 Qiu-yuan Xiang1
Qiu-yuan Xiang1 Ji-Hao Zhou
Ji-Hao Zhou- 1Department of Hematology, The Second Clinical Medical College of Jinan University, Shenzhen, China
- 2Department of Pathology, Shenzhen People’s Hospital (The Second Clinical Medical College of Jinan University; The First Affiliated Hospital of Southern University of Science and Technology), Shenzhen, China
- 3Department of Hematology, Shenzhen People’s Hospital (The Second Clinical Medical College of Jinan University; The First Affiliated Hospital of Southern University of Science and Technology), Shenzhen, China
Activated phosphoinositide 3-kinase δ syndrome (APDS), an inborn error of immunity associated with gain-of-function mutations in the PIK3CD gene, is characterized by dysregulated PI3Kδ signaling. The clinical spectrum commonly includes recurrent respiratory infections and lymphoproliferative manifestations. We present an adolescent male with APDS1 manifesting recurrent sinopulmonary infections, generalized lymphadenopathy, hepatosplenomegaly, gastrointestinal manifestations, and combined T-cell/B-cell lymphopenia, complicated by Epstein-Barr virus-positive diffuse large B-cell lymphoma (EBV+ DLBCL). Whole-exome sequencing identified a heterozygous PIK3CD variant (c.3061G>A p.Glu1021Lys), supporting the molecular diagnosis of APDS1. This case adds to emerging evidence linking APDS1 with EBV-driven lymphomagenesis, thereby further supporting the critical role of PI3K δ pathway dysregulation in promoting EBV-associated lymphoid malignancies.
Introduction
Activated phosphoinositide 3-kinase δ syndrome (APDS) is an autosomal dominant primary combined immunodeficiency disorder characterized by mutations in defined genes. APDS is classified into two subtypes: APDS1, caused by gain-of-function variants in the PIK3CD gene, and APDS2, associated with loss-of-function variants in the PIK3R1 gene (1). These genetic alterations lead to constitutive activation of the PI3K-AKT-mTOR signaling pathway, contributing to lymphoproliferative manifestations and combined immunodeficiency (2, 3). APDS1 specifically involves pathogenic variants in the PI3Kδ catalytic subunit encoded by PIK3CD. These variants promote enhanced PI3Kδ activity, elevating phosphatidylinositol (3,4,5)-trisphosphate (PIP3) levels and facilitating downstream AKT activation in lymphocytes (4). This signaling dysregulation is associated with functional deficits in T and B cell populations, correlating with compromised antibacterial immunity. To date, whole-exome sequencing studies have identified 11 recurrent PIK3CD variants linked to APDS1 (E1021K, E1025G, R929C, E81K, G124D, E525K, E525A, Y524N, N334K, R405C, C416R) (1, 3, 5–11). with the E1021K substitution (c.3061G>A) accounting for approximately 85% of documented cases (12). APDS demonstrates notable clinical heterogeneity, spanning from asymptomatic presentations to severe combined immunodeficiency. The clinical spectrum typically includes enhanced susceptibility to bacterial and viral pathogens, prominent lymphoproliferative manifestations, and predisposition to malignancy development. Autoimmune phenomena may coexist in a subset of cases (4, 10, 13). Recurrent infections represent the predominant clinical presentation, with the majority of patients developing early-onset and severe respiratory tract infections including sinusitis, pharyngitis, tonsillitis, pneumonia, and empyema (14). Persistent herpes viral infections are particularly noteworthy, with Epstein-Barr virus (EBV) (28%) and cytomegalovirus (CMV) (14.7%) infections being frequently reported in case series (15). Approximately three-quarters of APDS1 patients develop benign lymphoproliferation, manifesting as chronic/recurrent generalized lymphadenopathy accompanied by hepatosplenomegaly. Concurrent gastrointestinal mucosal lymphoid hyperplasia commonly leads to chronic diarrhea and/or hematochezia (13, 16). Patients with APDS commonly develop autoimmune complications emerging after the first decade of life, which typically manifest as cytopenia and glomerulonephritis (17, 18). The immunological profile of APDS is characterized by combined T- and B-cell lymphopenia, hypogammaglobulinemia affecting IgA with concomitant normal or elevated IgM levels, and selective IgG subclass deficiencies (19, 20). In this report, we describe a genetically confirmed APDS1 patient presenting with EBV-positive diffuse large B cell lymphoma (EBV+ DLBCL), highlighting this rare complication in the setting of congenital immunodeficiency.
Case presentation
A 23-year-old male presented with a six-year history of stable mild (≤1 cm) cervical lymphadenopathy first noted in 2015. Progressive nodal enlargement (1–2 cm) in December, 2021 prompted referral to our clinic. The patient’s medical history revealed childhood-onset bronchiectasis complicated by recurrent sinopulmonary infections, suggestive of underlying immunodeficiency.
The initial physical examination revealed marked craniofacial abnormalities, including mandibular prognathism with anterior crossbite and a disproportional facial structure, combined with concurrent underdevelopment of the vocal cords. Additionally, mobile submandibular lymphadenopathy was palpable bilaterally (1–2 cm in size), and splenomegaly was present, extending 4 cm below the left costal margin. Bilateral pulmonary crackles were also detected. Laboratory investigations revealed leukopenia (2.3×109/L), hypogammaglobulinemia (IgA: 0.27 g/L), and inverted CD4+/CD8+ ratio (0.20). Elevated β2-microglobulin (3.5 mg/L) and detectable whole blood EBV-DNA (6.62×10² copies/mL) were noted, though plasma EBV load remained negative. PET/CT imaging revealed multifocal hypermetabolic lymphadenopathy with SUVmax values ranging from 5.8 to 12.4, involving the cervical, axillary, retroperitoneal, and iliac lymph node stations. Concurrent findings included splenomegaly (16.3 cm) and consolidation in the right middle lobe. (Figure 1A).
