 Fei Yin1
Fei Yin1 Yangfang He
Yangfang He Yue Qiao
Yue Qiao- 1Department of Neurology, The Second Hospital of Jilin University, Changchun, China
- 2Department of Endocrinology and Metabolism, The Second Hospital of Jilin University, Changchun, China
- 3Department of Physical Examination Center, The Second Hospital of Jilin University, Changchun, China
- 4Department of Endocrinology, The Second Hospital of Jilin University, Changchun, China
Tumor-derived extracellular vesicles (TDEVs) represent a heterogeneous population of extracellular vesicles (EVs), including exosomes, microvesicles, and apoptotic bodies, which are essential for tumor growth. EVs function as natural carriers of bioactive molecules, including lipids, proteins, and nucleic acids, enabling them to influence and regulate complex cellular interactions within the tumor microenvironment (TME). The TDEVs mainly have immunosuppressive functions as a result of the inhibitory signals disrupting the immune cell anti-tumor activity. They enhance tumor progression and immune evasion by inhibiting the effector function of immune cells and by altering critical processes of immune cell recruitment, polarization, and functional suppression by different signaling pathways. In this sense, TDEVs modulate the NF-κB pathway, promoting inflammation and inducing immune evasion. The Janus kinase (JAK)-signal transducer and activator of transcription (STAT) signaling is required for TDEV-mediated immune suppression and the manifestation of tumor-supporting features. The phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) signaling, necessary for metabolic reprogramming, is orchestrated by TDEV to abrogate immune response and drive cancer cell proliferation. Finally, exosomal cargo can modulate the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome, activating pro-inflammatory responses that influence tumor development and immunomodulation. In this review, we take a deep dive into how TDEVs affect the immune cells by altering key signaling pathways. We also examine emerging therapeutic approaches aimed at disrupting EV-mediated pathways, offering promising avenues for the development of novel EV-based cancer immunotherapy.
1 Introduction
Tumor-derived extracellular vesicles (TDEVs) play a significant role in the tumor microenvironment (TME), impacting tumor progression, immune responses, and systemic physiology (1). These vesicles play a pivotal role in cell-to-cell communication by enabling continuous interactions between tumor cells and various elements of the immune system (2). TDEVs stimulate the secretion of tumor necrosis factor alpha (TNF-α) and Interleukin (IL)-1 beta (IL-1β) from CD14+ monocytes, leading to nuclear factor kappa B (NF-κB) activation (3). Popěna et al. (4) found that extracellular vesicles (EVs) secreted from colorectal cancer (CRC) modulate the immunophenotype of monocytes and macrophages, resulting in an increase in CD14+ in M0 and human leukocyte antigen-DR (HLA-DR) in M1 (pro-inflammatory) and M2 (anti-inflammatoryM2) macrophages. Another study found that EVs released from CRC cells containing high levels of miR-21-5p and miR-200a promote immune escape by enhancing the expression of programmed death (PD)-ligand 1 (PD-L1) in tumor-associated macrophages (TAMs), correlating with a poorer prognosis (5). EVs derived from glioblastoma that contain PD-L1 promote the development of immunosuppressive monocytes, which subsequently inhibit T-cell proliferation (6). Breast cancer cells, in response to endoplasmic reticulum (ER) stress, release exosomes enriched with miR-27a-3p, which are transferred to macrophages and facilitate tumor immune evasion by enhancing PD-L1 expression through the phosphatase and tensin homolog (PTEN)/phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) pathway (7).
Macrophage polarization is affected by significant intracellular metabolic alterations (8). Generally, M1 macrophages sustain their pro-inflammatory functions through aerobic glycolysis, whereas M2 macrophages depend primarily on oxidative phosphorylation (OXPHOS) to support their anti-inflammatory activities (9). In this regard, hypoxic TDEV-pyruvate kinase M2 (PKM2) has been shown to induce macrophage polarization toward the M2 phenotype via activation of Adenosine monophosphate (AMP)-activated protein kinase (AMPK) pathway, thereby promoting lung cancer progression and metastasis (10). TDEVs enhance macrophage glycolysis via the Toll-like receptor (TLR) 2-NF-κB, promoting an immunosuppressive phenotype characterized by high PD-L1 expression (11). PD-L1 upregulation mediated by melanoma-derived EVs involves TLR signaling via the Heat shock protein (HSP)86/TLR4 axis (12). The uptake of EVs by macrophages activates TLR2 and TLR4, leading to cytokine production (13, 14). Breast cancer-derived exosomes carrying miR-9 and miR-181a contribute to the expansion of myeloid-derived suppressor cells (MDSCs) by downregulating suppressor of cytokine signaling 3 (SOCS3) and protein inhibitor of activated signal transducer and activator of transcription 3 (STAT3) (PIAS3), respectively (15). This suppression leads to elevated IL-6 expression and hyperactivation of the JAK/STAT, primarily due to the loss of SOCS3-mediated regulation (15). Momen-Heravi et al. (16) showed that EVs released from head and neck squamous cell carcinoma (HNSCC) are internalized by monocytes, triggering activation of the NF-κB, which in turn enhances the production of matrix metalloproteinase (MMP) 9 and enhances cyclooxygenase-2 (COX-2)expression levels, prostaglandin E2 (PGE2), and vascular endothelial growth factor (VEGF) after stimulation of THP-1 cells. This review provides an in-depth overview of how TDEVs alter immune cell function via key signaling pathways, including NF-κB, PI3K/Akt/mammalian target of rapamycin (mTOR), JAK/STAT, and transforming growth factor-β (TGF-β). Additionally, we present novel therapies that target EV-mediated processes to provide further potential TDEV-based cancer immunotherapeutics.
2 Tumor-derived vesicle characterization and biogenesis
EVs comprise a diverse collection of membrane-bound vesicles essential for various physiological and pathological processes (17). They carry molecules such as proteins, lipids, and nucleic acids, and are associated with diverse functions (18). Exosomes and ectosomes are types of EVs secreted by normal and tumor cells into the extracellular microenvironment (19). TDEVs influence tumor development and metastasis by modulating immune, endothelial, and epithelial cells (18). They may contribute to tumor progression by enhancing tumor aggressiveness, invasiveness, angiogenesis, extracellular matrix remodeling, drug resistance, and immunosuppression (20, 21). The delivery of metastasis-associated components, such as proteins or microRNAs (miRNAs), can initiate and alter signaling pathways, thereby modifying the behavior and functions of target cells (22). EVs are categorized into three types according to their biogenesis pathway: exosomes, ectosomes, and apoptotic bodies (23). Exosomes are small vesicles, typically under 150 nanometers in size, formed when multivesicular bodies (MVBs) merge with the cell membrane and release their contents (24). Lastly, ectosomes, also referred to as microvesicles, are generated through outward budding from the plasma membrane and typically measure between 100 and 1,000 nanometers in diameter (25). The tetraspanin protein family is recognized as a crucial cellular effector in the biogenesis of vesicles, with distinct functions in exosome and ectosome formation (26). Apoptotic bodies are membrane-bound vesicles that are generated as a result of programmed cell death (apoptosis), encapsulating cellular components for clearance by phagocytes (27). The secretion of EVs into the extracellular matrix occurs as a result of the fusion between the outer membrane of MVBs with the plasma membrane of the originating cells, a process initiated by invagination of the plasma membrane during cell maturation, giving rise to endosomes, which are further categorized into early endosomes—mostly peripheral to the cell—and late endosomes—proximal to the nucleus (28, 29). Eventually, late lysosomes undergo a fusion process with the plasma membrane, resulting in the degradation of MVBs and the subsequent release of EVs into the extracellular environment (30). Figure 1 provides an overview of TDEV biogenesis.
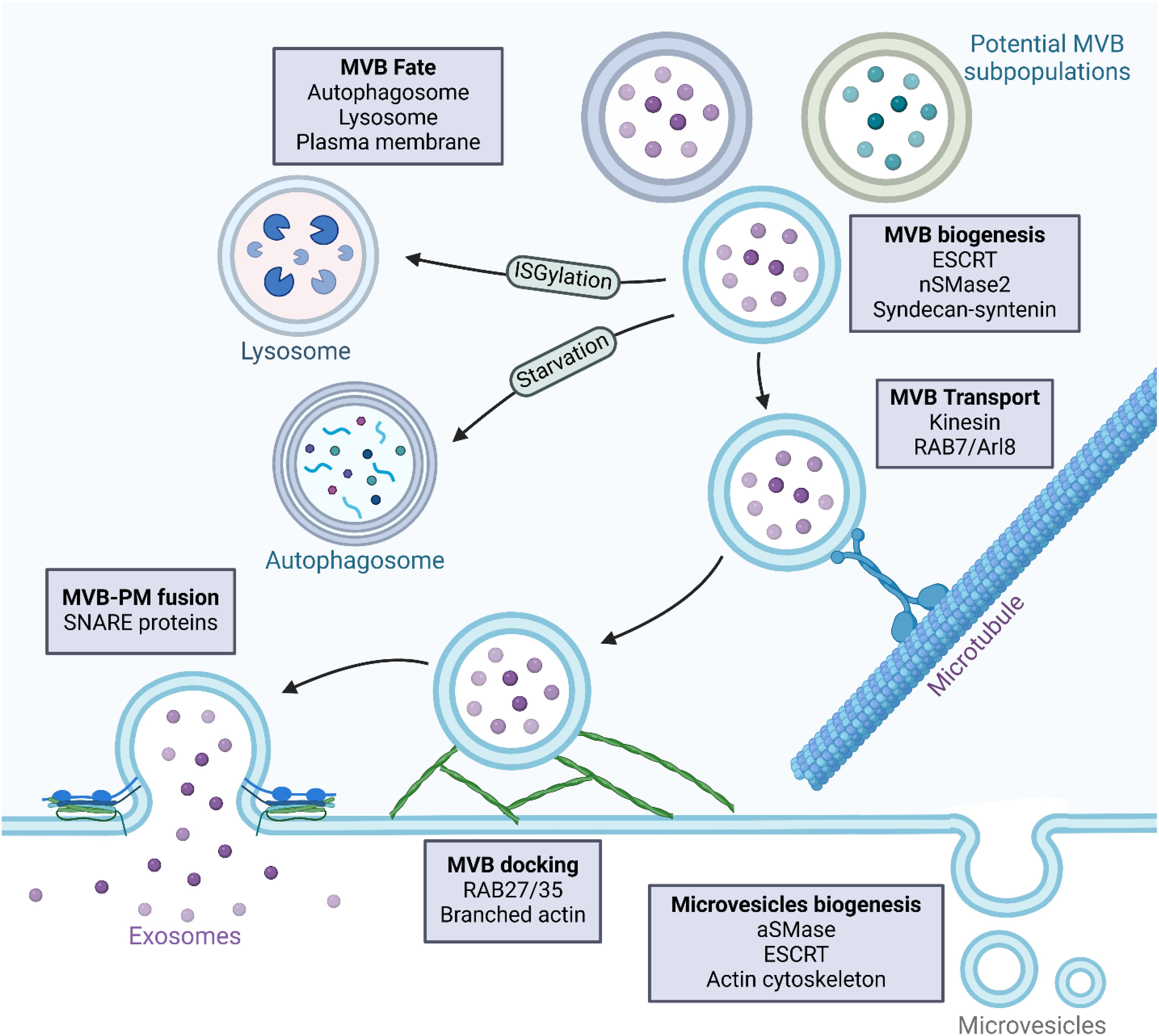
Figure 1. Biogenesis of tumor-derived vesicles. Tumor-derived vesicles are mainly composed of microvesicles and exosomes, which are different in their biogenesis and molecular composition. Microvesicles are formed by direct outward budding from the plasma membrane. This is initiated by plasma membrane remodeling, in which localized changes in lipid composition, along with cytoskeletal reorganization, cause membrane protrusions. Lipid raft domains and alterations in phospholipid asymmetry contribute to this process. Cytoskeletal rearrangements, mechanistically in large part mediated by actin and myosin-driven contractile forces, enable the pinching off of microvesicles from the plasma membrane. Key regulators include the Rho GTPases, ARF6, and the Endosomal Sorting Complex Required for Transport (ESCRT). Once formed, microvesicles bud and are released into the extracellular space, carrying molecular cargo that reflects the characteristics of the tumor cell of origin. In contrast, exosomes are derived from the endosomal pathway, specifically from multivesicular bodies (MVBs) containing intraluminal vesicles (ILVs). Their formation begins with the maturation of early endosomes into late endosomes, which is followed by the production of MVBs. The ESCRT machinery or lipid-mediated mechanisms, such as ceramide accumulation, drive the formation of ILVs within MVBs. Once formed, MVBs are subjected to several possible fates. They may fuse with the plasma membrane to release exosomes into the extracellular space, be directed to lysosomes for degradation, or interact with autophagosomes, although in most cases the mechanisms defining this fate are not well understood. MVBs that are destined for the secretion of exosomes are transported along microtubules toward the plasma membrane under the control of Rab GTPases like Rab11, Rab27a/b, and Rab35. Docking at the plasma membrane is a prerequisite for exosome release and is mediated by SNARE proteins that facilitate membrane fusion.
3 Tumor-derived vesicles and immune modulation
Emerging evidence indicates that EVs have a function in modulating immune responses, functioning either as immune suppressors or stimulators, depending on their intricate molecular composition (Table 1) (47). EVs derived from cancer cells assist in immune system evasion by strengthening the tumor’s ability to withstand immune-mediated responses (48). This effect depends on the specific immunosuppressive molecules present on the EVs and their interaction with related receptors on target immune cells (49). TDEVs contain not only immunosuppressive molecules but also costimulatory molecules, including major histocompatibility complex (MHC) I and II, growth-promoting cytokines, and various tumor-associated antigens (TAAs) (47).
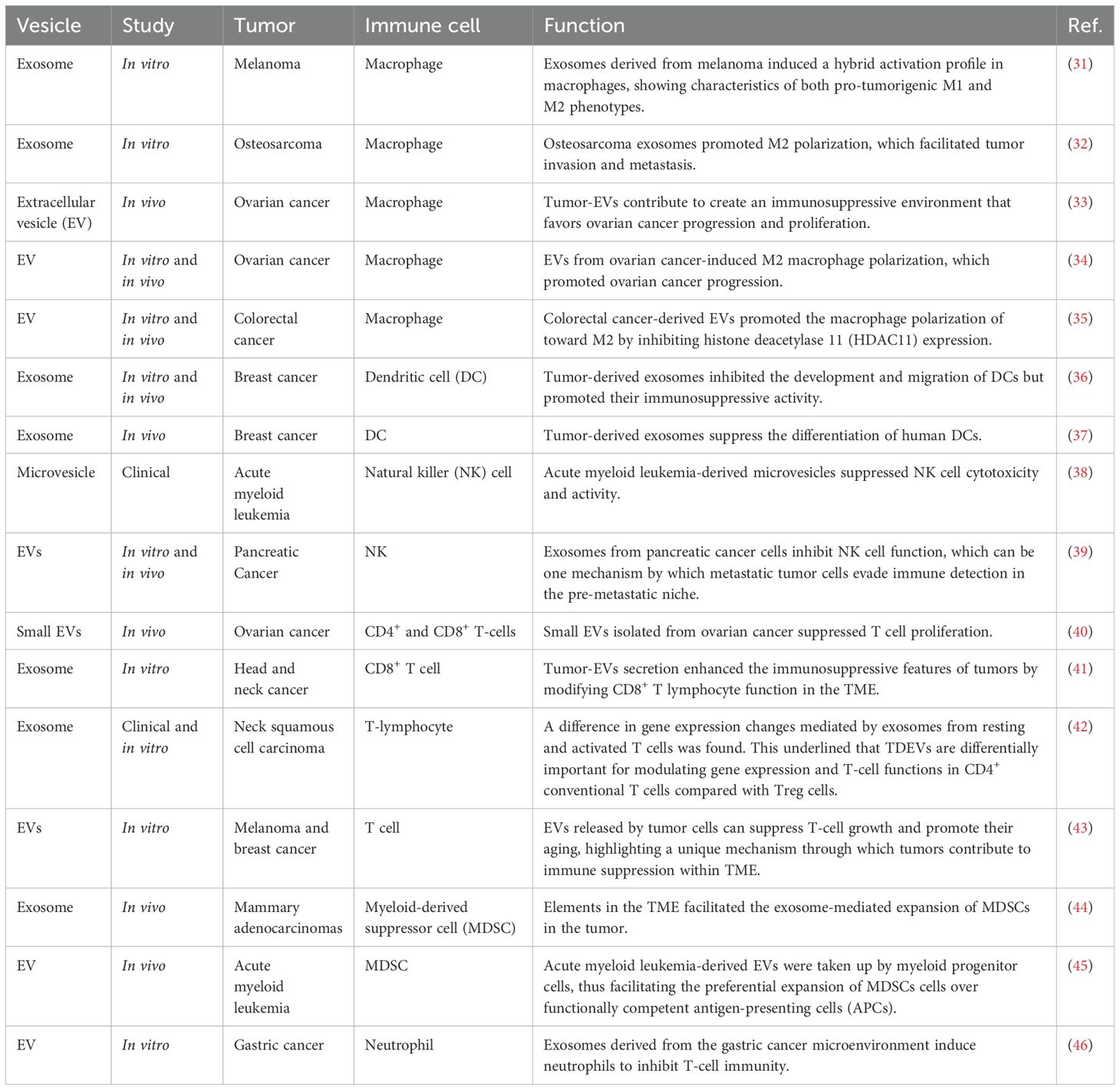
Table 1. Overview of tumor-derived vesicles in modulation of immune cells.
Indeed, many studies have suggested that TDEVs convey suppressive or activating molecular signals, which reprogram tumor-infiltrating immune cells (TIICs) to adopt crucial roles in tumor progression by manifesting immunosuppressive functions through the recruitment and differentiation of regulatory B (Breg) cells, regulatory T (Treg) cells, TAMs, MDSCs, and neutrophils (50). Hepatocellular carcinoma (HCC)-secreted exosomes contain high mobility group box 1 (HMGB1), which is responsible for the promotion of T-cell immunoglobulin and mucin domain 1 (TIM-1)+ Breg cells (51). These Breg cells help establish an immunosuppressive environment by producing IL-10 and suppressing the activity of CD8 T cells (51). Tumor-derived exosomal TGF-β promotes the differentiation of Treg cells from peripheral blood precursors, and these expanded Tregs subsequently secrete TGF-β to maintain their immunosuppressive function (52). One such mechanism involves tumor-derived exosomal surface HSP72 that, through the HSP72-TLR axis, initiates the signaling for the T cell-dependent immunosuppressive function of MDSCs, ultimately leading to STAT3 activation (53). Additionally, exosomes derived from tumors can drive monocyte differentiation into MDSC via TGF-β signaling, thereby inhibiting T cell proliferation and cytotoxic activity (54). The transfer of PKM2 from HCC-derived ectosomes to monocytes promotes their differentiation into M2-like TAMs (55). These macrophages contribute to HCC progression by releasing factors that support tumor growth through a process resembling regurgitation feeding (55). TDEVs play a significant role in tumor immune evasion and progression through the regulation and reprogramming of TIICs (56, 57). These vesicles influence the TME through various mechanisms, including the recruitment and promotion of immunosuppressive cell differentiation, such as Tregs, Bregs, TAMs, and neutrophils, thereby intensifying immune suppression within the TME (55, 58). TDEVs promote immune evasion by transferring immunosuppressive factors, including PD-L1 and Fas ligand, to immune cells, thereby inhibiting their anti-tumor functions (59, 60). Also, EVs produced by tumors can suppress the anti-tumor immune response in distant tissues, fostering an immunosuppressive microenvironment conducive to metastatic niche formation (61, 62). In summary, the findings show that TDEVs play a significant role in immune modulation depending on their molecular composition.
