 Min Deng
Min Deng Zhaohong Kong
Zhaohong Kong Yan Wang1
Yan Wang1 Tao Li
Tao Li- 1Renmin Hospital of Wuhan University, Wuhan, China
- 2Department of Gastrointestinal Surgery, Hubei Provincial Hospital of Traditional Chinese Medicine, Wuhan, Hubei, China
Guillain–Barré syndrome (GBS) is a rare neurological disorder characterized by muscle weakness and paralysis. Although the exact etiology remains unclear, the current standard treatments include intravenous immunoglobulin (IVIG) and plasma exchange (PLEX) therapy. While the majority of GBS patients respond well to immunotherapy, some severe cases can be fatal. Efgartigimod, an Fc receptor antagonist, has been utilized in the treatment of various autoimmune diseases. However, its clinical efficacy in acute GBS has been rarely documented. In this study, we administered intravenous efgartigimod to four patients with different subtypes of acute GBS, two of whom received efgartigimod monotherapy without concomitant glucocorticoids, IVIG, or PLEX. The treatment outcomes were favorable, suggesting that intravenous efgartigimod may represent a promising therapeutic option for acute GBS. Further research is warranted to validate these preliminary findings.
1 Introduction
Guillain-Barré syndrome (GBS) is an acute autoimmune polyradiculoneuropathy characterized by rapidly progressive, ascending flaccid paralysis. Worldwide, approximately 100,000 people develop this disease each year. While paralysis is a prominent feature, the syndrome encompasses a spectrum of neurological deficits, including sensory disturbances, cranial nerve involvement, and dysautonomia. The exact cause of GBS is unknown. However, about two-thirds of patients have symptoms of infection in the 6 weeks before illness onset (1). Approximately 20–30% of patients with GBS may have severe GBS with respiratory failure. Globally, the mortality rate of GBS patients is approximately 7.5% (1, 2). There is no significant difference in the incidence rate of 1–2 people per 100,000 per year between genders or countries (3).
Intravenous immunoglobulin (IVIG) and plasma exchange (PLEX) therapy are considered to be the best treatments. Efgartigimod (efgartigimod alfa-fcab, Vyvgart™) is an Fc receptor antagonist used to treat autoimmune diseases including myasthenia gravis. Subcutaneous efgartigimod has been FDA- approved in the USA for treatment of Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP), and that two more Fc receptor (FcRn) antagonists, rozanolixizumab and nipocalimab are also approved for generalized AchR and MuSK antibody positive myasthenia gravis (4). There are several ongoing clinical studies of efgartigimod for the treatment of other autoimmune diseases, including bullous pemphigoid, chronic inflammatory demyelinating polyradiculoneuropathy, immune thrombocytopenia, and autoimmune myositis. However, the clinical efficacy of efgartigimod in acute GBS has been documented in only a limited number of cases (5–7). In this study, we administered efgartigimod to four patients with different subtypes of acute GBS. Among them, two patients received efgartigimod as monotherapy, one was treated with a combination of IVIG and efgartigimod, and the remaining patient received glucocorticoids alongside efgartigimod. Notably, all four patients achieved favorable clinical outcomes, highlighting the potential therapeutic benefits of efgartigimod in acute GBS management.
2 Methods
2.1 Research design and data collection
Data from the current cases were systematically collected and analyzed to comprehensively evaluate the clinical characteristics, disease progression, and treatment responses associated with this condition. To identify factors influencing clinical outcomes, detailed analyses were performed on demographic information, prodromal symptoms, clinical manifestations, disease severity at peak and last follow-up, diagnostic findings, treatment strategies, and clinical outcomes.
Disease severity was assessed at peak disability and during each follow-up visit using standardized scales, including the Medical Research Council Scale (MRC) for muscle strength, the Guillain-Barré Syndrome Disability Scale (GBS-DS) for functional disability, and the Inflammatory Neuropathy Cause and Treatment Disability Scale (INCAT) for limb functionality. These scales provided a quantitative and qualitative framework to monitor disease progression and therapeutic efficacy.
In addition, electromyography and nerve conduction studies were performed before and after drug treatment to objectively evaluate changes in nerve conduction velocity, amplitude, and latency. These electrophysiological assessments served as critical biomarkers to complement clinical evaluations and further validate the effectiveness of the treatment interventions. Furthermore, longitudinal follow-up data were collected to assess long-term outcomes, including residual disability, quality of life, and potential complications.
3 Case presentations
3.1 Case 1
A 51-year-old female patient began to develop fever and cough in January 2024. One week later, she developed bilateral eyelid ptosis, accompanied by dyspnea and limb weakness. She was admitted to a community hospital and received antiviral treatment, but her symptoms did not resolve, so she attended our hospital and was admitted. She had a history of hypertension. She reported no history of diabetes, infectious diseases (such as hepatitis B and tuberculosis), surgical trauma, or poisoning, and no family history. Neurological examination revealed bilateral ptosis and ophthalmoplegia with the absence of light reflex. She could not show her teeth or puff her cheeks and she had loss of nasolabial folds and loss of forehead wrinkles, suggesting facial diplegia. Muscle strength in all four limbs was graded as Medical Research Council grade 4. The tendon reflexes in both the upper and lower extremities were diminished.
Nerve conduction studies demonstrated reduced motor nerve conduction velocity (MNCV) in the right median nerve. Compound motor action potential (CMAP) amplitudes were at the lower normal limits bilaterally in the median and ulnar nerves. Reduced sensory nerve action potential (SNAP) amplitudes with decreased sensory nerve conduction velocity (SNCV) were observed in bilateral median and ulnar nerves. Reduced CMAP amplitudes were noted in bilateral facial nerves. The blink reflex study revealed complete absence of R1, R2, and R2’ waveforms on the left side with sparse R1 and R2 waveforms on the right. Needle EMG detected minimal abnormal spontaneous activity in the left first dorsal interosseous muscle.