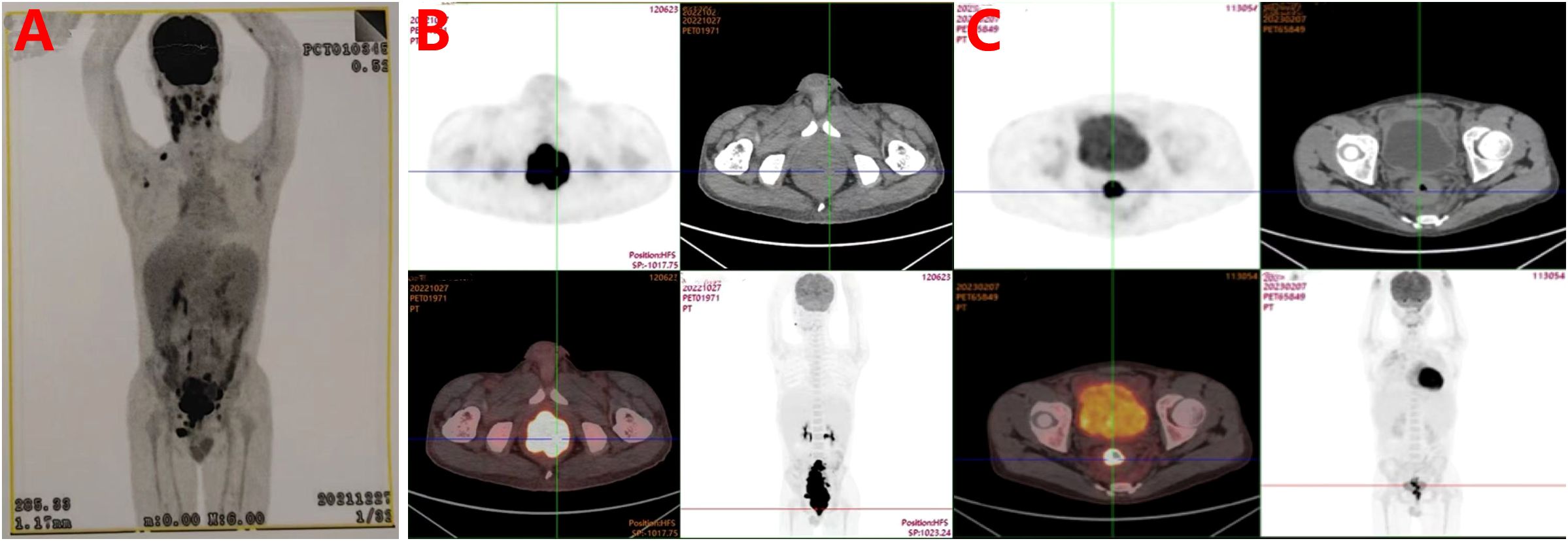
Figure 1. (A) Baseline whole-body PET-CT in December, 2021 revealed multistation hypermetabolic lymphadenopathy and rectal/ileocecal hypermetabolic lesions suggestive of lymphoma, with hepatosplenomegaly indicating reactive hyperplasia. Delayed rectal SUV retention and mild ileocecal tracer activity warrant clinicopathological correlation. (B) Diagnostic PET-CT in September, 2022 demonstrated a hypermetabolic sigmoidorectal mass (SUVmax=27.2) with disseminated lymphadenopathy meeting lymphoma criteria, accompanied by hepatosplenomegaly, sinusitis, and right lung collapse/bronchiectasis (our institution). (C) Interim PET-CT after four cycles of R-CHOPE demonstrated partial response with residual sigmoidorectal hypermetabolism (Deauville 5) and reduced nodal burden (size/number), though persistent nodal metabolic activity (Deauville 4) indicates incomplete remission.
Clinical progression occurred in September 2022 with development of daily high-volume diarrhea (10×/day), abdominal pain, and respiratory symptoms including productive cough with purulent sputum. Emergent evaluation revealed cachexia with characteristic facial dysmorphism (low nasal bridge, mandibular prognathism with anterior crossbite, a disproportional facial structure), persistent lymphadenopathy (largest 2×2 cm right cervical node), and left lower quadrant tenderness, Anthropometric parameters confirmed growth retardation (height 168 cm; weight 63.2 kg). Laboratory studies confirmed iron-deficiency anemia (positive fecal occult blood) with concurrent hypoalbuminemia and hypocalcemia. Marked inflammatory activation was evidenced by elevated CRP (65.52 mg/L), IL-6 (36.13 pg/ml), and procalcitonin (0.08 ng/ml). Lymphocyte subset profiling demonstrated a marked predominance of CD3+CD8+ suppressor/cytotoxic T cells (87.30%) alongside depleted CD3-CD19+ B cell populations (3.10%), with corresponding Th/Ts ratio elevation to 69.60%. The analysis further revealed diminished CD3+CD4+ helper/inducer T cell proportions (7.40%) coexisting with attenuated CD3-CD16+CD56+ NK cell representation (0.20%), while CD3+ T cells constituted 13.70% of the total lymphocyte pool, including a 3.1% subset of CD4+CD25+CD127- regulatory T cells.
Serial imaging revealed progressive pulmonary infiltrates with right middle lobe atelectasis, radiologically consistent with infectious etiology. In contrast, the newly identified colorectal wall thickening demonstrating intense FDG avidity (SUVmax 8.6) was characterized as suspicious for metastatic lymphoma involvement (Figure 1B). Histopathological evaluation of rectal biopsies revealed diffuse infiltration by atypical large lymphoid cells characterized by vesicular chromatin, prominent nucleoli, and frequent mitoses (Ki-67 index >70%). Immunophenotypic analysis demonstrated positivity for B-cell markers (CD20, CD79a, PAX-5).
