 Wenyi Nie
Wenyi Nie Yingbin Yue2,3†
Yingbin Yue2,3† Jingqing Hu
Jingqing Hu- 1School of Traditional Chinese Medicine, Changchun University of Chinese Medicine, Changchun, China
- 2Department of Pediatrics, The First Affiliated Hospital of Xinjiang Medical University, Urumqi, China
- 3State Key Laboratory of Pathogenesis, Prevention and Treatment of High Incidence Diseases in Central Asia, Xinjiang Medical University, Urumqi, China
- 4School of Traditional Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 5Institute of Basic Theory for Chinese Medicine, China Academy of Chinese Medical Sciences, Beijing, China
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by β-amyloid (Aβ) plaques, neurofibrillary tangles (NFTs), and neuroinflammation. Monocytes and macrophages, particularly microglia, play a dual role in AD pathogenesis. In the early stages, they delay disease progression by phagocytosing Aβ, but chronic activation leads to Aβ accumulation and exacerbated neuroinflammation. Monocyte chemoattractant protein 1 (MCP-1) is a key regulator in neuroinflammation, Aβ deposition, and tau pathology, making it a potential therapeutic target. Moreover, recent breakthroughs in fluid and imaging biomarkers and targeted immunomodulatory agents underscore the growing importance of early diagnostic and therapeutic interventions. This review explores the complex interplay between monocytes, macrophages, and AD pathology, highlighting their roles in neuroinflammation, Aβ metabolism, and tau phosphorylation. Understanding these mechanisms offers new insights into developing effective diagnostic biomarkers and therapeutic strategies for AD.
1 Introduction
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disorder, marked by progressive cognitive decline and the accumulation of neuropathological hallmarks, including extracellular β-amyloid (Aβ) plaques, intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau, and a sustained neuroinflammatory response (1). While Aβ and tau pathologies remain central to AD pathogenesis, mounting evidence implicates neuroinflammation as a key driver of disease progression, linking immune dysregulation to neuronal dysfunction and cognitive impairment (2).
Recent studies highlight the intricate and context-dependent roles of neuroinflammatory mechanisms in AD. Among the mediators of neuroinflammation, monocyte chemoattractant protein 1 (MCP-1) has emerged as a critical regulator, abundantly localized in Aβ plaques and associated with reactive microglia (3, 4). MCP-1 orchestrates monocyte recruitment to the brain, exacerbating neuroinflammation while also modulating Aβ clearance and tau hyperphosphorylation. Similarly, immune cells including microglia and infiltrating macrophages exhibit dual functions throughout disease progression. In early stages, microglia facilitate Aβ clearance, yet in chronic disease, they transition to a pro-inflammatory phenotype that perpetuates neuronal damage. Conversely, macrophages exhibit a complex interplay between Aβ/tau phagocytosis and the secretion of inflammatory cytokines, contributing to both protective and pathological outcomes (5, 6). These dynamic interactions underscore the necessity of delineating the precise mechanisms by which immune-inflammatory networks influence AD pathogenesis.
This review synthesizes recent advances in understanding MCP-1 and immune cells as pivotal mediators of AD pathology. We first examine the mechanisms by which MCP-1 bridges neuroinflammation with Aβ and tau aggregation. Further, we dissect the evolving roles of microglia and macrophages across distinct disease stages, emphasizing their dual neuroprotective and neurotoxic effects. Finally, we explore emerging therapeutic strategies targeting neuroinflammatory pathways, offering perspectives on potential interventions to mitigate AD progression.
2 Evolving paradigms in AD pathogenesis
2.1 Aβ dysregulation from homeostasis to pathology
The etiology of AD remains unclear, and its pathogenesis is extremely complex, primarily characterized by the abnormal deposition of Aβ in the brain and the presence of neurofibrillary tangles composed mainly of Tau protein. The mainstream theories of AD pathogenesis are the Aβ hypothesis and the Tau protein hypothesis, although other hypotheses have also emerged. Aβ is a metabolic product derived from the cleavage of amyloid precursor protein (APP) by β-secretase and γ-secretase. Under physiological conditions, Aβ is non-toxic, and its production and clearance are in dynamic equilibrium. However, in AD patients, increased generation or reduced clearance of Aβ leads to its abnormal deposition in the brain, forming insoluble amyloid plaques. These plaques activate microglia, triggering inflammatory responses that ultimately result in neuronal death and cognitive decline (2, 7). Research indicates that the formation of Aβ plaques is closely associated with abnormal processing of APP (8). APP is cleaved by β-secretase and γ-secretase to produce insoluble Aβ peptides, and post-translational modifications such as pyroglutamation and phosphorylation of Aβ reduce its degradation, leading to Aβ accumulation (9). The gradual accumulation of Aβ plaques impairs normal neuronal function and spreads to other brain regions, further exacerbating neuronal damage (10).
