 Lisha Li1,2,3†
Lisha Li1,2,3† Jun Liu1,2†Jiaye Lu1,2†
Jun Liu1,2†Jiaye Lu1,2† Junchao Wu1,2
Junchao Wu1,2 Xinyue Zhang1,2Tianyou Ma1,2Xiying Wu1,2Quangang Zhu1,2*
Xinyue Zhang1,2Tianyou Ma1,2Xiying Wu1,2Quangang Zhu1,2* Zhongjian Chen1,2*
Zhongjian Chen1,2* Zongguang Tai1,2*
Zongguang Tai1,2*- 1Shanghai Skin Disease Hospital, School of Medicine, Tongji University, Shanghai, China
- 2Shanghai Engineering Research Center of Topical Chinese Medicine, Shanghai, China
- 3Beijing Key Laboratory of Molecular Pharmaceutics and New Drug Delivery Systems, State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing, China
Intricate interactions between immune cells and cytokines define psoriasis, a chronic inflammatory skin condition that is immunological-mediated. Cytokines, including interleukins (ILs), interferons (IFNs), tumor necrosis factors (TNFs), chemokines, and transforming growth factor-β (TGF-β), are essential for controlling cellular activity and immunological responses, maintaining homeostasis and contributing to the pathogenesis of psoriasis. These molecules modulate the immune microenvironment by either promoting or suppressing inflammation, which significantly impacts therapeutic outcomes. Recent research indicates that treatment strategies targeting cytokines and chemokines have significant potential, offering new approaches for regulating the immune system, inhibiting the progression of psoriasis, and reducing adverse effects of traditional therapies. This review consolidates current knowledge on cytokine and chemokine signaling pathways in psoriasis and examines their significance in treatment. Specific attention is given to cytokines like IL-17, IL-23, and TNF-α, underscoring the necessity for innovative therapies to modulate these pathways and address inflammatory processes. This review emphasizes the principal part of cytokines in the -pathological process of psoriasis and explores the challenges and opportunities they present for therapeutic intervention. Furthermore, we examine recent advancements in targeted therapies, with a particular focus on monoclonal antibodies, in ongoing research and clinical trials.
1 Introduction
Psoriasis, as a chronic and persistently relapsing skin disease, has not yet been cured (1). Characterized by its unique clinical manifestations, this disease is known for the appearance of scaly erythematous or patchy skin lesions on the skin, nails, and joints, which not only affect the patient’s appearance but can also be accompanied by itching and pain, severely affecting the quality of life (2). Based on clinical presentation, psoriasis is divided into four types: guttate psoriasis, pustular psoriasis, plaque psoriasis, erythrodermic psoriasis and psoriatic arthritis (PsA) (3–5). Each type exhibits specific pathological features and clinical manifestations, but they all share a common pathological basis of abnormal keratinocyte (KC) proliferation and a hyperactive immune system (6). While the exact cause of psoriasis remains uncertain, recent research emphasizes the pivotal role of cytokines in its development.
Cytokines are diverse, low molecular weight proteins with pleiotropic biological activities, functioning in autocrine, paracrine, and endocrine manners. Produced by immune cells such as monocytes, macrophages, T cells, B cells, and natural killer cells, as well as non-immune cells like epithelial cells and fibroblasts, their production is triggered by external stimuli. By binding to their specific receptors, they regulate innate and adaptive immunity, hematopoiesis, cell growth, and tissue repair (7). Chemokines, a superfamily of small, secreted proteins within the cytokine family, are crucial for controlling inflammatory reactions because they attract immune cells to inflamed areas (8). In psoriasis, cytokines including tumor necrosis factor-alpha (TNF-α), interleukin-17 (IL-17), and IL-23, along with the chemokine CCL20, significantly contribute to disease initiation and progression (9–11). For instance, IL-17A can enhance the proliferative capacity of KCs, upregulate the expression of keratin 17 in KCs, while downregulating the production of filaggrin and diverse cell adhesion molecule-related genes, leading to structural damage of the epidermal barrier and triggering local psoriasis immune responses (12). Furthermore, IL-17A can stimulate KCs to express a variety of chemokines and upregulate the expression of IL-6, chemokine (C-X-C motif) ligand (CXCL) molecules in neutrophils, thereby chemoattracting a variety of immune cells to migrate to the psoriatic epidermal lesions (Figure 1) (13). It is evident that by inhibiting the activity of these molecules or blocking their signal transduction, inflammation can be effectively reduced, skin lesions decreased, and the quality of life for patients improved. The introduction of biologics, such as monoclonal antibodies targeting TNF-α, IL-17, and IL-23, has markedly improved psoriasis treatment by providing highly specific and effective treatment options. Nevertheless, some patients do not respond to these treatments, and prolonged use can lead to adverse effects, including a heightened risk of infections. Therefore, continued research and development of new targeted therapeutic strategies to improve efficacy, reduce side effects, and expand therapeutic options for individuals with psoriasis is a current focus of research.
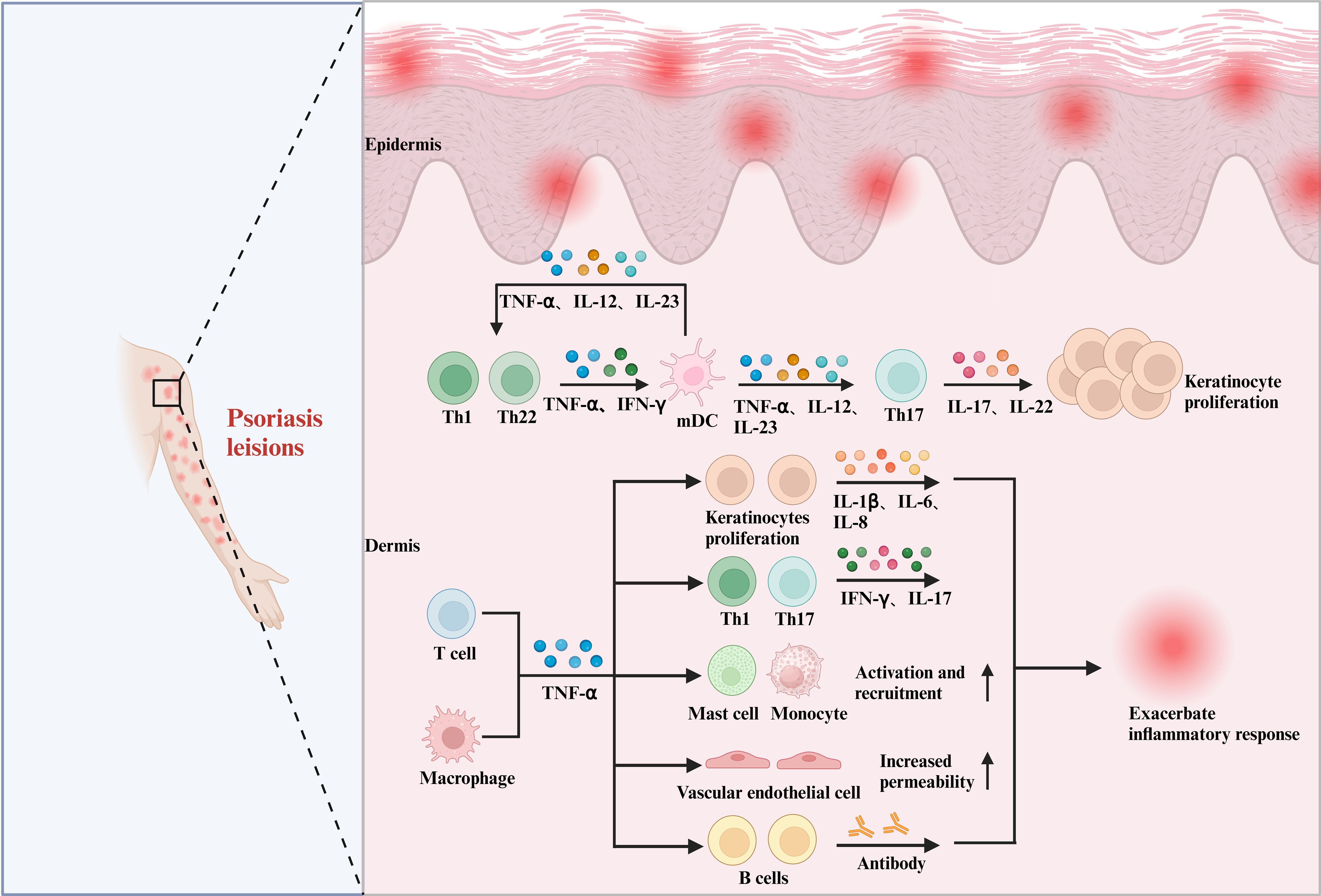
Figure 1. The pleiotropic effects of cytokines in psoriasis. In psoriasis, the role of TNF-α is equally significant and cannot be overlooked. Within psoriasis lesions, TNF-α secreted by T cells and macrophages interacts with DCs, keratinocytes, Th1 and Th17 cells, mast cells, monocytes, vascular endothelial cells, B cells, and others. This interaction promotes the downstream release of various inflammatory factors, including IL-17, IL-1β, and IL-6, which in turn leads to the abnormal proliferation of keratinocytes and exacerbates the inflammatory response.
This review offers a current and thorough investigation of cytokine and chemokine signaling pathways in psoriasis, highlighting their essential role in shaping therapeutic strategies amidst the rapidly changing landscape of psoriasis treatments. By discussing how these molecules influence immune responses in psoriasis, we have underscored the necessity of developing novel therapeutic agents capable of modulating these signaling pathways to effectively combat the inflammatory symptoms of psoriasis. The article also elaborates on the central role of cytokines in the pathological development of psoriasis, while analyzing the challenges and potential therapeutic opportunities they present. Furthermore, recent progress in targeted treatments is highlighted, particularly focusing on monoclonal antibodies currently under investigation in research and clinical trials, as well as promising targets in preclinical development. These studies significantly enhance our understanding of cytokines and chemokines in psoriasis treatment and support their potential for clinical application. The review provides a systematic summary and a comprehensive analysis, offering new insights and valuable references for the future development of specific therapeutic options for psoriasis.
2 Cytokine networks and their role in psoriasis
The immune system functions as an intricate network, encompassing lymphoid organs, immune cells, humoral factors, and cytokines. Cytokines play a pivotal role within this network as small molecular weight proteins secreted by cells (14). These molecules can exist in both secreted and membrane-bound forms, and they are instrumental in regulating cell behavior and facilitating communication between cells. This communication is crucial for regulating diverse biological processes such as immune responses, inflammation, cell proliferation, and apoptosis (Figure 2) (15).
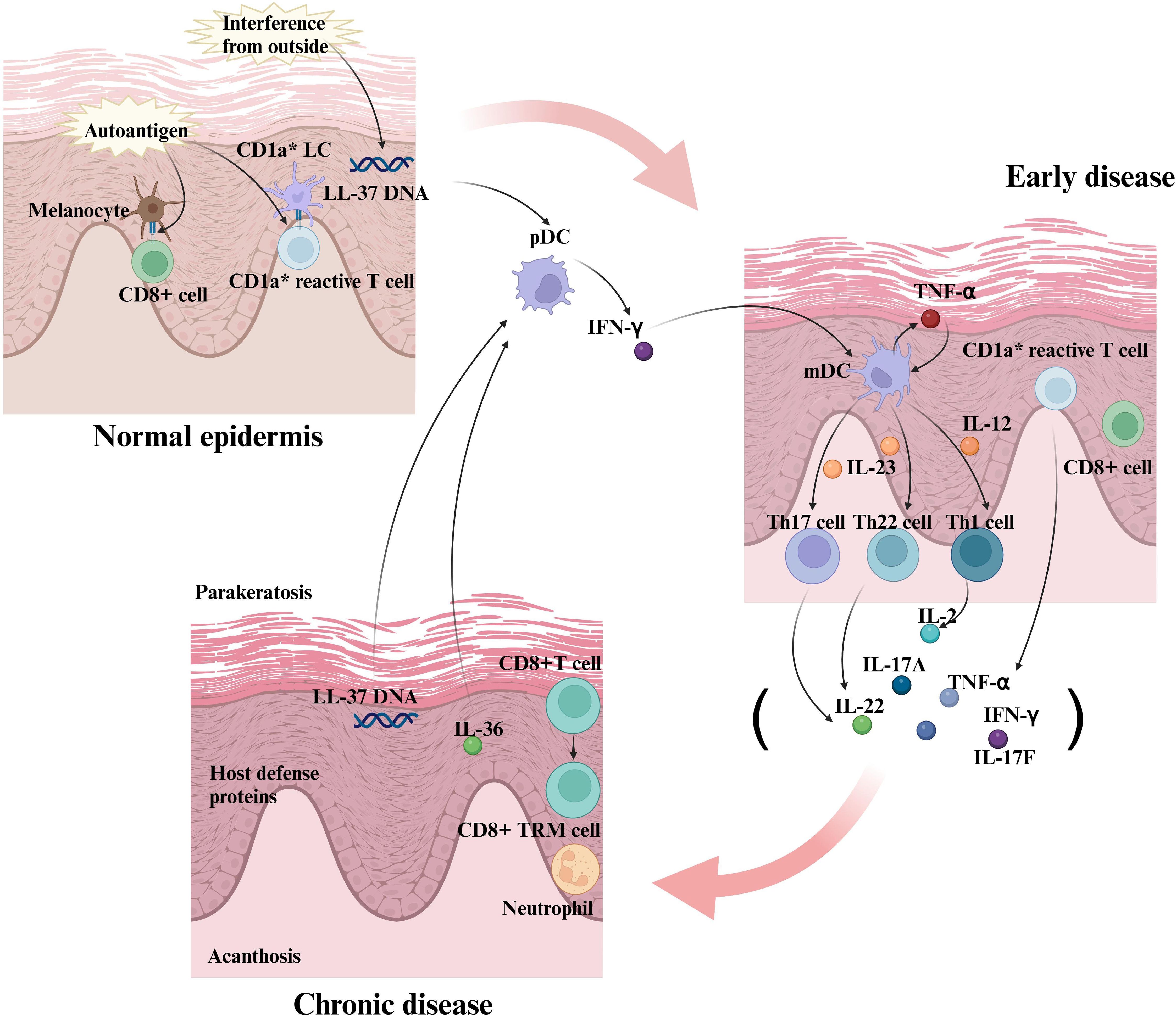
Figure 2. The formation of interactions between inflammatory factors and immune cells in the early stages of psoriasis and the creation of positive feedback loops. In the early stages of psoriasis, keratinocytes and immune cells (e.g., dendritic cells) release initial inflammatory factors. These inflammatory factors further activate and recruit immune cells, such as T cells, to the area of the skin lesion. In response to the inflammatory factors, the T cells are activated and differentiate into specific Th subpopulations, such as Th1, Th17, and Th22 cells. Th1 cells produce IL-2, Th17 cells produce IL-17, and Th22 cells produce IL-22, cytokines that further exacerbate the inflammatory response. Activated T cells interact with keratinocytes, endothelial cells, and dendritic cells to release more inflammatory factors, creating a positive feedback loop. Inflammatory factors such as TNF-α and IL-17 act directly on keratinocytes to promote their abnormal proliferation and differentiation, which are key features of lesion formation in psoriasis. In summary, the interactions between inflammatory factors and immune cells during the progression of psoriasis form a complex network that continually amplifies the inflammatory response through positive feedback mechanisms, leading to the development and persistence of skin lesions. Therapeutic strategies often target these key inflammatory factors and immune cells to reduce symptoms and control the disease.
Cytokines can be classified into distinct categories based on their origin, structure, and function, each playing a distinct role in immune regulation. ILs, for example, are essential in modulating white blood cell activity and are involved in both immune defense and inflammatory processes (15). Similarly, interferons (IFNs) are generated in response to viral infections, exhibiting antiviral, antiproliferative, and immunomodulatory effects (16). TNFs play a role in regulating cell death, inflammation, and immune cell activation. Colony-stimulating factors (CSFs) encourage the growth and differentiation of blood cell precursors, whereas growth factors facilitate cell division and tissue repair. Chemokines are crucial for directing immune cells to sites of inflammation or infection, significantly contributing to the immune response (14). The interaction between cytokines and their receptors is also vital, as these receptors are proteins that bind to cytokines and initiate signal transduction pathways. In addition to these, there are specialized cytokines such as Thrombopoietin (TPO) and leukemia inhibitory factor (LIF), which have specific roles in the regulation of blood cell production and the inhibition of leukemia cells, respectively. This intricate interplay of components within the immune system ensures a robust defense against pathogens and maintains overall health (15).
Cytokines are indispensable in the regulation as well as function within immunopathology, and targeting various cytokines has become a potent approach in treating immune diseases, ranging from autoimmune to allergic conditions (17). The inflammatory process in psoriasis engages both the innate and adaptive immune mechanisms. The characteristic lesion of psoriasis is the hyperproliferation and dysregulated differentiation of epidermal KCs, a process that involves the action of numerous cytokine pathways, thereby triggering and maintaining the inflammatory pathways of psoriasis. Plaque psoriasis, the most prevalent form, is linked to TNF-α, IL-23, and IL-17 pathways, while pustular psoriasis, a less common type, is associated with IL-36RN genetic mutations. In chronic psoriasis, the adaptive immune system sustains the disease via the TNF-α/IL-23/IL-17 axis. The interaction between IL-23 and IL-17 is central to psoriasis pathogenesis, sustaining Th17 cell activity, which mainly produces IL-17. Cytokines, such as IL-23, IL-17, IL-22, and TNF-α, play a role in the pathogenesis of PsA (9, 18, 19). In psoriasis lesions, TNF-α exhibits pro-inflammatory properties that stimulate IL-12 and IL-18 production, influencing Th1 immune responses. IL-12 and IL-18 are potent inducers of IFN-γ, playing a crucial part in regulating Th1 immune responses. TNF-α is instrumental in the IL-23/IL-17 axis, working in synergy with IL-17A to regulate cytokines and keratinocyte-associated genes involved in psoriasis. Multiple studies have demonstrated that IFN-γ serves as a prognostic factor for psoriasis, with the IFN signaling pathway contributing to its pathogenesis (19).
The interplay between cytokines forms a complex network that finely regulates immune responses and inflammatory processes through positive and negative feedback mechanisms. The cytokine network intricately regulates immune cell recruitment, activation, and proliferation, alongside the abnormal proliferation and differentiation of KCs. These interactions are vital in psoriasis pathogenesis, where the DC-IL-23-Th17 cell-IL-17-KC axis forms a positive feedback loop that advances the disease. Specifically, mature DCs (mDCs) release IL-23, which boosts Th17 cell development and proliferation. In turn, IL-17 produced by Th17 cells further stimulates the production of IL-23, creating a self-amplifying inflammatory cycle (19). IL-10, an anti-inflammatory cytokine, inhibits IL-23 and IL-17 secretion, creating a negative feedback loop to suppress inflammation (18). The balance between pro-inflammatory and anti-inflammatory cytokines is indispensable for maintaining immune homeostasis and preventing the worsening of inflammatory diseases. A foundational component of immune control is the delicate balance within the cytokine network, and knowledge of these mechanisms can help create tailored treatments for immunological-mediated diseases.
3 Immunometabolic regulation and environmental triggers in psoriasis
Keratinocytes and immune cells (such as Th17 and macrophages) in psoriasis lesions exhibit significant lipid metabolism abnormalities.
