 Salem Salman Almujri1,2*
Salem Salman Almujri1,2* Elena Stylianou1
Elena Stylianou1 Annalisa Nicastri1,3Iman Satti1
Annalisa Nicastri1,3Iman Satti1 Marcellus Korompis1
Marcellus Korompis1 Shuailin Li1
Shuailin Li1 Christopher J. De Voss1
Christopher J. De Voss1 Marco Polo Peralta Alvarez1
Marco Polo Peralta Alvarez1 Rachel Tanner1,4
Rachel Tanner1,4 Paulo J. G. Bettencourt1,5
Paulo J. G. Bettencourt1,5 Nicola Ternette1,6
Nicola Ternette1,6 Helen McShane1*
Helen McShane1*- 1The Jenner Institute, University of Oxford, Oxford, United Kingdom
- 2Department of Pharmacology, College of Pharmacy, King Khalid University, Asir-Abha, Saudi Arabia
- 3Chester Beatty Laboratories, The Institute Cancer Research, London, United Kingdom
- 4Department of Biology, University of Oxford, Oxford, United Kingdom
- 5Universidade Católica Portuguesa, Faculty of Medicine, Center for Interdisciplinary Research in Health, Lisbon, Portugal
- 6School of Life Sciences, University of Dundee, Dundee, United Kingdom
Introduction: Tuberculosis (TB), caused by Mycobacterium tuberculosis (MTB), remains a significant global health concern. The existing vaccine, Bacillus Calmette-Guérin (BCG), provides inconsistent protection, highlighting the pressing need for a more effective vaccine. We aimed to identify novel MTB antigens and assess their protective efficacy as TB vaccine candidates.
Methods: Using immunopeptidomics, we identified 64 and 80 unique mycobacterial antigens derived from BCG and MTB, respectively. We prioritised antigens based on HLA allele coverage through an immunoinformatics approach.
Results: The candidates, hisD, metE, and mmpL12, delivered as DNA vaccines, were evaluated for efficacy in mice using the ex vivo Mycobacterial Growth Inhibition Assay (MGIA) and metE was identified as a promising candidate. In vivo murine MTB challenge experiments confirmed the protective efficacy conferred by metE when formulated as recombinant protein with AS01™ or AddaS03™ adjuvants, compared to the naïve group. The immunogenic profiles of metE formulated in the two different adjuvants differed, with metE-AS01™ inducing antigen-specific IFN-γ, TNF-α, IL-2, IL-17, IgG1 and IgG2a-c, while metE-AddaS03™ induced TNF-α, IL-2, IL-17, IL-4, IgM, IgG1, IgG2b.
Conclusion: Our findings highlight metE as a promising protective antigen for future TB vaccine development.
1 Introduction
Tuberculosis (TB) remains a significant global health challenge despite being both preventable and treatable. According to the recent Global TB Report by the World Health Organisation, TB has regained its position as the leading cause of death from a single infectious agent, surpassing COVID-19. The causative agent, Mycobacterium tuberculosis (MTB), infected 10.8 million people and caused 1.25 million deaths in 2023 (1). Bacillus Calmette-Guérin (BCG), comprised of live attenuated Mycobacterium bovis, is the only licensed vaccine for TB, but has little protective effect against pulmonary disease in adolescents and adults (2, 3). There is an urgent need for the development of more effective preventive strategies.
The importance of cell-mediated immunity (CMI) in controlling TB, primarily through a T-helper 1 CD4+ T-cell response, has been demonstrated in HIV-infected individuals. These individuals experience a decrease in the number of CD4+ T-cells, making them highly susceptible to MTB infection and reactivation of latent MTB infection (LTBI) (4). In addition, accumulating evidence indicates that T-helper 17 (Th17) CD4+ T cells contribute to protective immunity against MTB (5). There is also evidence for a role for CD8+ T cells (6, 7), and emerging evidence that antibodies may play a role in immunity to TB (8). To present MTB-derived antigens to T cells, host immune cells such as macrophages and dendritic cells use major histocompatibility complex (MHC) molecules (9). CD4+ T cells recognize peptides presented by MHC class II (MHC-II) molecules, while CD8+ T cells recognize peptides presented by MHC class I (MHC-I) molecules (10). These interactions between T cells and MHC molecules are critical for activating and shaping the adaptive immune response to MTB infection. Since no single T cell subset has been definitively correlated with protection, inducing a diverse CD4+ T cell response remains a key objective in TB vaccine development, supported by strong and consistent evidence of CD4+ T cell-mediated protection in both preclinical and clinical studies (11–13). In contrast, CD8+ T cell responses have been more variable, and clinical evaluations of TB vaccine candidates have shown that CD8+ T cell responses were relatively poorly induced when compared to CD4+ T cell responses (14).
The discovery of novel protective antigens is key to the development of candidate subunit vaccines aimed at improving BCG efficacy. Subunit vaccines, composed of one or a few mycobacterial antigens delivered with a platform of choice, have better safety profiles than live attenuated vaccines (15). However, with over 4000 genes (16), the identification of mycobacterial antigens presented on MHC molecules has been challenging (17, 18). Immunopeptidomics, a mass spectrometry-based approach, is one of the methods used to identify potentially immunogenic mycobacterial antigens by analyzing peptides presented by MHC molecules in infected cells (19). By identifying pathogen peptides and their corresponding precursor antigens, it is possible to infer which proteins are processed and presented to the immune system and which are more likely to elicit a T-cell response. Since the 1990s, numerous research groups have applied immunopeptidomic techniques to discover novel antigens presented by MHC molecules in the context of cancers (20, 21) and infectious diseases including TB (22, 23). A study conducted by Flyer et al. identified three peptides associated with MHC-I, all derived from a single antigen (Rv0341), after infecting the U937 histiocytic lymphoma cell line with MTB (24). Similarly, another study identified 16 MTB antigens linked to MHC-I in primary human macrophages sourced from six donors with LTBI (25). Targeting non-classical MHC-I molecules also yielded 28 unique antigens (26). Recently, our group identified 94 mycobacterial peptides presented by MHC-II and 43 presented by MHC-I, in BCG-infected THP-1 cells, using immunopeptidomics. From these, three antigens [galactofuranosyl transferase (glfT2), isoniazid inductible gene (iniB) and probable fatty acid synthase (fas)] were expressed in viral vectors and evaluated as vaccine candidates in a murine aerosol MTB challenge model. When delivered as a boost to previous BCG vaccination, the combination of these vectors conferred significant protection in mice, compared to BCG alone (27).
To accelerate TB vaccine development, we employed a mass spectrometry-based immunopeptidomics approach to identify novel mycobacterial antigens. Analysis of peptides presented on MHC molecules in human macrophages (differentiated THP-1 cells) infected with either BCG or MTB led to the identification of 64 BCG-derived and 80 MTB-derived antigens. Because those identified antigens resulted from specific HLA alleles in THP-1 cells, we expanded the screening of T cell epitopes using the entire antigen sequence against the most frequent HLA alleles worldwide to maximize population coverage, which is important for vaccine development. To address this, immunoinformatic tools such as NetMHCpan and NetMHCIIpan were utilized to map T cell epitopes within a given antigen sequence (28). These tools were employed to rank antigens identified from immunopeptidomics data based on their T cell epitope richness. By predicting T cell epitopes against the most prevalent MHC alleles globally (29, 30), the development of vaccines capable of eliciting robust T-cell responses across diverse populations. Although our primary focus was on T cell epitope analysis, using the full-length antigenic sequence preserved the structural integrity of the antigens. This in turn, allowed for the potential induction of antibodies that recognize native conformations, thereby indirectly accounting for antibody responses as part of our overall vaccine design strategy.
A prioritization pipeline of identified antigens using immunopeptidomic and immunoinformatic tools highlighted candidate antigens for further assessment. From this list, three top candidates were selected for evaluation in mice using the ex vivo mycobacterial growth inhibition assay (MGIA), with one antigen, metE, emerging as a promising candidate due to its strong immunogenic profile. In murine MTB-challenge experiments, metE provided protection when administered as a recombinant protein formulated with either AS01™ or AddaS03™ adjuvants, with each formulation inducing distinct immune responses. These adjuvants were chosen for their clinical relevance and proven ability to elicit broad immune responses, including diverse effector T cell subsets and functional antibodies across multiple species (31, 32). AddaS03™ shares compositional similarities with AS03®, an adjuvant used in licensed influenza vaccines (33), while AS01™ is a component of the Shingrix vaccine (AS01B) and has demonstrated efficacy against TB in the M72/AS01E vaccine candidate (34). By using adjuvants with proven translational potential, minimizes the risk of generating protective responses in preclinical models that may not be reproducible in humans. Our findings indicate that metE is a promising novel antigen for inclusion in a subunit vaccine against TB.2 Results
2 Results
2.1 Identification and selection of mycobacterial antigens
Building on the work of Bettencourt et al. (27) which identified HLA-I and HLA-DR peptides from THP-1 cells infected with BCG using W6/32 and L243 antibodies respectively, resulting in the identification of 70 antigens (Figures 1A, B, dotted lines), remaining HLA molecules were investigated, specifically HLA-DP and HLA-DQ, using the same samples (Figures 1B, samples 1-6). To achieve this, antibodies targeting HLA-DP (B721), followed by a pan-HLA-II antibody (IVA12, non-selective HLA-II antibody) were employed to enrich for HLA-DQ and any remaining HLA-II molecules, particularly HLA-DR. This approach identified 39 BCG peptides associated with HLA-DP and 34 peptides associated with other class II molecules, resulting in a total of 64 unique antigens (Figure 1B; Supplementary Table 1). Interestingly, the number of identified peptides increased in samples treated with IFN-γ (Supplementary Figures 1C, D).
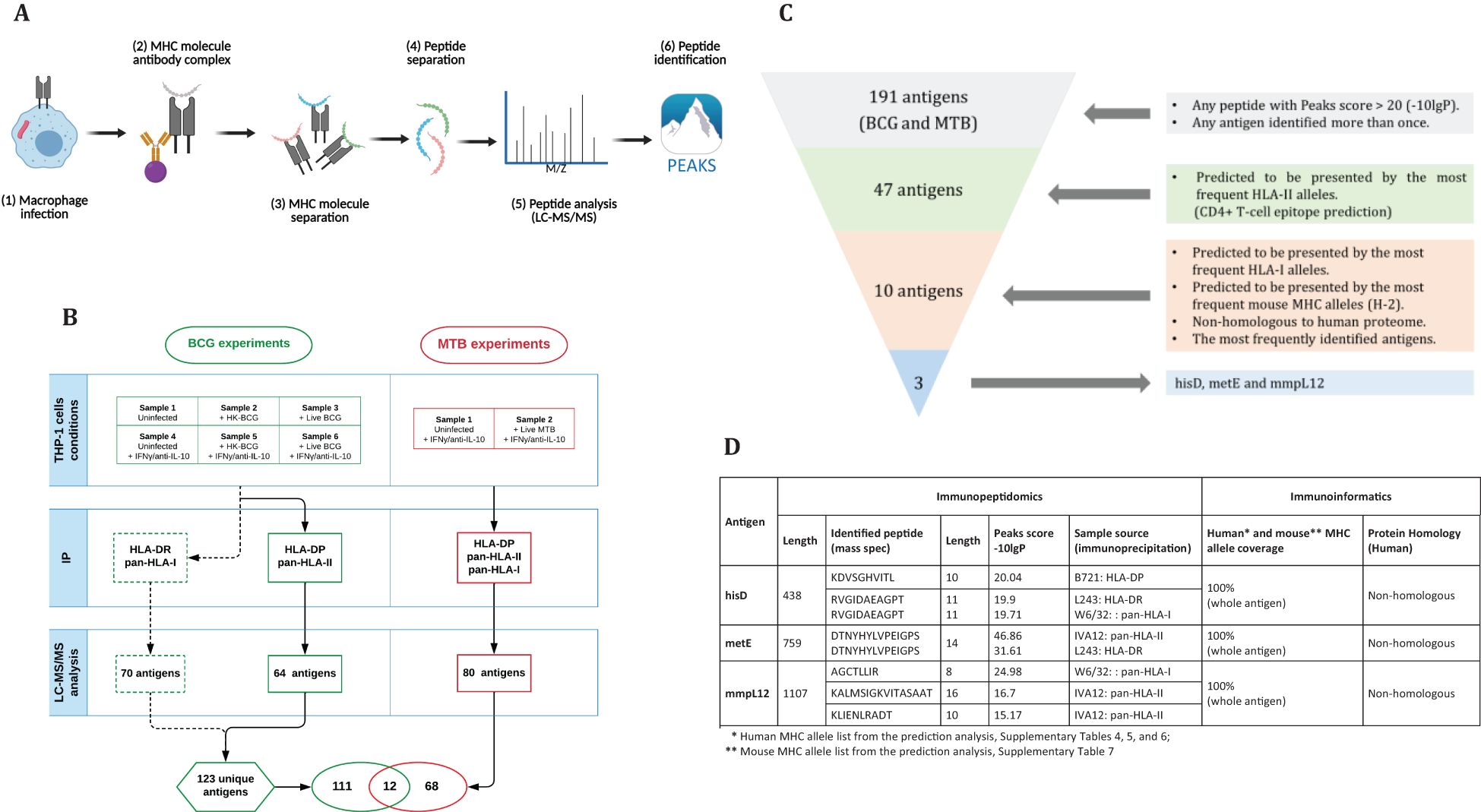
Figure 1. (A) Step-by-step procedure for antigen identification: THP-1 macrophages are infected with mycobacteria, harvested, lysed for immunoprecipitation, and peptides are eluted from MHC complexes (generated by BioRender). The peptides are separated by HPLC, analyzed via LC-MS/MS, and processed using Peaks software (v10). (B) Sample arrangement for immunopeptidomics analysis: In the BCG experiments, Bettencourt et al. (2020) prepared six samples, which underwent IP with L243 and W6/32 antibodies, identifying 70 antigens. Additional IP with B271 and IVA12 isolated HLA-DP and -DQ, identifying 64 antigens. In the MTB experiments, two samples underwent IP with three antibodies, leading to the identification of 80 antigens, totally 191 unique antigens. (C) Antigen selection criteria: selecting antigens with a Peaks score >20 or identified multiple times yielded 47 antigens out of 191. Antigens with full HLA-II coverage were narrowed down to 10. Finally, non-homologous antigens presented by common HLA-I alleles resulted in three candidates: hisD, metE, and mmpL12. (D) Characterization of candidate antigens using immunopeptidomics and immunoinformatics.
