 Tongwang Yang
Tongwang Yang Zhiyun Gu
Zhiyun Gu Juan Feng
Juan Feng Cheng Qian
Cheng Qian- Chongqing Key Laboratory of Translational Research for Cancer Metastasis and Individualized Treatment, Chongqing University Cancer Hospital, School of Medicine, Chongqing University, Chongqing, China
Non-neoplastic chronic liver diseases (CLDs), including alcoholic liver disease, metabolic-associated fatty liver disease, viral hepatitis, fibrosis, and cirrhosis, pose a global health challenge due to progressive fibro-inflammatory remodeling. Emerging evidence highlights the pivotal roles of non-parenchymal cells (NPCs)—liver sinusoidal endothelial cells (LSECs), hepatic stellate cells (HSCs), Kupffer cells (KCs), and innate immune lymphocytes such as natural killer (NK) and natural killer T (NKT) cells—in driving disease progression. Chronic liver injury triggers LSEC capillarization, HSC transdifferentiation into collagen-producing myofibroblasts, and KC polarization toward pro-inflammatory phenotypes, collectively exacerbating extracellular matrix deposition and immune dysregulation. Dysfunctional NK/NKT cells play dual roles in antiviral defense and fibrosis amplification through excessive cytokine production. This review summarizes recent advances in understanding NPC-driven mechanisms underlying chronic liver injury and fibrosis, with a focus on LSEC dysfunction, HSC activation, and inflammation mediated by KCs and NK/NKT cells. Furthermore, we delve into emerging therapeutic strategies aimed at targeting NPC-specific pathways, including mechanotransduction modulation in LSECs, metabolic reprogramming of HSCs, and regulation of KC polarization. These approaches provide valuable insights into halting CLD progression and advancing the development of innovative antifibrotic therapies.
Introduction
Chronic liver diseases (CLDs), including alcoholic liver disease (ALD), metabolic-associated fatty liver disease (MAFLD), viral hepatitis, fibrosis and cirrhosis, are key drivers of liver cancer development and contribute significantly to the high global mortality associated with liver diseases (1–4). There are regional variations in the etiology of liver cancer: chronic hepatitis B virus (HBV) infection remains the leading cause of hepatocellular carcinoma (HCC) in Asia (5), while metabolic dysfunction-associated steatohepatitis (MASH) has become the predominant risk factor in Western populations (6). Other contributing factors, such as aflatoxin exposure, alcohol misuse, tobacco use, obesity, and diabetes, further accelerate the progression of CLDs (7–10). If left unmanaged, these underlying conditions often lead to cirrhosis, with approximately 85% of cirrhotic patients ultimately progressing to HCC (11). Therefore, gaining deeper insights into the pathogenesis of CLDs is crucial for the early prevention of HCC.
The liver is a vital immune organ enriched with diverse cell populations (12). It comprises 80% parenchymal cells (hepatocytes) and 20% non-parenchymal cells (NPCs) (13). NPCs include liver sinusoidal endothelial cells (LSECs), hepatic stellate cells (HSCs), Kupffer cells (KCs), and innate lymphocytes such as NK and NKT cells (12, 14). Together, parenchymal and non-parenchymal cells form the structural basis of hepatic lobules: hepatocytes are organized into cords, while NPCs reside in the sinusoidal compartment. Specifically, LSECs line the sinusoids, KCs (liver-resident macrophages) are mainly localized within sinusoidal lumens, and HSCs occupy the perisinusoidal space of Disse (15). Interactions between these cells and sinusoid-dwelling NK/NKT cells are crucial for hepatic immunity and disease pathogenesis.
The development of CLD involves multifaceted cellular dysfunction. For instance, LSEC capillarization impairs their physiological functions (16), while HSC activation drives their transformation into extracellular matrix (ECM)-producing myofibroblasts (17). Concurrently, KCs polarize into M1 phenotypes, releasing mediators such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6 (18). NK and NKT cells further aggravate fibrosis and inflammation (19, 20). Among these, HSC activation is a pivotal event in CLD progression. Research indicates that activated HSCs release extracellular vesicles (EVs) during the early stages of activation, which induce pro-inflammatory KC polarization via Toll-like receptor 4 (TLR4) signaling (21). Interestingly, LSEC-derived EVs counteract fibrogenic phenotypes in activated HSCs and mitigate KC-mediated inflammation (22). Lipopolysaccharide (LPS) also activates LSECs to enhance IL-12/IL-18-driven interferon (IFN)-γ production in NK cells. IFN-γ promotes LSEC secretion of CXCL10, recruiting additional NK cells to amplify hepatic inflammation (16). Conversely, NK/NKT cell-derived IFN-γ can also attenuate liver injury (23). Modulating NK/NKT cell activity presents promising therapeutic opportunities. Activation of the E-prostanoid 3 receptor (EP3) enhances NK cell adhesion and cytotoxicity toward HSCs (24), while fasudil, a RhoA kinase inhibitor, attenuates fibrosis by activating NK cells and inhibiting HSC activation (25). These findings underscore the importance of understanding NPC interactions in CLD treatment.
This review highlights recent advances in elucidating the roles of liver-specific NPCs, including LSECs, HSCs, KCs, and innate lymphoid subsets (NK/NKT cells) in chronic liver injury, fibrosis, and cirrhosis. We emphasize their mechanistic contributions to CLD pathogenesis and explore emerging therapeutic strategies aimed at targeting these cells to reshape disease progression.
Liver sinusoidal endothelial cells
LSECs are a unique and highly specialized endothelial cell population, representing the most abundant NPC type that lining the sinusoidal capillaries of the liver, characterized by their plentiful fenestrae, absence of a continuous basement membrane, and lack of diaphragms. LSECs establish a highly permeable barrier between the bloodstream and hepatic parenchyma, playing a pivotal role in maintaining liver homeostasis (26). Under normal physiological conditions, LSECs regulate hepatic vascular tone and maintain the quiescent state of hepatic stellate cells, thereby inhibiting intrahepatic vasoconstriction and the development of fibrosis (27, 28). However, under pathological conditions, LSECs undergo capillarization, a process that promotes angiogenesis, vasoconstriction, and the release of vascular signaling molecules that drive the progression of liver fibrosis (16). For instance, in liver fibrotic models induced by carbon tetrachloride or bile duct ligation, levels of adipocyte fatty acid binding protein (A-FABP) derived from LSECs are elevated. This elevation activates the Hedgehog signaling pathway, facilitating LSEC capillarization (29). Capillarized LSECs undergo partial endothelial-to-mesenchymal transition, acquiring a myofibroblast-like phenotype that further promotes ECM synthesis and results in the deposition of perisinusoidal ECM, thereby exacerbating liver fibrosis (30). Recent studies have indicated that employing agents to inhibit ECM deposition can decrease ECM stiffness, potentially reversing LSEC capillarization. This finding opens new avenues for the treatment of diseases associated with liver fibrosis (31).
In addition to capillarization, the functional impairment of LSECs induced by liver injury is also a critical factor in the early development of liver diseases such as fibrosis and hepatitis. Aging is considered to increase susceptibility to CLD and accelerate the progression of liver fibrosis. Dai et al. found that aging leads to LSEC dysfunction by downregulating the expression of SIRT1 in hepatocytes, which subsequently activates HSCs and exacerbates the deterioration of liver fibrosis (32). Furthermore, LSECs are recognized as important regulators in the progression of liver disease. In pathological conditions of liver fibrosis or cirrhosis, LSECs respond significantly to the stimulation of TNF-α and transforming growth factor (TGF)-β, promoting the expression and secretion of the intermediate factor Midkine (MK) and its receptors, integrins α4 and α6. This process exacerbates LSEC dysfunction through a self-amplifying feedback mechanism, further facilitating the progression of hepatic vascular lesions (33). During intrahepatic sinusoidal remodeling induced by chronic liver injury, LSECs upregulate type IV collagen (COL4) in a TNF-α/NF-κB dependent manner. The deposition of COL4 promotes sinusoidal remodeling and the development of portal hypertension (PHTN) through the activation of angiogenic sprouting of LSECs (34). These findings suggest that restoring the normal function and phenotype of LSECs may represent an effective strategy for treating CLDs.
Targeting liver sinusoidal endothelial cells
Given that the transformation and dysfunction of LSECs are pivotal in the onset and progression of liver diseases, we explored the mechanisms of recently identified surface receptors on LSECs, along with other components expressed by these cells, in the context of LSEC-associated liver pathologies (Figure 1). Research has shown that LSEC capillarization induced by liver injury mediates the activation of HSCs, a key driver in the development of liver fibrosis (35). In murine models of diet-induced non-alcoholic steatohepatitis (NASH) and liver fibrosis caused by carbon tetrachloride, Guo et al. found that overexpression of LSEC VCAM-1 promotes LSEC capillarization, enhances HSC activation, and accelerates liver fibrosis by activating the Hippo pathway effector YAP1 (36). Furthermore, research has revealed that the expression of endothelial group IVA phospholipase A2 (IVA-PLA2) in LSECs drives sinusoidal capillarization, amplifies HSC activation and contributes to fibrosis, accelerating the progression of NASH. Therefore, developing inhibitors or siRNA-based therapeutics targeting LSEC-specific delivery systems may represent a pivotal strategy for treating NASH-related liver fibrosis (37). The ECM is a crucial component of the LSEC microenvironment, and its impact on LSEC function has drawn significant attention. Studies have demonstrated a mechano transduction mechanism between the ECM and LSECs, revealing that ECM stiffness induces LSEC capillarization via a NO-dependent pathway. Researchers propose that treatment with silybin might alleviate ECM stiffness in fibrotic liver tissue by modulating its biomechanical properties, thereby reducing liver sinusoid capillarization (31). Beyond addressing LSEC capillarization, targeting LSEC-mediated vascular signaling pathways holds potential for advancing liver disease therapies. For instance, TNF-α enhances the expression of CCL2 by promoting the interaction of p300 with NF-kB and BRD4 (38). Greuter et al. have uncovered that mechano transduction-induced glycolysis promotes NF-kB mediated expression of CXCL1, both of which exacerbate liver fibrosis and portal hypertension (39). Consequently, targeting p300 and its binding partners, along with intervention of the glycolytic pathway, may offer promising therapeutic strategies for liver disease. Previous research has identified that the deficiency of the sinusoidal master regulator GATA4 in LSECs drives a profibrotic angiocrine switch and activates HSC by upregulating the expression of the pro-fibrotic vascular secretion factor PDGFB and the transcription factor MYC. These findings highlight the potential therapeutic value of targeting endothelial GATA4 in preventing liver fibrosis and promoting hepatic regeneration (40). A deeper understanding of LSEC function and the underlying mechanisms will be pivotal in the development of novel treatments for liver diseases.
