 Jiayan Hu1,2†
Jiayan Hu1,2† Wenting Wang3†Muyuan Wang1†Chunye Wu2Yao Jiao1Yitong Li1
Wenting Wang3†Muyuan Wang1†Chunye Wu2Yao Jiao1Yitong Li1 Wenji Zhang1Chengtao Liang1Zhengdao Lin1Yitong Yu1Junxiang Li1*
Wenji Zhang1Chengtao Liang1Zhengdao Lin1Yitong Yu1Junxiang Li1* Tangyou Mao1*
Tangyou Mao1*- 1Dongfang Hospital, Beijing University of Chinese Medicine, Beijing, China
- 2Beijing University of Chinese Medicine, Beijing, China
- 3Beitaipingzhuang Community Health Service Center, Beijing, China
Tissue-resident memory T (TRM) cells are a type of tissue-restricted memory T cells with terminal differentiation and a memory function. They exist in mucosal tissues for a long period. In the absence of disease, TRM cells promote essential inflammation, which reinforces the intestinal barrier and prevents bacterial translocation. However, in inflammatory or autoimmune environments, TRM cells are hyperactivated. This heightened activity causes the host to release excessive pro-inflammatory cytokines, resulting in local immune imbalances and damage to the barrier, ultimately leading to tissue lesions. Numbers of studies have shown that TRM cells play a crucial role in the development and progression of inflammatory bowel disease (IBD), suggesting that targeted regulation of TRM cells homeostasis may be an important strategy for treating IBD. Here, we compiled the existing understanding of the role of TRM cells in IBD, with particular emphasis on the associated mechanisms and approaches for targeting TRM cells in IBD treatment. This review will serve as a foundation for a better understanding of IBD development and enhancing the effectiveness of clinical treatments for IBD.
1 Introduction
Inflammatory bowel disease (IBD), which includes Crohn’s disease (CD) and ulcerative colitis (UC), is a persistent inflammatory condition affecting the gastrointestinal tract, which is characterized by recurrent inflammation in local areas of the intestinal tract (1). In view of the chronic, early-onset and relatively low mortality of IBD, the prevalence of IBD has increased significantly over time with the increase of population aging. Studies utilizing the global burden of disease database indicate that the total number of cases is expected to rise from 298,412 in 1990-1994 to 490,887 by 2035-2039, with incidence rates consistently higher in men compared to women (2). The primary symptoms of IBD include abdominal pain, diarrhea, and hematochezia. While conventional treatment generally manages these symptoms, a complete cure remains elusive. A considerable number of patients may eventually necessitate surgical procedures and could encounter various complications, including extraintestinal manifestations, as well as disease-specific issues such as stenosis, fistulas, and abscesses (3). This has brought great pain to patients and affected their overall quality of life. Hence, it is crucial to investigate the underlying causes of IBD and create innovative and secure approaches for treatment.
The development of IBD is intricate and involves multiple factors, including genetic susceptibility, environmental factors, interactions between the microbiota and host immune system, and disrupted mucosal immunity (4). The intestinal immune system is a key factor that induces and maintains diseases (5–7). Despite genetic and environmental variations among patient populations, a dysfunctional immune response may ultimately lead to a common T cell-mediated inflammatory cascade that directly drives the development of IBD (8, 9). However, the mechanisms underlying abnormal intestinal immunity have not yet been fully elucidated. Various mucosal immune cells, including T helper cells (10), regulatory T cells (Treg) (11), innate lymphocytes (12–14), and macrophages (15), play a role in IBD development. However, the lifespan of most immune cells is relatively short, making it challenging to account for persistent immune abnormalities observed in IBD. Tissue-resident memory T (TRM) cells are generated from effector T cells and possess several key characteristics, including the abilities to continuously colonize tissues, secrete inflammatory cytokines, and self-proliferate, and a long lifespan. This aligns with the pathological features commonly observed in IBD (16). Recently, the important role of TRM cells in IBD has been consistently validated (17). TRM cells are terminally differentiated tissue-restricted memory T cells have a memory function and exist in mucosal tissues for a long period (18–20). TRM cells are distinguished by their surface markers, including CD103, CD69, and CD49a, and absence of L-selectin (CD62L) and chemokine receptor 7 (CCR7). These features ensure the stable and long-term presence of TRM cells within tissues. Their ability to contain and swiftly eliminate invading pathogens at the entry site is advantageous for the host, helping prevent tissue damage and systemic spread (21). Based on their phenotype and function, TRM are mainly divided into CD8+ and CD4+TRM cells. CD8+TRM cells are usually located in the epithelial layer of the barrier tissue. Functioning as sentinels, they initiate antigen-specific immune responses upon reinfection. CD4+TRM cells are typically situated beneath the epithelial layer, including within the basement membrane, and aggregate in lymphatic structures to enhance their interaction with antigen-presenting cells during reinfection (22). TRM cells mediate a protective response against microbial infection (23). Even in a quiescent state, TRM cells are capable of swiftly generating inflammatory mediators and stimulated surrounding tissues to upregulate defense mechanisms when stimulated (24, 25). Moreover, TRM cells can directly eliminate infected cells by releasing cytotoxic molecules, such as perforin and granzyme (26). Some pathogen-specific CD4+and CD8+TRM cells have powerful cytokine versatility, and produce high levels of interferon-γ (IFN-γ), interleukin-2 (IL-2), and tumor necrosis factor-α (TNF-α) (27, 28). TRM cells are enriched on mucosal surfaces with a heavy pathogen burden, such as on the skin, lungs, and gastrointestinal tract (29, 30). Nonetheless, TRM cells may undergo excessive activation in inflammatory or autoimmune contexts, leading to a ‘provocative state’ and resulting in pathogenic TRM cells. The gut serves as a barrier organ with the greatest exposure to antigens, therefore, TRM cells may play a vital role in facilitating localized immune responses specific to antigens in the gut (24), and may be involved in the development of IBD (31, 32). Furthermore, Zundler et al. (33) demonstrated that the presence of CD4+CD69+CD103+TRM cells could predict disease onset and depletion of these TRM cells limited colitis activity. While CD69 and CD103 are commonly used to identify TRM cells, it is important to note that TRM populations are heterogeneous, and some subsets may not express these markers (34). However, in the context of IBD, most studies have focused on CD69+ and/or CD103+ TRM subsets due to their prominence in mucosal tissues and established roles in immune surveillance and inflammation.
Here, we review the latest findings regarding the role of TRM cells in the pathogenesis of IBD. We highlight that the imbalance between pro-inflammatory and regulatory TRM cells, along with their intricate interactions within the immune network, constitutes a central pathological process. Therefore, we propose several therapeutic strategies aimed at targeting TRM cells, including the regulation of their proliferation, activation, homing, and apoptosis, while simultaneously enhancing the function of Treg cells. Finally, we highlight the essential avenues for future research to further progress the field of IBD treatment.
2 Relationship between TRM cells and IBD
2.1 Insights from clinical studies
2.1.1 CD4+TRM cells
Many clinical studies have confirmed that CD4+TRM cells may be involved in the pathogenesis of IBD. Zundler et al. (33) observed an increase in the number of CD69+CD103+TRM cells in the colons of patients with UC and CD. Importantly, they demonstrated that median flare-free survival in patients with high CD4+TRM cell frequency was significantly shorter than in patients with low CD4+TRM cell frequency (hazard ratio 3.39, 95% confidence interval 1.07-10.7). However, the exact mechanism still needs to be further elucidated. One possible mechanism could be related to the cytokine - mediated inflammatory cascade. CD4+CD69+T cells in the intestinal lamina propria of IBD patients produced significant amounts of pro-inflammatory cytokines, including IFN-γ, IL-13, IL-17A, and TNF-α. These cytokines play a role in tissue homeostasis and the innate immune response and have a profound impact on the intestinal microenvironment (35). For instance, IL-17A recruit neutrophils and promote the production of other pro-inflammatory mediators (36), which may disrupt the normal tissue homeostasis and contribute to the development of flares. Ohman L et al. (37) found that serum IL-17A levels of treatment-naive patients with UC reflect clinical disease severity at the onset of the disease and also predicted the disease course over the following 3 years. Consistent with this, Bishu et al. (32) analyzed the expression of related inflammatory factors in colon samples from patients with CD. They found that the levels of IFN-γ and IL-17A in CD4+TRM were significantly higher in CD patients compared to the control group, and CD4+TRM were identified as the primary subset of mucosal T cells producing TNF-α in these patients. High levels of CD103 in CD4+T cells correlate with increased production of pro-inflammatory cytokines and decreased expression of regulatory markers in individuals with UC (38). Yokoi et al. (39) found that CD103+CD4+TRM exhibiting an inflammatory phenotype were significantly increased in the intestines of patients with CD, but not in those with UC. They identified distinct CD4+TRM subsets with varying functions and transcriptional profiles in inflamed intestinal mucosa. These pathogenic CD4+TRM subsets, characterized by their unique properties, are specifically enriched in CD and play a crucial role in coordinating the local inflammatory response.
2.1.2 CD8+TRM cells
Bottomis et al (31). revealed that CD103+CD8+TRM cells in individuals with CD exhibited elevated expression of Th17-related genes, including IL-22 and IL-26, as well as genes encoding granzyme K, compared with the control group. The increased presence of Th17 TRM cells throughout the small intestine in CD contributes to disease pathogenesis by inducing IFN-γ and subsequently promoting chemokine production in myeloid cells (40). Therefore, TRM cells express more pro-inflammatory cytokines in patients with IBD, promoting the progress of IBD. In contrast, a recent study using single-cell RNA sequencing found that intestinal CD8+TRM cells in UC undergo considerable transformation into an inflammatory state, which is partially linked to the elevated expression of the transcription factor Eomes. Eomes enhances the expression of various downstream target genes, including cytokines (IFN-γ), cytolytic effectors (Gzma) (41). This leads to increased secretion of pro-inflammatory cytokines, contributing to the exacerbation of local inflammation. Eomes is highly homologous members of the T-box family of transcription factors and are highly expressed by activated CD8+T cells (42). During murine skin TRM cell differentiation following microbial infection, Eomes is initially upregulated; however, their expression decreases afterward to facilitate TGF-β signaling and support ongoing TRM cell development (43). In summary, the available clinical evidence suggests an important association between TRM cells and IBD pathogenesis, though their exact causal role requires further investigation. Table 1 presents an overview of clinical research on TRM cells and IBD.
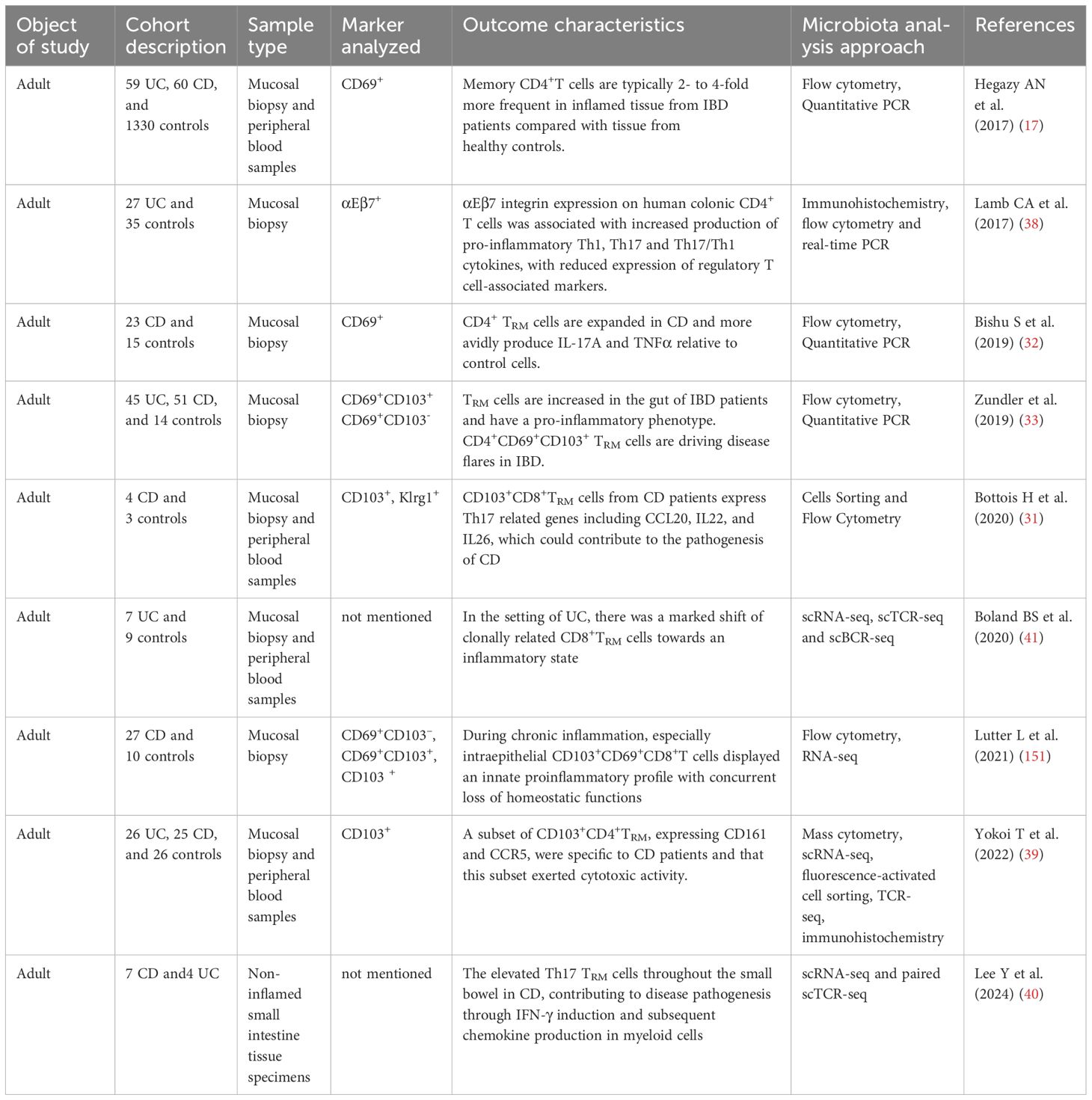
Table 1. Overview of clinical studies of TRM cells related to inflammatory bowel disease.
