 Nourhen Agrebi1
Nourhen Agrebi1 Rafah Mackeh1
Rafah Mackeh1 Mohamed Alsabbagh1
Mohamed Alsabbagh1 Asha Elmi1Amnah A. Al-Marri1
Asha Elmi1Amnah A. Al-Marri1 Satanay Z. Hubrack1
Satanay Z. Hubrack1 Saleema C. Purayil2Mohammed Yousuf Karim3,4Amel Hassan4,5*
Saleema C. Purayil2Mohammed Yousuf Karim3,4Amel Hassan4,5* Bernice Lo1,6*
Bernice Lo1,6*- 1Translational Medicine Department, Research Department, Sidra Medicine, Doha, Qatar
- 2Allergy & Immunology Division, Department of Medicine, Hamad Medical Corporation, Doha, Qatar
- 3Hematology, Immunology and Transfusion Division, Sidra Medicine, Doha, Qatar
- 4College of Medicine, Qatar University, Doha, Qatar
- 5Pediatric Allergy and Immunology Department, Sidra Medicine, Doha, Qatar
- 6College of Health and Life Sciences, Hamad Bin Khalifa University, Doha, Qatar
Background: Hyper-IgM syndrome (HIGM) is a genetic immunodeficiency characterized by elevated to normal IgM levels and decreased IgG, IgA, and IgE. The overlapping clinical presentations of different gene mutations complicate diagnosis and management.
Objective: This study aims to elucidate the clinical implications of concurrent AICDA and IKBKB homozygous variants in a pediatric patient diagnosed with hyper-IgM syndrome.
Methods: We present immunological and genetic analysis of a Tunisian patient with two homozygous variants of uncertain significance (VUSs) in the IKBKB and AICDA genes, suspected of causing hyper-IgM and immune deficiency. We conducted functional tests to ascertain the pathogenicity of IKBKB and AICDA mutations and to provide a definitive diagnosis and appropriate management.
Results: Genetic analysis identified two homozygous variants: AICDA (p.W80S) and IKBKB (p.R77Q). Immunophenotyping and functional studies found greatly reduced class-switched memory B cells and somatic hypermutations but normal T cell responses and NFkB activation.
Conclusion: The simultaneous presence of multiple homozygous VUSs emphasizes a major challenge in the genetic diagnosis of highly consanguinous patients. Functional workup as well as familial segregation studies are needed to clarify variant pathogenicity and provide a definitive diagnosis and tailored treatment strategies for these patients. Our studies suggest that the AICDA p.W80S variant is pathogenic, while the IKBKB p.R77Q variant is likely benign.
1 Introduction
Hyper-IgM syndrome is a rare genetic disorder resulting from B and, in some cases, T cell defects (1). Mutations in the activation-induced cytidine deaminase (AICDA) gene lead to autosomal recessive hyper-IgM syndrome type 2 (HIGM2) due to AID deficiency (2, 3). AID plays a crucial role in immunoglobulin class switch recombination (CSR) and somatic hypermutation (SHM) (2). Affected patients often present with recurrent bacterial infections, inflammation, gastrointestinal (GI) infections, lymphoid hyperplasia, and, in some cases, autoimmune cytopenias. However, they typically do not develop opportunistic infections and respond well to human immunoglobulin replacement therapy (4, 5).
Patients with AID deficiency have normal or elevated levels of IgM with low levels of IgG, IgA, and IgE, along with an intact T-cell immunity (1, 2). However, some patients with an increase in IgM and a decrease in other immunoglobulins have been reported with defects in NF-κB pathway components (such as NEMO and IKKβ) (6, 7). For example, Nielsen and colleagues reported a patient with features resembling hyper-IgM but was discovered to have IKKβ deficiency. This report illustrated how it can be challenging to distinguish between these disorders when diagnosing patients due to overlapping clinical presentations (7).
The IKKβ subunit is one of the two catalytic subunits of the IKK complex, which plays a central role in the NF-κB signaling pathway (8). The IKKβ subunit is responsible for phosphorylating the IκB proteins, leading to their degradation and the release of NF-κB for transcriptional activation (9). Inhibition or deficiency of the IKKβ subunit in the NF-κB signaling pathway leads to impaired IκBα phosphorylation and the subsequent inhibition of NF-κB activation (10). Patients with IKKβ deficiency do not respond sufficiently to treatment involving prophylactic antibiotic therapy or human immunoglobulin replacement. They generally require urgent hematopoietic stem cell transplantation (11). It is therefore important to carefully evaluate whether a patient has IKKβ deficiency versus other Inborn Errors of Immunity (IEI).
Herein, we describe a patient of consanguineous parents suspected of having hyper-IgM syndrome. Clinical genetic testing identified homozygous variants of uncertain significance (VUS) in two separate genes, in which pathogenic mutations cause two different IEIs (AID deficiency and IKKβ deficiency). This case led to further evaluation of the patient’s immune phenotype and the functional consequences of each variant to determine which variant might underlie the clinical presentation. Overall the study highlights the importance of functional studies in clarifying the pathogenicity of variants in IEI genes.
2 Materials and methods
2.1 Human subjects
This study was approved by the Sidra Medicine ethics committee (IRB protocol 1601002512). Informed consent was obtained from all individual participants included in the study and/or their parents.
2.2 Activation of T cells with anti-CD3 and anti-CD28 antibodies
Peripheral blood mononuclear cells (PBMCs) were purified by Ficoll density gradient centrifugation. PBMCs were stimulated with 1 μg/mL of anti-CD3 and anti-CD28 antibodies. The cells were cultured 3 days in complete RPMI-1640 medium with 10% FBS at 37°C with 5% CO2. Then the cells were cultured with 100 U/mL hIL-2 in fresh media, and fresh hIL-2-containing medium was supplemented every 2 days.
2.3 Establishment of EBV-transformed lymphoblasts cell culture
PBMCs of 1-2 million in 1 ml of RPMI 1640 medium with 20% FBS were cultured with cyclosporin A and B95-8 culture supernatant. Every following week, complete RPMI 1640 media with 20% FBS was added. After 3-5 weeks, once lymphoblast proliferation was established, the media was replaced with complete RPMI 1640 media with 10% FBS every two days for continued expansion of the cells or cryopreserved for subsequent culture and expansion.
2.4 DNA sequencing
Genomic DNA from blood of the patient was submitted to Invitae and sequenced using their Primary Immunodeficiency Panel, which included 207 genes at the time of analysis. The Invitae diagnostic testing results only found two variants as homozygous. The other VUS were heterozygous and not relevant to the disease phenotype. Familial segregation analysis was conducted by Sanger sequencing of these two homozygous variants. The following primers were used (IKBKB-F 5’-gggctctgtcttcctctgtt-3’, IKBKB-R 5’-gatctgagaggcaaagcagc-3’; AICDA-F 5’-ccagagccgcaataaaagtc-3’, AICDA-R 5’-gccttgtctctgagccattc-3’). IKBKB and AICDA PCRs were carried out using Taq PCR Master Mix Kit (Qiagen) on genomic DNA obtained from activated T cells and EBV-transformed lymphoblasts, respectively.
2.5 Isotype-switched memory B cells and numbers of CD45RO-positive memory T cells
PBMCs were stained in 1X PBS 0.3% BSA with appropriate antibodies for analysis of isotype-switched memory B cells (APC-CY7 anti-CD19, PE-Cy7 anti-CD27, FITC anti-IgM, PE anti-IgD) and CD45RO-BV605-positive memory T cells (FITC anti-CD3, APC anti-CD4, APC-CY7 anti-CD8). Flow cytometry data were acquired on the NovoCyte (ACEA Biosciences) and analyzed with FlowJo version 10.
2.6 Regulatory T cells levels
Cells were stained with surface marker antibodies (PE anti-CD25, FITC anti-CD3, and AF647 anti-CD127) for 45 minutes on ice. The cells were then washed, fixed/permeabilized using the eBioscience FoxP3 Transcription Factor Staining Buffer kit (ThermoFisher Scientific) and incubated overnight with V450 anti-CD4 and PE-Cy7 anti-FoxP3 (eBioscience) antibodies. Data were analyzed by FlowJo software version 10.
