 Yang Liu
Yang Liu Wanting Wang1†
Wanting Wang1† Chaofan Wang
Chaofan Wang Jun Deng
Jun Deng Yu Hu
Yu Hu Heng Mei
Heng Mei Shanshan Luo
Shanshan Luo- 1Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Key Laboratory of Molecular Biological Targeted Therapies of the Ministry of Education, Wuhan, China
Acute myeloid leukemia (AML) is a heterogeneously primary hematopoietic neoplasm characterized by uncontrolled proliferation of immature myeloid cells, which is characterized with poor outcomes. Despite tremendous advances in the treatment paradigm of AML in the past several decades, the cure and prognosis remain unfavorable. More effective treatments are therefore needed to improve the clinical outcomes. Among newly emerging immunotherapies, chimeric antigen receptor (CAR)-T cell immunotherapy is an exceedingly promising approach that has remarkably improved the overall survival for patients with AML. However, current CAR-T cell therapy for AML faces numerous significant challenges such as the identification of truly AML-specific surface antigens, the on-target/off-tumor toxicity, and the immunosuppressive microenvironment of AML. In order to conquer these limitations, novel strategies to advance CAR-T therapy are urgently needed. In this comprehensive review, we summarize the current status of immunotherapy, especially CAR-T cell therapy, highlight the outcomes of current trials and the limitations of CAR-T immunotherapy, hopefully to provide novel insights into the future directions of CAR-T cells in AML.
Introduction
In recent years, the occurrence of AML has been increased annually. It is reported that the incidence of AML in the US in 2023 is 20380 cases with approximately 11310 deaths (1, 2). Currently, the combination of cytarabine (ara-C) and anthracycline remains the standard induction chemotherapy for AML, which is commonly known as the “3 + 7” regimen, resulting in long-term cures of approximate 35% of the younger AML patients (3). Small molecular drugs that target specific molecules are gaining attention for their potential in treating AML. Among these, some are already used in clinic such as ivosidenib (IDH1 inhibitor) (4), enasidenib (IDH2 inhibitor) (5), gilteritinib (FLT3 inhibitor) (6), and venetoclax (BCL-2 inhibitor) (7). Despite the advancements of current available therapies, the majority of patients still have terrible prognosis due to the disease progression or recurrence, as a result of treatment resistance or adverse side effects (8, 9). Therefore, it is essential to identify and research potential novel therapeutic approaches for AML.
T-cell-based immunotherapy is an effective strategy, including the genetic modification and redirection of these cells to eradicate AML blasts (10). For example, CAR-T cell therapy is a relatively novel strategy, in which autologous/allogeneic T cells are collected and reprogrammed to express CARs that recognize tumor surface antigens specifically. The reprogrammed T cells can specifically identify tumor-associated targets and destroy these cells without the assistance of the major histocompatibility complex (11, 12). The treatment of hematological malignancies is the primary area for CAR-T cells, which has shown an impressive overall and complete response rate. This is because the adequate tumor antigen is easier to find and target in hematological malignancies compared with solid cancers (13, 14). However, due to the significant genetic and phenotypic heterogeneity, finding a true AML-specific antigen is challenging, which limits the successful application of CAR-T cell therapy in AML treatment (15). In addition, the expression of AML antigens on normal healthy tissues often causes variable degrees of toxicity due to the mistarget of the healthy tissues. The on-target/off-tumor toxicities are usually unavoidable for CAR-T therapy, such as the possibility of fatal myeloablation when targeting myeloid precursor cells (16). Here, we outline the progress achieved in the multiple categories of immunotherapeutic approaches for the management of AML, further discuss the particular mechanisms of CAR-T therapy, summarize the recent advances of CAR-T immunotherapy in AML, as well as the current limitations, hopefully providing some novel insights for the future research direction.
Overview of the current available immunotherapies for AML
In the management of AML, current available immunotherapy involves targeted antibodies, adoptive cell therapy (ACT), immune checkpoint inhibitors (ICIs), Hematopoietic stem cell transplantation (HSCT), and tumor vaccines. Among them, the allogeneic HSCT is still one of the most classic and effective immunotherapeutic approaches for hematological malignancies (17, 18). As the understanding of the genetic and phenotypic diversity of AML rapidly advances, immunological therapeutic targets have been revealed increasingly. Over the past 10–15 years, several small molecule targeting drugs have been successfully used for AML, either alone or in a combined form with other standard therapies (19, 20). For example, midostaurin which inhibits multiple tyrosine kinase receptors, is approved for FLT3-mutated AML alone or combined with chemotherapy (21). Cancer vaccines are a positive strategy to eliminate AML and prevent tumor recurrence, which stimulates a persistent immune response. Recently, researchers have developed the ECNV-αGC vaccination, demonstrating its efficacy in reducing the burden of AML. However, there is still a long way to go before cancer vaccines can be translated from the bench to the bedside (22). Despite the numerous ongoing trials, these immunotherapies for AML still have many limitations to overcome.
ACT has emerged as a widely applied immunotherapeutic strategy for patients with AML, including DC cell, TCR-T cell, cytokine-induced killer (CIK) cell and CAR-NK cell. Among them, CAR-T cells have become a promising candidate (23). The remarkable successful outcomes of CAR-T in other hematopoietic malignancies, such as acute lymphoblastic leukemia (ALL), have prompted their attempted application in AML (24). In a phase I clinical trial involving 10 cases, CLL-1 CAR-T cells demonstrated remarkable therapeutic potential that 7 of 10 R/R AML patients achieved CR/CRi (25). Although the application of CAR-T therapy in AML treatment remains in the early stages, trials to date have achieved encouraging initial outcomes just like in other hematological malignancies.
Principles of CAR-T cell therapies
Although the detail structures of each CAR construct are slightly different, the most commonly used CARs include an antigen binding domain, an activation domain from CD3z, an extracellular hinge domain and an intracellular costimulatory domain (Figure 1). When CAR-T cells recognize and bind to tumor-specific antigens, the intracellular structural domain initiates activation procedures via phosphorylation and subsequent signaling. Consequently, CAR-T cell specifically targets tumor-associated antigens with HLA independence (11). The primary cytotoxicity hinges on the cytokines secreted by CAR-T cells including granzymes and perforins (26). Furthermore, CAR-T cell triggers the apoptotic signaling cascade through the engagement of surface molecules, ultimately leading to the programmed death of cancer cells (27). The efficiency of the first generation was disappointing due to the absence of co-stimulatory signaling function. Therefore, the second‐generation CAR construct has one additional co‐stimulatory domain that differs from the first, respectively. Such bi-co-stimulatory domains enhance the activation and proliferation of CAR T cells. Furthermore, the structure of the third generation is similar to the second (28), while the fourth generation CAR-T has an additional inducible domain. When the domain is activated, CAR-T cells generate numerous cytokines displaying anti-tumor activity in local tumor tissue, leading to the improvement of durability and other comprehensive anti-tumor responses (29). The fifth generation incorporates an additional intracellular IL-2 receptor domain into the design of the second generation. This modification induces the production of memory T cells by the antigen-driven activation of the JAK/STAT signaling pathway, improving the therapeutic efficacy of CAR-T therapy. Genome editing technology is also used in the fifth generation, aiming to mitigate the risk of cytokine release syndrome (CRS) by inhibiting dominant negative receptors, including PD-1 and TGF-β (30).
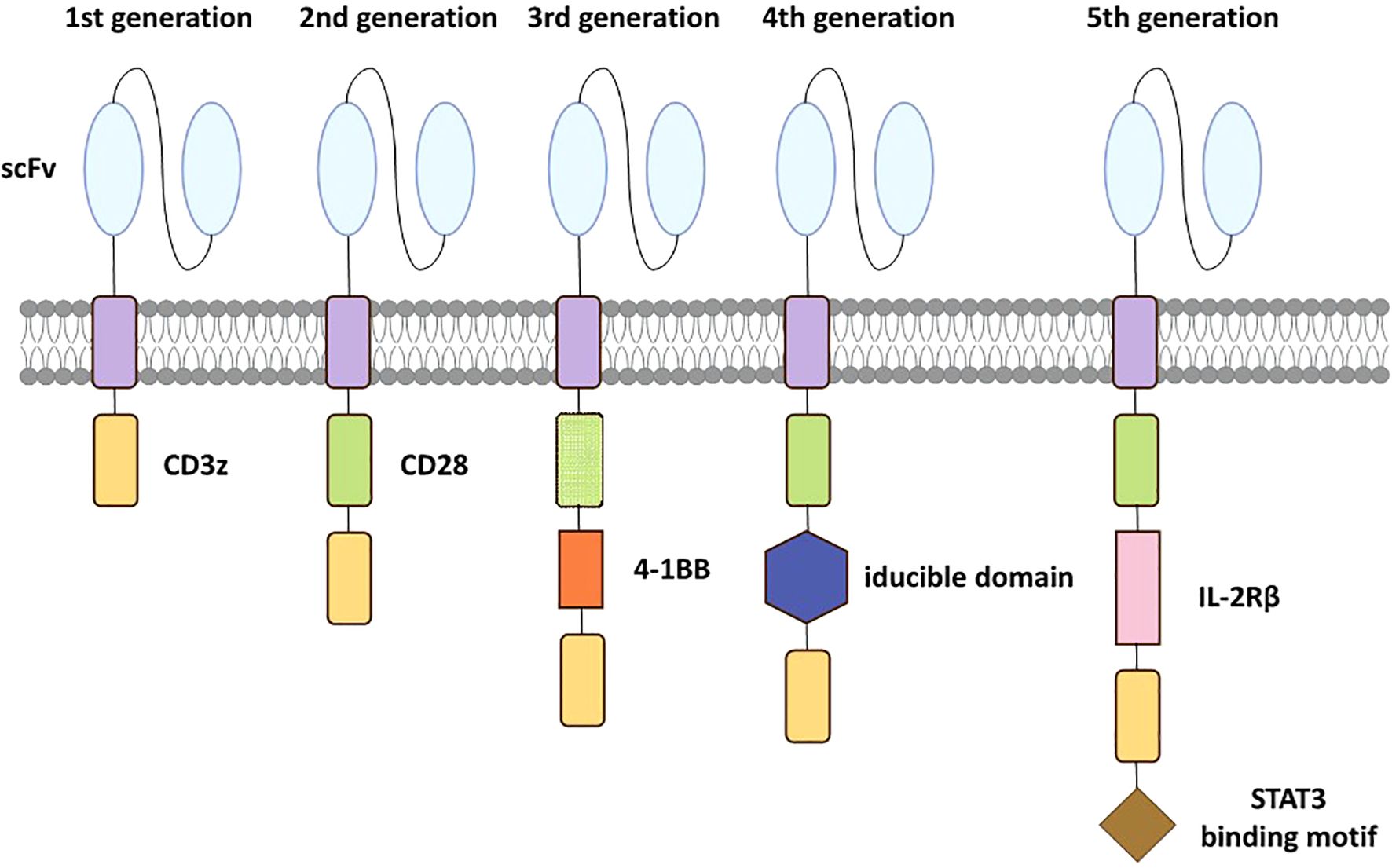
Figure 1. Evolution of the 5 generations CARs. The first-generation has only a CD3z signaling domain. The second-generation is characterized by an additional costimulatory domain based on the first-generation. The third-generation incorporates two costimulatory domains similarly. The fourth-generation has an additional inducible domain to induce the production of tumor-killing cytokines. The fifth-generation incorporates an intracellular IL-2Rβ domain with a STAT3 binding motif to activate the JAK-STAT path.