2.2 Insights from animal studies
2.2.1 CD4+TRM cells
Consistent with the findings of human clinical studies, preclinical studies have demonstrated a correlation between the presence of TRM cells and disease severity in animals with experimental colitis. DSS-induced colitis mice exhibited a notably increased number of CD4+ TRM cells in vivo compared with that in control mice, and these levels markedly decreased after treatment (44). Zundler et al. (33) found that the knockout of tissue-specific transcription factors like Hobit and Blimp-1, two essential transcription factors for TRM cells, inhibited the onset of colitis in several experimental mouse models (T cell transfer colitis, DSS-induced colitis, TNBS-induced colitis). Hobit (gene Zfp683), a homolog of Blimp1, suppresses genes involved in tissue egress (e.g., S1PR1, Tcf7, CCR7), thereby promoting retention in the small intestinal epithelium (45). Blimp1 itself downregulates Krüppel-like factor 2 (KLF2) and S1PR1 (46), which is critical for TRM cell retention. Hobit and Blimp-1 are pivotal not only in regulating tissue retention, but also in controlling the cytotoxic functions of TRM cells. In the absence of Blimp-1, the formation of TRM cells in gut shows defects in the production of granzyme B (47).Mice with a dual genetic deletion of Hobit and Blimp-1 in CD4+ T cells showed a decrease in the production of pro-inflammatory cytokines, including IFN-γ, IL-13, and IL-17A, and displayed a reduced influx of granulocytes and macrophages. Furthermore, depletion of TRM cells effectively prevented the onset of experimental colitis. These findings confirm that TRM cells play a crucial role in driving intestinal inflammation (31). Another study found that the number of CD4+TRM cells in DSS-induced colitis mice was significantly higher than that in normal mice, and the number of CD4+CD69+CD103-TRM cells was positively correlated with the disease activity index (48). Knockdown of TIGIT, a regulatory gene of CD4+TRM cells, reduced the number of CD4+CD69+CD103-TRM cells in the colon tissue and reduced the level of IL-17A. Furthermore, the transfer of intestinal CD4+TRM cells into RAG2−/− mice also induces experimental colitis (49). This suggests that CD4+TRM in the gut disseminate and cause local inflammation in UC. CD4+TRM cells were present in C. rodentium-infected mice, showing significant enrichment in the intestines and serving as the primary source of IL-17A (50). Paired immunoglobulin-like receptor B regulates the survival of CD4+IL-17A+T cells. Loss-of-function inhibits the differentiation and growth of CD4+IL-17A+TRM cells, thereby preventing the development of CD4+T cell-dependent colitis in a mouse model of T cell metastasis (35). Another study conducted in mice demonstrated that the insulin receptor present on gut T cells contributes to the differentiation of TRM cells, particularly CD4+TRM cells, by affecting the action of the Enhancer of Zeste Homolog 2 (EZH2). This process ultimately worsens intestinal inflammation by increasing the production of cytokines, such as TNF-α and IL-17 (51). Table 2 presents an overview of the animal studies related to TRM cells and IBD.
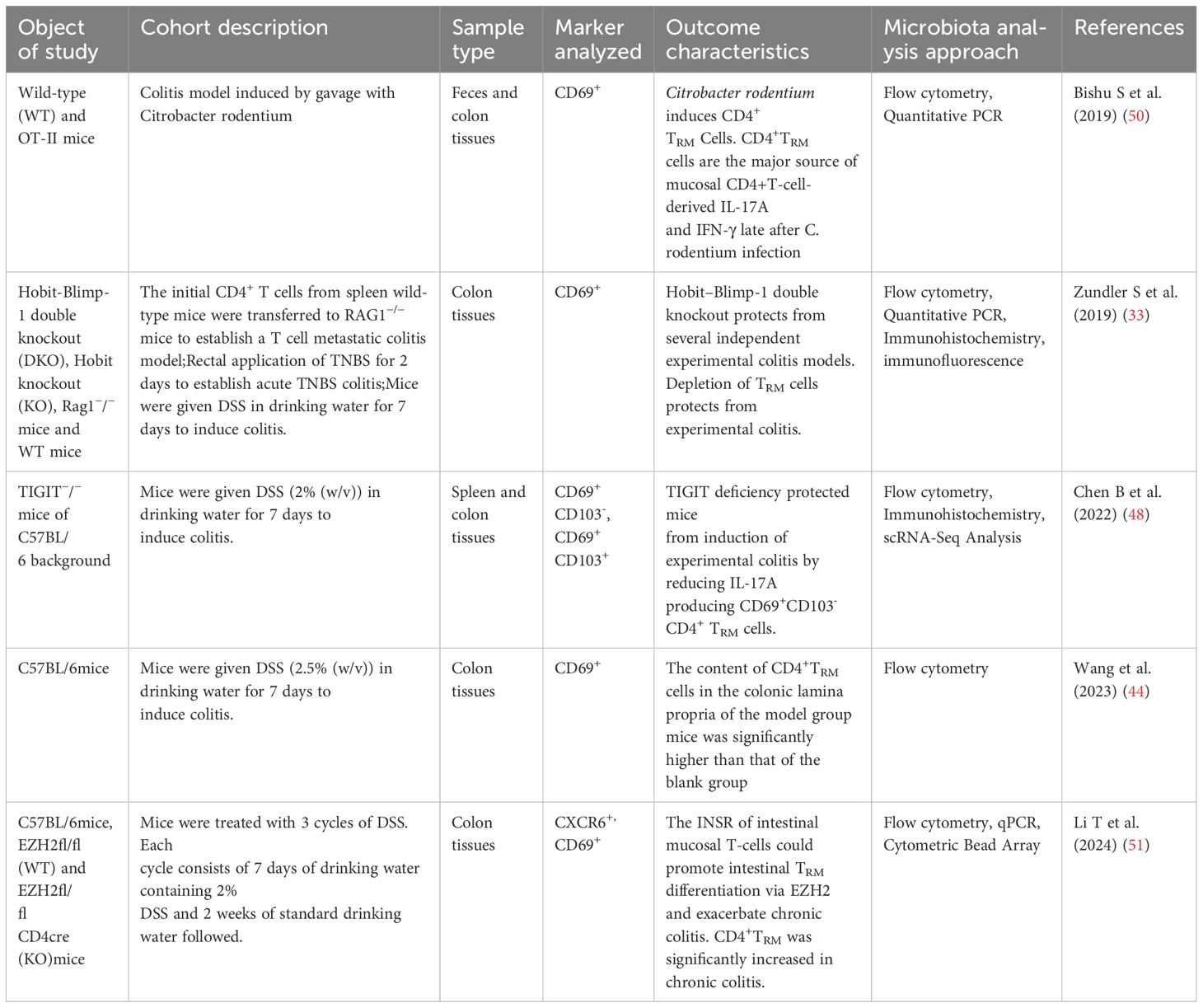
Table 2. Overview of animal studies of TRM cells related to inflammatory bowel disease.
2.3 Insights from single-cell and spatial transcriptomic
Recent advancements in single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics have provided unprecedented insights into the heterogeneity and compartmentalization of TRM cells in IBD. For instance, Boland et al. (41) identified CD8+ TRM cells localized to epithelial niches in UC patients, where they exhibit clonal expansion and Eomes-driven proinflammatory signatures, directly contributing to epithelial damage via IFN-γ and granzyme release. These cells were found to reside within specific epithelial niches, suggesting that they may contribute to local barrier disruption and tissue remodeling, which are central elements of IBD pathology. Similarly, Lutter et al. (52) demonstrated that intestinal CD4 T cells, including TRM cells, exhibit distinct transcriptional profiles based on their anatomical location (epithelium vs. lamina propria), with significant changes observed in the epithelium during inflammation. This compartmentalization underscores the importance of understanding how TRM cells interact with their local microenvironment to drive disease progression. In another study, Lee et al. (40) characterized Th17 TRM cells in the non-inflamed intestinal tissue of CD patients using scRNA-seq and paired T cell receptor sequencing. They found that Th17 TRM cells in CD exhibited a heightened expression of tissue-residency markers (ITGAE, ITGA1, and CXCR6) along with elevated levels of IL-17A, IL-22, CCR6, and CCL20. These cells also showed increased IFN-γ-related signatures, potentially linked to STAT1 activation, which could induce chemokines in myeloid cells and contribute to local inflammation. This finding highlights the role of Th17 TRM cells in maintaining chronic inflammation in CD and underscores the need for targeted therapeutic strategies. These studies collectively emphasize the importance of integrating single-cell and spatial transcriptomic data to elucidate the mechanisms by which TRM cells contribute to IBD pathogenesis. Understanding the specific transcriptional profiles and spatial distribution of TRM cells can provide critical insights into their role in local barrier disruption and tissue remodeling, potentially leading to the development of more effective targeted therapies.
There are extensive connections between TRM cells and IBD. However, TRM cells exhibit inconsistent behaviors in different locations of the intestine and under different conditions (active or remission phases of the disease). A comparative study of different anatomical locations of CD8+TRM cells in gut by John T. Chang’s research team has more comprehensively revealed the phenotypic and functional heterogeneity of CD8 +TRM cells in four intestinal compartments (53). Quantitatively, the order of intestinal CD8+TRM cell numbers was found to be intestinal epithelial cells(siIEL) > Lamina propria cells of small intestine(siLP) > colonic lamina propria (cLP) > colonic epithelial cells (cIEL) by both microscopic quantification and flow analysis. In terms of phenotypic heterogeneity, the majority of siIEL CD8+TRM cells express both CD69 and CD103, CD69 CD103 subset account differently in siLP, cIEL and cLP CD8+TRM cells. All in all, the heterogeneity of intestinal TRM cells still needs to be further elucidated.
3 The role of TRM cells in IBD
While TRM cells is primarily recognized for its protective role, pathogenic characteristics of TRM cells have also been linked to a range of diseases, such as autoimmune conditions like vitiligo, psoriasis, and cutaneous lupus (54).The gut is continually exposed to external antigens, including microorganisms and dietary components. While the specific antigens associated with IBD remain largely unidentified, these antigens may trigger localized, recurrent inflammation (55). Therefore, it is reasonable to infer that the immunological recall function of TRM cells, along with their ability to trigger local immune responses, contributes to the pathogenesis of IBD. Indeed, numerous studies have demonstrated that the generation and presence of TRM cells contribute to the development of IBD. (Figure 1).
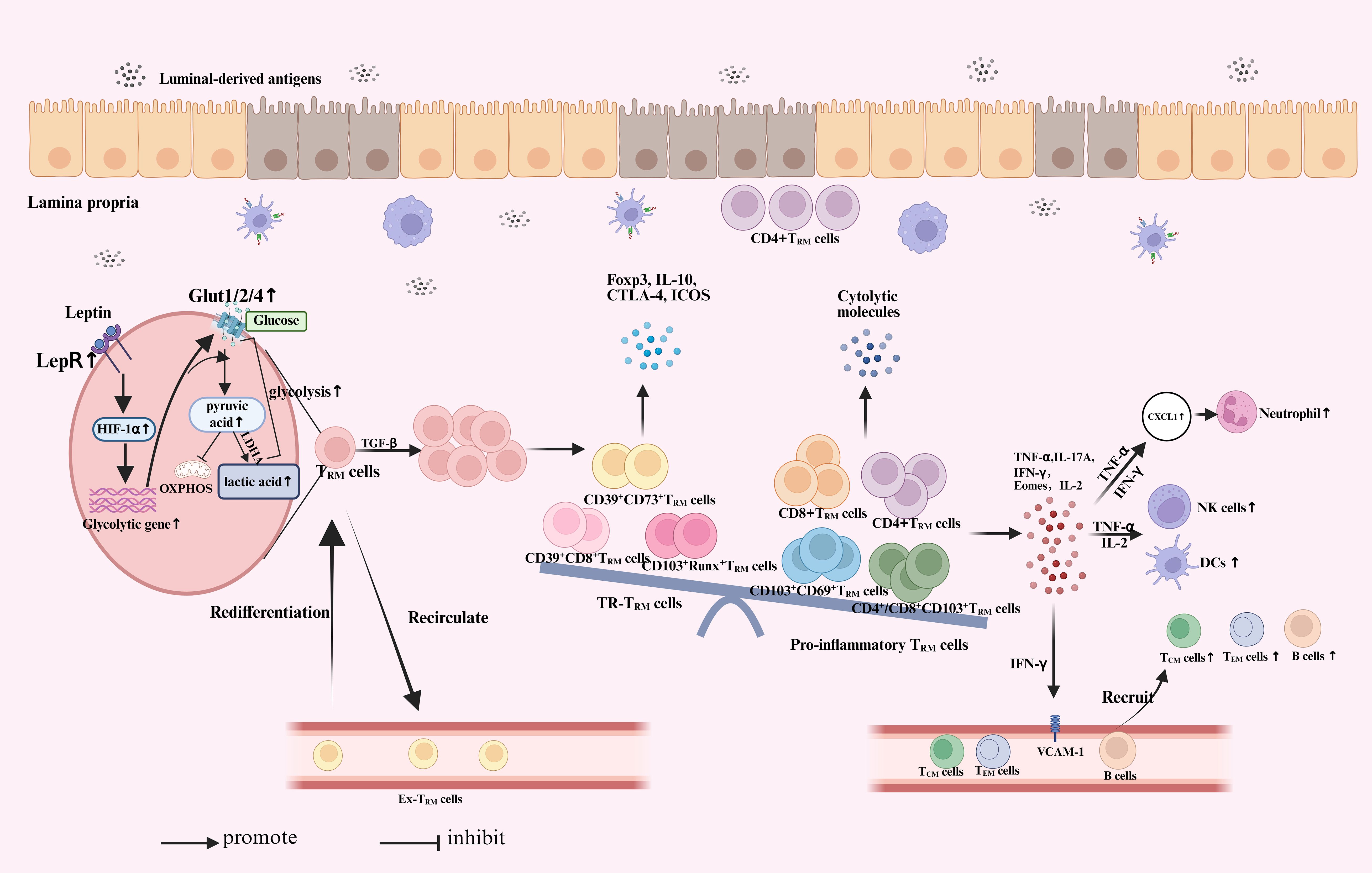
Figure 1. The role of TRM cells in inflammatory bowel disease. When the intestinal epithelial layer is compromised, antigens from the intestinal lumen, including microbiota and pathogens, can penetrate the barrier and access the intestinal lamina propria. Under stress, TRM cells upregulate LepR signaling and downstream HIF-1α expression to rapidly enhance energy production in response to antigen infection. This leads to a significant increase in Glut expression and glycolytic activity. With additional stimulation from TGF-β, pre-existing TRM cells undergo local proliferation and play a key role in effective recall responses. Some of these TRM cells may re-enter the circulation, showing a preference for migrating back and the potential to re-differentiate into TRM cells. A fraction of TRM cells undergo rapid proliferation and exhibit a greater propensity to differentiate into pro-inflammatory subsets, accompanied by the degradation of certain regulatory subsets. This change leads to an increased expression of inflammatory cytokines, chemokines, and cytotoxic granules. TRM cells secrete proinflammatory cytokines, including IFN-γ, IL-2, and TNF-α, which activate natural killer (NK) cells and dendritic cells (DC). This process enhances neutrophil infiltration and recruits additional immune cells by increasing the expression of VCAM-1 on endothelial cells. Ultimately resulting in inflammatory infiltration and causing the development of IBD. At the same time, CD4+TRM cells in the lamina propria are closely located to the intestinal epithelium, directly damage to intestinal epithelium. TRM, tissue-resident memory T cells; LepR, leptin receptor; HIF-1α, hypoxia-inducible factor 1 alpha; LDHA, lactate dehydrogenase; OXPHOS, oxidative phosphorylation; IFN, interferon; IL, Interleukin; DC, dendritic cells; TNF, tumor necrosis factor; TGF, transforming growth factor; NK, natural killer; VCAM, vascular cell adhesion molecule; ICOS, Inducible synergistic co-stimulation molecules; CTLA-4, Cytotoxic T-lymphocyte associated protein 4; Foxp3, Forkhead box protein P3.
