buffer. Jurkat cells were electroporated using 1325V, 10ms and three pulses and immediately placed in 0.5 ml pre-warmed RPMI, 10% FCS antibiotic-free media in a 24-well plate culture vessel. 24 h post-electroporation, GFP expression and cell viability were addressed by flow cytometry. LTR-GFP was used to address Tat transactivation and the pEGFP-N1 plasmid was used as an indicator of transfection efficiency.
A total of 2×106 CD4 cells and 7.5 μg of empty pcDNA3.1(+), pcDNA3.1(+) -Tat72-DYK, or pcDNA3.1(+) -Tat101-DYK DNA plasmids were electroporated in T buffer with a 100 μl NEON NXT tip using 2200V 20MS in one pulse (51). Immediately after the transfection, CD4+ T-cells were placed in pre-warmed RPMI, 10% FCS, antibiotic-free culture media. Transfection efficiency and viability was measured with a pEGFP-N1 plasmid as indicated above.
2.5 RNA isolation, RT-qPCR
Jurkat and electroporated CD4+ T-cell pellets were harvested and immediately frozen at the indicated times. Total RNA was isolated using the RNAeasy mini kit (Qiagen, Hilden, Germany) and the genomic DNA removed with an on-column DNAse incubation step (RNase-Free DNase Set; Qiagen). RNA concentrations were determined using an EzDrop 1000 Spectrophotometer (Blue-Ray Biotech) and integrity checked with a TapeStation RNA ScreenTape (Agilent, Madrid, Spain). Total RNA (0.5 μg for Jurkat cells; 0.15 μg for electroporated CD4+ T-cells) was transcribed to cDNA with a SuperScript™ IV VILO RT mastermix (Invitrogen) in a 20 μl reaction, following the manufacturer’s instructions. 1 μl from cDNA synthesis reaction and 5 μl Fast Advanced Taqman MasterMix were used in a total 10μl FAST-qPCR reaction in 0.1 ml MicroAmp Fast Optical 96-well plate (Applied Biosystems, Foster City, US) and fluorescence signal detected with a StepOne Plus instrument (Applied Biosystems). Predesigned FAM-MGB Taqman primers and probes (Applied Biosystems) were used to detect senescence markers and are listed in Table 1. The following custom FAM-MGB TaqMan primers and probe were synthesized (ThermoFisher) and used to detect both forms of HIV-TAT mRNA:
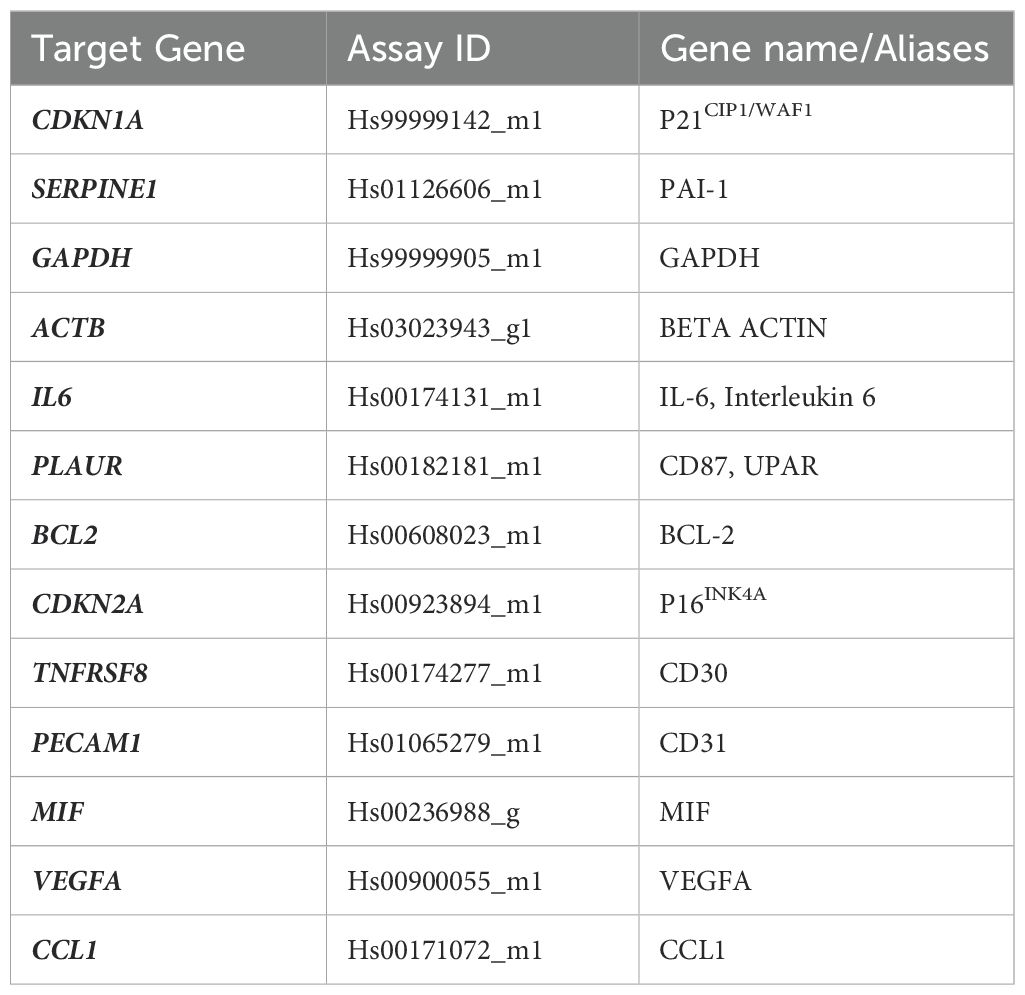
Table 1. Predesigned FAM-MGB Taqman primers and probes used in this study.
Forward 5’-TAGAGCCCTGGAAGCATCCAGGAAG-3’
Reverse 5’-CTATGCTCTGATAGAGAAGCT-3’
Probe: 5’-TGGCAGGAAGAAGCGGAGA-3’
The 2-ΔΔCt method was used to quantify relative mRNA changes against GAPDH. Data is represented as fold changes against TEToff control.
2.6 Droplet digital PCR
CD4+ T-cells were isolated from cryopreserved PBMCs from PWH with EasySep™ Human CD4+ T Cell Enrichment Kit (StemCell Technologies). Cell activation was performed with Dynabeads™ Human T-Expander CD3/CD28 (Gibco), using 10μl of beads per 1×106 cells, and 100U/ml IL-2. Retrotranscription of RNA and cDNA amplification was performed with the One-Step RT-ddPCR Advanced Kit for Probes (Bio-Rad). To detect Tat mRNA, we used the well characterized Tat/Rev primers and probe (FAM) (52) with an annealing temperature of 54°C. To detect Tat mRNA in stable Jurkats we used our Tat72 Taqman primers and probe (FAM), modified from (38) and described above. Every well contained a control of cellular mRNA presence for CD3 gene as housekeeping (HEX). Analysis was performed by adjusting ddPCR RNA raw data concentrations with RNA quantity and concentration in each well. The ddPCR was performed in a QX600 Droplet Digital PCR System (Bio-Rad).
2.7 SASP in cell supernatants
The different Jurkat cell lines were seeded at 0.5×106 cells/ml in a round bottom 96-well plate for 3 days. Plates were then centrifuged at 300 x g, 5 min and cell supernatant stored at -80°C for further use. Mediator levels of 105 human soluble cytokines were addressed in Jurkat supernatants using a Proteome Profiler Human XL Cytokine Array Kit (R&D Systems, Biotechne, Abingdon, UK), following the manufacturer’s instructions. The membranes were developed using SuperSignal™ West Atto reagents (Thermo) and immediately placed together in an Odyssey Fc instrument (LI-COR, Nebraska, USA), to generate a single image containing Tetoff, Tat72 and Tat101 cytokine arrays captured together at different time intervals. Files created by Image Studio acquisition software were analyzed in Empiria Studio 3.0 software (LI-COR), with the built-in signal analysis tool. The signal of each pair of spots was calculated and corrected by subtracting the mean signal intensity from the defined background spots. Then, the mean signal intensity of each cytokine was normalized to the mean signal intensity of the 6 reference spots distributed in each corresponding membrane.
For absolute quantification of sCD31, and sCD30 levels in cell-supernatants, a CD31 (PECAM-1) Human ProcartaPlex™ Simplex Kit, and a CD30 Human ProcartaPlex™ Simplex Kit were used, following the manufacturer’s instructions (Thermo Fisher Scientific). The assay was measured using a Luminex™ 200 Instrument System (Thermo Fisher Scientific).
2.8 Flow cytometry
4×105 Jurkat cells were grown in 1 ml R10 medium in 24-well plates for 24 h. Cells were collected in FACS tubes and washed 2 times with PBS before Live/Dead Near IR or V450 staining (Invitrogen). Live/Dead staining was included in all flow cytometry measurements. After washing with PBS and blocking Fc Receptors (Trustain X FC, Biolegend, San Diego, US), the expression of surface markers CD87, CD30 or CD31 was addressed with the indicated monoclonal antibodies (Table 2). To further assess the intracellular markers BCL2 and γH2AX, cells were fixed and permeabilized using the Cytofix/Cytoperm kit (BD biosciences) following the manufacturer’s instructions. To assess the levels of the proliferation marker Ki-67, after Live/Dead staining, cells were fixed with ice-cold 70% ethanol and placed at -20°C for a maximum of 1 week. Cells were then washed twice with 2 ml of PBS 0.1% bovine serum albumin (BSA, Sigma-Aldrich) and incubated with FxCycle™ Violet Stain (Invitrogen) in PBS 0.1% Triton X-100 (Sigma-Aldrich) for 15 min. Cells were immediately analyzed with a FACS CANTO II (BD Biosciences). To assess intracellular p21 levels in Jurkat cells, ice-cold 90% Methanol fixation was used to fix and permeabilize cells after live/dead staining. Cells were stored at -20° C up to a week, then washed twice in PBS 0.1% tween-20, blocked with 1% human AB Serum and stained with a primary, unlabeled rabbit anti-p21 antibody (Abcam). After washing twice with PBS 2% FCS, a secondary anti-Rabbit-Alexa Fluor 488 antibody (Invitrogen) was used. Cells were analyzed using a Gallios Instrument (Beckman Coulter, California, US).
Table 2. Antibodies used for flow cytometry.
Electroporated CD4+ T-cells were harvested at 48 or 72 h post electroporation and washed twice with PBS before Live/Dead Near IR staining. Cell membrane CD4, CD87, PD1 and intracellular BCL2, γH2AX, p21, p16, and FLAG (HIV-Tat) expression levels were determined using the corresponding antibodies (Table 2). The eBioscience Foxp3/Transcription Factor Staining Buffer Set (Invitrogen) was used to fix and permeabilize cells allowing the detection of intranuclear antigens. The acquisition was performed on an Aurora Spectral Flow Cytometry (Cytek Biosciences B.V., The Netherlands) with a 4 lasers configuration (16V-14B-10YG-8R).
2.9 SA-β-gal staining
SA-β-gal-positive cells were detected using SPiDER-βGal (DOJINDO, Kumamoto, Japan, SG03), according to the manufacturer’s instructions. Specifically, Jurkat cells were incubated with bafilomycin A1 for 1 h at 37 °C and 5% CO2. After washing with PBS 1X, cells were incubated with SPiDER-βGal for 30 min at 37 °C 5% CO2.
