 Yi Kang1,2,3†
Yi Kang1,2,3† Qian Jin3†Mengqi Zhou4†Huijuan Zheng1,2Danwen Li1,2,3Xuezhe Wang1,2,3Jingwei Zhou1,2Yaoxian Wang3*Jie Lv1*
Qian Jin3†Mengqi Zhou4†Huijuan Zheng1,2Danwen Li1,2,3Xuezhe Wang1,2,3Jingwei Zhou1,2Yaoxian Wang3*Jie Lv1*- 1Dongzhimen Hospital, Beijing University of Chinese Medicine, Beijing, China
- 2Key Laboratory of Chinese Internal Medicine of Ministry of Education, Beijing Dongzhimen Hospital, Beijing University of Chinese Medicine, Beijing, China
- 3Graduate School of Beijing University of Chinese Medicine, Beijing, China
- 4Department of Traditional Chinese Medicine, Beijing Puren Hospital, Beijing, China
Kidney diseases represent a diverse group of disorders with pathogenic mechanisms involving multiple pathological processes, including inflammation, immunity, and cell death. Neutrophils, as primary effector cells in inflammatory immune responses, participate in defending against renal infection and injury by releasing reactive oxygen species, proteases, and cytokines. However, persistent neutrophil activation is considered a crucial driver of kidney disease progression. Neutrophil apoptosis represents a critical turning point between inflammatory progression and resolution. Specialized pro-resolving mediators (SPMs) are endogenous anti-inflammatory mediators that play a critical role in resolving inflammation. They not only induce neutrophil programmed cell death and promote macrophage-mediated efferocytosis of apoptotic cells but also inhibit neutrophil infiltration and degranulation, ultimately facilitating the restoration of inflammatory microenvironment and tissue homeostasis. This review concentrates on elucidating the mechanisms by which SPMs regulate neutrophil apoptosis and systematically demonstrates their potential as novel therapeutic targets in kidney diseases.
1 Introduction
Kidney diseases represent a significant global public health challenge. The Global Burden of Disease study indicates that chronic kidney disease (CKD) has emerged as a leading contributor to rising global mortality rates. Once progressing to end-stage renal disease (ESRD), patients maintain poor long-term survival rates even with dialysis or transplantation (1, 2). While the precise pathogenic mechanisms of kidney diseases remain unclear, studies suggest their association with multiple factors including inflammation, autophagy, oxidative stress, and cell death. These factors may interact synergistically, ultimately leading to renal dysfunction (3, 4). Inflammation represents a crucial pathological mechanism in kidney diseases, and impaired inflammatory resolution may be a key factor driving irreversible disease progression (5). Deficiency or dysfunction of specialized pro-resolving mediators (SPMs) may result in persistent inflammation, closely associated with the chronic progression of kidney diseases (6).
Neutrophils serve as principal mediators in inflammatory responses, orchestrating defense mechanisms through the release of cytokines, chemokines, reactive oxygen species, and enzymes. While this process is essential for pathogen elimination, hyperactivation may initiate or aggravate tissue injury and chronic inflammation (7). Chronic inflammatory conditions are characterized by impaired or delayed neutrophil apoptosis (8), and the proper timing of neutrophil apoptosis has been demonstrated to be critical for inflammatory resolution across multiple inflammatory models (9). SPMs demonstrate therapeutic potential in attenuating the progression of neutrophil-mediated inflammation and chronic pathologies (including cardiovascular diseases, metabolic syndrome, and ischemia-reperfusion injury) through multiple mechanisms: reducing neutrophil recruitment, facilitating neutrophil apoptosis, and enhancing macrophage efferocytosis (10).
Based on these findings, we propose the following hypothesis: SPMs-regulated neutrophil apoptosis may influence the onset and progression of kidney diseases. This hypothesis not only explains the impact of persistent inflammation on kidney disease progression but also provides theoretical foundations for exploring novel therapeutic strategies. This review will examine in depth these mechanisms and research developments, aiming to offer new perspectives and approaches for the study and treatment of kidney diseases.
2 SPMs: key factors in kidney disease progression
Inflammation represents a complex defensive physiological response orchestrated by the immune system when the body encounters pathogenic infections, tissue damage, or harmful stimuli. Historically, inflammation resolution was considered a passive process, merely relying on the gradual degradation of pro-inflammatory mediators to inhibit further immune cell infiltration into damaged tissues. However, groundbreaking research by Charles Serhan’s team revealed that inflammation resolution is an actively regulated, highly programmed process involving multiple mechanisms and molecules, governed by a group of endogenous lipid mediators, which they designated as SPMs (11). Deficiency or dysfunction of SPMs may be involved in the occurrence and progression of various chronic inflammatory diseases, such as kidney disease, cardiovascular diseases, diabetes, neurodegenerative diseases, periodontitis, rheumatoid arthritis, and others (12–19). In the context of kidney disease pathogenesis and progression, SPMs demonstrate significant importance through their multifaceted actions: modulating inflammatory cell functions, maintaining homeostasis between pro- and anti-inflammatory factors, and providing protection to various renal cells, including tubular epithelial cells, glomerular mesangial cells, podocytes, endothelial cells, and fibroblasts (20).
2.1 Molecular identity and biological actions of SPMs
Inflammation resolution is a key phase in the regulation of the inflammatory response, with its mechanism fundamentally different from anti-inflammatory actions. From a pathophysiological standpoint, inflammation resolution begins with the peak recruitment of inflammatory cells to tissue cells, in which the body actively clears inflammatory mediators and promotes tissue homeostasis through programmed regulation (5). This process is initiated by the coordinated action of granulocytes and the monocyte/macrophage system recruited to the site of inflammation, where pro-inflammatory lipid mediators transition into pro-resolving lipid mediators, and SPMs begin to appear at the site of inflammation (21). SPMs are a class of endogenous regulatory molecules synthesized through the selective catalysis of enzymes such as lipoxygenases (LOX), cyclooxygenases (COX), and cytochrome P450 (CYP) from endogenous polyunsaturated fatty acids (PUFAs), including ω-3 fatty acids eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), docosapentaenoic acid (DPA), and ω-6 fatty acid arachidonic acid (AA) (6). Their biosynthesis depends on the cross-cellular collaboration between immune effector cells (such as neutrophils, monocytes/macrophages) and resident tissue cells (such as epithelial and endothelial cells), as these enzymes are differentially expressed in epithelial cells (15-LOX), endothelial cells (COX-2), granulocytes (5-LOX), and platelets (12-LOX) (11, 22), and are subsequently degraded by enzymes like 15-hydroxyprostaglandin dehydrogenase (15-PGDH) (23, 24). The SPMs discovered in mammals to date include four major lipids: Lipoxins (LXs), Resolvins (Rvs), Protectins (PDs), and Maresins (MaRs) (5, 23, 25), as well as non-lipid regulatory factors such as AnxA1, gaseous mediators, purines, and neuroregulatory substances (26).
SPMs promote inflammation resolution through G protein-coupled receptor (GPCR)-dependent signaling pathways, with different SPMs selectively activating their specific GPCRs, and their affinity for these GPCRs being in the low nanomolar range (25, 27). Although the understanding of the downstream targets following receptor activation by SPMs remains incomplete, it has been confirmed that several signaling pathways are involved, including the regulation of NF-κB, ERK, PI3K/AKT, p38 MAPK, and miRNA expression. Neutrophils, as core effector cells of the innate immune system, play a role throughout the entire process from inflammation initiation to resolution. SPMs exert pro-resolution effects by limiting neutrophil recruitment and activation, promoting neutrophil apoptosis, and facilitating macrophage-mediated efferocytosis to clear apoptotic neutrophils, among other mechanisms (12, 28, 29) (Figure 1). The mechanisms by which SPMs regulate inflammation resolution can be found in relevant reviews (28–31).
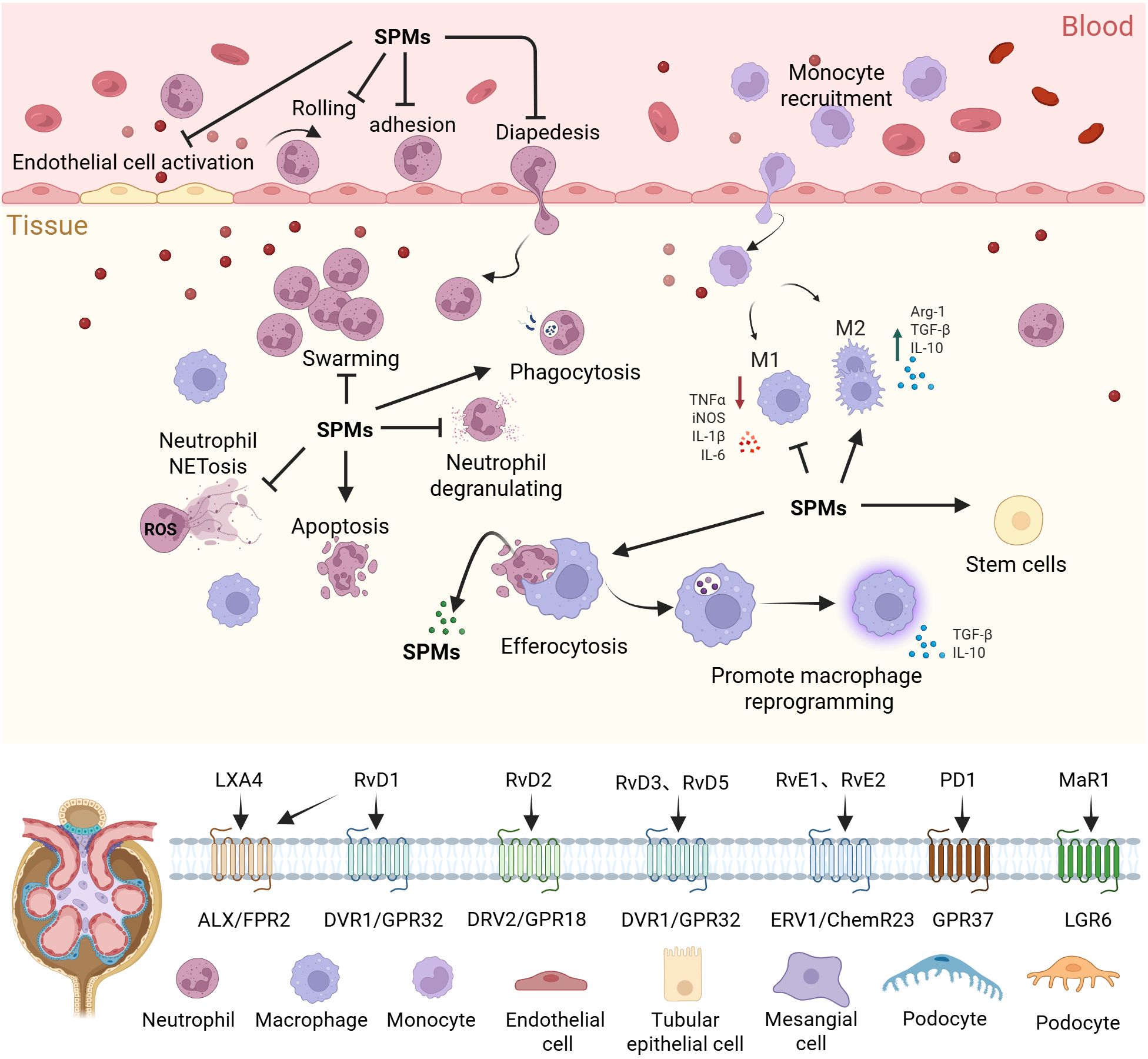
Figure 1. Regulation of inflammation by SPMs. SPMs regulate inflammation resolution and tissue repair through spatiotemporal dynamics. They limit the spread of inflammation by inhibiting excessive activation of endothelial cells and regulating the rolling-adhesion-transmigration cascade of neutrophils across the endothelium. Furthermore, SPMs restrict neutrophil degranulation, NETosis, and swarming, while promoting neutrophil apoptosis and enhancing phagocytosis of pathogens. SPMs also drive macrophage reprogramming, enhance efferocytosis, and facilitate the release of further SPMs. In synergy with stem cell activation, SPMs ultimately achieve the inflammation resolution and the restoration of tissue homeostasis, preventing chronic inflammatory damage. SPMs, Specialized pro-resolving mediators; TNF-α, Tumor Necrosis Factor Alpha; iNOS, Inducible Nitric Oxide Synthase; IL-1β - Interleukin 1 Beta; IL-6, Interleukin 6; Arg-1, Arginase-1; TGF-β, Transforming Growth Factor Beta; IL-10, Interleukin 10; NETosis, Neutrophil Extracellular Traps Formation; ROS, Reactive Oxygen Species.
2.2 SPMs dysregulation in kidney disease pathogenesis
Inflammation, as a core pathological feature of kidney disease, when unresolved, can drive changes such as glomerulosclerosis and tubular interstitial fibrosis (32, 33), and is a key driving factor in the progression of end-stage renal disease (34, 35). Kidney regulation of the inflammatory response is disease-specific: self-limiting diseases (such as post-streptococcal glomerulonephritis) can resolve inflammation through endogenous repair mechanisms (36), while other types of kidney diseases (such as diabetic nephropathy) are often associated with chronic inflammation and sustained activation, leading to severe organ damage (37). The deficiency or dysfunction of SPMs is considered closely related to the pathogenesis of chronic inflammatory diseases (38–40), and the kidney inflammation resolution mechanism is also a process finely regulated by SPMs, the mechanisms of which are just beginning to be understood (Table 1).
