 Hua Lu
Hua Lu Zhaojun Teng1
Zhaojun Teng1 Wenchao Zhang
Wenchao Zhang- 1Department of Urology, Qilu Hospital of Shandong University Dezhou Hospital, Dezhou, China
- 2Department of Oncology, Qilu Hospital of Shandong University Dezhou Hospital, Dezhou, China
Prostate cancer (PC) is one of the most common cancers that is diagnosed in about 10-15% of men in old age. It seems that the current treatments are not effective, and this leads to prostate cancer becoming the second-deadliest cancer. Treatments such as chemotherapy, radiotherapy, androgen deprivation therapy (ADT), and surgery are among these treatments. However, the possibility of disease recurrence after these treatments is high. Therefore, other methods have become necessary, and PC treatment is changing. One of the methods that has received much attention today is immunotherapy. Immunotherapy includes all interventions that help to treat cancer or any other disease by affecting the immune system’s responses. For this purpose, cytokines, cell therapy, and antibody-based methods can be used. Antibody-based treatments include immune checkpoint inhibitors (ICIs), and due to the high expression of immune checkpoint (ICP) molecules on the surface of prostate cancer cells and cancer stromal cells, these treatments have yielded promising results. Also, combining them with chemotherapy, surgery, and radiotherapy can help increase their efficiency. This review first updates standard treatments’ therapeutic efficacy and risk factors. Then, we will talk about different types of immunotherapies, emphasizing ICIs.
1 Introduction
Metastatic prostate cancer (PC) is a leading cause of morbidity and mortality (1). Androgen deprivation therapy (ADT) remains the cornerstone of treatment for non-metastatic prostate cancer (2). However, most of these patients will progress to CRPC, which is very difficult to treat (3). There is, however, an intermediate stage where, following ADT, patients have cancer progression without detectable metastasis, termed non-metastatic castration-resistant prostate cancer (nmCRPC) (4). Awareness of nmCRPC is increasing because of ADT’s increased utilization and failure. Men with nmCRPC have a high risk of progressing to metastatic castration-resistant prostate cancer (mCRPC) and thus have limited treatment options (5). However, some treatments have recently been found beneficial, including three nonsteroidal antiandrogen agents under phase III trials (6). These agents are now FDA-approved, offering effective options alongside ADT for nmCRPC patients. The treatment of nmCRPC has improved significantly in the last ten years, with three new nonsteroidal antiandrogen agents added to ADT (7, 8). Trials like ARAMIS, PROSPER, and SPARTAN showed better metastasis-free survival (MFS) for high-risk patients (9). Continuous ADT can lead to problems for patients, such as sexual dysfunction, low mood, acute renal injury, cardiovascular disease, and increased health costs (10, 11). Intermittent ADT allows recovery of testosterone levels and may reduce these issues (12, 13). Studies show no significant differences in cancer outcomes between intermittent and continuous ADT, but intermittent therapy usually has better sexual outcomes, less morbidity, and lower costs (14). Despite this, the best way to administer ADT is still unclear, and careful patient selection is essential for achieving benefits. Other types of PC treatments have also been used, such as chemotherapy, radiotherapy, and vaccines (Table 1). Various vaccines have been designed to treat PC; however, only dendritic cell (DCs) vaccines have significantly progressed, and only one has received FDA approval (Table 2).
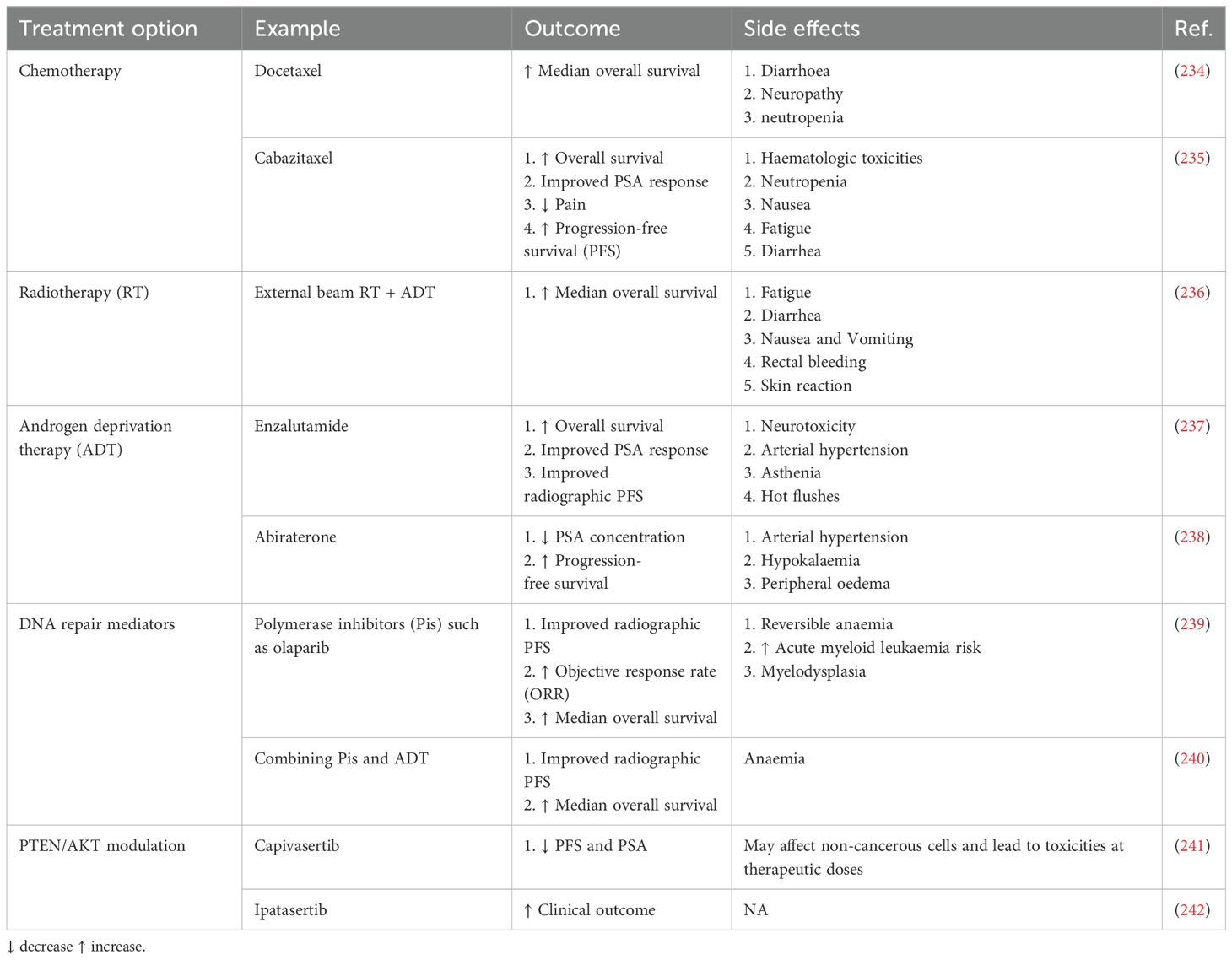
Table 1. Treatment options for prostate cancer treatment.
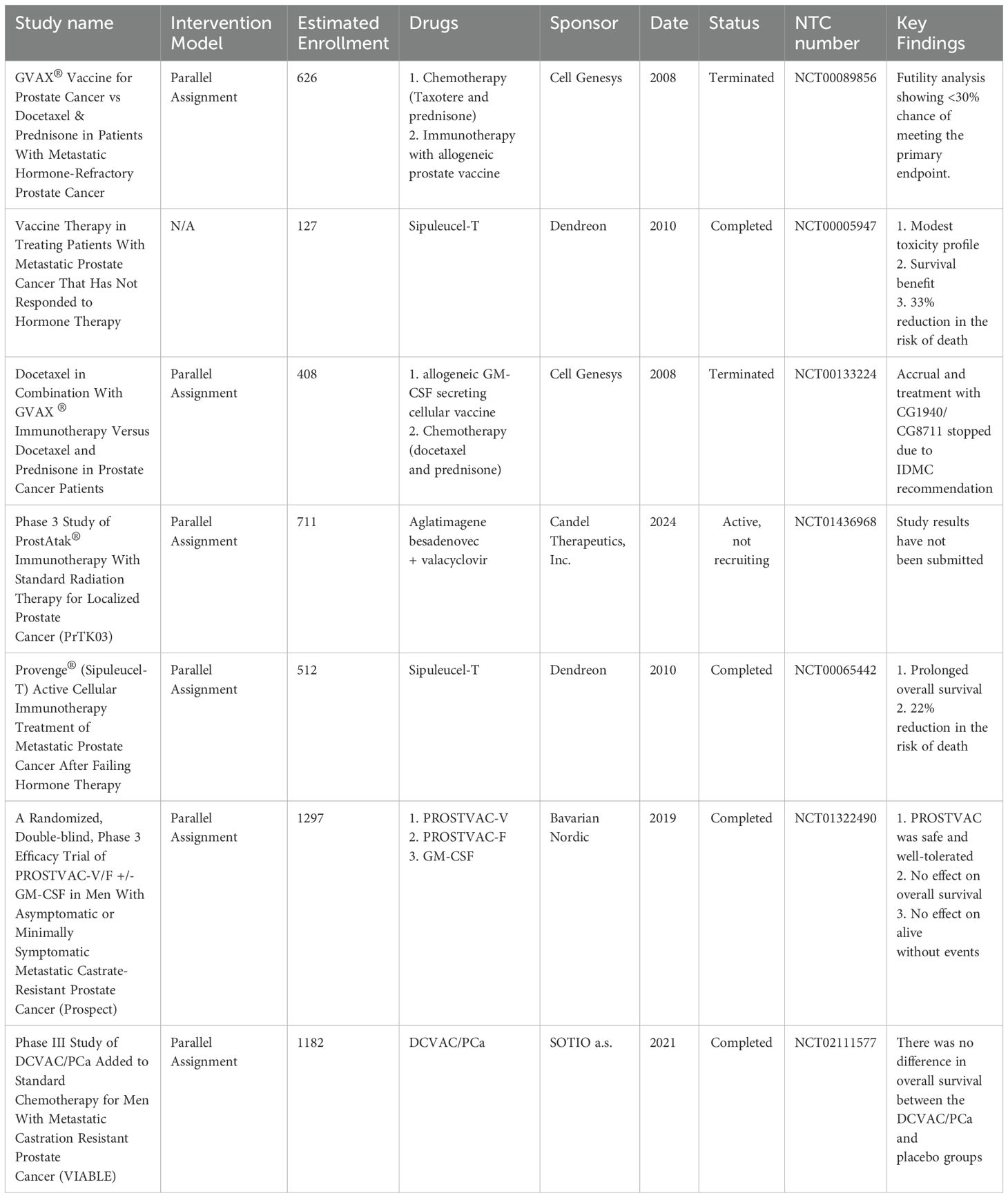
Table 2. Example of vaccine application for prostate cancer in phase 3 clinical trials.
Over the past decade, the standard of treatment has been immunotherapy, whereby various forms of immune response are employed to destroy the malignant cells (15). It has had favorable results in those suffering from aggressive forms of PC (like mCRPC), with some patients achieving permanent remission (16). Other therapies, including sipuleucel-T (17) and immune checkpoint inhibitors (ICIs) (18), have also emerged as alternatives to conventional ADT and chemotherapy for the management of CRPC.
Adoptive Cell Therapy (ACT) is also moderately successful in treating different cancers (19). It utilizes specially modified T-lymphocytes to target specific tumors effectively. Modifying patient T-lymphocytes with particular antigen receptors can generate an anticancer immune reaction against PC antigens (20). Chimeric antigen receptors (CAR) support the preparation of artificial T-cell receptors for ACT in PC patients (21).
However, immunotherapy tends to be less effective against prostate cancer than against other malignancies, such as non-small-cell lung cancer and melanoma, due to the suppressive nature of the tumor environment and lower T-cell content (22–24). Nevertheless, certain subsets of prostate cancer patients with specific characteristics have indeed responded well to ICIs (16). However, in many studies, ICIs have been used in combination with other therapies, such as chemotherapy, radiotherapy, and vaccines (25, 26). In this review, we first discuss the types of immunotherapies for prostate cancer and then explain them by classifying immunotherapy into two categories: cell therapy and antibody therapy. Then, we will discuss the limitations, challenges, and suggestions, and conclude.
2 Immunotherapy strategies
Immune surveillance failure contributes to tumor development (27) (Figure 1). Tumor cells can escape T-cell responses by several mechanisms. Active immunotherapy tries to augment the immune response against cancer (28). Prostate cancer is unique in that it is one of the tumors recognized by the immune system, as evidenced by tumor-infiltrating lymphocytes, and therefore is a candidate for immunotherapy (29). PC has defined antigens that enable targeting without inducing widespread autoimmune responses. There are several approaches to immunotherapy, and only Sipuleucel-T is an FDA-approved modality (17). Immunotherapy includes all interventions that increase the immune system’s potential to respond to cancer cells (30, 31). These types of interventions are categorized based on the type of intervention and the part of the affected immune system. In general, these types of treatments are divided into two categories: cell therapy and antibody therapy.
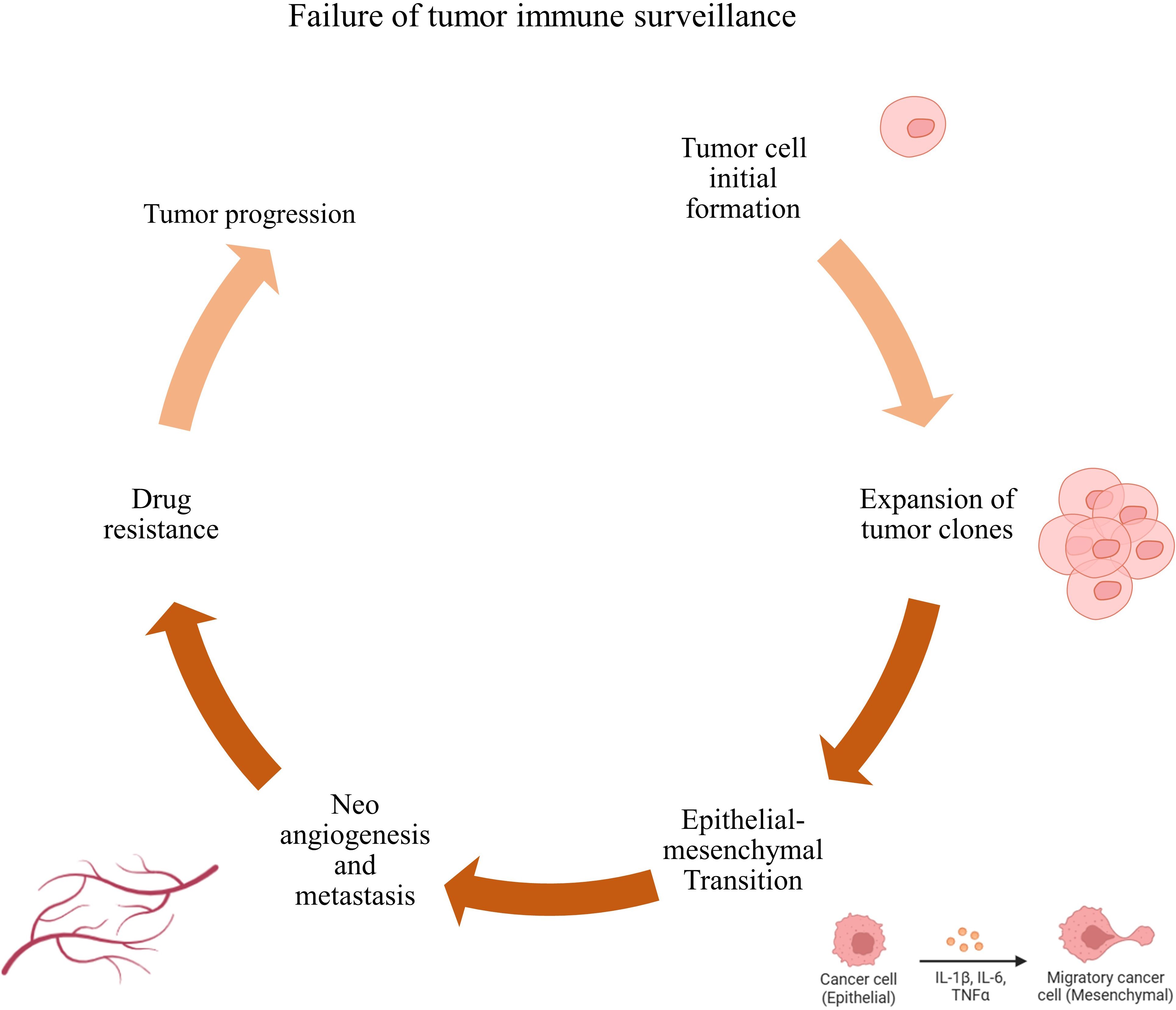
Figure 1. Failure of immune surveillance and tumor progression. Failure to kill tumor cells by immune cells leads to their rapid proliferation, increased invasion, metastasis, and resistance to tumor cell treatment, which in turn leads to tumor progression.
2.1 Cell therapy
In cell therapy for PC, different therapeutic strategies and cells are used. Among the most critical cells used in treating PC are DCs (32), tumor-infiltrating T cells (TILs) (33), and chimeric antigen receptor-expressing T cells (CAR-T) (34). Although the therapeutic uses of CAR-T cells seem to be confirmed for the treatment of blood cancers, their therapeutic efficacy for solid tumors is still under investigation.
2.1.1 Dendritic cell -based treatments
Vaccines have played a crucial role in eradicating various infectious diseases and saving millions of lives. These vaccines work through the induction of specific immune responses against attenuated or killed germs to protect against actual infections (35). So far, developing effective anti-tumor vaccines has been challenging and requires further improvement (36).
The DCs are one of the potent immune cells contributing to cancer immunotherapy by inducing specific immune responses (37). Various approaches have been tried to use DCs as therapeutic vaccines, employing either whole tumor cells or specific protein fragments.