2.2 Tau hyperphosphorylation and neuronal collapse
Tau protein is a crucial component of neuronal microtubules, participating in microtubule assembly and stability, and maintaining cytoskeletal integrity. In AD patients, tau protein undergoes hyperphosphorylation, losing its normal microtubule-binding capacity and aggregating into paired helical filaments and straight filaments, ultimately forming NFTs. The presence of NFTs disrupts neuronal structure and function, leading to synaptic loss, neuronal death, and cognitive decline (11). The hyperphosphorylation of tau protein is closely associated with the activation of cyclin-dependent kinase 5 (CDK5). Aβ activates calpain, which regulates p35, the primary activator of CDK5, causing its cleavage into p25 under high-calcium conditions, leading to excessive activation of CDK5. The overactivation of CDK5 further promotes the hyperphosphorylation of tau protein, forming NFTs and contributing to the neurodegenerative cascade in AD (12). Emerging biomarker p-Tau181 is linked to tau pathology, and neurofilament light chain (NfL) reflect axonal damage, when combined enhance diagnostic accuracy and provide a comprehensive understanding of AD pathology (13).
2.3 Neuroinflammation as a driver of AD progression
Neuroinflammation is a significant feature in the pathogenesis of AD. The presence of Aβ plaques and NFTs activates microglia, leading to the release of pro-inflammatory cytokines and chemokines (14), further exacerbating neuroinflammation and neuronal damage. Microglia, the resident macrophages in the CNS, are responsible for maintaining neuronal homeostasis. In the early stages of AD, microglia delay disease progression by phagocytosing Aβ. However, under chronic inflammatory conditions, the persistent activation of microglia leads to Aβ accumulation and exacerbation of neuroinflammation (15). The presence of Aβ plaques and NFTs causes synaptic damage and increased reactive oxidative stress, acting as pathological triggers that induce the sustained activation of Toll-like receptors 2 (TLR2), TLR4, TLR6, and their co-receptors on microglia, upregulating the secretion of pro-inflammatory cytokines and chemokines (16). The activation of cyclin-dependent kinases further promotes tau hyperphosphorylation and increases Aβ plaque formation, thereby worsening cognitive dysfunction in AD patients (17). In addition to the Aβ hypothesis, tau hypothesis, and neuroinflammation hypothesis, recent years have seen the proposal of viral, mitochondrial dysfunction, insulin signaling abnormalities, gut microbiota dysbiosis, excitatory amino acid toxicity, and cholinergic dysfunction hypotheses, further enriching the research on the pathogenesis of AD (6, 18–22). The pathogenesis of AD is extremely complex, with many influencing factors. A single hypothesis is difficult to explain the occurrence and development of AD. This also shows that its specific mechanism is still unclear and requires more basic research for in-depth exploration.
3 The role of MCP-1 in AD
3.1 MCP-1 origins, signaling pathways, and neuroinflammatory cascades
Abnormal levels of Aβ and tau protein phosphorylation in the brains of AD patients lead to plaque formation and NFTs, further activating microglia and inducing neuroinflammation, thereby promoting disease progression. Studies have found a significant correlation between MCP-1 gene polymorphisms and AD risk, and MCP-1 plays a crucial regulatory role in neuroinflammation, Aβ deposition, and tau phosphorylation (Table 1). MCP-1, also known as CC chemokine ligand 2 (CCL2), is primarily derived from epithelial cells, smooth muscle cells, fibroblasts, endothelial cells, mononuclear macrophages, astrocytes, and microglia. Its primary receptor is CC chemokine receptor 2 (CCR2). In the brain, MCP-1 is primarily produced by astrocytes and resident microglia, with a smaller contribution from endothelial cells. This chemokine-receptor interaction plays a critical role in mediating immune cell recruitment and inflammatory responses, highlighting its significance in various physiological and pathological processes. MCP-1 binds to CCR2, activating downstream signaling pathways and regulating the chemotaxis of monocytes, thereby participating in the development and progression of various diseases (23). In AD patients, the expression of MCP-1 is significantly elevated, particularly around senile plaques and in reactive microglia, indicating its important role in the neuroinflammatory and pathological processes of AD (5).

Table 1. MCP-1 in alzheimer’s disease: mechanisms and therapeutic implications.
Through binding to CCR2, MCP-1 triggers downstream signaling cascades, including the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) pathway and the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, both of which regulate immune cell infiltration and inflammation-related gene expression (16). In AD, these signaling events are closely tied to the disease’s neuroinflammatory landscape. Neuroinflammation is a central feature of AD pathogenesis, and MCP-1 directly contributes to its progression by recruiting monocytes that differentiate into macrophages within the CNS parenchyma, reinforcing the pro-inflammatory milieu (24). Elevated MCP-1 and its downstream mediators accumulate near senile plaques, promoting both local immune activation and further Aβ-driven inflammatory responses (25). Consequently, a feedforward cycle emerges in which MCP-1’s origins and receptor-mediated signaling drive robust neuroinflammatory cascades. The sustained infiltration of peripheral immune cells, coupled with microglial activation, ultimately fosters the chronic inflammatory state characteristic of AD. This persistent inflammation not only impairs Aβ clearance but also accelerates tau hyperphosphorylation, together exacerbating neuronal damage and cognitive decline.