2.7 T cell proliferation
PBMCs were incubated with 1 µM of CFSE, washed, and then cultured with beads conjugated to a combination of anti-CD28, -CD3, and -CD2 antibodies (T cell activation/expansion kit, Miltenyi Biotec) for 3 to 5 days. On days 3 and 5, cells underwent staining using Viability dye (LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit, ThermoFisher), V450 anti-CD4- and BV605 anti-CD8 (BD Biosciences) antibodies and analyzed on the flow cytometer.
2.8 Cytokines
Activated T cells were expanded in 100U/mL IL-2 and restimulated using 20 nM phorbol 12-myristate 13-acetate (PMA) and 1 mM ionomycin (Sigma-Aldrich) for 5 hours at 37°C. After the first hour of stimulation, Brefeldin-A (Invitrogen) was added to block cytokine secretion. After 5-hour stimulation, cells were washed and surface Fc receptors (Biolegend) were blocked for 15 min at 4°C. Cells were stained with APC-Cy7 anti-CD8, then fixed with BD Cytofix/Cytoperm (BD Bioscience) and stained with AF488 anti-CD4, PE anti-IFNγ, BV650 anti-TNFα, and APC anti-IL-2 (eBioscience).
2.9 Western blot
For AID protein expression, 2x106 EBV-transformed lymphoblasts were harvested, lysed in hot 1% SDS lysis buffer and sonicated. For IκBα and IKKβ protein analysis, 1x106 activated T cells were lysed. Both lysates were subjected to SDS-PAGE using NuPAGE 4–12% Bis-Tris Gels (Invitrogen). Primary antibodies used included AID monoclonal antibody (#4949S, Cell Signaling Technology), recombinant Anti-IKK beta antibody (ab124957, Abcam), recombinant Anti-IκBα antibody (#4814, Cell Signaling Technology) and Anti-β-Actin Rabbit Polyclonal Antibody (HRP) (VWR).
For the analysis of NFκB pathway proteins, anti-CD3/CD28 activated T cells of the patient, their two siblings, and two healthy unrelated controls (HDs) were expanded in the presence of IL-2 for at least 8 days in Advanced RPMI medium (ThermoFisher #12633012) supplemented with fetal bovine serum L-glutamine, streptomycin, and penicillin. 5x105 cells of each sample were stimulated with 20 ng/mL TNFα (Peprotech #300-01A) for the indicated times, after which the cells were washed twice with ice cold phosphate buffered saline. The samples were lysed in boiling lysis buffer containing 1% SDS 0.1 M Tris-HCl (pH 8.0) and then sonicated. The samples were resolved on 10% polyacrylamide gels at 140 V and transferred onto a PVDF membrane at 110 V for 1 hour on ice. The membrane was blocked with 5% milk in TBST and incubated with anti-phospho-p65 (Cell Signaling #3033) and anti-IκBα antibodies (Cell Signaling #4814) in 3% BSA TBST at 4°C overnight and then with goat anti-mouse (ThermoFisher #A-31553) and anti-rabbit (ThermoFisher #G-21234) peroxidase-conjugated secondary antibody (SeraCare #5220-0341) for 1 hour at room temperature. The membrane was treated with Pierce ECL Western Blotting Substrate (ThermoFisher #32106) and developed using ChemiDoc MP Imaging System (Bio-Rad).
2.10 TOPO TA cloning and sequencing of V3-23-Cμ transcripts
Total RNA was extracted from EBV-transformed lymphoblasts generated from the patient, her siblings, and unrelated healthy donors (HDs), including age-matched control. cDNA synthesis was performed as previously described (2, 12) with a Cμ A (5′-GAGGCAGCTCAGCAATC-3′) primer. PCR was performed with the following primers: V3-23 leader exon (5′-GGCTGAGCTGGCTTTTTCTTGTGG-3′) and Cμ B (5′-TCACAGGAGACGAGGGGGAA-3′) (2) (95°C, 5 min; 35 cycles *(94°C, 30sec; 68°C, 30sec; 72°C 45 sec); 72°C, 20 min). PCR products were cloned using the TOPO TA cloning kit (Invitrogen) and V3-23 positive colonies were sequenced. Following manual quality verification of the sequencing chromatograms using UGENE software to confirm single peaks and minimal read length of 292 bp, IgH sequences were aligned to germline sequence (NG_001019.6) and analyzed by Igblast using the default settings to calculate sequence similarity (13). Mutational Frequency was calculated as = 100 - (the number of mutations were divided by the number of nucleotides in the V3-23 region).
2.11 Statistical analysis
The statistical analysis was conducted using GraphPad Prism 9 with Mann-Whitney test.
3 Results
3.1 Clinical and laboratory findings
The present case is a Tunisian patient from consanguineous parents. She was seen aged 9 months in the emergency department with an episode of oral thrush and upper respiratory tract infection, but not admitted to hospital. She was first admitted to the pediatric ward, aged 2 years, with a history of fever, cough and shortness of breath. She was found to have bilateral pneumonia on chest X-ray. She deteriorated clinically and radiologically, and developed hypoxia with respiratory distress despite antibiotic treatment. She was admitted to the Pediatric Intensive Care Unit, treated with oxygen therapy, but did not need ventilation. She also had abdominal pain, constipation, and mild hepatomegaly was reported on ultrasound scan. She was found to have microcytic anemia, and noted to have failure to thrive - her weight was on the 3rd centile. Her microbiology was positive for respiratory syncytial virus, cytomegalovirus (CMV), and serology was IgM+ for herpes simplex virus. Extensive clinical laboratory evaluation revealed marked hypogammaglobulinemia with elevated IgM levels, high CRP, and high vitamin B12. Cellular immunological investigation was performed, and the percentage of T, B and NK cells were within age-adjusted ranges, while memory CD45RO+ cells were elevated for age (Table 1). She was within normal limits for lymphocyte proliferative responses to PHA, pokeweed and concanavalin A mitogens.

Table 1. Patient characteristics.
Based on these results, monthly intravenous immunoglobulin (IVIG) replacement therapy was commenced. Although the initial hepatic ultrasound showed mild hepatomegaly, this was repeated during follow-up and was normal. Liver function tests were performed and showed no evidence of transaminitis or elevated bilirubin. CMV PCR was positive at low levels initially (526 IU/mL), and then subsequently became negative within 6 weeks. The anemia was microcytic, and predominantly due to iron deficiency based on ferritin, transferrin saturation, and blood film appearances. The hemoglobin improved with oral iron supplementation. The patient’s overall condition improved with treatment, and she remains clinically stable under regular follow-up. She has not had any recurrent infections since commencement of IVIG, i.e. over 5 years of follow-up. She, her parents, and 2 sisters had an episode of COVID-19 infection in 2022. She receives trimethoprim/sulphamethoxazole prophylaxis twice daily, three times per week, and the family have been advised on the precautions against cryptosporidium infection.
The patient has dizygotic twin siblings, who are two years older and both currently in good health. One of them (Sibling-2) previously had poor weight gain and history of recurrent minor infections according to the mother. However, these infections did not require admission, and weight gain has improved with age.
3.2 Mutations detection
Due to the presence of marked hypogammaglobulinemia with elevated IgM, a genetic panel for primary immunodeficiencies was ordered to investigate a possible underlying inborn error of immunity. Clinical genetic testing using the Invitae PID panel identified two homozygous missense variants of uncertain significance (VUS) in two candidate genes: IKBKB c.230G>A, p.R77Q and AICDA c.239G>C, p.W80S. Familial segregation analysis by Sanger sequencing of the variants confirmed that the proband was homozygous for the IKBKB p.R77Q variant whereas both parents were heterozygous carriers, one sibling was homozygous for the reference allele, and the second sibling whose history included repeated infections and low weight gain was also homozygous for the p.R77Q variant. IKBKB encodes for IKKβ, and this mutation, which is located in the kinase domain, changes the IKKβ amino acid residue 77 from arginine, a basic and polar amino acid, to glutamine, a neutral and polar amino acid (p.Arg77Gln). According to the literature and population databases, this variant was novel at the time and has since been reported as a VUS in ClinVar.