2.10 Fluorescence microscopy
Jurkat E6.1 cells were treated 24h with 0,25 μM etoposide and washed with 1XPBS. After that, SA-βGal staining was performed as described above (2.8 section in methods). Cells were then laid on Superfrost ultra plus® microscope slides for 30 min at RT, salts were cleaned with deionized water and dried. Slides were cover-slipped with a drop of mounting medium containing DAPI for visualization of cell nuclei (ProLong Gold, Thermo Fisher Scientific). Finally, cells were observed using a 400x magnification under Nikon Eclipse E600 fluorescence microscope for green, red and blue fluorescence. Images were analyzed in ImageJ software.
2.11 Statistical analysis
Graphs were plotted using the GraphPad Prism 10.4.1 software (GraphPad Software, Inc., San Diego, California, USA). Data was subjected to normality tests. For multiple comparisons, a Repeated Measures One Way ANOVA followed by Tukey’s multiple comparison test was used. For all the tests used, a two tailed P value <0.05 was considered statistically significant.
3 Results
3.1 Tat101 expression increases cellular senescence protein biomarkers in Jurkat cells
To address whether HIV-Tat expression resulted in increased expression of canonical senescence biomarkers, we used a well-characterized Jurkat Tetoff model, stably expressing full-length Tat (Tat101), or Tat’s first exon (Tat72) (38, 41, 42, 49). As previously described (38, 41, 53), Jurkat Tat cell lines expressed comparable levels of TAT mRNA (Supplementary Figure S1). Interestingly, these levels were around 25-fold higher than activated CD4+ T-cells from PWH (Supplementary Figure S1). Despite being transformed cells, our Jurkat transfectants readily respond to senescence inducers (54) such as etoposide (ETO), that induce DNA damage. This results in increased γH2AX expression (55).Treating our cells with 0.25 μM ETO for 24h increased γH2AX, BCL2, and CD87 expression (Supplementary Figures S2A, B, C). SA-β-Gal staining after ETO treatment was also visualized in fluorescence microscopy (Supplementary Figure S3). The doses of ETO here used did not alter cell viability (Supplementary Figure S4).
Tat101 expression resulted in a statistically significant increase in the percentage of cells expressing CD87 compared to Tetoff controls and to Tat72-expressing cells (Figure 1A). Similarly, in all experiments the percentage of γH2AX+ and p21+ cells was higher in in Jurkat Tat101 than in Tetoff (p=0.0756; Figure 1B and p=0.0558; Figure 1C, respectively). Tat101 expression also increased BCL-2 geometric mean, compared to Tetoff and to Tat72 cells (Figure 1D). All experiments showed a trend towards a reduced expression of Ki-67 in Tat101 cells, compared to Tetoff (p=0.0769) (Figure 1E). Neither Tat72 nor Tat101 expression altered senescence-associated β-GAL activity (SA-β-Gal) (Figure 1F). There were no differences between Tat72 and Tetoff in any of these markers. Furthermore, DOX addition abrogated the Tat101-mediated increase in γH2AX+ cells (Figure 1B) and slightly reduced the percentage of CD87+ cells (p=0.0842) observed in Tat101-expressing cells (Figure 1A). Interestingly, DOX did not alter Tat101-mediated changes in BCL2 and only slightly reduced p21 levels. DOX addition did not alter cell viability at the times tested (Supplementary Figure S3).
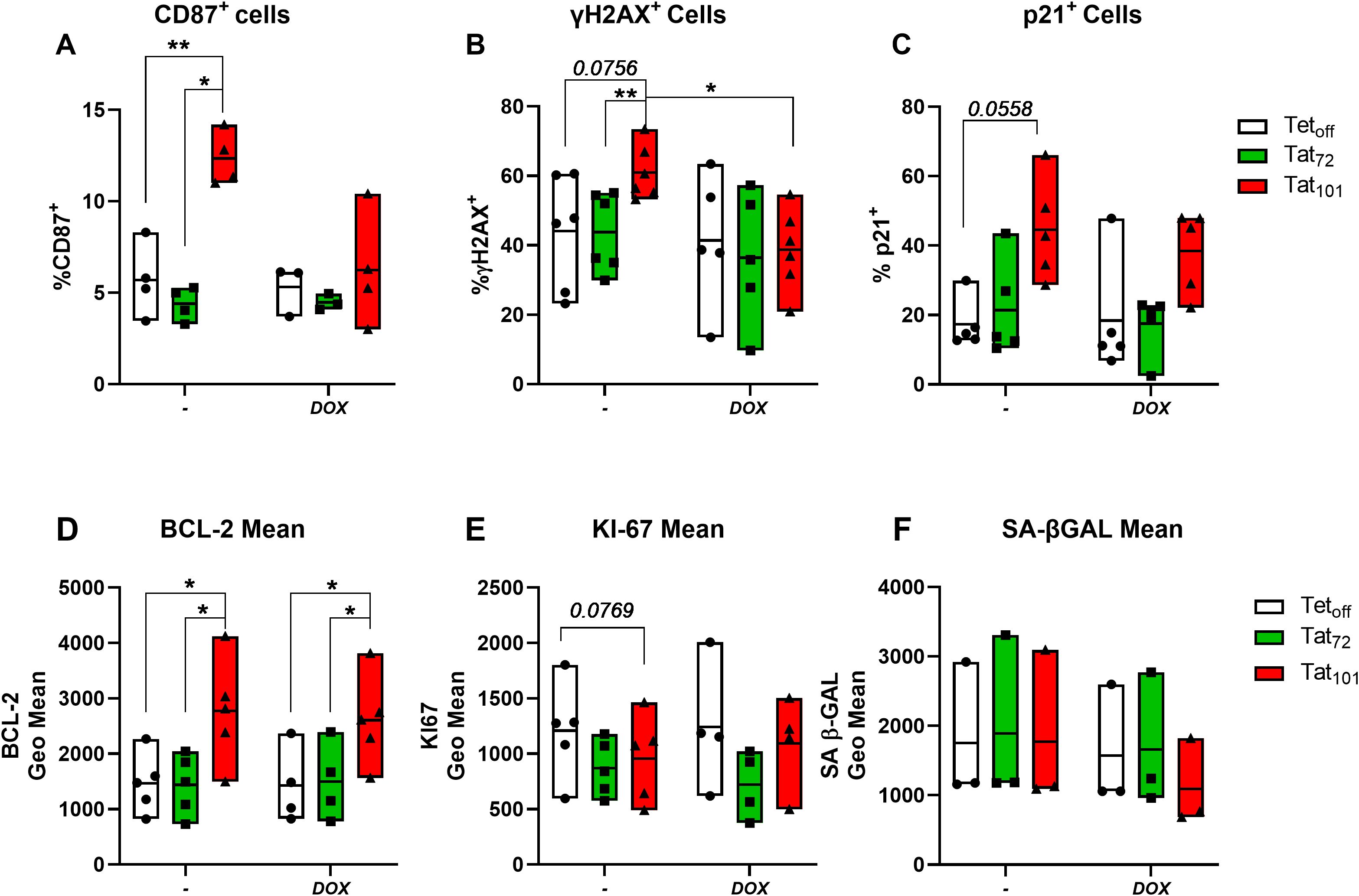
Figure 1. Full-length HIV-Tat increases the expression of senescence protein markers. Jurkat Tetoff T-cells stably expressing full-length HIV-Tat (Tat101) or first-exon Tat (Tat72) were treated with 1μg/ml Doxycycline for 48h (+DOX) or left untreated (-) and then the expression of senescence protein markers determined by flow cytometry. The percentage of live cells positive for CD87 (A), γH2AX (B), and p21 (C) and the geometric mean BCL-2 (D), KI67 (E), and Senescence associated β-GAL (SA β-GAL) (F) are shown. Symbols represent individual experiments (n=3-5) and floating bars indicate minimum to maximum values with a line at the mean value. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Statistical analysis was performed by one-way ANOVA with Tukey’s multiple comparisons test.
3.2 Full-length, intracellular HIV-Tat increases levels of senescence-associated secretory phenotype mediators
We analyzed the composition of the SASP using a membrane-based antibody array detecting 105 cytokines. Out of them, 43 were visually detectable and their mean relative levels for each cell line were calculated (Figure 2A, Supplementary Figure S5). 12 factors were upregulated at least 1.4-fold and 3 downregulated at least 0.7-fold in Tat101 cell supernatants compared to control Tetoff (Figure 2B). We found that CD30, PDGF-AA, and CD31 showed the highest increase over Tetoff, with a mean fold-change of 2.71, 2.4, and 1.93, respectively. Cystatin C and VEGF were the targets that showed the highest downregulation in Tat101 compared to Tetoff, displaying a mean fold change of 0.47 and 0.65 respectively (Figure 2B). Interestingly, 7 out of the 8 mediators upregulated at least 1.4-fold in Tat72 over Tetoff (Figure 2C) were the same as those seen in the Tat101 vs Tetoff comparison, but displaying a more modest increase. In Tat72, IL-3, GM-CSF and IL-2 showed the highest up-regulation with 1.57, 1.53 and 1.5-fold-change over Tetoff, respectively. Only Cystatin C showed a greater downregulation in Tat72 over control cells (0.38-fold-change) compared to full-length Tat (Figure 2C). Accordingly, CD30, PDGF-AA, IL-4 and Cystatin C were the only mediators that increased over 1.4-fold in Tat101 compared to Tat72 (Figure 2D). Importantly, 10 out of the 15 factors changed in Tat101 over Tetoff have well-defined roles in cellular senescence or aging (highlighted in bold in Figure 2C). Next, we validated and quantified the increase in soluble CD30 (sCD30) and CD31 (sCD31) levels in Tat101 and in Tat72 compared to Tetoff cells using a highly sensitive Luminex technique. Levels of sCD30 increased from 355.7 pg/ml (as a mean) in Tetoff cells to 619.8 in Tat72 and to 1636 pg/ml in Tat101 (Figure 2E). Regarding sCD31, levels increased from 800.7 pg/ml (as a mean) in Tetoff cell lines, to 1061 pg/ml in Tat72 and to 1408 pg/ml in Tat101 expressing cells (Figure 2F). We also addressed the expression of CD30 and CD31 in the cell membrane of Jurkat Tat101. Flow cytometry data showed a stark increase in the percentage of CD30+ cells (Figure 2G) and CD31+ cells (Figure 2H), from 18.78% and 86.6% of Tetoff cells to 94.2% and 99.86% in Tat101 cells, respectively. The geometric mean of these markers also increased, with a 5,02-fold increase in CD30 (Supplementary S5B) and a 6,8-fold increase in CD31 (Supplementary S5C) in Tat101 cells compared to Tetoff. Tat72 cells showed a modest increase over Tetoff in the percentage of cells expressing these markers (Figure 2F, H). Furthermore, combining both markers enables a clear identification of Tat101 cells as a separate population (Figure 2I). In this regard, 89,9% of Tat101 cells stain positive for both CD30 and CD31 whereas only 13,4% and 32,2% are double positive in Tetoff and Tat72, respectively.