3.2 MCP-1 in Aβ dynamics from clearance to accumulation
MCP-1 plays a significant role in Aβ deposition through several mechanisms, including the upregulation of apolipoprotein E (ApoE) expression, the accumulation of monocyte-derived macrophages in the brain, and a mutually reinforcing relationship with Aβ deposition. ApoE is the primary apolipoprotein in the CNS, mainly produced by astrocytes and microglia, and plays a crucial role in promoting the hydrolysis and clearance of Aβ. Research indicates that MCP-1 can enhance the expression of ApoE, thereby influencing Aβ deposition. In AD transgenic mouse models, the complete loss of ApoE significantly reduces Aβ deposition, while the upregulation of MCP-1 increases Aβ plaque formation (26). Additionally, MCP-1 disrupts the blood-brain barrier (BBB), promoting the migration and infiltration of peripheral monocytes into the brain. In the context of AD, sustained elevation of MCP-1 levels leads to BBB disruption, significantly increasing the infiltration of monocyte-derived macrophages and exacerbating Aβ deposition (27). The role of MCP-1 in tau pathology is gradually being uncovered. Studies have shown that MCP-1 upregulates phosphatase activity, inducing the hyperphosphorylation of tau protein and promoting the formation of NFTs. In mouse models of neuroinflammatory diseases, the overexpression of MCP-1 significantly increases the accumulation and phosphorylation levels of tau protein (28). Furthermore, MCP-1 activates microglia, promoting the spread and aggregation of tau protein, thereby exacerbating the pathological processes of AD.
4 Macrophage heterogeneity and functional dynamics in AD
4.1 Diverse macrophage subsets in the CNS
Macrophages, especially CNS-resident microglia (derived from monocytes), are crucial immune regulators with dual roles in AD pathogenesis (29). They influence AD progression via Aβ phagocytosis, neuroinflammation, and tau pathology exacerbation (30, 31). Early in AD, microglia clear soluble Aβ via TREM2/CD36, while peripheral macrophages aid Aβ removal (32, 33). Chronic Aβ exposure, however, shifts microglia toward a pro-inflammatory state (NLRP3 activation, IL-1β/TNF-α release), worsening synaptic dysfunction and neurodegeneration (34). Microglia also drive tau hyperphosphorylation (via GSK-3β/CDK5) and spread tau via exosomes (35), highlighting their shift from protective to detrimental roles. Macrophages within the CNS are stratified into parenchymal and non-parenchymal subsets (36). Parenchymal macrophages are exclusively represented by microglia, which originate from embryonic yolk sac progenitors and sustain neuronal homeostasis via synaptic refinement and metabolic support. Non-parenchymal populations include perivascular macrophages (PVM) localized at the BBB interface and monocyte-derived macrophages recruited during neuroinflammatory states (37, 38). Under physiological conditions, microglia dynamically patrol the CNS through ramified processes, while PVM regulate BBB permeability and participate in clearance of interstitial waste. In AD pathogenesis, both subsets initially engage in Aβ phagocytosis via TREM2-dependent mechanisms, but persistent Aβ exposure drives their transition toward a pro-inflammatory phenotype, exacerbating tau pathology and synaptic loss (39) (Table 2).
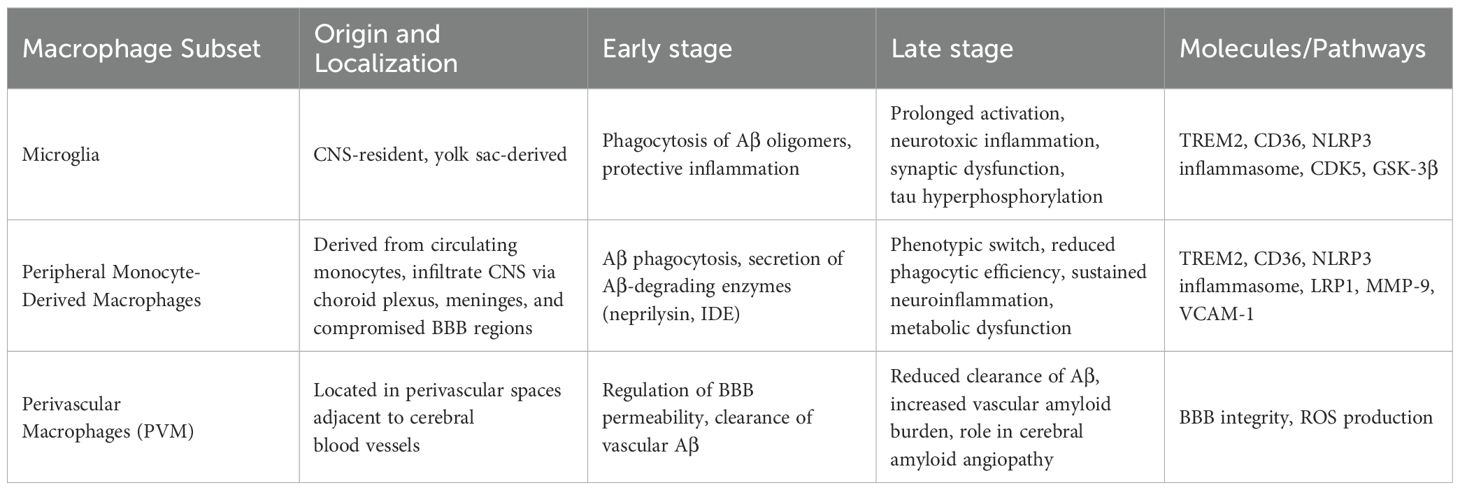
Table 2. Macrophage subsets and their functional dynamics in alzheimer’s disease.