EBV association was confirmed by EBER in situ hybridization (Figure 2). These findings established the diagnosis of EBV+ DLBCL. Whole-exome sequencing identified a pathogenic variant in the PIK3CD gene (c.3061G>A, p.Glu1021Lys), establishing a molecular diagnosis of APDS1. The treatment protocol commenced with dexamethasone bridging followed by a CHOP regimen (cyclophosphamide, doxorubicin, vincristine, prednisone) combined with etoposide, transitioning to rituximab-enhanced etoposide-modified CHOP (R-CHOPE: rituximab, etoposide, cyclophosphamide, pirarubicin, vindesine, dexamethasone) at cycle 2, which achieved transient EBV-DNA clearance (<4×10² copies/mL) in whole blood. Interim Positron Emission Tomography-Computed Tomography (PET-CT) at cycle 4 demonstrated partial metabolic response (Deauville 5-point scale score 3; Figure 1C). After the completion of 4 cycles of R-Chop chemotherapy, treatment was suspended for one cycle due to SARS-COV-2 infection. During this interim period, antiviral treatment was administered with Nirmatrelvir/Ritonavir (150 mg/100 mg twice daily). Considering the treatment-related myelosuppression following multi-agent chemotherapy, the dosing regimen of the PI3Kδ inhibitor was adjusted to Duvelisib (25 mg every 12 hours, for 14 days per month).
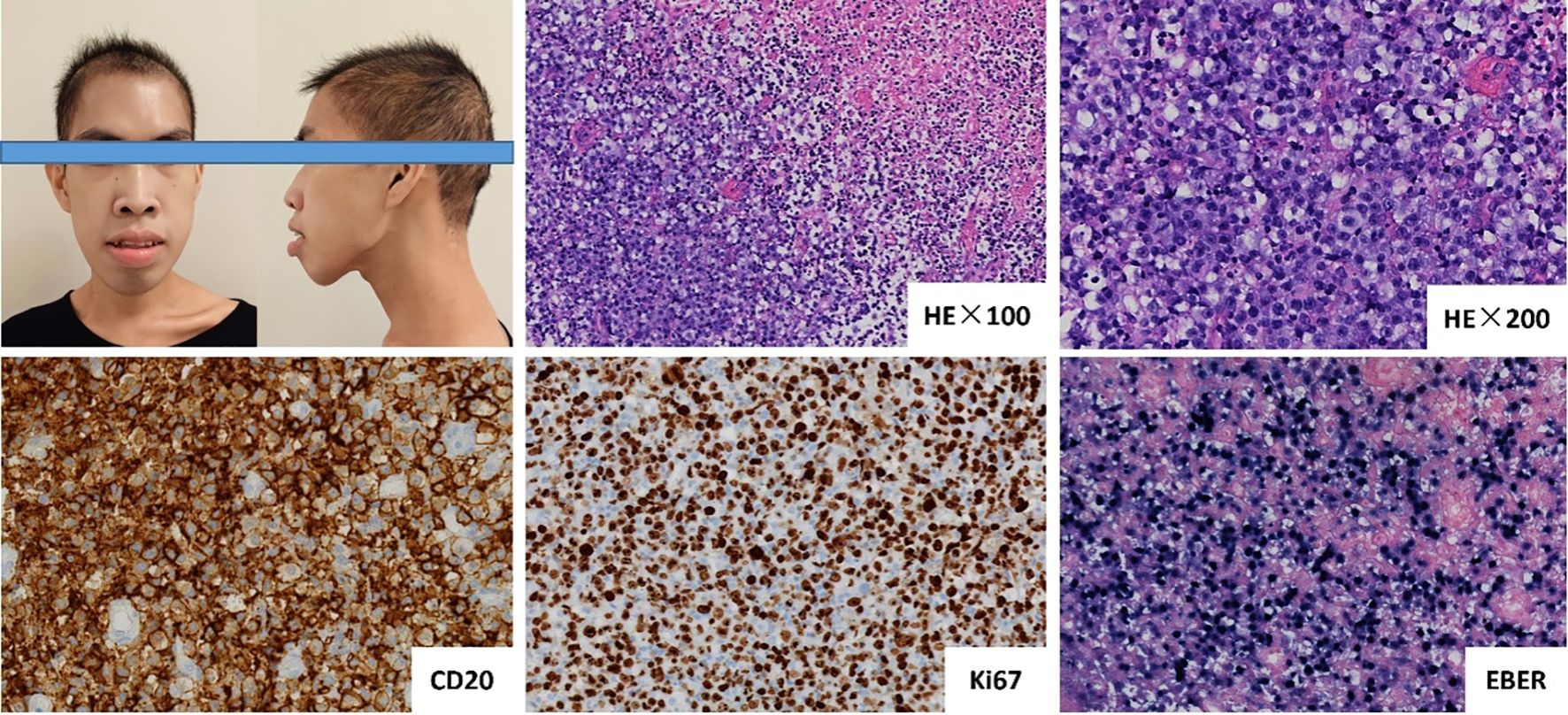
Figure 2. The patient exhibited distinct facial characteristics, including prominent mandible, and disharmonious facial proportions. Histopathological examination of rectal biopsies revealed diffuse infiltration by atypical large lymphoid cells, characterized by vesicular chromatin, prominent nucleoli, and frequent mitoses (Ki-67 index >70%). Immunophenotypic analysis demonstrated positivity for CD20, and Epstein-Barr virus (EBV) association was confirmed by EBER in situ hybridization.
Augmented R-CHOPE therapy incorporating programmed death-1 (PD-1) inhibitor (sintilimab, 200 mg every 21 days) from cycle 5 ultimately induced complete metabolic remission by cycle 8 after a one-month treatment interval, Post-treatment evaluation following duvelisib-combined chemotherapy demonstrated progressive nodal regression to near-normal dimensions on serial imaging, with concurrent sintilimab maintenance therapy exhibiting a favorable safety profile devoid of clinically significant adverse events. Post-chemotherapy lymphocyte subset analysis revealed complete B-cell depletion (CD3-CD19 + 0.0%), accompanied by a Th/Ts ratio of 0.67 (CD3+CD4 + 35.30%; CD3+CD8 + 52.90%), elevated CD3+ T-cell predominance (91.9%), CD3-CD16+CD56+ NK cells at 6.6%, and regulatory T-cell subsets (CD4+CD25+CD127-) accounting for 5.8%. Longitudinal imaging surveillance identified persistent lymphadenopathy and splenomegaly; however, PET-CT revealed no hypermetabolic activity, radiologically consistent with effective suppression of residual lymphoma lesions following therapeutic intervention. The patient declined allogeneic hematopoietic stem cell transplantation (allo-HSCT) and is currently under close surveillance, remaining in complete remission (CR) without EBV reactivation for over one year.
