 Weili Wang1†
Weili Wang1† Yilin Gao1†
Yilin Gao1† Yizhen Chen1
Yizhen Chen1 Meng Cheng2
Meng Cheng2 Yonghao Sang1Liuting Wei1
Yonghao Sang1Liuting Wei1 Rong Dai2
Rong Dai2 Yiping Wang2*
Yiping Wang2* Lei Zhang2*
Lei Zhang2*- 1First Clinical Medical College, Anhui University of Chinese Medicine, Hefei, China
- 2Department of Nephrology, The First Affiliated Hospital of Anhui University of Chinese Medicine, Hefei, China
Liver fibrosis is a core pathological process in the progression of chronic liver diseases to cirrhosis and hepatocellular carcinoma, characterized by abnormal deposition of extracellular matrix. Transforming growth factor-β (TGF-β), through classical small mothers against decapentaplegic (Smad)-dependent and non-Smad-dependent pathways, activates hepatic stellate cells to transdifferentiate into myofibroblasts, promotes extracellular matrix synthesis, and regulates immunity, serving as a key driver of fibrogenesis. This review systematically summarizes the role of TGF-β in liver fibrosis and details the research progress of TGF-β-targeted inhibitors. Studies show that TGF-β neutralizing antibodies, small molecule receptor antagonists, small molecule signaling inhibitors, and natural compounds and extracts significantly improve experimental liver fibrosis by inhibiting Smad or non-Smad pathways. In clinical trials, drugs such as Pirfenidone and Hydronidone have demonstrated potential for fibrosis reversal in patients with chronic hepatitis. Although TGF-β-targeted therapy has made breakthroughs in basic research and clinical translation, future studies need to focus on multi-target drug design, personalized treatment regimens, and novel delivery systems to accelerate the transition from preclinical research to clinical application, providing innovative therapeutic strategies for liver fibrosis and related liver diseases.
1 Introduction
Liver fibrosis is a pathological process involving structural changes in liver tissue and excessive extracellular matrix (ECM) deposition due to chronic injury and inflammation (1). Liver fibrosis is a common complication of various chronic liver diseases (CLDs), such as viral hepatitis, fatty liver disease, and alcoholic liver disease, and represents the early stage of liver cirrhosis (2, 3). The progression of liver fibrosis typically impairs liver function and may further lead to liver cirrhosis, failure, and cancer (4). Data show that from 1999 to 2016, the number of deaths due to liver cirrhosis in the United States increased by 65%, reaching 34,174. The number of deaths from hepatocellular carcinoma (HCC) more than doubled, reaching 11,073 (5). In 2017, approximately 1.5 billion people worldwide were affected by CLD; thus, it is a substantial global public health issue (6). Liver fibrosis is a critical stage in liver disease progression, and current treatments are focused primarily on managing underlying diseases; effective drugs that directly target fibrosis are scarce. Transforming growth factor-beta (TGF-β) is a representative member of the TGF-β family, which also includes activins, nodal factors, bone morphogenetic proteins (BMPs), growth and differentiation factors (GDFs), and other related factors (7). TGF-β is widely recognized as a key mediator of tissue fibrosis, primarily through the activation of its downstream Smad signaling pathway, triggering the overexpression of profibrotic genes and further inducing scar tissue formation (8). Numerous studies have demonstrated that dysregulation of the TGF-β pathway is a critical pathogenic mechanism in tissue fibrosis (9–12), playing a central role in its initiation and progression (13). TGF-β activates hepatic stellate cells (HSCs), causing their transdifferentiation into myofibroblasts (MFBs) and leading to the excessive accumulation of ECM components such as collagen in the liver, thereby driving the progression of liver fibrosis (14, 15). Additionally, the TGF-β signaling pathway is involved in liver repair, immune responses, and cell apoptosis (16). Given the critical role of TGF-β in liver fibrosis, the use of TGF-β inhibitors has become an important research focus in treating liver fibrosis. In recent years, numerous studies have validated the potential of TGF-β-targeted inhibitors in treating fibrotic diseases (17–22), indicating that TGF-β inhibitors are widely regarded as promising antifibrotic therapies. With a deeper understanding of the mechanisms of the TGF-β signaling pathway, researchers have developed various TGF-β-targeted inhibitors and achieved preliminary results in different animal experiments and clinical trials. This review focuses on the biological processes of TGF-β, its dual role in the development of liver fibrosis, and the application of TGF-β inhibitors in liver fibrosis treatment, exploring previous research progress, clinical achievements, and future development trends.
2 Relationship between the TGF-β signaling pathway and liver fibrosis
2.1 TGF-β family
In mammals, the 33 genes of the TGF-β family each encode a polypeptide comprising a secretory signal peptide, a pro-domain of 1–250 residues, and a growth factor domain of 1–110 residues (23, 24). The TGF-β superfamily includes TGF-β, BMPs, GDFs, activins, and nodal factors, with TGF-β being the prototype. The three primary TGF-β protein isoforms are TGF-β1, TGF-β2, and TGF-β3 (25). As key members of the TGF-β family, these isoforms are widely involved in critical physiological processes, such as embryonic development, cell differentiation, organ formation, and tissue repair (26–29). Additionally, TGF-β plays a significant role in immune regulation by inhibiting T-cell proliferation, promoting the generation of regulatory T cells, and modulating the differentiation and function of Th1/Th17 cells, thereby profoundly influencing immune tolerance, autoimmune diseases, and tumor immunity (30, 31). The expression of TGF-β is regulated by various factors, including proinflammatory cytokines, oxidative stress, Toll-like receptor (TLR) signaling, the ECM, and matrix metalloproteinases (MMPs) (32, 33). TGF-β, which plays important roles in numerous physiological and pathological processes, is primarily produced by macrophages and epithelial cells; platelets, T cells, fibroblasts, and mast cells can also secrete this cytokine (34, 35).
TGF-β is a key activator of fibroblasts and plays a central regulatory role in fibrotic responses. It directly promotes fibroblast activation and may further drive pathological progression by modulating the fibrotic phenotypes of immune cells and vascular cells (36, 37). Additionally, TGF-β stimulates the synthesis of ECM proteins and induces tissue fibrotic responses in vivo (38). In fibrotic diseases, TGF-β promotes the proliferation and activation of fibroblasts by inducing the transdifferentiation of HSCs, lung fibroblasts, and renal tubular epithelial cells into MFBs, leading to excessive collagen and ECM deposition, ultimately resulting in organ fibrosis. Therefore, TGF-β is considered a key pathogenic factor in fibrotic diseases of the liver, lungs, kidneys, and other organs (8, 39–41). Furthermore, TGF-β plays a dual role in the tumor microenvironment. In early tumorigenesis stages, TGF-β is suppressive; in the advanced stages, it promotes immune evasion, angiogenesis, and tumor metastasis, driving cancer progression (42, 43) (Figure 1).
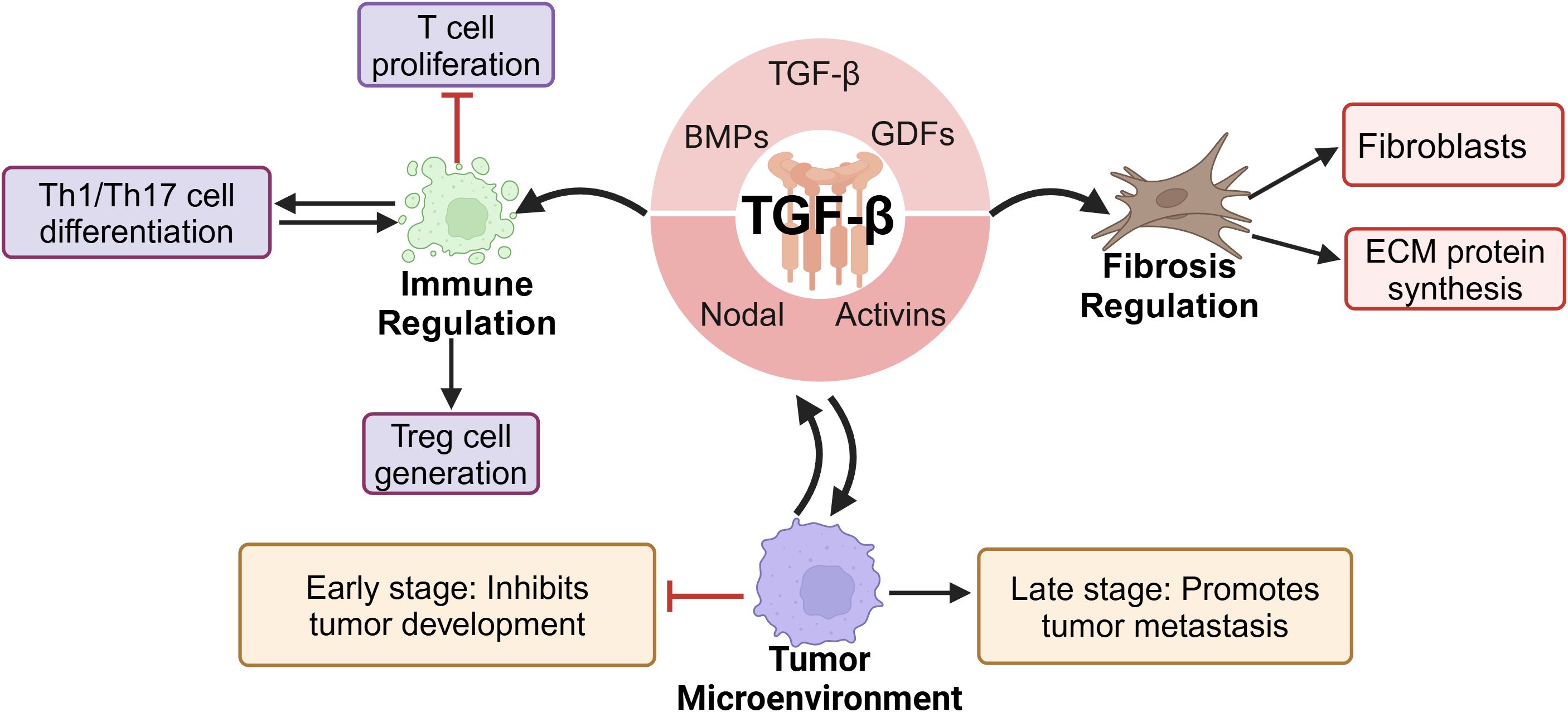
Figure 1. TGF-β family and its biological functions.
2.2 TGF-β signaling pathway
TGF-β ligands initiate downstream signaling by binding to their receptors, TGF-β receptor type I (TGF-βR I) and type II (TGF-βR II), which are primarily involving two pathways. Among these pathways, the canonical small mothers against decapentaplegic (Smad)-dependent pathway is crucial to TGF-β signaling. Smads are key transducers of the TGF-β family signaling pathway and are divided into three main subclasses: receptor-regulated Smads (R-Smads), common Smads (Co-Smads), and inhibitory Smads (I-Smads) (44, 45). R-Smads include Smad2, Smad3, Smad1, Smad5, and Smad8, with Smad2 and Smad3 being the key downstream mediators of TGF-β-induced tissue fibrosis (46). In this pathway, the TGF-β-activated receptor complex phosphorylates R-Smads, which then form a complex with Co-Smad4. This R-Smad–Co-Smad complex translocates to the nucleus and regulates the transcription of target genes (47, 48). Smad1, Smad5, and Smad8, which are R-Smads, primarily participate in BMP signaling, regulating osteoblast differentiation and bone and cartilage development (49, 50). Within the I-Smad family, Smad6 and Smad7 function as negative regulators of the TGF-β/Smad signaling pathway. Specifically, Smad7 inhibits pathway activation by binding to activated TGF-β type I receptors, thereby blocking Smad2 phosphorylation. Additionally, its MH2 domain (Mad Homology 2 Domain) mediates specific binding to TGF-β/BMP receptor complexes, further suppressing signaling from the TGF-β superfamily (51–53).
The other pathway is the non-Smad-dependent pathway, in which TGF-β activates signaling cascades through phosphorylation, acetylation, sumoylation, ubiquitination, and protein–protein interactions. These pathways include mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase/protein kinase B (PI3K/Akt), ras homolog family GTPase (Rho GTPase), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), wingless-related integration site/β-catenin (Wnt/β-catenin), mammalian target of rapamycin (mTOR) (54, 55). These non-Smad-dependent pathways interact with Smad signaling to jointly regulate fibroblast proliferation, fibrosis-related gene expression, and ECM remodeling, significantly driving fibrosis initiation, progression, and disease development (56, 57) (Figure 2).