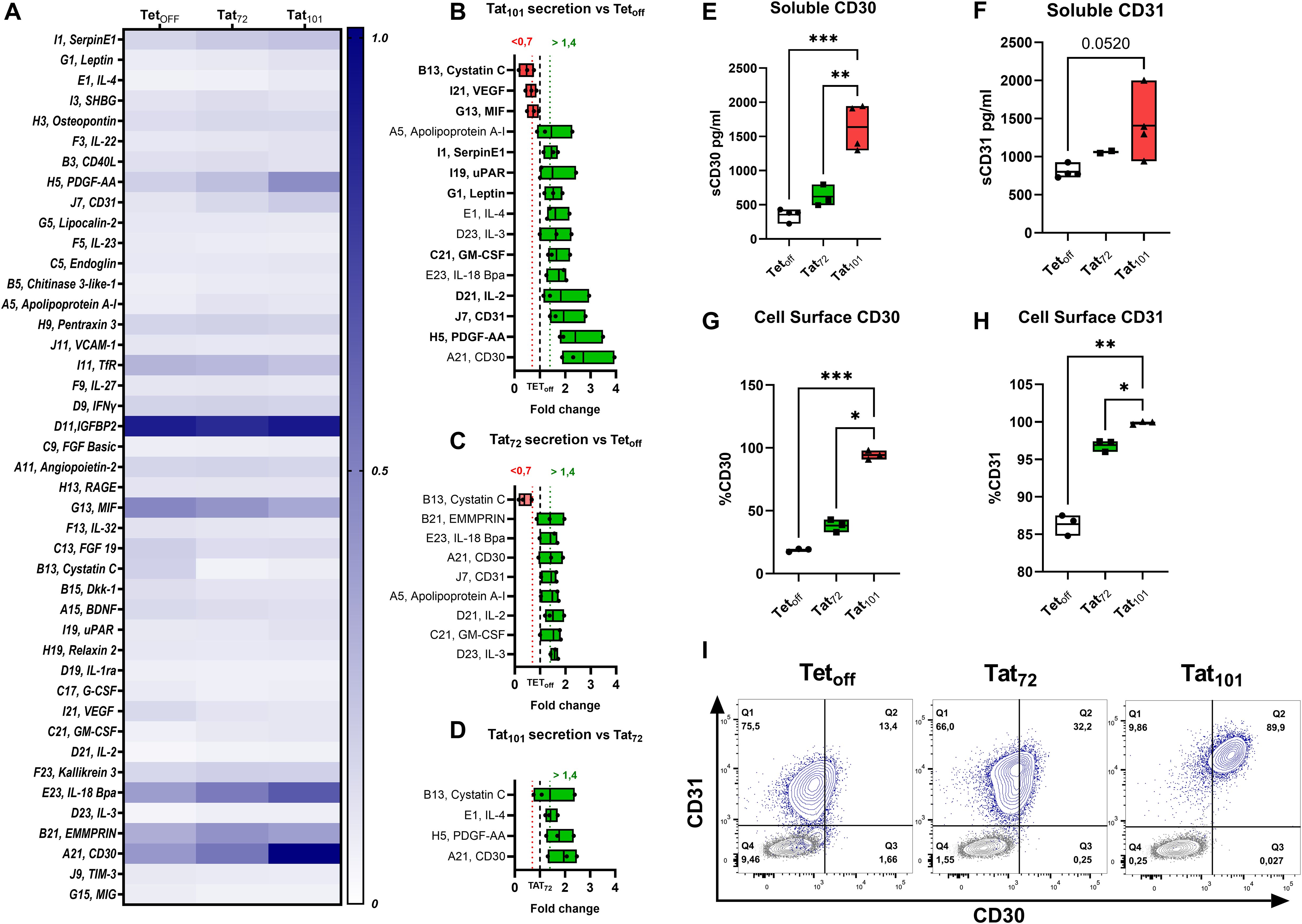
Figure 2. SASP Mediators in Jurkat cell line supernatants. Jurkat Tetoff, Tat72 or Tat101, cells were grown in round bottom 96-well plates at 5×105 cells/ml in 200 μl R10, and cell supernatants were collected after 72 hours. The expression levels of 105 cytokines were addressed with an antibody-based membrane array. (A) Heatmap indicates the mean intensity values of the 43 mediators with visible spots in the membrane after ELC incubation. Mediators with mean signals over 1.4 (green boxes) or below 0.7-fold change (red boxes) expression in Tat101 over Tetoff (dotted line) are shown in (B). Changes in Tat72 over Tetoff (C) and Tat101 over Tat72 (D) are also shown. (n=3). Secreted levels of CD30 (E) and CD31 (F) were evaluated in Tetoff (n=4), Tat72 (n=2-3) and Tat101 by Luminex on the same cell supernatants as (A). The percentage of cells expressing cell surface CD30 (G) or CD31 (H) was addressed by Flow Cytometry. Graphs indicate percentages of cells within the Live cell gate after 48h cell culture. CD30 and CD31 co-expression for each of the cell lines (blue contour plot) overlayed with autofluorescence controls (grey contour plot) from a representative experiment is shown in (I). Symbols represent individual experiments (n=3), and floating bars indicate minimum to maximum values with a line at the mean. Statistical analysis was performed by one-way ANOVA with Tukey’s multiple comparisons test *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
3.3 Tat101 increases the expression of senescence biomarkers at the mRNA level
We aimed to evaluate the expression of the previously addressed senescence biomarkers and additional SASP factors at the mRNA level, using a Taqman RT-qPCR approach. We found that BCL2, CDKN1A (p21), IL6, TNFRSF8 (CD30), PECAM1 (CD31), and CCL1 mRNA expression was significantly increased in Tat101 compared to Tetoff cell lines (Figures 3A–F). We noticed modest increases in PLAUR (CD87), SERPINE (PAI-1) and in VEGFA mRNA levels in Tat101 cell lines compared to Tetoff, without statistical significance (Figures 3G–I) due to higher intragroup dispersion. A trend (p=0.0516) towards a reduced MIF mRNA expression was observed in Tat101 compared to Tetoff. Tat72 expression did not significantly increase the senescence biomarkers here addressed when compared to Tetoff controls. The importance of the second exon is underscored by the fact that Tat101 expression significantly increased mRNA levels of BCL2, IL6, TNFRSF8 (CD30), PECAM1 (CD31) and PLAUR (CD87) mRNA over cells expressing Tat72. (Figures 3A, B, D, E). This data suggests that the second exon is important in inducing cellular senescence biomarkers at the transcriptional level.
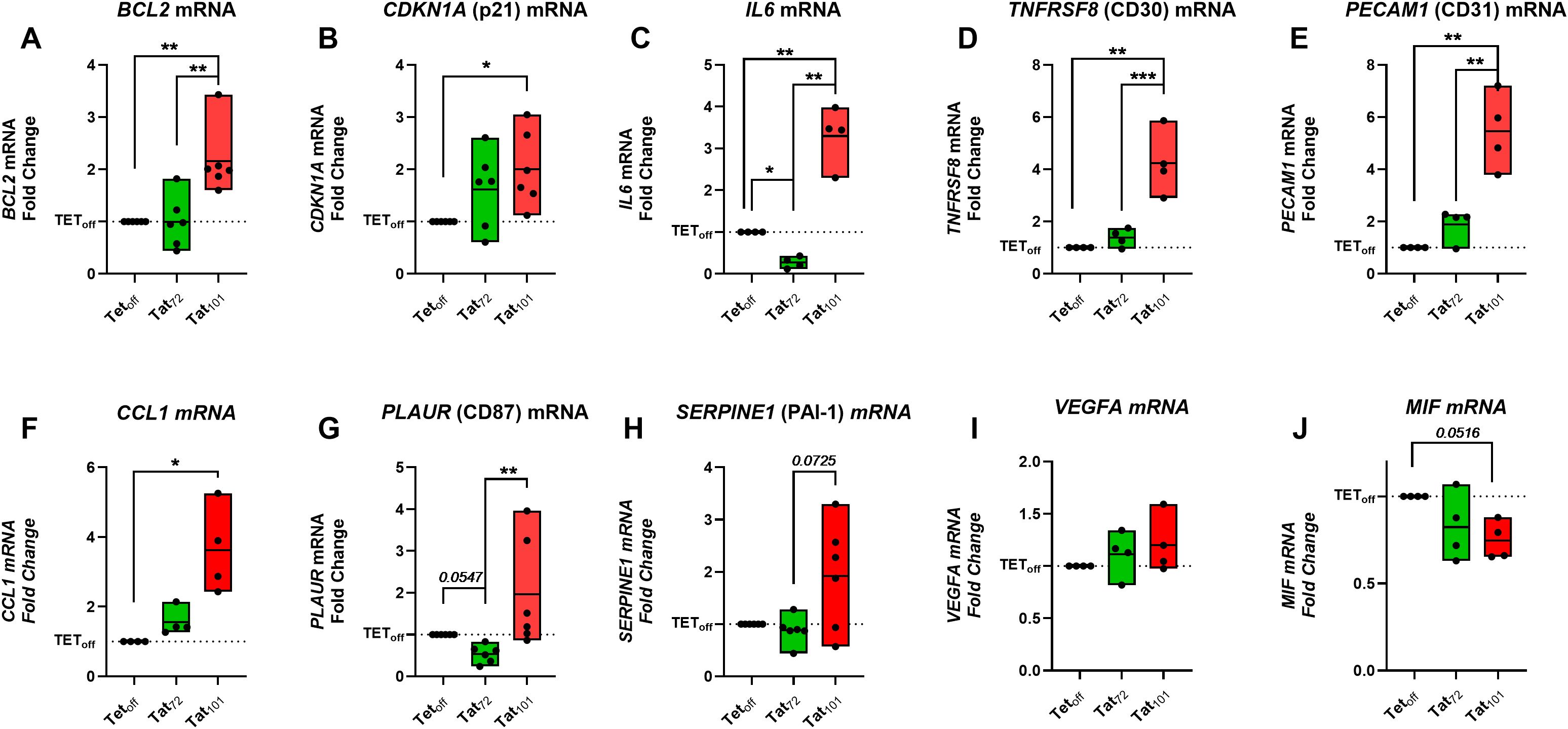
Figure 3. Full-length HIV-Tat increases several senescence markers at the mRNA level. Jurkat Tetoff, Tat72 or Tat101, cells were grown for 24h in 24-well plates at 4×105 cells/ml in 500 μl of R10. Cell pellets were collected, and mRNA levels of indicated senescent markers were determined by RT-qPCR (A-J). Symbols represent individual experiments (n=4-5) with values representing the Fold Change versus Jurkat TEToff control cell line (dotted line) following the 2-ΔΔCT calculation. Floating Bars indicate minimum to maximum values with a line at the mean value. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Statistical analysis was performed by one-way ANOVA with Tukey’s multiple comparisons test on ΔCT values.
To confirm and extend RNA results, we analyzed the expression of senescence associated-genes with RNA-Seq data generated in our companion paper (56). First, we tested well-known up-regulated (TNF) and down-regulated (CD1E) genes upon Tat expression (39, 44) (Figure 4A). TNF was upregulated 40-fold while CD1E was downregulated over 65-fold in Tat101 compared to Tetoff, confirming known Tat-derived effects on these cell lines. We also found that CCL1, CDKN1A, PECAM1, PDGFA, and TNFRSF8 were differentially expressed genes (DEG) in Tat101 compared to Tetoff (Figure 4B), supporting our previous mRNA and protein data. Next, DEG in the Tat101 vs Tetoff comparison, were compared to different senescence datasets available in the literature. We found that among the genes that comprise the SenMayo dataset (57), Tat101 expression upregulated TNF, CCL1, CDKN1A, PECAM1, and ITPKA mRNA levels while downregulated IFGBPT7, IGFBP2, TIMP2, AXL, and CD9 (Figure 4C). We further compared DEG to other senescence datasets like hsa04218 (Figure 4D) and R-HSA-2559583 (Figure 4E), showing increased levels of senescence hallmarks such as CCND2, CCND1, CCNE1, or AKT3 or decreased mRNA levels of PIK3R2 and RB1 in Tat101 cells. This information will prove valuable to understanding how full-length Tat alters cellular senescence pathways.