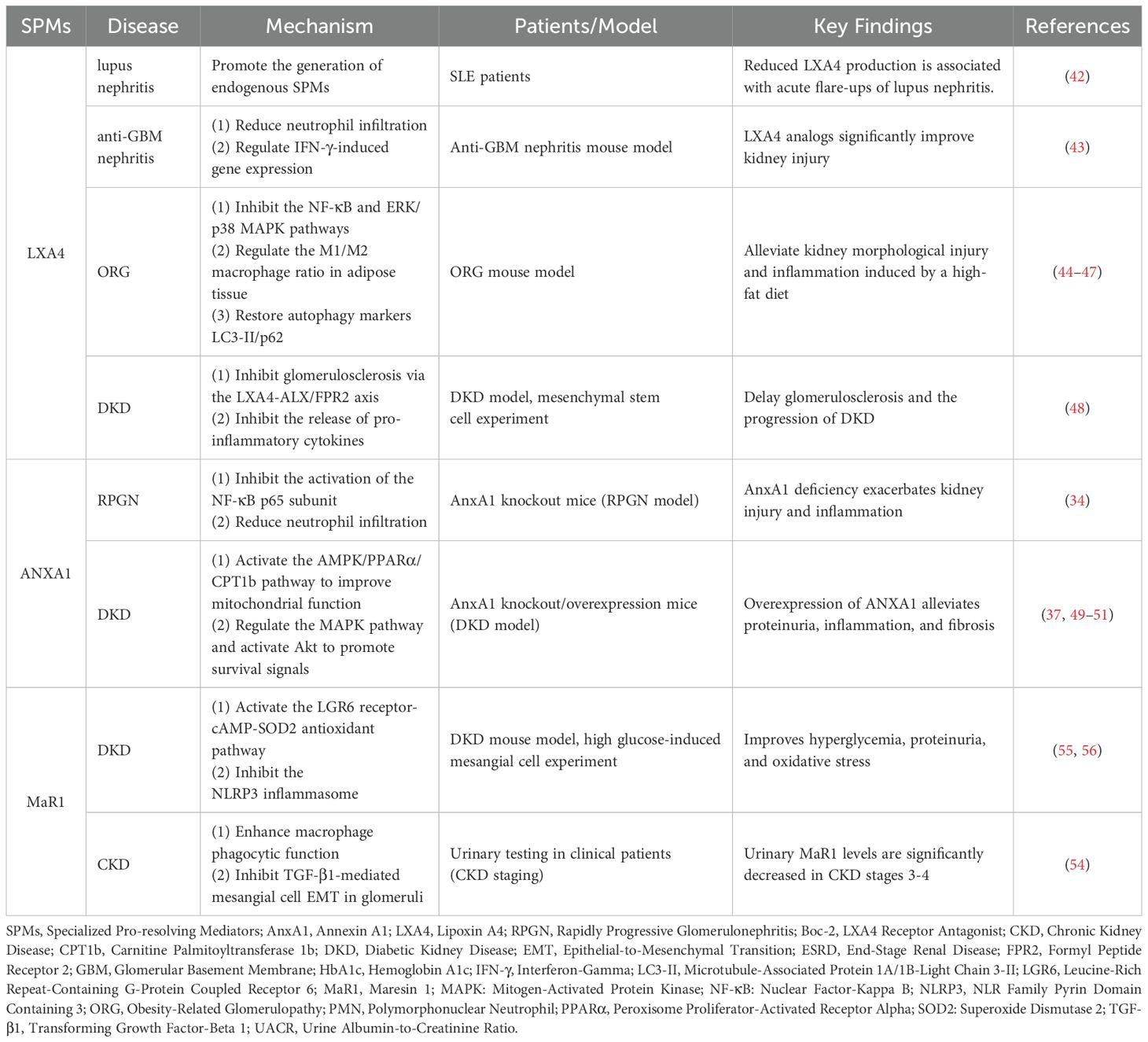
Table 1. Research on SPMs in kidney diseases.
2.2.1 Lipoxin A4
Lipoxins are the first mediators identified to specifically promote resolution (41) and have been studied in anti-glomerular basement membrane nephritis (GBM), lupus nephritis, obesity-related glomerulopathy (ORG), and DKD. In patients with Systemic Lupus Erythematosus, persistent inflammatory states may be related to a lack of endogenous SPMs, with decreased formation and release of LXA4 possibly directly associated with the development and acute flare-ups of lupus and its renal complications (42). In an anti-glomerular basement membrane nephritis (anti-GBM) mouse model, the LXA4 analog (15-epi-16-(FPhO)-LXA-Me) significantly improved renal injury in anti-GBM nephritis mice by reducing neutrophil infiltration, alleviating oxidative stress, and regulating IFN-γ-induced gene expression (43). ORG is characterized by glomerular enlargement and lipid deposition in renal cells, and is closely associated with the onset of ESRD (44). Uncontrolled inflammation is considered the pathological basis of obesity, with leukocytes from obese individuals showing impaired SPM characteristics (45). In the ORG model mouse, LXA4 injection significantly alleviated high-fat diet-induced renal morphological and functional damage, reduced the levels of pro-inflammatory cytokines and chemokines, and inhibited the activation of NF-κB and ERK/p38 MAPK pathways, with these effects significantly blocked by pretreatment with the LXA4 receptor antagonist Boc-2 (46). LXA4 and its synthetic analogs alleviate obesity-induced glomerular enlargement and mesangial matrix proliferation in mice by reducing adipose tissue inflammation, regulating the M1/M2 macrophage ratio in adipose tissue, and restoring the expression of autophagy markers LC3-II and p62 in adipose tissue (47). Notably, mesenchymal stem cells may inhibit glomerulosclerosis and pro-inflammatory cytokine release through the LXA4-ALX/FPR2 axis, thereby preventing the progression of DKD (48).
2.2.2 ANXA1
ANXA1 is a 37 kDa protein, also known as lipocortin 1, initially thought to be a mediator of the anti-inflammatory effects of glucocorticoids in the host defense system. Subsequent studies have shown that it promotes nearly all mechanisms of inflammation resolution (26). Rapidly Progressive Glomerulonephritis (RPGN) and DKD are common causes of end-stage renal disease. The lack of ANXA1 may be an important factor in the progression of RPGN and DKD. Compared to wild-type mice, AnxA1 knockout mice exhibit more severe kidney injury and inflammatory cell infiltration following RPGN induction. Endogenous AnxA1 exerts renal protective effects during RPGN by inhibiting pro-inflammatory signals and neutrophil infiltration (34). Failure of inflammation resolution is a major pathological factor in the progression of DKD. In diabetic mouse models, AnxA1 deficiency exacerbates kidney damage, as evidenced by more severe proteinuria, renal inflammation, and fibrosis. Overexpression of AnxA1 significantly alleviates kidney damage, possibly through the following mechanisms: AnxA1 binds to the p65 subunit of the transcription factor NF-κB, inhibiting its activation and thereby modulating the inflammatory state (37); at the same time, AnxA1 activates the AMPK/PPARα/CPT1b pathway, improving mitochondrial function and fatty acid oxidation, which in turn reduces renal inflammation and lipotoxicity in DKD (49); it may also be involved in AnxA1’s regulation of MAPK, activating pro-survival pathways (Akt), thereby preventing cardiac and renal dysfunction (50). Clinical studies have confirmed that serum ANXA1 levels are significantly lower in DKD patients compared to diabetic patients without renal lesions (51). The expression of ANXA1 in the kidneys is increased (37), which may be a compensatory response to inflammation.
2.2.3 Maresin 1
MaR1 is a potent anti-inflammatory and pro-resolving mediator synthesized by macrophages, and is the founding member of the maresins family. It effectively limits polymorphonuclear neutrophil (PMN) infiltration, enhances macrophage phagocytosis of apoptotic PMNs, promotes tissue regeneration, alleviates pain (52), regulates inflammation, and facilitates tissue repair. MaR1 has demonstrated significant protective effects in various diseases, including kidney disease, lung disease, liver disease, and vascular diseases (53). Studies have found that urinary MaR1 levels in stage 3–4 kidney disease subjects are significantly lower than in healthy individuals and stage 1–2 kidney disease patients (54). Serum MaR1 levels are significantly reduced in DKD patients and are negatively correlated with disease markers such as UACR, HbA1c, and neutrophil count. Animal experiments show that MaR1 injection improves hyperglycemia, reduces UACR, and alleviates renal pathological damage in DKD mice. It also activates the cAMP-SOD2 antioxidant pathway through the LGR6 receptor, inhibiting inflammation and oxidative stress. Cellular experiments further confirm the protective effects of MaR1. MaR1 holds promise as a predictor and therapeutic target for DKD, but its clinical application still requires further validation (55). Other studies have confirmed that MaR1 can inhibit NLRP3 inflammasome activation, reducing high glucose-induced epithelial-to-mesenchymal transition of glomerular mesangial cells mediated by TGF-β1 (56).
In conclusion, the renal protective effects of SPMs have been confirmed by multiple studies; however, there are still several scientific questions that need to be clarified. Taking DKD as an example, LXA4, ANXA1, and MaR1 are considered to play important roles in the pathogenesis of DKD. However, due to the limitations of current research, it remains unclear whether different SPMs exhibit synergistic effects. Is there an imbalance between SPMs during the disease progression, similar to what has been observed in other chronic inflammatory diseases (57)? Particularly, whether there are additive or antagonistic effects at different stages of the disease remains unsupported by systematic research evidence. Furthermore, during the resolution of inflammation, inflammatory immune cells such as neutrophils and macrophages are both the producers and effectors of SPMs. This suggests that SPMs may exert organ-protective effects indirectly through immune regulatory networks. However, current studies mostly focus on the direct protective mechanisms of SPMs on glomerular podocytes, mesangial cells, and renal tubular epithelial cells, while the analysis of the SPMs-immune cell interaction network is clearly insufficient. By drawing upon the research paradigm of ANXA1 in regulating tumor-associated macrophage efferocytosis to remodel the immune microenvironment in pancreatic cancer (58), a deeper understanding of the immune regulatory loops of SPMs and their interactions with parenchymal cells will help reveal the molecular landscape of SPMs-mediated renal protection. This will provide a theoretical foundation for developing precise therapeutic strategies based on the temporal regulation of SPMs.
2.3 SPMs: emerging potential and prospects for the treatment of kidney diseases
Resolution pharmacology represents an emerging therapeutic paradigm that aims to target acute and chronic inflammation through the utilization of SPMs (28). While traditional anti-inflammatory approaches are often limited by concerns about compromised host immunity and infection risks, SPMs-based therapeutic strategies not only reduce the overactivation of pro-inflammatory signals but also effectively avoid the risk of immunosuppression (26). However, endogenous SPMs are characterized by rapid degradation and high synthesis costs, which to some extent limit the potential of natural SPMs as candidate drugs for inflammatory diseases (23). In recent years, through methods such as chemical modification, several generations of SPMs mimetics (59) and their receptor agonists (60) have been successfully developed, significantly expanding the therapeutic potential of SPMs. Among them, receptor agonists such as ACT-389949 and BMS-986235 have entered Phase I clinical development (61), and several therapies based on SPMs have shown encouraging outcomes in preclinical models of both acute and CKD (Table 2).
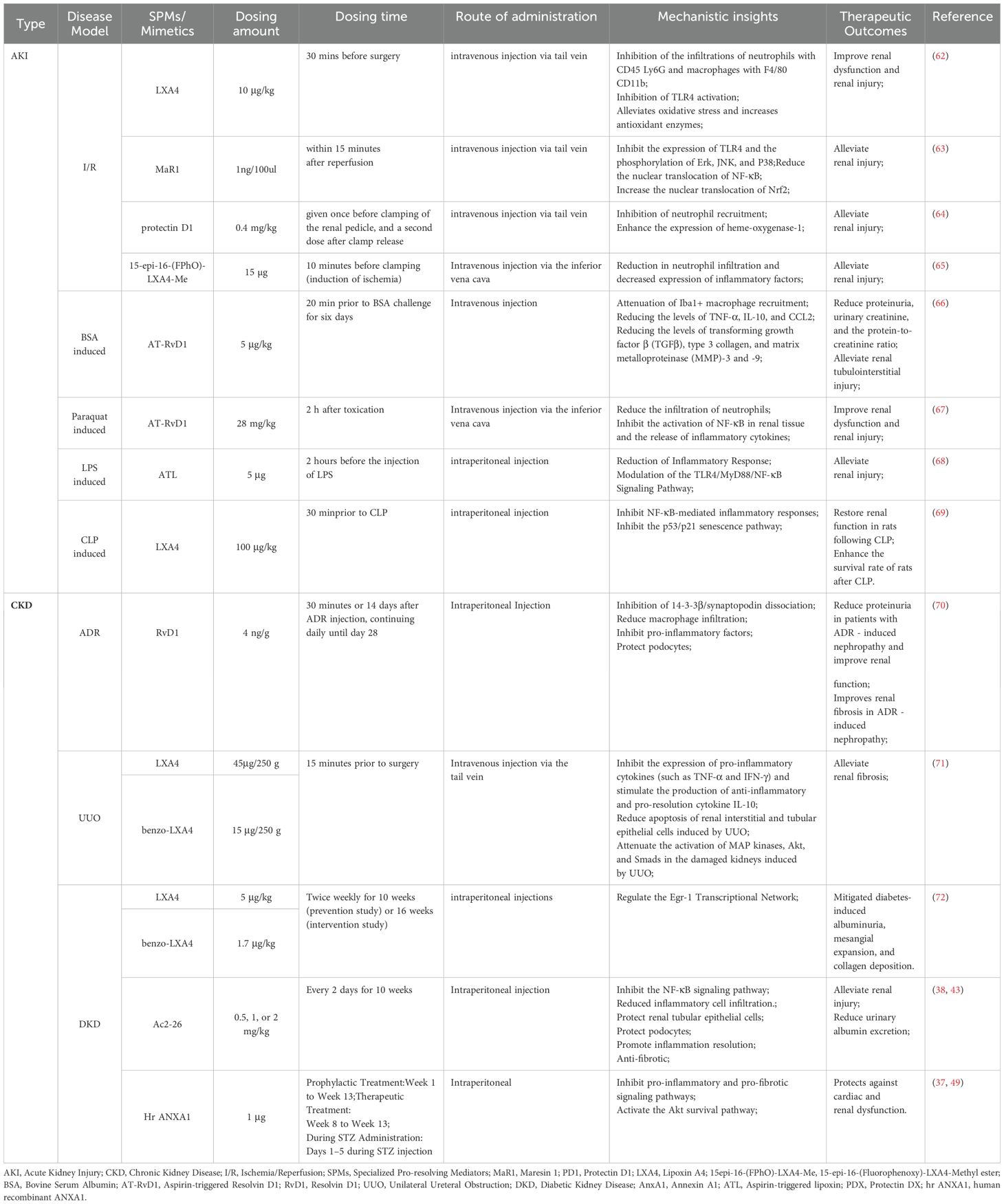
Table 2. Mechanistic studies of SPMs in the treatment of renal diseases.
2.3.1 Acute kidney injury models
In various AKI models, including ischemia-reperfusion Injury (I/R), nephrotoxic drugs, and sepsis-induced injury, SPMs treatments have demonstrated significant pro-resolving and renoprotective effects. In renal I/R models, treatments with MaR1, Protectin D1 (PD1), LXA4, and its analog 15epi-16-(FPhO)-LXA4-Me have shown to alleviate renal I/R injury through multiple mechanisms: reducing neutrophil infiltration, decreasing pro-inflammatory cytokine and chemokine expression, and inhibiting harmful lipid mediator production, thereby exerting potent anti-inflammatory and anti-oxidative stress effects (62–65). In AKI models induced by bovine serum albumin (BSA) and paraquat, RvD1 mimetics such as aspirin-triggered resolvin D1 (AT-RvD1) demonstrate therapeutic efficacy by reducing inflammatory cell infiltration in renal tissue, inhibiting NF-κB activation and inflammatory cytokine release, and activating the Nrf2 signaling pathway to upregulate antioxidant gene expression, thereby alleviating tubulointerstitial injury and fibrosis (66, 67). Furthermore, in sepsis-induced AKI models using LPS and Cecal Ligation and Puncture, pretreatment with Aspirin-triggered lipoxin (ATL) and LXA4 has shown remarkable therapeutic potential by significantly reducing serum creatinine, blood urea nitrogen, and urinary levels of kidney injury molecules and neutrophil gelatinase-associated lipocalin. These treatments also inhibited NF-κB pathway activation and decreased inflammatory cytokine levels, thereby significantly alleviating sepsis-induced AKI and improving rat survival rates (68, 69).