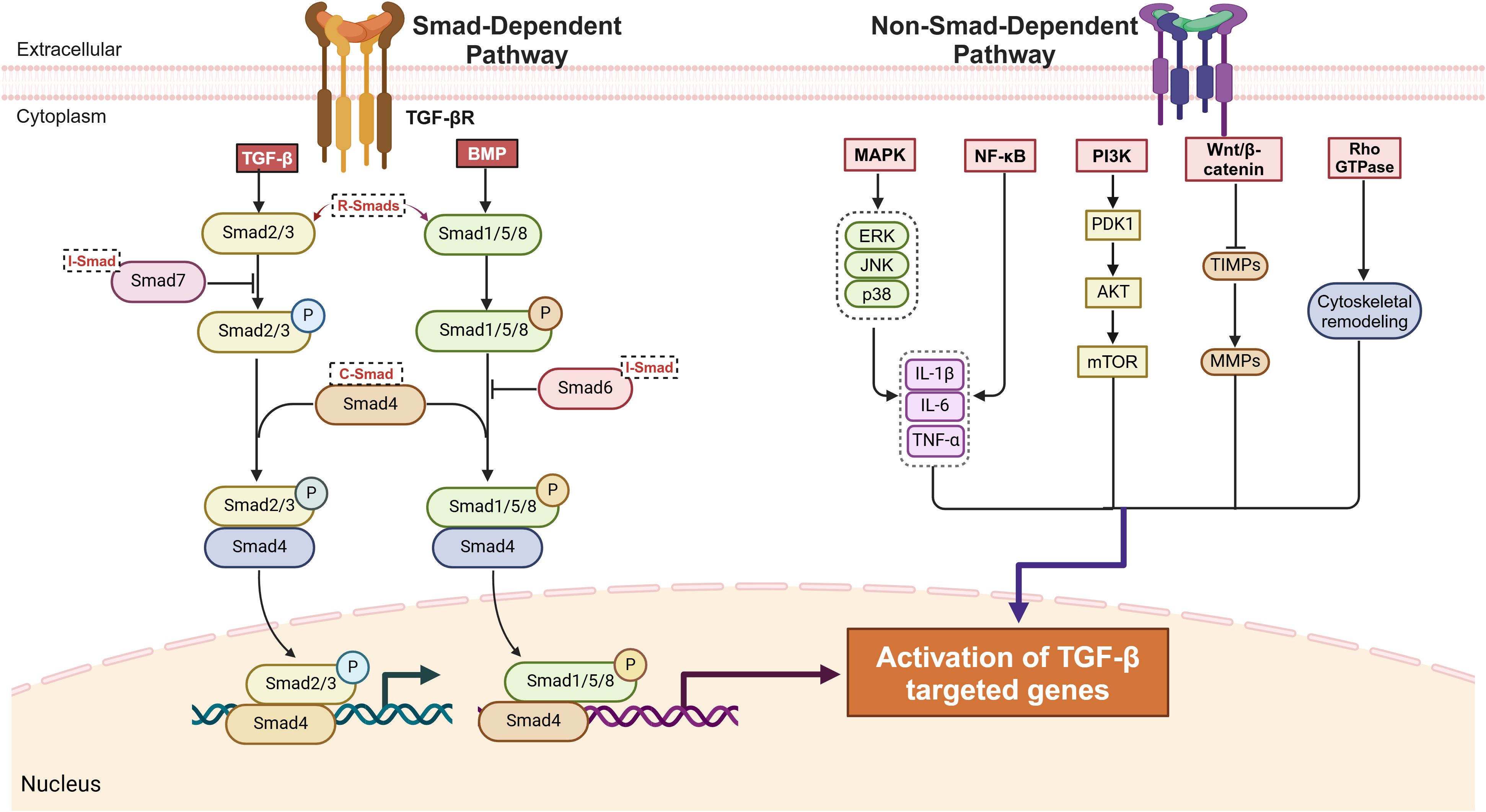
Figure 2. TGF-β signaling pathway: smad-dependent and non-smad-dependent mechanisms.
2.3 Pathogenesis of liver fibrosis
Liver fibrosis represents a reversible wound-healing response characterized by abnormal deposition of ECM triggered by chronic liver injury, which may progress to cirrhosis and even liver cancer (58, 59). While mild and transient liver injury allows the liver to restore normal structure through robust self-healing capacity, persistent damage induces chronic inflammation and excessive ECM accumulation, leading to gradual replacement of normal hepatic parenchyma by fibrotic scar tissue and eventual progression to cirrhosis (60). Hepatic parenchyma is composed primarily of hepatocytes and liver sinusoidal endothelial cells, whereas non-parenchymal cells include HSCs, Kupffer cells, and infiltrating immune cells. The development and progression of liver fibrosis depend fundamentally on intercellular interactions among these cell types (61).
Kupffer cells, as resident macrophages located on the surface of hepatic sinusoidal endothelial cells (62), collaborate with infiltrating inflammatory cells during liver injury to release proinflammatory cytokines that activate HSCs and induce their transdifferentiation into MFBs (63). Activated HSCs lose their original stellate morphology and intracellular vitamin A-laden lipid droplets, instead vigorously synthesizing ECM components and secreting proinflammatory mediators, thereby forming the core driver of fibrogenesis (64). Concurrently, capillarization of liver sinusoidal endothelial cells—characterized by basement membrane thickening and loss of fenestrated structures—diminishes their normal capacity to regulate HSCs quiescence (65), further exacerbating ECM deposition and fibrotic progression (Figure 3A). The stark contrast between the quiescent phenotype of HSCs (vitamin A storage, lipid droplet-rich) and their activated state (ECM production, proinflammatory signaling) during this process (Figure 3B) constitutes the central pathological mechanism underlying the initiation and development of liver fibrosis.
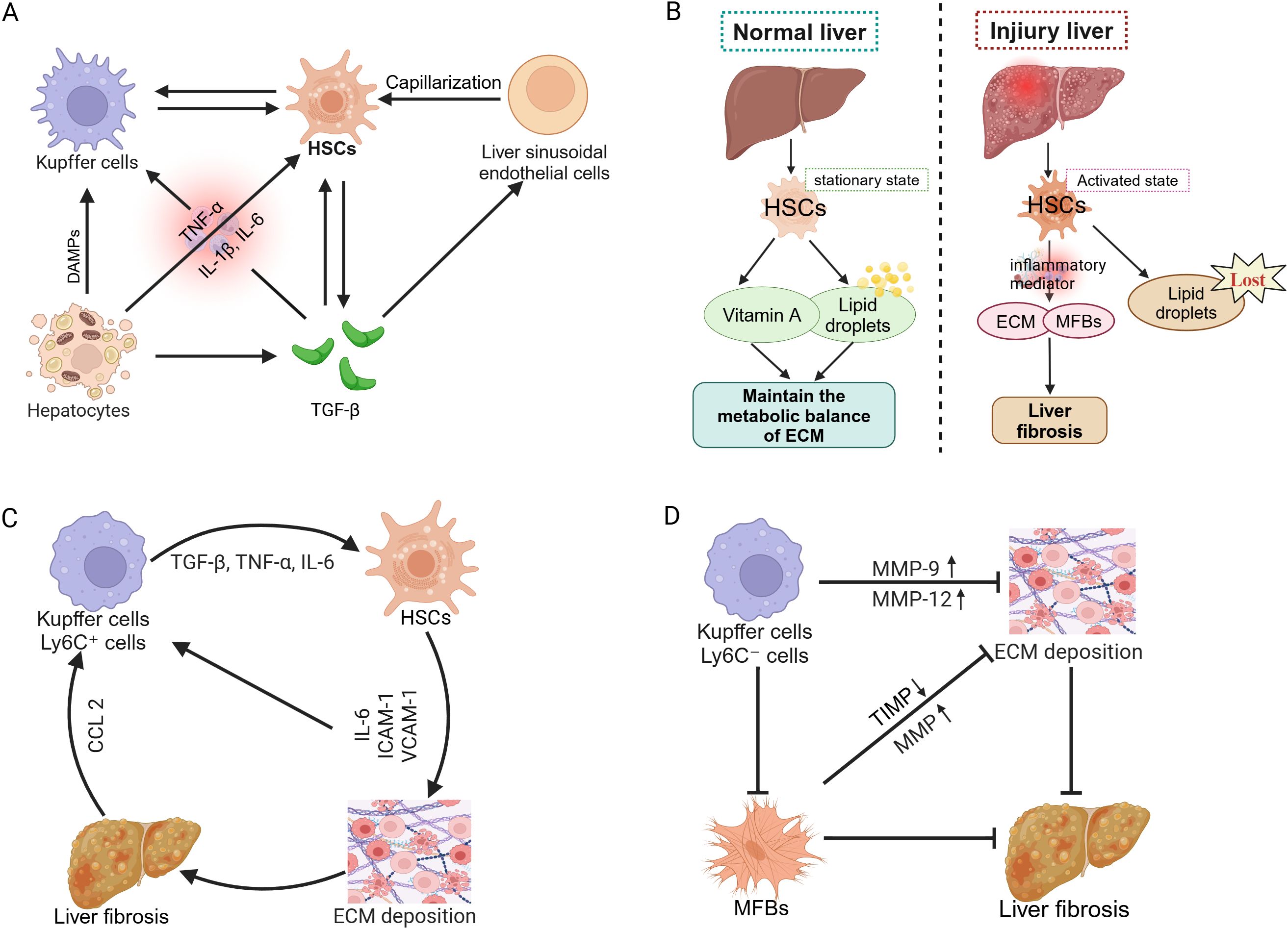
Figure 3. Pathogenesis of liver fibrosis. (A) Cellular interactions and progression mechanisms of liver fibrosis. (B) Role of HSCs in normal and injured liver. (C) Pro-inflammatory role of macrophages in liver fibrosis progression. (D) Macrophage-mediated reversal mechanism of liver fibrosis.
During fibrosis progression, large numbers of Kupffer cells and Ly6C+ proinflammatory monocyte-derived macrophages are recruited to the liver via C-C motif chemokine ligand 2 (CCL2)-mediated chemotaxis. These cells release proinflammatory factors such as TGF-β and tumor necrosis factor-alpha (TNF-α), further activating HSCs and accelerating ECM accumulation. Additionally, HSCs secrete interleukin-6 (IL-6), intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1), enhancing interactions with macrophages and T cells (66), thus amplifying profibrotic signaling and promoting disease progression (Figure 3C). However, during fibrosis regression, Kupffer cells and monocyte-derived macrophages transition into Ly6C− anti-inflammatory macrophages, releasing MMP-9 and MMP-12 and thus promoting ECM degradation and liver tissue repair (67). Simultaneously, the reduction in MFBs and TIMP levels further enhances MMP activity, accelerating ECM clearance and ultimately facilitating fibrosis reversal (68) (Figure 3D). In conclusion, macrophages play dual roles in the progression and regression of liver fibrosis.
2.4 Role of TGF-β in liver fibrosis
As a key regulatory factor in liver fibrosis, TGF-β drives the fibrogenic process through the TGF-β/Smad signaling pathway, which regulates DNA synthesis to promote the transformation of quiescent HSCs into MFBs. This process accelerates the excessive deposition of ECM and propels fibrosis progression. Concurrently, activated HSCs upregulate TGF-β1 expression via autocrine and paracrine mechanisms, forming a positive feedback loop that sustains the activation of the TGF-β signaling cascade (69). This process is mediated by the binding and activation of TβRI, which subsequently induces the phosphorylation of Smad2 and Smad3, leading to the formation of the Smad2/3/4 complex, which translocates to the nucleus. This complex triggers the transcription of fibrotic genes, including type I and type III collagens, ultimately resulting in ECM accumulation (39). However, Smad7, an endogenous inhibitor of the TGF-β signaling pathway, can competitively bind to TβRI, blocking TGF-β-mediated signaling and suppressing HSCs activation, thereby limiting fibrosis progression to some extent (70, 71). TGF-β also interacts with other signaling pathways, including the Notch, Wnt/β-catenin, and yes-associated protein/transcriptional co-activator with PDZ-binding motif (YAP/TAZ) pathways, further enhancing the profibrotic capacity of HSCs and exacerbating hepatocyte apoptosis and tissue damage (72–74).
