 Aya A. El-Taibany
Aya A. El-Taibany Parichehr Heydarian
Parichehr Heydarian Michael C. Seeds
Michael C. Seeds Anthony Atala
Anthony Atala- Wake Forest Institute for Regenerative Medicine, Wake Forest School of Medicine, Winston-Salem, NC, United States
Multiple sclerosis affects a significant portion of the world’s adult population and is the most common nontraumatic neuroimmunology disorder. Although the specific etiology of multiple sclerosis remains unknown, it has been associated with autoimmune components. While current treatment options relieve some symptoms in MS patients, most are immunosuppressive and only delay the progression of the disease without conferring definitive curative measures. Hence, a thorough understanding of disease pathobiology, the contribution of the neurovascular unit (NVU), and biological body-on-a-chip systems that replicate the blood–brain barrier may open new horizons for the discovery of potential therapeutics for MS.
Introduction
Multiple sclerosis is a chronic inflammatory demyelinating disorder of the CNS that affects adults aged 20–40 years and disproportionately affects the female population at a ratio of nearly 3:1 (1). One study reported that approximately 1.7 million people were diagnosed with MS globally in 2019, resulting in 22,438 deaths (2).
Many risk factors have been implicated both in the development of MS and in modulating the clinical severity of the disease. Most MS patients show evidence of previous Epstein–Barr virus (EBV) infection, which is reflected by increased IgG antibody titers to EBV nuclear antigens (EBNAs) (3–5), and the same association has been reported in meta-analysis studies (6–8). It has been proposed that EBV increases the risk of multiple sclerosis via molecular mimicry (9) and that EBNA-1-specific T cells are cross-reactive with myelin antigens (10). Another study linked an increased risk of MS to increased BBB permeability due to acute primary EBV infection (11). Furthermore, one study reported that B cells infiltrating the CNS were infected with EBV (12, 13), but the same finding was not confirmed in other studies (14). One review concluded that CNS infection with EBV has not been effectively proven (15).
Vitamin D has a well-established immunoregulatory function involving major histocompatibility complex (MHC) genes involved in antigen presentation that may impact MS (16–18). Additionally, the presence of vascular comorbidities worsens the progression of MS (19). Smoking has been identified as a risk factor for increased susceptibility to MS due to the release of carbon monoxide and nitric oxide (20, 21).
Other environmental factors that affect MS and may lead to exacerbations are high altitude, which has been implicated as a risk factor for MS due to changes in oxygen levels, cerebral vasoreactivity, and changes in the immune system (22). Latitude can affect the disease course of MS leading to earlier onset and higher prevalence. People living in high latitudes are more prone to develop MS. The prevalence is estimated to be a 10-fold increase between the equator and 60° north and south (23, 24).
Genetic susceptibility
Genetic susceptibility to MS has been described through family and twin-concordance studies where researchers reported a 25% recurrence risk in monozygotic twins and a 2–5% risk rate in dizygotic twins and first-degree relatives (25). Additionally, genome-wide association studies and the International MS Genetics Consortium have discovered 230 genetic susceptibility loci for MS (26). Most of these loci are linked to the immune system, and the strongest genetic association is linked to major histocompatibility complex II (MHC II) molecules, which are responsible for antigen presentation to T cells and their activation. Approximately 32 associations belong to the major histocompatibility complex (MHC). One highly associated link was HLA-DRB1*15, which confers a threefold increase in MS risk (27, 28).
A more recent GWAS study integrated with single cell accessibility data revealed the association of signals with B cell and monocyte/microglial cell-types (29).Transcriptional analysis studies are crucial to understanding the relation between genetic variants and gene expression. Transcriptional analysis helps decipher the pathogenic mechanisms in MS and identify potential therapeutic targets (30). Beyond GWAS and transcriptional analysis, genetic predispositions to MS have been revealed by variety of methods and thoroughly reviewed in other articles (31, 32).
Notably, some of these immune system gene variants have a potential link to disrupted NVU components that increase the complexity of disease etiology (33). This evidence implies that the integrity and function of the BBB warrant further investigation for its potential to ameliorate MS.
