 Alireza Tojjari
Alireza Tojjari Anwaar Saeed
Anwaar Saeed Ludimila Cavalcante3*
Ludimila Cavalcante3*- 1Department of Medicine, Division of Hematology & Oncology, University of Pittsburgh Medical Center (UPMC), Pittsburgh, PA, United States
- 2UPMC Hillman Cancer Center, Pittsburgh, PA, United States
- 3Department of Hematology and Medical Oncology, University of Virginia Comprehensive Cancer Center, Charlottesville, VA, United States
Recent progress in immunotherapy has significantly altered the therapeutic approach for gastrointestinal cancers, which are historically challenging due to their intricate pathologies and unfavorable outcomes. This review emphasizes the growing importance of immune checkpoints like TIGIT, VISTA, GITR, STING, and TIM-3 in the treatment of gastrointestinal oncology. These checkpoints are crucial elements within the tumor microenvironment, presenting new therapeutic possibilities. Studies show that TIGIT and GITR regulate the functions of T cells and NK cells, while the VISTA and STING pathways boost the body’s anti-tumor responses. TIM-3 is linked with T cell fatigue, highlighting its potential as a target to counteract immune evasion mechanisms. Integrating these immune checkpoints with traditional treatments could result in more customized and effective therapeutic approaches. This detailed review seeks to explore the changing field of immune checkpoint research, offering insights from molecular biology to clinical practice, and envisioning a future where advanced treatment methods greatly enhance patient outcomes in GI cancers.
1 Introduction
Immunotherapy has revolutionized cancer treatment, bringing new possibilities for patients whose conditions did not respond to traditional methods. This transformation is especially evident in gastrointestinal (GI) cancers. Gastrointestinal cancers remain notoriously resistant to immunotherapy, largely because their dense, immunosuppressive microenvironments and heterogeneous tumor biology blunt the effectiveness of conventional checkpoint inhibitors. T cell immunoreceptor with Ig and ITIM domains (TIGIT), V-domain Ig suppressor of T cell activation (VISTA) blockade, glucocorticoid-induced TNFR-related protein (GITR) agonism, Stimulator of Interferon Genes (STING) pathway activation, and T cell immunoglobulin and mucin-domain containing-3 (TIM-3) inhibition—has revealed strategies to both unleash CD8+ T-cell cytotoxicity and dismantle regulatory networks that foster tumor tolerance. By selectively enhancing T-cell receptor signaling, promoting type I interferon responses, and alleviating suppressive cues within the tumor niche, these emerging checkpoints hold promise for overcoming the core hurdles in GI cancer treatment (1) (2).
Enhancing this cadre of immune regulators is the TIM-3, a checkpoint that has emerged as a focal point of interest due to its association with T cell exhaustion and its potential as a cancer therapy target. TIM-3 is expressed on diverse immune cells, including T cells, natural killer cells, and dendritic cells. Its role in regulating immune responses and maintaining immune equilibrium is significant. The interaction between TIM-3 and its ligands, such as galectin-9, leads to the suppression of T-cell functionality, aiding in the immune evasion tactics of tumors. TIM-3 interactions extend beyond galectin-9 to include CEACAM-1, phosphatidylserine (PtdSer), and HMGB1. Recently, sabatolimab, a TIM-3 antibody, received FDA Fast Track designation (May 2021), emphasizing its potential clinical relevance (3). Recent investigations highlight TIM-3’s significance in the context of T cell exhaustion and its correlation with the efficacy of anti-PD-1 therapy, suggesting that targeting TIM-3 could represent a promising strategy in the realm of cancer immunotherapy (4).
This body of research underscores the need for a deep understanding of the tumor microenvironment (TME) and the complex roles played by immune checkpoints, including STING, in GI cancers. The nuanced expression and functional roles of TIGIT, VISTA, GITR,STING, and TIM-3 revealed through advanced genomic and immunological analyses provide a foundation for developing innovative combination therapies designed to overcome the limitations of existing treatments (5, 6).
Current standard immunotherapies for gastrointestinal (GI) cancers primarily involve the use of immune checkpoint inhibitors targeting the PD-1/PD-L1 axis, such as pembrolizumab and nivolumab. These therapies have significantly improved outcomes, especially in subsets of patients with microsatellite instability-high (MSI-H) or mismatch repair-deficient (dMMR) tumors, highlighting the therapeutic potential of immune modulation in GI oncology (1, 7).
As we move forward in the field of cancer immunotherapy, the exploration of TIGIT, VISTA, STING, GITR and TIM-3 within GI cancers expands our arsenal of therapeutic strategies and enhances our understanding of the intricate interactions between cancer and the immune system. This review explicitly aims to answer how targeting emerging checkpoints such as TIGIT, VISTA, GITR, STING, and TIM-3 can improve therapeutic outcomes in gastrointestinal oncology (5, 6, 8).
2 Background
Investigating immune checkpoint receptors like TIGIT, VISTA, GITR, TIM-3 and STING stands at the forefront of cancer research, unveiling new pathways to adjust the immune system’s reaction to tumors. These receptors are crucial in managing immune responses and frequently cross-regulate each other, marking them as valuable prospects for treatment strategies. It is essential to grasp the distinct roles and interactions these immune checkpoints have within the immune framework and their significance in the context of cancer treatment. This understanding could pave the way for groundbreaking approaches in immunotherapy, targeting these mechanisms to combat cancer more effectively.
2.1 TIGIT
TIGIT, an essential co-inhibitory receptor found on T cells and NK cells, is expressed on CD8+ T cells, NK cells, regulatory T cells (Tregs), CD4+ T cells, and dendritic cells. Studies have shown that it has an aiding impact on regenerative hyperplasia, indicating that liver regeneration is compromised in its absence in vivo (9). This is achieved by suppressing intracellular activities in NK and dendritic cells and curbing the growth and functionality of T cells (10). The engagement of TIGIT with its binding partners, poliovirus receptor (PVR) (CD155), nectin-2 (CD112), and nectin-4 (PVRL4)—plays a pivotal role in modulating immune reactions and the evolution of cancer. CD155, as the primary binding partner of TIGIT, predominantly initiates suppressive signals in the immune system and is linked with unfavorable outcomes in cancer cases (11–13). On the other hand, the interaction between TIGIT and CD226 attenuates immune operations, while nectin-2 and nectin-4 are identified as promising targets in cancer treatment due to their effects on the behavior of tumor cells (14, 15).
The presence of TIGIT on CD8+ T cells and NK cells is associated with a suppressive immune state and the production of cytokines, underscoring its significance in autoimmune diseases and various cancers like acute myeloid leukemia, chronic myelogenous leukemia, lung cancer, and melanoma (16, 17).
However, while targeting TIGIT presents a valuable opportunity in treating blood cancers, the potential for adverse side effects demands vigilant observation of patients. Reports from clinical studies of anti-TIGIT treatments have highlighted immune-related adverse events, emphasizing the importance of a thorough assessment of these therapies (7, 18). The CITYSCAPE study further corroborated these findings, with a significant majority of individuals treated with tiragolumab and atezolizumab reporting adverse immune responses, predominantly skin rashes, in addition to pancreatitis, underactive thyroid, colitis, and diabetes (19). These insights underscore the imperative for medical practitioners to diligently monitor for and mitigate the immunological side effects of TIGIT-targeting treatments(Figure 1A).
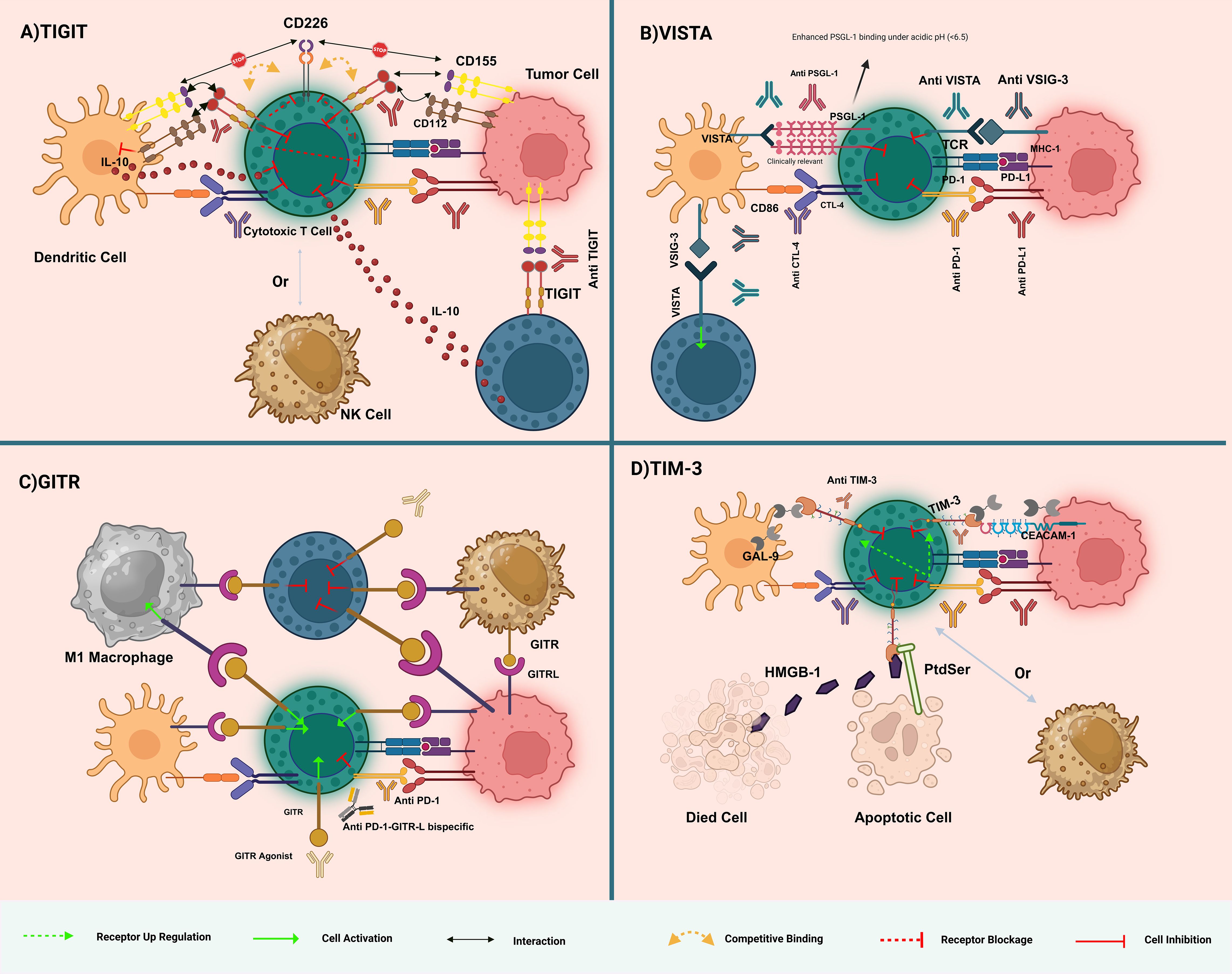
Figure 1. Overview of immune checkpoint interactions and therapeutic targets in immunotherapy. (A) TIGIT Pathway: TIGIT competes with CD226 for binding to CD155 on dendritic cells, leading to immune suppression. Anti-TIGIT antibodies block this inhibitory interaction, enhancing T-cell activation by allowing CD226 to bind CD155, thereby boosting anti-tumor immunity. (B) VISTA Pathway: VISTA interacts with VSIG-3 and B7 family members to suppress T-cell responses. VISTA binding to PSGL-1 is significantly enhanced under acidic conditions (pH < 6.5), as found in the tumor microenvironment. Anti-VISTA antibodies disrupt this suppression, restoring T-cell function. Combining anti-VISTA with other checkpoint inhibitors (e.g., anti-PD-1, anti-CTLA-4) can further enhance T-cell activation and reduce immune evasion by tumors. (C) GITR Pathway: GITR activation by agonistic antibodies or GITRL enhances T-cell cytokine production and immune response. GITR activation also depletes regulatory T cells (Tregs), which suppress immunity. Targeting GITR with bispecific antibodies like anti-PD-1-GITR-L aims to activate effector T cells and reduce Treg-mediated suppression, promoting tumor rejection. (D) TIM-3 Pathway: TIM-3 interacts with galectin-9, CEACAM-1, PtdSer, and HMGB1, contributing to T-cell exhaustion. Anti-TIM-3 antibodies block these inhibitory signals, reinvigorating T-cell function and promoting anti-tumor immunity. TIM-3 blockade is particularly effective when combined with PD-1/PD-L1 inhibitors to overcome multiple layers of immune suppression. Dashed arrows indicate reported cross-regulatory loops (e.g., TIGIT–PD-1 co-inhibition). The figure illustrates the dynamic complexity of immune checkpoint pathways, highlighting potential therapeutic strategies in cancer immunotherapy.