Currently, sipuleucel-T (sip-T) represents the only FDA-approved therapy for men with asymptomatic or minimally symptomatic metastatic castrate-resistant PC (38). Its toxicity profile is generally favorable, making it more attractive than docetaxel. In April 2010, the FDA approved sip-T for the treatment of PC. However, patient access to this type of treatment is limited, and side effects such as a fast heartbeat, fever, rash, and joint pain may occur following treatment (39).
Prostate acid phosphatase (PAP) is also used to design a therapeutic vaccine that elicits an immune response against prostate cancer cells and targets (40). Another DC-based vaccine was developed to generate T cells that recognize and kill cancer cells that express PAP, mainly from malignant prostate tissue post-surgery (41). A study used a recombinant fusion protein vaccine expressing PAP with GM-CSF (sip-T) to activate autologous peripheral blood mononuclear cells (PBMC). After processing, the cells containing activated APCs (especially DCs) are infused back into the patient (42). Given the success achieved in treating PC with this type of vaccine, many researchers have sought to increase the potential of this treatment. The results of studies have shown that this type of treatment can lead to increased migration of tumor antigen-specific T cells to tumor tissue (42). In another study by RK Pachynski et al., patients were divided into two groups after receiving sip-T. One group received recombinant human IL-7 (4 injections per week for 6 weeks); the other group received a placebo (43). The results of this study proved that IL-7 injection can increase the therapeutic efficacy of sip-T. In patients receiving IL-7, the number of CD8+, TCD4+ T cells, and NK cells was significantly increased compared to the control group (43). In this study, due to limited access to recombinant human IL-7, the number of patients decreased during the trial (from 80 to 54), and no association between immune findings and clinical outcomes was reported.
Another DC-based vaccine for PC is stapuldencel or DCVAC/PCa, and is obtained from autologous DCs derived from PBMC of patients exposed to a lysate of human prostate adenocarcinoma cell line (LNCaP) (44). Various studies show that this type of treatment is highly safe and well-tolerated. They can also lead to an increase in the population of prostate-specific Antigen (PSA)-specific T cells in patients (45). Different combinations of this cell therapy have also been used during various trials. For example, in a study by Vogelzang et al., Stapuldencel was used with docetaxel to treat metastatic PC refractory to treatment. This study’s results show that this combination can increase overall survival (OS) in patients compared to a single treatment (46). Also, in another Phase I/II study, it has been shown that the use of autologous DC vaccine as an adjuvant after prostatectomy can lead to a decrease in relapse in patients (47).
Although previous DC vaccine-based therapies have succeeded, given their high potential in PC treatment, studies on their therapeutic use are ongoing, and different antigens are used to prime them (48). A study used MAGE-A2 long peptide (LP) to induce maturation in DCs. The results of this study show that the use of DCs + MAGE-A2 LP leads to activated DCs and increases their ability to activate CD8+ T cells. Also, T cells’ cytotoxic ability and IFN-γ production are increased (49). NY-ESO-1 as an antigen has also been used to produce DC vaccines against prostate cancer (50). For this purpose, after isolating monocytes from the bone marrow of mice and inducing their differentiation into DC, a cytokine cocktail and a fusion protein containing NY-ESO-1, secretin-penetration, and ubiquitin (SNU) were used (51). The results show that this DC taxon effectively stimulates mouse PBMCs to produce inflammatory cytokines and increases their cytotoxic ability in co-culture with tumor cells. Overall, the observed cases increase the ability of T cells to mount a specific anti-tumor response and are a suitable option for transfer to clinical studies (51).
Therefore, identifying specific and dominant antigens related to PC and using them to prime T cells can help in its treatment. It is also suggested that DCs be primed with a specific antigen cocktail for PC and used for treatment. This proposed therapy can simultaneously stimulate different clones of PC-specific T cells, leading to tumor regression. An essential point about DC-based vaccines for prostate cancer is that this treatment is often used after surgery to prevent recurrence.
2.1.2 Tumor-infiltrated lymphocytes
Human tumors express antigens recognizable by T and B lymphocytes, and such recognition forms the basis for new immunotherapeutic approaches directed at tumor-associated antigens (TAA) (52). However, attempts to develop successful cancer vaccines have met with limited success. Although vaccination can expand tumor-reactive T lymphocytes, clinical responses have been seen in only a few patients. This is unsurprising because relatively few lymphocytes may reach the cancer, and tumors have found ways to evade the immunological attack (53, 54). Tumor cells can avoid or escape immune detection by losing antigens, producing immunosuppressive molecules, and recruiting suppressive immune cells (55, 56). Mixed responses have been frequently observed in patients receiving immune therapy, where some tumors regress while others progress. This is poorly understood, but differences in the tumor-immune cell interaction could be one factor (57). The tumor microenvironment (TME) generally does not support T lymphocyte activities, as various studies have documented that tumor-residing T lymphocytes often have impaired functionalities (58, 59) (Figure 2).
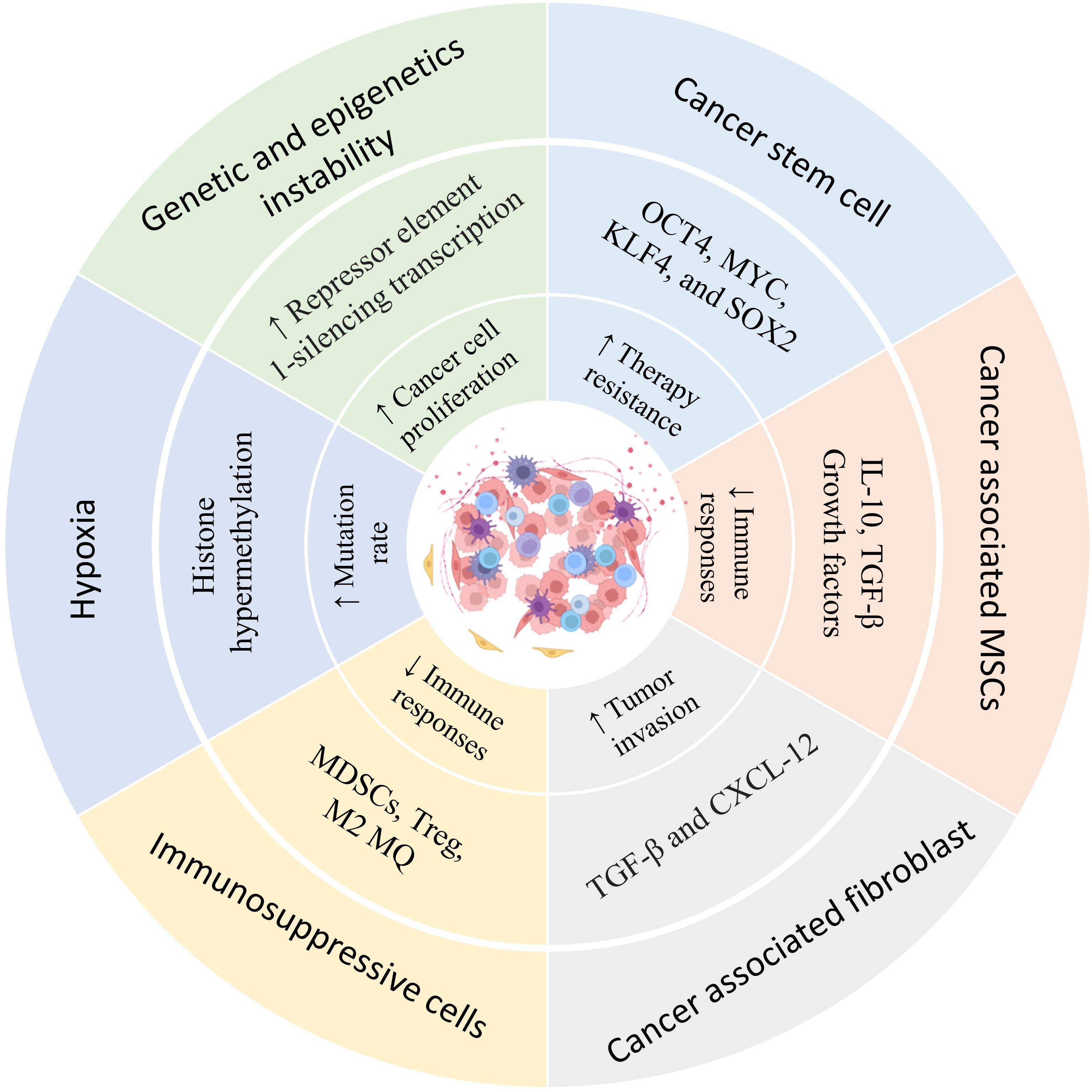
Figure 2. Heterogeneity of the prostate cancer microenvironment. The presence of different types of immune regulatory cells in the tumor tissue environment, hypoxia, mutations, and epigenetic changes resulting from food starvation, lead to changes in the characteristics of cells in the tumor microenvironment that contribute to tumor growth. Understanding this heterogeneity can help in tumor treatment and the selection of appropriate therapy.
Another essential immune regulatory aspect in the context of tumors is amino acid metabolism (60). For instance, it has been documented that tumors induce an enzyme that degrades tryptophan, which has been associated with immune evasion mechanisms (61). Inhibiting this enzyme in certain studies has helped reverse some tumor-induced immune dysfunctions (62). L-arginine metabolism is also altered in tumors. In immune cells, L-arginine is metabolized by several enzymes that affect immune responses (63). High activity of these enzymes has been associated with a range of cancers and appears to support tumor growth, yet they can also suppress T cell responses. It is, therefore, tempting to speculate that the tumor-associated metabolism of L-arginine inhibits T cell function, and indeed, inhibitors directed against these pathways seem to reconstitute T cell functions in PC (64).
Given the immunosuppressive environment in the TME, it appears that extracting tumor-specific lymphocytes from patients and reactivating them may help treat PC in two ways. First, cells expanded outside the body and no longer received inhibitory signals induced by tumor cells, which led to increased activation (65). Also, increasing the number of these cells outside the body and re-transfusing them into patients increases the number of activated tumor-specific lymphocytes and helps in tumor regression.
However, in the case of prostate cancer, various studies have shown that isolating and activating TILs from patients is challenging. However, a study by Sharon Yunger et al. has shown that TILs can be isolated from prostate tumor tissue. These cells also expanded in vitro after activation, and their ability to produce IFN-γ and tumor killing (in co-culture with tumor cells) was also increased (66).
2.1.3 Chimeric antigen receptor-expressing T cell
One of the techniques for empowering a patient’s T cells is to add a CAR-based receptor on their surface for specific recognition and destruction of cancer cells (67). These CAR T cells are often referred to as “living drugs” because they can grow and become long-lived memory cells (34). Unlike the regular T cells, which depend on the human leukocyte antigen (HLA) complex, CAR T cells recognize targets in an HLA-independent manner (68). When they bind to an antigen, they activate and release different substances, like cytokines, that destroy the target cells. The parts that make up CARs are numerous, including an antigen-binding domain and a linker region, which place the binding domain for better positioning in recognizing tumor cells (69). The type of linker and the length could influence the effectiveness of the CAR T cell.
Another part of a CAR is the transmembrane domain that can made of CD3, CD4, CD8, or CD28 transmembrane domains and anchor CARs in the cell membrane and contribute to sending signals for T-cell activation upon recognition of a target antigen (70). The final part is the intracellular domain of CARs, which has undergone the most variation over generations. First-generation CARs contained only the CD3 ζ-chain or FcϵRIγ as their signaling domain. These first-generation CARs demonstrated inferior T-cell activation (71, 72). Second- and third-generation CARs have been engineered by incorporating one or two co-stimulatory domains, commonly using CD28, 4-1BB, ICOS, or OX40 fragments (71). These co-stimulatory domains enhance T-cell activity and increase the efficacy of CAR T cells in clinical trials. The CD28 co-stimulatory CARs are more cytotoxic, while the 4-1BB CARs retain a more long-lasting memory function (73, 74).
To date, seven CAR T cell products have been approved by the FDA for treating lymphoid malignancies (75): YESCARTA for large B-cell lymphoma in 2017 (76), KYMRIAH for B-cell precursor acute lymphoblastic leukemia (ALL) in 2017 (77), TECARTUS for mantle cell lymphoma in 2020 (78), BREYANZI for large B-cell lymphomas in 2021 (79), ABECMA for multiple myeloma in 2021 (80), CARVYKTI for multiple myeloma in 2022 (81) and AUCATZYL for ALL in 2024 (82). Much less impressive results have so far emerged from CAR T cell therapy of solid tumors, where few complete responses have been seen in high-risk neuroblastoma or metastatic rhabdomyosarcoma. PSCA, PSMA, and EpCAM are currently target antigens for research in prostate cancer using CAR T cell approaches.
2.1.3.1 Prostate-specific membrane antigen
PSMA currently represents the most popular CAR T cell therapy target in prostate cancer (21). It is a glycoprotein in prostate cells, but is also expressed in other tissues, including salivary glands and the nervous system (83). PSMA is overexpressed on the surface of prostate cancer cells and relates to the aggressiveness and progression of the disease (84). Its presence in other tumors suggests that PSMA-targeting CAR T cells could also help treat these cancers. Several PSMA-specific monoclonal antibodies have been developed to generate these CAR T cells, and 3/F11-derived versions have demonstrated better activation and killing of prostate cancer cells (21, 85). Animal model studies showed that the earlier versions of CAR T cells had low effectiveness, but newer types demonstrated moderate activity. High-dose systemic administration had some effects, while direct administration of CAR T cells into the tumors completely eradicated the tumors (86). However, these study results are still preclinical; additional research must translate them to humans.
To prevent these excessive responses, the research group led by Gaia Zuccolotto et al. modified the CAR by adding a stimulatory domain to its cytoplasmic structure, so that excessive stimulation leads to the induction of cell death in these cells and prevents harmful responses in the body. On the other hand, studies have used suppression of transforming growth factor-β (TGF-β) signaling to increase the efficacy of PSAM-specific CAR T cells in the treatment of prostate cancer (87–89). These in vitro, preclinical, and clinical studies collectively indicate that this strategy can improve the therapeutic efficacy of PSMA-specific CAR T cells.
Many studies have utilized the combined use of PSMA-specific CAR T cells with chemotherapy to enhance the efficacy of these cells. In various studies, docetaxel was combined with PSAM-specific CAR T cells to treat mice with PC. The results showed that using this combination therapy could improve the condition of sick mice, their survival, and reduce the side effects of chemotherapy (90, 91). Another method to increase the efficacy of CAR T cells expressing the antigen against PSMA is using duo-CAR T cells. In a study by D Wang et al., CAR T cells expressing receptors for PSMA and IL-23 were used. The results show that this treatment can increase the recruitment of TCD8+ and TCD4+ cells to the tumor site (92).
2.1.3.2 Prostate stem cell antigen
Another target is PSCA, which is associated with various cancers, including prostate cancer (93). It is overexpressed in the advanced stages of the disease, and research indicates it may promote tumor growth (94). Second-generation CAR T cells targeting PSCA have been studied, showing that those with different co-stimulatory domains affect T cell performance differently (95). Intratumoral injections initially eliminated tumors, but afterward, the tumors relapsed because they lost PSCA. This implies that combinations with other therapies may improve efficacy. Overall, results suggested that too much activation of CAR T cells may harm their long-term tumor control. Another study used Vγ9Vδ2 TCR-enriched PSCA-specific CAR T cells expressing the receptor to treat PC. The γδ CAR-T cells in this study reduced prostate cancer bone metastasis (96).
2.1.3.3 Epithelial cell adhesion molecule
EpCAM is a type I glycoprotein expressed on epithelial cells and plays an essential role in cell adhesion, migration, and differentiation (97, 98). EpCAM is frequently overexpressed in prostate cancer tissues compared to benign tissues and healthy controls. However, studies on its association with clinical parameters in prostate cancer show conflicting results: while some report an association with poor prognosis, others show no such correlation. In one preclinical study, using EpCAM-targeting CAR T cells resulted in effective tumor control in a mouse model (99). Using EpCAM-targeting CAR T cells generated from peripheral blood T cells of prostate cancer patients improves OS. It reduces tumor size in PC3 (low EpCAM expression) and PC3M (high EpCAM expression)-induced prostate cancer mouse models. It seems that although PC3 tumor cells express low levels of EpCAM due to their high expression in cancer stem cells, using these CAR T cells can lead to improving symptoms in PC mouse models (99). However, some studies have shown that using this type of CAR T cell can cause side effects such as weight loss and cytokine release syndrome. Studies also show that due to the high level of EpCAM expression in lung tissue, using EpCAM-targeting CAR T cells can lead to pathogenesis and damage to lung tissue (100).
2.1.3.4 Natural killer group 2D ligand
Natural killer group 2D ligand (NKG2DL) is a potential target for CAR T cell therapy in prostate cancer. Studies in mice have shown that targeting NKG2DL with CAR T cells can control tumor growth and improve survival by over 80%. Combining CAR T cells with IL-7 enhanced effectiveness. However, high or repeated doses may be needed, which could be challenging in humans. The solid tumor microenvironment poses a significant obstacle; however, directly injecting CAR T cells into or near tumors could be a promising approach for patients. Combination strategies of this type of T cell work with IL-7 (101), IL-27 (102), and anti-PD-L1 monoclonal antibodies (103) have also shown therapeutic efficacy in in vitro and animal studies.
2.1.3.5 Other antigens
Six-transmembrane epithelial antigen of prostate-2 (STEAP2) is a protein highly expressed by prostate tumor cells, and its use for targeting CAR T cells can be effective (104). In the study by P. Zanvit et al., the receptor that binds to STEAP2 was used as the CAR. These cells were also engineered to express a trap receptor for TGF-β and prevent the induction of fatigue by this cytokine in the produced CAR T cells. The results show that these CAR T cells can decrease tumor cell growth (105).
