 Sirlaine Pio1
Sirlaine Pio1 Tatiana Prata Menezes1,2
Tatiana Prata Menezes1,2 Vitória Louise3
Vitória Louise3 Guilherme de Paula Costa1Daniel Malta Oliveira1Natiele Carlos1
Guilherme de Paula Costa1Daniel Malta Oliveira1Natiele Carlos1 Silvia Paula-Gomes4Luiza Oliveira Perucci5
Silvia Paula-Gomes4Luiza Oliveira Perucci5 André Talvani1,2,3*
André Talvani1,2,3*- 1Laboratory of Immunobiology of Inflammation, Department of Biological Science, Federal University of Ouro Preto, Ouro Preto, Minas Gerais, Brazil
- 2Graduate Program in Health and Nutrition, Federal University of Ouro Preto, Ouro Preto, Minas Gerais, Brazil
- 3Graduate Program in Health Sciences – Infectiology and Tropical Medicine, Federal University of Minas Gerais, Belo Horizonte, Minas Gerais, Brazil
- 4Laboratory of Biochemistry and Molecular Biology, Department of Biological Science, Federal University of Ouro Preto, Ouro Preto, Minas Gerais, Brazil
- 5School of Medicine, University of California, San Diego, San Diego, CA, United States
Introduction: During Trypanosoma cruzi infection, the immune system activates a robust inflammatory response, involving cytokines and chemokines like IFN-γ, TNF, IL-6, IL-1β, CCL2, and CCL5, to control parasite replication. The CX3CL1 chemokine and its receptor, CX3CR1, have been implicated in amplifying inflammation through pathways like NF-κB, MAPKs, STATs, TLRs, and NLRs, contributing to tissue damage. This study evaluated the effects of blocking CX3CR1 with the allosteric antagonist AZD8797 in a murine model of acute T. cruzi infection.
Methods: Male C57BL/6 mice were infected with 103 trypomastigote forms of T. cruzi (Y strain) and received AZD8797 (10 mg/kg) intraperitoneally for 10 days. On the 10th day, animals were euthanized and heart, skeletal muscle, and liver tissues were collected for CX3C L1 protein expression, biomarkers (IL-1β, IL-4, IL-6, IL-10, IL-15, IL-17, IFN-γ, TNF, and CCL2) quantified by Cytometric Bead Array and Enzyme Immunoassay.
Results: Treatment reduced spleen mass and cardiac levels of CCL2 and IL-15, with an increase of IL-4. Conversely, in skeletal muscle, TNF, IL-6, and IL-10 increased, while IL-15 decreased. Liver tissue showed reduced IL-15, IL-6, and IL-1β levels, alongside lowered plasma hepcidin and ferritin concentrations.
Discussion: These findings highlight CX3CL1’s site-specific role in modulating inflammation and iron metabolism during acute T. cruzi infection, suggesting its potential as a therapeutic target for infection management and disease prognosis.
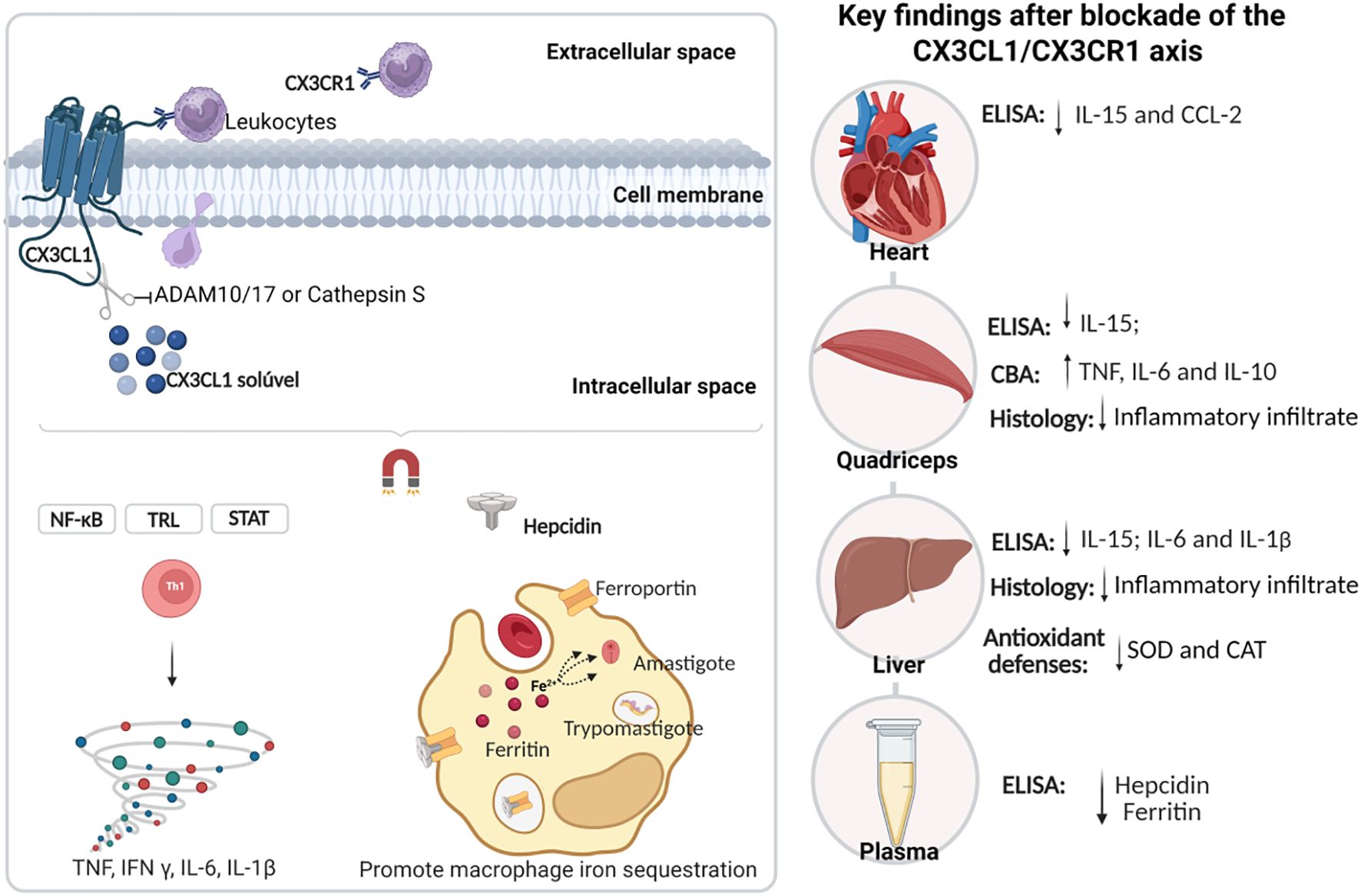
Graphical Abstract.
Introduction
The protozoan Trypanosoma cruzi triggers a systemic and multifaceted inflammatory response upon infecting vertebrate hosts, often culminating in chronic cardiomyopathy (1–3). This inflammatory cascade is initiated by parasite molecules, such as glycosylphosphatidylinositol (GPI)-anchored glycoconjugates and extracellular vesicles, which activate macrophages and other immune cells (4, 5). The resulting release of inflammatory mediators across tissues aims to control the parasite but also contributes to disease pathogenesis in both experimental models and humans (6–9).
Among these mediators, CX3CL1 has emerged as a dual-function chemokine, regulating inflammation and enhancing tissue repair (10) while exacerbating T. cruzi-induced cardiomyopathy (11). The CX3CL1-CX3CR1 axis has demonstrated therapeutic potential in modulating inflammatory responses in models of atherosclerosis and cardiac hypertrophy, by limiting monocyte recruitment and reducing tissue damage (12, 13). Additionally, this axis plays a role in iron regulation during inflammation, promoting hepcidin production (14, 15). This suggests a possible link between the CX3CL1-CX3CR1 axis, inflammation, and iron metabolism, particularly in cardiac diseases.
In T. cruzi infections, intracellular iron sequestration enhances parasite pathogenicity (16–18). Hepcidin, a hepatic hormone that regulates systemic iron levels, is upregulated by inflammatory mediators such as IL-6 and IL-1β (19, 20). Elevated hepcidin levels promote macrophage iron sequestration by degrading ferroportin and increasing ferritin storage (21). In vitro studies have shown that CX3CL1 can induce hepcidin expression in microglial cells, suggesting a potential link between this chemokine and iron homeostasis (14).
Despite these insights, the role of the CX3CL1-CX3CR1 axis in immune responses and iron metabolism during in vivo T. cruzi infection remains unexplored. This study investigates the modulatory effects of CX3CL1 via allosteric blockade of the CX3CR1 receptor (using AZD8797) on immune responses and iron metabolism during acute infection with the T. cruzi - Y strain.
Materials and methods
Animals
Twenty-four 10-week-old male C57BL/6 mice, weighing approximately 24 g, were obtained from the Center for Animal Science (CCA) at the Federal University of Ouro Preto (UFOP), Minas Gerais, Brazil. The mice were housed in an air-conditioned room maintained at 22.0 ± 2°C with a 12-hour light/dark cycle and had unrestricted access to water and standard chow. The study protocol was approved by the Ethics Commission on the Use of Animals (CEUA) at UFOP under protocol number 2025080822, in compliance with Resolution 196/96 of the National Health Council of the Brazilian Ministry of Health.
Trypanosoma cruzi infection
Experimental infection was initiated using bloodstream trypomastigote forms of the T. cruzi Y strain, harvested from Swiss mice seven days post-infection. The parasites were counted and adjusted in saline solution to a concentration of 10³ parasites per mouse in a 100 µL volume for intraperitoneal injection. Infection was confirmed by parasitemia analysis as described by Brener (22).
Treatment
Infected mice were treated daily with the CX3CL1 receptor antagonist AZD8797 (MedChemExpress, NJ, USA) or its vehicle via intraperitoneal injection. AZD8797 was administered at a dose of 10 mg/kg/day, solubilized in a vehicle solution containing 2% dimethyl sulfoxide (DMSO) in 0.9% NaCl, as per the manufacturer’s instructions. Treatment commenced 12 hours post-infection and continued for 10 consecutive days (Figure 1). A parallel uninfected control group received the same volume of either AZD8797 or the vehicle.