4.2 Microglia in AD shifting from protection to pathology
Microglia are unique tissue-resident macrophages (TRMs) in the CNS, functioning through self-renewal. Under homeostatic conditions, microglia remain in a resting state, but upon detecting threats to the CNS, they rapidly transition to an activated state, migrating to sites of injury to phagocytose pathogens (40). Microglial activation can be beneficial or detrimental, depending on disease stage and microenvironment. In early/moderate AD, some subsets promote Aβ clearance and suppress neurotoxic inflammation (41), whereas in advanced stages, chronic Aβ exposure and cytokine dysregulation drive a pro-inflammatory phenotype that exacerbates synaptic damage and neurodegeneration (42, 43). Single-cell studies reveal distinct microglial subsets with transcriptional profiles linked to protective or harmful roles (44–46), underscoring their context-dependent functions in AD. These divergent findings highlight ongoing debates, emphasizing that timing, location, and cellular milieu determine whether microglia aid or worsen AD progression. Inflammatory factors play vital roles in diseases progression (47–50). Molecular pathways driving this phenotypic shift include persistent TLR4/TREM2 signaling, which sustains inflammation, and NLRP3 inflammasome activation, promoting IL-1β/IL-18 release (51). Dysregulated calcium homeostasis and elevated ROS activate kinases like GSK-3β, amplifying pro-inflammatory genes while reducing Aβ-clearing receptors (52), collectively shifting microglia toward a neurotoxic state.
Microglia also drive AD-associated neuroinflammation. Aβ-activated microglia release pro-inflammatory cytokines, including interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6), as well as ROS and nitric oxide (NO) (53). While initially protective, chronic activation perpetuates synaptic dysfunction, neuronal loss, and Aβ deposition, creating a pathological feedback loop (54). Aβ-induced inflammasome upregulation (e.g., NLRP3) further amplifies inflammation and neuronal damage. Additionally, tau can activate microglia, and excessive Aβ stimulation induces immune dysfunction, triggering inflammatory responses (55). Activated microglia release neurotoxic mediators, damaging healthy neurons and promoting hyperphosphorylated tau production. This cycle impairs synaptic integrity, reduces excitatory potentials, and drives neurodegeneration, culminating in cognitive decline, motor deficits, and behavioral abnormalities characteristic of AD (56).
4.3 Peripheral monocyte-derived macrophages in Aβ clearance
Non-parenchymal macrophages, often termed peripheral-derived macrophages, constitute a ubiquitous immune population differentiated from circulating monocytes that transmigrate across vascular endothelia. These cells exhibit remarkable plasticity, adapting to tissue-specific microenvironments through epigenetic reprogramming and metabolic shifts (e.g., oxidative phosphorylation to glycolysis). In AD, monocyte-derived macrophages infiltrate the CNS via three primary routes: choroid plexus stromal channels (regulated by CCL2-CCR2 chemotaxis), leptomeningeal vasculature (guided by VCAM-1 integrin signaling), and compromised BBB regions (mediated by MMP-9-dependent endothelial remodeling). During early AD, these macrophages exert neuroprotective effects through TREM2/CD36-dependent phagocytosis of soluble Aβ species and secretion of Aβ-degrading enzymes (neprilysin, IDE). However, sustained exposure to Aβ oligomers induces a phenotypic switch characterized by phagocytic receptor downregulation (e.g., LRP1), lysosomal acidification failure, and NLRP3 inflammasome-driven IL-1β/IL-18 hypersecretion. This functional decline coincides with Aβ plaque maturation and microglial priming, creating a feedforward loop of neuroinflammation. Notably, single-cell transcriptomics reveals that AD-associated macrophages adopt a disease-associated metabolic profile (DAMP), marked by lipid droplet accumulation and impaired mitophagy, which correlates with their diminished Aβ clearance capacity in late-stage patients (57). Postmortem studies further confirm that while CD14+CD16+ monocyte-derived macrophages accumulate near cerebral amyloid angiopathy sites, their Aβ-binding receptors (SCARA1, RAGE) are internalized and degraded, rendering them functionally inert—a critical factor driving clinical disease progression (58).