Sanger sequencing also confirmed that the proband was homozygous for the AICDA p.W80S missense mutation, which changed an aromatic residue to a polar, non-charged residue. Both parents and siblings were heterozygous carriers. The mutation is in exon 3 and affects an amino acid within AID’s catalytic domain known to impact both CSR and SHM.
Pedigree structures for both mutations are shown in Figure 1 (Figures 1A, B).
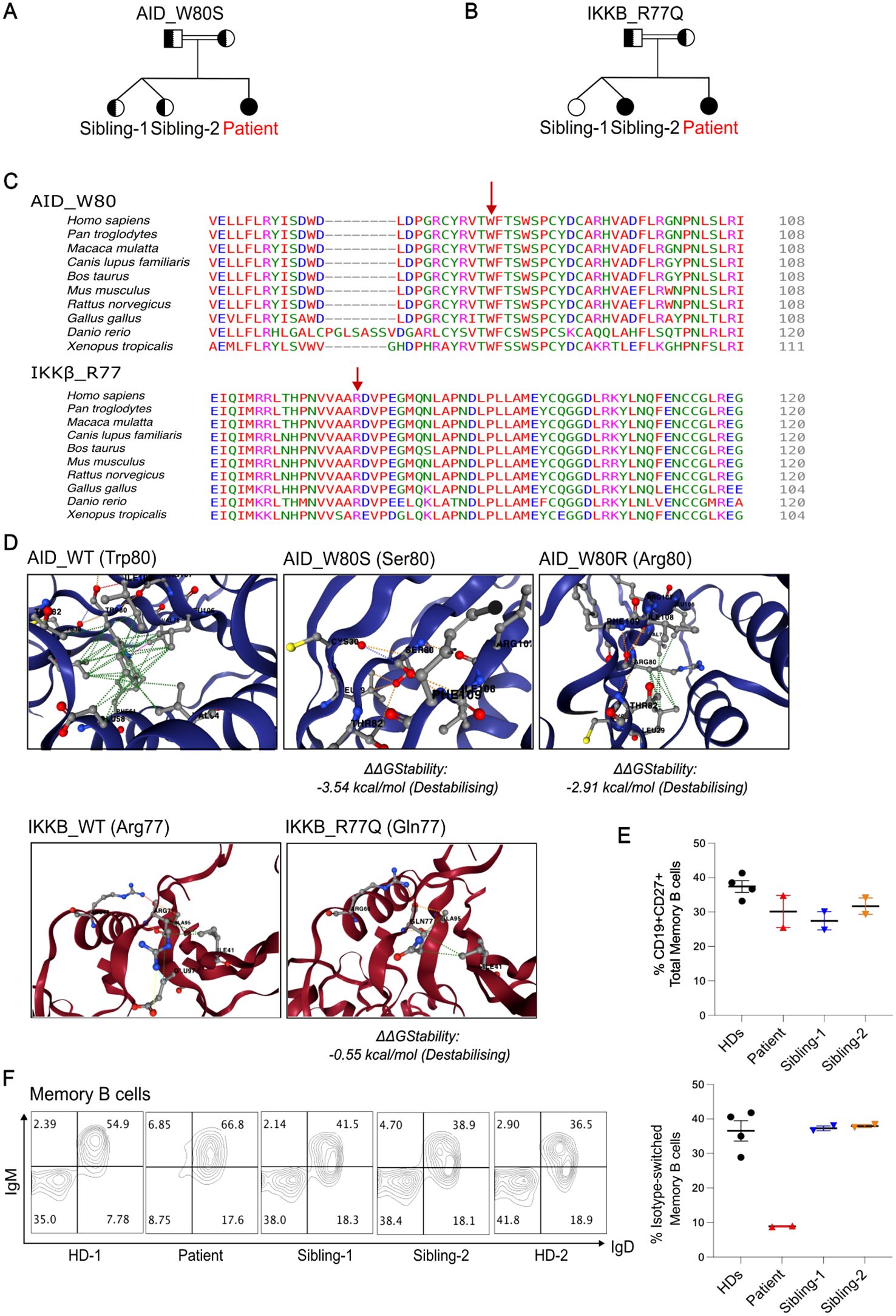
Figure 1. Two homozygous VUSs were identified in a hyper-IgM patient with reduced class-switched memory B cells. (A,B), Family pedigree showing genotypes of the indicated variants (full shaded = homozygous; half-shaded = heterozygous). (C), Multiple protein alignment demonstrating the degree of conservation of the residues AID p.W80 and IKKb p.R77 in several species. (D), Predicted stability change (ΔΔGStability) results of the genetic variants using DynaMut2. The AID p.W80R mutation was included as a disease control. (E), Quantification of total memory B cell percentages comparing Patient, Siblings and Healthy donors (HDs) from two independent experiments. (F), Representative flow plots showing percentages of non-switched and switched memory B cells within CD19+CD27+ B cells. Right panel, Quantification of class-switched memory B cells from 2 independent experiments. Class-switched memory B cells within CD19+CD27+ B cells normal range for patient age is 21.9-98.6%.
3.3 In silico characterization of identified mutations
Both variants were classified as VUS, and several in silico variant effect prediction tools indicated that both variants might be deleterious. AID p.W80 and IKKβ p.R77 residues were highly conserved between different species (Figure 1C). For the AID p.W80S variant, CADD score was 29.9, PolyPhen-2 score was 1.00 (probably damaging), SIFT score was 0.005 (damaging), MutationTaster predicted disease causing, and PrimateAI score was 0.9045 (damaging). For the IKKβ p.R77Q variant, CADD score was 25.4, PolyPhen score was 0.951 (possibly damaging), SIFT score was 0.336 (tolerated), MutationTaster predicted disease causing, and PrimateAI score was 0.5646 (damaging).
When comparing the wild-type proteins of AID and IKKβ to their mutant forms, structure-based prediction of the variants’ effect on protein stability using DYNAMUT2 revealed a folding free energy change for both structures (Figure 1D). Interestingly, we observed that AID p.W80S exhibits higher impact on the stability than the previously reported AID p.W80R mutation (14) (Figure 1D).
3.4 Class-switched memory B cells
A flow cytometric assessment of peripheral blood lymphocytes revealed normal percentage of total CD27+CD19+ memory B cells but a reduction of class-switched CD27+IgM-IgD- memory B cells compared to healthy donors (HDs) and siblings (Figures 1E, F).
3.5 Functional characterization of the IKKβ R77Q substitution
To investigate potential links between the R77Q IKKβ variant and a biological defect, we assessed Treg and memory T cell levels and function. We found a similar level of CD4+CD25+FOXP3+ Tregs in the patient compared to HDs (Figure 2A). We also observed no difference in frequency of CD45RO+ memory T cells between the proband and the siblings (Figure 2B). Evaluation of T cell cytokine production revealed a normal secretion of IL-2, TNFα and IFNγ (Figure 2C).