Cerebrospinal fluid (CSF) analysis showed normal intracranial pressure (130 mmH2O), normal leukocytes (3×106), normal protein (0.28 g/L), and normal glucose (3.8 mmol/L). The blood gas analysis results were as follows: pH of 7.32, oxygen partial pressure of 90 mmHg, carbon dioxide partial pressure of 55 mmHg, bicarbonate level of 26.0 mmol/L, and standard bicarbonate level of 28.0 mmol/L. Liver function, blood lipids, blood cell count, kidney function, procalcitonin, respiratory etiology indicators, electrolytes, blood glucose, myocardial infarction indicators, and coagulation function were normal. Chest X-ray, electrocardiogram, and cardiac color ultrasound were normal.
Peripheral neuropathy-associated antibodies in CSF and serum (GM1, GM2, GM3, GM4, GD1a, GD1b, GD2, GD3, GT1a, GT1b, GQ1b, sulfatide, NF155, NF186, CNTN1, CNTN2, CASPR1, and MAG) were assessed using immunofluorescence staining. No antibodies were detected in the CSF. GM2 IgM, GM1 IgM, and sulfatide IgM were positive in the serum. The patient was diagnosed with Miller–Fisher syndrome and Guillain–Barré overlap syndrome (MFS GBS overlap syndrome).
The patient received IVIG (0.4 g/kg/day for 5 days), ventilator-assisted respiratory support, and efgartigimod (10 mg/kg via weekly intravenous infusion for 3 weeks). She was discharged after dyspnea and peripheral facial paralysis improved.
Her extraocular movements and bilateral eyelid elevation had recovered, and she had no symptoms of unsteady gait at her follow-up examination 2 months after treatment. No adverse drug reactions occurred. The clinical manifestations, diagnosis, treatments, and follow-up of all four patients are detailed in Tables 1–3 and Figures 1, 2.
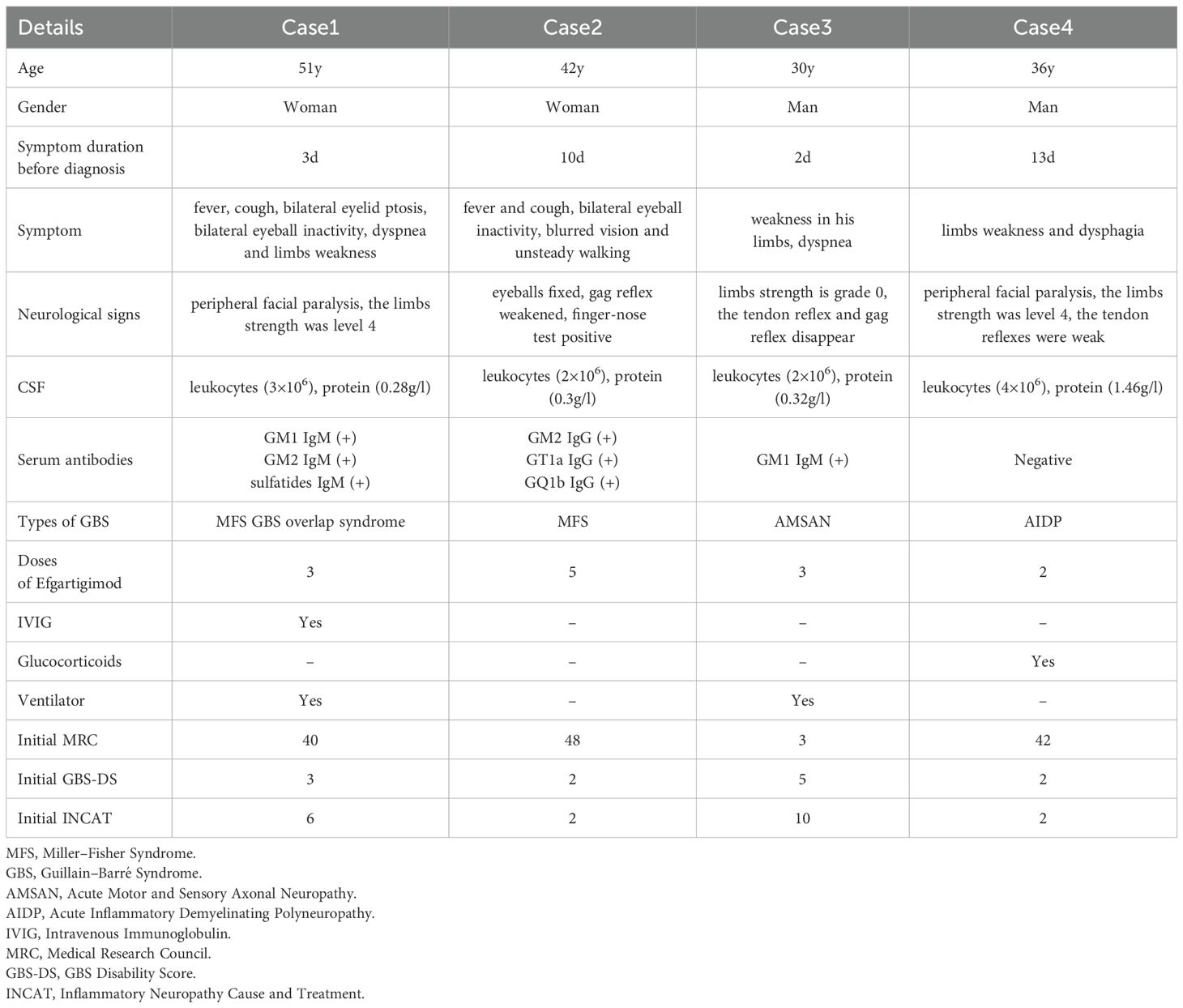
Table 1. Clinical manifestations and diagnosis of cases.