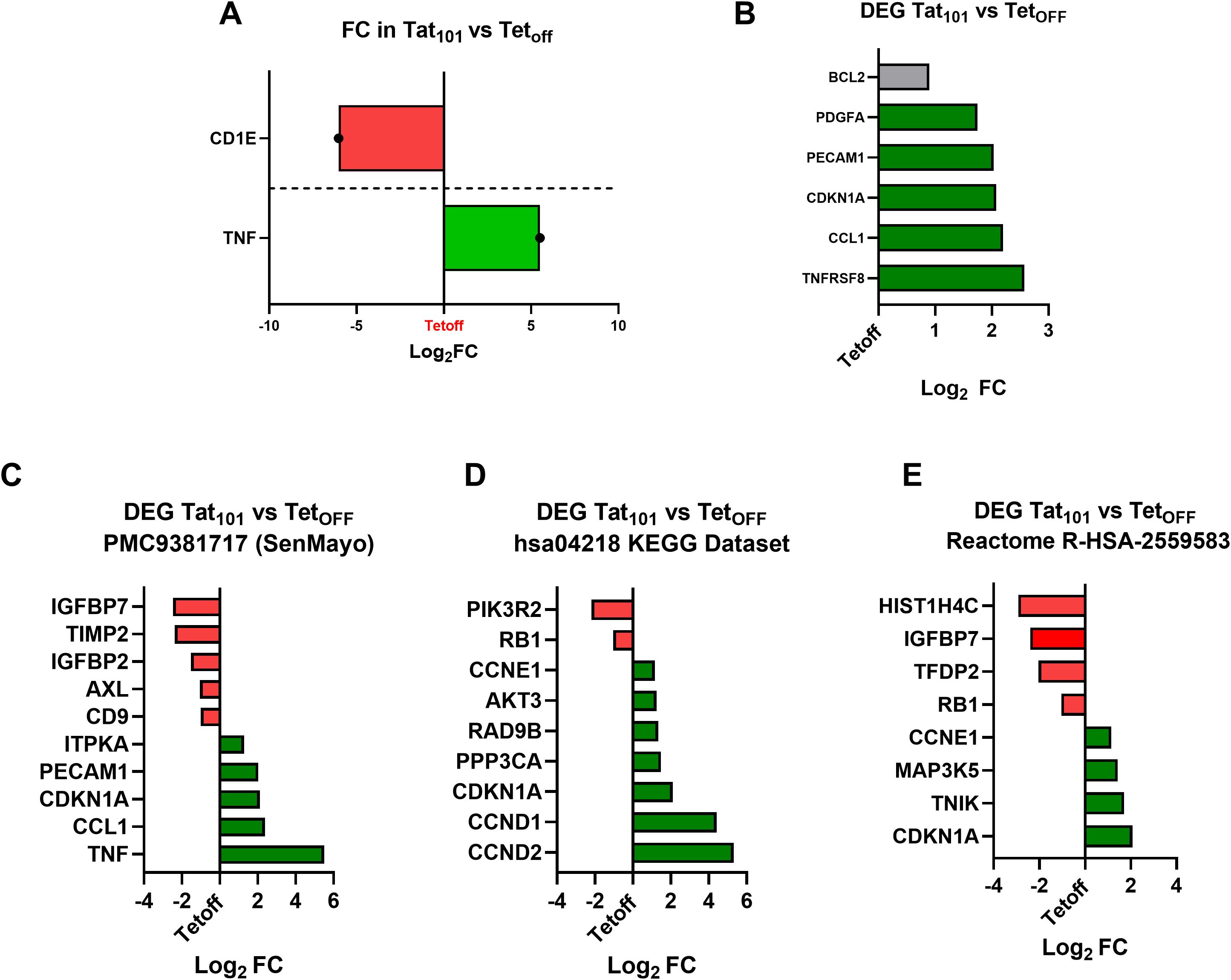
Figure 4. Full-length HIV-Tat increases transcripts involved in cellular senescence. An RNA-seq approach was used in the same Jurkat Tetoff, Tat72 or Tat101, cells used here (Rodríguez-Agustín, under revision). Bars indicate the Log2 Fold Change (FC) in relevant DEG (Differentially Expressed Genes) in the Tat101 vs Tetoff comparison. Green bars indicate upregulated DEG Genes (PPDE>0.95 and FC > 2), and red bars indicate downregulated DEG genes (PPDE>0.95, FC <0,5). (A) Tat101 stable expression increases TNF and decreases CD1E mRNA transcripts, well-known Tat target genes. (B) Senescence biomarkers and SASP targets addressed in this work that are also DEG in the RNA-seq experiment are shown. A grey bar indicates a DEG gene with an up-regulation below the 2-fold threshold (1, 8). Expression values (Log2FC) of Tat101 DEG genes present in the SenMayo dataset (C), in the hsa04218 KEGG Dataset (D) or in the Reactome R-HSA-2559583 dataset (E) are shown.
3.4 p21, p16 and γH2AX expression is increased in Tat-expressing (FLAG+) primary CD4+ T-cells
To address the role of HIV-Tat in cellular senescence in a non-transformed model, we used purified CD4+ T-cells, which are the primary target of HIV infection. Cells were electroporated with DNA plasmids (51) encoding full-length Tat or Tat’s first exon, fused to a C-terminal FLAG tag (DYK). Electroporation efficiency was around 16% at 24 hours post-electroporation (Supplementary Figure S6A) with cell viability at around 60% at this same time point (Supplementary Figure S6B). Nearly 3% of viable cells expressed Tat-DYK proteins at 24 hours post-electroporation, as measured by FLAG detection (Supplementary Figure S6C). This was reduced to a mean of 0.5% at 72 h post electroporation in Tat72-DYK (± 0.29%) and to around a mean of 1% in Tat101-DYK (± 0.7503%) (Supplementary Figure S6D). Cell viability was similar between Tat72-DYK (mean of 32.3% ± 9.1) and Tat101-DYK (mean of 33.73% ± 14.42) (Supplementary Figure S6E). Additionally, we verified Tat expression with a Taqman qPCR approach, confirming a high expression of Tat mRNA at 24 hours, with a lower but still abundant expression at 72 hours (data not shown).
Expressing either form of Tat in CD4+ T-cells resulted in a marked increase in the percentage of p21+, γH2AX+, and p16+ cells in the FLAG+, (Tat-expressing cells) compared to their FLAG- counterparts (Figures 5A–C). In addition, a similar trend was observed for the percentage of CD87+ cells (Figure 5D) but not reaching statistical significance. We also detected a marginal increase in BCL2 (Figure 5E), and a decrease in CD4 expression (Figure 5F) in FLAG+ cells compared to their FLAG- counterparts, but these changes did not reach statistical significance (p=0.1796 and p= 0.2104, respectively). Taken together, this data confirms that Tat upregulates important senescence biomarkers in primary CD4+ T-cells.
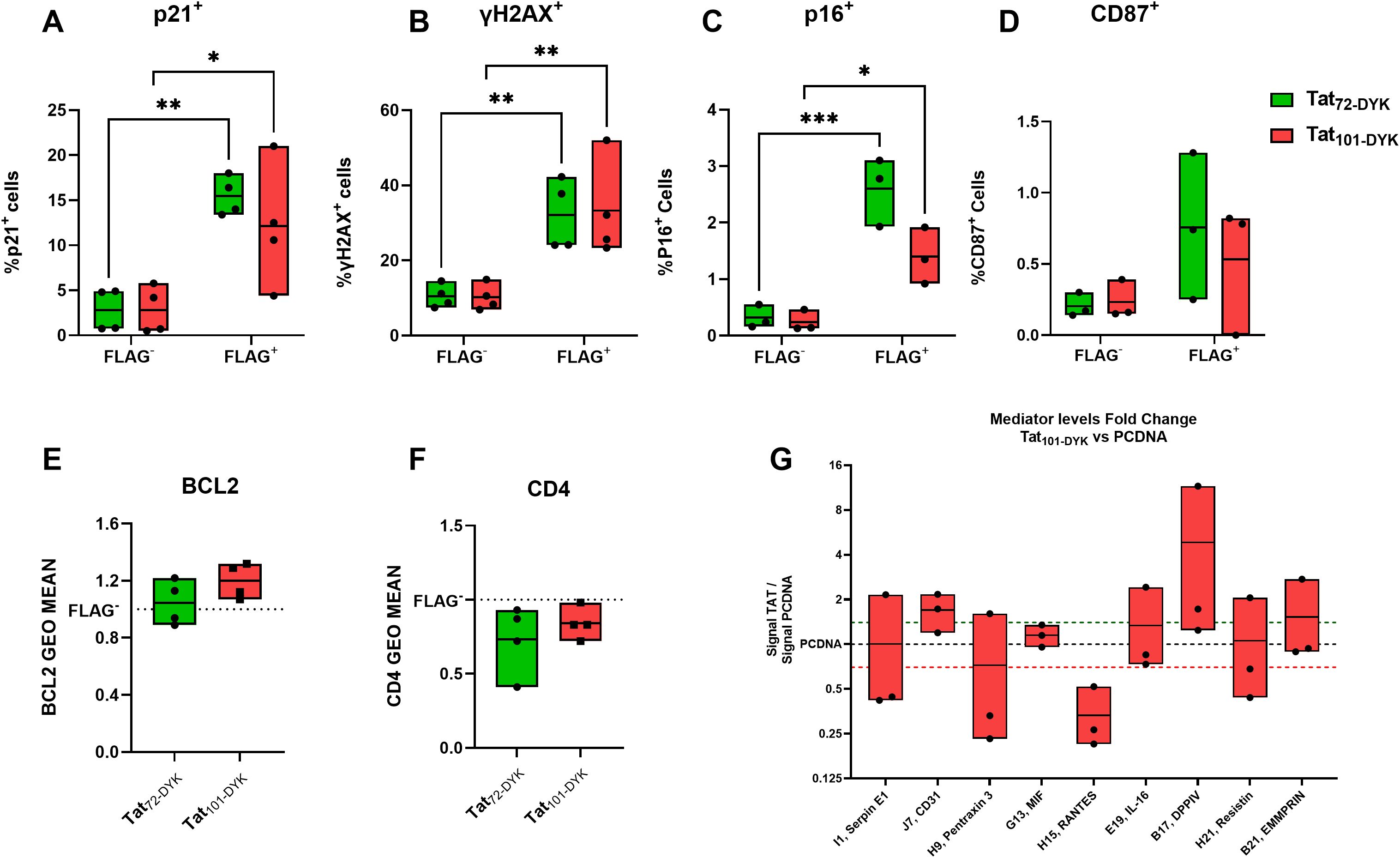
Figure 5. Tat-expressing CD4+ T-cells show increased levels of p21, γH2AX, and p16 senescence biomarkers. Resting CD4+T-cells isolated from buffy coats were transiently transfected with DNA plasmids encoding for Tat72-DYK, Tat101-DYK or a pcDNA empty vector backbone. An anti-FLAG antibody was used to detect Tat-expressing cells. The percentage of p21+ (A), γH2AX+ (B), p16+ (C) and CD87+ (D) cells within viable (Live Dead Negative) FLAG+ or FLAG- populations at 72hours post electroporation are shown. Fold change values in the geometric mean of BCL2 (E), and CD4 (F) in FLAG+ cells compared to FLAG- cells are indicated. An antibody membrane-based array was used to address changes in released mediators in cell supernatants. The levels (Fold-change) of detectable mediators in TAT101-DYK cell supernatants against pcDNA cell supernatants are shown in (G). Symbols represent individual experiments (n=3-4) and Floating Bars indicate minimum to maximum values with a line at the mean. A green dotted line indicates > 1.4-fold-change and a red dotted line indicates < 0.7-fold-change. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Statistical analysis performed by two-way ANOVA with Tukey’s multiple comparisons test.