Literature review
We report a male adolescent with APDS1 presenting with lymphadenopathy, hepatosplenomegaly, bronchiectasis complicated by recurrent pulmonary infections and severe gastrointestinal symptoms. Whole-exome sequencing revealed a pathogenic PIK3CD variant (E1021K, c.3061G>A). Although parental genetic testing was unavailable, the clinical presentation strongly supports congenital pathogenesis. The immunophenotype demonstrated helper T-cell and B-cell lymphopenia with hypogammaglobulinemia (reduced IgA), contrasting with classic APDS features of hyper-IgM, IgG and IgM levels remained within normal ranges. Notably, the patient developed severe gastrointestinal manifestations, with histologically confirmed EBV+ DLBCL identified via rectal biopsy. This case illustrates an uncommon presentation of gastrointestinal EBV-positive DLBCL in APDS1. The co-occurrence of this lymphoproliferative complication with characteristic immune dysregulation emphasizes the diagnostic challenges associated with APDS phenotypic variability, particularly when accompanied by atypical immunological features.
Regarding clinical distinctions between subtypes, studies indicate a significantly higher prevalence of autoimmune comorbidities in APDS2 compared to APDS1 (12, 42). APDS2 is more characteristically associated with a phenotypic triad comprising growth retardation, mild cognitive impairment, and facial dysmorphism (e.g., hypertelorism, depressed nasal bridge, retrognathia) (43, 44). Notably, our APDS1 patient manifested overlapping features classically linked to APDS2, including craniofacial abnormalities (depressed nasal bridge, mandibular prognathism with anterior crossbite, facial disproportionality), laryngeal hypoplasia, and growth delay. Although no intellectual disability was clinically apparent, the lack of formal neuropsychological evaluation introduces uncertainty regarding subtle cognitive deficits, highlighting the importance of comprehensive phenotyping in APDS diagnostics.
EBV, an oncogenic gammaherpes virus with near-ubiquitous distribution, demonstrates seroprevalence exceeding 90% in adult populations worldwide (21). In immunocompetent hosts, primary infection typically occurs asymptomatically during childhood or manifests as self-limiting infectious mononucleosis (IM) in adolescents, classically presenting with fever, pharyngitis, and lymphadenopathy. Following initial exposure, EBV establishes lifelong latency through sophisticated immune evasion strategies. However, immunocompromised individuals or those with genetic predispositions may develop persistent IM-like symptoms accompanied by sustained viremia (>104 EBV DNA copies/mL in peripheral blood), a clinical entity formally designated chronic active EBV infection (CAEBV) (22, 23). This case report delineates an atypical disease progression initially presenting with isolated lymphadenopathy that evolved into a complex syndrome featuring recurrent fever (>38.5°C), unintentional weight loss (>10% body mass index), multiple gastrointestinal ulcerations, and persistent high-grade viremia (EBV DNA load >105 copies/mL). The clinical trajectory ultimately culminated in EBV-driven lymphomagenesis, specifically DLBCL confirmed through histopathological examination.
The oncogenic potential of EBV stems from its complex interplay with host cell machinery. Post-infection, the virus establishes latency primarily in memory B-cells, retaining capacity for periodic reactivation through latent-lytic cycle switching (24). Central to its transforming capacity, the EBV-encoded latent membrane protein 1 (LMP-1) constitutively activates NF-κB and STAT3 signaling pathways through structural mimicry of activated CD40 receptors, thereby driving malignant B-cell proliferation and clonal expansion (25–27). Concurrently, EBV employs multiple immune evasion tactics including downregulation of MHC class I/II molecules and induction of PD-L1 overexpression on infected cells, effectively impairing T-cell recognition and promoting T-cell exhaustion (28). These mechanisms synergistically create an immunosuppressive tumor microenvironment further augmented by viral induction of pro-inflammatory cytokines (29).
First characterized as age-related EBV-associated B-cell lymphoproliferative disorder (aEBV-LPD) by Oyama et al. (2003) (30), EBV-positive DLBCL gained formal recognition in the 2008 WHO classification as a distinct clinicopathological entity. While predominantly affecting immunocompetent adults over 50 years, emerging evidence documents sporadic cases in younger demographics (31). Although current standard-of-care R-CHOP immunochemotherapy achieves 60% 10-year disease-free survival for DLBCL (32), recent studies have revealed significantly poorer survival outcomes in EBV+ DLBCL patients compared to their EBV-negative counterparts (33), highlighting the distinct biological behavior of this lymphoma subtype.
The treatment strategies for APDS encompass non-specific therapies, targeted therapies, and allo-HSCT. Non-specific therapies include long-term anti-infection treatment combined with IVIG supplementation and inhibition of the PI3K-AKT-mTOR pathway, such as corticosteroids, azathioprine, mycophenolate mofetil, and cyclosporine. Targeted therapies for APDS focus on inhibiting the PI3K-AKT-mTOR pathway, with mTOR inhibitors (e.g., rapamycin/sirolimus) and selective PI3Kδ inhibitors (e.g., idelalisib or duvelisib) being the primary options, Recent studies have highlighted the potential curative role of allo-HSCT in APDS. A retrospective international study reported 2-year overall survival (OS) and failure-free survival rates of 86% and 68%, respectively (34). Additional studies demonstrated that APDS1 patients undergoing allo-HSCT achieved comparable survival rates without severe graft-versus-host disease (GVHD) and showed significant improvement in humoral immunity (35, 36). However, the application of allo-HSCT is limited due to the lack of long-term follow-up data and the risk of significant complications and high mortality. In the case presented, although the patient achieved a brief CR after eight cycles of R-CHOPE chemotherapy, the underlying congenital immunodeficiency poses a risk of relapse. The use of the PI3K inhibitor duvelisib may partially suppress PI3K pathway activation and improve immunodeficiency, as evidenced by the patient’s disease-free survival without EBV reactivation. However, allo-HSCT remains the only definitive approach to reconstituting the immune system and achieving a cure for this disease.