In the context of immunoregulation, TGF-β regulates the progression of liver fibrosis in multiple stages. In the early stages of fibrosis, TGF-β promotes Kupffer cell and macrophage secretion of proinflammatory cytokines, such as IL-1β, TNF-α, and IL-6, and the chemokines CCL2 and CCL5. These factors activate paracrine protective or apoptotic signaling pathways in hepatocytes and recruit additional immune cells, thereby aggravating inflammation and exacerbating liver damage (75). As fibrosis progresses to the later stages, TGF-β induces M2 macrophage polarization, enhancing immune suppression and facilitating the occurrence of HCC (76). In the fibrosis resolution phase, TGF-β may promote the production of MMP-9 and MMP-12, accelerating ECM degradation and facilitating fibrosis regression (77, 78) (Figure 4). Given TGF-β’s involvement in key processes of liver fibrosis, including HSCs activation, ECM production, immunoregulation, and fibrosis reversal, TGF-β and its associated signaling pathways have emerged as critical therapeutic targets. Various interventions have been developed, such as TGF-β-neutralizing antibodies, small molecule TGF-β receptor antagonists, small molecule TGF-β signaling inhibitors, and natural compounds, all aiming to block its profibrotic effects, promote reversal of liver fibrosis, and reduce the risk of liver cancer.
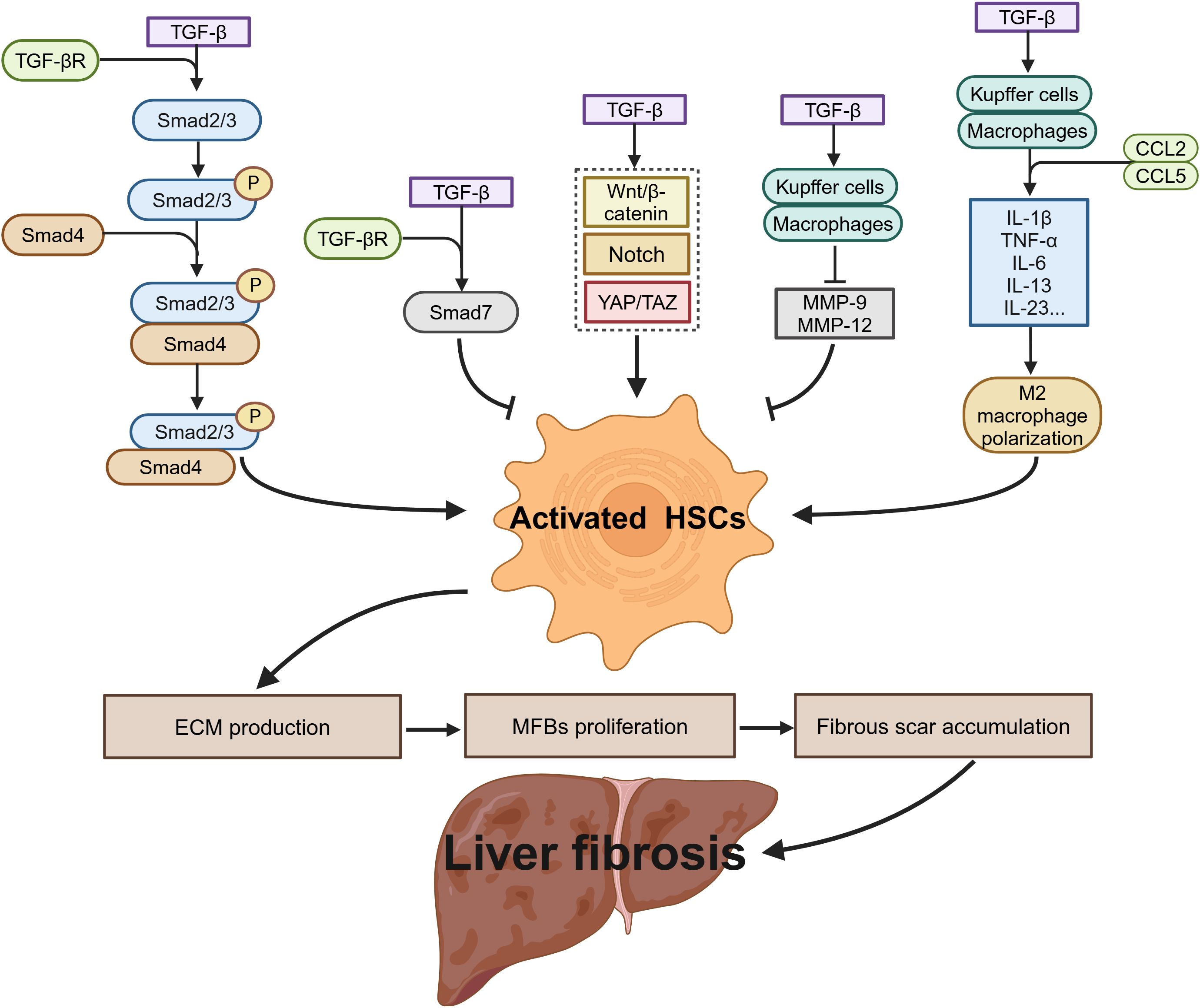
Figure 4. The role of TGF-β in liver fibrosis.
3 Research progress of TGF-β inhibitors in liver fibrosis
3.1 TGF-β inhibitors: types and molecular mechanism
3.1.1 TGF-β neutralizing antibodies
TGF-β neutralizing antibodies directly bind to TGF-β, blocking its interaction with receptors and inhibiting fibrosis progression (Table 1). For example, BI 113823, a B1R small-molecule nonpeptide orally active inhibitor, can inhibit TGF-β-induced activation, proliferation, migration, and fibrotic protein expression in human hepatic stellate cells (hHSCs) and suppress the activation of the PI3K/Akt signaling pathway (79). Additionally, peptide seq-1 targets HepG2 human hepatocellular carcinoma cells (HepG2 cells) to negatively regulate the TGF-β signaling pathway, reducing the synthesis of ECM and alleviating the imbalance of ECM-remodeling-related enzymes MMP-2 and TIMP-2. This effect subsequently inhibits the activation, oxidative stress, and fibrogenic responses of HSCs via paracrine signaling mediated by conditioned medium from seq-1-treated HepG2 cells (80). The bifunctional fusion protein Bintrafusp alfa simultaneously targets TGF-β and programmed death-ligand 1 (PD-L1), significantly promoting T cell activation, regulating ECM deposition, and inhibiting fibrogenesis in vitro. Studies have shown that bintrafusp alfa is more effective than monotherapy, as it can neutralize both circulating free TGF-β and locally activated TGF-β on target cell surfaces (81, 82).
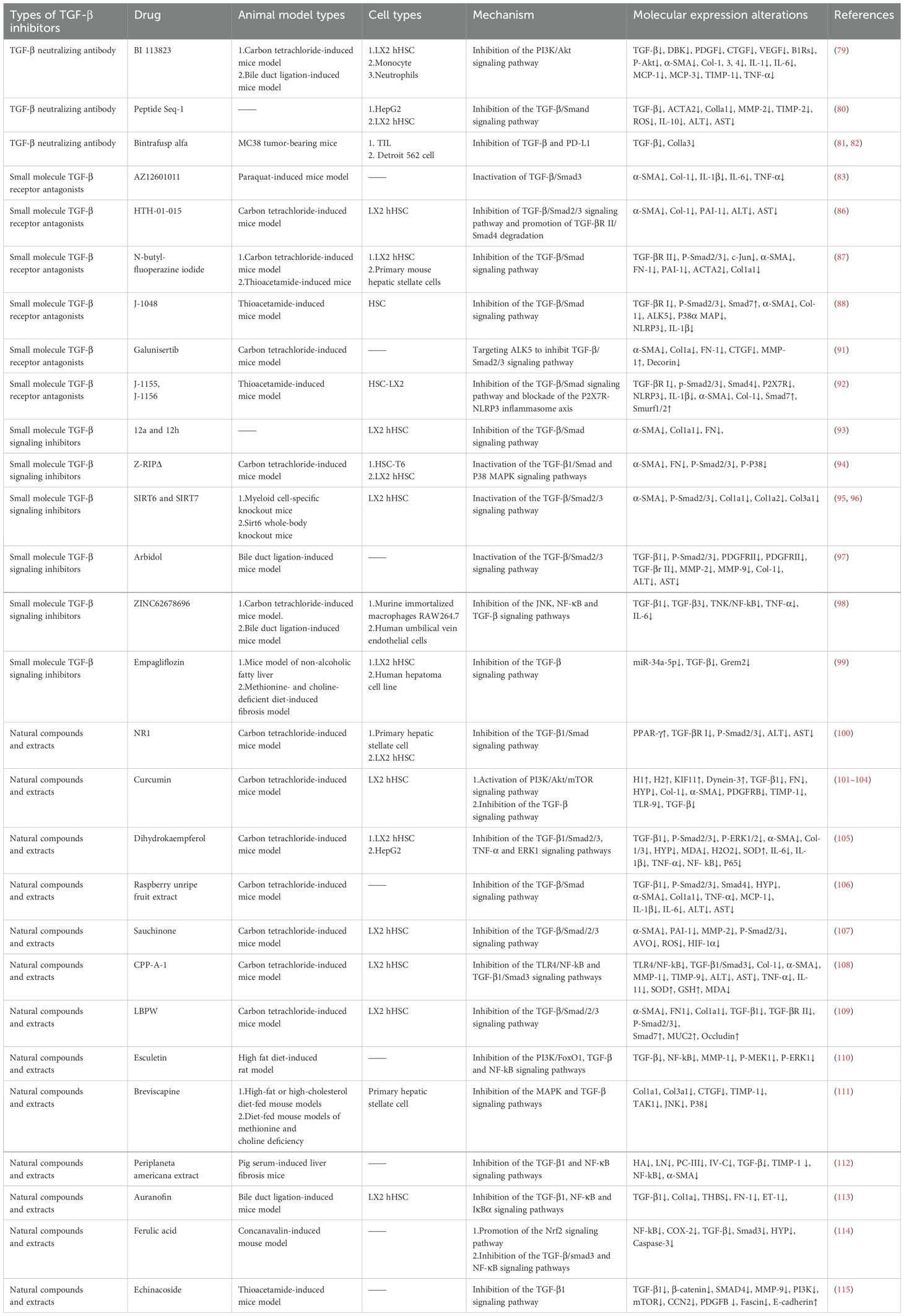
Table 1. Types and mechanism of action of TGF-β inhibitors.
3.1.2 Small molecule TGF-β receptor antagonists
Small molecule TGF-β receptor antagonists target TGF-β receptors to prevent receptor activation, thereby regulating Smad and non-Smad signaling pathways and influencing the progression of liver fibrosis (Table 1). AZ12601011 (a TGF-βR I inhibitor) directly binds to TGF-βR I, blocking Smad3 activation, inhibiting the TGF-β/Smad3 signaling pathway, and alleviating paraquat-induced liver fibrosis in mice (83). AMP-activated protein kinase 5 (ARK5) prevents the degradation of TGF-βR I and maternal Smad4 proteins by inhibiting the expression of Smad ubiquitin regulatory factor 2, thereby maintaining TGF-β signal transduction (84, 85), whereas the selective inhibitor HTH-01–015 can target ARK5, reducing carbon tetrachloride (CCl4)-induced liver fibrosis deposition in mice and the expression of fibrosis-related proteins such as α-SMA and type I collagen (Col-1) (86). N-butyl-fluoperazine iodide inhibits TGF-βR II, reducing the response of HSCs to TGF-β1, decreasing TGF-β/Smad signaling transduction and fibrotic gene expression, improving CCl4- or thioacetamide-induced liver fibrosis in mice in a dose-dependent manner, and alleviating liver function damage and tissue lesions (87). J-1048 (an ALK5 inhibitor, another name for a TGF-βR I inhibitor) reduces the phosphorylation levels of TGF-βR I and Smad2/3 and increases Smad7 expression in a dose-dependent manner, thereby regulating TGF-β signal transduction and inhibiting thioacetamide-induced liver fibrosis in mice (88). Galunisertib, an ALK5 inhibitor, exerts antifibrotic effects by targeting ALK5 downstream of TGF-β signaling, thereby impeding the expression of Smad2/3-regulated genes. In vivo, it reduces hepatic collagen deposition, inhibits HSCs activation, prevents steatosis, and restores proper liver lobular architecture in CCl4-induced liver fibrosis rat models (89–91). J-1155 and J-115, novel thiazole derivatives that selectively target and inhibit ALK5, effectively alleviate thioacetamide-induced liver fibrosis and associated inflammation in mice through dual inhibition of the TGF-β/Smad signaling pathway and blockade of the P2X7R-NLRP3 inflammasome axis (92).