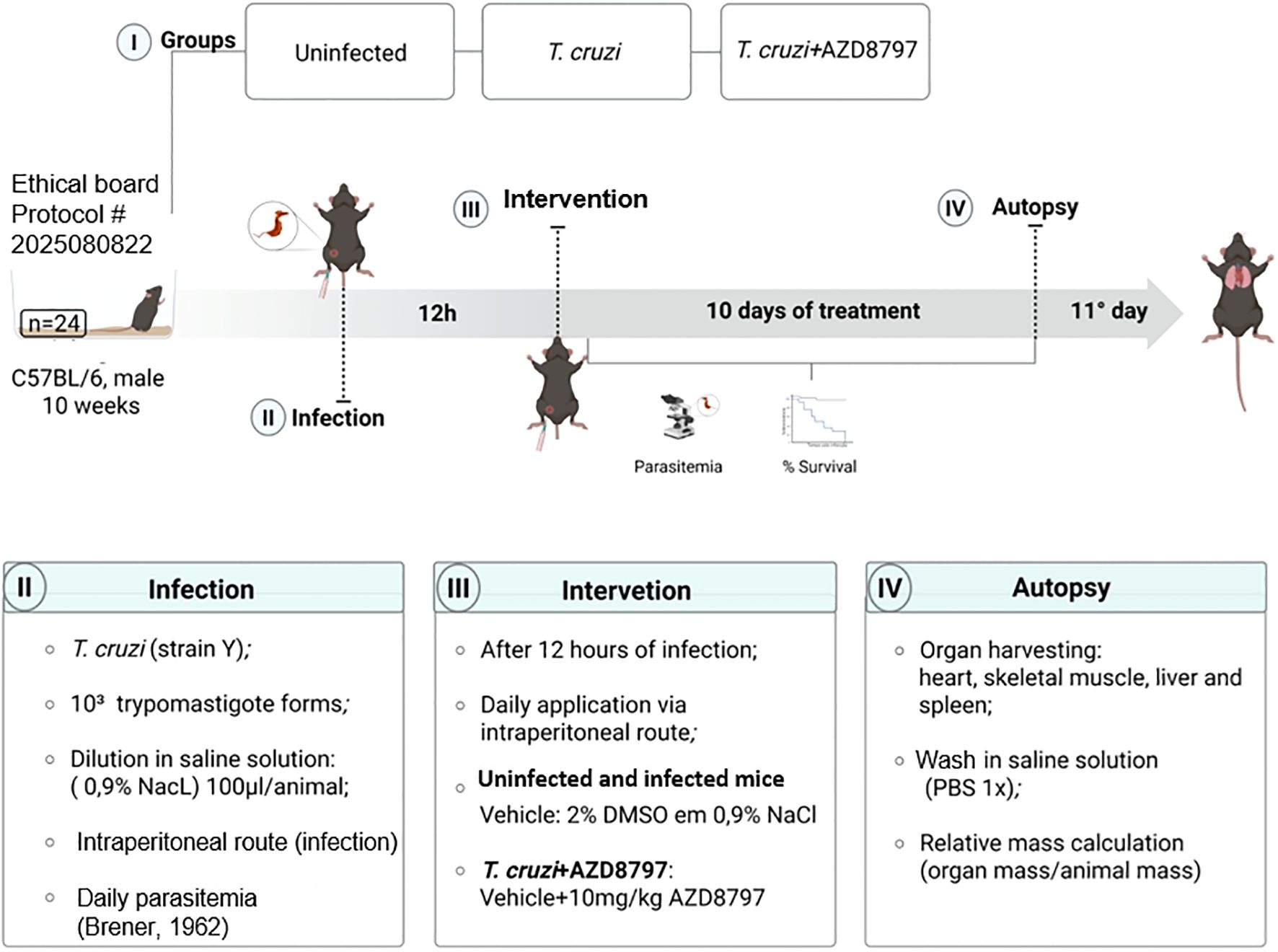
Figure 1. Timeline and experimental design. Male C57BL/6 mice (10 weeks old) were divided into three groups: Uninfected, T. cruzi-infected, and T. cruzi + AZD8797. Infection was performed using 10³ trypomastigotes (Y strain) via intraperitoneal injection. After 12 hours, treatment with AZD8797 (10 mg/kg/day) or vehicle was initiated and continued for 10 days. Parasitemia and survival were monitored during treatment. On day 11 post-infection, animals were euthanized, and organs (heart, skeletal muscle, liver, and spleen) were collected for analysis.
Western blot
Protein expression in heart tissue homogenates was analyzed using standard immunoblotting techniques. Protein concentrations were determined using the method described by Lowry et al. (23). Supernatants were mixed with Laemmli sample buffer (1:1), subjected to electrophoresis on a 10% SDS-PAGE gel, and transferred onto nitrocellulose membranes. The membranes were probed with anti-CX3CL1 (1:1000, #14-7986-81, eBioscience, USA) and anti-β-actin (1:1000, Cell Signaling Technology, MA, USA) primary antibodies. Secondary antibodies conjugated to peroxidase (1:5000, Cell Signaling Technology, MA, USA) were used for detection. Band intensities were quantified using ImageJ2 software (version 1.46m, NIH, USA).
Cytometric bead array immunoassay
Levels of inflammatory and regulatory cytokines, including IFN-γ, TNF, IL-4, IL-6, IL-10, and IL-17, were quantified using a mouse Th1/Th2/Th17 CBA kit (BD Biosciences, CA, USA). Cytokine levels were measured in supernatants of cardiac and skeletal muscle tissues using a BD FACSCalibur flow cytometer. Data were analyzed with CBA software and expressed in pg/mL, following the manufacturer’s protocol.
Enzyme-linked immunosorbent assays
Cytokines (IL-6, IL-1β, IL-10, IL-15) and the chemokine CCL2 were quantified in heart, skeletal muscle, and liver homogenates using commercially available ELISA kits (PeproTech®, Rocky Hill, NJ, USA). Hepcidin and ferritin levels in plasma samples from infected and treated animals were also measured using ELABScience kits (TX, USA). All procedures were performed in duplicate, adhering to the manufacturers’ instructions. Results were expressed as pg/mL.
Histological processing and number of infiltrated cells
Fragments of cardiac, skeletal muscle, and liver tissues were collected to assess cellular infiltration. These tissue samples were fixed in 10% buffered formalin for 24 hours, followed by dehydration and embedding in paraffin blocks. Thin sections (5 μm) were obtained using a rotary microtome equipped with a steel blade and subsequently stained with hematoxylin and eosin (HE). The number of infiltrated cells was quantified by analyzing randomly selected fields, covering a total area of 25,000 μm², at 40× magnification. Images were captured using an Axioscope A1 microscope (Carl Zeiss, Germany) and analyzed with ImageJ software (version 6.0, National Institutes of Health, USA).
Analysis of antioxidant defenses
Cardiac, skeletal muscle, and liver tissues were analyzed for oxidative damage by measuring the activity of key antioxidant enzymes. Superoxide dismutase (SOD) activity was assessed spectrophotometrically at 570 nm, following the method described by Marklund and Marklund (24). As SOD inhibits pyrogallol autoxidation, increased enzymatic activity indicates a reduction in pyrogallol oxidation. Catalase (CAT) activity was measured according to Aebi (25) by monitoring the decrease in hydrogen peroxide (H2O2) concentration at 240 nm over 60 seconds. Enzymatic activities were expressed as units per milligram of protein (U/mg).
Statistical analysis
Data analysis was conducted using GraphPad Prism software (v8.3, GraphPad Software, San Diego, CA, USA). Results are presented as mean ± standard deviation (SD). Normality was assessed using the Shapiro-Wilk test. For comparisons between two groups, a one-way Student’s t-test was used, and Tukey’s post hoc test was applied for multiple comparisons. Statistical significance was set at p< 0.05.
Results
In this study, parasitemia was monitored throughout the experiment, reaching its peak on the 8th day post-infection (Figure 2A). Daily administration of 10 mg/kg AZD8797 had no significant effect on the survival or mortality curves of infected mice (Figure 2B). Furthermore, AZD8797 treatment did not alter the relative mass of the heart or quadriceps muscle. However, T. cruzi infection led to a significant increase in the relative spleen mass, from 2.65 g ± 0.17 in uninfected mice to 22.40 g ± 1.54 in infected animals (p<0.0001). Notably, AZD8797 administration partially attenuated this increase, reducing the spleen mass of infected mice to 19.12 g ± 1.33 (p=0.005).
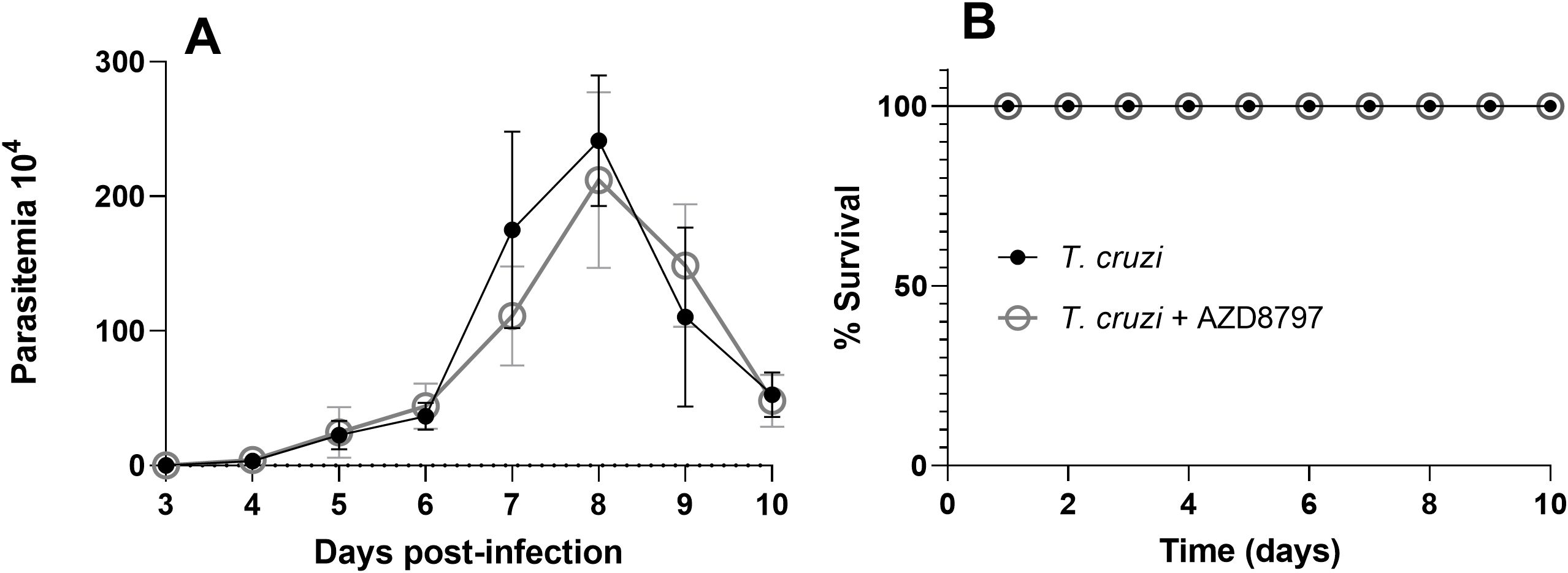
Figure 2. Parasitemia and survival curve. The level of parasitemia under CX3CL1 receptor blockade (A); the survival curve under CX3CL1 receptor blockade in mice infected with the T. cruzi Y strain during acute infection (B) (n=8). Data expressed as mean ± standard deviation (SD).