Given the limited documentation on secondary malignancies associated with APDS, this study conducted a systematic literature search across multiple databases including PubMed, Web of Science, and CNKI (China National Knowledge Infrastructure). The search strategy employed the following keywords: “APDS”, “PIK3CD hyperactivation syndrome”, and “PIK3CD”. Through comprehensive screening of relevant articles, we extracted and tabulated critical data elements encompassing APDS subtypes, tumor phenotypes, therapeutic interventions, and hematopoietic stem cell transplantation status (Table 1).
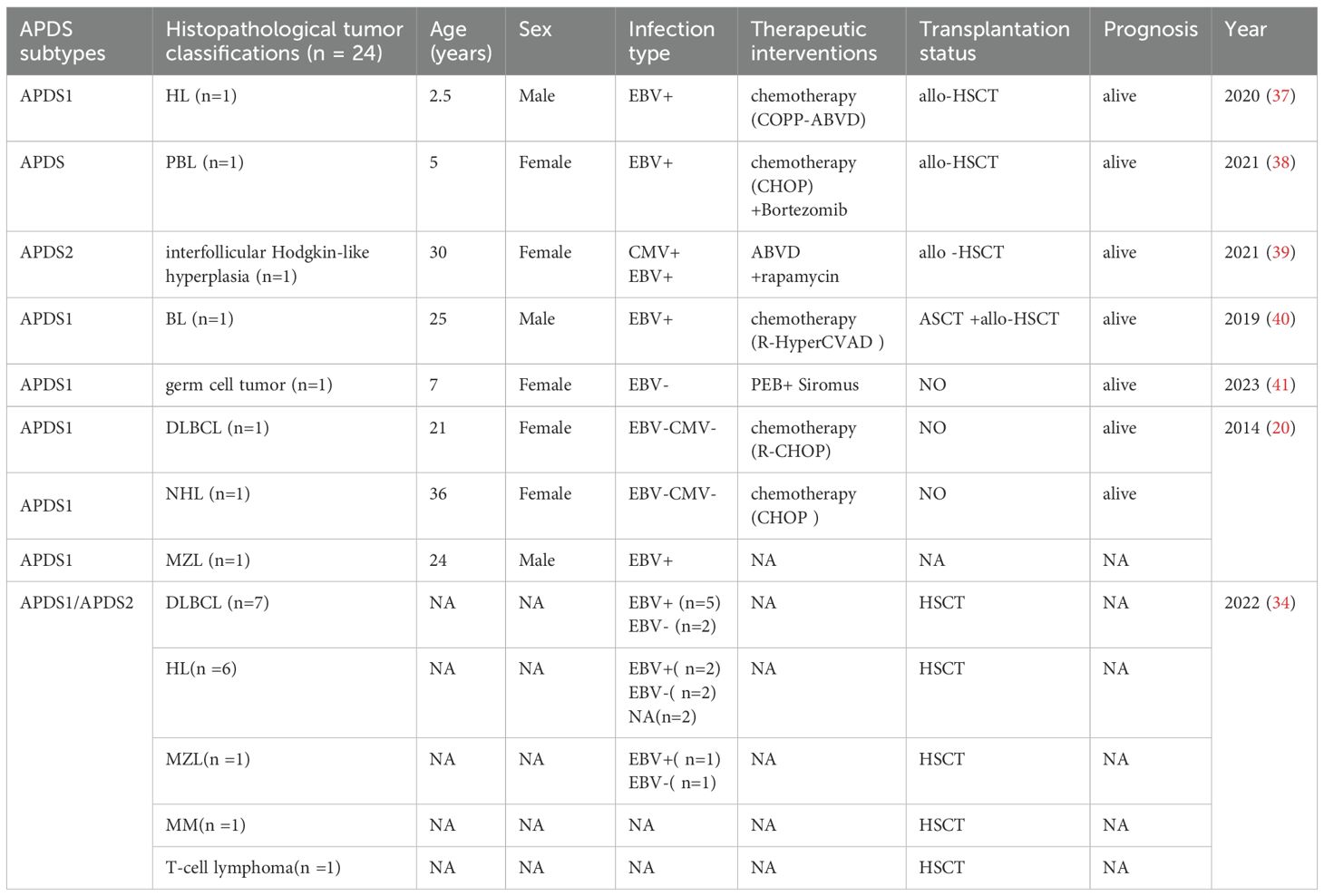
Table 1. Summary of case reports on secondary malignancies associated with APDS.
The study cohort comprised 23 malignancies including Hodgkin lymphoma (HL, n=7), diffuse large B-cell lymphoma (DLBCL, n=8), Burkitt lymphoma (BL, n=1), plasmablastic lymphoma (PBL, n=1), non-Hodgkin lymphoma (NHL, n=1), marginal zone lymphoma (MZL, n=2), multiple myeloma (MM, n=1), T-cell lymphoma (n=1), and germ cell tumor (n=1). Pooled analysis demonstrated EBV infection in 13/23 cases, including a subset with CMV coinfection. CR was achieved through allo-HSCT in virally coinfected patients, while chemotherapy alone proved effective for those without viral involvement. Notably, one case of interfollicular Hodgkin-like hyperplasia (n=1) was identified in a patient with inborn errors of immunity (IEI) (45). Initially misdiagnosed as HL, the diagnosis was revised following comprehensive evaluation. This patient achieved sustained remission through combined chemotherapy, rapamycin, and allo-HSCT, remaining alive at last follow-up.
These findings suggest that viral coinfections may amplify immune dysregulation in APDS. The diagnostic complexity is further compounded by lymphoproliferative manifestations in IEI, where lymphoma-mimicking lymphadenopathy creates significant challenges in differentiating benign hyperproliferation from hematologic malignancies. Notably, the observed therapeutic synergy between PI3Kδ inhibitors and chemotherapy demonstrates potential for reducing transplantation dependency, especially in clinically complex APDS presentations.