4 Tumor-derived vesicles and various aspects of immune modulation with a focus on the signaling pathways
TDEVs are crucial modulators of TME and also control many aspects of immunity (63). These vesicles have been regulating the immune responses through different signaling pathways (Figure 2). This section provides an in-depth view of the signaling pathways with an emphasis on the mechanism of action and the function of the TDEVs in the modulation of immune cells (Table 2).
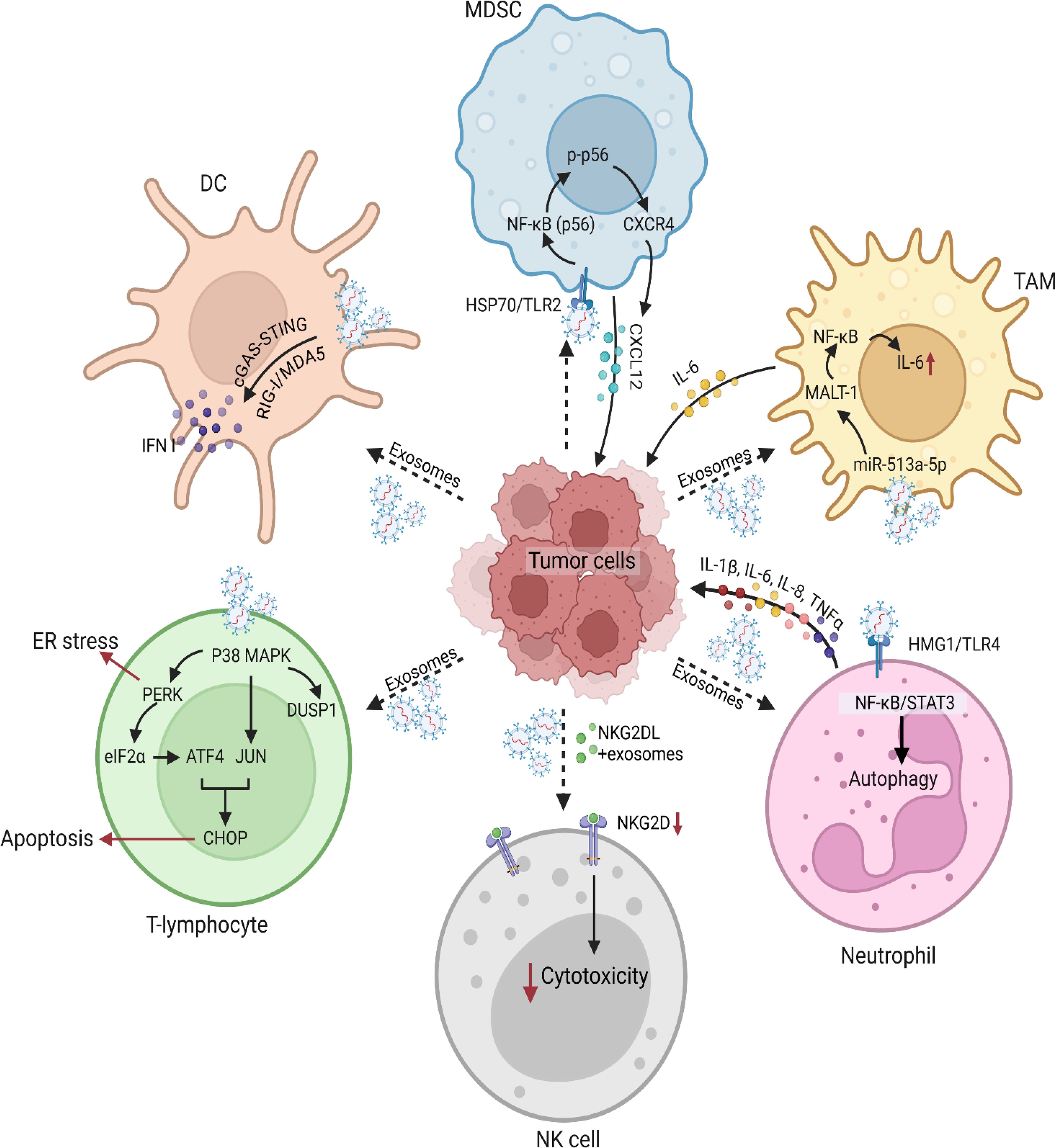
Figure 2. Tumor-derived vesicles and induced signaling modulation in various immune cells. Tumor-derived vesicles play a critical role in modulating immune signaling pathways. These vesicles carry a diverse array of molecules such as DNA, RNA, proteins, and lipids, which interact with various immune cell populations to alter their function and phenotype. Tumor-derived exosomes also exert immunosuppressive effects by interfering with immune responses and cytotoxic functions of immune cells. For instance, tumor cells secrete exosomes enriched with epidermal growth factor receptor (EGFR), which are taken up by macrophages. These EGFR+ exosomes modulate innate immune responses by deregulating interferon regulatory factor 3 (IRF3) through mitogen-activated protein kinase kinase 2 (MEKK2), thereby dampening immunity. Furthermore, tumor-derived microvesicles and exosomes carrying ligands for the NK group 2 member D (NKG2D) receptor downregulate NKG2D expression on DCs, impairing their cytotoxic activity. This suppression of immune effector functions contributes to immune escape and enhances tumor survival within the microenvironment. One of the key mechanisms by which tumor-derived vesicles impact immune function involves their interaction with DCs. Cancer cells release tumor-derived exosomes carrying double-stranded DNA (dsDNA), which are internalized by DCs, leading to their activation and the release of type I IFN. These findings highlight the role of tumor-derived vesicles in shaping DC-mediated immune responses, which can either enhance antitumor immunity or facilitate immune evasion. Another significant aspect of tumor-derived exosomes is their role in the recruitment and expansion of MDSCs, which are key players in tumor-induced immunosuppression. Prostate cancer-derived exosomes have been shown to promote the migration of MDSCs into the tumor microenvironment by upregulating CXCR4 via the TLR2/NF-κB signaling pathway. MDSCs exhibited enhanced migration toward exosome-enriched environments, an effect that was mitigated by the CXCR4-specific inhibitor AMD3100. Analyses further confirmed a marked increase in CXCR4 expression in MDSCs upon exosome stimulation, along with elevated TLR2 and NF-κB activation. Blocking TLR2 with a specific inhibitor (C29) significantly reduced the expression of phosphorylated p65 (p-p65) and CXCR4, reinforcing the notion that TLR2-mediated NF-κB activation is a key driver of MDSC recruitment via the CXCR4-CXCL12 axis. In addition to their effects on innate immune cells, tumor-derived exosomes have been implicated in modulating T lymphocyte function. Prostate cancer-derived exosomes have been shown to integrate with T lymphocytes, leading to the activation of the p38 mitogen-activated protein kinase (MAPK) pathway. Phosphorylated p38 MAPK subsequently triggers endoplasmic reticulum (ER) stress, activating the PERK-eIF2α-ATF4-CHOP signaling axis, which culminates in T cell apoptosis. Interestingly, the dual-specificity phosphatase 1 (DUSP1) was upregulated following p38 MAPK activation, suggesting a potential negative feedback mechanism to regulate MAPK signaling. Pharmacological inhibition of p38 MAPK using SB203580 attenuated ER stress, thereby protecting T lymphocytes from apoptosis. Further investigations into tumor-derived vesicle-mediated immune modulation have uncovered a link between IL-6 and the activation of the JAK2-STAT3 signaling pathway in tumor cells. Specifically, exosomes from TAMs were found to promote the expression of eukaryotic initiation factor 4A3 (EIF4A3) in breast cancer cells through JAK2-STAT3 activation. Gene set enrichment analysis (GSEA) confirmed the significant upregulation of genes associated with the JAK2-STAT3 pathway in breast cancer cells co-cultured with TAM-derived exosomes. Chromatin immunoprecipitation (ChIP) and luciferase reporter assays validated the direct interaction between STAT3 and the EIF4A3 promoter. In the context of neutrophil, gastric cancer-derived exosomes have been shown to prolong neutrophil survival and promote the expression of inflammatory factors, further enhancing gastric cancer cell migration. Gastric cancer-derived exosome -mediated neutrophil activation is primarily driven by the high mobility group box-1 (HMGB1) protein, which interacts with TLR4 to activate the NF-κB pathway. This interaction triggers an autophagic response in neutrophils, reinforcing their pro-tumor activity. Blocking HMGB1/TLR4 interactions, inhibiting the NF-κB pathway, or suppressing autophagy reversed gastric cancer-derived exosome-induced neutrophil activation. These findings suggest that gastric cancer-derived exosomes facilitate a tumor-supportive neutrophil phenotype through HMGB1/TLR4/NF-κB signaling, shedding light on the complex interplay between exosomes and immune cells in shaping the tumor microenvironment.
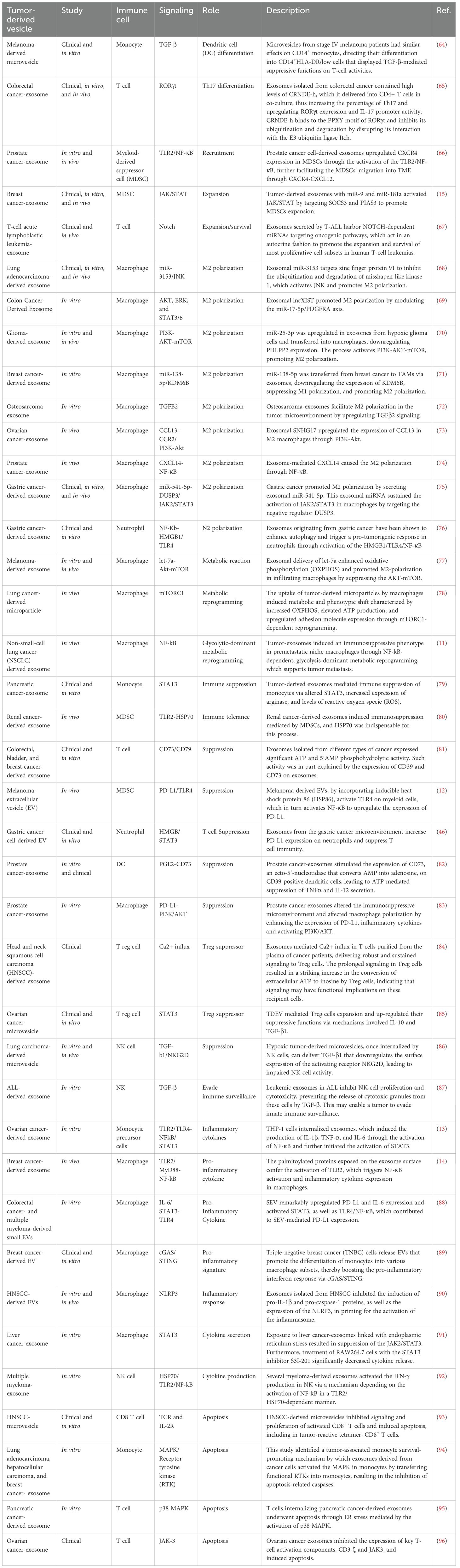
Table 2. Summary of tumor-derived vesicles and induced signaling modulation in immune cells.
4.1 Recruitment, differentiation, and expansion
During cancer progression, immune cells migrate from the bloodstream into the TME, where they trigger inflammation and contribute to the spread of the tumor (97). EVs secreted by pancreatic ductal adenocarcinoma (PDAC) promote the recruitment of bone marrow-derived cells, such as macrophages, to the liver by stimulating Kupffer cells to increase their secretion of TGF-β. EVs derived from myeloma cells that contain serglycin enhance macrophage migration more effectively than EVs lacking serglycin, following their uptake by the macrophages (98). Moreover, EVs derived from the breast cancer with high expression of signal-induced proliferation-associated 1 (SIPA1) enhance macrophage migration toward the tumor site (99). Recent findings show that exosome-like EVs promote dendritic cell (DC) migration by enhancing their response to chemokine (C–C motif) ligand 19 (CCL19) and CCL21 (100). This effect is mediated by membrane-bound C-X3-C motif chemokine ligand 1 (CX3CL1) on endothelial EVs, which activates G-protein-coupled receptor (GPCR) through the CX3CR1, inducing dynamic protrusions and directional movement toward CCL19 (100).
Genetic deletion of TLR2 in macrophages found to abolish NF-κB activation triggered by breast cancer secreated exosomes (14). Li et al. (66) demonstrated that tumor-derived exosomes enhance NF-κB activation and upregulate CXCR4 expression in MDSCs, promoting their expansion and migration (66). This effect was significantly reduced by preincubation with the TLR2 inhibitor C29, suggesting that exosomes act via TLR2 to activate NF-κB signaling and subsequently elevate CXCR4 levels. Another experiment demonstrated that tumor-derived exosomes may enhance CXCR4 expression, potentially through the increased phosphorylation of NF-κB, leading to the expansion and migration of MDSCs (66).
An increased frequency of circulating CD4+CD25+FOXP3 (forkhead box P3)+ Tregs is commonly observed in cancer patients (101). TDEVs have been shown to promote the transformation of human conventional CD4+CD25– T cells into CD4+CD25+FOXP3+ Tregs in a TGF–β1–dependent manner, resulting in elevated levels of phosphorylated Suppressor of Mothers Against Decapentaplegic (SMAD) 2/3 and phosphorylated STAT3 and enhanced Treg proliferation (85, 93). Moreover, Tregs co-cultivated with TDEVs expressed higher levels of FasL, TGF-β, IL-10, cytotoxic T-lymphocyte antigen 4 (CTLA4), granzyme B, and perforin, and stronger suppressor functions (85). Additionally, Tregs that proliferated in response to TDEVs exhibited complete resistance to TDEV-mediated apoptosis (85, 102). Recent studies have also reported similar effects of TDEV on enhancing Treg activity (102).
Sun et al. (65) reported that exosomes from CRC carry the long non-coding RNA Colorectal Neoplasia Differentially Expressed (CRNDE)-h and transfer it to CD4+ T cells, which in turn promote T helper (Th) 17 differentiation. They found that CRNDE-h might inhibit the ubiquitination and degradation of retinoic acid-related orphan receptor gamma t (RORγt) by blocking its interaction with Itch, thus promoting Th17 differentiation in CRC. RORγt is a transcription factor for Th17 differentiation; it binds to the IL-17 promoter to induce secreted IL-17 and promotes the naive CD4+ T cells differentiation of into Th17 (103, 104). The results of Sun et al. (65) demonstrated that CRNDE-depleted CRC exosomes failed to enhance RORγt expression and IL-17 promoter activity, in contrast to normal CRC exosomes. Assays demonstrated that CRNDE binds to the PPXY motif of RORγt. These findings suggest that CRNDE-h, delivered by CRC-derived exosomes, enhances RORγt expression by suppressing its ubiquitination and degradation mediated by the E3 ubiquitin ligase Itch. Furthermore, it has been proposed that natural killer (NK) cell differentiation is contingent upon IL-15, which significantly contributes to NK cell proliferation and enhances NK cell survival (105, 106). Szczepanski et al. (107) found that exogenous IL-15 safeguards NK from the inhibitory influence of tumor-microvesicles via modulating SMAD signaling and that IL-15 can avert microvesicle-induced down-regulation of NK group 2D (NKG2D) expression. Recent findings indicate that IL-15 serves as a potent antagonist of TGF-β by obstructing the production of SMAD-DNA complexes, hence mitigating the down-regulatory effects on T cells (107).
Valenti et al. (64) discovered a pathway in which human tumor cells release membrane vesicles that influence monocyte differentiation into DCs, thereby promoting the generation of myeloid cells. Monocytes treated with tumor-derived microvesicles in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4 retained CD14 but showed reduced surface expression of HLA II, while costimulatory molecules CD80 and CD86 were not upregulated (64). The CD14+HLA-DR phenotype that resulted was linked to suppressive activity on various T-cell functions in response to T-lymphocyte receptor (TCR) triggering, such as proliferation, and expression of cytotoxic molecules, and Interferon gamma (IFN-γ). Valenti et al. (64) provide evidence that tumor cells in cancer patients may promote an immunosuppressive microenvironment by altering the differentiation pathway of circulating monocytes. Instead of developing into fully mature immune cells, these monocytes are redirected toward a CD14+ immature myeloid subset characterized by TGF–β–dependent suppressive functions, which impair T-cell proliferation and activity.
In HCC, exosomal HMGB1 has been shown to stimulate B cells with immunosuppressive properties that inhibit CD8+ T cell responses. This process also facilitates the expansion of Bregs by the TLR2/4 and mitogen-activated protein kinase (MAPK) signaling (51). Shi et al. (46) demonstrated that gastric cancer-derived EVs shuttle HMGB1, which activates STAT3 and upregulates PD-L1 on neutrophils that suppress T-cell proliferation, activation, and function. Notably, blocking the HMGB1 pathway only partially reduced PD-L1 expression, suggesting that TDEVs may also engage additional immunosuppressive mechanisms (46).
4.2 Immune cell polarization
Within the TME, TDEVs play a pivotal role as communicators (108). Through the transmission of several bioactive chemicals, TDEVs alter the immune cell polarization and activity in a way that promotes tumor growth (109). In this context, tumor-derived miR-103a in exosomes was demonstrated to directly modulate the macrophage polarization Phosphatase And Tensin Homolog (PTEN) by binding to its untranslated regions (UTRs), ultimately resulting in reduced expression (110). The downregulation of PTEN activates the PI3K/Akt and STAT3 signaling, resulting in increased accumulation of cancer-promoting molecules like IL-10, CCL2, and VEGF-A, while simultaneously reducing the antitumor immune response (110). Recent studies indicated that oral squamous cell carcinoma (OSCC) cells secrete a substantial quantity of circulating exosomes, abundant in miRNAs (111, 112). Studies have reported increased levels of miR-29 in developing OSCC tissues (112–114). When OSCC-derived exosomes were co-cultured with naïve macrophages, they induced M2 polarization, as indicated by elevated expression of CD206, Arg1, and IL-10. This effect was mediated by exosomal miR-29a-3p, which suppressed SOCS1 expression and promoted STAT1 phosphorylation, involving in the immunosuppressive TME (111). Cai et al. (112) found that tumor-exosomal miR-23a-3p induced M2 polarization by activating the SOCS1/STAT1 pathway, thereby facilitating OSCC cell proliferation and invasion. Similarly, coculture experiments using nonpolarized macrophages and miRNA-containing exosomes derived from normoxic or hypoxic epithelial ovarian cancer (EOC) cells elicited macrophage polarization into tumor-supportive M2 phenotypes, further supported by evidence of increased expression of M2 markers such as CD163 and CD206 (112). These miRNA-enriched exosomes were internalized by macrophages and regulated the SOCS/STAT pathway in these cells (112). Exosomes derived from EOC, enriched with miR-222-3p, were shown to downregulate SOCS3 expression and promote STAT3 phosphorylation in macrophages, enhancing the expression of M2 markers including CD206, Arg1, and IL-10 and decreasing pro-inflammatory cytokines TNF-α and IL-12, thus promoting the differentiation of macrophages into the M2 phenotype (115).
Lin et al. (116) found that exosomes derived bladder cancer can polarize M0 macrophages into an M2phenotype, evidenced by the secretion of particular cytokines and alterations in surface marker phenotypes. Their findings indicate that the exosomes produced from cancer cells initiate the polarization mechanism in macrophages. Lin et al. (116) further demonstrated that reduced expression of miR-21 in macrophages inhibits the polarization of M0 macrophages into the M2 phenotype. Conversely, overexpression of miR-21 in M0 macrophages was showed to enhance STAT3 expression, an effect reversed by miR-21 suppression. Additionally, the STAT3 upregulation induced by miR-21 inhibition was diminished upon treatment with SF1670, suggesting that this regulatory effect is dependent on PI3K/Akt pathway activation (116). Research indicated that augmenting the NF-κB in macrophages can increase the synthesis and secretion of IL-6 in cells (117). Recent evidence indicates that IL-6 and STAT3 are pivotal in the advancement of ovarian cancer, potentially through the polarization of TAMs (118).