Subsequently, a second set of experiments was conducted using MTB as the infectious agent. Two samples were prepared under two conditions (Figures 1A, B) and subjected to sequential immunoprecipitation with antibodies specific for HLA-DP (B721), pan-HLA-II (IVA12) to enrich for HLA-DQ and HLA-DR, and pan-HLA-I (W6/32). This workflow identified 87 peptides derived from 80 unique MTB antigens (Figure 1B; Supplementary Table 2).
The immunopeptidomics analysis revealed distinct peptide length distributions for the two HLA classes. Peptides presented by MHC-I molecules were predominantly 8 to 10 amino acids long, consistent with the expected size for MHC-I-presented peptides; peptides presented by MHC-II molecules were primarily between 13 and 17 amino acids, characteristic of MHC-II-presented peptides (Supplementary Figure 2). Shorter peptides were also observed in the MHC-II immunoprecipitates, likely due to co-immunoprecipitation of MHC-I peptides and the intrinsic ability of MHC-II molecules to accommodate peptides of varying lengths, including shorter ones (35).
By combining the identified mycobacterial antigens from BCG (123 antigens, including those reported by Bettencourt et al. (27) and MTB (80 antigens), we assembled a dataset of 191 unique antigens, 12 of which were shared between the two pathogens (Figures 1B, C; Table 1). Given the substantial number of identified antigens, we developed a streamlined in silico approach to prioritize and select candidate antigens. This approach integrated immunopeptidomic and immunoinformatic parameters and involved a three-step selection process (Figures 1C). First, antigens were ranked based on their Peaks score, which reflects the accuracy of sequence identification. Second, MHC allele coverage was evaluated using NetMHCpan and NetMHCIIpan tools to map T-cell epitope-rich antigens against the most prevalent HLA alleles, ensuring broad immunogenic potential. Finally, homology between mycobacterial antigens and the human proteome was assessed using the Vaxign server. This server employs BLAST to identify sequence similarities, and proteins with an Expect value (E-value) less than 1 × 105 were considered homologous (36). Consequently, antigens exhibiting an E-value below this threshold were excluded to avoid potential risk of cross-reactive immune responses against self-antigens (37, 38). Through this rigorous selection process, we identified three top-ranked candidate antigens: hisD, metE, and mmpL12. These antigens demonstrated broad HLA coverage for both class I and II alleles, and exhibited low homology with the human proteome (Figure 1D; Supplementary Table 2).
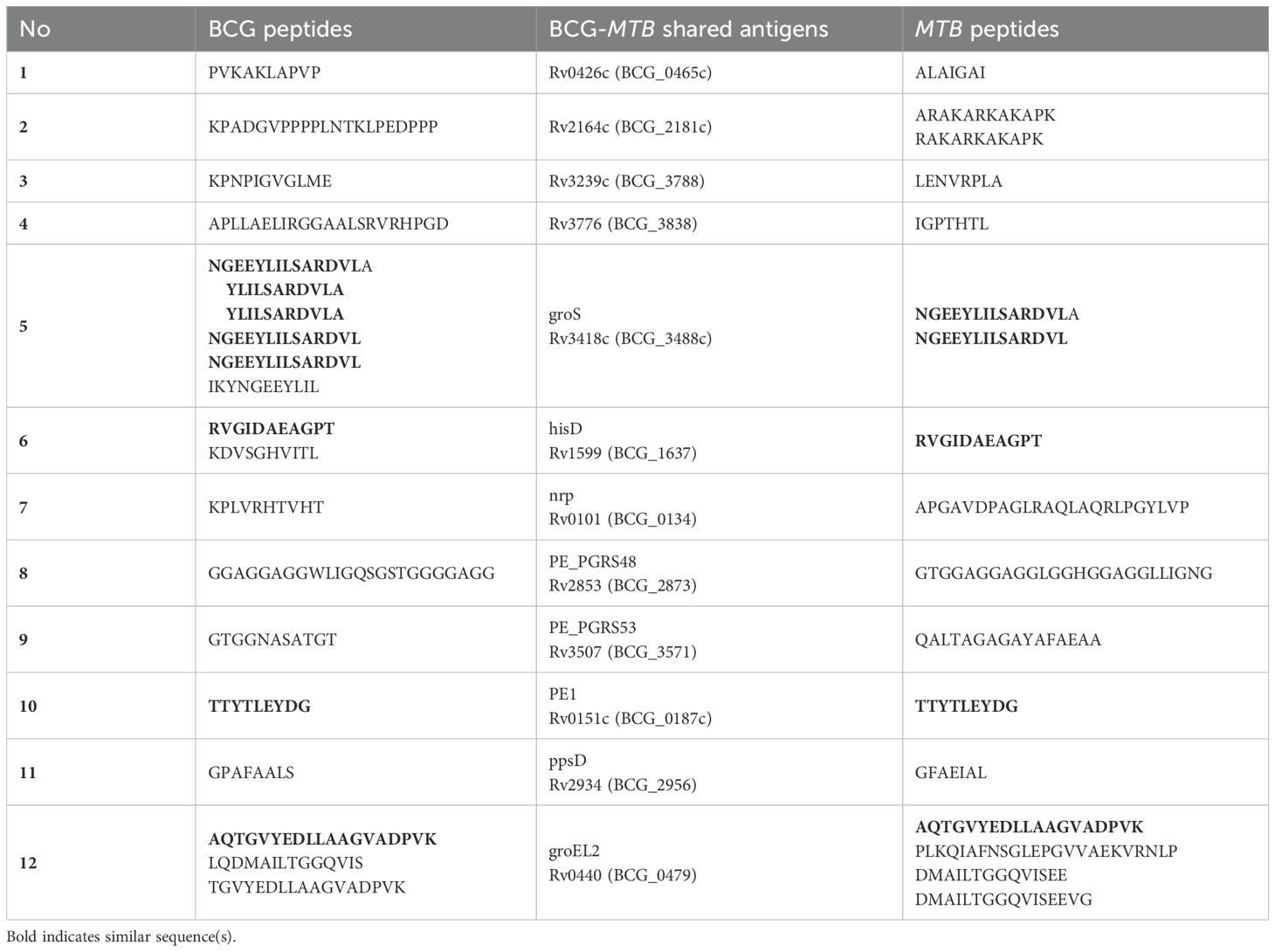
Table 1. Identified BCG and MTB peptides of shared antigens.
2.2 Confirmation of candidate antigen expression in DNA constructs
To evaluate the protective efficacy of the three candidate antigens, DNA constructs encoding each antigen were individually generated (Figure 2). Mice were immunized three times with the DNA constructs at two-week intervals. Two weeks following the final immunization, we employed the ex vivo Mycobacterial Growth Inhibition Assay (MGIA) as a surrogate of protective efficacy (39). The MGIA was performed using splenocytes collected from vaccinated and control animals to measure mycobacterial growth inhibition, as measured by Time to Detection (TTD) (Figure 3).
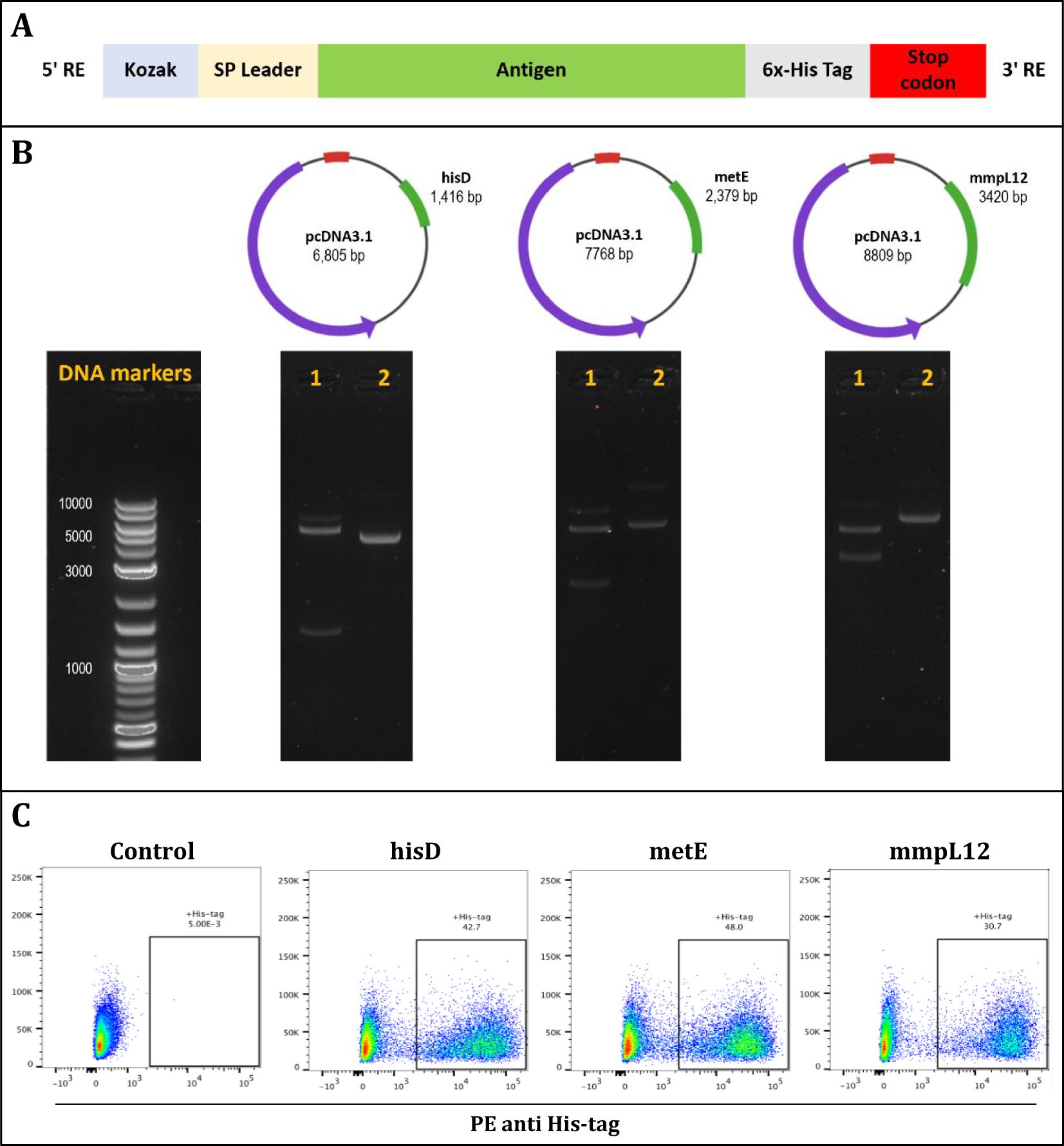
Figure 2. The design and validation of antigen inserts (hisD, metE, and mmpL12). (A) Schematic representation of the plasmid constructs showing insertion of Kozak sequences and signal peptides at the N-terminus, and a 6x-His tag followed by a stop codon at the C-terminus of each insert. (B) DNA agarose gels confirming successful plasmid assembly for hisD, metE, and mmpL12 constructs; lane 1 shows restriction enzyme-digested plasmids with bands at expected insert sizes, lane 2 shows uncut plasmids, and DNA markers are provided as size references. (C) Flow cytometry analysis confirming protein expression of each antigen (hisD, metE, mmpL12) following transfection of HEK293T cells, detected using an anti-6x-His antibody.