Notably, the largest cohort in this analysis derived from an international multicenter retrospective study involving 57 APDS patients, among whom 13 developed secondary malignancies. While this study primarily evaluated outcomes of allo-HSCT in APDS, it did not separately analyze prognosis in those with hematologic malignancies. However, the median follow-up of 26.3 months and a 2-year overall survival (OS) rate of 86% among transplanted patients underscore allo-HSCT as a viable strategy for prolonging survival. Furthermore, one case highlighted a patient with Burkitt lymphoma who relapsed after autologous stem cell transplantation (ASCT); subsequent genetic confirmation of APDS prompted salvage chemotherapy followed by allo-HSCT, achieving sustained CR. The sole germ cell tumor case achieved CR through surgical resection combined with immunotherapy, emphasizing the efficacy of multimodal approaches.
APDS is pathologically characterized by aberrant lymphocyte proliferation and heightened oncogenic predisposition. Mechanistic investigations reveal that PI3K signaling dysregulation drives tumorigenesis through pathological accumulation of phosphatidylinositol-3,4,5-trisphosphate (PtdInsP3) (46). This lipid secondary messenger activates the AKT/PKB-mTOR signaling axis, thereby mediating malignant phenotypes including uncontrolled proliferation, apoptosis resistance, metabolic reprogramming, and enhanced cellular migration (47). Notably, this constitutive pathway activation stems from functional loss of PTEN, a tumor suppressor that normally dephosphorylates PtdInsP3. Genetic mutations or epigenetic silencing of PTEN establishes a self-reinforcing oncogenic loop through sustained PI3K signaling (48, 49). Lineage-specific mechanistic analyses demonstrate distinct pathogenic consequences of PI3K hyperactivation: In T lymphocytes, aberrant PI3K signaling drives pathological expansion and survival programs, precipitating autoimmune manifestations and T-cell leukemogenesis (49). Conversely, B-cell PI3K dysregulation disrupts normal differentiation checkpoints while promoting proliferative signaling, thereby accelerating B-cell lymphoma progression (50, 51).
At the level of therapeutic intervention, selective PI3Kδ inhibitors (e.g., leniolisib) primarily modulate the AKT/PKB-mTOR signaling axis through competitive binding to the ATP-binding domain of the p110δ catalytic subunit. This mechanism effectively suppresses kinase activity and reduces pathological PtdInsP3 accumulation. Clinical studies have revealed that the co-occurrence of immunodeficiency and malignancy in APDS patients suggests multiple immune evasion mechanisms: (1) defective tumor antigen presentation mechanisms leading to immune recognition escape; (2) immunosuppressive microenvironments impairing effector cell function; and (3) aberrant activation of anti-apoptotic pathways conferring resistance to immune-mediated cytotoxicity (52). These synergistic mechanisms enable malignant cells to evade immune surveillance and sustain persistent proliferation. Recent phase III clinical trials have demonstrated sustained clinical benefits and favorable safety profiles of isoform-specific PI3Kδ inhibitors (e.g., leniolisib) in APDS management (45). However, incomplete blockade of tumor escape pathways by these agents may limit therapeutic efficacy in certain cases. In this context, allo-HSCT demonstrates unique advantages through dual mechanisms of action - hematopoietic system reconstitution and immune surveillance restoration - significantly mitigating risks of tumor immune evasion.
This study systematically analyzes the disease spectrum characteristics of secondary malignancies in APDS, validating the clinical value of combined allogeneic HSCT and precision targeted therapy. Our comprehensive evaluation not only enhances understanding of APDS pathogenesis but also establishes a theoretical framework for evidence-based management strategies in this rare immunodeficiency disorder.
Conclusion
APDS, an inborn error of immunity, is strongly associated with EBV-mediated lymphomagenesis. Here, we describe a rare case of APDS complicated by EBV-positive DLBCL. Current APDS management strategies involve antimicrobial prophylaxis, immunoglobulin supplementation, immunosuppressive regimens, PI3K inhibitors, and allo-HSCT. Although chemotherapy may induce partial remission in conventional DLBCL, immunodeficiency-associated DLBCL often demonstrates limited therapeutic responsiveness to standard treatment. In patients with concurrent inborn error of immunity and malignancy, allo-HSCT offers dual benefits: restoration of functional immunity and direct tumor suppression, potentially achieving complete remission. These observations suggest allo-HSCT may represent the sole potentially curative intervention for APDS patients with malignancy, particularly when conventional therapies fail to address the underlying immune dysfunction.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author/s.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
Q-YX: Data curation, Writing – original draft. MZ: Resources, Writing – original draft. J-HZ: Conceptualization, Supervision, Writing – review & editing. CF: Data curation, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fimmu.2025.1659197.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Angulo I, Vadas O, Garçon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science. (2013) 342:866–71. doi: 10.1126/science.1243292
2. Burke JE, Vadas O, Berndt A, Finegan T, Perisic O, and Williams RL. Dynamics of the Phosphoinositide 3-Kinase p110δ Interaction with p85α and Membranes Reveals Aspects of Regulation Distinct from p110α. Structure. (2011) 19:1127–37. doi: 10.1016/j.str.2011.06.003