Current CAR-T production methods mainly involve ex vivo strategy which needed isolation, genetic modification, and subsequent expansion of T cells outside the body, as well as in vivo strategy that engineered CAR-T cells directly within the body using delivery vehicles (31). Despite the pre-clinical results showing that in vivo production is less complicated than ex vivo production, in vivo CAR-T production is still under research, with no product receiving approval from the FDA compared with the mature ex vivo production strategy (32, 33).
Current CAR-T cell constructs for acute myeloid leukemia
CAR-T cell therapy is still in the early stages in AML compared with other hematopoietic malignancies. Currently, no CAR-T product is approved for clinical use in AML, however numerous AML-directed CAR-T cells are developed in preclinical and clinical trials. To designing an effective CAR, choosing an appropriate target is the most critical step. However, most of the targets identified in AML cells have not been ideal until now. The predominant targets of current clinical trials of AML are CD33 and CD123, however, these identified targets may also be expressed on healthy HSCs or may not be consistently presented in all AML cells. Currently, in a phase clinical I study, CLL-1 CAR-T cells in the R/R AML patients show a promising outcome with the CR/CRi rate 70% (n = 7/10), but off-target toxicity is quite concerning (25).
To overcome the limitations of CD33 and CD123 targets, researchers are exploiting other novel targets, such as NKG2DL, CLL-1, CD70, CLEAC12A and CD138, as well as dual antigen‐directed CARs. In the following sections, we will summarize the targets under current research (Table 1), and discuss the corresponding advances and the challenges of CAR-T cell therapy.
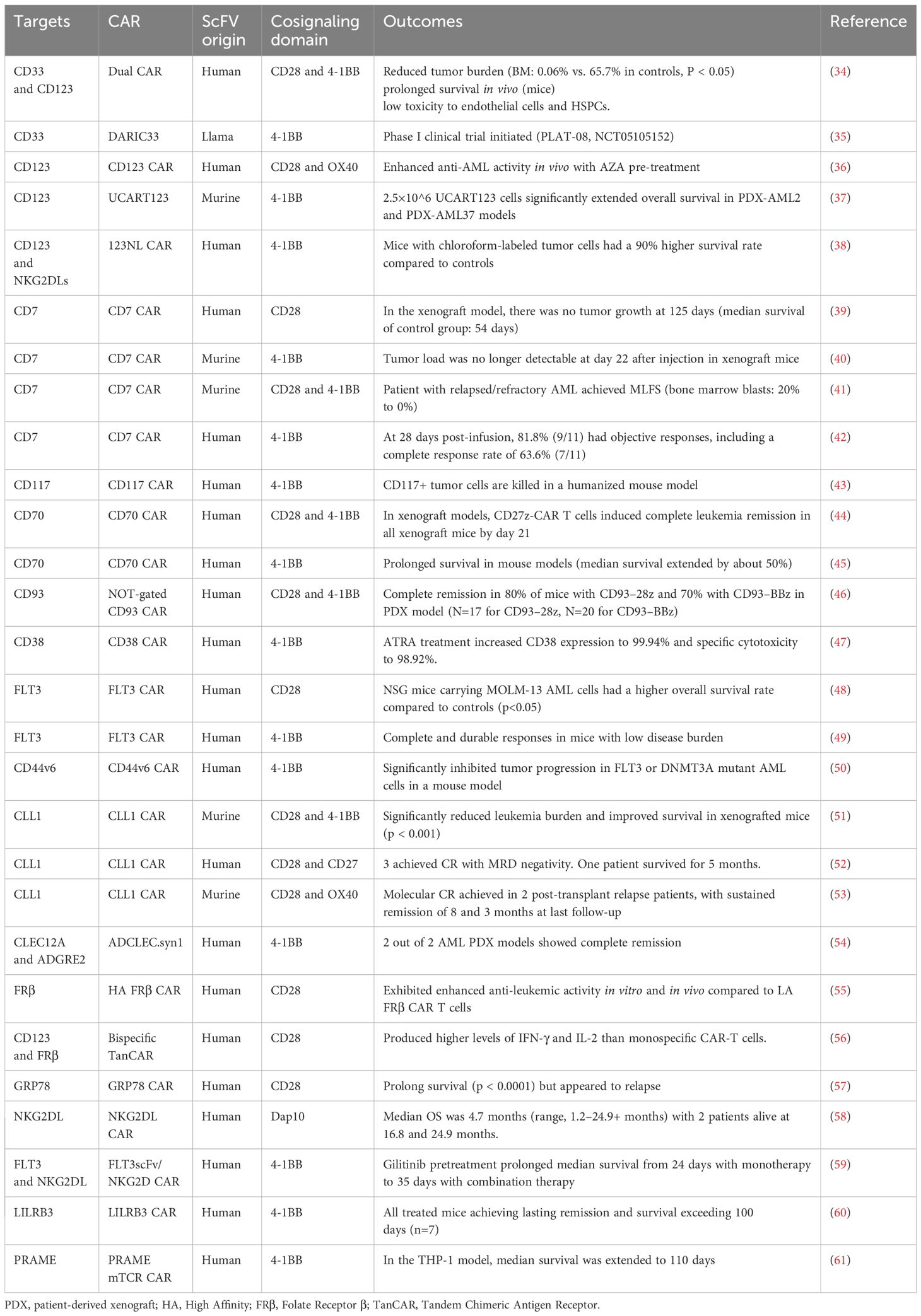
Table 1. Selected landmark clinical trials of chimeric antigen receptor T‐cell therapy in acute myeloid leukemia.
CD33
CD33 is an immunoglobulin-like lectin which expressed on cells of the monocytic and myeloid lineages, and is present in 87.8% of AML cases (62). However, the expression of CD33 in healthy HSCs was also detected, which could lead to off-target toxicity (63). Taking this into account, researchers performed an in vivo experiments through xenograft mouse model to selectively delete CD33 from normal HSCs to avoid undesired toxicity. Consequently, CAR-T cells targeting CD33 could efficiently destroy AML cells without myelotoxicity (64). Furthermore, a more commonly used strategy is to establish a balanced dual-CAR. Researchers established a balanced dual-CAR based on a low-affinity interleukin-3-zetakine (IL-3z) and a high specificity of CD33 to target AML cells. This CAR is designed without activating signaling domains in order to minimize off-target toxicity and maintain complete damaging capacity against AML cells (34). Meanwhile, the design of CAR that incorporates pharmacologic control is also considered. A clinical trial with rapamycin-regulated CD33 CAR-T cells has shown a controlled function in AML, demonstrating a promising prospect (35).
CD123
CD123, a cell membrane protein, is notably overexpressed on AML cells, but nearly absent on normal HSCs (65),unlike CD33, but CD123 expression on blood vessels leads to off-target toxicity (66). Researchers designed the CD123 CAR-T cells and analyzed the therapeutic effects on AML in a xenograft model. The data showed that these CAR-T cells exhibited anti-AML activity, importantly without toxicity to the hematopoietic system or other tissues (36). Furthermore, researchers found that 5′-Azacitidine (AZA) treatment could increase the density of CD123 on AML cells, therefore enhancing activity and abundance of CTLA-4neg CAR-T cells, revealing the potential benefits of combining CAR-T therapy with pharmaceuticals (36). A study evaluated the therapeutic effects of allogeneic gene-edited CAR-T cells (UCART123) targeting CD123 both in vitro and in vivo, displaying high efficacy to eliminate AML cell. In addition, the safety features are impressive by using genome editing technology to avoid graft versus host disease and expressing special antigens to eliminate CAR-T cells timely (37). Similarly, researchers also developed the bispecific CAR-T cell for both CD123 and NKG2DL, that not only eliminates AML cells but also targets immunosuppressive cells. Consequently, this dual-CAR strategy perfectly avoids antigen escape and counteracts the suppressive impacts of tumor microenvironment (TME) (38).