Clinical manifestations
MS manifests in four different clinical forms. Approximately 80% of patients present with relapsing–remitting MS (RRMS), where they experience flare-ups of the disease that coincide with the formation of new contrast-enhancing lesions in the brain and spinal cord followed by periods of remission where symptoms improve or disappear. Optic neuritis and brainstem and spinal cord syndromes are the most common manifestations, with cortical presentations being less common. After each relapse episode, there is a residual cumulative neurological deficit. After many relapses, remission tends to be incomplete. Approximately 50% of RRMS patients will develop secondary progressive MS (SPMS), in which the disease process becomes slowly progressive with or without periods of remission. However, 10–15% of patients will develop primary progressive MS (PPMS), in which the disease progressively worsens from the start with no remission. PPMS affects the spinal cord more with fewer lesions in the brain and commonly presents with progressive spastic paraparesis. The evidence of active lesions is much less common in PPRM than in SPMS. The progressive nature of the disease is reflected by the presence of the brain and spinal cord atrophy. An exceedingly rare form is progressive relapsing MS (PRMS), where the disease progresses from the start, and the patient can additionally suffer from periods of worsening symptoms (34–38).
These forms lack distinctive differential pathologies but represent a spectrum of disease progression starting from active infiltration and inflammation accompanied by demyelination to progressive irreversible damage to neurons and a decrease in the neurological reserve (35).
Neuropathology
The hallmarks of MS disease in the CNS are the formation of focal lesions of demyelination, the death of oligodendrocytes, astrogliosis, and the activation of microglia with infiltration of immune cells (39). These lesions are mostly found in the perivascular space and are localized mainly in the white matter, although they have been found to a lesser extent in the gray matter, deep brain stem nuclei and the spinal cord (40–42). These MS lesions show variable degrees of remyelination (43, 44). The lesions can be categorized into active lesions, chronic active lesions, and chronic inactive lesions (45, 46). The inactivity of the lesion is determined histologically by the absence of microglia and macrophages and the absence of myelin degradation. Active lesions are more highly expressed in acute relapsing patients, whereas chronic lesions are mostly found in patients with the progressive form of the disease (46). Focal white matter lesions may be less abundant or equal in PPMS than in SPMS (46–49). Both adaptive and innate immune system cells are found within lesions, but their composition varies with disease stage and activity.
Lesions in primary progressive MS show less immune cell infiltration than those in secondary progressive MS do (50), whereas active lesions show the most infiltration. In chronic progressive disease, the immune cell infiltrate is mostly composed of MHC class I-restricted CD8+ and, to a lesser degree, MHC class II-restricted CD4+ T cells (51–53), whereas the CD4+ T-cell population comprises the bulk of cells found in the active lesions.
CD20+ B cells can also be detected, especially in active lesions, with more plasma cells present in chronic progressive lesions (53, 66). The presence of oligoclonal immunoglobulin bands in the CSF of approximately 85% of MS patients supports the role of B cells in the pathogenesis of the disease (73).
Innate immune system cells, including activated microglia and macrophages, have been observed at CNS lesion sites in both acute and progressive MS but are present to a greater extent in the active disease state. Another form of aggregated immune cells is found in the Virchow Robin spaces of the periventricular veins and meninges, which, in severe cases, form tertiary lymphoid follicles (76, 77). These follicles have been reported to be associated with SPMS and rapidly progressive PPMS but not slowly progressive PPMS (78, 79).
Cortical demyelination and neurodegeneration have been reported in MS patients, especially those with SPMS and PPMS (80). These lesions have been observed in autopsy samples from early disease stages and increase in frequency and size in progressive stages (40, 81). These cortical lesions are associated with diffuse periventricular white matter abnormalities, which have not yet been well characterized but are likely associated with diffuse inflammation and secondary degeneration due to neuronal loss in the cortex (80). These lesions can also be detected in deep gray matter nuclei and spinal cord gray matter (82–84). Additionally, macroscopically normal white matter can show evidence of demyelination (80, 85), and MS severity is related to the severity of cortical demyelination (86). Cortical demyelination can be associated with meningeal inflammation, but meningeal inflammation by itself does not appear to be a prerequisite for cortical demyelination to occur (78, 79).
The diffusion of soluble neurotoxic factors may activate microglia and cause demyelination in the cortex or direct damage to myelin (40, 87). Some of these factors, including ceramide and semaphoring, have been previously characterized in MS patients (88, 89). In demyelinated cortical lesions, microglia are activated, which can be induced by soluble factors from B cells found in the meningeal cell infiltrate (68, 69).