Exosomal circFARSA (a circular RNA derived from the phenylalanyl-TRNA synthetase subunit alpha (FARSA) gene) generated from non-small-cell lung cancer (NSCLC) cells facilitates M2 polarization by enhancing PTEN ubiquitination and degradation, hence activating the PI3K/Akt pathway (119). The exosome derived circFARSA produced by NSCLC cells increased macrophage polarization towards the M2 phenotype, hence facilitating epithelial-mesenchymal transition (EMT) and migration. Chen et al. (119) discovered that exosome derived circFARSA, promotes PTEN ubiquitination and degradation in macrophages, consequently, facilitate macrophage polarization toward the M2 phenotype and activating the PI3K/Akt signaling.
Deng et al. (120) investigated the mechanism through which the exosome circATP8A1 facilitated M2 polarization. They revealed that STAT6 was activated following exosome treatment of macrophages, highlighting the significant involvement of STAT6 in gastric cancer (120). Prior research indicated that IL-4 facilitates M2 polarization primarily via STAT6 activation (121). M2 polarization entails tyrosine phosphorylation and STAT6 activation, which subsequently facilitates the transcriptional activation of M2 -specific genes, including Arg1 and mannose receptor 1 (Mrc1) (122). The findings of Deng et al. (120) validated that exosomal circATP8A1 produced from gastric cancer cells facilitates M2 polarization by influencing the STAT6. The ceRNA mechanism is a crucial pathway by which circRNAs exert their biological effects. The findings validated that the exosomes produced from gastric cancer cells, specifically circATP8A1, facilitate M2 macrophages polarization by competitively binding to miR-1-3p, hence activating the STAT6.
Tian et al. (74) demonstrated that exosomal CXCL14 facilitated M2 polarization via exosome-derived communication by NF-κB activation. As a member of the chemokine family, CXCL14 is essential for DC maturation, the induction of MHC I, immune cell infiltration, and cellular mobilization (123). Tian et al. (74) identified a strong correlation between the overexpression of CXCL14 and the expression of two M2 macrophage-associated markers, PD-L1 and IL-10, indicating CXCL14’s possible function in modulating M2 macrophage polarization. These findings indicated that CXCL14 facilitates macrophage polarization toward an M2-like phenotype. These findings demonstrated that NF-κB signaling was involved in the function of CXCL14 in M2 macrophage polarization in prostate cancer.
Research indicates that malignancies can elicit a pro-tumor phenotype in neutrophils (124). In a study by Zhang et al. (76), exosomes produced from gastric cancer cells stimulated autophagy in neutrophils by activating the NF-κB pathway via the HMGB1/TLR4 connection. Their findings elucidate a novel method for neutrophil regulation inside the TME and furnish additional data regarding the significant roles of exosomes in this context (76). G-CSF has been shown to activate autophagy in neutrophils, and the growth of neutrophils produced by G-CSF is compromised in the absence of autophagy (125). The findings indicate that exosomes derived from gastric cancer stimulated autophagy in neutrophils, resulting in the secretion of substances that enhanced the migratory capacity of gastric cancer. In summary, gastric cancer-exosomes promote autophagy and N2 neutrophil polarization via the HMGB1/TLR4/NF-кβ signaling.
4.3 Immunosuppression and immune evasion
TDEVs are essential for the ability of cancer cells to evade immune surveillance (126). These vesicles function as carriers of immunomodulatory molecules, thereby modifying the TME to promote immune evasion and suppress anti-tumor immunity (127). Previously, it was found that tumor-derived exosomes contain molecules from the TNF receptor (TNFR) superfamily, including FasL, which disrupt the function of T cells (96). Söderberg et al. (128) identified the presence of TNFR1 and TNFR2 in their study. Notably, TNFR2 has emerged as a key player in T cell activation and is thought to contribute to the tumor’s strategy to evade immune responses, a phenomenon often referred to as the tumor’s counterattack (129). Furthermore, the production of hydrogen peroxide (H2O2) in lymphocytes that internalized the exosomes highlights the functional importance of these vesicles (130, 131). Several lymphocyte receptors, including FasL, dopamine receptors, TLRs, and B-cell receptors, produce H2O2 as a signaling mediator (130, 131). Several studies have reported that reactive oxygen species (ROS) can lead to functional downregulation of TCR signaling by inactivating the CD3 zeta-chain (132). Melanoma-derived TNF displays dual functionality, acting as both a cytokine and growth factor by transferring to nearby melanoma cells via paracrine or juxtacrine routes, thereby activating NF-κB–mediated survival pathways, akin to IL-15 and IL-10 receptor signaling (133, 134). Exosomes play a key role in inducing oxidative stress, a condition recently shown to disrupt TCR signaling and reduce T cell numbers (135). The role of TNF in the TME of malignant melanoma may be partly explained by its involvement in promoting the secretion of ROS-generating exosomes, which contribute to immune evasion.
Chalmin et al. (53) discovered that MDSC expansion and activation are mediated by distinct molecular pathways. The ligand responsible for the activation and enhancement of the suppressive ability of TDEVs is Hsp72, which binds to TLR2 on MDSCs. Chalmin et al. (53) demonstrated that TDEVs possess immunosuppressive properties, which are activated by STAT3 phosphorylation in MDSCs. Their findings revealed that TDEVs trigger both STAT3 activation and the development of suppressive activity in MDSCs. Interestingly, the absence of PGE2 in these vesicles suggests that the Hsp72/TLR2 signaling axis may represent a previously unrecognized mechanism responsible for MDSC activation.
Jiang et al. (15) demonstrated that cancer exosome-miR-9 was strongly positively correlated with miR-181a and further upregulated miR-181a expression in MDSCs. Both miR-9 and miR-181a directly targeted SOCS3 and PIAS3, which are critical negative regulators of the JAK/STAT, leading to their suppressed expression. Notably, PIAS3 was upregulated when miR-181a levels were reduced and suppressed in MDSCs transfected with miR-181a mimics. The positive concordance observed between SOCS3 and PIAS3 in MDSCs hinted that miR-9 and miR-181a might act in a synergistic way. Jiang et al. (15) suggested that miR-9 and miR-181a independently regulated SOCS3 and PIAS3, they could coordinate with each other by regulating a synergetic ceRNA network to maintain the activation of the JAK/STAT.
Exosomes have an inhibitory effect on the maturation of DCs and the secretion of cytokines, as well as the differentiation of monocytes to DCs (82). DU145 exosomes drive DCs to express CD73 on their surface, creating a unique subset of DCs co-expressing CD39 and CD73. This phenotypic change allows the hydrolysis of adenosine triphosphate (ATP) into adenosine in the pericellular environment by these cells, thus contributing to immunosuppression (82). Adenosine has been demonstrated to have angiogenic, and metastasis-inducing effects under normal conditions. Additionally, it is a potent inhibitor of antitumor immune effector cells in the TME (136, 137). Salimu et al. (82) demonstrated that PGE2 is primarily found in the exosomal fraction of DU145 cell supernatants. They also found that the presence of ATP significantly reduced the ability of DCs treated with exosomes to secrete proinflammatory cytokines such as TNFα and IL-12 (82). These cytokines are essential for promoting anti-tumor immune responses, supporting DC maturation, and enhancing T cell activation (82). Inhibition of the PGE2 receptors EP2 and EP4 on DCs significantly reduced exosome-induced CD73 expression, suggesting that exosomal PGE2 engages in ligand–receptor interactions with DCs (138). PGE2 plays a critical role in upregulating C-C chemokine receptor type 7 (CCR7) and MMP-9 on DCs, thereby promoting their migration to lymph nodes (139, 140). PGE2 is a key driver of the expression of indoleamine 2,3-dioxygenase 1 (IDO1) on DCs, which attracts and induces Tregs. This would then lead to the suppression of CD8+ T cell responses—most likely creating an immunosuppressive environment (141). Salimu et al. (82) demonstrated that exosomal PGE2, through the induction of CD73 on DCs, played a role in the modulation of cytokine formation and T-cell activation. Notably, they assessed T-cell responses and showed that exosome-treated DCs suppressed T-cell activity in an adenosine-dependent manner, with the inhibitory effect intensifying in the presence of extracellular ATP or AMP.
TGF-β is a potent immunosuppressive factor that promotes tumor immune evasion by enhancing regulatory T cell activity, inhibiting T cell function, reducing IFN-γ production, and impairing antigen presentation (142, 143). TGF-β has been recently identified as a potent down-regulator of NKG2D-activating receptors on the surface of NK cells (144). Berchem et al. (86) demonstrated that hypoxic TDEVs significantly downregulate NKG2D expression on NK, suggesting a mechanism by which hypoxic TDEVs impair NK cytotoxicity, potentially through the delivery of TGF-β. Their findings support the notion that TDEVs reflect the hypoxic state of tumor cells by transporting immunosuppressive molecules that compromise NK function (86). Notably, NK from draining lymph nodes of mice treated with hypoxic TDEVs exhibited reduced levels of CD107a, IFN-γ, and Granzyme B compared to those treated with normoxic TDEVs (86).
Vulpis et al. (92) showed that the formation of IFN-γ on CD56 high NK is induced by MM-derived exosomes and is mediated by TLR2 engagement. NF-kB is involved in the activation of numerous cytokine genes, including IFN-γ, and is one of the primary pathways that TLRs activate. In this sense, exosome-induced IFN-γ production was determined to be contingent upon NF-kB signaling. A recent study proposed a model in which breast cancer-exosomes activate TLR2-mediated NF-κB signaling in human macrophages, leading to cytokine production, supporting and aligning with these findings (14). TLR2 recognizes damage-associated molecular patterns (DAMPs), including HSPs, which typically function intracellularly as molecular chaperones but act as potent danger signals when extracellular or membrane-bound, triggering immune activation (145, 146). EVs can associate with extracellular HSPs, as demonstrated by Vulpis et al. (92), who found elevated levels of HSP70 on the surface of multiple myeloma-exosomes. This HSP70 was suggested to play a role in stimulating IFN-γ production by NK in response to the exosomes.
Research has shown that infiltrating neutrophils express high levels of PD-L1, which suppresses T cell activation and proliferation (147). Shi et al. (46) showed a high PD-L1 expression on neutrophils, promoted by the gastric cancer microenvironment, thus suggesting that tumor signals may regulate neutrophils toward an immunosuppressive phenotype. They further show that gastric cancer-derived exosomes induce immunosuppressive neutrophils with elevated PD-1 expression via the activation of STAT3 (46). In conclusion, these results indicate that TDEVs are central mediators of tumor immune evasion, employing complex mechanisms to reduce immunity.
4.4 Inflammatory signatures
Cancer has been identified as an inflammatory disease (148, 149). Additionally, the immune system and inflammation have garnered a significant quantity of attention in relation to the development of cancer (149). In cancer cells, these exosomes have been found enriched with certain biomolecules such as NF-κB, STAT3, and pro-inflammatory cytokines (e.g., TNF-α, IL-1β, and IL-6); this means cancer cell-derived exosomes recruit immune cells to target sites (150). Bretz et al. (13) found that malignant ascites-derived ovarian cancer exosomes bind to TLR2 and TLR4 on THP-1 cells, thus eliciting the secretion of pro-inflammatory cytokines like IL-1β, TNF-α, IL-and 6, IL-8 by activation of NF-κB- and STAT3-dependent signaling. Other findings found that exosomes derived from breast and gastric cancer stimulate the production of pro-inflammatory cytokines (G-CSF, IL-1β, IL-6, IL-8, CCL2, and TNF-α) in M1 macrophages in an NF-κB-dependent manner (14). Chow et al. (14) demonstrated that cancer-derived exosomes interact with TLR2, leading to NF-κB activation in monocytes and macrophages, thereby highlighting the role of TLR2 in mediating the cross-talk between inflammation and cancer development. Moreover, they showed that exosomes from breast cancer can induce a pro-inflammatory response in macrophages, further supporting the link between tumor-derived exosomes and inflammation-driven tumor progression (14). These results were previously showed that exosomes activate NF-κB signaling and promote the secretion of inflammatory cytokines in monocytic cells (13). More recently, it was shown that membrane-associated Hsp72 from tumor exosomes activates STAT3 in MDSCs through a TLR2/Myeloid differentiation primary response 88 (MYD88)-dependent pathway mediated by the autocrine production of IL-6 (53). TNF-α secretion by myeloid cells is induced by the activation of TLR2:TLR6 complexes by cancer-secreted vesicles, thereby promoting the growth of metastatic tumors (151). Bretz et al. (13) showed that the treatment of THP-1 cells with tumor-derived exosomes triggers the activation of NF-κB and STAT3, which results in the formation of pro-inflammatory cytokines. They showed that tumor-derived exosomes promote the production of pro-inflammatory mediators, where NF-κB activation leads to IL-6 production, a prerequisite for subsequent STAT3 activation in an autocrine or paracrine way. Bretz et al. (13) showed this effect is mediated by TLR2/4, with cytokine induction reduced by their inhibition. MYD88 deficiency abolished this response, confirming TLR-dependent signaling. Finally, EVs can both activate and inhibit inflammasomes (152). Bottino et al. (90) found that HNSCC-derived EV-enriched vesicular secretome (VSF) inhibited NLRP3 inflammasome activation by reducing caspase-1 and IL-1β secretion in response to nigericin. This inhibition was linked to blocked NF-κB-mediated priming and may be due to TGF-β-related proteins in VSF. These findings suggest a tumor immune evasion mechanism via inflammasome suppression.
4.5 Metabolic reprogramming of immune cells
TDEVs reshape the immune environment by metabolically reprogramming immune cells to support immune evasion and tumor growth (1). Morrissey et al. (11) found that tumor exosomes activate NF-κB to drive an M1 macrophage and upregulate PD-L1 through both direct promoter binding and metabolic changes. They identified a new mechanism whereby NF-κB regulates PD-L1 expression, not only by direct binding to its promoter but also via metabolic reprogramming. NF-κB activation triggered two distinct pathways that enhanced glycolysis in response to TLR2/MyD88 signaling after tumor-derived exosome ligation. First, it upregulated Glucose transporter 1 (Glut1) expression via activation of Hypoxia-inducible factor 1α (HIF-1α), which increased glucose influx into macrophages. Second, activated Nitric oxide (NO) synthase-2 (NOS2) increased NO production, which inhibited complexes III and IV of the electron transport chain, driving pyruvate conversion to lactate. Morrissey et al. (11) also demonstrated that exogenous lactate activates NF-kB, resulting in the expression of PD-L1 on macrophages. The uptake of lactate intracellularly is required to promote PD-L1 expression, as evidenced by the reduction of lactate-induced PD-L1 expression on macrophages with the addition of the Monocarboxylate Transporter 1 (MCT-1) inhibitor AZ3965. In summary, Morrissey et al. (11) demonstrated that tumor-derived exosomes metabolically reprogram tissue-resident macrophages, promoting an immunosuppressive phenotype in the pre-metastatic niche.
Park et al. (77) documented an elevated level of macrophage chemotaxis toward exosomes generated by hypoxic tumors. Furthermore, hypoxic exosomes induced an increase in M2 polarization of myeloid cells and facilitated a modification in the immunometabolic profile of macrophages to improve OXPHOS activity (153, 154). These results are in accordance with observations that M2 macrophages promote tumor proliferation and increase angiogenesis by OXPHOS (155). Exosome-induced upregulation in the activity of OXPHOS in bone marrow-derived macrophages was associated with mTOR pathway inhibition and may thus hint at the possibility that exosomes could regulate the Akt-mTOR pathway by mechanisms of inhibition (77). Park et al. (77) demonstrated that let-7a miRNA is an epigenetic tumor suppressor and also a possible inhibitor of insulin-mediated mTOR signaling pathways. They showed that microenvironmental hypoxia downregulated let-7a levels in cancer cells and simultaneously enhanced exosome release (77). Moreover, the exosomal transfer of let-7a miRNA caused downregulation of insulin-mediated Akt-mTOR signaling, consequently promoting OXPHOS activity and M2 polarization of infiltrating macrophages (77).
Kersten et al. (78) showed that tumor-derived vesicles acutely activate mTORC1 in macrophages, which in turn promotes metabolic programs defined by increased OXPHOS and promotes anti-metastatic activity in the lung. The catalytic subunit mTOR, shared by both mTORC1 and mTORC2 complexes, is a key regulator of metabolism and found to promote mitochondrial biogenesis and oxidative activity (156, 157). An elaborate analysis of the mTORC1 pathway showed an upregulation of phospholipases D1 and D3 (Pld1 and Pld3) and genes involved in protein synthesis, such as Eukaryotic translation initiation factor 4E-binding protein 1 (Eif4ebp1), Protein Phosphatase 2 Phosphatase Activator (Ptpa), and Ribosomal Protein S23 (Rps23). In contrast, downregulation was noted for Prkab2, a gene encoding a positive regulator of the AMPK, an upstream negative regulator of mTOR, indicating repression of the AMPK in macrophages (158). Kersten et al. (78) showed that the uptake of tumor-derived microparticles activates mTORC1 signaling in macrophages, as demonstrated by the greatly increased levels of phosphorylated (p)4EBP1 and pS6, resulting in metabolic and phenotypic changes in non-resident macrophages in the early metastatic lung, which strongly depended on mTORC1 signaling.
Clayton et al. (81) found that cancer-derived exosomes convert ATP and AMP into adenosine, increasing adenosine levels in the TME and suppressing T-cell activity, highlighting a mechanism of immune evasion. They point out that the expression of catalytically active membrane-associated enzymes like CD73 is an intrinsic feature of exosomes since their release into the microenvironment enhances the surface area for enzyme activity severalfold. Clayton et al. (81) focused on the immunosuppressive consequences of exosomal CD39/CD73 in generating adenosine within the TME. This adenosine production initiates downstream signaling through the A2A and A2B receptors, which activates adenylyl cyclase and yields cAMP. Exosomes can sequentially hydrolyze ATP and AMP to produce adenosine, which acts on cAMP signaling in A2A receptor-positive cells to further support immunosuppression in the malignant microenvironment. Clayton et al. (81) also showed the impact of adenosine generation on T cell function and demonstrated that the production of inflammatory cytokines and the proliferative responses of T cells could be effectively suppressed by AMP as a substrate. This inhibition was eliminated by chemically inhibiting the hydrolytic function of CD73. In conclusion, these results suggest that exosome secretion by cancer is a mechanism that contributes to the increase in adenosine levels in the TME.
4.6 Immune cell apoptosis
TDEVs deliver molecular cargo that induces apoptosis in immune cells (159). In this regard, surface CD95 is expressed by CD8+ T lymphocytes in cancer patients, and a significant number of them express PD-1 (160, 161). Consequently, CD8+ T lymphocytes susceptible to apoptosis when TDEV carries the membrane form of FasL and PD-L1 (160, 161). The frequency of apoptosis-sensitive activated CD8+ T cells in cancer patients was correlated with the expression of these apoptosis-inducing molecules in TDEVs (162, 163). It is crucial to note that the disease stage and prognosis were significantly correlated with the spontaneous apoptosis of circulating CD8+ T cells (160, 162). TDEVs triggered apoptosis in activated CD8+ T cells, as evidenced by early membrane alterations such as annexin V binding, caspase-3 activation, cytochrome c (cyt c) release from mitochondria, disruption of mitochondrial membrane potential, and DNA fragmentation (164). Inactivated CD8+ T cells, the PI3K/Akt pathway is a critical target of TDEVs (164, 165). PTEN, a phosphatase known for its role in regulating the PI3K/Akt signaling pathway, was recently identified as part of the cargo carried by TDEVs, where it retains its phosphatase activity and functions within recipient cells (166). Incubating activated CD8+ T cells with TDEVs led to a significant, time-dependent decrease in Akt phosphorylation, accompanied by reduced expression of the anti-apoptotic proteins B-cell lymphoma (BCL)-2, BCL-xL, and MCL-1 (166). Simultaneously, TDEV increased the expression of the proapoptotic protein Bax (164, 167). Subsequently, TDEV induces apoptosis in activated CD8+ T cells by activating intrinsic and extrinsic apoptosis pathways (164).
