 Qinan Yin1†
Qinan Yin1† Youjin Huang2†Hulin Wang1†Yin Wang1
Youjin Huang2†Hulin Wang1†Yin Wang1 Xuefei Huang1Yujie Song1
Xuefei Huang1Yujie Song1 Yueyuan Wang1*Lizhu Han1*
Yueyuan Wang1*Lizhu Han1* Bian Yuan1*
Bian Yuan1*- 1Department of Pharmacy, Personalized Drug Research and Therapy Key Laboratory of Sichuan Province, Sichuan Provincial People's Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu, China
- 2Department of Vascular Surgery Center, Sichuan Academy of Medical Sciences & Sichuan Provincial People's Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu, China
The emergence of COVID-19 has been associated with an increased risk of arteriovenous thrombosis, with immune inflammation playing a significant role in the pathogenesis of thrombosis. Numerous drug-related clinical trials have been undertaken to prevent thrombosis, and guidelines for its prevention and treatment are continuously evolving as our understanding of the disease progresses. This article provides a comprehensive review of the mechanisms underlying thrombosis in COVID-19 patients, as well as the advancements in clinical trials and guidelines for thrombosis prevention with pharmacological interventions.
1 Introduction
COVID-19, caused by the coronavirus SARS-CoV-2, is primarily a respiratory viral infection; it has had a catastrophic impact on the world’s population, resulting in more than 6.5 million deaths worldwide. This represents the worst global health crisis since the 1918 influenza pandemic (1). The primary impact of SARS-CoV-2 infection is on the pulmonary system, however, emerging evidence suggests that it may also have implications for the vascularity of the extrapulmonary system, either through direct viral infection or indirectly through cytokine storms (2). Studies have shown that COVID-19 induces a prethrombotic state (3). Thrombosis of both microvessels and large vessels is prevalent among COVID-19 patients (4). Pulmonary embolism (PE) and deep vein thrombosis (DVT) are frequently observed thrombotic complications in patients with COVID-19. Arterial thrombosis is a notable occurrence in COVID-19 patients, leading to various complications, such as acute ischemic stroke, acute coronary syndrome (ACS), acute limb ischemia (ALI), mesenteric infarction, renal infarction, and spleen infarction (Figure 1) (5, 6).
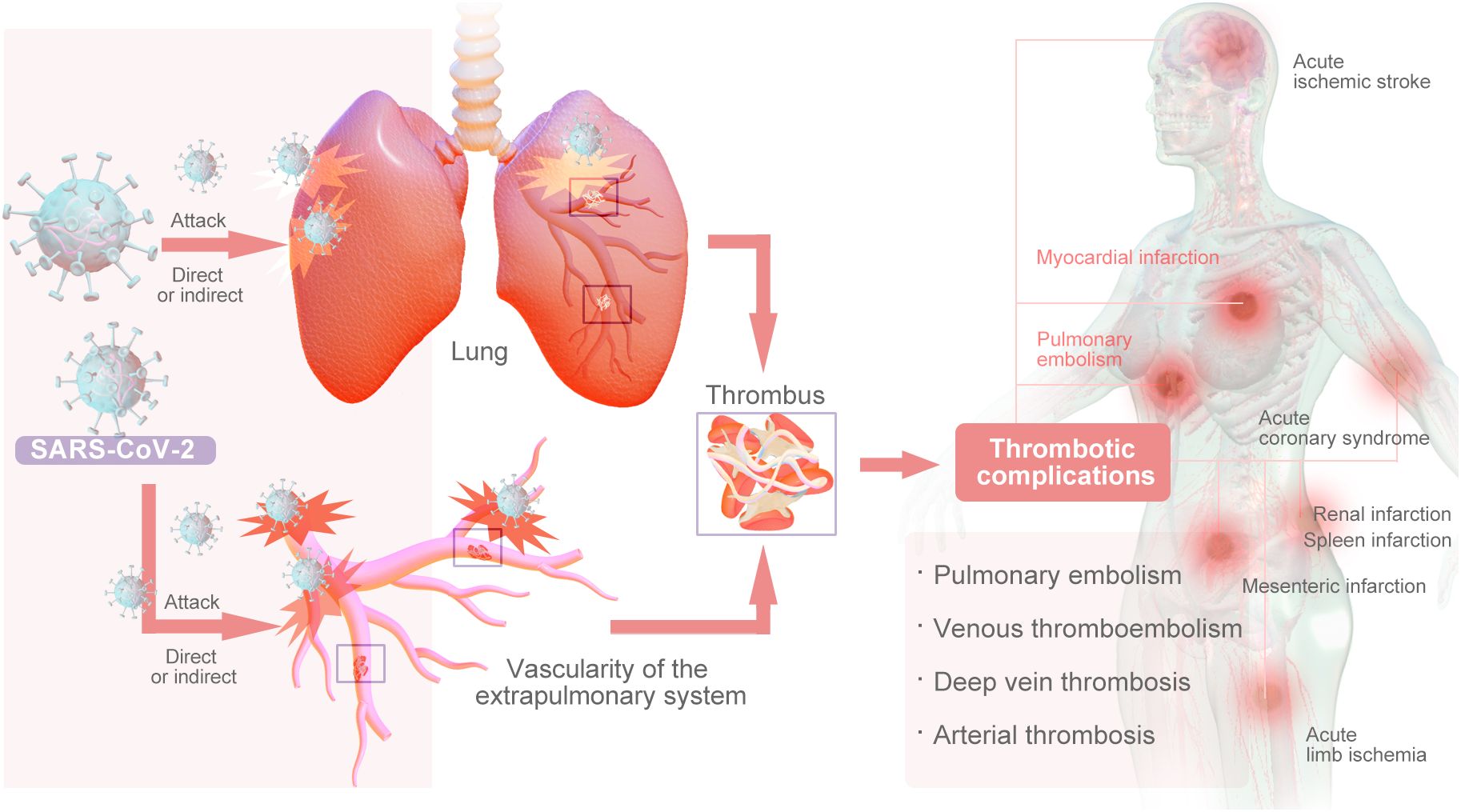
Figure 1. The Main Types of Thrombsiss Caused by COVID-19.
High mortality is associated with hypercoagulability in COVID-19 patients (7). A recent study revealed that patients with COVID-19 are more likely to develop venous thromboembolism (PE: 1.0–40.0%, DVT: 0.4–84%) than arterial thromboembolism (stroke: 0.5–15.2%, myocardial infarction: 0.8–8.7%). Finally, the all-cause mortality rate for COVID-19 patients ranges from 4.8% to 63%, whereas the mortality rate associated with thromboembolic complications ranges from 5% to 48% (8). A systematic review and meta-analysis of thromboembolic events in patients with COVID-19 from the start of the pandemic through August 31, 2021, involving 63 studies (104,920 patients with COVID-19) revealed an overall thrombotic rate of 21% (95% CI, 18%–25%). The deep vein thrombosis rate was 20% (95% CI, 16% ~ 25%), the pulmonary embolism rate was 8% (95% CI, 6% ~ 10%), and the arterial thrombosis rate was 5% (95% CI, 3% ~ 7%). The prevalence of all major outcomes in critically ill patients in the intensive care unit (ICU) significantly increased (P<0.05). The incidence of total thrombosis, pulmonary embolism and deep vein thrombosis in elderly patients significantly increased (P<0.05) (9). Furthermore, despite the implementation of anticoagulation prophylaxis, hospitalized individuals continue to face increased susceptibility to venous thromboembolism (VTE) (4). Almost all of the above findings are based on hospitalized COVID-19 patients. These findings are based on populations in an inpatient setting, but data on the incidence of venous thromboembolism events, such as acute pulmonary embolism (PE) and deep vein thrombosis (DVT), among individuals recovering from COVID-19 are limited. A systematic review and meta-analysis were conducted to assess the risk of acute PE and DVT in this population. The study included a total of 29,078,950 patients, with a mean age of 50.2 years, 63.9% of whom were male. Among these, 2,060,960 patients had been infected with COVID-19. The cumulative incidence rates of PE and DVT in patients who had recovered from COVID-19 were 1.2% (95% CI: 0.9–1.4, I: 99.8%) and 2.3% (95% CI: 1.7–3.0, I: 99.7%), respectively. During the same follow-up period, the hazard ratios for developing PE and DVT in recovered COVID-19 patients were 3.16 (95% CI: 2.63–3.79, I: 90.1%) and 2.55 (95% CI: 2.09–3.11, I: 92.6%), respectively (10). A separate study incorporated inpatient participants with primary care records from the Netherlands, Italy, Spain, and the United Kingdom, as well as outpatient specialist records from Germany. The 90-day cumulative incidence of venous thromboembolism (VTE) in COVID-19 patients ranged from 0.2% to 0.8%, increasing to as high as 4.5% among hospitalized patients. The incidence of VTE in COVID-19 patients was correlated with an elevated risk of mortality, with an adjusted hazard ratio (HR) of 4.42 [95% CI: 3.07–6.36] for nonhospitalized patients and 1.63 [95% CI: 1.39–1.90] for hospitalized patients. Similarly, the development of arterial thromboembolism was linked to an increased risk of death, with hazard ratios of 3.16 [95% CI: 2.65–3.75] for nonhospitalized patients and 1.93 [95% CI: 1.57-2.37] for hospitalized patients (11). Overall, the prevalence of COVID-19 was greater in women than in men. In a 2022 European network cohort study involving 909,473 COVID-19 patients and 32,329 COVID-19 hospitalizations, there were more COVID-19 cases in women than in men across all databases (11). A 2024 systematic review and meta-analysis also reported that the prevalence of post-COVID-19 syndrome by sex was 47.23% (95% CI: 44.03–50.42%) in men and 52.77% (95% CI: 49.58–55.97%) in women (12). For patients with COVID-19 and thrombosis, a prospective study identified 1106 patients with venous thromboembolism associated with COVID-19 (age, 62.3 ± 14.4 years; 62.9% male) (12, 13). Interestingly, women appear to be less likely to develop severe disease, despite the data collected thus far that more women are affected and that the mortality rate in women is lower than that in men. The role of female hormones in regulating inflammation may be the reason behind this sex difference.
At present, the association between COVID-19 and thrombosis and related anticoagulation therapy has received extensive attention. This article summarizes and explores the pathogenic mechanism of thrombosis induced by COVID-19 and evaluates current anticoagulation strategies for the prevention and management of COVID-19-associated thrombosis, providing a reference for the clinical formulation of scientific anticoagulation strategies.
2 Mechanism of thrombosis
Thrombosis is usually associated with vascular injury, blood stasis, and changes in blood properties. Coagulation involves a series of enzymatic reactions leading to thrombin production and fibrin formation (14). The coagulation pathways include two main pathways: the extrinsic pathway and the intrinsic pathway. In addition, the common pathway converges on factor X (FX). The coagulation pathway activates coagulation factors through a cascade of activation, also known as the coagulation cascade. The extrinsic pathway is the main structural element of the coagulation process; components of the extrinsic and common pathways are essential for coagulation, whereas components of the intrinsic pathway are needed to amplify thrombin production. Repeated activation and amplification trigger the intrinsic pathway driven by thrombin (under certain conditions driven by FXIIa) to accelerate thrombin and fibrin formation. Both the extrinsic and intrinsic pathways of the coagulation system are initially triggered by the release of active tissue factor (TF), the formation of TF complexes with FVIIa, and the subsequent activation of FX directly to FXa or through the FIXa pathway (15). Certain scholars posit that SARS-CoV-2 could exacerbate thrombosis by exerting various distinct and comprehensive influences on intravascular dysfunction, platelet activation and aggregation, the initiation of the coagulation cascade, the activation of eicosanoid pathways, and the disruption of fibrinolysis (16–20). Research has revealed that the levels of circulating inflammatory markers (CCL23 and IL-6) and vascular dysfunction markers (ACE-2 and TF) are significantly greater in individuals with severe illness than in those with mild illness (21). Recent research suggests that the mechanisms underlying thrombosis in individuals with COVID-19 are primarily initiated by vascular endothelial damage, dysregulation of the body’s coagulation system, the presence of viral particles, and subsequent immune responses. This process is multifactorial in nature (22, 23). In summary, inflammation facilitates thrombosis, which subsequently exacerbates the inflammatory response. Inflammation alters the functional phenotype of endothelial cells from an antithrombotic to a thrombotic state, thereby promoting the activation of the coagulation cascade. Thrombin generated during thrombotic events further amplifies inflammation by stimulating cells that express protease-activated receptors (PARs) to secrete cytokines. Furthermore, activated platelets engage with inflammatory and endothelial cells, enhancing neutrophil extracellular trap (NET) formation and cytokine release (24). The main mechanisms are shown in Figures 2, 3.
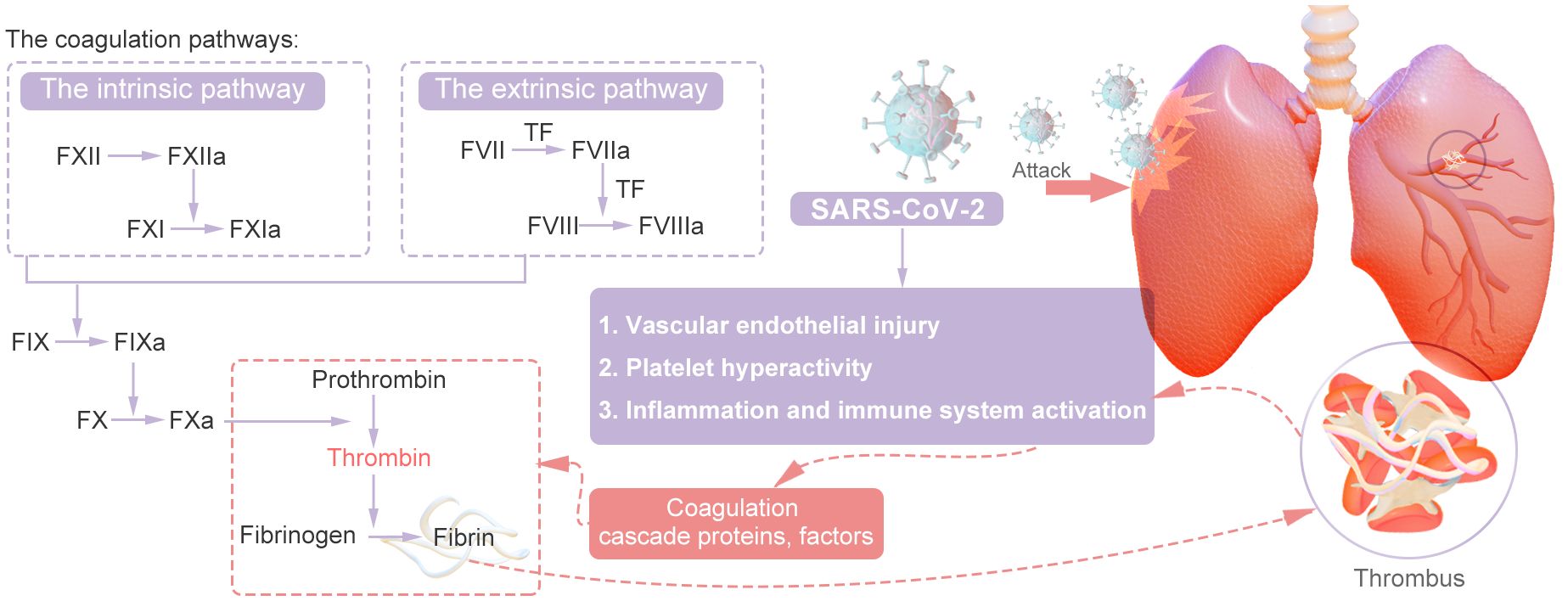
Figure 2. Simple Mechanism of COVID-19 Thrombosis.
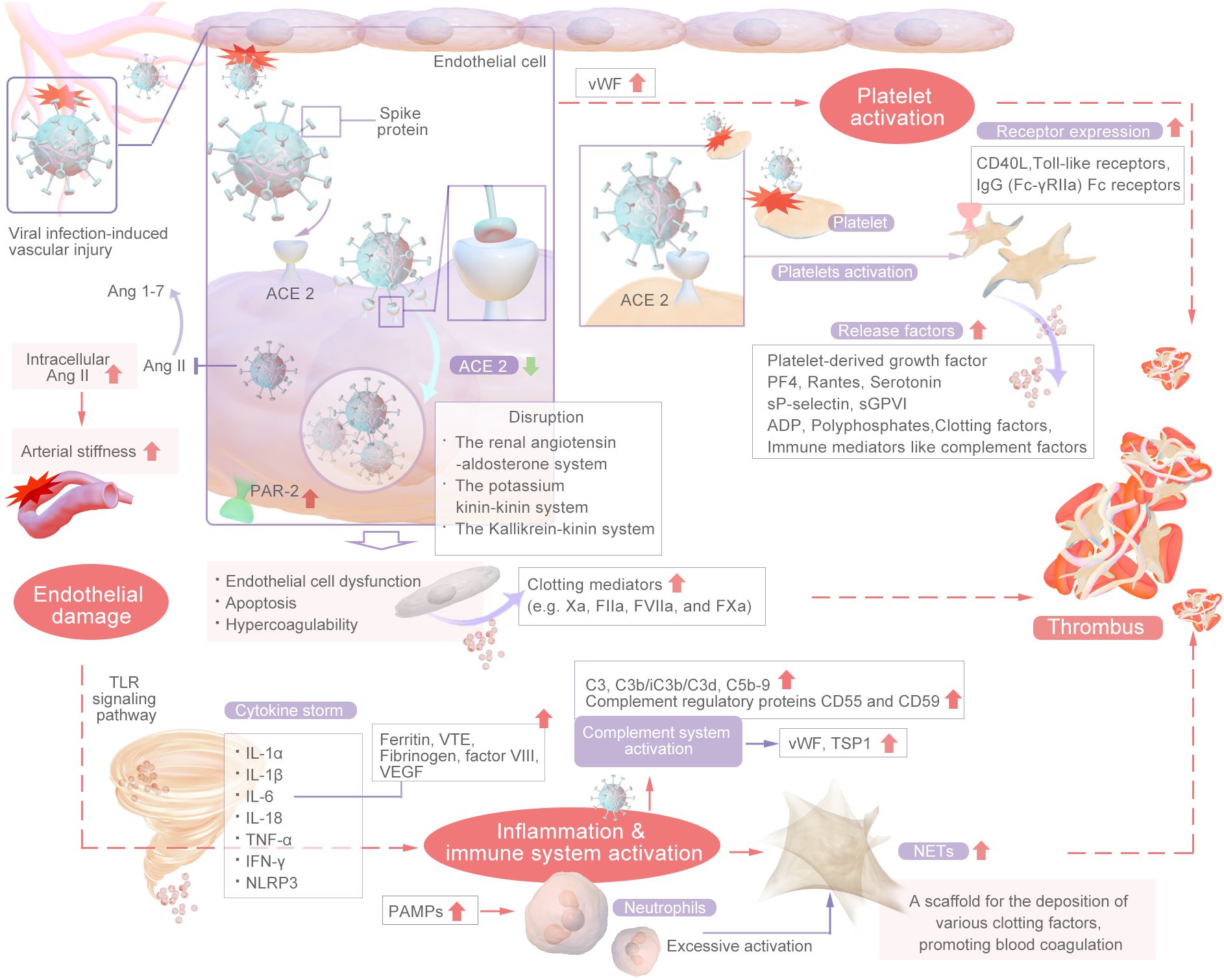
Figure 3. The Main Mechanism of COVID-related Thrombosis.
2.1 Vascular endothelial injury
SARS-CoV-2 affects endothelial cells through multiple mechanisms. Autopsies of patients with SARS-CoV-2 infection have shown evidence of endothelial cell apoptosis. The microvascular endothelial damage and microcirculation thrombus formation found in the autopsy assessment are consistent with the possible thrombotic microangiopathy that the patients may have (16). The study also found that circulating endothelial cells (CEC), a cellular biomarker derived from damaged blood vessels, were elevated in patients with severe COVID-19 and in those with underlying conditions. Notably, even patients who recovered from COVID-19 had significantly higher numbers of circulating endothelial cells than healthy individuals, suggesting that vascular dysfunction persists after recovery from COVID-19 (25).
The mechanism of vascular endothelial injury is shown in Figure 4. In the renin–angiotensin system (RAS), Ang I is converted into Ang II by ACE-2 (26). Ang II-mediated AT1R activation can increase platelet activation and prevent fibrinolysis, resulting in a hypercoagulable state (27). SARS-CoV-2 has been confirmed to have a direct effect on alveolar and systemic endothelial cells, causing endothelial dysfunction, altering the microvascular balance, and subsequently leading to organ ischemia, a procoagulation state, tissue inflammation and edema (28). SARS-CoV-2 exhibits a distinct affinity for the angiotensin-converting enzyme 2 (ACE2), which serves as its cellular receptor. This receptor is expressed by type II alveolar cells and is situated in close proximity to the pulmonary vascular network (29). The spike (S) proteins of SARS-CoV-2 bind to angiotensin-converting enzyme-2 (ACE-2) on endothelial cells, leading to endothelial cell damage (3). When SARS-CoV-2 binds to cells by ACE-2, the pulmonary expression of membrane-bound ACE-2 may be downregulated, leading to the accumulation of Ang II within cells. Ang II has various proinflammatory and prothrombotic effects and can be amplified in patients with COVID-19 (4). Thus, SARS-CoV-2 directly infects vascular endothelial cells, leading to cell damage and apoptosis, thereby reducing the antithrombotic activity of normal endothelial cells.
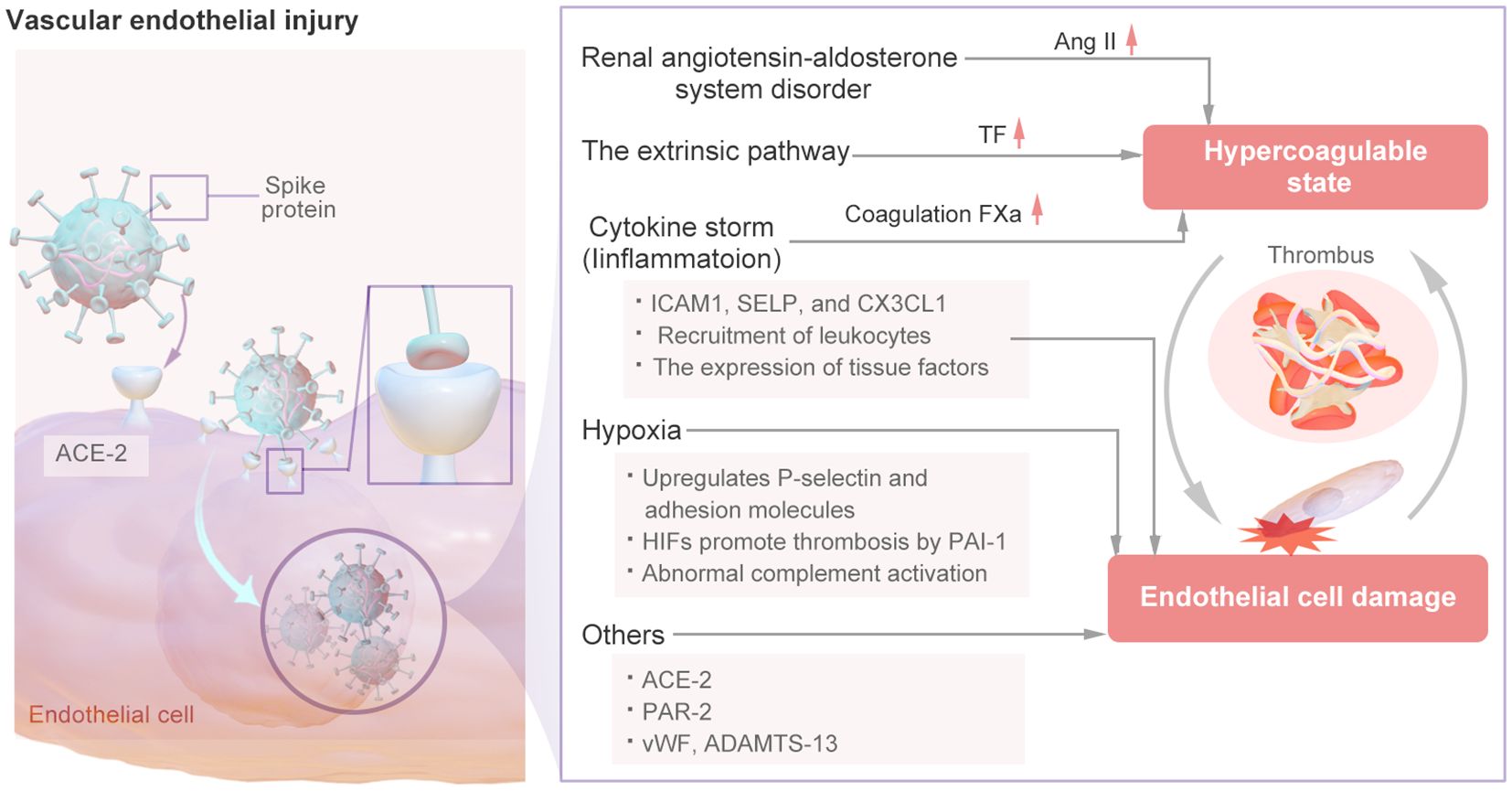
Figure 4. The Mechanism of Vascular Endothelial Injury.
Viral infection-induced vascular injury may stimulate endothelial cells to increase the expression of TF and activate the extrinsic coagulation pathway (30), Additional cytokines may contribute to the induction of tissue factor (TF) expression in endothelial cells derived from COVID-19 patients, thereby initiating the extrinsic coagulation pathway (31). The interaction between the viral spike protein and the cellular receptor can trigger the inflammatory pathway, alter the mitochondrial metabolism of microvascular endothelial cells, and induce significant production of coagulation factor Xa, resulting in a hypercoagulable state (32, 33). this also leads to excessive thrombin production and reduced fibrinolysis, and thrombin can cause further endothelial damage (34, 35). Moreover, The increased expression of ICAM1, SELP, and CX3CL1 may suggest that endothelial cells from the site of vascular injury are in a proinflammatory and procoagulant state (36). serum from patients with COVID-19 can modify the functional characteristics of endothelial cells through the activation of protease-activated receptor 2 (PAR-2), leading to increased apoptosis, compromised barrier integrity, and increased propensity for blood clotting (37). Moreover, endothelial dysfunction is facilitated by PAR-2, whereby certain clotting mediators (e.g., FIIa, FVIIa, and FXa) can trigger the production of proinflammatory cytokines, thus perpetuating a detrimental cycle (38). Endodermatitis induced by SARS-CoV-2 results in the release of elevated levels of vWF, a glycoprotein originating from activated endothelial cells, platelets, or subendothelial cells that plays a crucial role in mediating platelet adhesion and aggregation. The significant increase in vWF levels observed in individuals with COVID-19 signifies endothelial damage or impairment, promoting platelet aggregation at the site of injury and potentially leading to hyperactivation of the coagulation pathway, thereby initiating thrombotic events (39–41). Concurrently, a reduction in a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS-13) levels was noted, suggesting an imbalanced vWF: ADAMTS-13 ratio that could contribute to thrombotic occurrences. Both the vWF/ADAMTS-13 ratio and PF4 level are increased, indicating that prolonged endothelial cell activation leads to increased platelet aggregation after COVID-19 (42, 43).
COVID-19 patients often suffer from severe hypoxia, and hypoxia can also lead to endothelial dysfunction and coagulation. Hypoxia upregulates P-selectin and adhesion molecules (such as intracellular adhesion molecule-1) to induce the recruitment of platelets and leukocytes (44). Furthermore, hypoxia-inducible transcription factors (HIFs) promote thrombosis by increasing the release of the inflammatory cytokine PAI-1 by endothelial cells while downregulating thrombomodulin expression (45). Specifically, HIF-α downregulates the expression of the complement regulator CD55. This downregulation increases the release of C3a and the deposition of caspase 3 on endothelial cells, thereby further increasing complement-mediated endothelial damage in patients with COVID-19. Damaged endothelial cells subsequently trigger hypercoagulability (46). The abnormal activation of endothelial cells triggered by SARS-CoV-2 can further lead to cytokine storm and activation of the complement system, as described in the following sections.
2.2 Inflammation and immune system activation
COVID-19-associated coagulopathy may be a downstream consequence of the host inflammatory response to SARS-CoV-2 and of innate immune activation (4). During the course of COVID-19, the recruitment of immune cells and the subsequent production of proinflammatory cytokines serve as mediators of clotting activation in tissues affected by the virus. Viral infection triggers an inflammatory response in the host, leading to an increase in the production of proinflammatory cytokines. These cytokines have pleiotropic effects, including activation of coagulation and thrombin generation, and this phenomenon is known as thromboinflammation or immunothrombosis. The main mechanisms are cytokine storm, complement system activation and neutrophil extracellular traps (NETs), which constitute the “malignant triangle” of immune-related thrombosis. The inflammatory and immune thrombosis-related mechanisms is shown in Figure 5.
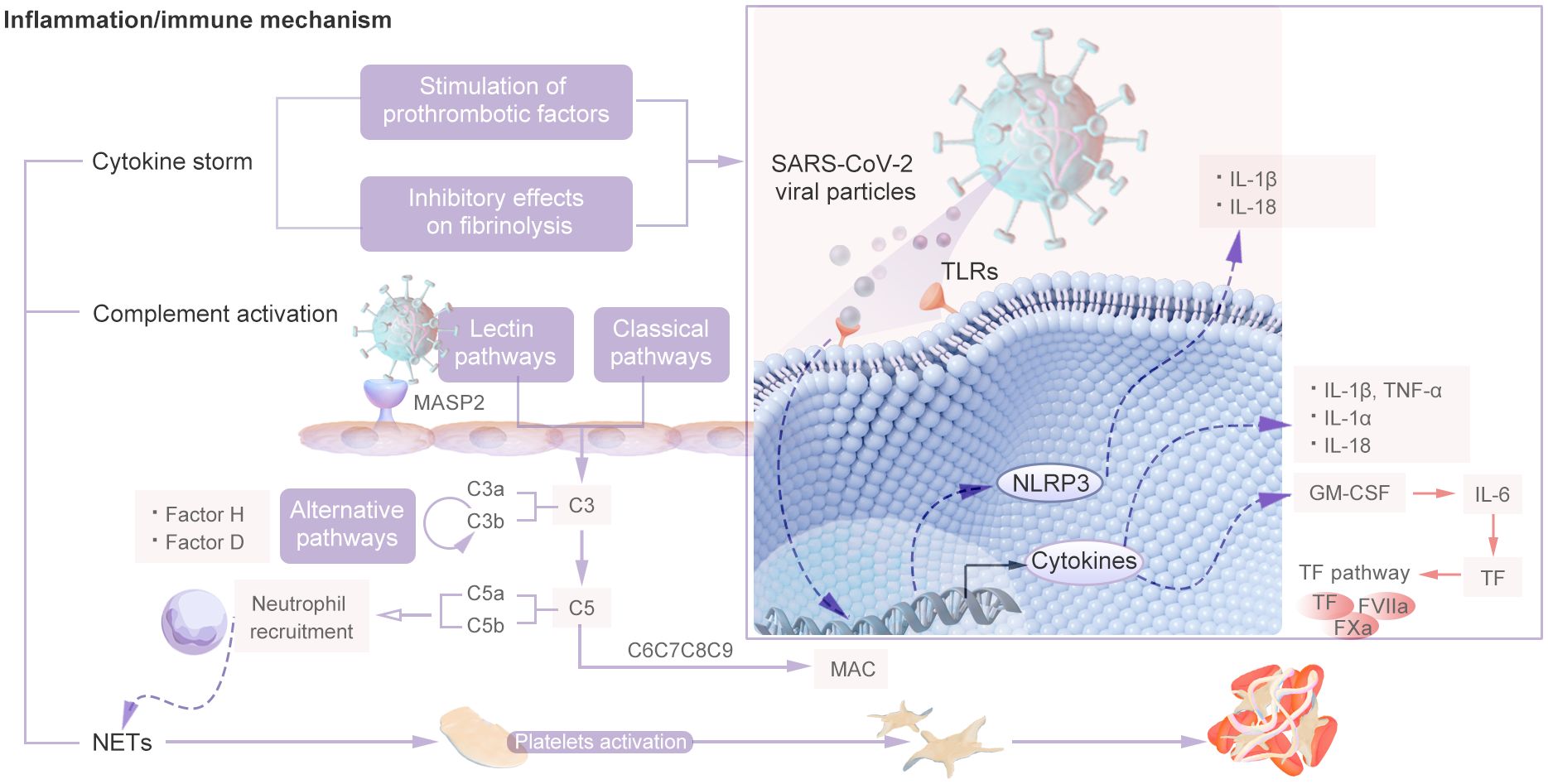
Figure 5. The Inflammatory and Immune Thrombosis Related Mechanisms.
2.2.1 Cytokine storm
Cytokine storm syndrome has been documented in 10-20% of patients with COVID-19 pneumonia and may result in severe outcomes, including multiorgan failure and mortality (47). The cytokine storm and the consequent hyperinflammatory state determine local and systemic consequences, inducing arterial and venous vasculopathy in the lung with thrombosis of the small vessels and progression toward serious lung lesions, acute respiratory distress syndrome (ARDS) and, in some cases, disseminated intravascular coagulation (DIC) (48, 49). SARS-CoV-2 infection in the host lungs, upon entry through type 2 alveolar epithelial cells and interaction with blood vessels at the pneumocyte–capillary interface, causes the activation of host innate immunity, leading to the release of proinflammatory cytokines, which eventually results in an uncontrolled and exaggerated response, the cytokine storm (50). Cytokine storm syndrome is associated with major thrombotic events in patients with COVID-19, and the mechanism is related primarily to the simultaneous stimulation of prothrombotic factors and inhibitory effects on fibrinolysis (28).
The activation of Toll-like receptors (TLRs) by molecules present in SARS-CoV-2 viral particles initiates the innate immune response. However, excessive activation of TLRs can disrupt intracellular signaling pathways, exacerbate the inflammatory response, and lead to an excessive and uncontrolled release of cytokines (51). Firstly, activated macrophages and monocytes release a large amount of pro-inflammatory cytokines such as IL-6, (IL)-1β, and TNF-α (52). In addition, Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a cytokine that facilitates the activation and mobilization of myeloid cells to inflammatory sites. It is synthesized by a variety of cell types. In patients with COVID-19, the production of GM-CSF initiates a positive feedback loop, leading to the overexpression of interleukin-6 (IL-6) and other proinflammatory factors, particularly by CD14+ and CD16+ inflammatory monocytes (53). In particular, TNF-α and IL-6 levels increase to levels that are not typically observed in patients with bacterial sepsis or influenza (54). In the context of inflammation, macrophages are stimulated to release TF in response to IL-6. The upregulation of the expression of IL-6 and its receptor in COVID-19 can lead to increased activation of endothelial cells, resulting in the excessive release of TF. This cascade of events can contribute to infection-induced coagulation dysfunction and increased platelet proliferation (55, 56). IL-6 facilitates the secretion of various cytokines, activates immune cells, and contributes to the synthesis of specific coagulation factors, including fibrinogen and factor VIII. (57). Moreover, IL-1α is involved in the activation of the inflammatory cascade in thrombotic pathology and serves as a crucial factor in thrombosis through the recruitment of granulocytes, prolonging clot lysis time and increasing platelet activity (50). Furthermore, the NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome is responsible for the generation of six primary proinflammatory cytokines belonging to the interleukin-1 family, namely, IL-1β and IL-18. NLRP3 inflammasome activation also increases immune thrombosis through mechanisms such as the release of neutrophil extracellular traps and TF by leukocytes, as well as the initiation of prothrombotic responses in platelets and the vascular endothelium (58). Therefore, it has been suggested that cytokine release may also be the result of high levels of extracellular neutrophil traps.
2.2.2 Complement activation
The complement system serves as the primary defense mechanism against pathogen invasion and damage to host cells and consists of a complex network that includes over 40 soluble and membrane-binding proteins. This finely tuned response is typically maintained at low levels of activity in individuals who are in good health, allowing for a rapid and localized response to insults as they arise. The complement system operates through three distinct pathways—classical, lectin, and alternative—to provide comprehensive protection against invading pathogens (59). The complement system serves as the primary immune response of the host to infection, but uncontrolled activation can lead to increased cellular damage, inflammation, and intravascular clotting and ultimately results in multiple organ failure and mortality (60).
patients with SARS-CoV-2 infection, complement system dysregulation has been observed, which is primarily characterized by disturbances in the terminal complement system and the persistent activation of both the alternative and classical complement pathways (39). The NP of SARS-CoV-2 can bind to mannose-binding lectin serine protease 2 (MASP 2), a key component of the complement system, which exacerbates tissue damage and leads to the release of inflammatory factors, thereby increasing cell adhesion and thrombosis during long-term COVID-19 (61). In instances of thrombotic inflammation, the complement pathway may initiate the coagulation cascade through the induction of tissue factor expression. The activation of the complement cascade may also recruit and activate white blood cells, leading to the local release of the proinflammatory cytokines IL-1, IL-6, IL-8 and interferon gamma, which are significantly amplified and subsequently cause microvascular damage (4). Compared with normal lung tissues, the lung tissues of COVID-19 patients display complement system hyperactivation, characterized by extensive deposition of C3, C3b/iC3b/C3d, and C5b-9 and the increased expression of the complement regulatory proteins CD55 and CD59 (62). In Addition, serine proteases within the lectin pathway are capable of cleaving prothrombin to generate activated thrombin. This mechanism is particularly evident in critically ill COVID-19 patients experiencing symptomatic thromboembolism, where there is a marked elevation of mannose-binding lectin (63). Research has demonstrated that the SARS-CoV-2 spike protein activates the complement alternative pathway by disrupting the function of complement factor H, a critical negative regulatory component of this pathway. Inhibitors targeting C5 and factor D have been shown to effectively prevent the accumulation of C5b-9 complexes induced by the SARS-CoV-2 spike protein (64). The activation of the complement stem in COVID-19 patients results in the formation of a terminal complement complex (TCC) consisting of C5b-9. Among them, C5b-9 is regarded as one of the key factors affecting the hemostasis system in the quantitative system immunology model (65). Furthermore, the insertion of the complement C5b–C7 complex into the endothelial cell membrane results in cellular damage, which subsequently triggers the release of vWF and thrombospondin-1 (TSP1). This process is highly prothrombotic, as the resulting vWF polymers facilitate platelet recruitment and thrombin generation while simultaneously inducing C3b binding and activating alternative complement pathways. This feedback mechanism perpetuates localized complement activation and inflammation and the formation of microthrombi (66). C5a plays a pivotal role in the recruitment and activation of neutrophils, monocytes, and macrophages, while simultaneously enhancing the expression of adhesion molecules on endothelial cells and platelets. This process can result in elevated production of interleukin-6 and CXCL8, subsequently initiating a robust procoagulant response. Consequently, this leads to the formation of neutrophil-derived tissue factors and the development of the neutrophil extracellular trap (67). Furthermore, the complement components C5a and the membrane attack complex facilitate platelet adhesion by inducing the secretion of endothelial P-selectin and vWF polymers, increasing the expression of TF and promoting platelet shedding, thereby contributing to thrombogenesis (68, 69).
2.2.3 Neutrophil extracellular traps
Neutrophils, also known as polymorphonuclear (PMN) cells, constitute the first line of defense in the innate immune system against invading pathogens. PMNs are the most abundant white blood cells in the human body and perform multiple protective functions, including phagocytosis of pathogens, degranulation, secretion of cytokines, and formation of NETs (70). Neutrophil extracellular traps (NETs) are complex, web-like structures consisting of DNA, histones, and antimicrobial proteins, which are extruded by activated neutrophils to ensnare and neutralize pathogens. Neutrophil elastase (NE), a constituent of neutrophil extracellular traps (NETs), plays a crucial role in platelet activation via the protease-activated receptor 4 (PAR4) and thrombin pathways, consequently altering platelet functionality and promoting fibrinogenesis. Additionally, myeloperoxidase (MPO), another integral component of NETs predominantly secreted by neutrophils, serves as a primary scavenger of the pro-oxidant hydrogen peroxide (71). The procoagulant properties of NETs are primarily attributed to their filamentous structures, which increase the adhesion of activated platelets and subsequently facilitate the entrapment of red blood cells (72).
Excessive NET formation is a process known as NETosis, which can occur in patients with COVID-19 and other viral infections, and can lead to hypercoagulation and thrombosis (73). Research findings spleen tyrosine kinase (Syk), in conjunction with C-type lectin member 2 (CLEC2), which is predominantly expressed in platelets and alveolar macrophages, is capable of directly interacting with the receptor binding domain (RBD) of the SARS-CoV-2 spike protein. In contrast to filamentous neutrophil extracellular traps (NETs), SARS-CoV-2 facilitates the formation of aggregated NETs in the presence of wild-type (WT) platelets; however, this aggregation is absent in CLEC2-deficient platelets. Subsequent investigations have demonstrated that a SARS-CoV-2 spike protein pseudolentivirus can induce NET formation via CLEC2, suggesting that the SARS-CoV-2 RBD promotes NET formation by binding to CLEC2, thereby activating platelets (74). High levels of extracellular neutrophil traps can also contribute to the release of cytokines, which play crucial roles in thrombotic events in critically ill patients (73, 75). Significantly, NETs exacerbate lung damage by triggering epithelial cell and endothelial cell death, leading to the secretion of cytokines such as IL-1β and IL-6, which initiate cytokine storms that exacerbate thrombosis (76). An intensified cycle of systemic and neutrophil-derived interleukin-8 (CXCL8/IL-8) dysregulation initiates and sustains neutrophil-driven immunopathology. This systemic positive feedback loop of neutrophil autocrine IL-8 results in an activated pre-thrombotic neutrophil phenotype, marked by degranulation and the formation of neutrophil extracellular traps (NETs). In severe cases of COVID-19, neutrophils directly trigger coagulation and complement cascade reactions, underscoring their association with the immune thrombotic status observed in these patients (77). A study examining NETs and platelet involvement in the autopsies of lungs from 3 COVID-19 patients revealed that guanidino histone H3+ neutrophils might undergo NETosis and colocalize with platelet-derived thrombogenic factors in pulmonary microthrombi (78). The findings indicate that the interaction between neutrophil extracellular traps (NETs) and platelets in patients with COVID-19 may initiate a thrombogenic cascade, culminating in a hypercoagulable state and subsequent thrombosis. Research has demonstrated that NETs play a role in promoting the thrombotic factor vWF in a platelet-dependent fashion and directly activate the clotting cascade through mechanisms including platelet adhesion activation, cell–fibrinogen binding, and von Willeophilia. Furthermore, NETs have been found to contribute to local thrombin production, thereby increasing the risk of thrombosis in addition to their effect on primary hemostasis (79). Furthermore, the colocalization of NETs with factor XII was confirmed, suggesting that NETs may serve as a platform for the activation of the intrinsic coagulation pathway involving factor XII (80).
2.3 Platelets
2.3.1 Platelet activation
Platelets have long been recognized for their crucial role in thrombi and the hemostatic system, as they rapidly respond to pathogens, signaling nearby immune cells and facilitating thrombosis and intravascular clot formation (81). The inflammation caused by viral infection can stimulate platelets to express TF, thereby promoting the interaction between platelets and monocytes and the formation of more platelet–monocyte aggregates that promote coagulation (82). Platelets from COVID-19 patients exhibit a hyperactive phenotype (83). Compared with those from healthy individuals, platelets from COVID-19 patients, including those with severe disease, are more sensitive to aggregation in response to low concentrations of agonists, such as thrombin and collagen (17). A high proportion, approximately 90%, of individuals with COVID-19 were shown to exhibit increased platelet aggregation, elevated platelet activation, platelet–monocyte aggregation and formation, and increased levels of fibrinogen and collagen diffusion (84). Therefore, SARS-CoV-2 infection induces greater platelet reactivity. The platelet hyperactivity mechanisms is shown in Figure 6. Research has indicated that hyperplatelet activity persists for a minimum of 40 days in the majority of COVID-19 patients, irrespective of the severity of the disease, following the resolution of acute inflammation. The sustained elevation in platelet activity after recovery from COVID-19 may predispose patients to thromboembolic complications (85).and results in an increased platelet reaction in the context of this particular illness, a phenomenon known as “fibrinolysis shutdown” (82).
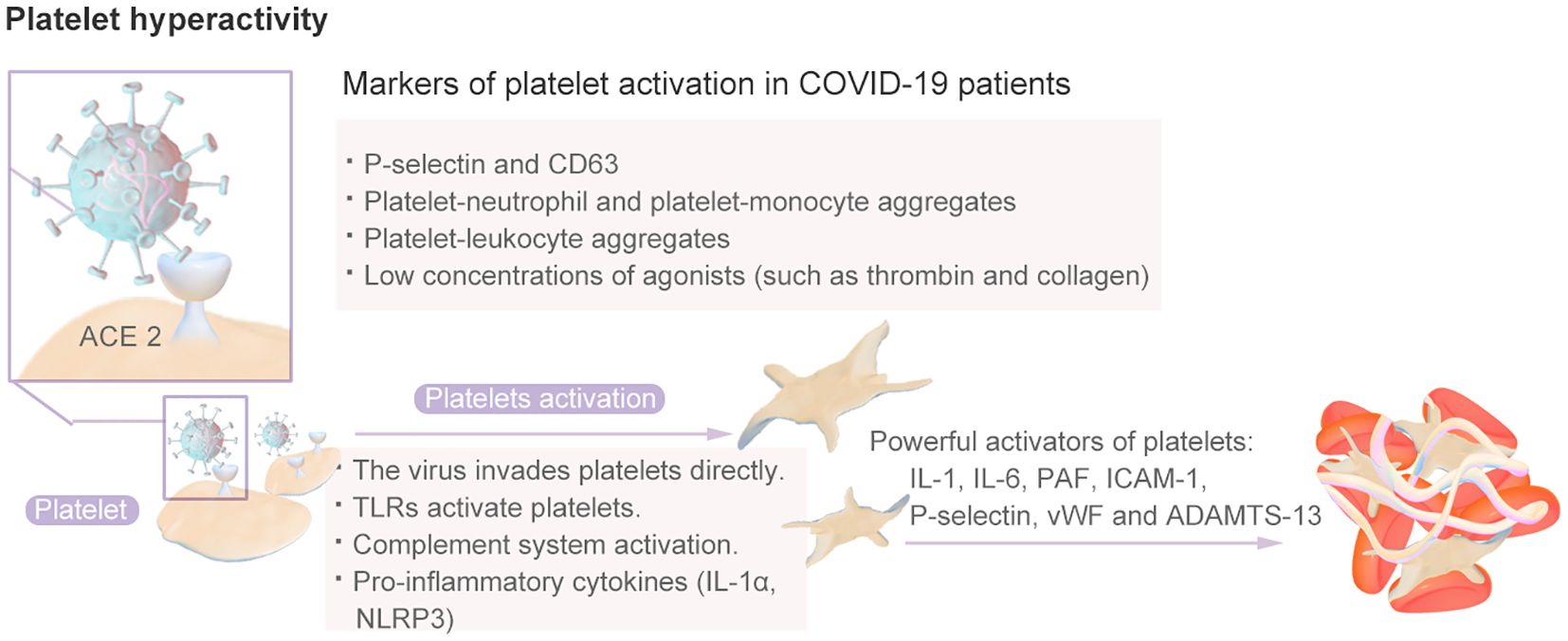
Figure 6. The Platelet Hyperactivity Mechanisms.
Various mechanisms explain platelet activation in patients with COVID-19. The first mechanism is related to the inflammatory response to SARS-CoV-2 infection and the resulting cytokine storm and endothelial dysfunction (82). IL-1, IL-6, platelet-activating factor (PAF), intercellular adhesion molecule 1 (ICAM-1), P-selectin and vWF released by endothelial cells are all powerful activators of platelets that increase the binding of platelets to endothelial cells, thereby further inducing platelet activation and aggregation and leading to a progressively stronger effect; this mechanism may even induce DIC and thrombocytopenia. SARS-CoV-2 can enter endothelial cells through the ACE-2 receptor, CD147, toll-like receptors (TLRs), or C-type lectin-like receptor 2 (CLEC-2) and platelets through the pericellular region. Subsequently, platelet activation leads to the release of platelet granules, the overexpression of TF, the formation of platelet leukocyte aggregates, and the formation of NETs (86).Moreover, the imbalance between vWF and ADAMTS-13 is related to platelet activation. An elevated vWF/ADAMTS-13 ratio may indicate an acquired ADAMTS-13 deficiency, which results in the retention of larger circulating vWF molecules and increases the risk of platelet binding and microthrombosis (82). Once activated, platelets secrete a vast repertoire of bioactive molecules, such as adenosine diphosphate (ADP), thromboxane A2 (TXA-2) and proinflammatory cytokines, from their intracellular granules that play essential roles in thrombus formation and inflammation (87). There is evidence that increased platelet response during SARS-CoV-2 infection is, at least in part, mediated by increased mitogen-activated protein kinase (MAPK) signaling. The activation of MAPK in platelets induces the activation of cytosolic phospholipase A2 (cPLA2), which results in thromboxane synthesis (88).
Sera from patients with severe COVID-19 possess the capability to activate platelets through the cross-linking of their FcγRIIa receptors. Such sera can induce a procoagulant phenotype in platelets derived from healthy donors. This mechanism can be triggered not only by antibody-mediated pathways but also by immune complexes (89). Immune complexes, which are soluble antigens formed through the binding of antibodies, serve as a crucial initial defense mechanism against pathogenic infections. In the context of COVID-19, these complexes consist of spike proteins from the virus and their corresponding antibodies. The interaction between immune complexes and platelet receptors triggers the activation of platelets and the release of intracellular molecules, including serotonin. This particular immune complex facilitates platelet activation and thrombosis in COVID-19, resembling the pathophysiological process observed in heparin-induced thrombocytopenia (90). Research has elucidated the mechanism by which the SARS-CoV-2 S1 spike protein (S1) induces platelet activation. Specifically, IgG antibodies bind to the S1 protein to form immune complexes, which enhance the expression of the FcγRIIa receptor on platelets, facilitating receptor cross-linking. This process initiates platelet activation and aggregation, leading to the formation of platelet-leukocyte aggregates (PLAs). Consequently, this mechanism directly contributes to the prothrombotic environment observed in COVID-19 (91).
2.3.2 Procoagulant platelets
Procoagulant platelets are a subset of activated platelets that expose phosphatidylserine (PS) and promote thrombin production (92). Studies have shown that the ΔΨm depolarization, cytosolic Ca2+ and PS externalization levels of platelets from COVID-19 patients in the ICU are high and that the expression of apoptosis markers is upregulated. Serum and immunoglobulin (IgG) fractions isolated from COVID-19 patients can induce the apoptosis of platelets from healthy donors, followed by changes in the coagulation system (93). Similarly, another study demonstrated a significant elevation in the levels of phosphorylated AKT in the platelets of COVID-19 patients, which was associated with CD62p expression and PS externalization. Notably, procoagulant thrombopoiesis induced by serum from COVID-19 patients in the ICU occurs independently of GPllb/llla. Furthermore, the inhibition of AKT and PI3K protein phosphorylation effectively prevented the production of procoagulant platelets. Thus, targeting PI3K/AKT phosphorylation may represent a promising therapeutic strategy to mitigate the risk of thrombosis in patients with severe COVID-19 (94).
2.4 Extracellular vesicle mechanism
Extracellular vesicles (EVs) are lipid bilayer-enclosed particles secreted by cells, capable of transferring proteins, lipids, metabolites, nucleic acids, and organelles to other cells, and are typically found in distant tissues throughout the body. EVs exhibit considerable heterogeneity, with sizes ranging from 20 nanometers (nm) to 1 micrometer. They can be categorized based on size, with exosomes representing the smaller end of the spectrum and microvesicles (formerly referred to as microparticles) representing the larger end. EVs are released by various cell types, including platelets, megakaryocytes, white blood cells, endothelial cells, and red blood cells, and these cells may be influenced by the effects mediated by EVs. EVs perform multiple functions, such as facilitating cell-cell communication, removing cellular waste or recycling molecules, and mediating host-pathogen interactions. Platelet-derived EVs may originate from the plasma membrane of resting platelets or from α granules following platelet activation. EVs derived from endothelial cells and monocytes express selectin, TF, vWF, other coagulation factors, and negatively charged phosphatidylserine, all of which can contribute to thrombosis. Additionally, circulating EVs may play a role in regulating inflammation and vascular permeability (95, 96).
Current studies have confirmed that extracellular vesicles (EVs) promote integrates the triple functions of “coagulation - inflammation - viral transmission”. Different from simple NETs or cytokine storm in COVID-19 (97). Studies have shown that the activity levels of EV-related TF in COVID-19 patients are higher than those in the control group (98). The activity of EV-related TF is closely related to the D-dimer level, which is a marker of thrombosis in this patient group. The EV level is also associated with prothrombin time, fibrinogen level, plasmin-antiplasmin complex, von Willebrand factor and ADAMTS13 (98). Another study compared severely ill COVID patients with non-COVID patients with septic shock and found that COVID-19 patients had a higher pro-coagulant effect, and the EV-TF activity was significantly higher (99).
EVs facilitate the coagulation process via the exposure of phosphatidylserine, providing a catalytic surface to facilitate the formation of the tenase (factors VIIIa, IXa, and X) and prothrombinase (factors Va, Xa, and II) complexes of the coagulation cascade (100). And EVs initiate clotting by surface expression of tissue factor (TF) under inflammatory conditions (101). Studies have characterized the expression of TF in clinical samples from patients with severe COVID-19, and it was found that the ratio of TF to EV was greater than 15% (101).
Moreover, Endothelial cells (EC) can release EVs (EC-EVs) that express endothelial markers CD31 (102). ECEC-EVs play a relevant role in regulating the survival of ECs, and they also participate in triggering coagulation and complement cascade reactions (103).
3 Prevention and treatment of thrombosis
Coagulation dysfunction and microvascular and macrovascular thrombotic events are common features of patients with acute COVID-19 in the early stages of the pandemic. Over the past four years, the incidence and manifestations of COVID-19-associated coagulopathy have evolved as a result of immunity acquired through natural infection and vaccination and the emergence of new SARS-CoV-2 variants. Therefore, there is a need to reevaluate diagnostic criteria and management strategies for COVID-19-associated coagulopathy on the basis of updated experiences and studies (104). The corresponding thrombus prevention and treatment options are also changing. Therefore, we include the review of clinical trials and guideline recommendations.
3.1 Clinical trials
Through a systematic search of the Clinical Trials.gov and PubMed databases, we identified a total of 14 completed clinical trials on the use of anticoagulant drugs in COVID-19 patients (Table 1). Among them, only one clinical trial (NCT04354155) (105) has been conducted in children. The remaining clinical trials were conducted in adult patients. The COVID-19 study population was divided into nonhospitalized and hospitalized patients on the basis of the severity of the infection. Hospitalized patients were divided into noncritically ill and critically ill COVID-19 patients. Most of the clinical trials used the anticoagulant drugs unfractionated heparin (UFH)/low-molecular-weight heparin (LMWH). Interestingly, these trials focused on thrombosis prevention.
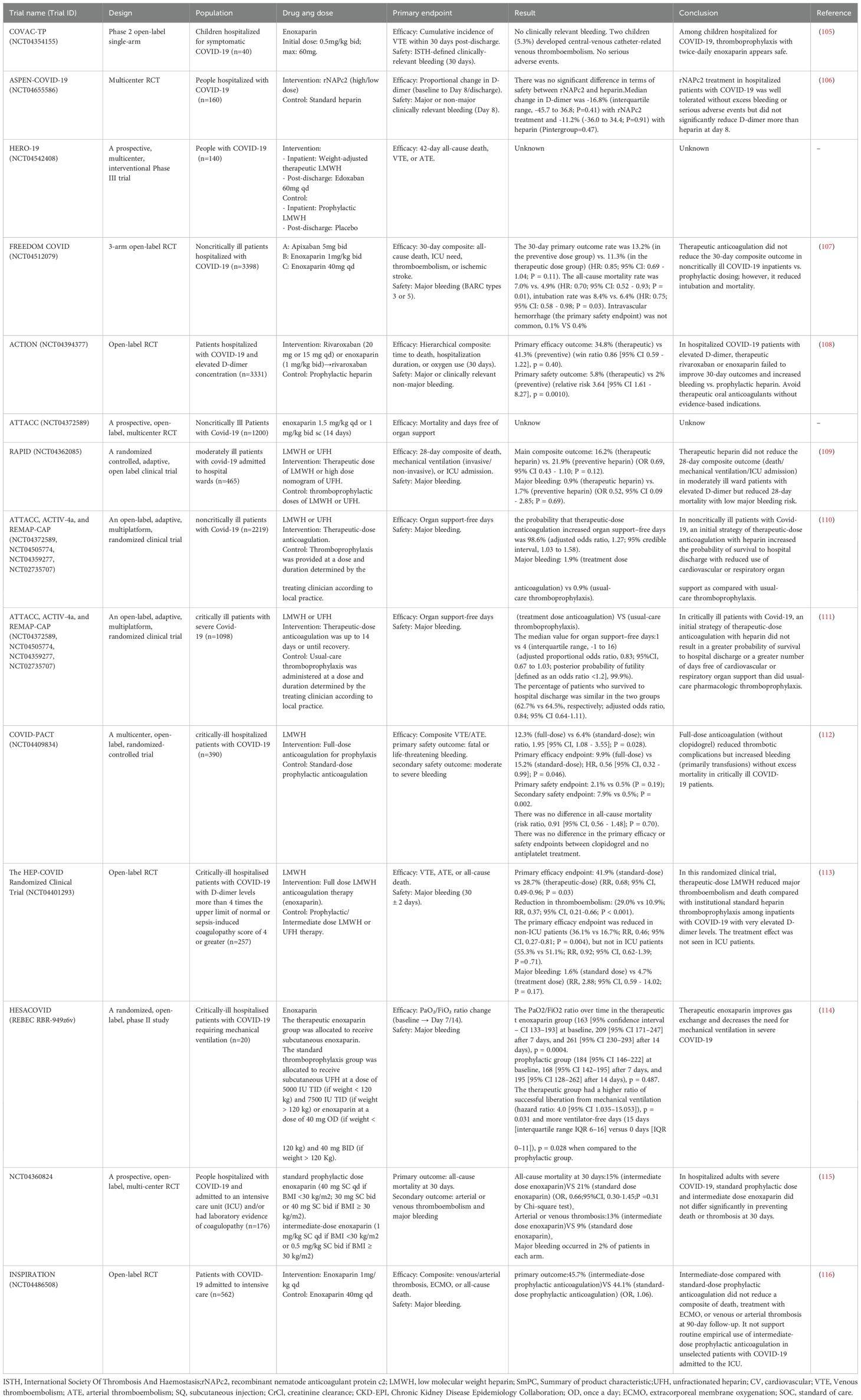
Table 1. Progress of clinical trials on prevention and treatment of thrombosis in patients with COVID-19.
In a single-arm clinical trial of enoxaparin for thrombosis prevention in hospitalized pediatric COVID-19 patients, the daily administration of enoxaparin for thrombosis prevention in children was found to be safe and worthy of further study to evaluate its efficacy (105).
In a clinical trial involving nonhospitalized COVID-19 patients, the incidence of cardiovascular events in the enoxaparin group was 0.9%, whereas that in the control group was 1.7% (relative risk 0.51; 95% confidence interval: 0.09–2.75). However, the early use of enoxaparin did not improve the course of the disease in patients with COVID-19 (117). In another nonhospitalized COVID-19 patient clinical trial, 8 patients who received enoxaparin experienced severe adverse reactions (118). A systematic review evaluated the efficacy of prolonged thromboprophylaxis in COVID-19 patients after discharge, including eight studies with a total of 10,148 patients. The results confirmed that prolonged thromboprophylaxis, mainly prophylactic use of anticoagulants for <35 days, was significantly associated with a poor overall prognosis in high-risk patients with COVID-19 after discharge (OR: 0.52; 95% CI: 0.41–0.67; P=0.000). Prolonged thromboprophylaxis did not increase the risk of major bleeding events (OR: 1.64; 95% CI: 0.95–2.82, P= 0.075) (119).
Most of the other clinical trials divided hospitalized patients into severe and nonsevere cases. One study compared two treatment regimens for noncritical hospitalized patients with COVID-19: one was a therapeutic dose of heparin anticoagulation therapy, and the other was a standard nursing drug for thrombosis prevention therapy. Among the 2219 patients ultimately included, initial therapeutic-dose heparin anticoagulant therapy improved the probability of survival to discharge while reducing the use of cardiovascular or respiratory support compared with conventional thromboprophylaxis. However, the incidence of major bleeding was greater in the group that received the therapeutic dose (1.9% vs. 0.9%) (110). Another trial compared patients with severe illness, with 1098 patients divided into 534 receiving treatment-dose anticoagulation and 564 receiving standard care thrombosis prophylaxis. The proportion of patients surviving to discharge was similar in both groups (62.7% and 64.5%, respectively; adjusted relative advantage, 0.84; 95% confidence interval, 0.64–1.11). A total of 3.8% of patients receiving treatment-dose anticoagulation had major bleeding, compared with 2.3% of patients receiving standard care thrombosis prophylaxis. In COVID-19 critically ill patients, the strategy of initial treatment-dose heparin anticoagulation compared with standard care pharmacological thrombosis prophylaxis did not result in a greater probability of survival to discharge or more days without cardiovascular or respiratory organ support (111). The NCT04360824 (115) and INSPIRATION (116) trials compared the efficacy and safety of different doses in critically ill patients and reported no significant difference in thrombotic or bleeding events between moderate and standard doses of enoxaparin. However, the therapeutic dose of enoxaparin in the HESACOVID trial (114) improved gas exchange and reduced the need for mechanical ventilation in patients with severe COVID-19. One meta-analysis included studies comparing therapeutic or intermediate anticoagulation versus prophylactic anticoagulation in COVID-19 patients, including 6 RCTs with 4678 patients and 4 cohort studies with 1080 patients. In randomized controlled trials, therapeutic anticoagulation or intermediate anticoagulation was associated with a significant reduction in thromboembolic events (5 studies, n=4,664; relative risk [RR], 0.72; P=0.01) and significantly increased bleeding events (5 studies, n=4,667; RR1.88; P= 0.004). In moderate patients, therapeutic or moderate anticoagulation was more beneficial than prophylactic anticoagulation for thromboembolic events, but the number of bleeding events was significantly greater. In severely ill patients, the incidence of thromboembolism and bleeding events occurs at or in the middle of treatment. It is suggested that patients with moderate and severe COVID-19 infection should be treated with preventive anticoagulation therapy (120). In another systematic review, which included 33 studies (11,387 patients), standard heparin prophylaxis versus high (intermediate or therapeutic) heparin regimens was evaluated in hospitalized patients with COVID-19. The incidence of VTE events was 5.2% and 8.2% with high- or standard-dose heparin prophylaxis, respectively (RR 0.71, 95% CI 0.55–0.90, 1248.8%). Major bleeding was significantly greater in patients receiving the high dose than in those receiving the standard dose (4.2% vs. 2.2%, RR 1.94, 95% CI 1.47–2.56, 1218.1%) (121).
Additionally, the COVID-PACT trial included critically ill patients and assessed the prevention of thrombosis with antiplatelet drugs. A total of 682 patients were enrolled, and this trial revealed that full-dose anticoagulation was superior to clopidogrel antiplatelet prevention for thrombosis, reducing the incidence of thrombotic events but increasing bleeding, which was mainly due to blood transfusions in patients with stable hemodynamics, with no significant increase in mortality. Owing to the decline in the incidence of ICU-level COVID-19, recruitment was discontinued in March 2022 (planned enrollment was 50%) (112). The HEP-COVID-19 multicenter randomized clinical trial assessed the efficacy of therapeutic doses of LMWH compared with standard prophylactic or medium-dose heparin in preventing COVID-19-related thrombosis in high-risk hospitalized patients. The trial also stratified patients on the basis of their ICU status, with 83 out of 257 randomized patients (32.8%) being in the ICU (113). Among the patients in the standard-dose group, 52 (41.9%) met the primary efficacy endpoint. In comparison, 37 (28.7%) patients in the therapeutic dose group met the primary efficacy endpoint. The incidence of thromboembolism decreased significantly from 29.0% to 10.9% (RR 0.37; 95% CI, 0.21–0.66; P < 0.001). Major bleeding occurred in 1.6% of the patients in the standard-dose heparin group and 4.7% of those in the therapeutic-dose heparin group (RR 2.88; 95% CI, 0.99–14.02; P = 0.17). The primary outcome measures were reduced in non-ICU patients (36.1% vs. 16.7%; RR 0.46; 95% CI, 0.27–0.81; P = 0.004), whereas no significant difference was observed in ICU patients (55.3% vs. 51.1%; RR 0.92; 95% CI, 0.62–1.39; P = 0.71). Compared with standard heparin thromboprophylaxis, therapeutic doses of low-molecular-weight heparin significantly reduced the incidence of major thromboembolism and mortality in hospitalized COVID-19 patients with markedly elevated D-dimer levels. Conversely, no therapeutic benefit was observed in patients admitted to the ICU.
Among the clinical trials described above, in addition to the heparin anticoagulant regimen, several trials evaluated the efficacy of new oral anticoagulants (NOACs) and a TF inhibitor in preventing blood clots in COVID-19 patients. rNAPc2 is a novel TF inhibitor with anticoagulant, anti-inflammatory, and potentially antiviral properties. Therefore, the ASPEN-COVID-19 trial utilized rNAPc2 to investigate whether TF could be a therapeutic target for COVID-19 thrombus prophylaxis. Among patients hospitalized with COVID-19, TF inhibition with rNAPc2 as the only anticoagulant was safe and well tolerated but did not significantly reduce D-dimer levels more than the heparin standard of care. The NCT04542408 trial evaluated edoxaban, but the trial data and results were not published. In the FREEDOM COVID anticoagulation strategy randomized trial, 3398 noncritically ill patients hospitalized for COVID-19 were randomly assigned to receive a prophylactic dose of enoxaparin (n=1141), a therapeutic dose of enoxaparin (n=1136), or a therapeutic dose of apixaban (n=1121). The results were similar in both treatment dose groups, with major bleeding uncommon in all three groups (107). ACTION is an open-label, multicenter, randomized, controlled trial: patients (aged ≥18 years) who were hospitalized with COVID-19 and had elevated D-dimer concentrations and symptoms of COVID-19 for up to 14 days prior to randomization were randomly assigned (1:1) to receive therapeutic or preventive anticoagulant therapy. Therapeutic anticoagulant therapy was oral rivaroxaban at hospitalization (20 mg or 15 mg daily) for stable patients and an initial subcutaneous injection of enoxaparin (1 mg/kg twice daily) or intravenous injection of UFH (target 0-3-0–7 IU/mL anti-Xa concentration), followed by rivaroxaban until day 30, for clinically unstable patients. There was no difference in primary efficacy outcomes among patients receiving therapeutic and preventive anticoagulant therapy. Compared with prophylactic anticoagulation, in-hospital anticoagulation treatment of up to 30 days of rivaroxaban or enoxaparin did not improve clinical outcomes, and bleeding increased (108).
It is perplexing that various anticoagulants exhibit differing therapeutic effects across patient subgroups stratified by disease severity. The author of this article hypothesizes that there are several possible reasons. The pathophysiological mechanism of the patients undergoes dynamic changes. For mild/moderate patients, it is mainly characterized by vascular endothelial micro-inflammation and local hypercoagulability, with limited thrombin production. Therefore, the effect of heparin-like drugs is limited. While for severe patients, “immune thrombosis” occurs (immunothrombosis), NETs are released and systemic coagulation activation takes place. In addition to their anticoagulant effect, using heparin can also neutralize the histones of NETs (through negative charge binding), exerting an anti-inflammatory effect. However, DOACs do not have this effect. From the perspective of pharmacokinetics, there are differences among patients with varying degrees of severity.
3.2 Related guidelines
To address the risk of thrombosis in patients with COVID-19, many academic groups in China and abroad have issued relevant guidelines for the prevention and treatment of thrombosis in COVID-19 patients. Most guidelines refer to anticoagulant therapy with heparin or low-molecular-weight heparin, but only a few guidelines mention direct oral anticoagulant (rivaroxaban) (122, 123).
Nonhospitalized COVID-19 patients: The ISTH guidelines define those who are “not hospitalized” as people with COVID-19 who live in the community and have not recently been hospitalized for COVID-19 (123). Guidelines suggest that initiating direct oral anticoagulant therapy or antiplatelet therapy does not effectively reduce the risk of hospitalization, arterial or venous thromboembolism, or death in symptomatic COVID-19 patients who are not hospitalized. In nonhospitalized COVID-19 patients at increased risk of disease progression, the initiation of oral sulodexide treatment within 3 days of symptom onset may be considered to reduce the risk of hospitalization. In asymptomatic COVID-19 patients, LMWH for preventing blood clots is not effective in reducing the risk of disease progression (123).
Noncritically ill patients hospitalized for COVID-19: For inpatients, most foreign guidelines classify hospitalized nonpregnant adult patients with COVID-19 without evidence of VTE into two categories: noncritically or acutely ill patients hospitalized for COVID-19 (in the general inpatient ward) and critically ill patients hospitalized for COVID-19 (in the ICU). Low-dose (prophylactic/standard) LMWH or UFH should be used in noncritically ill patients hospitalized for COVID-19 to reduce thromboembolism and the possible risk of death, and it is recommend that clinicians consider the therapeutic intensity of LMWH or UFH thromboprophylaxis in noncritically ill patients at increased risk of disease progression or thromboembolism and not at high risk of anticoagulation-related bleeding (123). In the Guidelines for diagnosis, prevention and treatment of COVID-19 in adults in China, anticoagulation prevention is also recommended for mild and general COVID-19 patients with medical or surgical conditions who are assessed as having a high or medium-high risk of VTE on the basis of the Padua score or Caprini score and who have no anticoagulation conjunctivitis (124). The ASH guidelines also recommend the use of anticoagulant therapy with standard prophylactic intensity over therapeutic intensity for patients with COVID-19-related acute illness without suspected or confirmed VTE or other anticoagulant indications (125). The NIH guidelines further recommend the use of therapeutic doses of heparin in patients with D-dimer levels above the upper limit of normal who require low oxygen flow and are not at increased risk of bleeding (126).
Critically ill hospitalized patients with COVID-19: In the absence of contraindications, all guidelines recommend anticoagulant prophylaxis in critically ill hospitalized patients with COVID-19. All guidelines (including domestic guidelines) recommend the use of a prophylactic dose for patients with COVID-19-related critical illness who do not have suspected or confirmed venous thromboembolism. For critically ill patients with COVID-19, the CHEST guidelines (127, 128) recommend against routine ultrasound screening for asymptomatic DVT and adding mechanical prophylaxis to drug thrombus prophylaxis. CHEST guidelines recommend mechanical thromboprophylaxis if drug thromboprophylaxis is contraindicated in critically ill COVID-19 patients. In addition, the domestic guidelines make other recommendations (129). For hospitalized patients with severe COVID-19, if there is a high risk of VTE and a low risk of bleeding and the disease has a tendency to worsen (such as a progressive increase in D-dimers and exacerbation of hypoxemia), the use of a therapeutic dose of LMWH/UFH anticoagulation is recommended. If bleeding is a high risk, mechanical prophylaxis is recommended. After the risk of bleeding is reduced, drug prophylaxis combined with mechanical prophylaxis should be carried out in time. The Anticoagulation Forum Guidelines (122) suggest that posthospital thromboprophylaxis with 10 mg of rivaroxaban daily for 35 days following hospitalization for COVID-19 may be considered in select patients at increased risk of thromboembolism (e.g., IMPROVE VTE score ≥ 4 or score 2–3 with elevated D-dimers at hospital discharge) and not at increased risk of bleeding, regardless of the intensity of their inpatient thromboprophylaxis.
Discharged patients: All guidelines recommend against routinely continuing VTE prophylaxis in patients with COVID-19 after hospital discharge unless they have another indication for anticoagulation. According to the Guidelines for Thrombosis Prevention and Anticoagulation Management in Hospitalized Patients with Novel Coronavirus Infection (129), if a high risk of VTE and low risk of bleeding are assessed before discharge, drug prophylaxis with NOACs can be recommended. If a high risk of VTE and a high risk of bleeding are assessed before discharge, mechanical prophylaxis (such as GCS) or early activity is recommended. For hospitalized patients with COVID-19, if out-of-hospital prophylaxis is assessed before discharge, prophylaxis should be prolonged depending on the risk of VTE or until the risk factors are removed. The ISTH guidelines (123) suggest that in select high-risk patients who have been hospitalized for COVID-19, postdischarge treatment with prophylactic-dose rivaroxaban for approximately 30 days may be considered to reduce the risk of major thromboembolism. In patients who have been hospitalized for COVID-19 and are not deemed at high risk for complications, routine postdischarge prophylactic doses of direct oral anticoagulants are not recommended to reduce the risk of death or thromboembolism.
For antithrombotic treatment, the CHEST guidelines (127, 128), Anticoagulation Forum guidelines (122) and ‘Guidelines for Thrombosis Prevention and Anticoagulation Management in Hospitalized Patients with Novel Coronavirus Infection’ refer to anticoagulation therapy for COVID-19 patients with thromboembolism. It is recommend that most patients who are diagnosed with VTE while hospitalized for COVID-19 receive anticoagulation therapy for a minimum of three to six months. For critically ill COVID-19 patients with proximal DVT or PE, we suggest parenteral rather than oral anticoagulant therapy. For critically ill COVID-19 patients with proximal DVT or PE who are treated with parenteral anticoagulation, we suggest LMWH or fondaparinux over UFH. In patients with COVID-19 and recurrent VTE despite anticoagulation with apixaban, dabigatran, rivaroxaban or edoxaban (and documented compliance) or vitamin K antagonist therapy (in the therapeutic range), we suggest switching treatment to therapeutic weight-adjusted LMWH.
4 Long COVID-19/postacute sequelae of COVID-19
COVID-19 can lead to a variety of pathophysiological changes and clinical symptoms. Although symptoms usually last only 2–3 weeks, after the acute phase, approximately 10% of patients develop persistent or new symptoms, a condition known as long COVID-19/postacute sequelae of COVID-19 (130). Long COVID-19 has the potential to impact multiple organ systems, resulting in significant and lasting functional impairments attributable to organ damage. The resulting burden of this disease on individuals, healthcare systems, and national economies is substantial (131). Cardiovascular complications (including thromboembolic diseases) have emerged as a key issue in PASC (132, 133). So far, the main potential pathophysiological mechanisms related to vascular complications include immune dysregulation, autoimmunity, endothelial dysfunction (ED), coagulopathy, etc. Although the acute phase of cardiovascular complications is characterized by excessive inflammation and coagulation dysfunction, the mechanisms driving the persistent cardiovascular dysfunction in PASC remain poorly understood (134). Furthermore, numerous procoagulant inflammatory molecules have been identified within microclots associated with long COVID-19. These molecules include α2-antifibrinolytic protein (α2AP), von Willebrand factor (vWF), platelet factor 4 (PF4), serum amyloid A (SAA), various fibrinogen chains, and a range of antibodies (1). Notably, circulating NET biomarkers do not return to normal levels until approximately 4 months after infection (135). In cases of long COVID-19, feedback loops involving NETs and thrombosis establish a cycle characterized by coagulation, inflammation, and localized hypoxia, which contributes to the severity of COVID-19 and the persistence of long-term symptoms.
New evidence suggests that mitochondrial dysfunction within immune cells may play a crucial role in the pathogenesis of PASC, especially in relation to cardiovascular sequelae (136). Mitochondria play a crucial role in the generation of cellular energy, the function of immune cells, regulation of oxidative phosphorylation (OXPHOS), production of reactive oxygen species (ROS), and cell apoptosis, among other processes. (137) Mitochondria are usually the targets of viral proteins, which can lead to disruptions in biological energy and changes in the immune response (138). Meanwhile, monocytes also rely heavily on mitochondrial function to maintain innate immunity and vascular homeostasis. Mitochondrial damage can also lead to a decline in monocyte function (139). In long-term COVID patients, the persistent oxidative stress leads to continuous mitochondrial damage and cellular dysfunction, thereby perpetuating the cycle of inflammation and tissue damage (140, 141). Furthermore, genetic factors and genetic polymorphisms may also contribute to an increased susceptibility of long COVID patients to vascular complications (142). For example, the polymorphisms of genes related to blood coagulation, F5 (R506Q) and F2 (G20210A), can increase the susceptibility to thromboembolic events by enhancing procoagulant activity or reducing anticoagulant regulation. In COVID-19, these polymorphisms work in synergy with a high inflammatory state, resulting in excessive thrombus formation in the pulmonary vessels, which leads to pulmonary thromboembolism (143, 144).
In patients with long-term COVID-19, the routine use of anticoagulants or antiplatelet drugs for thrombosis prevention is not recommended (145). A Korean guideline recommends treatment with anticoagulants or antiplatelet agents according to the relevant guidelines for patients with long COVID-19 diagnosed with thrombosis (146).
5 COVID-19 vaccine
Overall, the currently approved COVID-19 vaccines are effective in reducing the incidence and mortality rates associated with the disease (147). Real-world studies show that vaccination reduces the risk of COVID-19 hospitalization by 85-95% and the risk of severe disease by 91% (Delta variant era data) (148). Although vaccines may cause extremely rare blood clots (such as VITT, with an incidence of approximately 1 in 250,000 doses) (149), the risk of blood clot caused by COVID-19 itself is even higher (about 16.5% of hospitalized patients develop VTE) (150). Mathematical models show that vaccination can prevent more than 40 times the number of thrombosis-related deaths (151). A study analyzed the VTE cases of 792,010 patients who received the authorized COVID-19 vaccine in the United States. The results showed that there was no increase in the risk of VTE after vaccination compared to before vaccination (152). That is to say, the benefits of global vaccination for individuals and the public far outweigh the adverse effects of the vaccines (153).
Nevertheless, the potential risk of thromboembolic events associated with vaccination continues to warrant careful consideration. Studies have shown that patients who received SARS-CoV-2 vaccines (such as ChAdOx1 or nCoV-19) developed thrombosis or thrombocytopenia, which is referred to as vaccine-induced immune thrombotic thrombocytopenia (VITT) (154). As a result, many countries have restricted their use in young patients. The main mechanisms include that Anti-PF4 antibody-mediated platelet activation, vaccine components trigger immune responses, and adenovirus vector vaccines may release free DNA or negatively charged proteins (such as spike proteins), which combine with platelet factor 4 (PF4) to form PF4-polyanion complexes (155). Anti-PF4 antibodies can also activate platelets through the FcγRIIa receptor, leading to platelet aggregation, thrombosis, and secondary thrombocytopenia (156). In addition, autoantibodies are produced, and some individuals develop anti-PF4 antibodies (similar to heparin-induced thrombocytopenia, HIT), but unlike HIT, VITT patients usually have no history of heparin exposure (157). Activated platelets release procoagulant microparticles, and endothelial injury triggers the expression of TF, promoting widespread thrombosis (158).
The primary intervention for VITT management is immunomodulatory therapy with intravenous immunoglobulin (IVIG) (155). It can rapidly increase platelet count (usually within 24–48 hours) and reduce the risk of thrombosis progression (159). If IVIG is ineffective or in severe cases (such as multiple organ thrombosis), glucocorticoids can be used as an adjunctive treatment (160). Anticoagulation therapy should prefer non-heparin anticoagulants(typically argatroban infusion or fondaparinux) (161). Additionally, platelet transfusion should be avoided unless there is life-threatening bleeding, and it should be used after IVIG (17). For refractory VITT, intensified treatment can also consider plasma exchange (TPE) (161).
6 Discussion
In general, we have an increasing understanding of the mechanism of COVID-19-induced thrombosis from existing studies, which can provide more references for medical workers. The guidelines also have updated recommendations based on clinical studies. The authors of this review suggest that the prevention of thrombosis in COVID-19 patients requires the timely assessment of their thrombosis and bleeding risk, clarification of any anticoagulation contraindications, and the use of diagnostic tests to evaluate their hypercoagulable or fibrinolytic state to better apply treatment strategies. We also found several interesting points when conducting this review.
Besides COVID-19, infections resulting from other respiratory diseases may also increase the risk of thrombotic events. a case report of a 58-year-old man with confirmed influenza virus infection who presented with deep vein thrombosis and acute pulmonary embolism (162). A review of 58 patients with laboratory-confirmed influenza A or B and associated intravascular thrombosis revealed 21 cases of pulmonary embolism (36.2%), 12 cases of DVT (20.6%) and 3 cases of DVT with pulmonary embolism (5.1%) (163). Similar thrombotic complications were observed in patients with severe acute respiratory syndrome (SARS) (164). In a nationwide study conducted from 2012–2014, the incidence of thrombosis among patients hospitalized with acute viral respiratory illnesses, excluding COVID-19, was compared to the incidence among patients hospitalized with COVID-19 within a large health system in New York. That study revealed that 1.6% of patients with non-COVID-19 viral respiratory illnesses also experienced venous thromboembolism. Furthermore, from 2002 –2014, the proportion of hospitalized patients with viral respiratory diseases who developed thrombosis was significantly lower than that of patients with COVID-19 (5% versus 16%; P < 0.001) (165). A further literature search revealed many similarities and differences between influenza and COVID-19. Comorbidities such as cardiovascular disease, diabetes, and obesity were significantly more common in COVID-19 patients, whereas lung disease and immunocompromised status were significantly more common in influenza patients. In terms of mechanism, both influenza and COVID-19 can cause a cytokine storm, which further stimulates inflammatory immunity to promote thrombogenesis (166). That study revealed that TF expression was upregulated in lung epithelial cells, with concomitant increases in EVTF activity and coagulation activity in bronchoalveolar lavage fluid (BALF) following influenza A virus infection in murine models. Similarly, mice infected with MERS-CoV, SARS-CoV, or SARS-CoV-2 presented elevated lung TF expression. Furthermore, patients suffering from severe respiratory tract infections demonstrated high levels of TF expression, strongly indicating that this upregulation may contribute to the pathogenesis of thrombosis (167).
The multinational and multicenter data included in this review summarize the clinical trials of anticoagulant therapy for COVID-19 patients. The differences in the studies may result from the different definitions, diagnostic methods and preventive management methods of all the included studies. In clinical trials, low-molecular-weight heparin/common liver and kidney drugs are often used, and there are a few mentions of new oral anticoagulants. The reason might be that anticoagulants such as heparin, LMWH or UFH have anti-inflammatory properties, which can inhibit the formation of thrombin and reduce inflammatory responses (168). The expected effect of heparin may also disrupt the lung-protective process of blood clotting, which could increase the survival rate of the host in COVID-19 (168). Furthermore, the antiviral efficacy of heparin explains why it prevents entry of the SARS-CoV-2 virus by acting on the ACE-2 receptor and interacting with the SARS-CoV-2 spike glycoprotein (169).
In addition to clots caused by the previously described immunoinflammatory mechanisms, D-dimers are also of concern, with elevated D-dimers being the most common feature of clotting diseases associated with COVID-19. The value of D-dimers in predicting the risk of VTE in COVID-19 patients has been demonstrated by numerous clinical studies, such as a multicenter study in France that revealed that the cooccurrence of elevated D-dimers (>3000 μg/L) and elevated white blood cell count (≥12.0×109/L) was significantly associated with PE. Among many clinical laboratory indicators, D-dimers have the highest predictive value for VTE. In addition to the acute phase of infection, studies have shown that COVID-19 patients with higher peak D-dimer levels (>3000 μg/L) have an approximately fourfold increased risk of VTE after being cured and discharged (170). In clinical trials, some of which also included patients with moderate COVID-19, therapeutic heparin was not significantly associated with a reduction in the primary outcome in moderate patients hospitalized with COVID-19 and elevated D-dimer levels but was associated with a reduced chance of death within 28 days; in the trial, the risk of major bleeding appeared to be low (109). In addition to existing anticoagulant or antiplatelet agents, nematode anticoagulant protein c2 (rNAPc2), a potent TF inhibitor, was investigated in the ASPEN-COVID-19 trial. rNAPc2 was found to be well tolerated in hospitalized COVID-19 patients with no excessive bleeding or serious adverse events, but its ability to reduce the level of D-dimers was not significantly greater than that of heparin at day 8 (106). Fibrin amyloid microclots have been extensively observed in cases of long-term SARS-CoV-2 infection, which causes the disease known as long COVID-19. These microclots have the capacity to obstruct capillaries, consequently impeding the passage of red blood cells and hindering oxygen exchange. This obstruction can result in the formation of microthrombi. Consequently, the development and careful monitoring of anticoagulation and antiplatelet treatment regimens in randomized clinical trials could present a viable therapeutic strategy (171). In a similar manner, fibrin interacts with the SARS-CoV-2 spike proteins to form proinflammatory clots, which contribute to systemic thromboinflammation and neuropathology in COVID-19 patients. A monoclonal antibody that targets the inflammatory domain of fibrin has been shown to protect against microglial activation, neuronal injury, and thrombotic inflammation in the postinfection lung. Consequently, fibrin plays a pivotal role in the inflammation and neuropathology associated with SARS-CoV-2 infection, suggesting that fibrin-targeted immunotherapy could serve as a potential therapeutic intervention for patients experiencing both acute and long-term effects of COVID-19 (172). These findings also provide some ideas for our future research direction. We look forward to the results of more studies in the future and advancements in the guidelines.
Author contributions
QY: Writing – original draft, Writing – review & editing, Conceptualization, Methodology. YH: Methodology, Writing – original draft, Writing – review & editing. HW: Writing – original draft, Methodology, Writing – review & editing. YW: Writing – original draft, Methodology. XH: Investigation, Writing – original draft. YS: Investigation, Writing – original draft. BY: Writing – review & editing. YYW: Writing – review & editing. LH: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the National Key Research and Development Program of China (2020YFC2005500), the Sichuan Science and Technology Plan Project (2022NSFSC0818), and the Chinese Society of Research Hospitals (Y2022FH-YWPJ01-202).
Acknowledgments
The authors acknowledge the support received from the foundations.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Turner S, Khan MA, Putrino D, Woodcock A, Kell DB, and Pretorius E. Long COVID: pathophysiological factors and abnormalities of coagulation. Trends Endocrinol Metab. (2023) 34:321–44. doi: 10.1016/j.tem.2023.03.002
2. Xu SW, Ilyas I, and Weng JP. Endothelial dysfunction in COVID-19: an overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol Sin. (2023) 44:695–709. doi: 10.1038/s41401-022-00998-0
3. Connors JM and Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. (2020) 135:2033–40. doi: 10.1182/blood.2020006000
4. Hanff TC, Mohareb AM, Giri J, Cohen JB, and Chirinos JA. Thrombosis in COVID-19. Am J Hematol. (2020) 95:1578–89. doi: 10.1002/ajh.25982
5. Violi F, Pignatelli P, Cammisotto V, Carnevale R, and Nocella C. COVID-19 and thrombosis: Clinical features, mechanism of disease, and therapeutic implications. Kardiol Pol. (2021) 79:1197–205. doi: 10.33963/KP.a2021.0154
6. Louis DW, Saad M, Vijayakumar S, Ilyas S, Kokkirala A, and Aronow HD. The cardiovascular manifestations of COVID-19. Cardiol Clin. (2022) 40:277–85. doi: 10.1016/j.ccl.2022.03.001
7. Tang N, Li D, Wang X, and Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemostasis. (2020) 18:844–7. doi: 10.1111/jth.14768
8. Othman HY, Zaki IAH, Isa MR, Ming LC, and Zulkifly HH. A systematic review of thromboembolic complications and outcomes in hospitalised COVID-19 patients. BMC Infect Dis. (2024) 24:484. doi: 10.1186/s12879-024-09374-1
9. Bagheri B, Alipour A, Yousefi M, Jalalian R, Moghimi M, Mohammadi M, et al. Prevalence of thromboembolic events, including venous thromboembolism and arterial thrombosis, in patients with COVID-19: A systematic review with meta-analysis. J Tehran Heart Cent. (2023) 18:154–69. doi: 10.18502/jthc.v18i3.14110
10. Zuin M, Barco S, Giannakoulas G, Engelen MM, Hobohm L, Valerio L, et al. Risk of venous thromboembolic events after COVID-19 infection: a systematic review and meta-analysis. J Thromb Thrombolysis. (2023) 55:490–498. doi: 10.1007/s11239-022-02766-7
11. Burn E, Duarte-Salles T, Fernandez-Bertolin S, Reyes C, Kostka K, Delmestri A, et al. Venous or arterial thrombosis and deaths among COVID-19 cases: a European network cohort study. Lancet Infect Dis. (2022) 22:1142–52. doi: 10.1016/S1473-3099(22)00223-7
12. Sk Abd Razak R, Ismail A, Abdul Aziz AF, Suddin LS, Azzeri A, and Sha’ari NI. Post-COVID syndrome prevalence: a systematic review and meta-analysis. BMC Public Health. (2024) 24:1785. doi: 10.1186/s12889-024-19264-5
13. Jara-Palomares L, Bikdeli B, Jimenez D, Muriel A, Demelo-Rodriguez P, Moustafa F, et al. Risk of recurrence after discontinuing anticoagulation in patients with COVID-19- associated venous thromboembolism: a prospective multicentre cohort study. EClinicalMedicine. (2024) 73:102659. doi: 10.1016/j.eclinm.2024.102659
14. He S, Cao H, Thalin C, Svensson J, Blomback M, and Wallen H. The clotting trigger is an important determinant for the coagulation pathway in vivo or in vitro-inference from data review. Semin Thromb Hemost. (2021) 47:63–73. doi: 10.1055/s-0040-1718888
15. Yin Q, Zhang X, Liao S, Huang X, Wan CC, and Wang Y. Potential anticoagulant of traditional chinese medicine and novel targets for anticoagulant drugs. Phytomedicine. (2023) 116:154880. doi: 10.1016/j.phymed.2023.154880
16. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
17. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. (2020) 13:120. doi: 10.1186/s13045-020-00954-7
18. Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. (2020) 7:e575–82. doi: 10.1016/S2352-3026(20)30216-7
19. Smadja DM, Mentzer SJ, Fontenay M, Laffan MA, Ackermann M, Helms J, et al. COVID-19 is a systemic vascular hemopathy: insight for mechanistic and clinical aspects. Angiogenesis. (2021) 24:755–88. doi: 10.1007/s10456-021-09805-6
20. Levi M, Thachil J, Iba T, and Levy JH. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. (2020) 7:e438–40. doi: 10.1016/S2352-3026(20)30145-9
21. Goonewardena SN, Chen Q, Tate AM, Grushko OG, Damodaran D, Blakely PK, et al. Monocyte-mediated thrombosis linked to circulating tissue factor and immune paralysis in COVID-19. Arterioscler Thromb Vasc Biol. (2024) 44:1124–34. doi: 10.1161/ATVBAHA.122.318721
22. Valencia I, Lumpuy-Castillo J, Magalhaes G, Sanchez-Ferrer CF, Lorenzo O, and Peiro C. Mechanisms of endothelial activation, hypercoagulation and thrombosis in COVID-19: a link with diabetes mellitus. Cardiovasc Diabetol. (2024) 23:75. doi: 10.1186/s12933-023-02097-8
23. Ali EW and Ibrahim IK. Multi-factorial mechanism behind COVID-19 related thrombosis. Med Arch. (2022) 76:62–5. doi: 10.5455/medarh.2022.76.62-65
24. Ma L and Willey J. The interplay between inflammation and thrombosis in COVID-19: Mechanisms, therapeutic strategies, and challenges. Thromb Update. (2022) 8:100117. doi: 10.1016/j.tru.2022.100117
25. Ma Z, Yang KY, Huang Y, and Lui KO. Endothelial contribution to COVID-19: an update on mechanisms and therapeutic implications. J Mol Cell Cardiol. (2022) 164:69–82. doi: 10.1016/j.yjmcc.2021.11.010
26. Patel VB, Zhong J-C, Grant MB, and Oudit GY. Role of the ACE2/angiotensin 1–7 axis of the renin–angiotensin system in heart failure. Circulation research. (2016) 118:1313–26. doi: 10.1161/CIRCRESAHA.116.307708
27. Brown NJ and Vaughan DE. Prothrombotic effects of angiotensin. Advances in Internal Medicine. (2000) 45:419–29.
28. Wolf A, Khimani F, Yoon B, Gerhart C, Endsley D, Ray AK, et al. The mechanistic basis linking cytokine storm to thrombosis in COVID-19. Thromb Update. (2022) 8:100110. doi: 10.1016/j.tru.2022.100110
29. Bonaventura A, Vecchié A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. (2021) 21:319–29. doi: 10.1038/s41577-021-00536-9
30. Heidari Z, Naeimzadeh Y, Fallahi J, Savardashtaki A, Razban V, and Khajeh S. The role of tissue factor in signaling pathways of pathological conditions and angiogenesis. Curr Mol Med. (2023) 24:1135–51. doi: 10.2174/0115665240258746230919165935
31. Mackman N. Tissue factor and COVID-19 associated thrombosis. Arterioscler Thromb Vasc Biol. (2024) 44:523–9. doi: 10.1161/ATVBAHA.123.320144
32. Zanini G, Selleri V, Roncati L, Coppi F, Nasi M, Farinetti A, et al. Vascular “Long COVID”: A new vessel disease? Angiology. (2024) 75:8–14. doi: 10.1177/00033197231153204
33. Perico L, Benigni A, and Remuzzi G. SARS-CoV-2 and the spike protein in endotheliopathy. Trends Microbiol. (2024) 32:53–67. doi: 10.1016/j.tim.2023.06.004
34. Levi M and Van Der Poll T. Coagulation and sepsis. Thromb Res. (2017) 149:38–44. doi: 10.1016/j.thromres.2016.11.007
35. Schmitt FCF, Manolov V, Morgenstern J, Fleming T, Heitmeier S, Uhle F, et al. Acute fibrinolysis shutdown occurs early in septic shock and is associated with increased morbidity and mortality: results of an observational pilot study. Ann Intensive Care. (2019) 9:19. doi: 10.1186/s13613-019-0499-6
36. Marchetti M. COVID-19-driven endothelial damage: complement, HIF-1, and ABL2 are potential pathways of damage and targets for cure. Ann Hematol. (2020) 99:1701–7. doi: 10.1007/s00277-020-04138-8
37. Vieceli Dalla Sega F, Fortini F, Licastro D, Monego SD, Degasperi M, Ascierto A, et al. Serum from COVID-19 patients promotes endothelial cell dysfunction through protease-activated receptor 2. Inflammation Res. (2024) 73:117–30. doi: 10.1007/s00011-023-01823-y
38. Mohseni Afshar Z, Tavakoli Pirzaman A, Hosseinzadeh R, Babazadeh A, Taghizadeh Moghadam MA, Miri SR, et al. Anticoagulant therapy in COVID-19: A narrative review. Clin Transl Sci. (2023) 16:1510–25. doi: 10.1111/cts.13569
39. Cervia-Hasler C, Brüningk SC, Hoch T, Fan B, Muzio G, Thompson RC, et al. Persistent complement dysregulation with signs of thromboinflammation in active Long Covid. Science. (2024) 383:eadg7942. doi: 10.1126/science.adg7942
40. Sastry S, Cuomo F, and Muthusamy J. COVID-19 and thrombosis: The role of hemodynamics. Thromb Res. (2022) 212:51–7. doi: 10.1016/j.thromres.2022.02.016
41. Chen AT, Wang CY, Zhu WL, and Chen W. Coagulation disorders and thrombosis in COVID-19 patients and a possible mechanism involving endothelial cells: A review. Aging Dis. (2022) 13:144–56. doi: 10.14336/AD.2021.0704
42. Nicolai L, Kaiser R, and Stark K. Thromboinflammation in long COVID-the elusive key to postinfection sequelae? J Thromb Haemost. (2023) 21:2020–31. doi: 10.1016/j.jtha.2023.04.039
43. Fogarty H, Ward SE, Townsend L, Karampini E, Elliott S, Conlon N, et al. Sustained VWF-ADAMTS-13 axis imbalance and endotheliopathy in long COVID syndrome is related to immune dysfunction. J Thromb Haemost. (2022) 20:2429–38. doi: 10.1111/jth.15830
44. Chan CK and Vanhoutte PM. Hypoxia, vascular smooth muscles and endothelium. Acta Pharm Sin B. (2013) 3:1–7. doi: 10.1016/j.apsb.2012.12.007
45. Gupta N, Zhao YY, and Evans CE. The stimulation of thrombosis by hypoxia. Thromb Res. (2019) 181:77–83. doi: 10.1016/j.thromres.2019.07.013
46. Khan MA, Shamma T, Kazmi S, Altuhami A, Ahmed HA, Assiri AM, et al. Hypoxia-induced complement dysregulation is associated with microvascular impairments in mouse tracheal transplants. J Transl Med. (2020) 18:147. doi: 10.1186/s12967-020-02305-z
47. Ramiro S, Mostard RLM, Magro-Checa C, Van Dongen CMP, Dormans T, Buijs J, et al. Historically controlled comparison of glucocorticoids with or without tocilizumab versus supportive care only in patients with COVID-19-associated cytokine storm syndrome: results of the CHIC study. Ann Rheum Dis. (2020) 79:1143–51. doi: 10.1136/annrheumdis-2020-218479
48. Solomon A, Rosen N, Treister G, and Bartov E. Intravitreal suture: a complication of pterygium surgery. Ophthalmic Surg. (1991) 22:56–7. doi: 10.3928/1542-8877-19910101-16
49. Stasi C, Fallani S, Voller F, and Silvestri C. Treatment for COVID-19: an overview. Eur J Pharmacol. (2020) 889:173644. doi: 10.1016/j.ejphar.2020.173644
50. Savla SR, Prabhavalkar KS, and Bhatt LK. Cytokine storm associated coagulation complications in COVID-19 patients: Pathogenesis and Management. Expert Rev Anti Infect Ther. (2021) 19:1397–413. doi: 10.1080/14787210.2021.1915129
51. Avdonin PP, Blinova MS, Serkova AA, Komleva LA, and Avdonin PV. Immunity and coagulation in COVID-19. Int J Mol Sci. (2024) 25:11267. doi: 10.3390/ijms252011267
52. Soy M, Keser G, Atagunduz P, Tabak F, Atagunduz I, and Kayhan S. Cytokine storm in COVID-19: pathogenesis and overview of anti-inflammatory agents used in treatment. Clin Rheumatol. (2020) 39:2085–94. doi: 10.1007/s10067-020-05190-5
53. Charles J and Ploplis VA. COVID-19 induces cytokine storm and dysfunctional hemostasis. Curr Drug Targets. (2022) 23:1603–10. doi: 10.2174/1389450124666221025102929
54. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. (2020) 27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009
55. Gupta V, Acharya S, and Keerti A. Common coagulopathies associated with COVID-19 patients. Cureus. (2023) 15:e38067. doi: 10.7759/cureus.38067
56. Szilveszter M, Pal S, Simon-Szabo Z, Akacsos-Szasz OZ, Moldovan M, Reger B, et al. The management of COVID-19-related coagulopathy: A focus on the challenges of metabolic and vascular diseases. Int J Mol Sci. (2023) 24:12782. doi: 10.3390/ijms241612782
57. Teodoro AGF, Rodrigues WF, Farnesi-De-Assuncao TS, Borges A, Obata MMS, Neto J, et al. Inflammatory response and activation of coagulation after COVID-19 infection. Viruses. (2023) 15:938. doi: 10.3390/v15040938
58. Potere N, Garrad E, Kanthi Y, Di Nisio M, Kaplanski G, Bonaventura A, et al. NLRP3 inflammasome and interleukin-1 contributions to COVID-19-associated coagulopathy and immunothrombosis. Cardiovasc Res. (2023) 119:2046–60. doi: 10.1093/cvr/cvad084
59. Conway EM and Pryzdial ELG. Complement contributions to COVID-19. Curr Opin Hematol. (2022) 29:259–65. doi: 10.1097/MOH.0000000000000724
60. Hendaus MA and Jomha FA. From COVID-19 to clot: the involvement of the complement system. J Biomol Struct Dyn. (2022) 40:1909–14. doi: 10.1080/07391102.2020.1832919
61. Eltayeb A, Adilović M, Golzardi M, Hromić-Jahjefendić A, Rubio-Casillas A, Uversky VN, et al. Intrinsic factors behind long COVID: exploring the role of nucleocapsid protein in thrombosis. PeerJ. (2025) 13:e19429. doi: 10.7717/peerj.19429
62. Ge X, Yu Z, Guo X, Li L, Ye L, Ye M, et al. Complement and complement regulatory proteins are upregulated in lungs of COVID-19 patients. Pathol Res Pract. (2023) 247:154519. doi: 10.1016/j.prp.2023.154519
63. Gianni P, Goldin M, Ngu S, Zafeiropoulos S, Geropoulos G, and Giannis D. Complement-mediated microvascular injury and thrombosis in the pathogenesis of severe COVID-19: A review. World J Exp Med. (2022) 12:53–67. doi: 10.5493/wjem.v12.i4.53
64. Yu J, Yuan X, Chen H, Chaturvedi S, Braunstein EM, and Brodsky RA. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood. (2020) 136:2080–9. doi: 10.1182/blood.2020008248
65. Murad D, Paracha RZ, and Nisar M. Unravelling the impact of SARS-CoV-2 on hemostatic and complement systems: a systems immunology perspective. Front Immunol. (2024) 15:1457324. doi: 10.3389/fimmu.2024.1457324
67. Cesta MC, Zippoli M, Marsiglia C, Gavioli EM, Cremonesi G, Khan A, et al. Neutrophil activation and neutrophil extracellular traps (NETs) in COVID-19 ARDS and immunothrombosis. Eur J Immunol. (2023) 53:e2250010. doi: 10.1002/eji.202250010
68. Afzali B, Noris M, Lambrecht BN, and Kemper C. The state of complement in COVID-19. Nat Rev Immunol. (2022) 22:77–84. doi: 10.1038/s41577-021-00665-1
69. Torabizadeh M, Modaresi Asfeh H, Deris Zayeri Z, Dominique C, Kazemi H, and Saki N. Implications of complement imbalance in COVID-19: A molecular mechanistic discussion on the importance of complement balance. Iran J Immunol. (2023) 3:247–61. doi: 10.22034/iji.2023.97585.2522
70. Mayadas TN, Cullere X, and Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathology: Mech Dis. (2014) 9:181–218. doi: 10.1146/annurev-pathol-020712-164023
71. Enochs C, Colpo GD, Couture L, Baskin L, Cahuiche AE, Lee EA, et al. The contribution of neutrophil extracellular traps to coagulopathy in patients with COVID-19-related thrombosis. Viruses. (2024) 16:1677. doi: 10.3390/v16111677
72. Thierry AR and Salmon D. Inflammation-, immunothrombosis,- and autoimmune-feedback loops may lead to persistent neutrophil self-stimulation in long COVID. J Med Virol. (2024) 96:e29887. doi: 10.1002/jmv.29887
73. Serrano-Gonzalo I, Menéndez-Jandula B, Franco-García E, Arévalo-Vargas I, Lahoz-Gil C, Latre P, et al. Neutrophil extracellular traps and macrophage activation contibute to thrombosis and post-covid syndrome in SARS-CoV-2 infection. Front Immunol. (2025) 16:1507167. doi: 10.3389/fimmu.2025.1507167
74. Sung PS, Sun CP, Tao MH, and Hsieh SL. Inhibition of SARS-coV-2-mediated thromboinflammation by CLEC2.Fc. EMBO Mol Med. (2023) 15:e16351. doi: 10.15252/emmm.202216351
75. Xiao MT, Ellsworth CR, and Qin X. Emerging role of complement in COVID-19 and other respiratory virus diseases. Cell Mol Life Sci. (2024) 81:94. doi: 10.1007/s00018-024-05157-8
76. Ibrahim N, Eilenberg W, Neumayer C, and Brostjan C. Neutrophil extracellular traps in cardiovascular and aortic disease: A narrative review on molecular mechanisms and therapeutic targeting. Int J Mol Sci. (2024) 25:3983. doi: 10.3390/ijms25073983
77. Kaiser R, Leunig A, Pekayvaz K, Popp O, Joppich M, Polewka V, et al. Self-sustaining IL-8 loops drive a prothrombotic neutrophil phenotype in severe COVID-19. JCI Insight. (2021) 6:e150862. doi: 10.1172/jci.insight.150862
78. Middleton EA, He X-Y, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. (2020) 136:1169–79. doi: 10.1182/blood.2020007008
79. Li J, Zhang K, Zhang Y, Gu Z, and Huang C. Neutrophils in COVID-19: recent insights and advances. Virol J. (2023) 20:169. doi: 10.1186/s12985-023-02116-w
80. Sayyadi M, Hassani S, Shams M, and Dorgalaleh A. Status of major hemostatic components in the setting of COVID-19: the effect on endothelium, platelets, coagulation factors, fibrinolytic system, and complement. Ann Hematol. (2023) 102:1307–22. doi: 10.1007/s00277-023-05234-1
81. Sciaudone A, Corkrey H, Humphries F, and Koupenova M. Platelets and SARS-coV-2 during COVID-19: immunity, thrombosis, and beyond. Circ Res. (2023) 132:1272–89. doi: 10.1161/CIRCRESAHA.122.321930
82. He S, Blomback M, and Wallen H. COVID-19: Not a thrombotic disease but a thromboinflammatory disease. Ups J Med Sci. (2024) 129:e9863. doi: 10.48101/ujms.v129.9863
83. Taus F, Salvagno G, Cane S, Fava C, Mazzaferri F, Carrara E, et al. Platelets promote thromboinflammation in SARS-coV-2 pneumonia. Arterioscler Thromb Vasc Biol. (2020) 40:2975–89. doi: 10.1161/ATVBAHA.120.315175
84. Schieppati F. Post-COVID-19 thrombotic sequelae: The potential role of persistent platelet hyperactivity. Br J Haematol. (2024) 204:383–5. doi: 10.1111/bjh.19159
85. Nara N, Shimizu M, Yamamoto M, Nakamizo T, Hayakawa A, and Johkura K. Prolonged platelet hyperactivity after COVID-19 infection. Br J Haematol. (2024) 204:492–6. doi: 10.1111/bjh.19125
86. Theofilis P, Sagris M, Antonopoulos AS, Oikonomou E, Tsioufis C, and Tousoulis D. Inflammatory mediators of platelet activation: focus on atherosclerosis and COVID-19. International Journal of Molecular Sciences. (2021) 22:11170. doi: 10.3390/ijms222011170
87. Comer SP, Cullivan S, Szklanna PB, Weiss L, Cullen S, Kelliher S, et al. COVID-19 induces a hyperactive phenotype in circulating platelets. PloS Biol. (2021) 19:e3001109. doi: 10.1371/journal.pbio.3001109
88. Manne BK, Munzer P, Badolia R, Walker-Allgaier B, Campbell RA, Middleton E, et al. PDK1 governs thromboxane generation and thrombosis in platelets by regulating activation of Raf1 in the MAPK pathway. J Thromb Haemost. (2018) 16:1211–25. doi: 10.1111/jth.14005
89. Althaus K, Zlamal J, and Bakchoul T. Antibody-mediated platelet activation in COVID-19: A coincidence or a new mechanism of the dysregulated coagulation system? J Thromb Haemost. (2021) 19:1171–3. doi: 10.1111/jth.15275
90. Jevtic SD and Nazy I. The COVID complex: A review of platelet activation and immune complexes in COVID-19. Front Immunol. (2022) 13:807934. doi: 10.3389/fimmu.2022.807934
91. Petry J, Shoykhet M, Weiser T, Griesbaum L, Bashiri Dezfouli A, Verschoor A, et al. SARS-CoV-2 S1 protein induces IgG-mediated platelet activation and is prevented by 1.8-cineole. BioMed Pharmacother. (2025) 187:118100. doi: 10.1016/j.biopha.2025.118100
92. Millington-Burgess SL and Harper MT. A double-edged sword: antibody-mediated procoagulant platelets in COVID-19. Platelets. (2021) 32:579–81. doi: 10.1080/09537104.2021.1912315
93. Althaus K, Marini I, Zlamal J, Pelzl L, Singh A, Häberle H, et al. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood. (2021) 137:1061–71. doi: 10.1182/blood.2020008762
94. Pelzl L, Singh A, Funk J, Witzemann A, Marini I, Zlamal J, et al. Antibody-mediated procoagulant platelet formation in COVID-19 is AKT dependent. J Thromb Haemost. (2022) 20:387–98. doi: 10.1111/jth.15587
95. Becker RC, Sexton T, and Smyth S. COVID-19 and biomarkers of thrombosis: focus on von Willebrand factor and extracellular vesicles. J Thromb Thrombolysis. (2021) 52:1010–9. doi: 10.1007/s11239-021-02544-x
96. Schiavello M, Vizio B, Bosco O, Pivetta E, Mariano F, Montrucchio G, et al. Extracellular vesicles: new players in the mechanisms of sepsis- and COVID-19-related thromboinflammation. Int J Mol Sci. (2023) 24:1920. doi: 10.3390/ijms24031920
97. Qian H, Zang R, Zhang R, Zheng G, Qiu G, Meng J, et al. Circulating extracellular vesicles from severe COVID-19 patients induce lung inflammation. mSphere. (2024) 9:e0076424. doi: 10.1128/msphere.00764-24
98. Rosell A, Havervall S, Von Meijenfeldt F, Hisada Y, Aguilera K, Grover SP, et al. Patients with COVID-19 have elevated levels of circulating extracellular vesicle tissue factor activity that is associated with severity and mortality—Brief report. Arteriosclerosis Thrombosis Vasc Biol. (2021) 41:878–82. doi: 10.1161/ATVBAHA.120.315547
99. Guervilly C, Bonifay A, Burtey S, Sabatier F, Cauchois R, Abdili E, et al. Dissemination of extreme levels of extracellular vesicles: tissue factor activity in patients with severe COVID-19. Blood Adv. (2021) 5:628–34. doi: 10.1182/bloodadvances.2020003308
100. Tripisciano C, Weiss R, Eichhorn T, Spittler A, Heuser T, Fischer MB, et al. Different potential of extracellular vesicles to support thrombin generation: contributions of phosphatidylserine, tissue factor, and cellular origin. Sci Rep. (2017) 7:6522. doi: 10.1038/s41598-017-03262-2
101. Weiss R, Mostageer M, Eichhorn T, Huber S, Egger D, Spittler A, et al. The fluorochrome-to-protein ratio is crucial for the flow cytometric detection of tissue factor on extracellular vesicles. Sci Rep. (2024) 14:6419. doi: 10.1038/s41598-024-56841-5
102. Cappellano G, Raineri D, Rolla R, Giordano M, Puricelli C, Vilardo B, et al. Circulating platelet-derived extracellular vesicles are a hallmark of sars-cov-2 infection. Circulation. (2021) 10:85. doi: 10.3390/cells10010085
103. Guglielmetti G, Quaglia M, Sainaghi PP, Castello LM, Vaschetto R, Pirisi M, et al. War to the knife” against thromboinflammation to protect endothelial function of COVID-19 patients. Crit Care. (2020) 24:365. doi: 10.1186/s13054-020-03060-9
104. Iba T, Levy JH, Maier CL, Connors JM, and Levi M. Four years into the pandemic, managing COVID-19 patients with acute coagulopathy: what have we learned? J Thromb Haemost. (2024) 22:1541–9. doi: 10.1016/j.jtha.2024.02.013
105. Sochet AA, Morrison JM, Jaffray J, Godiwala N, Wilson HP, Thornburg CD, et al. Enoxaparin thromboprophylaxis in children hospitalized for COVID-19: A phase 2 trial. Pediatrics. (2022) 150:e2022056726. doi: 10.1542/peds.2022-056726
106. Hess CN, Hsia J, Carroll IA, Nehler MR, Ruf W, Morrow DA, et al. Novel tissue factor inhibition for thromboprophylaxis in COVID-19: primary results of the ASPEN-COVID-19 trial. Arterioscler Thromb Vasc Biol. (2023) 43:1572–82. doi: 10.1161/ATVBAHA.122.318748
107. Stone GW, Farkouh ME, Lala A, Tinuoye E, Dressler O, Moreno PR, et al. Randomized trial of anticoagulation strategies for noncritically ill patients hospitalized with COVID-19. J Am Coll Cardiol. (2023) 81:1747–62. doi: 10.1016/j.jacc.2023.02.041
108. Lopes RD, De Barros ESPGM, Furtado RHM, Macedo AVS, Bronhara B, Damiani LP, et al. Therapeutic versus prophylactic anticoagulation for patients admitted to hospital with COVID-19 and elevated D-dimer concentration (ACTION): an open-label, multicentre, randomised, controlled trial. Lancet. (2021) 397:2253–63. doi: 10.1016/S0140-6736(21)01203-4
109. Sholzberg M, Tang GH, Rahhal H, Alhamzah M, Kreuziger LB, Áinle FN, et al. Effectiveness of therapeutic heparin versus prophylactic heparin on death, mechanical ventilation, or intensive care unit admission in moderately ill patients with covid-19 admitted to hospital: RAPID randomised clinical trial. Bmj. (2021) 375:n2400. doi: 10.1136/bmj.n2400
110. Investigators A, Investigators AC-A, Investigators R-C, Lawler PR, Goligher EC, Berger JS, et al. Therapeutic anticoagulation with heparin in noncritically ill patients with covid-19. N Engl J Med. (2021) 385:790–802. doi: 10.1056/NEJMoa2105911
111. Investigators R-C, Investigators AC-A, Investigators A, Goligher EC, Bradbury CA, Mcverry BJ, et al. Therapeutic anticoagulation with heparin in critically ill patients with covid-19. N Engl J Med. (2021) 385:777–89. doi: 10.1056/NEJMoa2103417
112. Bohula EA, Berg DD, Lopes MS, Connors JM, Babar I, Barnett CF, et al. Anticoagulation and antiplatelet therapy for prevention of venous and arterial thrombotic events in critically ill patients with COVID-19: COVID-PACT. Circulation. (2022) 146:1344–56. doi: 10.1161/CIRCULATIONAHA.122.061533
113. Spyropoulos AC, Goldin M, Giannis D, Diab W, Wang J, Khanijo S, et al. Efficacy and safety of therapeutic-dose heparin vs standard prophylactic or intermediate-dose heparins for thromboprophylaxis in high-risk hospitalized patients with COVID-19: the HEP-COVID randomized clinical trial. JAMA Intern Med. (2021) 181:1612–20. doi: 10.1001/jamainternmed.2021.6203
114. Lemos ACB, Do Espírito Santo DA, Salvetti MC, Gilio RN, Agra LB, Pazin-Filho A, et al. Therapeutic versus prophylactic anticoagulation for severe COVID-19: A randomized phase II clinical trial (HESACOVID). Thromb Res. (2020) 196:359–66. doi: 10.1016/j.thromres.2020.09.026
115. Perepu US, Chambers I, Wahab A, Ten Eyck P, Wu C, Dayal S, et al. Standard prophylactic versus intermediate dose enoxaparin in adults with severe COVID-19: A multi-center, open-label, randomized controlled trial. J Thromb Haemost. (2021) 19:2225–34. doi: 10.1111/jth.15450
116. Bikdeli B, Talasaz AH, Rashidi F, Bakhshandeh H, Rafiee F, Rezaeifar P, et al. Intermediate-dose versus standard-dose prophylactic anticoagulation in patients with COVID-19 admitted to the intensive care unit: 90-day results from the INSPIRATION randomized trial. Thromb Haemost. (2022) 122:131–41. doi: 10.1055/a-1485-2372
117. Voci D, Gotschi A, Held U, Bingisser R, Colucci G, Duerschmied D, et al. Enoxaparin for outpatients with COVID-19: 90-day results from the randomised, open-label, parallel-group, multinational, phase III OVID trial. Thromb Res. (2023) 221:157–63. doi: 10.1016/j.thromres.2022.10.021
118. Barco S, Voci D, Held U, Sebastian T, Bingisser R, Colucci G, et al. Enoxaparin for primary thromboprophylaxis in symptomatic outpatients with COVID-19 (OVID): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet Haematol. (2022) 9:e585–93. doi: 10.1016/S2352-3026(22)00175-2
119. Dai MF, Xin WX, Kong S, Ding HY, and Fang L. Effectiveness and safety of extended thromboprophylaxis in post-discharge patients with COVID-19: A systematic review and meta-analysis. Thromb Res. (2023) 221:105–12. doi: 10.1016/j.thromres.2022.11.019
120. Lee HJ, Jang HJ, Choi WI, Joh J, Kim J, Park J, et al. Comparison of safety and efficacy between therapeutic or intermediate versus prophylactic anticoagulation for thrombosis in COVID-19 patients: a systematic review and meta-analysis. Acute Crit Care. (2023) 38:160–71. doi: 10.4266/acc.2022.01424
121. Vedovati MC, Graziani M, Agnelli G, and Becattini C. Efficacy and safety of two heparin regimens for prevention of venous thromboembolism in hospitalized patients with COVID-19: a meta-analysis. Intern Emerg Med. (2023) 18:863–77. doi: 10.1007/s11739-022-03159-7
122. Barnes GD, Burnett A, Allen A, Ansell J, Blumenstein M, Clark NP, et al. Thromboembolic prevention and anticoagulant therapy during the COVID-19 pandemic: updated clinical guidance from the anticoagulation forum. J Thromb Thrombolysis. (2022) 54:197–210. doi: 10.1007/s11239-022-02643-3
123. Schulman S, Arnold DM, Bradbury CA, Broxmeyer L, Connors JM, Falanga A, et al. 2023 ISTH update of the 2022 ISTH guidelines for antithrombotic treatment in COVID-19. J Thromb Haemost. (2024) 22:1779–97. doi: 10.1016/j.jtha.2024.02.011
124. Society, C. T and Physicians C. A. O. C. Guidelines for diagnosis, prevention and treatment of COVID-19 in adults in China. Chin Med J. (2021) 18:1293–356. doi: 10.3760/cma.j.cn112137-20210112-00090
125. Cuker A, Tseng EK, Nieuwlaat R, Angchaisuksiri P, Blair C, Dane K, et al. American Society of Hematology 2021 guidelines on the use of anticoagulation for thromboprophylaxis in patients with COVID-19. Blood Adv. (2021) 5:872–88. doi: 10.1182/bloodadvances.2020003763
126. Final coronavirus disease (COVID-19) treatment guidelines. Bethesda (MD): National Institutes of Health (2024).
127. Moores LK, Tritschler T, Brosnahan S, Carrier M, Collen JF, Doerschug K, et al. Thromboprophylaxis in patients with COVID-19: A brief update to the CHEST guideline and expert panel report. Chest. (2022) 162:213–25. doi: 10.1016/j.chest.2022.02.006
128. Moores LK, Tritschler T, Brosnahan S, Carrier M, Collen JF, Doerschug K, et al. Prevention, diagnosis, and treatment of VTE in patients with coronavirus disease 2019: CHEST guideline and expert panel report. Chest. (2020) 158:1143–63. doi: 10.1016/j.chest.2020.05.559
129. Working Group on Guidelines for Thromboprophylaxis and Management of Anticoagulation in Hospitalized Patients with COVID-19, Pulmonary Embolism & Pulmonary Vascular Diseases Group of the Chinese Thoracic Society, Pulmonary Embolism & Pulmonary Vascular Disease Working Committee of Chinese Association of Chest Physicians, National Cooperation Group on Prevention & Treatment of Pulmonary Embolism & Pulmonary Vascular Disease, and National Program Office for Prevention Treatment of Pulmonary Embolism & Deep Vein Thrombosis, China Grade Center. Practice guidelines of thromboprophylaxis and management of anticoagulation in hospitalized patients with COVID-19. Chin Med J. (2023) 103:1–23. doi: 10.3760/cma.j.cn112137-2023-01-20-00115
130. Ballering AV, Van Zon SKR, Olde Hartman TC, Rosmalen JGM, and Lifelines Corona Research, I. Persistence of somatic symptoms after COVID-19 in the Netherlands: an observational cohort study. Lancet. (2022) 400:452–61. doi: 10.1016/S0140-6736(22)01214-4
131. Greenhalgh T, Sivan M, Perlowski A, and Nikolich J. Long COVID: a clinical update. Lancet. (2024) 404:707–24. doi: 10.1016/S0140-6736(24)01136-X
132. Xie Y, Xu E, Bowe B, and Al-Aly Z. Long-term cardiovascular outcomes of COVID-19. Nat Med. (2022) 28:583–90. doi: 10.1038/s41591-022-01689-3
133. Soriano JB, Murthy S, Marshall JC, Relan P, and Diaz JV. A clinical case definition of post-COVID-19 condition by a Delphi consensus. Lancet Infect Dis. (2022) 22:e102–7. doi: 10.1016/S1473-3099(21)00703-9
134. Guzik TJ, Mohiddin SA, Dimarco A, Patel V, Savvatis K, Marelli-Berg FM, et al. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res. (2020) 116:1666–87. doi: 10.1093/cvr/cvaa106
135. Zhu Y, Chen X, and Liu X. NETosis and neutrophil extracellular traps in COVID-19: immunothrombosis and beyond. Front Immunol. (2022) 13:838011. doi: 10.3389/fimmu.2022.838011
136. Semo D, Shomanova Z, Sindermann J, Mohr M, Evers G, Motloch LJ, et al. Persistent monocytic bioenergetic impairment and mitochondrial DNA damage in PASC patients with cardiovascular complications. Int J Mol Sci. (2025) 26:4562. doi: 10.3390/ijms26104562
137. Weinberg SE, Sena LA, and Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. (2015) 42:406–17. doi: 10.1016/j.immuni.2015.02.002
138. Ajaz S, Mcphail MJ, Singh KK, Mujib S, Trovato FM, Napoli S, et al. Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. American Journal of Physiology-cell Physiology. (2021) 320:C57–65. doi: 10.1152/ajpcell.00426.2020
139. Ravi S, Mitchell T, Kramer PA, Chacko B, and Darley-Usmar VM. Mitochondria in monocytes and macrophages-implications for translational and basic research. Int J Biochem Cell Biol. (2014) 53:202–7. doi: 10.1016/j.biocel.2014.05.019
140. Delgado-Roche L and Mesta F. Oxidative stress as key player in severe acute respiratory syndrome coronavirus (SARS-coV) infection. Arch Med Res. (2020) 51:384–7. doi: 10.1016/j.arcmed.2020.04.019
141. Beltrán-García J, Osca-Verdegal R, Pallardó FV, Ferreres J, Rodríguez M, Mulet S, et al. Oxidative stress and inflammation in COVID-19-associated sepsis: the potential role of anti-oxidant therapy in avoiding disease progression. Antioxidants (2020) 9:936. doi: 10.3390/antiox9100936
142. Ayyoub S, Dhillon NK, and Tura-Ceide O. Genetics of long COVID: exploring the molecular drivers of persistent pulmonary vascular disease symptoms. Infect Dis Rep. (2025) 17:15. doi: 10.3390/idr17010015
143. Moness H, Mousa SO, Mousa SO, Adel NM, Ibrahim RA, Hassan EE, et al. Thrombophilia genetic mutations and their relation to disease severity among patients with COVID-19. PloS One. (2024) 19:e0296668. doi: 10.1371/journal.pone.0296668
144. Roach REJ, Roshani S, Meijer K, Hamulyák K, Lijfering WM, Prins MH, et al. Risk of cardiovascular disease in double heterozygous carriers and homozygous carriers of F5 R506Q (factor V Leiden) and F2 (prothrombin) G20210A: a retrospective family cohort study. British Journal of Haematology. (2011) 153:134–6. doi: 10.1111/j.1365-2141.2010.08529.x
145. Kim Y, Kim SE, Kim T, Yun KW, Lee SH, Lee E, et al. Preliminary guidelines for the clinical evaluation and management of long COVID. Infect Chemother. (2022) 54:566–97. doi: 10.3947/ic.2022.0141
146. Seo JW, Kim SE, Kim Y, Kim EJ, Kim T, Kim T, et al. Updated clinical practice guidelines for the diagnosis and management of long COVID. Infect Chemother. (2024) 56:122–57. doi: 10.3947/ic.2024.0024
147. Tan ST, Park HJ, Rodríguez-Barraquer I, Rutherford GW, Bibbins-Domingo K, Schechter R, et al. COVID-19 vaccination and estimated public health impact in california. JAMA Netw Open. (2022) 5:e228526. doi: 10.1001/jamanetworkopen.2022.8526
148. Tenforde MW, Olson SM, Self WH, Talbot HK, Lindsell CJ, Steingrub JS, et al. Effectiveness of pfizer-bioNTech and moderna vaccines against COVID-19 among hospitalized adults aged ≥65 years - United States, january-march 2021. MMWR Morb Mortal Wkly Rep. (2021) 70:674–9. doi: 10.15585/mmwr.mm7018e1
149. Engelen MM, Vandenbriele C, Balthazar T, Claeys E, Gunst J, Guler I, et al. Venous thromboembolism in patients discharged after COVID-19 hospitalization. Semin Thromb Hemost. (2021) 47:362–71. doi: 10.1055/s-0041-1727284
150. Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. (2020) 191:145–7. doi: 10.1016/j.thromres.2020.04.013
151. Sholzberg M, Tang GH, Negri E, Rahhal H, Kreuziger LB, Pompilio CE, et al. Coagulopathy of hospitalised COVID-19: A Pragmatic Randomised Controlled Trial of Therapeutic Anticoagulation versus Standard Care as a Rapid Response to the COVID-19 Pandemic (RAPID COVID COAG - RAPID Trial): A structured summary of a study protocol for a randomised controlled trial. Trials. (2021) 22:202. doi: 10.1186/s13063-021-05076-0
152. Houghton DE, Wysokinski W, Casanegra AI, Padrnos LJ, Shah S, Wysokinska E, et al. Risk of venous thromboembolism after COVID-19 vaccination. Journal of Thrombosis And Haemostasis. (2022) 20:1638–44. doi: 10.1111/jth.15725
153. Laverdure E, Sperlich C, and Fox S. Refractory immune TTP following Pfizer-BioNTech COVID-19 vaccine successfully salvaged with caplacizumab. J Thromb Haemost. (2022) 20:1696–8. doi: 10.1111/jth.15751
154. Schultz NH, Sørvoll IH, Michelsen AE, Munthe LA, Lund-Johansen F, Ahlen MT, et al. Thrombosis and Thrombocytopenia after ChAdOx1 nCoV-19 Vaccination. New England Journal of Medicine. (2021) 384:2124–30. doi: 10.1056/NEJMoa2104882
155. Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, and Eichinger S. Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. N Engl J Med. (2021) 384:2092–101. doi: 10.1056/NEJMoa2104840
156. Cines DB and Bussel JB. SARS-coV-2 vaccine-induced immune thrombotic thrombocytopenia. N Engl J Med. (2021) 384:2254–6. doi: 10.1056/NEJMe2106315
157. Scully M, Singh D, Lown R, Poles A, Solomon T, Levi M, et al. Pathologic Antibodies to Platelet Factor 4 after ChAdOx1 nCoV-19 Vaccination. N Engl J Med. (2021) 384:2202–11. doi: 10.1056/NEJMoa2105385
158. Ramacciotti E, Barile Agati L, Calderaro D, Aguiar VCR, Spyropoulos AC, De Oliveira CCC, et al. Rivaroxaban versus no anticoagulation for post-discharge thromboprophylaxis after hospitalisation for COVID-19 (MICHELLE): an open-label, multicentre, randomised, controlled trial. Lancet. (2022) 399:50–9. doi: 10.1016/S0140-6736(21)02392-8
159. Kang W, Peng K, Yan VKC, Al-Badriyeh D, Lee SF, Yiu HHE, et al. Direct oral anticoagulants versus low-molecular-weight heparin in patients with cancer-associated venous thrombosis: a cost-effectiveness analysis. J Pharm Policy Pract. (2024) 17:2375269. doi: 10.1080/20523211.2024.2375269
160. Pavord S, Scully M, Hunt BJ, Lester W, Bagot C, Craven B, et al. Clinical features of vaccine-induced immune thrombocytopenia and thrombosis. N Engl J Med. (2021) 385:1680–9. doi: 10.1056/NEJMoa2109908
161. Chevassut T, Hunt BJ, and Pavord S. VITT, COVID-19 and the Expert Haematology Panel: The story of how the UK responded to emerging cases of vaccine-induced immune thrombocytopenia and thrombosis during the vaccination programme. Clin Med (Lond). (2021) 21:e600–2. doi: 10.7861/clinmed.2021-0488
162. Ishiguro T, Matsuo K, Fujii S, and Takayanagi N. Acute thrombotic vascular events complicating influenza-associated pneumonia. Respir Med Case Rep. (2019) 28:100884. doi: 10.1016/j.rmcr.2019.100884
163. Rubino R, Imburgia C, Bonura S, Trizzino M, Iaria C, and Cascio A. Thromboembolic events in patients with influenza: A scoping review. Viruses. (2022) 14:2817. doi: 10.3390/v14122817
164. Wong RSM, Wu A, To KF, Lee N, Lam CWK, Wong CK, et al. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. Bmj-british Medical Journal. (2003) 326:1358–62. doi: 10.1136/bmj.326.7403.1358
165. Smilowitz NR, Subashchandran V, Yuriditsky E, Horowitz JM, Reynolds HR, Hochman JS, et al. Thrombosis in hospitalized patients with viral respiratory infections versus COVID-19. Am Heart J. (2021) 231:93–5. doi: 10.1016/j.ahj.2020.10.075
166. Pacheco-Hernández LM, Ramírez-Noyola JA, Gómez-García IA, Ignacio-Cortés S, Zúñiga J, and Choreño-Parra JA. Comparing the cytokine storms of COVID-19 and pandemic influenza. J Interferon Cytokine Res. (2022) 42:369–92. doi: 10.1089/jir.2022.0029
167. Mackman N, Grover SP, and Antoniak S. Tissue factor expression, extracellular vesicles, and thrombosis after infection with the respiratory viruses influenza A virus and coronavirus. J Thromb Haemost. (2021) 19:2652–8. doi: 10.1111/jth.15509
168. Hippensteel JA, Lariviere WB, Colbert JF, Langouët-Astrié CJ, and Schmidt EP. Heparin as a therapy for COVID-19: current evidence and future possibilities. American Journal of Physiology-lung Cellular And Molecular Physiology. (2020) 319:L211–7. doi: 10.1152/ajplung.00199.2020
169. Santoro F, Núñez-Gil IJ, Vitale E, Viana-Llamas MC, Romero R, Maroun Eid C, et al. Aspirin therapy on prophylactic anticoagulation for patients hospitalized with COVID-19: A propensity score-matched cohort analysis of the HOPE-COVID-19 registry. Journal of the American Heart Association. (2022) 11:. doi: 10.1093/eurheartj/ehac544.2715
170. Galland J, Thoreau B, Delrue M, Neuwirth M, Stepanian A, Chauvin A, et al. White blood count, D-dimers, and ferritin levels as predictive factors of pulmonary embolism suspected upon admission in noncritically ill COVID-19 patients: The French multicenter CLOTVID retrospective study. Eur J Haematol. (2021) 107:190–201. doi: 10.1111/ejh.13638
171. Kell DB, Laubscher GJ, and Pretorius E. A central role for amyloid fibrin microclots in long COVID/PASC: origins and therapeutic implications. Biochem J. (2022) 479:537–59. doi: 10.1042/BCJ20220016
Keywords: COVID-19, thrombosis, mechanism, anticoagulation, vaccine
Citation: Yin Q, Huang Y, Wang H, Wang Y, Huang X, Song Y, Wang Y, Han L and Yuan B (2025) COVID-19: a vascular nightmare unfolding. Front. Immunol. 16:1593885. doi: 10.3389/fimmu.2025.1593885
Received: 17 March 2025; Accepted: 10 July 2025;
Published: 01 August 2025.
Edited by:
Patricia Pia Wadowski, Medical University of Vienna, AustriaReviewed by:
Priya Ranjan, Centers for Disease Control and Prevention (CDC), United StatesParesh Kulkarni, Cleveland Clinic, United States
Copyright © 2025 Yin, Huang, Wang, Wang, Huang, Song, Wang, Han and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bian Yuan, Ymlhbnl1YW41NjdAMTI2LmNvbQ==; Yueyuan Wang, NTUwOTA5MjM3QHFxLmNvbQ==; Lizhu Han, eXVtbXlsaWFAMTYzLmNvbQ==
†These authors have contributed equally to this work