3.1 Lipid metabolism reprogramming drives amplification of inflammatory signaling
3.1.1 Cholesterol/fatty acid metabolism imbalance
Patients with psoriasis show elevated levels of oxidized low-density lipoprotein (oxLDL) in their serum. OxLDL activates the scavenger receptor CD36 on the surface of macrophages, triggering the assembly of the NLRP3 inflammasome and promoting the maturation and release of IL-1β (20) Additionally, the upregulation of fatty acid synthase (FASN) in keratinocytes leads to the accumulation of free fatty acids, which enhances IL-23 secretion via the TLR4/MyD88 pathway, thereby activating the IL-17 signaling axis (21).
3.1.2 PPARγ signaling inhibition
The key lipid metabolism regulator PPARγ’s expression is reduced in psoriasis patients’ epidermis. This reduction leads to decreased synthesis of anti-inflammatory lipid mediators (such as 15d-PGJ2), which fails to inhibit NF-κB-driven transcription of TNF-α (22).
3.1.3 SCD1 and lipotoxic inflammation
The expression of stearoyl-CoA desaturase 1 (SCD1) is significantly upregulated in psoriasis lesions. SCD1 catalyzes the conversion of saturated fatty acids (such as palmitic acid) to monounsaturated fatty acids (such as oleic acid). Accumulation of palmitic acid activates the endoplasmic reticulum stress (ERS) signaling pathway (PERK/eIF2α) in keratinocytes, inducing the expression of IL-23 and IL-17F and exacerbating epidermal hyperproliferation (23).
3.1.4 SREBP Pathway dysregulation
Sterol regulatory element-binding protein (SREBP1c) is abnormally activated in psoriasis, promoting the transcription of fatty acid synthesis-related genes (such as ACC and FASN), leading to excessive lipid droplet deposition. Lipid droplets release arachidonic acid derivatives (such as prostaglandin E2, PGE2), which activate the cAMP-PKA pathway dependent on the EP4 receptor, inhibiting Treg function and promoting Th17 differentiation (24).
3.2 Oxidative stress and inflammatory signaling in a vicious cycle
In psoriatic epidermis, the inhibition of the Nrf2-Keap1 pathway results in reduced nuclear translocation of Nrf2 and downregulation of antioxidant response element (ARE)-driven genes, such as HO-1 and NQO1 (25), leading to decreased reactive oxygen species (ROS) clearance capacity and further amplification of NF-κB and STAT3 signaling. Additionally, mitochondrial ROS cause oxidative damage to mitochondrial DNA (mtDNA), triggering its release into the cytoplasm. This mtDNA activates the cGAS-STING pathway, inducing the production of type I interferons (IFN-α/β) (26), which synergize with IL-17A to promote abnormal keratinocyte proliferation. Furthermore, ROS-induced inhibition of DNA methyltransferase (DNMT) activity leads to hypomethylation of the SOCS3 gene promoter, impairing its negative regulation of the JAK/STAT pathway and exacerbating IL-6/IL-23 signaling (26).
3.3 Environmental triggers and cytokine network interactions
3.3.1 Dysbiosis of the microbiome reconfigures immune-epidermal dialogue
In psoriasis, disruption of the gut-skin axis is characterized by an increased abundance of Prevotella in the gut microbiota and a corresponding reduction in short-chain fatty acids (SCFAs), leading to compromised intestinal barrier integrity. This facilitates the translocation of endotoxins into the bloodstream, activating monocytes via TLR2/4 and promoting IL-23 secretion (27). Meanwhile, the overproliferation of Malassezia spp. can degrade sebum to produce free fatty acids, such as oleic acid, disrupting the epidermal barrier and activating the CARD14 signaling pathway in keratinocytes. This leads to the release of IL-36γ, which recruits IL-17A+ γδ T cells (28). Besides, reduced colonization of Staphylococcus epidermidis weakens its ability to induce Tregs to produce IL-10, indirectly enhancing the pathogenic effects of Th17 cells via IL-17A (29).
3.3.2 Chronic stress exacerbates inflammation via the neuro-immune axis
Chronic stress contributes to the pathogenesis of psoriasis through several neuroimmune pathways. Activation of the sympathetic nerve system stimulates dermal DCs via the β2-adrenergic receptor (β2-AR), promoting IL-23 secretion while inhibiting IL-10 expression in Tregs, thereby creating a Th17-polarized microenvironment (30). Chronic psychological stress dysregulates the hypothalamic-pituitary-adrenal (HPA) axis, reducing the sensitivity of glucocorticoid receptors (GR) and impeding cortisol’s ability to effectively inhibit the production of TNF-α and IL-1β in macrophages (31). In addition, stress-induced overexpression of nerve growth factor (NGF) enhances the excitability of sensory neurons via the TrkA receptor, promoting the release of calcitonin gene-related peptide (CGRP). This directly stimulates mast cell degranulation, releasing TNF-α and IL-33, and amplifying the IL-17/IL-23 axis (31).
4 Cytokines in different types of psoriasis
As mentioned earlier, psoriasis can be divided into these subtypes: guttate psoriasis, pustular psoriasis, plaque psoriasis, erythrodermic psoriasis and psoriatic arthritis (PsA). There are specific differences in the cytokine-driven mechanisms of different psoriasis subtypes: each pathologic subtype corresponds to a characteristic cytokine profile. Their mediated immune signaling networks and tissue damage patterns show significant heterogeneity. This section focuses on the different psoriasis subtypes and their relationship to cytokines.
4.1 Guttate psoriasis
Guttate psoriasis presents as tear-drop erythematous scaly lesions scattered throughout the body and is most common in children and adolescents. Treatment is centered on long-term remission and lesion clearance is achieved through a stepwise regimen. This includes a combination of topical agents (glucocorticoids/retinoids) and oral immunomodulators; narrow-spectrum UVB or PUVA photochemotherapy to modulate aberrant epidermal proliferation; and novel biologics targeting key pathways such as TNF-α/IL-17/IL-23. The clinical pathway emphasizes the selection of individualized regimens based on age, disease duration and extent of skin lesions, combined with multimodal therapy to maintain long-term remission status (32). The disease exhibits genetic susceptibility, and enhanced polymorphisms in genes such as human leukocyte antigen (HLA)-Cw6 may be associated with an increased risk of immune abnormalities. HLA-Cw6 positivity has been linked to guttate psoriasis, while HLA-Cw1 positivity has been associated with erythrodermic psoriasis, pustular psoriasis, and axial psoriatic arthritis (33, 34). Notably, plaque psoriasis and guttate psoriasis exhibit significant differences in T cell phenotype and function. In the skin lesions and peripheral blood of patients with plaque psoriasis, the number of Foxp3+ Tregs is higher than in patients with guttate psoriasis. Conversely, patients with guttate psoriasis have a higher proportion of IL-17+ CD4+ cells compared to those with plaque psoriasis (35). Furthermore, IFN-γ and IL-17 derived from CD4+ T cells play a pivotal role in the pathogenesis of guttate psoriasis (36, 37). Interestingly, in HLA-Cw6+ patients experiencing guttate psoriasis flare-ups associated with pharyngitis, the Th17-associated response is more pronounced, leading to significantly elevated levels of IL-17A, IL-17F, and IL-6, which are involved in Th17 differentiation (38). Additional studies have indicated that IL-9 also plays a crucial role in guttate psoriasis by upregulating IL-17A through its impact on cutaneous lymphocyte-associated antigen (CLA)+ T cell viability, thereby promoting inflammatory responses in guttate psoriasis (34, 39).
4.2 Pustular psoriasis
Pustular psoriasis is a rare subtype of psoriasis marked by sterile neutrophilic pustules, which can be fatal in some cases (40). Traditional therapies include oral retinoids, cyclosporine, and methotrexate, however, all are based on plaque psoriasis experience (41). There are no GPP-specific drugs in the world (especially in Europe and the United States), and some countries have the conditions to use the existing therapies. Anti-TNF/IL-17A drugs (e.g., infliximab, sukinumab) are effective in only some patients, and IL-17 inhibitors combine ease of injection with low side effects (42). IL-36 (IL-1 family member) acts on keratinocytes, immune cells, etc. via an autocrine/paracrine mechanism, and its receptor complex (IL-36R/IL-1RAcP) triggers inflammatory signaling. Imbalance between IL-36 proinflammatory isoforms (α/β/γ) and the antagonist IL-36Ra drives an aberrant inflammatory cascade, inducing the release of proinflammatory factors, neutrophil chemotactic factors and antimicrobial peptide release. The simultaneous activation of dendritic cells and T cells creates a vicious cycle. Dysregulation of this pathway is a central mechanism of GPP pustule formation and tissue damage, and targeting IL-36R blockade is a potential therapeutic direction (42, 43). The IL-1β inhibitor gevokizumab demonstrated preliminary efficacy in 2 cases, suggesting its potential therapeutic value. Focusing on the IL-36/IL-1 pathway, we are promoting the clinical translation of precision biologics to break through the bottleneck of refractory GPP treatment (44).
4.3 Plaque psoriasis
Plaque psoriasis is the most common subtype of psoriasis (11). It is characterized by well-demarcated papular scaly lesions, which appear as round or oval infiltrated plaques covered with silvery-white mica-like scales. It is often accompanied by significant itching, which in severe cases may lead to epidermal cracking and spot bleeding (45). TNF-α, IL-17 and IL-23 constitute the core inflammatory axis in psoriasis. Genetic/environmental factors activate plasmacytoid dendritic cells (pDC), which release TNF-α, IFN-α/γ, and activate mDC, which produce large amounts of IL-12/IL-23 in psoriatic lesions, and accordingly drive the differentiation of Th1/Th17 cells. Th1 cells release TNF-α, and Th17 is the main source of IL-17 that directly stimulates the abnormal proliferation of KC. Activated KCs produce more IL-1β, TNF-α and chemokines, recruiting immune cells to form a self-sustained inflammatory network (18, 46, 47). In addition, it has been claimed that pathogenic memory T cells retain the ability to generate IL-17A for a long period, exacerbating the chronic course of the disease (48). Traditional targets have focused on TNF-α, IL-12, and IL-17/receptor inhibitors. More recently, biologics of IL-23 have emerged (49). There is evidence that drugs targeting the p19 subunit of IL-23 (e.g. Risankizumab) significantly enhance efficacy durability, with excellent 4-year drug survival data (50).
4.4 Erythrodermic psoriasis
Erythrodermic psoriasis is a rare and severe subtype of psoriasis that presents with generalized cutaneous inflammation and immune disorders with high mortality (51). Existing conventional treatments include glucocorticoids and cyclosporine, which are effective in the short term but are prone to relapse and have limited efficacy. Erythrodermic psoriasis shares the TNF/IL-17A inflammatory axis with plaque psoriasis, but there is a unique immunophenotype of Th2/Th17 coactivation in erythrodermic psoriasis. Th17 cell infiltration is predominant in the lesion area and IL-17A is the common core pathway (shared with plaque type). Some studies have shown significant elevation of serum IgE, IL-4, and IL-13, suggesting the involvement of Th2 immune abnormalities in the pathologic process. Biologic anti-TNF (e.g., Etanercept), anti-IL-17A (e.g., Skuticilumab), and anti-IL-12/23 agents (e.g., Ustekinumab) have demonstrated therapeutic potential in studies (52). In addition, Deucravacitinib is the world’s first oral highly selective TYK2 inhibitor, which provides a novel targeted treatment option for severe erythrodermic psoriasis by precisely inhibiting pathogenic cytokine signaling such as IL-12/IL-23 through targeting and regulating TYK2 kinase in the JAK-STAT signaling pathway (53).
4.5 Psoriatic arthritis
PsA is a chronic immune joint inflammatory disease involving the joints, skin, nails and spine and is a common comorbidity of psoriasis. PsA is associated with elevated cardiovascular risk. Approximately 30% of patients with psoriasis progress to PsA (54), and 40% have an inadequate response to conventional therapy (55). Joint inflammation is dominated by the IL-23/IL-17 axis, which is activated by IL-23 to activate Th17 cells that secrete IL-17/IL-22/TNF-α, triggering inflammation, bone destruction, and tissue damage (56). Skin/joint innate immune cells trigger the IL-12/IL-23 pathway, expanding Th1/Th17 cells and perpetuating the inflammatory cycle (55). IL-23/IL-17 pathway-targeted agents (e.g., Ustekinumab, Stavudineumab) are effective in psoriasis, but have relatively limited improvement in PsA (55, 57).
5 The interleukin family
5.1 IL-23
Monoclonal antibodies that specifically suppress IL-23 have been demonstrated to significantly reduce or even completely eradicate skin lesions in psoriasis patients, suggesting that IL-23 are fundamental to the development of psoriatic lesions (58). IL-23, a heterodimeric protein composed of p19 and p40 subunits linked by disulfide bonds, shares structural similarities with IL-12, leading to analogous roles in T-cell-mediated immune responses (59–61). Psoriatic lesions reveal significantly higher levels of IL-23 expression than normal skin, notably in the affected areas’ epidermis and dermis (62). IL-23 is primarily derived from immune cells within the dermis of psoriatic lesions, notably macrophages and bone mDCs, as well as other cellular subsets (63, 64). Thus, further exploration is necessary to elucidate how the function of these cells can be modulated to decrease IL-23 secretion and to discern the consequent effects on the disease evolution of psoriasis.
Th17 cells, a type of T lymphocyte pivotal for secreting inflammatory cytokines like IL-17, IL-22, IL-26, and TNF-α, are primarily activated by IL-23 to trigger inflammatory and immune responses. In patients with psoriasis, a significant elevation in the number of Th17 cells is observed within the skin tissue, and the inflammatory cytokines they secrete can induce the growth and proliferation of KCs, leading to the hyperplasia of the skin’s keratin layer and the formation of scaly lesions (65, 66). Under the influence of IL-23, Th17 cells generate an auto-amplifying feedforward inflammatory response in KCs and increase their number and vitality, accelerating the progression of skin lesions (63, 67–69). IL-23 promotes the infiltration and activation of inflammatory cells, worsening psoriasis, in addition to affecting Th17 cells. IL-23 promotes the infiltration and activation of various inflammatory cells, such as neutrophils, monocytes, macrophages, and DCs. Activated inflammatory cells secrete cytokines such as TNF-α, IL-1β, and IL-6, which enhance inflammatory and immune responses in the skin, leading to tissue damage and pathological changes (69–71).
IL-23 mediates the dysfunction of endothelial cells and KCs in psoriasis patients, accelerating KC proliferation and thereby contributing to disease progression. Psoriatic lesions are characterized by high microvascular permeability and angiogenesis, which are further promoted by IL-23 through the stimulation of endothelial cell proliferation and secretion, as well as neovascularization and vasodilation within the skin tissue, ultimately exacerbating the cutaneous inflammatory response and pathological changes (72). The application of selective VEGF inhibitors has notably improved skin lesions in mice with psoriasis (73). In contrast, introducing VEGF transgenically into mouse skin results in severe inflammatory dermatosis, resembling human psoriasis (74, 75). IL-23 also influences the cytoskeletal structure and intercellular junctions of KCs, inducing hyperproliferation and aberrant differentiation. These cellular alterations lead to stratum corneum thickening and keratinocyte accumulation, forming psoriasis’s characteristic lesions (9, 68, 76). Consequently, research targeting IL-23 has become a focal point in the current drug development strategy for psoriasis. Selective IL-23 inhibitors are increasingly used to treat psoriasis and other immune-mediated diseases. These therapeutics specifically target and inhibit IL-23 activity, thereby reducing Th17 cell differentiation and proliferation, and ultimately ameliorating psoriasis (76, 77). Furthermore, some novel IL-23 inhibitors are currently being investigated and hold promise for more effective outcomes in psoriasis treatment.
5.2 IL-17
Six members of the IL-17 family have been identified thus far, including IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F. Notably, IL-17A shares approximately 55% homology with IL-17F, is usually co-expressed, and possesses similar biological functions (78). IL-17A was traditionally thought to be primarily secreted by Th17 cells. Recent evidence indicates that various cells, such as mast cells, γδ T cells, αβ T cells, and innate lymphoid cells found in psoriasis skin lesions and synovial fluid, can also produce IL-17A (79, 80). IL-17RA acts as a shared receptor for IL-17A, IL-17F, IL-17C, and IL-17E, partnering with another receptor subunit (IL-17RB, IL-17RC, IL-17RD, or IL-17RE) to enable specific signal transduction and initiate diverse inflammatory responses. These reactions involve the recruitment of inflammatory cells and the production of pro-inflammatory cytokines, such as IL-6, IL-8, and TNF-α, which contribute to the development and progression of various inflammatory conditions (81, 82).
Numerous studies conducted over the last ten years have highlighted the significant function of IL-17A in regulating the adaptive as well as innate immune responses, distinguishing it as an imperative cytokine associated with the etiology of psoriasis and PsA (83, 84). Numerous cell types, such as fibroblasts, endothelial cells, and KCs, are triggered by IL-17A. In psoriasis patients, IL-17A levels are notably increased at skin lesions and nearby tissues, correlating positively with lesion severity (85) and KCs are the primary targets of IL-17A (86). Within KCs, IL-17A stimulates the production of various chemotactic factors, including CXCL1 and CXCL8, which recruit neutrophils, and CXCL20, which further attracts Th17 cells, Th22 cells, and mDCs. This process modulates immune cell function, contributing to the incidence and deterioration of psoriasis symptoms (87, 88). IL-17A not only enhances KC proliferation and keratinization but also stimulates immune cells to release inflammatory mediators such as TNF-α, IL-6, and IL-8, thereby amplifying inflammatory responses (84, 86, 89). Moreover, IL-17A principally drives the expression of four key cytokines: IL-36, IL-17C, IL-20, and IL-19, each possessing distinct functions in psoriasis (Figure 3). For instance, IL-36 and IL-17C mainly act synergistically to enhance tissue inflammation (90–92); overexpression of IL-20 can lead to epidermal changes characteristic of psoriasis (93–95); and IL-19 potentiates the effects of IL-17A via a positive feedback mechanism (96, 97). IL-17A enhances cutaneous inflammatory responses by activating several inflammation-related signaling pathways, such as STAT, NF-κB, and MAPK (98–101). The impact of IL-17A on psoriasis is modest by itself, but its significant influence in the disease’s development and progression is enhanced through synergy with TNF-α, IL-22, and IFN-γ (102). In psoriasis, peripheral neutrophils are elevated, recruited by IL-17E and CXCL8, and release IL-17A and extracellular vesicles, stimulating KCs to produce pro-inflammatory factors, enhance migration, and intensify inflammation. Infiltrating neutrophils highly express MMP-9, increasing vascular permeability and facilitating CD4+ T cell infiltration. While neutrophils do not directly express IL-17A, they can accumulate and release it via extracellular traps (103). The IL-23/IL-17 axis activates NF-κB, promotes the production of G-CSF, GM-CSF and chemokines (CXCL1, CXCL2, CXCL5, CXCL8), and facilitates neutrophil recruitment and mobilization (104). Neutrophil-derived IL-17 may be a target for IL-17A inhibitors that block its crosstalk with KCs and reduce inflammatory infiltration (79, 105). CXCL12+ Fibroblasts are located in the reticular dermis, express adipocyte markers, and respond to IL-17 and TNFα. The release of CXCR2 ligand and CXCL12 via the NFKBIZ pathway promotes neutrophil infiltration. Similar cells are highly expressive of neutrophil chemokines in human psoriatic skin and decrease after IL-17-targeted therapy (106).