The pathological hallmark of MS white matter lesions can be readily detected via conventional magnetic resonance imaging (MRI) (90, 91). However, the severity of these lesions has not been linked to neurological deficits or disability in MS patients. This is attributed in part to the presence of macroscopically undetected chronic injury in normal white matter, which can be detected by the magnetic resonance (MR) magnetization transfer ratio and diffusion tensor imaging (92–94). The definitive pathology of these lesion areas has not yet been fully elucidated but has been shown to include axonal injury, secondary demyelination, increased blood–brain barrier permeability, and microglial activation (95, 96). These normal-appearing white matter (NAWM) abnormalities have been linked to the presence of active focal lesions and demyelination that results from axonal loss, known as secondary Wallerian degeneration (97, 98).
NAWM changes have also been demonstrated to be associated with meningeal inflammation and are independent of focal lesion presence (99). Furthermore, metabolic disturbances in myelin phospholipids have been reported in areas of the NAWM (100, 101). These observations raise the possibility that MS could be a primary neurodegenerative disease that elicits immunological interactions in the CNS. It has been hypothesized that focal lesions in MS patients are only the “tip of the iceberg” of a more diffuse injury occurring in the CNS (102). Thus, elucidating the pathology of NAWM lesions and their associated BBB damage would provide insights into the pathogenesis of MS and aid in the development of therapeutics that target the disease process, as it might represent events prior to lesion formation (103, 104).
Blood–brain barrier pathology
The evolution of MS lesions in the CNS includes myelin autoreactive encephalitogenic CD4+ T cells breaching the blood–brain barrier (BBB) and gaining access to the CNS parenchyma to initiate the lesion.
The function of the BBB is to maintain CNS homeostasis by regulating the passage of molecules into the CNS and providing efflux of metabolic waste and harmful materials from the brain. The BBB is formed mainly by brain microvascular endothelial cells (BMECs) surrounded by pericytes and astrocyte-end feet and regulated by CNS parenchymal cells such as microglia, neurons, and oligodendrocytes in a structure known as the neurovascular unit (NVU) (105). BMECs acquire their barrier properties through the formation of interendothelial sealing tight junctions (TJs) and adherent junctions (AJs) formed by specialized junctional complexes and accessory proteins (106–109).
BMECs also possess a highly specialized membrane transport system (110), decreased pinocytic activity, and lack the transendothelial fenestrations observed in other capillary systems (105). As a result, BBB transendothelial electric resistance (TEER) ranges from 1,000 to 1,500 Ω/cm2 (111). Two major tight junction proteins expressed by BMECs are occludins (112) and claudins (113). The integrity of the BBB was shown to be maintained after occludin expression was knocked down, but the integrity of the BBB was lost after claudin expression was knocked down (114–116). Accordingly, there is increasing scientific awareness of the role of claudin-5 in the integrity of the BBB and its involvement in many neurological disorders (117). Consequently, this protein has been investigated for its therapeutic potential in many of these disorders, and its manipulation has been investigated as a vehicle for drug delivery into the CNS (118, 119). In addition to the main junctional complex proteins, accessory junctional proteins, such as the cytoplasmic zonula occludins proteins ZO-1, ZO-2, and ZO-3 (120), are membrane-associated guanylate kinases that interact with other intracellular molecules, including cingulin (121), the 7H6 antigen (122), and other cytoskeletal proteins. Additional proteins coexist with junctional proteins, including junctional adhesion molecules (JAMS) (123, 124) and platelet/endothelial cell adhesion molecule-1 (PECAM-1) or CD31 (125). PECAM-1 is involved in transendothelial transmigration, and PECAM-1-deficient mice exhibit increased BBB permeability (126).
Histopathological studies revealed abnormalities in the BBB in active as well as inactive lesions in multiple sclerosis (127, 128). More recently, BBB abnormalities have been reported in NAWM (102, 129). This finding suggests that such barrier disruption occurs even before obvious lesion formation and immune cell infiltration are evident, which makes the mechanisms of BBB disruption in diseases such as MS particularly important to study, as they might provide novel insights into the process of disease development. Whether BBB dysfunction is a consequence or cause of increased immune cell infiltration in the CNS is still debated, with evidence supporting both hypotheses (130–132).