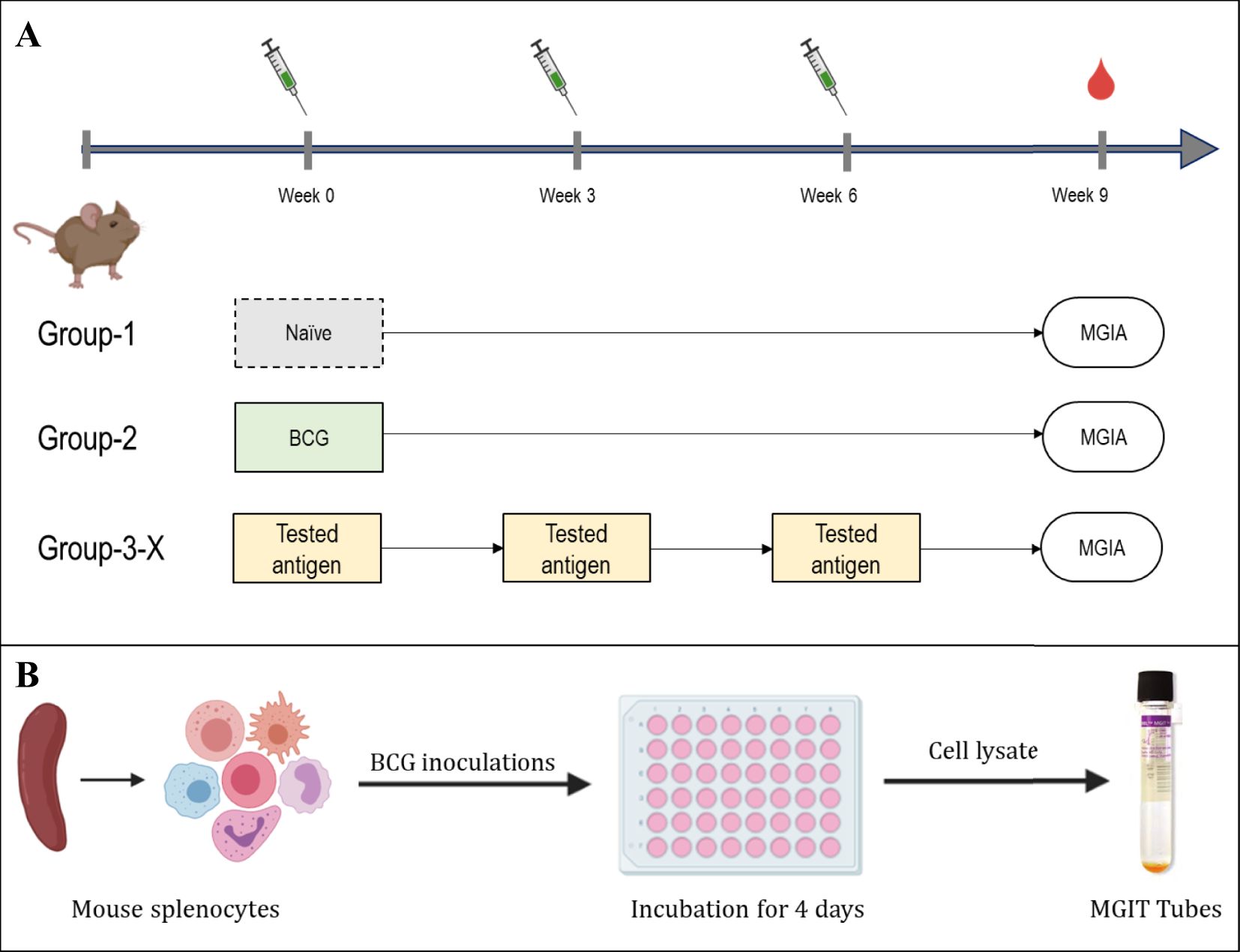
Figure 3. Experimental layout of MGIA experiments. (A) immunisation layout. CB6F1 mice were randomly divided into six groups, each containing eight mice. A single group was given a one-time intradermal BCG vaccination, while four groups designated for antigen testing received plasmid DNA vaccinations three times, with two-week intervals. One group of animals remained unvaccinated. (B) Schematic of the MGIA assay procedure; Splenocytes are co-cultured with approximately 300 or 500 CFU of BCG. After a 96-hour incubation period, cells were lysed, and both extracellular and intracellular bacteria were introduced to the BACTEC MGIT system and monitored until the time to detection (TTD) registered as positive.
Spleen cells (5 × 106 cells/well) were co-cultured with ~500 CFU of BCG. Splenocytes from mice vaccinated with BCG, metE DNA, or mmpL12 DNA showed statistically significant inhibition of mycobacterial growth compared to those from naïve mice (Figures 4A, B). The BCG group had the highest TTD (p = 0.0002), followed by the metE (p = 0.0017) and mmpL12 (p = 0.045) DNA vaccine groups. No significant inhibition of mycobacterial growth was noted in groups vaccinated with hisD (Figure 4A).
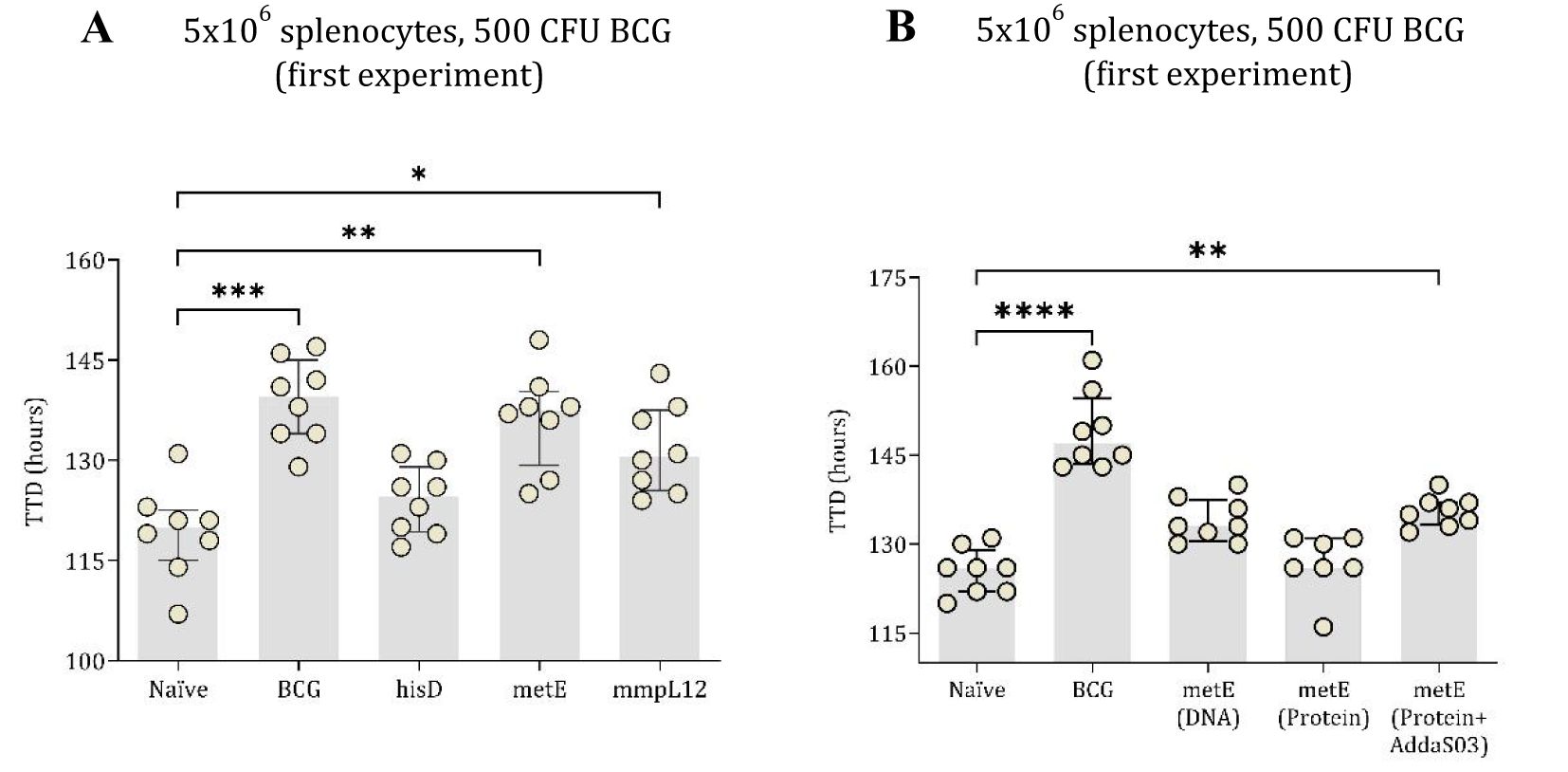
Figure 4. TTD values of mycobacterial growth as measured by MGIA. In the first experiment, mouse splenocytes, counted at 5x106 (A), were co-cultured with 500 CFU of BCG. These splenocytes were taken from both unvaccinated (naïve) mice and those vaccinated with BCG or DNA vaccines expressing hisD, metE or mmpL12. In the second experimental setup, TTD values were derived from 5x106 mouse splenocytes co-cultured with BCG, inoculated at ~500 CFU (B). Splenocytes originated from naïve mice and those vaccinated with BCG, metE, metE-AddaS03™, or metE-DNA. Plotted circles represent individual mice, lines representing median data, and whiskers indicating interquartile ranges (IQR). For identifying significant deviations compared to the naïve control, a combination of the Kruskal-Wallis and Dunn’s multiple comparisons tests was applied. Significance levels are denoted as * for P values <0.05, ** for P values <0.01, and *** indicating P values <0.001.
To further investigate the protective potential of the metE antigen, its efficacy as a protein-adjuvant vaccine was evaluated using the MGIA. In this experiment, 5 × 106 splenocytes were co-cultured with BCG at 500 CFU. Both the BCG-vaccinated group and the metE-AddaS03™ protein-adjuvant group significantly inhibited mycobacterial growth compared to the unvaccinated group (p = 0.0001 and p = 0.01, respectively; Figure 4B). The metE DNA vaccine group did not exhibit significant bacterial inhibition at 500 CFU (p = 0.0604; Figure 4B) but provided protection at BCG inoculum of 300 CFU (p = 0.0311; Supplementary Figure 3).
2.3 Assessment of metE antigen immunogenicity and efficacy using in vivo aerosol MTB challenge studies
Following promising results from the MGIA experiments, we then assessed whether the metE antigen, when delivered as a DNA vaccine or as a protein/adjuvant combination, could protect against an aerosol MTB challenge in mice (Figure 5A). The metE protein was formulated in two distinct adjuvants, AddaS03™ and AS01™, capable of inducing robust immune responses with a high safety profile (31, 40). Given the absence of definitive immune correlates of protection from TB, both formulations were evaluated. Additionally, recent clinical trials have demonstrated the promising efficacy of AS01™ in a candidate subunit TB vaccine (34), prompting us to assess the performance of metE combined with this adjuvant. Vaccinated and control animals were challenged with aerosolized MTB and lungs and spleens were collected for bacterial enumeration at the end of the study (Figure 5A).