3.1 Imbalance between pro-inflammatory TRM cells and regulatory TRM cells
In IBD, a sustained inflammatory response is a key factor that contributes to disease progression. TRM cells persist in the intestine for extended periods and are rapidly activated upon re-exposure to antigens, leading to a continued inflammatory response (21). This characteristic allows TRM cells to play a vital role in the chronic inflammatory processes associated with IBD. In patients with IBD, there is a notable increase in the number of pro-inflammatory TRM cells, coupled with a decrease in the number of regulatory subgroups. This shift may contribute to an immune imbalance in the intestine, exacerbating inflammation (56).
3.1.1 CD4+TRM cells
The pathological feature of IBD is excessive activation of innate immune cells in the intestine, leading to increased antigen presentation and secretion of inflammatory factors. TRM cells further exacerbate this process by recognizing antigens and releasing cytokines (39). Multiple studies have confirmed that the number of CD4+ and CD8+TRM cells in the intestines of patients with IBD significantly increases and produces large amounts of inflammatory factors, and the activation status of these cells is correlated with the severity of the disease (32, 33, 48). Moreover, CD4+TRM cells in the lamina propria are located near the intestinal epithelium, and this spatial arrangement may further contribute to epithelial damage. In addition, TRM cells may trigger or exacerbate inflammation by disrupting immune tolerance due to certain factors, such as genetics and the environment (57). Yokoi T et al. (39) has discovered that under pathological conditions, a specific group of CD103+CD4+TRM cells, which express CD161 and chemokine receptor 5 (CCR5), are the primary source of pro-inflammatory cytokines in the lamina propria region of IBD. Expanded CD4+TRM cells play an important role in the production of Th1 and Th17 cytokines by CD (39) and UC (38), highlighting the crucial role of this TRM subset in the development of these conditions. In patients with IBD, microbiota-reactive CD4+TRM cells also display a Th17-skewed phenotype, which may indicate the host’s protective response aimed at enhancing tissue integrity (17). In colon samples from patients with UC and CD, the number of CD103+CD69+cells increased compared to that of colon CD69-T cells, indicating increased mRNA expression of pro-inflammatory (IFN-γ, IL-17A, and TNF-a) (33). This suggests that the function of TRM cells is closely aligned with that of Th1 and Th17 cells. Simultaneously, human CD4+TRM cells were found to express specific molecules, including markers CCR8 and PPAR-γ, which are closely linked to pathogenic Th2 cells (58). Increased numbers of CD4+CD45RO+GPR15+ memory-type Th2 cells are found in the colon of patients with UC (59). On the other hand, in CD patients, CD4+TRM cells exhibit unique effector properties distinct from conventional T cell subsets. These cells possess an innate-like activation mechanism that operates independently of requiring T-cell receptor (TCR) engagement, characterized by transcriptional enrichment in cytotoxic pathways (e.g., granzyme-perforin axis) and IL-12 responsiveness typically associated with NK cells. This distinctive molecular signature enables spontaneous release of cytolytic molecules (granzyme A) and inflammatory cytokines (IFN-γ), even in the absence of antigenic stimulation (39, 60).
3.1.2 CD8+TRM cells
In addition to CD4+TRM cells, CD8+TRM cells in patients with IBD also affect disease progression. Bottois et al. (31) found that CD103+CD8+TRM cells in patients with CD express more Th17- (such as IL-22 and IL-26) and granzyme K-related genes than those in the control group. In UC, CD8+TRM exhibit increased expression of IL-26 and granzymes, potentially contributing to disease pathogenesis (61). The subset of CD8+TRM cells that express Eomes and are clonally expanded in UC are pro-inflammatory, displaying increased inflammatory traits (41).
3.1.3 Tissue-resident Treg cells
TRM cells possess regulatory functions, particularly in the human colon, where certain TRM cells express markers characteristic of Tregs. Currently, the specific markers for Tissue-resident Treg cells (TR-Tregs), especially those in the intestine, remain unclear. It is generally believed that these cells express CD39, CD73, and Foxp3 under such characteristics (62). TR-Tregs are retained within non-lymphoid tissues and capable of producing cytokines, such as IL-10, which have the ability to quell pro-inflammatory responses from tissue-resident T cells and actively facilitate tissue repair (63). Burton OT et al. (64) found that tissue residency was generally short, on the order of ∼3 weeks, and extracted tissue Treg cells were tissue-agnostic on re-entry. And their results suggest that TR-Tregs operate under a different residency paradigm from TRM cells, being characterized by slow percolation through multiple tissues in a pan-tissue adapted state. Intestinal GATA3+Helios+ and RORγt+ are two main subtypes of TR-Tregs (65). GATA3+Helios+ and RORγt+Tregs are mainly derived from thymus and play the key immunosuppressors during intestinal inflammation. Deletion of GATA3 in Tregs leads to the development of a spontaneous inflammatory disorder with impaired suppressive function, reduced Foxp3 expression, and the adoption of Th17 phenotypes (66). RORγt+ Tregs constitute the main colonic Tregs subset, promoted by the microbiota (64). RORγt enhances Foxp3 expression in colonic Tregs, partly by suppressing effector programs. When RORγt-deficient Tregs are transferred into Rag-/- mice with induced colitis, they are unable to provide protection against the disease because of their Th1-like effector characteristics and the resulting loss of suppressive function (67). Studies on IBD have revealed that CD4+αEβ7+ T cells from both healthy individuals and patients with UC exhibit decreased mRNA expression of regulatory T cell-related cytokines and surface molecules (such as Foxp3, IL-10, CTLA-4, and ICOS), when compared to CD4+αEβ7- T lymphocytes (40). Analysis of the intestinal mucosa of patients with IBD showed a decrease in the CD8+TRM expression of CD39 (62, 68). CD39 is known to degrade excess extracellular adenosine triphosphate (ATP) and adenosine diphosphate (ADP) into adenosine monophosphate, whereas intestinal extracellular ATP and ADP promote colitis (69). In addition, Treg function is mediated by CD39 (62) and a decrease in CD8+TRM expression of CD39 may exacerbate colitis. A reduction in the number of CD39+CD8+TRM cells has also been noted in children with active IBD (68). In mice with depleted circulating CD8+T cells, TRM cells assume effector cell functions when re-challenged with a virus. These TRM cells kill infected cells by releasing granzyme B and perforin, effectively controlling the pathogen independent of natural killer cells (70).
3.1.4 Natural killer T cells
Natural killer T (NKT) cells have been shown to be tissue resident. NKT cells are a special subset of T cells that simultaneously express the TCR and surface markers of NK cells. They have been proven to be able to colonize barrier tissues such as the intestine and liver, and exhibit the typical residence characteristics of TRM cells (23). Resident NKT cells display a distinct pattern of integrins that is integral to their long-term tissue retention (71).Melsen JE et al. (72) found that lymphoid tissue-resident CD69+CD8+TRM cells share a transcriptional and phenotypic profile with CD69+CXCR6+ lymphoid tissue (lt)NK cells by the use of published data. NKT cells exhibit both protective and harmful roles in inflammatory bowel disease (IBD). On one hand, when stimulated with α-galactosylceramide, NKT cells provide protective effects in theDSS-induced colitis model (73). Conversely, NKT cells can also contribute to detrimental inflammatory and immune responses in the gut, such as promoting oxazolone-induced colitis through the production of IL-13 (74).
Energy metabolism plays a crucial role in the immune function of TRM. Under aerobic conditions, normal cells generate energy through cytoplasmic glycolysis followed by mitochondrial oxidative phosphorylation. However, in hypoxic environments, such as during inflammation, cells shift their energy production to glycolysis, which does not require oxygen, to meet their energy demands (75). Activated effector T cells predominantly utilize glucose through the glucose transporter, Glut1. The expression of Glut1 is essential for glucose metabolism, proliferation, and production of inflammatory cytokines in effector T cells. Th1 and Th17 cells demand substantial amounts of glucose for aerobic glycolysis, which supports T-cell activity and generates the biosynthetic precursors necessary for cell growth and division. In contrast, Tregs do not depend on Glut1; they rely on oxidative metabolism for their energy needs and suppressive functions (76, 77). These findings indicate that different metabolic programs are necessary for the development of T cell subsets and these programs can be modulated in vivo to regulate the development of Tregs and pro-inflammatory T cells in inflammatory diseases. The leptin receptor (LepR) and hypoxia-inducible factor 1 alpha (HIF-1α) pathway play crucial roles in regulating glycolytic metabolism. HIF-1α activates the transcription of Glut1, Glut2, and Glut4, enabling the body to adapt to low-oxygen environments. Clinical studies have revealed abnormal upregulation of LepR and HIF-1α expression in patients with UC (78, 79). Wang et al. (42) found that the levels of LepR and HIF-1α genes in the colons of colitis mice were significantly elevated, along with abnormal overexpression of glucose transporters, such as Glut1. Additionally, the levels of key glycolytic enzymes, including hexokinase 1 and pyruvate kinase 2, were elevated compared with those in healthy mice. These findings suggested that the metabolic profile of colitis mice underwent reprogramming and shifted towards glycolysis. These results suggest that under hypoxic conditions, the glycolytic activity in the body is substantially increased. This increase may promote pro-inflammatory TRM cell differentiation, while inhibiting the differentiation of TR-TRM cells. This imbalance in the immune response may contribute to the development of intestinal inflammation.
In conclusion, the immune system is abnormally activated under the stimulation of various antigens, and the function of TRM cells is transformed to Th1 and Th17 cells, resulting in an imbalance between pro-inflammatory TRM and regulatory TRM. This will increase the expression of pro-inflammatory factors, such as TNF-α, IFN–γ, and IL-17, and reduce the expression of anti-inflammatory factors, such as IL-10, which will eventually induce local inflammatory response and cause or aggravate IBD.
3.2 Influence the behavior or function of other immune cells
TRM cells influence local immunity and inflammatory responses by directly interacting with other immune cells, such as macrophages and dendritic cells (DCs), thereby promoting their activation and function and enhancing local immune responses (80). TRM cells secrete pro-inflammatory cytokines, such as IFN-γ, IL-2, and TNF-α, which activate natural killer and DCs. They also promote the recruitment of additional immune cells by increasing the expression of vascular cell adhesion molecule-1 (VCAM-1) on endothelial cells, thereby boosting the local immune response (81). TRM cells also recruit and activate other immune cells, such as neutrophils and macrophages, leading to local immune disorders and further aggravation of intestinal inflammation (53). Zheng et al. (82) show that CD4+TRM cells enhance the expression of cytokines, IFN-γ and TNF-α, which may lead to increased chemokine CXCL1 expression. This process recruits neutrophils and contributes to the chronic recurrent inflammation observed in atopic dermatitis. Funch et al. (83) arrived at a similar conclusion, demonstrating that CD8+TRM cells induce the production of CXCL1 and CXCL2 in the skin, leading to the recruitment of neutrophils and subsequently triggering allergic contact dermatitis. Additionally, Stelekati et al. (84) discovered that activated CD8+T cells enhance the expression of MHC-1 and the costimulatory molecule, 4-1BB, on mast cells in an in vitro co-culture environment. However, the mechanism underlying this effect, whether mediated by T cell-derived chemokines or direct cell-to-cell interactions, remains unclear and requires further investigation.