2.2 VISTA
VISTA, or PD-1H, is predominantly expressed on myeloid cells and T-regulatory cells, including CD4+ and Foxp3+ subsets. VISTA is also expressed on activated T cells in specific immune contexts. Its presence on tumor-infiltrating lymphocytes (TILs) and macrophages, juxtaposed with its general absence in most tumor cell types, underpins its complex role in cancer (20). Notably, VISTA has been identified in varying proportions within tumor cells across a spectrum of cancers, such as NSCLC, hepatocellular carcinoma, ovarian and endometrial cancers, melanoma, gastric cancer, and breast cancer (21). Functioning as a negative regulator of T-cell activation, proliferation, and cytokine production, VISTA mainly suppresses CD4+ T-cell-mediated immune responses. This regulatory role is further complicated by the discovery of a fusion protein (VISTA-Ig) that acts as a ligand, illustrating VISTA’s dual function in potentially enhancing proliferation and cytokine production in CD4+ T-cells, thereby indicating its receptor functionality. Moreover, VISTA’s direct influence on the effector functions of myeloid cells underscores the necessity for an in-depth investigation into its multifaceted role in immune regulation, emphasizing its significance as a target in cancer immunotherapy (22, 23).
Adjacent to VISTA’s regulatory mechanisms, VSIG-3, or IGSF11, serves as a ligand for VISTA, implicated in cell adhesion processes and expressed predominantly in human tumor cell lines, testes, and ovaries, with lesser expression noted in the brain and kidneys (24). The interaction between VISTA and VSIG-3 curtails the production of IL-2, IFN cytokines, and various chemokines by activated T-cells and peripheral blood mononuclear cells, delineating its immunosuppressive capacity (25). The overexpression of VSIG-3, correlated with high tumor malignancy and poor prognosis, has been identified in cancers such as colorectal and hepatocellular carcinoma, often in association with PD-L1 and PD-1 expressions (26). Despite acknowledging its role in immunosuppression, the precise mechanisms by which VSIG-3 influences cancer pathogenesis remain elusive, with some reports, such as Johnston et al., challenging the specificity of the VISTA-VSIG-3 interaction (27). Despite acknowledgment of its role in immunosuppression, the precise mechanisms by which VSIG-3 influences cancer pathogenesis remain elusive, with some reports, such as Johnston et al., challenging the specificity of the VISTA-VSIG-3 interaction. Further complicating VISTA’s interaction network are its associations with galectin-9 and PSGL-1, particularly under the acidic conditions characteristic of the tumor microenvironment, which hint at novel therapeutic targets. These interactions, especially with PSGL-1 and galectin-9, illuminate VISTA’s critical position within the immune regulatory framework, advocating for extensive research to elucidate effective immunotherapeutic strategies (28, 29)(Figure 1B).
2.3 GITR
The GITR, known by aliases TNFRSF18, AITR, or CD357, is a critical member of the tumor necrosis factor receptor superfamily, pivotal in modulating immune responses (30). Initially identified in dexamethasone-treated T cells in mice, GITR is broadly expressed across a spectrum of immune cells, including CD4+ and CD8+ T cells, regulatory T cells (Tregs), natural killer (NK) cells, macrophages, and dendritic cells (31). The interaction between GITR and its ligand, which is present on antigen-presenting cells and various immune cells, plays a fundamental role in a range of immunological functions such as T cell activation, differentiation, survival, regulation of Treg function, and enhancement of effector T cell activities against tumors and infections.
Activation of GITR is instrumental in counteracting Treg-mediated suppression, enhancing effector T cell functions, and promoting anti-tumor immunity. This efficacy positions GITR as a promising target for immunotherapy. Furthermore, the interaction between GITR and its ligand (GITRL) on antigen-presenting cells highlights the complex signaling pathways mediated by GITR that regulate the balance between Treg and effector T cell activities (32).
GITR’s role extends beyond T cells, influencing NK cells, dendritic cells (DCs), and macrophages, indicating its extensive impact on the immune system. The therapeutic potential of modulating GITR-GITRL interactions is explored through agonistic approaches that enhance tumor immune responses and strategies to dampen excessive immune reactions in autoimmune and inflammatory diseases (33). This dual approach to manipulating GITR pathways underscores its significance in developing immunotherapeutic strategies and managing various immune-related conditions (Figure 1C).
2.4 TIM-3
Tim-3 was originally discovered as a cell surface molecule preferentially expressed on interferon-gamma (IFN-γ)-producing CD4+ Th1 and CD8+ T cells. It belongs to the TIM gene family, which are found in syntenic chromosomal regions linked with both allergy and autoimmune diseases (34). Tim-3 attracted interest when it was discovered as a T-cell inhibitory receptor, and the concept was further supported by the studies showing that in vivo administration of monoclonal antibody specific to Tim-3 (mAbs) ameliorated or exacerbated disease in models of autoimmunity. With the exception of lacking a canonical inhibitory signaling motif in its cytoplasmic tail, Tim-3 is considered an immune checkpoint because of the negative regulatory roles it performs (35). This association with diseases driven by hyperactive immune cells sets Tim-3 in place as an important modulator of immune response. Clinical trials with Tim-3 antibodies, either alone or in combination with another treatment, have yielded promising results in different tumors, thus supporting their potentiality as an agent within cancer immunotherapy (Figure 1D).
2.5 STING
The STING pathway plays a pivotal role in the body’s innate and cancer immune responses by detecting tumor-derived DNA. This detection triggers the activation of interferon genes, thereby mobilizing antitumor immunity. Advances in drug delivery technologies have facilitated the systemic administration of STING agonists, demonstrating a promising capacity for eliminating tumors in preclinical experiments (36). The innovation in the design of novel STING agonists, including non-cyclic dinucleotide (CDN) molecules such as amidobenzimidazole (ABZI) and its analogs, addresses prior challenges concerning stability and cellular uptake (37, 38). These breakthroughs herald a new era in the application of STING agonist immunotherapy for cancer, offering a comprehensive strategy for managing the disease (Figure 2).
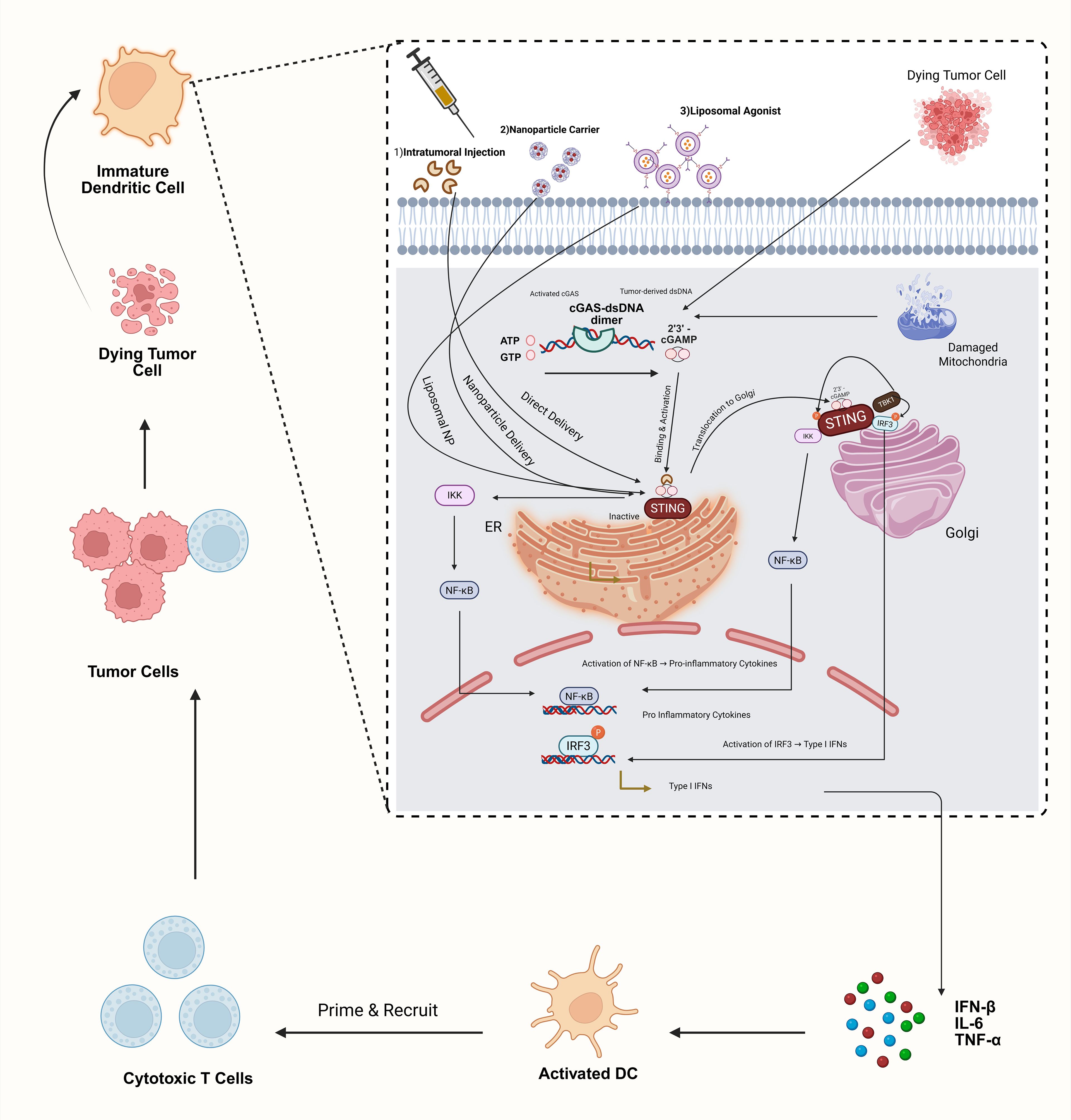
Figure 2. STING pathway activation with agonist delivery strategies. This schematic illustrates activation of the cGAS–STING axis in dendritic cells by (1) tumor-derived cytosolic dsDNA via cGAS conversion to 2′3′-cGAMP and (2) synthetic STING agonists delivered intratumorally, in polymeric nanoparticles, or in liposomal vesicles. Agonist binding induces STING translocation to the Golgi, recruitment of TBK1 and IKK, phosphorylation of IRF3, and NF-κB activation. The resulting type I interferon and pro-inflammatory cytokine production drive DC maturation and CD8+ T-cell priming. Clinical translation remains challenging due to inefficient agonist delivery and systemic toxicity.