B7-H3 (CD267) is also expressed as an immune checkpoint by tumor cells, especially prostate cancer stem cells (106). Yida Zhang et al. have investigated its effects in vivo and in vitro by producing B7-H3 CAR T cells. The results of this study suggest that fractionated irradiation (FIR) combined with B7-H3 CAR T can inhibit tumor growth in a mouse model (107).
2.2 Antibody-based immunotherapies
Antibodies are one of the primary therapeutic methods inhibiting the various pathways tumors use to escape the immune system (108). After production, antibodies can be efficiently isolated and efficiently isolated and, depending on the antibody-recognized antigen, used to treat various diseases. In the case of prostate cancer, growth-inhibitory antibodies, metastasis-inhibitory antibodies, and angiogenesis-inhibitory antibodies can be mentioned (109)Among the antigens for which antibodies have been produced to treat PC is PSMA. Additionally, due to their targeted function, antibodies are now used in engineered forms. Among the antibodies engineered to increase their therapeutic efficacy are Radionuclide-conjugated antibodies, Antibody-drug conjugates, and T–cell–recruiting bispecific engagers, which we will discuss in more detail below.
2.2.1 PSMA-antibody-based treatments
Antibodies J591 and J415 have been produced against PSMA, one of the main tumor-specific antigens in PC, which can bind to the extracellular domain of this antigen with high affinity (110). Therefore, they are commonly used in various applications, including those engineered for the treatment of PC (111). The first attempts to conduct studies with the 7E11 antibody (for the intercellular domain of PSMA) were unsuccessful, but the development of J591 targeting the extracellular part of PSMA was a big step forward. The studies performed with 177Lu-J591 (as β-emitting radiopharmaceuticals conjugated to J591) demonstrated effective targeting and better clinical responses (112). Trials performed with 225Ac-J591 (as α-emitting radiopharmaceuticals conjugated to J591) also demonstrated safety and preliminary signs of effectiveness (113).
The first PSMA antibody-drug conjugate (ADC) was MLN2704, using J591 to deliver drug maytansinoid 1 (DM1). Phase 1/2 studies demonstrated activity in metastatic castration-resistant prostate cancer; however, development was halted due to neurotoxicity (114). Another study used the microtubule-disrupting agent monomethyl auristatin E (MMAE) conjugated with anti-PSMA antibody and determined 2.5 mg/kg as the maximum tolerated dose (115). Several other PSMA-ADCs are in development because of the limited therapeutic window despite confirmations of safety and efficacy.
Pasotuxizumab is a bispecific T-cell engager (BiTE) immune therapy that engages T cells by binding to CD3, enabling them to attack prostate cancer cells that express PSMA. Studies have shown promise in activating T cells and delaying tumor growth. The study by Hummel et al. aims to assess the safety, tolerability, and maximum tolerated dose of pasotuxizumab as a single agent in patients with advanced PC through phase I (116). This study is the first report that shows that BiTE immune therapy is effective as a monotherapy against prostate cancer and, in fact, against any solid tumor. Early results in patients with advanced PC are encouraging. JNJ-081 is another BiTE that, like the previous study, was produced against CD3 T cells and PSMA tumor cells and has completed its phase 1 clinical trial (117).
On the other hand, the EN Glud and colleagues’ research group designed T-cell engagers that bind to CD3 on one side and PSMA on the other, with specificity and the ability to bind to FcRn. The results of this study showed that these T-cell engagers are recycled by FcRn and, based on the level of PSMA expression, have acceptable cytotoxicity on tumor cells by activating T-cells (118). Given that this type of treatment can produce large amounts of cytokines and the resulting side effects, the K Dang research group and colleagues solved this problem by lowering the binding affinity of the T-cell engager to CD3 (119).
2.2.2 Antibody against other antigens
The results of various studies show that the expression of N-cadherin by prostate cancer cells can have a direct relationship with the ability to metastasize and resistance to tumor treatment (120). For this reason, in the study conducted by H Tanaka et al., an antibody produced against the extracellular domain of N-cadherin was used to treat PC. The results of this study show that the antibody against N-cadherin is effective in vitro and in vivo, and in addition, it can reduce the growth of tumor cells and reduce the ability to metastasize and resist treatment. In addition, the establishment of tumor xenografts in an animal model was also reduced. Overall, the results of this study indicated that N-cadherin is one of the main factors involved in treatment resistance in castration-resistant prostate cancer (121).
Many studies have shown that prostate tumor cells express enolase-1, thereby increasing their migratory ability. In a study conducted by ML Chen et al., an antibody against enolase-1 was used to treat PC. The use of this antibody in mice resulted in the inhibition of tumor cell growth, specifically in the PC3 cell line. Further investigation to determine the exact effect of this antibody showed that the antibody against enolase-1 could exert its antitumor action by inhibiting tube formation (mediated by VEGF-A) and preventing bone metastasis (122).
As discussed throughout this section, different antibodies directed against different antigens can inhibit tumor growth and affect tumor cell function in PC models. However, none of the studies have compared the efficacy and safety of the discussed antibodies. When comparing antibody-based therapies, factors to consider include the level of target antigen expression on the surface of tumor cells, the functional activity involved after antibody binding, the time required for production, the cost of production, and the availability after clinical trials.
3 Immune checkpoint inhibitor-based treatments
The development of immune checkpoint inhibitors has transformed cancer treatment by permitting long-term survival in patients with advanced disease and new options in earlier stages of the disease (123).
Essential points that relate to ICIs’ use in prostate cancer include the fact that this disease has low tumor mutational burden (TMB)and a low somatic mutation frequency compared to other highly responsive diseases of this type of treatment, like melanoma or lung cancer (124); thus, relatively fewer immune cells, including tumor-specific T cells, would migrate toward the tumor. Moreover, hypoxic zones in the tumor site cause reduced T-cell infiltration by affecting factors such as low pH and nutrient deficiency (125). These hypoxic zones further lead to increased myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs), inhibiting the immune response (126, 127). The T cell population mainly includes immunosuppressive regulatory T cells (128). Other factors that impede anti-tumor responses are the loss of MHC class I and the frequent loss of PTEN, thereby decreasing the efficacy of immunotherapy in prostate cancer (129).
In addition to the above, studying tumor characteristics using biomarkers can be crucial and aid in selecting the appropriate type of immunotherapy. Today, many biomarkers such as hot or cold tumor, microRNAs, cell-free DNA, circulating tumor DNA (ctDNA), CD8/CD4 ratio, studying the population of CD8-positive anti-tumor specific T cells, and also exploring the altered microbiome of patients are mentioned (130, 131). Given that changes in the levels of DNA and extracellular RNA in the blood can indicate the level of malignancy, metastasis, resistance to treatment, and treatment selection, they are of great importance. It also seems that the altered microbiome (dysbiosis) can affect the initiation and spread of PC, and treatment and response to treatment through the production of various metabolites (132, 133).
Therefore, considering the immunosuppressive properties of the tumor microenvironment, the use of ICIs can compensate for the suppression induced by this environment and help treat prostate cancer.
3.1 Anti-programmed death-1/ligand antibody
PD-1 is expressed on activated T, B, and NK cells and has two ligands, PD-L1 and PD-L2 (134). The interaction of PD-L1 with PD-1 inhibits T cell activation and converts naive T cells into regulatory T cells that suppress excessive immune responses against normal cells during antigen-specific responses (134). Tumors exploit PD-L1 to evade T cell attack by inducing the energy or death of the effector T cells (Figure 3). PD-1 shares a structural similarity with the molecules CD28 and CTLA-4, also implicated in T cell activation and inhibition (135).
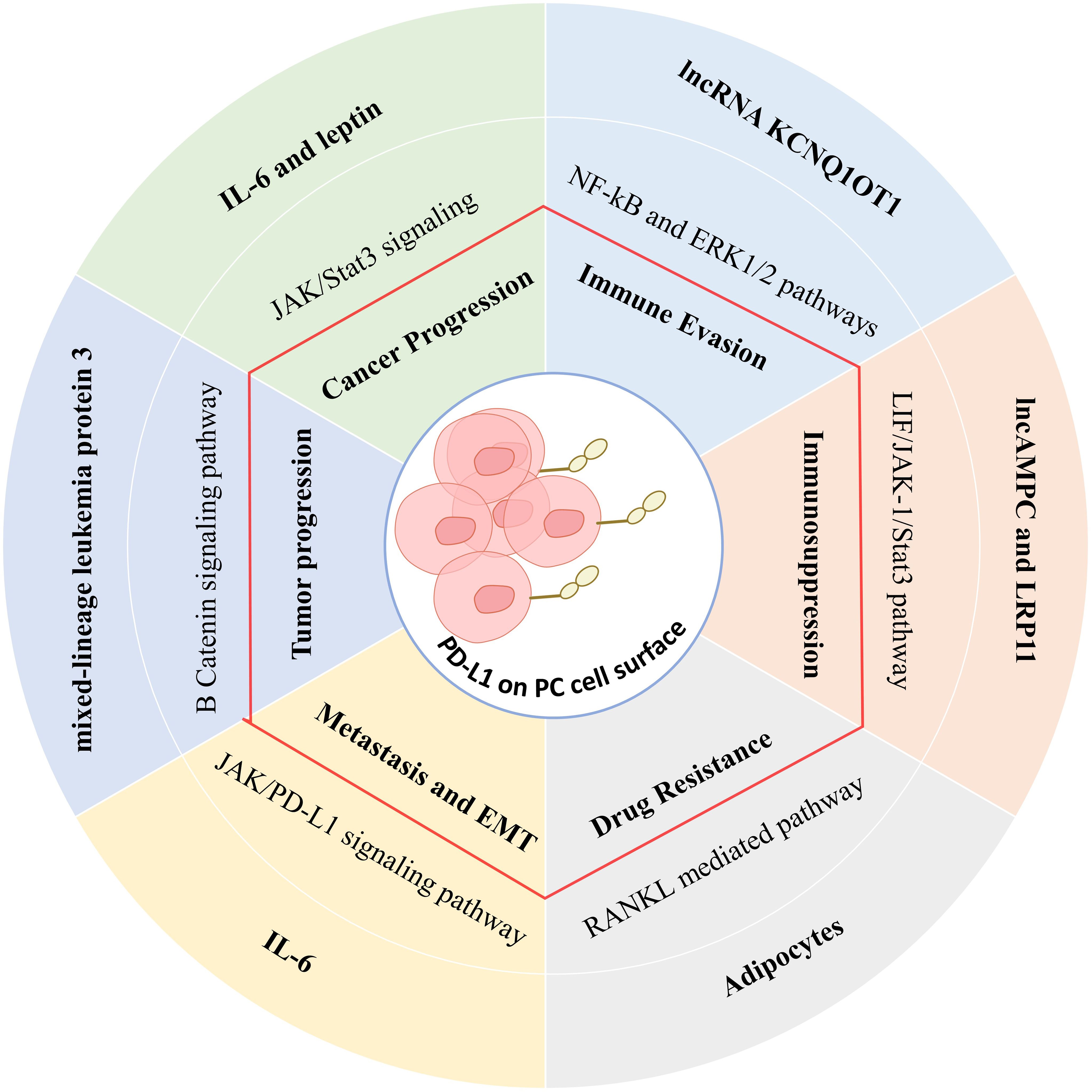
Figure 3. The effect of the tumor microenvironment on PDL-1 expression through various signaling pathways in tumor cells and its immunosuppressive effects.
High levels of PD-1/PD-L1 expression are associated with significant clinical features in prostate cancer (136). The promoter of PD-1 is hypermethylated in cancer tissues compared with normal prostate tissues, establishing a negative correlation between PD-1 methylation and PD-1 mRNA expression (137). Higher PD-1 methylation is associated with higher preoperative PSA scores, higher Gleason grades, and more advanced tumor categories, serving as an adverse prognostic factor for biochemical recurrence-free survival (RFS) (138). Methylation of PD-1 is also associated with androgen receptor activity and ERG gene fusion, suggesting a regulatory role in autoimmune responses (137). The study of PD-1 promoter methylation may help identify patients who could benefit from adjuvant therapy after surgery. Essential research evidence has shown that PD-L1 is highly expressed in prostate cancer tissues compared to normal tissues, with over half of the tested cases showing moderate to high expression (139, 140). The expression of PD-L1 in aggressive cancers correlates with tumor proliferation, Gleason scores, and androgen receptor expression (141). It is also known to be an independent negative predictor of biochemical recurrence. What is more, high-risk patients with highly expressed PD-L1 have an impaired prognosis after hormonal treatment following surgery. One study showed that PD-L1 was more frequently expressed in high-stage tumors or lymph-node-positive cases, whereas PD-1 expression was independent of tumor stage (142).
High PD-L1 expression is associated with unfavorable survival and, specifically, with aggressive prostate cancer (143). Moreover, PD-L1 is also expressed on both tumor-associated nerves and circulating tumor cells, and its tumor expression correlates with clinical progression risks (144). Finally, exosomal PD-L1 has not been fully elucidated regarding predicting the response to anti-PD-1 therapy in prostate cancer (145).
The preclinical studies related to PD-1/PD-L1 expression in prostate cancer have investigated the efficiency of checkpoint inhibitors. The results have been inconsistent, at least in part because the positivity criteria for PD-L1 have varied. One such study reported the presence of PD-L1 in 92% of prostatectomy specimens but without relation to cancer outcomes (146). Other studies showed variable rates of PD-L1 expression; some of these mentioned its possible prognostic significance for biochemical recurrence (147). The results also varied widely in different patient samples and settings.
A 17.4% objective response rate was demonstrated in the single-arm, phase Ib study, KEYNOTE-028, using pembrolizumab among 23 heavily treated mCRPC patients with measurable disease and a PD-L1 expression of at least 1% (148). Three of the four who showed partial response had an extraordinary decline in their PSA levels. The KEYNOTE-199 trial studied pembrolizumab as monotherapy in three separate groups within the mCRPC population according to their PD-L1 status (149). Median OS by blinded independent central review was 9.5 months for PD-L1-positive, 7.9 months for PD-L1-negative, and 14.1 months for non-measurable disease (149). As a single agent, atezolizumab demonstrated safety with clinical activity, including 55. 6% at a 12-month OS rate with a median OS not yet reached in this group of 15 heavily treated mCRPC (149). However, it is important to note that the lack of PD-L1 enrichment may have contributed to limited responses in this study.
Studies show that human Vγ2Vδ2 (or Vγ9Vδ2) T cells can recognize specific metabolites associated with isoprenoid biosynthesis and thus help to fight infections and tumors (150, 151). Bisphosphonate treatment enhances these metabolites, enabling Vγ2Vδ2 T cells to recognize tumors independently of MHC expression or mutation load (152). Immunotherapy with these T cells has been tested in more than 400 patients in trials and is associated with limited side effects but also limited remissions. This study is focused on adding PD-1 checkpoint blockade to Vγ2Vδ2 T cells in a PC-3 prostate tumor model in mice. Results show that blocking PD-1 improves the efficacy of T cells and causes a significant reduction in tumor volume (152).
3.2 Anti-CTL-associated antigen-4 antibody
T-cell activation requires specific antigenic peptide recognition by T-cell receptors and costimulatory signals (153). T cells express the CD28 and CTLA-4 receptors that interact with ligands on antigen-presenting cells (APCs) (154). While CD28 engagement activates T cells, CTLA-4 engagement inhibits them. Blockade of the CTLA-4 interactions has shown promise in enhancing immune responses in preclinical models (155). Indeed, in vivo studies have confirmed that CTLA-4 blockade can effectively induce tumor rejection in mouse PC models. In a model of prostate cancer, the administration of an anti-CTLA-4 antibody diminished the risk of metastatic relapse after tumor resection (156). Few toxicities from CTLA-4 blockade were observed, mainly confined to prostatitis and vitiligo, with no significant findings in primate or human tissue studies (157).
In a pilot trial, James P. Allison et al. used a single-dose anti-CTLA-4 antibody (ipilimumab) in metastatic PC to test its safety and efficacy (158). The goals of this trial included assessing safety, potential autoimmune toxicity, and changes in PSA levels. Generally, in metastatic PC patients, the therapy activity is well-gauged when PSA decline is considered equal to or higher than 50% (158). During such a study, 2–14 patients had declined at a rate above 50%. The contribution of CTLA-4 blockades and steroid therapy to this PSA decline is poorly defined. Recent data indicate that CTLA-4 blockade can induce anti-PSA antibodies that clear PSA (158).
Immunological studies show that using the CTLA-4 blocker can increase the number of TCD4+ ICOShi cells. These cells can produce IFN-γ and contribute to activating CTLs and NK cells and their antitumor function (159). However, anti-CTLA-4 immunotherapy does not remove FOXP3+ Treg cells in tumors, indicating that modifying mAbs or their combinations might improve efficacy (160).
3.3 Anti-TIM-3 antibody
T-cell immunoglobulin domain and mucin domain-containing molecule 3 (TIM-3) is a surface marker specific for Th1 CD4+ T cells. TIM-3 is expressed mainly on fully differentiated Th1 lymphocytes but not Th2 cells (161). TIM-3 may not affect the differentiation of T cells, but it plays an essential role in the transport of Th1 cells. It negatively regulates Th1 and Th17 responses and is critical in T-cell exhaustion, particularly in tumors such as NSCLC (162, 163). TIM-3 levels in patients may relate to PC patients’ survival. It showed higher TIM-3 expression on CD4+ and CD8+ T cells in PC patients’ blood and tumor tissue. These levels relate to the advanced disease stage, a factor of poor prognosis (164). It seems that the overall expression of TIM-3 by TCD8+ cells leads to dysfunction of these cells in prostate cancer (165).
The results of the study conducted by J. Harding et al. show that the use of LY3321367 as a monoclonal antibody blocking TIM-3 functions has safe and tolerable effects on patients (166). Further studies show that this treatment can increase the CD8 expression in patient biopsies. Given that CD8 is observed more often on T cells and also on DCs, it seems that this treatment can lead to an increase in the recruitment of immune cells involved in anti-tumor defense to the tumor site (166).