3.1.3 Small molecule TGF-β signaling inhibitors
Various small molecule inhibitors exert antifibrotic effects by targeting the TGF-β signaling pathway and its key proteins (Table 1). Studies have shown that rsodeoxycholic acid-amino pyrimidine hybrids (12a and 12h) significantly inhibit the migration of the human hepatic stellate cell line LX2 (HSC-LX2) by blocking the TGF-β/Smad signaling pathway (93). In vivo studies have demonstrated that Z-RIPΔ, a novel TGF-βR I mimetic peptide, specifically binds to TGF-β1-activated HSCs, inhibiting cell proliferation and migration while reducing the expression of the fibrosis markers α-SMA and fibronectin and the TGF-β1 pathway effectors phosphorylated-Smad2/3 and phosphorylated-P38 MAPK (94). Sirtuin 6 (SIRT6) and Sirtuin 7 (SIRT7) inhibit Smad2/3 transcriptional activity by inducing its deacetylation, thereby reducing HSCs activation (95, 96).
Arbidol reduces the mRNA expression levels of PDGDR, TGF-βR I, TGF-βR II, MMP-2, and MMP-9, inhibits the phosphorylation of Smad2/3 in TGF-β1-treated HSCs and bile duct ligation-induced mice, and ultimately alleviates collagen deposition, liver injury, and fibrosis (97). G protein-coupled receptor 65 (GPR65) is closely related to liver inflammation, injury, and fibrosis. The GPR65 inhibitor ZINC62678696 reduces the activation of HSCs and hepatocyte injury induced by bile duct ligation and CCl4 by inhibiting the c-Jun N-terminal kinase (JNK) and NF-κB pathways, thereby inducing downregulation of pro-inflammatory cytokines such as TNF-α, IL-6, and TGF-β (98). Empagliflozin, a sodium-glucose cotransporter 2 inhibitor, inhibits the TGF-β pathway in HSCs by downregulating miR-34a-5p expression and upregulating Gremlin 2 in the LX-2 human hepatic stellate cell line (LX-2 HSCs) and ob/ob mouse fibrosis models, thereby improving NAFLD-related fibrosis (99).
3.1.4 Natural compounds and extracts
Studies have shown that certain natural compounds and extracts inhibit CCl4-induced liver fibrosis in mice by regulating TGF-β/Smad and associated signaling pathways (Table 1). In vivo investigations demonstrate that notoginsenoside R1 (NR1), an extract derived from the traditional Chinese medicine Panax notoginseng, suppresses TGF-β1/Smads signaling by upregulating peroxisome proliferator-activated receptor gamma (PPAR-γ), thereby attenuating the activation of HSCs (100). Curcumin, a natural polyphenolic pigment extracted from Curcuma longa, exerts anti-fibrotic effects by inhibiting autophagy and inducing apoptosis in HSC-LX2 cells via activation of the PI3K/Akt/mTOR signaling pathway. Additionally, curcumin interacts with fibrosis-related proteins such as platelet-derived growth factor receptor B (PDGFRB), TIMP-1, TLR-9, and TGF-β, thereby blocking the TGF-β/Smad signaling pathway (101–104). Dihydrokaempferol (CAS 480-20-6), a natural compound related to flavonol in E. alatus, exerts its effects by inhibiting the PARP-1-regulated TGF-β1 pathway and TNF-α transcription, leading to reduced phosphorylation levels of Smad2/3 and ERK1 (105). Raspberry unripe fruit extract attenuates HSCs activation and proliferation by suppressing the TGF-β/Smads signaling pathway and downregulating the expression of related proteins, including TGF-β1, phosphorylated-Smad2/3, and Smad4 (106). Sauchinone, an active lignan found in Saururus chinensis, exerts anti-fibrotic effects in vivo by blocking TGF-β1-induced Smad2/3 phosphorylation and inhibiting the transcription of plasminogen activator inhibitor-1 (PAI-1) and MMP-2 (107). Codonopsis pilosula root polysaccharide (CPP-A-1), extracted from the traditional Chinese medicine Radix Codonopsis, downregulates Col-1 and α-SMA expression through the inhibition of the TLR-4/NF-κB and TGF-β1/Smad3 signaling pathways. Additionally, while also inhibits ECM production by restoring the balance between MMPs and TIMPs (108). A peptidoglycan isolated from the fruit of Lycium barbarum, designated as LBPW, upregulates Smad7—a negative regulator of the TGF-β/Smad signaling pathway—to delay the activation of HSCs (109). These studies collectively demonstrate that natural compounds and extracts exert antifibrotic effects in CCl4-induced liver fibrosis models by modulating the TGF-β/Smad pathway.
Esculetin, a coumarin compound belonging to the benzopyran derivative class, alleviates high-fat diet-induced liver fibrosis by activating Forkhead box O1 (FoxO1) through modulation of the Akt/PI3K/FoxO1 signaling pathway, thereby inhibiting TGF-β-mediated lipid peroxidation and ECM protein accumulation (110). Breviscapine, a flavonoid extract derived from the traditional Chinese herb Erigeron breviscapus (Vant.), mitigates lipid accumulation, inflammatory cell infiltration, liver injury, and fibrosis by directly binding to transforming growth factor-β-activated kinase 1 (TAK1) and inhibiting both MAPK and TGF-β signaling pathways (111). Periplaneta americana extract reduces hepatic collagen deposition and reverses liver fibrosis in vivo by inhibiting the expression of TGF-β1, NF-κB, TIMP-1, and α-SMA (112). Auranofin, a gold-based compound, reduces hepatic steatosis and fibrosis in vivo by decreasing TGF-β1-induced NF-κB and inhibitor of nuclear factor kappa B alpha (IκBα) levels (113). Ferulic acid, a naturally occurring phenolic acid compound, mitigates concanavalin-induced liver fibrosis by stimulating the Nrf2 signaling pathway while inhibiting NF-κB and the TGF-β/Smad3 signaling pathway (114). Echinacoside, a natural phenol belonging to the phenylpropanoid class of caffeic acid glycosides, suppresses liver fibrosis in vivo by reducing the expression of TGF-β1, β-catenin, Smad4, MMP-9, PI3K, mTOR, cellular communication network factor 2 (CCN2), platelet-derived growth factor-B (PDGFB) (115).
3.2 Clinical applications of TGF-β inhibitors
Several TGF-β inhibitors have been evaluated in clinical trials (Table 2). Pirfenidone (PFD), a broad-spectrum antifibrotic drug acting on the TGF-β target, blocks TGF-β signaling through dual action pathways. On one hand, it directly binds to TGF-β1 mRNA to inhibit its transcription; on the other hand, it suppresses Smad2/3 phosphorylation and blocks their nuclear translocation. These mechanisms collectively downregulate the mRNA and protein levels of TGF-β1, TGF-βR I, and TGF-βR II, ultimately inhibiting TGF-β-induced fibroblast proliferation, ECM synthesis, and the expression of fibrotic genes (116). A single-dose open-label study by a Mexican team demonstrated that in cirrhotic patients (8 cases each with Child-Pugh class A/B), oral administration of 1200 mg prolonged-release pirfenidone (PR-PFD) resulted in the area under the curve (AUC0-last and AUC0-α) and maximal concentration 3.6-fold and 4.4-fold higher than those in healthy controls, respectively—with more pronounced increases in Child-Pugh B patients—while maintaining good tolerability (117). Another multicenter study involving 122 patients with alcoholic liver fibrosis showed that 35% of those in the PR-PFD treatment group exhibited significant fibrosis reduction (vs. 4.1% in the control group), with a 29.7% improvement rate in Child-Pugh scores (118). Hydronidone, a novel structural modification of PFD designed to reduce hepatotoxicity, was tested in a Chinese Phase II double-blind trial: chronic hepatitis B patients receiving combination therapy of hydronidone (270 mg/day) and entecavir for 52 weeks showed the most significant histological improvement in liver fibrosis with favorable safety (119). Collectively, these three studies confirm the antifibrotic potential of PR-PFD and its derivatives, with efficacy demonstrating dose-dependent trends. However, PFD is associated with inherent limitations, including high-dose-specific toxic effects and off-target reactions, which restrict its broader clinical implementation (120, 121).
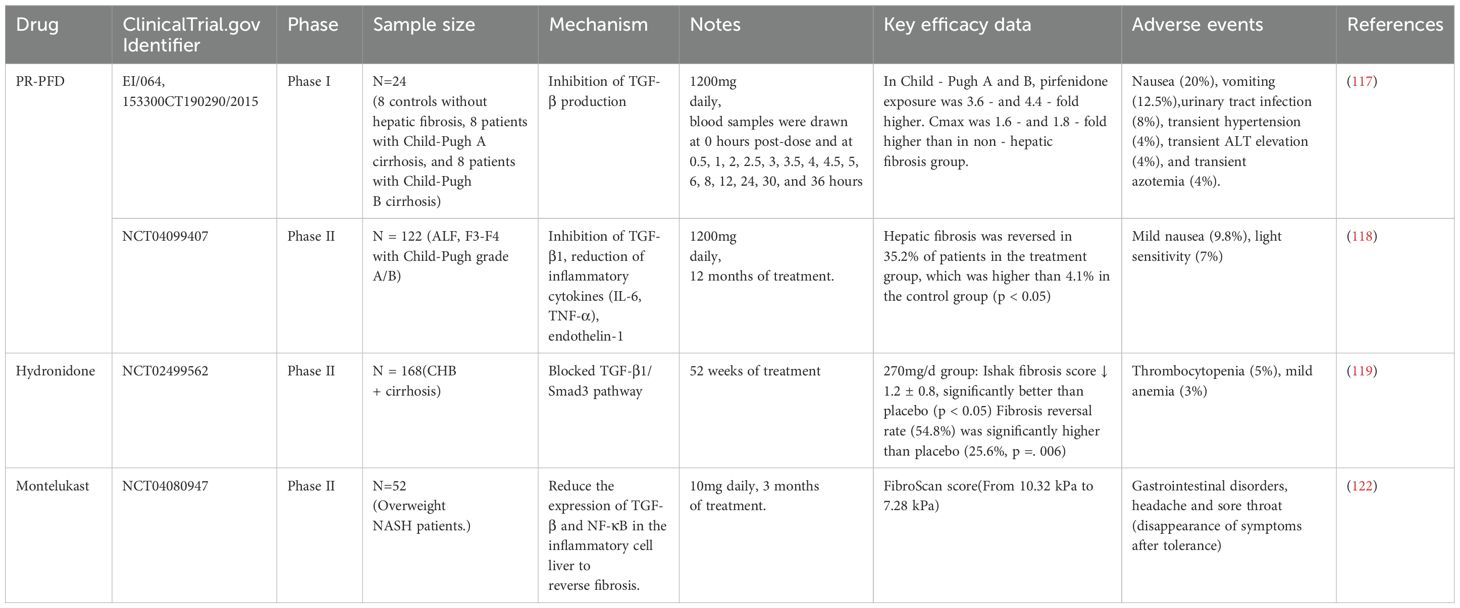
Table 2. Clinical application of TGF-β inhibitors.
Montelukast, a leukotriene receptor antagonist, is used in the treatment of non-alcoholic steatohepatitis. In a 12-week randomized, double-blind, placebo-controlled trial conducted in Egypt, 52 overweight non-alcoholic steatohepatitis patients were randomly assigned to receive either montelukast 10 mg once daily (n=26) or placebo (n=26). Results showed significant improvements in liver stiffness, liver enzymes, metabolic parameters, TNF-α, and liver fibrosis biomarkers (hyaluronic acid and TGF-β1) in the montelukast group, with good tolerability observed (122).