CD7
As a surface marker, CD7 is commonly expressed on T lymphocytes and natural killer cells (67). It is also expressed in about 30% of AML cases, but is absent on healthy myeloid cells, leading to high specificity and limited toxicity (68). Considering its expression on T cells has been proving fratricidal, researchers used genome editing technology to produce the CD7KO T cells that effectively eliminate CD7 AML cells but spare healthy myeloid cells (39). Later, the specific CD7 CAR-T cells which have a low expression of CD7 was further designed by transducing an anti-CD7 CAR into T blasts followed by the natural selection. These cells inhibited leukemia cell proliferation in a xenograft mouse model and efficiently killed CD7 AML cells of R/R AML patients in vitro, showing a novel effective strategy without expensive gene ablation (40, 69). A clinical trial reported a R/R AML patient who was treated with CD7 CAR-T cell therapy, displayed reduced tumor burden with controlled CAR-related toxicity (41). Another study also tested the therapeutic effects of CD7 CAR-T cells (RD13-01) in a R/R AML patient which achieved an MRD++− CR after CD7 CAR-T treatment (42).
CD117
CD117, also recognized as KIT, is categorized as a type III receptor tyrosine kinase, which is predominantly expressed on the majority of myeloid blasts and is crucial in the AML development. Nevertheless, the expression of CD117 is significantly elevated on healthy HSCs and most primary AML cells (70). Therefore, the CD117 CAR-T therapy which targets both healthy and cancerous cells at the same time to avoid additional myeloablative conditioning before HSCTs, holds promise as a transitional treatment of HSCTs. A study had successfully generated and examined CD117 CAR-T cells in a xenograft mouse model, showing an efficient clinical prospect (43).
CD70
CD70 is a tumor necrosis factor (TNF) superfamily member and is expressed on most leukemic blasts, but unlike CD33 and CD123, it is absent from normal HSCs (71). In a conducted research, researchers produced a series of CD70 CAR-T cells and evaluated their activity against AML both in vivo and in vitro. The findings indicated that the CAR utilizing the CD70 receptor CD27 had an enhanced anti-AML activity when compared to the conventional scFv-based CAR-T cells (44). In another study, another CD70-targeted CAR was designed which has a panel of hinge-modified regions. Functional analysis showed an enhanced ability to target tumor surface antigens (72). Researchers also evaluated CD70 targeted CAR-T in xenograft mouse model, exhibiting that such CAR-T displayed significant anti-AML activity and durability in an xenograft mouse model (45).
CD93
CD93 is a transmembrane glycoprotein, which is predominantly expressed in AML blasts and LSCs, but is absent in healthy HSCs (73).Importantly, CD93 expression is relatively stable and highly expressed in a significant proportion of relapsed AML patients. Therefore, CD93 is a perfect target for CAR-T cell therapy. Researchers have developed CD93 CAR-T cells utilizing a humanized CD93-specific binder which effectively targets and eliminates AML cells without side toxicity to HSCs. Furthermore, they introduced the NOT-gated CD93 CAR-T cells, aiming to overcome the undesired endothelial-targeting toxicity (46).
CD38
CD38 is expressed on most leukemic blasts and has successfully been harnessed in the treatment for various hematological malignancies, including multiple myeloma and ALL (62, 74). Considering the relatively low expression of CD38 in AML, a study was conducted to test its anti-AML effects when enhancing CD38 density on AML cells by combining all-trans retinoic acid (ATRA) with CD38 CAR-T cells. The research data demonstrated that ATRA significantly enhanced the anti-AML activity of CD38 CAR-T cells through elevating the CD38 surface expression levels (47). Recently, researchers developed and evaluated a novel CD38-targeting T-cell engager, which showed an enlightening outcome. The outcome demonstrated that this CD38-targeting T-cell engager could stimulate T cells to release IFN-γ and transform surrounding CD38neg cells into CD38pos cells when interacting with CD38pos AML cells, thereby efficiently eliminating AML. This strategy showed good application prospect, which may be used in the construction of CAR in the future (75).
FLT3
FMS-like tyrosine kinase 3 (FLT3) is typically expressed both on healthy HSCs and on AML blasts, with high specificity for FLT3 with internal tandem duplication (FLT3-ITD) (76). A study examined FLT3-specific CAR-T cells, revealing that these CAR-T cells could recognize and disrupt healthy HSCs in vitro and in vivo (48). Therefore, researchers further improved and developed an allogeneic CAR-T cell with the elimination of endogenous TCR, achieving a lower risk of alloreactivity and a more timely treatment with off-the-shelf CAR-T cells (49). In addition, considering about 37% AML patients have FLT3 mutations and a high expression of CD44v6, researchers constructed CD44v6 CAR-T cells to treat these patients with FLT3 mutations (50).
CLL1
The human C-type lectin-like molecule-1, identified as CLL-1 or CLEC12A, is primarily expressed on most AML blasts. Importantly, CLL-1 is expressed within LSCs contrasting with its absence in HSCs, much like CD123 (77). The CLL-1 CAR-T cells have been proven to specifically damage AML cells in vitro without toxicity to HSCs (51). A study also identified CD33/CLL1 as the preferred combinatorial targets for pediatric AML, which further expanded the potential clinical application of CAR-T (52, 78). 2 patients with R/R AML displayed successful outcomes when treated with PD-1 silenced CLL-1 CAR-T therapy, after the failure of HSCT and CD38 CAR-T therapy (53). In a clinical trial, CLL-1 CAR-T therapy showed positive efficacy and tolerable safety in R/R AML patients (25). Furthermore, researchers developed a special CAR that combined the CLL1 as a costimulatory receptor with the ADGRE2-CAR to specifically target ADGRE2pos and CLL1pos LSCs, while sparing the ADGRE2low and CLL1 neg healthy HSCs. Collectively, this combined targeting strategy could selectively eliminate AML cells and reduce hematological toxicity (54).
Folate Receptor β (FRβ)
In 2015, the first production of FRβ CAR-T cells which selectively disrupted AML cells, showed therapeutic potential. Furthermore, the application of ATRA resulted in improved elimination of AML cells with enhanced FRβ expression (79). A subsequent study proved that the high-affinity FRβ CAR-T cells displayed greatly enhanced anti-tumor activity compared with the low-affinity FRβ CAR-T cells (55). Furthermore, researchers generated a bispecific tandem CAR by combining FRβ with CD123 in the retroviral vector, proving to have an enhanced effect for AML (56).
GRP78
Glucose-regulated protein 78 (GRP78) is typically located within the endoplasmic reticulum (ER). However, when ER stress is elevated, the overexpressed GRP78 is transferred to the surface of tumor cells (80). Researchers designed T cells expressing a peptide-based CAR specifically targeting GRP78, and proved a decrease in fratricide treated with dasatinib during the production. In addition, the GRP78 CAR-T cells could effectively eliminate GRP78pos tumor cells without toxicity against HSCs (57).
NKG2DL
Natural killer group 2D ligand (NKG2DL) is widely expressed in various malignant neoplasms, but nearly absent in healthy tissues (81). In a phase I clinical trial, a single patient who received the maximum dose of NKG2D CAR-T cell therapy proved to have an improvement of blood cell counts and maintained clinical stability over several months without additional supplementary treatment. Since the endpoints of this clinical trial are assessing the feasibility and safety of a single injection of NKG2D CAR-T cells instead of stable disease, no objective clinical efficacy of CAR-T cells was proved. However, considering the outstanding safety and the unexpected disease stability of several patients during subsequent therapies, NKG2D CAR-T cells have shown potential therapeutic value in AML (58). Since high expression of NKG2DL can be induced by FLT3 inhibitors, researchers constructed dual-target FLT3scFv/NKG2D CAR-T cells, and examined the inhibitory effects in vitro, which showed the powerful ability to lyse AML cells and improvement by gilteritinib-pretreatment (59).
LILRB3
The members of LILR subfamily B (LILRB) are negative factors to regulate myeloid cell activation and are commonly expressed on myeloid and lymphocyte cells (82). In a recent study, CAR-T specifically targeting LILRB3 has exhibited remarkable anti-AML activity both in vivo and in vitro. Furthermore, LILRB receptors are upregulated in response to inflammatory stimulus and chemotherapy conditions, suggesting that combined CAR-T with specific stimulus can be applied by artificially regulating the tumor microenvironment to improve the LILRB3 CAR-T efficacy (60).
Intracellular targets: PRAME
PRAME is a melanoma-associated antigen overexpressed in a variety of hematologic malignancies, including AML and CML. Conventional CAR-T cells are unable to target PRAME because it is an intracellular antigen (83). Researchers developed a special CAR-T (PRAME mTCRCAR-T) by using a T-cell receptor mimic antibody that recognizes the complex constituent of HLA-A2 and PRAME ALY peptide, therefore achieving an effective anti-AML capacity upon applications of such CAR-T cells in vivo (61).
Limitations of CAR-T cell therapy in the treatment of AML
Despite the above-mentioned examples of CAR-T cells in AML, many challenges existing limit the clinical efficacy of CAR-T cells in AML. Limitations to effective CAR-T cell therapy include restricted anti-tumor efficacy, severe life-threatening toxicities, relapse and resistance. Furthermore, other unsolved challenges commonly exist. For example, the tumor microenvironment significantly influences the activity of CAR-T cells. The excessive period of waiting for treatment initiation, the requirement for an optimal CAR design, the design of the most effective intracellular costimulatory domains, and the determination of the optimal timing for CAR-T cell infusion are all critical and unsolved for CAR-T therapy in AML.
Restricted anti-tumor efficacy
Although AML has a modest mutational load in contrast to other malignancies like melanoma or lung cancer, the genetic diversity, epigenetic alterations, and clonal heterogeneity all contribute to the complexity of CAR-T therapy in AML. Among them, the genetic and phenotypic heterogeneity in the AML cells is the foremost challenge that limits the applicability of the universal CAR-T cells (84). Currently, the resistance mechanisms of CAR-T therapy remain largely elusive, only with some hypotheses including tumor heterogeneity, antigen escapes, and the exhaustion of T-cells, along with their diminished persistence. However, it is evident that the primary forms of resistance involve antigen-negative and antigen-positive relapses. These antigen-negative relapses are linked to a range of factors, such as CAR-T cell-induced mutations, alternative splicing, the masking of epitopes and low antigen density (85–87). In a study utilizing a mouse model of leukemia, it has been demonstrated that target antigens can be transferred to T cells via CAR T cell trogocytosis. This process results in a reduction in the density of target antigens on tumor cells, thereby promoting the exhaustion of CAR-T cells. Co-targeting different antigens may prove beneficial in addressing this type of antigen-negative relapses. Antigen-positive relapses are frequently attributed to inadequate persistence of CAR-T cells, which might be caused by several factors, including the immunogenicity of the CAR itself, the inherent quality of the T-cells, the initial phenotype of the T-cells, the co-stimulatory domain present within the CAR constructs, and the impact of the tumor microenvironment (88–90).