3.5 HIV-Tat101 expression in resting CD4+ T-cells increases soluble CD31 and CD26 secretion in cell supernatants
We next aimed to determine whether Tat expression also resulted in increased SASP mediator secretion. Only 18 out of 105 mediators were visually detectable. Importantly, increased levels of CD31 and DPP4 (CD26) were detected in all experiments performed (Figure 5G), with 1.69 and 4.8-fold-change increases over pCDNA, respectively. In addition, we observed a 0.333-fold change (3.03-fold reduction) in the levels of RANTES (Figure 5G).
4 Discussion
Recent studies show that a broad range of viral infections, including lentiviral infections, result in the onset of a canonical cellular senescence program (58), reminiscent of other cellular responses to stress, such as oncogene-induced senescence (11). Growing pieces of evidence suggest that HIV infection induces cellular senescence, as an in vitro HIV infection of primary human fetal microglia results in increased SA‐β‐gal and p21 expression and IL-6 and IL-8 secretion (59). In addition, in vitro HIV infection activates the DNA damage response (DDR), increasing γH2AX and 53BP1 staining in U2OS cells (60) and CD4+ T-cells (61). In ex vivo T-cells from PWH, an elevated percentage of p16INK4A -positive cells and reduced telomere length are observed in untreated PWH (19, 25, 26). We show here that full-length, intracellular Tat expression upregulates p21CIP, p16INK4A, CD87, γH2AX, and BCL-2 expression, which translates to increased expression of key cell cycle inhibitors and enhanced resistance to apoptosis. Additionally, the secretion of several SASP mediators is increased, shaping a pro-inflammatory secretome. These changes in Tat-expressing cells are characteristics of cellular senescence (13, 43). This supports the important role of Tat in the increased senescence markers observed in HIV infection and in PWH.
Our experimental model consists of a well-characterized Jurkat Tetoff cell line, stably expressing 1 or 2 exons forms of Tat (38, 39, 42, 49, 53). The 2 exons, 101aa form of Tat is the most common in clinical HIV isolates, while some strains encode for a 2 exon, 86aa Tat form (62). Exogenous delivery of an 86aa (32, 35) or a 101aa (31) form of soluble, recombinant HIV-Tat resulted in reduced cellular proliferation, increased SA‐β‐gal and p21 expression together with augmented IL-6 and IL-8 SASP secretion in cell cultures of human microglia (35), adipose tissue (32) and bone marrow mesenchymal stem cells (31). In our model, Tat is produced intracellularly, but we cannot exclude the possibility of Tat being released to the extracellular space. In our Jurkat model, we observe increased p21 protein and mRNA levels and elevated IL-6 mRNA production. In addition, we detect increased γH2AX in Tat101-expressing cells, which suggests a cellular response to DNA damage. Exogenous, recombinant HIV-Tat has been shown to increase ROS levels and increased DDR in primary B-cells, as detected by increased γH2AX levels (63). A similar finding is reported in an immortalized lymphoblastoid B cell line RPMI-8866 (64). Previous results showed increased ROS generation upon Tat101 expression in the same Jurkat cell model used here (41), which indicates an active DDR response. Importantly, the DDR response is known to elevate the transcriptional activity of CDKN1A (p21CIP1) and CDKN2A (p16INK4A) genes (15). We have also addressed the levels of CD87/uPAR, a novel, cell-surface biomarker of senescence (65), also secreted by senescent cells. Soluble uPAR/CD87 (suPAR) was among the first SASP factors identified (18). In vitro HIV infection leads to increased membrane and secreted CD87 expression (66, 67), and elevated levels of serum CD87 in PWH correlate with poor survival (68). Thus, increases in CD87 during HIV infection may be linked to Tat activity.
A critical feature of cellular senescence is SASP. Here, we report that Tat101 increases the secretion of known SASP mediators like PAI-1 (SerpinE1), suPAR, Leptin, GM-CSF and IL-2 (18, 69). In addition, CD30 and IL-18Bpa are considered markers of healthy aging (70). Out of the 105 mediators addressed, CD31, PDGF-AA and CD30 are the three more upregulated over Tetoff levels. We corroborate this finding at the mRNA level using a sensitive qPCR approach, with consistent results to previous gene microarray data, which also showed increased TNFRSF8 (CD30) and PECAM1 (CD31) mRNA levels on the same Jurkat Tat101 cells (38).
CD31 is a member of the immunoglobulin superfamily and influences leukocyte migration, angiogenesis, and integrin activation. PECAM1, is a member of the SenMAYO geneset, used to identify senescent cells across tissues (57). The expression of CD31 is high in naïve T-cells, especially in recent thymic emigrants, and decreases during T-cell activation (71). However, CD31 remains elevated during T-cell differentiation in HIV infection, which may result in exhausted T-cell functionality (72). We found PECAM1 as an upregulated DEG in the database of an RNA-seq approach performed on a Jurkat Teton model of Tat expression (44), which suggests that PECAM1 is upregulated early upon Tat expression and remains high upon chronic Tat exposure, as we see in our Jurkat Tetoff model. Importantly, elevated CD31 levels are found in the serum of PWH, which may contribute to blood-brain barrier permeability and neuro-AIDS (73).
CD30 is a TNF-receptor superfamily member, expressed in activated or in transformed cells, and very rarely on naïve T-cells (74). Importantly, CD4+ CD30+ cells are increased during HIV infection, both in c-ART treated or untreated individuals (75). Hogan et al. show that FACS-sorted CD4+ CD30+ from PWH express higher levels of HIV RNA than their CD30- counterparts, even in ART-suppressed individuals. Further, increased levels of cell surface CD30 are detected in CD4+ T-cells from PWH with a viral load rebound upon an analytical treatment interruption (ATI). Critically, changes in CD30 expression are detected even before changes in plasma viral load occur (76). Older studies had already shown that crosslinking cell surface CD30 in the chronically HIV-infected cell line ACH2 triggered HIV expression without cell proliferation and that NF-κB signaling was required for this process (77). As HIV-Tat is critical for HIV transcription and increases NF-κB signaling, it is plausible that changes in CD30 in those studies are linked to HIV-TAT activity. Increased serum levels of sCD30 are a predictor of AIDS progression (78). Importantly, the stark increase in cell surface CD30 and CD31 co-expression in Tat101 cells compared to Tetoff and to Tat72, allows the discrimination of full-length Tat-expressing cells. This would allow therapeutic targeting strategies such as chimeric antigen receptor T-cells or antibody-based approaches (65, 79).
Additionally, we found an important increase in the secretion of platelet-derived growth factor AA subunit (PDGF-AA). This growth factor is important in angiogenesis and wound healing and is a SASP factor secreted by senescent skin cells (80). Previous studies show that soluble HIV-Tat causes an upregulation of the related PDGF-BB in endothelial cells, astrocytes, and pericytes, with an important role for NF-κB and ROS signaling pathways in this effect (81). In addition, increased mRNA levels of the PDGF/VEGF growth factor family members (PDGFA, VEGFA-C) are reported in transformed human mammary cells after HIV-Tat addition (82). Increased levels of PDGF during HIV infection contribute to the loss of blood-brain barrier integrity and neurological manifestations of HIV infection (81).
We took advantage of an RNA-seq performed in our Jurkat-Tat model (56) to address additional senescence-related transcripts and pathways that may be altered by Tat101 expression. The pathway analysis performed by Rodríguez-Agustín et al. unveils that one of the most up-regulated pathways in Tat101 expressing cells is the p53 pathway and one of the most downregulated is ribosome biogenesis. These pathways are both linked to cellular senescence onset (69, 83). Here, we looked up specific DEG genes in the Tat101 vs Tetoff comparison that are present in different senescence databases. We validated some of the targets found in our work, such as CDKNA1 (p21), PECAM1 (CD31), PDGFA (PDGF-AA), CCL1, and TNFRSF8 (CD30). Importantly, CDKNA1 (p21) is a critical senescence initiator under p53 regulation, underscoring the importance of this pathway for Tat-mediated effects on cellular senescence. We also find a striking (38 and 20-fold) increase in CCND2 and CCND1 genes and a 2-fold increase in CCNE1. These genes encode for cyclins D and E, whose expression is increased in senescent cells (84, 85) and are among the top 20 (out of 504) ranked cellular senescence genes screened in a literature-based gene resource (86). In this same study, senescence-related genes (504) are over-represented in processes related to HIV, and the top 4 enriched diseases of all senescence genes are all related to HIV transcription from LTR promoter and Tat activity (86). Our analysis also uncovers a transcriptional downmodulation of targets such as IGFBP7, IGFBP2, HIST1H4C, RB1 or TIMP2, which are usually upregulated in senescent cells at the protein level (43, 69). This suggests that HIV-Tat increases some, but not all the senescence biomarkers addressed. These markers could be altered by other HIV-derived proteins or by HIV infection progression.
Our work shows that Tat full-length protein is required for most of the effects on cellular senescence targets addressed. The 72aa, one exon form of Tat, can transactivate the LTR promoter in Jurkat cells, albeit with a reduced efficiency (38). Introducing a stop codon in position 72 in the Tat open reading frame of the HIV 89.6 infectious clone, results in viruses expressing a 72aa form of Tat, which show a significantly reduced replication rate than its wild-type counterpart (40). Here we show that Tat72 modestly increases p21 in the Jurkat cell line, but does not change BCL2, γH2AX or CD87 expression. In addition, Tat72 had a limited effect on SASP secretion. For instance, the secretion of CD30 and CD31 was upregulated 2.71 and 1.94-fold respectively, in Tat101 over Tetoff, but only 1.42 and 1.44-fold respectively in Tat72 vs Tetoff. Previous work determined that the 86ESKKKVE92 domain located in Tat101 second exon is important for the increased NF-κB signaling and protection from FasL-mediated apoptosis observed in Jurkat Tat101 (42, 49). These studies showed that Jurkat Tat01 cells markedly increased CD69 and BCL-2 expression, resistance to apoptosis, and overall NF-κB signaling over Tetoff cells. Additionally, increased ROS generation and mitochondrial dysfunction were observed in such cells (41), now considered hallmarks of cellular senescence (43). Jurkat Tat72 only modestly increased these parameters, which indicates that Tat’s second exon, is important for the Tat-mediated induction of cellular senescence and that NF-κB signaling pathway is involved in this process. Recent studies also show that the addition of an HIV-Tat protein containing only 66aa of the first exon to latently infected CD4+ T-cells resulted in a latency reversal effect without significantly altering the CD4+ T-cell transcriptome (87).