Clinical trial results indicate that TGF-β inhibitors can slow or reverse liver fibrosis and improve liver function. However, their long-term safety and efficacy require further validation. Additionally, different stages and etiologies of liver fibrosis may influence the effectiveness of TGF-β inhibitors. Furthermore, hepatic and renal function can affect drug metabolism and clearance, impacting drug concentration and efficacy.
3.3 Combination therapy
Combination therapy has received widespread attention in liver fibrosis research, as studies have shown that different drugs can enhance antifibrotic effects through synergistic mechanisms (Table 3). Andrographolide, a diterpenoid compound extracted from the traditional Chinese herb Andrographis paniculata, has been demonstrated in preclinical studies to prevent hepatic inflammation and fibrosis (123). The combination of PFD and andrographolide inhibits the TGF-β/Smad signaling pathway, reduces Smad2/3 phosphorylation, and downregulates the expression of α-SMA, Col-1, connective tissue growth factor (CTGF), and inflammatory factors such as IL-1β, IL-6, and TNF-α. This effect suppresses HSCs activation and improves liver fibrosis (124). Curcumin 2005-8, a curcumin derivative, improves fatty liver disease through AMPK activation and autophagy regulation (125). EW-7197 (vactosertib), a small-molecule inhibitor of TGF-βR I, alleviates fibrosis by reducing reactive oxygen species via the classical Smad2/3 pathway (126). The combined application of curcumin 2005–8 and EW-7197 reduces liver fibrosis and steatohepatitis while maintaining the benefits of both drugs (127).
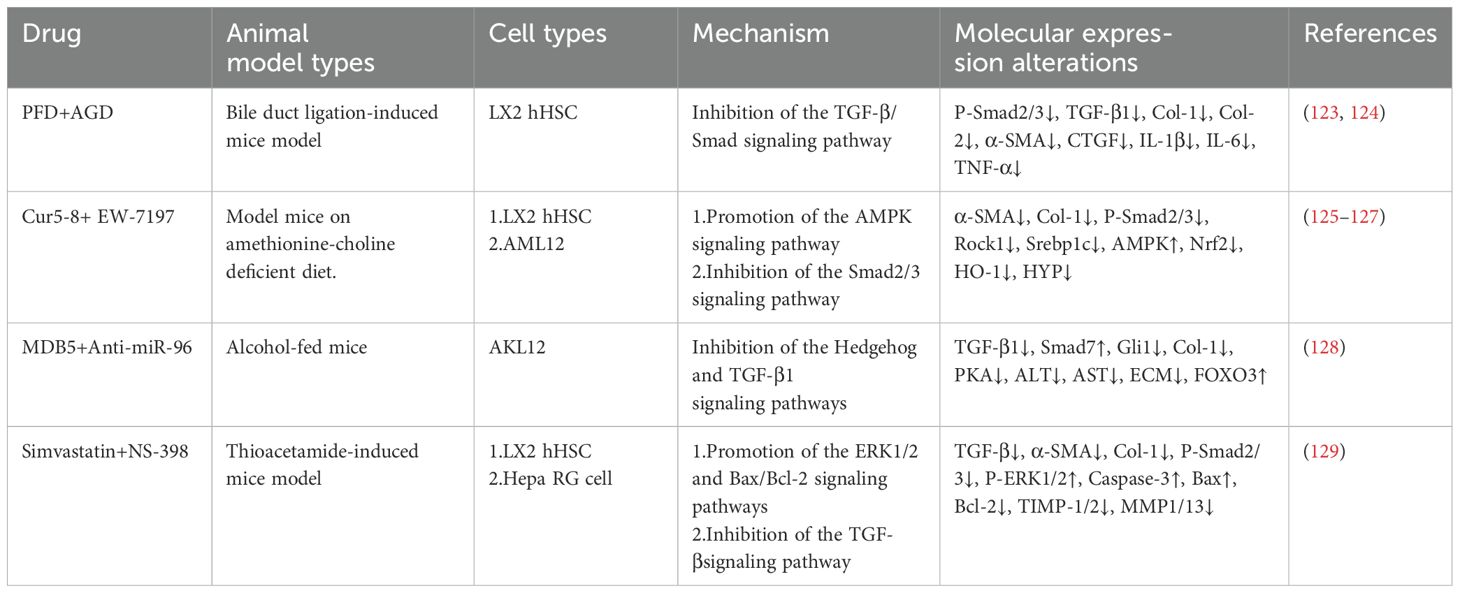
Table 3. The mechanism of combination therapy.
MDB5, a small molecule inhibitor targeting the Hedgehog pathway, blocks HSCs activation by inhibiting Gli1 transcriptional activity. Lipid nanoparticles co-loaded with MDB5 and anti-miR-96 suppress the Hedgehog pathway, reducing HSCs activation and ECM gene expression while upregulating forkhead box O3 (FOXO3) and Smad7. These actions collectively inhibit TGF-β1 signal transduction and collagen synthesis, reducing liver fibrosis (128). NS-398, a selective cyclooxygenase-2 (COX-2) inhibitor, alleviates inflammation-driven fibrosis by inhibiting the synthesis of prostaglandin E2. The combination of simvastatin and NS-398 exerts antifibrotic effects by activating the ERK1/2 and Bax/Bcl-2 signaling pathways, inhibiting the TGF-β pathway, and reducing TIMP-1 and TIMP-2 expression. This effect leads to decreased liver fibrosis and collagen deposition, ultimately suppressing HSCs activation (129).
These findings suggest that the rational development of multiple antifibrotic therapies, such as the combination of TGF-β inhibitors with natural compounds, has greater therapeutic potential for improving liver fibrosis.
3.4 Applications of TGF-β inhibitors in liver cancer treatment
In addition to treating liver fibrosis, inhibiting the TGF-β signaling pathway helps reduce tumor progression and metastasis. A nanoparticle-based drug (NCG) encapsulating the TGF-β receptor inhibitor galunisertib and the sonosensitizer chlorin e6 has been shown to inhibit the differentiation of myeloid-derived suppressor cells, induce M1-like polarization of tumor-associated macrophages, and disrupt the immunosuppressive barrier formed by tumor-associated fibroblasts. In a mouse model of colorectal cancer liver metastasis, combination therapy with NCG (+) and anti-PD-L1 effectively inhibited colorectal cancer liver metastasis (130). The C-C motif chemokine receptor 4 inhibitor C-021 or the TGF-βR I inhibitor galunisertib, when combined with anti-PD-L1 therapy, has been found to suppress SOX12-mediated HCC progression and metastasis (131). Similarly, the TGF-βR I inhibitor (vactosertib) or the C-X-C chemokine receptor 4 inhibitor (AMD3100), in combination with anti-PD-L1, has been shown to significantly inhibit SYR-related high-mobility group box 18-mediated HCC progression and metastasis (82). Moreover, microwave ablation in combination with the ALK5 inhibitor SB-525334 effectively inactivates the TGF-β1/Smad2/Smad3 pathway, reducing the survival rate of HCC cells and promoting apoptosis (132).
4 Conclusion and future prospects
The TGF-β signaling pathway plays a central role in the occurrence and progression of liver fibrosis. The processes it mediates, including HSCs activation, ECM deposition, and inflammation regulation, are key mechanisms driving fibrosis. In recent years, TGF-β inhibitors have emerged as potential antifibrotic therapeutic strategies, and significant progress has been made. Various interventions, including TGF-β neutralizing antibodies, TGF-β receptor antagonists, small-molecule inhibitors, and natural compounds, have been found to inhibit HSCs activation and reduce fibrosis marker expression in both in vitro cell experiments and animal models. Furthermore, several TGF-β inhibitors have entered clinical trials, with certain drugs (such as PFD and galunisertib) showing promising effects in improving liver fibrosis and liver function.
Although progress has been made in the use of TGF-β inhibitors for treating various fibrosis-related diseases (133–135), the multifaceted roles of TGF-β suggest that single-target inhibition may not fully address the complexities of fibrosis treatment. Therefore, future research should focus on developing multitarget therapeutic strategies, personalized treatment approaches, and novel drug delivery systems. For example, the combination of TGF-β inhibitors with anti-inflammatory agents, antioxidants, or immunomodulators may enhance antifibrotic efficacy. Integrating gene editing technologies and cell therapy also holds promise for advancing liver fibrosis treatment. Additionally, further clinical trials are essential to evaluate the long-term efficacy and safety of TGF-β inhibitors.
In conclusion, the use of TGF-β inhibitors represents a promising antifibrotic therapeutic strategy, demonstrating potential in both basic research and clinical trials. TGF-β-targeted therapy may become an important approach for treating liver fibrosis and related liver diseases in the future through approaches to optimize drug design, develop combination therapies, and advance precision medicine applications.
Author contributions
WW: Conceptualization, Writing – original draft. YG: Data curation, Writing – review & editing. YC: Investigation, Writing – review & editing. MC: Visualization, Writing – review & editing. YS: Data curation, Writing – original draft. LW: Investigation, Writing – review & editing. RD: Resources, Writing – original draft. YW: Writing – review & editing. LZ: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by grants from the National Natural Science Foundation of China (82274307), Hefei City Science and Technology Bureau of Life and Health Special Project (GJ2022SM03), 2022 Clinical Medical Research Translation Special Project (202104j07020014), Anhui Provincial Natural Science Foundation Facial Project (2308085MH292, 2208085MH269), Anhui Province Higher Education Institutions Scientific Research Key Project (2023AH050749, 2022AH050457, and 2022AH050465), and Research Project of the Center for Xin’an Medicine and Modernization of Traditional Chinese Medicine of IHM, Anhui University of Chinese Medicine, 2023 (No. 2023CXMMTCM018).