Of note, the immune response to tumor cells can be inhibited by exosomes released by cancer cells, particularly via apoptosis in T lymphocytes (168, 169). Shen et al. (95) found that exosomes from pancreatic cancer could significantly induce apoptosis of peripheral T lymphocytes following their internalization. They used Gene Set Enrichment Analysis (GSEA) to describe essential signaling pathways in this process and observed changes in the gene expression profiling of exosome-treated T cells (95). Notably, genes related to apoptosis and ER stress, such as p38 MAPK, JUN, eukaryotic translation initiation factor 2A (eIF2A), Protein Kinase RNA-Like ER Kinase (PERK), C/EBP homologous protein (CHOP), and Activating transcription factor 4 (ATF4), were altered, suggesting their roles in the induction of apoptosis (95). Phosphorylation of p38 MAPK was found to be a key driver of T lymphocyte apoptosis via ER stress. RNA sequencing of exosome-treated T cells identified 177 differentially expressed mRNAs, with 151 upregulated and 26 downregulated (95). Of these, apoptosis-related and unfolded protein response (ER stress) gene sets were strikingly enriched, suggesting that the ER stress signals are transduced to the nucleus by PERK-eIF2α-ATF4-CHOP and ultimately promote apoptosis (170). Shen et al. (95) further showed that pancreatic cancer-derived exosomes upregulated the expression of ATF4, PERK, and CHOP, accompanied by increased phosphorylation of eIF2α, indicative of ER stress signaling activation. The pro-apoptotic effects of ER stress were demonstrated to be finely modulated by the MAPK signaling. More precisely, p38 MAPK was shown to interact with CHOP, and the p38 MAPK inhibitor SB203580 efficiently disrupted this interaction. Shen et al. (95) found the involvement of p38 MAPK in ER stress-induced apoptosis and showed that its inhibition by SB203580 (a potent inhibitor of p38 MAPK activity) reduced the apoptotic effects. Pancreatic cancer derived exosomes, through upregulation of JUN, activated p38 MAPK, which mediated ER stress and apoptosis in T lymphocytes (95). Evidence also supported that p38 MAPK further induce JUN expression in T lymphocytes to reinforce the apoptotic signaling cascade (171, 172). SB203580 was able to inhibit this action, intriguingly, while exosome treatment resulted in an upregulation of DUSP1, a phosphatase that negatively modulates the MAPK; SB203580 reduced its levels (95). The alteration in p38 phosphorylation was in accordance with the alteration in DUSP1 expression. The results of Shen et al. (95) suggest that DUSP1 may serve as a critical mechanism to mitigate the excessive activity of p38 MAPK. In conclusion, the findings demonstrate that pancreatic cancer-derived exosomes are taken up by T lymphocytes, alter their gene expression and cytotoxic function, and induce ER stress-mediated apoptosis through activation of the p38 MAPK.
Abusamra et al. (173) demonstrated that the Fas–FasL pathway was essential for the apoptosis of T-cells mediated by cancer exosomes. Cancers have been observed to exhibit an elevated expression of FasL (174). The Fas–FasL pathway has also been extensively used to document the apoptosis of tumor-reactive T cells (175). Abusamra et al. (173) indicated that “membrane-bound” FasL, which is transported by exosomes, is present in the peripheral circulation. This tumor-derived circulating FasL has the potential to efficiently induce apoptosis by targeting circulating T cells at a distance (175). These results demonstrated that exosomes expressing FasL suppressed T-cell responses by inducing apoptosis. Finally, Huber et al. (169) demonstrated that CRC triggered apoptosis in T cells via the release of FasL and TNF-related apoptosis-inducing ligand (TRAIL)-bearing microvesicles. The expression of FasL, a key molecule in CRC progression, emerges in the early stages of colon carcinogenesis, correlates with tumor spread, and is associated with increased apoptosis of lymphocytes within the TME (169). The secretory pathway of proapoptotic microvesicles, common to various tumor histotypes including melanoma, constitutes an effective mechanism for CRC to transmit death signals to antitumor T lymphocytes without requiring direct cell-to-cell interaction. Collectively, the evidence underscores the pivotal role of TDEVs, including exosomes and microvesicles, in mediating immune evasion through the induction of T cell apoptosis. The findings highlight the multifaceted strategies employed by tumors to suppress antitumor immunity and emphasize the therapeutic potential of targeting TDEV-mediated immune suppression to restore effective T cell responses in cancer.
5 The function of TDEVs in immune signaling modulation caused tumor development, angiogenesis, and metastasis
One of the important roles of TDEVs is the regulation of immune signaling, which plays an essential role in tumor development, angiogenesis, and metastasis (Table 3). In this sense, TAMs, which are predominantly of the M2 phenotype, are significant contributors to pro-angiogenic activity in malignancies and, as such, play vital roles within the TME (191). TAMs involve in tumor angiogenesis by secreting several pro-angiogenic cytokines, including VEGFA, platelet-derived growth factor β (PDGF-β), angiogenin, placental growth factor (PlGF), TGF-β, and MMPs (192–195). In a coordinated manner, these factors promote the formation and remodeling of tumor-associated vasculature to feed tumor growth and progression (184, 196). Some works confirmed that the PI3K/AKT regulates numerous pro-angiogenic cytokines, including VEGFA and MMP9, which are involved in tumor angiogenesis (197). PTEN is a phosphatase that has the ability to dephosphorylate PIP3 into PIP2 and thereby directly inhibit the oncogenic PI3K/AKT (198). Esophageal squamous cell carcinoma (ESCC) cells secrete exosomes that induce M2 polarization through the PTEN/PI3K/AKT, as revealed by Shou et al. (184). Research has proven the role of TAMs in promoting tumor angiogenesis by showing that TAMs can enhance the migration, proliferation, and invasion of tumor endothelial cells (TECs) through the secretion of VEGFA and MMP9, contributing to the development and remodeling of tumor-associated vasculature to support angiogenesis and tumor progression. Shou et al. (184) identified miR-301a-3p as being significantly upregulated and demonstrated its positive correlation with Tumor, Node, Metastasis (TNM) stage, tumor invasion, and lymph node metastasis; therefore, it may regulate the pro-angiogenic switch of macrophages via PTEN/PI3K/Akt signaling. Their findings showed that exosomal miR-301a-3p derived from ESCC could induce M2 polarization via this pathway, thereby indirectly promoting angiogenesis and tumor progression.
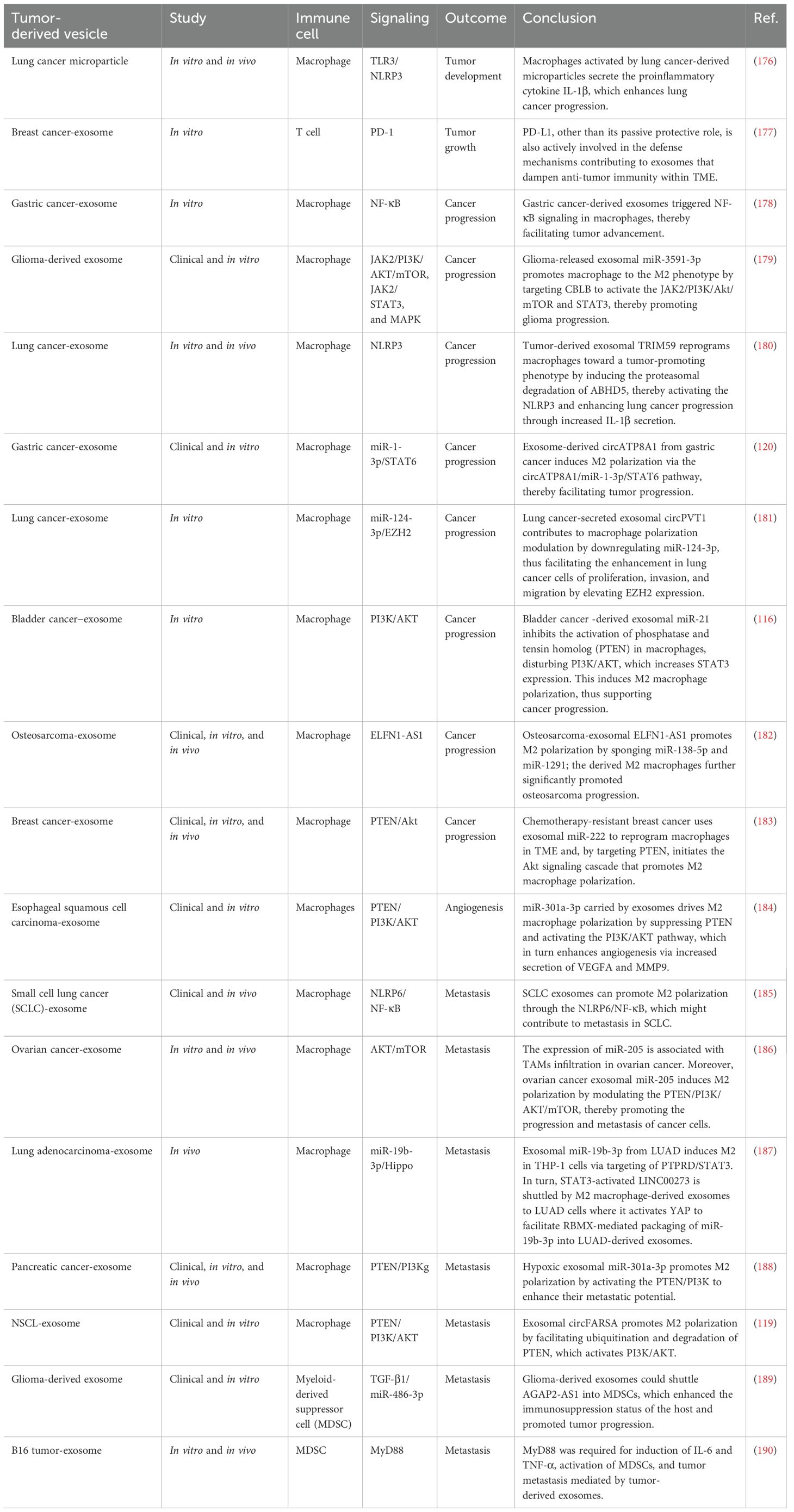
Table 3. Tumor-derived vesicles and immune signaling modulation caused tumor development and metastasis.
Wu et al. (178) found the function of exosomes in the activation of macrophages with regard to the NF-κB pathway. Their findings revealed that exosomes derived from gastric cancer could trigger NF-κB activation in human primary macrophages. This activation was significantly suppressed when macrophages were pretreated with an NF-κB inhibitor. To survey the function of exosome-activated macrophages on gastric cancer proliferation, migration, and invasion, Wu et al. (178) incubated MGC-803 cells with supernatant from cancer exosome-treated primary macrophages and the findings showed that the supernatant enhanced MGC-803 cell migration, colony formation, and invasion, effects that were reversed by NF-κB inhibitor pretreatment. Collectively, the study highlights that gastric cancer-derived exosomes activate macrophages through the NF-κB, thereby facilitating tumor progression.
Zhou et al. (199) revealed that mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) was expressed in tumor but vastly expressed in TAMs in these patients with breast cancer with highly expressed cirRNA cSERPINE2. They demonstrated that tumor exosomal cSERPINE2 from breast cancer could upregulate MALT1 in TAMs, which then activated the NF-κB and increased the secretion of IL-6 by TAMs. Blocking IL-6 could reverse the tumor-promoting effect caused by cSERPINE2 overexpression. In addition, the TAM-secreted IL-6 stimulated the JAK2-STAT3 pathway in tumor cells and then upregulated the expression of CCL2 and EIF4A3. The IL-6 secretion upregulated by tumor exosomal cSERPINE2-educated TAMs enhanced CCL2 expression, and thereby, TAMS infiltration into TME was enhanced.
Menghisteab et al. (200) identified a novel mechanism through which CRC-derived EVs stimulate macrophages to generate MMPs and promote invasion, contributing to the formation of a pre-metastatic niche. While further research is needed to confirm the exact mechanisms, their findings clearly indicate that CD147 on CRC-EVs plays a key role in activating MMP12 in macrophages, thereby enhancing tumor cell invasion. The results of Menghisteab et al. (200) indicated that CD147+ EVs are involved in the activation of MAPK in macrophages, which results in the expression of MMP and the enhancement of tumor cell invasion. They emphasized that CRC-EVs are major contributors to promoting an invasion-prone phenotype in macrophages, possibly via CD147. This exosome population can significantly contribute to the activities of macrophages, including extracellular matrix (ECM) degradation, VEGF release, recruitment of more cells in the polymorphonuclear leukocytes (PMNs), and, finally, metastasis enhancement through ECM degradation and modulation of the PMN. These findings suggest that CRC-EVs could become essential mediators in the metastatic process by modulating macrophage behavior.
Rao et al. (185) demonstrated that exosomes secreted from small cell lung cancer (SCLC) cells can polarize macrophages toward the M0 to M2 phenotype. Such phenotype switching induced by SCLC-derived exosomes can partly explain one way that exosomes promote distant metastasis in SCLC because switching to the M2 phenotype in other tissues can initiate metastasis. Metabolic stimuli and inflammatory signals, including Peroxisome proliferator-activated receptor gamma (PPAR-γ) and TNF-α, have been shown to modulate NLRP6 expression at transcriptional and post-translational levels (201). Through the inhibition of IL-6 signaling, NLRP6 safeguards against the development of colon tumors associated with inflammation (202). Rao et al. (185) demonstrated that NLRP6, which normally inhibits tumorigenicity in gastric cancer and CRC, acts as a promoter of metastasis in SCLC and also its expression was significantly upregulated. In addition, their study of SCLC-derived exosomes showed that the inhibition of exosome release markedly inhibited NLRP6 expression and the M0-M2 switching response. These results underscore the association of SCLC-derived exosomes with the M0 phenotype switch and that activation of the NLRP6/NF-κB pathway facilitated SCLC metastasis. Taken together, TDEVs play a central role in establishing an immunosuppressive TME, hence allowing tumors to survive and develop (Figure 3). Also, TDEVs actively support angiogenesis and are likely to contribute to the preparation of pre-metastatic niches, thus positioning them as real drivers of tumor progression and metastasis.
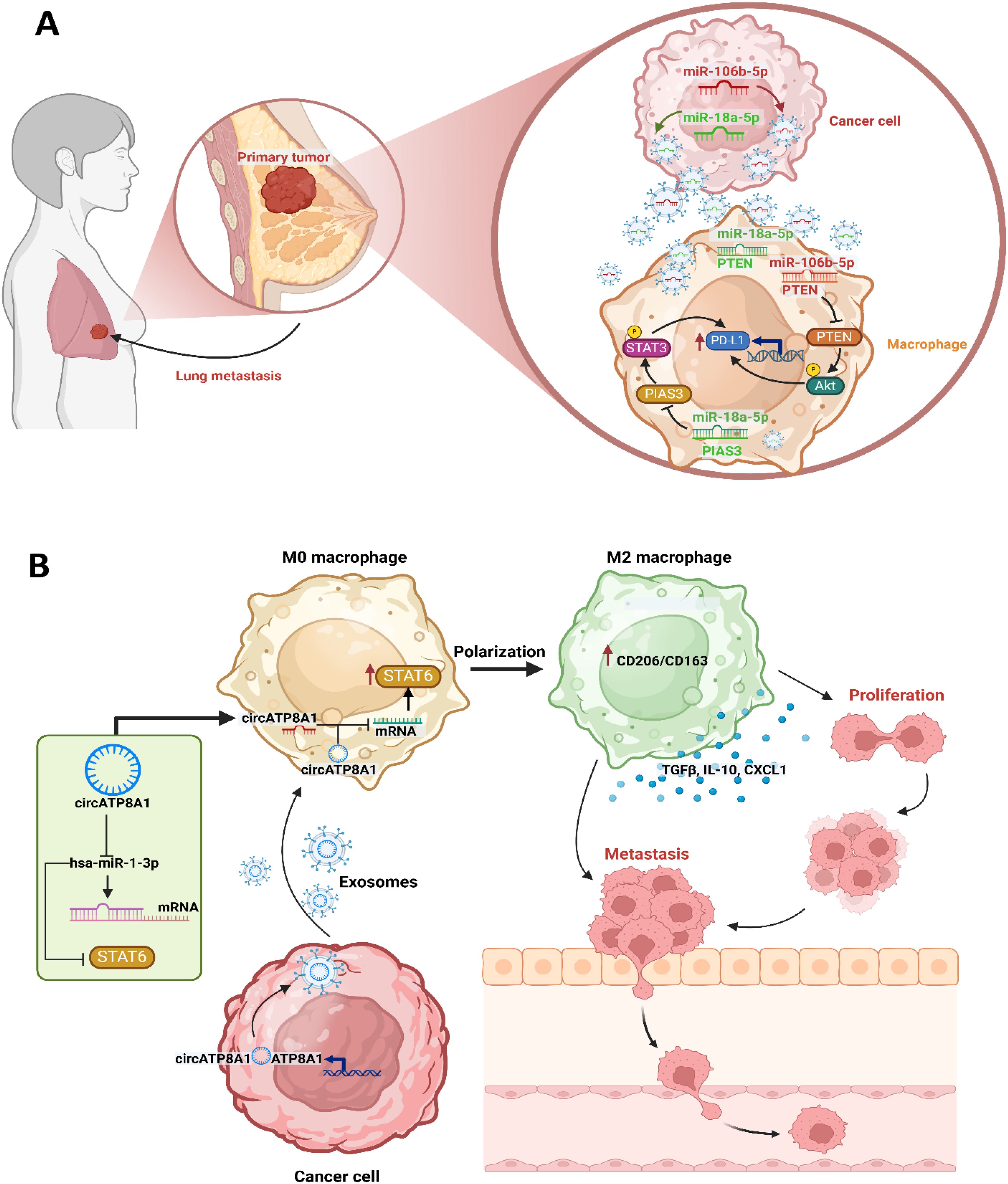
Figure 3. Immune signaling modulation by tumor-derived vesicles caused tumor development and metastasis. (A) In breast cancer, tumor-derived small EVs (sEVs) induce an immunosuppressive niche through the regulation of TAMs and increased expression of PD-L1. Especially, PD-L1 expression is higher in TAMs compared with cancer cells in breast, and a specific PD-L1+CD163+ TAM subpopulation is strongly associated with metastatic potential. Further, breast cancer-derived sEVs contribute to the establishment of this immunosuppressive environment by promoting PD-L1 expression and supporting M2 macrophage polarization, which allows immune evasion and leads to enhanced metastatic progression. Among the key molecular drivers of this process are sEV-associated miR-106b-5p and miR-18a-5p, whose expression is associated with poor prognosis and high metastatic potential in patients with breast cancer. In a synergistic manner, these miRNAs ensure the upregulation of PD-L1 in M2 TAMs by targeting the PTEN/AKT and PIAS3/STAT3. Through this orchestrated immune modulation, breast cancer cells acquire an invasive advantage by evading immune surveillance but favoring metastatic dissemination. (B) In gastric cancer, TDEV-mediated immune modulation is generally dominated by circATP8A1, a circular RNA that is highly enriched in exosomes derived from gastric cancer. Clinical studies have shown that high circATP8A1 levels correlate with advanced TNM staging and poor prognosis. Mechanistically, circATP8A1 acts through the induction of M2 macrophage polarization, associated with immunosuppression and tumor promotion. Unlike many other tumor-associated immune mechanisms that depend on STAT3, circATP8A1 specifically activates the STAT6 by competitively binding miR-1-3p. This sequestration of miR-1-3p blocks its inhibitory effects on STAT6, thus promoting M2 polarization. Functionally, exosome-treated M2 macrophages increase gastric cancer cell migration, which reinforces a pro-tumorigenic loop. Knockdown of circATP8A1 disturbs this axis and inhibits gastric cancer progression.