3 Methods
A systematic literature search was conducted using PubMed, Web of Science, and ClinicalTrials.gov databases from inception until April 2025. Inclusion criteria involved studies on TIGIT, VISTA, GITR, STING, and TIM-3 in gastrointestinal cancers. Exclusion criteria included non-English articles, reviews without original data, and animal studies without clinical relevance. We additionally performed a formal quality assessment of all included studies using the Cochrane Risk-of-Bias tool (Higgins 2011) to evaluate methodological rigor and potential sources of bias (39).
4 Preclinical insights into cancer immunotherapy: focusing on TIGIT, VISTA, GITR, STING, and TIM-3 pathways
In the vanguard of oncological research, particularly within the immunotherapeutic domain, a series of foundational preclinical investigations have cast a spotlight on the TIGIT, VISTA, GITR, STING and TIM-3 signaling conduits. These studies delineate these conduits as quintessential targets for the genesis of groundbreaking therapeutic modalities aimed at potentiating the immune apparatus’s prowess in eradicating malignancy.
4.1 TIGIT
4.1.1 Mechanism
TIGIT functions as an inhibitory receptor primarily expressed on CD8+ T cells, CD4+ T cells, Tregs, and NK cells. It competes with the co-stimulatory receptor CD226 (DNAM-1) for binding to shared ligands CD155 (PVR) and CD112 (PVRL2) on antigen-presenting cells and tumor cells. Upon engagement, TIGIT transmits an intracellular inhibitory signal via its ITIM and ITT-like motifs, leading to decreased AKT phosphorylation, suppression of TCR signalling, and reduced secretion of IL-2, IFN-γ, and TNF-α. It also enhances Treg suppressive capacity while impairing NK cell cytotoxicity and promoting immune exhaustion. TIGIT signalling thus establishes an immunosuppressive microenvironment favorable for tumour survival and metastasis (40) (41–44).
4.1.2 Preclinical evidence
Murine models of head and neck, lung, and colorectal cancers have shown TIGIT upregulation in CD8+ and CD4+ tumor-infiltrating lymphocytes (TILs), often co-expressed with PD-1 and LAG-3 (45–47) TIGIT-blocking antibodies like EOS-448 and bispecific agents such as D3L-002 significantly enhanced T and NK cell-mediated antitumor immunity, particularly when combined with PD-1/PD-L1 inhibitors (48–50). Ex vivo models using GI cancer specimens showed TIGIT antagonists improved cytotoxic T cell function and modulated regulatory T cell suppressive markers (6, 8, 51, 52). Importantly, TIGIT blockade also synergizes with dendritic cell–T cell crosstalk mechanisms via IFN-γ and IL-12 signaling (53).
4.1.3 Model limitations
Mouse models often lack the complexity of human TME, particularly in TIGIT’s expression on Tregs and its interplay with other checkpoints. Differences in cytokine milieus and receptor-ligand affinities may limit the translatability of these results to human tumors (54, 57).
Phase III trials SKYSCRAPER-01 (tiragolumab + atezolizumab), SKYSCRAPER-06, and KeyVibe-008 (vibostolimab) failed due to lack of efficacy, diminishing expectations for TIGIT-targeted therapies (54–56).
4.2 VISTA
4.2.1 Mechanism
VISTA, also known as PD-1H, is an inhibitory checkpoint receptor predominantly expressed on myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and Tregs. Unlike other checkpoints, VISTA can function as both a ligand and a receptor. When acting as a ligand, it binds to PSGL-1 and VSIG-3 on T cells, especially under acidic pH conditions of the tumor microenvironment, delivering a suppressive signal that reduces T cell proliferation and cytokine production. Notably, VISTA binding to PSGL-1 is significantly enhanced under acidic conditions typical of the tumor microenvironment, underscoring its dual inhibitory and stimulatory capacities dependent on local context (27).
As a receptor, VISTA directly transmits inhibitory signals into myeloid cells, reducing their antigen presentation capabilities and skewing them toward a tolerogenic phenotype. This dual functionality enables tumors to exploit VISTA to blunt both innate and adaptive immune responses (23, 57, 58).
4.2.2 Preclinical evidence
In murine models of colorectal and pancreatic cancer, VISTA blockade led to increased CD8+ T cell infiltration and enhanced IFN-γ production. Combination therapy with anti-VISTA and anti-PD-1 antibodies showed superior tumor control compared to monotherapy, suggesting synergistic immunomodulatory effects (59, 60).
4.2.3 Model limitations
VISTA’s expression profile and immune functions differ significantly between species. In mice, VISTA is predominantly expressed on granulocytic MDSCs, while in humans it is more prominent on monocytic subsets. Additionally, the lack of homologous antibodies that mimic human VISTA-targeting compounds limits the translatability of murine findings. Preclinical models often fail to account for the dynamic regulation of VISTA in chronic inflammation and its context-dependent role in tolerance versus activation (21, 61).
4.3 GITR
4.3.1 Mechanism
GITR is a co-stimulatory immune receptor found constitutively on Tregs and upregulated on activated CD4+ and CD8+ T cells. Its ligand, GITRL, is expressed on APCs such as dendritic cells and macrophages. Upon ligand engagement, GITR activates the NF-κB and MAPK pathways through TRAF2 and TRAF5 signaling adaptors, promoting survival, proliferation, and effector functions in CD8+ T cells and Th1/Th17 subsets. Simultaneously, GITR ligation impairs Treg suppressive capacity by destabilizing FOXP3 expression and disrupting IL-10/TGF-β signaling loops. This dual effect enhances overall immune activation, making GITR a compelling target for restoring anti-tumor immunity, especially in immunosuppressive microenvironments (31, 62, 63). GITR agonist monotherapies have largely failed clinically due to dose-limiting immune toxicity and insufficient stimulation of anti-tumor immunity (64).
4.3.2 Preclinical evidence
GITR agonists have demonstrated synergy with PD-1 and LAG-3 inhibitors in murine models of melanoma and NSCLC, leading to improved tumor control and survival (62, 63, 65, 66). In GI tumor ex vivo models, GITR agonists selectively enhanced cytotoxic CD8+ T cell responses (6, 8, 66). Despite promising preclinical synergy, early-phase clinical monotherapy trials of GITR agonists (e.g., REGN6569 + cemiplimab) have encountered dose-limiting toxicities and underwhelming antitumor activity in solid tumors (67).
4.3.3 Model limitations
Rodent Tregs differ functionally from human Tregs in response to GITR agonism, necessitating caution in interpretation (68).Moreover, dose-dependent overstimulation of the immune system can trigger paradoxical immune suppression or autoimmune-like pathologies in some preclinical models. Additionally, the translation of efficacy from murine systems to humans has been inconsistent due to interspecies variation in GITR/GITRL signaling cascades (63).
4.4 STING
4.4.1 Mechanism
STING (Stimulator of Interferon Genes) is a cytosolic adaptor protein that senses the presence of aberrant cytosolic DNA, either through direct binding to cyclic dinucleotides (CDNs) like cGAMP or downstream of DNA sensors like cGAS. Upon activation, STING translocates from the ER to the Golgi, initiating a cascade involving TBK1-mediated phosphorylation of IRF3, leading to transcription of type I interferons (IFN-α/β) and proinflammatory cytokines. This immune activation recruits dendritic cells and primes cytotoxic T lymphocytes against tumor antigens. In the TME, STING activation enhances antigen presentation, reverses immune suppression, and can modulate stromal components like CAFs, reducing desmoplasia and improving immune cell infiltration (36, 69) (70, 71). Clinical application of STING agonists remains challenging due to delivery limitations and systemic toxicity, complicating their translation into effective treatments (72).
4.4.2 Preclinical evidence
STING agonists (e.g., ADU-S100) in combination with TLR9 ligands enhanced immune infiltration and reduced CAFs in colon carcinoma models, improving survival. Activation of STING pathways also boosted NK cell responses and proinflammatory cytokine levels in adjacent spleen tissues (69, 70).
4.4.3 Model limitations
Murine STING exhibits different binding affinities and downstream activation profiles compared to human STING, which can lead to overestimation of therapeutic efficacy in preclinical. Additionally, several human tumors show low baseline STING expression or have mutations in STING-related pathways, which are not recapitulated in mouse models (71). However, systemic STING agonists continue to be hampered by cytokine-release toxicities and dose-limiting myelitis, as reported in early MIW815 (ADU-S100) trials (37).
4.5 TIM-3
4.5.1 Mechanism
TIM-3 (T cell immunoglobulin and mucin-domain containing-3) is an inhibitory receptor expressed on exhausted CD8+ T cells, Tregs, NK cells, and dendritic cells. It binds to multiple ligands: Galectin-9 (induces apoptosis of effector T cells), phosphatidylserine (mediates clearance of apoptotic cells), HMGB1 (inhibits innate immune activation), and CEACAM1 (regulates tolerance and exhaustion). Upon ligand binding, TIM-3 disrupts TCR signaling by interacting with Bat3 and Fyn, leading to suppression of Th1 cytokines and CTL activity. In dendritic cells, TIM-3 downregulates nucleic acid sensing, reducing IFN production. TIM-3 also maintains Treg stability and suppresses inflammation, making it a key regulator of T cell exhaustion and immune dysfunction in chronic tumors (73–76).
4.5.2 Preclinical evidence
Preclinical studies using radiolabeled anti-TIM-3 antibodies have demonstrated selective uptake in colon and breast cancer mouse models. Co-blockade of TIM-3 with PD-1 restored CD8+ T cell function and suppressed tumor growth. TIM-3 targeting has also shown promise in selectively eliminating leukemia stem cells (76).
4.5.3 Model limitations
TIM-3 exhibits heterogeneity in expression across immune cell subsets and cancer types, making it difficult to identify predictive biomarkers for response. Moreover, ligand-binding affinity and downstream signalling differ between mice and humans due to species-specific receptor-ligand interactions (77). Additionally, monoclonal antibodies targeting TIM-3 may bind distinct epitopes in murine versus human systems, affecting pharmacodynamics and efficacy. These discrepancies necessitate validation in humanized or patient-derived models before clinical translation (77).
These preclinical ventures provide critical insights into the intricate mechanisms underlying immunological evasion and identify novel therapeutic targets. The orchestration of therapies derived from these insights is poised to revolutionize oncological care, enhancing the immunological arsenal against neoplastic entities. Table 1 provides a summary of the preclinical evaluation of immunotherapy checkpoints in gastrointestinal cancer models.
