 Yinyu Chen
Yinyu Chen Shoupeng Wang1†
Shoupeng Wang1†- 1Department of Stomatology, Xuzhou Central Hospital, Xuzhou, Jiangsu, China
- 2School of Stomatology, Xuzhou Medical University, Xuzhou, China
- 3The Xuzhou Clinical College of Xuzhou Medical University, Xuzhou Medical University, Xuzhou, China
Background: A head and neck cancer organoid (HNCO) and peripheral blood T cell co-culture model was established to investigate whether HNCOs can induce the differentiation of peripheral blood T cells into tumor-reactive T cells. Additionally, this study seeks to explore the cytotoxicity of these T cells against autologous tumor organoids, providing theoretical and experimental evidence for the feasibility of this model as a platform for adoptive cell immunotherapy in head and neck cancer (HNC).
Methods: HNCO single cells were co-cultured with peripheral blood lymphocytes (PBLs) collected and isolated from patients with HNC. The culture supernatant was collected and assayed for interferon-gamma (IFN-γ) and tumor necrosis factor-α (TNF- α). The expression of T cell activation markers cluster of differentiation (CD)137 and CD107a was measured by flow cytometry to confirm tumor specificity and cytotoxicity. Additionally, the optimal effector-to-target (E/T) ratio was determined using the Cell Counting Kit-8 assay, and HNCO killing was quantified by fluorescent labeling.
Results: Of the 27 successfully established HNCO-T cell co-culture systems, 81.48% induced the in vitro differentiation and tumor-reactive CD8+ T cell expansion capable of mediating the killing of mature HNCOs.
Conclusion: The patient-derived HNCO-T cell co-culture model effectively induced PBL differentiation into tumor-reactive CD8+ T cells with enhanced tumor-killing activities. This model serves as a novel in vitro preclinical tool for advancing personalized adoptive immunotherapy in HNC.
1 Introduction
According to the Global Cancer Database, HNC is highly prevalent among men in developing countries, particularly in Central and South Asia, with its mortality ranking third among all cancers (1, 2). HNC is a heterogeneous disease affecting multiple sites in the head and neck region. Surgery, chemoradiotherapy, and other combination treatments have substantially halted disease progression and prolonged patient survival; nonetheless, they are often associated with adverse effects that worsen the patient’s quality of life (3). This necessitates safe and effective novel treatments for the clinical diagnosis and treatment of HNC.
Immunotherapy has emerged as a promising treatment modality for cancer. Adoptive T-cell therapy (ACT) is a highly personalized form of tumor immunotherapy that involves culturing the patient’s autologous T cells in the laboratory and reinfusing them into the patient to mediate immune responses (4–6). T-cell cytotoxicity is critical for effective immunotherapy. During in vitro culture, T cells can be specifically stimulated to differentiate into cytotoxic cluster of differentiation (CD)8+ T lymphocytes (CTLs), which mediate the recognition and killing of tumor cells. A higher proportion of CD8+ T cells is associated with a more robust response to tumor cells.
Three major modalities of ACT exist: tumor-infiltrating lymphocytes (TILs), chimeric antigen receptor T cells (CAR-T), and T cell receptor-engineered T cell (TCR-T) therapies. TIL therapy involves expanding heterogeneous populations of endogenous T cells from tumor tissues (7), whereas CAR-T and TCR-T therapies focus on expanding genetically engineered T cells that target specific antigens (8). ACT has demonstrated impressive clinical responses in hematologic malignancies; nonetheless, its application in solid tumors is under investigation (9, 10). However, HNCs are highly immunosuppressive because of the low number of T cells in the tumor microenvironment and inflammatory infiltrates that render existing TILs ineffective. This makes it difficult to obtain adequate tumor-reactive TILs from patients with HNC for ACT (11–13). Therefore, generating immune cells that can specifically recognize and kill tumor cells in vitro, as well as enhancing their anti-tumor responses, are key areas of intensive research. Upon reaching a sufficient quantity, these in vitro-cultured immune cells can be reinfused into the patient, where they mediate therapeutic effects as a cellular drug. Immune cell reinfusion exerts less impact on healthy tissues, compared with chemotherapy and other agents that directly kill cancer cells.
To obtain specific tumor-reactive immune cells in vitro, autologous peripheral blood lymphocytes (PBLs) were isolated from patients and used as the source of T cells. These cells were co-cultured with primary HNCOs in a three-dimensional culture. HNCOs effectively simulate the tumor microenvironment and accurately recapitulate tumor characteristics observed in patients, thereby supporting in-depth research on HNC and providing data for the development of personalized treatment regimens (14–16). The use of HNCOs helps to advance the understanding of tumor-immune interactions, which is crucial for tumor immunotherapy research and the clinical translation of personalized immunotherapy.
This study seeks to investigate the antitumor response of T cells derived from the organoid co-culture system by measuring cytokine secretion and the expression of cytotoxic markers.
2 Materials and methods
2.1 Sample source
Patients with HNC were recruited from the Xuzhou Central Hospital. The study protocol was reviewed and approved by the Biomedical Research Ethics Committee of Xuzhou Central Hospital. Informed consent was obtained from all patients and their families before specimen collection. The pathological type of the HNC samples was confirmed by the Pathology Department of Xuzhou Central Hospital before further processing.
2.2 Materials
Human fibroblast growth factor 10, recombinant human R-spodin-1 protein, recombinant human noggin protein, recombinant human Wnt-3A protein, N-acetylcysteine, A83-01, gastrin I, and nivolumab were purchased from MedChemExpress (Monmouth Junction, USA). Advance Dulbecco’s Modified Eagle’s Medium/F12 basal medium, Roswell Park Memorial Institute-1640 medium, N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid buffer, GlutaMAX™, B27 supplement, and N2 supplement were purchased from ThermoFisher (Waltham, MA USA). Nicotinamide, protablin E 2, and butylhydroxyanisole were purchased from Sigma. Rock Inhibitor (Y-27632) was purchased from Selleck Chemicals (Houston, USA). Human epidermal growth factor and recombinant human interleukin-2 were purchased from Peprotech (New Jersey, USA). Penicillin/streptomycin/amphotericin solution was purchased from Beyotime Biotech Inc. (Shanghai, China).
2.3 Establishing the HNCO model
Fresh HNC tissues—obtained by biopsy or surgical resection—were transported to the laboratory for organoid culture. Tumor tissue was minced and digested into a single-cell suspension. Specifically, epithelial-origin tumors were digested with collagenase I, whereas salivary gland tumors were digested with collagenase II. Single-cell suspensions were cultured in Matrigel to form organoids resembling the original tumors. The culture medium was changed twice weekly, and the organoids were passaged approximately every 2 weeks (17, 18).
2.4 Histology
Mature HNCOs were collected, centrifuged, and resuspended in liquid agarose. Once solidified, the organoids were dehydrated, cleared, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E). They were processed for immunohistochemistry (IHC) and mounted. The organoids were compared with the original tumor sections.
2.5 PBL isolation and T cell expansion
Fresh whole blood samples were collected and transported to the laboratory. The peripheral blood samples and tumor specimens used for co-culture model establishment were obtained from the same patients. PBLs were isolated using density gradient centrifugation with a lymphocyte separation solution under sterile conditions. After cell counting and viability assessment, PBLs were cultured in a T cell expansion medium at 37°C with 5% CO2 overnight.