6 Tumor-derived vesicles on immune signaling as a target for immunotherapy
TDEVs are under intense scrutiny in immunotherapeutic research due to several key attributes (203). First, their possible role in antigen presentation supports the development of anti-tumor vaccination strategies (204). Second, TDEVs’ inherent immunosuppressive properties shed light on tumor-immune interactions (59, 205). Third, based on their ability to be internalized by living cells, TDEVs become excellent biological carriers for delivering engineered therapeutic agents (206). Therefore, targeting key transcriptional networks regulating M2-polarized macrophages is a novel approach to reprogram the genetic profile of TAMs directly (207). STAT6 was selected for these experiments because of its pivotal role as a regulator of the macrophage M2 transcriptional program inside the TME across many malignancies (208). Besides its function in TAMs, STAT6 is crucial for CD4 Th2 responses and facilitates immunoglobulin (Ig) class switching to IgE and IgG1 in B cells, along with antigen presentation (209, 210).
Kamerkar et al. (207) revealed that exosomes facilitate the selective targeting of immune-suppressive TAMs and myeloid cells while minimally affecting lymphocytes and non-immune cells. In their study, augmented STAT6 suppression using an exosome therapeutic candidate delivering an antisense oligonucleotide (ASO) targeting STAT6 (exoASO-STAT6) led to significant macrophage reprogramming. ExoASOSTAT6 reprogramed macrophages in the presence of a combination of immunosuppressive cytokines such as TGF-β, IL-4, and IL-10 that often present in the TME (207). The administration of exoASO-STAT6 led to a significant reduction in the M2 macrophage phenotype and simultaneously, a strong activation of the M1 gene signature and proinflammatory cytokines was noted following exoASO-STAT6 therapy (207). The disparity between exoASO-STAT6 and free ASO is pronounced, evidenced by the significant tumor growth inhibition elicited by exoASO-STAT6, in contrast to the inactivity of free ASO at an equivalent dosage. Kamerkar et al. (207) presented the inaugural evidence of utilizing exosomes for the targeted delivery of ASOs, enhancing STAT6 knockdown in TAMs, and reprogramming macrophages amid immune-suppressive cytokines. Furthermore, the benefits of exosome-mediated delivery of ASO are evident in vivo, demonstrating that the effective antitumoral dosage of exoASO-STAT6 is 50–100 times lower than that reported in preclinical research involving untargeted ASO treatments (211). In conclusion, exosome-mediated delivery significantly enhances the potency of ASOs to inhibit STAT6 expression in TAMs and effectively reprograms them to an M1 phenotype. Consequently, exoASO-STAT6 therapy induces significant modification of the TME to a proinflammatory, antitumoral state and facilitates the formation of the CD8+ T cell-mediated response. The potency and specificity of exoASO-STAT6 yield significant single-agent antitumor efficacy, setting this innovative therapeutic strategy apart from conventional macrophage-targeting treatments and underscoring its clinical promise.
In addition to therapeutic delivery, TDEVs themselves modulate immune responses. During monocyte-to-macrophage differentiation, the downregulation of specific miRNAs leads to increased IKKα and NF-κB activation (212). Jang et al. (213) demonstrated that epigallocatechin gallate (EGCG) influences the tumor exosomes and enhances miR-16, which leads to the downregulation of IKKα and the inhibition of M2 polarization in TAM following EGCG treatment. Given that miR-16 may act as a tumor suppressor, it is plausible that elevated exosomal miR-16 could have influenced the survival and proliferation of tumor cells (213). NF-κB activation is recognized as a crucial factor for the differentiation of macrophages into M2 macrophages and has a role in tumor growth (214). Consequently, NF-κB in TAMs is regarded as a unique therapeutic target for cancer management. Jang et al. (213) noted the down-regulation of the NF-κB, specifically regarding IKKα and I-κB expression in TAM, induced by EGCG-treated exosomes (213). They identified this as the mechanism responsible for the EGCG-mediated inhibition of macrophage infiltration and differentiation into tumor-promoting M2 macrophages despite the absence of direct evidence.
It has been revealed that T-cells triggered by OKT3 (anti-CD3 antibody) incurred apoptosis, as seen by DNA fragmentation when treated with isolated fractions of TDEVs or CH-11 (anti-CD9) antibody, which cross-links Fas (213). The pretreatment of T cells with pan-caspase inhibitors [Z-VAD-FMK (benzyloxycarbonyl-Val-Ala-DL-Asp-fluoromethylketone)] slightly, however considerably, inhibited this effect (162). Only TDEVs, such as DCs or fibroblasts, triggered apoptosis in activated CD8+ T-cells (215). The capacity of TDEVs to provoke CD8+ T-cell apoptosis was attributed to the presence of membrane-bound FasL and MHC I in TDEVs (162, 163, 216). While the function of FasL in TDEV causing apoptosis in activated Fas+ CD8+ T-cells is unequivocal, the participation of MHC I remains conjectural. The direct interaction of MHC I with the CD8 receptor may activate the intrinsic Fas/FasL pathway, resulting in T-cell death (217, 218). The ex vivo investigations involving human T-cells and pure TDEVs unequivocally associate TDEVs with the decline of CD8+ T-cells noted in cancer patients, particularly pronounced following immunotherapy, during which CD8+ T-cells are stimulated and susceptible to apoptosis (217, 218). Kitai et al. (219) showed that topotecan (TPT) generates immunostimulatory DNA by TPT-mediated cell death, where the TPT-topoisomerase I-DNA complex activates the Stimulator of IFN genes (STING) pathway, likely through engagement with the DNA sensor cGAS. Kitai et al. (219) showed that TPT-treated cells activate DCs to produce inflammatory cytokines, chemokines, and type I IFNs in a STING-dependent manner (219). Moreover, TPT treatment elicited CD8+ T cell infiltration and activation at the tumor sites. Conditioned medium (CM) stimulation increased type I IFN production, STAT1 phosphorylation, and upregulation of cGAS, STING, and IFN regulatory factor 7 (IRF7). Type I IFN further stimulated DCs to upregulate the expression of genes, including 4-1BBL, CD30L, CD86, IL-6, IL-27, CXCL9, CXCL10, and CXCL11, suggesting that the cGAS-STING axis is essential in the immune responses elicited by TPT (219). TPT enhanced migration and expression of costimulatory molecules in unstimulated DCs but inhibited DC maturation induced by proinflammatory cytokines through suppression of NF-κB activity (219). Recently, it was demonstrated that TPT inhibits endotoxin-induced septic shock by suppressing pathogen-associated molecular pattern (PAMP)-induced inflammatory cytokine production by facilitating topoisomerase I-mediated nucleosome remodeling (220). In summary, Kitai et al. (219) showed that treatment with a high concentration of TPT only marginally increased IL-6 production by DCs. This may suggest that excessive TPT could suppress the innate immune activation either through the induction of excessive cell death and, as a result, a decrease in the availability of the immunostimulatory DNA or through the induction of topoisomerase I-mediated nucleosome remodeling, impairing immune response signaling.
As the general immunosuppressive activities of TDEVs involve the polarization of TAMs toward the M2 phenotype, suppression of CD8+ T cells, and modulation of DC function, therapies inhibiting the secretion of TDEVs from tumor cells may be one potential strategy to restore anti-tumor immunity (221, 222). These can include targeting key regulators of vesicle biogenesis (e.g., endosomal sorting complexes required for transport (ESCRT) complex, Rab proteins), vesicle trafficking inhibitors, or inhibitors of vesicle fusion with immune cells (223, 224). Inhibition of TDEV release can potentially suppress immune suppression in the TME, enhance CD8+ T cell survival, and increase the efficacy of checkpoint blockade therapies (225, 226). A balanced approach—targeting immunosuppressive vesicle subsets or their cargo modulation—thus offers a more promising therapeutic option. In conclusion, while exosome-based delivery systems like exoASO-STAT6 represent novel strategies for immunomodulation, the parallel development of inhibitors of the immunosuppressive functions or secretion of TDEVs would be complementary to current therapies. Exploring these possibilities has the potential to significantly enhance the translational potential of TDEV-targeted immunotherapies.
7 Future directions for targeting TDEV-mediated immune reprogramming
The ability of TDEVs to reprogram immune cells into immunosuppressive states has become a key factor in how tumors evade the immune system (227). To tackle this, future therapies should consider interactions between TDEVs and immune cells, especially those that influence crucial intracellular signaling pathways such as NF-κB, JAK/STAT, TGF-β, and PI3K/Akt/mTOR. Engineered EVs can be loaded with precision therapeutics, including antisense oligonucleotides (ASOs), siRNAs, CRISPR-Cas9 systems, or small-molecule inhibitors—to disrupt immunosuppressive pathways in the TME. These modified EVs selectively inhibit molecular programs driving M2-TAMs polarization while concurrently suppressing MDSC activity. Through these targeted actions, they enhance the antitumor functions of effector T and NK cells, effectively reprogramming the immune landscape toward tumor control (228–230). Moreover, using exosomes to deliver STING agonists or TLR ligands can boost the activity of antigen-presenting cells and reinstate type I IFN signaling within the immunosuppressive TME (231). This approach to exosome-based immunomodulation could be paired with immune checkpoint inhibitors (such as anti-PD-1/PD-L1 or anti-CTLA-4) to broaden and deepen the body’s antitumor immune response. Another intriguing therapeutic strategy is to inhibit the release of TDEVs (232). Several pharmacological agents have been discovered that can disrupt the pathways involved in exosome biogenesis and secretion (233). For example, the sphingomyelinase inhibitor GW4869 effectively blocks ceramide-dependent exosome formation, while inhibitors that target Rab27a/b GTPases can interfere with vesicle trafficking and fusion processes (234, 235). Additionally, suppressing components of the ESCRT machinery—like Alix and TSG101—has shown potential in reducing exosomal output (236, 237). However, these strategies face significant challenges, particularly regarding specificity. Many of the molecular targets involved in EV production are widely expressed, which complicates the development of targeted therapies. TDEVs are increasingly being valued as multifunctional platforms for therapeutic delivery with potential applications in gene editing, regenerative strategies, immunotherapy, and cancer vaccination (227, 238). The ability of EVs to modulate immune responses has opened up new avenues for the improvement of treatment outcomes, especially in the context of tumor immune evasion. However, the efficacy of EV-based therapeutics, and TDEVs more specifically, is largely dictated by the conditions under which they are produced. Parameters such as cell origin, culture conditions, and method of isolation can notably alter the composition and role of EVs, which is the key challenge for their standardization and clinical application.
There is still no consensus on a single protocol for isolation of homogeneous and functionally uniform EV populations (239). Traditional methods like ultracentrifugation, polymer-based precipitation, and column filtration can yield large quantities of EVs but lack the specificity to enrich for subtypes carrying specific molecular cargo (240). Newer approaches—like size-exclusion chromatography, immunoaffinity capture, and microfluidics-based isolation—offer greater specificity in targeting disease-relevant EV subsets and surface markers (241). An ideal platform for isolation needs to offer consistency in cargo composition, scalability, reproducibility, and compatibility with downstream clinical use. Such technical challenges have delayed the clinical implementation of EV-based immunotherapies, including for the reversal of tumor-induced immune reprogramming. While efforts have been underway to introduce Good Manufacturing Practice (GMP)-compliant workflows for the production of EVs, issues related to guaranteeing sterility, yield consistency, and therapeutic efficiency remain (242). Maybe most disturbing is the batch-to-batch heterogeneity that undermines the reproducibility that clinical-grade applications demand. These advances are particularly crucial for the use of TDEVs in precision oncology, where immune modulation must be targeted and consistent. Despite recent progress, large-scale manufacturing of EVs, particularly from tumor sources, is a major obstacle due to vesicle heterogeneity and the fact that it is hard to isolate them in therapeutic quantities (243). Consistent, clinically relevant doses of TDEVs require improvement in isolation technology and a greater understanding of EV biology. Moving forward, cross-disciplinary collaboration will be required to overcome these bottlenecks. The intersection of nanotechnology, systems biology, and bioengineering with artificial intelligence-driven analytics may pave the way for more streamlined EV production and purification techniques. Such advances will be critical to translating TDEV-based immune interventions from bench to bedside, enabling them to fulfill their full potential in next-generation cancer immunotherapies.
In the end, while the field has advanced in determining the impact of TDEVs on immune modulation, there is still work to be done in other areas. One promising area of study is examining GPI (glycosylphosphatidylinositol)-anchored proteins found in TDEVs (244). The existence of anchoring proteins and their roles in molecules of TDEV signaling pathways is not well documented, even though they are known to aid in important cellular adhesive, signaling, and immune system processes in cancer. Future investigations need to define the GPI-anchored roles of proteins in TDEVs. Also, investigate the possibility of those proteins assisting in the reprogramming of immune cells as well as determining if such proteins could be novel therapeutic targets. Developing more insight into these mechanisms may establish ways to target TDEV immunosuppressive control networks and increase mechanisms of effective anti-tumor immunity.
8 Conclusion
TDEVs serve as a complex and dynamic communication network between tumors and immune cells, playing a crucial role in shaping the immune environment of the TME. These vesicles are not just passive by-products of tumor activity; they actively influence immune responses, promoting immune suppression and aiding in tumor growth and spread. TDEVs can directly hinder the activation, differentiation, and cytotoxic functions of immune effector cells, or they can indirectly modulate immune responses by boosting the activity of immunosuppressive cells. TDEVs mediate multifaceted intercellular communication through their diverse molecular payload, which includes proteins, miRNAs, lipids, and metabolites. These vesicular components actively regulate critical immune pathways, notably NF-κB, PI3K/Akt/mTOR, JAK/STAT, and TGF-β signaling cascades—to coordinate fundamental biological processes. Through these mechanisms, TDEVs modulate cellular metabolism, direct immune cell differentiation, influence blood vessel formation, and promote tumor immune escape, demonstrating their central role in shaping the TME.
Even with significant progress in understanding the roles of TDEVs, several hurdles still hinder their clinical application. One major challenge is the diversity of EV populations and the absence of universally accepted markers that can reliably differentiate TDEVs from normal EVs. Current research faces two major technical challenges in studying TDEVs. First, the absence of standardized protocols for vesicle isolation, purification, and functional characterization hinders cross-study comparisons and data reproducibility. Second, the intricate signaling networks activated by TDEVs, with their frequent pathway crosstalk and redundant functions—create significant barriers to identifying viable therapeutic targets. These limitations collectively constrain both basic research and clinical translation in the TDEV field. Most of the insights we have come from in vitro or mouse models, and we still need to fully validate their relevance to human cancer. Looking to the future, research should concentrate on deciphering the specific functions of TDEV cargo and understanding how they exert their immunomodulatory effects across various cancer types and stages of disease.
Further investigation into the biology, heterogeneity, and therapeutic manipulation of TDEVs enhances our knowledge of cancer immunology and makes it easier to create novel, TDEV-targeted cancer treatment approaches.
Author contributions
FY: Writing – original draft. YH: Writing – review & editing. YQ: Writing – original draft. YY: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wang S, Sun J, Dastgheyb RM, and Li Z. Tumor-derived extracellular vesicles modulate innate immune responses to affect tumor progression. Front Immunol. (2022) 13:1045624. doi: 10.3389/fimmu.2022.1045624
2. Ma Y, Zhang X, Liu C, and Zhao Y. Extracellular vesicles in cancers: mechanisms, biomarkers, and therapeutic strategies. MedComm (2020). (2024) 5:e70009. doi: 10.1002/mco2.70009
3. Gärtner K, Battke C, Dünzkofer J, Hüls C, von Neubeck B, Kellner MK, et al. Tumor-derived extracellular vesicles activate primary monocytes. Cancer Med. (2018) 7:2013–20. doi: 10.1002/cam4.1465
4. Popēna I, Ābols A, Saulīte L, Pleiko K, Zandberga E, Jēkabsons K, et al. Effect of colorectal cancer-derived extracellular vesicles on the immunophenotype and cytokine secretion profile of monocytes and macrophages. Cell Commun Signal. (2018) 16:17. doi: 10.1186/s12964-018-0229-y
5. Yin Y, Liu B, Cao Y, Yao S, Liu Y, Jin G, et al. Colorectal cancer-derived small extracellular vesicles promote tumor immune evasion by upregulating pd-L1 expression in tumor-associated macrophages. Adv Sci (Weinh). (2022) 9:2102620. doi: 10.1002/advs.202102620
6. Himes BT, Peterson TE, de Mooij T, Garcia LMC, Jung M-Y, Uhm S, et al. The role of extracellular vesicles and pd-L1 in glioblastoma-mediated immunosuppressive monocyte induction. Neuro-Oncology. (2020) 22:967–78. doi: 10.1093/neuonc/noaa029
7. Yao X, Tu Y, Xu Y, Guo Y, Yao F, and Zhang X. Endoplasmic reticulum stress-induced exosomal mir-27a-3p promotes immune escape in breast cancer via regulating pd-L1 expression in macrophages. J Cell Mol Med. (2020) 24:9560–73. doi: 10.1111/jcmm.15367
8. Liu X, Xiang R, Fang X, Wang G, and Zhou Y. Advances in metabolic regulation of macrophage polarization state. Immunological Investigations. (2024) 53:416–36. doi: 10.1080/08820139.2024.2302828
9. de-Brito NM, Duncan-Moretti J, da-Costa HC, Saldanha-Gama R, Paula-Neto HA, Dorighello GG, et al. Aerobic glycolysis is a metabolic requirement to maintain the M2-like polarization of tumor-associated macrophages. Biochim Biophys Acta Mol Cell Res. (2020) 1867:118604. doi: 10.1016/j.bbamcr.2019.118604
10. Zhou S, Lan Y, Li Y, Li Z, Pu J, and Wei L. Hypoxic tumor-derived exosomes induce M2 macrophage polarization via pkm2/ampk to promote lung cancer progression. Cell Transplant. (2022) 31:09636897221106998. doi: 10.1177/09636897221106998
11. Morrissey SM, Zhang F, Ding C, Montoya-Durango DE, Hu X, Yang C, et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab. (2021) 33:2040–58.e10. doi: 10.1016/j.cmet.2021.09.002
12. Fleming V, Hu X, Weller C, Weber R, Groth C, Riester Z, et al. Melanoma extracellular vesicles generate immunosuppressive myeloid cells by upregulating pd-L1 via tlr4 signaling. Cancer Res. (2019) 79:4715–28. doi: 10.1158/0008-5472.Can-19-0053
13. Bretz NP, Ridinger J, Rupp AK, Rimbach K, Keller S, Rupp C, et al. Body fluid exosomes promote secretion of inflammatory cytokines in monocytic cells via toll-like receptor signaling. J Biol Chem. (2013) 288:36691–702. doi: 10.1074/jbc.M113.512806
14. Chow A, Zhou W, Liu L, Fong MY, Champer J, Van Haute D, et al. Macrophage immunomodulation by breast cancer-derived exosomes requires toll-like receptor 2-mediated activation of nf-Kb. Sci Rep. (2014) 4:5750. doi: 10.1038/srep05750
15. Jiang M, Zhang W, Zhang R, Liu P, Ye Y, Yu W, et al. Cancer exosome-derived mir-9 and mir-181a promote the development of early-stage mdscs via interfering with socs3 and pias3 respectively in breast cancer. Oncogene. (2020) 39:4681–94. doi: 10.1038/s41388-020-1322-4
16. Momen-Heravi F and Bala S. Extracellular vesicles in oral squamous carcinoma carry oncogenic mirna profile and reprogram monocytes via nf-Kb pathway. Oncotarget. (2018) 9:34838–54. doi: 10.18632/oncotarget.26208
17. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (Misev2018): A position statement of the international society for extracellular vesicles and update of the misev2014 guidelines. J extracellular vesicles. (2018) 7:1535750. doi: 10.1080/20013078.2018.1535750
18. Zhang C, Qin C, Dewanjee S, Bhattacharya H, Chakraborty P, Jha NK, et al. Tumor-derived small extracellular vesicles in cancer invasion and metastasis: molecular mechanisms, and clinical significance. Mol Cancer. (2024) 23:18. doi: 10.1186/s12943-024-01932-0
19. Spugnini EP, Logozzi M, Di Raimo R, Mizzoni D, and Fais S. A role of tumor-released exosomes in paracrine dissemination and metastasis. Int J Mol Sci. (2018) 19:3968. doi: 10.3390/ijms19123968
20. Tai Y-L, Chu P-Y, Lee B-H, Chen K-C, Yang C-Y, Kuo W-H, et al. Basics and applications of tumor-derived extracellular vesicles. J Biomed Sci. (2019) 26:35. doi: 10.1186/s12929-019-0533-x
21. Guo X, Song J, Liu M, Ou X, and Guo Y. The interplay between the tumor microenvironment and tumor-derived small extracellular vesicles in cancer development and therapeutic response. Cancer Biol Ther. (2024) 25:2356831. doi: 10.1080/15384047.2024.2356831
22. Fang T, Lv H, Lv G, Li T, Wang C, Han Q, et al. Tumor-derived exosomal mir-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat Commun. (2018) 9:191. doi: 10.1038/s41467-017-02583-0
23. Busatto S, Morad G, Guo P, and Moses MA. The role of extracellular vesicles in the physiological and pathological regulation of the blood–brain barrier. FASEB BioAdvances. (2021) 3:665. doi: 10.1096/fba.2021-00045
24. Mirzaei R, Babakhani S, Ajorloo P, Ahmadi RH, Hosseini-Fard SR, Keyvani H, et al. The emerging role of exosomal mirnas as a diagnostic and therapeutic biomarker in mycobacterium tuberculosis infection. Mol Med. (2021) 27:1–31. doi: 10.1186/s10020-021-00296-1
25. Yu J, Sane S, Kim JE, Yun S, Kim HJ, Jo KB, et al. Biogenesis and delivery of extracellular vesicles: harnessing the power of evs for diagnostics and therapeutics. Front Mol Biosci. (2023) 10:1330400. doi: 10.3389/fmolb.2023.1330400
26. Perez-Hernandez D, Gutiérrez-Vázquez C, Jorge I, López-Martín S, Ursa A, Sánchez-Madrid F, et al. The intracellular interactome of tetraspanin-enriched microdomains reveals their function as sorting machineries toward exosomes. J Biol Chem. (2013) 288:11649–61. doi: 10.1074/jbc.M112.445304
27. Nagata S. Apoptosis and clearance of apoptotic cells. Annu Rev Immunol. (2018) 36:489–517. doi: 10.1146/annurev-immunol-042617-053010
28. Zhang X, Liu D, Gao Y, Lin C, An Q, Feng Y, et al. The biology and function of extracellular vesicles in cancer development. Front Cell Dev Biol. (2021) 9:777441. doi: 10.3389/fcell.2021.777441
29. Harding C, Heuser J, and Stahl P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol. (1983) 97:329–39. doi: 10.1083/jcb.97.2.329
30. Taylor DD and Gercel-Taylor C. Retracted: microrna signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Elsevier (2008).