2.3.2 CKD models
In various CKD models, including those induced by adriamycin (ADR), unilateral ureteral obstruction (UUO), and in DKD models, SPMs treatments have demonstrated significant pro-resolving and renoprotective effects. In ADR-induced nephropathy mouse models, early intervention with RvD1 has shown remarkable efficacy by reducing renal macrophage infiltration, thereby preventing ADR-induced podocyte injury, reducing proteinuria, and maintaining podocyte structural stability (70). In UUO models, LXA4 and its synthetic analog benzo-LXA4 effectively inhibit renal fibrosis progression through multiple mechanisms: suppressing pro-inflammatory cytokine expression, promoting anti-inflammatory cytokine IL-10 production, and reducing collagen deposition and cellular apoptosis in renal tissue (71). In STZ-induced Type 1 diabetes mellitus (T1DM) models, hrAnxA1 treatment administered during weeks 8–13 demonstrates therapeutic potential by attenuating the increase in proteinuria and fractional sodium excretion, while also delaying the progression of cardiac dysfunction (50). Furthermore, in STZ-induced diabetes mellitus (DM) ApoE−/− mouse models, LXA4 and Benzo-LXA4 significantly ameliorate DM-induced proteinuria, glomerular expansion, and collagen deposition, demonstrating the ability to reverse established renal lesions and supporting the pro-resolving therapeutic paradigm (72). Notably, the ANAX1 mimetic Ac2–26 shows significant improvement in renal lipotoxicity in both ANAX1-/- DM mouse models (induced by STZ combined with high-fat diet) and db/db mouse models (37, 49), leading researchers to propose the ANAX1 mimetic peptide Ac2–26 as a promising therapeutic agent for DKD treatment (73).
2.3.3 Positive effects of dietary SPM precursors on kidney disease
The consumption of diets rich in SPMs precursors, particularly those containing high levels of ω-3 and ω-6 PUFAs, demonstrates significant therapeutic potential in alleviating kidney disease. Fish oil, characterized by its abundance of long-chain ω-3 PUFAs, has been extensively validated through clinical trials and experimental studies to enhance glucose and lipid metabolism while reducing oxidative stress and inflammatory responses (74–77). Notably, in CKD patients, ω-3 PUFA supplementation exhibits a strong correlation with elevated circulating SPMs levels (78). In an ESRD mouse model utilizing LDLr/ApoB-/-100/100 mice fed with a high-fat high-sucrose (HFHS) diet, dietary supplementation with long-chain ω-3 PUFAs resulted in increased Protectin DX (PDX) levels, effectively preventing ESRD and cardiac dysfunction. PDX-treated mice demonstrated significant improvements in renal fibrosis and glomerular expansion, alongside preserved cardiac function, manifested through enhanced ejection fraction, maintained heart rate, and increased cardiac output—effects potentially independent of glycemic regulation (79). Furthermore, in the 5/6 nephrectomy CKD model, ω-3 supplementation effectively attenuated oxidative stress, inflammation, and fibrotic responses in the remnant kidney (80). Extensive clinical and experimental evidence has established a robust connection between AKI and CKD (81, 82). Considering the pivotal role of SPMs in both acute and chronic kidney diseases, we posit that the progression of these conditions is intrinsically linked to unresolved primary renal injury and persistent inflammation. Neutrophils, serving as central mediators of inflammatory responses, can initiate or amplify tissue damage and chronic inflammation when aberrantly activated. The mechanistic underpinnings of how delayed or impaired neutrophil apoptosis contributes to kidney disease pathogenesis warrant further investigation.
3 Neutrophil apoptosis: a critical determinant in renal homeostasis and disease progression
Neutrophils are a crucial part of the innate immune system, constituting 50%-70% of human blood leukocytes. As primary responders during the initial phase of inflammation, these cells rapidly infiltrate inflamed tissues, serving as critical mediators in host defense against infection. Neutrophils exhibit remarkable plasticity in adapting to their microenvironment. Mature neutrophils are characterized by their relatively short half-lives in blood circulation and various tissues, though their longevity varies significantly across different tissue types (83) and can be modulated by local inflammatory signals (84, 85). Following the completion of their immune functions, neutrophils undergo programmed cell death (apoptosis) and are subsequently recognized, phagocytosed, and eliminated by macrophages. When neutrophil apoptosis is delayed or apoptotic neutrophils are not efficiently cleared, these cells continue to release substantial quantities of lysosomal enzymes, Reactive Oxygen Species (ROS), and inflammatory mediators, resulting in tissue damage and inflammatory amplification. In the context of kidney diseases, delayed neutrophil apoptosis can lead to persistent or chronic inflammation, subsequently exacerbating renal tissue injury (86–88).
3.1 Molecular orchestration of neutrophil apoptosis
The human bone marrow generates approximately 1011 neutrophils daily, with neutrophil apoptotic duration demonstrating remarkable plasticity. This temporal regulation can be extended to enhance microbial pathogen elimination or abbreviated to facilitate inflammation resolution, representing a critical mechanism in maintaining circulating neutrophil homeostasis and effectively resolving inflammatory responses (89). The process of neutrophil apoptosis is orchestrated through an intricate network of pro-survival and pro-apoptotic signaling pathways.
3.1.1 Core molecular mechanisms and regulatory pathways of neutrophil apoptosis
Neutrophil apoptosis takes place through two distinct mechanisms: the extrinsic death receptor pathway and the intrinsic mitochondrial pathway. Cysteinyl aspartate specific protease (Caspase), expressed as multiple cysteine family proteases in neutrophils, functions as the central executor of both apoptotic pathways (90). The extrinsic pathway begins with the activation of death receptors on the neutrophil surface. When ligands bind to these receptors, they initiate the recruitment of adaptor molecules and death domains, which ultimately result in the formation of death-inducing signaling complexes. These complexes interact with pro-caspase-8, facilitating its conversion to caspase-8 and subsequent activation of caspase-3, ultimately executing apoptosis through the activation of Caspase-Activated DNase (CAD) (91–94). In the intrinsic pathway, the dynamic interplay between Bcl-2 family members, including pro-survival proteins (Mcl-1, A1, Bcl-x) and pro-apoptotic proteins (Bad, Bax, Bak), is mediated through crucial dimerization processes. During apoptosis, pro-apoptotic proteins Bid and Bax translocate to mitochondria, enhancing outer membrane permeability and facilitating cytochrome c release. The released cytochrome C then binds to apoptosis protease-activating factor-1 (Apaf-1), triggering its oligomerization and subsequent activation of caspase-9 (95). This activation cascade proceeds through caspase-9-mediated activation of caspase-3, ultimately culminating in cellular apoptosis (96). This sophisticated process is intricately regulated by multiple signaling pathways and genes, including MAPK/ERK, PI3K, NF-κB, and JAK/STAT (97).
3.1.2 Cellular interactions and inflammatory mediator regulatory networks in delayed neutrophil apoptosis
Delayed neutrophil apoptosis is governed by both direct cellular interactions and indirect effects of soluble inflammatory mediators. The direct cellular interactions involve macrophages, NK cells, and endothelial cells, while the indirect effects are mediated through pro-inflammatory factors and SPMs. Macrophages orchestrate neutrophil recruitment to inflammatory sites through chemokine release and produce granulocyte-macrophage colony-stimulating factor (GM-CSF), which extends neutrophil survival (98). Recent investigations have revealed that during the non-phagocytic resolution phase, macrophage-derived IFN-β activates the STAT3 signaling pathway to promote neutrophil apoptosis, enhance macrophage phagocytosis of apoptotic neutrophils, and facilitate macrophage reprogramming toward anti-inflammatory and pro-resolving phenotypes. This creates a positive feedback loop promoting inflammation resolution, suggesting that delayed neutrophil apoptosis may result from insufficient macrophage IFN-β production (99). NK cells contribute to neutrophil survival through GM-CSF and IFN-γ secretion (100), while neutrophils reciprocally stimulate NK cell IFN-γ production, establishing a positive feedback loop under inflammatory conditions (101). Endothelial cells extend neutrophil lifespan through paracrine GM-CSF secretion (102). In addition to GM-CSF, low concentrations of TNF-α prolong the survival of cultured neutrophils (103). The deficiency or dysfunction of SPMs represents another significant factor in delayed neutrophil apoptosis (104) (Figure 2). Moreover, recent studies have demonstrated that some neutrophils can undergo reverse migration from inflammatory tissues back into circulation, a process that enhances their anti-apoptotic properties and consequently exacerbates tissue damage (105).
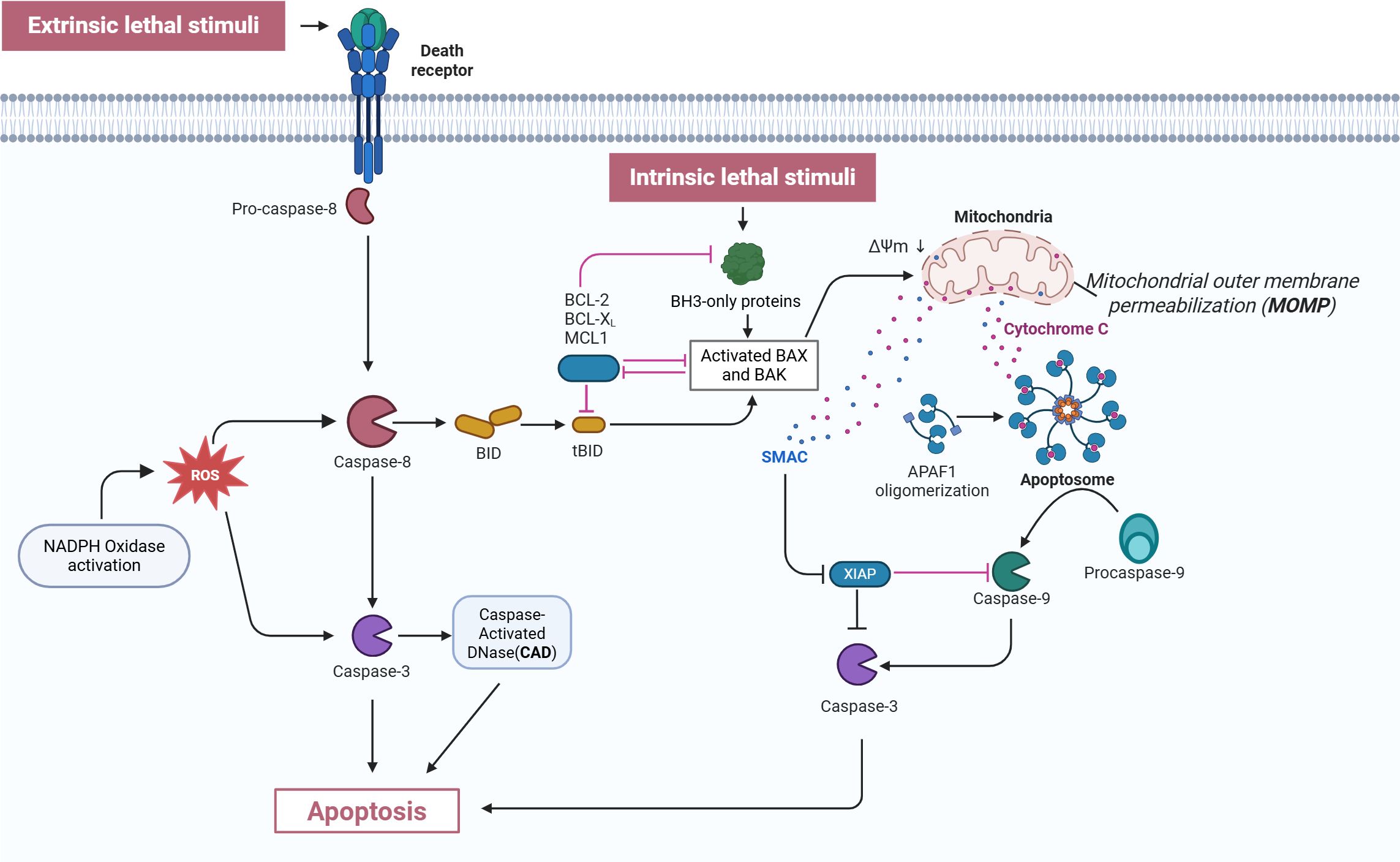
Figure 2. Mechanism of neutrophil apoptosis. This figure depicts both the extrinsic and intrinsic apoptotic pathways. In the extrinsic pathway, death receptors are triggered by external lethal signals, resulting in the activation of pro-caspase-8, which is then converted into active caspase-8. Activated caspase-8, on one hand, directly activates caspase-3, and on the other hand, cleaves BID protein into tBID. Simultaneously, NADPH oxidase is activated to generate ROS, which further activate caspase-3 and CAD. In the intrinsic pathway, intrinsic lethal stimuli activate BH3-only proteins, a process which is inhibited by BCL-2, BCL-XL, and MCL1. BH3-only proteins activate BAX and BAK, resulting in MOMP and the release of cytochrome C. The released cytochrome C binds to APAF1, forming the apoptosome, which then activates procaspase-9 into caspase-9. This process is inhibited by XIAP, while SMAC counteracts XIAP. Ultimately, caspase-9 activates caspase-3, triggering apoptosis. Both pathways ultimately execute the apoptosis program through the activation of effector caspase-3, illustrating the complex regulatory network of the apoptosis signaling pathway. CAD: Caspase-activated DNase; ROS, Reactive oxygen species; XIAP, X-linked Inhibitor of Apoptosis Protein; tBID, truncated BID; ΔΨm, mitochondrial membrane potential; APAF1, Apoptotic protease activating factor 1; MOMP, mitochondrial outer membrane permeabilization; SMAC, Second mitochondria-derived activator of caspases. This Figure adapted from Ona (2020); created with BioRender (biorender.com).