Other significant factors contributing to the diminished efficacy of CAR-T therapy are inadequate T cell proliferation and short-term T cell survival, which leads to a weak therapeutic response. It is widely believed that the immunosuppressive microenvironment created by AML contributes much to such restrictions. Among the critical elements for the suppressive tumor microenvironment, regulatory T (Treg) cells play a prominent role in inhibiting immune responses. First, Tregs with an overexpression of PD, OX40 and TIM3 have an increased frequency in the peripheral blood of AML patients (91). The engagement of PD-1 with PD-L1 or PD-L2 initiates a series of intracellular signals that inhibit T-cell activation. Furthermore, the expression of PD-1 on Tregs that migrate to the tumor microenvironment can stimulate these immunosuppressive cells, reinforcing their immunosuppressive functions (92, 93). Recently, the 123NL CAR-T cells likely targeting immunosuppressive cells through CD123 and NKG2DL, demonstrated a promising approach to overcome tumor microenvironment (38). In addition, researchers have proved that TP53 deficiency confers resistance of AML. Therefore, inhibiting the mevalonate pathway in TP53-deficient AML cells or enhancing the Wnt pathway in CAR-T cells in vitro restores the efficiency of CAR-T-cell-mediated AML cell lysis in a recent study. This data provided a novel insight into our understanding of CAR-T therapy resistance in terms of metabolic mechanisms (94). Lots of studies have been done to overcome these immune pathways which were hypothesized as the important barriers to T cell activation, but the conclusion remains uncertain.
Severe life-threatening toxicities
The frequently observed adverse effects for CAR-T therapies are CRS, neurotoxicity, cytopenias and infections. These adverse effects can range from minor symptoms to serious life-threatening situations. For example, ten R/R AML patients treated with CLL-1 CAR-T cells all suffered from CRS, among them, 4 cases of them were low-grade, the remaining 6 were considered high-grade (25). The precise mechanism of CRS remains elusive but is theorized that it may arise from the over-activation of T-cells, which subsequently leads to the emission of a variety of inflammatory cytokines such as TNF-α and interferon-gamma (95). Symptoms associated with CRS triggered by CAR-T cell therapy may include fever, tachycardia, headache, nausea, rash and shortness of breath. Furthermore, some severe cases could even lead to multiple organ failure (96). The standard approach to managing CRS typically involves supportive care alongside the administration of corticosteroids or tocilizumab as treatment options (95, 97).
Despite the enhanced targeting precision that CAR-based therapies present in comparison to conventional chemotherapy and radiotherapy, the on-target/off-tumor toxicity remains common and troublesome. Such type of toxicity is characterized by the devastation of health tissue when targeting tumor antigens due to the common expression of target tumor antigens on healthy cells. On-target/off-tumor toxicity is a common issue in CAR-T cell therapies, but many of its manifestations remain unidentified or are also obscured by other symptoms. For example, some toxicities are directly associated with the targeting effects by CAR-T cells, such as immune effector cell-associated neurotoxicity syndrome, hypogammaglobulinemia, hematologic toxicities; the others may be indirectly linked to the therapy-induced immunosuppression to the patients, such as infections, sepsis (98).
Potential effective strategies to improve CAR-T treatment, specially for failure and resistance
CAR-T cell therapy has achieved numerous encouraging progress in AML. This therapy has inimitable advantages compared with conventional chemotherapy and radiotherapy, but also has some limitations that need to be addressed. A variety of strategies including the combination of CAR-T cell therapy with other anti-AML approaches and the utilization of advanced CAR engineering techniques, have been put forward to enhance the therapeutic efficacy of CAR-T, mitigate adverse effects and broaden clinical applicability. In the following section, we describe some strategies for overcoming these limitations.
Optimizing CAR structure
The current clinical outcomes of CAR-T cells are unsatisfactory for AML therapy due to the modest cell responses. To improve it, researchers are extremely enthusiastic about modifying the structure of CAR. Recently, research has reported that a CD33 CAR targeting membrane-proximal epitopes outperforms the CAR targeting membrane-distal epitopes, emphasizing the importance of the antigen epitopes for optimizing CAR design (99). Researchers redesigned a series of hinge domains to reduce the proteolytic cleavage of the CD27 extracellular segment. This modification enhanced the binding stability between the CAR to CD70, thereby enhancing the binding ability and anti-tumor activity in vivo (72). Continuous optimization of the CAR structure might overcome the root cause of the undesired efficacy of CAR-T cells in AML. In addition, using different target-specific CARs coherently may also be a promising strategy. Researchers established a novel T-cell platform known as the Fab-based adapter CAR (AdCAR) which uses adapter molecules such as anti-CD33, anti-CD123, and anti-CLL1 to selectively target AML cells. The AdCAR platform overcomes the chronic T-cell exhaustion and antigen heterogeneity in AML, providing a more adaptable strategy against the complexities of AML (100).
Targeting at a single antigen may produce a selective pressure which potentially drives the evolution of cancer cells and results in the immune escape. Therefore, bispecific CAR-T cell is a promising therapeutic approach for AML. The bispecific CAR-T cells targeted both GRP78 and CD123 had been proven to successfully improve anti-AML activity compared to the CAR-T cells targeted GRP78/CD123 (101). A vitro study has demonstrated that CAR’TCR-T cells, engineered to co-express both a CD33-CAR and a transgenic dNPM1-TCR, exhibit enhanced and sustained anti-tumor efficacy. This is especially notable in a case where the target antigen density is extremely low, highlighting the potential value of this dual-expressing cell strategy (102). In addition, discovering novel CAR targets is also meaningful for the therapy efficacy of CAR-T. A study proposed an approach that evaluated many candidates simultaneously and applied some particular principles to guide combinatorial pairings (103). Structural surface-omic and Single-cell transcriptomic might also be helpful for discovering new targets (90, 104). Given the current lack of sufficiently suitable targets, a strategy of targeting multiple antigens may represent the optimal approach to overcome the heterogeneity of AML. Continuous refinement of CAR structures through bioengineering techniques, coupled with the potential of gene editing to enhance CAR-T efficiency, holds promise for overcoming AML heterogeneity.
Combining other therapies to improve the efficacy of CAR-T
Combining CAR-T cell therapy with other therapy is also necessary to improve the anti-tumor efficacy in AML. Research has confirmed that the pretreatment with rapamycin, which diminishes mTORC1 signaling, can attenuate the activity of CAR-T cells to infiltrate bone marrow by diminishing mTORC1 signaling. This intervention has been shown to intensify the eradication of AML cells within the bone marrow in mouse models of leukemia xenografts and may inspire us to combine other chemotherapeutic agents with CAR-T cells for AML treatment (105). Furthermore, a study reported a CD38-CD3 T-cell engager, named BN-CD38 was developed, which has demonstrated the ability to facilitate T-cell activation and proliferation, as well as contribute to the elimination of AML LSCs within an autologous context, offering a promising strategy for the targeted treatment of AML. Interestingly, IFN-γ–induced upregulation of CD38 may improve the CAR-T therapy through higher antigen density (75). Combining CAR-T therapy with AZA (demethylating agents) appears highly promising for clinical translation (36). Additionally, integrating CAR-T therapy with small molecule drugs such as venetoclax (BCL-2 inhibitors) and immune checkpoint inhibitors also offers significant potential for enhancing clinical efficacy (106).
Applying dual targets to avoid on-target/off-tumor effects
Because most target antigens are present on both AML cells and healthy tissues, CAR-T cell therapy often causes on-target/off-tumor effects inevitably, even threatening the patient’s life. In order to avoid these toxicities in CAR-T cell treatment of AML, researchers have proposed many strategies. For example, a universal CAR platform technology known as UniCAR divides conventional CARs into two distinct elements: a CAR for the non-specific manipulation of T cells and a targeting module for redirecting the activity of UniCAR T cells. This strategy reduces the risk of on-target side effects by allowing the precise activation and deactivation of CAR-T cells in a regulated method (107). Logic-gated CAR also shows a promising prospect, such as AND gates incorporating two individual receptors, OR gates based on dual or tandem CARs, and NOT gates with inhibitory signaling. In a study, the CAR-T cells that dual-target CD13 and TIM3 showed great specificity for eradicating both CD13pos and TIM3pos AML cells, with acceptable toxicity to the healthy cells with only expressing CD13 (108). A recent study repurposed cytosolic molecules into CARs by combining the LAT with SLP-76, and generated the AND-gate CAR-T cells which exhibited enhanced functionality and specificity (109). Furthermore, researchers developed a new platform named AbTCR-CSR, which combined an antibody-T-cell receptor (AbTCR) CAR with costimulatory signaling receptor (CSR), representing a similar AND gates strategy to avoid the toxicity of CAR-T therapy in AML (110). In the context of NOT-gated CAR-T cell therapy, these cells are engineered to express a secondary inhibitory CAR (iCAR) designed to recognize an antigen exclusively expressed in healthy cells, not present in tumor cells. This iCAR delivers an inhibitory signal that effectively counteracts the activation signal intended for the CAR-T cells, thus modulating their responses to prevent unintended interactions with healthy tissues. Researchers have tested the NOT-gated CD93 CAR-T in an in vitro model with a rewarding outcome and promising prospect (46).