2.6 Co-culture of T cells with HNCO single cells
HNCO single cells were collected, counted, and stimulated with interferon-gamma (IFN-γ) for 24 h before co-culture. Coated U-bottom 96-well plates with 5 μg/mL CD28 antibody.Isolated T cells were counted, mixed with HNCO single cells at a predefined effector-to-target (E/T) ratio, and seeded into 96-well U-shaped culture plates for co-culture, cultured in presence of 150 U/mL IL-2 and 20 μg/mL Nivolumab. T cells co-cultured with HNCO for 14 days were labeled as “CT,” whereas those at day 0 of co-culture were labeled as “UT.”
2.7 Enzyme-linked immunosorbent assay
Cytokine release from in vitro-activated T cells was quantified using the Human TNF-α and Human IFN-γ enzyme-linked immunosorbent assay (ELISA) kit (19, 20). Co-culture supernatants were collected, and mean IFN-γ and TNF-α concentrations were calculated from OD450 values measured by a microplate reader using a standard curve.
2.8 Flow cytometry
T-cell markers CD137 and CD107a were quantified using flow cytometry (21–24). Briefly, T cells were re-stimulated with single cells for 5 h, stained with anti-CD3-PerCP-Cy5.5, anti-CD4-FITC, anti-CD8-BV421, anti-CD137-APC, CD107a-PE, and near-infrared viability dye, and analyzed using a flow cytometer. Data were processed using FlowJo software (25).
2.9 Cell counting kit-8 assay
To determine the optimal E/T ratio, T cells were collected, counted, resuspended in a culture medium, and adjusted to a predefined density relative to HNCO (per single cell count). They were seeded into 96-well plates. After 24 h, diluted Cell Counting Kit-8 (CCK8) reagent was added, and the cells were incubated at 37°C for 2 h. OD450 was measured using a microplate reader to quantify viable tumor cells in each well.
2.10 Fluorescent labeling
Cells were stained using the Calcein-acetoxymethyl ester (AM)/propidium iodide (PI) kit, labeling live cells with Calcein-AM and dead cells with PI. Fluorescence was observed under an inverted fluorescence microscope, and fluorescence intensity was quantified using ImageJ.
2.11 Data analysis
Statistical analysis was conducted using GraphPad Prism. Independent t-tests were used to compare between two groups, whereas one-way analysis of variance was used to compare among multiple groups. A P-value <0.05 indicated statistical significance.
3 Results
3.1 Establishment and morphological validation of the HNCO model
A total of 27 patient-derived HNCO models were developed. Table 1 summarizes the clinical pathology data and passage of HNCOs, all enrolled patients were diagnosed with primary head and neck carcinoma and had not received any prior anti-tumor therapies (including radiotherapy, chemotherapy, immunotherapy or biological therapy) for their current tumors. Additionally, none of the patients presented with hematological disorders. HNCOs formed 100 to 150 µm spherical structures after 3 to 5 days of culture and were stably expanded for over four generations without morphological changes (Figures 1A, B).
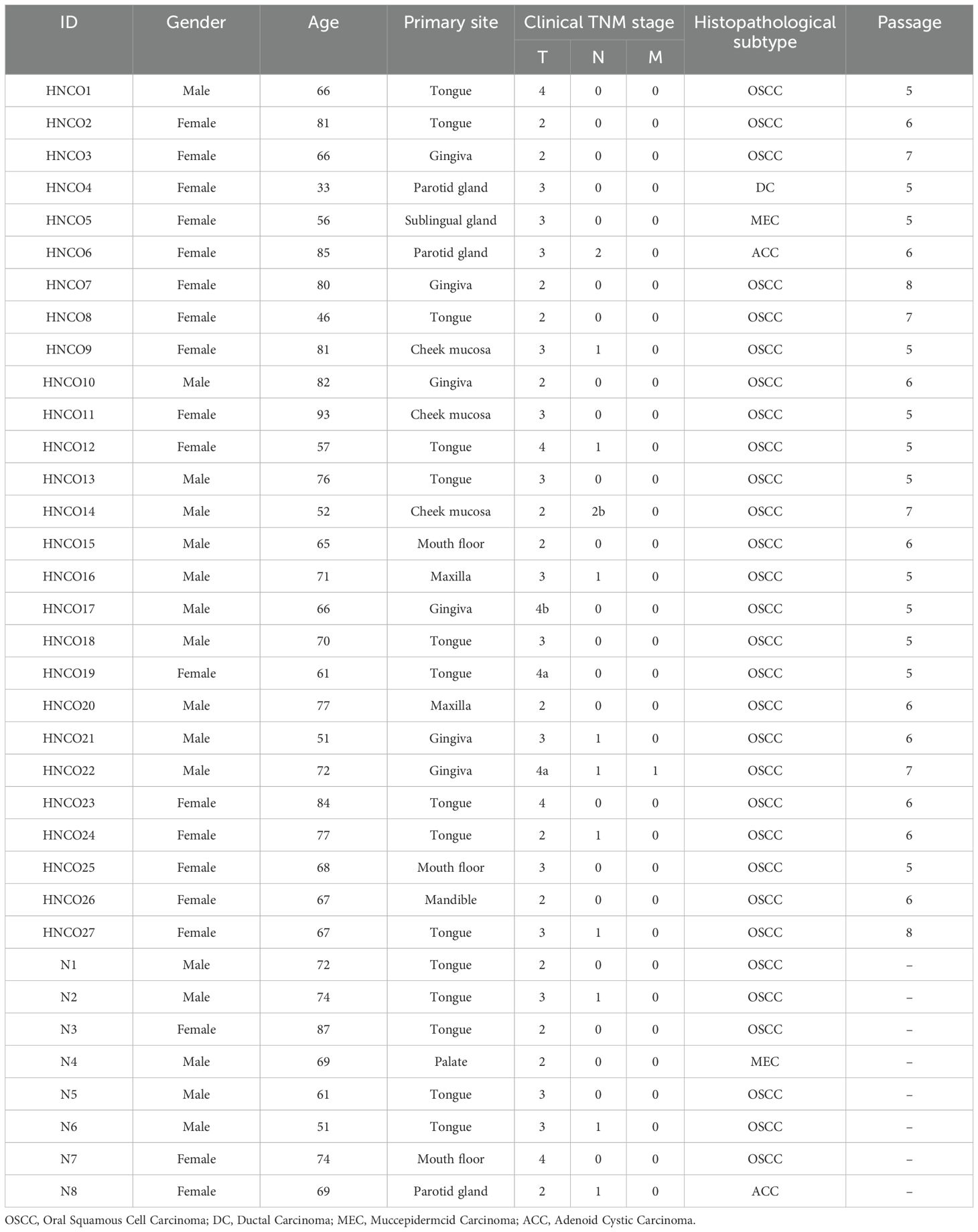
Table 1. Patient information.