4.4 Perivascular macrophages and amyloid clearance
PVM are a unique type of brain macrophage, closely associated with the cerebral vascular system, located in the perivascular spaces of cerebral resistance arteries. PVM play roles in brain infection, immune activation, AD, and vascular-cognitive dysfunction through interactions with brain endothelial cells, circulating macrophages, and the production of reactive oxygen species. Studies in AD mouse models have shown that a reduction in PVM significantly increases vascular Aβ levels, and modulating PVM density can affect the clearance of amyloid from cerebral vessels. Conversely, stimulating PVM turnover can reduce cerebral amyloid angiopathy burden independently of microglia clearance, highlighting the importance of PVM in AD (59). In summary, existing evidence supports that PVM are a crucial component of the brain-resident immune system and play a protective role in the pathogenesis of AD.
4.5 Therapeutic strategies targeting macrophage plasticity
Progression of AD and targeting macrophage mechanisms may represent a promising therapeutic strategy for AD, aiming to shift cellular phenotypes from neurotoxic to neuroprotective states. Animal studies have demonstrated that osteopontin (OPN) can accelerate the recruitment of monocyte-derived macrophages to the brains of AD mice, promoting macrophage polarization toward an anti-inflammatory, highly phagocytic phenotype. This process inhibits Aβ production and aggregation while enhancing Aβ clearance, thereby potentially preventing AD progression (60). Additionally, blocking the Smad2/3 signaling pathway has been shown to effectively reduce Aβ levels in peripheral organs and circulation, enhance Aβ efflux from the brain, and improve Aβ clearance by peripheral macrophages. These effects collectively reduce Aβ deposition, neuroinflammation, and cognitive deficits in the brain (61). These findings highlight the therapeutic potential of modulating macrophage function and signaling pathways to mitigate AD pathology. In this context, targeting MCP-1 represents a particularly compelling avenue for therapeutic intervention (62). Experimental approaches to reduce MCP-1 or block its receptor CCR2 have shown promise in dampening neuroinflammation and modulating macrophage phenotypes (63). By inhibiting the MCP-1/CCR2 axis, it may be possible to decrease the infiltration of pro-inflammatory monocytes into the brain and simultaneously promote a reparative or Aβ-clearing macrophage phenotype (63, 64), these findings underscore the potential for translating MCP-1 modulation into tangible AD treatments.
5 Conclusion
Monocytes and macrophages play a crucial role in the pathogenesis of AD. MCP-1 regulates the migration and infiltration of monocytes, participating in neuroinflammation, Aβ deposition, and tau pathology. Macrophages, particularly microglia, delay disease progression by phagocytosing Aβ in the early stages of AD, but their persistent activation under chronic inflammatory conditions leads to Aβ accumulation and exacerbation of neuroinflammation. Future research should further explore the specific molecular mechanisms of MCP-1 and macrophages in AD, particularly their interactions in neuroinflammation, Aβ metabolism, and tau pathology. Based on the regulatory mechanisms of MCP-1 and macrophages, the development of new therapeutic strategies and early diagnostic biomarkers holds promise for advancing the prevention and treatment of AD. Recent advances in single-cell RNA sequencing, spatial transcriptomics, and advanced imaging enable high-resolution spatiotemporal analysis of MCP-1 and monocyte-derived cells in Alzheimer’s disease, revealing their dynamic roles in neuroinflammation and offering insights for stage-specific interventions. These technologies map immune cell heterogeneity, migration, and activation, linking molecular profiles to AD pathology like Aβ plaques and tau deposits (13). In summary, monocytes and macrophages occupy a central position in the complex pathological network of AD, and future research should continue to explore their multifaceted roles, with the aim of developing more effective diagnostic and therapeutic strategies, offering new hope for AD patients.
Author contributions