To assess the cardiac production of the polypeptide CX3CL1 in both uninfected and T. cruzi-infected mice, western blot analysis was performed. CX3CL1 expression was found to be preserved and highly expressed in the cardiac tissue of both treated and untreated mice 10 days post-infection (Figure 3). A representative gel image depicting band intensities is shown on the right side of the graphic.
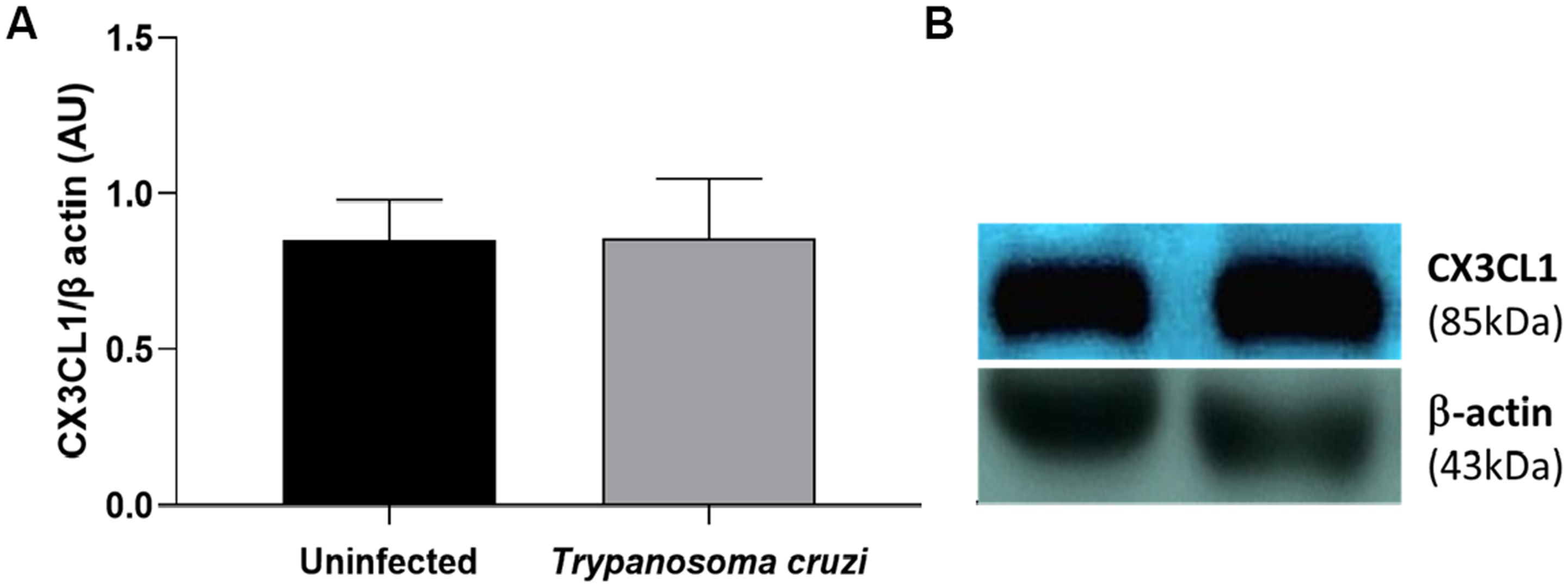
Figure 3. Expression of soluble CX3CL1 in the cardiac tissue. The western blot quantitative analysis of the soluble CX3CL1 protein levels in the cardiac tissue (A); the representative examples of the western blot analysis of the soluble CX3CL1 protein levels in the cardiac tissue (B). The soluble CXC3CL1 were determined by normalization with β-actin. The data are demonstrated as the mean ± standard deviation (SD) (n=6). AU, Arbitrary units.
After confirming that soluble CX3CL1 was preserved in both uninfected and infected mice, we evaluated the influence of the CX3CL1/CX3CR1 axis on the release of soluble biomarkers in cardiac and skeletal muscle. In the cardiac tissue, CCL2 (Figure 4A) and IL-15 (Figure 4B) productions were significantly elevated in response to T. cruzi infection, but both were reduced following AZD8797 administration. IFN-γ (Figure 4C), TNF (Figure 4D), IL-6 (Figure 4E), and IL-17A (Figure 4F) levels were also increased in infected cardiac tissue. However, AZD8797 did not reduce their production. Interestingly, AZD8797 treatment elevated IL-4 levels (Figure 4G) compared to uninfected mice, while IL-10 levels (Figure 4H) remained unchanged across all groups.
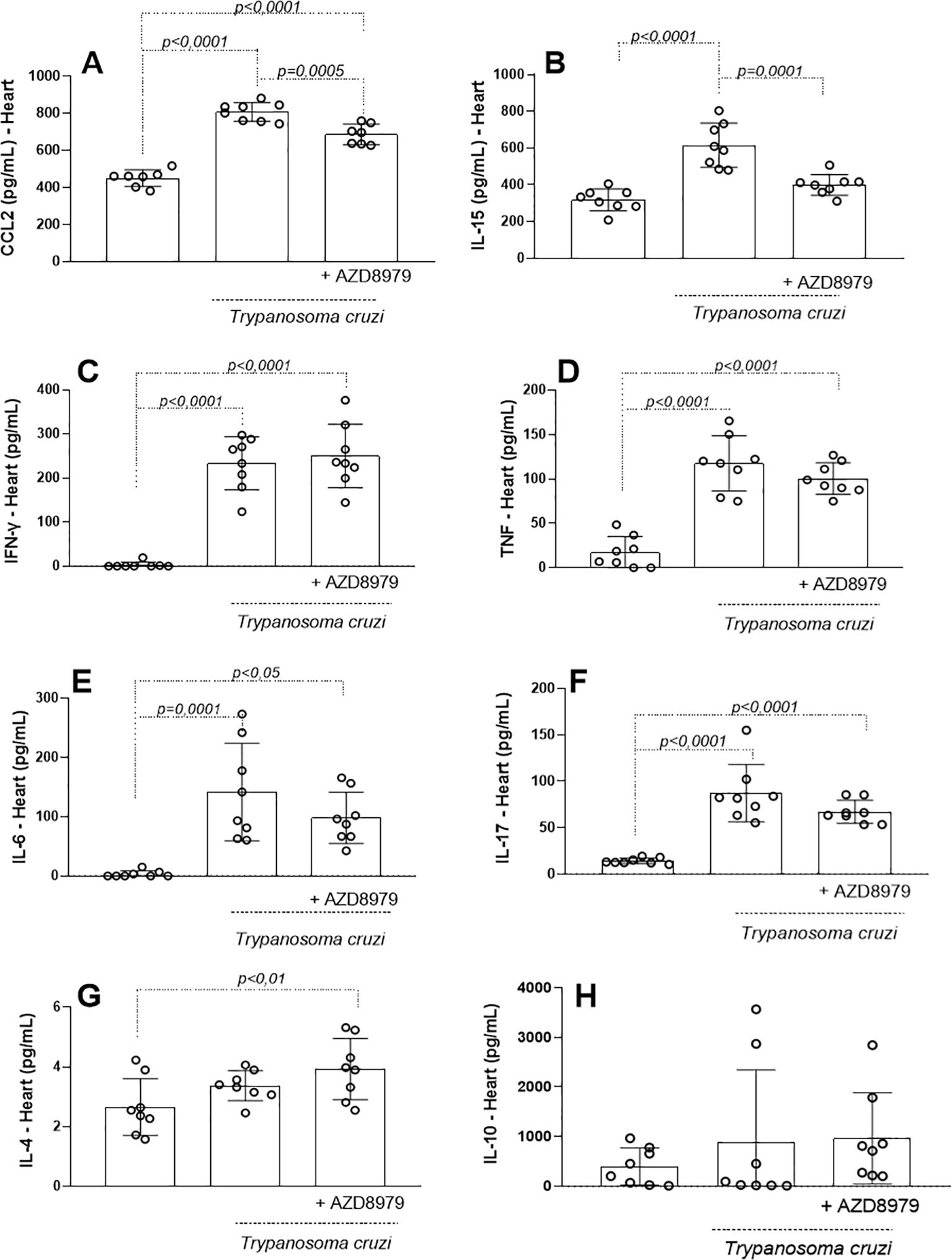
Figure 4. Inflammatory markers in the cardiac tissue. The concentration of inflammatory mediators in the cardiac tissue was detected in uninfected and infected-T. cruzi mice treated or not with CX3CL1 receptor blockade during 10 days. Cardiac tissue macerate was evaluated by ELISA as (A) CCL2, (B) IL-15, and by cytometric bead array (CBA) as (C) IFN-γ, (D) TNF, (E) IL-6, (F) IL-17, (G) IL-4 and (H) IL-10. Data expressed as mean ± standard deviation (SD) (n=8). The p-values were determined by one-way ANOVA with Tukey’s posttest and expressed in pg/mL.
In skeletal muscle, T. cruzi infection increased levels of CCL2 (Figure 5A), IL-15 (Figure 5B), and IFN-γ (Figure 5C). AZD8797 administration specifically reduced IL-15 levels. However, during the acute phase of infection, CX3CL1 receptor blockade increased TNF (Figure 5D) and IL-6 (Figure 5E) production in skeletal muscle. Levels of IL-17A (Figure 5F) and IL-4 (Figure 5G) were unaffected by infection or CX3CL1 receptor inhibition. Interestingly, IL-10 levels (Figure 5H) were significantly elevated in infected mice following AZD8797 treatment.
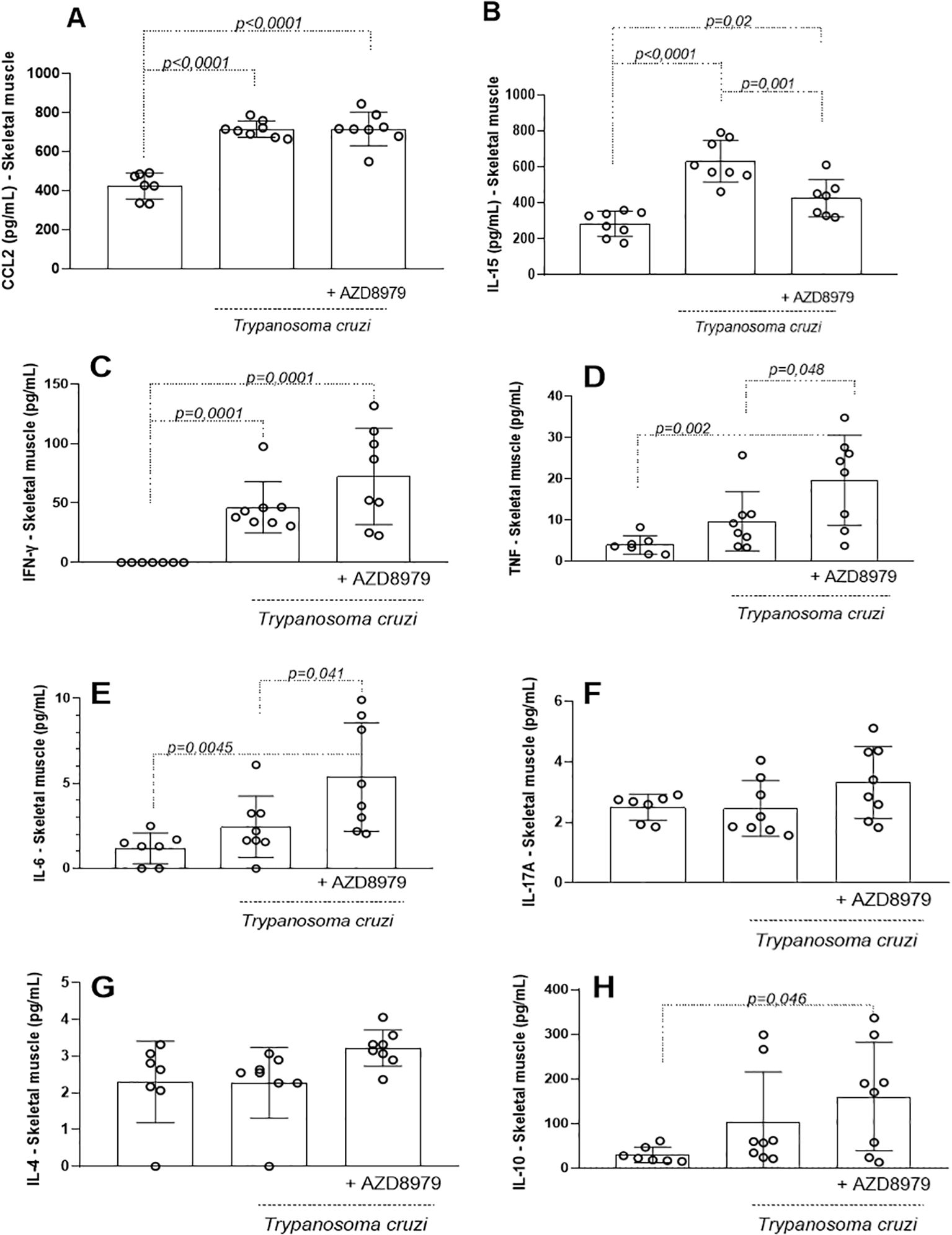
Figure 5. Inflammatory markers in the skeletal muscle. The concentration of the inflammatory markers in the skeletal tissue was detected in uninfected and infected-T. cruzi mice treated with CX3CL1 receptor blockade or vehicle for 10 days. Immunoassay was performed as (A) CCL2, (B) IL-15 and cytometric bead array (CBA) as (C) IFN- γ, (D) TNF, (E) IL-6, (F) IL-17, (G) IL-4 and (H) IL-10. Data expressed as mean ± standard deviation (SD) (n=8). The p values were determined by one-way ANOVA with Tukey’s posttest and expressed in pg/mL.
In the liver, T. cruzi infection resulted in elevated levels of CCL2 (Figure 6A), IL-15 (Figure 6B), IL-6 (Figure 6C), IL-1β (Figure 6D), and IL-10 (Figure 6E). AZD8797 treatment effectively reduced the production of IL-15, IL-6, and IL-1β, but not CCL2 or IL-10.
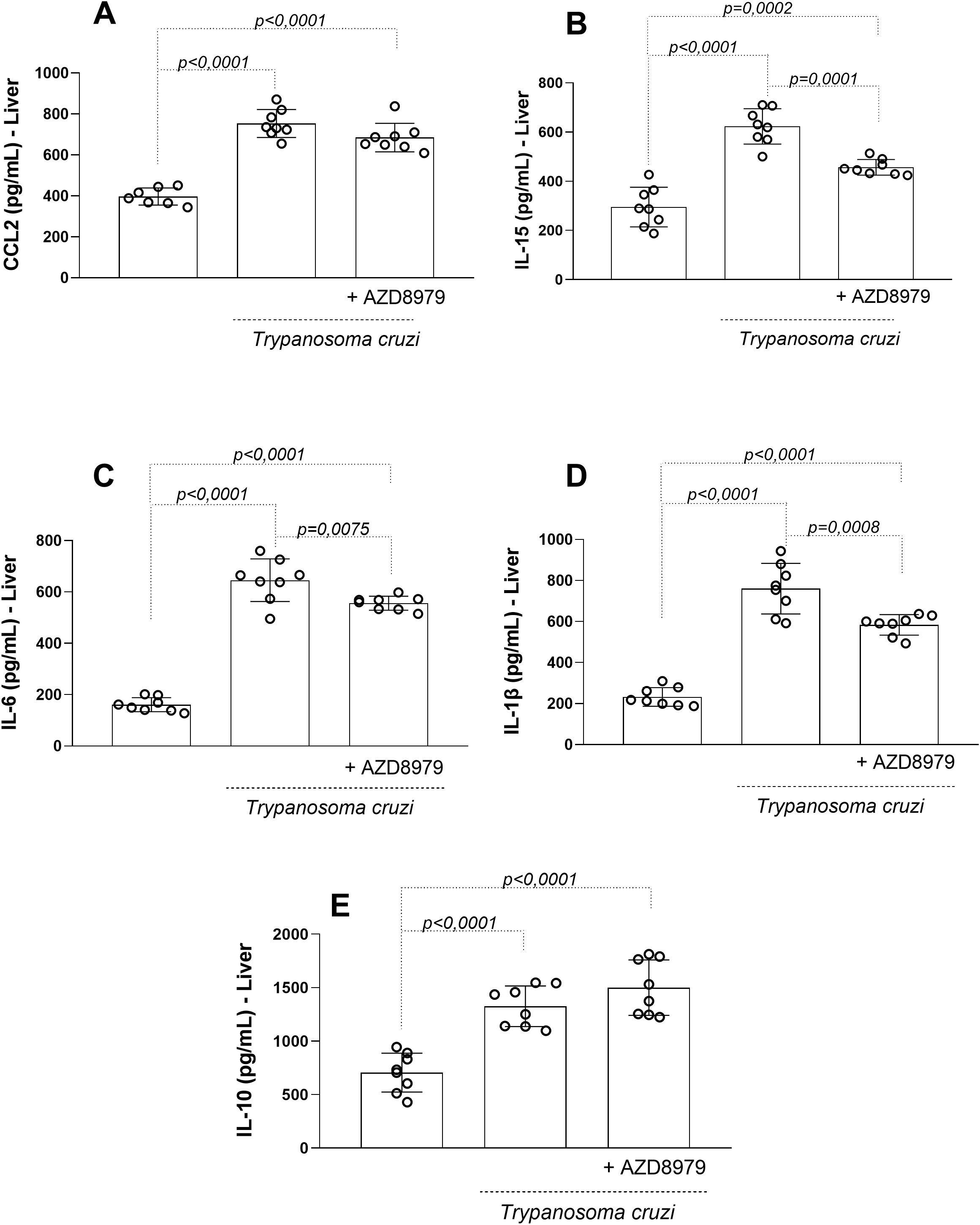
Figure 6. The liver levels of the inflammatory markers. The inflammatory markers (A) CCL2, (B) IL-15 (C) IL-6, (D) IL-1β, e (E) IL-10 in uninfected and infected T. cruzi, under CX3CL1 receptor blockade (or vehicle/control), were measured by enzyme-linked immunosorbent assay. Data expressed as mean ± standard deviation (SD) (n=8). The p values were determined by one-way ANOVA with Tukey’s posttest and expressed in pg/mL.
Together with inflammatory markers, the activity of enzymes SOD and CAT were also determined in the cardiac, skeletal muscle and liver to verify alterations in the antioxidant defense system. The SOD and CAT (Table 1) was higher in animals infected. The blockade of the CX3CL1 receptor via AZD8797 administration reduced both enzymes in the liver. However, there is an increased CAT in the skeletal muscle when using the blockade of the CX3CL1 receptor.
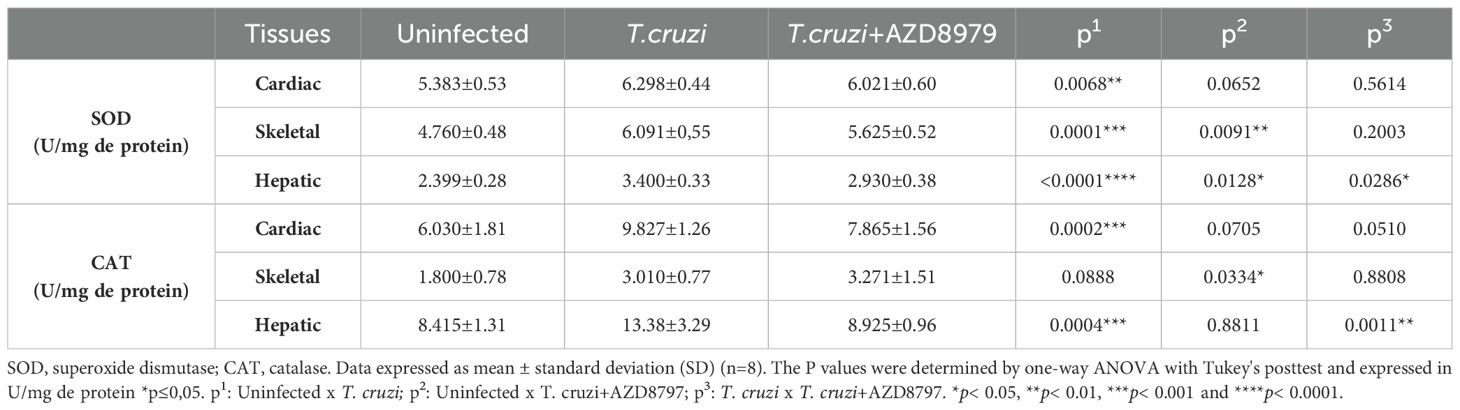
Table 1. Oxidative stress markers in cardiac and skeletal muscles and, in liver.
In this study, the CX3CL1/CX3CR1 axis was also evaluated contributing to ferric modulation. Infected mice exhibited significantly increased serum levels of hepcidin (Figure 7A) and ferritin (Figure 7B) during the acute phase of infection. Blockade of the CX3CL1 receptor via AZD8797 administration effectively reduced serum ferric element levels, highlighting a modulatory effect on iron metabolism during T. cruzi infection.
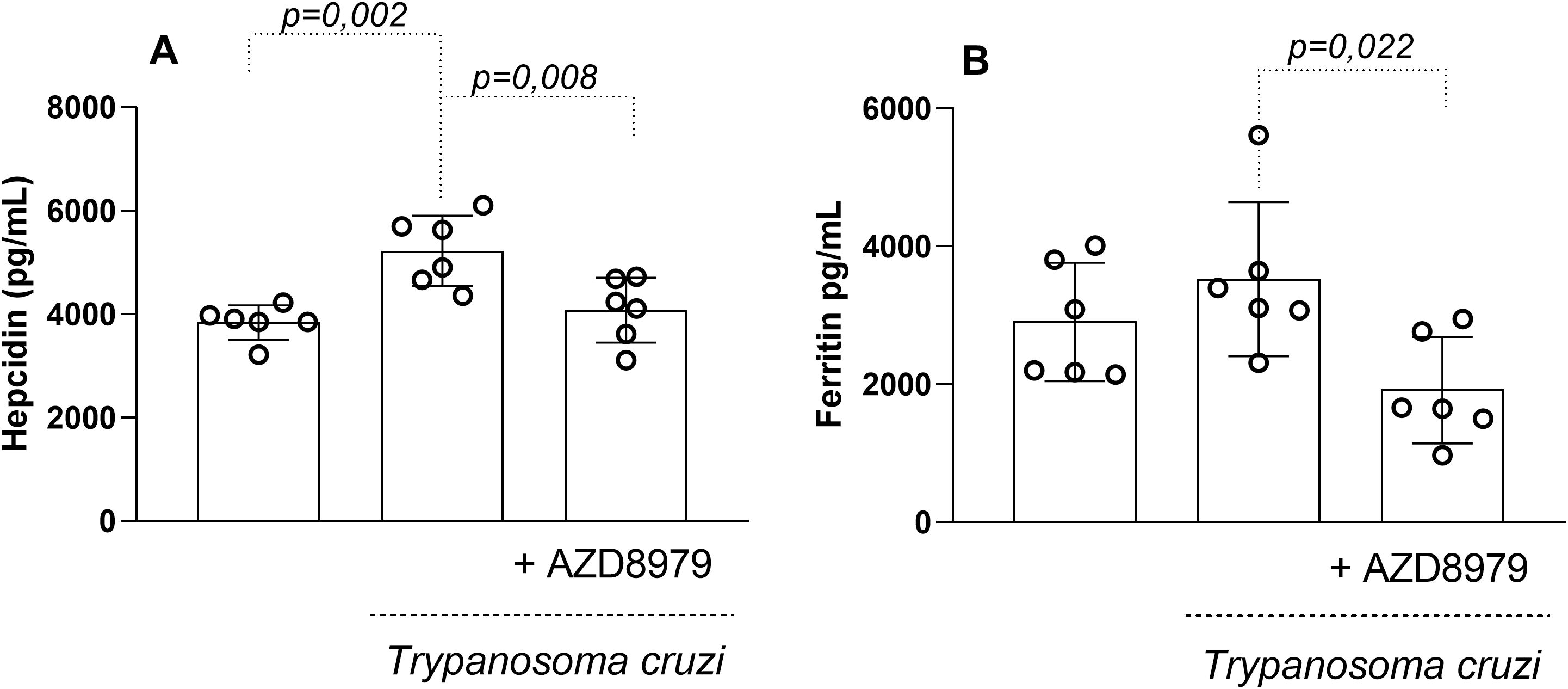
Figure 7. The serum levels of hepcidin and ferritin. The serum levels of the hepcdina (A), ferritina (B) in uninfected and T. cruzi-infected mice treated with CX3CL1 receptor blockade were measured by enzyme-linked immunosorbent assay. Data expressed as mean ± standard deviation (SD) (n=8). The p-values were determined by one-way ANOVA with Tukey’s posttest and expressed in pg/mL.
It is important to note that this study was conducted during the acute phase of experimental T. cruzi infection. Despite this, cardiac, skeletal muscle, and liver tissues were analyzed to quantify inflammatory infiltration following AZD8979 administration. T. cruzi infection led to an increase in the number of cellular nuclei (Figure 8A) and nucleus area (Figure 8B) in cardiac tissue. Interestingly, in liver tissue, T. cruzi infection resulted in both an increased number of cellular nuclei (Figure 9A) and an expanded nucleus area (Figure 9B). Treatment with the CX3CL1R blocker successfully reduced both inflammatory parameters. Finally, in skeletal muscle, infection also increased the number of cellular nuclei; however, AZD8979 effectively regulated this inflammatory response (Figure 10A), though it did not alter nucleus area (Figure 10B).
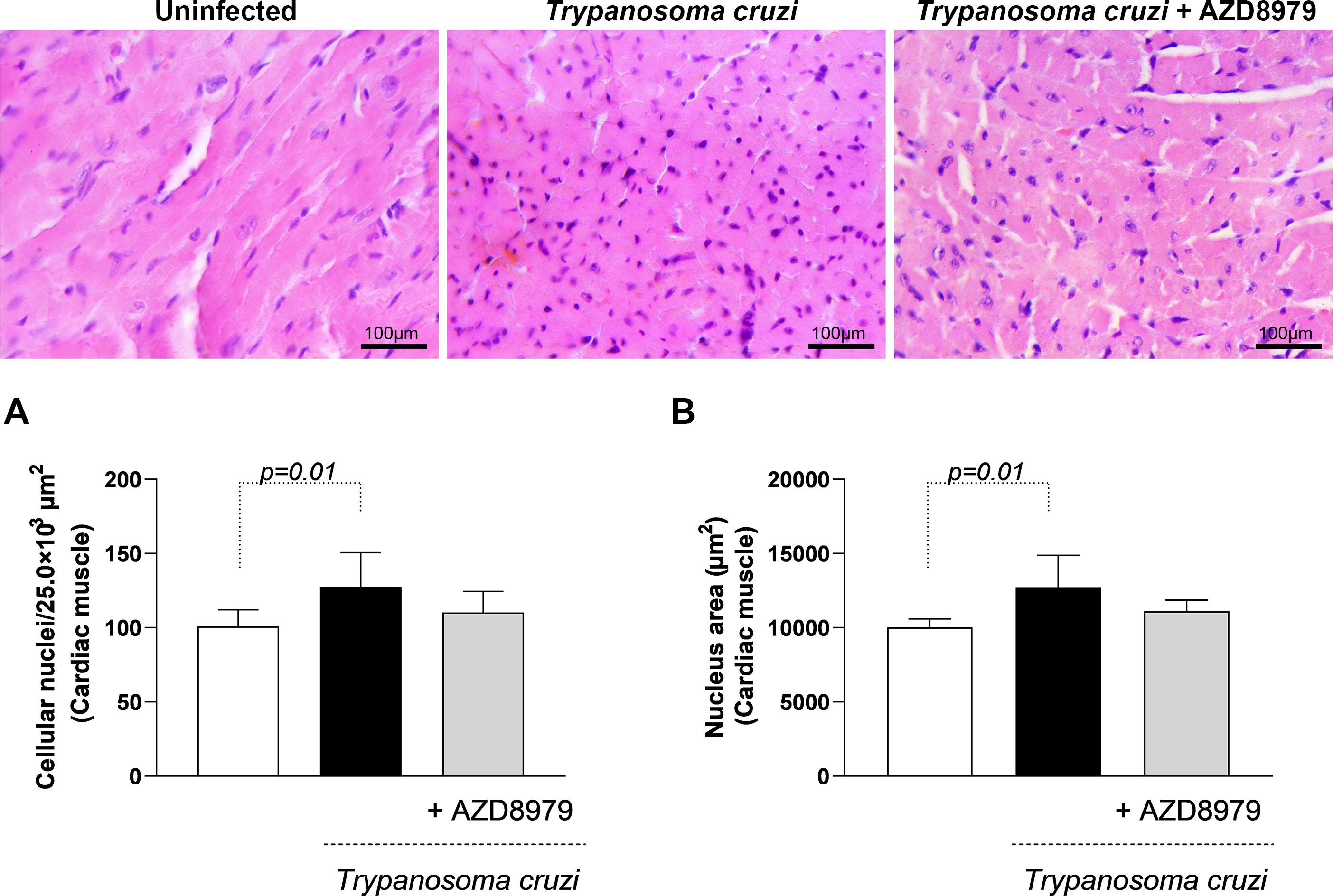
Figure 8. Morphological analysis of inflammatory infiltrate in cardiac muscle tissue. (A) Quantification of the number of cell nuclei per 25.0x10³ µm² of cardiac tissue; (B) Mean nuclear area (µm²) in cardiac tissue. Images of cardiac muscle tissue stained with H&E. Bar = 100µm, 400x magnification. Data were expressed as mean ± standard deviation and the p values (p<0.05) were determined by one-way ANOVA with Tukey’s posttest.
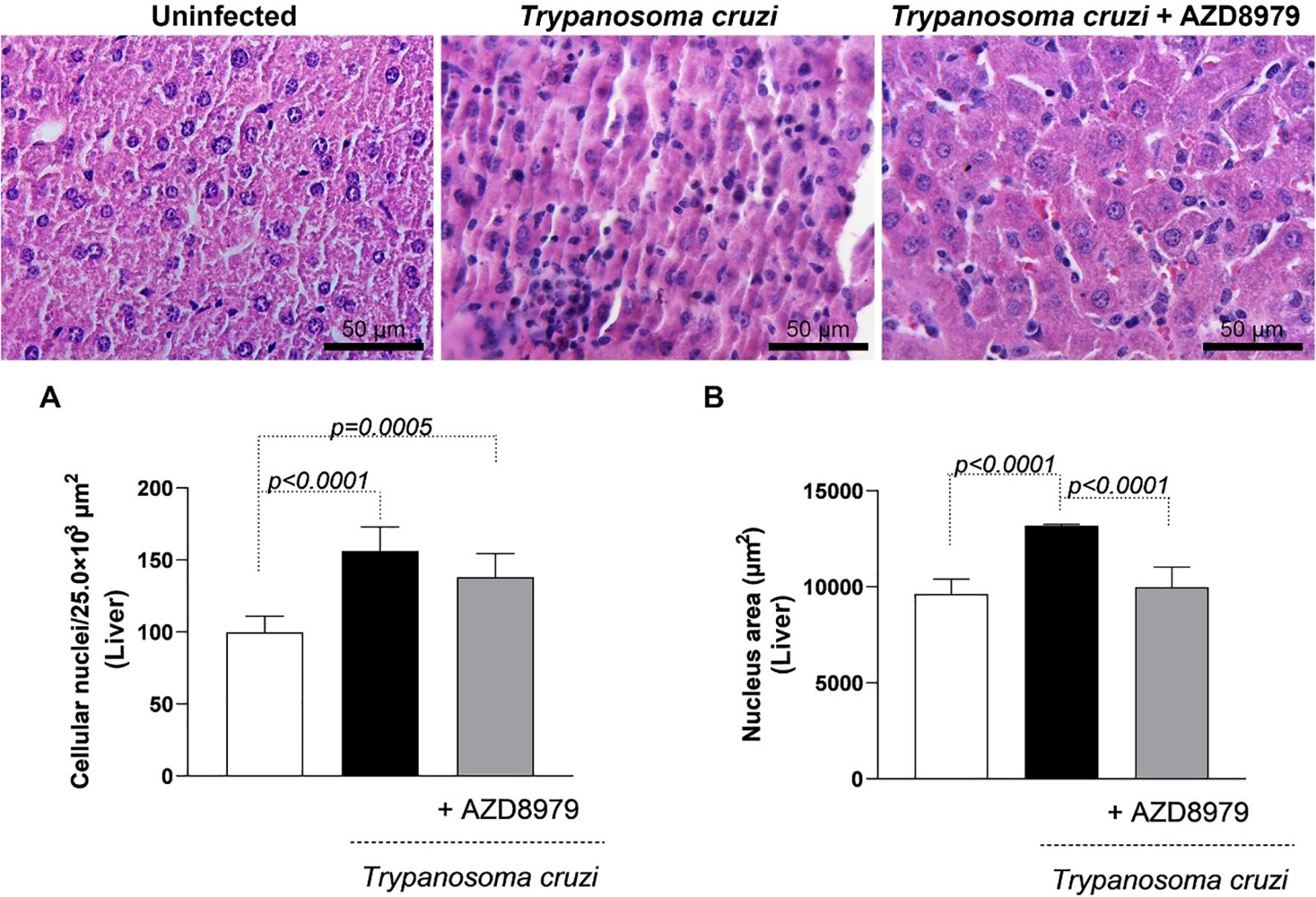
Figure 9. Morphological analysis of the inflammatory infiltrate in the liver. (A) Quantification of the number of cell nuclei per 25.0x10³ µm² of liver tissue; (B) Mean nuclear area (µm²) in liver tissue. Images of liver tissue stained with H&E. Bar = 50µm, 400x magnification. Data were expressed as mean ± standard deviation and the p values (p<0.05) were determined by one-way ANOVA with Tukey’s posttest.
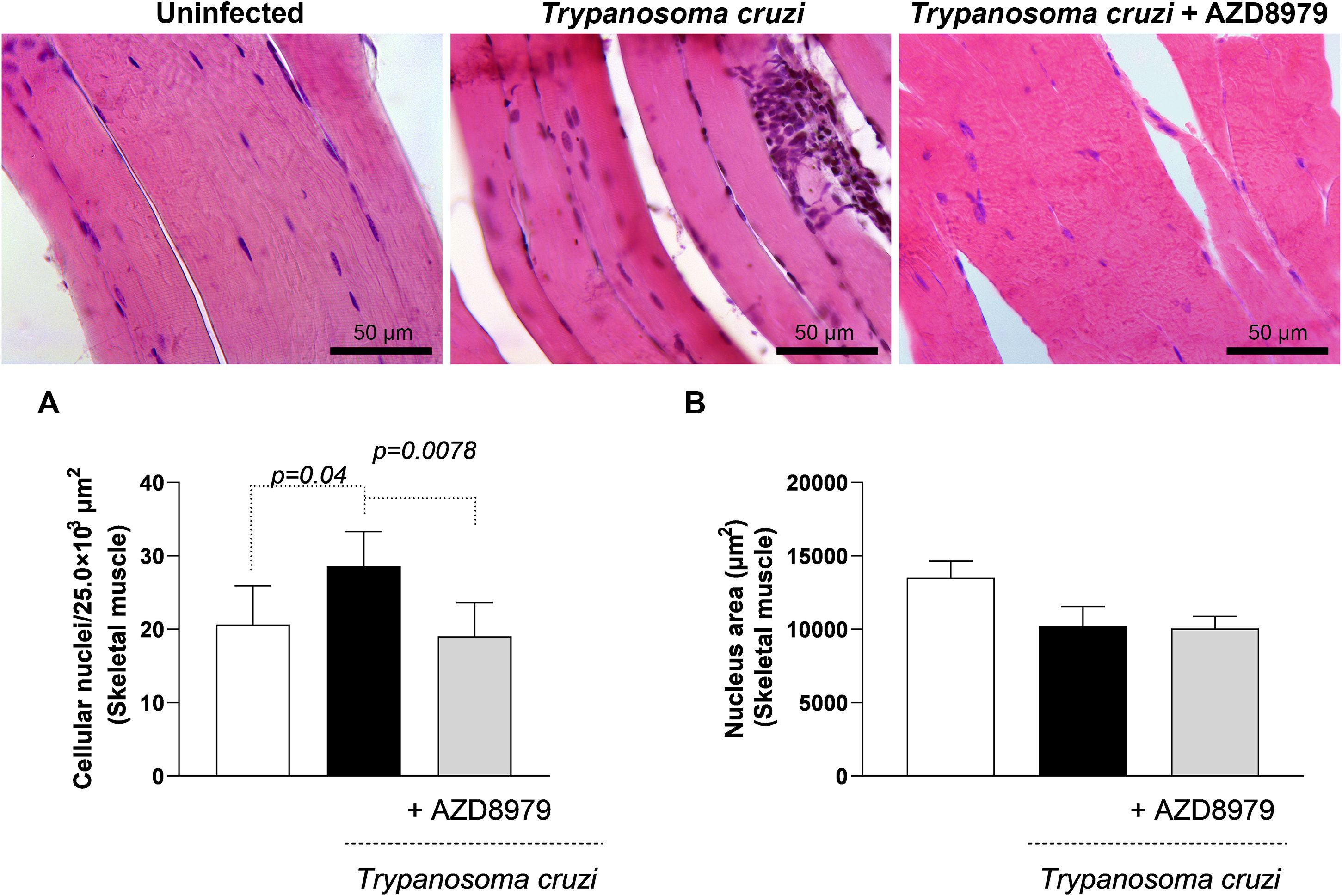
Figure 10. Morphological analysis of inflammatory infiltrate in skeletal muscle. (A) Quantification of the number of cell nuclei per 25.0x10³ µm² of skeletal muscle tissue; (B) Mean nuclear area (µm²) in skeletal muscle tissue. Images of skeletal muscle tissue stained with H&E. Bar = 50µm, 400x magnification. Data were expressed as mean ± standard deviation, and the p values <0.05 were determined by one-way ANOVA with Tukey’s posttest.
Discussion
The observed tissue-specific effects of CX3CR1 inhibition likely reflect the distinct immunological microenvironments and differential parasite loads in different tissues during acute T. cruzi infection, even without altering blood parasitemia. The heart, a major target of T. cruzi, typically sustains high levels of parasite persistence and inflammation, which may explain why CX3CR1 blockade led to a significant reduction in proinflammatory mediators such as CCL2 and IL-15, along with an increase in the anti-inflammatory cytokine IL-4. This suggests a potential protective modulation of cardiac inflammation. In contrast, the skeletal muscle exhibited elevated levels of TNF, IL-6, and IL-10 following treatment, indicating a shift toward a more complex inflammatory profile, suggestive of a compensatory response in this less severely affected tissue. Meanwhile, the liver, the main site of iron homeostasis and of T. cruzi-induced inflammation, showed a marked reduction in IL-6, IL-1β, and IL-15, consistent with the observed decline in circulating hepcidin and ferritin. These changes suggest that CX3CR1 signaling may contribute to hepatic inflammation and iron regulation during infection. These findings underscore a multifaceted and compartmentalized nature of C57BL/6 mice in response to T. cruzi, and highlight how CX3CL1/CX3CR1 axis modulation can yield divergent outcomes depending on tissue-specific dynamics of immune activation, parasite burden, and homeostatic demands.
Iron (Fe) is a critical micronutrient for T. cruzi and other trypanosomatids like Leishmania spp. However, the role of Fe in the life cycle and pathogenicity of these parasites remains incompletely understood. T. cruzi depends on Fe for vital metabolic functions, including DNA synthesis, heme biosynthesis, energy production via the electron transport chain, oxidative phosphorylation, and pathogenicity in mammalian hosts (16). The parasite hijacks host Fe-proteins to fulfill its metabolic needs. Within the host, free Fe must be stored or processed upon entering the cytosol to prevent the generation of reactive oxygen species, which can cause cellular damage (26). Previous in vivo evidence has suggested a key role for iron in providing energy for the T. cruzi life cycle and in the propagation of Chagas disease (27). In addition, Estevam and colleagues demonstrated, using portable X-ray fluorescence, that C57BL/6 mice exhibit reduced skin iron levels during acute infection, which correlates with lower parasite burden and increased oxidative stress (28). Notably, the parasite utilizes specific iron transporters, such as the 39-kDa protein TciT and the iron reductase TcFR, to internalize iron (29). There is growing evidence that TciT and TcFR may also participate in copper transport and intracellular distribution, another essential element for aerobic metabolism in trypanosomatids (30). These findings support the proposed role of iron regulation in determining disease outcomes and should be considered to strengthen the mechanistic rationale of iron involvement in T. cruzi infection.
During T. cruzi infection, CX3CL1 appears to extend its role beyond leukocyte recruitment, promoting increases in ferritin and hepcidin levels. Hepcidin, a key iron-regulatory hormone, modulates the body’s iron balance and plays a significant role in innate immunity by regulating Fe availability during infections (31). Ferritin, synthesized by nearly all living organisms, serves as a key intracellular and extracellular Fe storage protein, releasing Fe in a controlled manner to prevent toxicity (32). Disruption of the hepcidin/ferritin axis can increase host susceptibility to T. cruzi infection.
Under normal physiological conditions, the liver serves as the primary iron reservoir. However, iron is also distributed throughout other organs, where it is integrated into enzymes and structural proteins, particularly in the skeletal muscles, pancreas, brain, and heart. Given the close relationship between iron and hemoglobin in red blood cells, and considering that T. cruzi-induced inflammation enhances local tissue activity and blood flow, we hypothesize that CX3CR1 blockade may exertxorgan-specific effects on inflammatory cells activation and inflammatory mediator’s release. These effects are likely influenced by both the tissue microenvironment and the genetic background of the infecting T. cruzi strain (33, 34).
Previous studies have shown that elevated iron stores correlate with increased parasitemia and higher mortality in mice, a condition reversible by Fe chelation (16). Notably, the efficacy of benznidazole, the clinical drug of choice for treating Chagas disease, is enhanced in the presence of the Fe chelator desferrioxamine (18). Our findings suggest that, during the initial stage of T. cruzi infection, blocking the CX3CL1/CX3CR1 axis partially disrupts Fe metabolism, creating an environment less favorable for parasite survival and reducing its pathogenicity.
CX3CL1 is predominantly expressed by endothelial cells in myocardial ischemia and heart failure. It is an atypical chemokine that exists in a membrane-bound form and a cleaved soluble form (35). The membrane-bound form functions as an adhesion molecule, facilitating leukocyte adhesion to NK cells, monocytes, and specific T cell subsets via interaction with its receptor, CX3CR1. Since the CX3CL1/CX3CR1 axis is primarily expressed on endothelial and cardiac cells, it has been implicated in the athogenesis of diseases such as atherosclerosis and heart failure. Notably, the CX3CL1/CX3CR1 axis has been studied in coronary artery disease, where patients exhibit an expansion of the CX3CR1+CD3+CD8+ T cell subset, characterized by enhanced chemotactic, adhesive, and inflammatory responses to CX3CL1 (36).
In the context of cardiomyopathy induced by T. cruzi infection, the role of the CX3CL1/CX3CR1 axis was not explored until the late 1990s. Prior to this, other chemokines such as CCL2, CCL5, CXCL9, and CXCL10, along with their receptors, were well-documented as mediators of experimental pathogenesis and contributors to the worsening of clinical status in humans (37–39).
The involvement of CX3CL1 in T. cruzi infection was first highlighted in a model of low-protein diet-induced immunomodulation. Intriguingly, systemic CX3CL1 levels were elevated, but cardiac tissue levels were reduced, accompanied by decreased inflammatory infiltration, particularly of CD68+ and CD163+ macrophages (40). At the time, the differential inflammatory responses mediated by CX3CL1 across various tissues did not attract significant attention. However, in a subsequent study using a CX3CR1 inhibitor (AZD8797), distinct inflammatory profiles were observed in the cardiac, hepatic, and skeletal muscle tissues in response to T. cruzi infection. These findings are particularly relevant, given that advanced Chagas cardiomyopathy in humans is characterized by dilated cardiomyopathy, reduced ejection fraction, and ventricular enlargement (41).
A subsequent study reinforced the significance of CX3CL1 in T. cruzi infection. It demonstrated that intravenous administration of T. cruzi-derived neurotrophic factors in MyD88-knockout mice (deficient in TLR signaling) increased CX3CL1 and CCL2 levels, modulating inflammatory responses mediated by Trk signaling and reducing cardiac fibrosis (11). Another study investigating AngII-induced hypertension in the context of T. cruzi infection revealed a synergistic effect of hypertension and T. cruzi infection on TNF and CX3CL1 expression, leading to enhanced leukocyte recruitment into cardiac tissue (42). Interestingly, TNF and endothelin-1 are well-established biomarkers of pathogenesis in experimental T. cruzi infection and indicators of severity in Chagas cardiomyopathy (37, 43–45). The CX3CL1/CX3CR1 axis positively correlates with these biomarkers, exacerbating cardiac pathology in T. cruzi infection (11).
In a non-parasitic model, early β-adrenergic stimulation was shown to activate the cardiac CX3CL1/CX3CR1 axis, supporting transient concentric remodeling and delaying the progression to heart failure (46). This finding is relevant to Chagas cardiomyopathy, as adrenergic autoantibodies are known to contribute to the progression of disease (47–49). CX3CL1 and TNF, secreted by inflammatory and cardiac cells, may synergistically promote cardiomyocyte hypertrophy in chronic T. cruzi infection.
Altogether, there remains a promising avenue of investigation into the inhibition of the CX3CR1/CX3CL1 axis in the context of Chagas disease. Although clinical application in humans would require a carefully staged development process, this strategy represents a novel and rational shift from traditional antiparasitic multitherapy toward a more integrated immune-metabolic modulation. During T. cruzi infection, inflammation-driven upregulation of hepcidin promotes iron sequestration, particularly in macrophages and hepatic tissue, which may create a favorable environment for parasite persistence. The CX3CR1 inhibitor AZD8797 emerges as a potential host-targeted adjunctive therapy by disrupting this iron retention and concurrently mitigating tissue-specific inflammation, notably within the heart. Existing chemotherapeutic options for Chagas disease frequently fail to effectively control parasite replication during both the acute and chronic phases, which negatively impacts long-term cardiac outcomes (50). However, before advancing to human trials with AZD8797, it is critical to conduct comprehensive studies to determine human-specific dose-response relationships and assess the long-term safety of this approach, particularly given the potential effects on iron metabolism and the partial immune suppression associated with CX3CR1 inhibition.
In summary, our findings underscore the CX3CL1/CX3CR1 axis as a critical regulator at the intersection of immune and iron metabolism responses during T. cruzi infection. By influencing both inflammatory pathways and iron availability, both key factors in parasite persistence and tissue damage, this axis emerges as a strategic node for therapeutic intervention. Targeting this pathway may not only attenuate pathogenic inflammation but also limit iron-dependent parasite survival, offering a dual mechanism of action in the management of T. cruzi-induced heart disease.
Data availability statement
The immunological and biochemical data used to support the findings of this study are available from the corresponding author upon request.
Ethics statement
The animal study was approved by Ethics Commission on the Use of Animals (CEUA) at UFOP under protocol number 2025080822, in compliance with Resolution 196/96 of the National Health Council of the Brazilian Ministry of Health. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SP: Conceptualization, Formal analysis, Investigation, Methodology, Resources, Writing – original draft, Writing – review & editing. TM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Visualization, Writing – original draft, Writing – review & editing. VL: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. GC: Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. DO: Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. NC: Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. SP-G: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Project administration, Supervision, Writing – original draft, Writing – review & editing. LO: Conceptualization, Formal analysis, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AT: Conceptualization, Formal analysis, Funding acquisition, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by the National Council for Scientific and Technological Development (CNPq - #405946/2021-0 and DECIT/SECTICS/MS #442782/2024-1), the Foundation for Research Support of the State of Minas Gerais (FAPEMIG – APQ-02.029-21, APQ-00720-23, APQ-02511-21, APQ-02511-22 and BPD-00754-22), and FINEP (financial support to CCA/UFOP). This study was funded by the Federal University of Ouro Preto.
Acknowledgments
The authors would like to thank the Multiuser Laboratory of Advanced Microscopy and Microanalysis and the Flow Cytometry Laboratory of the Biological Sciences Research Center at the Federal University of Ouro Preto.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ribeiro AL, Nunes MP, Teixeira MM, and Rocha MO. Diagnosis and management of Chagas disease and cardiomyopathy. Nat Rev Cardiol. (2012) 9:576–89. doi: 10.1038/nrcardio.2012.109, PMID: 22847166
2. Talvani A and Teixeira MM. Inflammation and Chagas disease some mechanisms and relevance. Adv Parasitol. (2011) 76:171–94. doi: 10.1016/B978-0-12-385895-5.00008-6, PMID: 21884892
3. Esper L, Talvani A, Pimentel P, Teixeira MM, and MaChado FS. Molecular mechanisms of myocarditis caused by Trypanosoma cruzi. Curr Opin Infect Dis. (2015) 28:246–52. doi: 10.1097/QCO.0000000000000157, PMID: 25887609
4. Ropert C and Gazzinelli RT. Signaling of immune system cells by glycosylphosphatidylinositol (GPI) anchor and related structures derived from parasitic protozoa. Curr Opin Microbiol. (2000) 3:395–403. doi: 10.1016/s1369-5274(00)00111-9, PMID: 10972501
5. Coelho PS, Klein A, Talvani A, Coutinho SF, Takeuchi O, Akira S, et al. Glycosylphosphatidylinositol-anchored mucin-like glycoproteins isolated from Trypanosoma cruzi trypomastigotes induce in vivo leukocyte recruitment dependent on MCP-1 production by IFN-gamma-primed-macrophages. J Leukoc Biol. (2002) 71:837–44. doi: 10.1189/jlb.71.5.837, PMID: 11994509
6. Talvani A, Rocha MO, Cogan J, Maewal P, de Lemos J, Ribeiro AL, et al. Brain natriuretic peptide and left ventricular dysfunction in chagasic cardiomyopathy. Mem Inst Oswaldo Cruz. (2004) 99:645–9. doi: 10.1590/s0074-02762004000600020, PMID: 15558179
7. Guedes PM, Veloso VM, Talvani A, Diniz LF, Caldas IS, Do-Valle-Matta MA, et al. Increased type 1 chemokine expression in experimental Chagas disease correlates with cardiac pathology in beagle dogs. Vet Immunol Immunopathol. (2010) 138:106–13. doi: 10.1016/j.vetimm.2010.06.010, PMID: 20619467
8. Gibaldi D, Vilar-Pereira G, Pereira IR, Silva AA, Barrios LC, Ramos IP, et al. CCL3/Macrophage Inflammatory Protein-1α is dually Involved in parasite persistence and induction of a TNF and IFNγ-enriched inflammatory milieu in Trypanosoma cruzi-induced chronic cardiomyopathy. Front Immunol. (2020) 11:306. doi: 10.3389/fimmu.2020.00306, PMID: 32194558
9. Wang Y, Wessel N, Kohse F, Khan A, Schultheiss HP, Moreira MDCV, et al. Measurement of multiple cytokines for discrimination and risk stratification in patients with Chagas’ disease and idiopathic dilated cardiomyopathy. PloS Negl Trop Dis. (2021) 15:e0008906. doi: 10.1371/journal.pntd.0008906, PMID: 33755669
10. Salvador R, Aridgides D, and Pereira Perrin M. Parasite-derived neurotrophic factor/trans-sialidase of Trypanosoma cruzi links neurotrophic signaling to cardiac innate immune response. Infect Immun. (2014) 82:3687–96. doi: 10.1128/IAI.02098-14, PMID: 24935974
11. Menezes TP, MaChado BAM, Toledo DNM, Santos PV, and Talvani A. Insights into CX3CL1/Fractalkine during experimental Trypanosoma cruzi infection. Parasitol Int. (2022) 87:102530. doi: 10.1016/j.parint.2021.102530, PMID: 34929405
12. Saederup N, Chan L, Lira SA, and Charo IF. Fractalkine deficiency markedly reduces macrophage accumulation and atherosclerotic lesion formation in CCR2–/– mice: evidence for independent chemokine functions in atherogenesis. Circulation. (2008) 117:1642–8. doi: 10.1161/CIRCULATIONAHA.107.743872, PMID: 18165355
13. Nemska S, Gassmann M, Bang ML, Frossard N, and Tavakoli R. Antagonizing the CX3CR1 receptor markedly reduces development of cardiac hypertrophy after transverse aortic constriction in mice. J Cardiovasc Pharmacol. (2021) 78:792–801. doi: 10.1097/FJC.0000000000001130, PMID: 34882111
14. Pandur E, Tmasi K, Pap R, Varga E, Miseta A, and Sipos K. Fractalkine induces hepcidin expression of BV-2 microglia and causes iron accumulation in SH-SY5Y cells. Cell Mol Neurobiol. (2019) 39:985–1001. doi: 10.1007/s10571-019-00694-4, PMID: 31172340
15. Pandur E, Tamasi K, Pap R, Janosa G, and Sipos K. Modulatory effects of fractalkine on inflammatory response and iron metabolism of lipopolysaccharide and lipoteichoic acid-activated THP-1 macrophages. Int J Mol Sci. (2022) 23:2629. doi: 10.3390/ijms23052629, PMID: 35269771
16. Lalonde RG. Holbein BE Role of iron in Trypanosoma cruzi infection of mice. J Clin Invest. (1984) 73:470–6. doi: 10.1172/JCI111233, PMID: 6421877
17. Arantes JM, Pedrosa ML, Martins HR, Veloso VM, de Lana M, Bahia MT, et al. Trypanosoma cruzi: treatment with the iron chelator desferrioxamine reduces parasitemia and mortality in experimentally infected mice. Exp Parasitol. (2007) 117:43–50. doi: 10.1016/j.exppara.2007.03.006, PMID: 17521632
18. Francisco AF, de Abreu Vieira PM, Arantes JM, Pedrosa ML, Martins HR, Silva M, et al. Trypanosoma cruzi: effect of benznidazole therapy combined with the iron chelator desferrioxamine in infected mice. Exp Parasitol. (2008) 120:314–9. doi: 10.1016/j.exppara.2008.08.002, PMID: 18789321
19. Ganz T and Nemeth E. Iron homeostasis in host defence and inflammation. Nat Rev Immunol. (2015) 15:500–10. doi: 10.1038/nri3863, PMID: 26160612
20. Roth MP, Meynard D, and Coppin H. Regulators of hepcidin expression. Vitam Horm. (2019) 110:101–29. doi: 10.1016/bs.vh.2019.01.005, PMID: 30798807
21. Camaschella C, Nai A, and Silvestri L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica. (2020) 31:260–72. doi: 10.3324/haematol.2019.232124, PMID: 31949017
22. Brener Z. Therapeutic activity and criterion of cure of mice experimentally infected with Trypanosorna cruzi. Rev do Inst Med Trop São Paulo. (1962) 4:389–96., PMID: 14015230
23. Lowry OH, Rosebrough NJ, Farr AL, and Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. (1951) 193:265–75. doi: 10.1016/S0021-9258(19)52451-6
24. Marklund S and Marklund G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem. (1974) 47:469–74. doi: 10.1111/j.1432-1033.1974.tb03714.x, PMID: 4215654
25. Aebi H. Catalase in vitro. Methods Enzymol. (1984) 105:121–6. doi: 10.1016/s0076-6879(84)05016-3, PMID: 6727660
26. Taylor MC and Kelly JM. Iron metabolism in trypanosomatids, and its crucial role in infection. Parasitology. (2010) 137:899–917. doi: 10.1017/S0031182009991880, PMID: 20152063
27. Dick CF, Alcantara CL, Carvalho-Kelly LF, Lacerda-Abreu MA, Cunha-E-Silva NL, Meyer-Fernandes JR, et al. Iron uptake controls trypanosoma cruzi metabolic shift and cell proliferation. Antioxidants. (2023) 22:984. doi: 10.3390/antiox12050984, PMID: 37237850
28. Estevam M, Appoloni CR, Malvezi AD, Tatakihara VL, Panis C, Cecchini R, et al. Trypanosoma cruzi: in vivo evaluation of iron in skin employing X-ray fluorescence (XRF) in mouse strains that differ in their susceptibility to infection. FEMS Immunol Med Microbiol. (2012) 64:334–42. doi: 10.1111/j.1574-695X.2011.00917.x, PMID: 22136203
29. Dick CF, Rocco-MaChado N, Dos-Santos ALA, Carvalho-Kelly LF, Alcantara CL, Cunha-E-Silva NL, et al. An iron transporter is involved in iron homeostasis, energy metabolism, oxidative stress, and metacyclogenesis in trypanosoma cruzi. Front Cell Infect Microbiol. (2022) 11:789401. doi: 10.3389/fcimb.2021.789401, PMID: 35083166
30. Merli ML, Mediavilla MG, Zhu X, Cobine PA, and Cricco JA. Solving the puzzle of copper trafficking in Trypanosoma cruzi: candidate genes that can balance uptake and toxicity. FEBS J. (2025) 292:391–411. doi: 10.1111/febs.17340, PMID: 39639518
31. Ganz T. Hepcidin and iron regulation, 10 years later. Blood. (2011) 117:4425–33. doi: 10.1182/blood-2011-01-258467, PMID: 21346250
32. Zhang J, Chen X, Hong J, Tang A, Liu Y, Xie N, et al. Biochemistry of mammalian ferritins in the regulation of cellular iron homeostasis and oxidative responses. Sci China Life Sci. (2021) 64:352–62. doi: 10.1007/s11427-020-1795-4, PMID: 32974854
33. Guedes-da-Silva FH, Shrestha D, Salles BC, Figueiredo VP, Lopes LR, Dias L, et al. Trypanosoma cruzi antigens induce inflammatory angiogenesis in a mouse subcutaneous sponge model. Microvasc Res. (2015) 97:130–6. doi: 10.1016/j.mvr.2014.10.007, PMID: 25446369
34. Shrestha D, Bajracharya B, Paula-Costa G, Salles BC, Leite AL, Menezes AP, et al. Expression and production of cardiac angiogenic mediators depend on the Trypanosoma cruzi-genetic population in experimental C57BL/6 mice infection. Microvasc Res. (2017) 110:56–63. doi: 10.1016/j.mvr.2016.12.002, PMID: 27956355
35. Xuan W, Liao Y, Chen B, Huang Q, Xu D, Liu Y, et al. Detrimental effect of fractalkine on myocardial ischaemia and heart failure. Cardiovasc Res. (2011) 92:385–93. doi: 10.1093/cvr/cvr221, PMID: 21840883
36. Damås JK, Boullier A, Waehre T, Smith C, Sandberg WJ, Green S, et al. Expression of fractalkine (CX3CL1) and its receptor, CX3CR1, is elevated in coronary artery disease and is reduced during statin therapy. Arterioscler Thromb Vasc Biol. (2005) 25:2567–72. doi: 10.1161/01.ATV.0000190672.36490.7b, PMID: 16224053
37. Talvani A, Rocha MO, Barcelos LS, Gomes YM, Ribeiro AL, and Teixeira MM. Elevated concentrations of CCL2 and tumor necrosis factor-alpha in chagasic cardiomyopathy. Clin Infect Dis. (2004) 38:943–50. doi: 10.1086/381892, PMID: 15034825
38. Nogueira LG, Santos RH, Ianni BM, Fiorelli AI, Mairena EC, Benvenuti LA, et al. Myocardial chemokine expression and intensity of myocarditis in Chagas cardiomyopathy are controlled by polymorphisms in CXCL9 and CXCL10. PloS Negl Trop Dis. (2012) 6:e1867. doi: 10.1371/journal.pntd.0001867, PMID: 23150742
39. Frade AF, Pissetti CW, Ianni BM, Saba B, Lin-Wang HT, Nogueira LG, et al. Genetic susceptibility to Chagas disease cardiomyopathy: involvement of several genes of the innate immunity and chemokine-dependent migration pathways. BMC Infect Dis. (2013) 12:587. doi: 10.1186/1471-2334-13-587, PMID: 24330528
40. Martins RF, Martinelli PM, Guedes PMM, Padua BC, Santos FM, Silva ME, et al. Protein deficiency alters CX3CL1 and endothelin-1 in experimental Trypanosoma cruzi induced cardiomyopathy. Trop Med Int Health. (2013) 18:466–76. doi: 10.1111/tmi.12071, PMID: 23398597
41. Jeyalan V, Austin D, Loh SX, Wangsaputra VK, and Spyridopoulos I. Fractalkine/CX3CR1 in dilated cardiomyopathy: A potential future target for immunomodulatory therapy? Cells. (2023) 28:2377. doi: 10.3390/cells12192377, PMID: 37830591
42. Silva MC, Azevedo MA, Figueiredo VP, Moura Junior MR, Coelho Junior D, Martinelli PM, et al. Renovascular hypertension increases serum TNF and CX3CL1 in experimental Trypanosoma cruzi infection. Braz J Med Biol Res. (2018) 26:e6690. doi: 10.1590/1414-431X20186690, PMID: 29590257
43. Bilate AM, Salemi VM, Ramires FJ, de Brito T, Russo M, Fonseca SG, et al. TNF blockade aggravates experimental chronic Chagas disease cardiomyopathy. Microbes Infect. (2007) 9:1104–13. doi: 10.1016/j.micinf.2007.05.014, PMID: 17644389
44. Lula JF, Rocha MO, Nunes Mdo C, Ribeiro AL, Teixeira MM, Bahia MT, et al. Plasma concentrations of tumour necrosis factor-alpha, tumour necrosis factor-related apoptosis-inducing ligand, and FasLigand/CD95L in patients with Chagas cardiomyopathy correlate with left ventricular dysfunction. Eur J Heart Fail. (2009) 11:825–31. doi: 10.1093/eurjhf/hfp105, PMID: 19654138
45. Freeman BD, MaChado FS, Tanowitz HB, and Desruisseaux MS. Endothelin-1 and its role in the pathogenesis of infectious diseases. Life Sci. (2014) 24:110–9. doi: 10.1016/j.lfs.2014.04.021, PMID: 24780317
46. Flamant M, Mougenot N, Balse E, Le Fèvre L, Atassi F, Gautier EL, et al. Early activation of the cardiac CX3CL1/CX3CR1 axis delays β-adrenergic-induced heart failure. Sci Rep. (2021) 9:17982. doi: 10.1038/s41598-021-97493-z, PMID: 34504250
47. Borda ES and Sterin-Borda L. Antiadrenergic and muscarinic receptor antibodies in Chagas’ cardiomyopathy. Int J Cardiol. (1996) 54:149–56. doi: 10.1016/0167-5273(96)02592-2, PMID: 8803679
48. Joensen L, Borda E, Kohout T, Perry S, García G, and Sterin-Borda L. Trypanosoma cruzi antigen that interacts with the beta1-adrenergic receptor and modifies myocardial contractile activity. Mol BiochemParasitol. (2003) 127:169–77. doi: 10.1016/s0166-6851(03)00003-3, PMID: 12672526
49. Wallukat G, Botoni FA, Rocha MODC, Louise V, Müller J, and Talvani A. Functional antibodies against G-protein coupled receptors in Beagle dogs infected with two different strains of Trypanosoma cruzi. Front Immunol. (2022) 13:926682. doi: 10.3389/fimmu.2022.926682, PMID: 36389686
Keywords: inflammation, Trypanosoma cruzi, cardiac disease, CX3CR1, CX3CL1
Citation: Pio S, Menezes TP, Louise V, Costa GdP, Oliveira DM, Carlos N, Paula-Gomes S, Perucci LO and Talvani A (2025) Role of the CX3CL1/CX3CR1 axis in iron metabolism and immune regulation during acute Trypanosoma cruzi infection. Front. Immunol. 16:1585883. doi: 10.3389/fimmu.2025.1585883
Received: 01 March 2025; Accepted: 31 July 2025;
Published: 27 August 2025.
Edited by:
Abhay Satoskar, The Ohio State University, United StatesReviewed by:
Phileno Pinge-Filho, State University of Londrina, BrazilEmi Tsuru, Kochi University, Japan
Priscila Santos, Federal University of Sergipe, Brazil
Copyright © 2025 Pio, Menezes, Louise, Costa, Oliveira, Carlos, Paula-Gomes, Perucci and Talvani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: André Talvani, dGFsdmFuaUB1Zm9wLmVkdS5icg==