3. Burke JE, Perisic O, Masson GR, Vadas O, and Williams RL. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110α (PIK3CA). Proc Natl Acad Sci U S A. (2012) 109:15259–64. doi: 10.1073/pnas.1205508109
4. Thouenon R, Moreno-Corona N, Poggi L, Durandy A, and Kracker S. Activated PI3Kinase delta syndrome-A multifaceted disease. Front Pediatr. (2021) 9:652405. doi: 10.3389/fped.2021.652405
5. Dornan GL, Siempelkamp BD, Jenkins ML, Vadas O, Lucas CL, and Burke JE. Conformational disruption of PI3Kδ regulation by immunodeficiency mutations in PIK3CD and PIK3R1. Proc Natl Acad Sci U S A. (2017) 114:1982–7. doi: 10.1073/pnas.1617244114
6. Wang X, Qi X, Yang B, Chen S, and Wang J. Autophagy benefits the replication of egg drop syndrome virus in duck embryo fibroblasts. Front Microbiol. (2018) 9:1091. doi: 10.3389/fmicb.2018.01091
7. Dulau Florea AE, Braylan RC, Schafernak KT, Williams KW, Daub J, Goyal RK, et al. Abnormal B-cell maturation in the bone marrow of patients with germline mutations in PIK3CD. J Allergy Clin Immunol. (2017) 139:1032–1035.e6. doi: 10.1016/j.jaci.2016.08.028
8. Wentink M, Dalm V, Lankester AC, van Schouwenburg PA, Schölvinck L, Kalina T, et al. Genetic defects in PI3Kδ affect B-cell differentiation and maturation leading to hypogammaglobulineamia and recurrent infections. Clin Immunol. (2017) 176:77–86. doi: 10.1016/j.clim.2017.01.004
9. Tsujita Y, Mitsui-Sekinaka K, Imai K, Yeh TW, Mitsuiki N, Asano T, et al. Phosphatase and tensin homolog (PTEN) mutation can cause activated phosphatidylinositol 3-kinase δ syndrome–like immunodeficiency. J Allergy Clin Immunol. (2016) 138:1672–1680.e10. doi: 10.1016/j.jaci.2016.03.055
10. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol. (2014) 15:88–97. doi: 10.1038/ni.2771
11. Burke JE and Williams RL. Synergy in activating class I PI3Ks. Trends Biochem Sci. (2014) 40:88–100. doi: 10.1016/j.tibs.2014.12.003
12. Jamee M, Moniri S, Zaki-Dizaji M, Olbrich P, Yazdani R, Jadidi-Niaragh F, et al. Clinical, immunological, and genetic features in patients with activated PI3Kδ Syndrome (APDS): a systematic review. Clin Rev Allergy Immunol. (2020) 59:323–33. doi: 10.1007/s12016-019-08738-9
13. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: A large patient cohort study. J Allergy Clin Immunol. (2017) 139:597–606.e4. doi: 10.1016/j.jaci.2016.06.021
14. Condliffe AM and Chandra A. Respiratory manifestations of the activated phosphoinositide 3-kinase delta syndrome. Front Immunol. (2018) 9. doi: 10.3389/fimmu.2018.00338
15. Cohen JI. Herpesviruses in the activated phosphatidylinositol-3-kinase-δ Syndrome. Front Immunol. (2018) 9. doi: 10.3389/fimmu.2018.00237
16. Kim ES, Kim D, Yoon Y, Kwon Y, Park S, Kim J, et al. Needs for increased awareness of gastrointestinal manifestations in patients with human inborn errors of immunity. Front Immunol. (2021) 12:698721. doi: 10.3389/fimmu.2021.698721
17. Maccari ME, Abolhassani H, Aghamohammadi A, Aiuti A, Aleinikova O, Bangs C, et al. Disease evolution and response to rapamycin in activated phosphoinositide 3-kinase δ Syndrome: the european society for immunodeficiencies-activated phosphoinositide 3-kinase δ Syndrome registry. Front Immunol. (2018) 9:543. doi: 10.3389/fimmu.2018.00543
18. Narayan N, Hewins P, Lane P, and Rhodes B. Activated PI3 kinase delta syndrome: a rare cause of inflammatory disease. Rheumatol Adv Pract. (2018) 2(1):rky034.010. doi: 10.1093/rap/rky034.010
19. Bloomfield M, Klocperk A, Zachova R, Milota T, Kanderova V, and Sediva A. Natural course of activated phosphoinositide 3-kinase delta syndrome in childhood and adolescence. Front Pediatr. (2021) 9:697706. doi: 10.3389/fped.2021.697706
20. Crank MC, Grossman JK, Moir S, Pittaluga S, Buckner CM, Kardava L, et al. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol. (2014) 34:272–6. doi: 10.1007/s10875-014-0012-9
21. Thorley-Lawson DA and Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med. (2004) 350:1328–37. doi: 10.1056/NEJMra032015
22. Cohen JI, Kimura H, Nakamura S, Ko YH, and Jaffe ES. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8–9 September 2008. Ann Oncol. (2009) 20:1472–82. doi: 10.1093/annonc/mdp064
23. Kimura H and Cohen JI. Chronic active epstein-barr virus disease. Front Immunol. (2017) 8. doi: 10.3389/fimmu.2017.01867
24. Cohen JI. Epstein-Barr virus infection. N Engl J Med. (2000) 343:481–92. doi: 10.1056/NEJM200008173430707
25. Sukswai N, Lyapichev K, Khoury JD, and Medeiros LJ. Diffuse large B-cell lymphoma variants: an update. Pathology. (2020) 52:53–67. doi: 10.1016/j.pathol.2019.08.013
26. Shannon-Lowe C and Rickinson AB and AIB. Epstein-Barr virus-associated lymphomas. Philos Trans R Soc Lond B Biol Sci. (2017) 372(1732):20160271. doi: 10.1098/rstb.2016.0271
27. Kato H, Karube K, Yamamoto K, Takizawa J, Tsuzuki, et al. Gene expression profiling of Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly reveals alterations of characteristic oncogenetic pathways. Cancer Sci. (2014) 105:537–44. doi: 10.1111/cas.2014.105.issue-5
28. Jiang XN, Yu BH, Yan WH, Lee J, Zhou XY, and Li XQ. Epstein-Barr virus-positive diffuse large B-cell lymphoma features disrupted antigen capture/presentation and hijacked T-cell suppression. Oncoimmunology. (2019) 9(1):1683346. doi: 10.1080/2162402X.2019.1683346
29. Ferla V, Rossi FG, Goldaniga MC, et al. Biological difference between epstein–barr virus positive and negative post-transplant lymphoproliferative disorders and their clinical impact. Front Oncol. (2020) 10. doi: 10.3389/fonc.2020.00506