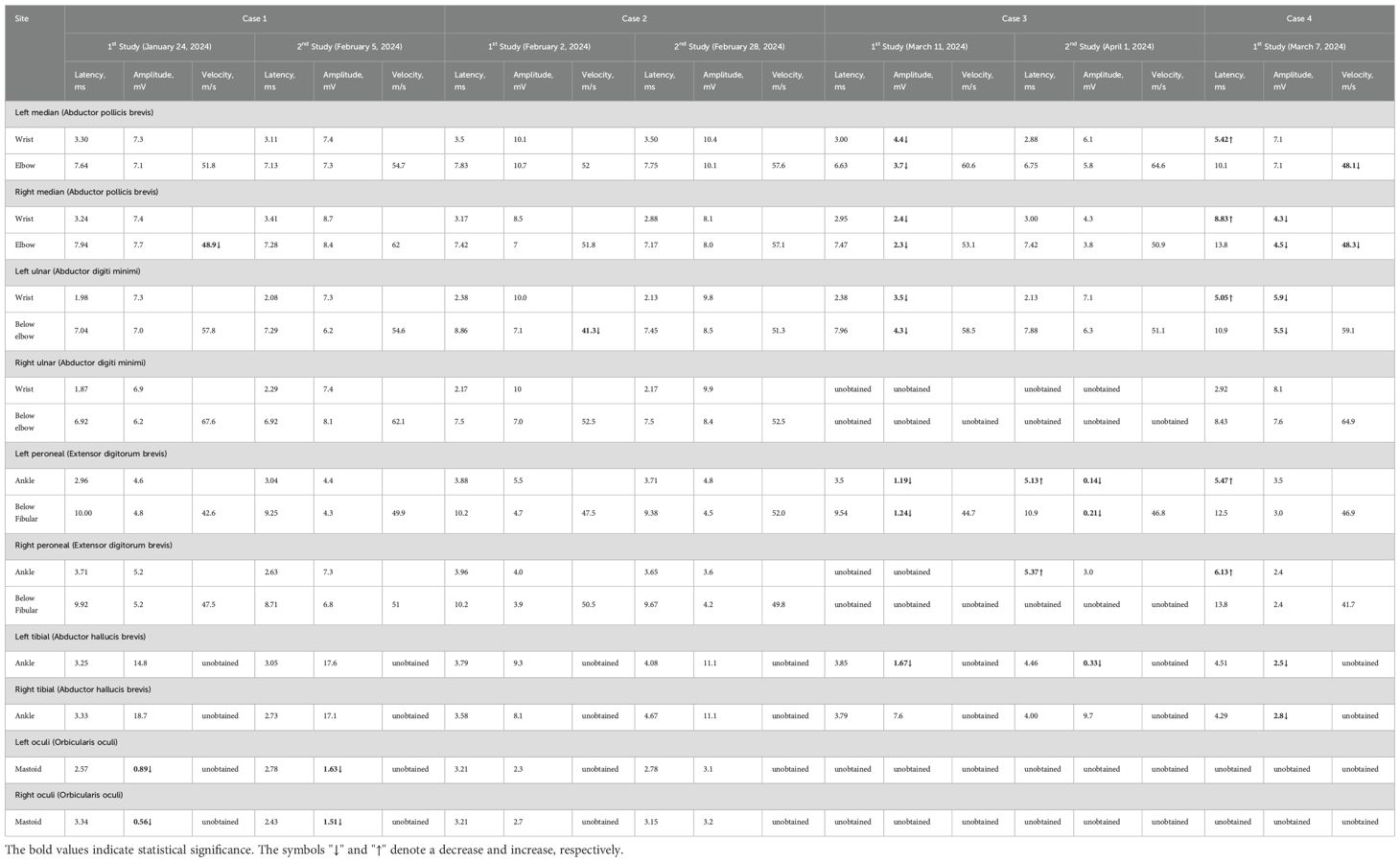
Table 2. Changes of motor nerve conduction before and after treatment.
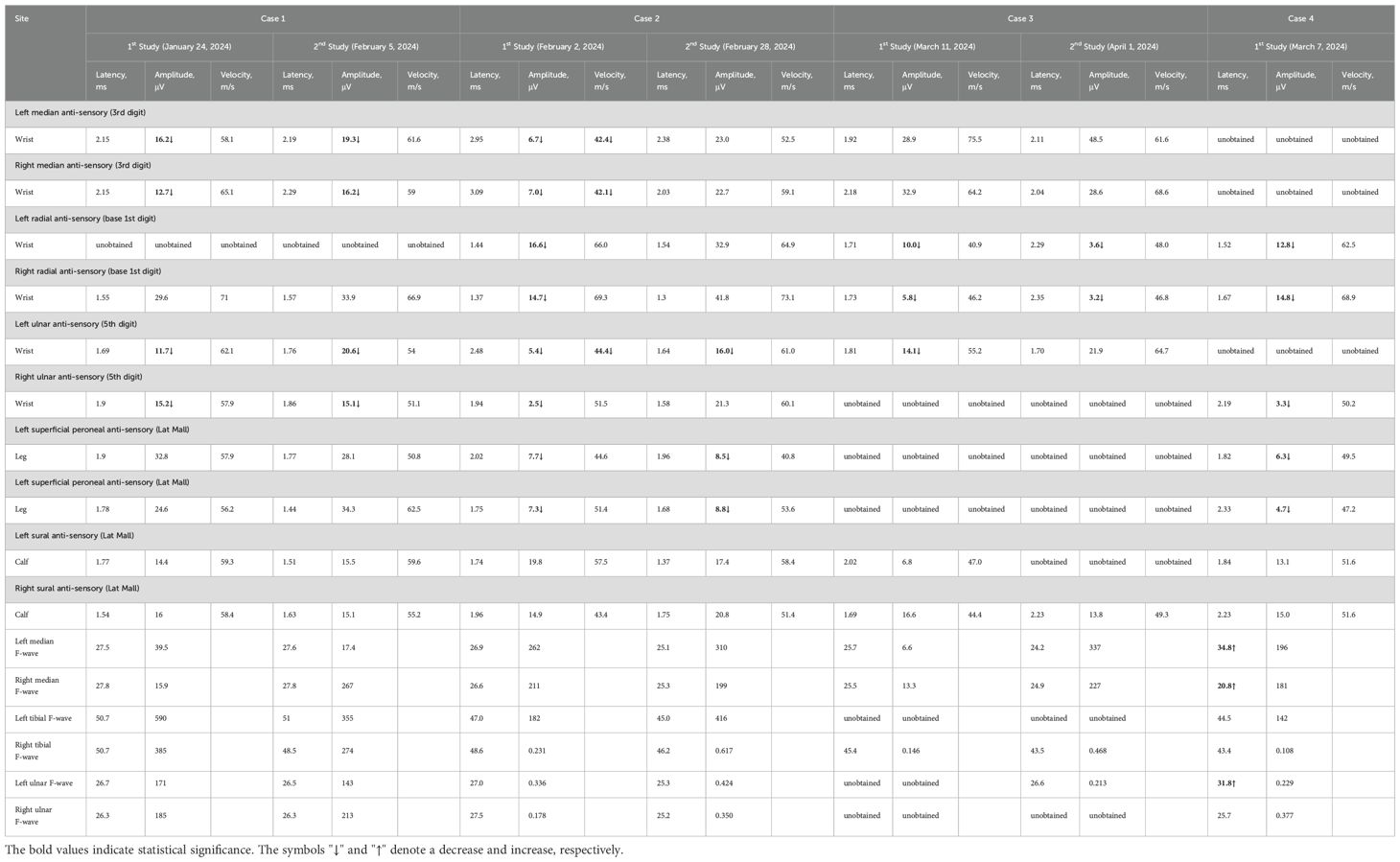
Table 3. Changes of sensory nerve conduction before and after treatment.
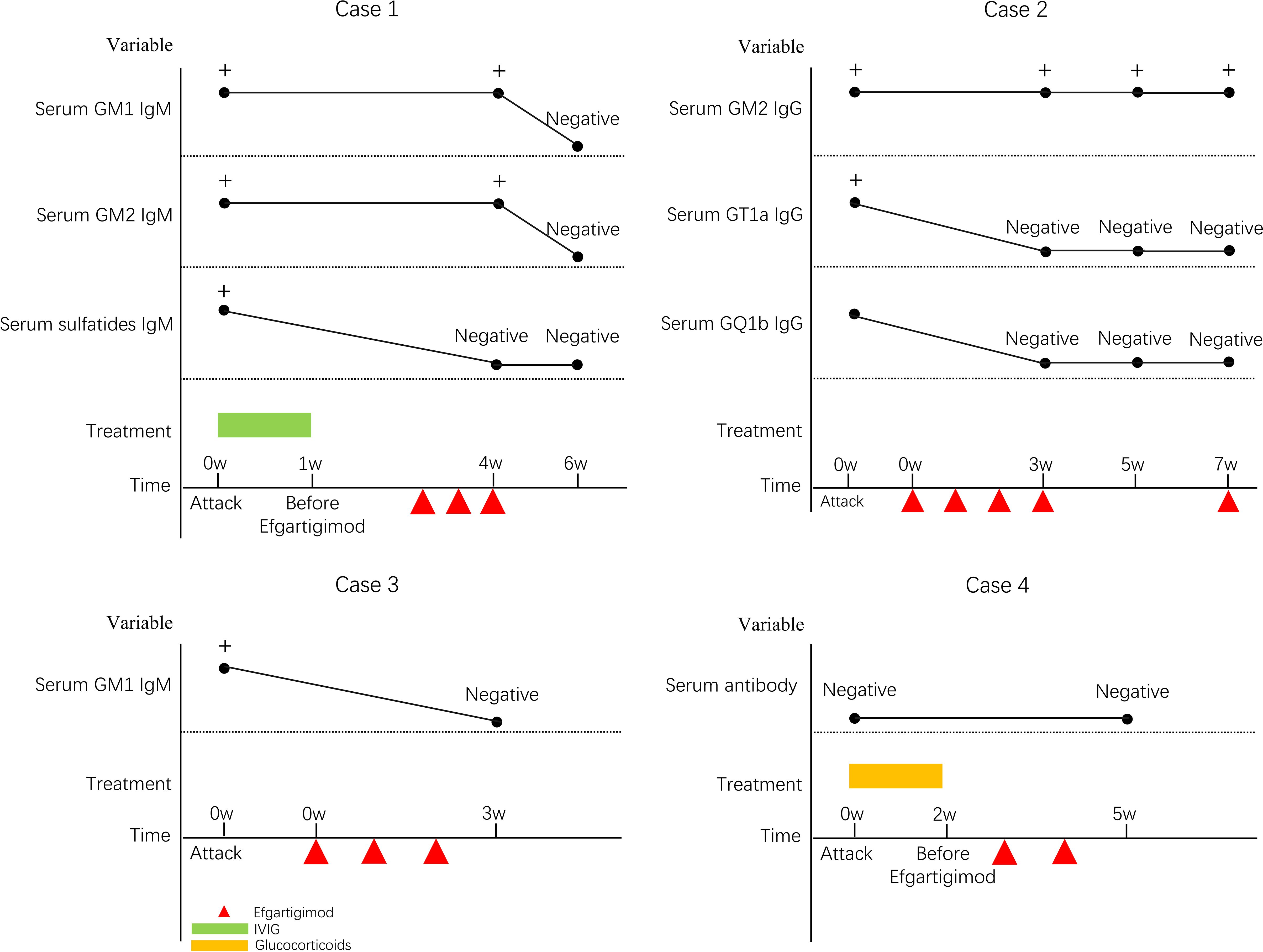
Figure 1. Changes in serum antibodies.