31. Bardi GT, Smith MA, and Hood JL. Melanoma exosomes promote mixed M1 and M2 macrophage polarization. Cytokine. (2018) 105:63–72. doi: 10.1016/j.cyto.2018.02.002
32. Cheng Z, Wang L, Wu C, Huang L, Ruan Y, and Xue W. Tumor-derived exosomes induced M2 macrophage polarization and promoted the metastasis of osteosarcoma cells through tim-3. Arch Med Res. (2021) 52:200–10. doi: 10.1016/j.arcmed.2020.10.018
33. Mittal S, Kadamberi IP, Gupta P, Kumar S, George J, Geethadevi A, et al. Abstract 2321: tumor-derived extracellular vesicles induce macrophages immunosuppressive polarization to promote ovarian cancer progression. Cancer Res. (2023) 83:2321–. doi: 10.1158/1538-7445.Am2023-2321
34. Tang X, Ma C, Wu Q, and Yu M. Ovarian cancer derived extracellular vesicles promote the cancer progression and angiogenesis by mediating M2 macrophages polarization. J Ovarian Res. (2024) 17:172. doi: 10.1186/s13048-024-01497-y
35. Shinohara H, Kuranaga Y, Kumazaki M, Sugito N, Yoshikawa Y, Takai T, et al. Regulated polarization of tumor-associated macrophages by mir-145 via colorectal cancer–derived extracellular vesicles. J Immunol. (2017) 199:1505–15. doi: 10.4049/jimmunol.1700167
36. Ning Y, Shen K, Wu Q, Sun X, Bai Y, Xie Y, et al. Tumor exosomes block dendritic cells maturation to decrease the T cell immune response. Immunol Lett. (2018) 199:36–43. doi: 10.1016/j.imlet.2018.05.002
37. Yu S, Liu C, Su K, Wang J, Liu Y, Zhang L, et al. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J Immunol. (2007) 178:6867–75. doi: 10.4049/jimmunol.178.11.6867
38. Szczepanski MJ, Szajnik M, Welsh A, Whiteside TL, and Boyiadzis M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica. (2011) 96:1302–9. doi: 10.3324/haematol.2010.039743
39. Zhao J, Schlößer HA, Wang Z, Qin J, Li J, Popp F, et al. Tumor-derived extracellular vesicles inhibit natural killer cell function in pancreatic cancer. Cancers. (2019) 11:874. doi: 10.3390/cancers11060874
40. Czystowska-Kuzmicz M, Sosnowska A, Nowis D, Ramji K, Szajnik M, Chlebowska-Tuz J, et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat Commun. (2019) 10:3000. doi: 10.1038/s41467-019-10979-3
41. Maybruck BT, Pfannenstiel LW, Diaz-Montero M, and Gastman BR. Tumor-derived exosomes induce cd8(+) T cell suppressors. J Immunother Cancer. (2017) 5:65. doi: 10.1186/s40425-017-0269-7
42. Muller L, Mitsuhashi M, Simms P, Gooding WE, and Whiteside TL. Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci Rep. (2016) 6:20254. doi: 10.1038/srep20254
43. Ma F, Wang Y, and Peng G. Effects of tumor-derived extracellular vesicles on T cell fate and function. J Immunol. (2022) 208:177. doi: 10.4049/jimmunol.208.Supp.177.18
44. Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer. (2009) 124:2621–33. doi: 10.1002/ijc.24249
45. Pyzer AR, Stroopinsky D, Rajabi H, Washington A, Tagde A, Coll M, et al. Muc1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood. (2017) 129:1791–801. doi: 10.1182/blood-2016-07-730614
46. Shi Y, Zhang J, Mao Z, Jiang H, Liu W, Shi H, et al. Extracellular vesicles from gastric cancer cells induce pd-L1 expression on neutrophils to suppress T-cell immunity. Front Oncol. (2020) 10:629. doi: 10.3389/fonc.2020.00629
47. Mittal S, Gupta P, Chaluvally-Raghavan P, and Pradeep S. Emerging role of extracellular vesicles in immune regulation and cancer progression. Cancers (Basel). 2020):12(12). doi: 10.3390/cancers12123563
48. Ahmadi M, Abbasi R, and Rezaie J. Tumor immune escape: extracellular vesicles roles and therapeutics application. Cell Communication Signaling. (2024) 22:9. doi: 10.1186/s12964-023-01370-3
49. Filipazzi P, Bürdek M, Villa A, Rivoltini L, and Huber V eds. Recent advances on the role of tumor exosomes in immunosuppression and disease progression. In: Seminars in cancer biology. Elsevier.
50. Hou P-P and Chen H-Z. Extracellular vesicles in the tumor immune microenvironment. Cancer Lett. (2021) 516:48–56. doi: 10.1016/j.canlet.2021.05.032
51. Ye L, Zhang Q, Cheng Y, Chen X, Wang G, Shi M, et al. Tumor-derived exosomal hmgb1 fosters hepatocellular carcinoma immune evasion by promoting tim-1+ Regulatory B cell expansion. J immunotherapy Cancer. (2018) 6:1–15. doi: 10.1186/s40425-018-0451-6
52. Yen E-Y, Miaw S-C, Yu J-S, and Lai I-R. Exosomal tgf-B1 is correlated with lymphatic metastasis of gastric cancers. Am J Cancer Res. (2017) 7:2199.
53. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, et al. Membrane-associated hsp72 from tumor-derived exosomes mediates stat3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest. (2010) 120:457–71. doi: 10.1172/jci40483
54. Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, et al. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-B–mediated suppressive activity on T lymphocytes. Cancer Res. (2006) 66:9290–8. doi: 10.1158/0008-5472.CAN-06-1819
55. Hou P-P, Luo L-J, Chen H-Z, Chen Q-T, Bian X-L, Wu S-F, et al. Ectosomal pkm2 promotes hcc by inducing macrophage differentiation and remodeling the tumor microenvironment. Mol Cell. (2020) 78:1192–206.e10. doi: 10.1016/j.molcel.2020.05.004
56. Chen J, Fei X, Wang J, and Cai Z. Tumor-derived extracellular vesicles: regulators of tumor microenvironment and the enlightenment in tumor therapy. Pharmacol Res. (2020) 159:105041. doi: 10.1016/j.phrs.2020.105041
57. Robbins PD and Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. (2014) 14:195–208. doi: 10.1038/nri3622
58. Liu Y, Gu Y, Han Y, Zhang Q, Jiang Z, Zhang X, et al. Tumor exosomal rnas promote lung pre-metastatic niche formation by activating alveolar epithelial tlr3 to recruit neutrophils. Cancer Cell. (2016) 30:243–56. doi: 10.1016/j.ccell.2016.06.021
59. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal pd-L1 contributes to immunosuppression and is associated with anti-pd-1 response. Nature. (2018) 560:382–6. doi: 10.1038/s41586-018-0392-8
60. Poggio M, Hu T, Pai C-C, Chu B, Belair CD, Chang A, et al. Suppression of exosomal pd-L1 induces systemic anti-tumor immunity and memory. Cell. (2019) 177:414–27. doi: 10.1016/j.cell.2019.02.016
61. Feng W, Dean DC, Hornicek FJ, Shi H, and Duan Z. Exosomes promote pre-metastatic niche formation in ovarian cancer. Mol Cancer. (2019) 18:124. doi: 10.1186/s12943-019-1049-4
62. Kong J, Tian H, Zhang F, Zhang Z, Li J, Liu X, et al. Extracellular vesicles of carcinoma-associated fibroblasts creates a pre-metastatic niche in the lung through activating fibroblasts. Mol Cancer. (2019) 18:1–16. doi: 10.1186/s12943-019-1101-4
63. Cavallari C, Camussi G, and Brizzi MF. Extracellular vesicles in the tumour microenvironment: eclectic supervisors. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21186768
64. Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, et al. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-beta-mediated suppressive activity on T lymphocytes. Cancer Res. (2006) 66:9290–8. doi: 10.1158/0008-5472.Can-06-1819
65. Sun J, Jia H, Bao X, Wu Y, Zhu T, Li R, et al. Tumor exosome promotes th17 cell differentiation by transmitting the lncrna crnde-H in colorectal cancer. Cell Death Dis. (2021) 12:123. doi: 10.1038/s41419-020-03376-y
66. Li N, Wang Y, Xu H, Wang H, Gao Y, and Zhang Y. Exosomes derived from rm-1 cells promote the recruitment of mdscs into tumor microenvironment by upregulating cxcr4 via tlr2/nf-Kb pathway. J Oncol. (2021) 2021:5584406. doi: 10.1155/2021/5584406
67. Colangelo T, Panelli P, Mazzarelli F, Tamiro F, Melocchi V, De Santis E, et al. Extracellular vesicle micrornas contribute to notch signaling pathway in T-cell acute lymphoblastic leukemia. Mol Cancer. (2022) 21:226. doi: 10.1186/s12943-022-01698-3
68. Xu L, Wang L, Yang R, Li T, and Zhu X. Lung adenocarcinoma cell-derived exosomes promote M2 macrophage polarization through transmission of mir-3153 to activate the jnk signaling pathway. Hum Mol Genet. (2023) 32:2162–76. doi: 10.1093/hmg/ddad052
69. Gao B, Wang L, Wen T, Xie X, Rui X, and Chen Q. Colon cancer-derived exosomal lncrna-xist promotes M2-like macrophage polarization by regulating pdgfra. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms252111433
70. Xue Z, Liu J, Xing W, Mu F, Wu Y, Zhao J, et al. Hypoxic glioma-derived exosomal mir-25-3p promotes macrophage M2 polarization by activating the pi3k-akt-mtor signaling pathway. J Nanobiotechnology. (2024) 22:628. doi: 10.1186/s12951-024-02888-5
71. Xun J, Du L, Gao R, Shen L, Wang D, Kang L, et al. Cancer-derived exosomal mir-138-5p modulates polarization of tumor-associated macrophages through inhibition of kdm6b. Theranostics. (2021) 11:6847–59. doi: 10.7150/thno.51864
72. Wolf-Dennen K, Gordon N, and Kleinerman ES. Exosomal communication by metastatic osteosarcoma cells modulates alveolar macrophages to an M2 tumor-promoting phenotype and inhibits tumoricidal functions. Oncoimmunology. (2020) 9:1747677. doi: 10.1080/2162402x.2020.1747677
73. Liang H, Geng S, Wang Y, Fang Q, Xin Y, and Li Y. Tumour-derived exosome snhg17 induced by oestrogen contributes to ovarian cancer progression via the ccl13-ccr2-M2 macrophage axis. J Cell Mol Med. (2024) 28:e18315. doi: 10.1111/jcmm.18315
74. Tian HY, Liang Q, Shi Z, and Zhao H. Exosomal cxcl14 contributes to M2 macrophage polarization through nf-Kb signaling in prostate cancer. Oxid Med Cell Longev. (2022) 2022:7616696. doi: 10.1155/2022/7616696
75. Xiao H, Fu J, Liu R, Yan L, Zhou Z, and Yuan J. Gastric cancer cell-derived exosomal mir-541-5p induces M2 macrophage polarization through dusp3/jak2/stat3 pathway. BMC Cancer. (2024) 24:957. doi: 10.1186/s12885-024-12672-1
76. Zhang X, Shi H, Yuan X, Jiang P, Qian H, and Xu W. Tumor-derived exosomes induce N2 polarization of neutrophils to promote gastric cancer cell migration. Mol Cancer. (2018) 17:146. doi: 10.1186/s12943-018-0898-6
77. Park JE, Dutta B, Tse SW, Gupta N, Tan CF, Low JK, et al. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microrna-mediated metabolic shift. Oncogene. (2019) 38:5158–73. doi: 10.1038/s41388-019-0782-x
78. Kersten K, You R, Liang S, Tharp KM, Pollack J, Weaver VM, et al. Uptake of tumor-derived microparticles induces metabolic reprogramming of macrophages in the early metastatic lung. Cell Rep. (2023) 42:112582. doi: 10.1016/j.celrep.2023.112582
79. Javeed N, Gustafson MP, Dutta SK, Lin Y, Bamlet WR, Oberg AL, et al. Immunosuppressive cd14(+)Hla-dr(Lo/neg) monocytes are elevated in pancreatic cancer and “Primed” by tumor-derived exosomes. Oncoimmunology. (2017) 6:e1252013. doi: 10.1080/2162402x.2016.1252013
80. Gao Y, Xu H, Li N, Wang H, Ma L, Chen S, et al. Renal cancer-derived exosomes induce tumor immune tolerance by mdscs-mediated antigen-specific immunosuppression. Cell Commun Signal. (2020) 18:106. doi: 10.1186/s12964-020-00611-z
81. Clayton A, Al-Taei S, Webber J, Mason MD, and Tabi Z. Cancer exosomes express cd39 and cd73, which suppress T cells through adenosine production. J Immunol. (2011) 187:676–83. doi: 10.4049/jimmunol.1003884
82. Salimu J, Webber J, Gurney M, Al-Taei S, Clayton A, and Tabi Z. Dominant immunosuppression of dendritic cell function by prostate-cancer-derived exosomes. J Extracell Vesicles. (2017) 6:1368823. doi: 10.1080/20013078.2017.1368823
83. Xu W, Lu M, Xie S, Zhou D, Zhu M, and Liang C. Endoplasmic reticulum stress promotes prostate cancer cells to release exosome and up-regulate pd-L1 expression via pi3k/akt signaling pathway in macrophages. J Cancer. (2023) 14:1062–74. doi: 10.7150/jca.81933
84. Muller L, Simms P, Hong CS, Nishimura MI, Jackson EK, Watkins SC, et al. Human tumor-derived exosomes (Tex) regulate treg functions via cell surface signaling rather than uptake mechanisms. Oncoimmunology. (2017) 6:e1261243. doi: 10.1080/2162402x.2016.1261243
85. Szajnik M, Czystowska M, Szczepanski MJ, Mandapathil M, and Whiteside TL. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PloS One. (2010) 5:e11469. doi: 10.1371/journal.pone.0011469
86. Berchem G, Noman MZ, Bosseler M, Paggetti J, Baconnais S, Le Cam E, et al. Hypoxic tumor-derived microvesicles negatively regulate nk cell function by a mechanism involving tgf-B and mir23a transfer. Oncoimmunology. (2016) 5:e1062968. doi: 10.1080/2162402x.2015.1062968
87. Yu H, Huang T, Wang D, Chen L, Lan X, Liu X, et al. Acute lymphoblastic leukemia-derived exosome inhibits cytotoxicity of natural killer cells by tgf-B Signaling pathway. 3 Biotech. (2021) 11:313. doi: 10.1007/s13205-021-02817-5
88. Pucci M, Raimondo S, Urzì O, Moschetti M, Di Bella MA, Conigliaro A, et al. Tumor-derived small extracellular vesicles induce pro-inflammatory cytokine expression and pd-L1 regulation in M0 macrophages via il-6/stat3 and tlr4 signaling pathways. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms222212118
89. Tkach M, Thalmensi J, Timperi E, Gueguen P, Névo N, Grisard E, et al. Extracellular vesicles from triple negative breast cancer promote pro-inflammatory macrophages associated with better clinical outcome. Proc Natl Acad Sci U S A. (2022) 119:e2107394119. doi: 10.1073/pnas.2107394119
90. Bottino L, Rodrigues-Junior DM, Farias IS, Branco LM, Iyer NG, de Albuquerque GE, et al. Extracellular vesicles derived from head and neck squamous cells carcinoma inhibit nlrp3 inflammasomes. Curr Res Immunol. (2021) 2:175–83. doi: 10.1016/j.crimmu.2021.10.005
91. He C, Hua W, Liu J, Fan L, Wang H, and Sun G. Exosomes derived from endoplasmic reticulum-stressed liver cancer cells enhance the expression of cytokines in macrophages via the stat3 signaling pathway. Oncol Lett. (2020) 20:589–600. doi: 10.3892/ol.2020.11609
92. Vulpis E, Cecere F, Molfetta R, Soriani A, Fionda C, Peruzzi G, et al. Genotoxic stress modulates the release of exosomes from multiple myeloma cells capable of activating nk cell cytokine production: role of hsp70/tlr2/nf-kb axis. Oncoimmunology. (2017) 6:e1279372. doi: 10.1080/2162402x.2017.1279372
93. Wieckowski EU, Visus C, Szajnik M, Szczepanski MJ, Storkus WJ, and Whiteside TL. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated cd8+ T lymphocytes. J Immunol. (2009) 183:3720–30. doi: 10.4049/jimmunol.0900970
94. Song X, Ding Y, Liu G, Yang X, Zhao R, Zhang Y, et al. Cancer cell-derived exosomes induce mitogen-activated protein kinase-dependent monocyte survival by transport of functional receptor tyrosine kinases. J Biol Chem. (2016) 291:8453–64. doi: 10.1074/jbc.M116.716316
95. Shen T, Huang Z, Shi C, Pu X, Xu X, Wu Z, et al. Pancreatic cancer-derived exosomes induce apoptosis of T lymphocytes through the P38 mapk-mediated endoplasmic reticulum stress. FASEB J. (2020) 34:8442–58. doi: 10.1096/fj.201902186R
96. Taylor DD and Gerçel-Taylor C. Tumour-derived exosomes and their role in cancer-associated T-cell signalling defects. Br J Cancer. (2005) 92:305–11. doi: 10.1038/sj.bjc.6602316
97. Wan-Jiao G, Jian-Xin L, Meng-Nan L, Yun-Da Y, Zhong-Qiu L, Liang L, et al. Macrophage 3d migration: A potential therapeutic target for inflammation and deleterious progression in diseases. Pharmacol Res. (2021) 167:105563. doi: 10.1016/j.phrs.2021.105563
98. Purushothaman A, Bandari SK, Chandrashekar DS, Jones RJ, Lee HC, Weber DM, et al. Chondroitin sulfate proteoglycan serglycin influences protein cargo loading and functions of tumor-derived exosomes. Oncotarget. (2017) 8:73723. doi: 10.18632/oncotarget.20564
99. Feng L, Weng J, Yao C, Wang R, Wang N, Zhang Y, et al. Extracellular vesicles derived from sipa1high breast cancer cells enhance macrophage infiltration and cancer metastasis through myosin-9. Biology. (2022) 11:543. doi: 10.3390/biology11040543
100. Brown M, Johnson LA, Leone DA, Majek P, Vaahtomeri K, Senfter D, et al. Lymphatic exosomes promote dendritic cell migration along guidance cues. J Cell Biol. (2018) 217:2205–21. doi: 10.1083/jcb.201612051
101. Whiteside TL. Immune responses to cancer: are they potential biomarkers of prognosis? Front Oncol. (2013) 3:107.
102. Mrizak D, Martin N, Barjon C, Jimenez-Pailhes A-S, Mustapha R, Niki T, et al. Effect of nasopharyngeal carcinoma-derived exosomes on human regulatory T cells. J Natl Cancer Institute. (2015) 107:dju363.