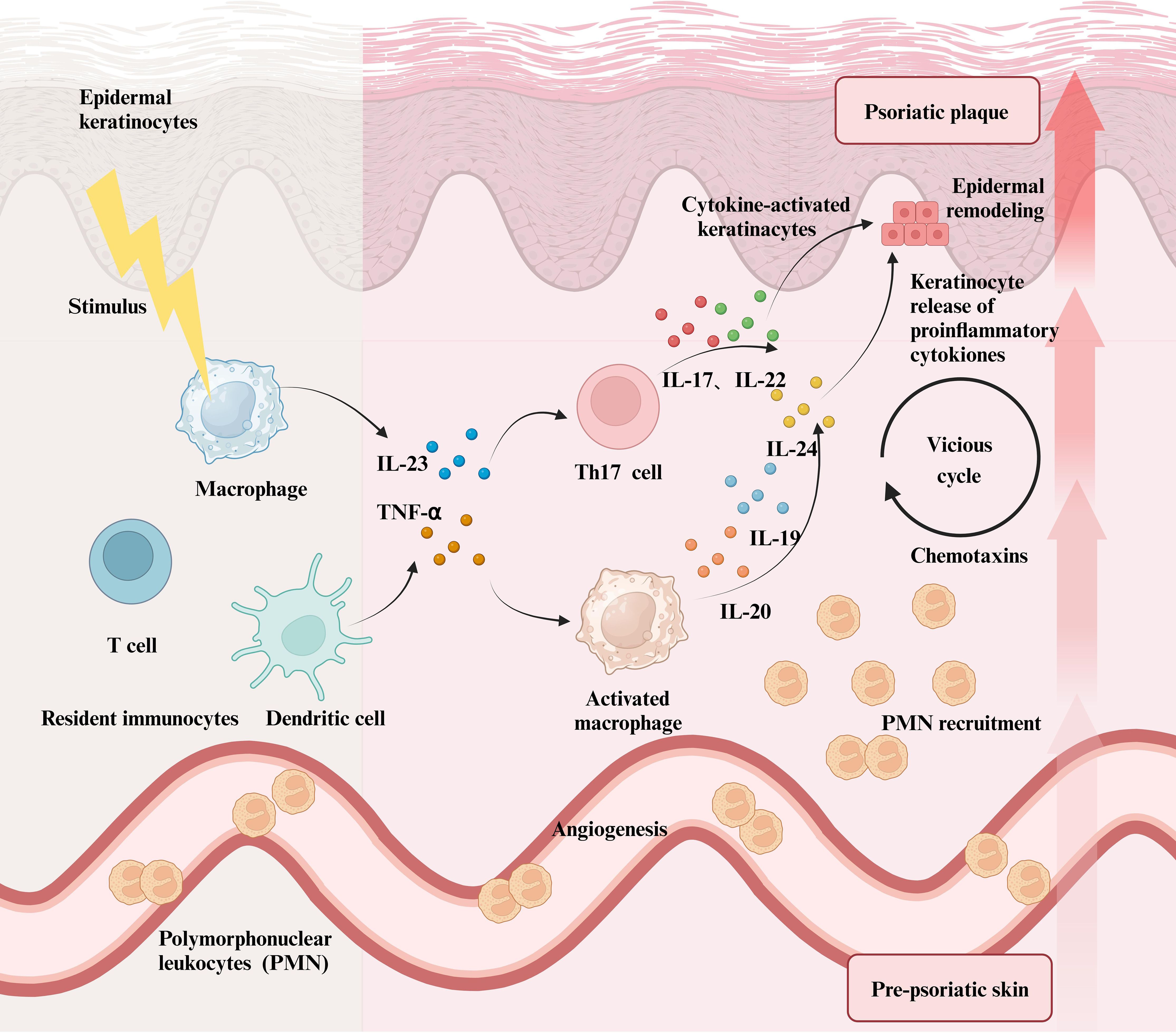
Figure 3. The role of immune cells in Psoriasis and the resulting inflammatory Cascade. When immune cells, including macrophages, DCs, and T cells, in healthy skin are stimulated by exogenous physicochemical factors, they secrete a significant number of inflammatory mediators such as TNF-α and IL-23. This not only activates macrophages, prompting them to release large quantities of inflammatory interleukins but also promotes the differentiation of T cells into Th17 cells, which in turn secrete IL-17, IL-22, and other cytokines. All of these inflammatory factors stimulate keratinocytes, leading to their abnormal proliferation and an inflammatory response in the skin. In psoriasis lesions, polymorphonuclear leukocytes further amplify local inflammatory reactions by releasing various inflammatory chemokines. Ultimately, the symptoms of psoriasis and the level of inflammation intensify, resulting in more severe lesions driven by multiple factors.
Targeting the pivotal role of IL-17 in psoriasis, a variety of drugs that focus on IL-17 are currently under intensive basic and clinical research. Monoclonal antibodies against IL-17A and IL-17RA have been developed and proven effective in alleviating psoriasis symptoms. Furthermore, small molecule inhibitors aimed at the IL-17 signaling pathway are currently being investigated, presenting potential novel therapeutic options for the disease (1, 9, 107). In conclusion, more investigation needs to be conducted to clarify the mechanisms in which IL-17A causes psoriasis, which will contribute to the creation of innovative remedies.
The roles of other IL-17 family members should not be neglected alongside IL-17A. Research suggests IL-17C could be an effective biomarker for systemic anti-psoriasis therapy in moderate to severe psoriasis patients (108). IL-17F is known to contribute to the onset of psoriasis, while other IL-17 family members show a weaker link to disease progression, necessitating further exploration (109). Understanding the various functions of the IL-17 cytokine family is pivotal for clarifying psoriasis pathogenesis and developing new drugs.
5.3 The interleukin-1 superfamily
5.3.1 IL-6
IL-6 plays a crucial part in inflammatory responses, with its biological activity being realized through binding to IL-6R and gp130 (110). IL-6 binding to IL-6R triggers gp130 homodimerization, resulting in the formation of an IL-6/IL-6R/gp130 complex. The complex then activates the Janus kinase (JAK) pathway, which subsequently triggers the STAT3, MAPK, and NF-κB pathways (111, 112). IL-6, a key inflammatory marker, is predominantly released by Th2 and antigen-presenting cells during immunological inflammation (113, 114). RORγt, a transcription factor specific to Th17 cells, plays a vital role in their differentiation and stabilization and IL-6 activates the STAT3 signaling pathway to enhance RORγt expression (115). Fibroblasts in psoriatic lesions release more IL-6 and mediate epidermal hyperplasia in conjunction with cytokines and growth factors such as IL-4 and IL-8 (116). Additionally, through suppressing the differentiation of RORγt in other T cell subsets, like Th 1 and Treg cells, IL-6 indirectly promotes the formation of Th17 cells and facilitates to preserve the relative preponderance of Th17 cells. The interaction between IL-6 and IL-17 in an inflammatory environment creates a positive feedback loop, enhancing the inflammatory response.IL-6 promotes IL-17A secretion, which subsequently triggers the release of inflammatory cytokines like IL-1β and TNF-α, intensifying inflammatory responses (117). Research suggests that IL-1β and TNF-α can promote Th17 cell differentiation facilitated by TGF-β and IL-6 (118). IL-6 enhances IL-23’s effect on Th17 cells, boosting IL-17 production and sustaining Th17 cell activity (119, 120). IL-6 is closely linked with IL-17A/Th17 in immune-mediated inflammation, affecting Th17 cell differentiation and inflammatory response progression through various pathways. This furthers our comprehension of its role in autoimmune disorders like psoriasis.
5.3.2 IL-36
IL-36, a member of the IL-1 family, is expressed and functions in an autocrine or paracrine manner on various cells. Their influence encompasses KCs (121), epithelial cells, and immune cells (122). The IL-36 pathway, which includes IL-36α, IL-36β, IL-36γ, and their antagonists IL-36Ra and IL-38, serves as a crucial regulatory mechanism (123). Under certain conditions, aberrant activation or signaling of IL-36 can result in exorbitant inflammatory responses and neutrophil aggregation (124). Dysregulated expression of components within the IL-36 pathway can potentially initiate a positive feedback loop. For example, the excessive production of IL-36 pathway components may stimulate immune cells to secrete inflammatory cytokines, thereby amplifying the inflammatory response (125–128). This can stimulate the expression of chemokines like CCL13, CCL19, CXCL1, and CXCL12, which direct immune cells, especially neutrophils, to the site of inflammation. Neutrophil influx and chemokine release can damage skin tissue, promoting psoriasis progression. The presence of this positive feedback loop can disrupt the balance of the immune response, thereby impacting skin health. GPP is mainly marked by the abnormal activation of IL-36 (129). Mutations in several genetic loci are associated with GPP, such as Caspase Recruitment Domain 14 (CARD14), Adaptor Protein 1 Complex Sigma 3 Subunit (AP1S3), TNFAIP3 Interacting Protein 1 (TNIP1), and Serpin Family A Member 3 (SERPINA3) (130, 131). These genes are involved in the IL-1 and IL-36 signaling pathways, highlighting the importance of IL-36 in GPP. The IL17D-DDX5-IL-36R regulatory mechanism includes IL-36 and involves IL-17D, a member of the IL-17 family, with DDX5 being a protein-coding gene from the DEAD-box protein family (132, 133) and IL-36R mediates signaling for IL-36α, IL-36β, and IL-36γ (134). In psoriatic lesions, the expression of IL-17D is markedly elevated, promoting IL-36R-mediated signaling by suppressing the expression of DDX5 and further selectively amplifying the inflammatory response mediated by IL-36 (132). In addition, tissue protease S expressed by SFRP2+ fibroblasts activates IL-36G in keratinocytes, further exacerbating inflammation (122). In summary, IL-36 cytokines exert a crucial function in skin immunity, particularly in maintaining the skin barrier and modulating inflammation.
5.3.3 IL-38
IL-38, a member of the IL-1 family, demonstrates anti-inflammatory effects by suppressing pro-inflammatory cytokine secretion (135). It regulates IL-36 via direct and indirect pathways. Direct actions involve suppressing IL-8 expression induced by IL-36 in PBMCs and reducing the pro-inflammatory effects of IL-36γ on KCs (136). Indirectly, IL-38 reduces IL-36γ release by modulating TLR signaling. Secondly, the downregulation of IL-17 and related mediators associated with Th38 differentiation increases the rebound effect of IL-36, meaning that the differentiation of Th38 may reduce the release of IL-36 (137). The X-linked receptor accessory protein-like 1 gene on the X chromosome is believed to mediate IL-38’s inhibitory effects on γδ T cells. Activation of γδT cells upregulates this gene, boosting IL-17 and IL-38 synthesis and indirectly reducing IL-36 release (138).
5.4 Other interleukin members
5.4.1 IL-9
IL-9, a cytokine synthesized by diverse immune cells, like T lymphocytes, B lymphocytes, mast cells, and macrophages, exhibits elevated expression levels in the skin tissue of psoriasis patients (139). This rise suggests a possible link between IL-9 and disease pathogenesis (140, 141). IL-9 is mainly produced by Th9 cells, Th17 cells, Treg, and type 2 innate lymphoid cells (ILC2). IL-9 possesses an essential task in controlling immunological responses, especially when it concerns T cell-mediated immunity (142). Th9 cells are the primary producers of IL-9, independently contributing to allergic inflammation. Th9 cell differentiation is mainly regulated by IL-4/STAT6, IL-2/STAT5, and TGF-β signaling pathways (143). In the advancement of psoriasis, the excessive activation and differentiation of Th17 cells are critical contributors. Through direct or indirect procedures, IL-9 may alter the emergence and activity of Th17 cells, consequently influencing the course of skin inflammation. A potential cause is that IL-9 binds to a receptor complex made up of IL-9RA and the common gamma chain, which engages the MAPK, phosphoinositide 3-kinase (PI3K), and Jak-STAT signaling networks (144). An increased inflammatory response may arise from the transformation and growth of Th17 cells being aided by the activation of multiple pathways of signaling. Additionally, IL-9 may indirectly influence Th17 cell activation by modulating the production of cytokines or signaling molecules from other immune cells. For instance, IL-9 stimulates neighboring cells to produce pro-inflammatory factors like IL-23, thereby augmenting the development and functionality of Th17 cells (145). Excessive production of IL-9 may be linked to the hyperactivation of immune cells and dysregulated inflammatory responses, therefore contributing to the etiology of inflammatory skin conditions, especially psoriasis.
5.4.2 IL-12
Macrophages, DCs, and fibroblasts produce the cytokines IL-12 and IL-23, both part of the IL-12 family (146). Their synthesis is regulated by multiple factors, such as gene expression variations, patterns of TLR expression, and the cross-regulation between different immune cell populations. This process also involves the secretion of other cytokines, like IL-10 and IFN-γ. IL-12, composed of IL-12α and IL-12β subunits, functions by interacting with IL-12Rβ1 and IL-12Rβ2 receptors. This interaction activates JAK2 and TYK2, resulting in the phosphorylation of STAT4 and other STAT family members, ultimately inducing IFN-γ secretion (146). IL-23, consisting of the IL-12β and IL-23α subunits, is elevated in psoriatic lesions compared to healthy skin (147). Although IL-12 and IL-23 are structurally similar, they have different functions. IL-23 interacts with the IL-12Rβ1 and IL-23R complex, primarily activating STAT3. This activation enhances the production of IL-17A, IL-17F, and IL-22, and supports the stabilization of Th17 cells (148, 149).
5.4.3 IL-22
Several immune cell sorts, involving Th17 cells, γδT cells, natural killer T cells (NKT cells), and macrophages, manufacture IL-22, with Th17 cells being the principal source (150). In the dermis of both humans and mice, γδ T cells expressing the T cell marker CD69 have been observed, contributing to the promotion of Th17 cell differentiation (151). Th17 cells produce various cytokines, including IL-17A, IL-17F, and IL-22 (152). IL-22R, the receptor for IL-22, is predominantly expressed on epithelial cells and primarily facilitates the IL-22 signaling cascade (153). Under physiological conditions, IL-22 is essential for preserving the integrity of the epidermal barrier while supporting healing processes. However, in individuals with psoriasis, IL-22 is believed to promote keratinocyte proliferation, leading to enhanced keratinization and the development of psoriatic lesions (94, 154, 155). Neutrophils also secrete IL-22 in psoriatic lesions, and normally IL-22 increases the proliferation of keratin-forming cells (105). The actions of IL-22 are mediated through the transmembrane receptor complex comprising IL-22R1 and IL-10R2, with IL-22R1 likely playing a significant role in the pathogenesis or exacerbation of psoriasis (156). IL-20 and IL-24 are recognized as receptor subunits of IL-22R1 (157, 158). Additionally, cytokines such as IL-17, TNF, and IL-1β can enhance the effects of IL-22. Targeting the interactions between IL-22R1 and cytokines such as IL-20, IL-24, IL-17, TNF, and IL-1β could improve clinical outcomes in psoriasis treatment (159).
5.4.4 TNF-α
TNF is a trimeric cytokine released by immune and epithelial cells during infections or tissue damage, interacting with two specific receptors: TNF receptor 1 (TNFR1) and TNFR2 (160). While TNFR1 is widely distributed across various cell types, TNFR2 exhibits high expression levels primarily in myeloid cells and lower levels in other tissues (161). The expression of TNFR2 can be upregulated by several factors, including IL-33, IFN-γ, IL-1, TL1A, and TNF itself (162). TNF, present in both membrane-bound and soluble forms, can bind to TNFR1 and TNFR2, with the membrane-bound form displaying a stronger affinity for TNFR2 (163). TNF-α, located on chromosome 6, is an important transmembrane protein with pro-inflammatory and immunomodulatory functions. TNF-α signals through two receptors, with TNFR1, also known as p55 or CD120a, being the primary receptor for TNF-α (164). The binding of TNF-α to TNFR1 is essential for regulating inflammatory signaling pathways and apoptosis. TNFR1 activates kinases including IκBα kinase-2 (IKK2), TBK1, and IKKϵ, initiating the NF-κB signaling pathway. This process triggers inflammatory cytokines such as TNF and IL-17, which initiate further inflammatory responses (Figure 4) (165). Unlike TNFR1, TNFR2 (also referred to as p75 or CD120b) has a lower affinity for TNF-α, yet it is capable of binding under specific conditions. Currently, TNF inhibitors primarily function by targeting TNFR1 (166). Five anti-TNF-α medications have been approved: infliximab (Remicade), adalimumab (Humira), golimumab (Simponi), certolizumab pegol (Cimzia), and etanercept (Enbrel) (167). Remicade, Humira, Simponi, and Cimzia manage autoimmune conditions like inflammatory bowel disease and psoriasis by inhibiting TNF-α and TNFR1 interaction, thereby moderating inflammation and modulating immune responses. Unlike other TNF inhibitors, Enbrel has a high affinity for TNFR2. It alleviates inflammation and immune responses by binding to and neutralizing free TNF-α in the body, and it is leveraged for various autoimmune conditions, including PsA and plaque psoriasis.
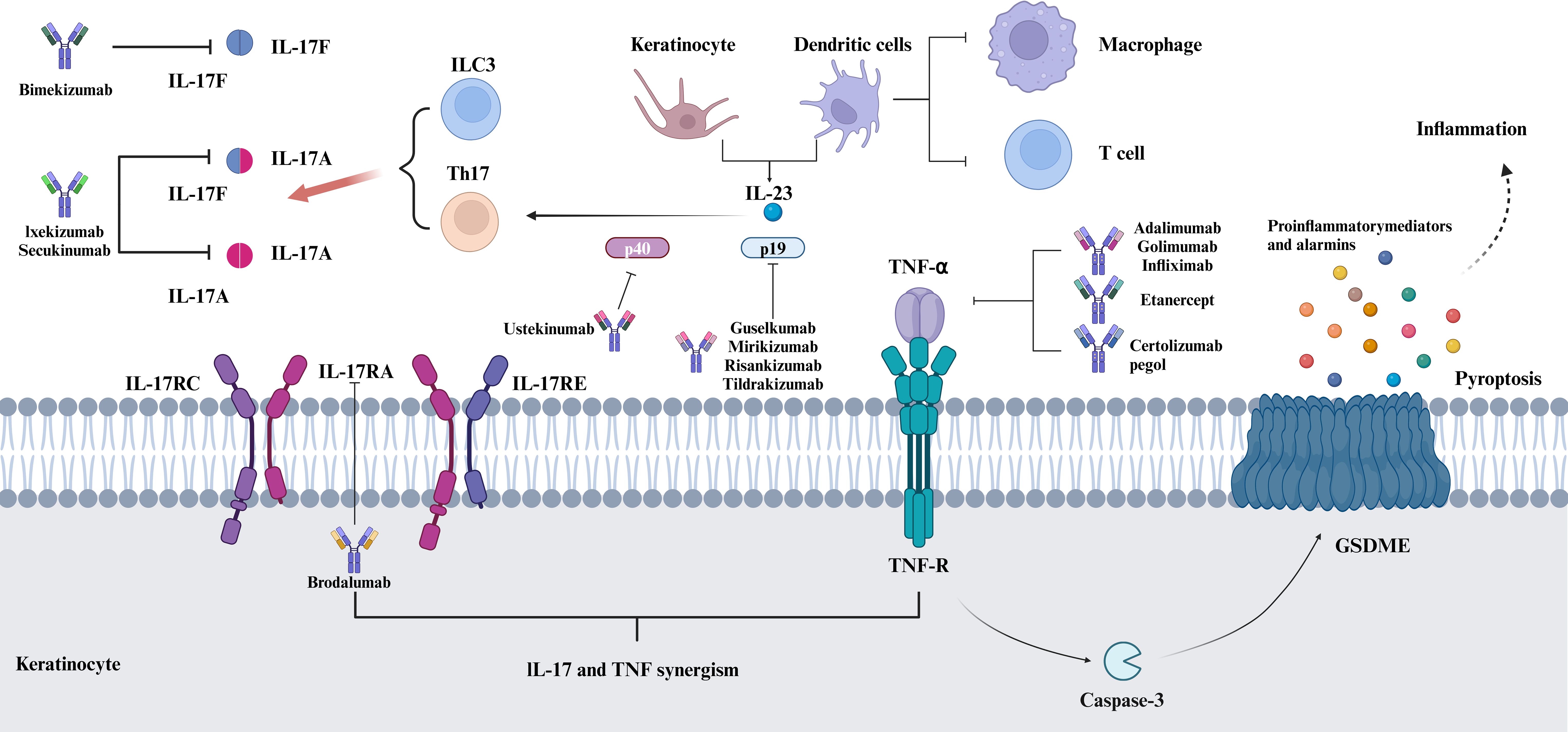
Figure 4. IL-17 and TNF-α: Key Psoriasis inflammation and therapeutic targets. Both IL-17 and TNF-α are key inflammatory mediators in psoriasis, and they are involved in the regulation of skin inflammation and immune responses. Both interact in the pathogenesis of psoriasis and together contribute to the inflammatory response and the development of skin lesions.IL-17 induces keratinocytes and other cell types to produce a variety of inflammatory factors, including TNF-α, which amplify the inflammatory response. Because of their role in psoriasis, IL-17 and TNF-α have become important targets for psoriasis treatment, and by inhibiting the activity of these cytokines, the symptoms of psoriasis can be effectively controlled. Biological agents targeting TNF-α and IL-17 have been widely used in the treatment of psoriasis.