A limitation of our study is the transformed nature of the Jurkat cell line, which is homozygously deleted for CDKN2A (p16INK4A) and presents point mutations in other relevant genes such as TP53 (p53) (88). Despite this, plenty of studies showed senescence induction in transformed cells and in the Jurkat cell line (89, 90). We have also tested that extremely low doses of etoposide can induce the upregulation of senescence biomarkers in our Jurkat model. We do not observe changes in SA‐β‐gal staining in Tat101-expressing Jurkat cells. Cellular senescence is highly heterogeneous and may vary due to different inducers on different cell types (91). We delivered DNA plasmids encoding HIV-Tat to primary CD4+ T-cells which are the main target of HIV infection, to validate our findings in a relevant model. This strategy allowed the identification of live, Tat-expressing CD4+ T-cells by flow cytometry and the subsequent expression of senescent biomarkers in such cells. We thus confirmed that Tat expression upregulates p21, γH2AX and, importantly, p16 levels compared to Tat- cells. This confirms our Jurkat cell data in a more physiological setting. Interestingly, previous work showed that p16 levels increased after exposing CD4+ T-cells to recombinant HIV-Tat for 24h (92). Authors suggest that this may be caused by the inhibition of telomerase activity (93) and reduced nuclear hTERT levels caused by HIV-Tat (92), further strengthening the idea that HIV-Tat induces cellular senescence in infected CD4+ T-cells.
Regarding other cell types, HIV-Tat has been shown to reduce phagocytosis in primary monocytes and macrophages, reducing its effectiveness against pathogens (94). Furthermore, HIV-Tat increases SA-βGAL, p21, IL6, IL8, and ROS generation in microglia (35, 59).Thus, we expect HIV-Tat to increase cellular senescence in other immune cells, like monocytes or macrophages. Interestingly, we observe that delivering Tat72 to CD4+ T-cells increases senescence biomarkers to a similar extent to Tat101. This could be explained by very high levels of HIV-Tat mRNA transcripts 24 hours after CD4+ T-cell electroporation (data not shown), presumably due to the strong CMV promoter in our DNA plasmids. These increased levels of Tat72 may be sufficient to increase senescence biomarkers. Our approach is based on resting T-cells, as a strong T-cell activation may modify senescence biomarker expression and difficult detection of Tat-mediated changes. As a consequence, the expression of some senescence biomarkers, such as CD87 is very low, and CD30 is nearly undetectable (75) in this model, so we could not fully assess changes in such markers. However, levels of secreted CD31 are increased in cell supernatants of Tat-transfected CD4+ cells, suggesting that Tat acts similarly to our Jurkat model. Both CD31 and CD26 levels are increased in serum from HIV-infected individuals (72). CD26 has been reported as a surface marker of senescent fibroblasts (95) and both CD31 and CD30 cleavage from cell surface leads to its presence in extracellular media. Importantly SASP expressing cells interacted with T-cells predominantly via the MHC-I, MIF and CD31 (PECAM1) pathways (57) underscoring the importance of these targets for senescent cells.
Recently, delivery of Tat RNA to CD4 T-cells by lipid nanoparticle constructs resulted in far superior efficiency and viability than traditional RNA transfection or soluble Tat addition (87, 96). We believe that this strategy deserves further consideration for future cellular senescence studies. An important point regarding the relevance of our findings is Tat mRNA in stable Jurkat cell lines is around 25-fold higher than in activated CD4+ T-cells. However, it should be taken into account that nearly all Jurkat cell lines express Tat, whereas approximately 1-10% of CD4 T-cells may be infected in the conditions tested. Accordingly, this data suggests that in a single CD4 lymphocyte actively replicating, HIV Tat expression may reach similar levels to Jurkat cells, which may lead to the induction of a senescence program in CD4 T cells. In summary, HIV-Tat increases senescence biomarkers in T-cells, possibly by increased DDR, p53, and NF-κB signaling. These effects and the increased SASP mediators released may have an important impact on chronic inflammation, blood-brain barrier permeability, neurological manifestations and other comorbidities seen in PWH, which may critically contribute to the premature aging phenotype of PWH. Importantly, the expression of Tat is largely unaffected by c-ART (97), and Tat is found in serum and cerebrospinal fluid of some ART-treated and virally suppressed individuals (45), which suggests that Tat may continue inducing senescence in those individuals, despite therapy. Thus, HIV-Tat is a relevant therapeutic target. Specific inhibitors of Tat exist (98, 99), which block the transactivation function of Tat. In addition, intradermal therapeutic vaccines using HIV-Tat as an immunogen have been quite successful at inducing HIV-Tat neutralizing antibody responses and reducing HIV proviral DNA in blood, even after 8 years of follow-up (100). It would be of great interest to evaluate senescence biomarkers in PWH with detectable levels of HIV-Tat in serum and those successfully responding to an HIV-Tat vaccine. All these data confirm HIV-Tat as a critical therapeutic target and an important driver of HIV pathogenesis and age-related pathologies in PWH.
Data availability statement
The data presented in the study are deposited in the GEO repository, accession number GSE282545.
Ethics statement
The studies involving humans were approved by Comitè d’ètica d’investigació amb medicaments (CEIm) de l’Hospital U. Clínic de Barcelona. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from a by-product of routine care or industry. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
VC: Conceptualization, Formal analysis, Investigation, Writing – original draft, Writing – review & editing. AR-A: Formal analysis, Investigation, Visualization, Writing – review & editing. RA-S: Data curation, Formal analysis, Writing – review & editing. ElM: Writing – review & editing, Methodology, Formal analysis, Investigation. MM: Investigation, Resources, Writing – review & editing. JM: Funding acquisition, Writing – review & editing. EsM: Writing – review & editing. SS-P: Resources, Writing – review & editing. JMM: Writing – review & editing. JA: Conceptualization, Funding acquisition, Supervision, Writing – review & editing. NC: Conceptualization, Formal analysis, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported through the generous donations of Mr. Javier Moreno (II040141). Research project has been carried out thanks to the grants from the “Programa de becas Gilead a la Investigación Biomédica GLD21_00111” and CIBERINFEC INFECG30 S.N./2024 from the “Instituto de Salud Carlos III”, Madrid, Spain. RA-S and AR-A are supported by private funding from AIDS and HIV Unit (Hospital Clínic Barcelona) and Fundació Clínic per la Recerca Biomèdica de Catalunya (FCRB). JMM received a personal 80:20 research grant from Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain, during 2017–25.
Acknowledgments
We sincerely thank Dr Mayte Coiras from the AIDS Immunopathogenesis and Reservoirs Unit in ISCIII, Madrid, for the generous provision of the Jurkat-Tat cell lines, which have been instrumental in advancing our research. We are indebted to the Cytometry and cell sorting core facility and Functional genomics core facility of the Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS) for their technical help. We also thank the Cytometry Facility and the Genomics facility of the CCiTUB (Parc Científic de Barcelona, Universitat de Barcelona) for their technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1568762/full#supplementary-material
References
1. Smit M, Brinkman K, Geerlings S, Smit C, Thyagarajan K, van Sighem A, et al. ATHENA observational cohort. Future challenges for clinical care of an ageing population infected with HIV: a modelling study. Lancet Infect Dis. (2015) 15:810–8. doi: 10.1016/S1473-3099(15)00056-0
2. Premeaux TA, Ndhlovu LC. Decrypting biological hallmarks of aging in people with HIV. Curr Opin HIV AIDS. (2023) 18:237–45. doi: 10.1097/COH.0000000000000810
3. Montano M, Oursler KK, Xu K, Sun YV, Marconi VC. Biological ageing with HIV infection: evaluating the geroscience hypothesis. Lancet Health Longevity. (2022) 3:e194–205. doi: 10.1016/S2666-7568(21)00278-6
4. Morales DR, Moreno-Martos D, Matin N, McGettigan P. Health conditions in adults with HIV compared with the general population: A population-based cross-sectional analysis. eClinicalMedicine. (2022) 47. doi: 10.1016/j.eclinm.2022.1013921
5. Maciel RA, Klück HM, Durand M, Sprinz E. Comorbidity is more common and occurs earlier in persons living with HIV than in HIV-uninfected matched controls, aged 50 years and older: A cross-sectional study. Int J Infect Dis. (2018) 70:30–5. doi: 10.1016/j.ijid.2018.02.009
6. Schouten J, Wit FW, Stolte IG, Kootstra NA, van der Valk M, Geerlings SE, et al. Cross-sectional comparison of the prevalence of age-associated comorbidities and their risk factors between HIV-infected and uninfected individuals: the AGEhIV cohort study. Clin Infect Dis. (2014) 59:1787–97. doi: 10.1093/cid/ciu701
7. De Francesco D, Wit FW, Bürkle A, Oehlke S, Kootstra NA, Winston A, et al. Do people living with HIV experience greater age advancement than their HIV-negative counterparts? AIDS. (2019) 33:259–68. doi: 10.1097/QAD.0000000000002063
8. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell. (2023) 186:243–78. doi: 10.1016/j.cell.2022.11.001
9. Cohen J, Torres C. HIV-associated cellular senescence: A contributor to accelerated aging. Ageing Res Rev. (2017) 36:117–24. doi: 10.1016/j.arr.2016.12.004
10. Rodés B, Cadiñanos J, Esteban-Cantos A, Rodríguez-Centeno J, Arribas JR. Ageing with HIV: challenges and biomarkers. eBioMedicine. (2022) 77:103896. doi: 10.1016/j.ebiom.2022.103896
11. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. (1997) 88:593–602. doi: 10.1016/S0092-8674(00)81902-9