4 TRM cell-based strategies in IBD treatment
Recently, TRM cells have become crucial for the occurrence and progression of IBD, and a focal point of research. These unique immune cells play pivotal roles in the regulation of intestinal inflammation. They participate in the inflammatory response and affect the process and severity of the disease. With further research on the immune mechanisms underlying IBD, increasing evidence suggests that targeting TRM cells may be a promising therapeutic strategy. Regulation of the activation and function of TRM cells may help alleviate the symptoms and pathological changes associated with IBD. Currently, some drugs and treatment methods have shown efficacy in clinical trials, showing potential for IBD treatment (Table 3).
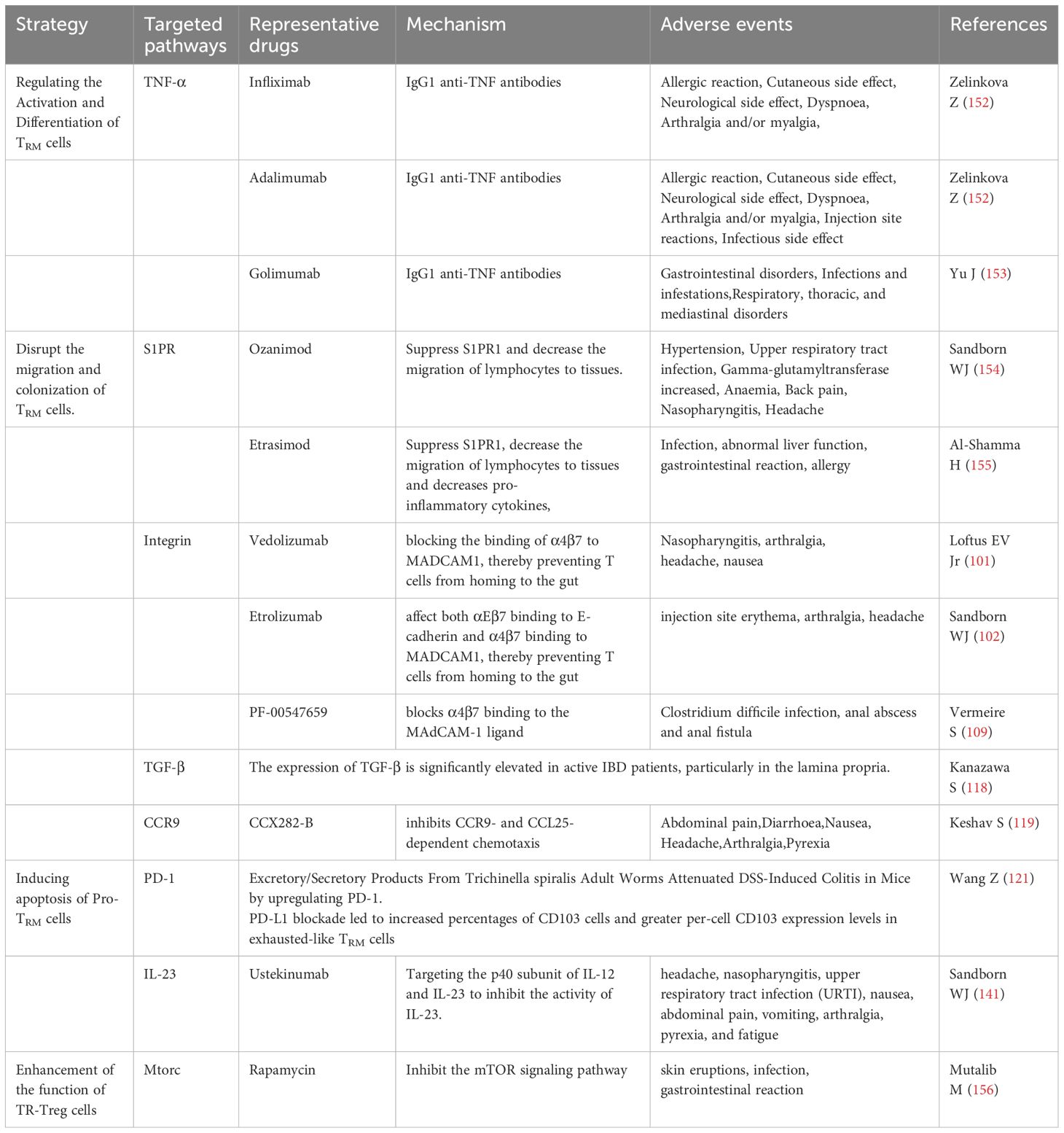
Table 3. Therapeutic targets against TRM cells in IBD.
4.1 Effect of the activation and differentiation of TRM cells
Cytokines, such as TGF-β, IL-6, and TNF-α, can promote TRM cell differentiation (56). TNF signaling not only promotes the expression of acute-phase proteins, but also affects key cellular behaviors, including migration, proliferation, and cell death, in a manner that is highly dependent on the context. Elevated levels of TNF-α promote cell proliferation, differentiation, and the enhancement of adhesion molecules on endothelial cells, facilitating increased cell migration to the inflamed site (85). TNF-α inhibitors, such as infliximab, adalimumab, and golimumab are used for IBD treatment. They mainly trigger apoptosis of CD4+T cells in vivo (86) by fostering and maintaining an anti-inflammatory IL-10+ phenotype, while also slowing down the activation, maturation, and proliferation of CD4+ T cells (87, 88). Atreya et al. (86) observed the occurrence of apoptosis in the lamina propria of six patients four weeks after initiating treatment with infliximab or adalimumab. By demonstrating a noteworthy surge in active caspase staining within the lamina propria, particularly in CD4+ T cells, they presented compelling evidence for the induction of T cell apoptosis by anti-TNF. In conclusion, TNF-α induces apoptosis in T cells, indicating its potential role in regulating the quantity and functional state of TRM cells. This study offers a novel perspective on IBD treatment. However, a comprehensive assessment of the specific effects of TNF-α inhibitors when administered to humans, including their therapeutic efficacy and potential adverse reactions, requires further rigorous and systematic investigation for validation.
4.2 Interference of the migration and colonization of TRM cells
To migrate to the intestine, primed T cells need to express specific migratory receptors. This involves the upregulation of CCR9, α4β7, CD103, and CD69, along with the downregulation of sphingosine-1-phosphate (S1P) receptor 1 (S1PR1) and CCR7 (89, 90).
4.2.1 Upregulation of S1PR1 signaling
Sphingosine-1-phosphate (S1P) is a membrane-derived lysophosphatide signaling molecule that regulates the immune response. S1P1 is the most common S1P receptor because it is expressed in endothelial cells and lymphocytes (91, 92). CD69 is a reliable marker of tissue residency, and inhibits exits from lymphoid organs and peripheral tissues by counteracting S1PR1, which has traditionally been viewed as having its primary role in the establishment of TRM cells (93). The expression of S1PR1 on T cells allows them to detect gradients of S1P concentration, facilitating the chemotactic migration of these cells and enabling the exit of T cells from lymphoid tissues (94). The CD69 surface marker is usually positive for intestinal TRM cells, but its target, S1P1R, is weakly expressed (95). Therefore, S1P1 negatively regulates the development of TRM cells. The interaction between S1P and S1P1 inhibits the outflow of lymphocytes from lymphoid organs by triggering the internalization and degradation of the S1P1 receptor to ensure that lymphocytes do not migrate to the tissues. This may impair the local production of intestinal TRM cells (96). Drugs that directly act on the S1P1 pathway, such as ozanimod and etrasimod, are currently being studied for IBD treatement (97). Ozanimod has shown superior efficacy compared with that of the placebo in treating patients with moderate-to-severe active UC for both induction and maintenance therapy, and has received approval from the FDA and EMA for UC. Results from two separate phase 3 trials demonstrated that etosimod (2 mg daily) was effective and well tolerated as an induction and maintenance therapy in patients with moderate-to-severe active UC. In the ELEVATE UC 52 trial, the clinical remission rate at week 12 was 27.0% for the etrasimod group compared with 7.4% for the placebo group, and at week 52, the rates were 32.1% and 6.7%, respectively. Similarly, the ELEVATE UC 12 trial demonstrated that etrasimod treatment significantly outperformed the placebo in various measures, including endoscopic improvement, symptomatic remission, and mucosal healing (98).
4.2.2 Targeted integrins
A research demonstrated that DCs from the Peyer Patches are especially effective in promoting gut-homing traits in T cells by increasing the expression of migratory receptors and integrin α4β7 (99). Additionally, the Listeria monocytogenes (Lm) infection model established by Sheridan et al. (20) confirmed that Lm infection and the migration of T cells to the intestinal lamina propria and epithelium were, at least partially, dependent on the expression of α4β7. CD103, predominantly expressed on intestinal CD8+TRM cells (29), is a well-established marker for TRM cells, particularly those residing at epithelial surfaces. By interacting with E-cadherin, CD103 facilitates the establishment and retention of intestinal TRM populations in both the small and large intestines (100). The interaction between integrins and their ligands governs the movement and retention of white blood cells in peripheral tissues, such as the intestine, and modulates the local inflammatory milieu. Therefore, integrin-targeting treatments, such as etrolizumab and vedolizumab, are emerging therapeutic targets for IBD (101, 102).
Etrolizumab selectively targets the β7 subunit of α4β7 and αEβ7 integrins. Sandborn et al. (102) completed a phase III clinical trial of etrolizumab for the treatment of patients with moderate-to-severe CD. They found that compared with the placebo, the proportion of patients with who achieved clinical remission and endoscopic improvement significantly increased in the etrolizumab-treated group during the maintenance period. This finding indicates that etrolizumab effectively treats UC, and its mechanism may involve regulating the inflammatory response and promoting mucosal healing (103). The mechanism of etrolizumab blocking αEβ7 and α4β7 integrin heterodimer in the treatment of IBD is explained by its impact on TRM cells. Post-hoc analysis of phase II trials in UC suggests that patients with high CD103 expression are more likely to respond to etrolizumab (104). Furthermore, etrolizumab disrupts the retention of epithelial cells of αEβ7-dependent CD8+T cells in the intestine (105). Gonzalez-Vivo et al. (106) discovered that patients in clinical remission have a higher baseline concentration of CD8 α4β7+ memory T cells than those not in remission. Furthermore, they identified CD8 α4β7+ memory T cell subpopulations as early indicators of remission in response to vedolizumab treatment for UC. Fischer et al. (107) injected human T cells or PBMCs into the NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mouse strain that lacks murine T cells, B cells and NK cells via the ileocolic artery. They demonstrated that vedolizumab selectively hindered the movement of Tregs from patients with UC in this model, without affecting the migration of effector T cells. And a meta-analysis shows that the overall safety of vedolizumab is comparable to that of infliximab (108).PF-547659 is a recombinant IgG-2 monoclonal antibody that targets MAdCAM-1 and inhibits the binding of α4β7 to its MAdCAM-1 ligand. Compared with the placebo group, the 12-week clinical remission rate of patients administered pf-547659 was significantly higher (109).
4.2.3 Targeted TGF-β
In the small intestine of mice, TGF-β induces the upregulation of CD103 and downregulates KLF2 expression in CD8+ T cells, both of which facilitate the retention of TRM cells in that region (110). TGF-β activates the integrin-linked kinase and protein kinase pathways, initiating inside-out signaling of integrins, which strengthens the interaction between CD103 and E-cadherin (111). In addition, TGF-β downregulates the expression of T-box transcription factors, Eomes and T-bet, with Eomes levels gradually decreasing during TRM cell development, while low T-bet levels enhance the expression of the IL-15Rβ chain (CD122), which is essential for the survival and function of TRM cells (112, 113). Wang L et al (114). found that TGF-β-dependent downregulation of T-bet expression is an early event in the differentiation of intestinal CD103+TRM cells. Moreover, the deletion of T-bet can partially bypass the blockade of CD103+TRM cells formation caused by the absence of TGF-β signaling, promoting the differentiation of CD103+TRM. However, it should be noted that these factors exhibit context- and dose-dependent regulators roles. Their functions may be changed depending on inflammatory conditions and cellular activation states. Taking CD8+ T cells as an example, after blocking the TGF-β signaling pathway, the expression of Eomes is significantly upregulated, while the expression of T-bet shows a downward trend, resulting in an increase in the Eomes/T-bet ratio. This is associated with the differentiation of memory T cells (115). In the TGF-β-dominated intestinal microenvironment, the coordinated suppression of these transcriptional regulators appears critical for stabilizing the tissue-resident program. Therefore, inhibiting the function of TGF-β is expected to reduce the intestinal residency of TRM cells and affect their function. Elevated levels of TGF-β expression have been observed in patients with active IBD, particularly in lamina propria lymphocytes (116). It is worth noting that previous research by Monteleone G et al. (117) has shown that T cells from IBD patients have a higher Smad7 expression. Smad7 is an inhibitor of the TGF-β signaling pathway, which adds complexity to the role of TGF-β in IBD. Kanazawa et al. (118) observed an increase in the expression of TGF-β2 and TGF-β3 in the lamina propria lymphocytes of patients with UC and CD. However, to date, no TGF-β inhibitors have been reported for the treatment of IBD. Conversely, mongersen, which restores TGF-β signaling, has been reported to effectively relieve the clinical symptoms of CD. Hence, further research is required to reveal the intricate relationship between TGF-β and IBD, with the aim of enhancing comprehension of its function in the disease.
Certain medications can influence the homing of TRM cells through other mechanisms. CCX282-B is a targeted orally-administered antagonist of the chemokine receptor, CCR9, which regulates the movement and activation of inflammatory cells within the intestine. Results from a randomized controlled trial revealed that a greater percentage of individuals achieved clinical remission (CDAI ≤ 150) in the CCX282-B group compared with that of the placebo group at the conclusion of the maintenance period (119).