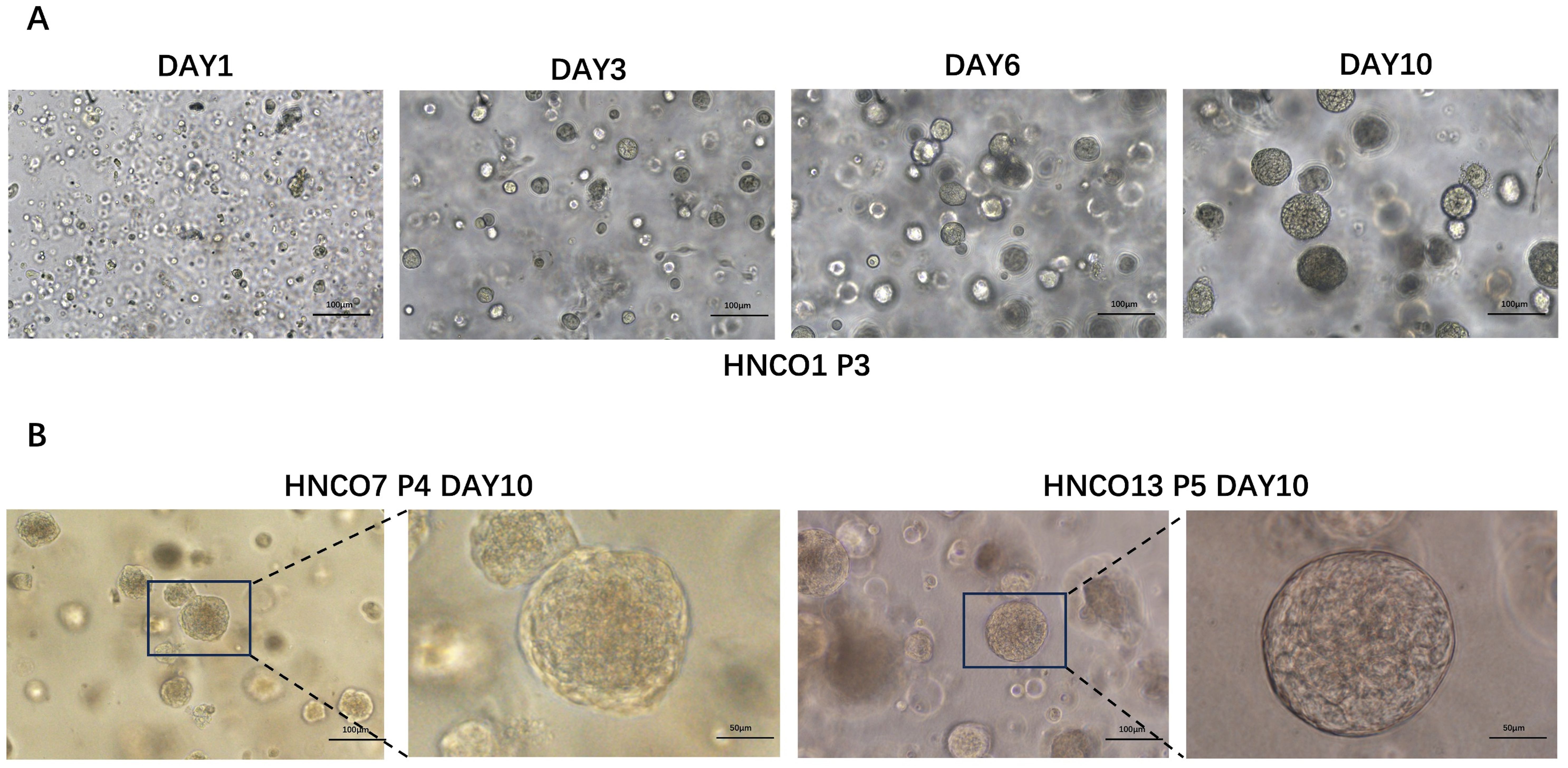
Figure 1. (A) Course of HNCO1 growth (100 um). (B) Microscopic images of HNCO7 and HNCO13 (100 um, 50 um) HNCO: head and neck cancer organoid.
The morphological and histological characteristics of HNCOs were compared with their parental tumors using H&E staining and IHC analysis. HNCOs closely resembled the parental tumor tissues, particularly in nuclear atypia. Furthermore, IHC analysis indicated canonical tumor markers, including the high expression of pan-cytokeratin, tumor protein p53, and Antigen Kiel 67, confirming the successful establishment of histopathologically consistent HNCOs (Figures 2A, B).
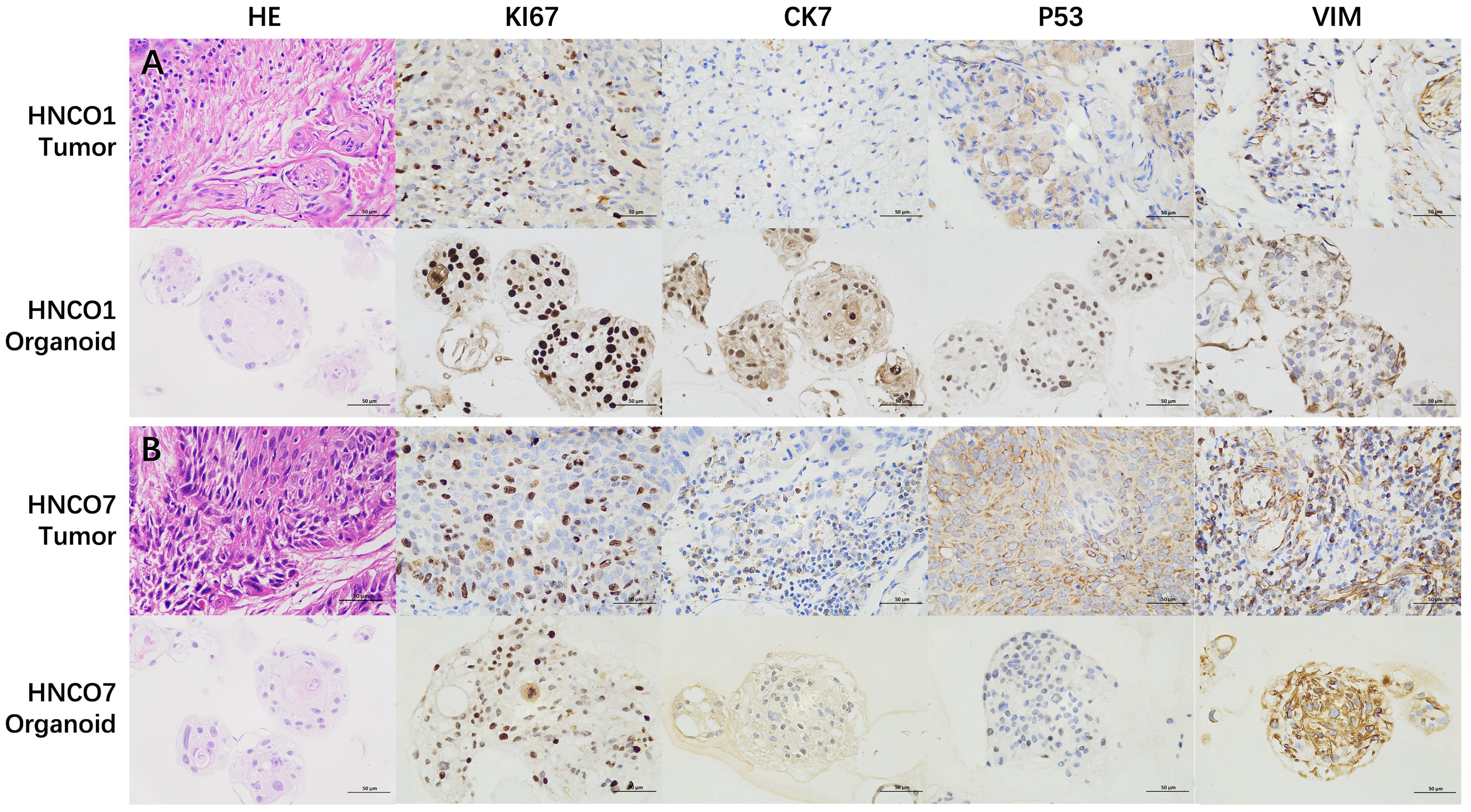
Figure 2. (A) H&E and IHC images of HNCO1 vs. parental tumor tissues (50 um). (B) H&E and IHC images of HNCO7 vs. parental tumor tissues (50 um). HNCO, head and neck cancer organoid; IHC, immunohistochemistry; and H&E, hematoxylin and eosin.