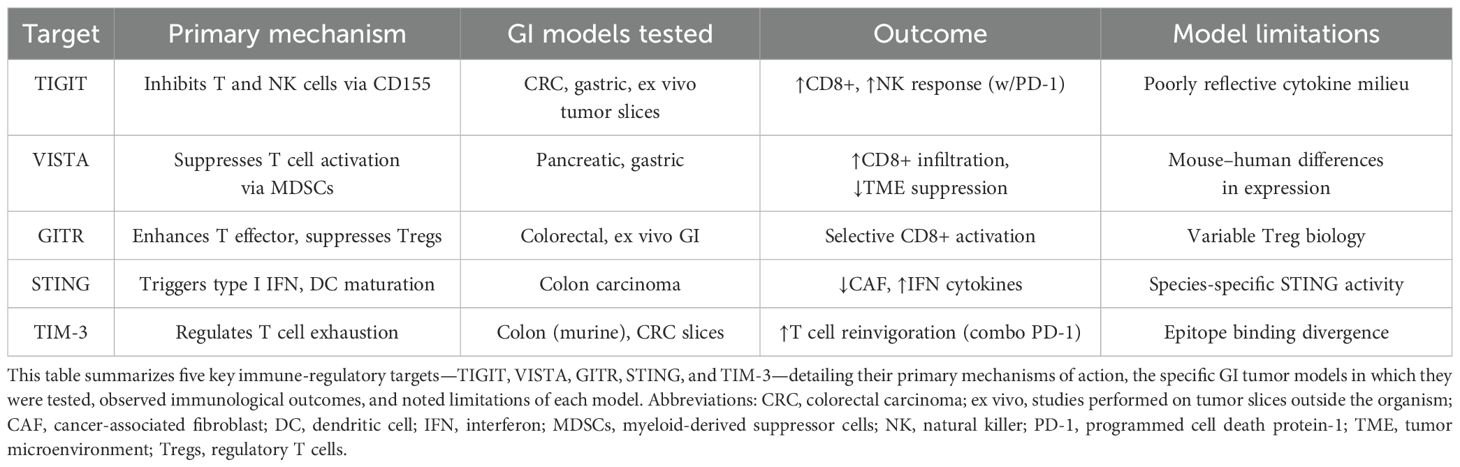
Table 1. Summary of preclinical evaluation of immunotherapy checkpoints in gastrointestinal cancer models.
5 Crosstalk, synergy and antagonism between emerging checkpoints
5.1 Crosstalk between TIGIT and the PD-1 axis
Functionally exhausted CD8+ TILs frequently co-express TIGIT and PD-1; dual blockade re-invigorates proliferation and cytokine release more potently than either antibody alone, as shown in murine head-and-neck and colorectal models and in ex-vivo GI-cancer slices where TIGIT+/PD-1+ T cells dominate (8, 78, 79).
5.2 GITR–TIGIT counter-regulation
GITR agonism destabilises FOXP3 in TIGIT^high Tregs, indirectly relieving TIGIT-mediated suppression of effector CD8+ cells, while TIGIT antagonists broaden the response spectrum to include dysfunctional CD8+ and TFH-like subsets (80, 81).
5.3 VISTA and PD-1 non-redundancy
In colorectal and pancreatic models, VISTA blockade heightens IFN-γ production, and the combination with anti-PD-1 produces superior tumour control versus monotherapy, reflecting parallel, non-overlapping immunosuppressive circuits (60, 82).
5.4 STING activation amplifies checkpoint inhibition
STING agonists up-regulate CXCL10 and CCL5, enhancing dendritic-cell priming and sensitising “cold” MSS-CRC and HCC to PD-1 blockade (83).
5.5 TIM-3 adaptive up-regulation post PD-1/TIGIT therapy
Preclinical data show compensatory TIM-3 expression after PD-1 ± TIGIT inhibition, supporting bispecific TIM-3/PD-1 antibodies now in phase I (84).
Figure 3 shows a schematic of these checkpoint interactions and STING-mediated activation in the tumor microenvironment.
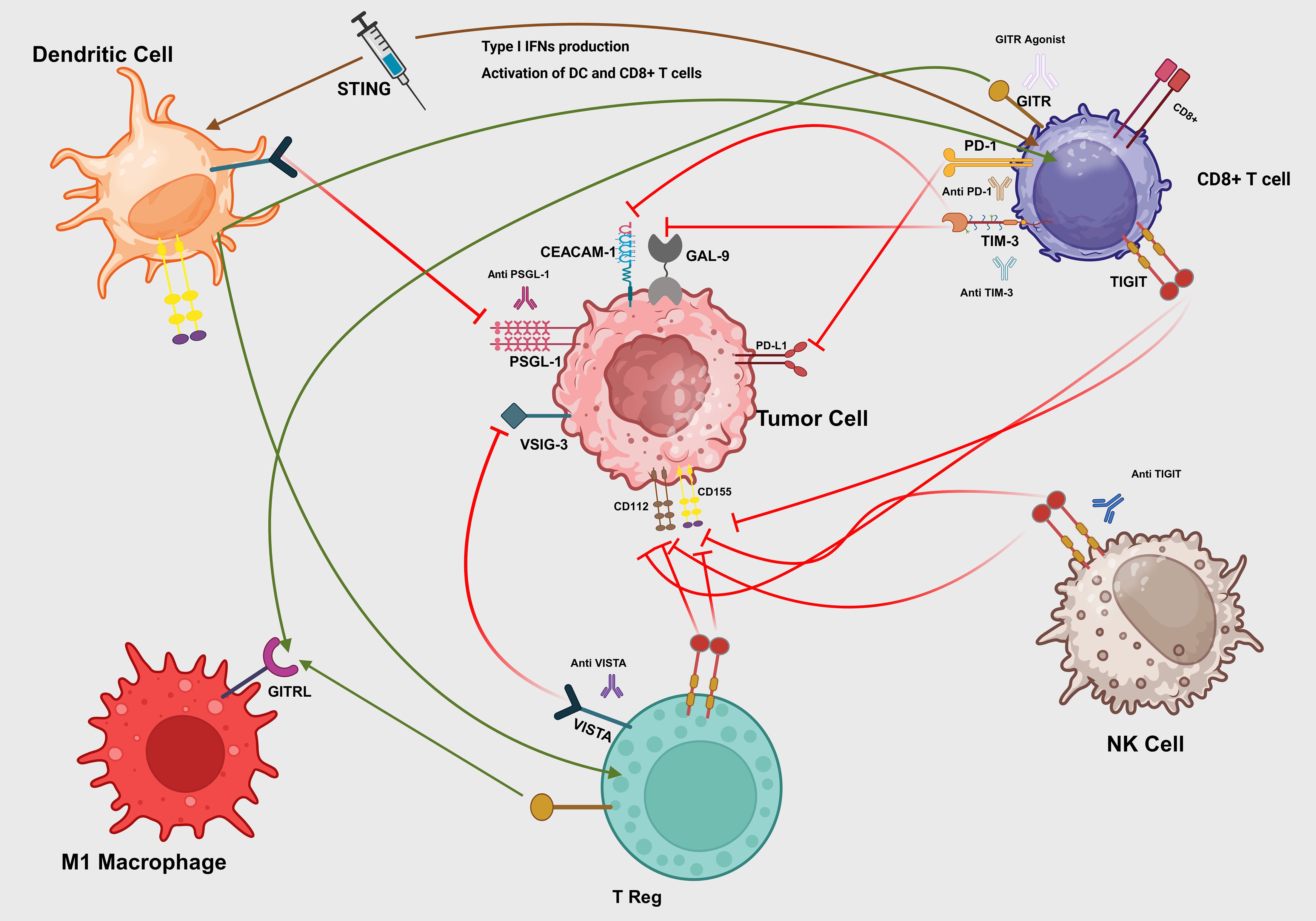
Figure 3. Interactions of immune checkpoints in the tumor microenvironment. A synthetic STING agonist (blue syringe) binds to the STING adaptor on immature dendritic cells (DCs), triggering production of type I interferons (IFN-β) that activate both DCs and CD8+ T cells (brown arrows). Mature DCs present tumor antigens and co-stimulatory signals to CD8+ T cells (green arrow), priming cytotoxic responses. Tumor-expressed PD-L1 engages PD-1 on CD8+ T cells (red T-bar), dampening TCR signaling. Galectin-9 (GAL-9) and CEACAM-1 on tumor cells bind TIM-3 on CD8+ T cells (red T-bars), driving exhaustion. Tumor ligands CD155 and CD112 engage TIGIT on NK cells (red T-bar), inhibiting cytotoxicity, which can be blocked by anti-TIGIT antibody. VSIG-3 and PSGL-1 on tumor cells interact with VISTA on Tregs and DCs (red T-bars), enforcing local immunosuppression, while VISTA blockade (green arrow) restores immune activation. M1 macrophage–expressed GITRL binds GITR on Tregs (green arrow), reinforcing suppression, but a GITR agonist antibody can convert this into co-stimulation on effector T cells. Therapeutic antibodies (anti-PD-1, anti-TIM-3, anti-VISTA, anti-PSGL-1) intercept their respective inhibitory axes to reinvigorate DC, T cell, and NK cell functions.
6 Integration and advancements in immuno-oncology clinical research
6.1 Insightful developments in targeting the TIGIT pathway
A pioneering investigation in Japan has showcased the potential of a novel therapeutic combination targeting advanced solid tumors. This approach, employing tiragolumab in conjunction with atezolizumab, has been characterized by its commendable safety profile, opening avenues for further large-scale evaluations. Grade ≥3 treatment-related AEs occurred in 15% of patients, chiefly hypertension and pruritus, and were comparable to atezolizumab-based historical controls (85). The consistency of therapeutic outcomes across various patient profiles underscores the promise of this regimen in future comprehensive trials aimed at establishing its efficacy (86). In line with these advancements, the MORPHEUS-EC trial is a phase Ib/II open-label, randomized study evaluating the efficacy and safety of the combination of tiragolumab (tira) and atezolizumab (atezo) with chemotherapy as a first-line treatment for patients with esophageal cancer. This trial aims to explore the potential benefits of adding immune checkpoint inhibitors to standard chemotherapy regimens, potentially improving patient outcomes in esophageal cancer (87). In this gastric cohort, domvanalimab + zimberelimab + FOLFOX produced an objective response rates (ORR) of 59% (69% in PD-L1-high patients) with a 12-month PFS rate of 58% (88).
Similarly, the MORPHEUS-Liver study focuses on a phase Ib/II randomized evaluation of tira, atezo, and bevacizumab in patients with unresectable, locally advanced, or metastatic hepatocellular carcinoma (uHCC). The primary goal of this study is to determine the efficacy and safety of this combination therapy in enhancing anti-tumor immune responses and improving overall survival rates in patients with advanced liver cancer (89). Across upper- and lower-GI malignancies, TIGIT- or TIM-3–directed combinations are now yielding a consistent response pattern. In PD-L1–high oesophageal and gastric cancers, ORR cluster around 55-70%, with 12-month progression-free survival (PFS) ≥ 50% as seen in MORPHEUS-EC and EDGE-Gastric (87, 90). The regimen’s safety mirrored PD-1 + chemo standards; most common any-grade AEs were neutropenia (61%), nausea (59%), and anemia (29%), with grade ≥3 immune-related events ≤ 2% (88).Hepatocellular carcinoma shows more modest activity (ORR ≈ 40%, median PFS ≈ 7 months), yet still clearly superior to historical PD-1 monotherapy benchmarks (85). By contrast, refractory colorectal cohorts seldom exceed single-digit ORR regardless of checkpoint targeted, mirroring the immunologically ‘cold’ micro-environment of most MSS CRC. This gradient underscores a tumour-intrinsic hierarchy of TIGIT-axis sensitivity across the GI spectrum and guides trial stratification moving forward.