4.3 Induced apoptosis of pro-TRM cells
Notably, CD4+and CD8+TRM cells also express a range of surface molecules associated with the suppression of T cell activity, such as PD-1 (21). This serves as a crucial marker of T cell depletion linked to chronic infection and functional hypo responsiveness in tumors (120). The presence of PD-1 may limit the ability of TRM cells to contribute to the tissue pathology. Research on lung infections revealed that PD-1 activation constrains the inflammatory action of TRM cells. However, blocking PD-1 could enhance the proliferation and restoration of TRM cells (121). Wang et al. (122) also discovered that excretory or secretory products from Trichinella spiralis adult worms (AES) exhibit a common property of mitigating DSS-induced colitis and this beneficial effect seems to be closely associated with an increase in PD-1 expression. Conversely, PD-1 deficiency compromised the therapeutic potential of AES in ameliorating DSS-induced colitis, highlighting the crucial role of PD-1 in mediating this therapeutic outcome. Blocking PD-L1 results in a high proportion of CD103+ cells and elevated CD103 expression levels per cell in exhausted TRM cells (121). The classic anti-PD-1 antibody, nivolumab, induces colitis, which is called immune checkpoint inhibitor-associated colitis (123). Reschke R et al (124). studied colon biopsies from patients with immune checkpoint inhibitor-associated colitis compared with those from healthy people using immunofluorescence, spatial transcriptomics, and RNA in situ hybridization. They found that immune checkpoint inhibitor-associated colitis are dominated by TRM cells and Th1/Tc1 cytokines. Emerging evidence suggests that PD-1 may differentially regulate TRM subsets in barrier versus tumor tissues. In the gut, PD-1 signaling likely restrains pro-inflammatory TRM activation to maintain mucosal tolerance (122). Blocking PD-1 could disrupt this balance, releasing a large amount of pro-inflammatory cytokines, leading to colitis. Conversely, in tumors, PD-1 inhibition may preferentially enhance anti-tumor TRM cytotoxicity (125). This tissue specificity and the bidirectional nature of TRM functions may explain why PD-1 blockade enhances TRM-mediated tumor immunity while triggering colitis.
IL-23 plays a vital role in the activation of TRM cells. Mechanistically, IL-23 signaling through its heterodimeric receptor (IL-23R/IL-12Rβ1) activates STAT3 phosphorylation (pSTAT3), which drive their commitment to a Th17-like pathogenic subset—a process critical for the production of IL-17A and IL-22 (126), which in turn enhance epithelial permeability and inflammatory infiltration in the gut (127). Whitley SK et al. found that Local IL-23 is required for proliferation and retention of skin-resident memory Th17 cells and Administration of anti-IL-23R antibody to mice resulted in loss of CD69+ CD103+ tissue resident memory Th17cells from skin (128).Yen et al. (129) presented data indicating that IL-23-induced activation of TRM cells contributes to chronic intestinal inflammation. Specifically, CD4+ TRM cells are the key targets of IL-23 in the progression of chronic intestinal inflammation. Their findings demonstrated that IL-23 enhances the production of IL-6 and IL-17 by memory-activated T cells, and inhibiting IL-6 and IL-17 can improve IBD. While IL-17 inhibition holds theoretical potential for IBD treatment, clinical trials targeting IL-17A (e.g. secukinumab, ixekizumab) have yielded mixed results. Secukinumab (anti-IL-17A) and ixekizumab (anti-IL-17A) demonstrated efficacy in psoriasis and ankylosing spondylitis, yet are ineffective in IBD and may even trigger IBD in patients (130, 131). A study based on literature and database analysis found that IL-17 inhibitor treatment is associated with exacerbation and new onset of IBD and colitis. Patients who receive IL-17 inhibitors (secukinumab, ixekizumab, brodalumab) may experience obvious abdominal pain, diarrhea, and bloody diarrhea (132). These results may be attributed to the pleiotropic effects of IL-17A and heterogeneity of Th17 cells (133). On the one hand, IL-17A induces inflammation but also promotes intestinal epithelial barrier function and repair (134), and participates in an autoregulatory loop to limit the pathogenicity of Th17 cells (135). The plasticity of Th17 cells, influenced by various factors including T cell-polarizing cytokines and the inflammatory tissue environment, is crucial in maintaining gut mucosal homeostasis. On the other hand, there are two different types of Th17 cells: pathogenic and homeostatic. Pathogenic Th17 cells drive tissue damage by secreting pro-inflammatory cytokines such as IL-17A, IL-17F, GM-CSF, and IFN-γ. In contrast, homeostatic Th17 cells secrete IL-10, IL-22, and antimicrobial peptides to maintain the integrity of the mucosal barrier, promote epithelial repair, and inhibit excessive inflammatory responses (136). For example, in patients with psoriasis, a significantly elevated level of IL-17A has been observed in the lesional skin area (137). In the skin of healthy individuals and the non-lesional skin area of psoriasis patients, a small number of homeostatic Th17 cells are present, and their IL-22 may be involved in normal epidermal renewal (138, 139). Therefore, IL-17A inhibitors may worsen IBD by indiscriminately blocking both pathogenic TRM-Th17 activity and protective IL-17A-mediated barrier repair. And the limited effectiveness of drugs targeting IL-17A may be linked to the presence of ex-Th17 cells that switch to producing IFN-γ (140). In contrast, drugs targeting the upstream factor IL-23 (such as ustekinumab) indirectly regulate the level of IL-17 by inhibiting the differentiation of Th17 while preserving its protective functions, thus demonstrating better efficacy and safety in the treatment of IBD. Medications that target IL-12/IL-23, which are currently approved or are undergoing clinical trials for treating IBD, consist of fully human monoclonal antibodies that specifically target the p40 subunit shared by IL-12 and IL-23. Ustekinumab and briakinumab are examples of these drugs. In particular, ustekinumab has been highlighted as an effective treatment option. Ustekinumab is well tolerated, and effective in inducing and sustaining remission in patients with moderate-to-severe IBD who respond positively to the initial treatment, with no significant adverse effects or events reported (141).
4.4 Enhancement of the function of regulatory TRM cells
The high number of Tregs present at the site of inflammation are still unable to protect against IBD. This indicates a potential compromise in Treg function in these patients (142). Conventional therapies for IBD exert beneficial effects on Tregs. Treatment of patients with UC using aminosalicylates or glucocorticoids elevate the frequency of CD4+CD45RO+CD25+T cells in the peripheral blood, a subpopulation that is abundant in Tregs (143). However, these methods potentially increase the risk of infection and lack specificity for exclusively targeting Tregs. Currently, therapies for IBD that target Tregs can be divided into two primary categories: cell-based treatments, which involve the transfer of in vitro expanded or stimulated Tregs to patients, and pharmacological approaches, which are designed to modify Tregs within the body (144). In a phase 1/2a clinical trial (145), ovalbumin-specific Tregs were effectively isolated from the peripheral blood mononuclear cells of patients with CD. These isolated cells underwent in vitro culture expansion before being intravenously reinfused into patients. Importantly, during the fifth and eighth weeks of the study, 40% of the participants showed a notable decrease of 100 points in their Crohn’s Disease Activity Index score. However, the transferred Tregs may fail to accurately migrate and localize within inflamed target tissues. An alternative strategy involves the use of drugs that modulate Tregs in the body. Janyst et al. (146) evaluated the effectiveness of different pharmacological agents believed to have immunomodulatory effects on Treg development. The researchers observed that rapamycin and prednisolone successfully increased the number of CD4+CD25highFoxp3high cells and enhanced the expression of Foxp3 in Treg cells (recognized as CD4+CD25highFoxp3high cells). Additionally, both medications extended the phenotypic stability of Tregs and stimulated the production of fully functional Tregs in functional assays. Rapamycin inhibits the glycolysis pathway and enhances the differentiation, proliferation, and distribution of Tregs, while inhibiting the formation of Th17 cells by targeting mTOR signaling. This process aids in reinstating the equilibrium between Th17 and Treg development, ultimately contributing to the treatment of IBD (147, 148). Rapamycin also increases the abundance of beneficial bacteria and decreases the abundance of harmful bacteria, extending experimental colitis (149). Goldberg R et al. (150) measured levels of the integrin α4β7 on Treg cells isolated from peripheral blood or lamina propria of patients with CD and healthy individuals (controls), and intervened with Treg cells in vitro using rapamycin. They found that incubation of patients’ ex vivo expanded Treg cells with rapamycin induced expression of α4β7, which might be developed for treatment of CD. Overall, the technology for utilizing Tregs to counteract TRM cells in IBD treatment is still in its early stages. To develop novel therapies that rely on Tregs, researchers must first delineate the characteristics of Tregs throughout disease progression and reveal the mechanisms that hinder their suppressive function in IBD. These insights will facilitate the development of therapeutic strategies aimed at increasing the quantity, efficacy, and stability of Tregs. Figure 2 illustrates a targeted therapeutic strategy using TRM cells for IBD treatment.

Figure 2. Targeted therapeutic strategies employing TRM cells in the treatment of IBD. CD127+KLRG-TEM cells can be activated into TRM cells in reaction to TNF-α and TGF-β. The inhibition of TNF-α activity (using Infliximab, Golimumab, and Adalimumab) effectively suppresses the activation of TRM cells. After activation, TRM cells migrate from the circulation to the intestinal mucosa by upregulating the expression of αEβ7, α4β7, and CCR9 while downregulating S1PR1 expression. Therefore, downregulating the expression of CCR9 (CCX282-B), αEβ7 (Etrolizumab), and α4β7 (Vedolizumab), inhibiting the binding of MAdCAM-1 to α4β7 (PF-00547659), and upregulating S1PR1 expression (Etrasimod, Ozanimod) can reduce TRM cell homing. The upregulation of PD-1 and the reduction of IL-23 (Ustekinumab) will promote the apoptosis of TRM cells. Finally, Rapamycin boosts the differentiation, proliferation, and distribution of Treg cells by inhibiting mTOR.
5 Conclusions and perspectives
In conclusion, the intricate relationship between TRM cells and IBD has been progressively revealed, highlighting the potential of TRM cells as biomarkers and therapeutic targets. The chronic inflammatory characteristics of IBD are significantly influenced by the activation and function of TRM cells within the intestinal mucosa. Although TRM cells play a vital role in protecting the immune system from pathogens, dysregulation of these cells can result in excessive inflammation and tissue damage in IBD. The heterogeneity of TRM cells and their plasticity in response to the intestinal microenvironment underscore the complexity of their role in IBD pathogenesis.
Despite these promising insights, several limitations and challenges remain. Firstly, the functions and mechanisms of action of TRM remain unclear. The strategies for targeting TRM cells to treat IBD discussed in the text remain merely theoretical possibilities. There is still a lack of direct evidence to prove their effectiveness, and the underlying mechanisms also await further clarification. Secondly, the heterogeneity of TRM across patients and within the same patient at different disease stages complicates the development of targeted therapies. Targeting TRM cells may disrupt the immune system balance. Finally, over-inhibition of TRM cell activity may increase the risk of infections or adverse reactions beyond the gut. Future studies should elucidate the development, function, and regulatory mechanisms of TRM cells to establish a robust theoretical foundation for targeted therapies. Additionally, understanding the role of TRM cells in IBD will enhance our understanding of disease pathogenesis. This investigation may involve the identification of specific markers, transcription factors, and signaling pathways associated with TRM cells, as well as their contribution to intestinal inflammation. The apparent contradictions between studies may reflect methodological differences in TRM identification rather than true biological discrepancies. Studies examining different marker combinations (e.g., CD69 vs CD103) likely capture distinct TRM subsets with divergent functions. Standardized multi-parameter phenotyping will be essential for future research.