Utilizing genetic engineering for CAR-T cells or healthy tissue cells
Gene editing can effectively avoid the T-cell exhaustion and enhance the activity of CAR-T cells. This strategy includes deleting negative regulatory molecules (111) or expressing specific transgenic molecules (112) to enhance anti-tumor activity, such strategy has been proved in many tumor models including AML. In addition, the gene editing strategy is also promising to avoid on-target/off-tumor toxicity. A study that deleted CD33 from normal HSCs in order to generate a hematopoietic system resistant to CAR-T cells showed the specific and effective killing capability of AML cells (64). However, the production of clinically compliant gene-edited CAR-T cells faces challenges including safety risks from off-target modifications causing genomic instability or oncogenic mutations, production efficiency concerns regarding CAR-T cell numbers and activity, and ethical issues in meeting global regulatory requirements (113, 114).
Attaching polyethylene glycol to the exterior of CAR-T cells
CRS and neurotoxicity are recognized as the predominant and distinctive adverse effects linked to CAR-T cell therapies. A recent study has demonstrated that in vivo attachment of PEG to the exterior of CAR-T cells significantly alleviates the incidence of CRS and neurotoxicity which are commonly induced by CAR-T cells. Importantly, this modification does not influence the capacity of CAR-7 to eliminate tumors, therefore preserving their therapeutic efficacy. This is because the CAR-T cell can block the interactions between CAR-T cells, tumor cells and monocytes, and decrease both monocyte overactivation and cytokine release in vitro. Furthermore, the gradual proliferation of CAR-T cells decreases PEG surface density and facilitates the re-establishment of interactions between CAR-T cells and AML cells, leading to robust anti-tumor responses, while reducing adverse effects (115).
Optimizing the production procedure of CAR-T
Despite some potential advantages, using autologous T cells to produce the CAR-T cells has complicated procedures which cost plenty of time, therefore prolonging the waiting time for treatment. The allogeneic FLT3 CAR-T cells is an off-the-shelf CAR-T therapy, showing the convenience and feasibility of, shortening the waiting time before treatment and increasing the odds for patients to acquire these therapies. Furthermore, the off-switch in the lead CAR mentioned in this study provides the possibility to modulate CAR-T cell activity and thus restores the functions of the hematopoietic system after the elimination of AML cells (49). Another limitation is that the intensive induction/salvage regimens, both for initial treatment and for rescuing purposes in AML, can significantly reduce the activity and the number of autologous T cells collected during leukapheresis. Consequently, producing CAR-T cells directly from AML patients would lead to the disappointing availability of CAR-T cells. Employing engineered CIK cell, genetically modified, as an allogeneic resource within the dual CAR strategy is promising for avoiding the decrease of autologous T cells activity (34).
Despite the many limitations in CAR-T therapy for AML, researchers have demonstrated numerous strategies to deal with them (Figure 2). These existing strategies provide valuable insights and inspiration for the subsequent research. For example, developing more effective CAR structure may be significant potential for advancement. Combing other therapies, such as rapamycin for pre-treatment, is also rewarding for overcoming these challenges.
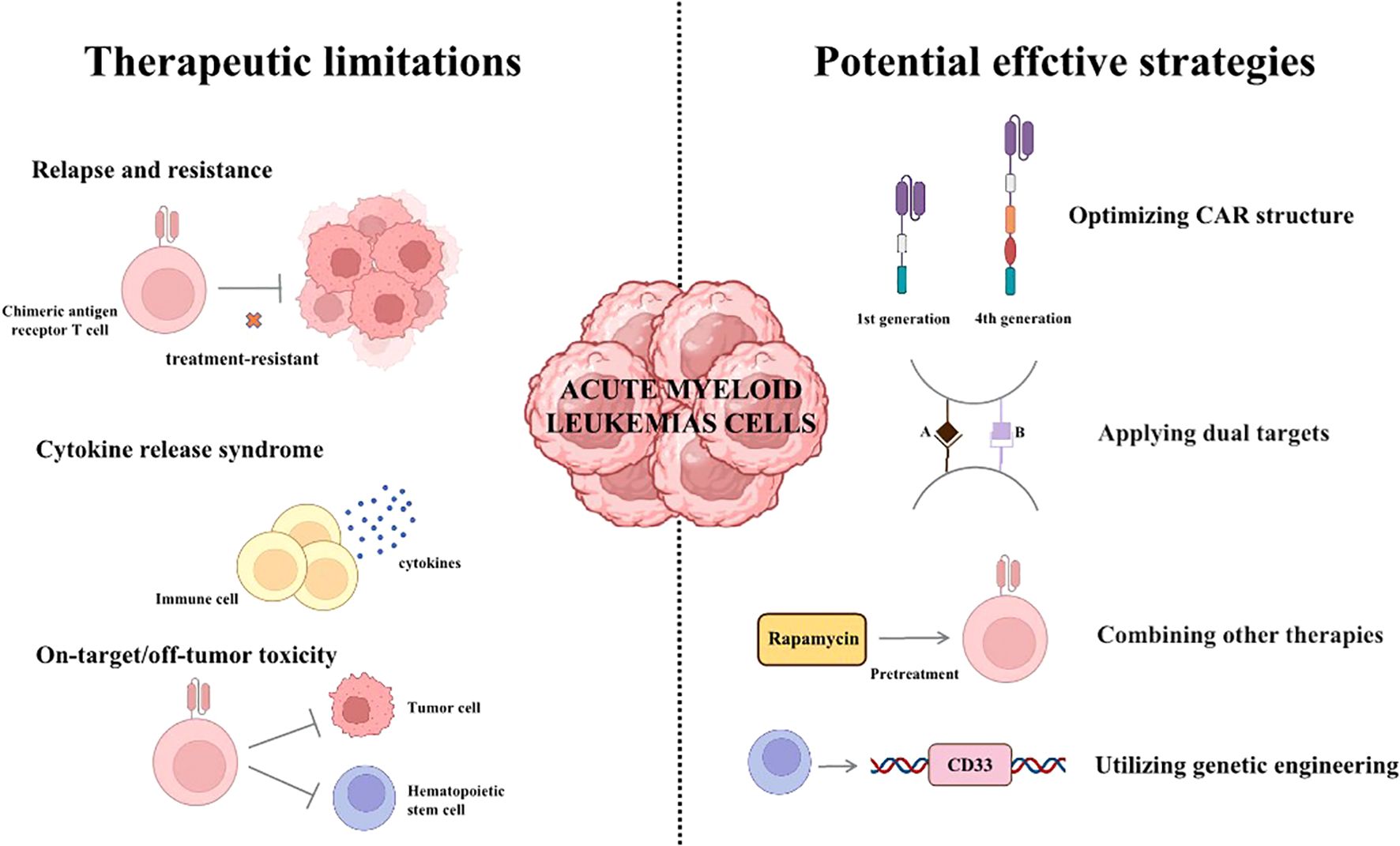
Figure 2. The therapeutic limitations and potential effective strategies of CAR-T therapy. (I) Relapse, resistance and adverse effects are significant challenges of CAR-T therapy currently. (II) Some strategies for overcoming the limitations of CAR-T therapy.
Translating these strategies into subsequent trials still faces numerous challenges. These challenges include the limitations of mouse models in accurately replicating the immunosuppressive and metabolic stress conditions of the human tumor microenvironment, the potential for relapse following treatment observed in clinical trials, the necessity for hematopoietic stem cell transplantation as a salvage therapy, the rapid disease progression characteristic of AML patients, and the intricate manufacturing process required for personalized CAR-T cell production. Collectively, these factors impose stringent requirements on the design and execution of subsequent clinical trials (116).
Conclusions
Compared to recent systematic evaluations or meta-analyses (e.g., Shahzad et al., 2023) (14), we discuss in greater depth the many barriers and limitations faced by CAR-T cell therapies for the treatment of AML as well as strategies for avoiding adverse effects and improving the efficacy of CAR-T therapies. In this review, we outlined the progress achieved in the multiple categories of immunotherapeutic approaches for the management of AML, discussed the particular mechanisms of CAR-T therapy, and further summarized the recent advances of CAR-T immunotherapy in AML, as well as the current limitations, hopefully providing some novel insights for the future research direction.
Despite more and more treatment options in recent years, AML still poses a serious threat to human health with approximately 50% of patients ultimately dying from the disease progression and relapse. Immunological treatments, particularly CAR-T therapy, have shown magnificent efficacy in eliciting responses among patients with AML, indicating the likelihood of CAR-T to improve AML patient’s prognosis in future clinical practice. In the last several years, various CARs have been engineered and further evaluated rigorously in the clinical trials of AML patients to clarify their safety and efficacy. In addition, significant advancements have been achieved in overcoming the resistance of R/R AML and avoiding CAR-T cell-associated adverse toxicities. Although the CAR-T therapy for AML remains immature compared with other hematological malignancies such as ALL, we believe that CAR-T therapy holds great potential benefits for AML patients in the future.
At present, CAR-T cells still display numerous limitations to be overcome in order to improve the prognosis of AML patients. The foremost challenge among these issues is the safety of CAR-T therapy, which needs further input into the better design of CAR and avoiding the cross-interaction of both AML cells and health tissues, thus emphasizing the importance of optimization of CAR structures or combination of CAR with other therapies. Resistance and relapse of AML are also a sticky challenge, therefore clarifying the genetic and phenotypic heterogeneity of AML cells and utilizing gene editing technology to modify the CAR-T cells might be highly appreciated. The combination of multiple strategies may be even the futural dominant approach. All in all, in spite of the limitations, CAR-T therapy enriches the toolbox of AML treatment currently and is worthy of much great research in the future.