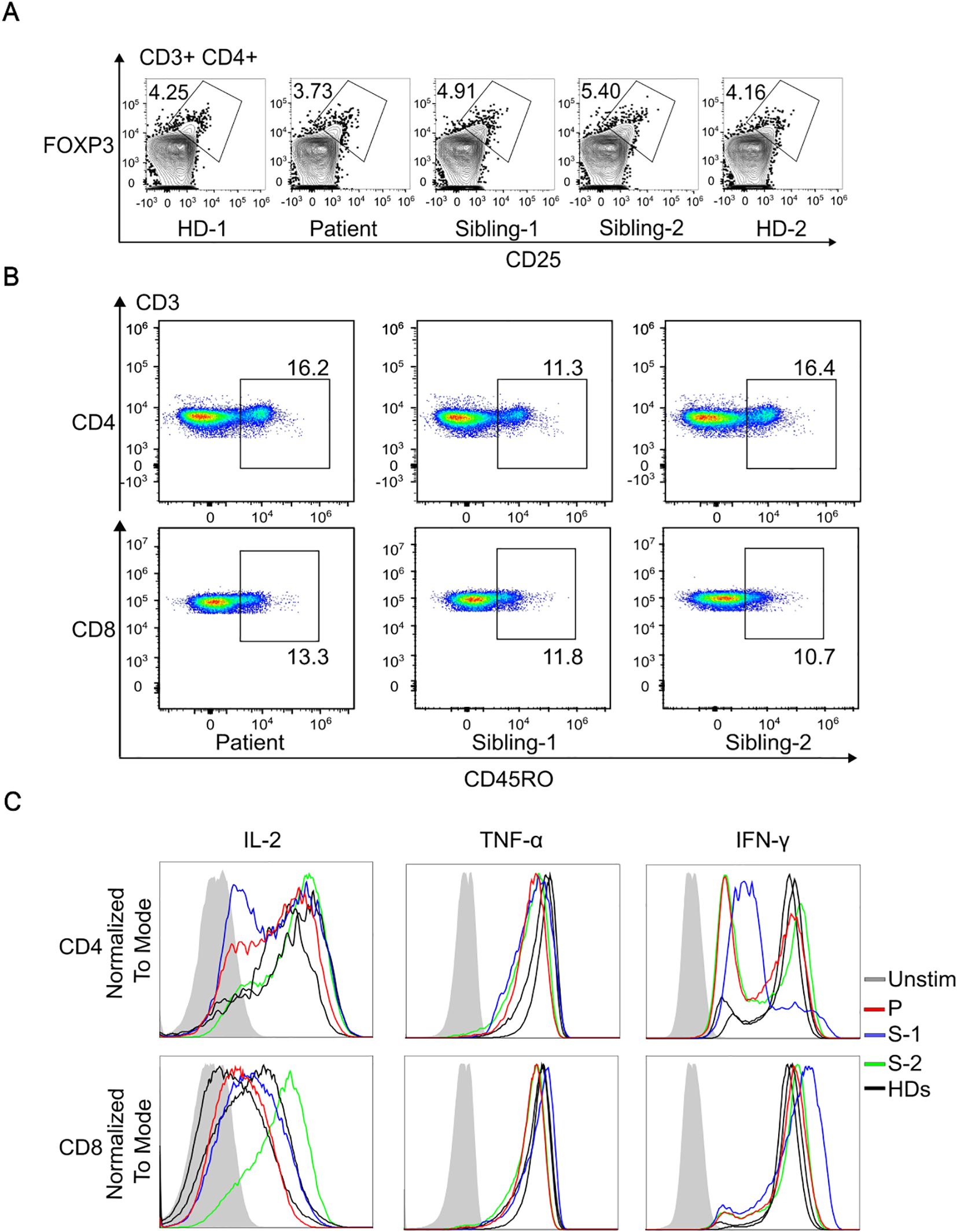
Figure 2. Immunophenotyping of T cells and assessment of IKKb expression. (A), Representative dot plots gated on CD3+CD4+ cells, illustrating the percentage of Tregs co-expressing CD25 and FoxP3. (B), Comparison of CD45RO+ frequency in CD4+ and in CD8+ T cells in patient and siblings. (C), Representative histogram of flow cytometry showing normal cytokine production comparing the patient, siblings and two healthy donors (HDs).
We assessed the IKKβ and IκBα protein levels in PBMCs and activated T cell blasts to help rule out a potential hypermorphic impact of the p.R77Q variant, although it would be unlikely due to the recessive mode of inheritance. The immunoblot analysis of PBMCs revealed that our patient had a slightly decreased IKKβ expression and normal IκBα levels compared to controls (Figure 3A). IKKβ and IκBα expression, however, was comparable to that of HDs in activated T cell blasts (Figure 3A). If there was a gain-of-function, we would expect a decrease in IκBα levels due to increased phosphorylation and degradation.
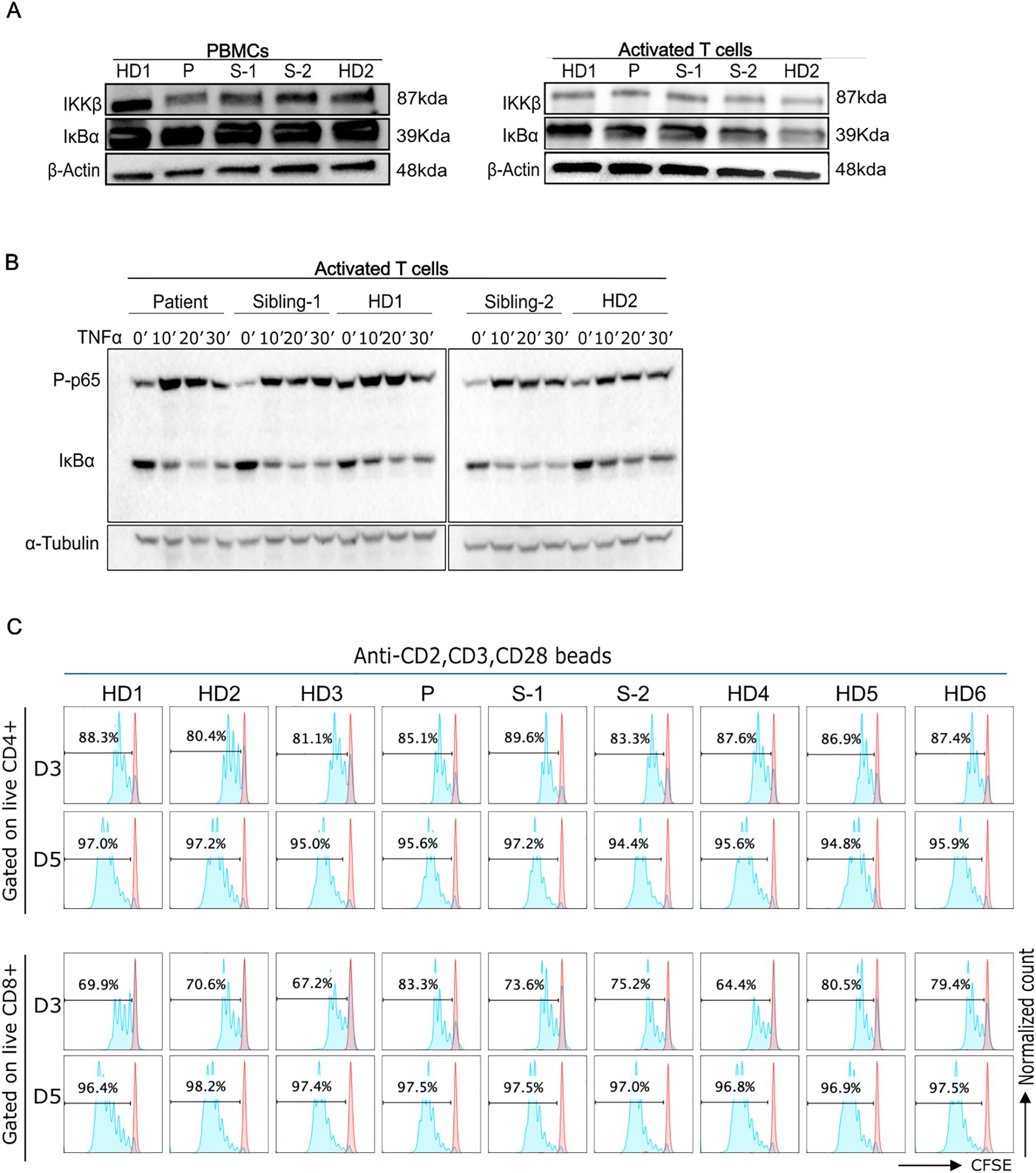
Figure 3. Functional characterizations of IKBKB p.R77Q variant. (A), Western blot analysis of PBMCs from the patient showing a lower level of the IKKβ protein (left panel). Protein levels of IkBα were normal as compared with control PBMCs. Right panel, Western blot analysis of activated T cells cultured for 6 days in the presence of IL-2 demonstrating normal levels of the IKKβ and IkBα proteins. Equal amounts of total protein derived from the lysed cells were loaded. (B), Levels of p65 phosphorylation and IkBα degradation at different time points (0-30min) post-TNFa stimulation. Left blots are representative of 3 independent experiments, and right blots are representative of 2 independent experiments. (C), Representative CFSE cell proliferation profiles for T cells are shown for days 3 and 5 as indicated for CD4 and CD8 T cells. Numbers above gate indicate the percent of cells divided in response to activation with anti-CD2, CD3, and CD28 beads. Data are representative of 4 independent experiments.