3.4 Anti-LAG3 antibody
LAG3 is a novel immune checkpoint expressed on the surface of CD4+ T, CD8+ T, NK cells, NKT cells, and regulatory T cells (167, 168). It can be rapidly induced to the cell surface upon T-cell activation. LAG3 has been reported to interact with MHC class II and other ligands such as FGL-1 and galectins (169). LAG3 is highly expressed on CD4+ T cells during bacterial infections and can restore their function upon blockade. LAG3 also modulates CD4+ T regulatory cells, a subset of T cells that suppress other immune responses (167, 170). Its specific role in cancer, however, remains unclear. The ligand for LAG3, FGL1, accumulates considerably in the prostate tumor tissues and supports rapid tumor growth (171). The expression of the inhibitory receptors, including LAG3, in various treatments is of interest in developing therapies against prostate cancer. A study by Xinyao Zhang et al. identified LAG3 upregulation in the CD4+ T cells from PC patients (172).
A DSW Tan et al. study has shown that ieramilimab, an antibody against LAG-3, can inhibit the interaction of LAG-3 with FGL-1. This antibody has been shown to have therapeutic efficacy in treating various cancers, including prostate cancer. The therapeutic use of ieramilimab in patients can increase the production of IFN-γ and the rise in the activity of T cells (173).
3.5 Anti-tyrosine-based inhibition motif domain antibody
TIGIT interacts with several PVR/NECTIN family ligands, among which CD155/PVR is considered the most prominent, followed by CD112/NECTIN-2 and CD113/NECTIN-3 (174). The same ligands are targeted by other inhibitory receptors, including the recently described CD96 and CD112R/PVRIG, which dampen NK and T cell activities (175). On the contrary, the costimulatory receptor CD226/DNAM-1 partially shares ligands with TIGIT (176). The expression patterns of TIGIT and CD226 parallel those of CTLA-4 and CD28, with CD226 present on naïve T cells and TIGIT increasing after activation. TIGIT exerts its immune suppressive effects through several mechanisms, including delivering negative signals, competing for ligands that bind with higher affinity to TIGIT, and modifying DCs function (177). TIGIT+ T cells are less active than TIGIT− T cells in chronic viral infections, whereas TIGIT+ regulatory T cells are more suppressive of effector T-cell activation (178). TIGIT is frequently expressed in human tumors at higher levels in tumor-infiltrating lymphocytes than in those in peripheral blood, suggesting that TIGIT contributes to generating an immune-suppressive tumor environment (179).
Since tumor cells often express TIGIT ligands, targeting TIGIT may be advantageous in cancer therapy. Indeed, studies have demonstrated that TIGIT blockade restrains tumor growth, especially when combined with other ICIs (174, 180). Results from various studies show that prostate cancer TME has high expression of TGIT-related ligands such as CD276, PVR, and NECTIN2 (181). Therefore, they can suppress T-cell responses in this way.
It was identified that the TIGIT monoclonal antibody vibostolimab increases NK cell function against PC by increasing key markers and cytokine production. The TIGIT blockade also increased NK cell signaling pathways and improved T cell attraction to the tumor site. These findings support using TIGIT antibodies and NK cell strategies for PC treatment (182).
Most studies now focus on anti-TIGIT or anti-LAG-3 alone and combination therapies with anti-PD-1 therapies (183, 184). These studies put forth that for therapy against cancer treatment, the combination of anti-TIGIT and anti-LAG-3 may provide a novel approach. A study by Dai et al. showed that ZGGS15 (IgG4 bispecific antibody targeting TIGIT and LAG-3) has potent functional binding and blocking activity against TIGIT and LAG-3. The results showed that ZGGS15 has a high affinity to human LAG-3 and TIGIT, specifically binding to activated CD4+ and CD8+ T cells with near-equal strength (184). Moreover, ZGGS15 outcompeted LAG-3 and MHC-II, as well as TIGIT and CD155, indicating potential advantages in patients with high levels of FGL1 who tend to have poor outcomes after receiving current treatments. It binds to prostate cancer cell-expressed MHC-II and CD155 and the inhibitory receptors LAG-3 and TIGIT on immune cells. Studies carried out in mouse models reveal that ZGGS15 improves responses by T cells and potentiates the anti-tumor efficacy (184).
4 ICIs combined treatments
4.1 Dual immune checkpoint inhibitor applications
Studies have shown that, under certain conditions, using a single ICI can lead to compensatory increased expression of other ICIs (185). For this reason, it is recommended to use a combination of two or three ICIs for the treatment of prostate cancer (186). Also, basic immunological studies indicate that immune checkpoint receptors are co-expressed in self-antigen tolerance, chronic infectious disease, and inflammation. In addition to lymphocyte-intrinsic inhibitory receptors, there are two presumed inhibitory ligands from the B7 family, B7-H3 and B7-H4, with no assigned receptor (187). Studies on mouse prostate cancer models have shown that inhibiting a particular ligand or its receptor improves anti-tumor immunity (185).
As mentioned earlier, several receptors, including LAG3, 2B4, BTLA, TIM3, and A2aR, negatively regulate lymphocyte activity and, under appropriate circumstances, anergic lymphocytes (188, 189). Antibody targeting of these receptors enhances anti-tumour immunity in tumor models. Since many tumor cells express more than one inhibitory ligand, several opportunities exist to enhance immune responses by dual or triple checkpoint blocking (190). However, it is essential to know that in some ICI combinations it is possible to overcome implications. For example, in the CheckMate 650 trial, combining anti-CTLA-4 and anti-PD-1 showed an objective rate of response of 25% and 10% in pre- and post-chemotherapy groups, respectively. But patients developed adverse effects in 42%-53% (dose dependent), necessitating dose adjustments (191). While human antibodies targeting some of these inhibitory receptors are being developed, none have yet reached clinical use. Most of these receptors are activated during T cell activation, suggesting they regulate T cell responses when their corresponding ligands are present (Table 3).
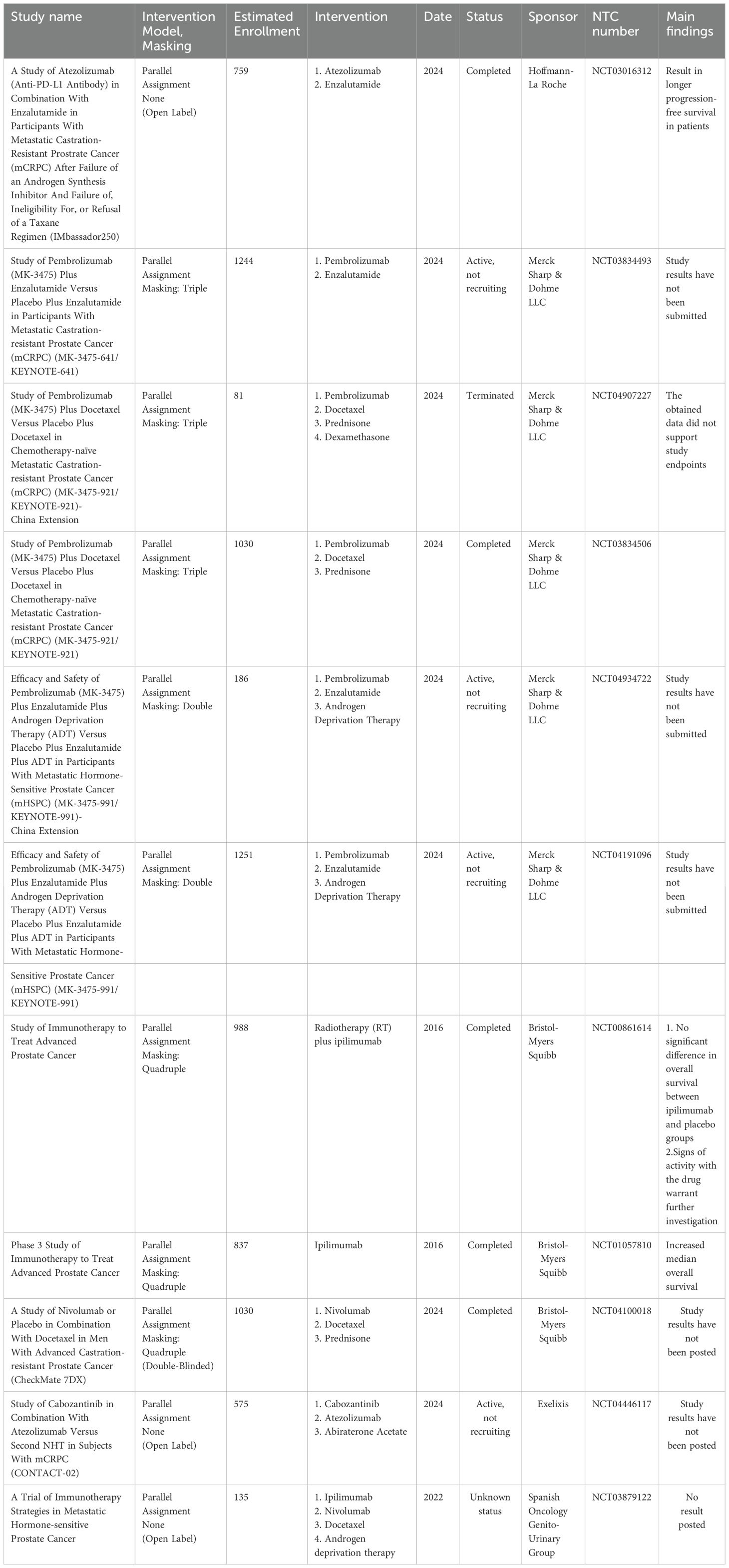
Table 3. Example of immune checkpoints inhibitors application in combination with other treatments for prostate cancer in phase 3 clinical trials.
4.2 ICIs combined with chemotherapy
Docetaxel is generally given after resistance develops to either abiraterone or enzalutamide in patients with metastatic PC (192). A phase 1b/2 study by Evan Y Yu et al. assessed the efficacy and safety of adding pembrolizumab (anti-PD-1 Ab) to a combination of docetaxel and prednisone (NCT02861573) (193). The measured outcomes of this study are safety, PSA response rate, and ORR. In 104 patients, the confirmed PSA response rate was 34%, and ORR was 23%. This combination regimen demonstrated antitumor activity with manageable safety in chemotherapy-naïve metastatic PC (194). In a phase 1/2 study, the bispecific antibody Vudalimab, which binds to CTLA-4 on one side and PD-1 on the other, was used in combination with docetaxel to treat metastatic prostate cancer (195).
Another study tested combined ICIs in patients with chemotherapy-naïve metastatic PC to the bone. Patients received tremelimumab (anti-CTLA-4 Ab) and durvalumab (anti-PD-1 Ab) every four weeks in combination with chemotherapy. Results showed that 42% experienced grade ≥3 treatment-related adverse events, and 24% had stable disease for over six months. Median overall survival was 28.1 months. The findings suggest that the combination may require additional treatments to address resistance in the bone environment (196).
4.3 ICIs combined with radiotherapy
Radiotherapy has been demonstrated to have both immune-enhancing and immunosuppressive effects at the tumor site and systemically (197). In the wake of several promising preclinical studies, several clinical trials were launched, testing the hypothesis that radiotherapy in combination with ICIs would augment anti-tumor immunity. However, many trials have not resulted in impressive gains (198).
Radium-223 dichloride is a treatment primarily directed at bone metastases that improves overall survival in patients with prostate cancer (PC). In one study, 44 patients received a combination of atezolizumab (anti-PD-L1 Ab) with radium-223 in a safety-testing exercise that did not return clinically meaningful benefits, as evidenced by an overall response rate of 6.8% and a median overall survival of 16.3 months (199).
Radiotherapy can cause tumor shrinkage in areas distant from the primary tumor, known as the abscopal effect (200). A study with 28 patients evaluated the combination of ipilimumab (anti-CTLA-4 Ab) and radiotherapy, resulting in one complete response and several stable disease cases (201). A larger trial compared ipilimumab to placebo in 799 patients, showing similar median survival rates. However, longer follow-ups indicated better survival rates with ipilimumab at 2 and 5 years. Adverse effects included severe gastrointestinal issues, and some deaths were due to the treatment (202).
In another study, 90Y-NM600 was used as a targeted radionuclide therapy (TRT) mediator in combination with an anti-PD-1 antibody to treat prostate cancer in an animal model. Surprisingly, that study showed that this combination was ineffective in treating PC because the anti-PD-1 antibody activated and expanded Treg cells (203). Indeed, the triple combination of anti-CTLA-4/anti-PD-1/radiotherapy showed the best survival and tumor growth delay compared to dual and single therapies. Thus, the preclinical model suggests that this combination is effective and must be pursued in a clinical setting for PC (204).
Therefore, although this combination presents challenges, such as the abscopal effect, and can lead to “off-target” anti-tumor effects, it is used in many clinical studies to treat PC. The clinical trial studies using this combination are NCT04446117, NCT04946370, NCT04931979, and NCT03543189. However, the results of these studies are not yet available.
4.4 Other combinations
4.4.1 Androgen deprivation therapy
One of the main treatments for advanced prostate cancer is ADT, which causes immune alterations in the tumor microenvironment. Thymic regeneration, reduced tolerance, and an improved adaptive immune response can facilitate immune cell infiltration after ADT (205). However, these positive effects are transient and may be counterbalanced by an increased immunosuppressive cell compartment. Thus, this provides a rationale for combining it with ADT. The PRIME-CUT Phase II trial is investigating the combination of ADT and cemiplimab (anti-PD-1 Ab) treatment in men with metastatic castration-sensitive prostate cancer (206, 207). The results of this study led to the introduction of a new concept of “Highly Active Anti-Tumor Therapy” HAATT, including vigorous early treatment to eradicate resistant tumor cells, enhance the function of CD8+ T immune cells, reduce regulatory T cells, and interfere with growth-promoting substances in the tumor environment (206).
4.4.2 Electroporation
Some studies have also used more creative methods to develop an ICI-based therapy. Studies have shown that focused electroporation at the tumor site can damage tumor tissue and kill tumor cells (207). These killed tumor cells release antigens that can activate immune system cells. For this reason, in a study by BJ Burbach et al., the ICI beta electroporation was used to treat PC. For this purpose, after tumor induction in mice by transplantation of TRAMP-C2 cells and treatment by electroporation, a stock solution containing three types of ICI (antibodies against PD-1, PD-L1, and CTLA-4) was used. The results of this study showed that this combination can lead to an increase in the prostate tumor-specific CD8+ T cell population (208).
4.4.3 STAT3
The STAT3 signaling pathway contributes to the development of immunosuppressive cells and represses DCs functions within tumors to support immune evasion of cancer (209). It controls immunosuppressive factors of tumor cells and is an attractive drug target, which may enhance the effects of immune checkpoint inhibitors (210).
Therefore, in the study by Kristina Witt et al., a combination of an antibody against CTLA-4 and an inhibitor of the STAT3 signaling pathway (GPB730) was used. The results of this study show that after combination treatment in the PC mouse model, the population of regulatory T cells within the tumor is significantly reduced compared to single treatments. Considering the pathological role of regulatory T cells in tumor progression, a reduction in the population of these cells leads to tumor improvement and a decrease in size (211). In another study, CFF-1 was also used as a treatment for PC. This treatment reduced PSA levels and improved life quality for patients with advanced prostate cancer. In a mouse model, PD-1/PD-L1 increased during tumor growth, reducing CD3+ T cell subsets. CFF-1 decreased PD-L1 expression dose-dependently, inhibited tumor growth, and helped recover CD3+ T lymphocytes (212). It also extended survival and reduced lung metastasis, especially when combined with docetaxel. CFF-1 can serve as an ICI and STAT3 inhibitor and shows promise as a treatment for prostate cancer by enhancing immune response (212).
4.4.4 Vaccine
Tumor vaccines typically require a booster treatment to improve their efficacy. For this reason, in many cases, adjuvants are used to increase immune responses. However, it seems that using ICI can lead to improved vaccine efficacy. For this reason, in the phase 2 study by G McNeel et al., the MVI-816 vaccine (antigen prostatic acid phosphatase (PAP)) was used in combination with pembrolizumab for the treatment of PC in patients (NCT02499835) (213). The results of this study show that the use of this combination therapy can lead to the expansion of Th1 cell responses, an increase in the response of specific CD8+ T cells, and an increase in the production of IFN-γ and granzyme B by these cells (213).
4.4.5 Cryotherapy
A small pilot study also utilized cryotherapy in combination with pembrolizumab (an anti-PD-1 Antibody). The median progression-free survival was 14 months, with no serious adverse effects (214).
5 Challenges, limitations, and future perspectives
ICI therapy has transformed the treatment landscape over the last decade and continues to evolve. Appreciating organ-specific toxicities will remain critical for the practicing oncologist and other specialists who contribute to patient care (215). Endocrine toxicities represent some of the most common adverse effects, and therefore, management by the endocrinologist becomes paramount (216). This awareness will further grow with the increased use of ICI regimens. Some of the research challenges concerning ICI-induced endocrine disorders are finding predictors and risk factors of these effects, such as pituitary dysfunction, which will help select cancer treatment and enhance monitoring and prevention strategies (217). The other challenge is investigating the mechanisms behind the endocrine syndromes associated with ICIs. Investigation into thyroid inflammation might provide some insights and further opportunities for study (218). Finally, investigating the relationship between thyroid dysfunction and improved survival in cancer is also warranted (216).
Also, the long-term effects of ICIs are becoming increasingly important (219). Whereas most studies have focused on short-term toxicities, new evidence suggests that long-term effects may be more common than previously thought. Chronic toxicities, in general, affect all organ systems, including endocrine and rheumatological issues (219). Given the potential for long-term survival, the development of fatal toxicities is 0.4–1. 2% of patients is not trivial. Moreover, ICIs touch many aspects of immune function, and their long-term impact on cancer survivors remains unknown mainly (220).