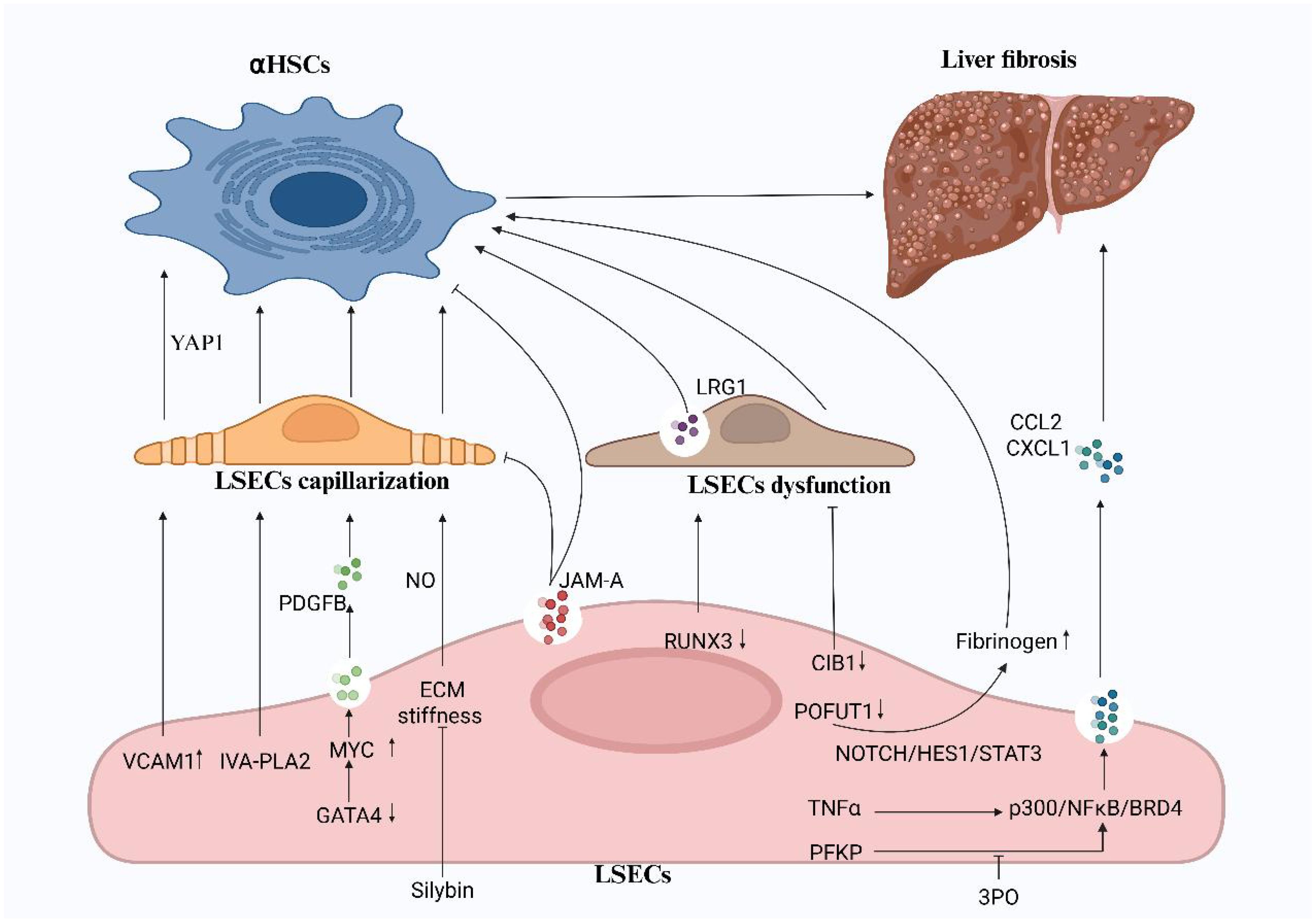
Figure 1. The capillarization and dysfunction of LSECs promote liver fibrosis. The activation of HSCs, driven by the capillarization and functional impairment of LSECs, is a key factor in the progression of liver fibrosis. Targeted inhibition of VCAM1, IVA-PLA2, and ECM stiffness, along with the maintenance of GATA4 expression, can alleviate LSEC capillarization. Silybins have emerged as an effective therapeutic agent in reducing ECM stiffness. while the glycolysis inhibitor 3PO can reduce early-stage liver fibrosis in vivo. CIB1, RUNX3, POFUT1, and JAM-A are important factors regulating LSEC function and represent promising targets for the treatment of liver fibrosis. VCAM-1, vascular cell adhesion molecule1; IVA-PLA2, IVA phospholipase A2; CIB1, calcium-and integrin-binding protein1; RUNX3, runt-related transcription factor 3; POFUT1, protein O-fucosyltransferase 1; JAM-A, junctional adhesion molecule A; PFKP, phosphofructokinase 1 isoform P; NF-kB, nuclear factor kappa B; BRD4, bromodomain-containing protein 4. (Created in BioRender).
Dysfunctional LSEC is also a key factor involved in the development and progression of liver fibrosis. Numerous factors, such as aging, increased liver stiffness, and abnormal expression of various transcriptional regulators and growth factors, contribute to the functional impairment of LSECs (32, 33, 41, 42). Recently, Wang et al. demonstrated that the expression of CIB1 is significantly increased in LSECs during cirrhosis. Knockdown of CIB1 can improve LSEC function by modulating intracellular tension and inhibiting inflammatory responses (41). Uttam et al. identified RUNX3 as a crucial regulator of the gatekeeping functions of LSECs. Deficiency in RUNX3 leads to LSEC dysfunction and promotes the production of a novel vascular secretion factor, LRG1, which activates HSCs via the TGFBR1-SMAD2/3 signaling pathway. Thus, LRG1 may serve as a potential therapeutic target for liver fibrosis (42). He et al. identified POFUT1 was another critical regulatory factor in LSECs, POFUT1 protects against injury-induced liver fibrosis by inhibiting the expression of fibrinogen (43). Furthermore, previous studies have revealed that the LSEC-specific regulatory factor JAM-A controls the formation of liver sinusoidal capillaries and maintains the quiescent state of HSCs (44). These findings offer valuable insights into the role of LSECs in liver fibrosis and propose potential therapeutic strategies targeting LSEC dysfunction.
In recent years, several drugs targeting LSECs have shown promising therapeutic effects in animal models. Asada and colleagues were the first to demonstrate that tofogliflozin alleviates the progression of perihilar ductal necrosis (PHDN), liver inflammation, and fibrosis by modulating the interplay between LSECs and HSCs in cirrhotic rat models (45). Furthermore, studies have shown that delivering simvastatin specifically to LSECs significantly reduces LSEC capillarization in mouse models. When combined with anti-PD-L1 antibodies, simvastatin nanoparticles showed notable therapeutic efficacy in HCC (46). Mishra and colleagues highlighted that a rationally designed protein ProAgio could induce apoptosis of activated HSCs (aHSCs) and capillarized LSECs, thereby reducing immune cell infiltration and showing potential value in treating advanced NASH and alcoholic hepatitis (AH) (47). In addition, traditional Chinese medicine has shown significant therapeutic effects in HCC progression. Fu et al. demonstrated that Dahuang Zhechong Pill (DHZCP) enhances the capillarization of liver sinusoids in patients with liver cirrhosis and HCC by inhibiting the MK/Itgα signaling pathway of LSECs (48). These studies have paved the way for novel therapeutic approaches to liver fibrosis and related diseases, particularly with a focus on LSECs.
In summary, although current mechanistic studies have identified critical targets that regulate LSEC functions and phenotypes, many therapeutic targets and signaling pathways exhibit multifunctionality across different cell types. Future research should focus on exploring LSEC-specific targets and understanding their interactions with other liver cells within the specialized liver microenvironment, which is crucial for advancing therapeutic strategies for liver fibrosis and related diseases. Additionally, the development of highly targeted nanoparticle-based drug delivery systems, combined with existing immunotherapies, holds the potential to significantly enhance therapeutic efficacy and offer innovative approaches for clinical application.
Hepatic stellate cells
Residing within the perisinusoidal space of Disse, a specialized microenvironment between sinusoidal endothelial cells and hepatocyte plates, HSCs are specialized resident mesenchymal cells that serve as central mediators of hepatic injury responses through their dual roles in vitamin A storage and fibrogenic activation (49). Under healthy conditions, HSCs remain in a quiescent state with low proliferation rates, primarily has function on the storage and transport of vitamin A, immune modulation, and the support of liver development and regeneration. In the context of chronic liver injury, like that caused by viral infections, ALD and non-alcoholic fatty liver disease (NAFLD), HSCs become activated, undergoing a series of phenotypic transformations. These include transdifferentiation into a myofibroblast-like phenotype, enhanced proliferation and migratory capabilities, and increased chemotaxis. Consequently, activated HSCs produce excessive ECM proteins and release pro-inflammatory mediators, contributing to the progression of liver fibrosis (50, 51). Emerging pathways regulate HSC activation, including autophagy, cellular stress responses, nuclear receptor signaling, energy metabolism, epigenetic modifications and receptor-mediated signaling. Additionally, cytokines and growth factors secreted by resident and infiltrating inflammatory cells within the liver, such as TNF-α, PDGF, and TGF-β1 contribute to HSC activation (49, 52, 53). Activated HSCs play a crucial role in the progression of liver fibrosis by inducing ECM accumulation and secreting various pro-inflammatory cytokines that mediate immune responses, thus perpetuating the inflammatory process (54). Additionally, HSCs promote their own pro-angiogenic phenotype and induce pro-inflammatory phenotypes in other cell types (21, 55). Therefore, strategies targeting the inactivation or apoptosis of activated HSCs hold significant promise for reversing fibrosis. In summary, a comprehensive understanding of the activation mechanisms of HSCs is crucial for developing therapeutic approaches aimed at modulating HSC activity. Progress in this field will provide new insights and strategies for the treatment of liver fibrosis and related diseases in the future.
Targeting hepatic stellate cells
Currently, there are no approved clinical drugs specifically targeting HSCs for the treatment and prevention of early liver disease. However, numerous studies have focused on the mechanisms of HSC activation and proposed corresponding therapeutic strategies (Figure 2). Recent studies have highlighted the significant therapeutic potential of traditional Chinese medicine and natural compounds in the treatment of liver diseases. Excessive release of TGF-β1 is one of the critical stimuli inducing HSC activation and their transdifferentiation into myofibroblasts. It has been reported that imperatorin (IMP) significantly inhibits HSC activity by downregulating TGF-β, resulting in reduced liver fibrosis and inflammation. Additionally, IMP has been shown to suppress angiogenesis and vascular remodeling, further contributing to its therapeutic effects (56). Additionally, Ye et al. demonstrated that Jiawei Taohe Chengqi Decoction (JTCD) effectively inhibits HSC activation and reverses liver fibrosis by suppressing the TGF-β1/CUGBP1 signaling pathway and activating the IFN-γ/Smad7 signaling pathway (57). VEGF plays a pivotal role in promoting angiogenesis in the liver. Xue et al. were the first to report the antifibrotic and anti-angiogenic effects of hydroxysafflor yellow A (HSYA), potentially achieved by inhibiting TGF-β1-induced HSC activation and reducing VEGF-A release through the miR-29a-3p/PDGFRB axis (58). Furthermore, studies have shown that Ficus hirta Vahl (FV) can induce HSCs ferroptosis through the glutathione (GSH)/glutathione peroxidase 4 (GPX4) pathway to alleviate liver fibrosis in mouse models (59). Cryptotanshinone (CTS) has been reported to inhibit the activation of mouse HSCs by suppressing STAT3/CPT1A-dependent fatty acid oxidation (FAO) (60). UDP-glucose ceramide glucosyltransferase (UGCG), a key enzyme regulating energy metabolism in HSC activation, has been shown to inhibit HSC activation through inhibiting UGCG by salvianolic acid B (SAB) (61). Another study revealed that protein tyrosine phosphatase (PTP) is involved in the regulation of HSC activation. Luteolin-7-diglucuronide (L7DG) effectively suppresses HSC activation by inhibiting PTP1B activity and activating the phosphorylation of AMP-activated protein kinase (AMPK) (53). Oxidative stress is a crucial factor leading to hepatic oxidative damage. Wang et al. found that glycyrrhizic acid (GA) could reverses the progression of liver fibrosis by inducing the expression of AKR7A2, thus inhibiting ROS-mediated oxidative stress in activated HSCs (62). These studies indicate that herbal medicines and natural compounds regulating HSC activity may represent a novel approach for the treatment of liver diseases.