3.2 Increase in IFN-γ and TNFα after co-culture
Effector cytokines IFN-γ and TNFα are central to the activation, differentiation, and tumor-killing activity of CTLs. To assess whether HNCOs could induce T cell-mediated anti-tumor response, single cells from the 27 HNCOs were co-cultured with autologous PBL-derived T cells (Figure 3). Before co-culture, HNCO single cells were pre-treated with IFN-γ and nivolumab (anti-PD-1 antibody) to enhance antigen presentation and counteract PD-L1-induced inhibition of T cell activation (26).
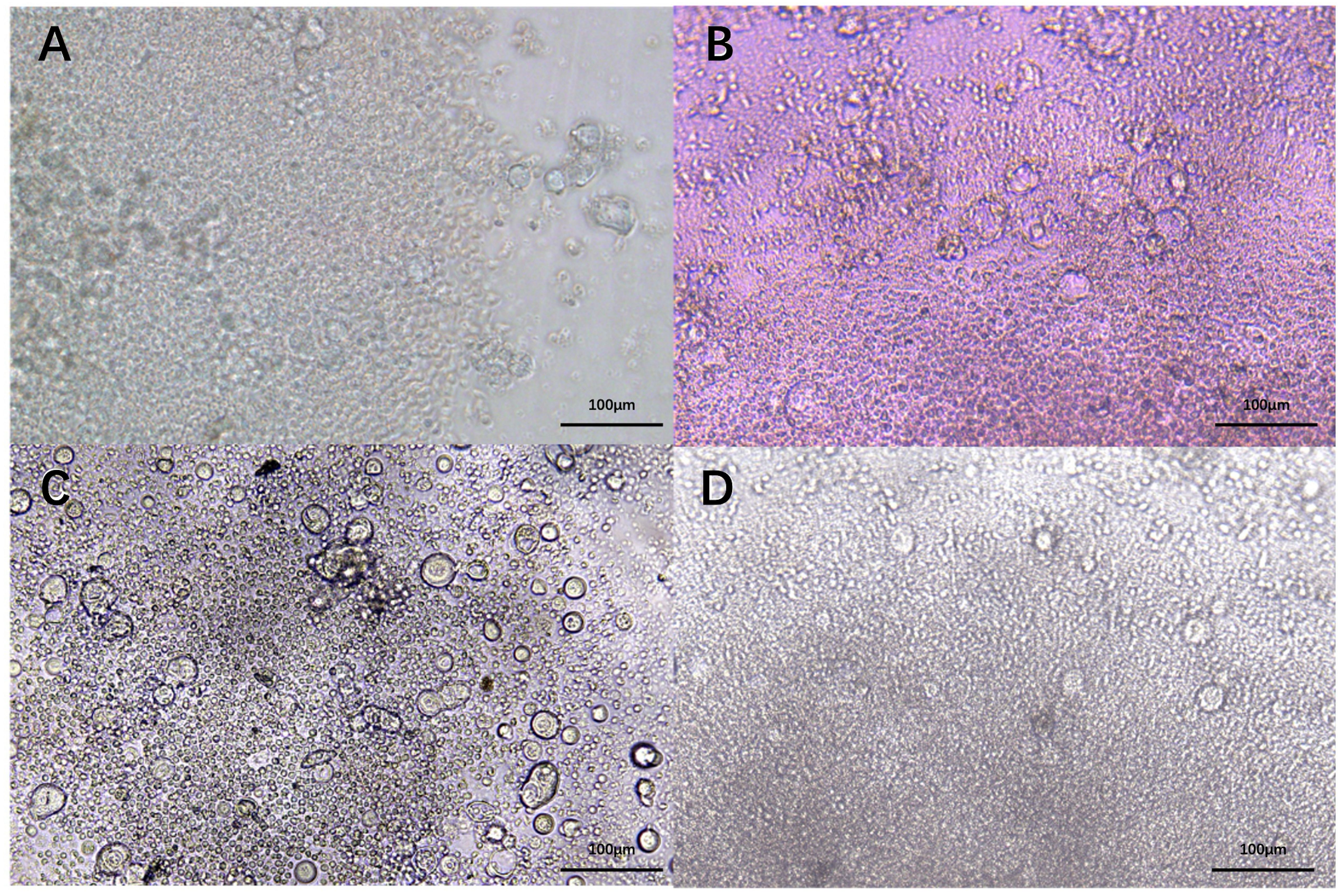
Figure 3. HNCO2 (A). HNCO6 (B), HNCO10 (C), and HNCO12 (D) single cells in co-culture with autologous PBLs (100 um). HNCO: head and neck cancer organoid; PBL: peripheral blood lymphocyte.
Supernatant samples were collected on day 0 and day 14 to quantify IFN-γ and TNF- α concentrations using ELISA kits. After 14 days of co-culture, IFN-γ and TNF-α levels significantly increased in 23 co-culture systems, whereas remained unchanged in the remaining 4 systems (Figure 4).