3.2 Aberrant neutrophil apoptosis in kidney diseases
3.2.1 Close association between kidney disease risk factors and neutrophil apoptosis
Hyperlipidemia, hyperglycemia, aging, and cardiovascular diseases (CVD) are common risk factors for kidney disease, and they are intricately linked to chronic inflammation and consequent tissue damage. These metabolic disturbances induce the production of pro-inflammatory neutrophils, perpetuating chronic inflammation (106). In the context of CKD and hemodialysis patients, high-density lipoprotein (HDL) demonstrates significant inhibitory effects on neutrophil apoptosis, potentially mediated through the activation of PI3K and Extracellular signal-Regulated Kinase (ERK) signaling pathways in Polymorphonuclear Neutrophils (PMN) (107). In type 2 diabetes mellitus (T2DM) patients, neutrophil apoptosis rates exhibit a significant correlation with Hemoglobin A1c (HbA1C) levels. The chronic hyperglycemic state suppresses natural neutrophil apoptotic processes, potentially exacerbating tissue damage and elevating the risk of microvascular complications (108). A distinct neutrophil subpopulation characterized by anti-apoptotic and pro-inflammatory features has been identified in T2DM patients, suggesting that reduced neutrophil apoptosis may contribute to the risk of CKD development in these patients (109). Notably, neutrophil count demonstrates a strong correlation with the occurrence of autoimmune DKD (110), and elevated neutrophil numbers may serve as an independent predictor of CKD progression in diabetic patients (111). Furthermore, under renal inflammatory conditions, pro-inflammatory factors, including GM-CSF and IL-8, can delay neutrophil apoptosis through multiple pathways (112). Aging is also accompanied by a decrease in the levels of SPMs (113, 114). With advancing age, neutrophil phagocytic activity, cytokine production, and reactive oxygen species (ROS) generation all decline, while the production of pro-inflammatory cytokines increases, leading to excessive formation of neutrophil extracellular traps (NETs) and exacerbating chronic inflammation (115). Inflammation is considered a key driver of many age-related kidney diseases (114). In CVD, impaired clearance of apoptotic neutrophils (116) and the release of activated NETs are important contributors to the inflammation in both CVD (117) and kidney disease (118). These findings highlight that neutrophil apoptosis and dysfunction serve as common hubs for kidney damage induced by metabolic disturbances, aging, and cardiovascular diseases.
3.2.2 Pathological mechanisms of neutrophil apoptosis in various kidney diseases
Neutrophil apoptosis serves as a critical determinant in kidney recovery, with its dysregulation potentially accelerating renal injury and disease progression (Table 3). Studies utilizing Bax gene knockout bone marrow chimeric mice during IRI recovery have demonstrated that impaired neutrophil clearance results in delayed functional and tissue recovery, elevated cytokine levels, and accelerated renal fibrosis (119). In antineutrophil cytoplasmic antibody-associated vasculitis (AAV), delayed neutrophil apoptosis emerges as a crucial factor in secondary renal injury (120). Renal biopsies from ANCA-positive RPGN patients reveal an imbalance between neutrophil apoptosis and proliferation, favoring cellular proliferation over apoptosis-mediated inflammation resolution (121). In anti-glomerular basement membrane glomerulonephritis, neutrophil-dependent kidney injury represents a fundamental pathogenic mechanism. Notably, CD44, a molecule involved in human monocyte recognition of apoptotic neutrophils, demonstrates the capacity to rapidly induce neutrophil apoptosis in vitro while inhibiting neutrophil-dependent kidney injury in vivo (122). In Poststreptococcal Glomerulonephritis (PSGN), neutrophil and macrophage infiltration and activation constitute primary drivers of renal injury (123). While PSGN typically follows a self-limiting course, some patients progress to chronic renal failure, potentially due to inadequate induction of cellular apoptosis, particularly in neutrophils (124). In lupus nephritis, insufficient clearance of apoptotic neutrophils leading to secondary necrosis and autoimmune responses may significantly contribute to disease activity (125). Research in immune complex glomerulonephritis (ICGN) models has revealed that less than 20% of neutrophils undergo apoptosis within glomeruli, with the majority exhibiting reverse migration into circulation (105). This reverse migration process enhances neutrophils’ anti-apoptotic characteristics, consequently exacerbating tissue damage (126). Clinical investigations have further demonstrated that neutrophil dysfunction and delayed apoptosis, induced by elevated serum immunoglobulin light chain (IgLC) levels in renal failure patients, represent significant factors in uremic patients’ susceptibility to infections. These infections often constitute critical determinants of disease progression and mortality in ESRD patients (127).
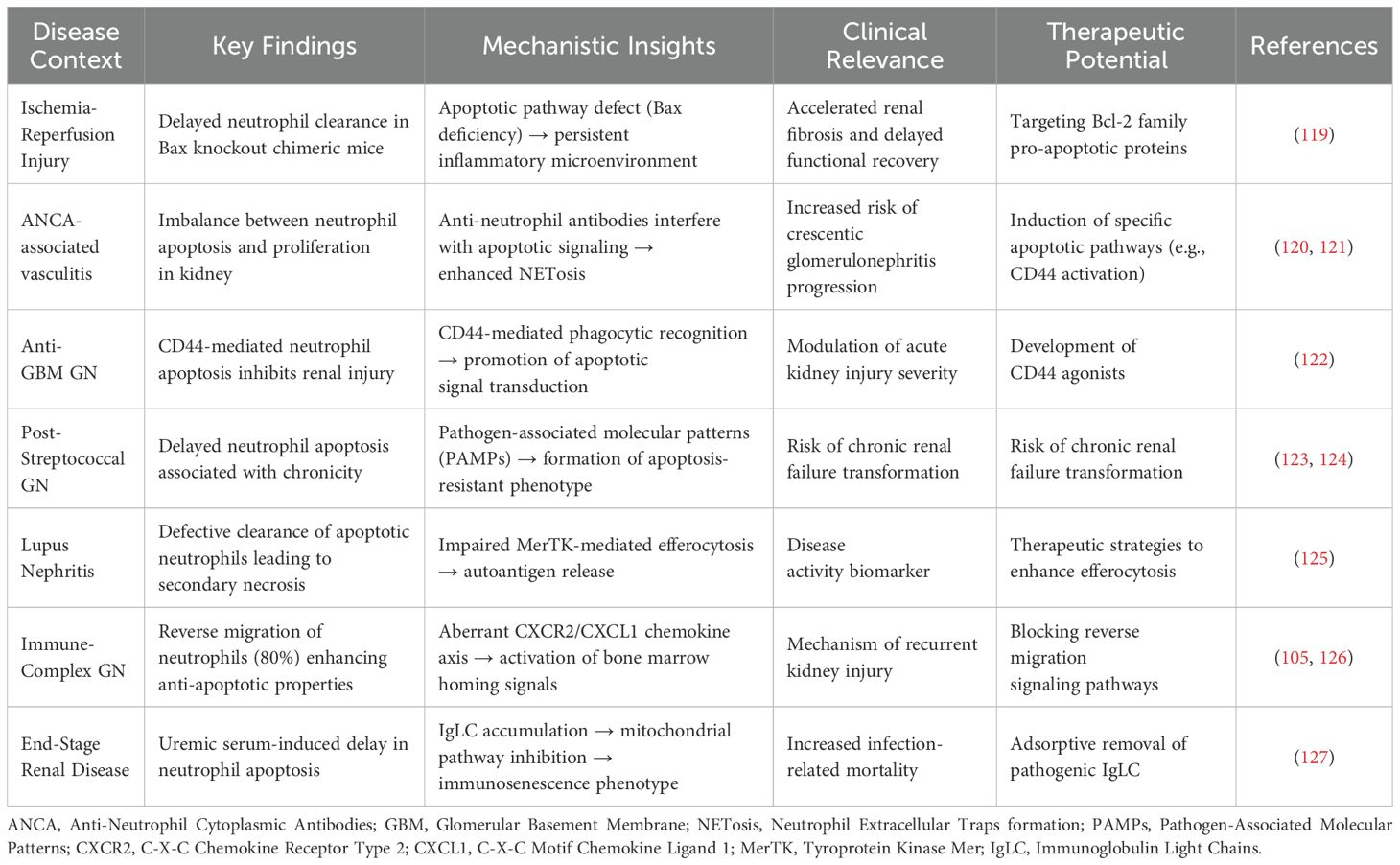
Table 3. Pathological roles and intervention strategies of aberrant neutrophil apoptosis in kidney diseases.
4 Synergistic therapeutic: inflammatory resolution mediators modulating neutrophil apoptosis in kidney disease
Neutrophil apoptosis serves as the initiating signal for inflammation resolution and marks a critical turning point between persistent inflammation and its resolution (106, 128). SPMs exhibit multiple regulatory functions, including the inhibition of neutrophil recruitment and activation while promoting apoptosis and efferocytosis. The therapeutic induction of cellular apoptosis has emerged as a central focus in inflammation resolution research.
4.1 SPMs and neutrophil apoptosis
Growing evidence underscores neutrophil apoptosis as a crucial determinant of inflammatory response outcomes and a promising therapeutic target (128–131). Notably, endogenous lipid mediators, such as LXA4 and Cyclin-Dependent Kinase (CDK) inhibitors (132), demonstrate the ability to promote neutrophil apoptosis in animal models while enhancing acute inflammation resolution. SPMs’ regulation of neutrophil apoptosis operates through multiple beneficial mechanisms: preventing excessive neutrophil activation and subsequent inflammatory exacerbation, avoiding secondary necrosis of apoptotic neutrophils and the associated release of tissue-damaging toxic contents, and promoting macrophage metabolic reprogramming (Figure 3).
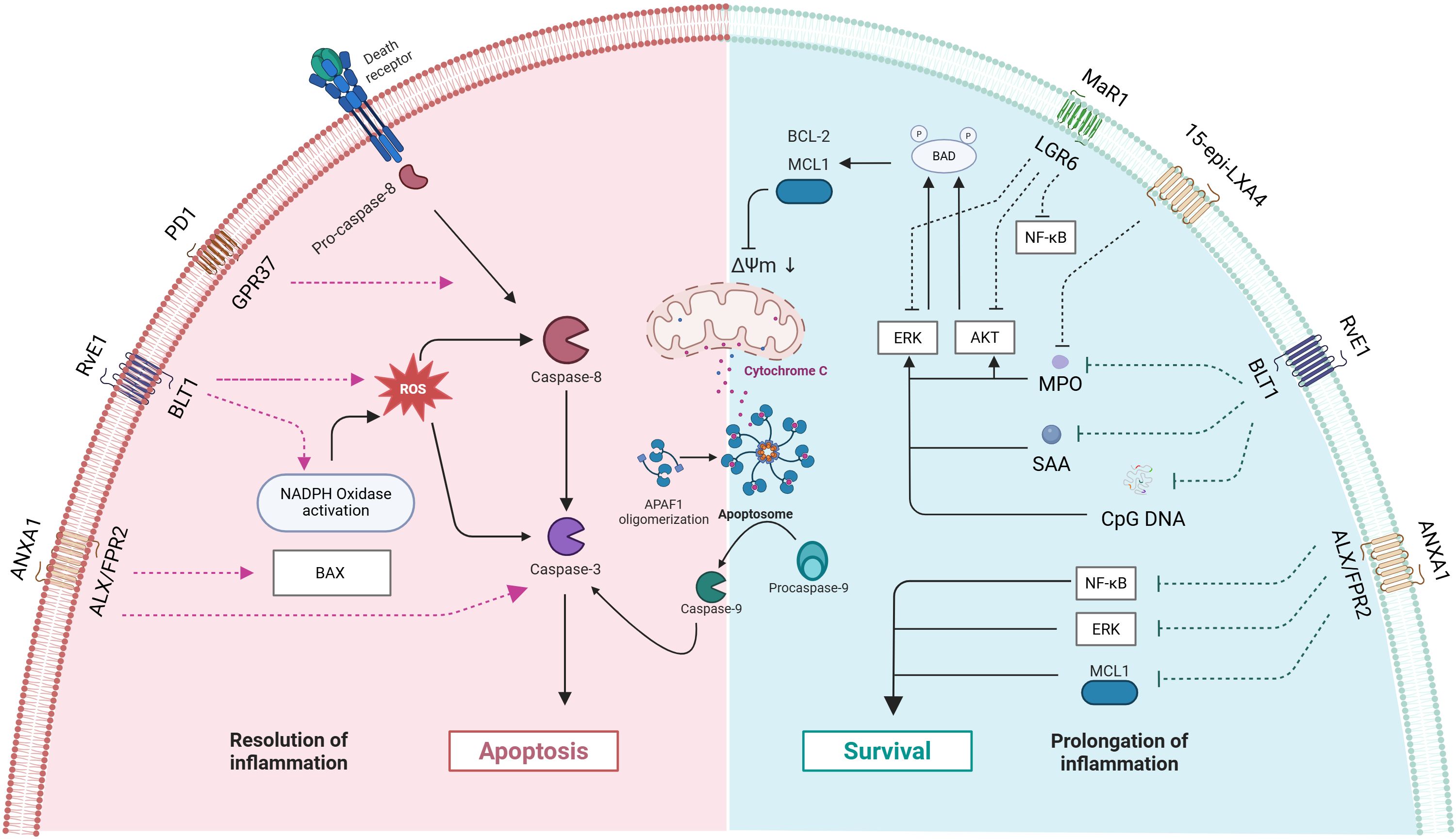
Figure 3. Mechanisms by which mediators of inflammation resolution regulate neutrophil apoptosis and survival. The image is divided into two regions: the left side (pink area) represents the pathway of inflammation resolution, while the right side (blue area) represents the pathways of cell survival and inflammation prolongation. The blue module depicts pro-survival mechanisms, including BCL-2 family anti-apoptotic proteins, survival signaling pathways (AKT, ERK, NF-κB), and the inflammation-prolonging states they mediate. The red module illustrates pro-apoptotic mechanisms, encompassing caspase cascade activation and mitochondrial apoptosis pathways. The interactions between various receptors and signaling molecules collectively determine the ultimate cell fate and the progression of inflammation. PD1, Protectin D1; GPR37, G Protein-Coupled Receptor 37; ANXA1 - Annexin A1; ROS, Reactive Oxygen Species; ΔΨm, mitochondrial membrane potential: APAF1, Apoptotic Protease Activating Factor 1; MPO, Myeloperoxidase; SAA, Serum Amyloid A; CpG DNA, Cyclodextrin-Glycosylated DNA.
4.1.1 Promote the release of SPMs
Neutrophils play a pivotal role in inflammation resolution as key immune cells involved in SPMs transcellular synthesis and express various SPMs receptors. The process of neutrophil apoptosis is characterized by the production of multiple SPMs (20, 23, 128, 129, 133, 134), including crucial intracellular proteins such as AnxA1 and calgranulins. AnxA1, a well-established pro-resolving factor, exhibits distinct distribution patterns in neutrophils: primarily cytoplasmic in resting cells, with significant presence in cytoplasmic granules and other organelles (135). Upon cellular activation and endothelial adhesion, AnxA1 undergoes rapid translocation to the cell surface (136), where it forms complexes with proteinase 3 and PLSCR1, facilitating phosphatidylserine externalization during apoptosis (137). Through interaction with formyl peptide receptor 2 (FPR2/ALX) on the membrane, it modulates leukocyte adhesion and migration (138, 139). The soluble AnxA1 released during apoptosis demonstrates multiple pro-resolving functions: inducing neutrophil apoptosis, regulating monocyte recruitment, and enhancing macrophage-mediated clearance of apoptotic cells. In the resolution phase of inflammation, neutrophils undergo a remarkable metabolic shift in their arachidonic acid pathway, transitioning from the production of pro-inflammatory leukotriene B4 to anti-inflammatory lipoxin A4 (140).