6.2 Enhancing immunotherapy efficacy through strategic combinations
The collaboration of vibostolimab with pembrolizumab represents a significant stride in immunotherapy, particularly for NSCLC. This regimen’s ability to produce positive outcomes, particularly in patients new to anti-PD-1/PD-L1 therapy, positions it as a potentially transformative option in cancer treatment. The nuanced effectiveness in different patient subsets highlights the importance of continued exploration and optimization of such therapeutic strategies (7).
6.3 Reevaluating GITR’s influence on gastric cancer immunity
The intricate dynamics of GITR within the gastric cancer immune environment suggest a pivotal yet complex role in shaping patient prognosis. The association of GITR expression with a more suppressive tumor microenvironment invites a rethinking of therapeutic approaches, advocating for personalized strategies that consider the multifaceted roles of immune checkpoints. This perspective encourages a deeper examination of GITR’s potential as a therapeutic target, aiming for a more targeted and effective cancer immunotherapy (91).
6.4 Prognostic implications of VISTA in colorectal cancer
The relationship between VISTA expression and colorectal cancer outcomes offers new insights into the prognostic landscape of this disease. The correlation of high VISTA levels with several markers of better prognosis highlights its utility as a therapeutic target and a guide for clinical decision-making. This understanding enriches the dialogue on immunotherapeutic innovations and their potential to redefine treatment paradigms (92).
6.5 Synergistic frontiers: unveiling the potential of sabatolimab and spartalizumab in advanced solid tumors
In a pioneering phase I/Ib clinical trial, Curigliano et al. explored the combination of Sabatolimab, a TIM-3 antibody, received FDA Fast Track designation in May 2024 and entered Phase II studies by June 2024, with preliminary safety data reported at ASCO24 (93). This study enrolled 219 patients, revealing a tolerable safety profile predominantly characterized by fatigue and identifying an optimal phase II dose for further investigation. Dose-escalation showed ≤ 15% grade ≥3 TRAEs, predominantly fatigue, and no dose-limiting immune-mediated toxicity (94). Notably, the combination therapy elicited partial responses in a subset of patients across various solid tumors, including colorectal cancer and NSCLC, with responses lasting between 12 to 27 months. These findings underscore the potential of targeting the TIM-3 and PD-1 pathways simultaneously, offering a new horizon for patients previously unresponsive to conventional treatments and signaling a significant step forward in the realm of immunotherapy. The trial’s insights into the synergistic efficacy of sabatolimab and spartalizumab pave the way for future explorations into combination therapies, heralding a promising direction for personalized cancer treatment strategie (93). Table 2 summarises the ongoing trials of TIGIT, VISTA, GITR, and STING.
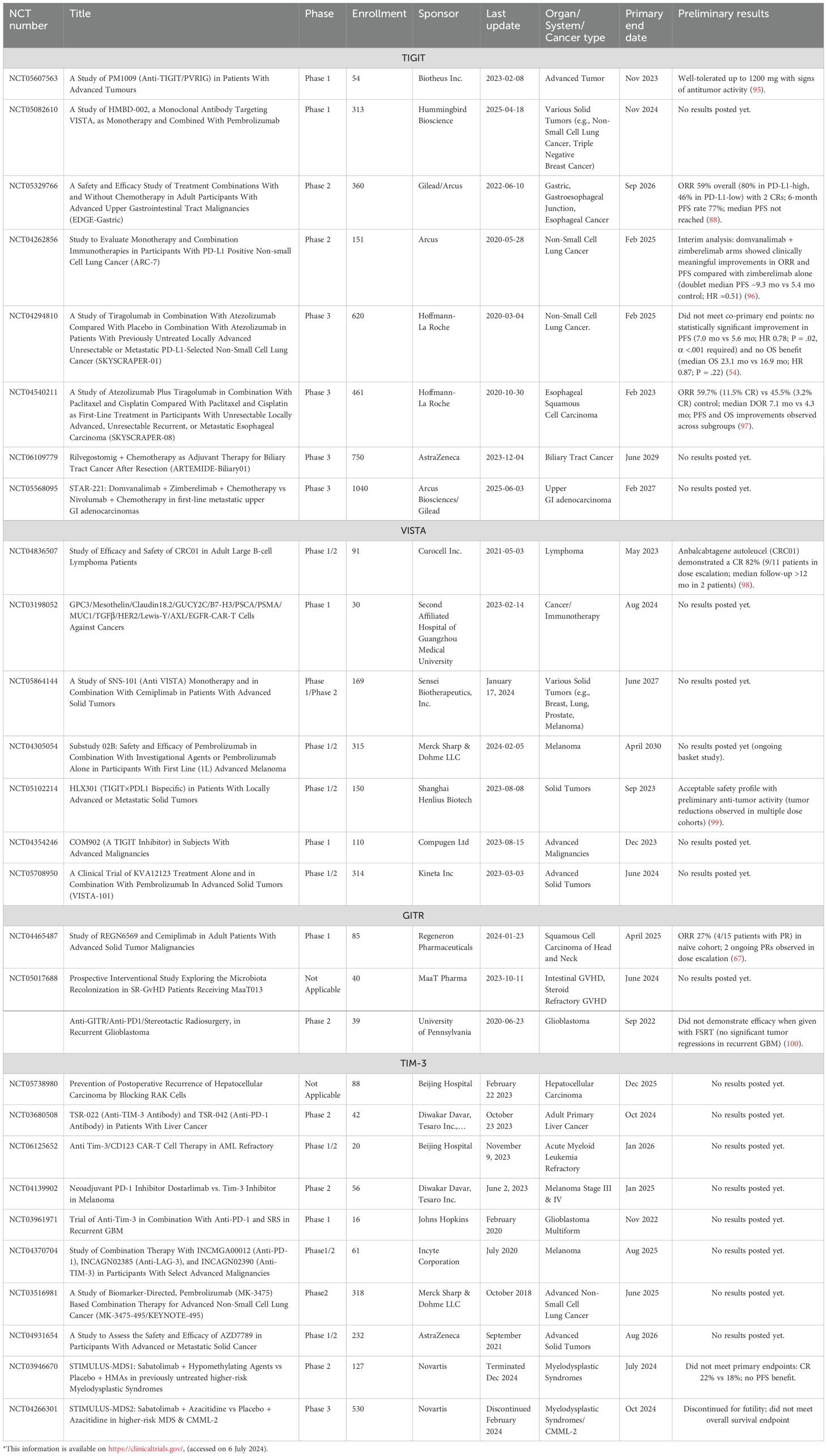
Table 2. Ongoing trials of TIGIT, VISTA, GITR and STING*.
7 Enhancing immunotherapy efficacy through strategic combinations
Recent studies have explored the potentiation of anti-PD-L1 and CD40-based immuno-chemotherapy combinations in heterogeneous pancreatic tumors through context-specific GITR agonism. Utilizing four representative immunocompetent mouse models, this research underscores the importance of GITR agonist therapy in enhancing the efficacy of existing immuno-chemotherapy strategies. This work provides a promising avenue for overcoming resistance mechanisms in pancreatic cancer, a malignancy known for its dismal prognosis (101).
Another significant contribution to GITR-focused preclinical trials is the study on the combination of PD1 inhibition and GITR agonism with fractionated stereotactic radiotherapy for treating recurrent glioblastoma. The combination of retifanlimab (anti-PD1) and INCAGN01876 (GITR agonist) may offer a novel therapeutic strategy for glioblastoma patients, suggesting an improvement in overcoming the high resistance of glioblastoma to conventional therapies (100).
Furthermore, some studies have focused on the heterogeneous cellular responses to GITR and TIGIT immunotherapy in the human gastrointestinal tumor microenvironment. Their findings emphasize the complexity of GITR’s role within the tumor microenvironment, suggesting that personalized approaches may be necessary to fully leverage the therapeutic potential of GITR agonism in gastrointestinal cancers (6).
8 Future perspectives in GI oncology: harnessing the potential of VISTA, TIGIT, GITR, STING and TIM-3
In the dynamic field of gastrointestinal (GI) cancer research, the emergence of innovative immunotherapy targets like VISTA, TIGIT, GITR, STING, and TIM-3 signals a groundbreaking shift in cancer care. This forward-looking perspective not only highlights the vast potential of these targets in scientific and clinical realms but also underlines the importance of adopting a holistic strategy to fully harness their advantages. Key to this endeavor is the leverage of advanced technologies, including next-generation sequencing and machine learning, to unveil novel targets and forecast treatment outcomes, facilitating the creation of efficient, tailored therapies.
8.1 Checkpoint modulation plus cellular or vaccine platforms
● Armoured CAR-T cells: CRISPR/Cas9 PD-1-knock-out mesothelin-CAR-T cells showed > 2-fold increased persistence and tumour clearance in gastric-cancer PDXs, and TIGIT-KO NK- or CAR-NK approaches further prevented fratricide (102).
● Checkpoint-enhanced cancer vaccines. Personalised neoantigen RNA vaccines combined with PD-1 and STING agonists elicited poly-epitope CD4+/CD8+ responses and durable regressions in refractory MSI-low colorectal cancer (103).
● Bispecific adaptor systems. A GITR-ligand/PD-1 bispecific adaptor fused into Claudin 18.2 CAR-T cells boosted IL-2 secretion and overcame the suppressive GI-TME in vitro and in murine orthotopic models (104).
These data suggest that coupling emerging checkpoints to cellular or vaccine platforms may convert transient responses into long-term disease control, a priority for hard-to-treat GI malignancies.
8.2 Prioritized combination strategies in the clinical pipeline
1) Dual TIGIT + PD-1 blockade in PD-L1-high oesophagogastric cancers, now yielding ORR ≈ 60% in phase II trials (88, 90). 2) STING agonists combined with PD-1 antibodies for microsatellite-stable colorectal and hepatocellular carcinomas to convert ‘cold’ tumours into inflamed phenotypes (105). 3) VISTA antagonism in myeloid-rich colorectal and pancreatic tumours where high VISTA expression drives immune escape (106). 4) GITR agonists paired with low-dose radiotherapy to expand effector CD8+ T cells while ablating intratumoural Tregs (107). 5) Bispecific TIM-3/PD-1 antibodies to forestall adaptive resistance seen with single-checkpoint inhibition (108). 6) Microbiome-modulating strategies (e.g., FMT) to amplify checkpoint efficacy across GI subtypes (109).
Actionable biomarkers should now extend beyond PD-L1 IHC. Multi-parameter panels already correlate with response: (i) tumour TIGIT-ligand (CD155) density together with TIGIT+CD8+ infiltration (101); (ii) VISTA-high MDSC score by CyTOF in colorectal cancer (109); (iii) circulating IFN-β-inducible chemokines (CXCL10, CCL5) as pharmacodynamic surrogates for STING agonism (105); (iv) baseline ctDNA mutation clearance at week 4 as an early on-treatment predictor of durable benefit. Prospective qualification of these signatures should be embedded in every registrational study.
A deep comprehension of the mechanisms behind therapy resistance is imperative for formulating approaches to maintain therapeutic efficacy. Customizing treatment regimens based on the patient’s genetic, molecular, and immune profiles will be a cornerstone in advancing personalized medicine within oncology.
Additionally, it’s paramount to assess the enduring safety and impact on the quality of life of these therapies, ensuring they are not only efficacious but also bearable for patients. Tackling regulatory and ethical issues, such as accessibility and fairness in treatment distribution, is crucial for the just dissemination of these innovations among varied patient demographics.