103. Klotz L and Knolle P. Nuclear receptors: th17 cell control from within. FEBS Lett. (2011) 585:3764–9. doi: 10.1016/j.febslet.2011.06.027
104. Withers DR, Hepworth MR, Wang X, Mackley EC, Halford EE, Dutton EE, et al. Transient inhibition of ror-Γt therapeutically limits intestinal inflammation by reducing th17 cells and preserving group 3 innate lymphoid cells. Nat Med. (2016) 22:319–23. doi: 10.1038/nm.4046
105. Boyiadzis M, Memon S, Carson J, Allen K, Szczepanski MJ, Vance BA, et al. Up-regulation of nk cell activating receptors following allogeneic hematopoietic stem cell transplantation under a lymphodepleting reduced intensity regimen is associated with elevated il-15 levels. Biol Blood Marrow Transplant. (2008) 14:290–300. doi: 10.1016/j.bbmt.2007.12.490
106. Carson WE, Fehniger TA, Haldar S, Eckhert K, Lindemann MJ, Lai C-F, et al. A potential role for interleukin-15 in the regulation of human natural killer cell survival. J Clin Invest. (1997) 99:937–43. doi: 10.1172/JCI119258
107. Benahmed M, Meresse B, Arnulf B, Barbe U, Mention JJ, Verkarre V, et al. Inhibition of tgf-B Signaling by il-15: A new role for il-15 in the loss of immune homeostasis in celiac disease. Gastroenterology. (2007) 132:994–1008. doi: 10.1053/j.gastro.2006.12.025
108. Mitchell MI and Loudig O. Communicator extraordinaire: extracellular vesicles in the tumor microenvironment are essential local and long-distance mediators of cancer metastasis. Biomedicines. (2023) 11. doi: 10.3390/biomedicines11092534
109. Xu Y, Feng K, Zhao H, Di L, Wang L, and Wang R. Tumor-derived extracellular vesicles as messengers of natural products in cancer treatment. Theranostics. (2022) 12:1683–714. doi: 10.7150/thno.67775
110. Baig MS, Roy A, Rajpoot S, Liu D, Savai R, Banerjee S, et al. Tumor-derived exosomes in the regulation of macrophage polarization. Inflammation Res. (2020) 69:435–51. doi: 10.1007/s00011-020-01318-0
111. Xiao M, Zhang J, Chen W, and Chen W. M1-like tumor-associated macrophages activated by exosome-transferred thbs1 promote Malignant migration in oral squamous cell carcinoma. J Exp Clin Cancer Res. (2018) 37:143. doi: 10.1186/s13046-018-0815-2
112. Cai J, Qiao B, Gao N, Lin N, and He W. Oral squamous cell carcinoma-derived exosomes promote M2 subtype macrophage polarization mediated by exosome-enclosed mir-29a-3p. Am J Physiol Cell Physiol. (2019) 316:C731–c40. doi: 10.1152/ajpcell.00366.2018
113. Wang JY, Zhang Q, Wang DD, Yan W, Sha HH, Zhao JH, et al. Mir-29a: A potential therapeutic target and promising biomarker in tumors. Biosci Rep. (2018) 38. doi: 10.1042/bsr20171265
114. Lu L, Xue X, Lan J, Gao Y, Xiong Z, Zhang H, et al. Microrna-29a upregulates mmp2 in oral squamous cell carcinoma to promote cancer invasion and anti-apoptosis. BioMed Pharmacother. (2014) 68:13–9. doi: 10.1016/j.biopha.2013.10.005
115. Chen X, Zhou J, Li X, Wang X, Lin Y, and Wang X. Exosomes derived from hypoxic epithelial ovarian cancer cells deliver micrornas to macrophages and elicit a tumor-promoted phenotype. Cancer Lett. (2018) 435:80–91. doi: 10.1016/j.canlet.2018.08.001
116. Lin F, Yin HB, Li XY, Zhu GM, He WY, and Gou X. Bladder cancer cell−Secreted exosomal mir−21 activates the pi3k/akt pathway in macrophages to promote cancer progression. Int J Oncol. (2020) 56:151–64. doi: 10.3892/ijo.2019.4933
117. Wang Y-C, Hu Y-W, Sha Y-H, Gao J-J, Ma X, Li S-F, et al. Ox-ldl upregulates il-6 expression by enhancing nf-Kb in an igf2-dependent manner in thp-1 macrophages. Inflammation. (2015) 38:2116–23. doi: 10.1007/s10753-015-0194-1
118. Ham S, Lima LG, Chai EPZ, Muller A, Lobb RJ, Krumeich S, et al. Breast cancer-derived exosomes alter macrophage polarization via gp130/stat3 signaling. Front Immunol. (2018) 9:871. doi: 10.3389/fimmu.2018.00871
119. Chen T, Liu Y, Li C, Xu C, Ding C, Chen J, et al. Tumor-derived exosomal circfarsa mediates M2 macrophage polarization via the pten/pi3k/akt pathway to promote non-small cell lung cancer metastasis. Cancer Treat Res Commun. (2021) 28:100412. doi: 10.1016/j.ctarc.2021.100412
120. Deng C, Huo M, Chu H, Zhuang X, Deng G, Li W, et al. Exosome circatp8a1 induces macrophage M2 polarization by regulating the mir-1-3p/stat6 axis to promote gastric cancer progression. Mol Cancer. (2024) 23:49. doi: 10.1186/s12943-024-01966-4
121. Duan Z and Luo Y. Targeting macrophages in cancer immunotherapy. Signal transduction targeted Ther. (2021) 6:127. doi: 10.1038/s41392-021-00506-6
122. Abdelaziz MH, Abdelwahab SF, Wan J, Cai W, Huixuan W, Jianjun C, et al. Alternatively activated macrophages; a double-edged sword in allergic asthma. J Trans Med. (2020) 18:1–12. doi: 10.1186/s12967-020-02251-w
123. Shabgah AG, Al-Qaim ZH, Markov A, Yumashev AV, Ezzatifar F, Ahmadi M, et al. Chemokine cxcl14; a double-edged sword in cancer development. Int Immunopharmacol. (2021) 97:107681. doi: 10.1016/j.intimp.2021.107681
124. Yu X, Li C, Wang Z, Xu Y, Shao S, Shao F, et al. Neutrophils in cancer: dual roles through intercellular interactions. Oncogene. (2024) 43:1163–77. doi: 10.1038/s41388-024-03004-5
125. Leveque-El Mouttie L, Vu T, Lineburg KE, Kuns RD, Bagger FO, Teal BE, et al. Autophagy is required for stem cell mobilization by G-csf. Blood J Am Soc Hematol. (2015) 125:2933–6. doi: 10.1182/blood-2014-03-562660
126. Moloudizargari M, Asghari MH, Jørgensen MM, Reiter RJ, and Kabelitz D. Editorial: extracellular vesicles in cancer immunosurveillance. Front Immunol. (2022) 13:993967. doi: 10.3389/fimmu.2022.993967
127. Reale A, Khong T, and Spencer A. Extracellular vesicles and their roles in the tumor immune microenvironment. J Clin Med. (2022) 11. doi: 10.3390/jcm11236892
128. Söderberg A, Barral AM, Söderström M, Sander B, and Rosén A. Redox-signaling transmitted in trans to neighboring cells by melanoma-derived tnf-containing exosomes. Free Radical Biol Med. (2007) 43:90–9. doi: 10.1016/j.freeradbiomed.2007.03.026
129. Kim EY and Teh HS. Critical role of tnf receptor type-2 (P75) as a costimulator for il-2 induction and T cell survival: A functional link to cd28. J Immunol. (2004) 173:4500–9. doi: 10.4049/jimmunol.173.7.4500
130. Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, et al. Ros-dependent activation of the traf6-ask1-P38 pathway is selectively required for tlr4-mediated innate immunity. Nat Immunol. (2005) 6:587–92. doi: 10.1038/ni1200
131. Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol. (2002) 3:1129–34. doi: 10.1038/ni1202-1129
132. Kono K, Salazar-Onfray F, Petersson M, Hansson J, Masucci G, Wasserman K, et al. Hydrogen peroxide secreted by tumor-derived macrophages down-modulates signal-transducing zeta molecules and inhibits tumor-specific T cell-and natural killer cell-mediated cytotoxicity. Eur J Immunol. (1996) 26:1308–13. doi: 10.1002/eji.1830260620
133. Aggarwal BB. Signalling pathways of the tnf superfamily: A double-edged sword. Nat Rev Immunol. (2003) 3:745–56. doi: 10.1038/nri1184
134. Dubois S, Mariner J, Waldmann TA, and Tagaya Y. Il-15ralpha recycles and presents il-15 in trans to neighboring cells. Immunity. (2002) 17:537–47. doi: 10.1016/s1074-7613(02)00429-6
135. Sander B and Boeryd B. Tumor necrosis factor-alpha expression in human primary Malignant malanoma and it relationship to tumor infiltration by cd3+ Cells. Int J Cancer. (1996) 66:42–7. doi: 10.1002/(sici)1097-0215(19960328)66:1<42::Aid-ijc8>3.0.Co;2-z
136. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2a adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci. (2006) 103:13132–7. doi: 10.1073/pnas.0605251103
137. Hoskin DW, Mader JS, Furlong SJ, Conrad DM, and Blay J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells. Int J Oncol. (2008) 32:527–35. doi: 10.3892/ijo.32.3.527
138. Cuenca-Escalona J, Bödder J, Subtil B, Sánchez-Sánchez M, Vidal-Manrique M, Sweep MWD, et al. Ep2/ep4 targeting prevents tumor-derived pge2-mediated immunosuppression in cdc2s. J Leukoc Biol. (2024) 116:1554–67. doi: 10.1093/jleuko/qiae164
139. Scandella E, Men Y, Gillessen S, Foürster R, and Groettrup M. Prostaglandin E2 is a key factor for ccr7 surface expression and migration of monocyte-derived dendritic cells. Blood J Am Soc Hematol. (2002) 100:1354–61. doi: 10.1182/blood-2001-11-0017
140. Yen J-H, Khayrullina T, and Ganea D. Pge2-induced metalloproteinase-9 is essential for dendritic cell migration. Blood J Am Soc Hematol. (2008) 111:260–70. doi: 10.1182/blood-2007-05-090613
141. Trabanelli S, Lecciso M, Salvestrini V, Cavo M, Očadlíková D, Lemoli RM, et al. Pge2-induced ido1 inhibits the capacity of fully mature dcs to elicit an in vitro antileukemic immune response. J Immunol Res. (2015) 2015:253191. doi: 10.1155/2015/253191
142. Gorelik L and Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-B Signaling in T cells. Nat Med. (2001) 7:1118–22. doi: 10.1038/nm1001-1118
143. Thomas DA and Massagué J. Tgf-B Directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. (2005) 8:369–80. doi: 10.1016/j.ccr.2005.10.012
144. Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, and Parsa AT. Tgf-B Downregulates the activating receptor nkg2d on nk cells and cd8+ T cells in glioma patients. Neuro-oncology. (2010) 12:7–13. doi: 10.1093/neuonc/nop009
145. Asea A, Rehli M, Kabingu E, Boch JA, Baré O, Auron PE, et al. Novel signal transduction pathway utilized by extracellular hsp70: role of toll-like receptor (Tlr) 2 and tlr4. J Biol Chem. (2002) 277:15028–34. doi: 10.1074/jbc.M200497200
146. Multhoff G. Heat shock protein 70 (Hsp70): membrane location, export and immunological relevance. Methods. (2007) 43:229–37. doi: 10.1016/j.ymeth.2007.06.006
147. He G, Zhang H, Zhou J, Wang B, Chen Y, Kong Y, et al. Peritumoural neutrophils negatively regulate adaptive immunity via the pd-L1/pd-1 signalling pathway in hepatocellular carcinoma. J Exp Clin Cancer Res. (2015) 34:1–11. doi: 10.1186/s13046-015-0256-0
148. Weber D, Wheat J, and Currie G. Inflammation and cancer: tumor initiation, progression and metastasis, and chinese botanical medicines. J Integr Med. (2010) 8:1006–13. doi: 10.3736/jcim20101101
149. Greten FR and Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
150. Othman N, Jamal R, and Abu N. Cancer-derived exosomes as effectors of key inflammation-related players. Front Immunol. (2019) 10:2103. doi: 10.3389/fimmu.2019.02103
151. Kim S, Takahashi H, Lin W-W, Descargues P, Grivennikov S, Kim Y, et al. Carcinoma-produced factors activate myeloid cells through tlr2 to stimulate metastasis. nature. (2009) 457:102–6. doi: 10.1038/nature07623
152. Mezzasoma L, Bellezza I, Romani R, and Talesa VN. Extracellular vesicles and the inflammasome: an intricate network sustaining chemoresistance. Front Oncol. (2022) 12:888135. doi: 10.3389/fonc.2022.888135
153. O’Neill LA and Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. (2016) 213:15–23. doi: 10.1084/jem.20151570
154. Galván-Peña S and O’Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol. (2014) 5:420. doi: 10.3389/fimmu.2014.00420
155. Wenes M, Shang M, Di Matteo M, Goveia J, Martín-Pérez R, Serneels J, et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab. (2016) 24:701–15. doi: 10.1016/j.cmet.2016.09.008
156. Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, and Puigserver P. Mtor controls mitochondrial oxidative function through a yy1–pgc-1α Transcriptional complex. nature. (2007) 450:736–40. doi: 10.1038/nature06322
157. Morita M, Gravel S-P, Chenard V, Sikström K, Zheng L, Alain T, et al. Mtorc1 controls mitochondrial activity and biogenesis through 4e-bp-dependent translational regulation. Cell Metab. (2013) 18:698–711. doi: 10.1016/j.cmet.2013.10.001
158. Weichhart T, Hengstschläger M, and Linke M. Regulation of innate immune cell function by mtor. Nat Rev Immunol. (2015) 15:599–614. doi: 10.1038/nri3901
159. Fernández-Delgado I, Calzada-Fraile D, and Sánchez-Madrid F. Immune regulation by dendritic cell extracellular vesicles in cancer immunotherapy and vaccines. Cancers (Basel). (2020) 12. doi: 10.3390/cancers12123558
160. Hoffmann TK, Dworacki G, Tsukihiro T, Meidenbauer N, Gooding W, Johnson JT, et al. Spontaneous apoptosis of circulating T lymphocytes in patients with head and neck cancer and its clinical importance. Clin Cancer Res. (2002) 8:2553–62.
161. Schuler PJ, Schilling B, Harasymczuk M, Hoffmann TK, Johnson J, Lang S, et al. Phenotypic and functional characteristics of cd4+ Cd39+ Foxp3+ and cd4+ Cd39+ Foxp3neg T-cell subsets in cancer patients. Eur J Immunol. (2012) 42:1876–85. doi: 10.1002/eji.201142347
162. Kim JW, Wieckowski E, Taylor DD, Reichert TE, Watkins S, and Whiteside TL. Fas ligand–positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res. (2005) 11:1010–20. doi: 10.1158/1078-0432.1010.11.3
163. Andreola G, Rivoltini L, Castelli C, Huber V, Perego P, Deho P, et al. Induction of lymphocyte apoptosis by tumor cell secretion of fasl-bearing microvesicles. J Exp Med. (2002) 195:1303–16. doi: 10.1084/jem.20011624
164. Czystowska M, Szczepanski MJ, Szajnik M, Quadrini K, Brandwein H, Hadden JW, et al. Mechanisms of T-cell protection from death by irx-2: A new immunotherapeutic. Cancer immunology immunotherapy. (2011) 60:495–506. doi: 10.1007/s00262-010-0951-9
165. Czystowska M, Han J, Szczepanski M, Szajnik M, Quadrini K, Brandwein H, et al. Irx-2, a novel immunotherapeutic, protects human T cells from tumor-induced cell death. Cell Death Differentiation. (2009) 16:708–18. doi: 10.1038/cdd.2008.197
166. Putz U, Howitt J, Doan A, Goh C-P, Low L-H, Silke J, et al. The tumor suppressor pten is exported in exosomes and has phosphatase activity in recipient cells. Sci Signaling. (2012) 5:ra70–ra. doi: 10.1126/scisignal.2003084
167. Taylor DD, Gercel-Taylor C, Lyons KS, Stanson J, and Whiteside TL. T-cell apoptosis and suppression of T-cell receptor/cd3-Z by fas ligand-containing membrane vesicles shed from ovarian tumors. Clin Cancer Res. (2003) 9:5113–9.