Developing approaches to predict the risk of immune-related adverse events (irAEs) is critical for optimizing ICI therapies or switching patients to less dangerous treatments (221). Some studies have demonstrated that cytokines may amplify the development of irAEs, and therapies targeting such cytokines have been used in clinical practice to manage severe irAEs (222). Most irAEs are mild, but early detection and treatment are crucial. Nurses are essential in identifying irAEs and educating patients on symptoms (223). In addition to monitoring, it is also essential to control these events in patients through pre-treatment examinations and post-treatment measures. The Common Terminology Criteria for Adverse Events (PRO-CTCAE) version for patient-reported outcomes effectively lowers hospitalization rates, improves quality of life, and facilitates survival (224). Limited data regarding its use in monitoring irAEs are available. In a trial with 16 PC patients, irAE-related items from NCI’s PRO-CTCAE were used alongside CTCAE (224). Symptoms like fatigue and rash were often reported, with greater irAE severity noted in PRO-CTCAE. Further study is needed on the role of monitoring programs in irAE management.
However, it is tough to establish a universal strategy for preventing irAEs since patients may respond differently. The gut microbiome and its metabolites have shown good interactions with ICI therapy and reduced side effects associated with such drugs. Both preclinical studies and clinical data warrant that the gut microbiome may help manage the development of irAEs (225).
The gut microbiota differed in mice lacking the PDCD-1 receptor, with a marked decrease in Lactobacillales (226). A model highlighted that Lactobacillus salivarius is a key species predictive of irAEs. Other anti-inflammatory probiotics include Bifidobacterium and Lactobacillus (227). Two species of Parabacteroides and two of Ruminococcus were identified as playing a key role in predicting the onset of irAEs (228). Lower menaquinone biosynthesis has been shown in patients with irAE compared to non-irAE (225). This may indicate the potential for either gut microbiome modulation or metabolites such as menaquinone to prevent irAE. Further research is needed to establish if low levels of menaquinone contribute to developing irAEs. It is still under debate whether treating irAEs diminishes the effectiveness of the immunotherapy itself. Biomarker identification regarding irAE could allow for better therapeutic efficacy while limiting adverse events. A better understanding of gut microbiome interactions and their roles in irAE will require more rigorous experimental designs and further validation in future studies.
Another point that is raised in the treatment of different cancers by ICIs is the variation in the efficacy of this type of treatment (229). Several factors can affect this efficacy and can include the ICIs surface expression level (like PDL1), the level of tumor mutational burdens, as well as the degree of penetration of the antibody into the tumor tissue (230). These factors lead to better responses in head and neck, lung, and gastro-esophageal junction cancers with elevated mutation burden (230). Main metastatic cancers also responded well because they typically contain high mutational burdens. This seems to be one of the reasons why the success rate of PC treatment using ICIs differs from other cancers (231). Given the variations seen in ICI treatment across different cancers, the Yoo and colleagues research group developed an AI-based algorithm to predict ICIs-based treatment outcomes (232). Predicting whether cancer patients will benefit from ICI without advanced testing is important. The group developed SCORPIO, a machine learning system that uses routine blood tests and patient data from 9,745 patients treated with ICI across 21 cancer types. SCORPIO was trained on 1,628 patients from Memorial Sloan Kettering Cancer Center (232). The method outperformed tumor mutational burden in predicting overall survival and clinical benefit in two sets of trials.
6 Conclusion
Given the impact of PC on patients’ quality of life and its mortality rate, there is a need for an effective treatment to prevent tumor progression. Current treatments are insufficient. Since the immune cell response can halt tumor growth, immunotherapy measures can assist in treating this disease. Immunotherapy is typically administered using cells or antibodies. With the advent of ICIs and their integration into tumor treatment, they have been extensively utilized in managing PC. Although the outcomes have been promising, some challenges include the induction of treatment resistance, the emergence of endorphin responses, and their toxicity in various tissues. Therefore, researchers have sought to combine different therapies with ICIs. To date, treatment modalities such as chemotherapy, radiotherapy, cryotherapy, PARP inhibitors, vaccines, ADTs, and several other therapies have been combined with ICIs to inhibit tumor growth in preclinical and clinical trials. Examining biomarkers such as microbiome composition, tumor gene mutation burden (TMB), microsatellite instability (MSI), and DNA-repair gene mutations that result in reduced DNA mismatch repair (MMR), as well as target protein expression in PC patients and tumor tissue, can significantly assist in selecting appropriate treatment based on ICIs. These biomarkers show us the potential for tumor cell transformation, and in general, it seems that MMR-deficient and MSI-high tumors are better responders to ICI treatment (233). Combination therapies prevent tumors from escaping treatment by targeting different mechanisms. However, research in this area is ongoing, and further studies are required to confirm an effective therapeutic combination with minimal side effects.
Author contributions
HL: Writing – original draft, Writing – review & editing. ZT: Writing – original draft, Writing – review & editing. JW: Writing – original draft, Writing – review & editing. WZ: Conceptualization, Methodology, Resources, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Vaccarella S., Li M., Bray F., Kvale R., Serraino D., Lorenzoni V., et al. Prostate cancer incidence and mortality in Europe and implications for screening activities: population based study. bmj. (2024) 386:1–13. doi: 10.1136/bmj-2023-077738
2. Kishan A.U., Sun Y., Hartman H., Pisansky T.M., Bolla M., Neven A., et al. Androgen deprivation therapy use and duration with definitive radiotherapy for localised prostate cancer: an individual patient data meta-analysis. Lancet Oncol. (2022) 23:304–16. doi: 10.1016/S1470-2045(21)00705-1
3. Lokeshwar SD, Klaassen Z, and Saad F. Treatment and trials in non-metastatic castration-resistant prostate cancer. Nat Rev Urol. (2021) 18:433–42. doi: 10.1038/s41585-021-00470-4
4. Hird A.E., Dvorani E., Saskin R., Emmenegger U., Herschorn S., Kodama R., et al. Prevalence and natural history of non-metastatic castrate resistant prostate cancer: a population-based analysis. Clin Genitourin Cancer. (2023) 21:e27–34. doi: 10.1016/j.clgc.2022.10.003
5. Shore N.D., Antonarakis E.S., Ross A.E., Marshall C.H., Stratton K.L., Ayanambakkam A., et al. A multidisciplinary approach to address unmet needs in the management of patients with non-metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. (2024), 1–10. doi: 10.1038/s41391-024-00803-5
6. Berruti A., Bracarda S., Caffo O., Cortesi E., R., Del Re M., et al. nmCRPC, a look in the continuous care of prostate cancer patients: state of art and future perspectives. Cancer Treat Rev. (2023) 115:102525. doi: 10.1016/j.ctrv.2023.102525
7. Cattrini C., Caffo O., De Giorgi U., Mennitto A., Gennari A., Olmos D., et al. Apalutamide, darolutamide and enzalutamide for nonmetastatic castration-resistant prostate cancer (nmCRPC). Cancers. (2022) 14:1–15. doi: 10.3390/cancers14071792
8. Brave M., Weinstock C., Brewer J.R., Chi D.-C., Suzman D.L., Cheng J., et al. An FDA review of drug development in nonmetastatic castration-resistant prostate cancer. Clin Cancer Res. (2020) 26:4717–22. doi: 10.1158/1078-0432.CCR-19-3835
9. Hadfield M.J., Lyall V., Holle L.M., and Dennison M.. Updates in the treatment of non-metastatic castrate-resistant prostate cancer: the benefit of second-generation androgen receptor antagonists. Ann Pharmacother. (2023) 57:1302–11. doi: 10.1177/10600280231155441
10. Ahmadi H and Daneshmand S. Androgen deprivation therapy: evidence-based management of side effects. BJU Int. (2013) 111:543–8. doi: 10.1111/j.1464-410X.2012.11774.x
11. Crawford ED and Moul JW. ADT risks and side effects in advanced prostate cancer: cardiovascular and acute renal injury. Oncology. (2015) 29:55–5.
12. Taylor LG, Canfield SE, and Du XL. Review of major adverse effects of androgen-deprivation therapy in men with prostate cancer. Cancer. (2009) 115:2388–99. doi: 10.1002/cncr.v115:11
13. Corona G., Gacci M., Baldi E., Mancina R., Forti G., Maggi M., et al. Androgen deprivation therapy in prostate cancer: focusing on sexual side effects. J Sexual Med. (2012) 9:887–902. doi: 10.1111/j.1743-6109.2011.02590.x
14. Perera M., Roberts M.J., Klotz L., Higano C.S., Papa N., Sengupta S., et al. Intermittent versus continuous androgen deprivation therapy for advanced prostate cancer. Nat Rev Urol. (2020) 17:469–81. doi: 10.1038/s41585-020-0335-7
15. Di Lorenzo G, Buonerba C, and Kantoff PW. Immunotherapy for the treatment of prostate cancer. Nat Rev Clin Oncol. (2011) 8:551–61. doi: 10.1038/nrclinonc.2011.72
16. Lu X., Horner J.W., Paul E., Shang X., Troncoso P., Deng P., et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. (2017) 543:728–32. doi: 10.1038/nature21676
17. Kantoff P.W., Higano C.S., Shore N.D., Berger E.R., Small E.J., Penson D.F., et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. New Engl J Med. (2010) 363:411–22. doi: 10.1056/NEJMoa1001294
18. Rebuzzi S.E., Rescigno P., Catalano F., Mollica V., Vogl U.M., Marandino L., et al. Immune checkpoint inhibitors in advanced prostate cancer: current data and future perspectives. Cancers. (2022) 14:1245. doi: 10.3390/cancers14051245
19. Wong Y.N.S., Joshi K., Pule M., Peggs K.S., Swanton C., Quezada S.A., et al. Evolving adoptive cellular therapies in urological Malignancies. Lancet Oncol. (2017) 18:e341–53. doi: 10.1016/S1470-2045(17)30327-3
20. Porter L., Zhu J., Lister N., Harrison S., Keerthikumar S., Goode D., et al. Low-dose carboplatin modifies the tumor microenvironment to augment CAR T cell efficacy in human prostate cancer models. Nat Commun. (2023) 14:5346. doi: 10.1038/s41467-023-40852-3
21. Wolf P., Alzubi J., Gratzke C., and Cathomen T.. The potential of CAR T cell therapy for prostate cancer. Nat Rev Urol. (2021) 18:556–71. doi: 10.1038/s41585-021-00488-8
22. Shiao SL, Chu GC-Y, and Chung LW. Regulation of prostate cancer progression by the tumor microenvironment. Cancer Lett. (2016) 380:340–8. doi: 10.1016/j.canlet.2015.12.022
23. Pasero C., Gravis G., Guerin M., Granjeaud S., Thomassin-Piana J., Rocchi P., et al. Inherent and tumor-driven immune tolerance in the prostate microenvironment impairs natural killer cell antitumor activity. Cancer Res. (2016) 76:2153–65. doi: 10.1158/0008-5472.CAN-15-1965
24. Bou-Dargham M.J., Sha L., Sang Q.-X.A., and Zhang J.. Immune landscape of human prostate cancer: immune evasion mechanisms and biomarkers for personalized immunotherapy. BMC Cancer. (2020) 20:1–10. doi: 10.1186/s12885-020-07058-y
25. Kon E and Benhar I. Immune checkpoint inhibitor combinations: Current efforts and important aspects for success. Drug Resist Updates. (2019) 45:13–29. doi: 10.1016/j.drup.2019.07.004
26. Burotto M., Singh N., Heery C.R., Gulley J.L., and Madan R.A.. Exploiting synergy: immune-based combinations in the treatment of prostate cancer. Front Oncol. (2014) 4:351. doi: 10.3389/fonc.2014.00351
27. Sfanos K.S., Yegnasubramanian S., Nelson W.G., and De Marzo A.M.. The inflammatory microenvironment and microbiome in prostate cancer development. Nat Rev Urol. (2018) 15:11–24. doi: 10.1038/nrurol.2017.167
28. Schuster M, Nechansky A, and Kircheis R. Cancer immunotherapy. Biotechnol J: Healthc Nutr Technol. (2006) 1:138–47. doi: 10.1002/biot.200500044
29. Drake CG. Prostate cancer as a model for tumour immunotherapy. Nat Rev Immunol. (2010) 10:580–93. doi: 10.1038/nri2817
30. Mellman I, Coukos G, and Dranoff G. Cancer immunotherapy comes of age. Nature. (2011) 480:480–9. doi: 10.1038/nature10673
31. Goldberg MS. Improving cancer immunotherapy through nanotechnology. Nat Rev Cancer. (2019) 19:587–602. doi: 10.1038/s41568-019-0186-9
32. Palucka K and Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. (2012) 12:265–77. doi: 10.1038/nrc3258
33. Creelan B.C., Wang C., Teer J.K., Toloza E.M., Yao J., Kim S., et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nat Med. (2021) 27:1410–8. doi: 10.1038/s41591-021-01462-y
34. Baker D.J., Arany Z., Baur J.A., Epstein J.A., and June C.H.. CAR T therapy beyond cancer: the evolution of a living drug. Nature. (2023) 619:707–15. doi: 10.1038/s41586-023-06243-w
35. Excler J.-L., Saville M., Berkley S., and Kim J.H.. Vaccine development for emerging infectious diseases. Nat Med. (2021) 27:591–600. doi: 10.1038/s41591-021-01301-0
36. Saxena M., van der Burg S.H., Melief C.J., and Bhardwaj N.. Therapeutic cancer vaccines. Nat Rev Cancer. (2021) 21:360–78. doi: 10.1038/s41568-021-00346-0
37. Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. (2008) 29:372–83. doi: 10.1016/j.immuni.2008.08.004
38. Handy CE and Antonarakis ES. Sipuleucel-T for the treatment of prostate cancer: novel insights and future directions. Future Oncol. (2017) 14:907–17. doi: 10.2217/fon-2017-0531
39. Hammerstrom A.E., Cauley D.H., Atkinson B.J., and Sharma P.. Cancer immunotherapy: sipuleucel-T and beyond. Pharmacother: J Hum Pharmacol Drug Ther. (2011) 31:813–28. doi: 10.1592/phco.31.8.813
40. Wargowski E., Johnson L.E., Eickhoff J.C., Delmastro L., Staab M.J., Liu G., et al. Prime-boost vaccination targeting prostatic acid phosphatase (PAP) in patients with metastatic castration-resistant prostate cancer (mCRPC) using Sipuleucel-T and a DNA vaccine. J Immunother Cancer. (2018) 6:1–12. doi: 10.1186/s40425-018-0333-y
41. Olson B.M., Jankowska-Gan E., Becker J.T., Vignali D.A., Burlingham W.J., McNeel D.G., et al. Human prostate tumor antigen–specific CD8+ regulatory T cells are inhibited by CTLA-4 or IL-35 blockade. J Immunol. (2012) 189:5590–601. doi: 10.4049/jimmunol.1201744
42. Fong L., Carroll P., Weinberg V., Chan S., Lewis J., Corman J., et al. Activated lymphocyte recruitment into the tumor microenvironment following preoperative sipuleucel-T for localized prostate cancer. J Natl Cancer Institute. (2014) 106:dju268. doi: 10.1093/jnci/dju268
43. Pachynski R.K., Morishima C., Szmulewitz R., Harshman L., Appleman L., Monk P., et al. IL-7 expands lymphocyte populations and enhances immune responses to sipuleucel-T in patients with metastatic castration-resistant prostate cancer (mCRPC). J ImmunoTher Cancer. (2021) 9:1–16. doi: 10.1136/jitc-2021-002903
44. Patel HD and Berg S. Active cellular immunotherapy in the desert of advanced prostate cancer. JAMA Oncol. (2022) 8:522–3. doi: 10.1001/jamaoncol.2021.7282
45. Podrazil M., Horvath R., Becht E., Rozkova D., Bilkova P., Sochorova K., et al. Phase I/II clinical trial of dendritic-cell based immunotherapy (DCVAC/PCa) combined with chemotherapy in patients with metastatic, castration-resistant prostate cancer. Oncotarget. (2015) 6:18192. doi: 10.18632/oncotarget.v6i20
46. Vogelzang N.J., Beer T.M., Gerritsen W., Oudard S., Wiechno P., Kukielka-Budny B., et al. Efficacy and safety of autologous dendritic cell–based immunotherapy, docetaxel, and prednisone vs placebo in patients with metastatic castration-resistant prostate cancer: The viable phase 3 randomized clinical trial. JAMA Oncol. (2022) 8:546–52. doi: 10.1001/jamaoncol.2021.7298
47. Tryggestad A.M., Axcrona K., Axcrona U., Bigalke I., Brennhovd B., Inderberg E.M., et al. Long-term first-in-man phase I/II study of an adjuvant dendritic cell vaccine in patients with high-risk prostate cancer after radical prostatectomy. Prostate. (2022) 82:245–53. doi: 10.1002/pros.24267
48. Fu C., Zhou L., Mi Q.-S., and Jiang A.. DC-based vaccines for cancer immunotherapy. Vaccines. (2020) 8:706. doi: 10.3390/vaccines8040706
49. Bakhshi P., Nourizadeh M., Sharifi L., Farajollahi M.M., and Mohsenzadegan M.. Development of dendritic cell loaded MAGE-A2 long peptide; a potential target for tumor-specific T cell-mediated prostate cancer immunotherapy. Cancer Cell Int. (2023) 23:270. doi: 10.1186/s12935-023-03108-0
50. Fosså A., Berner A., Fosså S.D., Hernes E., Gaudernack G., Smeland E.B., et al. NY-ESO-1 protein expression and humoral immune responses in prostate cancer. Prostate. (2004) 59:440–7. doi: 10.1002/pros.20025
51. Yang Y., Guo X., Hu B., He P., Jiang X., Wang Z., et al. Generated SecPen_NY-ESO-1_ubiquitin-pulsed dendritic cell cancer vaccine elicits stronger and specific T cell immune responses. Acta Pharm Sin B. (2021) 11:476–87. doi: 10.1016/j.apsb.2020.08.004
52. Hadaschik B., Su Y., Huter E., Ge Y., Hohenfellner M., Beckhove P., et al. Antigen specific T-cell responses against tumor antigens are controlled by regulatory T cells in patients with prostate cancer. J Urol. (2012) 187:1458–65. doi: 10.1016/j.juro.2011.11.083
53. Lanitis E., Dangaj D., Irving M., and Coukos G.. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann Oncol. (2017) 28:xii18–32. doi: 10.1093/annonc/mdx238
54. Li J., Byrne K.T., Yan F., Yamazoe T., Chen Z., Baslan T., et al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity. (2018) 49:178–193. e7. doi: 10.1016/j.immuni.2018.06.006
55. Brusa D., Simone M., Gontero P., Spadi R., Racca P., Micari J., et al. Circulating immunosuppressive cells of prostate cancer patients before and after radical prostatectomy: profile comparison. Int J Urol. (2013) 20:971–8. doi: 10.1111/iju.2013.20.issue-10
56. Krueger T.E., Thorek D.L., Meeker A.K., Isaacs J.T., and Brennen W.N.. Tumor-infiltrating mesenchymal stem cells: Drivers of the immunosuppressive tumor microenvironment in prostate cancer? Prostate. (2019) 79:320–30. doi: 10.1002/pros.23738
57. Xin S., Liu X., Li Z., Sun X., Wang R., Zhang Z., et al. ScRNA-seq revealed an immunosuppression state and tumor microenvironment heterogeneity related to lymph node metastasis in prostate cancer. Exp Hematol Oncol. (2023) 12:49. doi: 10.1186/s40164-023-00407-0
58. Novysedlak R., Guney M., Al Khouri M., Bartolini R., Koumbas Foley L., Benesova I., et al. The immune microenvironment in prostate cancer: A comprehensive review. Oncology. (2024), 1–37. doi: 10.1159/000541881
59. Kfoury Y., Baryawno N., Severe N., Mei S., Gustafsson K., Hirz T., et al. Human prostate cancer bone metastases have an actionable immunosuppressive microenvironment. Cancer Cell. (2021) 39:1464–1478. e8. doi: 10.1016/j.ccell.2021.09.005
60. Ling Z.-N., Jiang Y.-F., Ru J.-N., Lu J.-H., Ding B., Wu J., et al. Amino acid metabolism in health and disease. Signal Transduct Target Ther. (2023) 8:345. doi: 10.1038/s41392-023-01569-3
61. Yan J., Chen D., Ye Z., Zhu X., Li X., Jiao H., et al. Molecular mechanisms and therapeutic significance of Tryptophan Metabolism and signaling in cancer. Mol Cancer. (2024) 23:241. doi: 10.1186/s12943-024-02164-y
62. Liu X-H and Zhai X-Y. Role of tryptophan metabolism in cancers and therapeutic implications. Biochimie. (2021) 182:131–9. doi: 10.1016/j.biochi.2021.01.005
63. Matos A., Carvalho M., Bicho M., and Ribeiro R.. Arginine and arginases modulate metabolism, tumor microenvironment and prostate cancer progression. Nutrients. (2021) 13:4503. doi: 10.3390/nu13124503
64. Bronte V., Kasic T., Gri G., Gallana K., Borsellino G., Marigo I., et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med. (2005) 201:1257–68. doi: 10.1084/jem.20042028
65. Lee H., Kim K., Chung J., Hossain M., and Lee H.J.. Tumor-infiltrating lymphocyte therapy: Clinical aspects and future developments in this breakthrough cancer treatment. BioEssays. (2023) 45:2200204. doi: 10.1002/bies.202200204
66. Yunger S., Bar El A., Zeltzer L.-a., Fridman E., Raviv G., Laufer M., et al. Tumor-infiltrating lymphocytes from human prostate tumors reveal anti-tumor reactivity and potential for adoptive cell therapy. Oncoimmunology. (2019) 8:e1672494. doi: 10.1080/2162402X.2019.1672494
67. Labanieh L, Majzner RG, and Mackall CL. Programming CAR-T cells to kill cancer. Nat Biomed Eng. (2018) 2:377–91. doi: 10.1038/s41551-018-0235-9
68. Mansilla-Soto J., Eyquem J., Haubner S., Hamieh M., Feucht J., Paillon N., et al. HLA-independent T cell receptors for targeting tumors with low antigen density. Nat Med. (2022) 28:345–52. doi: 10.1038/s41591-021-01621-1
69. Bachmann M. The UniCAR system: a modular CAR T cell approach to improve the safety of CAR T cells. Immunol Lett. (2019) 211:13–22. doi: 10.1016/j.imlet.2019.05.003
70. Zhang C., Liu J., Zhong J.F., and Zhang X.. Engineering car-t cells. biomark Res. (2017) 5:1–6. doi: 10.1186/s40364-017-0081-z
71. Zheng Z., Li S., Liu M., Chen C., Zhang L., Zhou D., et al. Fine-tuning through generations: advances in structure and production of CAR-T therapy. Cancers. (2023) 15:3476. doi: 10.3390/cancers15133476
72. Abate-Daga D and Davila ML. CAR models: next-generation CAR modifications for enhanced T-cell function. Mol Therapy Oncol. (2016) 3. doi: 10.1038/mto.2016.14
73. Long A.H., Haso W.M., Shern J.F., Wanhainen K.M., Murgai M., Ingaramo M., et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
74. Philipson B.I., R.S., May M.J., June C.H., Albelda S.M., Milone M.C., et al. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-κB signaling. Sci Signaling. (2020) 13:eaay8248. doi: 10.1126/scisignal.aay8248
75. Goyco Vera D., Waghela H., Nuh M., Pan J., and Lulla P.. Approved CAR-T therapies have reproducible efficacy and safety in clinical practice. Hum Vaccines Immunother. (2024) 20:2378543. doi: 10.1080/21645515.2024.2378543
76. Weinstein B., Muresan B., Solano S., de Macedo A.V., Lee Y., Su Y.-C., et al. Efficacy and safety of innovative experimental chimeric antigen receptor (CAR) T-cells versus axicabtagene ciloleucel (Yescarta) for the treatment of relapsed/refractory large B-cell lymphoma (LBCL): matching adjusted indirect comparisons (MAICs) and systematic review. Innov Pharm. (2021) 12. doi: 10.24926/iip.v12i4.4345
77. Awasthi R., Maier H.J., Zhang J., and Lim S.. Kymriah®(tisagenlecleucel)–an overview of the clinical development journey of the first approved CAR-T therapy. Hum Vaccines Immunother. (2023) 19:2210046. doi: 10.1080/21645515.2023.2210046
78. Lovell A. Brexucabtagene Autoleucel (Tecartus™). Oncol Times. (2022) 44:10. doi: 10.1097/01.COT.0000872268.55217.67
79. Bogacz A., Bukowska A., Bukowska M., Olbromski K., Łaba A., Klupieć R., et al. Modern immunotherapy using CAR-T cells in haemato-oncology and solid tumors. Acta Haematol Polonica. (2024) 55:34–41. doi: 10.5603/ahp.97189
80. Robinson A. Idecabtagene Vicleucel (Abecma®). Oncol Times. (2021) 43:21. doi: 10.1097/01.COT.0000753336.18581.7d
81. Delforge M., Mazza I.A., Mateos M.-V., Carlson K., Qi K., Mendes J., et al. Efficacy of Carvykti in cartitude-4 versus alternative treatments from daratumumab clinical trials for the treatment of patients with lenalidomide-refractory multiple myeloma. Blood. (2024) 144:7085. doi: 10.1182/blood-2024-201606
82. Hoffman M FDA Approves Obe-Cel for Adults With R/R B-Cell Precursor Acute Lymphoblastic Leukemia. (2004)
83. Bakht MK and Beltran H. Biological determinants of PSMA expression, regulation and heterogeneity in prostate cancer. Nat Rev Urol. (2024), 1–20. doi: 10.1038/s41585-024-00900-z
84. Al Saffar H., Chen D.C., Delgado C., Ingvar J., Hofman M.S., Lawrentschuk N., et al. The current landscape of prostate-specific membrane antigen (psma) imaging biomarkers for aggressive prostate cancer. Cancers. (2024) 16:939. doi: 10.3390/cancers16050939
85. Zekri L., Vogt F., Osburg L., Müller S., Kauer J., Manz T., et al. An IgG-based bispecific antibody for improved dual targeting in PSMA-positive cancer. EMBO Mol Med. (2021) 13:e11902. doi: 10.15252/emmm.201911902
86. Porter L., Harrison S., Risbridger G., Lister N., and Taylor R.. Left out in the cold: moving beyond hormonal therapy for the treatment of immunologically cold prostate cancer with CAR T cell immunotherapies. J Steroid Biochem Mol Biol. (2024) 243:106571. doi: 10.1016/j.jsbmb.2024.106571
87. Kloss C, Lee J, and June C. 638. TGFBeta signaling blockade within PSMA targeted CAR human T cells for the eradication of metastatic prostate Cancer. Mol Ther. (2016) 24:S252–3. doi: 10.1016/S1525-0016(16)33446-3
88. Narayan V., Barber-Rotenberg J.S., Jung I.-Y., Lacey S.F., Rech A.J., Davis M.M., et al. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med. (2022) 28:724–34. doi: 10.1038/s41591-022-01726-1
89. Kloss C.C., Lee J., Zhang A., Chen F., Melenhorst J.J., Lacey S.F., et al. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. (2018) 26:1855–66. doi: 10.1016/j.ymthe.2018.05.003
90. Zhang X., Sun S., Miao Y., Yuan Y., Zhao W., Li H., et al. Docetaxel enhances the therapeutic efficacy of PSMA-specific CAR-T cells against prostate cancer models by suppressing MDSCs. J Cancer Res Clin Oncol. (2022) 148:3511–20. doi: 10.1007/s00432-022-04248-y
91. Alzubi J., Dettmer-Monaco V., Kuehle J., Thorausch N., Seidl M., Taromi S., et al. PSMA-directed CAR T cells combined with low-dose docetaxel treatment induce tumor regression in a prostate cancer xenograft model. Mol Therapy Oncol. (2020) 18:226–35. doi: 10.1016/j.omto.2020.06.014
92. Wang D., Shao Y., Zhang X., Lu G., and Liu B.. IL-23 and PSMA-targeted duo-CAR T cells in Prostate Cancer Eradication in a preclinical model. J Trans Med. (2020) 18:1–10. doi: 10.1186/s12967-019-02206-w
93. Zhigang Z and Wenlv S. Prostate stem cell antigen (PSCA) expression in human prostate cancer tissues and its potential role in prostate carcinogenesis and progression of prostate cancer. World J Surg Oncol. (2004) 2:1–7. doi: 10.1186/1477-7819-2-13
94. Gu Z., Thomas G., Yamashiro J., Shintaku I., Dorey F., Raitano A., et al. Prostate stem cell antigen (PSCA) expression increases with high gleason score, advanced stage and bone metastasis in prostate cancer. Oncogene. (2000) 19:1288–96. doi: 10.1038/sj.onc.1203426
95. Dorff T.B., Blanchard M.S., Adkins L.N., Luebbert L., Leggett N., Shishido S.N., et al. PSCA-CAR T cell therapy in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med. (2024), 1–9. doi: 10.1038/s41591-024-02979-8
96. Frieling J.S., Tordesillas L., Bustos X.E., Ramello M.C., Bishop R.T., Cianne J.E., et al. γδ-Enriched CAR-T cell therapy for bone metastatic castrate-resistant prostate cancer. Sci Adv. (2023) 9:eadf0108. doi: 10.1126/sciadv.adf0108
97. Baeuerle P and Gires O. EpCAM (CD326) finding its role in cancer. Br J Cancer. (2007) 96:417–23. doi: 10.1038/sj.bjc.6603494
98. Terris B, Cavard C, and Perret C. EpCAM, a new marker for cancer stem cells in hepatocellular carcinoma. J Hepatol. (2010) 52:280–1. doi: 10.1016/j.jhep.2009.10.026
99. Deng Z., Wu Y., Ma W., Zhang S., and Zhang Y.-Q.. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol. (2015) 16:1–9. doi: 10.1186/s12865-014-0064-x
100. Qin D., Li D., Zhang B., Chen Y., Liao X., Li X., et al. Potential lung attack and lethality generated by EpCAM-specific CAR-T cells in immunocompetent mouse models. Oncoimmunology. (2020) 9:1806009. doi: 10.1080/2162402X.2020.1806009
101. He C., Zhou Y., Li Z., Farooq M.A., Ajmal I., Zhang H., et al. Co-expression of IL-7 improves NKG2D-based CAR T cell therapy on prostate cancer by enhancing the expansion and inhibiting the apoptosis and exhaustion. Cancers. (2020) 12:1969. doi: 10.3390/cancers12071969
102. Li Z., Simin L., Jian K., Xin G., and Youlin K.. 4-1BB antibody enhances cytotoxic activity of natural killer cells against prostate cancer cells via NKG2D agonist combined with IL-27. Immunotherapy. (2022) 14:1043–53. doi: 10.2217/imt-2021-0232
103. Wang F., Wu L., Yin L., Shi H., Gu Y., Xing N., et al. Combined treatment with anti-PSMA CAR NK-92 cell and anti-PD-L1 monoclonal antibody enhances the antitumour efficacy against castration-resistant prostate cancer. Clin Trans Med. (2022) 12:e901. doi: 10.1002/ctm2.v12.6
104. Whiteland H., Spencer-Harty S., Morgan C., Kynaston H., Thomas D.H., Bose P., et al. A role for STEAP2 in prostate cancer progression. Clin Exp Metastasis. (2014) 31:909–20. doi: 10.1007/s10585-014-9679-9
105. Zanvit P., van Dyk D., Fazenbaker C., McGlinchey K., Luo W., Pezold J.M., et al. Antitumor activity of AZD0754, a dnTGF β RII-armored, STEAP2-targeted CAR-T cell therapy, in prostate cancer. J Clin Invest. (2023) 133:e169655.