5.4.5 IFN-γ
IFN-γ, an essential cytokine in the interferon family, is mainly produced by diverse immune cells, including T lymphocytes, NK cells, and macrophages. IFN-γ binds to its receptor, activating the JAK-STAT pathway and inducing the expression of responsive genes. This cytokine participates in modulating immune cell function and in initiating immune responses, making it essential for both innate and adaptive immunity. Consequently, dysregulated IFN-γ expression is strongly linked to the pathophysiology of assorted human illnesses (168, 169).
Recent studies have shown elevated levels of IFN-γ in psoriasis, with significant secretion primarily from T cells, including Th1, Th17, and Th22 subsets (63, 170). Moreover, the levels of IFN-γ in psoriatic lesions correlate closely with disease severity (170, 171). Notably, IFN-γ produced by CD4+ T cells has been recognized as a major contributor to the emergence of guttate psoriasis, while IFN-γ from CD8+ T cells appears to be more influential in the immunopathogenesis of plaque psoriasis (36). RNA sequencing and transcriptomic analyses of skin biopsies reveal increased IFN-γ signaling in non-pustular palmoplantar psoriasis, while palmoplantar pustular psoriasis is more associated with the IL-17 pathway and neutrophil-related genes like IL-36A (172). These findings suggest that IFN-γ levels may act as a biomarker for differentiating psoriasis subtypes and highlight its necessary role in the disease’s pathophysiology. Recent studies have demonstrated that IFN-γ can induce significant transcriptomic changes, such as the downregulation of miR-149, which increases KC sensitivity to the inflammatory cytokine TWEAK, thus exacerbating skin inflammation (173). Additionally, IFN-γ has been shown to modulate the activation and function of multiple immune cell populations, including T cells, macrophages, and DCs. For example, IFN-γ facilitates the transformation of human monocytes into inflammatory macrophages (174). In DCs, the inhibition of the STAT1/NF-κB signaling pathway by IFN-γ results in reduced production of pro-inflammatory cytokines and chemokines, including TNF, IL-1β, IFN-γ, and IL-17. This process impairs the development of effector CD8+ T cells, thereby reducing skin thickening and alleviating psoriatic symptoms like scaling and fissuring (175). These findings suggest that the downregulation of the JAK-STAT inflammatory signaling pathway downstream of IFN-γ may improve psoriasis-like lesions (175, 176).In vitro studies have shown that IFN-γ enhances the production of heat shock proteins HSP90α and HSP90β in HaCaT cells. Inhibition of IFN-γ resulted in alterations in the immune cell population and led to marked improvement in psoriatic lesions and symptoms (177). In summary, while the precise mechanisms of IFN-γ in psoriasis remain to be fully elucidated, current evidence underscores its involvement in the inflammatory response, abnormal KCs differentiation, and immune cell activation, revealing a critical regulatory role in the disease’s development and progression. Targeting IFN-γ and its downstream pathways could provide a promising therapeutic approach for future psoriasis management.
6 Other inflammatory factors
6.1 TGF-β
TGF-β exists in various isoforms in humans and participates in regulating physiological and pathological processes. These encompass development, immune system regulation, cell proliferation and differentiation, apoptosis, cell migration, and tissue repair (178). TGF-β dysregulation is linked to the onset and advancement of diseases such as cancer, inflammatory disorders, and fibrosis. It is widely acknowledged that psoriatic lesions exhibit downregulated levels of TGF-β, which typically functions to inhibit KC growth; thus, a decrease in TGF-β concentrations within the skin can facilitate KC uncontrolled proliferation. Researches reveal that Growth Differentiation Factor 11 (GDF11), part of the TGF-β superfamily, eases skin inflammation in a mouse model of psoriasis induced by imiquimod. GDF11 administration significantly improved symptoms like erythema, desquamation, and epidermal thickening, while reducing inflammatory cell infiltration, epithelial hyperplasia, and levels of inflammatory mediators such as IL-1β, TNFα, COX-2, iNOS, and IL-6 (179). Infusion of mouse mesenchymal stromal cells that secrete IL-6 and TGF-β has been illustrated to accelerate the resolution of psoriasis-like skin inflammation, marked by epidermal thinning and reduced CD3+ T cell infiltration. The upregulation of TGF-β and IL-17A in the skin likely inhibits T cell-mediated pathological processes (180). In contrast to previous findings, certain studies suggest a positive correlation between TGF-β levels and psoriasis development. In particular, TGF-β1 is significantly upregulated in psoriatic KCs and promotes the overexpression of keratin 17 and the proliferation of KCs through the TGF-β/SMAD/miR-486-3p signaling axis, thereby promoting the development of psoriasis (181). Similarly, GDF15, part of the TGF-β superfamily, presents decreased expression in the epidermis of psoriasis patients and animal models. It suppresses cytokine and chemokine production by keratinocytes and directly inhibits neutrophil adhesion and migration, spotlighting its anti-inflammatory effects that ameliorate psoriasis (182). In conclusion, TGF-β emerges as a crucial biological regulator that profoundly influences cellular and tissue functionality, alongside the onset and progression of diseases. However, elucidating the precise role of TGF-β in psoriasis, along with the specific mechanisms and regulatory networks involved, necessitates further investigation to achieve a comprehensive understanding.
6.2 CXCLs
Chemokines represent a specific category of cytokines that direct and activate the movement of immune cells and other cell types towards particular chemotactic gradients, thereby playing an essential role in immune system function. Abnormal chemokine expression during disease processes can initiate and exacerbate inflammatory disorders such as psoriasis. This study explores the regulatory roles and mechanisms of chemokines CXCL9, CXCL10, and CXCL11 in psoriasis. Multiple chemokines have higher expression levels within the lesion-prone skin of psoriasis sufferers, including the CXC chemokine family members CXCL9, CXCL10, and CXCL11, with increased expression of CXCL9/10 being characteristic of psoriasis (183). CXCL10 is suggested as a potential biomarker for psoriasis progression, but existing evidence is inadequate to confirm this, necessitating further validation (184). The effects of CXCL9, CXCL10, and CXCL11 are mediated through their specific receptor, CXCR3, with CXCL11 recognized as the primary agonist due to its superior chemotactic potency relative to CXCL10 and CXCL9 (185). CXCR3 is predominantly expressed on monocytes, endothelial cells, fibroblasts, and cancer cells. Activation of CXCR3 has been demonstrated to attract CD4+ T lymphocytes and CD8+ T lymphocytes to inflammatory sites while also facilitating their differentiation into more effective effector T cells (185). The accumulation of immune cells in affected areas can trigger inflammatory responses and subsequent tissue damage. These inflammatory processes, coupled with immune cell activation, further stimulate keratinization, leading to the abnormal growth and differentiation of skin cells that manifest as the characteristic scales and lesions of psoriasis. Endothelial cells, fibroblasts, and KCs release CXCL9, CXCL10, and CXCL11 in response to IFN-γ, with CXCL10 significantly influencing Th1 cell development and function (186, 187). In tissues, recruited Th1 cells can increase IFN-γ and TNF-α secretion, which in turn stimulates CXCL10 production by various cells, creating a feedback loop that sustains autoimmune processes (186, 187). Recent research suggests that estrogen affects the expression of immune cells (DCs and γδT cells), chemokines (CCL5 and CXCL10), and cytokines (TNF-α, IL-23, and IL-17 family), which worsens cutaneous inflammation in psoriasis and provides new insights for developing treatment strategies (188). It should be noted that psoriasis is a multifaceted disease involving multiple cytokines and signaling pathways in its development and progression (Figure 5). Chemokines constitute one facet of this complexity, contributing to the disease’s etiology by attracting and regulating the migration and activation of immune cells, promoting inflammatory responses, and interacting with various factors to collectively influence pathological processes. In-depth investigation of chemokine mechanisms is essential for enhancing our understanding of psoriasis pathogenesis and developing novel treatments.
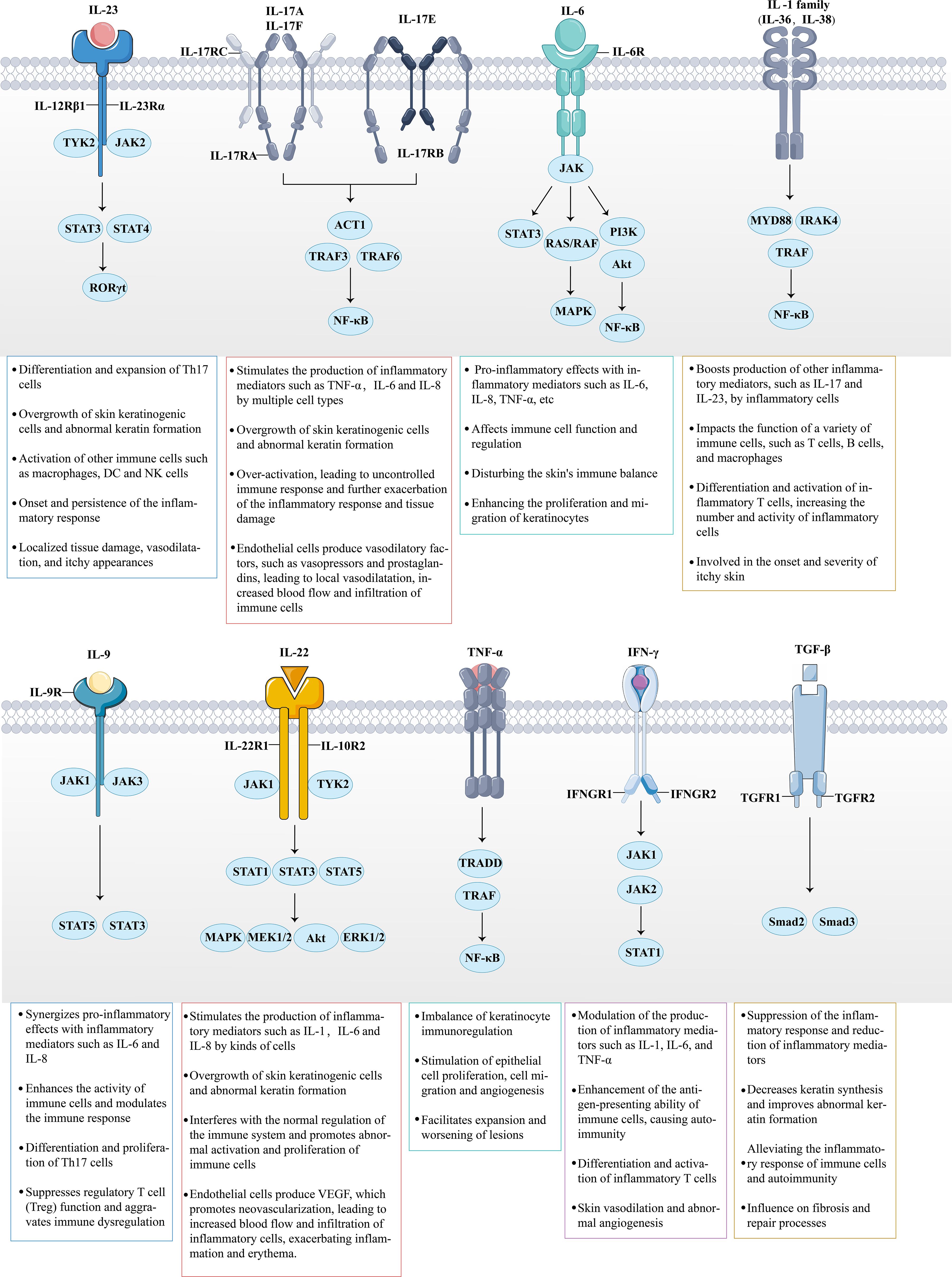
Figure 5. The role of multiple cytokines and signaling pathways in psoriasis is highly significant. Cytokines secreted by immune cells bind to their corresponding receptors. For instance, the massive release of IL-23, IL-6, IL-9, IL-22, and IFN-γ activates the intracellular JAK-STAT signaling pathway, thereby triggering downstream inflammatory responses. Meanwhile, IL-17, members of the IL-1 family, and TNF-α amplify the inflammatory response by activating the NF-κB signaling pathway. Under the combined influence of these inflammatory factors and signaling pathways, the inflammatory response in psoriasis continues to escalate, promoting the involvement of additional inflammatory cells (such as Th17 and DCs). This results in a multidimensional stimulation of keratinocytes, ultimately leading to further deterioration of the psoriasis condition.
7 Drugs targeting cytokines for psoriasis
Psoriasis pathogenesis is regulated by various immune cells and cytokines. As research in this area continues, treatments are becoming more diverse. Conventional therapies for psoriasis employ a wide range of anti-inflammatory and immunosuppressive treatments. These include topical corticosteroids for localized disease, as well as NSAIDs, vitamin D analogues (e.g., calcipotriol), and vitamin A analogues. Phototherapy options, such as broadband UVB, narrowband UVB, PUVA, and tanning, are also utilized. For refractory or extensive psoriasis, systemic therapies like methotrexate (MTX), retinoic acid, and cyclosporine are commonly prescribed (11, 189). However, these treatments have limited efficacy and may cause adverse reactions. They are also not suitable for most patients. UVB treatment reduces the infiltration of p38 pY323+ T cells in psoriatic lesions and suppresses IRF4 and IL-17 production, which may contribute to the therapeutic effects of phototherapy in psoriasis (190). UVB also induces CD95L expression in keratinocytes, leading to intradermal T-cell depletion. Additionally, it influences psoriasis manifestation by promoting vitamin D production and modulating immune responses to local antigens. However, prolonged use of UVB therapy increases the risk of photoaging and skin cancer (191, 192). Although MTX is a first-line treatment for moderate-to-severe psoriasis and PsA, its long-term use is limited by gastrointestinal toxicity, myelosuppression, and hepatic and pulmonary injury. For mild PsA cases, short-term use of nonsteroidal anti-inflammatory drugs may be appropriate, whereas oral glucocorticoids are not recommended (193, 194). Topical corticosteroids are often considered skin sensitizers, and prolonged use may cause adverse effects such as skin atrophy, osteoporosis, and contact dermatitis (195–197). In contrast, biologic therapies represent a new paradigm of targeted treatment modalities that offer enhanced safety, improved effectiveness, and a reduced incidence of side effects. These biologics exert their therapeutic effects by specifically modulating pivotal cytokines implicated in the immunopathogenesis of psoriasis (198). This article aims to systematically summarize and thoroughly review advancements in drug development strategies that target key cytokines associated with psoriasis. Our objective is to provide valuable insights and references to inform future endeavors in the creation of specific therapeutic agents for psoriasis (Table 1).
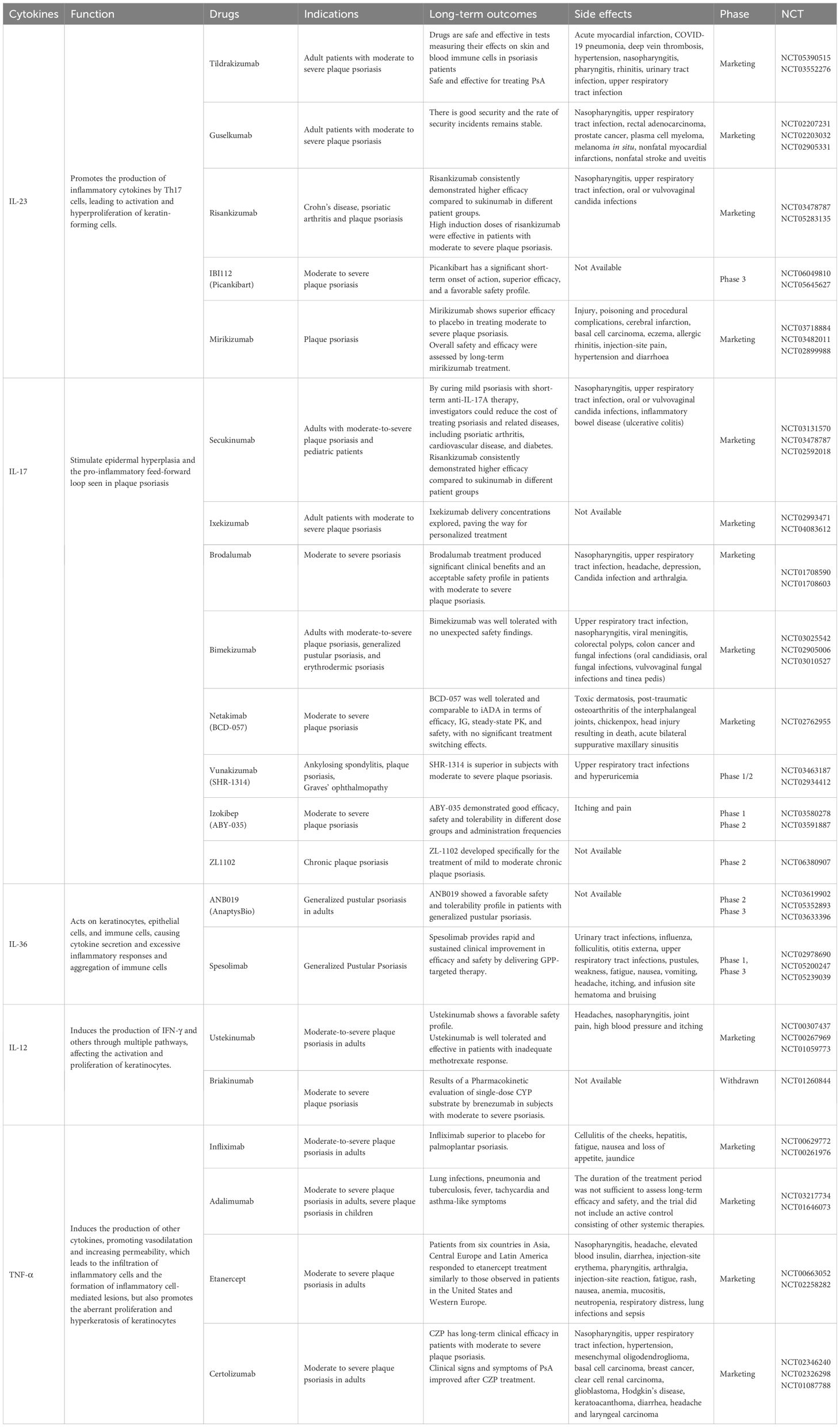
Table 1. Cytokine-based therapeutic drugs and clinical research.