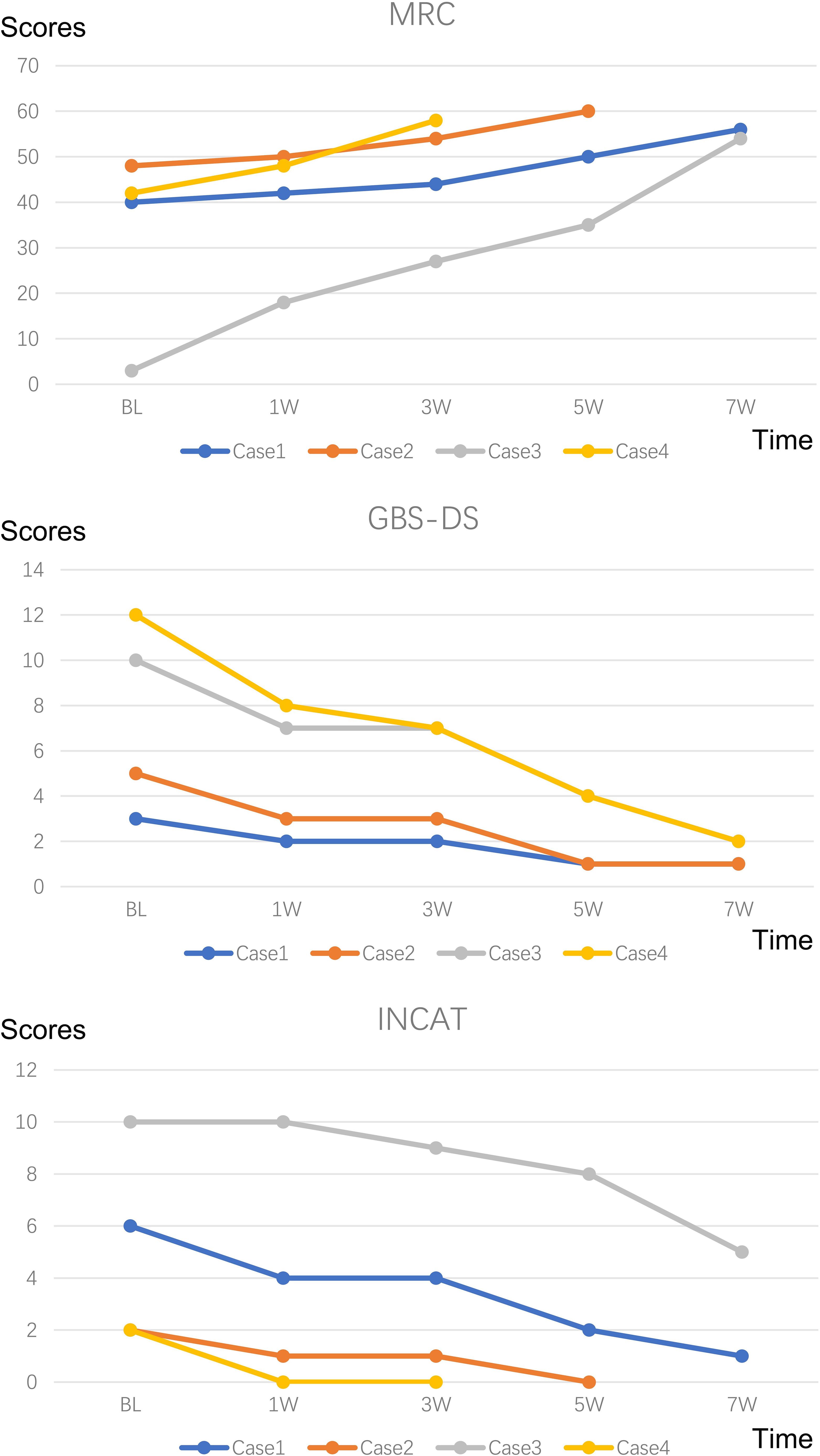
Figure 2. Changes in MRC, GBS-DS and INCAT score. MRC Sum Score: Quantifies muscle strength by summing scores from six bilateral limb muscles (three upper/three lower limbs), ranging from 60 (normal) to 0 (quadriplegia); GBS-DS: Functional disability scale graded 0-6 (0=healthy; 6=death); INCAT: Assesses limb functionality through validated mobility and arm function subscales.