106. Guo C., Figueiredo I., Gurel B., Neeb A., Seed G., Crespo M., et al. B7-H3 as a therapeutic target in advanced prostate cancer. Eur Urol. (2023) 83:224–38. doi: 10.1016/j.eururo.2022.09.004
107. Zhang Y., He L., Sadagopan A., Ma T., Dotti G., Wang Y., et al. Targeting radiation-resistant prostate cancer stem cells by B7-H3 CAR T cells. Mol Cancer Ther. (2021) 20:577–88. doi: 10.1158/1535-7163.MCT-20-0446
108. Chen W, Yuan Y, and Jiang X. Antibody and antibody fragments for cancer immunotherapy. J Controlled Release. (2020) 328:395–406. doi: 10.1016/j.jconrel.2020.08.021
109. Weiner LM, Murray JC, and Shuptrine CW. Antibody-based immunotherapy of cancer. Cell. (2012) 148:1081–4. doi: 10.1016/j.cell.2012.02.034
110. Bander N.H., Trabulsi E.J., Kostakoglu L., Yao D., Vallabhajosula S., Smith-Jones P., et al. Targeting metastatic prostate cancer with radiolabeled monoclonal antibody J591 to the extracellular domain of prostate specific membrane antigen. J Urol. (2003) 170:1717–21. doi: 10.1097/01.ju.0000091655.77601.0c
111. Nauseef JT, Bander NH, and Tagawa ST. Emerging prostate-specific membrane antigen-based therapeutics: small molecules, antibodies, and beyond. Eur Urol Focus. (2021) 7:254–7. doi: 10.1016/j.euf.2021.02.006
112. Tagawa S.T., Vallabhajosula S., Christos P.J., Jhanwar Y.S., Batra J.S., Lam L., et al. Phase 1/2 study of fractionated dose lutetium-177–labeled anti–prostate-specific membrane antigen monoclonal antibody J591 (177Lu-J591) for metastatic castration-resistant prostate cancer. Cancer. (2019) 125:2561–9. doi: 10.1002/cncr.32072
113. Nauseef J.T., Sun M.P., Thomas C., Bissassar M., Patel A., Tan A., et al. A phase I/II dose-escalation study of fractionated 225Ac-J591 for progressive metastatic castration-resistant prostate cancer (mCRPC) in patients with prior treatment with 177Lu-PSMA. Am Soc Clin Oncol. (2023) 41:1–14. doi: 10.1200/JCO.2023.41.6_suppl.TPS288
114. Milowsky M.I., Galsky M.D., Morris M.J., Crona D.J., George D.J., Dreicer R., et al. Phase 1/2 multiple ascending dose trial of the prostate-specific membrane antigen-targeted antibody drug conjugate MLN2704 in metastatic castration-resistant prostate cancer. Urol Oncol: Semin Orig Invest. (2016) 34:530.e15-530.e21. doi: 10.1016/j.urolonc.2016.07.005
115. Petrylak D.P., Kantoff P.W., Mega A.E., Vogelzang N.J., Stephenson J., Fleming M.T., et al. Prostate-specific membrane antigen antibody drug conjugate (PSMA ADC): A phase I trial in metastatic castration-resistant prostate cancer (mCRPC) previously treated with a taxane. Am Soc Clin Oncol. (2013) 31. doi: 10.1200/jco.2013.31.6_suppl.119
116. Hummel H.-D., Kufer P., Grüllich C., Seggewiss-Bernhardt R., Deschler-Baier B., Chatterjee M., et al. Pasotuxizumab, a BiTE® immune therapy for castration-resistant prostate cancer: Phase I, dose-escalation study findings. Immunotherapy. (2021) 13:125–41. doi: 10.2217/imt-2020-0256
117. Lim E.A., Schweizer M.T., Chi K.N., Aggarwal R., Agarwal N., Gulley J., et al. Phase 1 study of safety and preliminary clinical activity of JNJ-63898081, a PSMA and CD3 bispecific antibody, for metastatic castration-resistant prostate cancer. Clin Genitourinary Cancer. (2023) 21:366–75. doi: 10.1016/j.clgc.2023.02.010
118. Glud E.N., Rasmussen M., Zhang Y., Mandrup O.A., Salachan P.V., Borre M., et al. Identification of a high-risk immunogenic prostate cancer patient subset as candidates for T-cell engager immunotherapy and the introduction of a novel albumin-fused anti-CD3× anti-PSMA bispecific design. Br J Cancer. (2022) 127:2186–97. doi: 10.1038/s41416-022-01994-1
119. Dang K., Castello G., Clarke S.C., Li Y., Balasubramani A., Boudreau A., et al. Attenuating CD3 affinity in a PSMAxCD3 bispecific antibody enables killing of prostate tumor cells with reduced cytokine release. J Immunother Cancer. (2021) 9. doi: 10.1136/jitc-2021-002488
120. Gravdal K., Halvorsen O.J., Haukaas S.A., and Akslen L.A.. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin Cancer Res. (2007) 13:7003–11. doi: 10.1158/1078-0432.CCR-07-1263
121. Tanaka H., Kono E., Tran C.P., Miyazaki H., Yamashiro J., Shimomura T., et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med. (2010) 16:1414–20. doi: 10.1038/nm.2236
122. Chen M.-L., Yuan T.-T., Chuang C.-F., Huang Y.-T., Chung I.-C., Huang W.-C., et al. A novel enolase-1 antibody targets multiple interacting players in the tumor microenvironment of advanced prostate cancer. Mol Cancer Ther. (2022) 21:1337–47. doi: 10.1158/1535-7163.MCT-21-0285
123. Tison A., Garaud S., Chiche L., Cornec D., and Kostine M.. Immune-checkpoint inhibitor use in patients with cancer and pre-existing autoimmune diseases. Nat Rev Rheumatol. (2022) 18:641–56. doi: 10.1038/s41584-022-00841-0
124. Halbert B and Einstein DJ. Hot or not: tumor mutational burden (TMB) as a biomarker of immunotherapy response in genitourinary cancers. Urology. (2021) 147:119–26. doi: 10.1016/j.urology.2020.10.030
125. Marignol L., Coffey M., Lawler M., and Hollywood D.. Hypoxia in prostate cancer: a powerful shield against tumour destruction? Cancer Treat Rev. (2008) 34:313–27. doi: 10.1016/j.ctrv.2008.01.006
126. Chen F.-H., Chiang C.-S., Wang C.-C., Tsai C.-S., Jung S.-M., Lee C.-C., et al, et al. Radiotherapy decreases vascular density and causes hypoxia with macrophage aggregation in TRAMP-C1 prostate tumors. Clin Cancer Res. (2009) 15:1721–9. doi: 10.1158/1078-0432.CCR-08-1471
127. Jayaprakash P., Ai M., Liu A., Budhani P., Bartkowiak T., Sheng J., et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest. (2018) 128:5137–49. doi: 10.1172/JCI96268
128. Mortezaee K and Majidpoor J. The impact of hypoxia on immune state in cancer. Life Sci. (2021) 286:120057. doi: 10.1016/j.lfs.2021.120057
129. Lautert-Dutra W., Melo C.M., Chaves L.P., Crozier C., Saggioro F.P., dos Reis R.B., et al. Loss of heterozygosity impacts MHC expression on the immune microenvironment in CDK12-mutated prostate cancer. Mol Cytogenetics. (2024) 17:11. doi: 10.1186/s13039-024-00680-6
130. Deluce J.E., Cardenas L., Lalani A.-K., Maleki Vareki S., and Fernandes R.. Emerging biomarker-guided therapies in prostate cancer. Curr Oncol. (2022) 29:5054–76. doi: 10.3390/curroncol29070400
131. Olson P and Wagner J. Established and emerging liquid biomarkers for prostate cancer detection: A review. Urol Oncol: Semin Orig Invest. (2024) 43:3–14. doi: 10.1016/j.urolonc.2024.05.011
132. Zhong W., Wu K., Long Z., Zhou X., Zhong C., Wang S., et al. Gut dysbiosis promotes prostate cancer progression and docetaxel resistance via activating NF-κB-IL6-STAT3 axis. Microbiome. (2022) 10:94. doi: 10.1186/s40168-022-01289-w
133. Shyanti R.K., Greggs J., Malik S., and Mishra M.. Gut dysbiosis impacts the immune system and promotes prostate cancer. Immunol Lett. (2024) 268:106883. doi: 10.1016/j.imlet.2024.106883
134. Durvalumab N. Trends in clinical development for PD-1/PD-L1 inhibitors. Nat Rev Drug Discov. (2020) 19:163. doi: 10.1038/d41573-019-00182-w
135. Quezada S and Peggs K. Exploiting CTLA-4, PD-1 and PD-L1 to reactivate the host immune response against cancer. Br J Cancer. (2013) 108:1560–5. doi: 10.1038/bjc.2013.117
136. Gevensleben H., Dietrich D., Golletz C., Steiner S., Jung M., Thiesler T., et al. The immune checkpoint regulator PD-L1 is highly expressed in aggressive primary prostate cancer. Clin Cancer Res. (2016) 22:1969–77. doi: 10.1158/1078-0432.CCR-15-2042
137. Goltz D., Gevensleben H., Dietrich J., Ellinger J., Landsberg J., Kristiansen G., et al. Promoter methylation of the immune checkpoint receptor PD-1 (PDCD1) is an independent prognostic biomarker for biochemical recurrence-free survival in prostate cancer patients following radical prostatectomy. Oncoimmunology. (2016) 5:e1221555. doi: 10.1080/2162402X.2016.1221555
138. Gevensleben H., Holmes E.E., Goltz D., Dietrich J., Sailer V., Ellinger J., et al. PD-L1 promoter methylation is a prognostic biomarker for biochemical recurrence-free survival in prostate cancer patients following radical prostatectomy. Oncotarget. (2016) 7:79943. doi: 10.18632/oncotarget.13161
139. Palicelli A., Bonacini M., Croci S., Magi-Galluzzi C., Cañete-Portillo S., Chaux A., et al. What do we have to know about PD-L1 expression in prostate cancer? A systematic literature review. Part 1: Focus on immunohistochemical results with discussion of pre-analytical and interpretation variables. Cells. (2021) 10:3166. doi: 10.3390/cells10113166
140. Vicier C., Ravi P., Kwak L., Werner L., Huang Y., Evan C., et al. Association between CD8 and PD-L1 expression and outcomes after radical prostatectomy for localized prostate cancer. Prostate. (2021) 81:50–7. doi: 10.1002/pros.24079
141. Aditya G.A., Dany Y.A., Danarto R., Soeroharjo I., and Hendri A.Z.. PD-L1 overexpression in prostate cancer: a potential targeted therapy. Bali Med J. (2023) 12:2303–6. doi: 10.15562/bmj.v12i2.4506
142. Cooper W.A., Tran T., Vilain R.E., Madore J., Selinger C.I., Kohonen-Corish M., et al. PD-L1 expression is a favorable prognostic factor in early stage non-small cell carcinoma. Lung Cancer. (2015) 89:181–8. doi: 10.1016/j.lungcan.2015.05.007
143. He J., Yi M., Tan L., Huang J., and Huang L.. The immune checkpoint regulator PD-L1 expression are associated with clinical progression in prostate cancer. World J Surg Oncol. (2021) 19:1–8. doi: 10.1186/s12957-021-02325-z
144. Mo R.J., Han Z.D., Liang Y.K., Ye J.H., Wu S.L., Lin S.X., et al. Expression of PD-L1 in tumor-associated nerves correlates with reduced CD8+ tumor-associated lymphocytes and poor prognosis in prostate cancer. Int J Cancer. (2019) 144:3099–110. doi: 10.1002/ijc.v144.12
145. Li D., Zhou X., Xu W., Chen Y., Mu C., Zhao X., et al. Prostate cancer cells synergistically defend against CD8+ T cells by secreting exosomal PD-L1. Cancer Med. (2023) 12:16405–15. doi: 10.1002/cam4.v12.15
146. Lindh C., Kis L., Delahunt B., Samaratunga H., Yaxley J., Wiklund N.P., et al. PD-L1 expression and deficient mismatch repair in ductal adenocarcinoma of the prostate. Apmis. (2019) 127:554–60. doi: 10.1111/apm.2019.127.issue-8
147. Li Y., Huang Q., Zhou Y., He M., Chen J., Gao Y., et al. The clinicopathologic and prognostic significance of programmed cell death ligand 1 (PD-L1) expression in patients with prostate cancer: a systematic review and meta-analysis. Front Pharmacol. (2019) 9:1494. doi: 10.3389/fphar.2018.01494
148. Ott P.A., Elez E., Hiret S., Kim D.-W., Morosky A., Saraf S., et al. Pembrolizumab in patients with extensive-stage small-cell lung cancer: results from the phase Ib KEYNOTE-028 study. J Clin Oncol. (2017) 35:3823–9. doi: 10.1200/JCO.2017.72.5069
149. Antonarakis E.S., Piulats J.M., Gross-Goupil M., Goh J., Ojamaa K., Hoimes C.J., et al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: multicohort, open-label phase II KEYNOTE-199 study. J Clin Oncol. (2020) 38:395–405. doi: 10.1200/JCO.19.01638
150. Wang H., Sarikonda G., Puan K.-J., Tanaka Y., Feng J., Giner J.-L., et al. Indirect stimulation of human Vγ2Vδ2 T cells through alterations in isoprenoid metabolism. J Immunol. (2011) 187:5099–113. doi: 10.4049/jimmunol.1002697
151. Thompson K, Rojas-Navea J, and Rogers MJ. Alkylamines cause Vγ9Vδ2 T-cell activation and proliferation by inhibiting the mevalonate pathway. Blood. (2006) 107:651–4. doi: 10.1182/blood-2005-03-1025
152. Nada M.H., Wang H., Hussein A.J., Tanaka Y., and Morita C.T.. PD-1 checkpoint blockade enhances adoptive immunotherapy by human Vγ2Vδ2 T cells against human prostate cancer. Oncoimmunology. (2021) 10:1989789. doi: 10.1080/2162402X.2021.1989789
153. Chambers CA and Allison JP. Co-stimulation in T cell responses. Curr Opin Immunol. (1997) 9:396–404. doi: 10.1016/S0952-7915(97)80087-8
154. Alegre M-L, Frauwirth KA, and Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. (2001) 1:220–8. doi: 10.1038/35105024
155. Rudd CE, Taylor A, and Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. (2009) 229:12–26. doi: 10.1111/j.1600-065X.2009.00770.x
156. Kwon E.D., Foster B.A., Hurwitz A.A., Madias C., Allison J.P., Greenberg N.M., et al. Elimination of residual metastatic prostate cancer after surgery and adjunctive cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) blockade immunotherapy. Proc Natl Acad Sci. (1999) 96:15074–9. doi: 10.1073/pnas.96.26.15074
157. Di Giacomo AM, Biagioli M, and Maio M. The emerging toxicity profiles of anti–CTLA-4 antibodies across clinical indications. Semin Oncol. (2010) 37:499–507. doi: 10.1053/j.seminoncol.2010.09.007
158. Small E.J., Tchekmedyian N.S., Rini B.I., Fong L., Lowy I., Allison J.P., et al. A pilot trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory prostate cancer. Clin Cancer Res. (2007) 13:1810–5. doi: 10.1158/1078-0432.CCR-06-2318
159. Chen H., Liakou C.I., Kamat A., Pettaway C., Ward J.F., Tang D.N., et al. Anti-CTLA-4 therapy results in higher CD4+ ICOShi T cell frequency and IFN-γ levels in both nonmalignant and Malignant prostate tissues. Proc Natl Acad Sci. (2009) 106:2729–34. doi: 10.1073/pnas.0813175106
160. Sharma A., Subudhi S.K., Blando J., Scutti J., Vence L., Wargo J., et al. Anti-CTLA-4 immunotherapy does not deplete FOXP3+ regulatory T cells (Tregs) in human cancers. Clin Cancer Res. (2019) 25:1233–8. doi: 10.1158/1078-0432.CCR-18-0762
161. Khademi M., Illés Z., Gielen A.W., Marta M., Takazawa N., Baecher-Allan C., et al. T Cell Ig-and mucin-domain-containing molecule-3 (TIM-3) and TIM-1 molecules are differentially expressed on human Th1 and Th2 cells and in cerebrospinal fluid-derived mononuclear cells in multiple sclerosis. J Immunol. (2004) 172:7169–76. doi: 10.4049/jimmunol.172.11.7169
162. Hastings W.D., Anderson D.E., Kassam N., Koguchi K., Greenfield E.A., Kent S.C., et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. (2009) 39:2492–501. doi: 10.1002/eji.200939274
163. Gao X., Zhu Y., Li G., Huang H., Zhang G., Wang F., et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PloS One. (2012) 7:e30676. doi: 10.1371/journal.pone.0030676
164. Piao Y and Jin X. Analysis of Tim-3 as a therapeutic target in prostate cancer. Tumor Biol. (2017) 39:1010428317716628. doi: 10.1177/1010428317716628
165. Japp A.S., Kursunel M.A., Meier S., Mälzer J.N., Li X., Rahman N.A., et al. Dysfunction of PSA-specific CD8+ T cells in prostate cancer patients correlates with CD38 and Tim-3 expression. Cancer Immunol Immunother. (2015) 64:1487–94. doi: 10.1007/s00262-015-1752-y
166. Harding J.J., Moreno V., Bang Y.-J., Hong M.H., Patnaik A., Trigo J., et al. Blocking TIM-3 in treatment-refractory advanced solid tumors: a phase Ia/b study of LY3321367 with or without an anti-PD-L1 antibody. Clin Cancer Res. (2021) 27:2168–78. doi: 10.1158/1078-0432.CCR-20-4405
167. Huang C.-T., Workman C.J., Flies D., Pan X., Marson A.L., Zhou G., et al. Role of LAG-3 in regulatory T cells. Immunity. (2004) 21:503–13. doi: 10.1016/j.immuni.2004.08.010
168. Maruhashi T., Sugiura D., Okazaki I.-m., and Okazaki T.. LAG-3: from molecular functions to clinical applications. J Immunother Cancer. (2020) 8. doi: 10.1136/jitc-2020-001014
169. Aggarwal V, Workman CJ, and Vignali DA. LAG-3 as the third checkpoint inhibitor. Nat Immunol. (2023) 24:1415–22. doi: 10.1038/s41590-023-01569-z
170. Camisaschi C., Casati C., Rini F., Perego M., De Filippo A., Triebel F., et al. LAG-3 expression defines a subset of CD4+ CD25highFoxp3+ regulatory T cells that are expanded at tumor sites. J Immunol. (2010) 184:6545–51. doi: 10.4049/jimmunol.0903879
171. Shi A.-P., Tang X.-Y., Xiong Y.-L., Zheng K.-F., Liu Y.-J., Shi X.-G., et al. Immune checkpoint LAG3 and its ligand FGL1 in cancer. Front Immunol. (2022) 12:785091. doi: 10.3389/fimmu.2021.785091
172. Zhang X., Chen H., Han J., Wang Z., Guo Y., Zhou Z., et al. ATM-AMPKα mediated LAG-3 expression suppresses T cell function in prostate cancer. Cell Immunol. (2023) 393:104773. doi: 10.1016/j.cellimm.2023.104773
173. Tan D.S., Martin M., Ochoa de Olza Amat M., Sarantopoulos J., Carvajal R., Schoffski P., et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) ± anti-PD-1 spartalizumab (PDR001) in patients with advanced Malignancies. J Immunother Cancer. (2022) 10:e003776. doi: 10.1136/jitc-2021-003776
174. Harjunpää H and Guillerey C. TIGIT as an emerging immune checkpoint. Clin Exp Immunol. (2020) 200:108–19. doi: 10.1111/cei.13407
175. Dougall W.C., Kurtulus S., Smyth M.J., and Anderson A.C.. TIGIT and CD 96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. (2017) 276:112–20. doi: 10.1111/imr.2017.276.issue-1
176. Shibuya A and Shibuya K. DNAM-1 versus TIGIT: competitive roles in tumor immunity and inflammatory responses. Int Immunol. (2021) 33:687–92. doi: 10.1093/intimm/dxab085