7.1 IL-23 inhibitors
In the pathophysiology of psoriasis, IL-23 performs a crucial upstream regulatory position by encouraging Th17 cell activation, which results in the secretion of a variety of inflammatory cytokines. By activating and proliferating KCs, these types of cytokines collaborate in concert to exacerbate psoriatic skin disorders (60). Notably, the inhibition of IL-23 does not interfere with the normal physiological roles of other cytokines, such as IL-17, derived from different cellular sources, making IL-23 a highly attractive therapeutic target for psoriasis. Approved by the FDA in 2017, Guselkumab is the first biologic agent targeting IL-23 for treating moderate-to-severe plaque psoriasis in adults (199). Due to its enhanced safety and effectiveness, its application has been extended for treating PsA (200). Tildrakizumab, a humanized antibody with high affinity for the IL-23 p19 subunit, received FDA approval in 2018 for treating moderate-to-severe plaque psoriasis in adults. A Phase 3 clinical trial among Chinese patients confirmed its strong efficacy and safety in treating moderate-to-severe plaque psoriasis (201). Approved in 2019, Risankizumab is the third FDA-approved inhibitor of the IL-23 p19 subunit for treating moderate-to-severe plaque psoriasis in adults, demonstrating a favorable safety and tolerability profile (202). Risankizumab, approved in Japan for generalized pustular psoriasis, erythrodermic psoriasis, and PsA, also demonstrates potential for treating autoimmune diseases worldwide, including hidradenitis suppurativa, ulcerative colitis, Crohn’s disease, asthma, atopic dermatitis, and ankylosing spondylitis (203). Mirikizumab, the latest IL-23 inhibitor to be approved by the FDA in October 2023, focuses on inflammatory bowel disease rather than psoriasis in its approved indication. However, clinical evidence has demonstrated the efficacy of Mirikizumab in psoriasis, providing significant relief of psoriatic lesions, thereby underscoring its potential as a prominent treatment option for the condition (204, 205). Beyond the four FDA-approved IL-23 inhibitors, numerous other biologic agents targeting IL-23 are in clinical trials, underlining the potential of IL-23 as a therapeutic target for psoriasis.
7.2 IL-17 inhibitors
The IL-17 family, particularly IL-17A, plays a pivotal role in psoriasis by acting as a pro-inflammatory factor that induces the expression of chemokines and cytokines, recruits neutrophils and monocytes, and drives inflammation (79). The prominent role of the IL-17 pathway in psoriasis has led to blocking IL-17 signaling becoming a vital strategy for treating psoriasis. Presently, drug development for this purpose focuses on two primary strategies: targeting anti-IL-17A and IL-17RA. In 2015, the FDA approved Secukinumab, a fully human monoclonal antibody targeting IL-17A, for adults with moderate-to-severe plaque psoriasis unsuitable for phototherapy or systemic therapy. Secukinumab specifically targets IL-17A epitopes, acts quickly, is well-tolerated with few side effects, and minimizes anti-drug antibody production (206, 207). Similarly, Ixekizumab, another humanized monoclonal antibody targeting IL-17A, received FDA approval in 2016 based on its robust efficacy and safety profile, and has been widely marketed (208). Netakimab, a chimeric IgG1 monoclonal antibody against IL-17A, was developed in Russia and approved in 2019 for moderate-to-severe plaque psoriasis. In 2020, its utilization was extended to ankylosing spondylitis and PsA. Clinical trials are ongoing in the U.S. and China (209). Bimekizumab, a monoclonal antibody that targets IL-17A and IL-17F, received marketing approval in Europe in 2021 and in Japan in 2022 for the treatment of psoriasis and PsA (209). Unlike the four aforementioned drugs, Brodalumab, a humanized IgG2 monoclonal antibody targeting IL-17RA, has predominantly been utilized for the intervention of same indications since its approval in Europe in 2017 (210). Recent multicenter researches have confirmed Brodalumab’s efficacy and safety in both short-term and long-term psoriasis treatment (211). In addition to these five FDA-approved drugs, ongoing research and development efforts are exploring additional IL-17-targeting therapies, such as Vunakizumab, Izokibep (ABY-035), and ZL1102, all of which show promising potential for psoriasis treatment.
7.3 IL-36 inhibitors
Continuous abnormal activation of IL-36R leads to the production of chemokines and cytokines, which in turn encourages the infiltration and activation of different inflammatory cells such as macrophages, neutrophils, and Th17 cells. This activation induces cytokine release, including IL-17, TNF, and IL-22, which in turn enhances IL-36 expression by skin cells and DCs. Consequently, a feedback loop intensifies the inflammatory response, attracting numerous neutrophils and other immune cells to the site. This process leads to the formation of pustular patches and the onset of pustular psoriasis (123, 124). Therefore, the drugs currently undergoing clinical studies primarily target IL-36R. The main examples include Imsidolimab (ANB019) and Spesolimab.
7.4 IL-12 inhibitors
IL-12 and IL-23 are part of the same family, both containing a shared p40 subunit, while IL-12 uniquely includes a p35 subunit. The pathogenesis of psoriasis is influenced by IL-12/23, making it a crucial target for blocking the development of the condition (60). In 2009, the FDA approved Ustekinumab, a monoclonal antibody targeting the IL-12/IL-23 p40 subunit, for treating moderate-to-severe plaque psoriasis in adults (212). In recent years, its indications have expanded to include PsA and ulcerative colitis. Common adverse effects of Ustekinumab include mild nasopharyngitis and headaches; nonetheless, it generally exhibits a favorable safety profile (212).
7.5 TNF-α inhibitors
TNF-α, a key cytokine linking inflammation and immune responses, is released by activated macrophages, NK cells, and T lymphocytes. Its biological functions, including cell proliferation, metabolic activation, and inflammatory response, are substantial in psoriasis pathogenesis (213). The four approved and marketed drugs targeting TNF-α are Infliximab, Adalimumab, Etanercept, and Certolizumab. Their main indications include psoriasis, ankylosing spondylitis, inflammatory bowel disease, and other autoimmune diseases. However, the safety of TNF-α inhibitors requires further improvement. Studies have revealed that some female patients treated with TNF-α monoclonal antibodies for inflammatory bowel disease developed psoriasis, with the underlying mechanism remaining obscure and warranting further investigation (214). The core mechanism of action of the characteristic metabolite polycyclic polypentadienylacylated acyl resorcinols (PPAPs) from Hypericum perforatum includes selective inhibition of the TNF-α signaling pathway, which alleviates psoriasis-associated pathologic epidermal thickening through modulation of the epidermal cell proliferation cycle, and the compounds provide potential candidate molecules for psoriasis treatment through multi-targeted intervention in the TNF-α-associated signaling network (215).
7.6 Biomarkers for predicting response to therapy
There is currently no cure for psoriasis or PsA, however, biologic therapies targeting key inflammatory pathways have demonstrated significant efficacy. Identifying reliable biomarkers is crucial for predicting disease prognosis and treatment response, thereby aiding in the optimization of personalized therapeutic strategies. The primary biomarker categories identified to date include genetic, blood-based, tissue-derived, and transcriptional markers (216). Studies indicate that the response to anti-IL-17 therapy may be influenced by common genetic variants, HLA alleles, polygenic risk scores, and shared genetic susceptibility across multiple diseases (217). The key gene PYCARD, primarily involved in inflammatory pathways, is highly expressed in keratinocytes and significantly upregulated in psoriatic lesions. Studies have demonstrated its crucial role in the development of psoriatic lesions (218). The ABCC1 and ABCG2 SNPs are associated with a favorable response to methotrexate in psoriasis patients, while DHFR SNPs show similar effects in PsA. In PsA patients, the TNF gene promoter -308 G/G genotype exhibits greater responsiveness to anti-TNF therapy. After anti-TNF treatment, Th1 and Th17 cell frequencies decrease in psoriasis patients, reinforcing the critical role of the IL-23/Th17 axis in disease pathology. Additionally, IL-23, IL-23R, and Th17-related cytokines are elevated in psoriatic lesions compared to non-lesional skin. This upregulation is accompanied by increased expression of anti-apoptotic proteins (Bcl-X, Bax, and Bak), which correlate with response to anthracycline and anti-TNF therapy (219). The immune response in psoriasis involves inflammatory mediators that are strongly linked to vascular inflammation and the progression of atherosclerosis. Consequently, atherosclerosis-related biomarkers may also serve as therapeutic indicators for psoriasis (220). Deucravacitinib modulates inflammatory cytokines, including IL-23 and type I interferon, by targeting the Tyk2 signaling pathway. Additionally, it influences biomarkers related to collagen matrix turnover, which may serve as predictors of its therapeutic response in PsA (221). TNF-α is a key therapeutic target for PsA and has shown significant efficacy in clinical trials. Several proteins are crucial in inflammation regulation and immune response, including apolipoproteins, fibrinogen, type II collagen, serum amyloid A, conjugated bead proteins, 14-3-3 proteins, and S100 family proteins, particularly S100A8. Additionally, major drug-binding proteins such as human serum albumin and α-1-acid glycoprotein have been identified as potential predictors of response to anti-TNF-α therapy in PsA. Changes in their levels may be linked to TNF-α neutralization (222). Studies have shown that golimumab regulates acute-phase reactants, inflammatory markers, metabolic factors, and bone remodeling markers. Serum levels of CRP, VEGF, MMP-3, and ICAM-1 are strongly correlated with psoriasis-related scores, and some of these markers have been linked to clinical endpoints of anti-TNF therapy in RA studies. Additionally, IL-6, VEGF, MMP-3, and YKL-40 levels are significantly associated with the response to anti-TNFα therapy in patients with spondyloarthropathies (223).
7.7 Others
In addition to those previously noted, cytokines like IFN-γ, TGF-β, CXCL9, CXCL10, IL-33 and CXCL11 play a significant role in psoriasis pathogenesis. In addition, natural compounds can also effectively regulate the expression of cytokines. For example, natural products (e.g., the flavonoid tetramethoxy lignans/methlut) can inhibit IL-33 production and its signaling with the ST2 receptor; and reduce downstream inflammatory factor release by inhibiting IL-33/Substance P-mediated mast cell activation. It is a preferred lead compound for the development of drugs for the treatment of psoriasis (224). Curcumin inhibits psoriasis inflammation and keratinocyte proliferation through downregulation of pro-inflammatory cytokines and inhibition of IL17/IL23, NF-κB and STAT-3 signaling pathway activities (225, 226). However, their transdermal absorption is limited, compromising efficacy. To improve delivery efficiency, nanoparticles are used as curcumin carriers (216). Common formulations include nanosuspensions, nanoparticles, nanoemulsions, solid lipid particles, nanocapsules, nanospheres and liposomes (227).
It is crucial to acknowledge that, despite the continuous development of various therapeutic agents for psoriasis, the overall non-response rate to TNF-α inhibitors remains significantly high, exceeding 30% (228). Furthermore, approximately 10% to 20% of patients do not respond to biologics targeting IL-17 and IL-23 (206, 229, 230). The reasons for this lack of efficacy are multifactorial and may encompass several key factors: (1) Although recent studies have emphasized the predominant role of the Th17 pathway in mediating inflammation in psoriasis and psoriatic arthritis, other signaling pathways, such as TNF-α and IFN-γ, are also critically important (1). Therefore, reliance on a single IL-17, IL-23 and TNF-α inhibitor may not sufficiently address the diverse critical pathways involved in psoriasis, potentially resulting in suboptimal outcomes in targeted therapy. (2) Additionally, the IL-17 family comprises several members beyond IL-17A, with evidence indicating that IL-17F plays a significant role in the pathogenesis of psoriasis (1). Further research is warranted to explore the potential involvement of other IL-17 family members in similar mechanisms. (3) Moreover, polymorphisms within the IL-23 gene have been identified (231), suggesting that variants in the IL-23R gene may diminish the efficacy of currently available IL-23 inhibitors. (4) Recent studies have also increasingly highlighted a strong correlation between gut microbiota and the immune imbalance of T-cells in psoriasis, offering new insights into the disease’s underlying mechanisms (232–234).
Clinical trials are currently underway for drugs that target cytokines, and it is anticipated that these drugs will address the limitations of existing treatments and exhibit improved therapeutic effects. The introduction of these drugs holds the potential for revolutionary advancements in the future management of psoriasis.
8 Summary and prospects
Over recent decades, significant advancements have been made in understanding the intricate cytokine networks involved in psoriasis. These findings have significantly enhanced our understanding of psoriasis pathophysiology and laid a strong foundation for developing novel therapies, providing renewed hope for patients. The pathogenesis of psoriasis is driven by a complex interplay of cytokines, which collectively modulate immune responses and inflammatory processes through intricate positive and negative feedback loops. In particular, targeted therapeutic strategies aimed at these key cytokines have proven effective in controlling the abnormal immune response in psoriasis. Recognizing the pleiotropic and context-dependent roles of cytokines in psoriasis immunity is vitally indispensable for a comprehensive understanding. To achieve personalized and precise treatment, the development of biomarkers that can predict patients’ responses to specific cytokine-targeted therapies is crucial. This will help doctors tailor the most appropriate treatment plan for each patient, thereby maximizing therapeutic effects. The future of psoriasis therapy is evolving beyond monoclonal antibodies to include small-molecule inhibitors, gene-editing technologies, RNA-based therapies, and nanocarriers (235). Oral inhibitors like JAK-STAT (e.g., deucravacitinib) and PI3K/NF-κB blockers offer convenience and potential for cytokine modulation, though specificity and long-term safety remain challenges. Gene-editing tools such as CRISPR-Cas9 can precisely target cytokine-related genes (e.g., silencing IL-23R), with ex vivo T cell or stem cell editing promising personalized treatments. RNA therapies targeting IL-17C, IL-36γ, or TNF-α can suppress inflammation, boosted by nanoparticle delivery for stability and skin penetration. Engineered Tregs and CAR-T cells aim to restore immune balance by targeting Th17 activity or pathogenic cells. Nanocarriers, like curcumin-loaded particles, enhance topical drug delivery while minimizing systemic exposure. As biomedical research advances, a multimodal strategy integrating these innovations will drive precise, personalized treatments and improve outcomes for psoriasis patients. Furthermore, combining cytokine-targeted therapies with emerging CAR-T therapies and phototherapy may provide a more comprehensive treatment strategy for psoriasis patients. This integrated approach is expected to improve efficacy while reducing adverse effects and enhancing life satisfaction in patients. As biomedical research progresses, the treatment of psoriasis in the future will become more precise and personalized, offering a brighter outlook for patients.
Author contributions
LL: Conceptualization, Methodology, Visualization, Writing – original draft. JLi: Data curation, Software, Visualization, Writing – original draft. JLu: Conceptualization, Software, Validation, Writing – original draft. JW: Data curation, Investigation, Methodology, Writing – review & editing. XZ: Methodology, Software, Validation, Writing – review & editing. TM: Methodology, Visualization, Writing – review & editing. XW: Methodology, Writing – review & editing. QZ: Funding acquisition, Resources, Supervision, Writing – review & editing. ZC: Funding acquisition, Resources, Supervision, Writing – review & editing. ZT: Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82373274), Science and Technology Commission of Shanghai Municipality (22S21902700, 23S11901300, and 23S21900800), and Shanghai Municipal Health Commission (202240370).
Acknowledgments
We thank BioRender (www.biorender.com) for the convenience of drawing figures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AP1S3, Adaptor Protein 1 Complex Sigma 3 Subunit; CARD14, Caspase Recruitment Domain 14; CSFs, Colony-Stimulating Factors; CXCL, Chemokine (C-X-C motif) Ligand; DC, Dendritic cell; GDF11, Growth Differentiation Factor 11; GPP, Generalized Pustular Psoriasis; IFNs, Interferons; IKK2, IκBα Kinase-2; IL, Interleukin; IL-1F, IL-1 Family; IL1RAPL1, X-linked Receptor Accessory Protein-like 1; IL-36RN, IL-36 Receptor Antagonist; ILC2, Innate Lymphoid Cells Type 2; JAK, Janus Kinase; KC, Keratinocyte; LIF, Leukemia Inhibitory Factor; MAPK, Mitogen-activated Protein Kinase; mDC, Mature Dendritic Cells; NF-κB, Nuclear Factor Kappa-B; NKT cells, Natural Killer T cells; PBMCs, Peripheral Blood Mononuclear Cells; PI3K, Phosphoinositide 3-kinase; PsA, Psoriatic Arthritis; RORγt, RAR-related Orphan Receptor Gamma t; SERPINA3, Serpin Family A Member 3; STAT, Signal Transducer and Activator of Transcription; TGF-β, Transforming Growth Factor-β; Th, Helper T Cell; TLR, Toll-like Receptor; TNF-α, Tumor Necrosis Factor-alpha; TNFR1, Tumor Necrosis Factor Receptor 1; TNIP1, TNFAIP3 Interacting Protein 1; TPO, Thrombopoietin; Treg, Regulatory T Cells; VEGF, Vascular Endothelial Growth Factor; γδT cells, Gamma-delta T Cells.