WN: Writing – original draft. YY: Writing – original draft. JH: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study is funded by the Tianjin Top Scientists Program (25JRRCRC00010), and the Research and Development Fund of the China Academy of Chinese Medical Sciences (2023021), and the Research and Development Fund of the Institute of Basic Theory for Chinese Medicine, China Academy of Chinese Medical Sciences (KJX202404). State Key Laboratory of Pathogenesis, Prevention and Treatment of High Incidence Diseases in Central Asia Fund (SKL-HIDCA-2024- BC7), and Macrophage migration inhibitory factor (MIF) and immune thrombocytopenia pathogenesis, development and hormone resistance mechanism (2023YFY-QKMS-01).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hyman BT, Holtzman DM. Apolipoprotein E levels and Alzheimer risk. Ann Neurol. (2015) 77:204–5. doi: 10.1002/ana.24355
2. Passamonti L, Tsvetanov KA, Jones PS, Bevan-Jones WR, Arnold R, Borchert RJ, et al. Neuroinflammation and functional connectivity in Alzheimer’s disease: interactive influences on cognitive performance. J Neurosci. (2019) 39:7218–26. doi: 10.1523/JNEUROSCI.2574-18.2019
3. Zhang G, Wang Z, Hu H, Zhao M, Sun L. Microglia in Alzheimer’s disease: A target for therapeutic intervention. Front Cell Neurosci. (2021) 15:749587. doi: 10.3389/fncel.2021.749587
4. Cheng M, Yuan C, Ju Y, Liu Y, Shi B, Yang Y, et al. Quercetin attenuates oxidative stress and apoptosis in brain tissue of APP/PS1 double transgenic AD mice by regulating Keap1/Nrf2/HO-1 pathway to improve cognitive impairment. Behav Neurol. (2024) 2024:5698119. doi: 10.1155/2024/5698119
5. Rajesh Y, Kanneganti TD. Innate immune cell death in neuroinflammation and Alzheimer’s disease. Cells. (2022) 11:1885. doi: 10.3390/cells11121885
6. Mammana S, Fagone P, Cavalli E, Basile MS, Petralia MC, Nicoletti F, et al. The role of macrophages in neuroinflammatory and neurodegenerative pathways of Alzheimer’s disease, amyotrophic lateral sclerosis, and multiple sclerosis: pathogenetic cellular effectors and potential therapeutic targets. Int J Mol Sci. (2018) 19:831. doi: 10.3390/ijms19030831
7. Gomez-Gutierrez R, Ghosh U, Yau WM, Gamez N, Do K, Kramm C, et al. Two structurally defined Aβ polymorphs promote different pathological changes in susceptible mice. EMBO Rep. (2023) 24:e57003. doi: 10.15252/embr.202357003
8. Marin A, Budson AE. Recent advances in understanding Alzheimer’s Disease: diagnosis and management strategies. Fac Rev. (2023) 12:24. doi: 10.12703/r/12-24
9. Ao C, Li C, Chen J, Tan J, Zeng L. The role of Cdk5 in neurological disorders. Front Cell Neurosci. (2022) 16. doi: 10.3389/fncel.2022.951202
10. Zhang Y, Zhao Y, Ao X, Yu W, Zhang L, Wang Y, et al. The role of non-coding RNAs in Alzheimer’s disease: from regulated mechanism to therapeutic targets and diagnostic biomarkers. Front Aging Neurosci. (2021) 13:654978. doi: 10.3389/fnagi.2021.654978
11. Murray TE, Wenzel TJ, Simtchouk S, Greuel BK, Gibon J, Klegeris A. Extracellular cardiolipin modulates select immune functions of astrocytes in toll-like receptor (TLR) 4-dependent manner. Mediators Inflamm. (2022) 2022:9946439. doi: 10.1155/2022/9946439
12. Pozzi S, Scomparin A, Ben-Shushan D, Yeini E, Ofek P, Nahmad AD, et al. MCP-1/CCR2 axis inhibition sensitizes the brain microenvironment against melanoma brain metastasis progression. JCI Insight. (2022) 7:e154804. doi: 10.1172/jci.insight.154804
13. He Y, Lu W, Zhou X, Mu J, Shen W. Unraveling Alzheimer’s disease: insights from single-cell sequencing and spatial transcriptomic. Front Neurol. (2024) 15:1515981. doi: 10.3389/fneur.2024.1515981
14. Miao J, Ma H, Yang Y, Liao Y, Lin C, Zheng J, et al. Microglia in Alzheimer’s disease: pathogenesis, mechanisms, and therapeutic potentials. Front Aging Neurosci. (2023) 15:1201982. doi: 10.3389/fnagi.2023.1201982
15. Ball BK, Kuhn MK, Fleeman Bechtel RM, Proctor EA, Brubaker DK. Differential responses of primary neuron-secreted MCP-1 and IL-9 to type 2 diabetes and Alzheimer’s disease-associated metabolites. Sci Rep. (2024) 14:12743. doi: 10.1038/s41598-024-62155-3
16. Yang G, Hu Y, Qin X, Sun J, Miao Z, Wang L, et al. Micheliolide attenuates neuroinflammation to improve cognitive impairment of Alzheimer’s disease by inhibiting NF-κB and PI3K/Akt signaling pathways. Heliyon. (2023) 9:e17848. doi: 10.1016/j.heliyon.2023.e17848
17. Lin X, Zhao Q, Fu B, Xiong Y, Zhang S, Xu S, et al. ISOC1 modulates inflammatory responses in macrophages through the AKT1/PEX11B/peroxisome pathway. Molecules. (2022) 27:5896. doi: 10.3390/molecules27185896
18. Ganz T, Fainstein N, Ben-Hur T. When the infectious environment meets the AD brain. Mol Neurodegener. (2022) 17:53. doi: 10.1186/s13024-022-00559-3
19. Sun X, Chen WD, Wang YD. β-Amyloid: the key peptide in the pathogenesis of Alzheimer’s disease. Front Pharmacol. (2015) 6:221. doi: 10.3389/fphar.2015.00221
20. Lee JW, Lee IH, Iimura T, Kong SW. Two macrophages, osteoclasts and microglia: from development to pleiotropy. Bone Res. (2021) 9:11. doi: 10.1038/s41413-020-00134-w
21. van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci. (2020) 21:21–35. doi: 10.1038/s41583-019-0240-3
22. Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun. (2014) 2:135. doi: 10.1186/s40478-014-0135-5
23. Chen L, Qin Q, Huang P, Cao F, Yin M, Xie Y, et al. Chronic pain accelerates cognitive impairment by reducing hippocampal neurogenesis may via CCL2/CCR2 signaling in APP/PS1 mice. Brain Res Bull. (2023) 205:110801. doi: 10.1016/j.brainresbull.2023.110801
24. Conductier G, Blondeau N, Guyon A, Nahon JL, Rovère C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J Neuroimmunol. (2010) 224:93–100. doi: 10.1016/j.jneuroim.2010.05.010
25. Wang T, Yao Y, Han C, Li T, Du W, Xue J, et al. MCP-1 levels in astrocyte-derived exosomes are changed in preclinical stage of Alzheimer’s disease. Front Neurol. (2023) 14:1119298. doi: 10.3389/fneur.2023.1119298
26. Wojcieszak J, Kuczyńska K, Zawilska JB. Role of chemokines in the development and progression of Alzheimer’s disease. J Mol Neurosci. (2022) 72:1929–51. doi: 10.1007/s12031-022-02047-1
27. Xu L, Dong Q, Xu L, Zou W, Li H. The MCP-1 A-2518G polymorphism increases the risk of Alzheimer’s disease: A case-control study. Neurosci Lett. (2021) 749:135710. doi: 10.1016/j.neulet.2021.135710
28. Vaz M, Domingues C, Trindade D, Barra C, Oliveira JM, Rosa IM, et al. IL-8 and MCP-1 impact on tau phosphorylation and phosphatase activity. Curr Alzheimer Res. (2020) 17:985–1000. doi: 10.2174/1567205017666201130091129
29. Song WM, Colonna M. The identity and function of microglia in neurodegeneration. Nat Immunol. (2018) 19:1048–58. doi: 10.1038/s41590-018-0212-1
30. Dias D, Socodato R. Beyond amyloid and tau: the critical role of microglia in Alzheimer’s disease therapeutics. Biomedicines. (2025) 13:279. doi: 10.3390/biomedicines13020279
31. Zhang Q, Yang G, Luo Y, Jiang L, Chi H, Tian G. Neuroinflammation in Alzheimer’s disease: insights from peripheral immune cells. Immun Ageing. (2024) 21:38. doi: 10.1186/s12979-024-00445-0
32. Fruhwurth S, Zetterberg H, Paludan SR. Microglia and amyloid plaque formation in Alzheimer’s disease - Evidence, possible mechanisms, and future challenges. J Neuroimmunol. (2024) 390:578342. doi: 10.1016/j.jneuroim.2024.578342
33. Gate D, Rezai-Zadeh K, Jodry D, Rentsendorj A, Town T. Macrophages in Alzheimer’s disease: the blood-borne identity. J Neural Transm (Vienna). (2010) 117:961–70. doi: 10.1007/s00702-010-0422-7
34. Sun Z, Zhang X, So KF, Jiang W, Chiu K. Targeting microglia in Alzheimer’s disease: pathogenesis and potential therapeutic strategies. Biomolecules. (2024) 14:883. doi: 10.3390/biom14070833
35. Dyck PJ, Windebank AJ, Low PA, Baumann WJ. Blood nerve barrier in rat and cellular mechanisms of lead-induced segmental demyelination. J Neuropathol Exp Neurol. (1980) 39:700–9. doi: 10.1097/00005072-198011000-00009
36. Sun R, Jiang H. Border-associated macrophages in the central nervous system. J Neuroinflamm. (2024) 21:67. doi: 10.1186/s12974-024-03059-x
37. Guedes JR, Ferreira PA, Costa JM, Cardoso AL, Peça J. Microglia-dependent remodeling of neuronal circuits. J Neurochem. (2022) 163:74–93. doi: 10.1111/jnc.v163.2
38. Zheng L, Guo Y, Zhai X, Zhang Y, Chen W, Zhu Z, et al. Perivascular macrophages in the CNS: From health to neurovascular diseases. CNS Neurosci Ther. (2022) 28:1908–20. doi: 10.1111/cns.13954
39. Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci. (2011) 14:1227–35. doi: 10.1038/nn.2923
40. Wang J, Gu BJ, Masters CL, Wa YJ. A systemic view of Alzheimer disease - insights from amyloid-β metabolism beyond the brain. Nat Rev Neurol. (2017) 13:612–23. doi: 10.1038/nrneurol.2017.111
41. Nizami S, Hall-Roberts H, Warrier S, Cowley SA, Di Daniel E. Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br J Pharmacol. (2019) 176:3515–32. doi: 10.1111/bph.v176.18
42. Solito E, Sastre M. Microglia function in Alzheimer’s disease. Front Pharmacol. (2012) 3:14. doi: 10.3389/fphar.2012.00014
43. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y). (2018) 4:575–90. doi: 10.1016/j.trci.2018.06.014