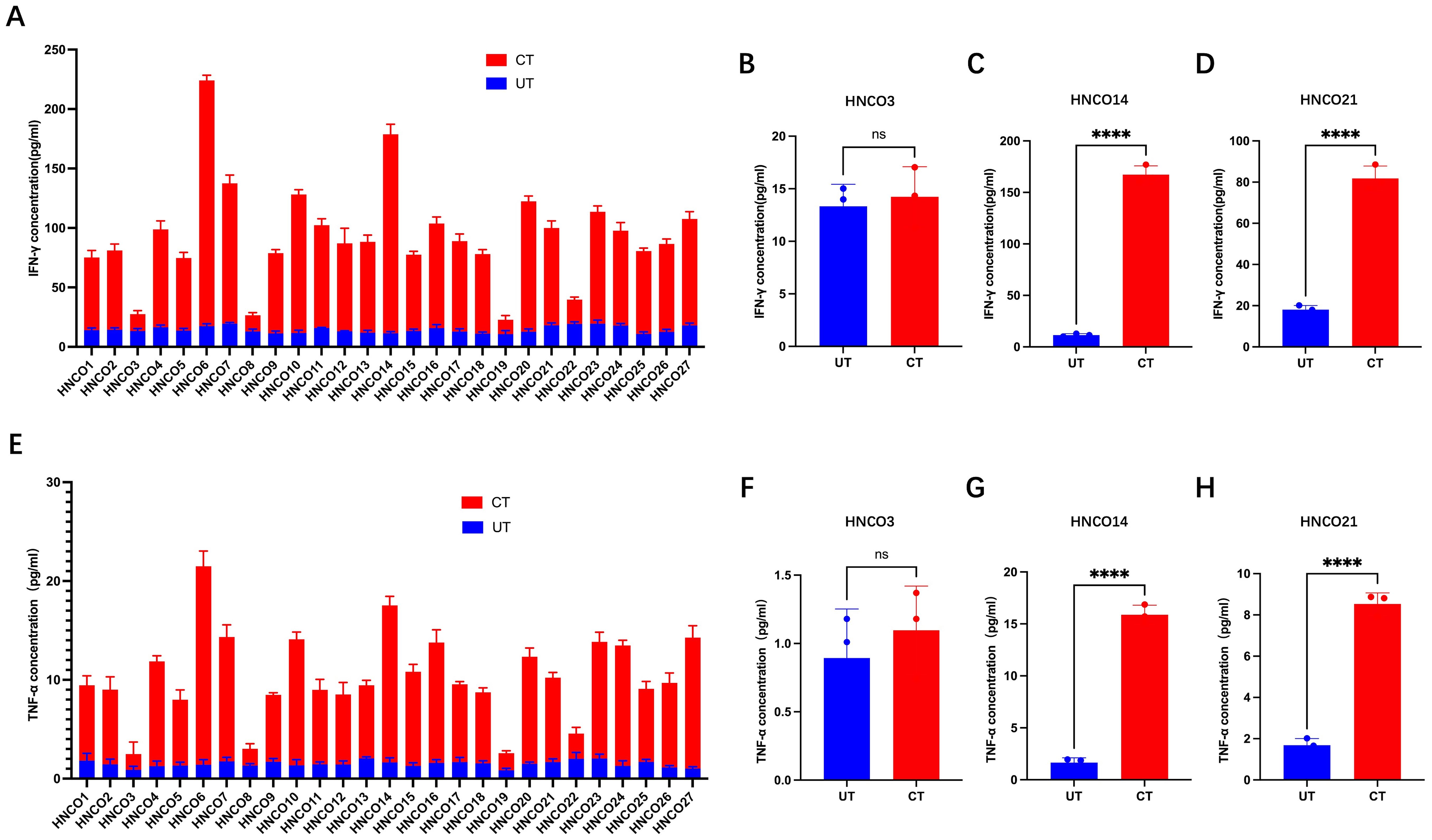
Figure 4. (A) IFN-g production in 27 co-culture models. IFN-g levels before and after co-culture with HNCO3 (B), HNCO14 (C), or HNCO21 (D), ns P>0.05, **: P<0.0001. (E) TNF-a production in 27 co-culture models. TNF-a before and after co-culture with HNCO3 (F), HNCO14 (G), or HNCO21 (H), ns: P>0.05, **P<0.0001. HNCO, head and neck cancer organoid; IN-g, interferon-gamma; and TNF-a, tumor necrosis factor alpha.
3.3 T cell activation and tumor reactivity
Activated and differentiated CD8+ T cells express specific markers, such as CD107a and CD137. Therefore, flow cytometry was used to assess the degranulation and cytotoxicity of CD107a and CD137 expression on CD8+ T cells after co-culture with HCNOs (Figure 5A).
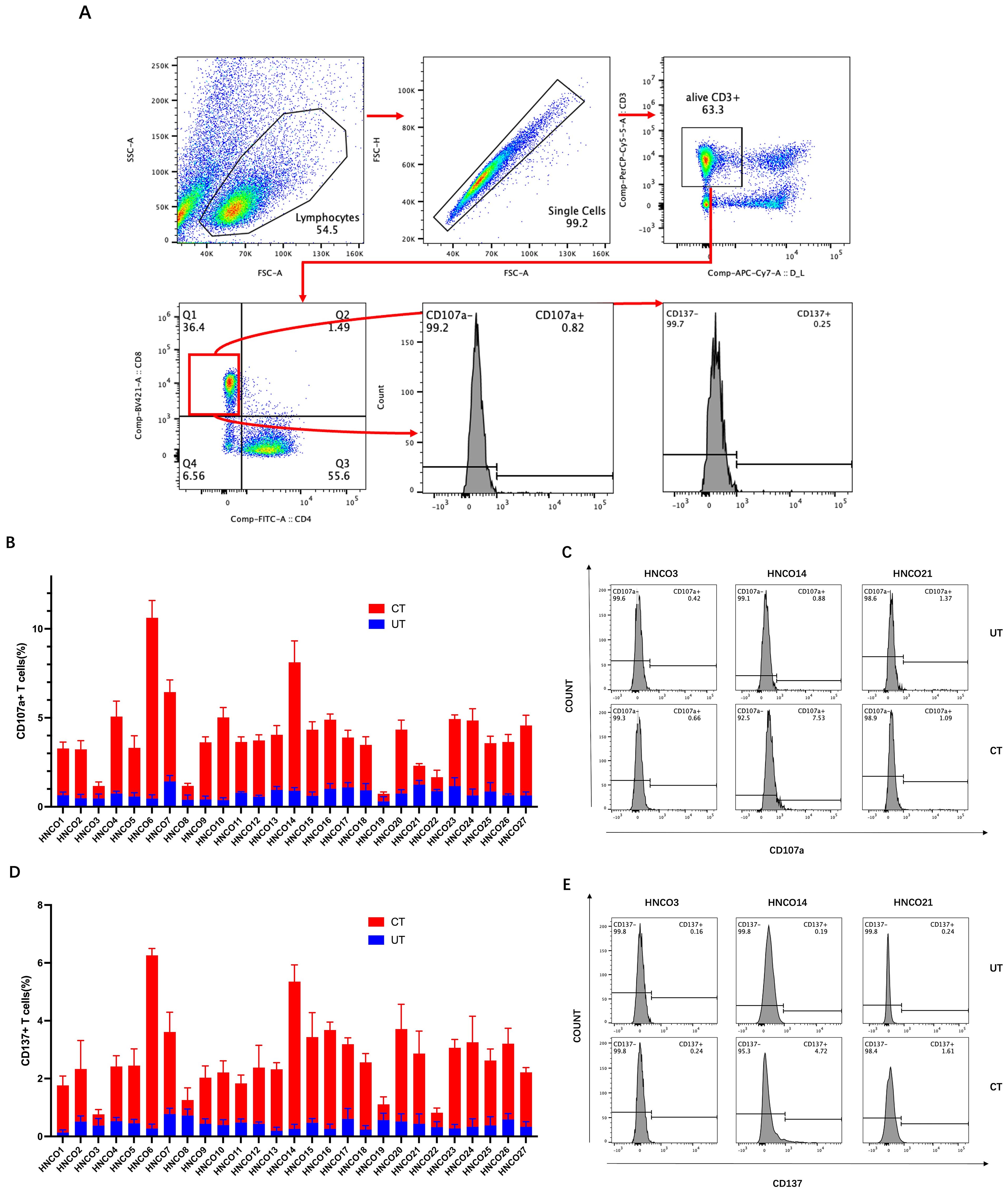
Figure 5. (A) Flow cytometry plots of CD107a and CD137 expression on CD8+ T cells. (B) Percentage of CD107a-positive cells after co-culture with 27 HCNOs. (C) Percentage of CD107a-positive cells after co-culture with HNCO3, HCNO14, and HCNO21. (D) Percentage of CD137-positive cells after co-culture with 27 HCNOs. (E) Percentage of CD137-positive cells after co-culture with HNCO3, HCNO14, and HCNO21. HNCO, head and neck cancer organoid; CD, cluster of differentiation.