Author contributions
JH: Conceptualization, Visualization, Writing – original draft. WW: Visualization, Writing – original draft. MW: Conceptualization, Methodology, Writing – review & editing. CW: Investigation, Validation, Writing – original draft. YJ: Resources, Visualization, Writing – original draft. YL: Formal analysis, Investigation, Writing – original draft. WZ: Methodology, Writing – original draft. CL: Conceptualization, Methodology, Writing – original draft. ZL: Conceptualization, Software, Writing – original draft. YY: Methodology, Writing – original draft. JL: Resources, Supervision, Writing – review & editing. TM: Conceptualization, Funding acquisition, Supervision, Visualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (NO.82374411), Capital’s Funds for Health Improvement and Research (NO. shoufa 2022-4-4205), National High Level Chinese Medicine Hospital Clinical Research Funding (NO.2024-DYZJ-003 and DFRCZY-2024GJRC010), Unveiling and Leading Projects of Beijing University of Chinese Medicine (NO.2023-JYB-JBQN-014), China Association of Chinese Medicine Young Talent Support Project (NO.CACM-2022-QNRC2-A02), Young Talents Program for Traditional Chinese Medicine Clinical Practice under the Eagle Plan of China Association of Chinese Medicine (NO.CYJH2024057), and Qihuang Excellent Youth Science and Technology Talent Cultivation Plan of Beijing University of Chinese Medicine (NO.K2023A01).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kaser A, Zeissig S, and Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. (2010) 28:573–621. doi: 10.1146/annurev-immunol-030409-101225
2. Li CJ, Wang YK, Zhang SM, Ren MD, and He SX. Global burden of inflammatory bowel disease 1990-2019: A systematic examination of the disease burden and twenty-year forecast. World J Gastroenterol. (2023) 29:5751–67. doi: 10.3748/wjg.v29.i42.5751
3. Shen B. Interventional ibd: the role of endoscopist in the multidisciplinary team management of ibd. Inflammation Bowel Dis. (2018) 24:298–309. doi: 10.1093/ibd/izx058
4. Chang JT. Pathophysiology of inflammatory bowel diseases. N Engl J Med. (2020) 383:2652–64. doi: 10.1056/NEJMra2002697
5. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. (2014) 14:329–42. doi: 10.1038/nri3661
6. Lee JC, Biasci D, Roberts R, Gearry RB, Mansfield JC, Ahmad T, et al. Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in crohn’s disease. Nat Genet. (2017) 49:262–8. doi: 10.1038/ng.3755
7. Ananthakrishnan AN, Bernstein CN, Iliopoulos D, Macpherson A, Neurath MF, Ali RAR, et al. Environmental triggers in ibd: A review of progress and evidence. Nat Rev Gastroenterol Hepatol. (2018) 15:39–49. doi: 10.1038/nrgastro.2017.136
8. Zundler S and Neurath MF. Pathogenic T cell subsets in allergic and chronic inflammatory bowel disorders. Immunol Rev. (2017) 278:263–76. doi: 10.1111/imr.12544
9. Imam T, Park S, Kaplan MH, and Olson MR. Effector T helper cell subsets in inflammatory bowel diseases. Front Immunol. (2018) 9:1212. doi: 10.3389/fimmu.2018.01212
10. Lee JY, Hall JA, Kroehling L, Wu L, Najar T, Nguyen HH, et al. Serum amyloid a proteins induce pathogenic th17 cells and promote inflammatory disease. Cell. (2020) 183:2036–9. doi: 10.1016/j.cell.2020.12.008
11. Negi S, Saini S, Tandel N, Sahu K, Mishra RPN, and Tyagi RK. Translating treg therapy for inflammatory bowel disease in humanized mice. Cells. (2021) 10:1847. doi: 10.3390/cells10081847
12. Zeng B, Shi S, Ashworth G, Dong C, Liu J, and Xing F. Ilc3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis. (2019) 10:315. doi: 10.1038/s41419-019-1540-2
13. Coman D, Coales I, Roberts LB, and Neves JF. Helper-like type-1 innate lymphoid cells in inflammatory bowel disease. Front Immunol. (2022) 13:903688. doi: 10.3389/fimmu.2022.903688
14. Schroeder JH, Howard JK, and Lord GM. Transcription factor-driven regulation of ilc1 and ilc3. Trends Immunol. (2022) 43:564–79. doi: 10.1016/j.it.2022.04.009
15. Koelink PJ, Bloemendaal FM, Li B, Westera L, Vogels EWM, van Roest M, et al. Anti-tnf therapy in ibd exerts its therapeutic effect through macrophage il-10 signalling. Gut. (2020) 69:1053–63. doi: 10.1136/gutjnl-2019-318264
16. Szabo PA, Miron M, and Farber DL. Location, location, location: tissue resident memory T cells in mice and humans. Sci Immunol. (2019) 4. doi: 10.1126/sciimmunol.aas9673
17. Hegazy AN, West NR, Stubbington MJT, Wendt E, Suijker KIM, Datsi A, et al. Circulating and tissue-resident cd4(+) T cells with reactivity to intestinal microbiota are abundant in healthy individuals and function is altered during inflammation. Gastroenterology. (2017) 153:1320–37.e16. doi: 10.1053/j.gastro.2017.07.047
18. Schenkel JM, Fraser KA, Vezys V, and Masopust D. Sensing and alarm function of resident memory cd8+ T cells. Nat Immunol. (2013) 14:509–13. doi: 10.1038/ni.2568
19. Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, and Kupper TS. Skin infection generates non-migratory memory cd8+ T(Rm) cells providing global skin immunity. Nature. (2012) 483:227–31. doi: 10.1038/nature10851
20. Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, and Lefrançois L. Oral infection drives a distinct population of intestinal resident memory cd8(+) T cells with enhanced protective function. Immunity. (2014) 40:747–57. doi: 10.1016/j.immuni.2014.03.007
21. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. (2017) 20:2921–34. doi: 10.1016/j.celrep.2017.08.078
22. Takamura S. Niches for the long-term maintenance of tissue-resident memory T cells. Front Immunol. (2018) 9:1214. doi: 10.3389/fimmu.2018.01214
23. Mackay LK and Kallies A. Transcriptional regulation of tissue-resident lymphocytes. Trends Immunol. (2017) 38:94–103. doi: 10.1016/j.it.2016.11.004
24. Park CO and Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med. (2015) 21:688–97. doi: 10.1038/nm.3883
25. Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, et al. T cell memory. Skin-resident memory cd8+ T cells trigger a state of tissue-wide pathogen alert. Science. (2014) 346:101–5. doi: 10.1126/science.1254803
26. Behr FM, Chuwonpad A, Stark R, and van Gisbergen K. Armed and ready: transcriptional regulation of tissue-resident memory cd8 T cells. Front Immunol. (2018) 9:1770. doi: 10.3389/fimmu.2018.01770
27. Romagnoli PA, Fu HH, Qiu Z, Khairallah C, Pham QM, Puddington L, et al. Differentiation of distinct long-lived memory cd4 T cells in intestinal tissues after oral listeria monocytogenes infection. Mucosal Immunol. (2017) 10:520–30. doi: 10.1038/mi.2016.66
28. Kiniry BE, Li S, Ganesh A, Hunt PW, Somsouk M, Skinner PJ, et al. Detection of hiv-1-specific gastrointestinal tissue resident cd8(+) T-cells in chronic infection. Mucosal Immunol. (2018) 11:909–20. doi: 10.1038/mi.2017.96
29. Schenkel JM and Masopust D. Tissue-resident memory T cells. Immunity. (2014) 41:886–97. doi: 10.1016/j.immuni.2014.12.007
30. Mueller SN and Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol. (2016) 16:79–89. doi: 10.1038/nri.2015.3
31. Bottois H, Ngollo M, Hammoudi N, Courau T, Bonnereau J, Chardiny V, et al. Klrg1 and cd103 expressions define distinct intestinal tissue-resident memory cd8 T cell subsets modulated in crohn’s disease. Front Immunol. (2020) 11:896. doi: 10.3389/fimmu.2020.00896
32. Bishu S, El Zaatari M, Hayashi A, Hou G, Bowers N, Kinnucan J, et al. Cd4+ Tissue-resident memory T cells expand and are a major source of mucosal tumour necrosis factor A in active crohn’s disease. J Crohns Colitis. (2019) 13:905–15. doi: 10.1093/ecco-jcc/jjz010
33. Zundler S, Becker E, Spocinska M, Slawik M, Parga-Vidal L, Stark R, et al. Hobit- and blimp-1-driven cd4(+) tissue-resident memory T cells control chronic intestinal inflammation. Nat Immunol. (2019) 20:288–300. doi: 10.1038/s41590-018-0298-5
34. Stolley JM, Johnston TS, Soerens AG, Beura LK, Rosato PC, Joag V, et al. Retrograde migration supplies resident memory T cells to lung-draining ln after influenza infection. J Exp Med. (2020) 217. doi: 10.1084/jem.20192197
35. Uddin J, Tomar S, Sharma A, Waggoner L, Ganesan V, Marella S, et al. Pir-B regulates cd4(+) il17a(+) T-cell survival and restricts T-cell-dependent intestinal inflammatory responses. Cell Mol Gastroenterol Hepatol. (2021) 12:1479–502. doi: 10.1016/j.jcmgh.2021.06.013
36. Zhang Y, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A, et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J Exp Med. (2020) 217. doi: 10.1084/jem.20190354
37. Ohman L, Dahlén R, Isaksson S, Sjöling A, Wick MJ, Sjövall H, et al. Serum il-17a in newly diagnosed treatment-naive patients with ulcerative colitis reflects clinical disease severity and predicts the course of disease. Inflammation Bowel Dis. (2013) 19:2433–9. doi: 10.1097/MIB.0b013e3182a563cb
38. Lamb CA, Mansfield JC, Tew GW, Gibbons D, Long AK, Irving P, et al. Aeβ7 integrin identifies subsets of pro-inflammatory colonic cd4+ T lymphocytes in ulcerative colitis. J Crohns Colitis. (2017) 11:610–20. doi: 10.1093/ecco-jcc/jjw189
39. Yokoi T, Murakami M, Kihara T, Seno S, Arase M, Wing JB, et al. Identification of a unique subset of tissue-resident memory cd4(+) T cells in crohn’s disease. Proc Natl Acad Sci U S A. (2023) 120:e2204269120. doi: 10.1073/pnas.2204269120
40. Lee Y, Baek J, Park S, Kim Y, Hwang SW, Lee JL, et al. Characterization of th17 tissue-resident memory cells in non-inflamed intestinal tissue of crohn’s disease patients. J Autoimmun. (2024) 145:103206. doi: 10.1016/j.jaut.2024.103206
41. Boland BS, He Z, Tsai MS, Olvera JG, Omilusik KD, Duong HG, et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci Immunol. (2020) 5. doi: 10.1126/sciimmunol.abb4432
42. Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, et al. Control of effector cd8+ T cell function by the transcription factor eomesodermin. Science. (2003) 302:1041–3. doi: 10.1126/science.1090148
43. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for cd103(+)Cd8+ Tissue-resident memory T cells of skin. Nat Immunol. (2013) 14:1294–301. doi: 10.1038/ni.2744
44. Muyuan W, Junxiang L, Xinyu L, Yali Y, Wenji Z, Yifei Y, et al. The regulatory effect of Qingchang Wenzhong Decoction on CD4+T cells in the tissue resident memory of ulcerative colitis mice. Chinese Journal of Integrated Traditional and Western Medicine on Digestion. (2023) 31:583–8.
45. Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, et al. Hobit and blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. (2016) 352:459–63. doi: 10.1126/science.aad2035
46. Wakim LM, Woodward-Davis A, and Bevan MJ. Memory T Cells Persisting within the Brain after Local Infection Show Functional Adaptations to Their Tissue of Residence. Proc Natl Acad Sci U S A. (2010) 107:17872–9. doi: 10.1073/pnas.1010201107
47. Kragten NAM, Behr FM, Vieira Braga FA, Remmerswaal EBM, Wesselink TH, Oja AE, et al. Blimp-1 induces and hobit maintains the cytotoxic mediator granzyme B in cd8 T cells. Eur J Immunol. (2018) 48:1644–62. doi: 10.1002/eji.201847771
48. Chen B, Ye B, Li M, Wang S, Li J, Lai Y, et al. Tigit deficiency protects mice from dss-induced colitis by regulating il-17a-producing cd4(+) tissue-resident memory T cells. Front Immunol. (2022) 13:931761. doi: 10.3389/fimmu.2022.931761
49. Takahara M, Nemoto Y, Oshima S, Matsuzawa Y, Kanai T, Okamoto R, et al. Il-7 promotes long-term in vitro survival of unique long-lived memory subset generated from mucosal effector memory cd4+ T cells in chronic colitis mice. Immunol Lett. (2013) 156:82–93. doi: 10.1016/j.imlet.2013.09.001
50. Bishu S, Hou G, El Zaatari M, Bishu SR, Popke D, Zhang M, et al. Citrobacter rodentium induces tissue-resident memory cd4(+) T cells. Infect Immun. (2019) 87:e00295–19. doi: 10.1128/iai.00295-19
51. Li T, Han B, Wang L, Sun L, Cai Y, Yu M, et al. Activation of mucosal insulin receptor exacerbates intestinal inflammation by promoting tissue resident memory T cells differentiation through ezh2. J Transl Med. (2024) 22:78. doi: 10.1186/s12967-023-04789-x
52. Lutter L, Ter Linde JJM, Brand EC, Hoytema van Konijnenburg DP, Roosenboom B, Horjus Talabur-Horje C, et al. Compartment-driven imprinting of intestinal cd4 T cells in inflammatory bowel disease and homeostasis. Clin Exp Immunol. (2023) 214:235–48. doi: 10.1093/cei/uxad095
53. Lin YH, Duong HG, Limary AE, Kim ES, Hsu P, Patel SA, et al. Small intestine and colon tissue-resident memory cd8(+) T cells exhibit molecular heterogeneity and differential dependence on eomes. Immunity. (2023) 56:207–23.e8. doi: 10.1016/j.immuni.2022.12.007