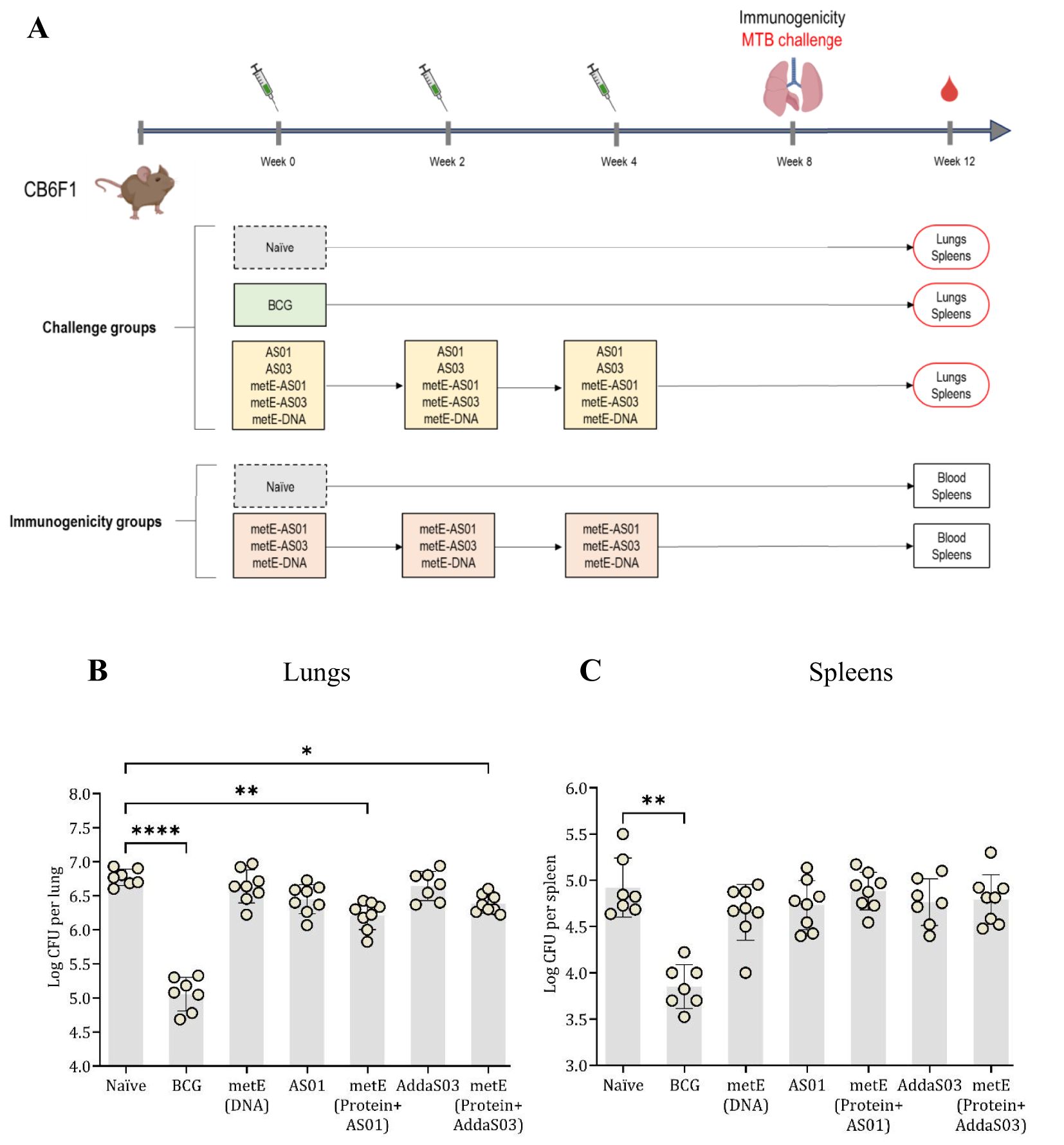
Figure 5. Aerosol MTB-Challenge and Immunogenicity Experiments. (A) MTB challenge, immunisation schedule and experimental groups. (B) lung and (C) spleen CFU counts. Plotted circles represent individual mice, lines representing median data, and whiskers indicating interquartile ranges (IQR). For identifying significant deviations compared to the naïve control, a combination of the Kruskal-Wallis and Dunn’s multiple comparisons tests was applied. Significance levels are denoted as * for P values <0.05, ** for P values <0.01, and **** indicating P values <0.0001.
Vaccination with metE administered as a protein with either adjuvant resulted in a statistically significant reduction in bacterial burden in the lungs compared to the unvaccinated groups (metE + AS01™, p = 0.0027; metE + AddaS03™, p = 0.0381). No protection was observed in the lungs when metE was administered as a DNA vaccine. In the spleen, vaccination with metE, whether administered as DNA or protein with adjuvant, did not significantly reduce bacterial load (Figures 5B, C). The BCG-vaccinated control group had a significant reduction in bacterial load in both lungs and spleen as expected (p = 0.0001 and p = 0.0017, respectively; Figures 5B. C).
Immunogenicity induced by the different formulations of metE antigen was also evaluated. Each formulation induced distinct humoral and cellular immune responses, reflecting a bias towards different T helper cell profiles (Figures 6A-C). MetE protein formulated with AS01™ adjuvant generated a robust Th1-biased CD4+ T cell response, characterized by antigen-specific IFN-γ, TNF-α, and IL-2 production (Figures 6A, B), alongside a broad humoral response with statistically significantly elevated IgG1, IgG2a, IgG2b, and IgG2c levels compared to naïve group (Figure 6C). In contrast, metE formulated with AddaS03™ elicited a mixed immune response with a moderate Th1 CD4+ T cell response marked by TNF-α and IL-2 expression and a prominent Th2-skewed response, as evidenced by significantly increased IL-4 production compared to naïve group (Figures 5A, B). The antibody response for the metE-AddaS03™ group was consistent with this Th2 bias, showing statistically significant IgM and IgG1 responses but lower levels of IgG2a and IgG2c (Figure 6C). The metE DNA vaccine primarily induced a Th1-biased immune response of CD4+ T cells producing IFN-γ, TNF-α, and IL-2, but failed to elicit a strong antibody response, with no statistically significant increase in any of the measured immunoglobulin subclasses compared to naïve group (Figures 6A–C). We did not detect a statistically significant CD8+ T cell cytokine response following metE vaccination (Supplementary Figure 4). This may be attributed to the stimulation conditions used in the ICS assay, where the native metE protein was applied instead of a peptide pool, which failed to elicit a response. Both the AddaS03™ and AS01™ metE vaccine formulations induced significantly elevated IL-17 production compared to the naïve group, as detected by ICS and ELISpot assays (Figures 6A, B).
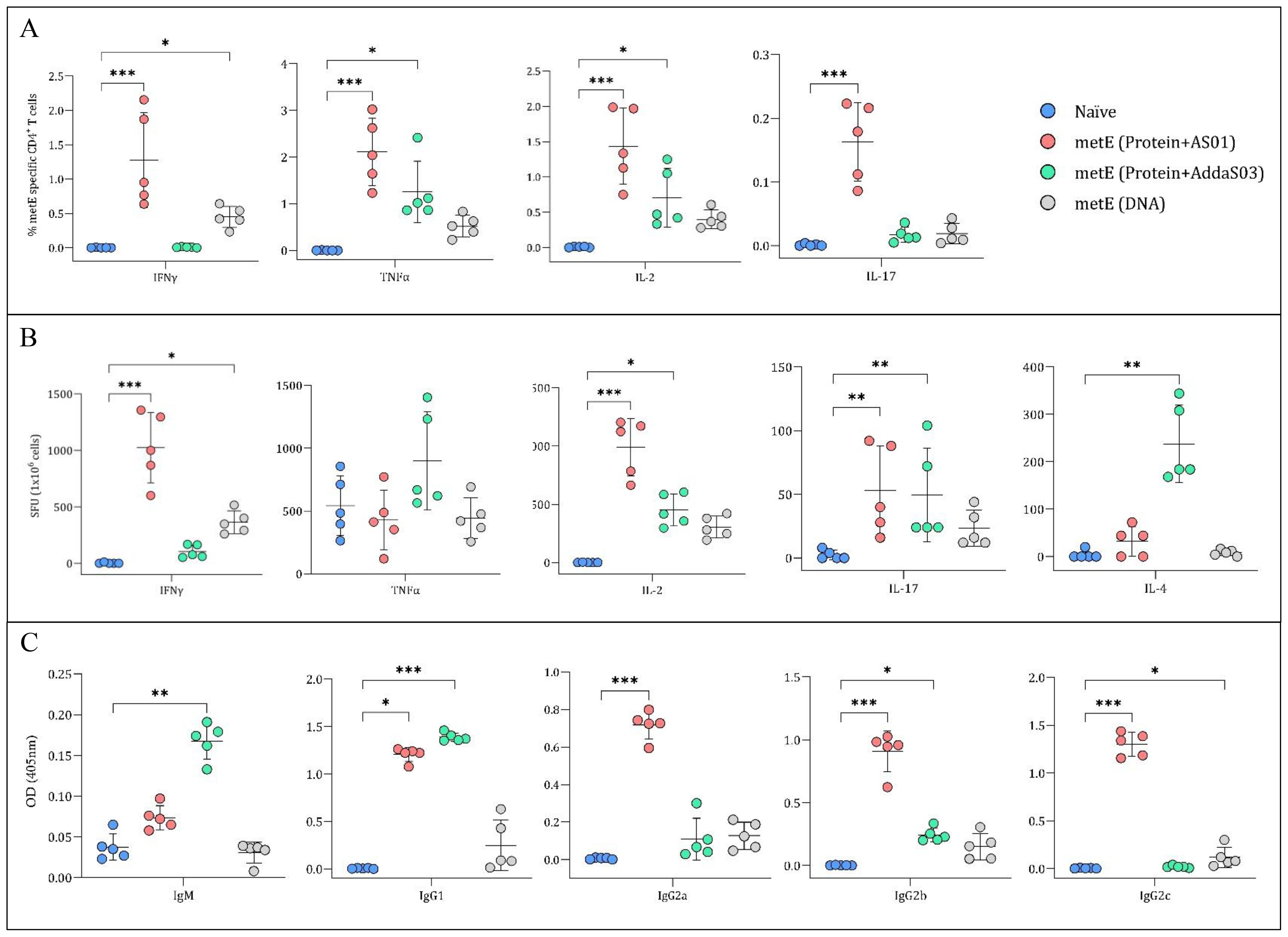
Figure 6. Analysis of Cytokine Secretion and metE-specific Antibody Titres. (A) Percentage of CD4+ T cells producing different cytokines. Mouse splenocytes were stained for CD4+ surface markers and cytokines (IFNγ, TNFα, IL-2, and IL-17) after overnight stimulation with metE protein. Cytokine staining was analysed by flow cytometry. (B) Cytokine secretion measured by ELISpot. Splenocytes were stimulated overnight with metE protein to measure the release of IFNγ, TNFα, IL-2, IL-4, and IL-17 cytokines. (C) MetE-specific antibody titers. Antibody titers (IgM, IgG1, IgG2a, IgG2b, and IgG2c) were analysed from mouse sera collected after the final vaccination by ELISA. Each plotted circle represents individual mice, with lines denoting median values. Significant differences compared to the naïve control were identified using the Kruskal-Wallis test with Dunn’s multiple comparisons test. Significance markers: * for P values <0.05, ** for P values <0.01, and *** for P values <0.001.
3 Discussion
Our study identified metE as a promising protective TB vaccine candidate through a combined immunopeptidomics and immunoinformatics approach. We demonstrated that metE, when formulated as a recombinant protein with either AS01™ or AddaS03™ adjuvants, provided significant protection against MTB in an in vivo challenge model, as evidenced by a reduction in bacterial burden compared to the naïve group. The metE-AS01™ formulation induced a robust Th1-skewed immune response, whereas the metE-AddaS03™ formulation elicited a mixed Th1/Th2 response. These findings suggest that metE could effectively enhance both cellular and humoral immune responses, both of which may contribute to protection against TB.
Building on our previous published work, we employed an immunopeptidomic approach to identify surface-presented mycobacterial antigens following infection of THP-macrophages with BCG or MTB (27). This approach led to the identification of 64 and 80 new mycobacterial antigens, respectively. We used an in silico-based approach to analyze the repertoire of MHC-bound peptides present in each identified antigen, allowing us to optimize candidate antigen selection based on epitope richness. As a result, three antigens (hisD, metE and mmpL12) were selected for protective efficacy evaluation. These antigens are present in both BCG and MTB, and their immunogenicity and protective efficacy has not been studied previously.