We additionally evaluated IKKβ activity by assessing IκBα degradation and p65 phosphorylation after NFkB pathway activation with TNFa. Both phospho-p65 and IκBα levels in patient activated T cells were comparable to that of the HDs following stimulation with TNFα. (Figure 3B, Supplementary Figure 1). Thus, the data does not support a possible hypermorphic nor hypomorphic function of the variant.
To further evaluate T cell function, we assessed the proliferative response of T cells to anti-CD2/CD3/CD28 beads after 3 or 5 days of culture. Our findings showed that patient and sibling-2 cells’ proliferative responses were comparable to HDs (Figure 3C, Supplementary Figure 2).
3.6 Functional characterization of the AID W80S substitution
We examined the levels of the AID protein in the EBV-transformed human B-cell lines of sibling-1, sibling-2, patient, and HDs to confirm the in silico predicted destabilizing effect of the W80S AID mutation. The findings showed that the patient cells displayed much lower AID expression than did controls and the heterozygous siblings’ cells (Figure 4A). Transcript levels of AICDA were not reduced in the patient compared to controls (Supplementary Figure 3).
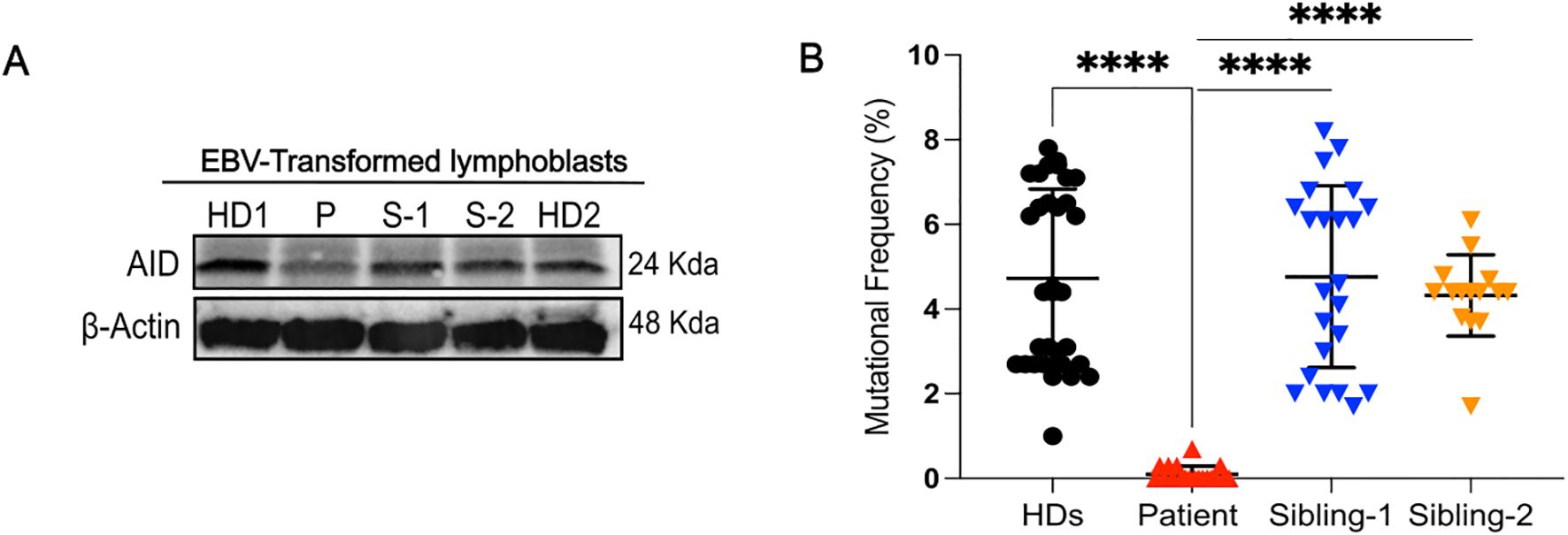
Figure 4. AID expression and analysis of somatic hypermutation. (A), Western blot analysis for AID in EBV-transformed lymphoblasts from healthy donors (HDs), patient, and siblings (S-1 and S-2). (B), Mutational frequency of the variable region of the IgM heavy chain of EBV lymphoblasts in different healthy donors (HDs) (32 clones from n=5 different controls), sibling-1 (23 clones), sibling-2 (15 clones) and patient (19 clones); each symbol represents one clone. Horizontal bars represent mean ± SD values of mutational frequency, Statistical analysis was performed using the Mann-Whitney U test; ****p<0.0001.
Therefore, we evaluated the functional impact of the W80S AID variant on SHM by comparing the mutational frequency within the variable region of the immunoglobulin heavy chain amplified from EBV-transformed B-cell lines from patient, siblings, and HDs. We found a near complete loss of SHM (0.10% ± 0.1915%) from the patient (Figure 4B, Supplementary Table 1).
3.7 ACMG classification
Variant classification was performed using the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines 2015 (15), and criteria were applied based on genetic, functional, and clinical data. These ACMG/AMP standards provide a systematic framework for interpreting sequence variants using defined criteria, which help determine the clinical significance of genetic alterations. For the IKKβ p.R77Q variant, careful review of the clinical history of the homozygous sibling found that she is currently healthy, which is inconsistent with the known fully penetrant phenotype of IKKβ deficiency, and meets the BS2 criterion. The normal NF-κB functional results were in favor of the BS3 criterion, resulting in classification to likely Benign/Benign. In contrast, classification of the AID p.W80S variant included moderate evidence from domain localization (PM1), functional impairment (PS3), and a strong phenotype match (PP4), leading to a classification of likely pathogenic/pathogenic (Table 2).
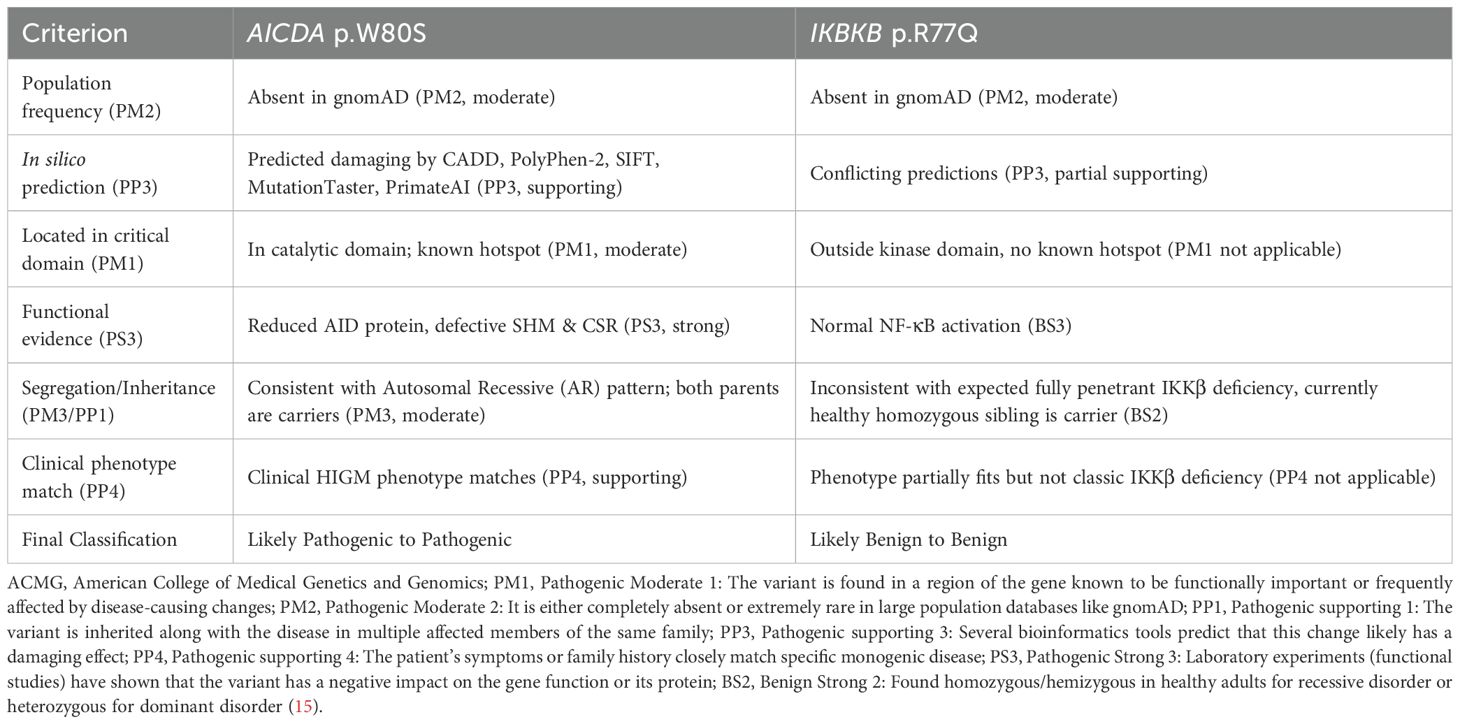
Table 2. ACMG classification of AICDA and IKBKB variants.