The transmigration of immune cells through the BBB occurs through a highly regulated series of sequential events. Initially, activated leukocytes are captured by endothelial cells via interactions between selectins and their receptors on the surface of the inflamed brain endothelium. Binding to selectins leads to slowing of immune cells (crawling) followed by the activation of leukocytes by chemokines through G-protein signaling and induces their firm adhesion to BMECs through the binding of endothelial cell adhesion molecules to their receptors on the surface of activated T cells (133). One of the most prominent inflammatory changes in the BMECs in MS is the upregulation of the selectin family of endothelial adhesion molecules; cell adhesion molecules such as ICAM-1, VCAM-1, MCAM, and ALCAM; chemokines on the luminal surface of brain endothelial cells; and the upregulation of class II MHC molecules (134), which contribute to the migration of immune cells into the brain.
Selectins are transmembrane glycoproteins. E-selectin and P-selectin are expressed on the surface of activated brain endothelial cells and are responsible for the initial adhesion of leukocytes to endothelial cells. L-selectin is expressed on leukocytes and is involved in directing leukocytes to inflammatory sites and binding leukocytes to other immune cells to recruit them. P-selectin glycoprotein ligand (PSGL-1) is the receptor for these three selectins and can also bind to CD44 (133, 135–137).
The intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) bind to their respective ligands, αLβ2 [lymphocyte function-associated antigen 1 (LFA-1)], and α4β1 [very late antigen 4 (VLA-4)] integrins, which are upregulated in encephalitogenic CD4+ T cells. It has been proposed that α4β1-integrin binding to VCAM-1 helps in the transmigration process in spinal cord microvessels, whereas LFA-1 binding to ICAM-1/2 regulates Th17 adhesion to the endothelial barrier in the brain ( (138, 139). Melanoma cell adhesion molecule (MCAM) (140, 141) is another adhesion molecule involved in T-cell transmigration, and its expression is a marker for GM-CSF, IL-22, and IL-17A/IFN-γ, which coproduce Th17 cells. Recently, αvβ3 integrin was shown to control Th17 transmigration, and depletion of the β3 subunit improved symptoms in an EAE model (142). In addition to this cellular upregulation, soluble forms of these adhesion molecules can be detected in patient sera and CSF, and their levels are associated with the severity of disease activity (143–145).
The factors that influence the pathway used by different immune cell subsets are still under investigation, although it has been demonstrated that the remodeling of certain junctional proteins influences transmigration routes, favoring either paracellular diapedesis (146, 147) or the transcellular route (148, 149), and that experimental interference via one route increases the utilization of the other commensurately. These findings indicate that BBB proteins are very important in determining the route of transmigration.
The activation of brain microvascular endothelial cells and disruption of the BBB in MS result either from the direct effects of cytokines secreted by activated myelin-specific T cells or indirectly from the effects of these cytokines on neurovascular unit (NVC) astrocytes and pericytes (129). Leaks in the BBB lead to increased infiltration of immune cells and their soluble immunomodulators; for instance, fibrinogen leakage across the BBB can be detected early in the disease process and signifies disruption of the BBB (150, 151).
Exposure to cytokines leads to many alterations in junctional complexes. Alterations in BBB TJs and AJs in MS have previously been described in many studies. These changes could result from various mechanisms, such as downregulation of expression, destruction of junctional proteins, their internalization, or changes in their binding affinities. As an example of these alterations, occludin expression decreases with exposure to IFN-δ alone or paired with TNF-α (152, 153), but TNF-α alone does not decrease occludin expression (154). TNF-α has also been shown to cause VE-cadherin phosphorylation (155) and induce the internalization of junctional proteins by upregulating NF-Kβ, which in turn induces the transcription of myosin light chain kinase (MLCK), which is responsible for this delocalization (156). IFN-δ can also affect ZO-1 through downregulating its expression and changing its subcellular localization (157, 158), as well as inducing endocytosis of occludin and claudin-1 (159). The downregulation of occludin and ZO-1 expression has also been reported in BMECs treated with IL-17 and IL-22 (160, 161). ZO-1, occludin, claudin-5, and junctional adhesion molecules can be cleaved by increased expression of MMP-9 (162, 163) induced by IL-1β (164). Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement (165). Other factors in addition to cytokines can lead to BBB disruption, such as oxidants, which cause ZO-1 and occludin breakdown, and VEGF, which induces the phosphorylation of tight junctions (166). These effects were demonstrated by studies in which MS patient sera were used to treat brain endothelial cells in vitro to identify soluble mediators that disrupt BBB integrity and learn how to counteract their effects (167, 168).