168. Van Niel G, Porto-Carreiro I, Simoes S, and Raposo G. Exosomes: A common pathway for a specialized function. J Biochem. (2006) 140:13–21. doi: 10.1093/jb/mvj128
169. Huber V, Fais S, Iero M, Lugini L, Canese P, Squarcina P, et al. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: role in immune escape. Gastroenterology. (2005) 128:1796–804. doi: 10.1053/j.gastro.2005.03.045
170. Ozcan L and Tabas I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu Rev Med. (2012) 63:317–28. doi: 10.1146/annurev-med-043010-144749
171. Zhang J, Gao JX, Salojin K, Shao Q, Grattan M, Meagher C, et al. Regulation of fas ligand expression during activation-induced cell death in T cells by P38 mitogen-activated protein kinase and C-jun nh2-terminal kinase. J Exp Med. (2000) 191:1017–30. doi: 10.1084/jem.191.6.1017
172. Zhang J, Salojin KV, Gao JX, Cameron MJ, Bergerot I, and Delovitch TL. P38 mitogen-activated protein kinase mediates signal integration of tcr/cd28 costimulation in primary murine T cells. J Immunol. (1999) 162:3819–29. doi: 10.4049/jimmunol.162.7.3819
173. Abusamra AJ, Zhong Z, Zheng X, Li M, Ichim TE, Chin JL, et al. Tumor exosomes expressing fas ligand mediate cd8+ T-cell apoptosis. Blood Cells Mol Dis. (2005) 35:169–73. doi: 10.1016/j.bcmd.2005.07.001
174. Houston A and O’Connell J. The fas signalling pathway and its role in the pathogenesis of cancer. Curr Opin Pharmacol. (2004) 4:321–6. doi: 10.1016/j.coph.2004.03.008
175. Abrahams VM, Kamsteeg M, and Mor G. The fas/fas ligand system and cancer. Mol Biotechnol. (2003) 25:19–30. doi: 10.1385/MB:25:1:19
176. Chen J, Sun W, Zhang H, Ma J, Xu P, Yu Y, et al. Macrophages reprogrammed by lung cancer microparticles promote tumor development via release of il-1β. Cell Mol Immunol. (2020) 17:1233–44. doi: 10.1038/s41423-019-0313-2
177. Yang Y, Li C-W, Chan L-C, Wei Y, Hsu J-M, Xia W, et al. Exosomal pd-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. (2018) 28:862–4. doi: 10.1038/s41422-018-0060-4
178. Wu L, Zhang X, Zhang B, Shi H, Yuan X, Sun Y, et al. Exosomes derived from gastric cancer cells activate nf-Kb pathway in macrophages to promote cancer progression. Tumour Biol. (2016) 37:12169–80. doi: 10.1007/s13277-016-5071-5
179. Li M, Xu H, Qi Y, Pan Z, Li B, Gao Z, et al. Tumor-derived exosomes deliver the tumor suppressor mir-3591-3p to induce M2 macrophage polarization and promote glioma progression. Oncogene. (2022) 41:4618–32. doi: 10.1038/s41388-022-02457-w
180. Liang M, Chen X, Wang L, Qin L, Wang H, Sun Z, et al. Cancer-derived exosomal trim59 regulates macrophage nlrp3 inflammasome activation to promote lung cancer progression. J Exp Clin Cancer Res. (2020) 39:176. doi: 10.1186/s13046-020-01688-7
181. Liu Y, Li L, and Song X. Exosomal circpvt1 derived from lung cancer promotes the progression of lung cancer by targeting mir-124-3p/ezh2 axis and regulating macrophage polarization. Cell Cycle. (2022) 21:514–30. doi: 10.1080/15384101.2021.2024997
182. Wang B, Wang X, Li P, Niu X, Liang X, Liu G, et al. Osteosarcoma cell-derived exosomal elfn1-as1 mediates macrophage M2 polarization via sponging mir-138-5p and mir-1291 to promote the tumorgenesis of osteosarcoma. Front Oncol. (2022) 12:881022. doi: 10.3389/fonc.2022.881022
183. Chen WX, Wang DD, Zhu B, Zhu YZ, Zheng L, Feng ZQ, et al. Exosomal mir-222 from adriamycin-resistant mcf-7 breast cancer cells promote macrophages M2 polarization via pten/akt to induce tumor progression. Aging (Albany NY). (2021) 13:10415–30. doi: 10.18632/aging.202802
184. Shou Y, Wang X, Chen C, Liang Y, Yang C, Xiao Q, et al. Exosomal mir-301a-3p from esophageal squamous cell carcinoma cells promotes angiogenesis by inducing M2 polarization of macrophages via the pten/pi3k/akt signaling pathway. Cancer Cell Int. (2022) 22:153. doi: 10.1186/s12935-022-02570-6
185. Rao X, Zhou X, Wang G, Jie X, Xing B, Xu Y, et al. Nlrp6 is required for cancer-derived exosome-modified macrophage M2 polarization and promotes metastasis in small cell lung cancer. Cell Death Dis. (2022) 13:891. doi: 10.1038/s41419-022-05336-0
186. He L, Chen Q, Wu D, Zhu W, Chen Q, and Wu X. Ovarian cancer cell-derived exosomal mir-205 promotes M2 macrophage polarization and ovarian cancer cell metastasis by activating the akt/mtor signalling pathway. (2021). doi: 10.21203/rs.3.rs-373729/v1
187. Chen J, Zhang K, Zhi Y, Wu Y, Chen B, Bai J, et al. Tumor-derived exosomal mir-19b-3p facilitates M2 macrophage polarization and exosomal linc00273 secretion to promote lung adenocarcinoma metastasis via hippo pathway. Clin Transl Med. (2021) 11:e478. doi: 10.1002/ctm2.478
188. Wang X, Luo G, Zhang K, Cao J, Huang C, Jiang T, et al. Hypoxic tumor-derived exosomal mir-301a mediates M2 macrophage polarization via pten/pi3kγ to promote pancreatic cancer metastasis. Cancer Res. (2018) 78:4586–98. doi: 10.1158/0008-5472.Can-17-3841
189. Tian Y, Gao X, Yang X, Chen S, and Ren Y. Glioma-derived exosome lncrna agap2-as1 promotes glioma proliferation and metastasis by mediating tgf-B1 secretion of myeloid-derived suppressor cells. Heliyon. (2024) 10:e29949. doi: 10.1016/j.heliyon.2024.e29949
190. Liu Y, Xiang X, Zhuang X, Zhang S, Liu C, Cheng Z, et al. Contribution of myd88 to the tumor exosome-mediated induction of myeloid derived suppressor cells. Am J Pathol. (2010) 176:2490–9. doi: 10.2353/ajpath.2010.090777
191. Fu L-Q, Du W-L, Cai M-H, Yao J-Y, Zhao Y-Y, and Mou X-Z. The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell Immunol. (2020) 353:104119. doi: 10.1016/j.cellimm.2020.104119
192. Zalpoor H, Aziziyan F, Liaghat M, Bakhtiyari M, Akbari A, Nabi-Afjadi M, et al. The roles of metabolic profiles and intracellular signaling pathways of tumor microenvironment cells in angiogenesis of solid tumors. Cell Communication Signaling. (2022) 20:186. doi: 10.1186/s12964-022-00951-y
193. Lugano R, Ramachandran M, and Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci. (2020) 77:1745–70. doi: 10.1007/s00018-019-03351-7
194. Lorenc P, Sikorska A, Molenda S, Guzniczak N, Dams-Kozlowska H, and Florczak A. Physiological and tumor-associated angiogenesis: key factors and therapy targeting vegf/vegfr pathway. Biomedicine Pharmacotherapy. (2024) 180:117585. doi: 10.1016/j.biopha.2024.117585
195. Krstic J and Santibanez JF. Transforming growth factor-beta and matrix metalloproteinases: functional interactions in tumor stroma-infiltrating myeloid cells. ScientificWorldJournal. (2014) 2014:521754. doi: 10.1155/2014/521754
196. Chanmee T, Ontong P, Konno K, and Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers. (2014) 6:1670–90. doi: 10.3390/cancers6031670
197. Jiang BH and Liu LZ. Pi3k/pten signaling in angiogenesis and tumorigenesis. Adv Cancer Res. (2009) 102:19–65. doi: 10.1016/S0065-230X(09)02002-8
198. Boosani CS, Gunasekar P, and Agrawal DK. An update on pten modulators–a patent review. Expert Opin Ther patents. (2019) 29:881–9. doi: 10.1080/13543776.2019.1669562
199. Zhou B, Mo Z, Lai G, Chen X, Li R, Wu R, et al. Targeting tumor exosomal circular rna cserpine2 suppresses breast cancer progression by modulating malt1-nf-?B-il-6 axis of tumor-associated macrophages. J Exp Clin Cancer Res. (2023) 42:48. doi: 10.1186/s13046-023-02620-5
200. Menghisteab I. Colorectal cancer small extracellular vesicles induce matrix metalloproteinase expression, an invasion-promoting phenotype, and cd147-dependent mapk/ap-1 activation in macrophages. (2020).
201. Zheng D, Kern L, and Elinav E. The nlrp6 inflammasome. Immunology. (2021) 162:281–9. doi: 10.1111/imm.v162.3
202. Hu B, Elinav E, Huber S, Strowig T, Hao L, Hafemann A, et al. Microbiota-induced activation of epithelial il-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc Natl Acad Sci. (2013) 110:9862–7. doi: 10.1073/pnas.1307575110
203. Whiteside TL. Biology of extracellular vesicles and the potential of tumor-derived vesicles for subverting immunotherapy of cancer. J Immunother Cancer. (2025) 13. doi: 10.1136/jitc-2024-010376
204. Pordanjani PM, Bolhassani A, Milani A, and Pouriayevali MH. Extracellular vesicles in vaccine development and therapeutic approaches for viral diseases. Process Biochem. (2023) 128:167–80. doi: 10.1016/j.procbio.2023.02.028
205. Ma F, Vayalil J, Lee G, Wang Y, and Peng G. Emerging role of tumor-derived extracellular vesicles in T cell suppression and dysfunction in the tumor microenvironment. J immunotherapy Cancer. (2021) 9:e003217. doi: 10.1136/jitc-2021-003217
206. Droste M, Thakur BK, and Eliceiri BP. Tumor-derived extracellular vesicles and the immune system-lessons from immune-competent mouse-tumor models. Front Immunol. (2020) 11:606859. doi: 10.3389/fimmu.2020.606859
207. Kamerkar S, Leng C, Burenkova O, Jang SC, McCoy C, Zhang K, et al. Exosome-mediated genetic reprogramming of tumor-associated macrophages by exoaso-stat6 leads to potent monotherapy antitumor activity. Sci Adv. (2022) 8:eabj7002. doi: 10.1126/sciadv.abj7002
208. Gordon S and Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. (2010) 32:593–604. doi: 10.1016/j.immuni.2010.05.007
209. Elo LL, Järvenpää H, Tuomela S, Raghav S, Ahlfors H, Laurila K, et al. Genome-wide profiling of interleukin-4 and stat6 transcription factor regulation of human th2 cell programming. Immunity. (2010) 32:852–62. doi: 10.1016/j.immuni.2010.06.011
210. Goenka S and Kaplan MH. Transcriptional regulation by stat6. Immunologic Res. (2011) 50:87–96. doi: 10.1007/s12026-011-8205-2
211. Proia TA, Singh M, Woessner R, Carnevalli L, Bommakanti G, Magiera L, et al. Stat3 antisense oligonucleotide remodels the suppressive tumor microenvironment to enhance immune activation in combination with anti–pd-L1. Clin Cancer Res. (2020) 26:6335–49. doi: 10.1158/1078-0432.CCR-20-1066
212. Li T, Morgan MJ, Choksi S, Zhang Y, Kim Y-S, and Liu Z-G. Micrornas modulate the noncanonical nf-Kb pathway by regulating ikkα Expression during macrophage differentiation. Nat Immunol. (2010) 11:799. doi: 10.1038/ni.1918
213. Jang JY, Lee JK, Jeon YK, and Kim CW. Exosome derived from epigallocatechin gallate treated breast cancer cells suppresses tumor growth by inhibiting tumor-associated macrophage infiltration and M2 polarization. BMC Cancer. (2013) 13:421. doi: 10.1186/1471-2407-13-421
214. Cornice J, Verzella D, Arboretto P, Vecchiotti D, Capece D, Zazzeroni F, et al. Nf-Kb: governing macrophages in cancer. Genes (Basel). (2024) 15. doi: 10.3390/genes15020197
215. Wieckowski E and Whiteside TL. Human tumor-derived vs dendritic cell-derived exosomes have distinct biologic roles and molecular profiles. Immunologic Res. (2006) 36:247–54. doi: 10.1385/IR:36:1:247
216. Abrahams VM, Straszewski SL, Kamsteeg M, Hanczaruk B, Schwartz PE, Rutherford TJ, et al. Epithelial ovarian cancer cells secrete functional fas ligand. Cancer Res. (2003) 63:5573–81.
217. Contini P, Ghio M, Poggi A, Filaci G, Indiveri F, Ferrone S, et al. Soluble Hla-a,-B,-C and-G Molecules Induce Apoptosis in T and Nk Cd8+ Cells and Inhibit Cytotoxic T Cell Activity through Cd8 Ligation. Eur J Immunol. (2003) 33:125–34. doi: 10.1002/immu.200390015
218. Contini P, Ghio M, Merlo A, Poggi A, Indiveri F, and Puppo F. Apoptosis of antigen-specific T lymphocytes upon the engagement of cd8 by soluble hla class I molecules is fas ligand/fas mediated: evidence for the involvement of P56lck, calcium calmodulin kinase ii, and calcium-independent protein kinase C signaling pathways and for nf-Kb and nf-at nuclear translocation. J Immunol. (2005) 175:7244–54. doi: 10.4049/jimmunol.175.11.7244
219. Kitai Y, Kawasaki T, Sueyoshi T, Kobiyama K, Ishii KJ, Zou J, et al. DNA-containing exosomes derived from cancer cells treated with topotecan activate a sting-dependent pathway and reinforce antitumor immunity. J Immunol. (2017) 198:1649–59. doi: 10.4049/jimmunol.1601694
220. Rialdi A, Campisi L, Zhao N, Lagda AC, Pietzsch C, Ho JSY, et al. Topoisomerase 1 inhibition suppresses inflammatory genes and protects from death by inflammation. Science. (2016) 352:aad7993. doi: 10.1126/science.aad7993
221. He Y, Hong Q, Chen S, Zhou J, and Qiu S. Reprogramming tumor-associated macrophages in gastric cancer: A pathway to enhanced immunotherapy. Front Immunol. (2025) 16:1558091. doi: 10.3389/fimmu.2025.1558091
222. Jiang J, Mei J, Ma Y, Jiang S, Zhang J, Yi S, et al. Tumor hijacks macrophages and microbiota through extracellular vesicles. Exploration. (2022) 2:20210144. doi: 10.1002/EXP.20210144
223. Gurung S, Perocheau D, Touramanidou L, and Baruteau J. The exosome journey: from biogenesis to uptake and intracellular signalling. Cell Communication Signaling. (2021) 19:47. doi: 10.1186/s12964-021-00730-1
224. Rezaie J, Akbari A, and Rahbarghazi R. Inhibition of extracellular vesicle biogenesis in tumor cells: A possible way to reduce tumorigenesis. Cell Biochem Funct. (2022) 40:248–62. doi: 10.1002/cbf.3695
225. Raskov H, Orhan A, Christensen JP, and Gögenur I. Cytotoxic cd8+ T cells in cancer and cancer immunotherapy. Br J Cancer. (2021) 124:359–67. doi: 10.1038/s41416-020-01048-4
226. Guo R and Wang P. Tumor-derived extracellular vesicles: hijacking T cell function through exhaustion. Pathol - Res Pract. (2025) 269:155948. doi: 10.1016/j.prp.2025.155948
227. Kumar MA, Baba SK, Sadida HQ, Marzooqi SA, Jerobin J, Altemani FH, et al. Extracellular vesicles as tools and targets in therapy for diseases. Signal Transduction Targeted Ther. (2024) 9:27. doi: 10.1038/s41392-024-01735-1
228. Aheget H, Mazini L, Martin F, Belqat B, Marchal JA, and Benabdellah K. Exosomes: their role in pathogenesis, diagnosis and treatment of diseases. Cancers (Basel). (2020) 13. doi: 10.3390/cancers13010084
229. Marima R, Basera A, Miya T, Damane BP, Kandhavelu J, Mirza S, et al. Exosomal long non-coding rnas in cancer: interplay, modulation, and therapeutic avenues. Non-coding RNA Res. (2024) 9:887–900. doi: 10.1016/j.ncrna.2024.03.014
230. Zhao M, Cheng X, Shao P, Dong Y, Wu Y, Xiao L, et al. Bacterial protoplast-derived nanovesicles carrying crispr-cas9 tools re-educate tumor-associated macrophages for enhanced cancer immunotherapy. Nat Commun. (2024) 15:950. doi: 10.1038/s41467-024-44941-9
231. McAndrews KM, Che SPY, LeBleu VS, and Kalluri R. Effective delivery of sting agonist using exosomes suppresses tumor growth and enhances antitumor immunity. J Biol Chem. (2021) 296:100523. doi: 10.1016/j.jbc.2021.100523
232. Catalano M and O’Driscoll L. Inhibiting extracellular vesicles formation and release: A review of ev inhibitors. J Extracell Vesicles. (2020) 9:1703244. doi: 10.1080/20013078.2019.1703244
233. Cesselli D, Parisse P, Aleksova A, Veneziano C, Cervellin C, Zanello A, et al. Extracellular vesicles: how drug and pathology interfere with their biogenesis and function. Front Physiol. (2018) 9:1394. doi: 10.3389/fphys.2018.01394
234. Irep N, Inci K, Tokgun PE, and Tokgun O. Exosome inhibition improves response to first-line therapy in small cell lung cancer. J Cell Mol Med. (2024) 28:e18138. doi: 10.1111/jcmm.18138
235. Kim JH, Lee C-H, and Baek M-C. Dissecting Exosome Inhibitors: Therapeutic Insights into Small-Molecule Chemicals against Cancer. Exp Mol Med. (2022) 54:1833–43. doi: 10.1038/s12276-022-00898-7
236. Ortega FG, Roefs MT, de Miguel Perez D, Kooijmans SA, de Jong OG, Sluijter JP, et al. Interfering with endolysosomal trafficking enhances release of bioactive exosomes. Nanomedicine: Nanotechnology Biol Med. (2019) 20:102014. doi: 10.1016/j.nano.2019.102014
237. Villarroya-Beltri C, Baixauli F, Gutiérrez-Vázquez C, Sánchez-Madrid F, and Mittelbrunn M. Sorting it out: regulation of exosome loading. Semin Cancer Biol. (2014) 28:3–13. doi: 10.1016/j.semcancer.2014.04.009
238. Yang M, Olaoba OT, Zhang C, Kimchi ET, Staveley-O’Carroll KF, and Li G. Cancer immunotherapy and delivery system: an update. Pharmaceutics. (2022) 14. doi: 10.3390/pharmaceutics14081630
239. Konoshenko MY, Lekchnov EA, Vlassov AV, and Laktionov PP. Isolation of extracellular vesicles: general methodologies and latest trends. BioMed Res Int. (2018) 2018:8545347. doi: 10.1155/2018/8545347
240. Zhao Z, Wijerathne H, Godwin AK, and Soper SA. Isolation and analysis methods of extracellular vesicles (Evs). Extracell Vesicles Circ Nucl Acids. (2021) 2:80–103. doi: 10.20517/evcna.2021.07
241. Sidhom K, Obi PO, and Saleem A. A review of exosomal isolation methods: is size exclusion chromatography the best option? Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21186466
242. Shimizu Y, Ntege EH, Inoue Y, Matsuura N, Sunami H, and Sowa Y. Optimizing mesenchymal stem cell extracellular vesicles for chronic wound healing: bioengineering, standardization, and safety. Regener Ther. (2024) 26:260–74. doi: 10.1016/j.reth.2024.06.001
243. Kimiz-Gebologlu I and Oncel SS. Exosomes: large-scale production, isolation, drug loading efficiency, and biodistribution and uptake. J Controlled Release. (2022) 347:533–43. doi: 10.1016/j.jconrel.2022.05.027
Keywords: TdEVs, immune modulation, signaling pathways, immunotherapy, tumor
Citation: Yin F, He Y, Qiao Y and Yan Y (2025) Tumor-derived vesicles in immune modulation: focus on signaling pathways. Front. Immunol. 16:1581964. doi: 10.3389/fimmu.2025.1581964
Received: 23 February 2025; Accepted: 28 April 2025;
Published: 15 May 2025.
Edited by:
Dr Anirban Ganguly, All India Institute of Medical Sciences Deoghar, IndiaReviewed by:
Prof Dwijendra k. Gupta, Allahabad University, IndiaYounghye Song, University of Arkansas, United States
Yun Ma, Chinese Academy of Sciences (CAS), China
Copyright © 2025 Yin, He, Qiao and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yangfang He, eWZoZUBqbHUuZWR1LmNu