Author contributions
YL: Writing – original draft. WW: Writing – review & editing. CW: Writing – review & editing. JD: Writing – review & editing. HM: Writing – review & editing. YH: Writing – review & editing. SL: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundations of China (No.82070136 to SL); and by National Key R&;D Program of China (No. 2022YFC2304600 to YHu).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA: A Cancer J clinicians. (2020) 70:7–30. doi: 10.3322/caac.21590
2. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA: A Cancer J clinicians. (2023) 73:17–48. doi: 10.3322/caac.21763
3. Short NJ, Rytting ME, Cortes JE. Acute myeloid leukaemia. Lancet (London England). (2018) 392:593–606. doi: 10.1016/s0140-6736(18)31041-9
4. Roboz GJ, DiNardo CD, Stein EM, de Botton S, Mims AS, Prince GT, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood. (2020) 135:463–71. doi: 10.1182/blood.2019002140
5. de Botton S, Montesinos P, Schuh AC, Papayannidis C, Vyas P, Wei AH, et al. Enasidenib vs conventional care in older patients with late-stage mutant-IDH2 relapsed/refractory AML: a randomized phase 3 trial. Blood. (2023) 141:156–67. doi: 10.1182/blood.2021014901
6. Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman EB, Paolini S, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. New Engl J Med. (2019) 381:1728–40. doi: 10.1056/NEJMoa1902688
7. DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. New Engl J Med. (2020) 383:617–29. doi: 10.1056/NEJMoa2012971
8. Bewersdorf JP, Abdel-Wahab O. Translating recent advances in the pathogenesis of acute myeloid leukemia to the clinic. Genes Dev. (2022) 36:259–77. doi: 10.1101/gad.349368.122
9. Short NJ, Daver N, DiNardo CD, Kadia T, Nasr LF, Macaron W. Azacitidine, venetoclax, and gilteritinib in newly diagnosed and relapsed or refractory FLT3-mutated AML. J Clin Oncol. (2024) 42:1499–508. doi: 10.1200/jco.23.01911
10. Daver N, Alotaibi AS, Bücklein V. & Subklewe, M. T-cell-based immunotherapy of acute myeloid leukemia: current concepts and future developments. Leukemia. (2021) 35:1843–63. doi: 10.1038/s41375-021-01253-x
11. Lu J, Jiang G. The journey of CAR-T therapy in hematological Malignancies. Mol Cancer. (2022) 21:194. doi: 10.1186/s12943-022-01663-0
12. Newell LF, Cook RJ. Advances in acute myeloid leukemia. BMJ (Clinical Res ed.). (2021) 375:n2026. doi: 10.1136/bmj.n2026
13. Landoni E, Savoldo B. Treating hematological Malignancies with cell therapy: where are we now? Expert Opin Biol Ther. (2018) 18:65–75. doi: 10.1080/14712598.2018.1384810
14. Shahzad M, Nguyen A, Hussain A, Ammad-Ud-Din M, Faisal MS, Tariq E, et al. Outcomes with chimeric antigen receptor t-cell therapy in relapsed or refractory acute myeloid leukemia: a systematic review and meta-analysis. Front Immunol. (2023) 14:1152457. doi: 10.3389/fimmu.2023.1152457
15. Biernacki MA, Bleakley M. Neoantigens in hematologic Malignancies. Front Immunol. (2020) 11:121. doi: 10.3389/fimmu.2020.00121
16. Baroni ML, Sanchez Martinez D, Gutierrez Aguera F, Roca Ho H, Castella M, Zanetti SR, et al. 41BB-based and CD28-based CD123-redirected T-cells ablate human normal hematopoiesis in vivo. J immunother Cancer. (2020) 8(1). doi: 10.1136/jitc-2020-000845
17. Vago L, Gojo I. Immune escape and immunotherapy of acute myeloid leukemia. J Clin Invest. (2020) 130:1552–64. doi: 10.1172/jci129204
18. Im A, Pavletic SZ. Immunotherapy in hematologic Malignancies: past, present, and future. J Hematol Oncol. (2017) 10:94. doi: 10.1186/s13045-017-0453-8
19. Maschmeyer G, Bullinger L, Garcia-Vidal C, Herbrecht R, Maertens J, Menna P, et al. Infectious complications of targeted drugs and biotherapies in acute leukemia. Clinical practice guidelines by the European Conference on Infections in Leukemia (ECIL), a joint venture of the European Group for Blood and Marrow Transplantation (EBMT), the European Organization for Research and Treatment of Cancer (EORTC), the International Immunocompromised Host Society (ICHS) and the European Leukemia Net (ELN). Leukemia. (2022) 36:1215–26. doi: 10.1038/s41375-022-01556-7
20. Döhner H, Wei AH, Löwenberg B. Towards precision medicine for AML. Nat Rev Clin Oncol. (2021) 18:577–90. doi: 10.1038/s41571-021-00509-w
21. Damiani D, Tiribelli M. Checkpoint inhibitors in acute myeloid leukemia. Biomedicines. (2023) 11(6):1724. doi: 10.3390/biomedicines11061724
22. Kuen DS, Hong J, Lee S, Koh CH, Kwak M, Kim BS, et al. A personalized cancer vaccine that induces synergistic innate and adaptive immune responses. Advanced materials (Deerfield Beach Fla.). (2023) 35:, e2303080. doi: 10.1002/adma.202303080
23. Kent A, Crump LS, Davila E. Beyond αβ T cells: NK, iNKT, and γδT cell biology in leukemic patients and potential for off-the-shelf adoptive cell therapies for AML. Front Immunol. (2023) 14:1202950. doi: 10.3389/fimmu.2023.1202950
24. Park JH, Rivière I, Gonen MG, Wang X, Sénéchal BS, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. New Engl J Med. (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
25. Jin X, Zhang M, Sun R, Lyu H, Xiao X, Zhang X, et al. First-in-human phase I study of CLL-1 CAR-T cells in adults with relapsed/refractory acute myeloid leukemia. J Hematol Oncol. (2022) 15:88. doi: 10.1186/s13045-022-01308-1
26. Dufva O, Koski J, Maliniemi P, Ianevski A, Klievink J, Leitner J, et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood. (2020) 135:597–609. doi: 10.1182/blood.2019002121
27. Singh N, Lee YG, Shestova O, Ravikumar P, Hayer KE, Hong SJ, et al. Impaired death receptor signaling in leukemia causes antigen-independent resistance by inducing CAR T-cell dysfunction. Cancer Discov. (2020) 10:552–67. doi: 10.1158/2159-8290.Cd-19-0813
28. Abbasi S, Asghari Totmaj M, Abbasi MA, Hajazimian S, Goleij P, Behroozi J, et al. Chimeric antigen receptor T (CAR-T) cells: Novel cell therapy for hematological Malignancies. Cancer Med. (2023) 12:7844–58. doi: 10.1002/cam4.5551
29. Chmielewski M, Abken H. TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv Cell Gene Ther. (2020) 3(3). doi: 10.1002/acg2.84
30. Yuti P, Sawasdee N, Natungnuy K, Rujirachaivej P, Luangwattananun P, Sujjitjoon J, et al. Enhanced antitumor efficacy, proliferative capacity, and alleviation of T cell exhaustion by fifth-generation chimeric antigen receptor T cells targeting B cell maturation antigen in multiple myeloma. Biomed pharmacother = Biomed pharmacotherapie. (2023) 168:115691. doi: 10.1016/j.biopha.2023.115691
31. Bui TA, Mei H, Sang R, Ortega DG, Deng W. Advancements and challenges in developing in vivo CAR T cell therapies for cancer treatment. EBioMedicine. (2024) 106:105266. doi: 10.1016/j.ebiom.2024.105266
32. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
33. Lee DW, Kochenderfer JN, Stetler-Stevenson MA, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (London England). (2015) 385:517–28. doi: 10.1016/s0140-6736(14)61403-3
34. Perriello VM, Rotiroti MC, Pisani I, Galimberti S, Alberti G, Pianigiani G, et al. IL-3-zetakine combined with a CD33 costimulatory receptor as a dual CAR approach for safer and selective targeting of AML. Blood Adv. (2023) 7:2855–71. doi: 10.1182/bloodadvances.2022008762
35. Appelbaum J, Price AE, Oda K, Zhang J, Leung WH, Tampella G, et al. Drug-regulated CD33-targeted CAR T cells control AML using clinically optimized rapamycin dosing. J Clin Invest. (2024) 134(9). doi: 10.1172/jci162593