Consistent with the ELISA results, CD107a and CD137 expression was upregulated in CD8+ T cells after co-culture with 22 patient-derived HCNOs, except for HNCO21, where no significant increase in CD107a expression was observed. Furthermore, CD107a and CD137 expression in CD8+T cells remained unchanged after co-culture with the remaining four HCNOs (Figures 5B–E).
In some patient samples, CD8+ T cells demonstrated enhanced tumor reactivity and cytotoxicity after 2 weeks of co-culture. For instance, in HNCO6, the mean IFN-γ concentration increased from 17.48 pg/mL before to 206.6 pg/mL (approximately 12 times) after 14 days of co-culture (Figure 6A). Similarly, the mean TNF-α concentration increased from 1.403 pg/mL before to 20.09 pg/mL after co-culture (approximately 14 times) (Figure 6B). Additionally, the percentages of CD107a+ cells increased from 0.45% before to 10.4% after co-culture and CD137+ CD8+ T cells increased from 0.20% before to 6.03% after co-culture (Figures 6C, D).
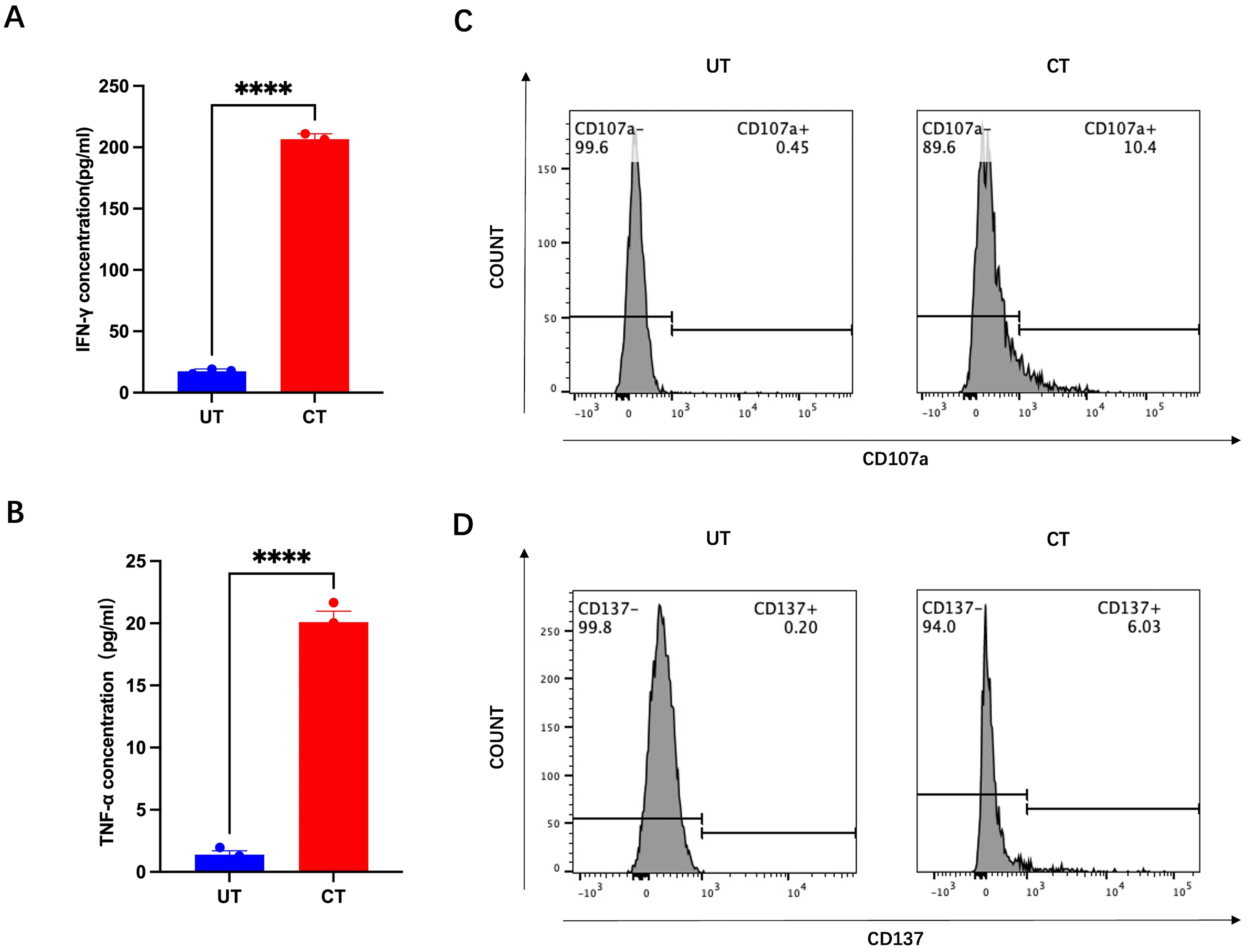
Figure 6. IFN-g (A), TNF-a (B), CD107a (C), and CD137 (D) expression before and after co-culture with HNCO6, ****P<0.0001. HNCO, head and neck cancer organoid; CD, cluster of differentiation; IFN-y, interferon-gamma; and TNF-a, tumor necrosis factor alpha.
Thus, some HNCOs effectively promote antigen-specific recognition and T-cell activation during co-culture.
3.4 Mature HNCOs are killed by induced CD8+ T cells
To determine the cytotoxicity of induced CD8+ T cells against autologous mature HNCOs, these cells were co-cultured for 24 h and tumor cell viability was assessed.
The association between CD8+ T cell cytotoxicity and the E/T ratio was assessed by measuring tumor cell viability after 24 h of co-culture at E/T ratios of 1:1, 2:1, 5:1, 10:1, and 20:1. Inverted microscopy indicated pronounced tumor cell lysis with increased E/T ratio (Figure 7A). Furthermore, the CCK-8 assay demonstrated that CD8+ T cells mediated tumor cell killing at all E/T ratios, with the tumor-killing effect increasing with the ratio (Figure 7B).