HisD (histidinol dehydrogenase) has been identified as a potential target for novel anti-TB drugs due to its essential role in histidine synthesis (41), countering immune cell-mediated histidine degradation to limit MTB proliferation (42). MetE (methionine synthase), found in cytosol, cell wall, cell membrane, and culture filtrate (43, 44), is essential for methionine synthesis, crucial for protein synthesis and cellular processes in MTB (45). Deleting the metE gene from MTB significantly hampers MTB growth (45, 46). Additionally, metE has been reported as a highly immunogenic antigen, playing a key role in the host immune response and being secreted at different stages of MTB growth (47). mmpL12 (mycobacterial membrane protein large) functions as a membrane transporter with broad substrate specificity, including glycolipid transportation (48). Although mmpL12 has been identified as a potential protective antigen through reverse vaccinology (49), which relies on computational tools to predict candidate antigens, its protective efficacy had not been tested prior to our study.
We focused first on evaluating the protective efficacy of these three antigens, and planned to perform detailed immunogenicity evaluation on any antigens found to be protective, given the lack of defined and validated immune correlates of protection for TB (50). Although antigen immunogenicity is important, it does not always translate to protection, as exemplified by numerous mycobacterial antigens that elicit IFNγ release but fail to confer protective effects against MTB challenge (51). Furthermore, CD4+ T cells can offer IFNγ-independent immunity against TB (52). The ex vivo MGIA provides an unbiased tool for high-throughput down-selection of promising protective antigens (53). The utility of the MGIA as a surrogate of protective efficacy has been demonstrated in mice, non-human primates and humans, allowing standardization and consistency in vaccine evaluation across different species (54–57). We demonstrated that mice immunized with metE, administered as a protein formulated with adjuvant, showed a significant protective effect compared to unvaccinated control mice.
Both metE-DNA and metE-AS01™ vaccine candidates induced a Th1-skewed phenotype, characterized by the expression of IFNγ, TNFα, and IL-2. In contrast, the metE-AddaS03™ vaccine candidate elicited a mixed Th1/Th2 immune response, marked by the production of TNFα, IL-2, and IL-4. The diverse CD4+ T cell phenotypes following metE immunization suggests multiple, potentially complementary, protective pathways, contingent on the delivery and adjuvant system. The consistent presence of TNFα and IL-2 indicates their significant roles in conferring protection independent of the presence of IFNγ (58).
The influence of the adjuvant system on the antibody response has also been observed. Studies in mice have shown that a high concentration of IgG2a is indicative of a Th1 response, while IgG1 presence suggests a Th2 response (59). This observation aligns with the Th1 and Th2 cellular responses triggered by metE-AS01™ and metE-AddaS03™ adjuvants, respectively. Similarly, in humans, the promising M72/AS01 subunit vaccine not only elicited effective T cell responses but also induced sustained production of IgG antibodies against the M72 antigen for up to three years (34, 60). This highlights the importance of choosing appropriate adjuvants in vaccine design to induce the desired immune response.
The challenges associated with interpreting murine spleen CFU data after aerosol challenge have previously been highlighted (61). It is unclear whether the mycobacteria are reaching the spleen through hematogenous disseminated infection or have disseminated into the lung capillaries and hence to the spleen. In our study, metE delivered as an adjuvanted protein conferred significant protection against pulmonary MTB infection, but this protection did not extend to the spleen. Several factors could potentially explain this difference. One explanation could be the susceptibility of the mice to MTB infection and/or the high dose of aerosol MTB used in our challenge, as evidenced by the CFU count of ~107 CFU in the naïve group. Similar observations reported previously suggested that the inherent replicative ability of BCG might confer a more robust and sustained protective immunity against extrapulmonary TB in contrast to subunit vaccines (62–65). However, the encouraging results in the lung indicate that metE delivered systemically as adjuvanted protein can effectively induce protective immunity at the primary site of infection, the lung. This is important as some candidates have demonstrated lung-specific protection exclusively when administered via the pulmonary route compared to the parenteral administration (66, 67).
Our findings suggest that the magnitude of the T cell response is a significant factor in the level of protection conferred against MTB challenge. This is evident from the contrasting results of metE-DNA vaccination and metE-protein-adjuvant vaccination in vivo. The latter elicited a stronger T cell and antibody response, and metE-DNA vaccination, which induced a lower magnitude of response, provided no protection in vivo against MTB challenge. This observation aligns with clinical data comparing various candidate TB vaccines (MVA85A, AERAS-402, H1:IC31, H56:IC31, M72/AS01, ID93/GLA-SE, and BCG) (14), where all vaccines triggered antigen-specific CD4+ T-cell responses, predominantly featuring Th1 cytokine production. However, the magnitude of Th1 cells generated was a key differentiator among the vaccines evaluated. Despite similar cytokine profiles across all vaccines, M72/AS01 was distinguished by its ability to elicit a more robust memory CD4+ T-cell response. This suggests that the magnitude and durability of the CD4+ T cell response, rather than the specific cytokine profile, might be a more reliable indicator of protection against MTB.
4 Conclusions
Our immunopeptidomic approach has led to the identification of novel protective antigens for TB, and demonstrated the importance of delivery system, as well as antigen, in protection conferred by a subunit vaccine. One of the antigens identified, metE, conferred significant protection against MTB in mice when delivered as a protein/adjuvant vaccine candidate, suggesting its potential for inclusion in the next-generation of TB vaccines. Further exploration of heterologous prime-boost immunization regimes and evaluation in other preclinical animal models will confirm the role of this novel antigen in a candidate TB vaccine regimen.
5 Methods
5.1 Cell culture
The THP-1 human leukemic monocytic cell line, obtained from the American Type Culture Collection, was cultured in RPMI-1640 medium (R0883-500ML, Sigma-Aldrich) supplemented with 10% heat-inactivated FCS, 2 mM L-glutamine, and 100 U/ml penicillin-streptomycin, and incubated at 37°C in a 5% CO2 atmosphere. BCG was grown in Middlebrook 7H9 broth (BD Biosciences, UK) with 50 μg/ml hygromycin (Merck Life Science UK Limited), 10% OADC enrichment (BD Biosciences, UK), 0.2% glycerol, and 0.05% tyloxapol (Sigma-Aldrich, UK), at 37°C with 200 rpm shaking. MTB was cultured under similar conditions but without shaking.
For infection experiments, 3 x 108 THP-1 cells were differentiated into macrophages using 20 nM Phorbol 12-Myristate Acetate (PMA, Sigma-Aldrich, UK) overnight, then treated with 50 ng/ml IFNγ (Immuno Tools, Germany) and 1 μg/ml anti-IL10 (BD Biosciences, UK) for one day. MTB in exponential growth phase was centrifuged at 3,000 g for 10 minutes, washed, and resuspended in antibiotic-free medium. Bacteria were de-clumped by centrifugation at 300 g for 2 minutes and sonicated for 10 minutes in 10 cycles (30 seconds in a sonication bath and 30 seconds in ice). On the day of infection, THP-1 cells were washed and infected with mycobacteria at an MOI of five, then washed thrice after 4 hours to remove excess bacteria and resuspended in fresh antibiotic-free medium. After 24 hours, cells were harvested and stored at -20˚C.
To obtain bacteriological sterility prior to conducting immunopeptidomics experiments, the cell pellet was lysed using 2x lysis buffer and complete™ protease inhibitor cocktail (Roche) for 30 minutes on ice, with periodic shaking every 10 minutes. Post-lysis, the mixture was centrifuged at 500 g for 10 minutes to separate nuclei. The resulting supernatant was then filtered twice through centrifugal filters (0.22 µm, hydrophilic PVDF, 0.5 mL, Millipore) at 2000 g for 5 minutes each. The clear filtrate was collected in a new 15 ml Falcon tube for immunoprecipitation (IP).
5.2 IP of peptide-MHC complex
Our previous study consisted of THP-1 cells infected with live BCG, treated with heat-killed BCG, and stimulated with a cocktail of IFN-γ and anti-IL10, to improve MHC-II antigen expression (27). The cell lysates were subjected to two immunoprecipitation procedures to isolate HLA-DR, DQ, DP and HLA-A, B, C, using the L243 and W6/32 antibodies, respectively. In the present study, we subjected the same cell lysates to two additional immunoprecipitation procedures. Given that L243 is well known to strongly bind to HLA-DR, we immunoprecipitated the lysates with HLA-DP-specific antibody (B721), followed by the use of another pan-class II antibody (IVA12) to isolate HLA-DQ molecules (1 mg each per 1x108 cells). Briefly, the antibodies were cross-linked to Protein A Sepharose beads with dimethyl pimelimidate (Sigma) and lysates were incubated overnight with each antibody. Beads were subsequently washed with 10 column volumes (cv) of 1x150 mM NaCl, 5 mM EDTA in 50 mM Tris, 1x150 mM NaCl in 50 mM Tris, 1x450 mM NaCl in 50 mM Tris and 50 mM Tris buffer without salt. Peptides bound to the HLA molecules were dissociated upon mild acid elution with 5 ml 10% acetic acid to denature α and β subunits of MHC molecules. The same protocol was applied to MTB IP with some modifications as follows. The IP order was B721 (HLA-DP), IVA12 (pan-MHC-II) and W632 (pan-MHC-I). A volume of 2 ml of Sepharose beads was used for 5 mg antibody per sample. Each sample was incubated with antibodies twice (first incubation overnight and second incubation for one hour).
HLA peptides were purified using reverse-phase high-performance liquid chromatography (RP-HPLC). Using the Ultimate 3000 HPLC system (Thermo Fisher Scientific), samples were injected into a 4.6x50 mm ProSwift RP-1S column (Thermo Fisher Scientific). They were then eluted over 10 minutes with a buffer gradient of 2% to 35% buffer B (0.1% TFA in acetonitrile) at a flow rate of 1000 µl/min. Fractions lacking ß2-microglobulin were pooled into two combined final fractions. These samples were dried, reconstituted in 20 µl of loading buffer (0.1% TFA, 1% acetonitrile), and stored at −20°C for subsequent mass spectrometry (MS) analysis.
For MS and data analysis, peptides were analyzed using nano-LC-MS-MS on an Ultimate 3000 RSLCnano System (PepMap C18 column, 2 µm, 75 µm × 50 cm; Thermo Fisher Scientific) connected to either an Orbitrap Fusion Lumos Tribrid or a Q Exactive HF-X mass spectrometer (both from Thermo Fisher Scientific). HLA-I peptides were separated with a 60-min gradient from 3% to 25% acetonitrile in 5% DMSO/0.1% formic acid, while HLA-II peptides used a gradient from 3% to 30% acetonitrile, both at 250nl/min flow rate.
For the Orbitrap Fusion Lumos Tribrid, full MS acquisition was at 120,000 resolution (300–1500 m/z range) with an AGC target of 400,000. Peptides were selected in a top-speed cycle of 2s, isolated at 1.2 amu, with an accumulation time of 120ms. MS-MS was at 30,000 resolution, AGC target of 300,000, using high-energy collisional dissociation (HCD) at 28V for peptides with 2–4 charge states and 32V for singly charged ions.
For the Q Exactive HF-X, full MS acquisition was at 120,000 resolution (320-1600m/z range) with an AGC target of 300,000. The top 20 precursor ions were fragmented at 1.6 amu isolation width for 120ms, resulting in a 2.5s cycle time. MS2 resolution was at 60,000 with an AGC target of 50,000, using HCD fragmentation at 25V collision energy.
For immunopeptidomics data analysis, MS data were processed using Peaks V8 and V10 (Bioinformatics Solutions). The spectra were matched against the proteomes of humans and BCG (Pasteur 1173P2) or MTB (H37Rv), both sourced from the UniProt database, to identify peptide spectrum matches (PSMs). Identified peptides and proteins were exported from Peaks with a minimum score threshold of -10logP=15. The exported data in Excel format were further analyzed to discern mycobacterial peptides and proteins, following these steps:
● Exclusion of all human peptides/proteins.
● Removal of any mycobacterial peptides/proteins found in the control sample from the infected samples.
● Detailed analysis of the mycobacterial peptides/proteins present in the infected samples.