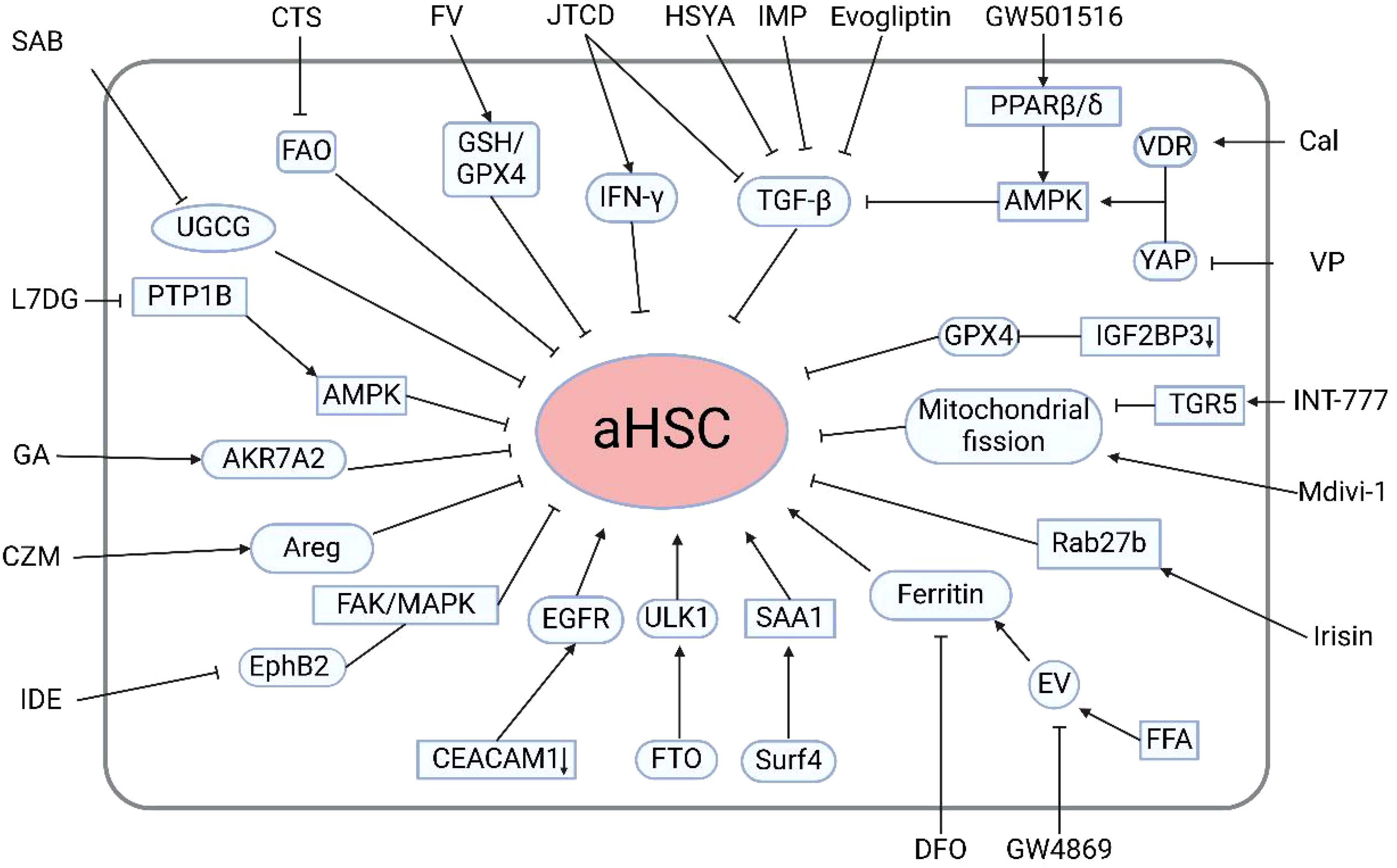
Figure 2. Regulatory mechanisms and therapeutic strategies for modulating HSC activation. Evogliptin, Imperatorin, Jiawei Taohe Chengqi Decoction, hydroxysafflor yellow A, Ficus hirta Vahl, Cryptotanshinone, salvianolic acid B, Luteolin-7-diglucuronide, glycyrrhizic acid, carfilzomib, Idebenone, irisin, INT-777, GW501516, calcipotriol, verteporfin, deferoxamine, and the extracellular vesicle inhibitor GW4869 can suppress hepatic stellate cell activation. Additionally, selective deletion of cell adhesion molecule 1 in HSCs, m6A demethylation of Unc-51-like autophagy activating kinase 1, and surfactant protein 4-mediated secretion of serum amyloid A1 from the liver promote HSC activation. aHSCs, activated HSCs; TGF-β1, transforming growth factor-beta1; FAO fatty, acid oxidation; UGCG, UDP-glucose ceramide glucosyltransferase; PTP1B, protein tyrosine phosphatase 1B; Areg, Amphiregulin; FTO, obesity-associated protein; ULK1, Unc-51-like autophagy activating kinase1; FFA, free fatty acids; PPAR, proliferator-activated receptor; AMPK, AMP-activated protein kinase; VDR, vitamin D receptor; GPX4, glutathione peroxidase 4. (Created in BioRender).
In addition to natural medicines, multiple studies have reported the potential role of chemical agents in the treatment of liver fibrosis. The proteasome inhibitor carfilzomib (CZM) could be a candidate agent to inhibit HSC activation by reducing the expression of the cytokine amphiregulin (Areg) without causing hepatic injury (63). Idebenone (IDE) significantly inhibits HSC activation. Mechanistically, idebenone inhibits EphB2, a receptor tyrosine kinase associated with liver fibrosis, through its antioxidant activity. The suppression of EphB2 in turn reduces the focal adhesion kinase (FAK)/mitogen-activated protein kinase (MAPK) signaling pathway, further diminishing HSC activation (64). Prior research has indicated that the dipeptidyl peptidase-4 (DPP4) inhibitor evogliptin can markedly suppress the expression of pro-inflammatory cytokines, including IL-1α, IL-1β, TNF-α, IL-6 and TGF-β. These collective actions inhibit HSC activation and mitigate liver fibrosis (65).
Beyond pharmacological treatments, current research is increasingly focused on targeting specific genes, proteins, and associated signaling pathways that mediate HSC activation. Notably, the selective deletion of cell adhesion molecule 1 (CEACAM1) in HSCs induces HSCs activation and promote HSCs transdifferentiation into myofibroblasts through the activation of the EGFR (66). Additionally, autophagy is recognized as a significant contributor to HSC activation and fibrosis. Huang et al. discovered that the obesity-associated protein FTO mediates the m6A demethylation of ULK1, thereby enhancing its expression, increasing autophagy, and ultimately promoting HSC activation (67). Wang et al. recently identified two novel therapeutic targets for liver fibrosis: Surf4 and SAA1. They found Surf4 facilitates the secretion of SAA1 from the liver, thereby activating HSCs and exacerbating fibrosis (68). EVs are critical participants for intercellular communication within the context of NAFLD. Sun et al. demonstrated that treatment with free fatty acids (FFAs) induces EVs and facilitates the release of ferritin from these cells via EVs. Subsequently, the ferritin contained within the EVs is taken up by HSCs, leading to accelerated activation of HSCs. The combination therapy of deferoxamine (DFO) and GW48699, an EV inhibitor, significantly inhibits the progression of NAFLD (69). Conversely, Liao et al. discovered that the cleavage fragment of FNDC5, known as irisin, can inhibit the release of fibrosis-associated EVs and HSC activation by promoting the ubiquitination and degradation of Rab27b (70). Mitochondrial fission has been identified as a key regulatory factor in liver fibrosis. Sun et al. revealed that the TGR5 agonist INT-777 activates TGR5, a bile acid membrane receptor, to suppress mitochondrial fission, thereby inhibiting the activation of HSCs that overexpress hepatitis B virus X protein (71). Additionally, Zhang et al. reported that the PPAR β/δ agonist GW501516 prevents HSC activation and fibrosis. GW501516 acts by activating AMPK, which subsequently inhibits ERK1/2, leading to a reduction in TGF-β1-mediated HSC activation and fibrosis (72). Interestingly, another study reported that the combination of VDR agonist calcipotriol (Cal) and the YAP inhibitor verteporfin (VP) synergistically inhibits HSC activation via AMPK activation (73). Recently, a novel regulatory factor for liver fibrosis, IGF2BP3 has been identified. Specific deletion of IGF2BP3 in HSCs suppresses the expression of GPX4, inducing ferroptosis of HSCs (74). Furthermore, Li et al. designed a prodrug nanoparticle system CREKA-CS-RA (CCR) that targets multiple signaling pathways, effectively delivering inhibitors to the Golgi apparatus of activated HSCs. This system disrupts HSC function and inhibits the Hedgehog signaling pathway, resulting in significant improvement of liver fibrosis (75).
The reversibility of activated HSCs to a quiescent phenotype has emerged as a promising therapeutic strategy for hepatic fibrosis. Emerging evidence demonstrates that multiple pharmacological and molecular interventions can effectively modulate HSC activation dynamics. Yang et al. revealed that astaxanthin exhibits dual therapeutic effects by inhibiting HSC activation and promoting phenotypic reversion of activated HSCs to their quiescent state (76). Miranda et al. demonstrated that triclosan-mediated suppression of fatty acid synthase (FASN) activity activates pro-survival metabolic pathways. This metabolic reprogramming triggers a cascade of cellular and molecular adaptations that facilitate phenotypic reversion of HSCs toward a quiescent-like state (77). Given the pivotal role of TGF-β1 in HSC activation, Cierpka et al. demonstrated that overexpression of PLIN5 facilitates the reversion of activated HSC to a quiescent phenotype by inhibiting the TGF-β1-SMAD2/3 and SNAIL signaling pathways, as well as by reducing the activity of the transcription factor STAT3 (78). Furthermore, Chen et al. developed decellularized ECM scaffolds mimicking the matrix stiffness observed at different fibrosis stages. Their findings indicate that low-stiffness scaffolds effectively promote the reversion of activated HSCs (79).
Future research should focus on elucidating the intricate interplay among HSC-specific genetic regulators, enzymatic networks, and receptor signaling cascades, while simultaneously advancing phenotypic reversal strategies to uncover the molecular mechanisms underlying HSC transdifferentiation and fibrogenesis. This research paradigm must be integrated with the development of multi-target therapeutic platforms for CLDs, particularly given that current HSC-targeted interventions remain largely confined to preclinical models, underscoring the critical need for clinical translation of these experimental agents.
Kupffer cells
Kupffer cells, the most abundant resident macrophages in the liver, are strategically located within the hepatic sinusoids. These cells exhibit remarkable plasticity, playing dual roles in maintaining liver homeostasis and driving pathological processes (80, 81). Under physiological conditions, KCs act as sentinels of hepatic immunity by phagocytosing pathogens and apoptotic debris, thereby mitigating systemic inflammation. They also secrete anti-inflammatory cytokines such as IL-10 and TGF-β, which promote immune tolerance and support hepatocyte differentiation (82–84). For instance, a recent study revealed that Ca2+-activated lipid scramblase TMEM16F (also known as anoctamin 6; ANO6), regulates lipid metabolism to suppress excessive inflammation during Listeria monocytogenes infection (85). Additionally, the erythroblast membrane-associated glycoprotein (ERMAP) on KC surfaces interacts with galectin-9, enhancing their ability to recognize and eliminate cancer cells through lectin-mediated pathways (83).
However, persistent insults like chronic viral infections, alcohol abuse, or lipid overload disrupt this equilibrium. Chronic liver injury promotes the M1 polarization of KCs, characterized by excessive production of TNF-α, IL-6, and IL-1β, alongside ROS and lipid peroxides (86–88). These mediators induce hepatocyte apoptosis and recruit neutrophils and monocytes, amplifying the inflammatory cascade. Furthermore, activated KCs secrete chemokines that promote HSC activation and ECM deposition, directly driving fibrogenesis (89). Pathological inflammation is further exacerbated by gut-derived endotoxins, such as LPS, which bind to TLR4 on KCs, creating a self-perpetuating cycle of injury and fibrosis (90).
Given the multiple roles of KCs in the pathogenesis of chronic liver disease, a deeper investigation into KC-related pro-inflammatory pathways or therapeutic approaches to restore immune homeostasis will become a potential strategy for CLD treatment.
Targeting Kupffer cells
Acute liver injury and the chronic inflammation are significant contributors to the development of CLDs. The role of KCs in this process is complex: they can alleviate liver damage by releasing anti-inflammatory factors. when excessively activated, they may exacerbate injury by producing pro-inflammatory mediators (91). Therefore, targeting KCs to block their pro-inflammatory activation is of significant importance for the treatment of liver diseases. Recently, numerous publications have reported important regulatory targets for their activity and function (Table 1). Research by Zhang and colleagues demonstrated that the deficiency of the inflammation-associated deubiquitinase BRISC (BRCC3 isopeptidase complex) in KCs significantly reduces LPS-induced NF-κB activation, thus decreasing the release of pro-inflammatory cytokines TNF-α, IL-6, and IL-1β. This finding offers novel therapeutic strategies for inhibiting KC-driven inflammation (92). Li et al. reported that silence of the E3 ubiquitin ligase Mid1 resulted in reduced secretion of chemokines CXCL1, CXCL2, CXCL6, and CXCL8, as well as inflammatory factors TNF-α, IL-1β, and IL-6 in KCs by inhibiting the NF-κB, c-JNK, and p38 signaling pathways. This intervention alleviated hepatic ischemia-reperfusion injury (HIRI) (93). Moreover, research by Chao et al. revealed the protective role of KCs against acetaminophen (APAP)-induced acute liver injury, primarily through the inhibition of necroptosis in hepatocytes (94). Additionally, studies using mouse models demonstrated that prolonged exposure to polystyrene (PS) microplastics promotes Kupffer cell pyroptosis. However, knockout of Gsdmd or treatment with the Gsdmd inhibitor necrosulfonamide (NSA) significantly suppressed pyroptosis of KCs, leading to reduced liver inflammatory responses (95).