Acknowledgments
We thank LetPub (www.letpub.com.cn) for its linguistic assistance during the preparation of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
ECM: Extracellular matrix
CLD: Chronic liver disease
HCC: Hepatocellular carcinoma
TGF-β: Transforming growth factor-beta
BMP: Bone morphogenetic protein
GDF: Growth and differentiation factor
HSCs: Hepatic stellate cells
MFBs: Myofibroblasts
MMP: Matrix metalloproteinase
TGF-βR I: Transforming growth factor beta receptor I
TGF-βR II: Transforming growth factor beta receptor II
Smad: Small mothers against decapentaplegic
R-Smad: Receptor-regulated small mothers against decapentaplegic
Co-Smad: Common small mothers against decapentaplegic
I-Smad: Inhibitory small mothers against decapentaplegic
MAPK: Mitogen-activated protein kinase
PI3K/Akt: Phosphoinositide 3-kinase/protein kinase B
NF-κB: Nuclear factor kappa-light-chain-enhancer of activated B cells
Wnt/β-catenin: Wingless-related integration site/β-catenin
mTOR: Mammalian target of rapamycin
NAFLD: Nonalcoholic fatty liver disease
TIMP: Tissue inhibitors of metalloproteinase
α-SMA: α-smooth muscle actin
CCL2: C-C motif chemokine ligand 2
TNF-α: Tumor necrosis factor-alpha
IL-6: Interleukin-6
HepG2 cell: Human hepatocellular carcinoma cell line HepG2
hHSCs: Human hepatic stellate cells
PD-L1: Programmed death-ligand 1
ARK5: AMP-activated protein kinase 5
CCl4: Carbon tetrachloride
Col-1: Type I collagen
HSC-LX2: Hepatic stellate cell line LX2
GPR65: G protein-coupled receptor 65
JNK: C-Jun N-terminal kinase
LX-2 HSCs: LX-2 human hepatic stellate cell line
NR1: Notoginsenoside R1
PPAR-γ: Peroxisome proliferator-activated receptor gamma
PDGFRB: Platelet-derived growth factor receptor B
PAI-1: Plasminogen activator inhibitor-1
CPP-A-1: Codonopsis pilosula root polysaccharide
TAK1: Transforming growth factor-β-activated kinase 1
IκBα: Inhibitor of nuclear factor kappa B alpha
CCN2: Cellular communication network factor 2
PDGFB: Platelet-derived growth factor B
TLR: Toll-like receptor
FoxO1: Forkhead box O1
PFD: Pirfenidone
PR-PFD: Prolonged-release pirfenidone formulation
CTGF: Connective tissue growth factor
COX-2: Cyclooxygenase-2
NCG: Nanoparticle-based drug
References
1. Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. (2007) 117:539–48. doi: 10.1172/JCI30542
2. Ekstedt M, Franzén LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. (2006) 44:865–73. doi: 10.1002/hep.21327
3. Cheemerla S and Balakrishnan M. Global epidemiology of chronic liver disease. Clin Liver Dis. (2021) 17:365–70. doi: 10.1002/cld.1061
4. Asrani SK, Devarbhavi H, Eaton J, and Kamath PS. Burden of liver diseases in the world. J Hepatol. (2019) 70:151–71. doi: 10.1016/j.jhep.2018.09.014
5. Tapper EB and Parikh ND. Mortality due to cirrhosis and liver cancer in the United States, 1999-2016: observational study. BMJ. (2018) 362:k2817. doi: 10.1136/bmj.k2817
6. GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. (2018) 392:1789–858. doi: 10.1016/S0140-6736(18)32279-7
7. David CJ and Massagué J. Contextual determinants of TGF-β action in development, immunity and cancer. Nat Rev Mol Cell Biol. (2018) 19:419–35. doi: 10.1038/s41580-018-0007-0
8. Lan HY. Diverse roles of TGF-β/Smads in renal fibrosis and inflammation. Int J Biol Sci. (2011) 7:1056–67. doi: 10.7150/ijbs.7.1056
9. Cao M, Zhao Q, Xia H, Lyu S, Luo J, Fu K, et al. Intracellular and extracellular Cyclophilin a promote cardiac fibrosis through TGF-β signaling in response to angiotensin II. Biochem Pharmacol. (2024) 225:116271. doi: 10.1016/j.bcp.2024.116271
10. Wang Y, Ping Z, Gao H, Liu Z, Xv Q, Jiang X, et al. LYC inhibits the AKT signaling pathway to activate autophagy and ameliorate TGFB-induced renal fibrosis. Autophagy. (2024) 20:1114–33. doi: 10.1080/15548627.2023.2287930
11. Trinh-Minh T, Chen CW, Tran Manh C, Li YN, Zhu H, Zhou X, et al. Noncanonical WNT5A controls the activation of latent TGF-β to drive fibroblast activation and tissue fibrosis. J Clin Invest. (2024) 134:e159884. doi: 10.1172/JCI159884
12. Humeres C, Shinde AV, Tuleta I, Hernandez SC, Hanna A, Huang S, et al. Fibroblast Smad7 induction protects the remodeling pressure-overloaded heart. Circ Res. (2024) 135:453–69. doi: 10.1161/CIRCRESAHA.123.323360
13. Sakai K, Jawaid S, Sasaki T, Bou-Gharios G, and Sakai T. Transforming growth factor-β–independent role of connective tissue growth factor in the development of liver fibrosis. Am J Pathol. (2014) 184:2611–7. doi: 10.1016/j.ajpath.2014.06.009
14. Fabregat I and Caballero-Díaz D. Transforming growth factor-β-induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front Oncol. (2018) 8:357. doi: 10.3389/fonc.2018.00357
15. Fu X, Qie J, Fu Q, Chen J, Jin Y, and Ding Z. miR-20a-5p/TGFBR2 axis affects pro-inflammatory macrophages and aggravates liver fibrosis. Front Oncol. (2020) 10:107. doi: 10.3389/fonc.2020.00107
16. Tsuchida T and Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. (2017) 14:397–411. doi: 10.1038/nrgastro.2017.38
17. Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, et al. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. (2002) 106:130–5. doi: 10.1161/01.cir.0000020689.12472.e0
18. Meng L, Lu Y, Wang X, Cheng C, Xue F, Xie L, et al. NPRC deletion attenuates cardiac fibrosis in diabetic mice by activating PKA/PKG and inhibiting TGF-β1/Smad pathways. Sci Adv. (2023) 9:eadd4222. doi: 10.1126/sciadv.add4222
19. Qiu Y, Song X, Liu Y, Wu Y, Shi J, Zhang F, et al. Application of recombinant TGF-β1 inhibitory peptide to alleviate isoproterenol-induced cardiac fibrosis. Appl Microbiol Biotechnol. (2023) 107:6251–62. doi: 10.1007/s00253-023-12722-x
20. Fukasawa H, Yamamoto T, Suzuki H, Togawa A, Ohashi N, Fujigaki Y, et al. Treatment with anti-TGF-beta antibody ameliorates chronic progressive nephritis by inhibiting Smad/TGF-beta signaling. Kidney Int. (2004) 65:63–74. doi: 10.1111/j.1523-1755.2004.00393.x
21. Liang X, Schnaper HW, Matsusaka T, Pastan I, Ledbetter S, and Hayashida T. Anti-TGF-β antibody, 1D11, ameliorates glomerular fibrosis in mouse models after the onset of proteinuria. PloS One. (2016) 11:e0155534. doi: 10.1371/journal.pone.0155534
22. McGaraughty S, Davis-Taber RA, Zhu CZ, Cole TB, Nikkel AL, Chhaya M, et al. Targeting anti–TGF-β therapy to fibrotic kidneys with a dual specificity antibody approach. J Am Soc Nephrol. (2017) 28:3616–26. doi: 10.1681/ASN.2017010013
23. Hinck AP, Mueller TD, and Springer TA. Structural biology and evolution of the TGF-β family. Cold Spring Harb Perspect Biol. (2016) 8:a022103. doi: 10.1101/cshperspect.a022103
24. Dong X, Zhao B, Iacob RE, Zhu J, Koksal AC, Lu C, et al. Force interacts with macromolecular structure in activation of TGF-β. Nature. (2017) 542:55–9. doi: 10.1038/nature21035
25. Clark DA and Coker R. Molecules in focus tansforming growth factor-beta (TGF-β). Int J Biochem Cell Biol. (1998) 30:293–8. doi: 10.1016/S1357-2725(97)00128-3
26. Chang Y, Bach L, Hasiuk M, Wen L, Elmzzahi T, Tsui C, et al. TGF-β specifies TFH versus TH17 cell fates in murine CD4+ T cells through c-Maf. Sci Immunol. (2024) 9:eadd4818. doi: 10.1126/sciimmunol.add4818
27. Haider S, Lackner AI, Dietrich B, Kunihs V, Haslinger P, Meinhardt G, et al. Transforming growth factor-β signaling governs the differentiation program of extravillous trophoblasts in the developing human placenta. Proc Natl Acad Sci USA. (2022) 119:e2120667119. doi: 10.1073/pnas.2120667119
28. Tsukui T, Wolters PJ, and Sheppard D. Alveolar fibroblast lineage orchestrates lung inflammation and fibrosis. Nature. (2024) 631:627–34. doi: 10.1038/s41586-024-07660-1
29. Geng K, Ma X, Jiang Z, Gu J, Huang W, Wang W, et al. WDR74 facilitates TGF-β/Smad pathway activation to promote M2 macrophage polarization and diabetic foot ulcer wound healing in mice. Cell Biol Toxicol. (2023) 39:1577–91. doi: 10.1007/s10565-022-09748-8
30. Chen W. TGF-β regulation of T cells. Annu Rev Immunol. (2023) 41:483–512. doi: 10.1146/annurev-immunol-101921-045939
31. Moreau JM, Velegraki M, Bolyard C, Rosenblum MD, and Li Z. Transforming growth factor–β1 in regulatory T cell biology. Sci Immunol. (2022) 7:eabi4613. doi: 10.1126/sciimmunol.abi4613
32. Chang C. Agonists and antagonists of TGF-β family ligands. Cold Spring Harb Perspect Biol. (2016) 8:a021923. doi: 10.1101/cshperspect.a021923
33. Morikawa M, Derynck R, and Miyazono K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. (2016) 8:a021873. doi: 10.1101/cshperspect.a021873
34. Frangogiannis NG. Transforming growth factor–β in tissue fibrosis. J Exp Med. (2020) 217:e20190103. doi: 10.1084/jem.20190103
35. Blakytny R, Ludlow A, Martin GEM, Ireland G, Lund LR, Ferguson MWJ, et al. Latent TGF-β1 activation by platelets. J Cell Physiol. (2004) 199:67–76. doi: 10.1002/jcp.10454
36. Huang H, Wang Z, Zhang Y, Pradhan RN, Ganguly D, Chandra R, et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell. (2022) 40:656–673.e7. doi: 10.1016/j.ccell.2022.04.011
37. Frangogiannis NG. TGF-β as a therapeutic target in the infarcted and failing heart: cellular mechanisms, challenges, and opportunities. Expert Opin Ther Targets. (2024) 28:45–56. doi: 10.1080/14728222.2024.2316735
38. Miyazawa K, Shinozaki M, Hara T, Furuya T, and Miyazono K. Two major Smad pathways in TGF-beta superfamily signalling. Genes Cells. (2002) 7:1191–204. doi: 10.1046/j.1365-2443.2002.00599.x
39. Dewidar B, Meyer C, Dooley S, and Meindl-Beinker N. TGF-β in hepatic stellate cell activation and liver fibrogenesis—updated 2019. Cells. (2019) 8:1419. doi: 10.3390/cells8111419
40. Chanda D, Otoupalova E, Smith SR, Volckaert T, De Langhe SP, and Thannickal VJ. Developmental pathways in the pathogenesis of lung fibrosis. Mol Aspects Med. (2019) 65:56–69. doi: 10.1016/j.mam.2018.08.004
41. Isaka Y. Targeting TGF-β signaling in kidney fibrosis. Int J Mol Sci. (2018) 19:2532. doi: 10.3390/ijms19092532
42. Principe DR, Doll JA, Bauer J, Jung B, Munshi HG, Bartholin L, et al. TGF-β: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst. (2014) 106:djt369. doi: 10.1093/jnci/djt369
43. Lebrun JJ. The dual role of TGFβ in human cancer: from tumor suppression to cancer metastasis. ISRN Mol Biol. (2012) 2012:381428. doi: 10.5402/2012/381428
44. Wrana JL, Attisano L, Wieser R, Ventura F, and Massagué J. Mechanism of activation of the TGF-beta receptor. Nature. (1994) 370:341–7. doi: 10.1038/370341a0
45. Kawabata M and Miyazono K. Signal transduction of the TGF-beta superfamily by Smad proteins. J Biochem. (1999) 125:9–16. doi: 10.1093/oxfordjournals.jbchem.a022273