5.3 T-cell MHC-II prediction
We adopted an in-silico approach utilizing immunopeptidomics and immunoinformatics tools to efficiently shortlist potential antigen candidates. The CD4+ T-cell epitopes were predicted using the NetMHCIIpan server (https://services.healthtech.dtu.dk/) with default settings for 15-mer peptides. The selected antigens were analyzed based on their entire sequences to determine which antigens contained a high number of T-cell epitopes. Binding affinity thresholds of 1% and 5% identified strong and weak binders, respectively. Predictions were made against 27 human HLA (DRB1*0101, DRB1*0301, DRB1*0401, DRB1*0405, DRB1*0701, DRB1*0802, DRB1*0901, DRB1*1101, DRB1*1201, DRB1*1302, DRB1*1501, DRB3*0101, DRB3*0202, DRB4*0101, DRB5*0101, DQA1*0501/DQB1*0201, DQA1*0501/DQB1*0301, DQA1*0301/DQB1*0302, DQA1*0401/DQB1*0402, DQA1*0101/DQB1*0501, DQA1*0102/DQB1*0602, DPA1*0201/DPB1*0101, DPA1*0103/DPB1*0201, DPA1*0103/DPB1*0401, DPA1*0301/DPB1*0402, DPA1*0201/DPB1*0501 and DPA1*0201/DPB1*1401). Given the relevance of mouse models at contributing to antigen presentation in the TB field, we included eight mouse MHC alleles (H-2-Dd, H-2-Db, H-2-Kd, H-2-Ld, H-2-Iad, H-2-IEd, H-2-Kb and H-2-IAb) relevant to the CB6F1 mouse strain. Mycobacterial proteins with a wide range of epitopes covering all these alleles were chosen for further analysis. HLA allele coverage was calculated by mapping the predicted epitopes against the aforementioned list of MHC alleles. Coverage for each antigen was determined based on the presence of predicted epitopes, categorized as either strong or weak binders, across the tested alleles. An antigen was considered to have 100% coverage if it contained epitopes for all tested alleles. For antigens with epitopes covering fewer alleles, coverage was calculated using the following equation:
Mycobacterial peptides derived from BCG and MTB were predicted against THP-1 cell HLA alleles, as reported by Bettencourt et al. (27), to predict the most likely associated allele, which is described as the “best hit.”
5.4 Non-homologous protein identification
To identify mycobacterial proteins that are non-homologous to the human proteome, each mycobacterial protein sequence was analyzed using the Vaxign server (https://violinet.org/vaxign/) for similarity with human proteins. The homology between sequences was assessed using the BLAST protein database, with smaller E-values indicating higher similarity.
5.5 Escherichia coli transformation
Competent E. coli bacteria were mixed with 5 μl DNA (~100ng, synthesized by Biomatik, USA) and incubated on ice for 20–30 minutes, heat-shocked at 42°C for ~45 seconds, and then cooled on ice for 2 minutes. Cultured in 500μl LB or SOC medium (New England Biolabs) at 37°C with shaking for 45 minutes, bacteria were then plated on LB agar with carbenicillin, using direct and streak-plate techniques. A selected colony was incubated in 10ml LB with carbenicillin at 37°C overnight with vigorous shaking. The starter culture was diluted 1:500 in fresh LB with carbenicillin and further incubated at 37°C overnight. Harvesting involved centrifuging at 4000g for 15 minutes and discarding the supernatant, with an option to freeze the pellet at -20°C.
5.6 Plasmid purification
Following the manufacturer’s instructions from the PureLink™ HiPure Expi Plasmid Gigaprep Kit (Thermo Fisher Scientific), bacterial pellets were processed with resuspension, lysis, and precipitation buffers. After centrifugation, the supernatant underwent filtration and vacuum application. Wash and endotoxin removal steps were followed by DNA binding and washing using a DNA-binding cartridge. The plasmids were then eluted with endotoxin-free buffer, precipitated with isopropanol, washed with ethanol, and dried. Finally, the DNA was resuspended in endotoxin/DNase-free water and its concentration measured using a NanoDrop™ UV-Vis Spectrophotometer (Thermo Fisher Scientific), with the purified DNA stored at -20°C.
5.7 Agarose gel electrophoresis
Agarose gel electrophoresis was performed to verify DNA insert sizes. Digested DNA in 0.5ml tubes was incubated at enzyme-specific temperatures for 5–15 minutes. A 1% agarose gel with 10μl of SYBR Safe stain was set in a comb-equipped tray and solidified for 15–20 minutes. The gel, placed in a tank with 1x TAE buffer, was loaded with the DNA mixture (25μl sample + 5μl 6x loading dye) and 5μl ladder, then run at 100V for 50 minutes. Post-electrophoresis, the gel was imaged using the Gel Doc EZ System (Bio-Rad).
5.8 Flow cytometry
To verify intracellular expression of antigens by the pDNA3.1 plasmid human embryonic kidney cells (293T) were cultured in six-well plates in RPMI-1640 medium (R0883-500ML, Sigma-Aldrich) supplemented with 10% heat-inactivated FCS, 2mM L-glutamine, and 100 U/ml penicillin-streptomycin at 37°C until they reached 70-80% confluency. Then, the cells were transfected with 1 µg of pDNA3.1 plasmid using a 1:3 DNA: PEI ratio for 2–3 hours. Post-transfection, cells were incubated for 24 hours in complete RPMI medium. Cells were harvested with 0.5 ml trypsin, fixed in 100 μl of 4% paraformaldehyde, and permeabilized with 100 μl of 0.1% saponin. They were stained with PE-anti-his-tag antibody (1:50 dilution) and washed. The analysis was conducted using an LSRII™ flow cytometer (BD Biosciences), with data processed in FlowJo® software.
5.9 Animal work
All procedures were performed in accordance with the UK Animals (Scientific Procedures) Act 1986, under project license number 30/2889 and P9804B4F1, granted by the UK Home Office. All procedures followed institutional and national ethical guidelines, approved by the University of Oxford’s Animal Welfare and Ethical Review Board (AWERB), and adhered to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.
5.9.1 Immunization experiments
Specific-pathogen-free female CB6F1 mice (6–9 weeks old, Charles River UK Ltd.) were used for all experiments. All injections were performed under short-term anesthesia using vaporized isoflurane. Mice were humanely euthanized by cervical dislocation at the end of each experiment. BCG (Pasteur strain) used for vaccination was prepared in-house in 7H9 broth supplemented with 10% ADC enrichment medium and 0.05% Tween 80 (Becton Dickinson, UK). All DNA vaccines were synthesized by Biomatik (USA), and MetE protein was synthesized by BiologicsCorp (USA). AddaS03™ (InvivoGen, UK) and AS01 (obtained from SHINGRIX vaccine, Oxford University Hospitals) were used as adjuvants.
In the first experiment MGIA, mice were divided into six groups of eight. One group received 50 µL intradermal BCG (3.5 × 105 CFU/mouse), administered once to the ears (25 µL per ear). Four groups received DNA vaccines (hisD, MetE, mmpL12; 100 µg/100 µL phosphate-buffered saline [PBS]/mouse), delivered intramuscularly into each thigh (50 µL per thigh) three times at two-week intervals. One group served as an unvaccinated control. Spleens and blood were collected two weeks after the final vaccination.
In the second MGIA experiment, mice were divided into five groups of eight. One group received 50 µL intradermal BCG (3.5 × 105 CFU/mouse), administered once to the ears (25 µL per ear). Three groups received intramuscular MetE vaccinations: metE protein alone (1 µg/100 µL PBS/mouse), metE-DNA (100 µg/100 µL PBS/mouse), and metE protein combined with AddaS03™ adjuvant at a 1:1 volume ratio (1 µg/100 µL PBS/mouse). Vaccinations were administered into each thigh (50 µL per thigh) three times at three-week intervals. One group served as an unvaccinated control. Spleens and blood were collected three weeks after the final vaccination.
In the Immunogenicity experiment, mice were divided into four groups of five. Three groups were vaccinated three times at three-week intervals with the following formulations: metE-DNA (100 µg/100 µL PBS/mouse), MetE protein alone (1 µg/100 µL PBS/mouse), and 1 µg metE protein combined with AddaS03™ adjuvant at a 1:1 volume ratio in a total of 100 µL PBS/mouse, and AS01 adjuvant (5 µg monophosphoryl lipid A [MPL]/5 µg QS-21 in a total of 100 µL PBS/mouse, representing 1/10th of the human dose of AS01). Vaccinations were delivered into each thigh (50 µL per thigh). One group served as an unvaccinated control. Spleens and blood were collected three weeks after the final vaccination.
In the MTB-Challenge experiment, mice were divided into six groups of eight. One group received 50 µL intradermal BCG (Pasteur strain, 3.5 × 105 CFU/mouse), administered to the ears (25 µL per ear). Five additional groups were vaccinated three times at three-week intervals with the following formulations: AddaS03™ adjuvant at a 1:1 volume ratio in a total of 100 µL PBS/mouse, AS01 adjuvant (5 µg MPL/5 µg QS-21 in a total of 100 µL PBS/mouse), metE-DNA (100 µg/100 µL PBS/mouse), 1 µg metE protein combined with AddaS03™ adjuvant at a 1:1 volume ratio in a total of 100 µL PBS/mouse, and AS01 adjuvant (5 µg MPL/5 µg QS-21 in a total of 100 µL PBS/mouse). Vaccinations were delivered into each thigh (50 µL per thigh). One group served as an unvaccinated control. Four weeks after the final vaccination, all mice were infected with 1 × 106 CFU/ml of MTB Erdman K01 (TMC107, BEI Resources, USA) using a Biaera Aero-MP-controlled nebulizer (Biaera Technologies, USA). The bacterial load in the lungs was determined to be 50–100 CFU one hour post-infection in two mice. At the end of the experiment, four weeks post-infection, lungs and spleens were collected and homogenized in reinforced tubes (Stretton Scientific) using a Precellys 24 homogenizer. Homogenates were serially diluted in PBS, plated on Modified 7H11 plates (Animal and Plant Health Agency, UK), incubated at 37°C for 4–6 weeks, and then enumerated for CFUs.
5.9.2 MGIA
Splenocytes were isolated by mashing spleens in a six-well plate, rinsing with PBS, and filtering through a 70 μm strainer into a 50 ml Falcon tube. Post-centrifugation at 350 g for 5 minutes, cells were lysed in 3 ml ACK buffer for 5 minutes, then diluted to 35 ml with RPMI with HEPES buffer and without antibiotics (RPMI-MGIA) and centrifuged again. The pellet was resuspended in 2 ml RPMI-MGIA, adjusting splenocyte concentration to 3x106 or 5x106 in 600μl for co-culture with BCG (300 or 500 CFU) in 48-well plates, and incubated for 96 hours at 37°C with CO2.
Well contents were transferred to 2 ml tubes and centrifuged at 12,000 rpm for 10 minutes. Concurrently, 500 µl sterile water was used to lyse remaining cells in each well. Supernatants were discarded, and water lysates were combined with the tube contents. After pulse-vortexing, the mixture was transferred to BACTEC mycobacteria growth indicator tubes (MGITs) with PANTA (polymyxin-B, amphotericin-B, nalidixic acid, trimethoprim, azilocillin) antibiotics and Oleic Albumin Dextrose Catalase (OADC) enrichment broth (Becton Dickinson, UK) until positivity was reached and expressed as TTD.
5.9.3 Enzyme-linked immunosorbent assay
Blood samples from mice were collected into 2 ml tubes, stored overnight at 4°C, and centrifuged at 2000 g for 10 minutes. Serum was extracted and stored at -20°C. Maxisorp 96-well plates (Thermo Fisher Scientific) were coated with 2.5 µg/ml recombinant metE in PBS and incubated overnight at room temperature. After washing with PBS/Tween-20 and blocking with casein buffer for 1–2 hours, diluted mouse sera (initial dilution 1:100) were serially diluted and incubated for 2 hours. Plates were washed and treated with alkaline phosphatase-conjugated anti-mouse secondary antibodies (BioRad) at a 1:3000 ratio for 1 hour. Development buffer (1mg/ml 4-nitrophenylphosphate in diethanolamine buffer) was added, and optical density was measured at 405nm using a BioTeK Microplate Reader. Adjusted optical densities were obtained by subtracting blank well averages from sample averages.
5.9.4 Enzyme-linked immunospot
Splenocytes were used in ELISpot assays to measure cytokine secretion upon metE stimulation. MultiScreen-IP filter plates (Millipore) were coated with 50 μl of 10 μg/ml anti-mouse cytokine antibodies (IFNγ, IL-2, IL-4, IL-17; Mabtech) and stored at 4°C overnight. After washing and blocking with complete media for 2 hours at 37°C, 50 μl of splenocyte suspensions (2.5 x 105 cells) and 50 µl of either media (negative control) or 10 μg/ml metE (dissolved in the same media) were added to each well. Following overnight incubation, plates were washed and treated with 50 µl/well of 1:1000 diluted biotinylated goat anti-mouse IgG (Mabtech) for 2 hours. After subsequent washing, 50μl/well of 1:1000 diluted streptavidin-ALP (Mabtech) was added, incubated for 1 hour, and then developed with 50 μl/well of AP Conjugate sub kit (BioRad) for 5–10 minutes. Post-wash, spots were counted using an ELISpot reader (AID Germany), with results reported as spot forming units (SFU) per 1x106 cells, adjusting for unstimulated cell counts.