Key development hurdles remain. Species-specific differences in STING and TIM-3 epitopes complicate murine-to-human translation, delaying candidate selection. Dose-limiting cytokine-release with systemic STING agonists and myelitis seen with GITR agonism highlight the narrow therapeutic window. Tumour heterogeneity—the coexistence of immune-inflamed and immune-excluded niches within the same lesion—undermines single-biopsy biomarker reliability (110). Finally, real-world access is threatened by the high manufacturing cost of multi-antibody regimens and by region-specific reimbursement delays; modelling suggests a three-year lag between FDA approval and universal Canadian coverage (111). Addressing these issues will be as decisive as scientific progress itself.
Promoting international cooperation and the exchange of data among scientific and medical communities is essential for expediting discoveries and their clinical implementation, dismantling conventional obstacles and promoting a united effort to enhance patient care in GI oncology. This inclusive strategy, which considers technological, scientific, ethical, and social factors, lays the groundwork for revolutionary advancements in GI cancer treatment, offering improved outcomes and heralding an era where personalized medicine becomes the standard for patients worldwide.
9 Conclusion
Recent clinical and preclinical investigations of VISTA, TIGIT, GITR, STING, and TIM-3 in gastrointestinal oncology have yielded heterogeneous outcomes. While TIGIT and TIM-3 inhibitors demonstrated limited efficacy in randomized phase III trials (e.g., SKYSCRAPER-01, SKYSCRAPER-06, KeyVibe-008), early-phase studies of GITR agonists and STING agonists have shown manageable safety profiles but inconsistent antitumor activity. VISTA-targeting agents remain under evaluation, with no published efficacy readouts to date. Combination strategies—for example, dual TIGIT/PD-1 blockade or STING agonists plus PD-1 inhibitors—have generated higher response rates in select PD-L1–high and MSI-high cohorts but also highlight tumor-intrinsic resistance mechanisms in “cold” microsatellite-stable colorectal cancers.
Moving forward, rigorous biomarker-driven trial designs are essential for identifying patient subsets most likely to benefit, and the systematic incorporation of robust translational endpoints (e.g., on-treatment ctDNA clearance, STING pathway activation signatures) will guide adaptive treatment refinements. Realistic appraisal of dose-limiting toxicities and delivery hurdles—particularly for STING and GITR agents—must inform dosing and administration schedules. Ultimately, incremental advances grounded in controlled, evidence-based assessments will be necessary to transition these emerging checkpoints from early promise to reproducible clinical benefit in GI cancer.
Author contributions
AT: Writing – original draft, Writing – review & editing, Methodology, Project administration, Resources, Software, Validation, Visualization. AS: Supervision, Validation, Writing – review & editing. LC: Conceptualization, Investigation, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
Figures were created with BioRender.com.
Conflict of interest
AS reports research grants to institution from AstraZeneca, Bristol Myers Squibb, Merck, Clovis, Exelixis, Actuate Therapeutics, Incyte Corporation, Daiichi Sankyo, Five Prime Therapeutics, Amgen, Innovent Biologics, Dragonfly Therapeutics, KAHR Medical, BioNTech, and advisory board fees from Merck, AstraZeneca, Bristol Myers Squibb, Exelixis, Taiho, and Pfizer; Ludimila Cavalcante Consulting or Advisory Role: Pliant Therapeutics, Janssen, and CDR-Life. Stock and Other Ownership Interests: Actuate Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ammannagari N and Atasoy A. Current status of immunotherapy and immune biomarkers in gastro-esophageal cancers. J gastrointestinal Oncol. (2018) 9:196. doi: 10.21037/jgo.2017.06.12
2. Tian Z, Zeng Y, Peng Y, Liu J, and Wu F. Cancer immunotherapy strategies that target the cGAS-STING pathway. Front Immunol. (2022) 13:996663. doi: 10.3389/fimmu.2022.996663
3. Novartis. Novartis receives FDA fast track designation for sabatolimab (MBG453) in myelodysplastic syndromes. Novartis News Releases. (2021).
4. He Y, Cao J, Zhao C, Li X, Zhou C, and Hirsch FR. TIM-3, a promising target for cancer immunotherapy. OncoTargets Ther. (2018) 11:7005–9. doi: 10.2147/OTT.S170385
5. Desai A, Subbiah V, Dimou A, Deshane J, Goliwas K, Ponnazhagan S, et al. Exploring the potential of combination immune checkpoint strategies in non-small cell lung cancer (NSCLC). J Clin Oncol. (2023) 41(16_suppl):9037. doi: 10.1200/JCO.2023.41.16_suppl.9037
6. Sathe A, Ayala C, Bai X, Grimes SM, Shelton A, Lee B, et al. Abstract 4121: Heterogenous cellular responses to GITR and TIGIT immunotherapy in the human gastrointestinal tumor microenvironment. Cancer Res. (2023) 83:4121. doi: 10.1158/1538-7445.AM2023-4121
7. Niu J, Maurice-Dror C, Lee D, Kim D-W, Nagrial A, Voskoboynik M, et al. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer☆. Ann Oncol. (2022) 33:169–80. doi: 10.1016/j.annonc.2021.11.002
8. Sathe A, Ayala C, Bai X, Grimes SM, Lee B, Kin C, et al. GITR and TIGIT immunotherapy provokes divergent multicellular responses in the tumor microenvironment of gastrointestinal cancers. Genome Med. (2023) 15:100. doi: 10.1186/s13073-023-01259-3
9. Bi J, Zheng X, Chen Y, Wei H, Sun R, and Tian Z. TIGIT safeguards liver regeneration through regulating natural killer cell-hepatocyte crosstalk. Hepatology. (2014) 60:1389–98. doi: 10.1002/hep.27245
10. Harjunpää H and Guillerey C. TIGIT as an emerging immune checkpoint. Clin Exp Immunol. (2019) 200:108–19. doi: 10.1111/cei.13407
11. Molfetta R, Zitti B, Lecce M, Milito ND, Stabile H, Fionda C, et al. CD155: A multi-functional molecule in tumor progression. Int J Mol Sci. (2020) 21:922. doi: 10.3390/ijms21030922
12. Lozano E, Dominguez-Villar M, Kuchroo V, and Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol. (2012) 188:3869–75. doi: 10.4049/jimmunol.1103627
13. Sun Y, Luo J, Chen Y, Cui J, Lei Y, Cui Y, et al. Combined evaluation of the expression status of CD155 and TIGIT plays an important role in the prognosis of LUAD (lung adenocarcinoma). Int Immunopharmacology. (2020) 80:106198. doi: 10.1016/j.intimp.2020.106198
14. Sethy C, Goutam K, Nayak D, Pradhan R, Molla S, Chatterjee S, et al. Clinical significance of a pvrl 4 encoded gene Nectin-4 in metastasis and angiogenesis for tumor relapse. J Cancer Res Clin Oncol. (2020) 146:245–59. doi: 10.1007/s00432-019-03055-2
15. Yeo J, Ko M, Lee D-H, Park Y, and Jin H-S. TIGIT/CD226 axis regulates anti-tumor immunity. Pharmaceuticals. (2021) 14:200. doi: 10.3390/ph14030200
16. Schorer M, Rakebrandt N, Lambert K, Hunziker A, Pallmer K, Oxenius A, et al. TIGIT limits immune pathology during viral infections. Nat Commun. (2020) 11:1288. doi: 10.1038/s41467-020-15025-1
17. Mao L, Hou H, Wu S, Zhou Y, Wang J, Yu J, et al. TIGIT signalling pathway negatively regulates CD4+ T-cell responses in systemic lupus erythematosus. Immunology. (2017) 151:280–90. doi: 10.1111/imm.2017.151.issue-3
18. Frentzas S, Meniawy T, Kao SC-H, Wang R, Zuo Y, Zheng H, et al. AdvanTIG-105: Phase 1 dose-escalation study of anti-TIGIT monoclonal antibody ociperlimab (BGB-A1217) in combination with tislelizumab in patients with advanced solid tumors. J Clin Oncol. (2021) 39:2583. doi: 10.1200/JCO.2021.39.15_suppl.2583
19. Rodriguez-Abreu D, Johnson ML, Hussein MA, Cobo M, Patel AJ, Secen NM, et al. Primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-selected NSCLC (CITYSCAPE). J Clin Oncol. (2020) 38(15_suppl):9503. doi: 10.1200/JCO.2020.38.15_suppl.9503
20. Wang G, Tai R, Wu Y, Yang S, Wang J, Yu X, et al. The expression and immunoregulation of immune checkpoint molecule VISTA in autoimmune diseases and cancers. Cytokine Growth Factor Rev. (2020) 52:1–14. doi: 10.1016/j.cytogfr.2020.02.002
21. Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O’Connell S, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. (2014) 74:1924–32. doi: 10.1158/0008-5472.CAN-13-1504
22. Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res. (2014) 74:1933–44. doi: 10.1158/0008-5472.CAN-13-1506
23. Xu W, Dong J, Zheng Y, Zhou J, Yuan Y, Ta HM, et al. Immune-checkpoint protein VISTA regulates antitumor immunity by controlling myeloid cell–mediated inflammation and immunosuppression. Cancer Immunol Res. (2019) 7:1497–510. doi: 10.1158/2326-6066.CIR-18-0489
24. Harada H, Suzu S, Hayashi Y, and Okada S. BT-IgSF, a novel immunoglobulin superfamily protein, functions as a cell adhesion molecule. J Cell Physiol. (2005) 204:919–26. doi: 10.1002/jcp.v204:3
25. Wang J, Wu G, Manick B, Hernandez V, Renelt M, Erickson C, et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology. (2019) 156:74–85. doi: 10.1111/imm.2019.156.issue-1
26. Ghouzlani A, Rafii S, Karkouri M, Lakhdar A, and Badou A. The promising IgSF11 immune checkpoint is highly expressed in advanced human gliomas and associates to poor prognosis. Front Oncol. (2021) 10:608609. doi: 10.3389/fonc.2020.608609
27. Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A, et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature. (2019) 574:565–70. doi: 10.1038/s41586-019-1674-5
28. Yasinska IM, Meyer NH, Schlichtner S, Hussain R, Siligardi G, Casely-Hayford M, et al. Ligand-receptor interactions of galectin-9 and VISTA suppress human T lymphocyte cytotoxic activity. Front Immunol. (2020) 11:580557. doi: 10.3389/fimmu.2020.580557
29. DeRogatis JM, Viramontes KM, Neubert EN, and Tinoco R. PSGL-1 immune checkpoint inhibition for CD4+ T cell cancer immunotherapy. Front Immunol. (2021) 12:636238. doi: 10.3389/fimmu.2021.636238