4.1.2 Inhibit inflammatory damage caused by cell necrosis
Apoptosis is considered an ideal form of cell death as it effectively prevents inflammation spread by containing cellular contents within membranes. However, during inflammatory responses, other cell death mechanisms (autophagy, necrosis, and NETs deposition) may also be activated (141). Non-apoptotic cell death can promote inflammatory responses through delayed clearance processes, as described in NETosis (142). Studies have shown that SPMs (including AnxA1, Resolvin E1, Protectin D1, Maresin 1, NO, 15-epi-LXA4, 17-epi-RvD1, etc.) participate in dual regulation of pro-apoptotic and anti-apoptotic pathways through binding to receptors such as ALX/FPR2, ChemR23, and BLT1. AnxA1 regulates both natural and glucocorticoid-induced inflammation resolution by inducing neutrophil apoptosis, activating pro-apoptotic pathways (including Bax and caspase-3 activation), and inhibiting survival pathways (including suppression of Mcl-1, ERK1/2, and NF-κB) (143). RvE1 induces apoptosis by promoting neutrophil phagocytosis, enhancing NADPH oxidase-generated ROS and activation of caspase-8 and caspase-3. Through binding to BLT1 receptor, it inhibits anti-apoptotic signals produced by inflammatory mediators such as myeloperoxidase (MPO), serum amyloid A (SAA), and bacterial DNA (CpG DNA) (which inhibit neutrophil apoptosis by activating ERK and Akt signaling pathways and increasing Mcl-1 expression), disrupts mitochondrial transmembrane potential (ΔΨm), thereby promoting neutrophil apoptosis (144). Protectin D1 promotes neutrophil apoptosis by enhancing caspase-dependent pathways, thereby facilitating inflammation resolution in LPS-induced acute lung injury mouse models (145). Maresin 1 promotes resolution of LPS-induced acute lung injury inflammation by inhibiting AKT, ERK, and p38 phosphorylation, downregulating Mcl-1 and Bcl-2 expression, and ultimately mediating neutrophil apoptosis through caspase-3 activation (146). In cerebral ischemia-reperfusion models, macrophage-derived maresin 1 inhibits NF-κB pathway activation, and suppressing production of pro-inflammatory mediators such as IL-1β, and TNF-α (147). Through the ALX/FPR2 receptor, 15-epi-LXA4 and 17-epi-RvD1 can antagonize CpG DNA and mitochondrial DNA (mtDNA) signals, maintain C5aR expression, restore impaired phagocytosis, and improve bacterial clearance while promoting phagocytosis-induced neutrophil apoptosis, thereby alleviating E. coli-induced lung injury (148). 15-epi-LXA4 facilitates human neutrophil apoptosis by inhibiting MPO-induced extracellular signal-regulated kinase and Akt-mediated Bad phosphorylation, decreasing the expression of the anti-apoptotic protein Mcl-1, and downregulating β2 integrin Mac-1 signaling. Furthermore, treatment with 15-epi-LXA4 effectively promotes the resolution of inflammation by accelerating neutrophil apoptosis and decreasing the release of inflammatory mediators (149).
4.1.3 Promoting clearance of apoptotic neutrophils
The efficient phagocytosis of apoptotic neutrophils, a process termed efferocytosis, represents a crucial endpoint in cellular apoptosis that effectively minimizes the release of cytotoxic contents and subsequent tissue injury. Macrophage recognition of apoptotic cells initiates feedback mechanisms that program these cells toward an enhanced efferocytosis phenotype (106). Significantly, efferocytosis promotes macrophage polarization toward anti-inflammatory and pro-homeostatic phenotypes (150). Macrophage lysosomes process phagocytosed apoptotic cells, liberating metabolic substrates, including lipid components from apoptotic cell membranes and SPMs. These substrates directly influence macrophage polarization potential and modulate their functional properties (151–153). During efferocytosis, neutrophils orchestrate their own clearance through the presentation of “find-me” and “eat-me” signals. The “find-me” signals serve as chemoattractants for scavenger cells, while the “eat-me” signals, expressed on the apoptotic cell surface, initiate the phagocytic process. AnxA1, one of several molecular mediators involved in macrophage recognition of apoptotic cells (106), undergoes redistribution to the cell membrane during neutrophil activation or apoptosis, facilitating macrophage recognition and subsequent clearance (154–156). 17R-RvD1 enhances macrophage-mediated phagocytosis of damaged or apoptotic erythrocytes and polymorphonuclear leukocytes while simultaneously suppressing NF-κB activation and reducing the expression of inflammatory cytokines and vascular activation markers, thereby attenuating inflammation and renal vascular dysfunction in patients with sickle cell disease (157). Resolvin E1 and PD1 enhance macrophage efferocytosis through activation of cellular ChemR23 and GPR37 receptors (158–161). ChemR23 receptor activation promotes both macrophage efferocytosis and neutrophil apoptosis, accelerating the resolution of acute inflammation and triggering inflammatory resolution in chronic colitis models (162). The anti-inflammatory mediators released during macrophage efferocytosis, particularly TGFβ and IL-10, likely serve as principal drivers of M2 polarization (163).
4.2 Therapeutic approaches targeting SPMs and neutrophil apoptosis
While research examining the role of SPMs in regulating neutrophil apoptosis in kidney disease remains limited, existing studies have identified pathological conditions characterized by impaired SPMs-mediated regulation of neutrophil apoptosis. Recent investigations have revealed that CKD patients exhibit increased intracellular AnxA1 expression in neutrophils, coupled with decreased membrane-bound and secreted AnxA1, suggesting compromised utilization of AnxA1’s anti-inflammatory and pro-resolving functions (164). AnxA1 deficiency, resulting in delayed neutrophil apoptosis and impaired macrophage efferocytosis, may contribute to persistent and exacerbated inflammatory responses in glomerulonephritis (165). In AAV-induced renal injury, delayed neutrophil apoptosis represents a critical pathogenic factor, where increased AnxA1 localization on apoptotic cell membranes, forming membrane protein platforms with proteinase 3 and phospholipid scramblase 1, may interfere with the normal clearance of apoptotic neutrophils (120). Studies utilizing RPGN models in AnxA1-deficient and wild-type mice have demonstrated AnxA1’s renoprotective effects through reduced neutrophil infiltration and enhanced neutrophil apoptosis (34). In investigations of cardiorenal syndrome following myocardial infarction, RvD1 administration effectively modulated inflammatory responses by promoting neutrophil clearance and increasing reparative macrophages, thereby limiting both cardiac and renal inflammatory injury (166).
Regulating neutrophil apoptosis has been shown in various diseases (e.g., cystic fibrosis (CF), acute lung injury, acute respiratory distress syndrome, myocardial infarction) to protect the body from damage by reducing NET levels and decreasing the release of pro-inflammatory mediators (167–171). Notably, similar to kidney diseases, the biosynthesis of SPMs, such as ANXA1, NO, and LXA4, is reduced in CF patients, and the levels of SPMs in the respiratory tract are correlated with lung function in CF patients (172). Experimental studies have confirmed that RvE1 can induce neutrophil apoptosis through phagocytosis and alleviate the exacerbation of acute lung inflammation caused by enhanced anti-apoptotic signals (144). In the context of myocardial infarction, defects in the synthesis of SPMs (LXA4, RvD1) are clearly associated with poor prognosis (173, 174). SPMs are potential targets for regulating neutrophil apoptosis, and the modulation of SPMs to control neutrophil apoptosis could facilitate the resolution of inflammation, offering promising therapeutic avenues for the aforementioned diseases.
Although “Resolution pharmacology” research primarily relies on in vitro cellular and in vivo disease models, stable SPMs analogs have demonstrated therapeutic efficacy in clinical trials for asthma and eczema (175, 176). Drug-loaded nanoparticles and formulations demonstrate the ability to selectively target inflammatory neutrophils in situ and induce neutrophil apoptosis through intracellular delivery, thereby mitigating inflammatory injury (177–179). Nano-pro-resolving medicines enhance human macrophage phagocytosis and bacterial/debris clearance, significantly abbreviating inflammation resolution time and reducing PMN infiltration, thus suppressing inflammation and promoting tissue repair (113, 180, 181). This suggests that nanotechnology may have significant translational medical value for targeting SPMs regulatory systems in inducing neutrophil apoptosis, offering new hope for developing precise strategies to modulate the inflammatory microenvironment in kidney diseases. However, the feasibility of this therapy needs to be carefully evaluated. Nanomedicines still face technical challenges such as premature drug release and insufficient targeting efficiency, which hinder their clinical translation. The use of SPMs analogs with significantly extended half-lives could introduce risks of side effects. The sustained suppression of inflammatory responses by such long-acting compounds may impair the body’s ability to activate necessary defense mechanisms, whereas moderate inflammatory responses play a protective role in disease progression. Thus, a balance between efficacy and risk needs to be carefully considered, and further investigation is needed. Elucidating the mechanisms linking kidney dysfunction with abnormalities in SPMs biosynthesis could potentially allow therapeutic goals to be achieved by modulating endogenous SPMs levels. Due to the complexity of the pathological environment of kidney diseases and individual differences, the development of universal therapeutic strategies still requires extensive clinical and basic research validation. Nevertheless, this approach holds significant clinical value.
5 Conclusion
Unresolved inflammation serves as a critical determinant in the progression of renal failure and fibrosis. The process of neutrophil apoptosis represents a pivotal turning point between inflammatory resolution and progression, with SPMs emerging as promising novel targets for regulating neutrophil apoptosis and clearance, demonstrating substantial therapeutic potential in kidney diseases. While the dysregulation of inflammatory resolution mechanisms in kidney diseases presents new therapeutic opportunities for drug development, comprehensive research is essential to elucidate the underlying mechanisms, facilitate clinical investigations, and optimize therapeutic applications. Potential therapeutic strategies may include selective targeting, precise intracellular delivery of SPMs, and controlled induction of programmed neutrophil apoptosis to modulate immune homeostasis and inflammatory responses. The monitoring of SPM biomarkers and neutrophil functional status holds significant promise in evaluating disease severity, prognosis, and outcomes in kidney diseases. Moreover, these findings indicate that therapeutic approaches to inflammatory diseases should extend beyond conventional inhibitors to encompass agonists such as SPMs, which activate endogenous inflammatory resolution processes and establish a new homeostatic balance, potentially enabling earlier therapeutic intervention and mitigating chronic disease progression.
Author contributions
YK: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. QJ: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Supervision, Validation, Writing – original draft, Writing – review & editing. MZ: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. HZ: Conceptualization, Investigation, Software, Writing – original draft, Writing – review & editing. DL: Conceptualization, Data curation, Methodology, Software, Writing – original draft. XW: Data curation, Methodology, Supervision, Writing – original draft. JZ: Conceptualization, Data curation, Formal analysis, Methodology, Writing – review & editing. YW: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. JL: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the National Natural Science Foundation of China (grant numbers 82174342, 82074361), the National Administration of Traditional Chinese Medicine Inheritance and Innovation “Hundreds and Thousands of Talents” Project (No. (2018)12 issued by the National Administration of Traditional Chinese Medicine); Central Universities’ Fundamental Research Funds (2023-JYB-JBQN-020) and joint Project of the China Association of Chinese Medicine (2023DYPLHGG-11).