3.2 Case 2
A 42-year-old female patient began to develop fever and cough in January 2024. One week later, she developed bilateral complete opthalmoplegia, blurred vision, and unsteady walking. She received antiviral treatment at a community hospital, but her symptoms did not resolve, so she attended our hospital and was admitted. She reported no history of chronic diseases (such as hypertension and diabetes), infectious diseases (such as hepatitis B and tuberculosis), surgical trauma, or poisoning, and no family history. Neurological examination showed that she could not move her eyeballs bilaterally, her gag reflex was weakened, and she exhibited instability in the finger-to-nose and finger-to-finger tests. She exhibited normal tendon reflexes in upper and lower extremities.
Nerve conduction studies demonstrated decreased MNCV in the left ulnar nerve, reduced SNAP amplitudes bilaterally in median, ulnar, radial, superficial peroneal and tibial nerves, and decreased SNCV bilaterally in median and tibial nerves with left ulnar involvement; in the blink reflex study, prolonged latencies and low amplitudes were observed for bilateral R1/R2 waveforms. On needle EMG, minimal abnormal spontaneous activity was detected in the left first dorsal interosseous. CSF analysis showed normal intracranial pressure (155 mmH2O), normal leukocytes (2×106), normal protein (0.3 g/L), and normal glucose (3.2 mmol/L). Liver function tests showed elevated alanine aminotransferase (143.00 U/L) and aspartate aminotransferase (120.00 U/L). Lipid tests showed elevated cholesterol (5.65 mmol/L), triacylglycerol (3.14 mmol/L), and low-density lipoprotein cholesterol (3.61 mmol/L). Blood cell count, kidney function, procalcitonin, respiratory etiology indicators, blood gas analysis results, electrolytes, blood glucose, myocardial infarction indicators, and coagulation function were normal. Head and spinal cord magnetic resonance imaging (MRI) scans, chest computed tomography (CT) scan, electrocardiogram, and cardiac color ultrasound were normal.
Peripheral neuropathy-associated antibodies in CSF and serum were assessed using immunofluorescence staining. No antibodies were detected in the CSF. GM2 IgG, GT1a IgG, and GQ1b IgG were positive in the serum. The patient was diagnosed with Miller–Fisher syndrome (MFS).
She received five doses of Efgartigimod (10 mg/kg intravenous infusion), administered weekly for the first four doses, followed by a fifth dose one month later. She was discharged after her eyeball movements returned to normal.
She showed no gait instability at the 7-week follow-up, but serum GM2 IgG antibodies remained positive. No adverse drug reactions occurred. The clinical manifestations, diagnosis, treatments, and follow-up of all four patients are detailed in Tables 1–3 and Figures 1, 2.
3.3 Case 3
A 30-year-old male patient attended our hospital and was admitted in February 2024 due to sudden slurred speech and difficulty breathing. A CT scan of his head showed brainstem bleeding, and he received symptomatic treatment, including treatment to control his blood pressure. He developed sudden weakness in his limbs 25 days after hospitalization. He reported no history of chronic diseases (such as hypertension and diabetes), infectious diseases (such as hepatitis B and tuberculosis), surgical trauma, or poisoning, and no family history. Neurological examination showed grade 0/5 strength in both proximal and distal muscles of the upper and lower limbs and absent tendon reflexes.
Nerve conduction studies revealed: Unelicitable CMAP in the right ulnar nerve; Reduced CMAP amplitudes bilaterally in median and ulnar nerves, with left tibial and peroneal nerve involvement; Unelicitable SNAP in superficial peroneal nerves; Reduced SNAP amplitudes bilaterally in radial nerves, and in left ulnar/sural nerves; Absent F-waves bilaterally in ulnar nerves and left tibial nerve. On needle EMG, significant abnormal spontaneous activities were observed in: bilateral first dorsal interosseous, extensor digitorum communis, tibialis anterior, gastrocnemius (medial heads), quadriceps (vastus lateralis), and left biceps brachii.
CSF analysis showed normal intracranial pressure (130 mmH2O), normal leukocytes (2×106), normal protein (0.32 g/L), and normal glucose (3 mmol/L). Liver function, blood lipids, blood cell count, kidney function, procalcitonin, electrolytes, and blood glucose were normal. A head CT scan showed bleeding in the pons. Chest CT scan, electrocardiogram, and cardiac color ultrasound were normal.
Peripheral neuropathy-associated antibodies in CSF and serum were assessed using immunofluorescence staining. No antibodies were detected in the CSF. GM1 IgM was positive in serum. The patient was diagnosed with acute motor and sensory axonal neuropathy (AMSAN).
He received ventilator-assisted respiratory therapy, anti-infective treatment (ceftazidime), and efgartigimod (10 mg/kg via weekly intravenous infusion for 3 weeks). When muscle strength in both upper and lower limbs reached grade 3 (MRC scale), the patient was transferred to the rehabilitation department for continued therapy.
At the 6 months follow-up assessment, muscle strength had recovered to grade 5/5 in all extremities. No adverse drug reactions occurred. The clinical manifestations, diagnosis, treatments, and follow-up of all four patients are detailed in Tables 1–3 and Figures 1, 2.
3.4 Case 4
A 36-year-old male patient suddenly developed limb weakness and dysphagia without any obvious cause in February 2024. He attended our hospital 4 days later. He reported no history of hypertension, diabetes, infectious diseases, surgical trauma, or poisoning, and no family history. Neurological examination showed droopy bilateral eyelids, shallow bilateral frontal lines and nasolabial folds, weak gag and tendon reflexes, and grade 4/5 strength in both proximal and distal muscles of the upper and lower limbs.
Nerve conduction studies demonstrated: Prolonged distal motor latencies with reduced CMAP amplitudes in bilateral median nerves (right amplitude reduction) and left ulnar nerve; Reduced CMAP amplitudes in bilateral tibial nerves; Prolonged motor distal latencies in bilateral common peroneal nerves; Unelicitable SNAPs in bilateral median and left ulnar nerves; Reduced SNAP amplitudes in bilateral radial, superficial peroneal, and right ulnar nerves; Prolonged F-wave latencies in bilateral median and left ulnar nerves. In the blink reflex study, markedly prolonged latencies were observed for bilateral R1 (Reference value 10–13 ms) and R2 (Reference value 30–40 ms) waveforms.
CSF analysis showed normal intracranial pressure (140 mmH2O), normal leukocytes (4×106), elevated protein (1.46 g/L), and normal glucose (3.5 mmol/L). These findings are consistent with albuminocytological dissociation. Liver function, blood lipids, blood cell count, kidney function, procalcitonin, electrolytes, and blood glucose were normal. Head MRI scan, chest CT scan, electrocardiogram, and cardiac color ultrasound were normal.
Peripheral neuropathy-associated antibodies in CSF and serum were assessed using immunofluorescence staining. No antibodies were detected in the CSF or serum. The patient was diagnosed with acute inflammatory demyelinating polyneuropathy (AIDP).
Due to the combined pressures of financial constraints and treatment accessibility challenges, both IVIG and PLEX were declined by the patient and their family. After being informed of the risks associated with corticosteroid therapy, they provided written consent for a course of methylprednisolone treatment (initial 500mg dose tapered by half every 3 days over 15 days). As this proved ineffective, the patient subsequently received efgartigimod (10 mg/kg via weekly intravenous infusion for 2 weeks).
The peripheral facial palsy resolved completely, accompanied by restoration of muscle strength to MRC grade 5/5 in all four extremities. No adverse drug reactions occurred. The clinical manifestations, diagnosis, treatments, and follow-up of all four patients are detailed in Tables 1–3 and Figures 1, 2.
4 Discussion
Our case series demonstrates that efgartigimod therapy is effective and safe across GBS variants, with sustained clinical improvement observed in all subtypes. GBS is the most common cause of acute flaccid paralysis worldwide.
Diagnosis of typical cases is usually straightforward. However, current diagnostic criteria have limitations that may result in missing atypical cases. Biomarkers that are used to diagnose typical cases may not apply to most GBS variants (8). One of the four cases was antibody negative, but his clinical symptoms supported a diagnosis of AIDP and he had increased protein in his CSF with a normal number of cells. In this study, normal CSF protein levels were observed in 3 out of 4 patients, which may be attributable to the early timing of lumbar puncture. All procedures were performed within the first week of admission, prior to the typical peak of albuminocytological dissociation. The higher antibody positivity rate (75%) may reflect the small cohort size (n=4) rather than true population prevalence, as limited samples can magnify statistical variability.
The detection of IgM anti-GM1 and anti-GM2 antibodies in case 1 patient with MFS-GBS overlap syndrome represents a departure from the classic serological profile of isolated MFS, which is dominated by IgG anti-GQ1b antibodies. While these IgM antibodies are typically associated with acute motor axonal neuropathy or motor-predominant GBS, their presence here may reflect an expanded autoimmune response targeting shared ganglioside epitopes within peripheral nerve complexes. Mechanistically, molecular mimicry between GM1/GM2 and GQ1b-containing glycolipid clusters could permit cross-reactive binding, while the potent complement-activating capacity of IgM may exacerbate axonal injury aligning with our electrophysiological findings of reduced CMAP amplitudes and prolonged distal latencies. Although causality cannot be definitively established, the temporal correlation of peak antibody titers with progressive limb weakness (exceeding typical MFS) supports a potential contributory role in disease pathogenesis, consistent with prior reports of anti-GM antibodies amplifying motor deficits in GBS. Further studies profiling ganglioside-specific IgM in overlap syndromes are warranted to validate their pathophysiological significance. Approximately one quarter of GBS patients require artificial ventilation (9). Many patients develop autonomic disorders and their symptoms typically peak within 4 weeks, followed by a weakening of the immune response and repair of peripheral nerves (10). The recovery period from this disease can last months or years.
Most people with GBS do well with immunotherapy, but some severe cases can be fatal. Additionally, most people are able to walk again 6 months after symptoms first appear, but some people remain disabled (1). Therefore, it is crucial that patients receive treatment as soon as possible, especially patients with rapidly progressive GBS.
Conventional treatments for acute GBS include IVIG and PLEX. At present, there is no clear understanding of how IVIG inhibits harmful inflammatory reactions, but it is generally believed to be related to the inhibition of Fc receptors. Beyond FcRn saturation mediated IgG half-life extension, IVIG’s anti-idiotypic antibodies directly neutralize pathogenic autoantibodies by sterically blocking their binding to neural targets. Subsequently, the formation of immune complexes facilitates accelerated clearance of circulating autoantibodies via FcγRIIB-dependent phagocytosis (11, 12). Concurrently, IVIG disrupts the complement cascade at critical junctures: specifically, it inhibits C3 convertase assembly to prevent amplification, while simultaneously binding C5b-7 complexes to obstruct membrane attack complex deposition on nerve membranes. Neonatal FcRn binds to IgG and protects it from lysosomal degradation. Efgartigimod is a IgG1 Fc fragment with strong affinity for FcRn (stronger than that of regular IgG), allowing it to compete with IgG for FcRn binding and reduce IgG recycling. Of the four cases, one was treated with IVIG and efgartigimod, one with glucocorticoids and efgartigimod, and two with efgartigimod only. Effective recovery of neurological function was observed in all cases. No drug-related adverse reactions occurred in any case. We regularly measured pathogenic antibodies in serum to assess the efficacy and safety of efgartigimod. After several efgartigimod injections, the pathogenic antibodies became negative in three cases, while serum GM2 IgG antibodies remained in one case.
This study, along with three previous case reports, evaluates the efficacy of efgartigimod in GBS, collectively supporting its potential as an effective and safe novel therapeutic option. The literature includes three pivotal case reports demonstrating efgartigimod’s therapeutic potential across neurological autoimmune conditions. Zhou et al. documented a 58-year-old AMAN patient with suboptimal response to IVIg/PLEX who achieved significant clinical and electrophysiological improvement after once-weekly infusions (10 mg/kg) over 4 weeks, establishing its efficacy in refractory axonal GBS (6). Zhang et al. reported two GBS cases where accelerated dosing (two 10 mg/kg doses within 5 days) enabled independent ambulation by 4 weeks, highlighting rapid functional recovery with transient adverse events (5). Watanabe et al. described an 84-year-old with anti-GQ1b syndrome overlapping myasthenia gravis, in whom efgartigimod induced ventilator weaning within 7 days and restored mobility after conventional therapies failed, proving its value in complex, treatment-resistant disorders (7). In contrast to prior single-case reports describing combination therapy (administered following failure of IVIG or PLEX), the current work presents a case series (n=4). Notably, it includes two monotherapy cases (without concurrent IVIG or PLEX), encompasses diverse GBS subtypes, demonstrates acute-phase application, and reports well-defined favorable treatment outcomes.
5 Conclusion
The rapid elimination of pathogenic antibodies is crucial for the effective treatment of GBS. Efgartigimod, an FcRn antagonist, accelerates antibody clearance by inhibiting the recycling of IgG into the bloodstream. Four patients treated with intravenous efgartigimod exhibited favorable clinical outcomes, suggesting its ability to rapidly reduce GBS-related antibodies and alleviate symptoms. These findings indicate that efgartigimod may be a promising therapeutic option for acute GBS. Further studies are needed to confirm its broader applicability.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
This study was approved by the ethics committee of Wuhan University Peoples Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
MD: Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Software, Writing – original draft. ZK: Investigation, Software, Writing – original draft. YW: Methodology, Writing – original draft. XW: Formal analysis, Investigation, Writing – original draft. TL: Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by the Natural Science Foundation of Hubei Province of China (2020CFB278) and the Independent Research Project of Wuhan University (2042021kf0126).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chung A and Deimling M. Guillain-barre syndrome. Pediatr Rev. (2018) 39:53–4. doi: 10.1542/pir.2017-0189
2. Doets AY, Verboon C, Van Den Berg B, Harbo T, Cornblath DR, Willison HJ, et al. Regional variation of Guillain-Barré syndrome. Brain. (2018) 141:2866–77. doi: 10.1093/brain/awy232
3. van den Berg B, Bunschoten C, van Doorn PA, and Jacobs BC. Mortality in guillain-barre syndrome. Neurology. (2013) 80:1650–4. doi: 10.1212/WNL.0b013e3182904fcc
5. Zhang H, Ma J, Feng Y, Ma H, Liu D, Pang X, et al. Efgartigimod in the treatment of Guillain-Barre syndrome. J Neurol. (2024) 271:3506–11. doi: 10.1007/s00415-024-12321-4
6. Zhou J, Yu W, Ding S, Shi C, and Liang H. Resolution of acute motor axonal neuropathy in a patient after treatment with efgartigimod: A case report. Med (Baltimore). (2024) 103:e40700. doi: 10.1097/MD.0000000000040700
7. Watanabe K, Takahashi S, Kanda A, Watanabe T, Kakinuma Y, Yano S, et al. Case Report: Therapeutic effect of efgartigimod in refractory anti-GQ1b antibody syndrome coexisting with myasthenia gravis. Front Immunol. (2025) 16:1605985. doi: 10.3389/fimmu.2025.1605985
8. Leonhard SE, Mandarakas MR, Gondim FA, Bateman K, Ferreira ML, Cornblath DR, et al. Diagnosis and management of Guillain–Barré syndrome in ten steps. Nat Rev Neurol. (2019) 15:671–83. doi: 10.1038/s41582-019-0250-9
9. Nabi S, Khattak S, and Irshad Awan M. Clinical predictors of mechanical ventilation in Guillain-Barré syndrome (GBS). Pakistan J Neurol Sci (PJNS). (2014) 9:1385945. doi: 10.3389/fneur.2024.1385945
10. van Doorn PA. Diagnosis, treatment and prognosis of Guillain-Barré syndrome (GBS). La Presse Médicale. (2013) 42:e193–201. doi: 10.1016/j.lpm.2013.02.328
11. Samuelsson A, Towers TL, and Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. (2001) 291:484–6. doi: 10.1126/science.291.5503.484
Keywords: Guillain-Barré syndrome, efgartigimod, Miller-Fisher syndrome, acute motor and sensory axonal neuropathy, acute inflammatory demyelinating polyneuropathy
Citation: Deng M, Kong Z, Wang Y, Wang X and Li T (2025) Efgartigimod in the treatment of Guillain-Barré syndrome: case report. Front. Immunol. 16:1586663. doi: 10.3389/fimmu.2025.1586663
Received: 05 March 2025; Accepted: 25 August 2025;
Published: 04 September 2025.
Edited by:
Augusto Vaglio, University of Florence, ItalyReviewed by:
Georgios E. Manousakis, University of Minnesota, United StatesMurat Mert Atmaca, University of Health Sciences, Türkiye
Copyright © 2025 Deng, Kong, Wang, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xufeng Wang, eXd4ZjEwMTFAMTYzLmNvbQ==; Tao Li, dGFvbGkuc3VubnlAd2h1LmVkdS5jbg==