177. Manieri NA, Chiang EY, and Grogan JL. TIGIT: a key inhibitor of the cancer immunity cycle. Trends Immunol. (2017) 38:20–8. doi: 10.1016/j.it.2016.10.002
178. Riquelme P., Haarer J., Kammler A., Walter L., Tomiuk S., Ahrens N., et al. TIGIT+ iTregs elicited by human regulatory macrophages control T cell immunity. Nat Commun. (2018) 9:2858. doi: 10.1038/s41467-018-05167-8
179. Preillon J., Cuende J., Rabolli V., Garnero L., Mercier M., Wald N., et al. Restoration of T-cell effector function, depletion of Tregs, and direct killing of tumor cells: the multiple mechanisms of action of a-TIGIT antagonist antibodies. Mol Cancer Ther. (2021) 20:121–31. doi: 10.1158/1535-7163.MCT-20-0464
180. Rousseau A, Parisi C, and Barlesi F. Anti-TIGIT therapies for solid tumors: a systematic review. ESMO Open. (2023) 8:101184. doi: 10.1016/j.esmoop.2023.101184
181. Papanicolau‐Sengos A., Yang Y., Pabla S., Lenzo F.L., Kato S., Kurzrock R., et al. Identification of targets for prostate cancer immunotherapy. Prostate. (2019) 79:498–505. doi: 10.1002/pros.23756
182. Wang F., Liu S., Liu F., Xu T., Ma J., Liang J., et al. TIGIT immune checkpoint blockade enhances immunity of human peripheral blood NK cells against castration-resistant prostate cancer. Cancer Lett. (2023) 568:216300. doi: 10.1016/j.canlet.2023.216300
183. Cai L., Li Y., Tan J., Xu L., and Li Y.. Targeting LAG-3, TIM-3, and TIGIT for cancer immunotherapy. J Hematol Oncol. (2023) 16:101. doi: 10.1186/s13045-023-01499-1
184. Dai T., Sun H., Liban T., Vicente-Suarez I., Zhang B., Song Y., et al. A novel anti-LAG-3/TIGIT bispecific antibody exhibits potent anti-tumor efficacy in mouse models as monotherapy or in combination with PD-1 antibody. Sci Rep. (2024) 14:10661. doi: 10.1038/s41598-024-61477-6
185. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
186. Topalian S.L., Taube J.M., Anders R.A., and Pardoll D.M.. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. (2016) 16:275–87. doi: 10.1038/nrc.2016.36
187. Yi KH and Chen L. Fine tuning the immune response through B7-H3 and B7-H4. Immunol Rev. (2009) 229:145–51. doi: 10.1111/j.1600-065X.2009.00768.x
188. Chen C and Li Y. Predictive value of co-expression patterns of immune checkpoint molecules for clinical outcomes of hematological Malignancies. Chin J Cancer Res. (2023) 35:245. doi: 10.1158/1538-7445.AM2023-LB245
189. Mollavelioglu B., Cetin Aktas E., Cabioglu N., Abbasov A., Onder S., Emiroglu S., et al. High co-expression of immune checkpoint receptors PD-1, CTLA-4, LAG-3, TIM-3, and TIGIT on tumor-infiltrating lymphocytes in early-stage breast cancer. World J Surg Oncol. (2022) 20:349. doi: 10.1186/s12957-022-02810-z
190. Roy D., Gilmour C., Patnaik S., and Wang L.L.. Combinatorial blockade for cancer immunotherapy: targeting emerging immune checkpoint receptors. Front Immunol. (2023) 14:1264327. doi: 10.3389/fimmu.2023.1264327
191. Sharma P., Pachynski R.K., Narayan V., Fléchon A., Gravis G., Galsky M.D., et al. Nivolumab plus ipilimumab for metastatic castration-resistant prostate cancer: preliminary analysis of patients in the CheckMate 650 trial. Cancer Cell. (2020) 38:489–499. e3. doi: 10.1016/j.ccell.2020.08.007
192. Pienta KJ. Preclinical mechanisms of action of docetaxel and docetaxel combinations in prostate cancer. Semin Oncol. (2001) 28:3–7. doi: 10.1016/S0093-7754(01)90148-4
193. Yu E.Y., Joshua A.M., Shore N.D., Kramer G., Hu H., Poehlein C.H., et al. Phase 1b/2 study of pembrolizumab plus belzutifan and belzutifan alone in patients with docetaxel-treated metastatic castration-resistant prostate cancer (mCRPC): KEYNOTE-365 cohort J. Am Soc Clin Oncol. (2024) 42 . doi: 10.1200/JCO.2024.42.4_suppl.TPS250
194. Evan Y.Y., Kolinsky M.P., Berry W.R., Retz M., Mourey L., Piulats J.M., et al. Pembrolizumab plus docetaxel and prednisone in patients with metastatic castration-resistant prostate cancer: long-term results from the phase 1b/2 KEYNOTE-365 cohort B study. Eur Urol. (2022) 82:22–30. doi: 10.1016/j.eururo.2022.02.023
195. Stein M.N., Dorff T.B., Goodman O.B., Thomas R.A., Silverman M.H., Guo M., et al. A phase 2, multicenter, parallel-group, open-label study of vudalimab (XmAb20717), a PD-1 x CTLA-4 bispecific antibody, alone or in combination with chemotherapy or targeted therapy in patients with molecularly defined subtypes of metastatic castration-resistant prostate cancer. Am Soc Clin Oncol. (2022) 40. doi: 10.1200/JCO.2022.40.16_suppl.TPS5097
196. Subudhi S.K., Siddiqui B.A., Aparicio A.M., Yadav S.S., Basu S., Chen H., et al. Combined CTLA-4 and PD-L1 blockade in patients with chemotherapy-naïve metastatic castration-resistant prostate cancer is associated with increased myeloid and neutrophil immune subsets in the bone microenvironment. J Immunother Cancer. (2021) 9. doi: 10.1136/jitc-2021-002919
197. Zaorsky N.G., Harrison A.S., Trabulsi E.J., Gomella L.G., Showalter T.N., Hurwitz M.D., et al. Evolution of advanced technologies in prostate cancer radiotherapy. Nat Rev Urol. (2013) 10:565–79. doi: 10.1038/nrurol.2013.185
198. Lynch C, Pitroda SP, and Weichselbaum RR. Radiotherapy, immunity, and immune checkpoint inhibitors. Lancet Oncol. (2024) 25:e352–62. doi: 10.1016/S1470-2045(24)00075-5
199. Morris M.J., Fong L., Petrylak D.P., Sartor A.O., Higano C.S., Pagliaro L.C., et al. Safety and clinical activity of atezolizumab (atezo)+ radium-223 dichloride (r-223) in 2L metastatic castration-resistant prostate cancer (mCRPC): Results from a phase Ib clinical trial. Am Soc Clin Oncol. (2020). doi: 10.1200/JCO.2020.38.15_suppl.5565
200. Siva S., MacManus M.P., Martin R.F., and Martin O.A.. Abscopal effects of radiation therapy: a clinical review for the radiobiologist. Cancer Lett. (2015) 356:82–90. doi: 10.1016/j.canlet.2013.09.018
201. Dewan M.Z., Galloway A.E., Kawashima N., Dewyngaert J.K., Babb J.S., Formenti S.C., et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti–CTLA-4 antibody. Clin Cancer Res. (2009) 15:5379–88. doi: 10.1158/1078-0432.CCR-09-0265
202. Kwon E.D., Drake C.G., Scher H.I., Fizazi K., Bossi A., Van den Eertwegh A.J., et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. (2014) 15:700–12. doi: 10.1016/S1470-2045(14)70189-5
203. Potluri H.K., Ferreira C.A., Grudzinski J., Massey C., Aluicio-Sarduy E., Engle J.W., et al. Antitumor efficacy of 90Y-NM600 targeted radionuclide therapy and PD-1 blockade is limited by regulatory T cells in murine prostate tumors. J Immunother Cancer. (2022) 10:1–14. doi: 10.1136/jitc-2022-005060
204. Eximond M, Wang J, and Kirschner A. Dual immune checkpoint therapy combined with radiotherapy treats castration-resistant prostate cancer. Int J Radiat Oncol Biol Phys. (2023) 117:e229. doi: 10.1016/j.ijrobp.2023.06.1141
205. Tang S., Moore M.L., Grayson J.M., and Dubey P.. Increased CD8+ T-cell function following castration and immunization is countered by parallel expansion of regulatory T cells. Cancer Res. (2012) 72:1975–85. doi: 10.1158/0008-5472.CAN-11-2499
206. Hawley J.E., Obradovic A.Z., Dallos M.C., Lim E.A., Runcie K., Ager C.R., et al. Anti-PD-1 immunotherapy with androgen deprivation therapy induces robust immune infiltration in metastatic castration-sensitive prostate cancer. Cancer Cell. (2023) 41:1972–1988. e5. doi: 10.1016/j.ccell.2023.10.006
207. Frandsen S.K., Gissel H., Hojman P., Tramm T., Eriksen J., Gehl J., et al. Direct therapeutic applications of calcium electroporation to effectively induce tumor necrosis. Cancer Res. (2012) 72:1336–41. doi: 10.1158/0008-5472.CAN-11-3782
208. Burbach B.J., S.D., Shao Q., Young K.M., Slaughter J.R., Rollins M.R., et al. Irreversible electroporation augments checkpoint immunotherapy in prostate cancer and promotes tumor antigen-specific tissue-resident memory CD8+ T cells. Nat Commun. (2021) 12:3862. doi: 10.1038/s41467-021-24132-6
209. Rébé C and Ghiringhelli F. STAT3, a master regulator of anti-tumor immune response. Cancers. (2019) 11:1280. doi: 10.3390/cancers11091280
210. Johnson DE, O’Keefe RA, and Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. (2018) 15:234–48. doi: 10.1038/nrclinonc.2018.8
211. Witt K., Evans-Axelsson S., Lundqvist A., Johansson M., Bjartell A., Hellsten R., et al. Inhibition of STAT3 augments antitumor efficacy of anti-CTLA-4 treatment against prostate cancer. Cancer Immunol Immunother. (2021) 70:1–12. doi: 10.1007/s00262-021-02915-6
212. Zhang Y., Wei Y., Jiang S., Dang Y., Yang Y., Zuo W., et al. Traditional Chinese medicine CFF-1 exerts a potent anti-tumor immunity to hinder tumor growth and metastasis in prostate cancer through EGFR/JAK1/STAT3 pathway to inhibit PD-1/PD-L1 checkpoint signaling. Phytomedicine. (2022) 99:153939. doi: 10.1016/j.phymed.2022.153939
213. McNeel D.G., Eickhoff J.C., Wargowski E., Johnson L.E., Kyriakopoulos C.E., Emamekhoo H., et al. Phase 2 trial of T-cell activation using MVI-816 and pembrolizumab in patients with metastatic, castration-resistant prostate cancer (mCRPC). J Immunother Cancer. (2022) 10:1–12. doi: 10.1136/jitc-2021-004198
214. Ross A.E., Hurley P.J., Tran P.T., Rowe S.P., Benzon B., Neal T.O., et al. A pilot trial of pembrolizumab plus prostatic cryotherapy for men with newly diagnosed oligometastatic hormone-sensitive prostate cancer. Prostate Cancer Prostatic Dis. (2020) 23:184–93. doi: 10.1038/s41391-019-0176-8
215. Wang Q, Fang Y., Li C., Leong T. L., Provencio M., Oh I-J., et al. Differential organ-specific tumor response to first-line immune checkpoint inhibitor therapy in non-small cell lung cancer—a retrospective cohort study. Trans Lung Cancer Res. (2023) 12:312. doi: 10.21037/tlcr-23-83
216. Wright JJ, Powers AC, and Johnson DB. Endocrine toxicities of immune checkpoint inhibitors. Nat Rev Endocrinol. (2021) 17:389–99. doi: 10.1038/s41574-021-00484-3
217. Al Ashi S.I., Thapa B., Flores M., Ahmed R., Rahim S.E.G., Amir M., et al. Endocrine toxicity and outcomes in patients with metastatic Malignancies treated with immune checkpoint inhibitors. J Endocr Soc. (2021) 5:bvab100. doi: 10.1210/jendso/bvab100
218. Kotwal A., Gustafson M.P., Bornschlegl S., Kottschade L., Delivanis D.A., Dietz A.B., et al. Immune checkpoint inhibitor-induced thyroiditis is associated with increased intrathyroidal T lymphocyte subpopulations. Thyroid. (2020) 30:1440–50. doi: 10.1089/thy.2020.0075
219. Tong J., Kartolo A., Yeung C., Hopman W., and Baetz T.. Long-term toxicities of immune checkpoint inhibitor (ICI) in melanoma patients. Curr Oncol. (2022) 29:7953–63. doi: 10.3390/curroncol29100629
220. Johnson D.B., Nebhan C.A., Moslehi J.J., and Balko J.M.. Immune-checkpoint inhibitors: long-term implications of toxicity. Nat Rev Clin Oncol. (2022) 19:254–67. doi: 10.1038/s41571-022-00600-w
221. Blum SM, Rouhani SJ, and Sullivan RJ. Effects of Immune-related adverse events (irAEs) and their treatment on antitumor immune responses. Immunol Rev. (2023) 318:167–78. doi: 10.1111/imr.v318.1
222. Kumar V., Chaudhary N., Garg M., Floudas C.S., Soni P., Chandra A.B., et al. Current diagnosis and management of immune related adverse events (irAEs) induced by immune checkpoint inhibitor therapy. Front Pharmacol. (2017) 8:49. doi: 10.3389/fphar.2017.00049
223. Davies M and Duffield EA. Safety of checkpoint inhibitors for cancer treatment: strategies for patient monitoring and management of immune-mediated adverse events. ImmunoTargets Ther. (2017) 6:51–71. doi: 10.2147/ITT.S141577
224. de Almeida D.V.P., Anderson J.M., Danila D.C., Morris M.J., Slovin S.F., Abida W., et al. Evaluating immune-related adverse events using PRO-CTCAE in a phase II study of ipilimumab for hormone-sensitive prostate cancer. J Immunother Precis Oncol. (2023) 6:162–9. doi: 10.36401/JIPO-23-9
225. Hu M., Lin X., Sun T., Shao X., Huang X., Du W., et al. Gut microbiome for predicting immune checkpoint blockade-associated adverse events. Genome Med. (2024) 16:16. doi: 10.1186/s13073-024-01285-9
226. Tan B., Liu Y.x., Tang H., Chen D., Xu Y., Chen M.j., et al. Gut microbiota shed new light on the management of immune-related adverse events. Thorac Cancer. (2022) 13:2681–91. doi: 10.1111/1759-7714.14626
227. Les I., Martinez M., Perez-Francisco I., Cabero M., Teijeira L., Arrazubi V., et al. Predictive biomarkers for checkpoint inhibitor immune-related adverse events. Cancers. (2023) 15:1629. doi: 10.3390/cancers15051629
228. Hamada K., Isobe J., Hattori K., Hosonuma M., Baba Y., Murayama M., et al. Turicibacter and Acidaminococcus predict immune-related adverse events and efficacy of immune checkpoint inhibitor. Front Immunol. (2023) 14:1164724. doi: 10.3389/fimmu.2023.1164724
229. Longo V., Brunetti O., Azzariti A., Galetta D., Nardulli P., Leonetti F., et al. Strategies to improve cancer immune checkpoint inhibitors efficacy, other than abscopal effect: A systematic review. Cancers. (2019) 11:539. doi: 10.3390/cancers11040539
230. Fitzsimmons T.S., Singh N., Walker T.D., Newton C., Evans D.G., Crosbie E.J., et al. Immune checkpoint inhibitors efficacy across solid cancers and the utility of PD-L1 as a biomarker of response: a systematic review and meta-analysis. Front Med. (2023) 10:1192762. doi: 10.3389/fmed.2023.1192762
231. Wu C., Ke Y., Wan L., and Xie X.. Efficacy of immune checkpoint inhibitors differs in various status of carcinoma: a study based on 29 cohorts with 3255 participants. Cancer Immunol Immunother. (2024) 73:79. doi: 10.1007/s00262-024-03663-z
232. Yoo S.-K., Fitzgerald C.W., Cho B.A., Fitzgerald B.G., Han C., Koh E.S., et al. Prediction of checkpoint inhibitor immunotherapy efficacy for cancer using routine blood tests and clinical data. Nat Med. (2025), 1–12. doi: 10.1038/s41591-024-03398-5
233. Li M, Kaili D, and Shi L. Biomarkers for response to immune checkpoint inhibitors in gastrointestinal cancers. World J Gastrointest Oncol. (2022) 14:19. doi: 10.4251/wjgo.v14.i1.19
234. Berthold D.R., Pond G.R., Soban F., De Wit R., Eisenberger M., Tannock I.F., et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. J Clin Oncol. (2008) 26:242–5. doi: 10.1200/JCO.2007.12.4008
235. Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A., et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
236. Boevé L.M., Hulshof M.C., Vis A.N., Zwinderman A.H., Twisk J.W., Witjes W.P., et al. Effect on survival of androgen deprivation therapy alone compared to androgen deprivation therapy combined with concurrent radiation therapy to the prostate in patients with primary bone metastatic prostate cancer in a prospective randomised clinical trial: data from the HORRAD trial. Eur Urol. (2019) 75:410–8. doi: 10.1016/j.eururo.2018.09.008
237. Scher H.I., Fizazi K., Saad F., Taplin M.-E., Sternberg C.N., Miller K., et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. New Engl J Med. (2012) 367:1187–97. doi: 10.1056/NEJMoa1207506
238. Ryan C.J., Smith M.R., Fizazi K., Saad F., Mulders P.F., Sternberg C.N., et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. (2015) 16:152–60. doi: 10.1016/S1470-2045(14)71205-7
239. Mateo J., de Bono J.S., Fizazi K., Saad F., Shore N., Sandhu S., et al. Olaparib for the treatment of patients with metastatic castration-resistant prostate cancer and alterations in BRCA1 and/or BRCA2 in the PROfound Trial. J Clin Oncol. (2024) 42:571–83. doi: 10.1200/JCO.23.00339
240. Saad F., Clarke N. W., Oya M., Shore N., Procopio G., and Guedes J. D. Olaparib plus abiraterone versus placebo plus abiraterone in metastatic castration-resistant prostate cancer (PROpel): final prespecified overall survival results of a randomised, double-blind, phase 3 trial. Lancet Oncol. (2023) 24:1094–108. doi: 10.1016/S1470-2045(23)00382-0
241. Crabb S.J., Griffiths G., Marwood E., Dunkley D., Downs N., Martin K., et al. Pan-AKT inhibitor capivasertib with docetaxel and prednisolone in metastatic castration-resistant prostate cancer: a randomized, placebo-controlled phase II trial (ProCAID). J Clin Oncol. (2021) 39:190–201. doi: 10.1200/JCO.20.01576
242. Sweeney C., Bracarda S., Sternberg C.N., Chi K.N., Olmos D., Sandhu S., et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. Lancet. (2021) 398:131–42. doi: 10.1016/S0140-6736(21)00580-8
Keywords: prostate cancer, immunotherapy, immune checkpoint inhibitors, treatment, combined therapy
Citation: Lu H, Teng Z, Wang J and Zhang W (2025) Prostate cancer immunotherapy-based strategies: an updated review emphasizing immune checkpoint inhibitors. Front. Immunol. 16:1583363. doi: 10.3389/fimmu.2025.1583363
Received: 25 February 2025; Accepted: 20 May 2025;
Published: 18 June 2025.
Edited by:
Feifei Sun, Shandong University, ChinaReviewed by:
Guanwen Zhou, Shandong University, ChinaLin Zhang, Binzhou Center for Disease Control and Prevention, China
Copyright © 2025 Lu, Teng, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenchao Zhang, endlbmNoYW8wMTA4QGdtYWlsLmNvbQ==