12. Lee S, Schmitt CA, Reimann M. The Myc/macrophage tango: Oncogene-induced senescence, Myc style. Semin Cancer Biol. (2011) 21:377–84. doi: 10.1016/j.semcancer.2011.10.002
13. McHugh D, Durán I, Gil J. Senescence as a therapeutic target in cancer and age-related diseases. Nat Rev Drug Discov. (2024) 24:57–71. doi: 10.1038/s41573-024-01074-4
14. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. (1965) 37:614–36. doi: 10.1016/0014-4827(65)90211-9
15. di Fagagna Fd’A, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. (2003) 426:194–8. doi: 10.1038/nature02118
16. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. (2014) 28:99–114. doi: 10.1101/gad.235184.113
17. Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol: Mech Dis. (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
18. Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PloS Biol. (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
19. Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. (2012) 11:345–9. doi: 10.1111/j.1474-9726.2012.00795.x
20. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. (2013) 15:978–90. doi: 10.1038/ncb2784
21. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. (2007) 445:656–60. doi: 10.1038/nature05529
22. Effros RB, Allsopp R, Chiu CP, Hausner MA, Hirji K, Wang L, et al. Shortened telomeres in the expanded CD28-CD8+ cell subset in HIV disease implicate replicative senescence in HIV pathogenesis. AIDS. (1996) 10:F17–22. doi: 10.1097/00002030-199607000-00001
23. Bestilny LJ, Gill MJ, Mody CH, Riabowol KT. Accelerated replicative senescence of the peripheral immune system induced by HIV infection. AIDS. (2000) 14:771–80. doi: 10.1097/00002030-200005050-00002
24. Nelson JAE, Krishnamurthy J, Menezes P, Liu Y, Hudgens MG, Sharpless NE, et al. Expression of p16 as a biomarker of T-cell aging in HIV-infected patients prior to and during antiretroviral therapy. Aging Cell. (2012) 11:916–8. doi: 10.1111/j.1474-9726.2012.00856.x
25. Pathai S, Lawn SD, Gilbert CE, McGuinness D, McGlynn L, Weiss HA, et al. Accelerated biological ageing in HIV-infected individuals in South Africa: a case-control study. AIDS. (2013) 27:2375–84. doi: 10.1097/QAD.0b013e328363bf7f
26. Ribeiro SP, Milush JM, Cunha-Neto E, Kallas EG, Kalil J, Passero LFD, et al. p16INK4a expression and immunologic aging in chronic HIV infection. PloS One. (2016) 11:e0166759. doi: 10.1371/journal.pone.0166759
27. Chandrasekar AP, Badley AD. Prime, shock and kill: BCL-2 inhibition for HIV cure. Front Immunol. (2022) 13:1033609. doi: 10.3389/fimmu.2022.1033609
28. Schank M, Zhao J, Moorman JP, Yao ZQ. The impact of HIV- and ART-induced mitochondrial dysfunction in cellular senescence and aging. Cells. (2021) 10:174. doi: 10.3390/cells10010174
29. Hijmans JG, Stockleman K, Reiakvam W, Levy MV, Brewster LM, Bammert TD, et al. Effects of HIV-1 gp120 and tat on endothelial cell sensescence and senescence-associated microRNAs. Physiol Rep. (2018) 6:e13647. doi: 10.14814/phy2.13647
30. Hijmans JG, Stockelman K, Levy M, Brewster LM, Bammert TD, Greiner JJ, et al. Effects of HIV-1 gp120 and TAT-derived microvesicles on endothelial cell function. J Appl Physiol. (2019) 126:1242–9. doi: 10.1152/japplphysiol.01048.2018
31. Beaupere C, Garcia M, Larghero J, Fève B, Capeau J, Lagathu C. The HIV proteins Tat and Nef promote human bone marrow mesenchymal stem cell senescence and alter osteoblastic differentiation. Aging Cell. (2015) 14:534–46. doi: 10.1111/acel.12308
32. Gorwood J, Ejlalmanesh T, Bourgeois C, Mantecon M, Rose C, Atlan M, et al. SIV infection and the HIV proteins tat and nef induce senescence in adipose tissue and human adipose stem cells, resulting in adipocyte dysfunction. Cells. (2020) 9:854. doi: 10.3390/CELLS9040854
33. Yuan Y, Zhao S, Wang X, Teng Z, Li D, Zeng Y. HIV-1 p55-gag protein induces senescence of human bone marrow mesenchymal stem cells and reduces their capacity to support expansion of hematopoietic stem cells. vitro Cell Biol Int. (2017) 41:969–81. doi: 10.1002/cbin.10791
34. Fiume G, Vecchio E, De Laurentiis A, Trimboli F, Palmieri C, Pisano A, et al. Human immunodeficiency virus-1 Tat activates NF-κB via physical interaction with IκB-α and p65. Nucleic Acids Res. (2012) 40:3548–62. doi: 10.1093/nar/gkr1224
35. Thangaraj A, Chivero ET, Tripathi A, Singh S, Niu F, Guo M-L, et al. HIV TAT-mediated microglial senescence: Role of SIRT3-dependent mitochondrial oxidative stress. Redox Biol. (2021) 40:101843. doi: 10.1016/j.redox.2020.101843
36. Gatignol A, Jeang KT. Tat as a transcriptional activator and a potential therapeutic target for HIV-1. Adv Pharmacol. (2000) 48:209–27. doi: 10.1016/s1054-3589(00)48007-5
37. Rayne F, Debaisieux S, Yezid H, Lin Y-L, Mettling C, Konate K, et al. Phosphatidylinositol-(4,5)-bisphosphate enables efficient secretion of HIV-1 Tat by infected T-cells. EMBO J. (2010) 29:1348–62. doi: 10.1038/emboj.2010.32
38. López-Huertas MR, Callejas S, Abia D, Mateos E, Dopazo A, Alcamí J, et al. Modifications in host cell cytoskeleton structure and function mediated by intracellular HIV-1 Tat protein are greatly dependent on the second coding exon. Nucleic Acids Res. (2010) 38:3287–307. doi: 10.1093/nar/gkq037
39. Coiras M, Camafeita E, Ureña T, López JA, Caballero F, Fernández B, et al. Modifications in the human T cell proteome induced by intracellular HIV-1 Tat protein expression. Proteomics. (2006) 6 Suppl 1:S63–73. doi: 10.1002/pmic.200500437
40. Mahlknecht U, Dichamp I, Varin A, Van Lint C, Herbein G. NF-kappaB-dependent control of HIV-1 transcription by the second coding exon of Tat in T cells. J Leukoc Biol. (2008) 83:718–27. doi: 10.1189/jlb.0607405
41. Rodríguez-Mora S, Mateos E, Moran M, Martín MÁ, López JA, Calvo E, et al. Intracellular expression of Tat alters mitochondrial functions in T cells: a potential mechanism to understand mitochondrial damage during HIV-1 replication. Retrovirology. (2015) 12:78. doi: 10.1186/s12977-015-0203-3
42. López-Huertas MR, Li J, Zafar A, Rodríguez-Mora S, García-Domínguez C, Mateos E, et al. PKCθ and HIV-1 transcriptional regulator tat co-exist at the LTR promoter in CD4(+) T cells. Front Immunol. (2016) 7:69. doi: 10.3389/fimmu.2016.00069
43. Suryadevara V, Hudgins AD, Rajesh A, Pappalardo A, Karpova A, Dey AK, et al. SenNet recommendations for detecting senescent cells in different tissues. Nat Rev Mol Cell Biol. (2024) 1–23. doi: 10.1038/s41580-024-00738-8
44. Reeder JE, Kwak Y-T, McNamara RP, Forst CV, D’Orso I. HIV Tat controls RNA Polymerase II and the epigenetic landscape to transcriptionally reprogram target immune cells. eLife. (2015) 4:e08955. doi: 10.7554/eLife.08955
45. Shmakova A, Tsimailo I, Kozhevnikova Y, Gérard L, Boutboul D, Oksenhendler E, et al. HIV-1 Tat is present in the serum of people living with HIV-1 despite viral suppression. Int J Infect Dis. (2024) 142:106994. doi: 10.1016/j.ijid.2024.106994
46. Henderson LJ, Johnson TP, Smith BR, Reoma LB, Santamaria UA, Bachani M, et al. Presence of Tat and Trans-activation response (TAR) element in spinal fluid despite antiretroviral therapy. AIDS. (2019) 33:S145–57. doi: 10.1097/QAD.0000000000002268
47. Dickens AM, Yoo SW, Chin AC, Xu J, Johnson TP, Trout AL, et al. Chronic low-level expression of HIV-1 Tat promotes a neurodegenerative phenotype with aging. Sci Rep. (2017) 7:7748. doi: 10.1038/s41598-017-07570-5
48. Qrareya AN, Mahdi F, Kaufman MJ, Ashpole NM, Paris JJ. Age-related neuroendocrine, cognitive, and behavioral co-morbidities are promoted by HIV-1 Tat expression in male mice. Aging (Albany NY). (2022) 14:5345–65. doi: 10.18632/aging.204166
49. López-Huertas MR, Mateos E, Sánchez Del Cojo M, Gómez-Esquer F, Díaz-Gil G, Rodríguez-Mora S, et al. The presence of HIV-1 Tat protein second exon delays fas protein-mediated apoptosis in CD4+ T lymphocytes: a potential mechanism for persistent viral production. J Biol Chem. (2013) 288:7626–44. doi: 10.1074/jbc.M112.408294
50. Bachelerie F, Alcami J, Arenzana-Seisdedos F, Virelizier JL. HIV enhancer activity perpetuated by NF-kappa B induction on infection of monocytes. Nature. (1991) 350:709–12. doi: 10.1038/350709a0
51. Aksoy P, Aksoy BA, Czech E, Hammerbacher J. Viable and efficient electroporation-based genetic manipulation of unstimulated human T cells. (2019), 466243. doi: 10.1101/466243
52. Yukl SA, Kaiser P, Kim P, Telwatte S, Joshi SK, Vu M, et al. HIV latency in isolated patient CD4+ T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci Trans Med. (2018) 10:eaap9927. doi: 10.1126/scitranslmed.aap9927
53. Sánchez-Del-Cojo M, López-Huertas MR, Díez-Fuertes F, Rodríguez-Mora S, Bermejo M, López-Campos G, et al. Changes in the cellular microRNA profile by the intracellular expression of HIV-1 Tat regulator: A potential mechanism for resistance to apoptosis and impaired proliferation in HIV-1 infected CD4+ T cells. PloS One. (2017) 12:e0185677. doi: 10.1371/journal.pone.0185677
54. Mansilla S, Piña B, Portugal J. Daunorubicin-induced variations in gene transcription: commitment to proliferation arrest, senescence and apoptosis. Biochem J. (2003) 372:703–11. doi: 10.1042/bj20021950
55. Maskey D, Yousefi S, Schmid I, Zlobec I, Perren A, Friis R, et al. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat Commun. (2013) 4:2130. doi: 10.1038/ncomms3130
56. Rodríguez-Agustín A, Ayala-Suárez R, Díez-Fuertes F, Maleno MJ, de Villasante I, Merkel A, et al. Intracellular HIV-1 Tat regulator induces epigenetic changes in the DNA methylation landscape. Front Immunol. (2025) 16:1532692. doi: 10.3389/fimmu.2025.1532692
57. Saul D, Kosinsky RL, Atkinson EJ, Doolittle ML, Zhang X, LeBrasseur NK, et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat Commun. (2022) 13:4827. doi: 10.1038/s41467-022-32552-1
58. Lee S, Yu Y, Trimpert J, Benthani F, Mairhofer M, Richter-Pechanska P, et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature. (2021) 599:283–9. doi: 10.1038/S41586-021-03995-1
59. Chen NC, Partridge AT, Tuzer F, Cohen J, Nacarelli T, Navas-Martín S, et al. Induction of a senescence-like phenotype in cultured human fetal microglia during HIV-1 infection. Journ Gerontol: Ser A. (2018) 73:1187–96. doi: 10.1093/gerona/gly022