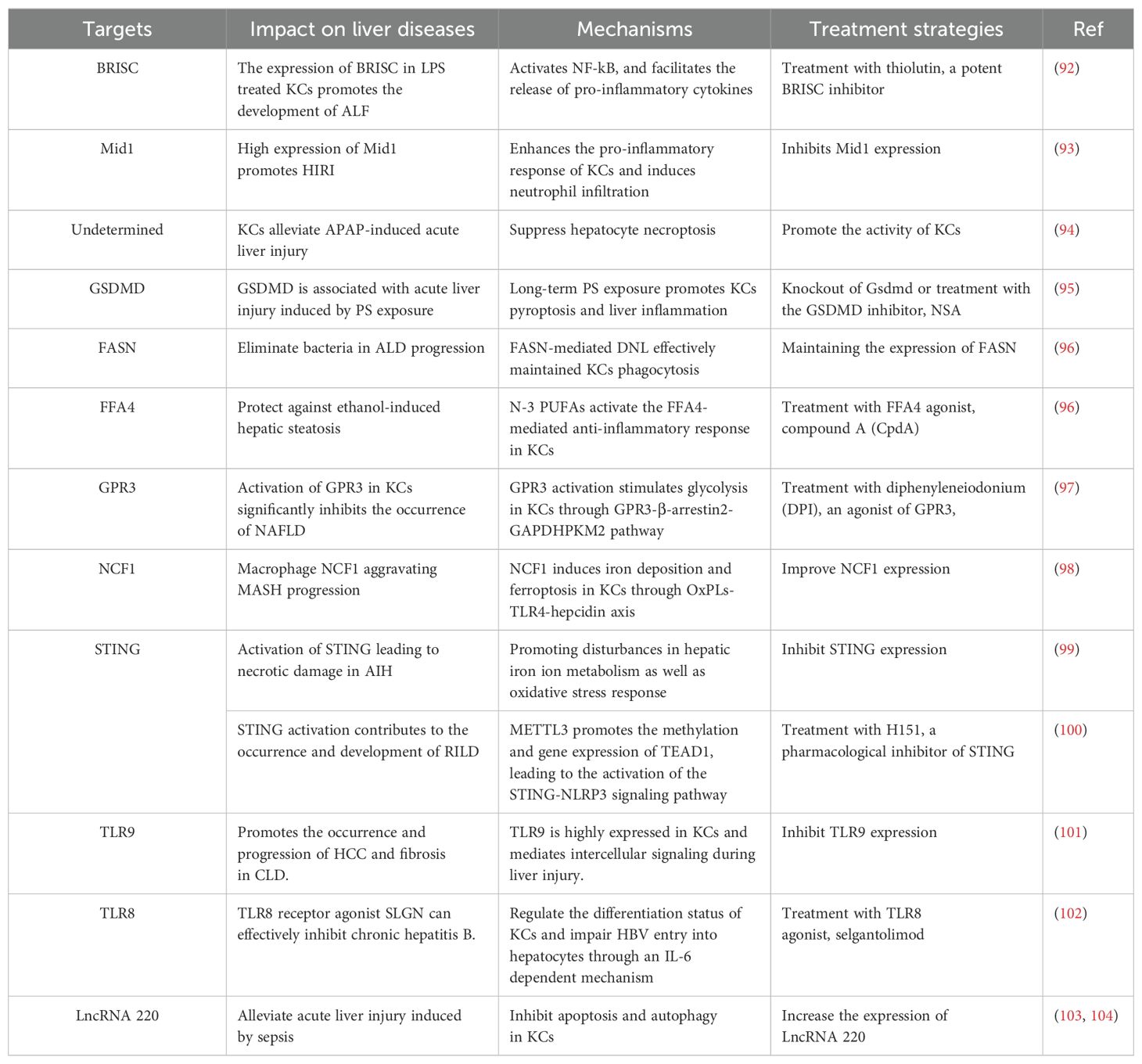
Table 1. Targets of kupffer cells: Their roles and therapeutic strategies in liver diseases.
In addition to their potential therapeutic roles in acute liver inflammation, the mechanisms by which metabolic reprogramming of KCs contribute to fatty liver disease and alcohol-associated liver disease also warrant further investigation. A recent study indicated that alcohol exposure inhibits FASN-mediated de novo lipogenesis (DNL) in KCs, thereby impairing their phagocytic capacity and inducing apoptosis, which promotes the progression of alcoholic liver disease (96). Additionally, Kang et al. identified the free fatty acid receptor 4 (FFA4) as another therapeutic target for alcoholic liver steatosis. Mechanistically, omega-3 polyunsaturated fatty acids (n-3 PUFAs) protect against alcoholic liver steatosis via activating the anti-inflammatory actions of FFA4 on KCs (105). Furthermore, Dong et al. found that activation of the G protein-coupled receptor3 (GPR3) in KCs mediates metabolic reprogramming through the GPR3-β-arrestin2-PKM2 pathway. This finding may provide new avenues for the treatment of NAFLD and obesity (97). Reports also suggest that impaired self-renewal of KCs is a critical factor in the progression of MASH. Zhang et al. found that neutrophil cytosolic factor 1 (NCF1) induces the production of hepcidin in hepatocytes by oxidizing phospholipids, thereby promoting KC iron deposition and MASH (98).
Kupffer cells, as a fundamental component of the liver immune system, exhibit considerable therapeutic potential through their key regulatory factors, which warrant careful consideration. Recent studies have indicated that the stimulator of interferon genes (STING) plays a crucial role in maintaining iron homeostasis in macrophages. Zhao et al. confirmed that activation of STING in KCs promotes dysregulation of iron metabolism, subsequently inducing the development of autoimmune hepatitis (AIH). Ferrostatin-1 (Fer-1) and desferoxamine (DFO) were found to significantly inhibit STING expression (99). Similarly, Wang et al. identified that methyltransferase-like 3 (METTL3) in KCs promotes the methylation and expression of TEAD1, thereby activating the STING-NLRP3 signaling pathway and driving hepatocyte apoptosis. This mechanism offers new therapeutic strategies for radiation-induced liver diseases (RILDs), with STING inhibitors such as H151 demonstrating promising efficacy (100). Furthermore, the Toll-like receptor family serves as important regulators of innate immunity in the liver. Recent studies have revealed elevated expression levels of TLR9 in KCs. The deletion of TLR9 can significantly reduce liver fibrosis by affecting intercellular communication during liver injury (101). Another study demonstrated that the TLR8 agonist selgantolimod (SLGN) can regulate the differentiation status of KCs and impairs HBV entry into hepatocytes via an IL-6-dependent mechanism (102). Interestingly, research conducted by Yang and colleagues has revealed that lncRNA220 acts as a novel modulator of KCs. Mechanistically, lncRNA220 regulates apoptosis and autophagy in LPS-treated KCs through the PI3K-AKT-mTORC1 pathway and the miR-5101/PI3K/AKT/mTOR axis, suggesting that lncRNA220 may serve as a molecular target with clinical, diagnostic, and therapeutic implications in septic acute liver injury (103, 104).
In recent years, the use of nanomaterials as drug delivery carriers in the treatment of various cancers, including HCC, has garnered significant attention (106, 107). Given that KCs are the first line of defense in innate immunity, it is crucial to explore the immune-regulating effects of nanodrugs on KCs and their underlying mechanisms. Recent findings by Jiang et al. elucidated the significant roles of KCs in determining the intrahepatic trafficking of PEGylated liposomal doxorubicin, providing critical insights for further development of nanodrugs (108). Additionally, Ji and colleagues discovered that clodronate-nintedanib-loaded exosome-liposome hybridization enhances the targeted delivery of antifibrotic agents while inhibiting the phagocytic activity of KCs, leading to a reduction in the release of inflammatory cytokines and thereby enhancing the effectiveness of fibrosis treatment (109). Moreover, research by Ding et al. demonstrated that polyethylene glycol-modified graphene oxide (GO-PEG) facilitates the polarization of KCs from M1 to M2, attenuating KC immune activation, which represents an effective strategy for treating liver inflammation (110). Recent studies have also identified that certain natural extracts can regulate the activation and polarization of KCs, influencing the progression of CLDs. For instance, artesunate has been shown to affect M1 macrophage polarization in KCs (111), while gossypetin inhibits their activation (112).
In summary, Kupffer cells, as the first line of defense in the liver immune system, exhibit tremendous potential in the treatment and prevention of liver diseases. Future research should further investigate the specific regulatory mechanisms of KC activity under different liver disease conditions and develop controllable nanoparticle combinations with anti-inflammatory drugs. This approach is anticipated to provide a significant and effective therapeutic strategy for improving treatment outcomes in patients with liver diseases.
Other innate immune lymphocytes
The liver plays a crucial role in the immune system, serving as a hub for a diverse population of immune cells, including innate immune cells such as NK cells, NKT cells, macrophages, and neutrophils, along with adaptive immune cells like T and B cells. Notably, NK and NKT cells account for approximately one-third of the total lymphocyte population in the liver, and are indispensable for antiviral and anticancer immunity (14, 23). Therefore, exploring the therapeutic potential of NK and NKT cells in the context of liver diseases is of significant importance.
NK cells
NK cells are a prominent class of non-parenchymal cells (NPCs) within the hepatic immune microenvironment, comprising approximately half of the total lymphocytes in the human liver. These cells play a vital role in maintaining liver immune homeostasis, providing defense against viral infections, and influencing the pathogenesis of various CLDs (113). During HBV infection, NK cells exhibit cytotoxic activity by lysing infected cells and secreting substantial amounts of antiviral cytokines, such as IFN-γ and TNF-α, which contribute to the elimination of tumor cells. Among these cytokines, IFN-γ is recognized as a critical factor for controlling HBV infection (114, 115). It has been reported that HBV infection is closely associated with diabetes-related liver cirrhosis, where the accumulation of S100A8/A9 in patients with cirrhosis and diabetes activates NK cells through receptor for advanced RAGE-mediated p38 MAPK signaling. This activation leads to the secretion of IFN-γ and subsequent necrotic apoptosis of β cells (116). Furthermore, in the liver, the biological activities of different NK cell subsets may have contrasting effects. For instance, persistent high lipid levels reduce UCP1, promoting necrotic apoptosis of NK cells and facilitating the progression of NASH to fibrosis (117). However, recent studies have identified that NK cells are activated in NASH mouse models, secreting a plethora of pro-inflammatory cytokines and chemokines, including IFN-γ, IL-1α, IL-1β, IL-12, GM-CSF, CCL3, CCL4, and CCL5. Simultaneously, they upregulate the JAK-STAT signaling pathway, contributing to hepatocyte injury and the progression of NASH (20). Therefore, specifically regulating the biological activity of NK cells to enhance their cytotoxicity may represent an effective strategy for the treatment of liver diseases.
NKT cells
NKT cells, a unique subset of T cells that exhibit characteristics of both T cells and NK cells, play significant roles in innate immune responses. Based on the characteristics of their T cell receptors (TCRs), NKT cells can be classified into invariant (iNKT) and non-invariant NKT cell subsets (118). NKT cells respond rapidly to a variety of stimuli and can be effectively activated during immune-mediated liver injury, contributing to the maintenance of immune microenvironment homeostasis and regulating immune functions in a multifaceted way. Specifically, while NKT cells enhance the immune response against tumors and infections, their excessive activation can lead to the release of numerous inflammatory cytokines, triggering a cascade of inflammatory responses and affecting the functions of other immune cells (23, 119). Furthermore, studies indicate that NKT cells play critical roles in the onset and progression of various liver diseases, including ALD, NAFLD, and MAFLD (120–122). Therefore, exploring factors that influence NKT cell function will provide new insights and a solid foundation for designing NKT cell-based immunotherapeutic strategies. This approach is expected to facilitate more precise and personalized immune interventions in the treatment of liver diseases, ultimately improving clinical outcomes for patients.
Targeting NK cells and NKT cells
In recent years, a range of targeted therapeutic strategies for NK cells has been developed, shedding light on their role in the mechanisms of liver diseases (Table 2). Modulating NK cell activity at different stages of liver disease may represent an effective therapeutic approach. For instance, in the context of NASH mediated by NK cell necroptosis induced by high fatty acid levels, Gu et al. found that poly I:C, a TLR3 agonist, effectively reverse cell death and restore UCP1 expression in UCP1−/− NK cells, thereby inhibiting the progression of NASH to fibrosis (117). Wang et al. demonstrated that depleting NK cells, either through NK cell deficiency or the NK cell-neutralizing antibody PK136, alleviates NASH at its early stages (20). Co-infection with HBV and HDV is a significant factor in severe viral hepatitis, and innovative antiviral therapies targeting NK cells have shown notable efficacy. For instance, Groth et al. demonstrated that HDV infection activates NK cells via IFN-β, allowing activated NK cells to eliminate HDV-infected hepatic cell lines via the TRAIL/TRAIL-R2 axis (123). Additionally, co-stimulation of monocytes promotes NK cell release of IFN-γ and TNF-α to combat HBV infection (124). Regulating NK cell-mediated immune homeostasis within the immune microenvironment also presents a viable therapeutic strategy. Guo et al. found that nifedipine increases NK and NKT cell numbers while inhibiting the proliferation of HSCs, impacting liver fibrosis development (129). Immunostimulatory therapies aimed at enhancing the anti-inflammatory cytokine IL-10 and reducing the pro-inflammatory cytokine IL-33 have also been proposed to augment NK cell function (130, 131). Research suggests that reducing the ratio of the inhibitory receptor NKG2A to the activating receptor NKG2D significantly enhances both NK cell immune suppression and anticancer activity (132). Beyond their therapeutic potential, recent studies indicate that the profiling of NK cells may serve as a rapid and valuable tool for assessing the risk of HCC development in hepatitis C virus (HCV) chronically infected patients with cirrhotic liver following HCV cure (133).
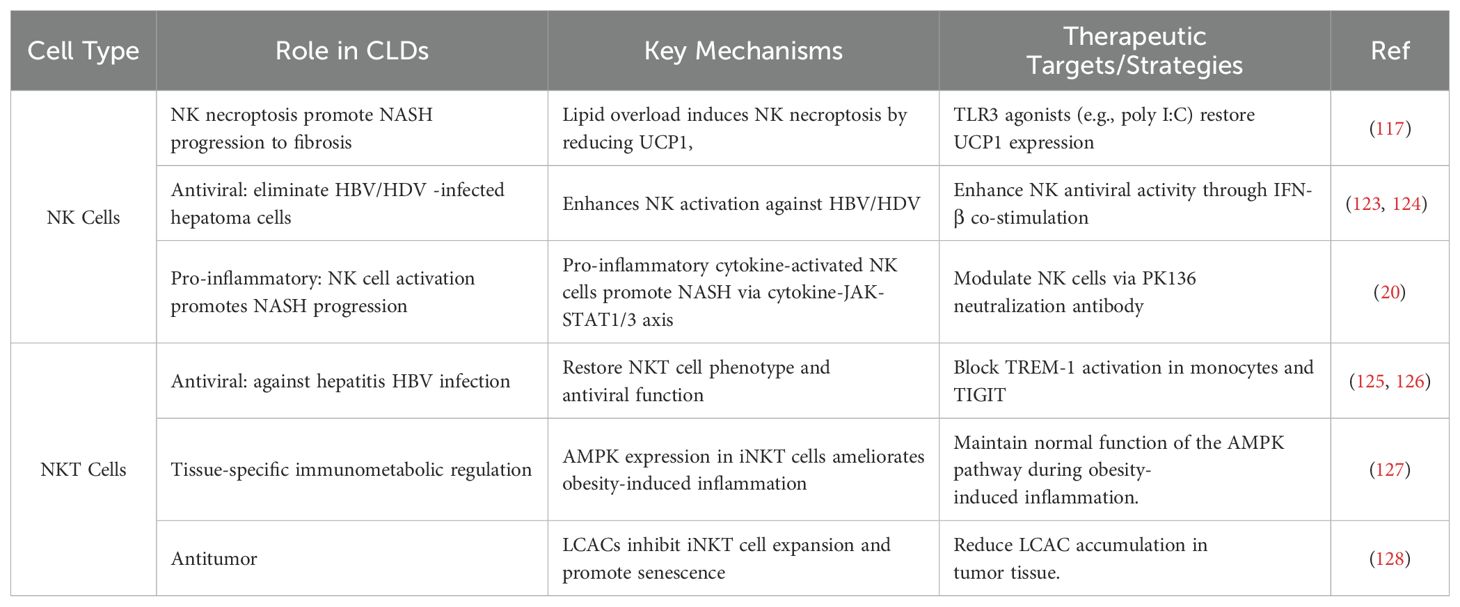
Table 2. The dual roles of NK and NKT Cells in CLDs and its therapeutic strategies.
Maintaining the activity and function of NKT cells and their subpopulations is considered an ideal strategy for treating liver diseases. For instance, Wu et al. discovered that blocking the activation of triggering receptor expressed on myeloid cells-1 (TREM-1) in monocytes can promote the elimination of HBV by inhibiting pyroptosis in iNKT cells and restoring their functionality (125). Yu et al. found that NKT-like cells express high levels of T-cell immunoreceptor with Ig and ITIM domains (TIGIT), and the blockade of TIGIT can enhance the antiviral capacity of NKT-like cells (126). Excessive activation of the mTORC1/SREBP2 signaling cascade induced by obesity-related NAFLD results in hepatic cholesterol accumulation, which in turn contributes to NKT cell depletion and dysfunction, serving as a key driver for the development of obesity-related HCC. Research indicates that cholesterol-lowering rosuvastatin can restore NKT cell-mediated liver immuno surveillance and prevent obesity-related HCC (134). Moreover, studies have shown that tissue-resident iNKT cells possess unique transcriptional and metabolic characteristics, with AMPK playing a crucial role in ameliorating obesity-induced inflammation in adipose tissue (127). Furthermore, NKT cells are closely linked to inflammation-driven liver damage; recent investigations revealed that T-cell immunoglobulin and mucin domain-containing molecule 4 (Tim-4) serve as an important regulatory factor for maintaining the homeostasis and function of NKT cells under inflammatory stimulation (135). Previous studies suggested that adrenergic signaling can mitigate liver damage caused by iNKT cell activation without affecting their anticancer activity (136). Additionally, Cheng et al. unveiled that the accumulation of long-chain acylcarnitines (LCAC) within tumor tissues can promote the senescence of iNKT cells, and reprogramming LCAC metabolism to restore their anticancer function may represent a novel therapeutic strategy for inflammation-associated tumors (128).
In summary, orchestrating the homeostasis and functions of NK cells and NKT cells may represent a promising strategy for treating liver inflammation and metabolic-related diseases. Future research should delve into the interplay between these cells and the impact of metabolic factors on their functions to uncover the immune mechanisms underlying liver disease, thereby facilitating the development of preventive and therapeutic strategies for metabolic disorders. Additionally, novel immunotherapies targeting NK and NKT cells should be developed to enhance antiviral and anticancer responses.
Conclusion
CLDs arise from a complex interplay of cellular dysfunction, immune dysregulation, and fibro-inflammatory remodeling, predominantly driven by NPCs. LSECs, HSCs, KCs, and innate lymphoid cells (NK/NKT cells) collectively orchestrate the transition from acute injury to chronic fibrosis and cirrhosis. LSECs initiate sinusoidal dysfunction and HSC activation through capillarization and angiocrine signaling, while HSCs transdifferentiate into collagen-producing myofibroblasts, exacerbating ECM deposition. KCs polarized toward pro-inflammatory phenotypes aggravate hepatocyte injury and promote fibrogenesis via cytokine storms and chemokine secretion. NK/NKT cells exhibit dual roles, balancing antiviral defense with the exacerbation of fibrosis through excessive cytokine production.
Emerging therapeutic strategies targeting NPC-specific pathways, such as modulation of LSEC mechanotransduction, metabolic interventions to inhibit or reverse HSC activation, regulation of KC polarization, and regulation of NK/NKT cell activity. Pharmacological agents (e.g., silybin, fasudil), nanoparticle-based delivery systems, and immunotherapies (e.g., TLR agonists, STING inhibitors) have demonstrated preclinical efficacy in restoring NPC homeostasis. However, challenges persist, including the pleiotropic roles of therapeutic targets across diverse cell populations and the limited clinical translation of preclinical findings. Future research should focus on developing cell-specific interventions, unraveling NPC crosstalk within the hepatic microenvironment, and leveraging multi-omics approaches to refine precision therapies. By aligning mechanistic insights with translational innovation, NPC-targeted strategies offer a promising paradigm to arrest the progression of CLD and mitigate their global burden.
Author contributions
YT: Writing – original draft, Writing – review & editing. ZG: Writing – original draft, Writing – review & editing. JF: Writing – original draft. JS: Writing – review & editing. CQ: Writing – original draft, Writing – review & editing. NZ: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Natural Science Foundation of Chongqing, grant number (CSTB2024NSCO-MSX0416); National Natural Science Foundation of China, grant number (82101931 and 82120108019).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CLD, Chronic liver disease; EVs, Extracellular vesicles; MASH, Metabolic dysfunction-associated steatohepatitis; MAFLD, Metabolic-associated fatty liver disease; HCC, Hepatocellular carcinoma; NPC, Non-parenchymal cell; LSEC, Liver sinusoidal endothelial cell; HSC, Hepatic stellate cell; ALD, Alcoholic liver disease; NAFLD, Non-alcoholic fatty liver disease; KC, Kupffer cell; NKT, Natural killer T cell; NK, Natural killer cells; HBV, Hepatitis B virus; IL, Interleukin; IFN-γ, Interferon-γ; A-FABP, Adipocyte fatty acid binding protein; ECM, Extracellular matrix; COL4, Collagen Type IV; TNF-α, Tumor Necrosis Factor-alpha; NASH, Non-alcoholic steatohepatitis; MK, Midkine; UGCG, UDP-glucose ceramide glucosyltransferase; FASN, fatty acid synthase; FAO, Fatty acid oxidation; PTP, Protein tyrosine phosphatase; AMPK, AMP-activated protein kinase; MAPK, Mitogen-activated protein kinase; TGF-β, Transforming growth factor-β; GPX4, Glutathione peroxidase 4; LPS, Lipopolysaccharide; STING, Stimulator of interferon genes; TLR, Toll-like receptor.
References
1. Asrani SK, Devarbhavi H, Eaton J, and Kamath PS. Burden of liver diseases in the world. J Hepatol. (2019) 70:151–71. doi: 10.1016/j.jhep.2018.09.014
2. Wang F, Fan J, Zhang Z, Gao B, and Wang H. The global burden of liver disease: The major impact of China. Hepatology. (2014) 60:2099–108. doi: 10.1002/hep.27406
3. Yan M, Cui Y, and Xiang Q. Metabolism of hepatic stellate cells in chronic liver diseases: emerging molecular and therapeutic interventions. Theranostics. (2025) 15:1715–40. doi: 10.7150/thno.106597
4. Zeng X, Yuan X, Cai Q, Tang C, and Gao J. Circular RNA as an epigenetic regulator in chronic liver diseases. Cells. (2021) 10:1945. doi: 10.3390/cells10081945
5. Lin Y, Zheng L, Fang K, Zheng Y, Wu J, and Zheng M. Proportion of liver cancer cases and deaths attributable to potentially modifiable risk factors in China. Int J Epidemiol. (2023) 52:1805–14. doi: 10.1093/ije/dyad100
6. Makarova-Rusher OV, Altekruse SF, McNeel TS, Ulahannan S, Duffy AG, Graubard BI, et al. Population attributable fractions of risk factors for hepatocellular carcinoma in the United States. Cancer. (2016) 122:1757–65. doi: 10.1002/cncr.v122.11
7. Liu Y and Wu F. Global burden of aflatoxin-induced hepatocellular carcinoma: A risk assessment. Environ Health Perspect. (2010) 118:818–24. doi: 10.1289/ehp.0901388
8. Global Burden of Disease Liver Cancer Collaboration, Akinyemiju T, Abera S, Ahmed M, Alam N, Alemayohu MA, et al. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: results from the global burden of disease study 201510.1001/jamaoncol.2017.3055. JAMA Oncol. (2017) 3:1683. doi: 10.1001/jamaoncol.2017.3055
9. Liu Z, Jiang Y, Yuan H, Fang Q, Cai N, Suo C, et al. The trends in incidence of primary liver cancer caused by specific etiologies: Results from the Global Burden of Disease Study 2016 and implications for liver cancer prevention. J Hepatol. (2019) 70:674–83. doi: 10.1016/j.jhep.2018.12.001
10. Kanwal F, Khaderi S, Singal AG, Marrero JA, Loo N, Asrani SK, et al. Risk factors for HCC in contemporary cohorts of patients with cirrhosis. Hepatology. (2023) 77:997–1005. doi: 10.1002/hep.32434
11. Ioannou GN, Splan MF, Weiss NS, McDonald GB, Beretta L, and Lee SP. Incidence and predictors of hepatocellular carcinoma in patients with cirrhosis. Clin Gastroenterol Hepatol. (2007) 5:938–945.e4. doi: 10.1016/j.cgh.2007.02.039
12. Gao B, Jeong WI, and Tian Z. Liver: An organ with predominant innate immunity. Hepatology. (2008) 47:729–36. doi: 10.1002/hep.22034
13. Blouin A, Bolender RP, and Weibel ER. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J Cell Biol. (1977) 72:441–55. doi: 10.1083/jcb.72.2.441
14. Racanelli V and Rehermann B. The liver as an immunological organ. Hepatology. (2006) 43:S54–62. doi: 10.1002/hep.21060
15. Hellerbrand C. Hepatic stellate cells—the pericytes in the liver. Pflüg Arch - Eur J Physiol. (2013) 465:775–8. doi: 10.1007/s00424-012-1209-5
16. Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, et al. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J Hepatol. (2017) 66:212–27. doi: 10.1016/j.jhep.2016.07.009
17. Puche JE, Saiman Y, and Friedman SL. Hepatic stellate cells and liver fibrosis. In: Terjung R, editor. Comprehensive physiology, 1st ed. Division of Liver Diseases, Icahn School of Medicine at Mount Sinai Hospital, New York, New York: Comprehensive Psychology (2013). p. 1473–92. doi: 10.1002/cphy.c120035
18. Wu L, Deng H, Feng X, Xie D, Li Z, Chen J, et al. Interferon-γ+ Th1 activates intrahepatic resident memory T cells to promote HBsAg loss by inducing M1 macrophage polarization. J Med Virol. (2024) 96:e29627. doi: 10.1002/jmv.29627
19. Nilsson J, Hörnberg M, Schmidt-Christensen A, Linde K, Nilsson M, Carlus M, et al. NKT cells promote both type 1 and type 2 inflammatory responses in a mouse model of liver fibrosis. Sci Rep. (2020) 10:21778. doi: 10.1038/s41598-020-78688-2
20. Wang F, Zhang X, Liu W, Zhou Y, Wei W, Liu D, et al. Activated natural killer cell promotes nonalcoholic steatohepatitis through mediating JAK/STAT pathway. Cell Mol Gastroenterol Hepatol. (2022) 13:257–74. doi: 10.1016/j.jcmgh.2021.08.019
21. Geng Y, Wang J, Serna-Salas SA, Villanueva AH, Buist-Homan M, Arrese M, et al. Hepatic stellate cells induce an inflammatory phenotype in Kupffer cells via the release of extracellular vesicles. J Cell Physiol. (2023) 238:2293–303. doi: 10.1002/jcp.v238.10
22. Wang J, Wu Z, Xia M, Salas SS, Ospina JA, Buist-Homan M, et al. Extracellular vesicles derived from liver sinusoidal endothelial cells inhibit the activation of hepatic stellate cells and Kupffer cells in vitro. Biochim Biophys Acta BBA - Mol Basis Dis. (2024) 1870:167020. doi: 10.1016/j.bbadis.2024.167020
23. Nakashima H and Kinoshita M. Antitumor immunity exerted by natural killer and natural killer T cells in the liver. J Clin Med. (2023) 12:866. doi: 10.3390/jcm12030866
24. Tao X, Zhang R, Du R, Yu T, Yang H, Li J, et al. EP3 enhances adhesion and cytotoxicity of NK cells toward hepatic stellate cells in a murine liver fibrosis model. J Exp Med. (2022) 219:e20212414. doi: 10.1084/jem.20212414
25. Han QJ, Mu YL, Zhao HJ, Zhao RR, Guo QJ, Su YH, et al. Fasudil prevents liver fibrosis via activating natural killer cells and suppressing hepatic stellate cells. World J Gastroenterol. (2021) 27:3581–94. doi: 10.3748/wjg.v27.i24.3581
26. Pandey E, Nour AS, and Harris EN. Prominent receptors of liver sinusoidal endothelial cells in liver homeostasis and disease. Front Physiol. (2020) 11:873. doi: 10.3389/fphys.2020.00873
27. Deleve LD, Wang X, and Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatol Baltim Md. (2008) 48:920–30. doi: 10.1002/hep.22351
28. Gracia-Sancho J, Laviña B, Rodríguez-Vilarrupla A, García-Calderó H, Fernández M, Bosch J, et al. Increased oxidative stress in cirrhotic rat livers: A potential mechanism contributing to reduced nitric oxide bioavailability. Hepatol Baltim Md. (2008) 47:1248–56. doi: 10.1002/hep.22166
29. Wu X, Shu L, Zhang Z, Li J, Zong J, Cheong LY, et al. Adipocyte fatty acid binding protein promotes the onset and progression of liver fibrosis via mediating the crosstalk between liver sinusoidal endothelial cells and hepatic stellate cells. Adv Sci. (2021) 8:2003721. doi: 10.1002/advs.202003721
30. Ruan B, Duan JL, Xu H, Tao KS, Han H, Dou GR, et al. Capillarized liver sinusoidal endothelial cells undergo partial endothelial-mesenchymal transition to actively deposit sinusoidal ECM in liver fibrosis. Front Cell Dev Biol. (2021) 9:671081. doi: 10.3389/fcell.2021.671081
31. Wu Q, Sun Q, Zhang Q, Wang N, Lv W, and Han D. Extracellular matrix stiffness-induced mechanotransduction of capillarized liver sinusoidal endothelial cells. Pharmaceuticals. (2024) 17:644. doi: 10.3390/ph17050644
32. Dai Q, Qing X, Jiang W, Wang S, Liu S, Liu X, et al. Aging aggravates liver fibrosis through downregulated hepatocyte SIRT1-induced liver sinusoidal endothelial cell dysfunction. Hepatol Commun. (2024) 8:e0350. doi: 10.1097/HC9.0000000000000350
33. Wu L, Chen H, Fu C, Xing M, Fang H, Yang F, et al. Midkine mediates dysfunction of liver sinusoidal endothelial cells through integrin α4 and α6. Vascul Pharmacol. (2022) 147:107113. doi: 10.1016/j.vph.2022.107113
34. Gan C, Yaqoob U, Lu J, Xie M, Anwar A, Jalan-Sakrikar N, et al. Liver sinusoidal endothelial cells contribute to portal hypertension through collagen type IV–driven sinusoidal remodeling. JCI Insight. (2024) 9:e174775. doi: 10.1172/jci.insight.174775
35. Furuta K, Guo Q, Pavelko KD, Lee JH, Robertson KD, Nakao Y, et al. Lipid-induced endothelial vascular cell adhesion molecule 1 promotes nonalcoholic steatohepatitis pathogenesis. J Clin Invest. (2021) 131:e143690. doi: 10.1172/JCI143690
36. Guo Q, Furuta K, Islam S, Caporarello N, Kostallari E, Dielis K, et al. Liver sinusoidal endothelial cell expressed vascular cell adhesion molecule 1 promotes liver fibrosis. Front Immunol. (2022) 13:983255. doi: 10.3389/fimmu.2022.983255
37. Kawashita E, Ozaki T, Ishihara K, Kashiwada C, and Akiba S. Endothelial group IVA phospholipase A2 promotes hepatic fibrosis with sinusoidal capillarization in the early stage of non-alcoholic steatohepatitis in mice. Life Sci. (2022) 294:120355. doi: 10.1016/j.lfs.2022.120355
38. Gao J, Wei B, Liu M, Hirsova P, Sehrawat TS, Cao S, et al. Endothelial p300 promotes portal hypertension and hepatic fibrosis through C-C motif chemokine ligand 2–mediated angiocrine signaling. Hepatology. (2021) 73:2468–83. doi: 10.1002/hep.31617
39. Greuter T, Yaqoob U, Gan C, Jalan-Sakrikar N, Kostallari E, Lu J, et al. Mechanotransduction-induced glycolysis epigenetically regulates a CXCL1-dominant angiocrine signaling program in liver sinusoidal endothelial cells in vitro and in vivo. J Hepatol. (2022) 77:723–34. doi: 10.1016/j.jhep.2022.03.029
40. Winkler M, Staniczek T, Kürschner SW, Schmid CD, Schönhaber H, Cordero J, et al. Endothelial GATA4 controls liver fibrosis and regeneration by preventing a pathogenic switch in angiocrine signaling. J Hepatol. (2021) 74:380–93. doi: 10.1016/j.jhep.2020.08.033
41. Wang C, Felli E, Selicean S, Nulan Y, Lozano JJ, Guixé-Muntet S, et al. Role of calcium integrin-binding protein 1 in the mechanobiology of the liver endothelium. J Cell Physiol. (2024) 239:e31198. doi: 10.1002/jcp.v239.5
42. Ojha U, Kim S, Rhee CY, You J, Choi YH, Yoon SH, et al. Endothelial RUNX3 controls LSEC dysfunction and angiocrine LRG1 signaling to prevent liver fibrosis. Hepatology. (2024) 81:1228–43. doi: 10.1097/HEP.0000000000001018
43. He S, Luo Y, Ma W, Wang X, Yan C, Hao W, et al. Endothelial POFUT1 controls injury-induced liver fibrosis by repressing fibrinogen synthesis. J Hepatol. (2024) 81:135–48. doi: 10.1016/j.jhep.2024.02.032
44. Brozat JF, Brandt EF, Stark M, Fischer P, Wirtz TH, Flaßhove A, et al. JAM-A is a multifaceted regulator in hepatic fibrogenesis, supporting LSEC integrity and stellate cell quiescence. Liver Int. (2022) 42:1185–203. doi: 10.1111/liv.15187
45. Asada S, Kaji K, Nishimura N, Koizumi A, Matsuda T, Tanaka M, et al. Tofogliflozin delays portal hypertension and hepatic fibrosis by inhibiting sinusoidal capillarization in cirrhotic rats. Cells. (2024) 13:538. doi: 10.3390/cells13060538
46. Yu Z, Guo J, Liu Y, Wang M, Liu Z, Gao Y, et al. Nano delivery of simvastatin targets liver sinusoidal endothelial cells to remodel tumor microenvironment for hepatocellular carcinoma. J Nanobiotechnology. (2022) 20:9. doi: 10.1186/s12951-021-01205-8
47. Mishra F, Yuan Y, Yang JJ, Li B, Chan P, and Liu Z. Depletion of activated hepatic stellate cells and capillarized liver sinusoidal endothelial cells using a rationally designed protein for nonalcoholic steatohepatitis and alcoholic hepatitis treatment. Int J Mol Sci. (2024) 25:7447. doi: 10.3390/ijms25137447
48. Fu C, Zhang Y, Xi WJ, Xu K, Meng F, Ma T, et al. Dahuang Zhechong pill attenuates hepatic sinusoidal capillarization in liver cirrhosis and hepatocellular carcinoma rat model via the MK/integrin signaling pathway. J Ethnopharmacol. (2023) 308:116191. doi: 10.1016/j.jep.2023.116191
49. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. (2008) 88:125–72. doi: 10.1152/physrev.00013.2007
50. Lee YA, Wallace MC, and Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. (2015) 64:830–41. doi: 10.1136/gutjnl-2014-306842
51. Cordero-Espinoza L and Huch M. The balancing act of the liver: tissue regeneration versus fibrosis. J Clin Invest. (2018) 128:85–96. doi: 10.1172/JCI93562
52. Zisser A, Ipsen DH, and Tveden-Nyborg P. Hepatic stellate cell activation and inactivation in NASH-fibrosis-roles as putative treatment targets? Biomedicines. (2021) 9:365. doi: 10.3390/biomedicines9040365
53. Tang BX, Zhang Y, Sun DD, Liu QY, Li C, Wang PP, et al. Luteolin-7-diglucuronide, a novel PTP1B inhibitor, ameliorates hepatic stellate cell activation and liver fibrosis in mice. Acta Pharmacol Sin. (2024) 46:122–33. doi: 10.1038/s41401-024-01351-3
54. Schwabe RF, Tabas I, and Pajvani UB. Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology. (2020) 158:1913–28. doi: 10.1053/j.gastro.2019.11.311
55. Hammerich L and Tacke F. Hepatic inflammatory responses in liver fibrosis. Nat Rev Gastroenterol Hepatol. (2023) 20:633–46. doi: 10.1038/s41575-023-00807-x
56. Zhao Z, Fan Q, Zhang C, Zheng L, Lin J, Chen M, et al. Imperatorin attenuates CCl4-induced cirrhosis and portal hypertension by improving vascular remodeling and profibrogenic pathways. Eur J Pharmacol. (2024) 980:176833. doi: 10.1016/j.ejphar.2024.176833
57. Ye L, Huang J, Liang X, Guo W, Sun X, Shao C, et al. Jiawei Taohe Chengqi Decoction attenuates CCl4 induced hepatic fibrosis by inhibiting HSCs activation via TGF-β1/CUGBP1 and IFN-γ/Smad7 pathway. Phytomedicine. (2024) 133:155916. doi: 10.1016/j.phymed.2024.155916
58. Xue X, Li Y, Zhang S, Yao Y, Peng C, and Li Y. Hydroxysafflor yellow A exerts anti-fibrotic and anti-angiogenic effects through miR-29a-3p/PDGFRB axis in liver fibrosis. Phytomedicine. (2024) 132:155830. doi: 10.1016/j.phymed.2024.155830
59. Yang Y, Chen Y, Feng D, Wu H, Long C, Zhang J, et al. Ficus hirta Vahl. ameliorates liver fibrosis by triggering hepatic stellate cell ferroptosis through GSH/GPX4 pathway. J Ethnopharmacol. (2024) 334:118557. doi: 10.1016/j.jep.2024.118557
60. Li Z, Zheng Y, Zhang L, and Xu E. Cryptotanshinone alleviates liver fibrosis via inhibiting STAT3/CPT1A-dependent fatty acid oxidation in hepatic stellate cells. Chem Biol Interact. (2024) 399:111119. doi: 10.1016/j.cbi.2024.111119
61. Li ZB, Jiang L, Ni JD, Xu YH, Liu F, Liu WM, et al. Salvianolic acid B suppresses hepatic fibrosis by inhibiting ceramide glucosyltransferase in hepatic stellate cells. Acta Pharmacol Sin. (2023) 44:1191–205. doi: 10.1038/s41401-022-01044-9
62. Wang Q, Lu T, Song P, Dong Y, Dai C, Zhang W, et al. Glycyrrhizic acid ameliorates hepatic fibrosis by inhibiting oxidative stress via AKR7A2. Phytomedicine. (2024) 133:155878. doi: 10.1016/j.phymed.2024.155878
63. Fujiwara A, Takemura K, Tanaka A, Matsumoto M, Katsuyama M, Okanoue T, et al. Carfilzomib shows therapeutic potential for reduction of liver fibrosis by targeting hepatic stellate cell activation. Sci Rep. (2024) 1):19288. doi: 10.1038/s41598-024-70296-8
64. Han Y, Gao Q, Xu Y, Chen K, Li R, Guo W, et al. Cysteine sulfenylation contributes to liver fibrosis via the regulation of EphB2-mediated signaling. Cell Death Dis. (2024) 15:602. doi: 10.1038/s41419-024-06997-9
65. Seo HY, Lee SH, Han E, Hwang JS, Han S, Kim MK, et al. Evogliptin directly inhibits inflammatory and fibrotic signaling in isolated liver cells. Int J Mol Sci. (2022) 23:11636. doi: 10.3390/ijms231911636
66. Muturi HT, Ghadieh HE, Asalla S, Lester SG, Belew GD, Zaidi S, et al. Conditional deletion of CEACAM1 in hepatic stellate cells causes their activation. Mol Metab. (2024) 88:102010. doi: 10.1016/j.molmet.2024.102010
67. Huang T, Zhang C, Ren J, Shuai Q, Li X, Li X, et al. FTO-mediated m 6A demethylation of ULK1 mRNA promotes autophagy and activation of hepatic stellate cells in liver fibrosis. Acta Biochim Biophys Sin. (2024) 56:1509–20. doi: 10.3724/abbs.2024098
68. Wang B, Li H, Gill G, Zhang X, Tao G, Liu B, et al. Hepatic surf4 deficiency impairs serum amyloid A1 secretion and attenuates liver fibrosis in mice. Research. (2024) 7:0435. doi: 10.34133/research.0435
69. Sun M, Tang M, Qian Y, Zong G, Zhu G, Jiang Y, et al. Extracellular vesicles-derived ferritin from lipid-induced hepatocytes regulates activation of hepatic stellate cells. Heliyon. (2024) 10:e33741. doi: 10.1016/j.heliyon.2024.e33741
70. Liao X, Luo Y, Gu F, Song W, Nie X, and Yang Q. Therapeutic role of FNDC5/irisin in attenuating liver fibrosis via inhibiting release of hepatic stellate cell-derived exosomes. Hepatol Int. (2023) 17:1659–71. doi: 10.1007/s12072-023-10523-y
71. Sun L, Shao Y, Zhuang Z, Liu Z, Liu M, Qu C, et al. Targeting TGR5 to mitigate liver fibrosis: Inhibition of hepatic stellate cell activation through modulation of mitochondrial fission. Int Immunopharmacol. (2024) 140:112831. doi: 10.1016/j.intimp.2024.112831
72. Zhang M, Barroso E, Peña L, Rada P, Valverde ÁM, Wahli W, et al. PPARβ/δ attenuates hepatic fibrosis by reducing SMAD3 phosphorylation and p300 levels via AMPK in hepatic stellate cells. BioMed Pharmacother. (2024) 179:117303. doi: 10.1016/j.biopha.2024.117303
73. Wang P, Li J, Ji M, Pan J, Cao Y, Kong Y, et al. Vitamin D receptor attenuates carbon tetrachloride-induced liver fibrosis via downregulation of YAP. J Hazard Mater. (2024) 478:135480. doi: 10.1016/j.jhazmat.2024.135480
74. Li X, Li Y, Zhang W, Jiang F, Lin L, Wang Y, et al. The IGF2BP3/Notch/Jag1 pathway: A key regulator of hepatic stellate cell ferroptosis in liver fibrosis. Clin Transl Med. (2024) 14:e1793. doi: 10.1002/ctm2.v14.8
75. Li Y, Zhang T, Zhang J, Liu Q, Jia Q, Chen W, et al. Dually fibronectin/CD44-mediated nanoparticles targeted disrupt the Golgi apparatus and inhibit the hedgehog signaling in activated hepatic stellate cells to alleviate liver fibrosis. Biomaterials. (2023) 301:122232. doi: 10.1016/j.biomaterials.2023.122232
76. Yang Y, Bae M, Kim B, Park YK, Koo SI, and Lee JY. Astaxanthin prevents and reverses the activation of mouse primary hepatic stellate cells. J Nutr Biochem. (2016) 29:21–6. doi: 10.1016/j.jnutbio.2015.11.005
77. Miranda JF, Scarinci LD, Ramos LF, Silva CM, Gonçalves LR, de Morais PF, et al. The modulatory effect of triclosan on the reversion of the activated phenotype of LX-2 hepatic stellate cells. J Biochem Mol Toxicol. (2020) 34:e22413. doi: 10.1002/jbt.22413
78. Cierpka R, Weiskirchen R, and Asimakopoulos A. Perilipin 5 ameliorates hepatic stellate cell activation via SMAD2/3 and SNAIL signaling pathways and suppresses STAT3 activation. Cells. (2021) 10:2184. doi: 10.3390/cells10092184
79. Chen G, Deng Y, Xia B, and Lv Y. In situ regulation and mechanisms of 3D matrix stiffness on the activation and reversion of hepatic stellate cells. Adv Healthc Mater. (2023) 12:e2202560. doi: 10.1002/adhm.202202560
80. Dixon LJ, Barnes M, Tang H, Pritchard MT, and Nagy LE. Kupffer cells in the liver. In: Prakash YS, editor. Comprehensive physiology, 1st ed. Liver Disease Research Center, Cleveland, Ohio, USA: Comprehensive Psychology (2013). p. 785–97. doi: 10.1002/cphy.c120026
81. Protzer U, Maini MK, and Knolle PA. Living in the liver: hepatic infections. Nat Rev Immunol. (2012) 12:201–13. doi: 10.1038/nri3169
82. Peiseler M, Araujo David B, Zindel J, Surewaard BGJ, Lee WY, Heymann F, et al. Kupffer cell–like syncytia replenish resident macrophage function in the fibrotic liver. Science. (2023) 381:eabq5202. doi: 10.1126/science.abq5202
83. Li J, Liu XG, Ge RL, Yin YP, Liu YD, Lu WP, et al. The ligation between ERMAP, galectin-9 and dectin-2 promotes Kupffer cell phagocytosis and antitumor immunity. Nat Immunol. (2023) 24:1813–24. doi: 10.1038/s41590-023-01634-7
84. Sasaki K, Rooge S, Gunewardena S, Hintz JA, Ghosh P, Pulido Ruiz IA, et al. Kupffer cell diversity maintains liver function in alcohol-associated liver disease. Hepatology. (2025) 81:870–87. doi: 10.1097/HEP.0000000000000918
85. Tang J, Song H, Li S, Lam SM, Ping J, Yang M, et al. TMEM16F expressed in kupffer cells regulates liver inflammation and metabolism to protect against listeria monocytogenes. Adv Sci. (2024) 11:2402693. doi: 10.1002/advs.202402693
86. Shan Z and Ju C. Hepatic macrophages in liver injury. Front Immunol. (2020) 11:322. doi: 10.3389/fimmu.2020.00322
87. Yan J, Li S, and Li S. The role of the liver in sepsis. Int Rev Immunol. (2014) 33:498–510. doi: 10.3109/08830185.2014.889129
88. Krenkel O and Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. (2017) 17:306–21. doi: 10.1038/nri.2017.11
89. Tsuchida T and Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. (2017) 14:397–411. doi: 10.1038/nrgastro.2017.38
90. Rivera CA, Adegboyega P, Van Rooijen N, Tagalicud A, Allman M, and Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. (2007) 47:571–9. doi: 10.1016/j.jhep.2007.04.019
91. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. (2017) 66:1300–12. doi: 10.1016/j.jhep.2017.02.026
92. Zhang W, Liu K, Ren GM, Wang Y, Wang T, Liu X, et al. BRISC is required for optimal activation of NF-κB in Kupffer cells induced by LPS and contributes to acute liver injury. Cell Death Dis. (2023) 14:743. doi: 10.1038/s41419-023-06268-z
93. Li J, Jin C, Li Y, and Liu H. Mid1 aggravates hepatic ischemia-reperfusion injury by inducing immune cell infiltration. FASEB J Off Publ Fed Am Soc Exp Biol. (2024) 38:e23823. doi: 10.1096/fj.202400843R
94. Chao S, Shan S, Liu Z, Liu Z, Wang S, Qiang Y, et al. Both TREM2-dependent macrophages and Kupffer cells play a protective role in APAP-induced acute liver injury. Int Immunopharmacol. (2024) 141:112926. doi: 10.1016/j.intimp.2024.112926
95. Qian X, Jin P, Fan K, Pei H, He Z, Du R, et al. Polystyrene microplastics exposure aggravates acute liver injury by promoting Kupffer cell pyroptosis. Int Immunopharmacol. (2024) 126:111307. doi: 10.1016/j.intimp.2023.111307