This BBB pathology extends to astrocytes, which exhibit astrogliosis, an inability to upregulate AQP4, and astrocytic end-feet retraction from the glia limitans (169, 170). The basement membrane (BM) also shows irregularity and deposition of its degraded components, mostly because of the secretion of MMPs and other enzymes by immune cells that cleave the BM, especially those associated with active lesions (171, 172). Some studies have concluded that the volume of Virchow Robin spaces (VRSs) is greater in MS patients than in controls, as shown via MRI; this increase in VRSs is associated with white matter and gray matter lesions, and VRSs accumulate immune cells that participate in the neurodegenerative process in MS, as previously mentioned (173, 174).
All of the described changes lead to increased BBB permeability, reflected by leakage of gadolinium contrast material during MRI examination, and consequently lead to increased solute permeability and infiltration of immune cells into the CNS (175).
Chemokines play an effector role in guiding immune cell adherence and infiltration across the BBB. They are secreted by many cells, such as microglia, astrocytes, and immune cells. Cytokines might also be potent contributors to chemokine production. Many chemokines are upregulated on the surface of brain endothelial cells during MS, and research is ongoing to identify key chemokines in MS that could be potential targets for treatment. For example, CCL19, CCL21 and their receptor CCR7 are upregulated Th1 in inflamed BMECs. The knockdown or inhibition of these cytokines leads to decreased myelin-specific T-cell adhesion to BMECs (176). Similarly, CXCL13 is increased in the CSF of MS patients compared with normal controls, and its level is correlated with increased immune cell infiltration. The interaction of chemokines with their receptors changes the low-affinity selectin-mediated interaction of leukocytes with the brain endothelium to a more potent integrin-mediated interaction (177). Chemokines bind to transmembrane G protein-coupled receptors on leukocyte surfaces and mediate the upregulation of integrins through G protein signaling (178). Presenting further detail about these chemokines is beyond the scope of this article but can be found in detail in other reviews (179).
Immunopathogenesis
T cells
CD4+ T cells are the main initiators of MS lesions in the CNS on the basis of histopathological examination, disease modeling from in vivo experimental autoimmune encephalomyelitis (EAE) animal models, and the association of MS with variants in MHC class II genes and regulatory molecules involved in their interactions (54). CD4+ cells are either Th1 or Th17 CD4+ T- cells. Th1 cells secrete TNF-α and IFN-γ, and their differentiation is dependent on T-cell transcription factor (TBET) (58–61). Th17 T cells mainly secrete IL-17, and their differentiation is driven by IL-23 (62–65). Both Th1 and Th17 cytokines are highly elevated in the patient’s plasma before active disease and decrease with remission (180). It has been proposed that activated myelin-specific Th1 cells lead to spinal cord inflammation, whereas Th17 cells induce inflammation in the brainstem, cerebellum, and cerebrum (181, 182).
CD4+ T cells and CD8+ T cells in MS lesions show evidence of clonal expansion, targeting myelin autoantigens. This clonal expansion implies that they are activated by specific antigens, although despite decades of research, these antigens remain unidentified (55, 56). Some studies have suggested that these antigen-specific T cells could serve as brain-resident T cells against neurotropic viruses that are activated by specific cytokines released from sources or events that are not specific to their antigens (57).
The activation of myelin-specific T cells in the peripheral circulation by molecular mimicry, where T cells are activated by viral or bacterial antigens that share homologous sequences with CNS antigens, has been a long-standing theory for peripheral activation of the immune system in MS (183, 184). Recent evidence suggests that the gut and lymphoid tissue contribute to the activation of these cells (185, 186). Both activated myelin autoreactive T cells and those reactive to antigens other than neural antigens can cross the BBB. However, only those specific to myelin are able to induce lesions in the CNS because of their reactivation by antigen-presenting cells inside the CNS (187, 188). There is no definite evidence for any difference in the frequency of myelin-specific T cells between MS patients and normal controls (189–192). Some studies have demonstrated functional differences in the increased secretion of IFN-α, IL-17, and GM-CSF by myelin-reactive T cells between MS patients and healthy controls (193). Additional functional differences include the suppressive ability of regulatory T cells in RRMS patients compared with controls and the resistance of effector T cells in MS patients to regulatory T-cell suppression (194, 195). It has also been suggested that peripheral activation of myelin-reactive T cells yields an autoproliferative ability that bypasses the need to be activated and maintains the capacity to produce IFN-α (196, 197). Another study hypothesized that myelin-specific T cells have what is called ‘T-cell degeneracy’, which means that they can be activated by many ligands even if they do not share homology with the original stimulus (198).
After crossing the BBB, inflammatory T cells cross the basement membrane (BM) via the binding of α6β1 integrin on the leukocyte surface to laminin α4 in the BM (199). Finally, immune cells reach the perivascular space, where they recognize their specific autoantigens via antigen-presenting cells and become reactivated to augment the immune response (200, 201). Ultimately, the entry of these activated immune cells into the CNS parenchyma requires passing through the glia limitans, which are enriched with laminin α1 and α2 (183, 202–204). Passage is achieved by the effect of secreted MMPs, whose level is correlated with disease activity. MMPs also target β-dystroglycan, a receptor that anchors astrocytic end feet to the parenchymal basement membrane, the disruption of which activates astrocytes and increases their chemokine secretion (183).
Other immune cells
B cells are also indirectly involved in the pathogenesis of MS lesions through their antigen-presentation capabilities (67) and are directly involved through their suggested role in producing factors that trigger demyelination and neurodegeneration (68, 69). The role of CD20+ B cells in MS has been highlighted by the success of rituximab (a therapeutic antibody against CD20), which decreases patient disease progression (70–72). The extent of increase in B cells correlates with the clinical severity of MS in patients (86, 205). Myelin-specific antibodies may contribute to lesional demyelination, likely by binding to target antigens and activating the complement system. Additionally, increased oligoclonal bands in patients with clinically isolated syndrome (CIS) are highly predictive of an increased risk of conversion to MS (206).
Innate immune cells as macrophages and microglia interact with adaptive immune T and B cells and can induce direct damage to myelin and neuronal axons. This damaging effect has been proposed to be mediated by the production of reactive oxygen and nitrogen species (74, 75). The microglia and macrophages phagocytose myelin debris from the lesion, and the presence of degradation products correlates with the activity of the lesion in terms of demyelination and neurodegeneration.
The inside-outside theory
Failure to cure or arrest disease progression in RRMS patients treated with current immunosuppressive and immunomodulatory drugs and failure to identify specific autoantigens against which autoreactive T cells are especially prevalent in MS despite decades of research poses many questions regarding the nature of MS disease (57, 207). An existing hypothesis is that MS originates from a disease process inside the CNS itself that leads to the activation of resident immune cells, the microglia, which in turn could lead to BBB disruption and peripheral immune system activation. In this context, immune cell infiltration is recognized as a secondary response to a primary event inside the brain rather than immune system activation outside the CNS. The presence of some areas in MS brains that show loss of oligodendrocytes with minimal immune cell infiltration is suggested to represent an early prephagocytic phase. This could favor the interpretation of the disease having an intrinsic origin within the CNS (103, 208, 209).
The inside-out theory of the CNS implies the presence of a draining lymphatic system with reciprocal access to the brain, which contradicts the old concept that the CNS is an immune-privileged organ (11, 210–213). Connection of the CNS with the cervical lymph nodes has been demonstrated, through which CNS antigens can be processed and presented to peripheral immune system cells through CNS antigen-presenting cells. Moreover, the CSF represents a draining system for CNS antigens. Brain microvascular endothelial cells have been implicated in antigen presentation to immune cells (214). One study demonstrated the ability of BMECs to support and promote the proliferation of CD8+ T cells through T-cell receptor and co-stimulation, and another suggested that myelin/MHC II complexes on the inflamed brain endothelium are recognized by myelin-reactive T cells and aid their transmigration (215, 216). Despite this, the presence of primary oligodendrocyte pathology is not supported by genome-wide association studies that found no MS variants related to neuro-glial units, but loci of genetic susceptibility were detected in the MHC locus and immune cell loci. Furthermore, secondary immune cell infiltration of the CNS is not achieved in genetic animal models of primary oligodendrocyte death, which results in the activation of microglia, suggesting that a primary defect in oligodendrocytes cannot induce an autoimmune reaction (217). Furthermore, immune cell infiltration is not a common feature of primary neurodegenerative disorders (218). Transient CNS infections may damage oligodendrocytes and cause the release of myelin epitopes with subsequent activation of myelin-reactive T cells, but in the Theiler’s murine encephalitis model of MS (Theiler’s Murine Encephalitis Virus-Induced Demyelinating Disease (TMEV-IDD)), immune-induced damage depends on the persistence of the virus and its clearance leads to disease subsidence (219).
The search for the true etiology of MS pathological events seeks to fill gaps in the existing body of knowledge, including the failure of current pharmacotherapeutics to halt the progress of MS disease in RRMS (130), the presence of diffusely abnormal white matter changes, axon death and demyelination with minimal immune cell infiltration, and the presence of the same pathology in pre-phagocytic lesions, together with the accumulating evidence of BBB disruption before lesion formation and in areas of NAWM (220, 221). In studies examining changes in the level of myelin in MS, myelin abnormalities were detected in the inner myelin sheath, which does not support the idea that myelin injury is immune- or antibody-mediated. Genome-wide association studies revealed a strong link between the MHC cluster and immune cell polymorphisms in patients with the RRMS variant of MS owing to its greater prevalence. For these patients, the immune system reaction to a primary event in the brain could be overwhelming and primarily reflect an increased immunogenetic predisposition to the unknown CNS disease process releasing autoantigens. This, in turn, could explain the wide spectrum of disease variants, which may reflect different degrees of the immune system response to the primary degenerative event in the brain.
Microglia are the resident CNS immune cells and account for 12–16% of the total human parenchymal CNS cells (222). They originate from erythro-myeloid progenitors of yolk sac (mesodermal) origin; they are self-renewing and are not replaced by blood-derived monocytes (223–225). Microglia proliferate and increase in number when activated, and this activation has been discovered in many neurodegenerative disorders, including MS, Parkinson’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis (226). In contrast, the choroid plexus and meningeal and perivascular macrophages are collectively known as border-associated macrophages (BAMs). These cells are nonparenchymal and reside at the interface of the CNS and blood–brain barrier (227, 228).
Under normal physiological conditions, microglia have important functions in CNS development (229), including synaptic pruning, remodeling, myelination (200, 230, 231) and the modulation of synaptic plasticity (200). In addition, microglia contribute to surveillance of the CNS microenvironment via the expression of pattern recognition receptors, such as Toll-like receptors (TLRs), the lipid and phosphatidylserine receptor TREM2, complement receptor 3, and the C-type lectin receptor DC-SIGN87. In response to any change in the CNS microenvironment, microglia can proliferate, change their morphology, present antigens, phagocytize macromolecular agonists, and secrete cytokines and chemokines (232). Microglia are traditionally classified as M1 proinflammatory microglia or M2 anti-inflammatory microglia. Recently, there has been increasing evidence for different subtypes of microglia with different regulatory functions and characteristics that form a vast phenotypic spectrum (233, 234).
In MS, activated microglia can be found in active lesions as well as in normal white matter (209, 235, 236). Microglia are also found in areas without inflammatory infiltration, and these areas are preactive lesions (235). Furthermore, microglial activation was shown to precede immune cell infiltration from the peripheral circulation in MS mouse models of Theiler’s murine encephalomyelitis, virus-induced demyelinating disease, and experimental autoimmune encephalomyelitis (EAE). In EAE mice, microglia first take up myelin antigens and present them to T cells through MHC II and costimulatory molecules (237), and the infiltration of inflammatory cells coincides with microglial activation (238).