References
1. Griffiths CEM, Armstrong AW, Gudjonsson JE, Barker J. Psoriasis. Lancet. (2021) 397:1301–15. doi: 10.1016/s0140-6736(20)32549-6
2. Hwang JK, Grover C, Iorizzo M, Lebwohl MG, Piraccini BM, Rigopoulos DG, et al. Nail psoriasis and nail lichen planus: Updates on diagnosis and management. J Am Acad Dermatol. (2024) 90:585–96. doi: 10.1016/j.jaad.2023.11.024
3. Mossner R, Wilsmann-Theis D, Oji V, Gkogkolou P, Lohr S, Schulz P, et al. The genetic basis for most patients with pustular skin disease remains elusive. Br J Dermatol. (2018) 178:740–8. doi: 10.1111/bjd.15867
4. Francis L, Capon F, Smith CH, Haniffa M, Mahil SK. Inflammatory memory in psoriasis: From remission to recurrence. J Allergy Clin Immunol. (2024) 154:42–50. doi: 10.1016/j.jaci.2024.05.008
5. Raychaudhuri SK, Maverakis E, Raychaudhuri SP. Diagnosis and classification of psoriasis. Autoimmun Rev. (2014) 13:490–5. doi: 10.1016/j.autrev.2014.01.008
6. Zhou Y, Xu F, Chen XY, Yan BX, Wang ZY, Chen SQ, et al. The epidermal immune microenvironment plays a dominant role in psoriasis development, as revealed by mass cytometry. Cell Mol Immunol. (2022) 19:1400–13. doi: 10.1038/s41423-022-00940-8
7. Yi M, Li T, Niu M, Zhang H, Wu Y, Wu K, et al. Targeting cytokine and chemokine signaling pathways for cancer therapy. Signal Transduct Target Ther. (2024) 9:176. doi: 10.1038/s41392-024-01868-3
8. Cohen C, Mhaidly R, Croizer H, Kieffer Y, Leclere R, Vincent-Salomon A, et al. WNT-dependent interaction between inflammatory fibroblasts and FOLR2+ macrophages promotes fibrosis in chronic kidney disease. Nat Commun. (2024) 15:743. doi: 10.1038/s41467-024-44886-z
9. Ghoreschi K, Balato A, Enerback C, Sabat R. Therapeutics targeting the IL-23 and IL-17 pathway in psoriasis. Lancet. (2021) 397:754–66. doi: 10.1016/S0140-6736(21)00184-7
10. Elnabawi YA, Garshick MS, Tawil M, Barrett TJ, Fisher EA, Lo Sicco K, et al. CCL20 in psoriasis: A potential biomarker of disease severity, inflammation, and impaired vascular health. J Am Acad Dermatol. (2021) 84:913–20. doi: 10.1016/j.jaad.2020.10.094
11. Armstrong AW, Read C. Pathophysiology, clinical presentation, and treatment of psoriasis: A review. Jama. (2020) 323:1945–60. doi: 10.1001/jama.2020.4006
12. Li L, Wu X, Wu J, Zhang X, Miao F, Wang J, et al. Transdermal delivery of Fn14 siRNA using a novel composite ionic liquid for treatment of psoriasis-like skin lesions. J Control Release. (2024) 365:818–32. doi: 10.1016/j.jconrel.2023.12.009
13. Adachi A, Honda T, Egawa G, Kanameishi S, Takimoto R, Miyake T, et al. Estradiol suppresses psoriatic inflammation in mice by regulating neutrophil and macrophage functions. J Allergy Clin Immunol. (2022) 150:909–919 e908. doi: 10.1016/j.jaci.2022.03.028
14. Liu C, Chu D, Kalantar-Zadeh K, George J, Young HA, Liu G, et al. Cytokines: from clinical significance to quantification. Adv Sci (Weinh). (2021) 8:e2004433. doi: 10.1002/advs.202004433
15. Cui A, Huang T, Li S, Ma A, Perez JL, Sander C, et al. Dictionary of immune responses to cytokines at single-cell resolution. Nature. (2024) 625:377–84. doi: 10.1038/s41586-023-06816-9
16. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discovery. (2020) 6:36. doi: 10.1038/s41421-020-0167-x
17. Dong C. Cytokine regulation and function in T cells. Annu Rev Immunol. (2021) 39:51–76. doi: 10.1146/annurev-immunol-061020-053702
18. Singh R, Koppu S, Perche PO, Feldman SR. The cytokine mediated molecular pathophysiology of psoriasis and its clinical implications. Int J Mol Sci. (2021) 22:12793. doi: 10.3390/ijms222312793
19. Guo J, Zhang H, Lin W, Lu L, Su J, Chen X. Signaling pathways and targeted therapies for psoriasis. Signal Transduct Target Ther. (2023) 8:437. doi: 10.1038/s41392-023-01655-6
20. He Y, Liu T. Oxidized low-density lipoprotein regulates macrophage polarization in atherosclerosis. Int Immunopharmacol. (2023) 120:110338. doi: 10.1016/j.intimp.2023.110338
21. Xiao Y, Yang Y, Xiong H, Dong G. The implications of FASN in immune cell biology and related diseases. Cell Death Dis. (2024) 15:88. doi: 10.1038/s41419-024-06463-6
22. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. (2013) 19:557–66. doi: 10.1038/nm.3159
23. Sun Q, Xing X, Wang H, Wan K, Fan R, Liu C, et al. SCD1 is the critical signaling hub to mediate metabolic diseases: Mechanism and the development of its inhibitors. BioMed Pharmacother. (2024) 170:115586. doi: 10.1016/j.biopha.2023.115586
24. Shen S, Shen M, Kuang L, Yang K, Wu S, Liu X, et al. SIRT1/SREBPs-mediated regulation of lipid metabolism. Pharmacol Res. (2024) 199:107037. doi: 10.1016/j.phrs.2023.107037
25. Liu S, Pi J, Zhang Q. Signal amplification in the KEAP1-NRF2-ARE antioxidant response pathway. Redox Biol. (2022) 54:102389. doi: 10.1016/j.redox.2022.102389
26. Zhang G, Wei H, Zhao A, Yan X, Zhang X, Gan J, et al. Mitochondrial DNA leakage: underlying mechanisms and therapeutic implications in neurological disorders. J Neuroinflamm. (2025) 22:34. doi: 10.1186/s12974-025-03363-0
27. Kortekaas Krohn I, Callewaert C, Belasri H, De Pessemier B, Diez Lopez C, Mortz CG, et al. The influence of lifestyle and environmental factors on host resilience through a homeostatic skin microbiota: An EAACI Task Force Report. Allergy. (2024) 79:3269–84. doi: 10.1111/all.16378
28. Bingöl FG, Ağagündüz D, Budán F. Probiotic Bacterium-Derived p40, p75, and HM0539 Proteins as Novel Postbiotics and Gut-Associated Immune System (GAIS) Modulation: Postbiotic-Gut-Health Axis. Microorganisms. (2024) 13:23. doi: 10.3390/microorganisms13010023
29. Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. (2007) 449:819–26. doi: 10.1038/nature06246
30. Zhang H, Wang M, Zhao X, Wang Y, Chen X, Su J, et al. Role of stress in skin diseases: A neuroendocrine-immune interaction view. Brain Behav Immun. (2024) 116:286–302. doi: 10.1016/j.bbi.2023.12.005
31. Ayasse MT, Buddenkotte J, Alam M, Steinhoff M. Role of neuroimmune circuits and pruritus in psoriasis. Exp Dermatol. (2020) 29:414–26. doi: 10.1111/exd.14071
32. Maruani A, Samimi M, Stembridge N, Abdel Hay R, Tavernier E, Hughes C, et al. Non-antistreptococcal interventions for acute guttate psoriasis or an acute guttate flare of chronic psoriasis. Cochrane Database Syst Rev. (2019) 4:Cd011541. doi: 10.1002/14651858.CD011541.pub2
33. Huang YW, Tsai TF. HLA-cw1 and psoriasis. Am J Clin Dermatol. (2021) 22:339–47. doi: 10.1007/s40257-020-00585-1
34. De-Jesús-Gil C, Ruiz-Romeu E, Ferran M, Chiriac A, Deza G, Hóllo P, et al. CLA(+) T cell response to microbes in psoriasis. Front Immunol. (2018) 9:1488. doi: 10.3389/fimmu.2018.01488
35. Yan KX, Fang X, Han L, Zhang ZH, Kang KF, Zheng ZZ, et al. Foxp3+ regulatory T cells and related cytokines differentially expressed in plaque vs. guttate psoriasis vulgaris. Br J Dermatol. (2010) 163:48–56. doi: 10.1111/j.1365-2133.2010.09742.x
36. Yan K, Han L, Deng H, Fang X, Zhang Z, Huang G, et al. The distinct role and regulatory mechanism of IL-17 and IFN-γ in the initiation and development of plaque vs guttate psoriasis. J Dermatol Sci. (2018) 92:106–13. doi: 10.1016/j.jdermsci.2018.07.001
37. De-Jesús-Gil C, Sans-de San Nicolás L, Ruiz-Romeu E, Ferran M, Soria-Martinez L, Chiriac A, et al. Specific igA and CLA(+) T-cell IL-17 response to streptococcus pyogenes in psoriasis. J Invest Dermatol. (2020) 140:1364–1370.e1361. doi: 10.1016/j.jid.2019.12.022
38. Ruiz-Romeu E, Ferran M, Sagristà M, Gómez J, Giménez-Arnau A, Herszenyi K, et al. Streptococcus pyogenes-induced cutaneous lymphocyte antigen-positive T cell-dependent epidermal cell activation triggers TH17 responses in patients with guttate psoriasis. J Allergy Clin Immunol. (2016) 138:491–499.e496. doi: 10.1016/j.jaci.2016.02.008
39. Ruiz-Romeu E, Ferran M, de Jesús-Gil C, García P, Sagristà M, Casanova JM, et al. Microbe-dependent induction of IL-9 by CLA(+) T cells in psoriasis and relationship with IL-17A. J Invest Dermatol. (2018) 138:580–7. doi: 10.1016/j.jid.2017.08.048
40. Armstrong AW, Elston CA, Elewski BE, Ferris LK, Gottlieb AB, Lebwohl MG. Generalized pustular psoriasis: A consensus statement from the National Psoriasis Foundation. J Am Acad Dermatol. (2024) 90:727–30. doi: 10.1016/j.jaad.2023.09.080
41. Prinz JC, Choon SE, Griffiths CEM, Merola JF, Morita A, Ashcroft DM, et al. Prevalence, comorbidities and mortality of generalized pustular psoriasis: A literature review. J Eur Acad Dermatol Venereol. (2023) 37:256–73. doi: 10.1111/jdv.18720
42. Uppala R, Tsoi LC, Harms PW, Wang B, Billi AC, Maverakis E, et al. Autoinflammatory psoriasis”-genetics and biology of pustular psoriasis. Cell Mol Immunol. (2021) 18:307–17. doi: 10.1038/s41423-020-0519-3
43. Marrakchi S, Puig L. Pathophysiology of generalized pustular psoriasis. Am J Clin Dermatol. (2022) 23:13–9. doi: 10.1007/s40257-021-00655-y
44. Mansouri B, Richards L, Menter A. Treatment of two patients with generalized pustular psoriasis with the interleukin-1β inhibitor gevokizumab. Br J Dermatol. (2015) 173:239–41. doi: 10.1111/bjd.13614
45. Armstrong AW, Gooderham M, Warren RB, Papp KA, Strober B, Thaçi D, et al. Treatment of plaque psoriasis with deucravacitinib (POETYK PSO-1 study): a plain language summary. Immunotherapy. (2023) 15:963–73. doi: 10.2217/imt-2023-0061
46. Korman NJ. Management of psoriasis as a systemic disease: what is the evidence? Br J Dermatol. (2020) 182:840–8. doi: 10.1111/bjd.18245
47. Krueger JG, Wharton KA Jr, Schlitt T, Suprun M, Torene RI, Jiang X, et al. IL-17A inhibition by secukinumab induces early clinical, histopathologic, and molecular resolution of psoriasis. J Allergy Clin Immunol. (2019) 144:750–63. doi: 10.1016/j.jaci.2019.04.029
48. Benezeder T, Wolf P. Resolution of plaque-type psoriasis: what is left behind (and reinitiates the disease). Semin Immunopathol. (2019) 41:633–44. doi: 10.1007/s00281-019-00766-z
49. Salimi S, Yamauchi PS, Thakur R, Weinberg JM, Kircik L, Abdelmaksoud A, et al. Interleukin 23p19 inhibitors in chronic plaque psoriasis with focus on mirikizumab: A narrative review. Dermatol Ther. (2020) 33:e13800. doi: 10.1111/dth.13800
50. Gargiulo L, Ibba L, Malagoli P, Balato A, Bardazzi F, Burlando M, et al. Drug survival of IL-12/23, IL-17 and IL-23 inhibitors for moderate-to-severe plaque psoriasis: a retrospective multicenter real-world experience on 5932 treatment courses - IL PSO (Italian landscape psoriasis). Front Immunol. (2023) 14:1341708. doi: 10.3389/fimmu.2023.1341708
51. Song B, Wu C, Liu W, Wang Y, Ning X, Guo L, et al. Integrative serum proteomics analysis reveals distinct immune and cardiovascular profile dysregulation in erythrodermic psoriasis. Br J Dermatol. (2023) 189:769–71. doi: 10.1093/bjd/ljad283
52. Shao S, Wang G, Maverakis E, Gudjonsson JE. Targeted treatment for erythrodermic psoriasis: rationale and recent advances. Drugs. (2020) 80:525–34. doi: 10.1007/s40265-020-01283-2
53. Hoy SM. Deucravacitinib: first approval. Drugs. (2022) 82:1671–9. doi: 10.1007/s40265-022-01796-y
54. Scher JU, Ogdie A, Merola JF, Ritchlin C. Preventing psoriatic arthritis: focusing on patients with psoriasis at increased risk of transition. Nat Rev Rheumatol. (2019) 15:153–66. doi: 10.1038/s41584-019-0175-0
55. Veale DJ, Fearon U. The pathogenesis of psoriatic arthritis. Lancet. (2018) 391:2273–84. doi: 10.1016/s0140-6736(18)30830-4
56. Poddubnyy D, Jadon DR, Van den Bosch F, Mease PJ, Gladman DD. Axial involvement in psoriatic arthritis: An update for rheumatologists. Semin Arthritis Rheum. (2021) 51:880–7. doi: 10.1016/j.semarthrit.2021.06.006
57. Gottlieb AB, Merola JF. Axial psoriatic arthritis: An update for dermatologists. J Am Acad Dermatol. (2021) 84:92–101. doi: 10.1016/j.jaad.2020.05.089
58. Kopp T, Riedl E, Bangert C, Bowman EP, Greisenegger E, Horowitz A, et al. Clinical improvement in psoriasis with specific targeting of interleukin-23. Nature. (2015) 521:222–6. doi: 10.1038/nature14175
59. Tait Wojno ED, Hunter CA, Stumhofer JS. The immunobiology of the interleukin-12 family: room for discovery. Immunity. (2019) 50:851–70. doi: 10.1016/j.immuni.2019.03.011
60. Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. (2015) 21:719–29. doi: 10.1038/nm.3895
61. Chyuan IT, Lai JH. New insights into the IL-12 and IL-23: From a molecular basis to clinical application in immune-mediated inflammation and cancers. Biochem Pharmacol. (2020) 175:113928. doi: 10.1016/j.bcp.2020.113928
62. Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. (2004) 199:125–30. doi: 10.1084/jem.20030451
63. Lowes MA, Suarez-Farinas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. (2014) 32:227–55. doi: 10.1146/annurev-immunol-032713-120225
64. Diani M, Altomare G, Reali E. T cell responses in psoriasis and psoriatic arthritis. Autoimmun Rev. (2015) 14:286–92. doi: 10.1016/j.autrev.2014.11.012
65. Yang J, Sundrud MS, Skepner J, Yamagata T. Targeting Th17 cells in autoimmune diseases. Trends Pharmacol Sci. (2014) 35:493–500. doi: 10.1016/j.tips.2014.07.006
66. Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discovery. (2012) 11:763–76. doi: 10.1038/nrd3794
67. Hu P, Wang M, Gao H, Zheng A, Li J, Mu D, et al. The role of helper T cells in psoriasis. Front Immunol. (2021) 12:788940. doi: 10.3389/fimmu.2021.788940
68. Lowes MA, Russell CB, Martin DA, Towne JE, Krueger JG. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol. (2013) 34:174–81. doi: 10.1016/j.it.2012.11.005
69. Hawkes JE, Yan BY, Chan TC, Krueger JG. Discovery of the IL-23/IL-17 signaling pathway and the treatment of psoriasis. J Immunol. (2018) 201:1605–13. doi: 10.4049/jimmunol.1800013
70. Singh TP, Zhang HH, Borek I, Wolf P, Hedrick MN, Singh SP, et al. Monocyte-derived inflammatory Langerhans cells and dermal dendritic cells mediate psoriasis-like inflammation. Nat Commun. (2016) 7:13581. doi: 10.1038/ncomms13581
71. Wang Y, Edelmayer R, Wetter J, Salte K, Gauvin D, Leys L, et al. Monocytes/Macrophages play a pathogenic role in IL-23 mediated psoriasis-like skin inflammation. Sci Rep. (2019) 9:5310. doi: 10.1038/s41598-019-41655-7
72. Detmar M, Brown LF, Claffey KP, Yeo KT, Kocher O, Jackman RW, et al. Overexpression of vascular permeability factor/vascular endothelial growth factor and its receptors in psoriasis. J Exp Med. (1994) 180:1141–6. doi: 10.1084/jem.180.3.1141
73. Schonthaler HB, Huggenberger R, Wculek SK, Detmar M, Wagner EF. Systemic anti-VEGF treatment strongly reduces skin inflammation in a mouse model of psoriasis. Proc Natl Acad Sci U.S.A. (2009) 106:21264–9. doi: 10.1073/pnas.0907550106
74. Xia YP, Li B, Hylton D, Detmar M, Yancopoulos GD, Rudge JS. Transgenic delivery of VEGF to mouse skin leads to an inflammatory condition resembling human psoriasis. Blood. (2003) 102:161–8. doi: 10.1182/blood-2002-12-3793
75. Canavese M, Altruda F, Ruzicka T, Schauber J. Vascular endothelial growth factor (VEGF) in the pathogenesis of psoriasis–a possible target for novel therapies? J Dermatol Sci. (2010) 58:171–6. doi: 10.1016/j.jdermsci.2010.03.023
76. Gupta RK, Gracias DT, Figueroa DS, Miki H, Miller J, Fung K, et al. TWEAK functions with TNF and IL-17 on keratinocytes and is a potential target for psoriasis therapy. Sci Immunol. (2021) 6:eabi8823. doi: 10.1126/sciimmunol.abi8823
77. Zhou L, Wang Y, Wan Q, Wu F, Barbon J, Dunstan R, et al. A non-clinical comparative study of IL-23 antibodies in psoriasis. MAbs. (2021) 13:1964420. doi: 10.1080/19420862.2021.1964420
78. Brembilla NC, Senra L, Boehncke WH. The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Front Immunol. (2018) 9:1682. doi: 10.3389/fimmu.2018.01682
79. Blauvelt A, Chiricozzi A. The immunologic role of IL-17 in psoriasis and psoriatic arthritis pathogenesis. Clin Rev Allergy Immunol. (2018) 55:379–90. doi: 10.1007/s12016-018-8702-3
80. Matos TR, O'Malley JT, Lowry EL, Hamm D, Kirsch IR, Robins HS, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing alphabeta T cell clones. J Clin Invest. (2017) 127:4031–41. doi: 10.1172/JCI93396
81. Monin L, Gaffen SL. Interleukin 17 family cytokines: signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harb Perspect Biol. (2018) 10:a028522. doi: 10.1101/cshperspect.a028522
82. Raychaudhuri SP. Role of IL-17 in psoriasis and psoriatic arthritis. Clin Rev Allergy Immunol. (2013) 44:183–93. doi: 10.1007/s12016-012-8307-1