Acknowledgments
We would like to express our sincere gratitude to all the participants and researchers who contributed to this study. Figures were created with biorender.com (www.biorender.com).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tamargo C, Hanouneh M, and Cervantes CE. Treatment of acute kidney injury: A review of current approaches and emerging innovations. J Clin Med. (2024) 13(9):2455. doi: 10.3390/jcm13092455
2. Chen TK, Hoenig MP, Nitsch D, and Grams ME. Advances in the management of chronic kidney disease. Bmj. (2023) 383:e074216. doi: 10.1136/bmj-2022-074216
3. Sanz AB, Sanchez-Niño MD, Ramos AM, and Ortiz A. Regulated cell death pathways in kidney disease. Nat Rev Nephrol. (2023) 19:281–99. doi: 10.1038/s41581-023-00694-0
4. Xie T, Yao L, and Li X. Advance in iron metabolism, oxidative stress and cellular dysfunction in experimental and human kidney diseases. Antioxidants (Basel Switzerland). (2024) 13(6):659. doi: 10.3390/antiox13060659
5. Vartak T, Godson C, and Brennan E. Therapeutic potential of pro-resolving mediators in diabetic kidney disease. Adv Drug Delivery Rev. (2021) 178:113965. doi: 10.1016/j.addr.2021.113965
6. Giardini E, Moore D, Sadlier D, Godson C, and Brennan E. The dual role of lipids in chronic kidney disease: Pathogenic culprits and therapeutic allies. Atherosclerosis. (2024) 398:118615. doi: 10.1016/j.atherosclerosis.2024.118615
7. Filep JG. Targeting neutrophils for promoting the resolution of inflammation. Front Immunol. (2022) 13:866747. doi: 10.3389/fimmu.2022.866747
8. Duffin R, Leitch AE, Fox S, Haslett C, and Rossi AG. Targeting granulocyte apoptosis: mechanisms, models, and therapies. Immunological Rev. (2010) 236:28–40. doi: 10.1111/j.1600-065X.2010.00922.x
9. Leitch AE, Lucas CD, and Rossi AG. Editorial: Neutrophil apoptosis: hot on the TRAIL of inflammatory resolution. J leukocyte Biol. (2011) 90:841–3. doi: 10.1189/jlb.0511222
10. Ghodsi A, Hidalgo A, and Libreros S. Lipid mediators in neutrophil biology: inflammation, resolution and beyond. Curr Opin Hematol. (2024) 31:175–92. doi: 10.1097/MOH.0000000000000822
11. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. (2014) 510:92–101. doi: 10.1038/nature13479
12. Brennan E, Kantharidis P, Cooper ME, and Godson C. Pro-resolving lipid mediators: regulators of inflammation, metabolism and kidney function. Nat Rev Nephrol. (2021) 17:725–39. doi: 10.1038/s41581-021-00454-y
13. Gila-Diaz A, Carrillo GH, Singh P, and Ramiro-Cortijo D. Specialized pro-resolving lipid mediators in neonatal cardiovascular physiology and diseases. Antioxidants (Basel Switzerland). (2021) 10(6):933. doi: 10.3390/antiox10060933
14. Halade GV, Kain V, Dillion C, Beasley M, Dudenbostel T, Oparil S, et al. Race-based and sex-based differences in bioactive lipid mediators after myocardial infarction. ESC Heart failure. (2020) 7:1700–10. doi: 10.1002/ehf2.12730
15. Wang YF, Zhu XT, and Hu ZP. Decreased plasma lipoxin A4, resolvin D1, protectin D1 are correlated with the complexity and prognosis of coronary heart disease: A retrospective cohort study. Prostaglandins other Lipid Mediators. (2025) 178:106990. doi: 10.1016/j.prostaglandins.2025.106990
16. de Gaetano M, McEvoy C, Andrews D, Cacace A, Hunter J, Brennan E, et al. Specialized pro-resolving lipid mediators: modulation of diabetes-associated cardio-, reno-, and retino-vascular complications. Front Pharmacol. (2018) 9:1488. doi: 10.3389/fphar.2018.01488
17. Ponce J, Ulu A, Hanson C, Cameron-Smith E, Bertoni J, Wuebker J, et al. Role of specialized pro-resolving mediators in reducing neuroinflammation in neurodegenerative disorders. Front Aging Neurosci. (2022) 14:780811. doi: 10.3389/fnagi.2022.780811
18. Hasturk H, Schulte F, Martins M, Sherzai H, Floros C, Cugini M, et al. Safety and preliminary efficacy of a novel host-modulatory therapy for reducing gingival inflammation. Front Immunol. (2021) 12:704163. doi: 10.3389/fimmu.2021.704163
19. Gomez EA, Colas RA, Souza PR, Hands R, Lewis MJ, Bessant C, et al. Blood pro-resolving mediators are linked with synovial pathology and are predictive of DMARD responsiveness in rheumatoid arthritis. Nat Commun. (2020) 11:5420. doi: 10.1038/s41467-020-19176-z
20. Brennan EP, Cacace A, and Godson C. Specialized pro-resolving mediators in renal fibrosis. Mol aspects Med. (2017) 58:102–13. doi: 10.1016/j.mam.2017.05.001
21. Serhan CN and Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. (2018) 128:2657–69. doi: 10.1172/JCI97943
22. Romano M, Cianci E, Simiele F, and Recchiuti A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur J Pharmacol. (2015) 760:49–63. doi: 10.1016/j.ejphar.2015.03.083
23. Byrne L and Guiry PJ. Advances in the chemistry and biology of specialised pro-resolving mediators (SPMs). Molecules (Basel Switzerland). (2024) 29(10):2233. doi: 10.3390/molecules29102233
24. Tisi A, Carozza G, Leuti A, Maccarone R, and Maccarrone M. Dysregulation of resolvin E1 metabolism and signaling in a light-damage model of age-related macular degeneration. Int J Mol Sci. (2023) 24(7):6749. doi: 10.3390/ijms24076749
25. Chiang N and Serhan CN. Specialized pro-resolving mediator network: an update on production and actions. Essays Biochem. (2020) 64:443–62. doi: 10.1042/EBC20200018
26. Perretti M and Dalli J. Resolution pharmacology: focus on pro-resolving annexin A1 and lipid mediators for therapeutic innovation in inflammation. Annu Rev Pharmacol Toxicol. (2023) 63:449–69. doi: 10.1146/annurev-pharmtox-051821-042743
27. Zhang J, Liu S, Ding W, Wan J, Qin JJ, and Wang M. Resolution of inflammation, an active process to restore the immune microenvironment balance: A novel drug target for treating arterial hypertension. Ageing Res Rev. (2024) 99:102352. doi: 10.1016/j.arr.2024.102352
28. Fullerton JN and Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov. (2016) 15:551–67. doi: 10.1038/nrd.2016.39
29. Sugimoto MA, Vago JP, Perretti M, and Teixeira MM. Mediators of the resolution of the inflammatory response. Trends Immunol. (2019) 40:212–27. doi: 10.1016/j.it.2019.01.007
30. Freire MO and Van Dyke TE. Natural resolution of inflammation. Periodontology 2000. (2013) 63:149–64. doi: 10.1111/prd.2013.63.issue-1
31. Panigrahy D, Gilligan MM, Serhan CN, and Kashfi K. Resolution of inflammation: An organizing principle in biology and medicine. Pharmacol Ther. (2021) 227:107879. doi: 10.1016/j.pharmthera.2021.107879
32. Rabb H, Griffin MD, McKay DB, Swaminathan S, Pickkers P, Rosner MH, et al. Inflammation in AKI: current understanding, key questions, and knowledge gaps. J Am Soc Nephrology: JASN. (2016) 27:371–9. doi: 10.1681/ASN.2015030261
33. Andrade-Oliveira V, Foresto-Neto O, Watanabe IKM, Zatz R, and Câmara NOS. Inflammation in renal diseases: new and old players. Front Pharmacol. (2019) 10:1192. doi: 10.3389/fphar.2019.01192
34. Labes R, Dong L, Mrowka R, Bachmann S, von Vietinghoff S, and Paliege A. Annexin A1 exerts renoprotective effects in experimental crescentic glomerulonephritis. Front Physiol. (2022) 13:984362. doi: 10.3389/fphys.2022.984362
35. Andrews D and Godson C. Lipoxins and synthetic lipoxin mimetics: Therapeutic potential in renal diseases. Biochim Biophys Acta Mol Cell Biol Lipids. (2021) 1866:158940. doi: 10.1016/j.bbalip.2021.158940
36. Wu SH, Liao PY, Yin PL, Zhang YM, and Dong L. Elevated expressions of 15-lipoxygenase and lipoxin A4 in children with acute poststreptococcal glomerulonephritis. Am J Pathol. (2009) 174:115–22. doi: 10.2353/ajpath.2009.080671
37. Wu L, Liu C, Chang DY, Zhan R, Sun J, Cui SH, et al. Annexin A1 alleviates kidney injury by promoting the resolution of inflammation in diabetic nephropathy. Kidney Int. (2021) 100:107–21. doi: 10.1016/j.kint.2021.02.025
38. Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. (2016) 7:12859. doi: 10.1038/ncomms12859
39. Norling LV, Headland SE, Dalli J, Arnardottir HH, Haworth O, Jones HR, et al. Proresolving and cartilage-protective actions of resolvin D1 in inflammatory arthritis. JCI Insight. (2023) 8(3):e168728. doi: 10.1172/jci.insight.168728
40. Kooij G, Troletti CD, Leuti A, Norris PC, Riley I, Albanese M, et al. Specialized pro-resolving lipid mediators are differentially altered in peripheral blood of patients with multiple sclerosis and attenuate monocyte and blood-brain barrier dysfunction. Haematologica. (2020) 105:2056–70. doi: 10.3324/haematol.2019.219519
41. Serhan CN, Hamberg M, and Samuelsson B. Trihydroxytetraenes: a novel series of compounds formed from arachidonic acid in human leukocytes. Biochem Biophys Res Commun. (1984) 118:943–9. doi: 10.1016/0006-291X(84)91486-4
42. Das UN. Lipoxins as biomarkers of lupus and other inflammatory conditions. Lipids Health Dis. (2011) 10:76. doi: 10.1186/1476-511X-10-76
43. Ohse T, Ota T, Kieran N, Godson C, Yamada K, Tanaka T, et al. Modulation of interferon-induced genes by lipoxin analogue in anti-glomerular basement membrane nephritis. J Am Soc Nephrology: JASN. (2004) 15:919–27. doi: 10.1097/01.ASN.0000119962.69573.CC
44. Martínez-Montoro JI, Morales E, Cornejo-Pareja I, Tinahones FJ, and Fernández-García JC. Obesity-related glomerulopathy: Current approaches and future perspectives. Obesity reviews: an Off J Int Assoc Study Obesity. (2022) 23(7):e13450. doi: 10.1111/obr.13450
45. López-Vicario C, Titos E, Walker ME, Alcaraz-Quiles J, Casulleras M, Durán-Güell M, et al. Leukocytes from obese individuals exhibit an impaired SPM signature. FASEB journal: Off Publ Fed Am Societies Exp Biol. (2019) 33:7072–83. doi: 10.1096/fj.201802587R
46. Guo YP, Jiang HK, Jiang H, Tian HY, and Li L. Lipoxin A4 may attenuate the progression of obesity-related glomerulopathy by inhibiting NF-κB and ERK/p38 MAPK-dependent inflammation. Life Sci. (2018) 198:112–8. doi: 10.1016/j.lfs.2018.02.039
47. Börgeson E, Johnson AM, Lee YS, Till A, Syed GH, Ali-Shah ST, et al. Lipoxin A4 attenuates obesity-induced adipose inflammation and associated liver and kidney disease. Cell Metab. (2015) 22:125–37. doi: 10.1016/j.cmet.2015.05.003
48. Bai Y, Wang J, He Z, Yang M, Li L, and Jiang H. Mesenchymal stem cells reverse diabetic nephropathy disease via lipoxin A4 by targeting transforming growth factor β (TGF-β)/smad pathway and pro-inflammatory cytokines. Med Sci monitor: Int Med J Exp Clin Res. (2019) 25:3069–76. doi: 10.12659/MSM.914860
49. Wu L, Liu C, Chang DY, Zhan R, Zhao M, Man Lam S, et al. The attenuation of diabetic nephropathy by annexin A1 via regulation of lipid metabolism through the AMPK/PPARα/CPT1b pathway. Diabetes. (2021) 70:2192–203. doi: 10.2337/db21-0050
50. Purvis GSD, Chiazza F, Chen J, Azevedo-Loiola R, Martin L, Kusters DHM, et al. Annexin A1 attenuates microvascular complications through restoration of Akt signalling in a murine model of type 1 diabetes. Diabetologia. (2018) 61:482–95. doi: 10.1007/s00125-017-4469-y
51. Pietrani NT, Ferreira CN, Rodrigues KF, Bosco AA, Oliveira MC, Teixeira AL, et al. Annexin A1 concentrations is decreased in patients with diabetes type 2 and nephropathy. Clinica chimica acta; Int J Clin Chem. (2014) 436:181–2. doi: 10.1016/j.cca.2014.05.027
52. Serhan CN, Dalli J, Karamnov S, Choi A, Park CK, Xu ZZ, et al. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB journal: Off Publ Fed Am Societies Exp Biol. (2012) 26:1755–65. doi: 10.1096/fj.11-201442
53. Li QF, Hao H, Tu WS, Guo N, and Zhou XY. Maresins: anti-inflammatory pro-resolving mediators with therapeutic potential. Eur Rev Med Pharmacol Sci. (2020) 24(13):7442–53. doi: 10.26355/eurrev_202007_21913
54. Morita Y, Kurano M, Sakai E, Sawabe M, Aoki J, and Yatomi Y. Simultaneous analyses of urinary eicosanoids and related mediators identified tetranor-prostaglandin E metabolite as a novel biomarker of diabetic nephropathy. J Lipid Res. (2021) 62:100120. doi: 10.1016/j.jlr.2021.100120
55. Li X, Xu B, Wu J, Pu Y, Wan S, Zeng Y, et al. Maresin 1 Alleviates Diabetic Kidney Disease via LGR6-Mediated cAMP-SOD2-ROS Pathway. Oxid Med Cell Longevity. (2022) 2022:7177889. doi: 10.1155/2022/7177889
56. Tang S, Gao C, Long Y, Huang W, Chen J, Fan F, et al. Maresin 1 mitigates high glucose-induced mouse glomerular mesangial cell injury by inhibiting inflammation and fibrosis. Mediators Inflammation. (2017) 2017:2438247. doi: 10.1155/2017/2438247
57. Tobón-Arroyave SI, Isaza-Guzmán DM, Gómez-Ortega J, and Flórez-Alzate AA. Salivary levels of specialized pro-resolving lipid mediators as indicators of periodontal health/disease status. J Clin periodontology. (2019) 46(10):978–90. doi: 10.1111/jcpe.13173
58. Hou Z, Lu F, Lin J, Wu Y, Chen L, Fang H, et al. Loss of Annexin A1 in macrophages restrains efferocytosis and remodels immune microenvironment in pancreatic cancer by activating the cGAS/STING pathway. J immunotherapy Cancer. (2024) 12(9):e009318. doi: 10.1136/jitc-2024-009318
59. Fu T, Mohan M, Brennan EP, Woodman OL, Godson C, Kantharidis P, et al. Therapeutic potential of lipoxin A(4) in chronic inflammation: focus on cardiometabolic disease. ACS Pharmacol Trans Sci. (2020) 3:43–55. doi: 10.1021/acsptsci.9b00097
60. Maciuszek M, Cacace A, Brennan E, Godson C, and Chapman TM. Recent advances in the design and development of formyl peptide receptor 2 (FPR2/ALX) agonists as pro-resolving agents with diverse therapeutic potential. Eur J medicinal Chem. (2021) 213:113167. doi: 10.1016/j.ejmech.2021.113167
61. Peng C, Vecchio EA, Nguyen ATN, De Seram M, Tang R, Keov P, et al. Biased receptor signalling and intracellular trafficking profiles of structurally distinct formylpeptide receptor 2 agonists. Br J Pharmacol. (2024) 181:4677–92. doi: 10.1111/bph.v181.22
62. Tie H, Kuang G, Gong X, Zhang L, Zhao Z, Wu S, et al. LXA4 protected mice from renal ischemia/reperfusion injury by promoting IRG1/Nrf2 and IRAK-M-TRAF6 signal pathways. Clin Immunol (Orlando Fla). (2024) 261:110167. doi: 10.1016/j.clim.2024.110167
63. Qiu Y, Wu Y, Zhao H, Sun H, and Gao S. Maresin 1 mitigates renal ischemia/reperfusion injury in mice via inhibition of the TLR4/MAPK/NF-κB pathways and activation of the Nrf2 pathway. Drug Des Devel Ther. (2019) 13:739–45. doi: 10.2147/DDDT.S188654