36. El Khawanky N, Hughes A, Yu W, Myburgh R, Matschulla T, Taromi S, et al. Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat Commun. (2021) 12:6436. doi: 10.1038/s41467-021-26683-0
37. Sugita M, Galetto R, Zong H, Ewing-Crystal N, Trujillo-Alonso V, Mencia-Trinchant N, et al. Allogeneic TCRαβ deficient CAR T-cells targeting CD123 in acute myeloid leukemia. Nat Commun. (2022) 13:2227. doi: 10.1038/s41467-022-29668-9
38. Jin X, Xie D, Sun R, Lu W, Xiao X, Yu Y, et al. CAR-T cells dual-target CD123 and NKG2DLs to eradicate AML cells and selectively target immunosuppressive cells. Oncoimmunology. (2023) 12:2248826. doi: 10.1080/2162402x.2023.2248826
39. Gomes-Silva D, Atilla E, Ataca Atilla P, Mo F, Tashiro H, Srinivasan M, et al. CD7 CAR T cells for the therapy of acute myeloid leukemia. Mol therapy: J Am Soc Gene Ther. (2019) 27:272–80. doi: 10.1016/j.ymthe.2018.10.001
40. Lu Y, Liu Y, Wen SP, Kuang N, Zhang XJ, Li JQ, et al. Naturally selected CD7 CAR-T therapy without genetic editing demonstrates significant antitumour efficacy against relapsed and refractory acute myeloid leukaemia (R/R-AML). J Trans Med. (2022) 20:600. doi: 10.1186/s12967-022-03797-7
41. Cao X, Dai HP, Cui QY, Li Z, Shen WH, Pan JL, et al. CD7-directed CAR T-cell therapy: a potential immunotherapy strategy for relapsed/refractory acute myeloid leukemia. Exp Hematol Oncol. (2022) 11:67. doi: 10.1186/s40164-022-00318-6
42. Hu Y, Zhou Y, Zhang MM, Zhao H, Wei GQ, Ge WG, et al. Genetically modified CD7-targeting allogeneic CAR-T cell therapy with enhanced efficacy for relapsed/refractory CD7-positive hematological Malignancies: a phase I clinical study. Cell Res. (2022) 32:995–1007. doi: 10.1038/s41422-022-00721-y
43. Myburgh R, Kiefer JD, Russkamp NF, Magnani CF, Nuñez N, Simonis A, et al. Anti-human CD117 CAR T-cells efficiently eliminate healthy and Malignant CD117-expressing hematopoietic cells. Leukemia. (2020) 34:2688–703. doi: 10.1038/s41375-020-0818-9
44. Sauer T, Parikh K, Sharma S, Omer B, Sedloev D, Chen Q, et al. CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood. (2021) 138:318–30. doi: 10.1182/blood.2020008221
45. Wu G, Guo SS, Luo Q, Wang XX, Deng WH, Ouyang GF, et al. Preclinical evaluation of CD70-specific CAR T cells targeting acute myeloid leukemia. Front Immunol. (2023) 14:1093750. doi: 10.3389/fimmu.2023.1093750
46. Richards RM, Zhao F, Freitas KA, Parker KR, Xu P, Fan A, et al. NOT-gated CD93 CAR T cells effectively target AML with minimized endothelial cross-reactivity. Blood Cancer Discov. (2021) 2:648–65. doi: 10.1158/2643-3230.Bcd-20-0208
47. Yoshida T, Mihara KC, Takei Y, Yanagihara K, Kubo T, Bhattacharyya J, et al. All-trans retinoic acid enhances cytotoxic effect of T cells with an anti-CD38 chimeric antigen receptor in acute myeloid leukemia. Clin Trans Immunol. (2016) 5:e116. doi: 10.1038/cti.2016.73
48. Jetani H, Garcia-Cadenas I, Nerreter T, Thomas S, Rydzek J, Briones Meijide J, et al. CAR T-cells targeting FLT3 have potent activity against FLT3(-)ITD(+) AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia. (2018) 32:1168–79. doi: 10.1038/s41375-018-0009-0
49. Sommer C, Cheng HY, Nguyen D, Dettling D, Yeung YA, Sutton J, et al. Allogeneic FLT3 CAR T cells with an off-switch exhibit potent activity against AML and can be depleted to expedite bone marrow recovery. Mol therapy: J Am Soc Gene Ther. (2020) 28:2237–51. doi: 10.1016/j.ymthe.2020.06.022
50. Tang L, Huang HM, Tang YT, Li Q, Wang J, Li DJ, et al. CD44v6 chimeric antigen receptor T cell specificity towards AML with FLT3 or DNMT3A mutations. Clin Trans Med. (2022) 12:e1043. doi: 10.1002/ctm2.1043
51. Wang J, Chen SY, Xiao W, Li W, Wang L, Yang S, et al. CAR-T cells targeting CLL-1 as an approach to treat acute myeloid leukemia. J Hematol Oncol. (2018) 11:7. doi: 10.1186/s13045-017-0553-5
52. Zhang H, Wang PF, Li Z, He YY, Gan W, Jiang H, et al. Anti-CLL1 chimeric antigen receptor T-cell therapy in children with relapsed/refractory acute myeloid leukemia. Clin Cancer Res. (2021) 27:3549–55. doi: 10.1158/1078-0432.Ccr-20-4543
53. Ma YJ, Dai HP, Cui QY, Cui W, Zhu WJ, Qu CJ, et al. Successful application of PD-1 knockdown CLL-1 CAR-T therapy in two AML patients with post-transplant relapse and failure of anti-CD38 CAR-T cell treatment. Am J Cancer Res. (2022) 12:615–21.
54. Haubner S, Mansilla-Soto J, Nataraj S, Kogel F, Chang Q, de Stanchina E, et al. Cooperative CAR targeting to selectively eliminate AML and minimize escape. Cancer Cell. (2023) 41:1871–1891.e1876. doi: 10.1016/j.ccell.2023.09.010
55. Lynn RC, Feng Y, Schutsky K, Poussin M, Kalota A, Dimitrov DS, et al. High-affinity FRβ-specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia. (2016) 30:1355–64. doi: 10.1038/leu.2016.35
56. Ghamari A, Pakzad P, Majd A, Ebrahimi M, Hamidieh AA. Design and Production An Effective Bispecific Tandem Chimeric Antigen Receptor on T Cells against CD123 and Folate Receptor ß towards B-Acute Myeloid Leukaemia Blasts. Cell J. (2021) 23:650–7. doi: 10.22074/cellj.2021.7314
57. Hebbar N, Epperly R, Vaidya A, Thanekar U, Moore SE, Umeda M, et al. CAR T cells redirected to cell surface GRP78 display robust anti-acute myeloid leukemia activity and do not target hematopoietic progenitor cells. Nat Commun. (2022) 13:587. doi: 10.1038/s41467-022-28243-6
58. Baumeister SH, Murad J, Werner L, Daley H, Trebeden-Negre H, Gicobi JK, et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol Res. (2019) 7:100–12. doi: 10.1158/2326-6066.Cir-18-0307
59. Li KX, Wu HY, Pan WY, Guo MQ, Qiu DZ, He YJ, et al. A novel approach for relapsed/refractory FLT3(mut+) acute myeloid leukaemia: synergistic effect of the combination of bispecific FLT3scFv/NKG2D-CAR T cells and gilteritinib. Mol Cancer. (2022) 21:66. doi: 10.1186/s12943-022-01541-9
60. Mai S, Hodges A, Chen HM, Zhang J, Wang YL, Liu Y, et al. LILRB3 modulates acute myeloid leukemia progression and acts as an effective target for CAR T-cell therapy. Cancer Res. (2023) 83:4047–62. doi: 10.1158/0008-5472.Can-22-2483
61. Kirkey DC, Loeb AM, Castro S, Nourigat McKay C, Perkins LK, Pardo L, et al. Therapeutic targeting of PRAME with mTCRCAR T cells in acute myeloid leukemia. Blood Adv. (2023) 7:1178–89. doi: 10.1182/bloodadvances.2022008304
62. Ehninger A, Kramer M, Röllig C, Thiede C, Bornhäuser M, von Bonin M, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. (2014) 4:e218. doi: 10.1038/bcj.2014.39
63. Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. (2005) 106:4086–92. doi: 10.1182/blood-2005-03-1072
64. Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. (2018) 173:1439–1453.e1419. doi: 10.1016/j.cell.2018.05.013
65. Pelosi E, Castelli G, Testa U. CD123 a therapeutic target for acute myeloid leukemia and blastic plasmocytoid dendritic neoplasm. Int J Mol Sci. (2023) 24(3):2718. doi: 10.3390/ijms24032718
66. Boucher JC, Shrestha B, Vishwasrao P, Leick M, Cervantes EV, Ghafoor T, et al. Bispecific CD33/CD123 targeted chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Mol Ther oncolytics. (2023) 31:100751. doi: 10.1016/j.omto.2023.100751
67. Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell Malignancies. Blood. (2017) 130:285–96. doi: 10.1182/blood-2017-01-761320
68. Rai S, Singh S, Gupta R. Prognostic significance of CD56 and CD7 in acute myeloid leukaemia and their outcome. Am J Blood Res. (2020) 10:109–17.
69. Lu P, Liu Y, Yang JF, Zhang X, Yang X, Wang H, et al. Naturally selected CD7 CAR-T therapy without genetic manipulations for T-ALL/LBL: first-in-human phase 1 clinical trial. Blood. (2022) 140:321–34. doi: 10.1182/blood.2021014498
70. Advani A, et al. c-kit (CD117) expression is a poor prognostic factor for relapse and overall survival in patients with newly diagnosed AML. Blood. (2006) 108:4510–0. doi: 10.1182/blood.V108.11.4510.4510
71. Jacobs J, Deschoolmeester V, Zwaenepoel K, Rolfo C, Silence K, Rottey S, et al. CD70: An emerging target in cancer immunotherapy. Pharmacol Ther. (2015) 155:1–10. doi: 10.1016/j.pharmthera.2015.07.007
72. Leick MB, Silva H, Scarfò I, Larson R, Choi BD, Bouffard AA, et al. Non-cleavable hinge enhances avidity and expansion of CAR-T cells for acute myeloid leukemia. Cancer Cell. (2022) 40:494–508.e495. doi: 10.1016/j.ccell.2022.04.001
73. Jia J, Liu B, Wang D, Wang XH, Song LR, Ren YZ, et al. CD93 promotes acute myeloid leukemia development and is a potential therapeutic target. Exp Cell Res. (2022) 420:113361. doi: 10.1016/j.yexcr.2022.113361
74. Morandi F, Horenstein AL, Costa F, Giuliani N, Pistoia V, Malavasi F, et al. CD38: A target for immunotherapeutic approaches in multiple myeloma. Front Immunol. (2018) 9:2722. doi: 10.3389/fimmu.2018.02722
75. Murtadha M, Park M, Zhu Y, Caserta E, Napolitano O, Tandoh TT, et al. A CD38-directed, single-chain T-cell engager targets leukemia stem cells through IFN-γ-induced CD38 expression. Blood. (2024) 143:1599–615. doi: 10.1182/blood.2023021570
76. Burchert A. Maintenance therapy for FLT3-ITD-mutated acute myeloid leukemia. Haematologica. (2021) 106:664-670. doi: 10.3324/haematol.2019.240747
77. Bakker AB, van den Oudenrijn S, Bakker AQ, Feller N, van Meijer M, Bia JA, et al. C-type lectin-like molecule-1: a novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. (2004) 64:8443–50. doi: 10.1158/0008-5472.Can-04-1659
78. Willier S, Rothämel P, Hastreiter M, Wilhelm J, Stenger D, Blaeschke F, et al. CLEC12A and CD33 coexpression as a preferential target for pediatric AML combinatorial immunotherapy. Blood. (2021) 137:1037–49. doi: 10.1182/blood.2020006921
79. Lynn RC, Poussin MP, Kalota AK, Feng Y, Low PS, Dimitrov DS, et al. Targeting of folate receptor β on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood. (2015) 125:3466–76. doi: 10.1182/blood-2014-11-612721
80. Tsai YL, Ha DP, Zhao H, Carlos AJ, Wei S, Pun TK, et al. Endoplasmic reticulum stress activates SRC, relocating chaperones to the cell surface where GRP78/CD109 blocks TGF-β signaling. Proc Natl Acad Sci United States America. (2018) 115:E4245–e4254. doi: 10.1073/pnas.1714866115
81. Duan S, Guo W, Xu Z, He Y, Liang C, Mo Y, et al. Natural killer group 2D receptor and its ligands in cancer immune escape. Mol Cancer. (2019) 18:29. doi: 10.1186/s12943-019-0956-8
82. van der Touw W, Chen HM, Pan PY, Chen SH. LILRB receptor-mediated regulation of myeloid cell maturation and function. Cancer immunol immunother: CII. (2017) 66:1079–87. doi: 10.1007/s00262-017-2023-x
83. Xu Y, Zou R, Wang J, Wang ZW, Zhu X. The role of the cancer testis antigen PRAME in tumorigenesis and immunotherapy in human cancer. Cell proliferation. (2020) 53:e12770. doi: 10.1111/cpr.12770
84. Rabilloud T, Potier D, Pankaew S, Nozais M, Loosveld M, Payet-Bornet D, et al. Single-cell profiling identifies pre-existing CD19-negative subclones in a B-ALL patient with CD19-negative relapse after CAR-T therapy. Nat Commun. (2021) 12:865. doi: 10.1038/s41467-021-21168-6
85. Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. (2019) 568:112–6. doi: 10.1038/s41586-019-1054-1
86. Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. (2018) 24:1504–6. doi: 10.1038/s41591-018-0146-z
87. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. (2015) 5:1282–95. doi: 10.1158/2159-8290.Cd-15-1020
88. Wagner DL, Fritsche E, Pulsipher MA, Ahmed N, Hamieh M, Hegde M, et al. Immunogenicity of CAR T cells in cancer therapy. Nat Rev Clin Oncol. (2021) 18:379–93. doi: 10.1038/s41571-021-00476-2
89. Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. (2015) 28:415–28. doi: 10.1016/j.ccell.2015.09.004
90. Mumme H, Thomas BE, Bhasin SS, Krishnan U, Dwivedi B, Perumalla P, et al. Single-cell analysis reveals altered tumor microenvironments of relapse- and remission-associated pediatric acute myeloid leukemia. Nat Commun. (2023) 14:6209. doi: 10.1038/s41467-023-41994-0
91. Williams P, Basu S, Garcia-Manero G, Hourigan CS, Oetjen KA, Cortes JE, et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer. (2019) 125:1470–81. doi: 10.1002/cncr.31896
92. Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. (2009) 114:1545–52. doi: 10.1182/blood-2009-03-206672
93. Dong Y, Han Y, Huang Y, Jiang SF, Huang Z, Chen RR, et al. PD-L1 is expressed and promotes the expansion of regulatory T cells in acute myeloid leukemia. Front Immunol. (2020) 11:1710. doi: 10.3389/fimmu.2020.01710
94. Mueller J, Schimmer RR, Koch C, Schneiter F, Fullin J, Lysenko V, et al. Targeting the mevalonate or Wnt pathways to overcome CAR T-cell resistance in TP53-mutant AML cells. EMBO Mol Med. (2024) 16:445–74. doi: 10.1038/s44321-024-00024-2
95. Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. (2022) 22:85–96. doi: 10.1038/s41577-021-00547-6
96. Santomasso BD, Nastoupil LJ, Adkins S, Lacchetti C, Schneider BJ, Anadkat M, et al. Management of immune-related adverse events in patients treated with chimeric antigen receptor T-cell therapy: ASCO guideline. J Clin Oncol. (2021) 39:3978–92. doi: 10.1200/jco.21.01992
97. Xiao X, Huang SK, Chen S, Wang Y, Sun Q, Xu XJ, et al. Mechanisms of cytokine release syndrome and neurotoxicity of CAR T-cell therapy and associated prevention and management strategies. J Exp Clin Cancer research: CR. (2021) 40:367. doi: 10.1186/s13046-021-02148-6
98. Thompson JA, Schneider BJ, Brahmer J, Achufusi A, Armand P, Berkenstock MK, et al. Management of immunotherapy-related toxicities, version 1.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Network: JNCCN. (2022) 20:387–405. doi: 10.6004/jnccn.2022.0020
99. Freeman R, Shahid S, Khan AG, Mathew SC, Souness S, Burns ER, et al. Developing a membrane-proximal CD33-targeting CAR T cell. J immunother Cancer. (2024) 12(5). doi: 10.1136/jitc-2024-009013
100. Nixdorf D, Sponheimer M, Berghammer D, Engert F, Bader U, Philipp N, et al. Adapter CAR T cells to counteract T-cell exhaustion and enable flexible targeting in AML. Leukemia. (2023) 37:1298–310. doi: 10.1038/s41375-023-01905-0
101. Zoine JT, Immadisetty K, Ibanez-Vega J, Moore SE, Nevitt C, Thanekar U, et al. Peptide-scFv antigen recognition domains effectively confer CAR T cell multiantigen specificity. Cell reports Med. (2024) 5:101422. doi: 10.1016/j.xcrm.2024.101422
102. Teppert K, Ogusuku IEY, Brandes C, Herbel V, Winter N, Werchau N, et al. CAR’TCR-T cells co-expressing CD33-CAR and dNPM1-TCR as superior dual-targeting approach for AML treatment. Mol Ther Oncol. (2024) 32:200797. doi: 10.1016/j.omton.2024.200797
103. Perna F, Berman SH, Soni RK, Mansilla-Soto J, Eyquem J, Hamieh M, et al. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell. (2017) 32:506–519.e505. doi: 10.1016/j.ccell.2017.09.004
104. Mandal K, Wicaksono GW, Yu C, Adams JJ, Hoopmann MR, Temple WC, et al. Structural surfaceomics reveals an AML-specific conformation of integrin β(2) as a CAR T cellular therapy target. Nat Cancer. (2023) 4:1592–609. doi: 10.1038/s43018-023-00652-6
105. Nian Z, Zheng X, Dou YC, Du XH, Zhou L, Fu BQ, et al. Rapamycin pretreatment rescues the bone marrow AML cell elimination capacity of CAR-T cells. Clin Cancer Res. (2021) 27:6026–38. doi: 10.1158/1078-0432.Ccr-21-0452
106. Restelli C, Ruella M, Paruzzo L, Tarella C, Pelicci PG, Colombo E, et al. Recent advances in immune-based therapies for acute myeloid leukemia. Blood Cancer Discov. (2024) 5:234–48. doi: 10.1158/2643-3230.Bcd-23-0202
107. Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, et al. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. (2016) 6:e458. doi: 10.1038/bcj.2016.61
108. He X, Feng Z, Ma J, Ling SB, Cao Y, Gurung B, et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood. (2020) 135:713–23. doi: 10.1182/blood.2019002779
109. Tousley AM, Rotiroti MC, Labanieh L, Rysavy LW, Kim WJ, Lareau CL, et al. Co-opting signalling molecules enables logic-gated control of CAR T cells. Nature. (2023) 615:507–16. doi: 10.1038/s41586-023-05778-2
110. Dao T, Xiong G, Mun SS, Meyerberg J, Korontsvit T, Xiang JY, et al. A dual-receptor T-cell platform with Ab-TCR and costimulatory receptor achieves specificity and potency against AML. Blood. (2024) 143:507–21. doi: 10.1182/blood.2023021054
111. Carnevale J, Shifrut E, Kale N, Nyberg WA, Blaeschke F, Chen YY, et al. RASA2 ablation in T cells boosts antigen sensitivity and long-term function. Nature. (2022) 609:174–82. doi: 10.1038/s41586-022-05126-w
112. Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. (2019) 576:293–300. doi: 10.1038/s41586-019-1805-z
113. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. (2022) 21:78. doi: 10.1186/s12943-022-01559-z
114. McPhedran SJ, Carleton GA, Lum JJ. Metabolic engineering for optimized CAR-T cell therapy. Nat Metab. (2024) 6:396–408. doi: 10.1038/s42255-024-00976-2
115. Gong N, Han XX, Xue L, El-Mayta R, Metzloff AE, Billingsley MM, et al. In situ PEGylation of CAR T cells alleviates cytokine release syndrome and neurotoxicity. Nat materials. (2023) 22:1571–80. doi: 10.1038/s41563-023-01646-6
Keywords: acute myeloid leukemia, chimeric antigen receptor T cell, immunotherapy, adoptive cell therapy, immunosuppressive microenvironment
Citation: Liu Y, Wang W, Wang C, Deng J, Hu Y, Mei H and Luo S (2025) Recent advances of chimeric antigen receptor T‐cell therapy for acute myeloid leukemia. Front. Immunol. 16:1572407. doi: 10.3389/fimmu.2025.1572407
Received: 07 February 2025; Accepted: 11 April 2025;
Published: 02 May 2025.
Edited by:
Laura Belver, Josep Carreras Leukaemia Research Institute (IJC), SpainReviewed by:
Mauro Di Ianni, University of Studies G. d’Annunzio Chieti and Pescara, ItalyNadia El Khawanky, Technical University of Munich, Germany
Copyright © 2025 Liu, Wang, Wang, Deng, Hu, Mei and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shanshan Luo, MjAxOFhIMDI1OEBodXN0LmVkdS5jbg==
†These authors have contributed equally to this work