83. Canavan TN, Elmets CA, Cantrell WL, Evans JM, Elewski BE. Anti-IL-17 medications used in the treatment of plaque psoriasis and psoriatic arthritis: A comprehensive review. Am J Clin Dermatol. (2016) 17:33–47. doi: 10.1007/s40257-015-0162-4
84. Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol. (2017) 140:645–53. doi: 10.1016/j.jaci.2017.07.004
85. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. (2009) 361:496–509. doi: 10.1056/NEJMra0804595
86. Moos S, Mohebiany AN, Waisman A, Kurschus FC. Imiquimod-induced psoriasis in mice depends on the IL-17 signaling of keratinocytes. J Invest Dermatol. (2019) 139:1110–7. doi: 10.1016/j.jid.2019.01.006
87. Harper EG, Guo C, Rizzo H, Lillis JV, Kurtz SE, Skorcheva I, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol. (2009) 129:2175–83. doi: 10.1038/jid.2009.65
88. Furue M, Furue K, Tsuji G, Nakahara T. Interleukin-17A and keratinocytes in psoriasis. Int J Mol Sci. (2020) 21:1275. doi: 10.3390/ijms21041275
89. Ekman AK, Bivik Eding C, Rundquist I, Enerback C. IL-17 and IL-22 promote keratinocyte stemness in the germinative compartment in psoriasis. J Invest Dermatol. (2019) 139:1564–1573 e1568. doi: 10.1016/j.jid.2019.01.014
90. Mahil SK, Catapano M, Di Meglio P, Dand N, Ahlfors H, Carr IM, et al. An analysis of IL-36 signature genes and individuals with IL1RL2 knockout mutations validates IL-36 as a psoriasis therapeutic target. Sci Transl Med. (2017) 9:eaan2514. doi: 10.1126/scitranslmed.aan2514
91. Johnston A, Fritz Y, Dawes SM, Diaconu D, Al-Attar PM, Guzman AM, et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol. (2013) 190:2252–62. doi: 10.4049/jimmunol.1201505
92. Bridgewood C, Fearnley GW, Berekmeri A, Laws P, Macleod T, Ponnambalam S, et al. IL-36gamma is a strong inducer of IL-23 in psoriatic cells and activates angiogenesis. Front Immunol. (2018) 9:200. doi: 10.3389/fimmu.2018.00200
93. Blumberg H, Conklin D, Xu WF, Grossmann A, Brender T, Carollo S, et al. Interleukin 20: discovery, receptor identification, and role in epidermal function. Cell. (2001) 104:9–19. doi: 10.1016/s0092-8674(01)00187-8
94. Wolk K, Haugen HS, Xu W, Witte E, Waggie K, Anderson M, et al. IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-gamma are not. J Mol Med (Berl). (2009) 87:523–36. doi: 10.1007/s00109-009-0457-0
95. Sabat R, Wolk K. Research in practice: IL-22 and IL-20: significance for epithelial homeostasis and psoriasis pathogenesis. J Dtsch Dermatol Ges. (2011) 9:518–23. doi: 10.1111/j.1610-0387.2011.07611.x
96. Witte E, Kokolakis G, Witte K, Philipp S, Doecke WD, Babel N, et al. IL-19 is a component of the pathogenetic IL-23/IL-17 cascade in psoriasis. J Invest Dermatol. (2014) 134:2757–67. doi: 10.1038/jid.2014.308
97. Xu X, Prens E, Florencia E, Leenen P, Boon L, Asmawidjaja P, et al. Interleukin-17A drives IL-19 and IL-24 expression in skin stromal cells regulating keratinocyte proliferation. Front Immunol. (2021) 12:719562. doi: 10.3389/fimmu.2021.719562
98. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. (2009) 9:556–67. doi: 10.1038/nri2586
99. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. (2019) 50:892–906. doi: 10.1016/j.immuni.2019.03.021
100. Tseng JC, Chang YC, Huang CM, Hsu LC, Chuang TH. Therapeutic development based on the immunopathogenic mechanisms of psoriasis. Pharmaceutics. (2021) 13:1064. doi: 10.3390/pharmaceutics13071064
101. Johansen C, Mose M, Ommen P, Bertelsen T, Vinter H, Hailfinger S, et al. IkappaBzeta is a key driver in the development of psoriasis. Proc Natl Acad Sci U.S.A. (2015) 112:E5825–5833. doi: 10.1073/pnas.1509971112
102. Sabat R, Wolk K, Loyal L, Docke WD, Ghoreschi K. T cell pathology in skin inflammation. Semin Immunopathol. (2019) 41:359–77. doi: 10.1007/s00281-019-00742-7
103. Sieminska I, Pieniawska M, Grzywa TM. The immunology of psoriasis-current concepts in pathogenesis. Clin Rev Allergy Immunol. (2024) 66:164–91. doi: 10.1007/s12016-024-08991-7
104. Suzuki E, Mellins ED, Gershwin ME, Nestle FO, Adamopoulos IE. The IL-23/IL-17 axis in psoriatic arthritis. Autoimmun Rev. (2014) 13:496–502. doi: 10.1016/j.autrev.2014.01.050
105. Dyring-Andersen B, Honoré TV, Madelung A, Bzorek M, Simonsen S, Clemmensen SN, et al. Interleukin (IL)-17A and IL-22-producing neutrophils in psoriatic skin. Br J Dermatol. (2017) 177:e321–2. doi: 10.1111/bjd.15533
106. Cavagnero KJ, Li F, Dokoshi T, Nakatsuji T, O'Neill AM, Aguilera C, et al. CXCL12+ dermal fibroblasts promote neutrophil recruitment and host defense by recognition of IL-17. J Exp Med. (2024) 221:e20231425. doi: 10.1084/jem.20231425
107. Boehncke WH, Schon MP. Psoriasis. Lancet. (2015) 386:983–94. doi: 10.1016/S0140-6736(14)61909-7
108. Wang X, Kaiser H, Kvist-Hansen A, McCauley BD, Skov L, Hansen PR, et al. IL-17 pathway members as potential biomarkers of effective systemic treatment and cardiovascular disease in patients with moderate-to-severe psoriasis. Int J Mol Sci. (2022) 23:555. doi: 10.3390/ijms23010555
109. Tollenaere MAX, Hebsgaard J, Ewald DA, Lovato P, Garcet S, Li X, et al. Signalling of multiple interleukin (IL)-17 family cytokines via IL-17 receptor A drives psoriasis-related inflammatory pathways. Br J Dermatol. (2021) 185:585–94. doi: 10.1111/bjd.20090
110. Rose-John S, Jenkins BJ, Garbers C, Moll JM, Scheller J. Targeting IL-6 trans-signalling: past, present and future prospects. Nat Rev Immunol. (2023) 23:666–81. doi: 10.1038/s41577-023-00856-y
111. Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci (Lond). (2012) 122:143–59. doi: 10.1042/CS20110340
112. Kang S, Narazaki M, Metwally H, Kishimoto T. Historical overview of the interleukin-6 family cytokine. J Exp Med. (2020) 217:e20190347. doi: 10.1084/jem.20190347
113. Blauvelt A. IL-6 differs from TNF-alpha: unpredicted clinical effects caused by IL-6 blockade in psoriasis. J Invest Dermatol. (2017) 137:541–2. doi: 10.1016/j.jid.2016.11.022
114. Chen Y, Miao X, Xiang Y, Kuai L, Ding X, Ma T, et al. Qinzhu Liangxue inhibits IL-6-induced hyperproliferation and inflammation in HaCaT cells by regulating METTL14/SOCS3/STAT3 axis. J Ethnopharmacol. (2023) 317:116809. doi: 10.1016/j.jep.2023.116809
115. Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. (2012) 181:8–18. doi: 10.1016/j.ajpath.2012.03.044
116. Barygina V, Becatti M, Prignano F, Lotti T, Taddei N, Fiorillo C. Fibroblasts to keratinocytes redox signaling: the possible role of ROS in psoriatic plaque formation. Antioxid (Basel). (2019) 8:566. doi: 10.3390/antiox8110566
117. Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. (2008) 454:350–2. doi: 10.1038/nature07021
118. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. (2006) 24:179–89. doi: 10.1016/j.immuni.2006.01.001
119. McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. (2009) 10:314–24. doi: 10.1038/ni.1698
120. Veldhoen M, Hocking RJ, Flavell RA, Stockinger B. Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat Immunol. (2006) 7:1151–6. doi: 10.1038/ni1391
121. Goldstein JD, Bassoy EY, Caruso A, Palomo J, Rodriguez E, Lemeille S, et al. IL-36 signaling in keratinocytes controls early IL-23 production in psoriasis-like dermatitis. Life Sci Alliance. (2020) 3:e202000688. doi: 10.26508/lsa.202000688
122. Ma F, Plazyo O, Billi AC, Tsoi LC, Xing X, Wasikowski R, et al. Single cell and spatial sequencing define processes by which keratinocytes and fibroblasts amplify inflammatory responses in psoriasis. Nat Commun. (2023) 14:3455. doi: 10.1038/s41467-023-39020-4
123. Madonna S, Girolomoni G, Dinarello CA, Albanesi C. The significance of IL-36 hyperactivation and IL-36R targeting in psoriasis. Int J Mol Sci. (2019) 20:3318. doi: 10.3390/ijms20133318
124. Iznardo H, Puig L. Exploring the role of IL-36 cytokines as a new target in psoriatic disease. Int J Mol Sci. (2021) 22:4344. doi: 10.3390/ijms22094344
125. Hojen JF, Kristensen MLV, McKee AS, Wade MT, Azam T, Lunding LP, et al. IL-1R3 blockade broadly attenuates the functions of six members of the IL-1 family, revealing their contribution to models of disease. Nat Immunol. (2019) 20:1138–49. doi: 10.1038/s41590-019-0467-1
126. Hu X, Qi C, Feng F, Wang Y, Di T, Meng Y, et al. Combining network pharmacology, RNA-seq, and metabolomics strategies to reveal the mechanism of Cimicifugae Rhizoma - Smilax glabra Roxb herb pair for the treatment of psoriasis. Phytomedicine. (2022) 105:154384. doi: 10.1016/j.phymed.2022.154384
127. Frey S, Derer A, Messbacher ME, Baeten DL, Bugatti S, Montecucco C, et al. The novel cytokine interleukin-36alpha is expressed in psoriatic and rheumatoid arthritis synovium. Ann Rheum Dis. (2013) 72:1569–74. doi: 10.1136/annrheumdis-2012-202264
128. Wolk K, Brembach TC, Simaite D, Bartnik E, Cucinotta S, Pokrywka A, et al. Activity and components of the granulocyte colony-stimulating factor pathway in hidradenitis suppurativa. Br J Dermatol. (2021) 185:164–76. doi: 10.1111/bjd.19795
129. Elewski BE, Lebwohl MG, Anadkat MJ, Barker J, Ghoreschi K, Imafuku S, et al. Rapid and sustained improvements in Generalized Pustular Psoriasis Physician Global Assessment scores with spesolimab for treatment of generalized pustular psoriasis flares in the randomized, placebo-controlled Effisayil 1 study. J Am Acad Dermatol. (2023) 89:36–44. doi: 10.1016/j.jaad.2023.02.040
130. Arakawa A, Vollmer S, Besgen P, Galinski A, Summer B, Kawakami Y, et al. Unopposed IL-36 activity promotes clonal CD4(+) T-cell responses with IL-17A production in generalized pustular psoriasis. J Invest Dermatol. (2018) 138:1338–47. doi: 10.1016/j.jid.2017.12.024
131. Akiyama M. Pustular psoriasis as an autoinflammatory keratinization disease (AiKD): Genetic predisposing factors and promising therapeutic targets. J Dermatol Sci. (2022) 105:11–7. doi: 10.1016/j.jdermsci.2021.11.009
132. Ni X, Xu Y, Wang W, Kong B, Ouyang J, Chen J, et al. IL-17D-induced inhibition of DDX5 expression in keratinocytes amplifies IL-36R-mediated skin inflammation. Nat Immunol. (2022) 23:1577–87. doi: 10.1038/s41590-022-01339-3
133. Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. (2003) 14:155–74. doi: 10.1016/s1359-6101(03)00002-9
134. Hovhannisyan Z, Liu N, Khalil-Aguero S, Panea C, VanValkenburgh J, Zhang R, et al. Enhanced IL-36R signaling promotes barrier impairment and inflammation in skin and intestine. Sci Immunol. (2020) 5:eaax1686. doi: 10.1126/sciimmunol.aax1686
135. Han MM, Yuan XR, Shi X, Zhu XY, Su Y, Xiong DK, et al. The pathological mechanism and potential application of IL-38 in autoimmune diseases. Front Pharmacol. (2021) 12:732790. doi: 10.3389/fphar.2021.732790
136. Boutet MA, Nerviani A, Pitzalis C. IL-36, IL-37, and IL-38 cytokines in skin and joint inflammation: A comprehensive review of their therapeutic potential. Int J Mol Sci. (2019) 20:1257. doi: 10.3390/ijms20061257
137. Puig L, Costanzo A, Munoz-Elias EJ, Jazra M, Wegner S, Paul CF, et al. The biological basis of disease recurrence in psoriasis: a historical perspective and current models. Br J Dermatol. (2022) 186:773–81. doi: 10.1111/bjd.20963
138. Garraud T, Harel M, Boutet MA, Le Goff B, Blanchard F. The enigmatic role of IL-38 in inflammatory diseases. Cytokine Growth Factor Rev. (2018) 39:26–35. doi: 10.1016/j.cytogfr.2018.01.001
139. Perumal NB, Kaplan MH. Regulating Il9 transcription in T helper cells. Trends Immunol. (2011) 32:146–50. doi: 10.1016/j.it.2011.01.006
140. Midde HS, Priyadarssini M, Rajappa M, Munisamy M, Raj Mohan PS, Singh S, et al. Interleukin-9 serves as a key link between systemic inflammation and angiogenesis in psoriasis. Clin Exp Dermatol. (2021) 46:50–7. doi: 10.1111/ced.14335
141. Ciccia F, Guggino G, Ferrante A, Raimondo S, Bignone R, Rodolico V, et al. Interleukin-9 overexpression and th9 polarization characterize the inflamed gut, the synovial tissue, and the peripheral blood of patients with psoriatic arthritis. Arthritis Rheumatol. (2016) 68:1922–31. doi: 10.1002/art.39649
142. Angkasekwinai P, Dong C. IL-9-producing T cells: potential players in allergy and cancer. Nat Rev Immunol. (2021) 21:37–48. doi: 10.1038/s41577-020-0396-0
143. Kaplan MH, Hufford MM, Olson MR. The development and in vivo function of T helper 9 cells. Nat Rev Immunol. (2015) 15:295–307. doi: 10.1038/nri3824
144. Kamiya S, Ikegami I, Yanagi M, Takaki H, Kamekura R, Sato T, et al. Functional interplay between IL-9 and peptide YY contributes to chronic skin inflammation. J Invest Dermatol. (2022) 142:3222–3231 e3225. doi: 10.1016/j.jid.2022.06.021
145. Feng B, Pan B, Huang J, Du Y, Wang X, Wu J, et al. PDE4D/cAMP/IL-23 axis determines the immunotherapy efficacy of lung adenocarcinoma via activating the IL-9 autocrine loop of cytotoxic T lymphocytes. Cancer Lett. (2023) 565:216224. doi: 10.1016/j.canlet.2023.216224
146. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. (2003) 3:133–46. doi: 10.1038/nri1001
147. Jin J, Xie X, Xiao Y, Hu H, Zou Q, Cheng X, et al. Epigenetic regulation of the expression of Il12 and Il23 and autoimmune inflammation by the deubiquitinase Trabid. Nat Immunol. (2016) 17:259–68. doi: 10.1038/ni.3347
148. Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. (2002) 168:5699–708. doi: 10.4049/jimmunol.168.11.5699
149. Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O'Shea JJ. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev. (2004) 202:139–56. doi: 10.1111/j.0105-2896.2004.00211.x
150. Cibrian D, Saiz ML, Fuente H, Sanchez-Diaz R, Moreno-Gonzalo O, Jorge I, et al. Erratum: CD69 controls the uptake of L-tryptophan through LAT1-CD98 and AhR-dependent secretion of IL-22 in psoriasis. Nat Immunol. (2016) 17:1235. doi: 10.1038/ni1016-1235c
151. Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suarez-Farinas M, Cardinale I, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. (2008) 159:1092–102. doi: 10.1111/j.1365-2133.2008.08769.x
152. Rutz S, Noubade R, Eidenschenk C, Ota N, Zeng W, Zheng Y, et al. Transcription factor c-Maf mediates the TGF-beta-dependent suppression of IL-22 production in T(H)17 cells. Nat Immunol. (2011) 12:1238–45. doi: 10.1038/ni.2134
153. Kotenko SV, Izotova LS, Mirochnitchenko OV, Esterova E, Dickensheets H, Donnelly RP, et al. Identification of the functional interleukin-22 (IL-22) receptor complex: the IL-10R2 chain (IL-10Rbeta) is a common chain of both the IL-10 and IL-22 (IL-10-related T cell-derived inducible factor, IL-TIF) receptor complexes. J Biol Chem. (2001) 276:2725–32. doi: 10.1074/jbc.M007837200
154. Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. (2005) 174:3695–702. doi: 10.4049/jimmunol.174.6.3695
155. Sa SM, Valdez PA, Wu J, Jung K, Zhong F, Hall L, et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J Immunol. (2007) 178:2229–40. doi: 10.4049/jimmunol.178.4.2229
156. Dumoutier L, Van Roost E, Colau D, Renauld JC. Human interleukin-10-related T cell-derived inducible factor: molecular cloning and functional characterization as an hepatocyte-stimulating factor. Proc Natl Acad Sci U.S.A. (2000) 97:10144–9. doi: 10.1073/pnas.170291697
157. Romer J, Hasselager E, Norby PL, Steiniche T, Thorn Clausen J, Kragballe K. Epidermal overexpression of interleukin-19 and -20 mRNA in psoriatic skin disappears after short-term treatment with cyclosporine a or calcipotriol. J Invest Dermatol. (2003) 121:1306–11. doi: 10.1111/j.1523-1747.2003.12626.x
158. Kunz S, Wolk K, Witte E, Witte K, Doecke WD, Volk HD, et al. Interleukin (IL)-19, IL-20 and IL-24 are produced by and act on keratinocytes and are distinct from classical ILs. Exp Dermatol. (2006) 15:991–1004. doi: 10.1111/j.1600-0625.2006.00516.x
159. Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discovery. (2014) 13:21–38. doi: 10.1038/nrd4176
160. Beutler B, Cerami A. The biology of cachectin/TNF–a primary mediator of the host response. Annu Rev Immunol. (1989) 7:625–55. doi: 10.1146/annurev.iy.07.040189.003205
161. Medler J, Wajant H. Tumor necrosis factor receptor-2 (TNFR2): an overview of an emerging drug target. Expert Opin Ther Targets. (2019) 23:295–307. doi: 10.1080/14728222.2019.1586886
162. Siegmund D, Wajant H. TNF and TNF receptors as therapeutic targets for rheumatic diseases and beyond. Nat Rev Rheumatol. (2023) 19:576–91. doi: 10.1038/s41584-023-01002-7
163. Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. (1995) 83:793–802. doi: 10.1016/0092-8674(95)90192-2
164. Sheehan KC, Pinckard JK, Arthur CD, Dehner LP, Goeddel DV, Schreiber RD. Monoclonal antibodies specific for murine p55 and p75 tumor necrosis factor receptors: identification of a novel in vivo role for p75. J Exp Med. (1995) 181:607–17. doi: 10.1084/jem.181.2.607