64. Hassan IR and Gronert K. Acute changes in dietary omega-3 and omega-6 polyunsaturated fatty acids have a pronounced impact on survival following ischemic renal injury and formation of renoprotective docosahexaenoic acid-derived protectin D1. J Immunol (Baltimore Md: 1950). (2009) 182:3223–32. doi: 10.4049/jimmunol.0802064
65. Leonard MO, Hannan K, Burne MJ, Lappin DW, Doran P, Coleman P, et al. 15-Epi-16-(para-fluorophenoxy)-lipoxin A(4)-methyl ester, a synthetic analogue of 15-epi-lipoxin A(4), is protective in experimental ischemic acute renal failure. J Am Soc Nephrology: JASN. (2002) 13:1657–62. doi: 10.1097/01.ASN.0000015795.74094.91
66. Silva J, Calcia TBB, Silva CP, Guilherme RF, Almeida-Souza F, Lemos FS, et al. ATRvD1 attenuates renal tubulointerstitial injury induced by albumin overload in sepsis-surviving mice. Int J Mol Sci. (2021) 22(21):11634. doi: 10.3390/ijms222111634
67. Hu X, Liang Y, Zhao H, and Zhao M. Effects of AT-RvD1 on paraquat-induced acute renal injury in mice. Int Immunopharmacol. (2019) 67:231–8. doi: 10.1016/j.intimp.2018.12.029
68. Zhang P, Peng H, Gao C, Fan Z, and Xia Z. Aspirin-triggered lipoxin protects lipopolysaccharide-induced acute kidney injury via the TLR4/myD88/NF-κB pathway. Saudi J Kidney Dis Transpl. (2020) 31(5):937–45. doi: 10.4103/1319-2442.301200
69. Chen C, Qiu R, Yang J, Zhang Q, Sun G, Gao X, et al. Lipoxin A4 restores septic renal function via blocking crosstalk between inflammation and premature senescence. Front Immunol. (2021) 12:637753. doi: 10.3389/fimmu.2021.637753
70. Zhang X, Qu X, Sun YB, Caruana G, Bertram JF, Nikolic-Paterson DJ, et al. Resolvin D1 protects podocytes in adriamycin-induced nephropathy through modulation of 14-3-3β acetylation. PloS One. (2013) 8:e67471. doi: 10.1371/journal.pone.0067471
71. Börgeson E, Docherty NG, Murphy M, Rodgers K, Ryan A, O’Sullivan TP, et al. Lipoxin A4 and benzo-lipoxin A4 attenuate experimental renal fibrosis. FASEB J. (2011) 25(9):2967–79. doi: 10.1096/fj.11-185017
72. Brennan EP, Mohan M, McClelland A, Tikellis C, Ziemann M, Kaspi A, et al. Lipoxins regulate the early growth response-1 network and reverse diabetic kidney disease. J Am Soc Nephrology: JASN. (2018) 29:1437–48. doi: 10.1681/ASN.2017101112
73. Pan Y, Zhang MZ, and Harris RC. Annexin 1 mimetic ac2–26 holds promise for the treatment of diabetic nephropathy. Diabetes. (2021) 70:2183–4. doi: 10.2337/dbi21-0030
74. de Assis AM, Rech A, Longoni A, da Silva Morrone M, de Bittencourt Pasquali MA, Perry ML, et al. Dietary n-3 polyunsaturated fatty acids revert renal responses induced by a combination of 2 protocols that increase the amounts of advanced glycation end product in rats. Nutr Res (New York NY). (2015) 35:512–22. doi: 10.1016/j.nutres.2015.04.013
75. Soleimani A, Taghizadeh M, Bahmani F, Badroj N, and Asemi Z. Metabolic response to omega-3 fatty acid supplementation in patients with diabetic nephropathy: A randomized, double-blind, placebo-controlled trial. Clin Nutr (Edinburgh Scotland). (2017) 36:79–84. doi: 10.1016/j.clnu.2015.11.003
76. Elajami TK, Alfaddagh A, Lakshminarayan D, Soliman M, Chandnani M, and Welty FK. Eicosapentaenoic and docosahexaenoic acids attenuate progression of albuminuria in patients with type 2 diabetes mellitus and coronary artery disease. J Am Heart Assoc. (2017) 6(7):e004740. doi: 10.1161/JAHA.116.004740
77. Vitlov Uljević M, Starčević K, Mašek T, Bočina I, Restović I, Kević N, et al. Dietary DHA/EPA supplementation ameliorates diabetic nephropathy by protecting from distal tubular cell damage. Cell Tissue Res. (2019) 378:301–17. doi: 10.1007/s00441-019-03058-y
78. Mas E, Barden A, Burke V, Beilin LJ, Watts GF, Huang RC, et al. A randomized controlled trial of the effects of n-3 fatty acids on resolvins in chronic kidney disease. Clin Nutr (Edinburgh Scotland). (2016) 35:331–6. doi: 10.1016/j.clnu.2015.04.004
79. Perazza LR, Mitchell PL, Lizotte F, Jensen BAH, St-Pierre P, Trottier J, et al. Fish oil replacement prevents, while docosahexaenoic acid-derived protectin DX mitigates end-stage-renal-disease in atherosclerotic diabetic mice. FASEB journal: Off Publ Fed Am Societies Exp Biol. (2021) 35:e21559. doi: 10.1096/fj.202100073R
80. An WS, Kim HJ, Cho KH, and Vaziri ND. Omega-3 fatty acid supplementation attenuates oxidative stress, inflammation, and tubulointerstitial fibrosis in the remnant kidney. Am J Physiol Renal Physiol. (2009) 297:F895–903. doi: 10.1152/ajprenal.00217.2009
81. Belayev LY and Palevsky PM. The link between acute kidney injury and chronic kidney disease. Curr Opin Nephrol hypertension. (2014) 23:149–54. doi: 10.1097/01.mnh.0000441051.36783.f3
82. Hsu CY. Yes, AKI truly leads to CKD. J Am Soc Nephrology: JASN. (2012) 23:967–9. doi: 10.1681/ASN.2012030222
83. Ballesteros I, Rubio-Ponce A, Genua M, Lusito E, Kwok I, Fernández-Calvo G, et al. Co-option of neutrophil fates by tissue environments. Cell. (2020) 183:1282–1297.e1218. doi: 10.1016/j.cell.2020.10.003
84. Fox S, Leitch AE, Duffin R, Haslett C, and Rossi AG. Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J innate Immun. (2010) 2:216–27. doi: 10.1159/000284367
85. Geering B and Simon HU. Peculiarities of cell death mechanisms in neutrophils. Cell Death differentiation. (2011) 18:1457–69. doi: 10.1038/cdd.2011.75
86. Wang C, Zhang Y, Shen A, Tang T, Li N, Xu C, et al. Mincle receptor in macrophage and neutrophil contributes to the unresolved inflammation during the transition from acute kidney injury to chronic kidney disease. Front Immunol. (2024) 15:1385696. doi: 10.3389/fimmu.2024.1385696
87. Heinzelmann M, Mercer-Jones MA, and Passmore JC. Neutrophils and renal failure. Am J Kidney diseases: Off J Natl Kidney Foundation. (1999) 34:384–99. doi: 10.1016/S0272-6386(99)70375-6
88. Jonsson H, Allen P, and Peng SL. Inflammatory arthritis requires Foxo3a to prevent Fas ligand-induced neutrophil apoptosis. Nat Med. (2005) 11:666–71. doi: 10.1038/nm1248
89. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, and Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. (2010) 31:318–24. doi: 10.1016/j.it.2010.05.006
90. Maianski NA, Maianski AN, Kuijpers TW, and Roos D. Apoptosis of neutrophils. Acta haematologica. (2004) 111:56–66. doi: 10.1159/000074486
91. Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, et al. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. (1998) 17:1675–87. doi: 10.1093/emboj/17.6.1675
92. Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, and Peter ME. Apoptosis signaling by death receptors. Eur J Biochem. (1998) 254:439–59. doi: 10.1046/j.1432-1327.1998.2540439.x
93. Hirata H, Takahashi A, Kobayashi S, Yonehara S, Sawai H, Okazaki T, et al. Caspases are activated in a branched protease cascade and control distinct downstream processes in Fas-induced apoptosis. J Exp Med. (1998) 187:587–600. doi: 10.1084/jem.187.4.587
94. Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death–inducing signaling complex. Cell. (1996) 85:817–27. doi: 10.1016/S0092-8674(00)81266-0
95. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. (1997) 91:479–89. doi: 10.1016/S0092-8674(00)80434-1
96. Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, et al. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. (1999) 144:281–92. doi: 10.1083/jcb.144.2.281
97. Noseykina EM, Schepetkin IA, and Atochin DN. Molecular mechanisms for regulation of neutrophil apoptosis under normal and pathological conditions. J evolutionary Biochem Physiol. (2021) 57:429–50. doi: 10.1134/S0022093021030017
98. Zahran N, Sayed A, William I, Mahmoud O, Sabry O, and Rafaat M. Neutrophil apoptosis: impact of granulocyte macrophage colony stimulating factor on cell survival and viability in chronic kidney disease and hemodialysis patients. Arch Med science: AMS. (2013) 9:984–9. doi: 10.5114/aoms.2013.39789
99. Kumaran Satyanarayanan S, El Kebir D, Soboh S, Butenko S, Sekheri M, Saadi J, et al. IFN-β is a macrophage-derived effector cytokine facilitating the resolution of bacterial inflammation. Nat Commun. (2019) 10:3471. doi: 10.1038/s41467-019-10903-9
100. Bhatnagar N, Hong HS, Krishnaswamy JK, Haghikia A, Behrens GM, Schmidt RE, et al. Cytokine-activated NK cells inhibit PMN apoptosis and preserve their functional capacity. Blood. (2010) 116:1308–16. doi: 10.1182/blood-2010-01-264903
101. Costantini C, Calzetti F, Perbellini O, Micheletti A, Scarponi C, Lonardi S, et al. Human neutrophils interact with both 6-sulfo LacNAc+ DC and NK cells to amplify NK-derived IFN{gamma}: role of CD18, ICAM-1, and ICAM-3. Blood. (2011) 117:1677–86. doi: 10.1182/blood-2010-06-287243
102. Hind LE, Ingram PN, Beebe DJ, and Huttenlocher A. Interaction with an endothelial lumen increases neutrophil lifetime and motility in response to P aeruginosa. Blood. (2018) 132:1818–28. doi: 10.1182/blood-2018-05-848465
103. Kolman JP, Pagerols Raluy L, Müller I, Nikolaev VO, Trochimiuk M, Appl B, et al. NET release of long-term surviving neutrophils. Front Immunol. (2022) 13:815412. doi: 10.3389/fimmu.2022.815412
104. El Kebir D and Filep JG. Role of neutrophil apoptosis in the resolution of inflammation. TheScientificWorldJournal. (2010) 10:1731–48. doi: 10.1100/tsw.2010.169
105. Buckley CD, Ross EA, McGettrick HM, Osborne CE, Haworth O, Schmutz C, et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J leukocyte Biol. (2006) 79:303–11. doi: 10.1189/jlb.0905496
106. Soehnlein O, Steffens S, Hidalgo A, and Weber C. Neutrophils as protagonists and targets in chronic inflammation. Nat Rev Immunol. (2017) 17:248–61. doi: 10.1038/nri.2017.10
107. Raupachova J, Kopecky C, and Cohen G. High-density lipoprotein from chronic kidney disease patients modulates polymorphonuclear leukocytes. Toxins. (2019) 11(2):73. doi: 10.3390/toxins11020073
108. Sudo C, Ogawara H, Saleh AW, Nishimoto N, Utsugi T, Ooyama Y, et al. Clinical significance of neutrophil apoptosis in peripheral blood of patients with type 2 diabetes mellitus. Lab hematology: Off Publ Int Soc Lab Hematol. (2007) 13:108–12. doi: 10.1532/LH96.07003
109. Homilius M, Zhu W, Eddy SS, Thompson PC, Zheng H, Warren CN, et al. Perturbational phenotyping of human blood cells reveals genetically determined latent traits associated with subsets of common diseases. Nat Genet. (2024) 56:37–50. doi: 10.1038/s41588-023-01600-x
110. Yu Y, Lin Q, Ye D, Wang Y, He B, Li Y, et al. Neutrophil count as a reliable marker for diabetic kidney disease in autoimmune diabetes. BMC endocrine Disord. (2020) 20:158. doi: 10.1186/s12902-020-00597-2
111. Zhang R, Chen J, Xiong Y, Wang L, Huang X, Sun T, et al. Increased neutrophil count Is associated with the development of chronic kidney disease in patients with diabetes. J Diabetes. (2022) 14:442–54. doi: 10.1111/1753-0407.13292
112. Klein JB, Rane MJ, Scherzer JA, Coxon PY, Kettritz R, Mathiesen JM, et al. Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways. J Immunol (Baltimore Md: 1950). (2000) 164:4286–91. doi: 10.4049/jimmunol.164.8.4286
113. Arnardottir HH, Dalli J, Colas RA, Shinohara M, and Serhan CN. Aging delays resolution of acute inflammation in mice: reprogramming the host response with novel nano-proresolving medicines. J Immunol (Baltimore Md: 1950). (2014) 193:4235–44. doi: 10.4049/jimmunol.1401313
114. Doyle R, Sadlier DM, and Godson C. Pro-resolving lipid mediators: Agents of anti-ageing? Semin Immunol. (2018) 40:36–48. doi: 10.1016/j.smim.2018.09.002
115. Liu Y, Xiang C, Que Z, Li C, Wang W, Yin L, et al. Neutrophil heterogeneity and aging: implications for COVID-19 and wound healing. Front Immunol. (2023) 14:1201651. doi: 10.3389/fimmu.2023.1201651
116. Adkar SS and Leeper NJ. Efferocytosis in atherosclerosis. Nat Rev Cardiol. (2024) 21:762–79. doi: 10.1038/s41569-024-01037-7
117. Klopf J, Brostjan C, Eilenberg W, and Neumayer C. Neutrophil extracellular traps and their implications in cardiovascular and inflammatory disease. Int J Mol Sci. (2021) 22(2):559. doi: 10.3390/ijms22020559
118. Nakazawa D, Masuda S, Nishibata Y, Watanabe-Kusunoki K, Tomaru U, and Ishizu A. Neutrophils and NETs in kidney disease. Nat Rev Nephrol. (2025). doi: 10.1038/s41581-025-00944-3
119. Cho W, Song JY, Oh SW, Kim MG, Ko YS, Lee HY, et al. Fate of neutrophils during the recovery phase of ischemia/reperfusion induced acute kidney injury. J Korean Med Sci. (2017) 32:1616–25. doi: 10.3346/jkms.2017.32.10.1616
120. Everts-Graber J, Martin KR, Thieblemont N, Mocek J, Roccabianca A, Chafey P, et al. Proteomic analysis of neutrophils in ANCA-associated vasculitis reveals a dysregulation in proteinase 3-associated proteins such as annexin-A1 involved in apoptotic cell clearance. Kidney Int. (2019) 96:397–408. doi: 10.1016/j.kint.2019.02.017
121. Kettritz R, Wilke S, von Vietinghoff S, Luft F, and Schneider W. Apoptosis, proliferation and inflammatory infiltration in ANCA-positive glomerulonephritis. Clin Nephrol. (2006) 65:309–16. doi: 10.5414/CNP65309
122. Takazoe K, Tesch GH, Hill PA, Hurst LA, Jun Z, Lan HY, et al. CD44-mediated neutrophil apoptosis in the rat. Kidney Int. (2000) 58:1920–30. doi: 10.1111/j.1523-1755.2000.00364.x
123. Khalighi MA and Chang A. Infection-related glomerulonephritis. Glomerular Dis. (2021) 1:82–91. doi: 10.1159/000515461
124. Mosquera-Sulbaran JA, Pedreañez A, Vargas R, and Hernandez-Fonseca JP. Apoptosis in post-streptococcal glomerulonephritis and mechanisms for failed of inflammation resolution. Pediatr Nephrol (Berlin Germany). (2024) 39:1709–24. doi: 10.1007/s00467-023-06162-y
125. Saisorn W, Saithong S, Phuengmaung P, Udompornpitak K, Bhunyakarnjanarat T, Visitchanakun P, et al. Acute kidney injury induced lupus exacerbation through the enhanced neutrophil extracellular traps (and apoptosis) in fcgr2b deficient lupus mice with renal ischemia reperfusion injury. Front Immunol. (2021) 12:669162. doi: 10.3389/fimmu.2021.669162
126. Hughes J, Johnson RJ, Mooney A, Hugo C, Gordon K, and Savill J. Neutrophil fate in experimental glomerular capillary injury in the rat. Emigration exceeds in situ clearance by apoptosis. Am J Pathol. (1997) 150(1):223–34.