54. Ryan GE, Harris JE, and Richmond JM. Resident memory T cells in autoimmune skin diseases. Front Immunol. (2021) 12:652191. doi: 10.3389/fimmu.2021.652191
55. Martini GR, Tikhonova E, Rosati E, DeCelie MB, Sievers LK, Tran F, et al. Selection of cross-reactive T cells by commensal and food-derived yeasts drives cytotoxic T(H)1 cell responses in crohn’s disease. Nat Med. (2023) 29:2602–14. doi: 10.1038/s41591-023-02556-5
56. Chen K, Gu X, Yang S, Tao R, Fan M, Bao W, et al. Research progress on intestinal tissue-resident memory T cells in inflammatory bowel disease. Scand J Immunol. (2023) 98:e13332. doi: 10.1111/sji.13332
57. Tang YY, Lou GX, and He WF. Analysis of the Development Mechanism of Cytokine Storm in Severe Burn Patients Complicated with Infection Sichuan Da Xue Xue Bao Yi Xue Ban (2025) 52(1):16–21. doi: 10.16761/j.cnki.1000-4963.2025.02.015
58. Kokubo K, Onodera A, Kiuchi M, Tsuji K, Hirahara K, and Nakayama T. Conventional and pathogenic th2 cells in inflammation, tissue repair, and fibrosis. Front Immunol. (2022) 13:945063. doi: 10.3389/fimmu.2022.945063
59. Nguyen LP, Pan J, Dinh TT, Hadeiba H, O’Hara E 3rd, Ebtikar A, et al. Role and species-specific expression of colon T cell homing receptor gpr15 in colitis. Nat Immunol. (2015) 16:207–13. doi: 10.1038/ni.3079
60. Asada N, Ginsberg P, Paust HJ, Song N, Riedel JH, Turner JE, et al. The integrated stress response pathway controls cytokine production in tissue-resident memory cd4(+) T cells. Nat Immunol. (2025) 26:557–66. doi: 10.1038/s41590-025-02105-x
61. Corridoni D, Antanaviciute A, Gupta T, Fawkner-Corbett D, Aulicino A, Jagielowicz M, et al. Single-cell atlas of colonic cd8(+) T cells in ulcerative colitis. Nat Med. (2020) 26:1480–90. doi: 10.1038/s41591-020-1003-4
62. Noble A, Durant L, Hoyles L, McCartney AL, Man R, Segal J, et al. Deficient resident memory T cell and cd8 T cell response to commensals in inflammatory bowel disease. J Crohns Colitis. (2020) 14:525–37. doi: 10.1093/ecco-jcc/jjz175
63. McGee MC, Zhang T, Magazine N, Islam R, Carossino M, and Huang W. Pd-1 and icos counter-regulate tissue resident regulatory T cell development and il-10 production during flu. Front Immunol. (2022) 13:984476. doi: 10.3389/fimmu.2022.984476
64. Burton OT, Bricard O, Tareen S, Gergelits V, Andrews S, Biggins L, et al. The tissue-resident regulatory T cell pool is shaped by transient multi-tissue migration and a conserved residency program. Immunity. (2024) 57:1586–602.e10. doi: 10.1016/j.immuni.2024.05.023
65. Cho I, Lui PP, and Ali N. Treg regulation of the epithelial stem cell lineage. J Immunol Regener Med. (2020) 8:100028. doi: 10.1016/j.regen.2020.100028
66. Wang Y, Su MA, and Wan YY. An essential role of the transcription factor gata-3 for the function of regulatory T cells. Immunity. (2011) 35:337–48. doi: 10.1016/j.immuni.2011.08.012
67. Bhaumik S, Mickael ME, Moran M, Spell M, and Basu R. Rorγt promotes foxp3 expression by antagonizing the effector program in colonic regulatory T cells. J Immunol. (2021) 207:2027–38. doi: 10.4049/jimmunol.2100175
68. Huang B, Chen Z, Geng L, Wang J, Liang H, Cao Y, et al. Mucosal profiling of pediatric-onset colitis and ibd reveals common pathogenics and therapeutic pathways. Cell. (2019) 179:1160–76.e24. doi: 10.1016/j.cell.2019.10.027
69. Kayama H, Okumura R, and Takeda K. Interaction between the microbiota, epithelia, and immune cells in the intestine. Annu Rev Immunol. (2020) 38:23–48. doi: 10.1146/annurev-immunol-070119-115104
70. Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, and Masopust D. T cell memory. Resident memory cd8 T cells trigger protective innate and adaptive immune responses. Science. (2014) 346:98–101. doi: 10.1126/science.1254536
71. Thomas SY, Scanlon ST, Griewank KG, Constantinides MG, Savage AK, Barr KA, et al. Plzf induces an intravascular surveillance program mediated by long-lived lfa-1-icam-1 interactions. J Exp Med. (2011) 208:1179–88. doi: 10.1084/jem.20102630
72. Melsen JE, Lugthart G, Vervat C, Kielbasa SM, van der Zeeuw SAJ, Buermans HPJ, et al. Human bone marrow-resident natural killer cells have a unique transcriptional profile and resemble resident memory cd8(+) T cells. Front Immunol. (2018) 9:1829. doi: 10.3389/fimmu.2018.01829
73. Saubermann LJ, Beck P, De Jong YP, Pitman RS, Ryan MS, Kim HS, et al. Activation of natural killer T cells by alpha-galactosylceramide in the presence of cd1d provides protection against colitis in mice. Gastroenterology. (2000) 119:119–28. doi: 10.1053/gast.2000.9114
74. Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, and Strober W. Oxazolone colitis, a th2 colitis model resembling ulcerative colitis, is mediated by il-13-producing nk-T cells. Immunity. (2002) 17:629–38. doi: 10.1016/s1074-7613(02)00453-3
75. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. (2021) 20:28. doi: 10.1186/s12943-021-01316-8
76. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory cd4+ T cell subsets. J Immunol. (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
77. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter glut1 is selectively essential for cd4 T cell activation and effector function. Cell Metab. (2014) 20:61–72. doi: 10.1016/j.cmet.2014.05.004
78. Tuzun A, Uygun A, Yesilova Z, Ozel AM, Erdil A, Yaman H, et al. Leptin levels in the acute stage of ulcerative colitis. J Gastroenterol Hepatol. (2004) 19:429–32. doi: 10.1111/j.1440-1746.2003.03300.x
79. Yu S, Li B, Hao J, Shi X, Ge J, and Xu H. Correlation of hypoxia-inducible facto-1α and C-reactive protein with disease evaluation in patients with ulcerative colitis. Am J Transl Res. (2020) 12:7826–35.
80. Rodger B, Stagg AJ, and Lindsay JO. The role of circulating T cells with a tissue resident phenotype (Ex-T(Rm)) in health and disease. Front Immunol. (2024) 15:1415914. doi: 10.3389/fimmu.2024.1415914
81. Lyu Y, Zhou Y, and Shen J. An overview of tissue-resident memory T cells in the intestine: from physiological functions to pathological mechanisms. Front Immunol. (2022) 13:912393. doi: 10.3389/fimmu.2022.912393
82. Zheng C, Cao T, Ye C, and Zou Y. Neutrophil recruitment by cd4 tissue-resident memory T cells induces chronic recurrent inflammation in atopic dermatitis. Clin Immunol. (2023) 256:109805. doi: 10.1016/j.clim.2023.109805
83. Funch AB, Mraz V, Gadsbøll A, Jee MH, Weber JF, Ødum N, et al. Cd8(+) tissue-resident memory T cells recruit neutrophils that are essential for flare-ups in contact dermatitis. Allergy. (2022) 77:513–24. doi: 10.1111/all.14986
84. Stelekati E, Bahri R, D’Orlando O, Orinska Z, Mittrücker HW, Langenhaun R, et al. Mast cell-mediated antigen presentation regulates cd8+ T cell effector functions. Immunity. (2009) 31:665–76. doi: 10.1016/j.immuni.2009.08.022
85. Fakhoury M, Negrulj R, Mooranian A, and Al-Salami H. Inflammatory bowel disease: clinical aspects and treatments. J Inflammation Res. (2014) 7:113–20. doi: 10.2147/jir.S65979
86. Atreya R, Zimmer M, Bartsch B, Waldner MJ, Atreya I, Neumann H, et al. Antibodies against tumor necrosis factor (Tnf) induce T-cell apoptosis in patients with inflammatory bowel diseases via tnf receptor 2 and intestinal cd14+ Macrophages. Gastroenterology. (2011) 141:2026–38. doi: 10.1053/j.gastro.2011.08.032
87. Roberts CA, Durham LE, Fleskens V, Evans HG, and Taams LS. Tnf blockade maintains an il-10(+) phenotype in human effector cd4(+) and cd8(+) T cells. Front Immunol. (2017) 8:157. doi: 10.3389/fimmu.2017.00157
88. Povoleri GAM, Lalnunhlimi S, Steel KJA, Agrawal S, O’Byrne AM, Ridley M, et al. Anti-tnf treatment negatively regulates human cd4(+) T-cell activation and maturation in vitro, but does not confer an anergic or suppressive phenotype. Eur J Immunol. (2020) 50:445–58. doi: 10.1002/eji.201948190
89. Cheng L and Becattini S. Intestinal cd8(+) tissue-resident memory T cells: from generation to function. Eur J Immunol. (2022) 52:1547–60. doi: 10.1002/eji.202149759
90. Masopust D and Soerens AG. Tissue-resident T cells and other resident leukocytes. Annu Rev Immunol. (2019) 37:521–46. doi: 10.1146/annurev-immunol-042617-053214
91. Parigi TL, Roda G, Argollo M, Gilardi D, and Danese S. Is there a role for therapeutic sphingolipids in inflammatory bowel disease? Expert Rev Gastroenterol Hepatol. (2020) 14:47–54. doi: 10.1080/17474124.2020.1709446
92. Scott FL, Clemons B, Brooks J, Brahmachary E, Powell R, Dedman H, et al. Ozanimod (Rpc1063) is a potent sphingosine-1-phosphate receptor-1 (S1p1) and receptor-5 (S1p5) agonist with autoimmune disease-modifying activity. Br J Pharmacol. (2016) 173:1778–92. doi: 10.1111/bph.13476
93. Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, et al. Cutting edge: cd69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol. (2015) 194:2059–63. doi: 10.4049/jimmunol.1402256
94. Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, and Cyster JG. Lymphocyte sequestration through S1p lyase inhibition and disruption of S1p gradients. Science. (2005) 309:1735–9. doi: 10.1126/science.1113640
95. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, and Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory cd8+ T cells. Nat Immunol. (2013) 14:1285–93. doi: 10.1038/ni.2745
96. McGinley MP and Cohen JA. Sphingosine 1-phosphate receptor modulators in multiple sclerosis and other conditions. Lancet. (2021) 398:1184–94. doi: 10.1016/s0140-6736(21)00244-0
97. Sandborn WJ, Feagan BG, D’Haens G, Wolf DC, Jovanovic I, Hanauer SB, et al. Ozanimod as induction and maintenance therapy for ulcerative colitis. N Engl J Med. (2021) 385:1280–91. doi: 10.1056/NEJMoa2033617
98. Sandborn WJ, Vermeire S, Peyrin-Biroulet L, Dubinsky MC, Panes J, Yarur A, et al. Etrasimod as induction and maintenance therapy for ulcerative colitis (Elevate): two randomised, double-blind, placebo-controlled, phase 3 studies. Lancet. (2023) 401:1159–71. doi: 10.1016/s0140-6736(23)00061-2
99. Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, et al. Selective imprinting of gut-homing T cells by peyer’s patch dendritic cells. Nature. (2003) 424:88–93. doi: 10.1038/nature01726
100. Gebhardt T, Whitney PG, Zaid A, Mackay LK, Brooks AG, Heath WR, et al. Different patterns of peripheral migration by memory cd4+ and cd8+ T cells. Nature. (2011) 477:216–9. doi: 10.1038/nature10339
101. Loftus EV Jr., Feagan BG, Panaccione R, Colombel JF, Sandborn WJ, Sands BE, et al. Long-term safety of vedolizumab for inflammatory bowel disease. Aliment Pharmacol Ther. (2020) 52:1353–65. doi: 10.1111/apt.16060
102. Sandborn WJ, Panés J, Danese S, Sharafali Z, Hassanali A, Jacob-Moffatt R, et al. Etrolizumab as induction and maintenance therapy in patients with moderately to severely active crohn’s disease (Bergamot): A randomised, placebo-controlled, double-blind, phase 3 trial. Lancet Gastroenterol Hepatol. (2023) 8:43–55. doi: 10.1016/s2468-1253(22)00303-x
103. Lasa JS, Olivera PA, Danese S, and Peyrin-Biroulet L. Efficacy and safety of biologics and small molecule drugs for patients with moderate-to-severe ulcerative colitis: A systematic review and network meta-analysis. Lancet Gastroenterol Hepatol. (2022) 7:161–70. doi: 10.1016/s2468-1253(21)00377-0
104. Tew GW, Hackney JA, Gibbons D, Lamb CA, Luca D, Egen JG, et al. Association between response to etrolizumab and expression of integrin Ae and granzyme a in colon biopsies of patients with ulcerative colitis. Gastroenterology. (2016) 150:477–87.e9. doi: 10.1053/j.gastro.2015.10.041
105. Zundler S, Schillinger D, Fischer A, Atreya R, López-Posadas R, Watson A, et al. Blockade of Aeβ7 integrin suppresses accumulation of cd8(+) and th9 lymphocytes from patients with ibd in the inflamed gut in vivo. Gut. (2017) 66:1936–48. doi: 10.1136/gutjnl-2016-312439
106. Gonzalez-Vivo M, Lund Tiirikainen MK, Andreu M, Fernandez-Clotet A, López-García A, Murciano Gonzalo F, et al. Memory T cell subpopulations as early predictors of remission to vedolizumab in ulcerative colitis. Front Med (Lausanne). (2022) 9:837294. doi: 10.3389/fmed.2022.837294
107. Fischer A, Zundler S, Atreya R, Rath T, Voskens C, Hirschmann S, et al. Differential effects of A4β7 and gpr15 on homing of effector and regulatory T cells from patients with uc to the inflamed gut in vivo. Gut. (2016) 65:1642–64. doi: 10.1136/gutjnl-2015-310022