4 Discussion
Overlapping symptoms and biomarkers in well-known IEI is an emerging diagnostic and therapeutic challenge, which is further complicated by the identification of VUS, especially multiple VUS, during clinical genetic testing. In this article, we provide a comprehensive immunological and genetic study of a patient with two homozygous VUSs in the IKBKB and AICDA genes suspected to be responsible for the hyper-IgM and immune deficiency observed in a Tunisian patient treated at Sidra Medicine in Qatar.
To date only 7 nonsense (7, 16–21), 8 missense (22–29), and 2 duplication (11, 30) IKBKB mutations have been described. IKKβ deficient patients presented with life-threatening bacterial and viral infections, oral candidiasis, and failure to thrive (30). In addition to the episode of acute severe respiratory infection aged 2 years, our patient had an episode of oral thrush during infancy. Clinically, the patient also was found to have iron deficiency anemia and failure to thrive. Although opportunistic infections have been described in IKKβ deficient patients, it is unusual for AID deficiency (31). Thus, we considered IKKβ R77Q as a potential candidate along with the AID W80S variant, especially since sibling-2, who was also homozygous for the IKBKB variant, had a prior history of recurrent infections. Since IKKβ and AID deficiency are associated with different disease prognoses and therapeutic strategies, it was important to distinguish which disease, if not both, did the patient have. Multiple in silico tools indicated that the variants could potentially be deleterious and might impact the structure folding free energy. Both variants affected residues that were highly conserved between various species. In silico tools alone were insufficient to guide diagnosis, thus functional studies were performed.
The proband was found to exhibit a significant deficiency of class-switched CD27+IgM-IgD- memory B cells and this is consistent with AID and IKKβ deficiencies, as a reduction in switched memory B cells is a feature of both diseases (7, 32). However, the normal level of total memory B cells is characteristic of AID deficiency but not IKKβ deficiency. Since T cell defects and deficiency of Tregs are a hallmark of previously reported homozygous IKBKB mutations in patients with severe early-onset immune deficiency (17, 30), we also examined T cell phenotype and function. Cardinez et al. (28) had previously reported that a hypermorphic variant of IKBKB had normal Treg and memory T cell subsets (28). In our patient, functional studies revealed no major impairments in T cell activation or proliferation, including comparable IκBα degradation and NF-κB p65 phosphorylation following stimulation. These results, together with normal IKKβ and IκBα levels in activated T cell blasts, argue against a gain- or loss-of-function mechanism. Taken together, the findings indicate that the IKKβ variant is unlikely to be disease-causative.
Of interest, AID deficiency is the most prevalent underlying molecular cause of immunoglobulin class-switch recombination defects in Tunisian individuals (68%) (33). The patient was diagnosed as having elevated IgM and decreased levels of the other immunoglobulin classes, which is indicative of a CSR defect (Table 1). According to earlier research, both CSR and SHM may be abolished by mutations in AID’s catalytic domain (14, 34), suggesting that W80 might be a key residue for AID enzymatic activity. Structurally, AID is a 198-amino-acid protein with a conserved catalytic core containing a zinc-binding motif necessary for the deamination of cytosine to uracil in single-stranded DNA (35). The N-terminal region aids in targeting immunoglobulin gene loci, while the C-terminal domain is crucial for interactions with DNA repair proteins such as UNG and MSH2/MSH6, which are particularly important for CSR (36, 37). The W80S mutation identified in our patient resides in exon 3, within the catalytic domain, a region with the highest degree of conservation (especially residue positions 75-87) and where the most damaging missense mutations per length have been reported (38). Tryptophan 80 is a conserved residue believed to contribute to structural stability and proper positioning of the catalytic site (39). Substitution with a polar residue like serine might disrupt the local hydrophobic environment, likely imparing AID activity. Our results indicate that the W80S mutation impairs both somatic hypermutation and class-switch recombination, resulting clinically in AID deficiency (40).
In a cohort of HIGM patients described by Revy et al. in 2000, a mutation causing the amino acid substitution of tryptophan (W) to arginine (R) at the same position (residue 80) was identified in a Turkish family with AID deficiency (2). Interestingly, the W80S mutation resulted in a reduced level of AID protein in the patient’s EBV-transformed lymphoblasts, in contrast to this previously reported p.W80R mutation, which exhibited a normal AID expression in transfected 293T cells (14). In fact, the in silico study of the ΔΔG values for both mutations supported the hypothesis that the W80S mutation may be more disruptive than the W80R variant. Importantly, the patient (P16) with the W80R variant had low serum IgG and IgA concentrations (14), whereas the patient described here with the W80S variant had IgG and IgA levels that were undetectable, thus further indicating that the W80S variant may be more damaging than W80R.
The previously reported pathogenic H56Y and C87R AID missense mutations, neighboring the W80 residue, affect conserved residues in the AID’s catalytic domain, yet they appear to differ in their biochemical and clinical consequences (41). A previous comprehensive structural analysis of AID mutations, was conducted by Mu et al. (41), in which the authors generated a structural model of AID using Apo3G-CD2 as a template and analyzed various mutations associated with Hyper-IgM syndrome type 2. They highlighted that residues such as H56 and C87 are involved in coordinating the zinc ion, which is crucial for AID’s catalytic activity. Mutations at these sites were shown to be important for catalysis and potentially disruptive to the enzyme’s function (41). The C87R mutation, located within the zinc-coordinating domain essential for AID’s catalytic function, has been associated with a severe immunodeficiency phenotype and complete loss of class-switch recombination (42). In contrast to our case, p.C87R transfected in 293T cells express normal amount of AID protein (14). H56Y was markedly reduced by Western blot, suggesting that most likely H56Y impairs protein stability or expression (14, 43).
Abolhassani et al. reported that 100% of missense mutations located in exon 3 of the AICDA gene led to fungal infections and susceptibility to cancer (38). These findings appear consistent with our patient’s experience with fungal infection and further suggest the need to closely monitor the patient for the development of cancer.
Given the consanguineous background and the patient’s Tunisian origin, the AID p.W80S variant could represent either an isolated mutation or a founder mutation. Although we did not perform haplotype analysis, the absence of this variant in large public databases such as gnomAD or in studies with Tunisian patients (33, 43, 44), suggests that it is extremely rare or potentially specific to some families. Consanguinity rates in Tunisia remain high, contributing to the expression of rare autosomal recessive mutations (up to 30–40%) (45, 46). Previous studies from Tunisian cohorts have highlighted the emergence of founder mutations in other immunodeficiencies, particularly in autosomal recessive forms of Hyper-IgM syndrome and other IEIs (33, 47–49). Future regional sequencing efforts could clarify whether the p.W80S mutation represents a founder or a novel unique variant in this population.
In summary, the patient’s clinical, laboratory, and immunological phenotype was consistent with a defect in both CSR and SHM and with the diagnosis of hyper-IgM syndrome type 2. The patient is now symptomatically better and continues to receive monthly IVIG replacement therapy. She is still being closely monitored.
This study highlights the challenges inherent in diagnosing immune deficiencies. The overlapping symptoms presented by many of these disorders complicate diagnosis, and the identification of multiple VUSs adds another layer of complexity. In silico predictions can provide valuable insights, however, they are insufficient on their own to guide diagnosis. Functional studies are needed to clarify the pathogenicity of VUSs identified in clinical genetic testing, which is important in order to provide a definitive diagnosis for treatment guidance, especially in consanguineous patients who may have multiple homozygous rare VUSs.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Sidra Medicine ethics committee (IRB protocol 1601002512). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
NA: Methodology, Conceptualization, Validation, Investigation, Writing – review & editing, Data curation, Formal analysis, Writing – original draft, Software, Visualization. RM: Methodology, Investigation, Visualization, Writing – review & editing. MA: Methodology, Visualization, Writing – review & editing. AE: Resources, Writing – review & editing, Data curation. AA: Methodology, Writing – review & editing, Investigation. SH: Methodology, Writing – review & editing. SP: Resources, Methodology, Writing – review & editing. MK: Data curation, Validation, Investigation, Resources, Writing – review & editing. AH: Investigation, Resources, Funding acquisition, Writing – review & editing, Conceptualization. BL: Data curation, Validation, Formal analysis, Methodology, Supervision, Conceptualization, Project administration, Investigation, Resources, Writing – review & editing, Funding acquisition, Visualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by a Path towards Personalized Medicine grant (PPM 04-0128-200015) from the Qatar National Research Fund (QNRF) and research funds from Sidra Medicine, Doha, Qatar.
Acknowledgments
The authors thank the patient’s family for their participation in the study. We thank Jonamae Dioso for technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1594636/full#supplementary-material
Abbreviations
AID, Activation-Induced Cytidine Deaminase; CSR, Class Switch Recombination; SHM, Somatic Hypermutation; HIGM, Hyper-IgM Syndrome; VUS, Variants of Uncertain Significance; GI, Gastrointestinal; PID, Primary Immunodeficiency.
References
1. Yazdani R, Fekrvand S, Shahkarami S, Azizi G, Moazzami B, Abolhassani H, et al. The hyper IgM syndromes: Epidemiology, pathogenesis, clinical manifestations, diagnosis and management. Clin Immunol. (2019) 198:19–30. doi: 10.1016/j.clim.2018.11.007
2. Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. (2000) 102:565–75. doi: 10.1016/S0092-8674(00)00079-9
3. Trotta L, Hautala T, Hamalainen S, Syrjanen J, Viskari H, Almusa H, et al. Enrichment of rare variants in population isolates: single AICDA mutation responsible for hyper-IgM syndrome type 2 in Finland. Eur J Hum Genet. (2016) 24:1473–8. doi: 10.1038/ejhg.2016.37
4. Etzioni A and Ochs HD. The hyper IgM syndrome–an evolving story. Pediatr Res. (2004) 56:519–25. doi: 10.1203/01.PDR.0000139318.65842.4A
5. Minegishi Y, Lavoie A, Cunningham-Rundles C, Bedard PM, Hebert J, Cote L, et al. Mutations in activation-induced cytidine deaminase in patients with hyper IgM syndrome. Clin Immunol. (2000) 97:203–10. doi: 10.1006/clim.2000.4956
6. Jain A, Ma CA, Liu S, Brown M, Cohen J, and Strober W. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol. (2001) 2:223–8. doi: 10.1038/85277
7. Nielsen C, Jakobsen MA, Larsen MJ, Muller AC, Hansen S, Lillevang ST, et al. Immunodeficiency associated with a nonsense mutation of IKBKB. J Clin Immunol. (2014) 34:916–21. doi: 10.1007/s10875-014-0097-1
8. Zandi E, Rothwarf DM, Delhase M, Hayakawa M, and Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. (1997) 91:243–52. doi: 10.1016/S0092-8674(00)80406-7
9. DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, and Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. (1997) 388:548–54. doi: 10.1038/41493
10. Adli M, Merkhofer E, Cogswell P, and Baldwin AS. IKKalpha and IKKbeta each function to regulate NF-kappaB activation in the TNF-induced/canonical pathway. PloS One. (2010) 5:e9428. doi: 10.1371/journal.pone.0009428
11. Cuvelier GDE, Rubin TS, Junker A, Sinha R, Rosenberg AM, Wall DA, et al. Clinical presentation, immunologic features, and hematopoietic stem cell transplant outcomes for IKBKB immune deficiency. Clin Immunol. (2019) 205:138–47. doi: 10.1016/j.clim.2018.10.019
12. Levy Y, Gupta N, Le Deist F, Garcia C, Fischer A, Weill JC, et al. Defect in IgV gene somatic hypermutation in common variable immuno-deficiency syndrome. Proc Natl Acad Sci U S A. (1998) 95:13135–40. doi: 10.1073/pnas.95.22.13135
13. Ye J, Ma N, Madden TL, and Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. (2013) 41:W34–40. doi: 10.1093/nar/gkt382
14. Ta VT, Nagaoka H, Catalan N, Durandy A, Fischer A, Imai K, et al. AID mutant analyses indicate requirement for class-switch-specific cofactors. Nat Immunol. (2003) 4:843–8. doi: 10.1038/ni964
15. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
16. Burns SO, Plagnol V, Gutierrez BM, Al Zahrani D, Curtis J, Gaspar M, et al. Immunodeficiency and disseminated mycobacterial infection associated with homozygous nonsense mutation of IKKbeta. J Allergy Clin Immunol. (2014) 134:215–8. doi: 10.1016/j.jaci.2013.12.1093
17. Mousallem T, Yang J, Urban TJ, Wang H, Adeli M, Parrott RE, et al. A nonsense mutation in IKBKB causes combined immunodeficiency. Blood. (2014) 124:2046–50. doi: 10.1182/blood-2014-04-571265
18. Al-Shamsi A, Hertecant JL, Souid AK, and Al-Jasmi FA. Whole exome sequencing diagnosis of inborn errors of metabolism and other disorders in United Arab Emirates. Orphanet J Rare Dis. (2016) 11:94. doi: 10.1186/s13023-016-0474-3
19. Arce-Estrada GE, Rodriguez-Morales M, Scheffler-Mendoza SC, Saez-de-Ocariz M, Berron-Ruiz L, Espinosa-Padilla SE, et al. Case Report: A novel IKBKB variant (c.1705G>T) is associated with immune dysregulation and disseminated tuberculosis. Front Immunol. (2025) 16:1541899. doi: 10.3389/fimmu.2025.1541899
20. Alsum Z, AlZahrani MS, Al-Mousa H, Alkhamis N, Alsalemi AA, Shamseldin HE, et al. Multiple family members with delayed cord separtion and combined immunodeficiency with novel mutation in IKBKB. Front Pediatr. (2020) 8:9. doi: 10.3389/fped.2020.00009
21. Taylor MG, Nicholas SK, Forbes Satter LR, Martinez C, and Cameron LH. Plasma metagenomic sequencing expedites diagnosis of disseminated BCG in an infant with IKBKB mutation. Pediatr Infect Dis J. (2022) 41:430–5. doi: 10.1097/INF.0000000000003465