44. Sun N, Victor MB, Park YP, Xiong X, Scannail AN, Leary N, et al. Human microglial state dynamics in Alzheimer’s disease progression. Cell. (2023) 186:4386–4403 e29. doi: 10.1016/j.cell.2023.08.037
45. Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun. (2020) 11:6129. doi: 10.1038/s41467-020-19737-2
46. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. (2017) 169:1276–1290 e17. doi: 10.1016/j.cell.2017.05.018
47. Zhai X, Zhang H, Xia Z, Liu M, Du G, Jiang Z, et al. Oxytocin alleviates liver fibrosis via hepatic macrophages. JHEP Rep. (2024) 6:101032. doi: 10.1016/j.jhepr.2024.101032
48. Xiao J, Lin H, Liu B, Xia Z, Zhang J, Jin J, et al. Decreased S1P and SPHK2 are involved in pancreatic acinar cell injury. Biomark Med. (2019) 13:627–37. doi: 10.2217/bmm-2018-0404
49. Xiao J, Huang K, Lin H, Xia Z, Zhang J, Li D, et al. Mogroside II(E) inhibits digestive enzymes via suppression of interleukin 9/interleukin 9 receptor signalling in acute pancreatitis. Front Pharmacol. (2020) 11:859. doi: 10.3389/fphar.2020.00859
50. Zhang H, Xia T, Xia Z, Zhou H, Li Z, Wang W, et al. KIF18A inactivates hepatic stellate cells and alleviates liver fibrosis through the TTC3/Akt/mTOR pathway. Cell Mol Life Sci. (2024) 81:96. doi: 10.1007/s00018-024-05114-5
51. Li T, Lu L, Pember E, Li X, Zhang B, Zhu Z. New insights into neuroinflammation involved in pathogenic mechanism of Alzheimer’s disease and its potential for therapeutic intervention. Cells. (2022) 11:1925. doi: 10.3390/cells11121925
52. LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci. (2002) 3:862–72. doi: 10.1038/nrn960
53. Muzio L, Viotti A, Martino G. Microglia in neuroinflammation and neurodegeneration: from understanding to therapy. Front Neurosci. (2021) 15. doi: 10.3389/fnins.2021.742065
54. Cai Y, Liu J, Wang B, Sun M, Yang H. Microglia in the neuroinflammatory pathogenesis of Alzheimer’s disease and related therapeutic targets. Front Immunol. (2022) 13:856376. doi: 10.3389/fimmu.2022.856376
55. Piccioni G, Mango D, Saidi A, Corbo M, Nisticò R. Targeting microglia-synapse interactions in Alzheimer’s disease. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22052342
56. Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, et al. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron. (2016) 90:724–39. doi: 10.1016/j.neuron.2016.05.003
57. Guo H, Zhao Z, Zhang R, Chen P, Zhang X, Cheng F, et al. Monocytes in the peripheral clearance of amyloid-β and Alzheimer’s disease. J Alzheimers Dis. (2019) 68:1391–400. doi: 10.3233/JAD-181177
58. Zhou J, Benoit M, Sharoar MG. Recent advances in pre-clinical diagnosis of Alzheimer’s disease. Metab Brain Dis. (2022) 37:1703–25. doi: 10.1007/s11011-021-00733-4
59. Faraco G, Park L, Anrather J, Iadecola C. Brain perivascular macrophages: characterization and functional roles in health and disease. J Mol Med (Berl). (2017) 95:1143–52. doi: 10.1007/s00109-017-1573-x
60. Rentsendorj A, Sheyn J, Fuchs DT, Daley D, Salumbides BC, Schubloom HE, et al. A novel role for osteopontin in macrophage-mediated amyloid-β clearance in Alzheimer’s models. Brain Behav Immun. (2018) 67:163–80. doi: 10.1016/j.bbi.2017.08.019
61. Xu L, Pan CL, Wu XH, Song JJ, Meng P, Li L, et al. Inhibition of Smad3 in macrophages promotes Aβ efflux from the brain and thereby ameliorates Alzheimer’s pathology. Brain Behav Immun. (2021) 95:154–67. doi: 10.1016/j.bbi.2021.03.013
62. Huang J, Wang Y, Stein TD, Ang TFA, Zhu Y, Tao Q, et al. The impact of blood MCP-1 levels on Alzheimer’s disease with genetic variation of UNC5C and NAV3 loci. Res Sq. (2023). doi: 10.21203/rs.3.rs-3376348/v1
63. Arfaei R, Mikaeili N, Daj F, Boroumand A, Kheyri A, Yaraghi P, et al. Decoding the role of the CCL2/CCR2 axis in Alzheimer’s disease and innovating therapeutic approaches: Keeping All options open. Int Immunopharmacol. (2024) 135:112328. doi: 10.1016/j.intimp.2024.112328
Keywords: Alzheimer’s disease, monocytes, macrophages, neuroinflammation, β-Amyloid, MCP-1
Citation: Nie W, Yue Y and Hu J (2025) The role of monocytes and macrophages in the progression of Alzheimer’s disease. Front. Immunol. 16:1590909. doi: 10.3389/fimmu.2025.1590909
Received: 10 March 2025; Accepted: 07 April 2025;
Published: 29 April 2025.
Edited by:
Yongfu Shao, Ningbo University, ChinaReviewed by:
Wei Xu, The First Affiliated Hospital of Soochow University, ChinaCopyright © 2025 Nie, Yue and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingqing Hu, aHVqcUBqaWljbS5vcmcuY24=
†These authors have contributed equally to this work