46. Chen SJ, Yuan W, Lo S, Trojanowska M, and Varga J. Interaction of smad3 with a proximal smad-binding element of the human alpha2(I) procollagen gene promoter required for transcriptional activation by TGF-beta. J Cell Physiol. (2000) 183:381–92. doi: 10.1002/(SICI)1097-4652(200006)183:3<381::AID-JCP11>3.0.CO;2-O
47. Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. (1997) 16:5353–62. doi: 10.1093/emboj/16.17.5353
48. Shi Y and Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. (2003) 113:685–700. doi: 10.1016/s0092-8674(03)00432-x
49. Retting KN, Song B, Yoon BS, and Lyons KM. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development. (2009) 136:1093–104. doi: 10.1242/dev.029926
50. Wang B, Zhao Q, Gong X, Wang C, Bai Y, Wang H, et al. Transmembrane anterior posterior transformation 1 regulates BMP signaling and modulates the protein stability of SMAD1/5. J Biol Chem. (2022) 298:102684. doi: 10.1016/j.jbc.2022.102684
51. Imamura T, Takase M, Nishihara A, Oeda E, Hanai J, Kawabata M, et al. Smad6 inhibits signalling by the TGF-beta superfamily. Nature. (1997) 389:622–6. doi: 10.1038/39355
52. Mochizuki T, Miyazaki H, Hara T, Furuya T, Imamura T, Watabe T, et al. Roles for the MH2 domain of Smad7 in the specific inhibition of transforming growth factor-β superfamily signaling. J Biol Chem. (2004) 279:31568–74. doi: 10.1074/jbc.M313977200
53. Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, et al. The MAD-related protein smad7 associates with the TGFβ Receptor and functions as an antagonist of TGFβ Signaling. Cell. (1997) 89:1165–73. doi: 10.1016/S0092-8674(00)80303-7
54. Zhang YE. Non-Smad pathways in TGF-β signaling. Cell Res. (2009) 19:128–39. doi: 10.1038/cr.2008.328
55. Zhang YE. Non-smad signaling pathways of the TGF-β Family. Cold Spring Harb Perspect Biol. (2017) 9:a022129. doi: 10.1101/cshperspect.a022129
56. Luo K. Signaling cross talk between TGF-β/smad and other signaling pathways. Cold Spring Harb Perspect Biol. (2017) 9:a022137. doi: 10.1101/cshperspect.a022137
57. Zhuang C, Guo Z, Zhu J, Wang W, Sun R, Qi M, et al. PTEN inhibitor attenuates cardiac fibrosis by regulating the M2 macrophage phenotype via the PI3K/AKT/TGF-β/Smad 2/3 signaling pathway. Int J Cardiol. (2022) 356:88–96. doi: 10.1016/j.ijcard.2022.04.007
58. Mallat A and Lotersztajn S. Cellular Mechanisms of Tissue Fibrosis. 5. Novel insights into liver fibrosis. Am J Physiol Cell Physiol. (2013) 305:C789–99. doi: 10.1152/ajpcell.00230.2013
59. Parola M and Pinzani M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med. (2019) 65:37–55. doi: 10.1016/j.mam.2018.09.002
60. Ginès P, Krag A, Abraldes JG, Solà E, Fabrellas N, and Kamath PS. Liver cirrhosis. Lancet. (2021) 398:1359–76. doi: 10.1016/S0140-6736(21)01374-X
61. Kumar S, Duan Q, Wu R, Harris EN, and Su Q. Pathophysiological communication between hepatocytes and non-parenchymal cells in liver injury from NAFLD to liver fibrosis. Adv Drug Delivery Rev. (2021) 176:113869. doi: 10.1016/j.addr.2021.113869
62. Bouwens L, Baekeland M, De Zanger R, and Wisse E. Quantitation, tissue distribution and proliferation kinetics of Kupffer cells in normal rat liver. Hepatology. (1986) 6:718–22. doi: 10.1002/hep.1840060430
63. Elpek GÖ. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J Gastroenterol. (2014) 20:7260. doi: 10.3748/wjg.v20.i23.7260
64. Blaner WS, O’Byrne SM, Wongsiriroj N, Kluwe J, D’Ambrosio D, Jiang H, et al. Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage. Biochim Biophys Acta. (2009) 1791:467–73. doi: 10.1016/j.bbalip.2008.11.001
65. McConnell MJ, Kostallari E, Ibrahim SH, and Iwakiri Y. The evolving role of liver sinusoidal endothelial cells in liver health and disease. Hepatology. (2023) 78:649–69. doi: 10.1097/HEP.0000000000000207
66. Koyama Y and Brenner DA. Liver inflammation and fibrosis. J Clin Invest. (2017) 127:55–64. doi: 10.1172/JCI88881
67. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A. (2012) 109:E3186–95. doi: 10.1073/pnas.1119964109
68. Tacke F and Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatology. (2014) 60:1090–6. doi: 10.1016/j.jhep.2013.12.025
69. Higashi T, Friedman SL, and Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Advanced Drug delivery Rev. (2017) 121:27. doi: 10.1016/j.addr.2017.05.007
70. Xu F, Liu C, Zhou D, and Zhang L. TGF-β/Smad pathway and its regulation in hepatic fibrosis. J Histochem Cytochem. (2016) 64:157–67. doi: 10.1369/0022155415627681
71. Dooley S, Hamzavi J, Breitkopf K, Wiercinska E, Said HM, Lorenzen J, et al. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology. (2003) 125:178–91. doi: 10.1016/S0016-5085(03)00666-8
72. Zhang K, Han X, Zhang Z, Zheng L, Hu Z, Yao Q, et al. The liver-enriched lnc-LFAR1 promotes liver fibrosis by activating TGF-β and Notch pathways. Nat Commun. (2017) 8:144. doi: 10.1038/s41467-017-00204-4
73. Wang JN, Li L, Li LY, Yan Q, Li J, and Xu T. Emerging role and therapeutic implication of Wnt signaling pathways in liver fibrosis. Gene. (2018) 674:57–69. doi: 10.1016/j.gene.2018.06.053
74. Liu H, Huang H, Liu Y, Yang Y, Deng H, Wang X, et al. Adipose-derived mesenchymal stem cells inhibit hepatic stellate cells activation to alleviate liver fibrosis via Hippo pathway. Stem Cell Res Ther. (2024) 15:378. doi: 10.1186/s13287-024-03988-7
75. Weiskirchen R, Weiskirchen S, and Tacke F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol Aspects Med. (2019) 65:2–15. doi: 10.1016/j.mam.2018.06.003
76. Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang Y, et al. Macrophage polarization and its role in liver disease. Front Immunol. (2021) 12:803037. doi: 10.3389/fimmu.2021.803037
77. Pellicoro A, Aucott RL, Ramachandran P, Robson AJ, Fallowfield JA, Snowdon VK, et al. Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology. (2012) 55:1965–75. doi: 10.1002/hep.25567
78. Feng M, Ding J, Wang M, Zhang J, Zhu X, and Guan W. Kupffer-derived matrix metalloproteinase-9 contributes to liver fibrosis resolution. Int J Biol Sci. (2018) 14:1033–40. doi: 10.7150/ijbs.25589
79. Rampa DR, Feng H, Allur-Subramaniyan S, Shim K, Pekcec A, Lee D, et al. Kinin B1 receptor blockade attenuates hepatic fibrosis and portal hypertension in chronic liver diseases in mice. J Transl Med. (2022) 20:590. doi: 10.1186/s12967-022-03808-7
80. Calixto-Tlacomulco S, Luna-Reyes I, Delgado-Coello B, Gutiérrez-Vidal R, Reyes-Grajeda JP, and Mas-Oliva J. CETP-derived peptide seq-1, the key component of HB-ATV-8 vaccine prevents stress responses, and promotes downregulation of pro-fibrotic genes in hepatocytes and stellate cells. Arch Med Res. (2024) 55:102937. doi: 10.1016/j.arcmed.2023.102937
81. Lan Y, Yeung TL, Huang H, Wegener AA, Saha S, Toister-Achituv M, et al. Colocalized targeting of TGF-β and PD-L1 by bintrafusp alfa elicits distinct antitumor responses. J Immunother Cancer. (2022) 10:e004122. doi: 10.1136/jitc-2021-004122
82. Chen J, Feng W, Sun M, Huang W, Wang G, Chen X, et al. TGF-β1-induced SOX18 elevation promotes hepatocellular carcinoma progression and metastasis through transcriptionally upregulating PD-L1 and CXCL12. Gastroenterology. (2024) 167:264–80. doi: 10.1053/j.gastro.2024.02.025
83. Zhang H, Yang H, Liu X, Ying J, Zu T, Jiang J, et al. Targeted inhibition of transforming growth factor-β type I receptor by AZ12601011 improves paraquat poisoning-induced multiple organ fibrosis. Pestic Biochem Physiol. (2024) 200:105831. doi: 10.1016/j.pestbp.2024.105831
84. Ye Z, He Q, Wang Q, Lin Y, Cen K, and Chen X. LINC00922 promotes the proliferation, migration, invasion and EMT process of liver cancer cells by regulating miR-424-5p/ARK5. Mol Cell Biochem. (2021) 476:3757–69. doi: 10.1007/s11010-021-04196-0
85. Liu L, Ulbrich J, Müller J, Wüstefeld T, Aeberhard L, Kress TR, et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature. (2012) 483:608–12. doi: 10.1038/nature10927
86. You Y, Gao C, Wu J, Qu H, Xiao Y, Kang Z, et al. Enhanced expression of ARK5 in hepatic stellate cell and hepatocyte synergistically promote liver fibrosis. Int J Mol Sci. (2022) 23:13084. doi: 10.3390/ijms232113084
87. Shen D, Cheng H, Cai B, Cai W, Wang B, Zhu Q, et al. N-n-butyl haloperidol iodide ameliorates liver fibrosis and hepatic stellate cell activation in mice. Acta Pharmacol Sin. (2022) 43:133–45. doi: 10.1038/s41401-021-00630-7
88. Yang HX, Guo FY, Lin YC, Wu YL, Nan JX, Jin CH, et al. Synthesis of and anti-fibrotic effect of pyrazole derivative J-1048: inhibition of ALK5 as a novel approach to liver fibrosis targeting inflammation. Bioorg Chem. (2023) 139:106723. doi: 10.1016/j.bioorg.2023.106723
89. Peterson JM, Jay JW, Wang Y, Joglar AA, Prasai A, Palackic A, et al. Galunisertib exerts antifibrotic effects on TGF-β-induced fibroproliferative dermal fibroblasts. Int J Mol Sci. (2022) 23:6689. doi: 10.3390/ijms23126689
90. van Leeuwen LL, Ruigrok MJR, Kessler BM, Leuvenink HGD, and Olinga P. Targeted delivery of galunisertib using machine perfusion reduces fibrogenesis in an integrated ex vivo renal transplant and fibrogenesis model. Br J Pharmacol. (2024) 181:464–79. doi: 10.1111/bph.16220
91. Panzarini E, Leporatti S, Tenuzzo BA, Quarta A, Hanafy NAN, Giannelli G, et al. Therapeutic effect of polymeric nanomicelles formulation of LY2157299-galunisertib on CCl4-induced liver fibrosis in rats. J Pers Med. (2022) 12:1812. doi: 10.3390/jpm12111812
92. Jiang XL, Liu C, Zhan ZY, Lan XQ, Wu YL, Nan JX, et al. Thiazole isomers as potential ALK5 inhibitors alleviate P2X7R-mediated inflammation during liver fibrosis. Int Immunopharmacol. (2025) 153:114472. doi: 10.1016/j.intimp.2025.114472
93. Hu Y, Li L, Tian Y, Xiao Y, Tang J, Zeng S, et al. Design, synthesis and evaluation of novel UDCA-aminopyrimidine hybrids as ATX inhibitors for the treatment of hepatic and pulmonary fibrosis. Eur J Med Chem. (2024) 264:116029. doi: 10.1016/j.ejmech.2023.116029
94. Liu X, Wang X, Xu L, Fan J, Yuan Q, Zhang F, et al. Targeting delivery of a novel TGF-β type I receptor-mimicking peptide to activated hepatic stellate cells for liver fibrosis therapy via inhibiting the TGF-β1/smad and p38 MAPK signaling pathways. Eur J Pharmacol. (2024) 977:176708. doi: 10.1016/j.ejphar.2024.176708
95. Ding C. SIRT7 protects against liver fibrosis by suppressing stellate cell activation via TGF-β/SMAD2/3 pathway. BioMed Pharmacother. (2024) 180:117477. doi: 10.1016/j.biopha.2024.117477
96. Dong XC. Sirtuin 6—a key regulator of hepatic lipid metabolism and liver health. Cells. (2023) 12:663. doi: 10.3390/cells12040663
97. Ren Y, Chen Y, Tang EH, Hu Y, Niu B, Liang H, et al. Arbidol attenuates liver fibrosis and activation of hepatic stellate cells by blocking TGF-β1 signaling. Eur J Pharmacol. (2024) 967:176367. doi: 10.1016/j.ejphar.2024.176367