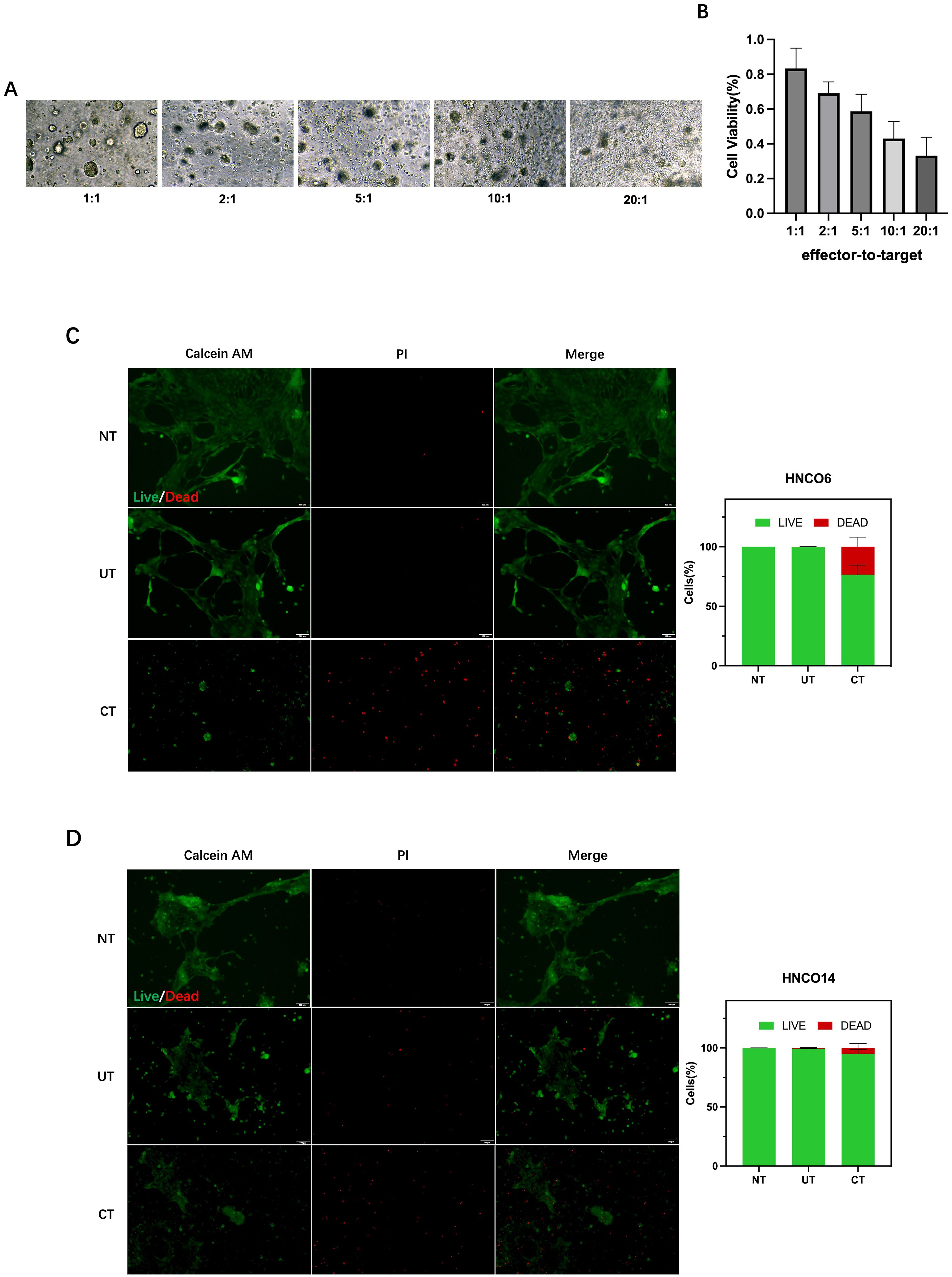
Figure 7. (A) HNCO-CD8+ T cell co-culture at various E/T ratios. (B) Percentage of HNCO kiling by CD8+ T cels after co-culture at diferent E/ratios measured using the CCK8 assay. (C) HNCO6-CD8+ T cell co-culture and celviability. (D) HNCO14-CD8+ T cell co-culture andcell viability. HNCO, head and neck cancer organoid; CD, cluster of diferentiation; E/T, effector-to-target; and CCK8, Cell Counting kit-8.
Tumor killing activity of CD8+ T cells was further investigated in the HNCO6 and HNCO14 co-culture models at a 20:1 E/T ratio. Controls comprised media without T cells (NT) and T cells without prior induction (UT). The co-culture of CD8+ T cells (CT) with mature HNCOs resulted in 23.5% cell death for HNCO6 (Figure 7C) and 5.07% for HNCO14 (Figure 7D), significantly surpassing the control groups. Thus, induced CD8+ T cells mediate the specific killing of mature HNCOs.
4 Discussion
Immunotherapy—particularly immune checkpoint inhibitors—has emerged as a mainstay treatment for cancer. Anti-PD-1 inhibitors are the first-line treatment for HNC, despite low response rates (20%) (4, 27). ACT using tumor-reactive T cells holds promise; however, research is still in the preclinical stages for HNC.
HNC is highly immunosuppressive, characterized by limited TILs. Patients with HNC often lack sufficient tumor-reactive T cells required for ACT (5, 6). Consequently, the in vitro culture and expansion of tumor-reactive T cells from PBLs represent a crucial strategy for advancing ACT in HNC because of their accessibility and availability. Moreover, they remain unaffected by the tumor type or volume (28). This study elucidates the feasibility of using HNCOs as a co-culture system for expanding tumor-reactive T cells in vitro and evaluating their cytotoxicity.
HNC displays high inter-individual variability, with T cell-mediated tumor killing influenced by various factors, highlighting the potential of personalized cancer treatment (15). Organoids preserve the heterogeneity of the parental tumors and replicate the tumor microenvironment, making them valuable models for personalized therapy. In this study, personalized HNCO-T cell co-culture models were established by pairing organoids with autologous peripheral blood T cells. HNCOs provide a personalized in vitro model for studying tumor-T cell interactions and assessing patient sensitivity to immunotherapy (29).
Of the 27 HNCO-T cell co-culture models established, T cells from 22 patients were successfully activated by autologous HNCO single cells, as evidenced by increased cytokine production and upregulated cytotoxicity-associated markers. In vitro validation using two HNCO co-culture models confirmed enhanced T cell-mediated cytotoxicity. However, the antitumor response of tumor-reactive T cells obtained using this model may exhibit variability, and the overall success rate remains uncertain. Additionally, the potential for tumor immunosuppression during this cell therapy warrants further investigation.
This study presents a novel approach and experimental foundation for expanding effector cells for ACT in HNC, despite unclear clinical efficacy. The model entirely recapitulates neither TILs or the tumor microenvironment nor their impact on T cell activation and cytotoxicity during ACT, such as immune checkpoint inhibition, which may generate different clinical outcomes. Additionally, the use of non-humanized materials, such as Matrigel—a murine-derived complex of the extracellular membrane with batch-to-batch variation—may affect experimental repeatability and stability, lowering the predictive accuracy of immunotherapy sensitivity assay in clinical settings (30).
Whereas this study elucidates the cytotoxicity of T cells in vitro, it remains elusive whether these cytotoxic T cells can be successfully enriched in HNC lesions and directly kill tumor cells after reinfusion. Moreover, the cell preparation process did not adhere to Good Clinical Practice standards for clinical application, warranting further research using animal models.