30. Nocentini G and Riccardi C. GITR: a modulator of immune response and inflammation. Ther Targets TNF Superfamily. (2009) 647:156–73. doi: 10.1007/978-0-387-89520-8_11
31. Stephens GL, McHugh RS, Whitters MJ, Young DA, Luxenberg D, Carreno BM, et al. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol. (2004) 173:5008–20. doi: 10.4049/jimmunol.173.8.5008
32. Liao G, O’Keeffe MS, Wang G, van Driel B, de Waal Malefyt R, Reinecker H-C, et al. Glucocorticoid-induced TNF receptor family-related protein ligand is requisite for optimal functioning of regulatory CD4+ T cells. Front Immunol. (2014) 5:35. doi: 10.3389/fimmu.2014.00035
33. Nocentini G, Ronchetti S, Petrillo MG, and Riccardi C. Pharmacological modulation of GITRL/GITR system: therapeutic perspectives. Br J Pharmacol. (2012) 165:2089–99. doi: 10.1111/j.1476-5381.2011.01753.x
34. McIntire JJ, Umetsu SE, Akbari O, Potter M, Kuchroo VK, Barsh GS, et al. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nat Immunol. (2001) 2:1109–16. doi: 10.1038/ni739
35. Sánchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, et al. Tim-3 inhibits T helper type 1–mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. (2003) 4:1093–101. doi: 10.1038/ni987
36. Corrales L, McWhirter SM, Dubensky TW, and Gajewski TF. The host STING pathway at the interface of cancer and immunity. J Clin Invest. (2016) 126:2404–11. doi: 10.1172/JCI86892
37. Meric-Bernstam F, Sweis RF, Hodi FS, Messersmith WA, Andtbacka RHI, Ingham M, et al. Phase I dose-escalation trial of MIW815 (ADU-S100), an intratumoral STING agonist, in patients with advanced/metastatic solid tumors or lymphomas. Clin Cancer Res. (2022) 28:677–88. doi: 10.1158/1078-0432.CCR-21-1963
38. Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang S-Y, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. (2018) 564:439–43. doi: 10.1038/s41586-018-0705-y
39. Higgins JP, Altman DG, Gøtzsche PC, Jüni P, Moher D, Oxman AD, et al. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. bmj. (2011) 343. doi: 10.1136/bmj.d5928
40. Wu L, Mao L, Liu JF, Chen L, Yu GT, Yang LL, et al. Blockade of TIGIT/CD155 signaling reverses T-cell exhaustion and enhances antitumor capability in head and neck squamous cell carcinoma. Cancer Immunol Res. (2019) 7:1700–13. doi: 10.1158/2326-6066.CIR-18-0725
41. Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. (2018) 7:e1466769. doi: 10.1080/2162402X.2018.1466769
42. Sunseri N, Chen X, Wald N, Preillon J, Smith SM, Driessens G, et al. Beyond PD-1: investigating the therapeutic potential of TIGIT blockade in DLBCL. Blood. (2019) 134:391. doi: 10.1182/blood-2019-131493
43. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell. (2014) 26:923–37. doi: 10.1016/j.ccell.2014.10.018
44. Yu X, Harden K C, Gonzalez L, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. (2009) 10:48–57. doi: 10.1038/ni.1674
45. Zhou X, Ding X, Li H, Yang C, Ma Z, Xu G, et al. Upregulation of TIGIT and PD-1 in colorectal cancer with mismatch-repair deficiency. Immunol Investigations. (2021) 50:338–55. doi: 10.1080/08820139.2020.1758130
46. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
47. Hansen K, Kumar S, Logronio K, Whelan S, Qurashi S, Cheng H-Y, et al. COM902, a novel therapeutic antibody targeting TIGIT augments anti-tumor T cell function in combination with PVRIG or PD-1 pathway blockade. Cancer Immunology Immunother. (2021) 70:3525–40. doi: 10.1007/s00262-021-02921-8
48. Preillon J, Cuende J, Rabolli V, Garnero L, Mercier M, Wald N, et al. Restoration of T-cell effector function, depletion of tregs, and direct killing of tumor cells: the multiple mechanisms of action of a-TIGIT antagonist antibodies. Mol Cancer Ther. (2021) 20:121–31. doi: 10.1158/1535-7163.MCT-20-0464
49. Ge Z, Zhou G, Carrascosa LC, Gausvik E, Boor PP, Noordam L, et al. TIGIT and PD1 co-blockade restores ex vivo functions of human tumor-infiltrating CD8+ T cells in hepatocellular carcinoma. Cell Mol Gastroenterol Hepatology. (2021) 12:443–64. doi: 10.1016/j.jcmgh.2021.03.003
50. Han D, Xu Y, Zhao X, Mao Y, Kang Q, Wen W, et al. A novel human anti-TIGIT monoclonal antibody with excellent function in eliciting NK cell-mediated antitumor immunity. Biochem Biophys Res Commun. (2021) 534:134–40. doi: 10.1016/j.bbrc.2020.12.013
51. Chu X, Tian W, Wang Z, Zhang J, and Zhou R. Co-inhibition of TIGIT and PD-1/PD-L1 in cancer immunotherapy: mechanisms and clinical trials. Mol Cancer. (2023) 22:93. doi: 10.1186/s12943-023-01800-3
52. Rui H, Yang B, Yang X, Zheng Z, Chen Z, Lu J, et al. Preclinical characterization of D3L-002, a novel cancer immunotherapy bispecific antibody that elicits potent anti-tumor efficacy via co-blocking TIGIT and PVRIG. J ImmunoTherapy Cancer. (13832023) 11:A1538–A. doi: 10.1136/jitc-2023-SITC2023.1383
53. Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, et al. Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity. (2018) 49:1148–61. e7. doi: 10.1016/j.immuni.2018.09.024
54. Peters S, Herbst R, Horinouchi H, Paz-Ares L, Johnson M, Solomon B, et al. Abstract CT051: SKYSCRAPER-01: A phase III, randomized trial of tiragolumab (tira) + atezolizumab (atezo) versus placebo (pbo) + atezo in patients (pts) with previously-untreated PD-L1-high, locally advanced unresectable/metastatic NSCLC. Cancer Res. (2025) 85:CT051–CT. doi: 10.1158/1538-7445.AM2025-CT051
55. Roche. Roche provides update on phase II/III SKYSCRAPER-06 study in metastatic non-squamous non-small cell lung cancer. Roche Media Releases. (2024).
56. Sands J, Lu S, Aerts JG, Reck M, Navarro A, Shentzer T, et al. Coformulated vibostolimab/pembrolizumab plus chemotherapy as first-line therapy vs atezolizumab plus chemotherapy for extensive-stage small-cell lung cancer (ES-SCLC): randomized, phase 3 KEYVIBE-008. J ImmunoTherapy Cancer. (14632024) 12:A1693–A4. doi: 10.1136/jitc-2024-SITC2024.1463
57. Lines JL, Sempere LF, Broughton T, Wang L, and Noelle R. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol Res. (2014) 2:510–7. doi: 10.1158/2326-6066.CIR-14-0072
58. ElTanbouly MA and Noelle RJ. Rethinking peripheral T cell tolerance: checkpoints across a T cell’s journey. Nat Rev Immunol. (2021) 21:257–67. doi: 10.1038/s41577-020-00454-2
59. DiMascio L, Thakkar D, Gandhi N, Guan S, Rowinsky EK, Ingram P, et al. HMBD-002 is a novel, neutralizing, anti-VISTA antibody exhibiting strong preclinical efficacy and safety, being developed as a monotherapy and in combination with pembrolizumab. J Clin Oncol. (2021) 39(15_suppl):e14569. doi: 10.1200/JCO.2021.39.15_suppl.e14569
60. Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci. (2015) 112:6682–7. doi: 10.1073/pnas.1420370112
61. ElTanbouly MA, Zhao Y, Nowak E, Li J, Schaafsma E, Le Mercier I, et al. VISTA is a checkpoint regulator for naïve T cell quiescence and peripheral tolerance. Science. (2020) 367:eaay0524. doi: 10.1126/science.aay0524
62. Yan S, Lam S, and Ho J. EP16. 01–024 differential efficacy of combined GITR co-stimulation with PD-1 blockade in preclinical non-small cell lung cancer models. J Thorac Oncol. (2022) 17:S567.
63. Schaer DA, Budhu S, Liu C, Bryson C, Malandro N, Cohen A, et al. GITR pathway activation abrogates tumor immune suppression through loss of regulatory T-cell lineage stability. Cancer Immunol Res. (2013) 1:320–31. doi: 10.1158/2326-6066.CIR-13-0086
64. Jhajj H. Discovery and development of agonist antibodies for T cell receptors. J Immunother Cancer. (2022) 10(Suppl 2):A881.
65. Maniyar R, Zappasodi R, Elhanati Y, St Jean S, Carrasco S, Greenbaum B, et al. 843 Lag3 blockade in combination with GITR improves anti-tumor immune responses in a preclinical melanoma model. BMJ Specialist Journals. (2022). doi: 10.1136/jitc-2022-SITC2022.0843
66. Davar D, Zappasodi R, Wang H, Naik GS, Sato T, Bauer T, et al. Phase IB study of GITR agonist antibody TRX518 singly and in combination with gemcitabine, pembrolizumab, or nivolumab in patients with advanced solid tumors. Clin Cancer Res. (2022) 28:3990–4002. doi: 10.1158/1078-0432.CCR-22-0339
67. Lakhani NJ, Hamid O, Brana I, Lostes-Bardaji MJ, Gajate P, Lopez-Criado MP, et al. A phase 1 study of REGN6569, a GITR mAb, in combination with cemiplimab in patients (pts) with advanced solid tumor Malignancies: Initial dose-escalation results. J Clin Oncol. (2024) 42:2650. doi: 10.1200/JCO.2024.42.16_suppl.2650
68. Vence L, Bucktrout SL, Fernandez Curbelo I, Blando J, Smith BM, Mahne AE, et al. Characterization and comparison of GITR expression in solid tumors. Clin Cancer Res. (2019) 25:6501–10. doi: 10.1158/1078-0432.CCR-19-0289
69. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. (2015) 15:760–70. doi: 10.1038/nri3921
70. Hajiabadi S, Alidadi S, Farahi ZM, Seno MMG, Farzin H, and Haghparast A. Immunotherapy with STING and TLR9 agonists promotes synergistic therapeutic efficacy with suppressed cancer-associated fibroblasts in colon carcinoma. Front Immunol. (2023) 14. doi: 10.3389/fimmu.2023.1258691
71. Shih AY, Damm-Ganamet KL, and Mirzadegan T. Dynamic structural differences between human and mouse STING lead to differing sensitivity to DMXAA. Biophys J. (2018) 114:32–9. doi: 10.1016/j.bpj.2017.10.027
72. Nelson BE, Naing A, Fu S, Sheth RA, Murthy R, and Piha-Paul S. Potentiating intratumoral therapy with immune checkpoint inhibitors: shifting the paradigm of multimodality therapeutics. Immuno-Oncology Technology. (2024) 101040. doi: 10.1016/j.iotech.2024.101040
73. Tao J, Zeng Z, He C, Meng L, Zhou W, Ren Yn, et al. Construction and preclinical evaluation of 124I/125I-labeled antibody targeting T cell immunoglobulin and mucin domain-3. Mol Pharmaceutics. (2024) 21(2):944–56. doi: 10.1021/acs.molpharmaceut.3c01046
74. Anderson AC, Joller N, and Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
75. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. (2005) 6:1245–52. doi: 10.1038/ni1271
76. Ma S, Tian Y, Peng J, Chen C, Peng X, Zhao F, et al. Identification of a small-molecule Tim-3 inhibitor to potentiate T cell–mediated antitumor immunotherapy in preclinical mouse models. Sci Trans Med. (2023) 15:eadg6752. doi: 10.1126/scitranslmed.adg6752
77. Gandhi AK, Kim WM, Sun Z-YJ, Huang Y-H, Bonsor DA, Sundberg EJ, et al. High resolution X-ray and NMR structural study of human T-cell immunoglobulin and mucin domain containing protein-3. Sci Rep. (2018) 8:17512. doi: 10.1038/s41598-018-35754-0
78. Pawłowska A, Skiba W, Suszczyk D, Kuryło W, Jakubowicz-Gil J, Paduch R, et al. The dual blockade of the TIGIT and PD-1/PD-L1 pathway as a new hope for ovarian cancer patients. Cancers (Basel). (2022) 14. doi: 10.3390/cancers14235757
79. Le X, Dang M, Hegde VL, Jiang B, Slay R, Xiao W, et al. TIGIT as a therapeutic target of HPV-positive head and neck squamous cell carcinomas. medRxiv. (2021) 2021.12.02.21266776. doi: 10.1101/2021.12.02.21266776
80. Gruber T, Kremenovic M, Sadozai H, Rombini N, Baeriswyl L, Maibach F, et al. IL-32γ potentiates tumor immunity in melanoma. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.138772
81. Chen B-J, Zhao J-W, Zhang D-H, Zheng A-H, and Wu G-Q. Immunotherapy of cancer by targeting regulatory T cells. Int Immunopharmacology. (2022) 104:108469. doi: 10.1016/j.intimp.2021.108469
82. Martin AS, Molloy M, Ugolkov A, von Roemeling RW, Noelle RJ, Lewis LD, et al. VISTA expression and patient selection for immune-based anticancer therapy. Front Immunol. (2023) 14:1086102. doi: 10.3389/fimmu.2023.1086102
83. Banta KL, Xu X, Chitre AS, Au-Yeung A, Takahashi C, O’Gorman WE, et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8+ T cell responses. Immunity. (2022) 55:512–26. e9. doi: 10.1016/j.immuni.2022.02.005
84. Mao L, Xiao Y, Yang Q-C, Yang S-C, Yang L-L, and Sun Z-J. TIGIT/CD155 blockade enhances anti-PD-L1 therapy in head and neck squamous cell carcinoma by targeting myeloid-derived suppressor cells. Oral Oncol. (2021) 121:105472. doi: 10.1016/j.oraloncology.2021.105472
85. Finn RS, Ryoo BY, Hsu CH, Li D, Burgoyne AM, Cotter C, et al. Tiragolumab in combination with atezolizumab and bevacizumab in patients with unresectable, locally advanced or metastatic hepatocellular carcinoma (MORPHEUS-Liver): a randomised, open-label, phase 1b-2, study. Lancet Oncol. (2025) 26:214–26. doi: 10.1016/S1470-2045(24)00679-X
86. Yamamoto N, Koyama T, Sato J, Yoshida T, Sudo K, Iwasa S, et al. Phase I study of the anti-TIGIT antibody tiragolumab in combination with atezolizumab in Japanese patients with advanced or metastatic solid tumors. Cancer Chemother Pharmacol. (2024) 94(1):109–15. doi: 10.1007/s00280-023-04627-3
87. Sun J-M, Chao Y, Kim S-B, Rha SY, Evans TJ, Strickland A, et al. MORPHEUS-EC: A phase Ib/II open-label, randomized study of first-line tiragolumab (tira)+ atezolizumab (atezo)+ chemotherapy (CT) in patients (pts) with esophageal cancer (EC). J Clin Oncol. (2024) 42(3_suppl):324. doi: 10.1200/JCO.2024.42.3_suppl.324
88. Janjigian YY, Oh D-Y, Pelster M, Wainberg ZA, Sison EAR, Scott JR, et al. EDGE-Gastric Arm A1: Phase 2 study of domvanalimab, zimberelimab, and FOLFOX in first-line (1L) advanced gastroesophageal cancer. J Clin Oncol. (2023) 41(36_suppl):433248. doi: 10.1200/JCO.2023.41.36_suppl.433248
89. Finn RS, Ryoo B-Y, Hsu C-H, Li D, Burgoyne A, Cotter C, et al. Results from the MORPHEUS-liver study: Phase Ib/II randomized evaluation of tiragolumab (tira) in combination with atezolizumab (atezo) and bevacizumab (bev) in patients with unresectable, locally advanced or metastatic hepatocellular carcinoma (uHCC). J Clin Oncol. (2023) 41(16_suppl):4010. doi: 10.1200/JCO.2023.41.16_suppl.4010
90. Rha S, Oh D, Pelster M, Wainberg Z, Sison A, Scott J, et al. 130MO EDGE-Gastric Arm A1: Phase II study of domvanalimab (D), zimberelimab (Z), and FOLFOX in first-line (1L) advanced gastroesophageal (GE) cancer. Ann Oncol. (2024) 35:S1455. doi: 10.1016/j.annonc.2024.10.156
91. Ke S, Xie F, Guo Y, Chen J, Wang Z, Yu Y, et al. High-level of intratumoral GITR+ CD4 T cells associate with poor prognosis in gastric cancer. Iscience. (2022) 25. doi: 10.1016/j.isci.2022.105529
92. Zong L, Yu S, Mo S, Zhou Y, Xiang Y, Lu Z, et al. High VISTA expression correlates with a favorable prognosis in patients with colorectal cancer. J Immunother. (2021) 44:22–8. doi: 10.1097/CJI.0000000000000343
93. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/Ib clinical trial of sabatolimab, an anti–TIM-3 antibody, alone and in combination with spartalizumab, an anti–PD-1 antibody, in advanced solid tumors. Clin Cancer Res. (2021) 27:3620–9. doi: 10.1158/1078-0432.CCR-20-4746
94. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/ib clinical trial of sabatolimab, an anti-TIM-3 antibody, alone and in combination with spartalizumab, an anti-PD-1 antibody, in advanced solid tumors. Clin Cancer Res. (2021) 27:3620–9. doi: 10.1158/1078-0432.CCR-20-4746
95. Xue L, Wu L, Tang W, Wang Z, Xue J, Li Q, et al. Phase I safety and preliminary efficacy of PM1009, a bispecific antibody targeting TIGIT and PVRIG, in patients with advanced solid tumors. J Clin Oncol. (2024)(16_suppl):e14695. doi: 10.1200/JCO.2024.42.16_suppl.e14695
96. Johnson ML, Fox W, Lee Y-G, Lee KH, Ahn HK, Kim Y-C, et al. ARC-7: Randomized phase 2 study of domvanalimab + zimberelimab ± etrumadenant versus zimberelimab in first-line, metastatic, PD-L1-high non-small cell lung cancer (NSCLC). J Clin Oncol. (2022) 40:397600. doi: 10.1200/JCO.2022.40.36_suppl.397600
97. Hsu C-H, Lu Z, Gao S, Wang J-Y, Sun J-M, Liu T, et al. SKYSCRAPER-08: A phase III, randomized, double-blind, placebo-controlled study of first-line (1L) tiragolumab (tira) + atezolizumab (atezo) and chemotherapy (CT) in patients (pts) with esophageal squamous cell carcinoma (ESCC). J Clin Oncol. (2024) 42:245. doi: 10.1200/JCO.2024.42.3_suppl.245
98. Kim WS, Kim SJ, Yoon SE, and Kim JR. S214: PHASE 1/2 STUDY OF ANBAL-CEL, NOVEL ANTI-CD19 CAR-T THERAPY WITH DUAL SILENCING OF PD-1 AND TIGIT IN RELAPSED OR REFRACTORY LARGE B CELL LYMPHOMA. HemaSphere. (2022) 6:115–6. doi: 10.1097/01.HS9.0000843748.64635.ac
99. Rohrberg KS, Brandão M, Castanon Alvarez E, Felip E, Gort EH, Hiltermann TJJN, et al. Safety, pharmacokinetics (PK), pharmacodynamics (PD) and preliminary efficacy of AZD2936, a bispecific antibody targeting PD-1 and TIGIT, in checkpoint inhibitor (CPI)-experienced advanced/metastatic non-small-cell lung cancer (NSCLC): First report of ARTEMIDE-01. J Clin Oncol. (2023) 41(16_suppl):9050. doi: 10.1200/JCO.2023.41.16_suppl.9050
100. Bagley SJ, Mathew D, Desai AS, Chen K, Long Q, Shabason JE, et al. PD1 inhibition and GITR agonism in combination with fractionated stereotactic radiotherapy for the treatment of recurrent glioblastoma: A phase 2, multi-arm study. J Clin Oncol. (2023) 41:2004. doi: 10.1200/JCO.2023.41.16_suppl.2004
101. Ragulan C, Desai K, Lawrence PV, Ikami Y, Aalam MM, Ps H, et al. Context-specific GITR agonism potentiates anti-PD-L1 and CD40-based immuno-chemotherapy combination in heterogeneous pancreatic tumors. bioRxiv. (2023). 2023.06.16.545301. doi: 10.1101/2023.06.16.545301
102. Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol Immunol. (2021) 18:2188–98. doi: 10.1038/s41423-021-00749-x
103. Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. (2017) 547:222–6. doi: 10.1038/nature23003
104. Chan S, Belmar N, Ho S, Rogers B, Stickler M, Graham M, et al. An anti-PD-1-GITR-L bispecific agonist induces GITR clustering-mediated T cell activation for cancer immunotherapy. Nat Cancer. (2022) 3:337–54. doi: 10.1038/s43018-022-00334-9
105. Liu C, Tang L, Yang W, Gu Y, Xu W, Liang Z, et al. cGAS/STING pathway and gastrointestinal cancer: Mechanisms and diagnostic and therapeutic targets. Oncol Rep. (2025) 53:1–14. doi: 10.3892/or.2024.8848
106. Chmiel P, Gęca K, Michalski A, Kłosińska M, Kaczyńska A, Polkowski WP, et al. Vista of the future: novel immunotherapy based on the human V-set immunoregulatory receptor for digestive system tumors. Int J Mol Sci. (2023) 24:9945. doi: 10.3390/ijms24129945
107. Schoenhals JE, Cushman TR, Barsoumian HB, Li A, Cadena AP, Niknam S, et al. Anti-glucocorticoid-induced tumor necrosis factor–related protein (GITR) therapy overcomes radiation-induced treg immunosuppression and drives abscopal effects. Front Immunol. (2018) 9:2170. doi: 10.3389/fimmu.2018.02170
108. Wen J, Cui W, Yin X, Chen Y, Liu A, Wang Q, et al. Application and future prospects of bispecific antibodies in the treatment of non-small cell lung cancer. Cancer Biol Med. (2025) 22:348–75. doi: 10.20892/j.issn.2095-3941.2024.0470
109. Zhang RJ and Kim TK. VISTA-mediated immune evasion in cancer. Exp Mol Med. (2024) 56:2348–56. doi: 10.1038/s12276-024-01336-6
110. Liu K, Yuan S, Wang C, and Zhu H. Resistance to immune checkpoint inhibitors in gastric cancer. Front Pharmacol. (2023) 14:1285343. doi: 10.3389/fphar.2023.1285343
Keywords: immune checkpoints, gastrointestinal oncology, TIGIT, GITR, VISTA, STING, TIM-3, cancer immunotherapy
Citation: Tojjari A, Saeed A and Cavalcante L (2025) Emerging IO checkpoints in gastrointestinal oncology. Front. Immunol. 16:1575713. doi: 10.3389/fimmu.2025.1575713
Received: 12 February 2025; Accepted: 07 July 2025;
Published: 24 July 2025.
Edited by:
Tonya J Webb, University of Maryland, United StatesReviewed by:
Duanwu Zhang, Fudan University, ChinaGuillaume Beyrend, Université de Lille, France
Chao Yang, Wuhan University, China
Copyright © 2025 Tojjari, Saeed and Cavalcante. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ludimila Cavalcante, bWtqNGdyQHV2YWhlYWx0aC5vcmc=