98. Zhang K, Zhang MX, Meng XX, Zhu J, Wang JJ, He YF, et al. Targeting GPR65 alleviates hepatic inflammation and fibrosis by suppressing the JNK and NF-κB pathways. Mil Med Res. (2023) 10:56. doi: 10.1186/s40779-023-00494-4
99. Shen Y, Cheng L, Xu M, Wang W, Wan Z, Xiong H, et al. SGLT2 inhibitor empagliflozin downregulates miRNA-34a-5p and targets GREM2 to inactivate hepatic stellate cells and ameliorate non-alcoholic fatty liver disease-associated fibrosis. Metabolism. (2023) 146:155657. doi: 10.1016/j.metabol.2023.155657
100. Guo C, Lai L, Ma B, Huang Q, and Wang Z. Notoginsenoside R1 targets PPAR-γ to inhibit hepatic stellate cell activation and ameliorates liver fibrosis. Exp Cell Res. (2024) 437:113992. doi: 10.1016/j.yexcr.2024.113992
101. Ceccherini E, Signore G, Tedeschi L, Vozzi F, Di Giorgi N, Michelucci E, et al. Proteomic modulation in TGF-β-treated cholangiocytes induced by curcumin nanoparticles. Int J Mol Sci. (2023) 24:10481. doi: 10.3390/ijms241310481
102. Gabr SA, Elsaed WM, Eladl MA, El-Sherbiny M, Ebrahim HA, Asseri SM, et al. Curcumin modulates oxidative stress, fibrosis, and apoptosis in drug-resistant cancer cell lines. Life. (2022) 12:1427. doi: 10.3390/life12091427
103. Shu Y, He Y, Ye G, Liu X, Huang J, Zhang Q, et al. Curcumin inhibits the activity and induces apoptosis of activated hepatic stellate cell by suppressing autophagy. J Cell Biochem. (2023) 124:1764–78. doi: 10.1002/jcb.30487
104. Elzoheiry A, Ayad E, Omar N, Elbakry K, and Hyder A. Anti-liver fibrosis activity of curcumin/chitosan-coated green silver nanoparticles. Sci Rep. (2022) 12:18403. doi: 10.1038/s41598-022-23276-9
105. Huang H, Wei S, Wu X, Zhang M, Zhou B, Huang D, et al. Dihydrokaempferol attenuates CCl4-induced hepatic fibrosis by inhibiting PARP-1 to affect multiple downstream pathways and cytokines. Toxicol Appl Pharmacol. (2023) 464:116438. doi: 10.1016/j.taap.2023.116438
106. Wu J, Zhang D, Zhu B, Wang S, Xu Y, Zhang C, et al. Rubus chingii hu. unripe fruits extract ameliorates carbon tetrachloride-induced liver fibrosis and improves the associated gut microbiota imbalance. Chin Med. (2022) 17:56. doi: 10.1186/s13020-022-00607-6
107. Lee JH, Jang EJ, Seo HL, Ku SK, Lee JR, Shin SS, et al. Sauchinone attenuates liver fibrosis and hepatic stellate cell activation through TGF-β/Smad signaling pathway. Chem-Biol Interact. (2014) 224:58–67. doi: 10.1016/j.cbi.2014.10.005
108. Meng X, Kuang H, Wang Q, Zhang H, Wang D, and Kang T. A polysaccharide from codonopsis pilosula roots attenuates carbon tetrachloride-induced liver fibrosis via modulation of TLR4/NF-κB and TGF-β1/Smad3 signaling pathway. Int Immunopharmacol. (2023) 119:110180. doi: 10.1016/j.intimp.2023.110180
109. Nie YM, Zhou WQ, Niu T, Mao MF, Zhan YX, Li Y, et al. Peptidoglycan isolated from the fruit of Lycium barbarum alleviates liver fibrosis in mice by regulating the TGF-β/Smad7 signaling and gut microbiota. Acta Pharmacol Sin. (2025) 46:1329–44. doi: 10.1038/s41401-024-01454-x
110. Pandey A, Raj P, Goru SK, Kadakol A, Malek V, Sharma N, et al. Esculetin ameliorates hepatic fibrosis in high fat diet induced non-alcoholic fatty liver disease by regulation of FoxO1 mediated pathway. Pharmacol Rep. (2017) 69:666–72. doi: 10.1016/j.pharep.2017.02.005
111. Lan T, Jiang S, Zhang J, Weng Q, Yu Y, Li H, et al. Breviscapine alleviates NASH by inhibiting TGF-β-activated kinase 1-dependent signaling. Hepatology. (2022) 76:155–71. doi: 10.1002/hep.32221
112. Li D, Ma D, Liu Y, Liu L, Chen Y, Liu H, et al. Extracts of periplaneta americana alleviate hepatic fibrosis by affecting hepatic TGF-β and NF-κB expression in rats with pig serum-induced liver fibrosis. Folia Histochem Cytobiol. (2022) 60:125–35. doi: 10.5603/FHC.a2022.0011
113. Lee SM, Koh DH, Jun DW, Roh YJ, Kang HT, Oh JH, et al. Auranofin attenuates hepatic steatosis and fibrosis in nonalcoholic fatty liver disease via NRF2 and NF- κB signaling pathways. Clin Mol Hepatol. (2022) 28:827–40. doi: 10.3350/cmh.2022.0068
114. Elazab ST and Hsu WH. Ferulic acid ameliorates concanavalin a-induced hepatic fibrosis in mice via suppressing TGF-β/smad signaling. Toxicol Appl Pharmacol. (2024) 492:117099. doi: 10.1016/j.taap.2024.117099
115. Albalawi AZ, Alatawi AS, Al-Atwi SM, Alhwyty LS, Alharbi KM, Alshehri SA, et al. EChinacoside ameliorates hepatic fibrosis and tumor invasion in rats with thioacetamide-induced hepatocellular carcinoma. Biomol Biomed. (2024) 24:1186–98. doi: 10.17305/bb.2024.10367
116. Xie Y, Jiang H, Zhang Q, Mehrotra S, Abel PW, Toews ML, et al. Upregulation of RGS2: a new mechanism for pirfenidone amelioration of pulmonary fibrosis. Respir Res. (2016) 17:103. doi: 10.1186/s12931-016-0418-4
117. Poo JL, Aguilar JR, Bernal-Reyes R, Alonso-Campero R, Gasca F, Hernández L, et al. Prolonged release pirfenidone pharmacokinetics is modified in cirrhosis GENESIS study. BioMed Pharmacother. (2023) 168:115712. doi: 10.1016/j.biopha.2023.115712
118. Poo JL, Torre A, Aguilar-Ramírez JR, Cruz M, Mejía-Cuán L, Cerda E, et al. Benefits of prolonged-release pirfenidone plus standard of care treatment in patients with advanced liver fibrosis: PROMETEO study. Hepatol Int. (2020) 14:817–27. doi: 10.1007/s12072-020-10069-3
119. Cai X, Liu X, Xie W, Ma A, Tan Y, Shang J, et al. Hydronidone for the treatment of liver fibrosis related to chronic hepatitis B: A phase 2 randomized controlled trial. Clin Gastroenterol Hepatol. (2023) 21:1893–1901.e7. doi: 10.1016/j.cgh.2022.05.056
120. Kuronuma K, Susai N, Kuroita T, Yamamoto H, Yoshioka T, Kaneko S, et al. Analysis of real-world data and a mouse model indicates that pirfenidone causes pellagra. ERJ Open Res. (2022) 8:00245–2022. doi: 10.1183/23120541.00245-2022
121. Cottin V, Koschel D, Günther A, Albera C, Azuma A, Sköld CM, et al. Long-term safety of pirfenidone: results of the prospective, observational PASSPORT study. ERJ Open Res. (2018) 4:00084–2018. doi: 10.1183/23120541.00084-2018
122. Abdallah MS, Eldeen AH, Tantawy SS, and Mostafa TM. The leukotriene receptor antagonist montelukast in the treatment of non-alcoholic steatohepatitis: A proof-of-concept, randomized, double-blind, placebo-controlled trial. Eur J Pharmacol. (2021) 906:174295. doi: 10.1016/j.ejphar.2021.174295
123. Lin L, Li R, Cai M, Huang J, Huang W, Guo Y, et al. Andrographolide ameliorates liver fibrosis in mice: involvement of TLR4/NF-κB and TGF-β1/smad2 signaling pathways. Oxid Med Cell Longev. (2018) 2018:7808656. doi: 10.1155/2018/7808656
124. Xu G, Ma T, Zhou C, Zhao F, Peng K, and Li B. Combination of pirfenidone and andrographolide ameliorates hepatic stellate cell activation and liver fibrosis by mediating TGF- β/smad signaling pathway. Anal Cell Pathol. (2024) 2024:2751280. doi: 10.1155/2024/2751280
125. Lee ES, Kwon MH, Kim HM, Woo HB, Ahn CM, and Chung CH. Curcumin analog CUR5–8 ameliorates nonalcoholic fatty liver disease in mice with high-fat diet-induced obesity. Metabolism. (2020) 103:154015. doi: 10.1016/j.metabol.2019.154015
126. Kim BG, Choi SH, Luo G, Sergeeva O, Lee Z, Driscoll J, et al. Vactosertib, a TGF-ß receptor I kinase/ALK5 inhibitor, diminishes tumor progression and bone disease in a mouse model of multiple myeloma and overcomes resistance to proteasome inhibitors. Blood. (2018) 132:1918. doi: 10.1182/blood-2018-99-117852
127. Ha KB, Lee ES, Park NW, Jo SH, Shim S, Kim D-K, et al. Beneficial effects of a curcumin derivative and transforming growth factor-β receptor I inhibitor combination on nonalcoholic steatohepatitis. Diabetes Metab J. (2023) 47:500–13. doi: 10.4093/dmj.2022.0110
128. Kumar V, Sethi B, Staller DW, Xin X, Ma J, Dong Y, et al. Anti-miR-96 and hh pathway inhibitor MDB5 synergistically ameliorate alcohol-associated liver injury in mice. Biomaterials. (2023) 295:122049. doi: 10.1016/j.biomaterials.2023.122049
129. Kang SH, Yim HJ, Hwang J, Kim M, Lee Y-S, Jung YK, et al. Improved anti-fibrotic effects by combined treatments of simvastatin and NS-398 in experimental liver fibrosis models. Korean J Intern Med. (2022) 37:745–56. doi: 10.3904/kjim.2021.138
130. Huang S, Ding D, Lan T, He G, Ren J, Liang R, et al. Multifunctional nanodrug performs sonodynamic therapy and inhibits TGF-β to boost immune response against colorectal cancer and liver metastasis. Acta Biomater. (2023) 164:538–52. doi: 10.1016/j.actbio.2023.04.001
131. Luo X, Huang W, Li S, Sun M, Hu D, Jiang J, et al. SOX12 facilitates hepatocellular carcinoma progression and metastasis through promoting regulatory T-cells infiltration and immunosuppression. Adv Sci. (2024) 11:2310304. doi: 10.1002/advs.202310304
132. Xiang S, Li J, and Zhang M. TGF-β1 inhibitor enhances the therapeutic effect of microwave ablation on hepatocellular carcinoma. Int J Hyperthermia. (2024) 41:2359496. doi: 10.1080/02656736.2024.2359496
133. Gobbo F, Martelli F, Di Virgilio A, Demaria E, Sarli G, and Migliaccio AR. The variation in the traits ameliorated by inhibitors of JAK1/2, TGF-β, P-selectin, and CXCR1/CXCR2 in the Gata1low model suggests that myelofibrosis should Be treated by these drugs in combination. Int J Mol Sci. (2024) 25:7703. doi: 10.3390/ijms25147703
134. Liu CH, Lee HS, Liou JP, Hua HS, Cheng WH, Yuliani FS, et al. MPT0E028, a novel pan-HDAC inhibitor, prevents pulmonary fibrosis through inhibition of TGF-β-induced CTGF expression in human lung fibroblasts: Involvement of MKP-1 activation. Eur J Pharmacol. (2024) 977:176711. doi: 10.1016/j.ejphar.2024.176711
Keywords: liver fibrosis, TGF-β signaling pathway, TGF-β inhibitors, hepatic stellate cells, antifibrotic therapy
Citation: Wang W, Gao Y, Chen Y, Cheng M, Sang Y, Wei L, Dai R, Wang Y and Zhang L (2025) TGF-β inhibitors: the future for prevention and treatment of liver fibrosis? Front. Immunol. 16:1583616. doi: 10.3389/fimmu.2025.1583616
Received: 26 February 2025; Accepted: 03 June 2025;
Published: 27 June 2025.
Edited by:
Natalie Nieuwenhuizen, Julius Maximilian University of Würzburg, GermanyReviewed by:
V. Nathan Subramaniam, Queensland University of Technology, AustraliaHiba Al Lawati, Queensland University of Technology, Australia, in collaboration with reviewer VNS
Alix Kay Berglund, University of Maryland, College Park, United States
Copyright © 2025 Wang, Gao, Chen, Cheng, Sang, Wei, Dai, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Zhang, emhhbmcwNTUxbGVpQDE2My5jb20=; Yiping Wang, d3lwd3lwNTRAYWxpeXVuLmNvbQ==
†These authors have contributed equally to this work and share first authorship