108. Peyrin-Biroulet L, Arkkila P, Armuzzi A, Danese S, Guardiola J, Jahnsen J, et al. Comparative efficacy and safety of infliximab and vedolizumab therapy in patients with inflammatory bowel disease: A systematic review and meta-analysis. BMC Gastroenterol. (2022) 22:291. doi: 10.1186/s12876-022-02347-1
109. Vermeire S, Sandborn WJ, Danese S, Hébuterne X, Salzberg BA, Klopocka M, et al. Anti-madcam antibody (Pf-00547659) for ulcerative colitis (Turandot): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet. (2017) 390:135–44. doi: 10.1016/s0140-6736(17)30930-3
110. Crowl JT, Heeg M, Ferry A, Milner JJ, Omilusik KD, Toma C, et al. Tissue-resident memory cd8(+) T cells possess unique transcriptional, epigenetic and functional adaptations to different tissue environments. Nat Immunol. (2022) 23:1121–31. doi: 10.1038/s41590-022-01229-8
111. Boutet M, Gauthier L, Leclerc M, Gros G, de Montpreville V, Théret N, et al. Tgfβ Signaling intersects with cd103 integrin signaling to promote T-lymphocyte accumulation and antitumor activity in the lung tumor microenvironment. Cancer Res. (2016) 76:1757–69. doi: 10.1158/0008-5472.Can-15-1545
112. Mami-Chouaib F, Blanc C, Corgnac S, Hans S, Malenica I, Granier C, et al. Resident memory T cells, critical components in tumor immunology. J Immunother Cancer. (2018) 6:87. doi: 10.1186/s40425-018-0399-6
113. Yuan R, Yu J, Jiao Z, Li J, Wu F, Yan R, et al. The roles of tissue-resident memory T cells in lung diseases. Front Immunol. (2021) 12:710375. doi: 10.3389/fimmu.2021.710375
114. Wang L, Mishra S, Fan KK, Quon S, Li G, Yu B, et al. T-bet deficiency and hic1 induction override tgf-B-dependency in the formation of cd103(+) intestine-resident memory cd8(+) T cells. Cell Rep. (2024) 43:114258. doi: 10.1016/j.celrep.2024.114258
115. Takai S, Schlom J, Tucker J, Tsang KY, and Greiner JW. Inhibition of tgf-B1 signaling promotes central memory T cell differentiation. J Immunol. (2013) 191:2299–307. doi: 10.4049/jimmunol.1300472
116. Babyatsky MW, Rossiter G, and Podolsky DK. Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterology. (1996) 110:975–84. doi: 10.1053/gast.1996.v110.pm8613031
117. Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, and MacDonald TT. Blocking smad7 restores tgf-beta1 signaling in chronic inflammatory bowel disease. J Clin Invest. (2001) 108:601–9. doi: 10.1172/jci12821
118. Kanazawa S, Tsunoda T, Onuma E, Majima T, Kagiyama M, and Kikuchi K. Vegf, basic-fgf, and tgf-beta in crohn’s disease and ulcerative colitis: A novel mechanism of chronic intestinal inflammation. Am J Gastroenterol. (2001) 96:822–8. doi: 10.1111/j.1572-0241.2001.03527.x
119. Keshav S, Vaňásek T, Niv Y, Petryka R, Howaldt S, Bafutto M, et al. A randomized controlled trial of the efficacy and safety of ccx282-B, an orally-administered blocker of chemokine receptor ccr9, for patients with crohn’s disease. PloS One. (2013) 8:e60094. doi: 10.1371/journal.pone.0060094
120. Dolina JS, Van Braeckel-Budimir N, Thomas GD, and Salek-Ardakani S. Cd8(+) T cell exhaustion in cancer. Front Immunol. (2021) 12:715234. doi: 10.3389/fimmu.2021.715234
121. Wang Z, Wang S, Goplen NP, Li C, Cheon IS, Dai Q, et al. Pd-1(Hi) cd8(+) resident memory T cells balance immunity and fibrotic sequelae. Sci Immunol. (2019) 4. doi: 10.1126/sciimmunol.aaw1217
122. Wang Z, Hao C, Zhuang Q, Zhan B, Sun X, Huang J, et al. Excretory/secretory products from trichinella spiralis adult worms attenuated dss-induced colitis in mice by driving pd-1-mediated M2 macrophage polarization. Front Immunol. (2020) 11:563784. doi: 10.3389/fimmu.2020.563784
123. Cañete F, Mañosa M, Lobatón T, Mesonero F, Rodríguez-Lago I, Cabré E, et al. Nivolumab-induced immune-mediated colitis: an ulcerative colitis look-alike-report of new cases and review of the literature. Int J Colorectal Dis. (2019) 34:861–5. doi: 10.1007/s00384-019-03268-4
124. Reschke R, Shapiro JW, Yu J, Rouhani SJ, Olson DJ, Zha Y, et al. Checkpoint blockade-induced dermatitis and colitis are dominated by tissue-resident memory T cells and th1/tc1 cytokines. Cancer Immunol Res. (2022) 10:1167–74. doi: 10.1158/2326-6066.Cir-22-0362
125. Xiao Z, Yan R, Liu H, Huang X, Liang Z, An G, et al. Preventive treatment with pd-1 antibody increases tissue-resident memory T cells infiltration and delays esophageal carcinogenesis. Cancer Prev Res (Phila). (2023) 16:669–79. doi: 10.1158/1940-6207.Capr-23-0196
126. Hou Y, Zhu L, Tian H, Sun HX, Wang R, Zhang L, et al. Il-23-induced macrophage polarization and its pathological roles in mice with imiquimod-induced psoriasis. Protein Cell. (2018) 9:1027–38. doi: 10.1007/s13238-018-0505-z
127. Glal D, Sudhakar JN, Lu HH, Liu MC, Chiang HY, Liu YC, et al. Atf3 sustains il-22-induced stat3 phosphorylation to maintain mucosal immunity through inhibiting phosphatases. Front Immunol. (2018) 9:2522. doi: 10.3389/fimmu.2018.02522
128. Whitley SK, Li M, Kashem SW, Hirai T, Igyártó BZ, Knizner K, et al. Local il-23 is required for proliferation and retention of skin-resident memory T(H)17 cells. Sci Immunol. (2022) 7:eabq3254. doi: 10.1126/sciimmunol.abq3254
129. Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, et al. Il-23 is essential for T cell-mediated colitis and promotes inflammation via il-17 and il-6. J Clin Invest. (2006) 116:1310–6. doi: 10.1172/jci21404
130. Fauny M, Moulin D, D’Amico F, Netter P, Petitpain N, Arnone D, et al. Paradoxical gastrointestinal effects of interleukin-17 blockers. Ann Rheum Dis. (2020) 79:1132–8. doi: 10.1136/annrheumdis-2020-217927
131. Schreiber S, Colombel JF, Feagan BG, Reich K, Deodhar AA, McInnes IB, et al. Incidence rates of inflammatory bowel disease in patients with psoriasis, psoriatic arthritis and ankylosing spondylitis treated with secukinumab: A retrospective analysis of pooled data from 21 clinical trials. Ann Rheum Dis. (2019) 78:473–9. doi: 10.1136/annrheumdis-2018-214273
132. Deng Z, Wang S, Wu C, and Wang C. Il-17 inhibitor-associated inflammatory bowel disease: A study based on literature and database analysis. Front Pharmacol. (2023) 14:1124628. doi: 10.3389/fphar.2023.1124628
133. Mills KHG. Il-17 and il-17-producing cells in protection versus pathology. Nat Rev Immunol. (2023) 23:38–54. doi: 10.1038/s41577-022-00746-9
134. Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, et al. Interleukin-23-independent il-17 production regulates intestinal epithelial permeability. Immunity. (2015) 43:727–38. doi: 10.1016/j.immuni.2015.09.003
135. Chong WP, Mattapallil MJ, Raychaudhuri K, Bing SJ, Wu S, Zhong Y, et al. The cytokine il-17a limits th17 pathogenicity via a negative feedback loop driven by autocrine induction of il-24. Immunity. (2020) 53:384–97.e5. doi: 10.1016/j.immuni.2020.06.022
136. Schnell A, Littman DR, and Kuchroo VK. T(H)17 cell heterogeneity and its role in tissue inflammation. Nat Immunol. (2023) 24:19–29. doi: 10.1038/s41590-022-01387-9
137. Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, Abbondanzo S, et al. Il-23 stimulates epidermal hyperplasia via tnf and il-20r2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. (2006) 203:2577–87. doi: 10.1084/jem.20060244
138. Fukaya T, Fukui T, Uto T, Takagi H, Nasu J, Miyanaga N, et al. Pivotal role of il-22 binding protein in the epithelial autoregulation of interleukin-22 signaling in the control of skin inflammation. Front Immunol. (2018) 9:1418. doi: 10.3389/fimmu.2018.01418
139. Fitch E, Harper E, Skorcheva I, Kurtz SE, and Blauvelt A. Pathophysiology of psoriasis: recent advances on il-23 and th17 cytokines. Curr Rheumatol Rep. (2007) 9:461–7. doi: 10.1007/s11926-007-0075-1
140. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate mapping of il-17-producing T cells in inflammatory responses. Nat Immunol. (2011) 12:255–63. doi: 10.1038/ni.1993
141. Sandborn WJ, Feagan BG, Danese S, O’Brien CD, Ott E, Marano C, et al. Safety of ustekinumab in inflammatory bowel disease: pooled safety analysis of results from phase 2/3 studies. Inflammation Bowel Dis. (2021) 27:994–1007. doi: 10.1093/ibd/izaa236
142. Mayne CG and Williams CB. Induced and natural regulatory T cells in the development of inflammatory bowel disease. Inflammation Bowel Dis. (2013) 19:1772–88. doi: 10.1097/MIB.0b013e318281f5a3
143. Takahashi M, Nakamura K, Honda K, Kitamura Y, Mizutani T, Araki Y, et al. An inverse correlation of human peripheral blood regulatory T cell frequency with the disease activity of ulcerative colitis. Dig Dis Sci. (2006) 51:677–86. doi: 10.1007/s10620-006-3191-2
144. Clough JN, Omer OS, Tasker S, Lord GM, and Irving PM. Regulatory T-cell therapy in crohn’s disease: challenges and advances. Gut. (2020) 69:942–52. doi: 10.1136/gutjnl-2019-319850
145. Desreumaux P, Foussat A, Allez M, Beaugerie L, Hébuterne X, Bouhnik Y, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory crohn’s disease. Gastroenterology. (2012) 143:1207–17.e2. doi: 10.1053/j.gastro.2012.07.116
146. Janyst M, Kaleta B, Janyst K, Zagożdżon R, Kozlowska E, and Lasek W. Comparative study of immunomodulatory agents to induce human T regulatory (Treg) cells: preferential treg-stimulatory effect of prednisolone and rapamycin. Arch Immunol Ther Exp (Warsz). (2020) 68:20. doi: 10.1007/s00005-020-00582-6
147. Yin H, Li X, Zhang B, Liu T, Yuan B, Ni Q, et al. Sirolimus ameliorates inflammatory responses by switching the regulatory T/T helper type 17 profile in murine colitis. Immunology. (2013) 139:494–502. doi: 10.1111/imm.12096
148. Xia Y, Zhang L, Ocansey DKW, Tu Q, Mao F, and Sheng X. Role of glycolysis in inflammatory bowel disease and its associated colorectal cancer. Front Endocrinol (Lausanne). (2023) 14:1242991. doi: 10.3389/fendo.2023.1242991
149. Guo X, Xu J, Huang C, Zhang Y, Zhao H, Zhu M, et al. Rapamycin extenuates experimental colitis by modulating the gut microbiota. J Gastroenterol Hepatol. (2023) 38:2130–41. doi: 10.1111/jgh.16381
150. Goldberg R, Scotta C, Cooper D, Nissim-Eliraz E, Nir E, Tasker S, et al. Correction of defective T-regulatory cells from patients with crohn’s disease by ex vivo ligation of retinoic acid receptor-A. Gastroenterology. (2019) 156:1775–87. doi: 10.1053/j.gastro.2019.01.025
151. Lutter L, Roosenboom B, Brand EC, Ter Linde JJ, Oldenburg B, van Lochem EG, et al. Homeostatic function and inflammatory activation of ileal cd8(+) tissue-resident T cells is dependent on mucosal location. Cell Mol Gastroenterol Hepatol. (2021) 12:1567–81. doi: 10.1016/j.jcmgh.2021.06.022
152. Zelinkova Z, Bultman E, Vogelaar L, Bouziane C, Kuipers EJ, and van der Woude CJ. Sex-dimorphic adverse drug reactions to immune suppressive agents in inflammatory bowel disease. World J Gastroenterol. (2012) 18:6967–73. doi: 10.3748/wjg.v18.i47.6967
153. Yu J, Park SJ, Kim HW, Lim YJ, Park J, Cha JM, et al. Effectiveness and safety of golimumab in patients with ulcerative colitis: A multicenter, prospective, postmarketing surveillance study. Gut Liver. (2022) 16:764–74. doi: 10.5009/gnl210335
154. Sandborn WJ, Feagan BG, Hanauer S, Vermeire S, Ghosh S, Liu WJ, et al. Long-term efficacy and safety of ozanimod in moderately to severely active ulcerative colitis: results from the open-label extension of the randomized, phase 2 touchstone study. J Crohns Colitis. (2021) 15:1120–9. doi: 10.1093/ecco-jcc/jjab012
155. Al-Shamma H, Lehmann-Bruinsma K, Carroll C, Solomon M, Komori HK, Peyrin-Biroulet L, et al. The selective sphingosine 1-phosphate receptor modulator etrasimod regulates lymphocyte trafficking and alleviates experimental colitis. J Pharmacol Exp Ther. (2019) 369:311–7. doi: 10.1124/jpet.118.254268
Keywords: inflammatory bowel disease, tissue-resident memory T cells, immune balance, mechanisms, strategies
Citation: Hu J, Wang W, Wang M, Wu C, Jiao Y, Li Y, Zhang W, Liang C, Lin Z, Yu Y, Li J and Mao T (2025) Immunological pathogenesis of inflammatory bowel disease: focus on tissue resident memory T cells. Front. Immunol. 16:1591584. doi: 10.3389/fimmu.2025.1591584
Received: 11 March 2025; Accepted: 14 May 2025;
Published: 03 June 2025.
Edited by:
Xuefeng Wang, Soochow University, ChinaReviewed by:
Jan-Hendrik Schroeder, King’s College London, United KingdomNathan Schuldt, University of Minnesota Twin Cities, United States
Tariq Ahmad Najar, New York University, United States
Copyright © 2025 Hu, Wang, Wang, Wu, Jiao, Li, Zhang, Liang, Lin, Yu, Li and Mao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tangyou Mao, bWFvdGFuZ3lvdXF1bkAxMjYuY29t; Junxiang Li, bGlqdW54aWFuZzEyMjZAMTYzLmNvbQ==
†These authors have contributed equally to this work