127. Cohen G. Immunoglobulin light chains in uremia. Kidney Int Supplement. (2003) 84:S15–18. doi: 10.1046/j.1523-1755.63.s84.8.x
128. Mázló A, Tang Y, Jenei V, Brauman J, Yousef H, Bácsi A, et al. Resolution potential of necrotic cell death pathways. Int J Mol Sci. (2022) 24(1):16. doi: 10.3390/ijms24010016
129. Ortega-Gómez A, Perretti M, and Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med. (2013) 5:661–74. doi: 10.1002/emmm.201202382
130. Headland SE and Norling LV. The resolution of inflammation: Principles and challenges. Semin Immunol. (2015) 27:149–60. doi: 10.1016/j.smim.2015.03.014
131. Chello M, Anselmi A, Spadaccio C, Patti G, Goffredo C, Di Sciascio G, et al. Simvastatin increases neutrophil apoptosis and reduces inflammatory reaction after coronary surgery. Ann thoracic Surg. (2007) 83:1374–80. doi: 10.1016/j.athoracsur.2006.10.065
132. Leitch AE, Lucas CD, Marwick JA, Duffin R, Haslett C, and Rossi AG. Cyclin-dependent kinases 7 and 9 specifically regulate neutrophil transcription and their inhibition drives apoptosis to promote resolution of inflammation. Cell Death differentiation. (2012) 19:1950–61. doi: 10.1038/cdd.2012.80
133. Abdulnour RE, Dalli J, Colby JK, Krishnamoorthy N, Timmons JY, Tan SH, et al. Maresin 1 biosynthesis during platelet-neutrophil interactions is organ-protective. Proc Natl Acad Sci United States America. (2014) 111:16526–31. doi: 10.1073/pnas.1407123111
134. Dalli J and Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. (2012) 120:e60–72. doi: 10.1182/blood-2012-04-423525
135. Perretti M, Christian H, Wheller SK, Aiello I, Mugridge KG, Morris JF, et al. Annexin I is stored within gelatinase granules of human neutrophil and mobilized on the cell surface upon adhesion but not phagocytosis. Cell Biol Int. (2000) 24:163–74. doi: 10.1006/cbir.1999.0468
136. Tang C, Wang C, Zhang Y, Xue L, Li Y, Ju C, et al. Recognition, intervention, and monitoring of neutrophils in acute ischemic stroke. Nano Lett. (2019) 19:4470–7. doi: 10.1021/acs.nanolett.9b01282
137. Thieblemont N, Wright HL, Edwards SW, and Witko-Sarsat V. Human neutrophils in auto-immunity. Semin Immunol. (2016) 28:159–73. doi: 10.1016/j.smim.2016.03.004
138. Dalli J, Norling LV, Renshaw D, Cooper D, Leung KY, and Perretti M. Annexin 1 mediates the rapid anti-inflammatory effects of neutrophil-derived microparticles. Blood. (2008) 112:2512–9. doi: 10.1182/blood-2008-02-140533
139. Perretti M, Chiang N, La M, Fierro IM, Marullo S, Getting SJ, et al. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med. (2002) 8:1296–302. doi: 10.1038/nm786
140. Serhan CN, Chiang N, and Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. (2008) 8:349–61. doi: 10.1038/nri2294
141. Tu H, Ren H, Jiang J, Shao C, Shi Y, and Li P. Dying to defend: neutrophil death pathways and their implications in immunity. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2024) 11:e2306457. doi: 10.1002/advs.202306457
142. Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology (Baltimore Md). (2015) 62:600–14. doi: 10.1002/hep.27841
143. Vago JP, Nogueira CR, Tavares LP, Soriani FM, Lopes F, Russo RC, et al. Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J leukocyte Biol. (2012) 92:249–58. doi: 10.1189/jlb.0112008
144. El Kebir D, Gjorstrup P, and Filep JG. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci United States America. (2012) 109:14983–8. doi: 10.1073/pnas.1206641109
145. Li X, Li C, Liang W, Bi Y, Chen M, and Dong S. Protectin D1 promotes resolution of inflammation in a murine model of lipopolysaccharide-induced acute lung injury via enhancing neutrophil apoptosis. Chin Med J (Engl). (2014) 127:810–4. doi: 10.3760/cma.j.issn.0366-6999.20131104
146. Gong J, Liu H, Wu J, Qi H, Wu ZY, Shu HQ, et al. MARESIN 1 PREVENTS LIPOPOLYSACCHARIDE-INDUCED NEUTROPHIL SURVIVAL AND ACCELERATES RESOLUTION OF ACUTE LUNG INJURY. Shock (Augusta Ga). (2015) 44:371–80. doi: 10.1097/SHK.0000000000000434
147. Xian W, Wu Y, Xiong W, Li L, Li T, Pan S, et al. The pro-resolving lipid mediator Maresin 1 protects against cerebral ischemia/reperfusion injury by attenuating the pro-inflammatory response. Biochem Biophys Res Commun. (2016) 472:175–81. doi: 10.1016/j.bbrc.2016.02.090
148. Sekheri M, El Kebir D, Edner N, and Filep JG. 15-Epi-LXA(4) and 17-epi-RvD1 restore TLR9-mediated impaired neutrophil phagocytosis and accelerate resolution of lung inflammation. Proc Natl Acad Sci United States America. (2020) 117:7971–80. doi: 10.1073/pnas.1920193117
149. El Kebir D, József L, Pan W, Wang L, Petasis NA, Serhan CN, et al. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am J Respir Crit Care Med. (2009) 180:311–9. doi: 10.1164/rccm.200810-1601OC
150. Stunault MI, Bories G, Guinamard RR, and Ivanov S. Metabolism plays a key role during macrophage activation. Mediators Inflammation. (2018) 2018:2426138. doi: 10.1155/2018/2426138
151. Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S, et al. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature. (2011) 477:220–4. doi: 10.1038/nature10340
152. Zhang S, Weinberg S, DeBerge M, Gainullina A, Schipma M, Kinchen JM, et al. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. (2019) 29:443–456.e445. doi: 10.1016/j.cmet.2018.12.004
153. Ariel A and Serhan CN. New lives given by cell death: macrophage differentiation following their encounter with apoptotic leukocytes during the resolution of inflammation. Front Immunol. (2012) 3:4. doi: 10.3389/fimmu.2012.00004
154. Perretti M and Solito E. Annexin 1 and neutrophil apoptosis. Biochem Soc Trans. (2004) 32:507–10. doi: 10.1042/bst0320507
155. Arur S, Uche UE, Rezaul K, Fong M, Scranton V, Cowan AE, et al. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev Cell. (2003) 4:587–98. doi: 10.1016/S1534-5807(03)00090-X
156. Scannell M, Flanagan MB, deStefani A, Wynne KJ, Cagney G, Godson C, et al. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol (Baltimore Md: 1950). (2007) 178:4595–605. doi: 10.4049/jimmunol.178.7.4595
157. Matte A, Recchiuti A, Federti E, Koehl B, Mintz T, El Nemer W, et al. Resolution of sickle cell disease-associated inflammation and tissue damage with 17R-resolvin D1. Blood. (2019) 133:252–65. doi: 10.1182/blood-2018-07-865378
158. Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med. (2005) 201:713–22. doi: 10.1084/jem.20042031
159. Ohira T, Arita M, Omori K, Recchiuti A, Van Dyke TE, and Serhan CN. Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J Biol Chem. (2010) 285:3451–61. doi: 10.1074/jbc.M109.044131
160. Schwab JM, Chiang N, Arita M, and Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. (2007) 447:869–74. doi: 10.1038/nature05877
161. Bang S, Xie YK, Zhang ZJ, Wang Z, Xu ZZ, and Ji RR. GPR37 regulates macrophage phagocytosis and resolution of inflammatory pain. J Clin Invest. (2018) 128:3568–82. doi: 10.1172/JCI99888
162. Trilleaud C, Gauttier V, Biteau K, Girault I, Belarif L, Mary C, et al. Agonist anti-ChemR23 mAb reduces tissue neutrophil accumulation and triggers chronic inflammation resolution. Sci Adv. (2021) 7(14):eabd1453. doi: 10.1126/sciadv.abd1453
163. Mehrotra P and Ravichandran KS. Drugging the efferocytosis process: concepts and opportunities. Nat Rev Drug Discov. (2022) 21:601–20. doi: 10.1038/s41573-022-00470-y
164. Scharf P, Sandri S, Rizzetto F, Xavier LF, Grosso D, Correia-Silva RD, et al. GPCRs overexpression and impaired fMLP-induced functions in neutrophils from chronic kidney disease patients. Front Immunol. (2024) 15:1387566. doi: 10.3389/fimmu.2024.1387566
165. Angeletti A, Bruschi M, Kajana X, Spinelli S, Verrina E, Lugani F, et al. Mechanisms limiting renal tissue protection and repair in glomerulonephritis. Int J Mol Sci. (2023) 24(9):8318. doi: 10.3390/ijms24098318
166. Halade GV, Kain V, and Serhan CN. Immune responsive resolvin D1 programs myocardial infarction-induced cardiorenal syndrome in heart failure. FASEB journal: Off Publ Fed Am Societies Exp Biol. (2018) 32:3717–29. doi: 10.1096/fj.201701173RR
167. Gray RD, Hardisty G, Regan KH, Smith M, Robb CT, Duffin R, et al. Delayed neutrophil apoptosis enhances NET formation in cystic fibrosis. Thorax. (2018) 73:134–44. doi: 10.1136/thoraxjnl-2017-210134
168. Song C, Li H, Mao Z, Peng L, Liu B, Lin F, et al. Delayed neutrophil apoptosis may enhance NET formation in ARDS. Respir Res. (2022) 23:155. doi: 10.1186/s12931-022-02065-y
169. Kurdowska AK and Florence JM. Promoting neutrophil apoptosis to treat acute lung injury. Am J Respir Crit Care Med. (2019) 200:399–400. doi: 10.1164/rccm.201903-0707LE
170. Kolpakov MA, Guo X, Rafiq K, Vlasenko L, Hooshdaran B, Seqqat R, et al. Loss of protease-activated receptor 4 prevents inflammation resolution and predisposes the heart to cardiac rupture after myocardial infarction. Circulation. (2020) 142:758–75. doi: 10.1161/CIRCULATIONAHA.119.044340
171. Bao L, Dou G, Tian R, Lv Y, Ding F, Liu S, et al. Engineered neutrophil apoptotic bodies ameliorate myocardial infarction by promoting macrophage efferocytosis and inflammation resolution. Bioactive materials. (2022) 9:183–97. doi: 10.1016/j.bioactmat.2021.08.008
172. Briottet M, Shum M, and Urbach V. The role of specialized pro-resolving mediators in cystic fibrosis airways disease. Front Pharmacol. (2020) 11:1290. doi: 10.3389/fphar.2020.01290
173. Chen R, Li J, Zhou J, Wang Y, Zhao X, Li N, et al. Prognostic impacts of Lipoxin A4 in patients with acute myocardial infarction: A prospective cohort study. Pharmacol Res. (2023) 187:106618. doi: 10.1016/j.phrs.2022.106618
174. Karayiğit O, Nurkoç SG, Başyiğit F, and Kızıltunç E. The role of serum resolvin D1 levels in determining the presence and prognosis of ST-segment elevation myocardial infarction. Med principles practice: Int J Kuwait University Health Sci Centre. (2022) 31(6):548–54. doi: 10.1159/000527064
175. Kong X, Wu SH, Zhang L, and Chen XQ. Pilot application of lipoxin A(4) analog and lipoxin A(4) receptor agonist in asthmatic children with acute episodes. Exp Ther Med. (2017) 14:2284–90. doi: 10.3892/etm.2017.4787
176. Wu SH, Chen XQ, Liu B, Wu HJ, and Dong L. Efficacy and safety of 15(R/S)-methyl-lipoxin A(4) in topical treatment of infantile eczema. Br J Dermatol. (2013) 168:172–8. doi: 10.1111/j.1365-2133.2012.11177.x
177. Zhang CY, Dong X, Gao J, Lin W, Liu Z, and Wang Z. Nanoparticle-induced neutrophil apoptosis increases survival in sepsis and alleviates neurological damage in stroke. Sci Adv. (2019) 5:eaax7964. doi: 10.1126/sciadv.aax7964
178. Liu X, Ou X, Zhang T, Li X, Qiao Q, Jia L, et al. In situ neutrophil apoptosis and macrophage efferocytosis mediated by Glycyrrhiza protein nanoparticles for acute inflammation therapy. J Controlled release: Off J Controlled Release Soc. (2024) 369:215–30. doi: 10.1016/j.jconrel.2024.03.029
179. Su R, Wang H, Xiao C, Tao Y, Li M, and Chen Z. Venetoclax nanomedicine alleviates acute lung injury via increasing neutrophil apoptosis. Biomaterials Sci. (2021) 9:4746–54. doi: 10.1039/D1BM00481F
180. Norling LV, Spite M, Yang R, Flower RJ, Perretti M, and Serhan CN. Cutting edge: Humanized nano-proresolving medicines mimic inflammation-resolution and enhance wound healing. J Immunol (Baltimore Md: 1950). (2011) 186:5543–7. doi: 10.4049/jimmunol.1003865
Keywords: specialized pro-resolving mediators, neutrophils, apoptosis, efferocytosis, kidney diseases
Citation: Kang Y, Jin Q, Zhou M, Zheng H, Li D, Wang X, Zhou J, Wang Y and Lv J (2025) Specialized pro-resolving mediators in neutrophil apoptosis regulation: unlocking novel therapeutic potential in kidney diseases. Front. Immunol. 16:1589923. doi: 10.3389/fimmu.2025.1589923
Received: 08 March 2025; Accepted: 22 April 2025;
Published: 15 May 2025.
Edited by:
Sofie Struyf, KU Leuven, BelgiumReviewed by:
Sean Eddy, University of Michigan, United StatesIrma Husain, Duke University, United States
Copyright © 2025 Kang, Jin, Zhou, Zheng, Li, Wang, Zhou, Wang and Lv. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaoxian Wang, dGNtd3l4MTIzQDEyNi5jb20=; Jie Lv, bHZqaWVidWNtQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship