 Mengxue Wang1†
Mengxue Wang1† Bing Yu
Bing Yu Qingqing Dai
Qingqing Dai- 1Department of Critical Care Medicine, The Obstetrics & Gynecology Hospital of Fudan University, Shanghai, China
- 2Department of Critical Care Medicine, The Obstetrics & Gynecology Hospital of Fudan University, Yangtze River Delta Integration Demonstration Zone (QingPu), Shanghai, China
- 3Department of Cell Biology, Navy Medical University, Shanghai, China
Pulmonary fibrosis is a disease that severely affects the patients’ life quality, characterized by lung tissue remodeling and functional impairment. Recent research has found that the NLRP3 inflammasome plays an important role in the pathogenesis of pulmonary fibrosis. Although existing researches have revealed the potential role of NLRP3 in pulmonary fibrosis, many mysteries still remain regarding its specific mechanisms and clinical applications. This article aims to review the mechanisms of action of NLRP3 in pulmonary fibrosis, related signaling pathways, and the latest research progress on its potential as a therapeutic target, in hopes of providing new ideas and directions for future clinical treatment.
1 Introduction
Pulmonary fibrosis (PF) is an interstitial lung disease characterized by progressive fibrosis and structural remodeling. Pulmonary fibrosis is categorized into idiopathic and secondary types, both of which are associated with triggers such as environmental exposures, infections, medications, and genetic predisposition. The distinction lies in that the etiology of secondary pulmonary fibrosis is well-established, whereas the underlying causes of idiopathic pulmonary fibrosis (IPF) remain elusive (1–4). As fibrosis progresses, it not only severely affects patients’ quality of life but also increases the risk of early death (5). So far, therapeutic options for pulmonary fibrosis remain limited, as conventional anti-inflammatory agents and immunosuppressants have demonstrated little to no significant efficacy in its treatment (6, 7). Currently, two advanced therapeutic drugs nintedanib and pirfenidone, have been approved for the management of pulmonary fibrosis; Nintedanib is a tyrosine kinase inhibitor that targets vascular endothelial growth factor, fibroblast growth factor, and platelet-derived growth factor signaling pathways. Pirfenidone has antifibrotic, anti-inflammatory and antioxidant effects, although its exact mechanisms are not fully elucidated. However, their efficacy is limited to decelerating the progression of fibrosis, with little potential to achieve a definitive cure (8, 9). In contrast, therapeutics targeting the NLRP3 inflammasome represent a novel approach that directly addresses the inflammatory cascade at its source, potentially offering more comprehensive control of both the inflammatory and fibrotic processes in PF. This innovative mechanism differs fundamentally from current approved therapies by intervening at the level of inflammatory initiation rather than downstream pathway inhibition. Lung transplantation also remains the ultimate treatment method to improve prognosis (8).
The mechanism of pulmonary fibrosis remains complex and mainly involves inflammatory responses, the activation and transformation of fibroblasts, cytokine signaling pathways, oxidative stress, epithelial-mesenchymal transition (EMT), and so on (7). Damage factors cause alveolar epithelial cells to undergo EMT and release pro-inflammatory and pro-fibrotic mediators, which further stimulate the proliferation and differentiation of fibroblasts and the activation of myofibroblasts, ultimately leading to excessive deposition of extracellular matrix (ECM) proteins (10, 11). The inflammatory cascade is central to pulmonary fibrosis pathogenesis, with various cytokines orchestrating this process. In the bleomycin (BLM)-induced pulmonary fibrosis model, alveolar damage leads to the recruitment of inflammatory cells, including macrophages, neutrophils, and lymphocytes. These cells produce key pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α, which further amplify the inflammatory response. (12). Subsequently, overabundant inflammatory responses release a large number of pro-inflammatory cytokines and growth factors, particularly IL-1β, not only perpetuate inflammation but also directly promote fibroblast activation (10). Furthermore, researches indicate that macrophages play a central role in the development of IPF (13). M1 macrophages induce inflammation by secreting pro-inflammatory cytokines (such as TNF-α, IL-6, and IL-1β), while M2 macrophages promote fibroblast proliferation and differentiation by secreting pro-fibrotic factors, such as transforming growth factor β (TGF-β) and platelet-derived growth factor (13, 14). This cytokine network creates a pro-fibrotic environment where TGF-β, produced by M2 macrophages, serves as a master regulator of fibrosis by inducing fibroblast differentiation into myofibroblasts. In addition, T cells contribute to the inflammatory response and subsequent fibrosis process through immune regulation, while activated B cells release various cytokines and metalloproteinases, leading to dysregulation during the resolution phase of inflammation and excessive extracellular matrix deposition (6). Therefore, the progression of pulmonary fibrosis involves both inflammatory responses and immunologic mechanisms.
NOD-like receptor (NLR) family pyrin domain-containing 3 (NLRP3) inflammasome is a cytoplasmic multiprotein complex that plays a critical role in regulating inflammation and immune responses (15, 16). It is not only present in macrophages but also in epithelial cells and myofibroblasts (17, 18). Its primary function is to recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), initiating innate immune responses, activating inflammatory reactions, and promoting the secretion of IL-1β and IL-18, which further drive downstream pathways and molecular changes (19). The NLRP3 inflammasome plays a dual role in various diseases, protecting the host from infection while potentially causing tissue damage under pathological conditions such as chronic inflammation and fibrosis. Increasing evidence suggests that activation of the NLRP3 inflammasome is closely associated with the progression of pulmonary fibrosis. Studies on the genetic polymorphism of pulmonary fibrosis have found that the NLRP3 rs35829419 variant allele is associated with an increased risk of asbestos-related PF (20). Elevated levels of NLRP3 have been observed in the bronchoalveolar lavage fluid (BALF) of rheumatoid arthritis-associated interstitial lung disease (RA-ILD) patients (21). Aberrant activation of the NLRP3 inflammasome has also been detected in animal models of pulmonary fibrosis induced by PM2.5, silica dust, asbestos, and BLM (8, 22–25). Moreover, inhibition of the NLRP3 inflammasome has been shown to attenuate the progression of pulmonary fibrosis (26).
Therefore, the NLRP3 inflammasome not only plays a role in inducing inflammatory responses and promoting the fibrotic process in pulmonary fibrosis, but it may also be an effective target for the treatment of pulmonary fibrosis. This review systematically summarizes the role and related mechanisms of the NLRP3 inflammasome in pulmonary fibrosis and integrates the latest research progress on NLRP3 inflammasome-targeted therapies in pulmonary fibrosis, hoping to provide new ideas and directions for clinical treatment.
2 The structure and activation of NLRP3 inflammasome
2.1 Inflammasome structure and assembly
The NLRP3 inflammasome is a sophisticated multiprotein complex with distinct structural components that determine its function. The NLRP3 inflammasome consists of NLRP3 protein, apoptosis-associated speck-like protein (ASC), and precursor of cysteine-aspartic protease-1 (pro-caspase-1) (27). Structurally, NLRP3 is characterized by a C-terminal leucine-rich repeat (LRR) domain, a central nucleotide oligomerization domain (named NACHT), and an N-terminal pyrin domain (PYD) (28). The NACHT domain includes a nucleotide-binding domain (NBD) and a winged-helix domain positioned between two helical domains (designated HD1 and HD2) (28, 29). The NBD contains highly conserved Walker A and Walker B motifs essential for ATP binding and hydrolysis, respectively (30).
The adaptor protein ASC contains both a PYD and a caspase activation and recruitment domain (CARD), allowing it to bridge NLRP3 and pro-caspase-1 through homotypic domain interactions. Upon activation, NLRP3 oligomerizes and interacts with ASC via PYD-PYD interactions, leading to ASC oligomerization into a large speck-like structure. This ASC speck serves as a platform that recruits multiple pro-caspase-1 molecules through CARD-CARD interactions, facilitating proximity-induced auto-activation of caspase-1 (11, 16).
Research has identified NIMA-related kinase 7 (NEK7) as a critical binding partner that directly interacts with NLRP3 to mediate inflammasome assembly and activation (31–33). This interaction occurs in a cell cycle-dependent manner, linking inflammasome activation to the cell cycle status. The NEK7-NLRP3 interaction is required downstream of potassium efflux, suggesting that NEK7 functions as a switch that permits NLRP3 activation only under specific cellular conditions.
2.2 Inflammasome activation
As the core sensor of the inflammasome, NLRP3 activation generally requires two signals: the first signal is the initial activation through pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), leading to NF-κB-mediated upregulation of NLRP3 and pro-IL-1β expression. This priming step not only increases the expression of inflammasome components but also induces post-translational modifications of NLRP3 that license its activation. The second signal involves diverse cellular stressors that converge on common pathways to trigger NLRP3 conformational changes and assembly. Key activation factors include:
2.2.1 Ionic flux
Potassium efflux represents a universal trigger for NLRP3 activation across diverse stimuli (34). Experimental evidence shows that elevated extracellular potassium concentrations inhibit NLRP3 activation, while agents that induce potassium efflux, such as ATP and nigericin, are potent NLRP3 activators. Calcium signaling also plays a crucial role, with calcium mobilization from both extracellular spaces and intracellular stores contributing to inflammasome activation. Additionally, chloride efflux acts as a specific signal for ASC oligomerization and inflammasome assembly (35, 36).
2.2.2 Mitochondrial dysfunction
Damaged mitochondria release mitochondrial reactive oxygen species (mtROS), mitochondrial DNA, and cardiolipin, which can directly or indirectly activate NLRP3 (8, 37). The mitochondria-associated ER membrane (MAM) serves as a platform for NLRP3 recruitment and activation, highlighting the interorganelle communication in inflammasome regulation (38).
2.2.3 Lysosomal damage
Particulate matter such as crystalline silica, asbestos, and PM2.5—all implicated in pulmonary fibrosis—can destabilize lysosomes, leading to the release of lysosomal contents including cathepsins that activate NLRP3 (39–41).
In addition, some studies have shown that the cytoskeleton and Golgi apparatus are also involved in the activation process of NLRP3, but the specific mechanism remains to be explored (42–44). Upon activation, NLRP3 binds to ASC via PYD-PYD interactions. Multiple ASC molecules then oligomerize into speck-like structures, and ASC further recruits pro-caspase-1 into a complex through CARD-CARD interactions. Studies have demonstrated that chloride efflux acts as a signal for ASC oligomerization and plays a pivotal role in NLRP3 inflammasome assembly (35, 36). Additionally, NLRP3 activation is regulated by multiple post-translational modifications, such as SUMOylation and ubiquitination, which affect NLRP3 stability and function, subsequently influencing inflammasome activation. Ubiquitination typically inhibits NLRP3 by promoting its degradation, while deubiquitination by enzymes like BRCC3 is required for activation. Phosphorylation can either activate or inhibit NLRP3 depending on the specific residues modified and the kinases involved. SUMOylation of NLRP3 maintains it in an inactive state, and deSUMOylation is necessary for activation.
3 The NLRP3 inflammasome as a central node connecting inflammation and fibrosis in PF
Although the precise pathogenesis of pulmonary fibrosis remains unclear, substantial research has highlighted the involvement of key signaling pathways and biological mechanisms. As the NLRP3 inflammasome is investigated, its role in the progression of pulmonary fibrosis is progressively being elucidated. In the progression of pulmonary fibrosis, various cell types participate in disease development through the NLRP3 inflammasome. Type II alveolar epithelial cells (ATII) are pivotal both for NLRP3 expression and fibrotic progression; under endoplasmic reticulum stress and oxidative stress conditions, NLRP3 activation in ATII cells leads to IL-1β production and pyroptosis (45), while epithelial-mesenchymal transition (EMT) of ATII cells directly promotes fibrosis (46). Alveolar macrophages, as the predominant immune cells in the lungs, represent a major source of NLRP3 inflammasome activation and IL-1β production (14), with their polarization state (M1/M2) being regulated by NLRP3 and subsequently influencing the fibrotic process (47, 48). The following sections will delve into the complex interactions between the NLRP3 inflammasome and various signaling pathways and cellular mechanisms in pulmonary fibrosis (Figure 1).
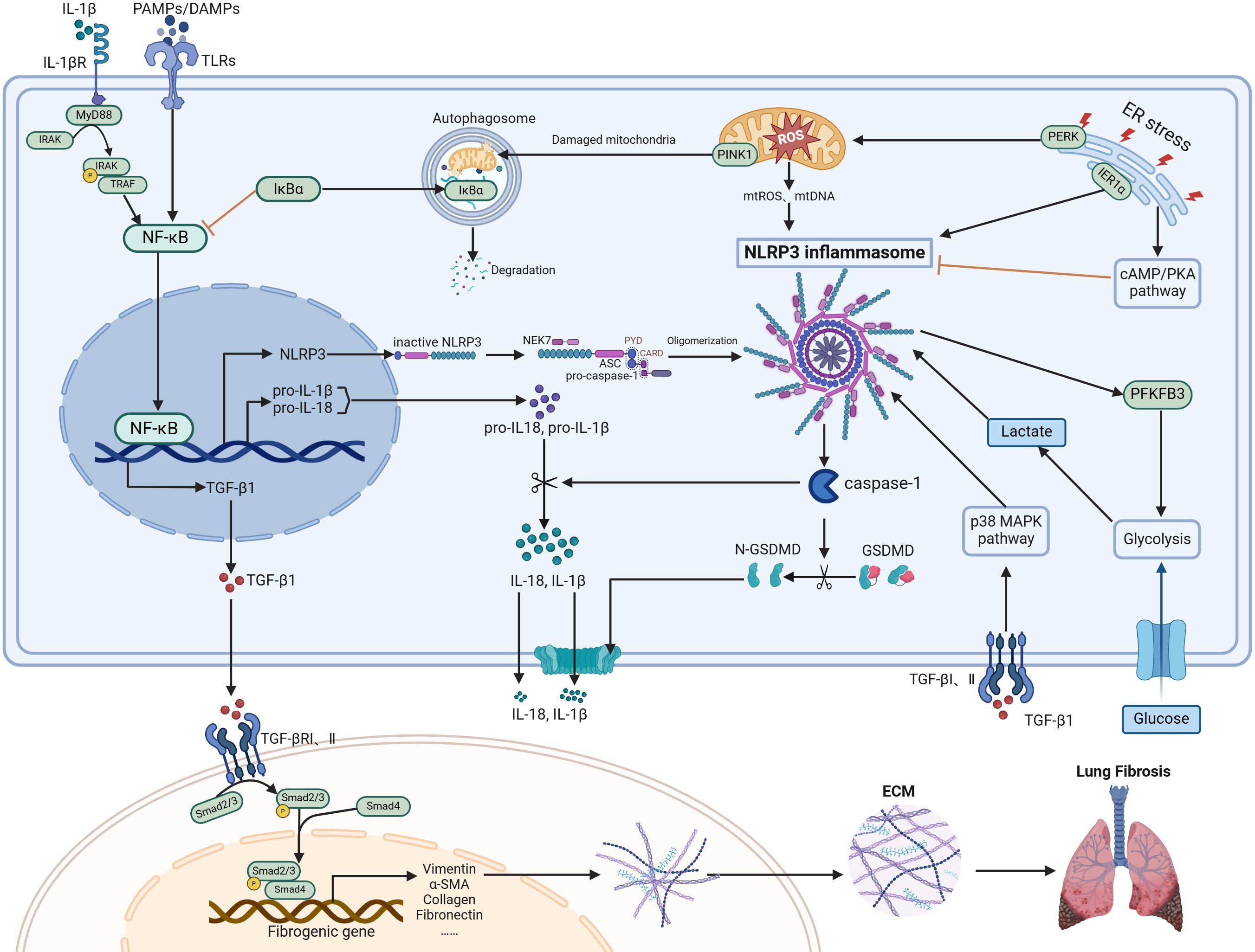
Figure 1. Regulatory mechanisms of NLRP3 inflammasome in PF. The NLRP3 inflammasome, mainly consisting of NLRP3, ASC, and pro-caspase-1, is supplied with components by the NF-κB signaling pathway. This complex can be activated in various cell types, particularly macrophages and type II alveolar epithelial cells. Activation of the inflammasome causes caspase-1 activation, leading to the maturation and release of IL-1β/IL-18 and GSDMD-mediated pyroptosis. A positive feedback loop between the NLRP3 inflammasome and NF-κB is established through IL-1β. ER stress, oxidative stress, autophagy, and metabolic changes such as glycolysis are all involved in regulating the levels of the NLRP3 inflammasome. TGF-β1 and NLRP3 inflammasome mutually enhance each other, collectively driving the progression of pulmonary fibrosis by promoting fibroblast differentiation into myofibroblasts and subsequent ECM accumulation. Figure was created with biorender.com.
3.1 The NLRP3 inflammasome and inflammatory signaling cascades: NF-κB/NLRP3/IL-1β pathway
In the initial stage of lung injury caused by environmental factors, bacterial and viral infections, and drugs that lead to the damage of epithelial cells and cause the inflammation of alveolar macrophages. PRRs recognize PAMPs and DAMPs to initiate the innate immune response, activating the NF-κB signaling pathway. Then, the expression of NLRP3, pro-IL-1β, and pro-IL-18 were upregulated, providing the necessary components for inflammasome activation (49). Subsequently, variable factors such as potassium efflux, calcium influx, mitochondrial reactive oxygen species (ROS) generation, and lysosomal disruption may trigger the assembly and activation of the NLRP3 inflammasome (50–52). After the NLRP3 inflammasome is activated, active caspase-1 further cleaves pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, which are then released (51). IL-1β, as a pro-inflammatory cytokine, plays a multifaceted role in the formation of pulmonary fibrosis. Early research indicates that IL-1β promotes pulmonary fibrosis in vivo by activating IL-1 receptor-dependent signaling pathways (11). Additionally, preclinical models of pulmonary fibrosis show that the expression levels of MyD88 in lung tissue increase after exposure to various profibrotic stimuli (53). Studies have demonstrated that the activated NLRP3 inflammasome can promote pulmonary EMT via the IL-1β/IL-1Rs/MyD88/NF-κB signaling pathway, leading to pulmonary fibrosis (14). After IL-1β binds to the IL-1R1 receptor on the cell surface, it recruits MyD88 and activates the IL-1 receptor-associated kinases (IRAK). These kinases, once phosphorylated, dissociate from MyD88 and bind to the tumor necrosis factor receptor-associated factor (TRAF), further inducing the activation of the transcription factor NF-κB (54). Then, the activated NF-κB upregulates the expression of NLRP3 and IL-1β again, forming a positive feedback loop that triggers an inflammatory cascade, ultimately leading to pulmonary fibrosis (8, 55). Also, the activated NF-κB pathway drives the expression of pro-inflammatory factors such as TNF-α, IL-6, and chemokines (like IL-8), resulting in an amplified inflammatory effect (56). In mice with IL-1R1 and MyD88 gene knockout, BLM administration did not provoke a fibrotic response (8). It can be seen that in the process of pulmonary fibrosis, the NF-κB signaling pathway is not only the initiating signal for the activation of the NLRP3 inflammasome but also has a positive feedback mechanism with NLRP3, leading to the persistent progression of pulmonary fibrosis. The activation of NF-κB not only directly promotes the expression and activation of the NLRP3 inflammasome, but may also be regulated by various cellular stress responses, which play important roles in the occurrence and development of pulmonary fibrosis.
3.2 The upstream regulatory factors of the NF-κB/NLRP3/IL-1β pathway in PF: the oxidative stress, ER stress, autophagy, and metabolism
3.2.1 Oxidative stress
Oxidative stress results from an imbalance between the production of oxidants and the cell’s antioxidant capacity, leading to the generation of ROS including hydrogen peroxide (H2O2), superoxide anion (O2-), hydroxyl radical (OH), hypochlorous acid (HOCl), and peroxynitrite (ONOO) (57). Mitochondrial dysfunction is associated with oxidative stress and releases signals such as excessive mtROS, translocation of mtDNA to the cytoplasm, or repositioning of mitochondria, all of which are considered direct activators of the NLRP3 inflammasome (58). In pulmonary fibrosis, excessive production of ROS can trigger the death of phagocytic cells, recruitment of inflammatory cells, and permanent lung damage. Previous studies have shown that the interaction between fibrogenic inducers and pulmonary macrophages stimulates the production of a large amount of ROS and activates the intracellular NF-κB signaling pathway, leading to the activation of the NLRP3 inflammasome and the secretion of IL-1β. IL-1β further promotes the transformation of fibroblasts and may in turn increase the production of ROS, leading to the continuous progression of pulmonary fibrosis (59). Early research has proven that antioxidant regulator Nrf2 is one of the key downstream mechanisms involved in mitochondrial dysfunction (60). It not only links to the expression of many genes required for mitochondrial respiratory function but also plays critical roles in enhancing mtDNA levels, oxidative phosphorylation (OXPHOS) activity, and mitochondrial protein import and assembly (61). In the IL-33 and LPS/IL-4-induced pulmonary fibrosis cell model, ROS production and mtDNA accumulation, key upstream events of NLRP3 activation in macrophages, were observed, along with a reduction in Nrf2 mRNA levels. When Nrf2 expression is absent, over-activation of NLRP3 leads to the release of a large amount of pro-inflammatory factors such as IL-1β and IL-18 (13). During another study, oxidative stress-associated protein high mobility group box 1 (HMGB1) can activate NLRP3 inflammasomes and promote PF by inhibiting the Nrf2/HO-1 pathway in BLM-induced PF model (62). Additionally, studies have shown that enhanced ROS levels can exacerbate the development of PF through NLRP3-mediated cellular senescence, and that the acceleration of PF by ROS is positively correlated with cellular senescence (63).
3.2.2 ER stress
The ER plays a crucial role in maintaining protein homeostasis (64). However, factors such as aging, hypoxia, oxidative stress, or inflammation can disrupt this balance, leading to the accumulation of misfolded proteins in ER, which activate the unfolded protein response (UPR), and thereby triggering ER stress and apoptosis (65). Relevant studies have shown that ER stress plays a key role in the occurrence and progression of pulmonary fibrosis (66, 67). ER stress affects cellular behavior and function through abnormal activation of UPR signaling pathways, including inositol-requiring enzyme 1 (IRE1), protein kinase R-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6) signaling pathways, which lead to cell apoptosis, EMT, fibroblast differentiation, and macrophage polarization (65). A substantial amount of research indicates that NLRP3 is involved in ER stress within ATII cells during pulmonary fibrosis. In ATII cells of patients with IPF, a large accumulation of unfolded or misfolded proteins has been observed. These abnormal proteins dephosphorylate the ER transmembrane receptor IRE1α. The dephosphorylation of IRE1α can further activate the NLRP3 inflammasome, thereby promoting the expression of IL-1β and IL-18 (68). Additionally, some studies have found that sustained ER stress activates the PERK pathway, disrupts mitochondrial homeostasis, and leads to the release of mitochondrial damage-associated molecular patterns (mt-DAMPs). These mt-DAMPs (such as mitochondrial DNA and ROS) activate the NLRP3 inflammasome either within the cell or outside the cell, further driving the release of profibrotic factors (such as IL-1β and IL-18) (69). Simultaneously, in vitro pulmonary fibrosis models have shown that ER stress promotes the activation of the cAMP/PKA pathway, which may inhibit the activation of the NLRP3 inflammasome in AECs II induced by ER stress. Therefore, the cAMP/PKA pathway has a certain protective effect against pulmonary fibrosis (45).
Cellular stress including oxidative stress and ER stress not only directly affects the activation of the NLRP3 inflammasome but may also indirectly influence inflammasome function by regulating the autophagy process, forming a complex regulatory network (70).
3.2.3 Autophagy
Autophagy is a lysosome-dependent cellular self-degradation process that plays a critical role in clearing damaged or excess organelles and proteins, thereby maintaining homeostasis within the organism (71, 72). Increasing evidence suggests that autophagy is closely associated with the process of pulmonary fibrosis. For instance, exposure to silica nanoparticles activates autophagy, ultimately leading to endothelial dysfunction in silicosis (73). Another study found that insufficient autophagy results in the senescence of lung epithelial cells and the differentiation of fibroblasts into myofibroblasts in IPF (74). In an LPS-induced pulmonary fibrosis model, impaired autophagy was observed alongside NLRP3 inflammasome activation. Treatment with oridonin significantly inhibited the activation of the NLRP3 inflammasome and reversed autophagy levels (75). In a PM2.5-induced pulmonary fibrosis model, PM2.5 was shown to downregulate ALKBH5 expression, promote m6A modifications at specific sites of Atg13 mRNA, and activate ULK complex (composed of ULK1, Atg13, FIP200, and Atg101)-mediated autophagy. This autophagy further mediated the degradation of IκB-α (a NF-κB inhibitor), allowing NF-κB to translocate into the nucleus, which subsequently promoted the expression of NLRP3 and its downstream inflammatory factors (23). Moreover, studies have demonstrated that autophagy interacts with the pyroptosis pathway through the NLRP3 inflammasome. In the silica-induced fibrosis model, PTEN-induced kinase 1 (PINK1)-mediated mitophagy facilitates the clearance of damaged mitochondria, thereby negatively regulating NLRP3 inflammasome-associated pyroptosis. When inhibitors targeting NLRP3, caspase-1, and GSDMD were used to restrict the pyroptotic cascade, mitochondrial autophagy was enhanced (76). In addition to autophagy regulating NLRP3 inflammasome activity, metabolic alterations can also modulate NLRP3 activity.
3.2.4 Metabolism
In recent years, the metabolic alterations associated with pulmonary fibrosis have garnered growing recognition and academic interest (77). In the lung tissue of IPF patients, 25 metabolic features have been identified, which suggest alterations in metabolic pathways, including glycolysis, glutathione biosynthesis, adenosine triphosphate (ATP) degradation, and ornithine transaminase pathways (78). Current research has found that the NLRP3 inflammasome is involved in metabolic abnormalities associated with PF. In lung tissue from a silica dust-induced PF model, overexpression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) and an increase in lactate have been observed (7). PFKFB3 is a key enzyme in glycolysis, and lactate is a crucial substrate for histone lactylation. Both of them are involved in the process of fibrosis (79–81). In the activation of fibroblasts induced in vitro, the upregulation of PFKFB3 and an increase in extracellular lactate were also observed, along with the upregulation of the expression of NLRP3, ASC, and the activated caspase-1 p20 subunit. A selective NLRP3 inflammasome inhibitor MCC950 not only suppressed the activation of the NLRP3 inflammasome but also reduced the upregulation of PFKFB3 and the expression of fibrotic markers α-smooth muscle actin (α-SMA) and collagen I. At the same time, the study also found that lactate activated the NLRP3 inflammasome by increasing the level of histone lactylation (7). In another study, the glycolysis inhibitor (2-DG) alleviated CS-induced NLRP3 inflammasome activation and macrophage pyroptosis. Therefore, the activation of the NLRP3 inflammasome in fibroblasts is associated with glycolysis. Purine metabolism also participates in the progression of pulmonary fibrosis through the NLRP3 inflammasome. In BLM-induced PF model, uric acid released from injured cells activates the NLRP3 inflammasome, leading to IL-1β production. Reduction of uric acid levels using the inhibitor of uric acid synthesis allopurinol or uricase leads to a decrease in BLM-induced IL-1β production, lung inflammation, repair, and fibrosis (82). In addition, other studies have found that disorders in lipid metabolism may trigger NLRP3 inflammasome-related inflammatory cascade reactions, but the exact mechanism remains to be further studied (83). Metabolic alterations promote the activation of the NLRP3 inflammasome, which further induces pyroptosis, releases inflammatory cytokines, and forms a pro-fibrotic microenvironment.
3.3 The role of pyroptosis mediated by NLRP3 inflammasome in PF
Pyroptosis is a type of cell death that triggers an inflammatory response and plays an important role in PF (84). The NLRP3 inflammasome plays a significant role in the initiation of pyroptosis (50, 77). Upon activation, the NLRP3 inflammasome drives the activation of caspase-1. Once activated, caspase-1 not only processes pro-IL-1β and pro-IL-18 into their biologically active forms but also cleaves gasdermin D (GSDMD), inducing the oligomerization of its N-terminal fragments (GSDMD-NT). These fragments assemble into 21-nanometer pores within the plasma membrane, facilitating the extracellular release of IL-1β and IL-18, compromising membrane integrity, and ultimately orchestrating the execution of pyroptosis (85). The intense inflammatory environment triggered by pyroptosis can activate myofibroblasts, leading to pulmonary fibrosis (83). Studies have shown that NLRP3-mediated pyroptosis in PF is closely related to TLR9 (86), which is an important member of the TLR family that primarily recognizes DNA containing unmethylated CpG sequences and plays a significant role in the pathogenesis of various diseases (87–89). In pulmonary fibrosis, self-DNA released from damaged cells (such as mitochondrial DNA and chromatin DNA) can activate TLR9 (88). Activated TLR9 promotes NF-κB activation, increasing NLRP3 and pro-IL-1β expression (88). Simultaneously, TLR9 signaling also promotes ROS production (90) and calcium ion change (91), thereby facilitating NLRP3 inflammasome assembly and activation. In mice with BLM-induced PF, it has been found that TLR9 expression is elevated, which increases the activation of the NLRP3/caspase-1 inflammasome pathway, thereby promoting pyroptosis of alveolar epithelial cells. In TLR9 knockdown mice, pulmonary fibrosis was alleviated and cellular pyroptosis was reduced. However, treatment with an NLRP3 activator reversed the levels of fibrosis and pyroptosis in the lung tissue of TLR9 knockout mice (86). Additionally, TLR9 can indirectly influence the activation state of the NLRP3 inflammasome by regulating the inflammatory responses of dendritic cells and macrophages (92). The cell death and more cytokine release mediated by pyroptosis not only directly promote inflammatory responses but also enhance TGF-β1 expression through augmentation of the NF-κB pathway, thereby driving the fibrotic process.
3.4 The NLRP3 inflammasome promote fibrotic signaling: TGF-β1 pathway
TGF-β1 is considered as a key mediator that drives the differentiation of fibroblasts into myofibroblasts and induces the expression of fibrosis-related genes (10). Elevated levels of TGF-β1 are detected in the BALF and lung tissue of IPF patients (93). Additionally, a genetic link has been discovered, with polymorphisms in the TGF-β1 gene associated with an increased susceptibility to IPF (94). Research has found that TGF-β1 is involved in the process of pulmonary fibrosis by activating both the canonical (SMAD-dependent) and non-canonical (SMAD-independent) pathways, with the SMAD signaling pathway being the primary one (51, 95). In the canonical pathway, active TGF-β is released and binds to the TGF-β type II receptor (TβRII), recruiting the TGF-β type I receptor (TβRI). TβRI is activated under the phosphorylation of TβRII, initiating kinase activity, which further leads to the phosphorylation of R-Smads (Smad2 and Smad3) (96). There is an evidence to suggest that the phosphorylation of Smad2/3 is a key profibrotic signal mediated by TGF-β1 (8). Activated Smad2 and Smad3 form a complex with Co-Smad (Smad4) and were translocated into the nucleus to regulate the expression of multiple profibrotic genes (97), promoting the transdifferentiation of fibroblasts into myofibroblasts and EMT (98). The non-canonical pathways include p38 mitogen-activated protein kinase (p38 MAPK), c-Jun N-terminal kinase (c-JNK), phosphatidylinositol 3-kinase (PI3K)-Akt-mTOR, NF-κB, transforming growth factor-β activated kinase 1 (TAK1), Janus kinase 2 (JAK2), signal transducer and activator of transcription 3 (STAT3), Raf-MEK1/2-ERK1/2, and Rho-associated kinase (ROCK) (95). These non-canonical pathways, in conjunction with the canonical pathway, promote the occurrence of fibrosis at multiple levels.
Recent studies have shown that NLRP3 inflammasome plays an important role in the fibrotic process induced by TGF-β1 (8). Compared to the control group, the levels of TGF-β1, phosphorylated Smad2/3 proteins, and the mRNA levels of NLRP3, caspase-1, and ASC in the lung tissues of mice exposed to BLM were all significantly increased. Concurrently, in vitro experiments found that the upregulation of NLRP3 reversed the inhibition of the TGF-β1/Smad2/3 pathway (51). Similarly, in a rat model exposed to silica, the expressions of NLRP3, TGF-β1, and IL-1β were all increased, and the activated NLRP3 inflammasome promoted the secretion of inflammatory cytokines IL-1β and TGF-β (10). Furthermore, it was found that after silencing the expression of NLRP3 in vitro, the protein level of TGF-β1 was significantly reduced (26). Early studies have found that in lung cells (A549), IL-1β stimulates TGF-β1 transcription through temporal regulation by NF-κB and AP-1 (99). As noted earlier, NF-κB plays a pivotal role in the assembly and activation of the NLRP3 inflammasome as well as the production of IL-1β. Therefore, it is plausible that the NLRP3 inflammasome promotes TGF-β1 expression via the NF-κB signaling pathway. The exact regulatory mechanisms in pulmonary fibrosis remain to be further investigated. In another study, to simulate intercellular interactions in pulmonary fibrosis, alveolar epithelial cells and macrophages were co-cultured under hypoxic conditions. This co-culture resulted in increased expression of NLRP3, TGF-β1, and TGFBRs, alongside a significant downregulation of SMAD7, a negative regulator of TGF-β signaling, thereby promoting EMT. Notably, silencing TGF-β1 or applying TGF-β1 inhibitors, TβRI kinase inhibitors, or p38 MAPK inhibitors effectively suppressed the upregulation of NLRP3 expression. However, treatment with a SMAD3 phosphorylation inhibitor did not lead to significant changes in NLRP3 expression levels. These findings indicate that TGF-β1 regulates NLRP3-mediated EMT primarily through the p38 MAPK pathway rather than the SMAD3 phosphorylation pathway (100).
In conclusion, the role of NLRP3 inflammasome in PF constitutes a comprehensive signaling network. This begins with PAMPs/DAMPs-mediated NF-κB activation as the upstream trigger, continues through cellular stress (oxidative stress and ER stress), autophagy, and metabolic reprogramming as regulatory mechanisms, and proceeds to NLRP3-mediated pyroptosis which serves as a positive feedback amplifying inflammatory responses. Ultimately, the NLRP3 inflammasome enhances the TGF-β1 signaling pathway, leading to ECM deposition and tissue fibrosis. This complete signaling cascade demonstrates the NLRP3 inflammasome as a central node in the pathological process of pulmonary fibrosis, connecting inflammatory responses and fibrotic progression, thus providing a theoretical basis for targeted therapeutic strategies.
4 Targeting NLRP3 inflammasome for the treatment of PF
As mentioned above, NLRP3 inflammasome plays a critical role in the pathological mechanism of PF. Therefore, targeting the NLRP3 inflammasome for the treatment of PF has become one of the current research hotspots. Several drugs have been developed to inhibit NLRP3 inflammasome in different disease models, such as ZYIL1, DFV890, VTX(2735, 3232), emlenoflast, selnoflast, NT(0796, 0527, 0249), dapansutrile, tranilast, MCC950, 3,4-Methylenedioxy-β-nitrostyrene, CY-09, RRx-001, and HT-6184, some of which are currently undergoing clinical trials (17, 101). Table 1 summarizes several drugs targeting NLRP3 inflammasome in PF models.
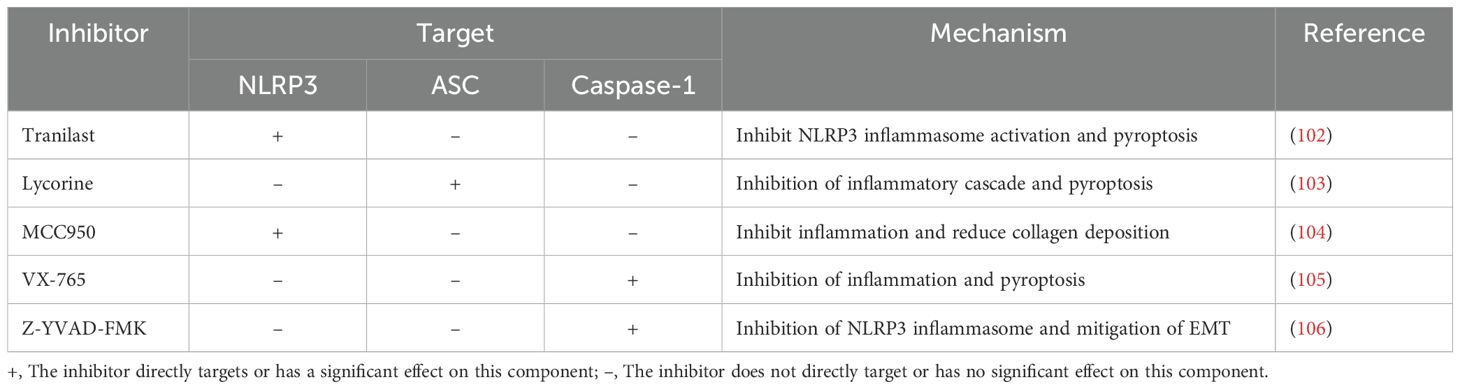
Table 1. Inhibitors targeting NLRP3 inflammasome and their mechanism in PF models.
Tranilast (N-(3’,4’-dimethoxycinnamoyl)-anthranilic acid), a tryptophan metabolite analog, binds to the NACHT domain of NLRP3 and disrupts the NLRP3-ASC interaction without affecting NLRP3-NEK7 binding (17, 101). It has been approved in South Korea and Japan for the treatment of asthma, keloids, and hypertrophic scars (102). In vivo and in vitro experiments demonstrate that tranilast protects against acute respiratory distress syndrome and early pulmonary fibrosis induced by smoke inhalation (107). In a case report of severe COVID-19 pneumonia with secondary pulmonary fibrosis, six months of tranilast treatment significantly improved lung fibrosis and respiratory function (102).
Lycorine, a natural alkaloid extracted from the amaryllidaceae family, was initially reported to inhibit the growth and cell division of yeasts, algae, and higher plants (108). In recent years, studies have demonstrated its antitumor, anti-inflammatory, antioxidant, and antifibrotic effects. In the BLM-induced pulmonary fibrosis model, lycorine could disrupt the interaction of NLRP3 with ASC by targeting the PYD domain at Leu9, Leu50, and Thr53, thereby inhibiting NLRP3 inflammasome activation and pyroptosis, ultimately ameliorating BLM-induced pulmonary fibrosis (103).
MCC950 (CP-456,773 or CRID3) is one of the most extensively studied highly selective NLRP3 inflammasome inhibitors. Its exact mechanism of action remains unclear, but it has been applied in animal models of various diseases, including pulmonary fibrosis (33). A study demonstrated that MCC950 inhibits NLRP3 activation by converting its active conformation into an inactive state (109). Another study showed that MCC950 directly targets the Walker B motif in the NACHT domain of NLRP3, thereby blocking ATP hydrolysis and inhibiting NLRP3 activation and inflammasome formation (110). In BLM-induced PF model, MCC950 treatment inhibited the NLRP3 inflammasome, alleviated alveolar hemorrhage and alveolitis, and reduced collagen fiber deposition, thereby improving pulmonary fibrosis (104).
VX-765 is a specific caspase-1 inhibitor that has been proven in animal models to alleviate Alzheimer’s disease, epilepsy, and cardiovascular diseases (111). In the silica-induced pulmonary fibrosis model, VX-765 reduced the expression of inflammatory cytokines, including IL-1β, TNF-α, IL-6, CCL2, and CCL3, downregulated endogenous DAMPs and inflammation-related pattern recognition receptors TLR4 and NLRP3, inhibited pyroptosis of alveolar macrophages, and decreased α-SMA, collagen, and fibronectin levels, thereby alleviating pulmonary fibrosis (105).
Z-YVAD-FMK, another caspase-1 inhibitor, has been utilized in various disease models, including ischemic stroke, neuroblastoma, epilepsy, myocardial infarction, and fibrosis (112–116). In a PM2.5-induced pulmonary fibrosis model, treatment with Z-YVAD-FMK effectively inhibited IL-1β secretion and the overexpression of ASC and NLRP3 proteins (18). Similarly, in a silicosis-induced pulmonary fibrosis model, Z-YVAD-FMK suppressed the activation of the NLRP3 inflammasome and mitigated silica-induced EMT (106).
Other drugs and molecules can also influence NLRP3 inflammasome in pulmonary fibrosis models. Nintedanib and pirfenidone both suppressed NLRP3 inflammasome activation in pulmonary fibrosis models, exerting anti-inflammatory effects (117, 118). Asiatic acid (AA), isolated from Centella Asiatica, has been shown to reduce NLRP3 expression in PF model, although the exact mechanism remains to be further investigated (119). Exosomes derived from mesenchymal stem cells and small RNA molecules may also alleviate pulmonary fibrosis by modulating the NLRP3 inflammasome (104, 120). In addition, several studies have identified potential targets, such as the autophagy adapter P62/SQSTM1, which can inhibit the excessive activation of the NLRP3 inflammasome by transporting ubiquitinated ASC to autophagosomes for degradation and through a positive feedback loop with Nrf2-ARE (121). The overexpression of membrane protein Caveolin-1 can suppress the NLRP3 inflammasome and its associated expression of interleukin-1β (IL-1β), thereby hindering the progression of fibrosis (122).
In preclinical studies of pulmonary fibrosis therapeutics, many laboratories utilize the bleomycin-induced pulmonary fibrosis mouse model (123). While mouse models, particularly the BLM-induced model, provide valuable tools for studying PF, it is important to note that these models exhibit significant differences from human IPF (124). The BLM model primarily represents fibrotic responses following acute lung injury, rather than the slowly progressive course characteristic of human IPF. The inflammatory response induced by BLM is typically more pronounced than that observed in human IPF (125). Additionally, species differences between mice and humans in NLRP3 regulation and inflammatory responses may affect the clinical translatability of research findings. Therefore, when evaluating NLRP3-targeted strategies, animal model data should be interpreted cautiously, and more clinically relevant experimental systems should be actively explored, such as humanized mouse models, human lung tissue organoids, and other approaches that more closely approximate human pathology.
5 Conclusions and future perspectives
PF is an interstitial lung disease with an unclear pathogenesis, presenting significant challenges for its treatment. The NLRP3 inflammasome has been extensively studied in various diseases, and its role in pulmonary fibrosis has been increasingly explored in recent years. This review discusses the role of the NLRP3 inflammasome in pulmonary fibrosis and its potential mechanisms. The NLRP3 inflammasome participates in inflammatory responses and immune regulation in the body, forms a positive feedback loop with the NF-κB pathway during the progression of pulmonary fibrosis, and interacts with the TGF-β1 signaling pathway to promote EMT, myofibroblast formation, and extracellular matrix accumulation, thereby driving fibrotic processes. Furthermore, the NLRP3 inflammasome is involved in the progression of PF through mechanisms such as oxidative stress, ER stress, pyroptosis, autophagy, and metabolism. Thus, the NLRP3 inflammasome is an important regulatory factor in PF. In addition, the potential of the NLRP3 inflammasome as a therapeutic target has gradually emerged. Several drugs targeting the NLRP3 inflammasome have already been developed, with some demonstrating promising therapeutic effects in pulmonary fibrosis models. However, additional in-depth research is required before these drugs can be translated into clinical applications. Consequently, future investigations should prioritize a comprehensive understanding of the mechanistic role of the NLRP3 inflammasome in pulmonary fibrosis, rigorously evaluate the efficacy and safety profiles of targeted therapies, and advance the development of personalized treatment strategies. Moreover, the integration of advanced drug delivery systems (DDS), including nano-delivery, hydrogel delivery, and biological carrier delivery, holds great potential in minimizing side effects and enhancing drug bioavailability (126). As such, the future application of DDS to specifically target the NLRP3 inflammasome could significantly improve the therapeutic outcomes in pulmonary fibrosis management.
Additionally, studies have revealed a close association between PF and lung cancer, with IPF patients exhibiting a 2.4-7.5-fold increased risk of developing lung cancer compared to the general population (127). The NLRP3 inflammasome appears to function as a molecular bridge in this transformation from IPF to cancer (103). Mechanistically, the chronic inflammatory microenvironment maintained by persistent NLRP3 activation promotes genomic instability and DNA damage (128, 129), while downstream inflammatory mediators such as IL-1β facilitate tumor cell proliferation and angiogenesis (130, 131). Furthermore, the extensively remodeled extracellular matrix and altered growth factor signaling characteristic of fibrotic tissues create a conducive microenvironment for tumor cell survival and migration. This dual involvement of the NLRP3 inflammasome in both IPF pathogenesis (14) and malignant transformation (132) positions it as a promising therapeutic target with potential to simultaneously address both serious pulmonary conditions.
In conclusion, the NLRP3 inflammasome provides a new perspective for understanding the pathogenesis of pulmonary fibrosis and offers novel therapeutic strategies for its treatment.
Author contributions
MW: Writing – review & editing, Investigation, Conceptualization, Software, Writing – original draft, Supervision, Project administration. YX: Writing – original draft, Writing – review & editing, Conceptualization, Investigation, Project administration, Supervision. YC: Methodology, Supervision, Investigation, Conceptualization, Writing – review & editing. BY: Investigation, Supervision, Software, Writing – review & editing. QD: Writing – review & editing, Funding acquisition, Project administration, Supervision, Investigation, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the National Key Research and Development Program of China (2021YFC2501800), the National Natural Science Foundation of China (82002077), and the 6th Shanghai Three-year Action Plan to Strengthen the Construction of Public Health System (GWVI-2.1-3).
Acknowledgments
We would like to express our sincere gratitude to all colleagues who contributed to this review. We would especially like to thank the constructive comments from our team members that helped improve the quality and clarity of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Naik PK, Moore BB. Viral infection and aging as cofactors for the development of pulmonary fibrosis. Expert Rev Respir Med. (2010) 4:759–71. doi: 10.1586/ers.10.73
2. Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med. (2018) 378:1811–23. doi: 10.1056/NEJMra1705751
3. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2022) 205:e18–47. doi: 10.1164/rccm.202202-0399ST
4. Poerio A, Carlicchi E, Zompatori M. Diagnosis of interstitial lung disease (ILD) secondary to systemic sclerosis (SSc) and rheumatoid arthritis (RA) and identification of ‘progressive pulmonary fibrosis’ using chest CT: a narrative review. Clin Exp Med. (2023) 23:4721–8. doi: 10.1007/s10238-023-01202-1
5. Alsomali H, Palmer E, Aujayeb A, Funston W. Early diagnosis and treatment of idiopathic pulmonary fibrosis: A narrative review. Pulm Ther. (2023) 9:177–93. doi: 10.1007/s41030-023-00216-0
6. Enzel D, Kriventsov M, Sataieva T, Malygina V. Cellular and molecular genetic mechanisms of lung fibrosis development and the role of vitamin D: A review. IJMS. (2024) 25:8946. doi: 10.3390/ijms25168946
7. Liu C, Zhang Q, Zhou H, Jin L, Liu C, Yang M, et al. GLP-1R activation attenuates the progression of pulmonary fibrosis via disrupting NLRP3 inflammasome/PFKFB3-driven glycolysis interaction and histone lactylation. J Transl Med. (2024) 22:954. doi: 10.1186/s12967-024-05753-z
8. Gairola S, Sinha A, Kaundal RK. Linking NLRP3 inflammasome and pulmonary fibrosis: mechanistic insights and promising therapeutic avenues. Inflammopharmacol. (2024) 32:287–305. doi: 10.1007/s10787-023-01389-5
9. Yun C-X, Huan M-L, Zhu X, Wan Y-H, Zou J-B, Zhang B-L. Construction of the pulmonary bio-adhesive delivery system of nintedanib nanocrystalline for effective treatment of pulmonary fibrosis. Int J Pharmaceut. (2024) 660:124302. doi: 10.1016/j.ijpharm.2024.124302
10. Mina S, Elfeky DM, Kabel AM, Hedya SE. Ameliorative potential of donepezil with or without prednisolone in bleomycin-induced pulmonary fibrosis in rats: involvement of the anti-inflammatory, antioxidant, and the antifibrotic pathways. Medicina. (2023) 59:980. doi: 10.3390/medicina59050980
11. Nguyen T-H, Nguyen H-H-N, Nguyen T-D, Tran VT-H, Nguyen H-A, Pham D-V. NLRP3 inflammasome activation contributes to the development of the pro-fibrotic phenotype of lung fibroblasts. Biochem Pharmacol. (2024) 229:116496. doi: 10.1016/j.bcp.2024.116496
12. Kabel AM. Targeting oxidative stress, proinflammatory cytokines, apoptosis and toll like receptor 4 by empagliflozin to ameliorate bleomycin-induced lung fibrosis. Respir Physiol Neurobiol. (2020) 273:103316. doi: 10.1016/j.resp.2019.103316
13. Nie Y, Li J, Zhai X, Wang Z, Wang J, Wu Y, et al. Elamipretide(SS-31) attenuates idiopathic pulmonary fibrosis by inhibiting the nrf2-dependent NLRP3 inflammasome in macrophages. Antioxidants. (2023) 12:2022. doi: 10.3390/antiox12122022
14. Wang H, Wen Y, Wang L, Wang J, Chen H, Chen J, et al. DDR1 activation in macrophage promotes IPF by regulating NLRP3 inflammasome and macrophage reaction. Int Immunopharmacol. (2022) 113:109294. doi: 10.1016/j.intimp.2022.109294
15. Guo H, Callaway JB, Ting JP-Y. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. (2015) 21:677–87. doi: 10.1038/nm.3893
16. Xu J, Núñez G. The NLRP3 inflammasome: activation and regulation. Trends Biochem Sci. (2023) 48:331–44. doi: 10.1016/j.tibs.2022.10.002
17. Colunga Biancatelli RML, Solopov PA, Catravas JD. The inflammasome NLR family pyrin domain-containing protein 3 (NLRP3) as a novel therapeutic target for idiopathic pulmonary fibrosis. Am J Pathol. (2022) 192:837–46. doi: 10.1016/j.ajpath.2022.03.003
18. Zheng R, Tao L, Jian H, Chang Y, Cheng Y, Feng Y, et al. NLRP3 inflammasome activation and lung fibrosis caused by airborne fine particulate matter. Ecotoxicol Environ Saf. (2018) 163:612–9. doi: 10.1016/j.ecoenv.2018.07.076
19. Kodi T, Sankhe R, Gopinathan A, Nandakumar K, Kishore A. New insights on NLRP3 inflammasome: mechanisms of activation, inhibition, and epigenetic regulation. J Neuroimmune Pharmacol. (2024) 19:7. doi: 10.1007/s11481-024-10101-5
20. Kukkonen MK, Vehmas T, Piirilä P, Hirvonen A. Genes involved in innate immunity associated with asbestos-related fibrotic changes. Occup Environ Med. (2013) 71:48–54. doi: 10.1136/oemed-2013-101555
21. Lasithiotaki I, Giannarakis I, Tsitoura E, Samara KD, Margaritopoulos GA, Choulaki C, et al. NLRP3 inflammasome expression in idiopathic pulmonary fibrosis and rheumatoid lung. Eur Respir J. (2016) 47:910–8. doi: 10.1183/13993003.00564-2015
22. Cao W, Wang X, Li J, Yan M, Chang CH, Kim J, et al. NLRP3 inflammasome activation determines the fibrogenic potential of PM2.5 air pollution particles in the lung. J Environ Sci. (2022) 111:429–41. doi: 10.1016/j.jes.2021.04.021
23. Ning J, Pei Z, Wang M, Hu H, Chen M, Liu Q, et al. Site-specific Atg13 methylation-mediated autophagy regulates epithelial inflammation in PM2.5-induced pulmonary fibrosis. J Hazard Mater. (2023) 457:131791. doi: 10.1016/j.jhazmat.2023.131791
24. Tian J, Song D, Peng Y, Zhang J, Ma L, Chen Z, et al. Silica-induced macrophage pyroptosis propels pulmonary fibrosis through coordinated activation of relaxin and osteoclast differentiation signaling to reprogram fibroblasts. Ecotoxicol Environ Saf. (2024) 273:116106. doi: 10.1016/j.ecoenv.2024.116106
25. Wang H, Wang G, Meng Y, Liu Y, Yao X, Feng C. Modified Guo-Min decoction ameliorates PM2.5-induced lung injury by inhibition of PI3K-AKT and MAPK signaling pathways. Phytomedicine. (2024) 123:155211. doi: 10.1016/j.phymed.2023.155211
26. Tian R, Zhu Y, Yao J, Meng X, Wang J, Xie H, et al. NLRP3 participates in the regulation of EMT in bleomycin-induced pulmonary fibrosis. Exp Cell Res. (2017) 357:328–34. doi: 10.1016/j.yexcr.2017.05.028
27. Zhao C, Zhao W. NLRP3 inflammasome—A key player in antiviral responses. Front Immunol. (2020) 11:211. doi: 10.3389/fimmu.2020.00211
28. Xiao L, Magupalli VG, Wu H. Cryo-EM structures of the active NLRP3 inflammasome disc. Nature. (2022) 613:595–600. doi: 10.1038/s41586-022-05570-8
29. Alehashemi S, Goldbach-Mansky R. Human autoinflammatory diseases mediated by NLRP3-, pyrin-, NLRP1-, and NLRC4-inflammasome dysregulation updates on diagnosis, treatment, and the respective roles of IL-1 and IL-18. Front Immunol. (2020) 11:1840. doi: 10.3389/fimmu.2020.01840
30. Liao Y, Kong Y, Chen H, Xia J, Zhao J, Zhou Y. Unraveling the priming phase of NLRP3 inflammasome activation: Molecular insights and clinical relevance. Int Immunopharmacol. (2025) 146:113821. doi: 10.1016/j.intimp.2024.113821
31. He Y, Zeng MY, Yang D, Motro B, Núñez G. Nek7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. (2016) 530:354–7. doi: 10.1038/nature16959
32. Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. (2016) 17:250–8. doi: 10.1038/ni.3333
33. Wu D, Chen Y, Sun Y, Gao Q, Li H, Yang Z, et al. Target of MCC950 in inhibition of NLRP3 inflammasome activation: a literature review. Inflammation. (2020) 43:17–23. doi: 10.1007/s10753-019-01098-8
34. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ Efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
35. Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun. (2017) 8:202. doi: 10.1038/s41467-017-00227-x
36. Green JP, Yu S, Martín-Sánchez F, Pelegrin P, Lopez-Castejon G, Lawrence CB, et al. Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc Natl Acad Sci USA. (2018) 115:E9371–80. doi: 10.1073/pnas.1812744115
37. Billingham LK, Stoolman JS, Vasan K, Rodriguez AE, Poor TA, Szibor M, et al. Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat Immunol. (2022) 23:692–704. doi: 10.1038/s41590-022-01185-3
38. Zhang Z, Meszaros G, He W, Xu Y, Magliarelli H, de F, et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J Exp Med. (2017) 214(9):2671–93. doi: 10.1084/jem.20162040
39. Song ZS, Shao H, Chen YQ, Zhang R. Expression and significance of NLRP3/IL-1β/TGF-β(1) signal axis in rat model of silicosis pulmonary fibrosis. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. (2018) 36:819–23. doi: 10.3760/cma.j.issn.1001-9391.2018.11.005
40. Liu X, Wang J, Dou P, Zhang X, Ran X, Liu L, et al. The ameliorative effects of arctiin and arctigenin on the oxidative injury of lung induced by silica via TLR-4/NLRP3/TGF-β Signaling pathway. Oxid Med Cell Longev. (2021) 2021:5598980. doi: 10.1155/2021/5598980
41. Zhang J, Zhang J, Yao Z, Shao W, Song Y, Tang W, et al. GAMG ameliorates silica-induced pulmonary inflammation and fibrosis via the regulation of EMT and NLRP3/TGF-β1/Smad signaling pathway. Ecotoxicol Environ Saf. (2024) 285:117124. doi: 10.1016/j.ecoenv.2024.117124
42. Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. (2013) 14:454–60. doi: 10.1038/ni.2550
43. Pandey A, Shen C, Feng S, Man SM. Cell biology of inflammasome activation. Trends Cell Biol. (2021) 31:924–39. doi: 10.1016/j.tcb.2021.06.010
44. Chen J, Chen ZJ. PtdIns4P on dispersed trans-golgi network mediates NLRP3 inflammasome activation. Nature. (2022) 564:71–6. doi: 10.1038/s41586-018-0761-3
45. Hong Q, Zhang Y, Lin W, Wang W, Yuan Y, Lin J, et al. Negative feedback of the cAMP/PKA pathway regulates the effects of endoplasmic reticulum stress-induced NLRP3 inflammasome activation on type II alveolar epithelial cell pyroptosis as a novel mechanism of BLM-induced pulmonary fibrosis. J Immunol Res. (2022) 2022:1–13. doi: 10.1155/2022/2291877
46. Zhang Y, Liang J, Cao N, Gao J, Song L, Tang X. Coal dust nanoparticles induced pulmonary fibrosis by promoting inflammation and epithelial-mesenchymal transition via the NF-κB/NLRP3 pathway driven by IGF1/ROS-mediated AKT/GSK3β signals. Cell Death Discov. (2022) 8:500. doi: 10.1038/s41420-022-01291-z
47. Zhang J, Liu X, Wan C, Liu Y, Wang Y, Meng C, et al. NLRP3 inflammasome mediates M1 macrophage polarization and IL-1β production in inflammatory root resorption. J Clinic Periodontol. (2020) 47:451–60. doi: 10.1111/jcpe.13258
48. Liu T, Wang L, Liang P, Wang X, Liu Y, Cai J, et al. USP19 suppresses inflammation and promotes M2-like macrophage polarization by manipulating NLRP3 function via autophagy. Cell Mol Immunol. (2021) 18:2431–42. doi: 10.1038/s41423-020-00567-7
49. Abd-Elmawla MA, Ghaiad HR, Gad ES, Ahmed KA, Abdelmonem M. Suppression of NLRP3 inflammasome by ivermectin ameliorates bleomycin-induced pulmonary fibrosis. J Zhejiang Univ Sci B. (2023) 24:723–33. doi: 10.1631/jzus.B2200385
50. Ma L, Han Z, Yin H, Tian J, Zhang J, Li N, et al. Characterization of cathepsin B in mediating silica nanoparticle-induced macrophage pyroptosis via an NLRP3-dependent manner. JIR Volume. (2022) 15:4537–45. doi: 10.2147/JIR.S371536
51. Min L, Shu-Li Z, Feng Y, Han H, Shao-Jun L, Sheng-Xiong T, et al. NecroX-5 ameliorates bleomycin-induced pulmonary fibrosis via inhibiting NLRP3-mediated epithelial–mesenchymal transition. Respir Res. (2022) 23:128. doi: 10.1186/s12931-022-02044-3
52. Zheng R, Song P, Wu Y, Wang Y, Han X, Yan J, et al. Property-activity relationship between physicochemical properties of PM2.5 and their activation of NLRP3 inflammasome. NanoImpact. (2022) 25:100380. doi: 10.1016/j.impact.2022.100380
53. Liu Y, Zhao C, Meng J, Li N, Xu Z, Liu X, et al. Galectin-3 regulates microglial activation and promotes inflammation through TLR4/MyD88/NF-kB in experimental autoimmune uveitis. Clin Immunol. (2022) 236:108939. doi: 10.1016/j.clim.2022.108939
54. Ding N, Wei B, Fu X, Wang C, Wu Y. Natural products that target the NLRP3 inflammasome to treat fibrosis. Front Pharmacol. (2020) 11:591393. doi: 10.3389/fphar.2020.591393
55. Song C, He L, Zhang J, Ma H, Yuan X, Hu G, et al. Fluorofenidone attenuates pulmonary inflammation and fibrosis via inhibiting the activation of NALP 3 inflammasome and IL -1β/IL -1R1/MyD88/NF -κB pathway. J Cell Mol Med. (2016) 20:2064–77. doi: 10.1111/jcmm.12898
56. Middleton EA, He X-Y, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. (2020) 136:1169–79. doi: 10.1182/blood.2020007008
57. Phan THG, Paliogiannis P, Nasrallah GK, Giordo R, Eid AH, Fois AG, et al. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell Mol Life Sci. (2020) 78:2031–57. doi: 10.1007/s00018-020-03693-7
58. Gurung P, Lukens JR, Kanneganti T-D. Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med. (2015) 21:193–201. doi: 10.1016/j.molmed.2014.11.008
59. Hindman B, Ma Q. Carbon nanotubes and crystalline silica stimulate robust ROS production, inflammasome activation, and IL-1β secretion in macrophages to induce myofibroblast transformation. Arch Toxicol. (2019) 93:887–907. doi: 10.1007/s00204-019-02411-y
60. Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. (2005) 97:673–83. doi: 10.1002/jcb.20743
61. Sekine H, Motohashi H. Roles of CNC transcription factors NRF1 and NRF2 in cancer. Cancers. (2021) 13:541. doi: 10.3390/cancers13030541
62. Huang Y, Wang A, Jin S, Liu F, Xu F. Activation of the NLRP3 inflammasome by HMGB1 through inhibition of the Nrf2/HO-1 pathway promotes bleomycin-induced pulmonary fibrosis after acute lung injury in rats. Allergol Immunopathol (Madr). (2023) 51:56–67. doi: 10.15586/aei.v51i3.668
63. Feng J, Liu H, Jiang K, Gong X, Huang R, Zhou C, et al. Enhanced oxidative stress aggravates BLM-induced pulmonary fibrosis by promoting cellular senescence through enhancing NLRP3 activation. Life Sci. (2024) 358:123128. doi: 10.1016/j.lfs.2024.123128
64. Luna-Marco C, Ubink A, Kopsida M, Heindryckx F. Endoplasmic reticulum stress and metabolism in hepatocellular carcinoma. Am J Pathol. (2023) 193:1377–88. doi: 10.1016/j.ajpath.2022.09.012
65. Sun Z, He W, Meng H, Li P, Qu J. Endoplasmic reticulum stress in acute lung injury and pulmonary fibrosis. FASEB J. (2024) 38:e70232. doi: 10.1096/fj.202401849RR
66. Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol Mech Dis. (2015) 10:173–94. doi: 10.1146/annurev-pathol-012513-104649
67. Yu Q-Q, Zhang H, Guo Y, Han B, Jiang P. The intestinal redox system and its significance in chemotherapy-induced intestinal mucositis. Oxid Med Cell Longevity. (2022) 2022:1–29. doi: 10.1155/2022/7255497
68. Katzen J, Rodriguez L, Tomer Y, Babu A, Zhao M, Murthy A, et al. Disruption of proteostasis causes IRE1 mediated reprogramming of alveolar epithelial cells. Proc Natl Acad Sci USA. (2022) 119:e2123187119. doi: 10.1073/pnas.2123187119
69. Dong Z, Wang X, Wang P, Bai M, Wang T, Chu Y, et al. Idiopathic pulmonary fibrosis caused by damaged mitochondria and imbalanced protein homeostasis in alveolar epithelial type II cell. Adv Biol. (2024) 9:2400297. doi: 10.1002/adbi.202400297
70. Zhu S, Li X, Dang B, Wu F, Wang C, Lin C. Lycium Barbarum polysaccharide protects HaCaT cells from PM2.5-induced apoptosis via inhibiting oxidative stress, ER stress and autophagy. Redox Rep. (2022) 27:32–44. doi: 10.1080/13510002.2022.2036507
71. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. (2011) 147:728–41. doi: 10.1016/j.cell.2011.10.026
72. Ding S, Wang H, Wang M, Bai L, Yu P, Wu W. Resveratrol alleviates chronic “real-world” ambient particulate matter-induced lung inflammation and fibrosis by inhibiting NLRP3 inflammasome activation in mice. Ecotoxicol Environ Saf. (2019) 182:109425. doi: 10.1016/j.ecoenv.2019.109425
73. Yang P, Song R, Li N, Sun K, Shi F, Liu H, et al. Silica dust exposure induces autophagy in alveolar macrophages through switching Beclin1 affinity from Bcl-2 to PIK3C3. Environ Toxicol. (2020) 35:758–67. doi: 10.1002/tox.22910
74. Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. (2013) 304:L56–69. doi: 10.1152/ajplung.00213.2012
75. Yang H, Wang L, Yang M, Hu J, Zhang E, Peng L. Oridonin attenuates LPS-induced early pulmonary fibrosis by regulating impaired autophagy, oxidative stress, inflammation and EMT. Eur J Pharmacol. (2022) 923:174931. doi: 10.1016/j.ejphar.2022.174931
76. Wei Y, You Y, Zhang J, Ban J, Min H, Li C, et al. Crystalline silica-induced macrophage pyroptosis interacting with mitophagy contributes to pulmonary fibrosis via modulating mitochondria homeostasis. J Hazard Mater. (2023) 454:131562. doi: 10.1016/j.jhazmat.2023.131562
77. Wei Y, Liu C, Li L. Geniposide improves bleomycin-induced pulmonary fibrosis by inhibiting NLRP3 inflammasome activation and modulating metabolism. J Funct Foods. (2023) 104:105503. doi: 10.1016/j.jff.2023.105503
78. Kang YP, Lee SB, Lee J, Kim HM, Hong JY, Lee WJ, et al. Metabolic profiling regarding pathogenesis of idiopathic pulmonary fibrosis. J Proteome Res. (2016) 15:1717–24. doi: 10.1021/acs.jproteome.6b00156
79. Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu R-M, et al. Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am J Respir Crit Care Med. (2015) 192:1462–74. doi: 10.1164/rccm.201504-0780OC
80. Cui H, Xie N, Banerjee S, Ge J, Jiang D, Dey T, et al. Lung myofibroblasts promote macrophage profibrotic activity through lactate-induced histone lactylation. Am J Respir Cell Mol Biol. (2021) 64:115–25. doi: 10.1165/rcmb.2020-0360OC
81. Rho H, Terry AR, Chronis C, Hay N. Hexokinase 2-mediated gene expression via histone lactylation is required for hepatic stellate cell activation and liver fibrosis. Cell Metab. (2023) 35:1406–1423.e8. doi: 10.1016/j.cmet.2023.06.013
82. Gasse P, Riteau N, Charron S, Girre S, Fick L, Pétrilli V, et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med. (2009) 179:903–13. doi: 10.1164/rccm.200808-1274OC
83. Ouyang B, Deng L, Yang F, Shi H, Wang N, Tang W, et al. Albumin-based formononetin nanomedicines for lung injury and fibrosis therapy via blocking macrophage pyroptosis. Mater Today Bio. (2023) 20:100643. doi: 10.1016/j.mtbio.2023.100643
84. Feng Y, Li M, Yangzhong X, Zhang X, Zu A, Hou Y, et al. Pyroptosis in inflammation-related respiratory disease. J Physiol Biochem. (2022) 78:721–37. doi: 10.1007/s13105-022-00909-1
85. Toldo S, Abbate A. The role of the NLRP3 inflammasome and pyroptosis in cardiovascular diseases. Nat Rev Cardiol. (2023) 21:219–37. doi: 10.1038/s41569-023-00946-3
86. Ren C, Wang Q, Fan S, Mi T, Zhang Z, He D. Toll-like receptor 9 aggravates pulmonary fibrosis by promoting NLRP3-mediated pyroptosis of alveolar epithelial cells. Inflammation. (2024) 47:1744–61. doi: 10.1007/s10753-024-02006-5
87. Lyu A, Zhu S, Chen J, Zhao Y, Pu D, Luo C, et al. Inhibition of TLR9 attenuates skeletal muscle fibrosis in aged sarcopenic mice via the p53/SIRT1 pathway. Exp Gerontol. (2019) 122:25–33. doi: 10.1016/j.exger.2019.04.008
88. Lu P, Zheng H, Meng H, Liu C, Duan L, Zhang J, et al. Mitochondrial DNA induces nucleus pulposus cell pyroptosis via the TLR9-NF-κB-NLRP3 axis. J Transl Med. (2023) 21:389. doi: 10.1186/s12967-023-04266-5
89. Luo X, Zhai Z, Lin Z, Wu S, Xu W, Li Y, et al. Cyclophosphamide induced intestinal injury is alleviated by blocking the TLR9/caspase3/GSDME mediated intestinal epithelium pyroptosis. Int Immunopharmacol. (2023) 119:110244. doi: 10.1016/j.intimp.2023.110244
90. Alekseeva A, Kameneva LV, Kostyuk SV, Veiko NN. Multiple ways of cfDNA reception and following ROS production in endothelial cells. In: Gahan PB, Fleischhacker M, Schmidt B, editors. Circulating Nucleic Acids in Serum and Plasma – CNAPS IX. Springer International Publishing, Cham (2016). p. 127–31. doi: 10.1007/978-3-319-42044-8_25
91. Kang Y-Q, Yuan X-H, Li Z-Z, Wang H, Zhou X-F, Wang X-X, et al. Antishock characteristics of erythrocyte-mediated endoplasmic reticulum stress in macrophages in severe hemorrhagic shock environment based on TLR9-cGAS-STING-IFN signal axis. Cell Transplant. (2020) 29:1–8. doi: 10.1177/0963689720950218
92. Shaker ME, Trawick BN, Mehal WZ. The novel TLR9 antagonist COV08–0064 protects from ischemia/reperfusion injury in non-steatotic and steatotic mice livers. Biochem Pharmacol. (2016) 112:90–101. doi: 10.1016/j.bcp.2016.05.003
93. Wang Q, Xie Z, Wan N, Yang L, Jin Z, Jin F, et al. Potential biomarkers for diagnosis and disease evaluation of idiopathic pulmonary fibrosis. Chin Med J. (2023) 136:1278–90. doi: 10.1097/CM9.0000000000002171
94. Bahia W, Soltani I, Abidi A, Mahdhi A, Mastouri M, Ferchichi S, et al. Structural impact, ligand–protein interactions, and molecular phenotypic effects of TGF-β1 gene variants: In silico analysis with implications for idiopathic pulmonary fibrosis. Gene. (2024) 922:148565. doi: 10.1016/j.gene.2024.148565
95. Li X, Ding Z, Wu Z, Xu Y, Yao H, Lin K. Targeting the TGF-β signaling pathway for fibrosis therapy: a patent review, (2015–2020. Expert Opin Ther Patents. (2021) 31:723–43. doi: 10.1080/13543776.2021.1896705
96. Hussein ZA, Abu-Raghif AR, Tahseen NJ, Rashed KA, Shaker NS, Fawzi HA. Vinpocetine alleviated alveolar epithelial cells injury in experimental pulmonary fibrosis by targeting PPAR-γ/NLRP3/NF-κB and TGF-β1/Smad2/3 pathways. Sci Rep. (2024) 14:11131. doi: 10.1038/s41598-024-61269-y
97. Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol Cancer. (2022) 21:104. doi: 10.1186/s12943-022-01569-x
98. Guo H, Jian Z, Liu H, Cui H, Deng H, Fang J, et al. TGF-β1-induced EMT activation via both Smad-dependent and MAPK signaling pathways in Cu-induced pulmonary fibrosis. Toxicol Appl Pharmacol. (2021) 418:115500. doi: 10.1016/j.taap.2021.115500
99. Lee K-Y, Kazuhiro I, Hayashi R, Jazrawi EPI, Barnes PJ, Adcock IM. NF-κB and activator protein 1 response elements and the role of histone modifications in IL-1β-induced TGF-β1 gene transcription. J Immunol. (2006) 176:603–15. doi: 10.4049/jimmunol.176.1.603
100. Furukawa S, Matsuda K, Sugano M, Uehara T, Honda T. NLRP3 upregulation in A549 cells co-cultured with THP-1 macrophages under hypoxia via deregulated TGF-β signaling. Exp Cell Res. (2019) 383:111506. doi: 10.1016/j.yexcr.2019.111506
101. Vande Walle L, Lamkanfi M. Drugging the NLRP3 inflammasome: from signalling mechanisms to therapeutic targets. Nat Rev Drug Discov. (2023) 23:43–66. doi: 10.1038/s41573-023-00822-2
102. Oshitani N, Watanabe K, Sakuma T, Matsuda M, Oyama Y. Tranilast, an antifibrotic agent and COVID-19-induced pulmonary fibrosis. Qjm-int J Med. (2022) 115:249–50. doi: 10.1093/qjmed/hcac069
103. Liang Q, Cai W, Zhao Y, Xu H, Tang H, Chen D, et al. Lycorine ameliorates bleomycin-induced pulmonary fibrosis via inhibiting NLRP3 inflammasome activation and pyroptosis. Pharmacol Res. (2020) 158:104884. doi: 10.1016/j.phrs.2020.104884
104. Wu L, Pu L, Zhuang Z. miR-155-5p/FOXO3a promotes pulmonary fibrosis in rats by mediating NLRP3 inflammasome activation. Immunopharmacol Immunotoxicol. (2023) 45:257–67. doi: 10.1080/08923973.2022.2115923
105. Tao H, Zhao H, Mo A, Shao L, Ge D, Liu J, et al. VX-765 attenuates silica-induced lung inflammatory injury and fibrosis by modulating alveolar macrophages pyroptosis in mice. Ecotoxicol Environ Saf. (2023) 249:114359. doi: 10.1016/j.ecoenv.2022.114359
106. Li X, Yan X, Wang Y, Wang J, Zhou F, Wang H, et al. NLRP3 inflammasome inhibition attenuates silica-induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells. Exp Cell Res. (2018) 362:489–97. doi: 10.1016/j.yexcr.2017.12.013
107. Cui P, Tang Z, Zhan Q, Deng C, Lai Y, Zhu F, et al. In vitro and vivo study of tranilast protects from acute respiratory distress syndrome and early pulmonary fibrosis induced by smoke inhalation. Burns. (2022) 48:880–95. doi: 10.1016/j.burns.2022.03.010
108. Alkreathy HM, Esmat A. Lycorine ameliorates thioacetamide-induced hepatic fibrosis in rats: emphasis on antioxidant, anti-inflammatory, and STAT3 inhibition effects. Pharmaceuticals. (2022) 15:369. doi: 10.3390/ph15030369
109. Tapia-Abellán A, Angosto-Bazarra D, Martínez-Banaclocha H, De Torre-Minguela C, Cerón-Carrasco JP, Pérez-Sánchez H, et al. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat Chem Biol. (2019) 15:560–4. doi: 10.1038/s41589-019-0278-6
110. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol. (2019) 15:556–9. doi: 10.1038/s41589-019-0277-7
111. Jin Y, Liu Y, Xu L, Xu J, Xiong Y, Peng Y, et al. Novel role for caspase 1 inhibitor VX765 in suppressing NLRP3 inflammasome assembly and atherosclerosis via promoting mitophagy and efferocytosis. Cell Death Dis. (2022) 13:512. doi: 10.1038/s41419-022-04966-8
112. Liu W, Sun J, Guo Y, Liu N, Ding X, Zhang X, et al. Calhex231 ameliorates myocardial fibrosis post myocardial infarction in rats through the autophagy-NLRP3 inflammasome pathway in macrophages. J Cell Mol Med. (2020) 24:13440–53. doi: 10.1111/jcmm.15969
113. Zhang H, Yu S, Xia L, Peng X, Wang S, Yao B. NLRP3 inflammasome activation enhances ADK expression to accelerate epilepsy in mice. Neurochem Res. (2021) 47:713–22. doi: 10.1007/s11064-021-03479-8
114. Zhang Y, Yao Z, Xiao Y, Zhang X, Liu J. Downregulated XBP-1 rescues cerebral ischemia/reperfusion injury-induced pyroptosis via the NLRP3/caspase-1/GSDMD axis. Mediators Inflammation. (2022) 2022:1–17. doi: 10.1155/2022/8007078
115. Wang C, Wang L, Huang C, Liu Y, Liu J, Kuang H, et al. Involvement of NLRP3/Caspase-1/GSDMD-Dependent pyroptosis in BPA-Induced apoptosis of human neuroblastoma cells. Biochem Pharmacol. (2022) 200:115042. doi: 10.1016/j.bcp.2022.115042
116. Wu B, Zhao S, Zhang J, Liu Y, Bai J, Wang G, et al. PD-1 inhibitor aggravate irradiation-induced myocardial fibrosis by regulating TGF-β1/smads signaling pathway via GSDMD-mediated pyroptosis. Inflammation. (2024) 48:181–98. doi: 10.1007/s10753-024-02056-9
117. Kim H-Y, Kim M-S, Kim S-H, Joen D, Lee K. Protective effects of nintedanib against polyhexamethylene guanidine phosphate-induced lung fibrosis in mice. Molecules. (2018) 23:1974. doi: 10.3390/molecules23081974
118. Li Y, Li H, Liu S, Pan P, Su X, Tan H, et al. Pirfenidone ameliorates lipopolysaccharide-induced pulmonary inflammation and fibrosis by blocking NLRP3 inflammasome activation. Mol Immunol. (2018) 99:134–44. doi: 10.1016/j.molimm.2018.05.003
119. Dong S-H, Liu Y-W, Wei F, Tan H-Z, Han Z-D. Asiatic acid ameliorates pulmonary fibrosis induced by bleomycin (BLM) via suppressing pro-fibrotic and inflammatory signaling pathways. Biomed Pharmacother. (2017) 89:1297–309. doi: 10.1016/j.biopha.2017.03.005
120. Sun L, Zhu M, Feng W, Lin Y, Yin J, Jin J, et al. Exosomal miRNA Let-7 from Menstrual Blood-Derived Endometrial Stem Cells Alleviates Pulmonary Fibrosis through Regulating Mitochondrial DNA Damage. Oxid Med Cell Longevity. (2019) 2019:1–17. doi: 10.1155/2019/4506303
121. Meng Y, Pan M, Zheng B, Chen Y, Li W, Yang Q, et al. Autophagy attenuates angiotensin II-induced pulmonary fibrosis by inhibiting redox imbalance-mediated NOD-like receptor family pyrin domain containing 3 inflammasome activation. Antioxid Redox Signaling. (2019) 30:520–41. doi: 10.1089/ars.2017.7261
122. Menzel V, Ziegler M, Hante N, Sake JA, Santos-Martinez MJ, Ehrhardt C, et al. Fyn-kinase and caveolin-1 in the alveolar epithelial junctional adherence complex contribute to the early stages of pulmonary fibrosis. Eur J Pharm Sci. (2022) 175:106236. doi: 10.1016/j.ejps.2022.106236
123. Moeller A, Ask K, Warburton D, Gauldie J, Kolb M. The bleomycin animal model: A useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol. (2008) 40:362–82. doi: 10.1016/j.biocel.2007.08.011
124. Jenkins RG, Moore BB, Chambers RC, Eickelberg O, Königshoff M, Kolb M, et al. An official american thoracic society workshop report: use of animal models for the preclinical assessment of potential therapies for pulmonary fibrosis. Am J Respir Cell Mol Biol. (2017) 56:667–79. doi: 10.1165/rcmb.2017-0096ST
125. Ye X, Zhang M, Gu H, Liu M, Zhao Y, Shi Y, et al. Animal models of acute exacerbation of pulmonary fibrosis. Respir Res. (2023) 24:296. doi: 10.1186/s12931-023-02595-z
126. Zhang X, Zhang L, Tian J, Li Y, Wu M, Zhang L, et al. The application and prospects of drug delivery systems in idiopathic pulmonary fibrosis. Biomater Adv. (2025) 168:214123. doi: 10.1016/j.bioadv.2024.214123
127. Junyao L, Ming Y, Ping L, Zhenzhong S, Peng G, Jie Z. Idiopathic pulmonary fibrosis will increase the risk of lung cancer. Chin Med J. (2014) 127(17):3142–9.
128. Ahechu P, Zozaya G, Martí P, Hernández-Lizoáin JL, Baixauli J, Unamuno X, et al. NLRP3 inflammasome: A possible link between obesity-associated low-grade chronic inflammation and colorectal cancer development. Front Immunol. (2018) 9:2918. doi: 10.3389/fimmu.2018.02918
129. Cox LA Jr. Dose-response modeling of NLRP3 inflammasome-mediated diseases: asbestos, lung cancer, and Malignant mesothelioma as examples. Crit Rev Toxicol. (2019) 49:614–35. doi: 10.1080/10408444.2019.1692779
130. Jin C, Lagoudas GK, Zhao C, Bullman S, Bhutkar A, Hu B, et al. Commensal microbiota promote lung cancer development via γδ T cells. Cell. (2019) 176:998–1013.e16. doi: 10.1016/j.cell.2018.12.040
131. Zhang J, Veeramachaneni N. Targeting interleukin-1β and inflammation in lung cancer. biomark Res. (2022) 10:5. doi: 10.1186/s40364-021-00341-5
Keywords: NLRP3 inflammasome, pulmonary fibrosis, cellular signaling, inhibitors of NLRP3 inflammasome, therapeutic strategies
Citation: Wang M, Xie Y, Cao Y, Yu B and Dai Q (2025) Pulmonary fibrosis through the prism of NLRP3 inflammasome: mechanistic pathways and prospective therapeutic innovations. Front. Immunol. 16:1593729. doi: 10.3389/fimmu.2025.1593729
Received: 14 March 2025; Accepted: 14 April 2025;
Published: 05 May 2025.
Edited by:
Chaofeng Han, Second Military Medical University, ChinaReviewed by:
Zhaocai Zhang, Zhejiang University, ChinaXiaoyang Zhang, The University of Utah, United States
Copyright © 2025 Wang, Xie, Cao, Yu and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingqing Dai, ZGFpcWluZ3Fpbmc4ODhAMTI2LmNvbQ==
†These authors have contributed equally to this work