60. Li D, Lopez A, Sandoval C, Nichols Doyle R, Fregoso OI. HIV vpr modulates the host DNA damage response at two independent steps to damage DNA and repress double-strand DNA break repair. mBio. (2020) 11:e00940–20. doi: 10.1128/mBio.00940-20
61. Cao D, Khanal S, Wang L, Li Z, Zhao J, Nguyen LN, et al. A matter of life or death: productively infected and bystander CD4 T cells in early HIV infection. Front Immunol. (2021) 11:626431. doi: 10.3389/fimmu.2020.626431
62. Marcello A, Zoppé M, Giacca M. Multiple modes of transcriptional regulation by the HIV-1 Tat transactivator. IUBMB Life. (2001) 51:175–81. doi: 10.1080/152165401753544241
63. El-Amine R, Germini D, Zakharova VV, Tsfasman T, Sheval EV, Louzada RAN, et al. HIV-1 Tat protein induces DNA damage in human peripheral blood B-lymphocytes via mitochondrial ROS production. Redox Biol. (2018) 15:97–108. doi: 10.1016/j.redox.2017.11.024
64. Akbay B, Germini D, Bissenbaev AK, Musinova YR, Sheval EV, Vassetzky Y, et al. HIV-1 tat activates akt/mTORC1 pathway and AICDA expression by downregulating its transcriptional inhibitors in B cells. IJMS. (2021) 22:1588. doi: 10.3390/ijms22041588
65. Amor C, Feucht J, Leibold J, Ho Y-J, Zhu C, Alonso-Curbelo D, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. (2020) 583:127–32. doi: 10.1038/s41586-020-2403-9
66. Speth C, Pichler I, Stöckl G, Mair M, Dierich MP. Urokinase plasminogen activator receptor (uPAR; CD87) expression on monocytic cells and T cells is modulated by HIV-1 infection. Immunobiology. (1998) 199:152–62. doi: 10.1016/S0171-2985(98)80071-5
67. Nebuloni M, Zawada L, Ferri A, Tosoni A, Zerbi P, Resnati M, et al. HIV-1 infected lymphoid organs upregulate expression and release of the cleaved form of uPAR that modulates chemotaxis and virus expression. PloS One. (2013) 8:e70606. doi: 10.1371/journal.pone.0070606
68. Sidenius N, Sier CF, Ullum H, Pedersen BK, Lepri AC, Blasi F, et al. Serum level of soluble urokinase-type plasminogen activator receptor is a strong and independent predictor of survival in human immunodeficiency virus infection. Blood. (2000) 96:4091–5. doi: 10.1182/blood.V96.13.4091
69. Kumari R, Jat P. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol. (2021) 9:645593. doi: 10.3389/fcell.2021.645593
70. Tanaka T, Biancotto A, Moaddel R, Moore AZ, Gonzalez-Freire M, Aon MA, et al. Plasma proteomic signature of age in healthy humans. Aging Cell. (2018) 17:e12799. doi: 10.1111/acel.12799
71. Junge S, Kloeckener-Gruissem B, Zufferey R, Keisker A, Salgo B, Fauchere J-C, et al. Correlation between recent thymic emigrants and CD31+ (PECAM-1) CD4+ T cells in normal individuals during aging and in lymphopenic children. Eur J Immunol. (2007) 37:3270–80. doi: 10.1002/eji.200636976
72. Briceño O, Peralta-Prado A, Garrido-Rodríguez D, Romero-Mora K, Chávez-Torres M, de la Barrera C-A, et al. Characterization of CD31 expression in CD4+ and CD8+T cell subpopulations in chronic untreated HIV infection. Immunol Lett. (2021) 235:22–31. doi: 10.1016/j.imlet.2021.04.004
73. Eugenin EA, Gamss R, Buckner C, Buono D, Klein RS, Schoenbaum EE, et al. Shedding of PECAM-1 during HIV infection: a potential role for soluble PECAM-1 in the pathogenesis of NeuroAIDS. J Leukoc Biol. (2006) 79:444–52. doi: 10.1189/jlb.0405215
74. Chiarle R, Podda A, Prolla G, Gong J, Thorbecke GJ, Inghirami G. CD30 in normal and neoplastic cells. Clin Immunol. (1999) 90:157–64. doi: 10.1006/clim.1998.4636
75. Hogan LE, Vasquez J, Hobbs KS, Hanhauser E, Aguilar-Rodriguez B, Hussien R, et al. Increased HIV-1 transcriptional activity and infectious burden in peripheral blood and gut-associated CD4+ T cells expressing CD30. PloS Pathog. (2018) 14:e1006856. doi: 10.1371/journal.ppat.1006856
76. Prator CA, Thanh C, Kumar S, Pan T, Peluso MJ, Bosch R, et al. Circulating CD30+CD4+ T cells increase before human immunodeficiency virus rebound after analytical antiretroviral treatment interruption. J Infect Dis. (2020) 221:1146–55. doi: 10.1093/infdis/jiz572
77. Biswas P, Smith CA, Goletti D, Hardy EC, Jackson RW, Fauci AS. Cross-linking of CD30 induces HIV expression in chronically infected T cells. Immunity. (1995) 2:587–96. doi: 10.1016/1074-7613(95)90003-9
78. Pizzolo G, Vinante F, Morosato L, Nadali G, Chilosi M, Gandini G, et al. High serum level of the soluble form of CD30 molecule in the early phase of HIV-1 infection as an independent predictor of progression to AIDS. AIDS. (1994) 8:741–5. doi: 10.1097/00002030-199406000-00003
79. Kim KM, Noh JH, Bodogai M, Martindale JL, Yang X, Indig FE, et al. Identification of senescent cell surface targetable protein DPP4. Genes Dev. (2017) 31:1529–34. doi: 10.1101/gad.302570.117
80. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. (2014) 31:722–33. doi: 10.1016/j.devcel.2014.11.012
81. Niu F, Yao H, Liao K, Buch S. HIV Tat 101-mediated loss of pericytes at the blood-brain barrier involves PDGF-BB. Ther Targets Neurol Dis. (2015) 2:e471. doi: 10.14800/ttnd.471
82. Bettaccini AA, Baj A, Accolla RS, Basolo F, Toniolo AQ. Proliferative activity of extracellular HIV-1 Tat protein in human epithelial cells: expression profile of pathogenetically relevant genes. BMC Microbiol. (2005) 5:20. doi: 10.1186/1471-2180-5-20
83. Lessard F, Igelmann S, Trahan C, Huot G, Saint-Germain E, Mignacca L, et al. Senescence-associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nat Cell Biol. (2018) 20:789–99. doi: 10.1038/s41556-018-0127-y
84. Lucibello FC, Sewing A, Brüsselbach S, Bürger C, Müller R. Deregulation of cyclins D1 and E and suppression of cdk2 and cdk4 in senescent human fibroblasts. J Cell Sci. (1993) 105:123–33. doi: 10.1242/jcs.105.1.123
85. Leontieva OV, Demidenko ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ. (2013) 20:1241–9. doi: 10.1038/cdd.2013.86
86. Zhao M, Chen L, Qu H. CSGene: a literature-based database for cell senescence genes and its application to identify critical cell aging pathways and associated diseases. Cell Death Dis. (2016) 7:e2053–3. doi: 10.1038/cddis.2015.414
87. Van Gulck E, Pardons M, Nijs E, Verheyen N, Dockx K, Van Den Eynde C, et al. A truncated HIV Tat demonstrates potent and specific latency reversal activity. Antimicrob Agents Chemother. (2023) 67:e00417–23. doi: 10.1128/aac.00417-23
88. Gioia L, Siddique A, Head SR, Salomon DR, Su AI. A genome-wide survey of mutations in the Jurkat cell line. BMC Genomics. (2018) 19:334. doi: 10.1186/s12864-018-4718-6
89. Gonzalez LC, Ghadaouia S, Martinez A, Rodier F. Premature aging/senescence in cancer cells facing therapy: good or bad? Biogerontology. (2016) 17:71–87. doi: 10.1007/s10522-015-9593-9
90. Hasegawa H, Yamada Y, Iha H, Tsukasaki K, Nagai K, Atogami S, et al. Activation of p53 by Nutlin-3a, an antagonist of MDM2, induces apoptosis and cellular senescence in adult T-cell leukemia cells. Leukemia. (2009) 23:2090–101. doi: 10.1038/leu.2009.171
91. Hernandez-Segura A, De Jong TV, Melov S, Guryev V, Campisi J, Demaria M. Unmasking transcriptional heterogeneity in senescent cells. Curr Biol. (2017) 27:2652–2660.e4. doi: 10.1016/j.cub.2017.07.033
92. Comandini A, Naro C, Adamo R, Akbar AN, Lanna A, Bonmassar E, et al. Molecular mechanisms involved in HIV-1-Tat mediated inhibition of telomerase activity in human CD4(+) T lymphocytes. Mol Immunol. (2013) 54:181–92. doi: 10.1016/j.molimm.2012.12.003
93. Franzese O, Comandini A, Adamo R, Sgadari C, Ensoli B, Bonmassar E. HIV-Tat down-regulates telomerase activity in the nucleus of human CD4+ T cells. Cell Death Differ. (2004) 11:782–4. doi: 10.1038/sj.cdd.4401346
94. Debaisieux S, Lachambre S, Gross A, Mettling C, Besteiro S, Yezid H, et al. HIV-1 Tat inhibits phagocytosis by preventing the recruitment of Cdc42 to the phagocytic cup. Nat Commun. (2015) 6:6211. doi: 10.1038/ncomms7211
95. Psaroudis RT, Singh U, Lora M, Jeon P, Boursiquot A, Stochaj U, et al. CD26 is a senescence marker associated with reduced immunopotency of human adipose tissue-derived multipotent mesenchymal stromal cells. Stem Cell Res Ther. (2022) 13:358. doi: 10.1186/s13287-022-03026-4
96. Pardons M, Cole B, Lambrechts L, van Snippenberg W, Rutsaert S, Noppe Y, et al. Potent latency reversal by Tat RNA-containing nanoparticle enables multi-omic analysis of the HIV-1 reservoir. Nat Commun. (2023) 14:8397. doi: 10.1038/s41467-023-44020-5
97. Johnson TP, Patel K, Johnson KR, Maric D, Calabresi PA, Hasbun R, et al. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc Natl Acad Sci U S A. (2013) 110:13588–93. doi: 10.1073/pnas.1308673110
98. Mousseau G, Clementz MA, Bakeman WN, Nagarsheth N, Cameron M, Shi J, et al. An analog of the natural steroidal alkaloid cortistatin A potently suppresses tat-dependent HIV transcription. Cell Host Microbe. (2012) 12:97–108. doi: 10.1016/j.chom.2012.05.016
99. Moranguinho I, Valente ST. Block-and-lock: new horizons for a cure for HIV-1. Viruses. (2020) 12:1443. doi: 10.3390/v12121443
Keywords: HIV-Tat, HIV, cellular senescence, SASP, aging, CD4+ T-Cell
Citation: Casanova V, Rodríguez-Agustín A, Ayala-Suárez R, Moraga E, Maleno MJ, Mallolas J, Martínez E, Sánchez-Palomino S, Miró JM, Alcamí J and Climent N (2025) HIV-Tat upregulates the expression of senescence biomarkers in CD4+ T-cells. Front. Immunol. 16:1568762. doi: 10.3389/fimmu.2025.1568762
Received: 30 January 2025; Accepted: 31 March 2025;
Published: 24 April 2025.
Edited by:
Aurelio Cafaro, National Institute of Health (ISS), ItalyReviewed by:
Nina Stoyanova Yancheva-Petrova, Medical University Sofia, BulgariaCollin Kieffer, University of Illinois at Urbana-Champaign, United States
Uma Maheswari Deshetty, University of Nebraska Medical Center, United States
Copyright © 2025 Casanova, Rodríguez-Agustín, Ayala-Suárez, Moraga, Maleno, Mallolas, Martínez, Sánchez-Palomino, Miró, Alcamí and Climent. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Núria Climent, bmNsaW1lbnRAcmVjZXJjYS5jbGluaWMuY2F0; José Alcamí, QUxDQU1JQGNsaW5pYy5jYXQ=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share senior authorship