30. Oyama T, Ichimura K, Suzuki R, Suzumiya J, Ohshima K, Yatabe Y, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. (2003) 27(1):16–26. doi: 10.1097/00000478-200301000-00003
31. Harkins RA, Chang A, Patel SP, et al. Remaining challenges in predicting patient outcomes for diffuse large B-cell lymphoma. Expert Rev Hematol. (2019) 12:959–73. doi: 10.1080/17474086.2019.1660159
32. Coiffier B, Thieblemont C, Van Den Neste E, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood. (2010) 116:2040–5. doi: 10.1182/blood-2010-03-276246
33. Park S, Lee J, Ko YH, Han A, Jun HJ, Lee SC, et al. The impact of Epstein-Barr virus status on clinical outcome in diffuse large B-cell lymphoma. Blood. (2007) 110:972–8. doi: 10.1182/blood-2007-01-067769
34. Dimitrova D, Nademi Z, Maccari ME, et al. International retrospective study of allogeneic hematopoietic cell transplantation for activated PI3K-delta syndrome. J Allergy Clin Immunol. (2022) 149:410–421.e7. doi: 10.1016/j.jaci.2021.04.036
35. Okano T, Imai K, Tsujita Y, et al. Hematopoietic stem cell transplantation for progressive combined immunodeficiency and lymphoproliferation in patients with activated phosphatidylinositol-3-OH kinase δ syndrome type 1. J Allergy Clin Immunol. (2019) 143:266–75. doi: 10.1016/j.jaci.2018.04.032
36. Nademi Z, Slatter MA, Dvorak CC, et al. Hematopoietic stem cell transplant in patients with activated PI3K delta syndrome. J Allergy Clin Immunol. (2017) 139:1046–9. doi: 10.1016/j.jaci.2016.09.040
37. Cansever M, Zietara N, Chiang SCC, et al. A rare case of activated phosphoinositide 3-kinase delta syndrome (APDS) presenting with hemophagocytosis complicated with hodgkin lymphoma. J Pediatr Hematol Oncol. (2020) 42:156–9. doi: 10.1097/MPH.0000000000001487
38. Yin Z, Tian X, Zou R, et al. Case report: first occurrence of plasmablastic lymphoma in activated phosphoinositide 3-kinase δ Syndrome. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.813261
39. Conti F, Catelli A, Cifaldi C, et al. Case report: hodgkin lymphoma and refractory systemic lupus erythematosus unveil activated phosphoinositide 3-kinase-δ Syndrome 2 in an adult patient. Front Pediatr. (2021) 9. doi: 10.3389/fped.2021.702546
40. Penther D, Viailly PJ, Latour S, et al. A recurrent clonally distinct Burkitt lymphoma case highlights genetic key events contributing to oncogenesis. Genes Chromosomes Cancer. (2019) 58:595–601. doi: 10.1002/gcc.22743
41. Yang PY, Jin M, Zhou YC, Duan C, Mao HW, Zhang R, et al. Activated PI3Kδ syndrome caused by PIK3CD gene mutation complicated with germ cell tumor in a child. Zhonghua Er Ke Za Zhi. (2023) 61:562–4. doi: 10.3760/cma.j.cn112140-20221012-00864
42. Nguyen T, Lau A, Bier J, Cooke KC, Lenthall H, Ruiz-Diaz S, et al. Human PIK3R1 mutations disrupt lymphocyte differentiation to cause activated PI3Kδ syndrome 2. J Exp Med. (2023) 220(6):e20221020. doi: 10.1084/jem.20221020
43. Bravo García-Morato M, García-Miñaúr S, Molina Garicano J, et al. Mutations in PIK3R1 can lead to APDS2, SHORT syndrome or a combination of the two. Clin Immunol. (2017) 179:77–80. doi: 10.1016/j.clim.2017.03.004
44. Notarangelo LD and Fleisher TA. Targeted strategies directed at the molecular defect: Toward precision medicine for select primary immunodeficiency disorders. J Allergy Clin Immunol. (2017) 139:715–23. doi: 10.1016/j.jaci.2017.01.004
45. Barzaghi F, Moratti M, Panza G, Rivalta B, Giardino G, De Rosa A, et al. Report of the italian cohort with activated phosphoinositide 3-kinase δ Syndrome in the target therapy era. J Clin Immunol. (2024) 45(1):58. doi: 10.1007/s10875-024-01835-1
46. Maehama T and Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. (1999) 9:125–8. doi: 10.1016/S0962-8924(99)01519-6
47. Cantley LC and Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. (1999) 96:8. doi: 10.1073/pnas.96.8.4240
48. Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, and Pandolfi PP. Impaired Fas response and autoimmunity in Pten+/- mice. Science Science. (1999) 285:2122–5. doi: 10.1126/science.285.5436.2122
49. Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, et al. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity. (2001) 14:523–34. doi: 10.1016/s1074-7613(01)00134-0
50. Marshall AJ, Niiro H, Lerner CG, et al. A novel B lymphocyte-associated adaptor protein, Bam32, regulates antigen receptor signaling downstream of phosphatidylinositol 3-kinase. J Exp Med. (2000) 8:1319–32. doi: 10.1084/jem.191.8.1319
51. Suzuki H, Matsuda S, Terauchi Y, et al. PI3K and Btk differentially regulate B cell antigen receptor-mediated signal transduction. Nat Immunol. (2003) 4:280–6. doi: 10.1038/ni890
Keywords: activated phosphoinositide 3-kinase δ syndrome, Epstein-Barr virus, diffuse large B-cell lymphoma, Epstein-Barr virus-positive diffuse large B-cell lymphoma (EBV+ DLBCL), inborn error of immunity
Citation: Xiang Q-y, Zuo M, Zhou J-H and Feng C (2025) EBV-positive diffuse large B cell lymphoma secondary to activated phosphoinositide 3 kinase δ syndrome type 1 (APDS1): a case report and literature review. Front. Immunol. 16:1583405. doi: 10.3389/fimmu.2025.1583405
Received: 25 February 2025; Accepted: 11 April 2025;
Published: 08 May 2025; Corrected: 15 July 2025.
Edited by:
Francisco Javier Espinosa-Rosales, Fundación Mexicana para Niñas y Niños con Inmunodeficiencias (FUMENI), MexicoReviewed by:
Francesca Conti, University of Bologna, ItalyMarco Antonio Yamazaki-Nakashimada, National Institute of Pediatrics (Mexico), Mexico
Copyright © 2025 Xiang, Zuo, Zhou and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-Hao Zhou, emhvdWppaGFvQGFsaXl1bi5jb20=; Chun Feng, ZmVuZ2NoOEBxcS5jb20=