ACT is a key focus in immunotherapy with substantial implications for cancer treatment. Standardizing the T cell culture protocol is essential for clinical translation, facilitating the re-infusion of successfully induced T cells into patients and providing a novel immunotherapy option for HNC.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Xuzhou Central Hospital Biomedical Research Ethics Review Committee. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
YC: Formal Analysis, Investigation, Methodology, Software, Writing – original draft. SW: Funding acquisition, Methodology, Supervision, Visualization, Writing – review & editing. LZ: Investigation, Methodology, Writing – original draft. LC: Conceptualization, Investigation, Writing – original draft. FX: Methodology, Supervision, Writing – review & editing. SG: Methodology, Supervision, Writing – review & editing. JM: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by grants from the Geriatric Health Research Project of Jiangsu Provincial Health Commission (Grant No. LKM2024031), the National Clinical Research Center for Oral Diseases Open Project (Grant No. NCRCO-202101)and Development Fund Project of Affiliated Hospital of Xuzhou Medical University(Grant No. XYFY202207).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Giaquinto AN, and Jemal A. Cancer statistic. CA: A Cancer J Clin. (2024) 74:12–49. doi: 10.3322/caac.21820
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
3. Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, and Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. (2020) 6:92. doi: 10.1038/s41572-020-00224-3
4. Economopoulou P, Agelaki S, Perisanidis C, Giotakis EI, and Psyrri A. The promise of immunotherapy in head and neck squamous cell carcinoma. Ann Oncol. (2016) 27:1675–85. doi: 10.1093/annonc/mdw226
5. Ferris RL. Immunology and immunotherapy of head and neck cancer. J Clin Oncol. (2015) 33:3293–304. doi: 10.1200/JCO.2015.61.1509
6. Moskovitz JM and Ferris RL. Tumor immunology and immunotherapy for head and neck squamous cell carcinoma. J Dental Res. (2018) 97:622–6. doi: 10.1177/0022034518759464
7. Wu R, Forget M-A, Chacon J, Bernatchez C, Haymaker C, Chen JQ, et al. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: current status and future outlook. Cancer J (Sudbury Mass.). (2012) 18:160–75. doi: 10.1097/PPO.0b013e31824d4465
8. Guedan S, Ruella M, and June CH. Emerging cellular therapies for cancer. Annu Rev Immunol. (2019) 37:145–71. doi: 10.1146/annurev-immunol-042718-041407
9. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. New Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
10. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. New Engl J Med. (2020) 382:1331–42. doi: 10.1056/NEJMoa1914347
11. Watermann C, Pasternack H, Idel C, Ribbat-Idel J, Brägelmann J, Kuppler P, et al. Recurrent HNSCC harbor an immunosuppressive tumor immune microenvironment suggesting successful tumor immune evasion. Clin Cancer Res. (2021) 27:632–44. doi: 10.1158/1078-0432.CCR-20-0197
12. Canning M, Guo G, Yu M, Myint C, Groves MW, Byrd JK, et al. Heterogeneity of the head and neck squamous cell carcinoma immune landscape and its impact on immunotherapy. Front Cell Dev Biol. (2019) 7:52. doi: 10.3389/fcell.2019.00052
13. Peltanova B, Raudenska M, and Masarik M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: A systematic review. Mol Cancer. (2019) 18:63. doi: 10.1186/s12943-019-0983-5
14. Cattaneo CM, Dijkstra KK, Fanchi LF, Kelderman S, Kaing S, van Rooij N, et al. Tumor organoid – T cell co-culture systems. Nat Protoc. (2020) 15:15–39. doi: 10.1038/s41596-019-0232-9
15. Choi SY, Shim J, Gu D, Kim SY, Kim HJ, Shin D-Y, et al. Clonal evolution of long-term expanding head and neck cancer organoid: Impact on treatment response for personalized therapeutic screening. Oral Oncol. (2023) 146:106571. doi: 10.1016/j.oraloncology.2023.106571
16. Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell. (2018) 174:1586–1598.e12. doi: 10.1016/j.cell.2018.07.009
17. Driehuis E, Kretzschmar K, and Clevers H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat Protoc. (2020) 15:3380–409. doi: 10.1038/s41596-020-0379-4
18. Jacob F, Salinas RD, Zhang DY, Nguyen PTT, Schnoll JG, Wong SZH, et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell. (2020) 180:188–204.e22. doi: 10.1016/j.cell.2019.11.036
19. Bhat P, Leggatt G, Waterhouse N, and Frazer IH. Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. (2017) 8:e2836. doi: 10.1038/cddis.2017.67
20. Manohar SM. At the crossroads of TNF α Signaling and cancer. Curr Mol Pharmacol. (2023) 17:1–20. doi: 10.2174/1874467217666230908111754
21. Aktas E, Kucuksezer UC, Bilgic S, Erten G, and Deniz G. Relationship between CD107a expression and cytotoxic activity. Cell Immunol. (2009) 254:149–54. doi: 10.1016/j.cellimm.2008.08.007
22. Etxeberria I, Glez-Vaz J, Teijeira Á, and Melero I. New emerging targets in cancer immunotherapy: CD137/4-1BB costimulatory axis. ESMO Open. (2019) 4:e000733. doi: 10.1136/esmoopen-2020-000733
23. Krzewski K, Gil-Krzewska A, Nguyen V, Peruzzi G, and Coligan JE. LAMP1/CD107a is required for efficient perforin delivery to lytic granules and NK-cell cytotoxicity. Blood. (2013) 121:4672–83. doi: 10.1182/blood-2012-08-453738
24. Zapata JM, Perez-Chacon G, Carr-Baena P, Martinez-Forero I, Azpilikueta A, Otano I, et al. CD137 (4-1BB) signalosome: complexity is a matter of TRAFs. Front Immunol. (2018) 9:2618. doi: 10.3389/fimmu.2018.02618
25. Olivo Pimentel V, Yaromina A, Marcus D, Dubois LJ, and Lambin P. A novel co-culture assay to assess anti-tumor CD8+ T cell cytotoxicity via luminescence and multicolor flow cytometry. J Immunol Methods. (2020) 487:112899. doi: 10.1016/j.jim.2020.112899
26. Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. New Engl J Med. (2016) 375:1856–67. doi: 10.1056/NEJMoa1602252
27. Jenkins RW, Barbie DA, and Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. (2018) 118:9–16. doi: 10.1038/bjc.2017.434
28. Wei T, Leisegang M, Xia M, Kiyotani K, Li N, Zeng C, et al. Generation of neoantigen-specific T cells for adoptive cell transfer for treating head and neck squamous cell carcinoma. OncoImmunology. (2021) 10:1929726. doi: 10.1080/2162402X.2021.1929726
29. Zhou Z, van der Jeught K, Li Y, Sharma S, Yu T, Moulana I, et al. A T cell-engaging tumor organoid platform for pancreatic cancer immunotherapy. Advanced Sci. (2023) 10:2300548. doi: 10.1002/advs.202300548
Keywords: adoptive cell therapy, peripheral blood T cells, head and neck cancer, organoid, co-culture
Citation: Chen Y, Wang S, Zheng L, Chen L, Xu F, Guo S and Meng J (2025) Generating tumor-specific T cells based on a head and neck cancer organoid for adoptive cell therapy. Front. Immunol. 16:1573965. doi: 10.3389/fimmu.2025.1573965
Received: 16 February 2025; Accepted: 26 May 2025;
Published: 12 June 2025.
Edited by:
Shang Xie, Peking University, ChinaReviewed by:
Zhi Li, Shanghai University of Traditional Chinese Medicine, ChinaZhongcheng Gong, First Affiliated Hospital of Xinjiang Medical University, China
Copyright © 2025 Chen, Wang, Zheng, Chen, Xu, Guo and Meng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Meng, bXJvY2tldEAxMjYuY29t
†These authors have contributed equally to this work and share first authorship