96. Xie L, Wu B, Fan Y, Tao Y, Jiang X, Li Q, et al. Fatty acid synthesis is indispensable for Kupffer cells to eliminate bacteria in ALD progression. Hepatol Commun. (2024) 8:e0522. doi: 10.1097/HC9.0000000000000522
97. Dong T, Hu G, Fan Z, Wang H, Gao Y, Wang S, et al. Activation of GPR3-β-arrestin2-PKM2 pathway in Kupffer cells stimulates glycolysis and inhibits obesity and liver pathogenesis. Nat Commun. (2024) 15:807. doi: 10.1038/s41467-024-45167-5
98. Zhang J, Wang Y, Fan M, Guan Y, Zhang W, Huang F, et al. Reactive oxygen species regulation by NCF1 governs ferroptosis susceptibility of Kupffer cells to MASH. Cell Metab. (2024) 36:1745–1763.e6. doi: 10.1016/j.cmet.2024.05.008
99. Zhao J, Yi Z, Deng G, Li Y, Li J, Qin M, et al. STING modulates iron metabolism to promote liver injury and inflammation in acute immune hepatitis. Free Radic Biol Med. (2024) 210:367–77. doi: 10.1016/j.freeradbiomed.2023.11.038
100. Wang B, Zhang Y, Niu H, Zhao X, Chen G, Zhao Q, et al. METTL3-mediated STING upregulation and activation in kupffer cells contribute to radiation-induced liver disease via pyroptosis. Int J Radiat Oncol Biol Phys. (2024) 119:219–33. doi: 10.1016/j.ijrobp.2023.10.041
101. Hatten H, Colyn L, Volkert I, Gaßler N, Lammers T, Hofmann U, et al. Loss of Toll-like receptor 9 protects from hepatocellular carcinoma in murine models of chronic liver disease. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:167321. doi: 10.1016/j.bbadis.2024.167321
102. Roca Suarez AA, Plissonnier ML, Grand X, Michelet M, Giraud G, Saez-Palma M, et al. TLR8 agonist selgantolimod regulates Kupffer cell differentiation status and impairs HBV entry into hepatocytes via an IL-6-dependent mechanism. Gut. (2024) 73:2012–22. doi: 10.1136/gutjnl-2023-331396
103. Yang Y, Tian T, Wang Z, Li S, Li N, Luo H, et al. LncRNA 220, a newly discovered long non-conding RNA inhibiting apoptosis and autophagy in Kupffer cells in LPS-induced endotoxemic mice through the XBP1u-PI3K-AKT pathway. Int Immunopharmacol. (2024) 128:111497. doi: 10.1016/j.intimp.2024.111497
104. Yang Y, Tian T, Li S, Li N, Luo H, and Jiang Y. LncRNA 220: A Novel Long Non-Coding RNA Regulates Autophagy and Apoptosis in Kupffer Cells via the miR-5101/PI3K/AKT/mTOR Axis in LPS-Induced Endotoxemic Liver Injury in Mice. Int J Mol Sci. (2023) 24:11210. doi: 10.3390/ijms241311210
105. Kang S, Koh JM, and Im DS. N-3 polyunsaturated fatty acids protect against alcoholic liver steatosis by activating FFA4 in kupffer cells. Int J Mol Sci. (2024) 25:5476. doi: 10.3390/ijms25105476
106. Yin X, Rong J, Shao M, Zhang S, Yin L, He Z, et al. Aptamer-functionalized nanomaterials (AFNs) for therapeutic management of hepatocellular carcinoma. J Nanobiotechnology. (2024) 22:243. doi: 10.1186/s12951-024-02486-5
107. Kong FH, Ye QF, Miao XY, Liu X, Huang SQ, Xiong L, et al. Current status of sorafenib nanoparticle delivery systems in the treatment of hepatocellular carcinoma. Theranostics. (2021) 11:5464–90. doi: 10.7150/thno.54822
108. Jiang K, Tian K, Yu Y, Wu E, Yang M, Pan F, et al. Kupffer cells determine intrahepatic traffic of PEGylated liposomal doxorubicin. Nat Commun. (2024) 15:6136. doi: 10.1038/s41467-024-50568-7
109. Ji K, Fan M, Huang D, Sun L, Li B, Xu R, et al. Clodronate-nintedanib-loaded exosome–liposome hybridization enhances the liver fibrosis therapy by inhibiting Kupffer cell activity. Biomater Sci. (2022) 10:702–13. doi: 10.1039/D1BM01663F
110. Ding X, Pang Y, Liu Q, Zhang H, Wu J, Lei J, et al. GO-PEG represses the progression of liver inflammation via regulating the M1/M2 polarization of kupffer cells. Small Weinh Bergstr Ger. (2024) 20:e2306483. doi: 10.1002/smll.202306483
111. Yang Z, Xia H, Lai J, Qiu L, and Lin J. Artesunate alleviates sepsis-induced liver injury by regulating macrophage polarization via the lncRNA MALAT1/PTBP1/IFIH1 axis. Diagn Microbiol Infect Dis. (2024) 110:116383. doi: 10.1016/j.diagmicrobio.2024.116383
112. Xu C, Tai H, Chu Y, Liu Y, He J, Wang Y, et al. Gossypetin targets the liver-brain axis to alleviate pre-existing liver fibrosis and hippocampal neuroinflammation in mice. Front Pharmacol. (2024) 15:1385330. doi: 10.3389/fphar.2024.1385330
113. Mikulak J, Bruni E, Oriolo F, Di Vito C, and Mavilio D. Hepatic natural killer cells: organ-specific sentinels of liver immune homeostasis and physiopathology. Front Immunol. (2019) 10:946. doi: 10.3389/fimmu.2019.00946
114. Wang R, Jaw JJ, Stutzman NC, Zou Z, and Sun PD. Natural killer cell-produced IFN-γ and TNF-α induce target cell cytolysis through up-regulation of ICAM-1. J Leukoc Biol. (2011) 91:299–309. doi: 10.1189/jlb.0611308
115. Hillaire MLB, Lawrence P, and Lagrange B. IFN-γ: A crucial player in the fight against HBV infection? Immune Netw. (2023) 23:e30. doi: 10.4110/in.2023.23.e30
116. Li X, Hong L, Ru M, Cai R, Meng Y, Wang B, et al. S100A8/A9-activated IFNγ+ NK cells trigger β-cell necroptosis in hepatitis B virus-associated liver cirrhosis. Cell Mol Life Sci. (2024) 81:345. doi: 10.1007/s00018-024-05365-2
117. Gu M, Zhang Y, Lin Z, Hu X, Zhu Y, Xiao W, et al. Decrease in UCP1 by sustained high lipid promotes NK cell necroptosis to exacerbate nonalcoholic liver fibrosis. Cell Death Dis. (2024) 15:518. doi: 10.1038/s41419-024-06910-4
118. Wang H and Yin S. Natural killer T cells in liver injury, inflammation and cancer. Expert Rev Gastroenterol Hepatol. (2015) 9:1077–85. doi: 10.1586/17474124.2015.1056738
119. Swain MG. Natural killer T cells within the liver: conductors of the hepatic immune orchestra. Dig Dis. (2010) 28:7–13. doi: 10.1159/000282059
120. Zheng S, Yang W, Yao D, Tang S, Hou J, and Chang X. A comparative study on roles of natural killer T cells in two diet-induced non-alcoholic steatohepatitis-related fibrosis in mice. Ann Med. (2022) 54:2232–44. doi: 10.1080/07853890.2022.2108894
121. Eom JA, Jeong JJ, Han SH, Kwon GH, Lee KJ, Gupta H, et al. Gut-microbiota prompt activation of natural killer cell on alcoholic liver disease. Gut Microbes. (2023) 15:2281014. doi: 10.1080/19490976.2023.2281014
122. Cuño-Gómiz C, De Gregorio E, Tutusaus A, Rider P, Andrés-Sánchez N, Colell A, et al. Sex-based differences in natural killer T cell-mediated protection against diet-induced steatohepatitis in Balb/c mice. Biol Sex Differ. (2023) 14:85. doi: 10.1186/s13293-023-00569-w
123. Groth C, Maric J, Garcés Lázaro I, Hofman T, Zhang Z, Ni Y, et al. Hepatitis D infection induces IFN-β-mediated NK cell activation and TRAIL-dependent cytotoxicity. Front Immunol. (2023) 14:1287367. doi: 10.3389/fimmu.2023.1287367
124. Kupke P, Brucker J, Wettengel JM, Protzer U, Wenzel JJ, Schlitt HJ, et al. Cytokine response of natural killer cells to hepatitis B virus infection depends on monocyte co-stimulation. Viruses. (2024) 16:741. doi: 10.3390/v16050741
125. Wu X, Zhao W, Miao Q, Shi S, Wei B, Luo L, et al. CCR2+TREM-1+ monocytes promote natural killer T cell dysfunction contributing towards HBV disease progression. Immunol Res. (2024) 72:938–47. doi: 10.1007/s12026-024-09495-4
126. Yu X, Zheng Y, Huang R, Dai X, Kang G, Wang X, et al. Restoration of CD3 + CD56 + NKT-like cell function by TIGIT blockade in inactive carrier and immune tolerant patients of chronic hepatitis B virus infection. Eur J Immunol. (2024) 54:2451046. doi: 10.1002/eji.202451046
127. Aguiar CF, Corrêa-da-Silva F, Gonzatti MB, Angelim MK, Pretti MA, Davanzo GG, et al. Tissue-specific metabolic profile drives iNKT cell function during obesity and liver injury. Cell Rep. (2023) 42:112035. doi: 10.1016/j.celrep.2023.112035
128. Cheng X, Tan X, Wang W, Zhang Z, Zhu R, Wu M, et al. Long-chain acylcarnitines induce senescence of invariant natural killer T cells in hepatocellular carcinoma. Cancer Res. (2023) 83:582–94. doi: 10.1158/0008-5472.CAN-22-2273
129. Guo Q, Yang A, Zhao R, Zhao H, Mu Y, Zhang J, et al. Nimodipine ameliorates liver fibrosis via reshaping liver immune microenvironment in TAA-induced in mice. Int Immunopharmacol. (2024) 138:112586. doi: 10.1016/j.intimp.2024.112586
130. Chen Y, Huang Y, Huang R, Chen Z, Wang X, Chen F, et al. Interleukin-10 gene intervention ameliorates liver fibrosis by enhancing the immune function of natural killer cells in liver tissue. Int Immunopharmacol. (2024) 127:111341. doi: 10.1016/j.intimp.2023.111341
131. Imaoka Y, Ohira M, Imaoka K, Bekki T, Nakano R, Yano T, et al. Interleukin-33 and liver natural killer cells: A novel perspective on antitumor activity in liver fibrosis. Hepatol Res. (2024). hepr.14102. doi: 10.1111/hepr.14102
132. Yu L, Sun L, Liu X, Wang X, Yan H, Pu Q, et al. The imbalance between NKG2A and NKG2D expression is involved in NK cell immunosuppression and tumor progression of patients with hepatitis B virus-related hepatocellular carcinoma. Hepatol Res Off J Jpn Soc Hepatol. (2023) 53:417–31. doi: 10.1111/hepr.13877
133. Engelskircher SA, Chen PC, Strunz B, Oltmanns C, Ristic T, Owusu Sekyere S, et al. Impending HCC diagnosis in patients with cirrhosis after HCV cure features a natural killer cell signature. Hepatology. (2024) 80:202–22. doi: 10.1097/HEP.0000000000000804
134. Tang W, Zhou J, Yang W, Feng Y, Wu H, Mok MTS, et al. Aberrant cholesterol metabolic signaling impairs antitumor immunosurveillance through natural killer T cell dysfunction in obese liver. Cell Mol Immunol. (2022) 19:834–47. doi: 10.1038/s41423-022-00872-3
135. Wang Y, Wang Y, Ge Y, Wu Z, Yue X, Li C, et al. Tim-4 alleviates acute hepatic injury by modulating homeostasis and function of NKT cells. Clin Exp Immunol. (2024) 218:101–10. doi: 10.1093/cei/uxae063
Keywords: chronic liver disease, non-parenchymal cells, innate immunity, therapeutic targeting, liver fibrosis
Citation: Yang T, Gu Z, Feng J, Shan J, Qian C and Zhuang N (2025) Non-parenchymal cells: key targets for modulating chronic liver diseases. Front. Immunol. 16:1576739. doi: 10.3389/fimmu.2025.1576739
Received: 14 February 2025; Accepted: 13 May 2025;
Published: 11 June 2025.
Edited by:
Amanda Maree Clark, University of Pittsburgh, United StatesReviewed by:
Francisco G. Vázquez-Cuevas, National Autonomous University of Mexico, MexicoJieyun Jiang, University of Kentucky, United States
Copyright © 2025 Yang, Gu, Feng, Shan, Qian and Zhuang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Na Zhuang, emh1YW5nbmEwNjE4QDE2My5jb20=; Cheng Qian, Y3FpYW44NjM0QGdtYWlsLmNvbQ==
†These authors have contributed equally to this work