22. Abbott J, Ehler AC, Jayaraman D, Reynolds PR, Otsu K, Manka L, et al. Heterozygous IKKbeta activation loop mutation results in a complex immunodeficiency syndrome. J Allergy Clin Immunol. (2021) 147:737–40 e6. doi: 10.1016/j.jaci.2020.06.007
23. Al-Herz W, Chou J, Delmonte OM, Massaad MJ, Bainter W, Castagnoli R, et al. Comprehensive genetic results for primary immunodeficiency disorders in a highly consanguineous population. Front Immunol. (2018) 9:3146. doi: 10.3389/fimmu.2018.03146
24. van Schouwenburg PA, Davenport EE, Kienzler AK, Marwah I, Wright B, Lucas M, et al. Application of whole genome and RNA sequencing to investigate the genomic landscape of common variable immunodeficiency disorders. Clin Immunol. (2015) 160:301–14. doi: 10.1016/j.clim.2015.05.020
25. Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. (2014) 515:216–21. doi: 10.1038/nature13908
26. Qin T, Jia Y, Liu Y, Dai R, Zhou L, Okada S, et al. A novel homozygous mutation destabilizes IKKbeta and leads to human combined immunodeficiency. Front Immunol. (2020) 11:517544. doi: 10.3389/fimmu.2020.517544
27. Sacco K, Kuehn HS, Kawai T, Alsaati N, Smith L, Davila B, et al. A heterozygous gain-of-function variant in IKBKB associated with autoimmunity and autoinflammation. J Clin Immunol. (2023) 43:512–20. doi: 10.1007/s10875-022-01395-2
28. Cardinez C, Miraghazadeh B, Tanita K, da Silva E, Hoshino A, Okada S, et al. Gain-of-function IKBKB mutation causes human combined immune deficiency. J Exp Med. (2018) 215:2715–24. doi: 10.1084/jem.20180639
29. Hanitsch LG, Muche M, Radbruch H, Hofmann J, and Corman VM. Fatal disseminated hepatitis E in an adult patient with IKBKB GOF mutation. J Clin Immunol. (2024) 44:120. doi: 10.1007/s10875-024-01721-w
30. Pannicke U, Baumann B, Fuchs S, Henneke P, Rensing-Ehl A, Rizzi M, et al. Deficiency of innate and acquired immunity caused by an IKBKB mutation. N Engl J Med. (2013) 369:2504–14. doi: 10.1056/NEJMoa1309199
31. Durandy A, Cantaert T, Kracker S, and Meffre E. Potential roles of activation-induced cytidine deaminase in promotion or prevention of autoimmunity in humans. Autoimmunity. (2013) 46:148–56. doi: 10.3109/08916934.2012.750299
32. Notarangelo LD, Lanzi G, Peron S, and Durandy A. Defects of class-switch recombination. J Allergy Clin Immunol. (2006) 117:855–64. doi: 10.1016/j.jaci.2006.01.043
33. Ouadani H, Ben-Mustapha I, Ben-ali M, Ben-khemis L, Largueche B, Boussoffara R, et al. Novel and recurrent AID mutations underlie prevalent autosomal recessive form of HIGM in consanguineous patients. Immunogenetics. (2016) 68:19–28. doi: 10.1007/s00251-015-0878-6
34. Durandy A, Peron S, Taubenheim N, and Fischer A. Activation-induced cytidine deaminase: structure-function relationship as based on the study of mutants. Hum Mutat. (2006) 27:1185–91. doi: 10.1002/(ISSN)1098-1004
35. Conticello SG, Thomas CJ, Petersen-Mahrt SK, and Neuberger MS. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol Biol Evol. (2005) 22:367–77. doi: 10.1093/molbev/msi026
36. Barreto V, Reina-San-Martin B, Ramiro AR, McBride KM, and Nussenzweig MC. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Mol Cell. (2003) 12:501–8. doi: 10.1016/S1097-2765(03)00309-5
37. Choi JE, Matthews AJ, Michel G, and Vuong BQ. AID phosphorylation regulates mismatch repair-dependent class switch recombination and affinity maturation. J Immunol. (2020) 204:13–22. doi: 10.4049/jimmunol.1900809
38. Abolhassani H, Marcotte H, Fang M, and Hammarstrom L. Clinical implications of experimental analyses of AID function on predictive computational tools: Challenge of missense variants. Clin Genet. (2020) 97:844–56. doi: 10.1111/cge.13737
39. Kohli RM, Maul RW, Guminski AF, McClure RL, Gajula KS, Saribasak H, et al. Local sequence targeting in the AID/APOBEC family differentially impacts retroviral restriction and antibody diversification. J Biol Chem. (2010) 285:40956–64. doi: 10.1074/jbc.M110.177402
40. Durandy A, Revy P, Imai K, and Fischer A. Hyper-immunoglobulin M syndromes caused by intrinsic B-lymphocyte defects. Immunol Rev. (2005) 203:67–79. doi: 10.1111/j.0105-2896.2005.00222.x
41. Mu Y, Prochnow C, Pham P, Chen XS, and Goodman MF. A structural basis for the biochemical behavior of activation-induced deoxycytidine deaminase class-switch recombination-defective hyper-IgM-2 mutants. J Biol Chem. (2012) 287:28007–16. doi: 10.1074/jbc.M112.370189
42. Quartier P, Bustamante J, Sanal O, Plebani A, Debre M, Deville A, et al. Clinical, immunologic and genetic analysis of 29 patients with autosomal recessive hyper-IgM syndrome due to Activation-Induced Cytidine Deaminase deficiency. Clin Immunol. (2004) 110:22–9. doi: 10.1016/j.clim.2003.10.007
43. Ouadani H, Ben-Mustapha I, Ben-Ali M, Largueche B, Jovanic T, Garcia S, et al. Activation induced cytidine deaminase mutant (AID-His130Pro) from Hyper IgM 2 patient retained mutagenic activity on SHM artificial substrate. Mol Immunol. (2016) 79:77–82. doi: 10.1016/j.molimm.2016.09.025
44. Fiorini C, Jilani S, Losi CG, Silini A, Giliani S, Ferrari S, et al. A novel activation-induced cytidine deaminase gene mutation in a Tunisian family with hyper IgM syndrome. Eur J Pediatr. (2004) 163:704–8. doi: 10.1007/s00431-004-1540-8
45. Barbouche MR, Galal N, Ben-Mustapha I, Jeddane L, Mellouli F, Ailal F, et al. Primary immunodeficiencies in highly consanguineous North African populations. Ann N Y Acad Sci. (2011) 1238:42–52. doi: 10.1111/j.1749-6632.2011.06260.x
46. Ben Halim N, Ben Alaya Bouafif N, Romdhane L, Kefi Ben Atig R, Chouchane I, Bouyacoub Y, et al. Consanguinity, endogamy, and genetic disorders in Tunisia. J Community Genet. (2013) 4:273–84. doi: 10.1007/s12687-012-0128-7
47. Ben-Khemis L, Mekki N, Ben-Mustapha I, Rouault K, Mellouli F, Khemiri M, et al. A founder mutation underlies a severe form of phosphoglutamase 3 (PGM3) deficiency in Tunisian patients. Mol Immunol. (2017) 90:57–63. doi: 10.1016/j.molimm.2017.06.248
48. Ben-Mustapha I, Ben-Ali M, Mekki N, Patin E, Harmant C, Bouguila J, et al. A 1,100-year-old founder effect mutation in IL12B gene is responsible for Mendelian susceptibility to mycobacterial disease in Tunisian patients. Immunogenetics. (2014) 66:67–71. doi: 10.1007/s00251-013-0739-0
Keywords: AICDA, diagnosis strategies, hyper-IgM syndrome, IKBKB, VUS
Citation: Agrebi N, Mackeh R, Alsabbagh M, Elmi A, Al-Marri AA, Hubrack SZ, Purayil SC, Karim MY, Hassan A and Lo B (2025) Dual variants of uncertain significance in a case of hyper-IgM syndrome: implications for diagnosis and management. Front. Immunol. 16:1594636. doi: 10.3389/fimmu.2025.1594636
Received: 16 March 2025; Accepted: 15 May 2025;
Published: 02 June 2025.
Edited by:
Samuel Cern Cher Chiang, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Krzysztof Piotr Michalak, Adam Mickiewicz University, PolandGabriel Emmanuel Arce-Estrada, Instituto Nacional de Pediatria, Mexico
Copyright © 2025 Agrebi, Mackeh, Alsabbagh, Elmi, Al-Marri, Hubrack, Purayil, Karim, Hassan and Lo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amel Hassan, YW1lbC5oQHF1LmVkdS5xYQ==; Bernice Lo, YmxvQHNpZHJhLm9yZw==