5.9.5 Intracellular cytokine staining
Splenocytes were prepared and stimulated in 96-well U-bottom plates with 1 million cells/well. Stimulation involved 10 μg/ml metE protein for metE vaccinated groups, 10 μg/ml tuberculin-purified protein derivative (PPD) for the BCG vaccinated group, and complete RPMI-1640 medium for unstimulated controls. This was accompanied by anti-CD28 and anti-CD49d (1 μg/ml each, BioLegend Inc.). Plates were incubated for 2 hours at 37°C with 5% CO2, followed by 4–6 hours with 1 μl/ml GolgiPlug (BD Biosciences), and then overnight at 4°C.
Post-incubation, cells were centrifuged, washed with PBS, and stained. Unstained wells received 10μl PBS, while others received 10 μl live/dead stain (1:200, Invitrogen, UK) for 10 minutes. Surface staining was conducted with 40 μl of antibody-cocktail 1 (Table 2), followed by a 30-minute incubation at 4°C. After washing with BSA/PBS, cells were fixed with Cytofix/Cytoperm (BD Biosciences) for 10 minutes and permeabilized with Perm/Wash solution.
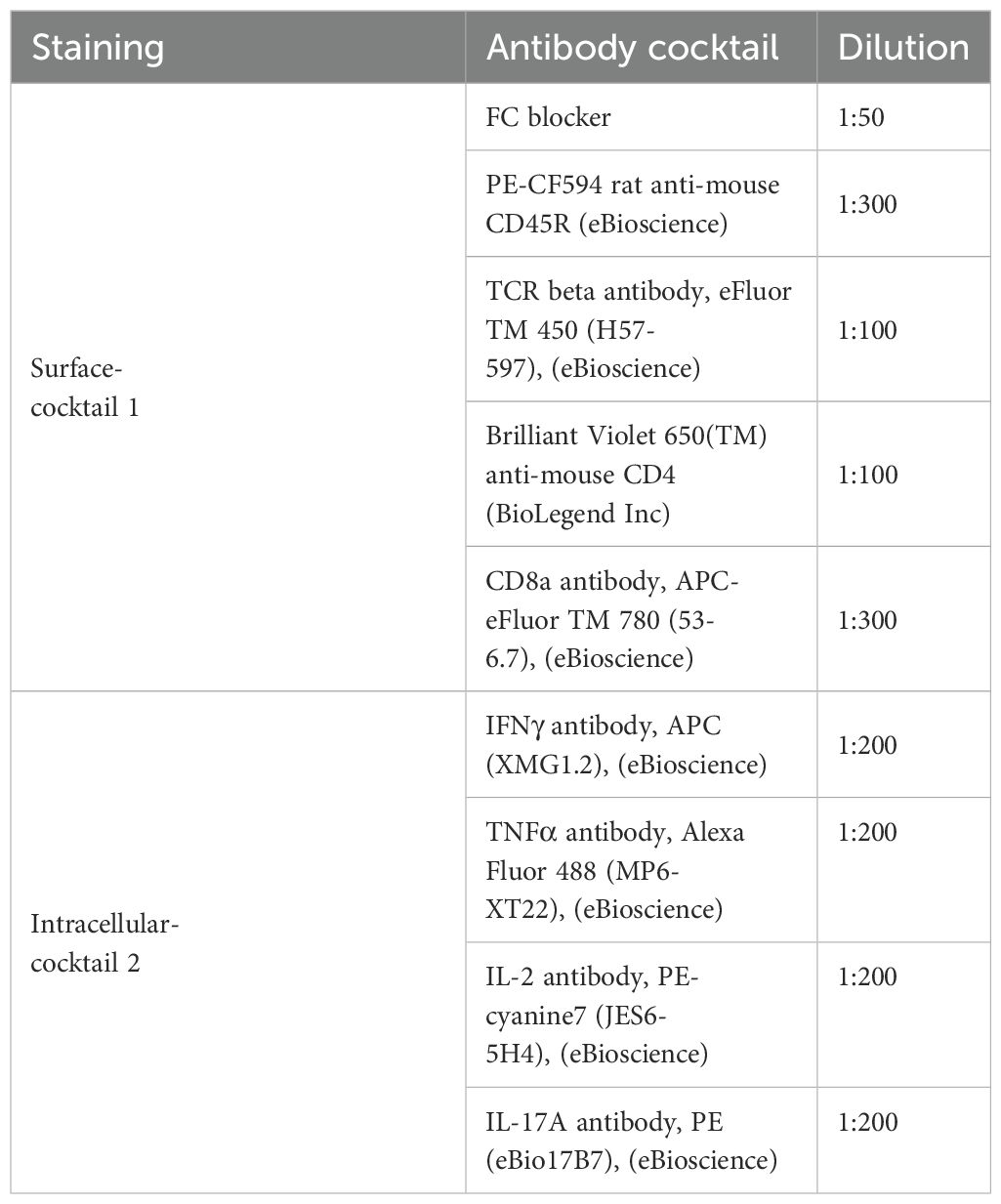
Table 2. Composition and dilution of antibodies used for surface and intracellular staining.
Intracellular Cytokine Staining (ICS):
Post-permeabilization, cells underwent intracellular cytokine staining using 40 μl of antibody-cocktail 2 (Table 2) in Perm/Wash solution, incubated for 30 minutes at 4°C. Following two washes with Perm/Wash and PBS/BSA solution, cells were resuspended in 80 μl PBS/BSA and analyzed on an LSR-II™ flow cytometer with FACSDiva™ software (BD Biosciences). Data were processed using FlowJo® software, applying the gating strategy outlined in Supplementary Figure 5.
5.10 Statistical analyses
Statistical analysis was conducted using GraphPad Prism version 8. The Mann-Whitney test compared two groups, while the Kruskal-Wallis test with Dunn’s multiple comparison post-test was utilized for analyses involving multiple groups. A p-value of less than 0.05 was considered statistically significant.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by the University of Oxford’s Animal Welfare and Ethical Review Board (AWERB), and adhered to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SA: Conceptualization, Visualization, Methodology, Software, Validation, Resources, Writing – original draft, Formal analysis, Funding acquisition, Investigation, Writing – review & editing, Data curation. ES: Validation, Investigation, Supervision, Writing – review & editing, Resources, Methodology. AN: Formal analysis, Writing – review & editing, Data curation, Methodology, Software. IS: Visualization, Project administration, Writing – review & editing, Investigation, Supervision. MK: Writing – review & editing, Resources, Data curation, Methodology. SL: Formal analysis, Software, Data curation, Writing – review & editing, Methodology. CV: Writing – review & editing, Investigation, Methodology. MP: Resources, Formal analysis, Methodology, Writing – review & editing. RT: Investigation, Resources, Writing – review & editing, Methodology. PB: Supervision, Visualization, Resources, Methodology, Investigation, Writing – review & editing, Conceptualization. NT: Data curation, Formal analysis, Writing – review & editing, Methodology, Investigation, Supervision, Conceptualization, Software. HM: Project administration, Visualization, Supervision, Conceptualization, Writing – review & editing, Funding acquisition, Investigation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded in part by the Wellcome Trust. HMcS is a Wellcome Trust Investigator [grant code WT 206331/Z/17/Z]. For the purpose of open access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission. This study was funded by the National Institute for Health and Care Research (NIHR) Oxford Biomedical Research Centre (BRC). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. This study was also supported by the Vice Presidency for Graduate Studies and Scientific Research at King Khalid University, Saudi Arabia.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1593263/full#supplementary-material
References
1. Global tuberculosis report 2024. Geneva: World Health Organization (2024). Available online at: https://www.who.int/publications/i/item/9789240101531.
2. Mangtani P, Abubakar I, Ariti C, Beynon R, Pimpin L, Fine PE, et al. Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clin Infect Dis. (2014) 58:470–80. doi: 10.1093/cid/cit790
3. Rieder HL. International Union against Tuberculosis and Lung Disease, IUATLD. @ in Interventions for tuberculosis control and elimination. Paris: International Union Against Tuberculosis and Lung Disease Paris (2002).
4. Bell LC and Noursadeghi M. Pathogenesis of HIV-1 and Mycobacterium tuberculosis co-infection. Nat Rev Microbiol. (2018) 16:80–90. doi: 10.1038/nrmicro.2017.128
5. Lin Y, Slight SR, and Khader SA. Th17 cytokines and vaccine-induced immunity. In: Seminars in immunopathology ser. Berlin: Springer (2010). p. 32.
6. Lin PL and Flynn JL. CD8 T cells and Mycobacterium tuberculosis infection. In: Seminars in immunopathology ser, vol. 37. Berlin: Springer (2015).
7. Serbina NV and Flynn JL. CD8 T cells participate in the memory immune response to Mycobacterium tuberculosis. Infect Immun. (2001) 69:4320–8. doi: 10.1128/IAI.69.7.4320-4328.2001
8. Lu LL, Chung AW, Rosebrock TR, Ghebremichael M, Yu WH, Grace PS, et al. A functional role for antibodies in tuberculosis. Cell. (2016) 167:433–443. e14. doi: 10.1016/j.cell.2016.08.072
9. Krogsgaard M and Davis MM. How T cells’ see’antigen. Nat Immunol. (2005) 6:239–45. doi: 10.1038/ni1173
10. Shao W, Pedrioli PG, Wolski W, Scurtescu C, Schmid E, Vizcaíno JA, et al. The SysteMHC atlas project. Nucleic Acids Res. (2018) 46:D1237–47. doi: 10.1093/nar/gkx664
11. Sakai S, Mayer-Barber KD, and Barber DL. Defining features of protective CD4 T cell responses to Mycobacterium tuberculosis. Curr Opin Immunol. (2014) 29:137–42. doi: 10.1016/j.coi.2014.06.003
12. Lewinsohn DA, Lewinsohn DM, and Scriba TJ. Polyfunctional CD4 T cells as targets for tuberculosis vaccination. Front Immunol. (2017) 8:1262. doi: 10.3389/fimmu.2017.01262
13. Aagaard C, Hoang TTKT, Izzo A, Billeskov R, Troudt J, Arnett K, et al. Protection and polyfunctional T cells induced by Ag85B-TB10. 4/IC31® against Mycobacterium tuberculosis is highly dependent on the antigen dose. . PloS One. (2009) 4:e5930. doi: 10.1371/journal.pone.0005930
14. Rodo MJ, Rozot V, Nemes E, Dintwe O, Hatherill M, Little F, et al. A comparison of antigen-specific T cell responses induced by six novel tuberculosis vaccine candidates. PloS Pathog. (2019) 15:e1007643. doi: 10.1371/journal.ppat.1007643
15. Duong VT, Skwarczynski M, and Toth I. Towards the development of subunit vaccines against tuberculosis: The key role of adjuvant. Tuberculosis. (2023) 139:102307. doi: 10.1016/j.tube.2023.102307
16. Cole S, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. (1998) 396:190. doi: 10.1038/24206
17. Faridi P, Purcell AW, and Croft NP. In immunopeptidomics we need a sniper instead of a shotgun. Proteomics. (2018) 18:1700464. doi: 10.1002/pmic.201700464
18. Dudek NL, Croft NP, Schittenhelm RB, Ramarathinam SH, and Purcell AW. A systems approach to understand antigen presentation and the immune response. Proteomics Syst Biology: Methods Protoc. (2016) 2016:189–209. doi: 10.1007/978-1-4939-3341-9_14
19. Purcell AW, Ramarathinam SH, and Ternette N. Mass spectrometry–based identification of MHC-bound peptides for immunopeptidomics. Nat Protoc. (2019) 14:1687–707. doi: 10.1038/s41596-019-0133-y
20. Chen R, Fulton KM, Twine SM, and Li J. Identification of MHC peptides using mass spectrometry for neoantigen discovery and cancer vaccine development. Mass Spectrom. Rev. (2021) 40:110–25. doi: 10.1002/mas.21619
21. Bassani-Sternberg M and Coukos G. Mass spectrometry-based antigen discovery for cancer immunotherapy. Curr Opin Immunol. (2016) 41:9–17. doi: 10.1016/j.coi.2016.04.005
22. Leddy OK, White FM, and Bryson BD. Leveraging immunopeptidomics to study and combat infectious disease. Msystems 6 10.1128/msystems. 00310–21. (2021) 6(4):10.1128/msystems. 00310–21. doi: 10.1128/msystems.00310-21
23. Mayer RL and Impens F. Immunopeptidomics for next-generation bacterial vaccine development. Trends Microbiol. (2021) 29:1034–45. doi: 10.1016/j.tim.2021.04.010
24. Flyer DC, Ramakrishna V, Miller C, Myers H, McDaniel M, Root K, et al. Identification by mass spectrometry of CD8 -T-cell Mycobacterium tuberculosis epitopes within the Rv0341 gene product. Infect Immun. (2002) 70:2926–32. doi: 10.1128/IAI.70.6.2926-2932.2002
25. Leddy O, White FM, and Bryson BD. Immunopeptidomics reveals determinants of Mycobacterium tuberculosis antigen presentation on MHC class I. Elife. (2023) 12:e84070. doi: 10.7554/eLife.84070
26. McMurtrey C, Harriff MJ, Swarbrick GM, Duncan A, Cansler M, Null M, et al. T cell recognition of Mycobacterium tuberculosis peptides presented by HLA-E derived from infected human cells. PloS One. (2017) 12:e0188288. doi: 10.1371/journal.pone.0188288
27. Bettencourt P, Muller J, Nicastri A, Cantillon D, Madhavan M, Charles PD, et al. Identification of antigens presented by MHC for vaccines against tuberculosis. NPJ Vaccines. (2020) 5:2. doi: 10.1038/s41541-019-0148-y
28. Reynisson B, Alvarez B, Paul S, Peters B, and Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. (2020) 48:W449–54. doi: 10.1093/nar/gkaa379
29. Greenbaum J, Sidney J, Chung J, Brander C, Peters B, Sette A, et al. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics. (2011) 63:325–35. doi: 10.1007/s00251-011-0513-0
30. Weiskopf D, Angelo MA, de Azeredo EL, Sidney J, Greenbaum JA, Fernando AN, et al. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8 T cells. Proc Natl Acad Sci. (2013) 110:E2046–53. doi: 10.1073/pnas.1305227110
31. Roman F, Burny W, Ceregido MA, Laupèze B, Temmerman ST, Warter L, et al. Adjuvant system AS01: from mode of action to effective vaccines. Expert Rev Vaccines. (2024) 23:715–29. doi: 10.1080/14760584.2024.2382725
32. Huang Z, Gong H, Sun Q, Yang J, Yan X, Xu F, et al. Research progress on emulsion vaccine adjuvants. Heliyon 10. (2024) 10(3):e24662. doi: 10.1016/j.heliyon.2024.e24662
33. Wilkins AL, Kazmin D, Napolitani G, Clutterbuck EA, Pulendran B, Siegrist C, et al. AS03-and MF59-adjuvanted influenza vaccines in children. Front Immunol. (2017) 8:1760. doi: 10.3389/fimmu.2017.01760
34. Tait DR, Hatherill M, Van Der Meeren O, Ginsberg AM, Van Brakel E, Salaun B, et al. Final analysis of a trial of M72/AS01E vaccine to prevent tuberculosis. N Engl J Med. (2019) 381:2429–39. doi: 10.1056/NEJMoa1909953
35. Partridge T, Nicastri A, Kliszczak AE, Yindom L, Kessler BM, Ternette N, et al. Discrimination between human leukocyte antigen class I-bound and co-purified HIV-derived peptides in immunopeptidomics workflows. Front Immunol. (2018) 9:912. doi: 10.3389/fimmu.2018.00912
36. Ong E, Cooke MF, Huffman A, Xiang Z, Wong MU, Wang H, et al. Vaxign2: the second generation of the first Web-based vaccine design program using reverse vaccinology and machine learning. Nucleic Acids Res. (2021) . 49:W671–8. doi: 10.1093/nar/gkab279
37. Kohm AP, Fuller KG, and Miller SD. Mimicking the way to autoimmunity: an evolving theory of sequence and structural homology. Trends Microbiol. (2003) 11:101–5. doi: 10.1016/S0966-842X(03)00006-4
38. Ebringer A and Wilson C. HLA molecules, bacteria and autoimmunity. J Med Microbiol. (2000) 49:305–11. doi: 10.1099/0022-1317-49-4-305
39. Painter H, Harriss E, Fletcher HA, McShane H, and Tanner R. Development and application of the direct mycobacterial growth inhibition assay: a systematic review. Front Immunol. (2024) 15:1355983. doi: 10.3389/fimmu.2024.1355983
40. Cohet C, van der Most R, Bauchau V, Bekkat-Berkani R, Doherty TM, Schuind A, et al. Safety of AS03-adjuvanted influenza vaccines: A review of the evidence. Vaccine. (2019) 37:3006–21. doi: 10.1016/j.vaccine.2019.04.048
41. Agüero F, Al-Lazikani B, Aslett M, Berriman M, Buckner FS, Campbell RK, et al. Genomic-scale prioritization of drug targets: the TDR Targets database. Nat Rev Drug Discovery. (2008) 7:900–7.
42. Dwivedy A, Ashraf A, Jha B, Kumar D, Agarwal N, and Biswal BK. De novo histidine biosynthesis protects Mycobacterium tuberculosis from host IFN-γ mediated histidine starvation. Commun Biol. (2021) 4:410.
43. De Souza GA, Leversen NA, Målen H, and Wiker HG. Bacterial proteins with cleaved or uncleaved signal peptides of the general secretory pathway. J Proteomics. (2011) 75:502–10.
44. Mawuenyega KG, Forst CV, Dobos KM, Belisle JT, Chen J, Bradbury EM, et al. Mycobacterium tuberculosis functional network analysis by global subcellular protein profiling. Mol Biol Cell. (2005) 16:396–404.
45. Warner DF, Savvi S, Mizrahi V, and Dawes SS. A riboswitch regulates expression of the coenzyme B12-independent methionine synthase in Mycobacterium tuberculosis: implications for differential methionine synthase function in strains H37Rv and CDC1551. J Bacteriol. (2007) 189:3655–9.
46. Gonzalo-Asensio J, Campos-Pardos E, and Uranga S. Dependency on the host vitamin B12 has shaped the Mycobacterium tuberculosis Complex evolution. Washington, DC, USA: American Society for Microbiology (2023).
47. Iacobino A, Teloni R, Mancone C, Facchiano F, Di Giamberardino A, Senatore C, et al. Identification of Rv1133c (MetE) as a marker of Mycobacterium tuberculosis replication and as a highly immunogenic antigen with potential immunodiagnostic power. Front Immunol. (2024) 15:1464923.
48. Melly G and Purdy GE. MmpL Proteins in Physiology and Pathogenesis of M. tuberculosis. Microorganisms. (2019) 7:70.
49. Teahan B, Ong E, and Yang Z. Identification of Mycobacterium tuberculosis Antigens with Vaccine Potential Using a Machine Learning-Based Reverse Vaccinology Approach. Vaccines. (2021) 9:1098.
50. Satti I and McShane H. Current approaches toward identifying a correlate of immune protection from tuberculosis. Expert Rev Vaccines. (2019) 18:43–59.
51. Bertholet S, Ireton GC, Kahn M, Guderian J, Mohamath R, Stride N, et al. Identification of human T cell antigens for the development of vaccines against Mycobacterium tuberculosis. J Immunol. (2008) 181:7948–57.
52. Gallegos AM, van Heijst JW, Samstein M, Su X, Pamer EG, and Glickman MS. A gamma interferon independent mechanism of CD4 T cell mediated control of M. tuberculosis infection vivo. PloS Pathog. (2011) 7:e1002052.
53. Tanner R, Satti I, Harris SA, O'Shea MK, Cizmeci D, O'Connor D, et al. Tools for assessing the protective efficacy of TB vaccines in humans: in vitro mycobacterial growth inhibition predicts outcome of in vivo mycobacterial infection. Front Immunol. (2020) 10:2983.
54. Tanner R, White AD, Boot C, Sombroek CC, O’Shea MK, Wright D, et al. A non-human primate in vitro functional assay for the early evaluation of TB vaccine candidates. NPJ Vaccines. (2021) 6:3.
55. Pepponi I, Khatri B, Tanner R, Villarreal-Ramos B, Vordermeier M, and McShane H. A mycobacterial growth inhibition assay (MGIA) for bovine TB vaccine development. Tuberculosis. (2017) 106:118–22.
56. Marsay L, Matsumiya M, Tanner R, Poyntz H, Griffiths KL, Stylianou E, et al. Mycobacterial growth inhibition in murine splenocytes as a surrogate for protection against Mycobacterium tuberculosis (M. tb). Tuberculosis. (2013) 93:551–7.
57. Parra M, Yang AL, Lim J, Kolibab K, Derrick S, Cadieux N, et al. Development of a murine mycobacterial growth inhibition assay for evaluating vaccines against Mycobacterium tuberculosis. Clin Vaccine Immunol. (2009) 16:1025–32.
58. Cowley SC and Elkins KL. CD4 T cells mediate IFN-γ-independent control of Mycobacterium tuberculosis infection both in vitro and in vivo. J Immunol. (2003) 171:4689–99.
59. Haile M, Schröder U, Hamasur B, Pawlowski A, Jaxmar T, Källenius G, et al. Immunization with heat-killed Mycobacterium bovis bacille Calmette–Guerin (BCG) in Eurocine™ L3 adjuvant protects against tuberculosis. Vaccine. (2004) 22:1498–508.
60. Melkie ST, Arias L, Farroni C, Makek MJ, Goletti D, and Vilaplana C. The role of antibodies in tuberculosis diagnosis, prophylaxis and therapy: a review from the ESGMYC study group. Eur Respir Rev. (2022) 31:1498–508.
61. Orme IM. Preclinical testing of new vaccines for tuberculosis: a comprehensive review. Vaccine. (2006) 24:2–19.
62. Lu J, Chen B, Wang G, Fu L, Shen X, Su C, et al. Recombinant tuberculosis vaccine AEC/BC02 induces antigen-specific cellular responses in mice and protects guinea pigs in a model of latent infection. J Microbiology Immunol Infection. (2015) 48:597–603.
63. Larsen SE, Reese VA, Pecor T, Berube BJ, Cooper SK, Brewer G, et al. Subunit vaccine protects against a clinical isolate of Mycobacterium avium in wild type and immunocompromised mouse models. Sci Rep. (2021) 11:9040.
64. Olsen AW, van Pinxteren LA, Okkels LM, Rasmussen PB, and Andersen P. Protection of mice with a tuberculosis subunit vaccine based on a fusion protein of antigen 85b and esat-6. Infect Immun. (2001) 69:2773–8.
65. Chen S, Quan DH, Wang XT, Sandford S, Kirman JR, Britton WJ, et al. Particulate mycobacterial vaccines induce protective immunity against tuberculosis in mice. Nanomaterials. (2021) 11:2060.
66. Counoupas C, Ferrell KC, Ashhurst A, Bhattacharyya ND, Nagalingam G, Stewart EL, et al. Mucosal delivery of a multistage subunit vaccine promotes development of lung-resident memory T cells and affords interleukin-17-dependent protection against pulmonary tuberculosis. NPJ Vaccines. (2020) 5:105.
67. Santosuosso M, Zhang X, McCormick S, Wang J, Hitt M, and Xing Z. Mechanisms of mucosal and parenteral tuberculosis vaccinations: adenoviral-based mucosal immunization preferentially elicits sustained accumulation of immune protective CD4 and CD8 T cells within the airway lumen. J Immunol. (2005) 174:7986–94.
Keywords: tuberculosis, mycobacterium tuberculosis, vaccines, immunopeptidomics, mass spectrometry, immunoinformatics, antigen discovery, HLA/MHC
Citation: Almujri SS, Stylianou E, Nicastri A, Satti I, Korompis M, Li S, De Voss CJ, Polo Peralta Alvarez M, Tanner R, Bettencourt PJG, Ternette N and McShane H (2025) MetE: a promising protective antigen for tuberculosis vaccine development. Front. Immunol. 16:1593263. doi: 10.3389/fimmu.2025.1593263
Received: 13 March 2025; Accepted: 26 June 2025;
Published: 21 July 2025.
Edited by:
Rajko Reljic, University of London, United KingdomReviewed by:
Angelo Izzo, Royal Prince Alfred Hospital, AustraliaAngel Alejandro Oñate, University of Concepcion, Chile
Premanshu Dandapat, Indian Council of Agricultural Research (ICAR), India
Copyright © 2025 Almujri, Stylianou, Nicastri, Satti, Korompis, Li, De Voss, Polo Peralta Alvarez, Tanner, Bettencourt, Ternette and McShane. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Salem Salman Almujri, c3NhbG11anJpQGtrdS5lZHUuc2E=; Helen McShane, aGVsZW4ubWNzaGFuZUBuZG0ub3guYWMudWs=