165. Li P, Zheng Y, Chen X. Drugs for autoimmune inflammatory diseases: from small molecule compounds to anti-TNF biologics. Front Pharmacol. (2017) 8:460. doi: 10.3389/fphar.2017.00460
166. Fischer R, Kontermann RE, Pfizenmaier K. Selective targeting of TNF receptors as a novel therapeutic approach. Front Cell Dev Biol. (2020) 8:401. doi: 10.3389/fcell.2020.00401
167. Leone GM, Mangano K, Petralia MC, Nicoletti F, Fagone P. Past, present and (Foreseeable) future of biological anti-TNF alpha therapy. J Clin Med. (2023) 12:1630. doi: 10.3390/jcm12041630
168. Ding H, Wang G, Yu Z, Sun H, Wang L. Role of interferon-gamma (IFN-γ) and IFN-γ receptor 1/2 (IFNγR1/2) in regulation of immunity, infection, and cancer development: IFN-γ-dependent or independent pathway. BioMed Pharmacother. (2022) 155:113683. doi: 10.1016/j.biopha.2022.113683
169. Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. (2018) 18:545–58. doi: 10.1038/s41577-018-0029-z
170. Dai H, Adamopoulos IE. Psoriatic arthritis under the influence of IFNγ. Clin Immunol. (2020) 218:108513. doi: 10.1016/j.clim.2020.108513
171. Della Bella C, Corrà A, Mantengoli E, Galano A, Benagiano M, Bonciani D, et al. Skin IL-17A and IFN-γ Production correlate with disease severity in patients with psoriasis and streptococcal infection. J Invest Dermatol. (2023) 143:925–32. doi: 10.1016/j.jid.2022.10.025
172. Wang CQ, Haxhinasto S, Garcet S, Kunjravia N, Cueto I, Gonzalez J, et al. Comparison of the inflammatory circuits in psoriasis vulgaris, non–Pustular palmoplantar psoriasis, and palmoplantar pustular psoriasis. J Invest Dermatol. (2023) 143:87–97.e14. doi: 10.1016/j.jid.2022.05.1094
173. Srivastava A, Luo L, Lohcharoenkal W, Meisgen F, Pasquali L, Pivarcsi A, et al. Cross-talk between IFN-γ and TWEAK through miR-149 amplifies skin inflammation in psoriasis. J Allergy Clin Immunol. (2021) 147:2225–35. doi: 10.1016/j.jaci.2020.12.657
174. Luque-Martin R, Angell DC, Kalxdorf M, Bernard S, Thompson W, Eberl HC, et al. IFN-γ Drives human monocyte differentiation into highly proinflammatory macrophages that resemble a phenotype relevant to psoriasis. J Immunol. (2021) 207:555–68. doi: 10.4049/jimmunol.2001310
175. Clement CC, D'Alessandro A, Thangaswamy S, Chalmers S, Furtado R, Spada S, et al. 3-hydroxy-L-kynurenamine is an immunomodulatory biogenic amine. Nat Commun. (2021) 12:4447. doi: 10.1038/s41467-021-24785-3
176. Li Y, Cui H, Li S, Li X, Guo H, Nandakumar KS, et al. Kaempferol modulates IFN-γ induced JAK-STAT signaling pathway and ameliorates imiquimod-induced psoriasis-like skin lesions. Int Immunopharmacol. (2023) 114:109585. doi: 10.1016/j.intimp.2022.109585
177. Lv J, Zhou D, Wang Y, Sun W, Zhang C, Xu J, et al. Effects of luteolin on treatment of psoriasis by repressing HSP90. Int Immunopharmacol. (2020) 79:106070. doi: 10.1016/j.intimp.2019.106070
178. Morikawa M, Derynck R, Miyazono K. TGF-β and the TGF-β Family: context-dependent roles in cell and tissue physiology. Cold Spring Harbor Perspect Biol. (2016) 8:a021873. doi: 10.1101/cshperspect.a021873
179. Machelak W, Szczepaniak A, Jacenik D, Zielińska M. The role of GDF11 during inflammation – An overview. Life Sci. (2023) 322:121650. doi: 10.1016/j.lfs.2023.121650
180. Cuesta-Gomez N, Medina-Ruiz L, Graham GJ, Campbell JDM. IL-6 and TGF-β-secreting adoptively-transferred murine mesenchymal stromal cells accelerate healing of psoriasis-like skin inflammation and upregulate IL-17A and TGF-β. Int J Mol Sci. (2023) 24:10132. doi: 10.3390/ijms241210132
181. Yang L, Zhang S, Wang G. Keratin 17 in disease pathogenesis: from cancer to dermatoses. J Pathol. (2018) 247:158–65. doi: 10.1002/path.5178
182. Zhang J, He L, Wang Z, Shao S, Qiao P, Zhang J, et al. Decreasing GDF15 promotes inflammatory signals and neutrophil infiltration in psoriasis models. J Invest Dermatol. (2023) 143:419–430.e418. doi: 10.1016/j.jid.2022.07.026
183. He H, Bissonnette R, Wu J, Diaz A, Saint-Cyr Proulx E, Maari C, et al. Tape strips detect distinct immune and barrier profiles in atopic dermatitis and psoriasis. J Allergy Clin Immunol. (2021) 147:199–212. doi: 10.1016/j.jaci.2020.05.048
184. Ramessur R, Corbett M, Marshall D, Acencio ML, Barbosa IA, Dand N, et al. Biomarkers of disease progression in people with psoriasis: a scoping review. Br J Dermatol. (2022) 187:481–93. doi: 10.1111/bjd.21627
185. Karin N. CXCR3 ligands in cancer and autoimmunity, chemoattraction of effector T cells, and beyond. Front Immunol. (2020) 11:976. doi: 10.3389/fimmu.2020.00976
186. Antonelli A, Ferrari SM, Giuggioli D, Ferrannini E, Ferri C, Fallahi P. Chemokine (C–X–C motif) ligand (CXCL)10 in autoimmune diseases. Autoimmun Rev. (2014) 13:272–80. doi: 10.1016/j.autrev.2013.10.010
187. Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev. (2018) 63:40–7. doi: 10.1016/j.ctrv.2017.11.007
188. Wu H, Zeng L, Ou J, Wang T, Chen Y, Nandakumar KS. Estrogen acts through estrogen receptor-β to promote mannan-induced psoriasis-like skin inflammation. Front Immunol. (2022) 13:818173. doi: 10.3389/fimmu.2022.818173
189. Lebwohl M, Ting PT, Koo JY. Psoriasis treatment: traditional therapy. Ann Rheum Dis. (2005) 64 Suppl 2:ii83–86. doi: 10.1136/ard.2004.030791
190. Alam MS, Gaida MM, Ogawa Y, Kolios AG, Lasitschka F, Ashwell JD. Counter-regulation of T cell effector function by differentially activated p38. J Exp Med. (2014) 211:1257–70. doi: 10.1084/jem.20131917
191. Gutierrez-Steil C, Wrone-Smith T, Sun X, Krueger JG, Coven T, Nickoloff BJ. Sunlight-induced basal cell carcinoma tumor cells and ultraviolet-B-irradiated psoriatic plaques express Fas ligand (CD95L). J Clin Invest. (1998) 101:33–9. doi: 10.1172/jci1165
192. Hart PH, Gorman S, Finlay-Jones JJ. Modulation of the immune system by UV radiation: more than just the effects of vitamin D? Nat Rev Immunol. (2011) 11:584–96. doi: 10.1038/nri3045
193. Gossec L, Kerschbaumer A, Ferreira RJO, Aletaha D, Baraliakos X, Bertheussen H, et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2023 update. Ann Rheum Dis. (2024) 83:706–19. doi: 10.1136/ard-2024-225531
194. Du H, Liu P, Zhu J, Lan J, Li Y, Zhang L, et al. Hyaluronic acid-based dissolving microneedle patch loaded with methotrexate for improved treatment of psoriasis. ACS Appl Mater Interf. (2019) 11:43588–98. doi: 10.1021/acsami.9b15668
195. Mercader-García P, Silvestre JF, Navarro-Triviño FJ, Giménez-Arnau AM, Pastor-Nieto MA, Cordoba-Guijarro S, et al. A re-assessment of the value of markers of corticosteroid contact allergy in the Spanish baseline series: Clobetasol propionate in the spotlight. Contact Dermatitis. (2024) 91:228–36. doi: 10.1111/cod.14639
196. Tuckermann JP, Kleiman A, Moriggl R, Spanbroek R, Neumann A, Illing A, et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J Clin Invest. (2007) 117:1381–90. doi: 10.1172/jci28034
197. Tatu AL. Topical steroid induced facial rosaceiform dermatitis. Acta Endocrinol (Buchar). (2016) 12:232–3. doi: 10.4183/aeb.2016.232
198. Tokuyama M, Mabuchi T. New treatment addressing the pathogenesis of psoriasis. Int J Mol Sci. (2020) 21:7488. doi: 10.3390/ijms21207488
199. Blauvelt A, Papp KA, Griffiths CE, Randazzo B, Wasfi Y, Shen YK, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: Results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. (2017) 76:405–17. doi: 10.1016/j.jaad.2016.11.041
200. Mease PJ, McInnes IB, Tam LS, Rajalingam R, Peterson S, Hassan F, et al. Comparative effectiveness of guselkumab in psoriatic arthritis: updates to a systematic literature review and network meta-analysis. Rheumatol (Oxford). (2023) 62:1417–25. doi: 10.1093/rheumatology/keac500
201. Yu C, Geng S, Yang B, Deng Y, Li F, Kang X, et al. Tildrakizumab for moderate-to-severe plaque psoriasis in Chinese patients: A 12-week randomized placebo-controlled phase III trial with long-term extension. Chin Med J (Engl). (2024) 137:1190–8. doi: 10.1097/cm9.0000000000002873
202. Gordon KB, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. (2018) 392:650–61. doi: 10.1016/s0140-6736(18)31713-6
203. Gooderham M, Pinter A, Ferris LK, Warren RB, Zhan T, Zeng J, et al. Long-term, durable, absolute Psoriasis Area and Severity Index and health-related quality of life improvements with risankizumab treatment: a post hoc integrated analysis of patients with moderate-to-severe plaque psoriasis. J Eur Acad Dermatol Venereol. (2022) 36:855–65. doi: 10.1111/jdv.18010
204. Blauvelt A, Kimball AB, Augustin M, Okubo Y, Witte MM, Capriles CR, et al. Efficacy and safety of mirikizumab in psoriasis: results from a 52-week, double-blind, placebo-controlled, randomized withdrawal, phase III trial (OASIS-1). Br J Dermatol. (2022) 187:866–77. doi: 10.1111/bjd.21743
205. Papp K, Warren RB, Green L, Reich K, Langley RG, Paul C, et al. Safety and efficacy of mirikizumab versus secukinumab and placebo in the treatment of moderate-to-severe plaque psoriasis (OASIS-2): a phase 3, multicentre, randomised, double-blind study. Lancet Rheumatol. (2023) 5:e542–52. doi: 10.1016/s2665-9913(23)00120-0
206. Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med. (2014) 371:326–38. doi: 10.1056/NEJMoa1314258
207. Reich K, Sullivan J, Arenberger P, Jazayeri S, Mrowietz U, Augustin M, et al. Secukinumab shows high and sustained efficacy in nail psoriasis: 2.5-year results from the randomized placebo-controlled TRANSFIGURE study. Br J Dermatol. (2021) 184:425–36. doi: 10.1111/bjd.19262
208. Griffiths CE, Reich K, Lebwohl M, van Kerkhof P, Paul C, Menter A, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. (2015) 386:541–51. doi: 10.1016/s0140-6736(15)60125-8
209. Kostareva O, Svoeglazova A, Kolyadenko I, Nikulin A, Evdokimov S, Dzhus U, et al. Two epitope regions revealed in the complex of IL-17A and anti-IL-17A V(H)H domain. Int J Mol Sci. (2022) 23:14904. doi: 10.3390/ijms232314904
210. Lebwohl M, Strober B, Menter A, Gordon K, Weglowska J, Puig L, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. (2015) 373:1318–28. doi: 10.1056/NEJMoa1503824
211. von Kiedrowski R, Hinz T, Mauer G, Schwinn A, Timmel A, Hutt HJ, et al. Management of moderate to severe psoriasis with brodalumab-Real-world evidence from the LIBERO study. J Eur Acad Dermatol Venereol. (2024) 38:2156–66. doi: 10.1111/jdv.19974
212. Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet. (2008) 371:1665–74. doi: 10.1016/s0140-6736(08)60725-4
213. Grine L, Dejager L, Libert C, Vandenbroucke RE. An inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev. (2015) 26:25–33. doi: 10.1016/j.cytogfr.2014.10.009
214. Mylonas A, Conrad C. Psoriasis: classical vs. Paradoxical. The yin-yang of TNF and type I interferon. Front Immunol. (2018) 9:2746. doi: 10.3389/fimmu.2018.02746
215. Duan Y, Sun W, Li Y, Shi Z, Li L, Zhang Y, et al, Spirohypertones A. and B as potent antipsoriatics: Tumor necrosis factor-α inhibitors with unprecedented chemical architectures. Acta Pharm Sin B. (2024) 14:2646–56. doi: 10.1016/j.apsb.2024.02.002
216. Kolahdooz H, Khori V, Erfani-Moghadam V, Livani F, Mohammadi S, Memarian A. Niosomal curcumin suppresses IL17/IL23 immunopathogenic axis in skin lesions of psoriatic patients: A pilot randomized controlled trial. Life (Basel). (2023) 13:1076. doi: 10.3390/life13051076
217. Zhang C, Shestopaloff K, Hollis B, Kwok CH, Hon C, Hartmann N, et al. Response to anti-IL17 therapy in inflammatory disease is not strongly impacted by genetic background. Am J Hum Genet. (2023) 110:1817–24. doi: 10.1016/j.ajhg.2023.08.010
218. Xu B, Yu B, Xu Z, Ye S, Qing Y, Sun H, et al. Investigation and confirmation of PYCARD as a potential biomarker for the management of psoriasis disease. J Inflammation Res. (2024) 17:6415–37. doi: 10.2147/jir.S468746
219. Villanova F, Di Meglio P, Nestle FO. Biomarkers in psoriasis and psoriatic arthritis. Ann Rheum Dis. (2013) 72 Suppl 2:ii104–110. doi: 10.1136/annrheumdis-2012-203037
220. Garshick MS, Ward NL, Krueger JG, Berger JS. Cardiovascular risk in patients with psoriasis: JACC review topic of the week. J Am Coll Cardiol. (2021) 77:1670–80. doi: 10.1016/j.jacc.2021.02.009
221. FitzGerald O, Gladman DD, Mease PJ, Ritchlin C, Smolen JS, Gao L, et al. Phase 2 trial of deucravacitinib in psoriatic arthritis: biomarkers associated with disease activity, pharmacodynamics, and clinical responses. Arthritis Rheumatol. (2024) 76:1397–407. doi: 10.1002/art.42921
222. Ademowo OS, Hernandez B, Collins E, Rooney C, Fearon U, van Kuijk AW, et al. Discovery and confirmation of a protein biomarker panel with potential to predict response to biological therapy in psoriatic arthritis. Ann Rheum Dis. (2016) 75:234–41. doi: 10.1136/annrheumdis-2014-205417
223. Wagner CL, Visvanathan S, Elashoff M, McInnes IB, Mease PJ, Krueger GG, et al. Markers of inflammation and bone remodelling associated with improvement in clinical response measures in psoriatic arthritis patients treated with golimumab. Ann Rheum Dis. (2013) 72:83–8. doi: 10.1136/annrheumdis-2012-201697
224. Chan BCL, Lam CWK, Tam LS, Wong CK. IL33: roles in allergic inflammation and therapeutic perspectives. Front Immunol. (2019) 10:364. doi: 10.3389/fimmu.2019.00364
225. Zhou T, Zhang S, Zhou Y, Lai S, Chen Y, Geng Y, et al. Curcumin alleviates imiquimod-induced psoriasis in progranulin-knockout mice. Eur J Pharmacol. (2021) 909:174431. doi: 10.1016/j.ejphar.2021.174431
226. Huh JR, Leung MW, Huang P, Ryan DA, Krout MR, Malapaka RR, et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature. (2011) 472:486–90. doi: 10.1038/nature09978
227. Laurindo LF, de Carvalho GM, de Oliveira Zanuso B, Figueira ME, Direito R, de Alvares Goulart R, et al. Curcumin-based nanomedicines in the treatment of inflammatory and immunomodulated diseases: an evidence-based comprehensive review. Pharmaceutics. (2023) 15:229. doi: 10.3390/pharmaceutics15010229
228. Menter A, Korman NJ, Elmets CA, Feldman SR, Gelfand JM, Gordon KB, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 6. Guidelines of care for the treatment of psoriasis and psoriatic arthritis: case-based presentations and evidence-based conclusions. J Am Acad Dermatol. (2011) 65:137–74. doi: 10.1016/j.jaad.2010.11.055
229. Wu D, Hailer AA, Wang S, Yuan M, Chan J, El Kurdi A, et al. A single-cell atlas of IL-23 inhibition in cutaneous psoriasis distinguishes clinical response. Sci Immunol. (2024) 9:eadi2848. doi: 10.1126/sciimmunol.adi2848
230. Papp KA, Blauvelt A, Bukhalo M, Gooderham M, Krueger JG, Lacour JP, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. (2017) 376:1551–60. doi: 10.1056/NEJMoa1607017
231. Meglio P, Di Cesare A, Laggner U, Chu CC, Napolitano L, Villanova F, et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PloS One. (2011) 6:e17160. doi: 10.1371/journal.pone.0017160
232. Ramón-Vázquez A, Flood P, Cashman TL, Patil P, Ghosh S. T lymphocyte plasticity in chronic inflammatory diseases: The emerging role of the Ikaros family as a key Th17-Treg switch. Autoimmun Rev. (2025) 24:103735. doi: 10.1016/j.autrev.2024.103735
233. Olejniczak-Staruch I, Ciążyńska M, Sobolewska-Sztychny D, Narbutt J, Skibińska M, Lesiak A. Alterations of the skin and gut microbiome in psoriasis and psoriatic arthritis. Int J Mol Sci. (2021) 22:3998. doi: 10.3390/ijms22083998
234. Buhaş MC, Gavrilaș LI, Candrea R, Cătinean A, Mocan A, Miere D, et al. Gut microbiota in psoriasis. Nutrients. (2022) 14:2970. doi: 10.3390/nu14142970
Keywords: psoriasis, inflammatory skin disease, immune cells, cytokines, cytokine network
Citation: Li L, Liu J, Lu J, Wu J, Zhang X, Ma T, Wu X, Zhu Q, Chen Z and Tai Z (2025) Interventions in cytokine signaling: novel horizons for psoriasis treatment. Front. Immunol. 16:1573905. doi: 10.3389/fimmu.2025.1573905
Received: 10 February 2025; Accepted: 27 March 2025;
Published: 15 April 2025.
Edited by:
Raouia Fakhfakh, Centre de Recherche du CHU de Québec, CanadaReviewed by:
Saeed Mohammadi, Golestan University of Medical Sciences, IranAlin Laurentiu Tatu, Dunarea de Jos University, Romania
Copyright © 2025 Li, Liu, Lu, Wu, Zhang, Ma, Wu, Zhu, Chen and Tai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quangang Zhu, emh1cWdAc2hza2luLmNvbQ==; Zhongjian Chen, YWFqaWFuODE4QDE2My5jb20=; Zongguang Tai, dGFpem9uZ2d1YW5nQDEyNi5jb20=
†These authors have contributed equally to this work