During MS, activated microglia secrete nitric oxide (NO) and reactive oxygen species (ROS) that damage myelin and oligodendrocyte progenitor cells (239). Activated microglia also produce proinflammatory cytokines (TNF-α, IFN-δ, and IL-1β) and chemokines (MCP-1), which damage the BBB and downregulate VE-cadherin, occludin, and claudin-5 proteins in the BBB (240). Microglia secrete MMPs that contribute to the breakdown of the blood–brain-barrier basement membrane in multiple sclerosis (241, 242). One of the more interesting pathways through which microglia are activated during brain injury is the production of danger-associated molecular patterns (DAMPs), which activate microglia and initiate neuroinflammation (243–245). In MS brains and EAE brains, activated microglia and T cells are usually closely associated, especially at sites of demyelination (74, 81, 246), and their presence is correlated with axonal damage (34, 247). Microglia aid in recruiting T cells into brain tissue (248, 249) and act as antigen-presenting cells by upregulating the expression of class I and II MHC molecules and coexpressing costimulatory molecules (250).
Conversely, microglia can also act protectively in MS and help remyelinate CNS cells through the secretion of neuroprotective molecules and anti-inflammatory cytokines (251), assist in oligodendrocyte proliferation, and phagocytize myelin debris (252). For example, previous studies in CX3CR1 knockout mice revealed reduced myelin debris clearance and remyelination due to the absence of phagocytic function of microglia (253). Microglia can increase the production of neuroprotective substances such as brain-derived neurotrophic factor and neurotrophin when exposed to MBP-primed Th2 cells (254).
The potential of some drugs to modulate the activation of microglia and hence their damaging effects in experimental models of MS or on the BBB suggests that microglia are a central contributor to inflammation in MS brains. This makes microglia an attractive therapeutic target for MS; this opportunity may be especially applicable in the progressive form of the disease for which no therapeutics are currently available. Glatiramer acetate, a drug approved for treating relapsing MS, has a demonstrated neuroprotective effect, which is thought to be mediated by activated M2 microglia (255). Other drugs that have been shown to be effective at modulating the severity of EAE in an animal model of MS through effects on microglia or macrophages include forskolin (233), bryostatin-1 (256), and ethyl pyruvate (257). The inhibition of microglia by minocycline reduces their deleterious effects on the BBB, supports the differentiation of oligodendrocyte precursors into immature oligodendrocytes and facilitates remyelination (258–261). Additionally, dipyridamole reduces microglial activation and cytokine secretion (262). Microglia can also be skewed toward an M2 anti-inflammatory phenotype by targeting AMP-activated protein kinase (AMPK) (263). In vitro models incorporating microglia have become advantageous for testing therapeutic targets that engage with microglia to develop MS therapeutics.
Treatments
An evolving demand to investigate other potential therapeutics for MS that target different arms of the disease process, such as BBB dysfunction and microglial activation as more central processes orchestrating inflammation, is imposed by the failure of current therapeutics to treat the disease or prevent progression of the associated disability, as well as the lack of available drugs to treat the chronic progressive form of MS.
In vivo and in vitro modeling systems are important in the processes of drug discovery and translation of research outcomes to the clinic. It is therefore important to look critically into the model systems available and their ability to mimic complex inflammatory processes and the diversity of cells and pathways involved. It is also important to consider how well models account for human genetic makeup differences.
New treatments for multiple sclerosis have been investigated intensely for more than 30 years (Table 1). The first drug to be approved for the treatment of RRMS was injectable INF-β, an anti-inflammatory cytokine, in 1993 (264–266), followed by the approval of another injectable anti-inflammatory drug, glatiramer acetate. Clinical trials have demonstrated the success of these compounds in altering MS disease progression and severity in patients as disease-modifying therapies (DMTs). This was followed by the FDA approval of the first humanized monoclonal antibody, natalizumab, which targets the α-4 integrin component of very late antigen-4 (VLA-4) on the surface of leukocytes and thus prevents their adhesion to VCAM-1 on the surface of endothelial cells (267). The first oral drug to be approved was fingolimod, an analog of sphingosine 1-phosphate (S1P) that acts as an S1P antagonist to block the flow of T cells from secondary lymph organs into the peripheral circulation (268, 269). For pediatric MS, fingolimod is the only approved oral DMT drug (270). Each of these drugs has been approved for RRMS but not for progressive MS. Siponimod, a selective S1P1 and S1P5 modulator, is approved for treating patients with relapsing forms of MS, including RRMS and active SPMS (271). Ozanimod and ponesimod are other S1P modulators that are approved for RRMS (270, 272).
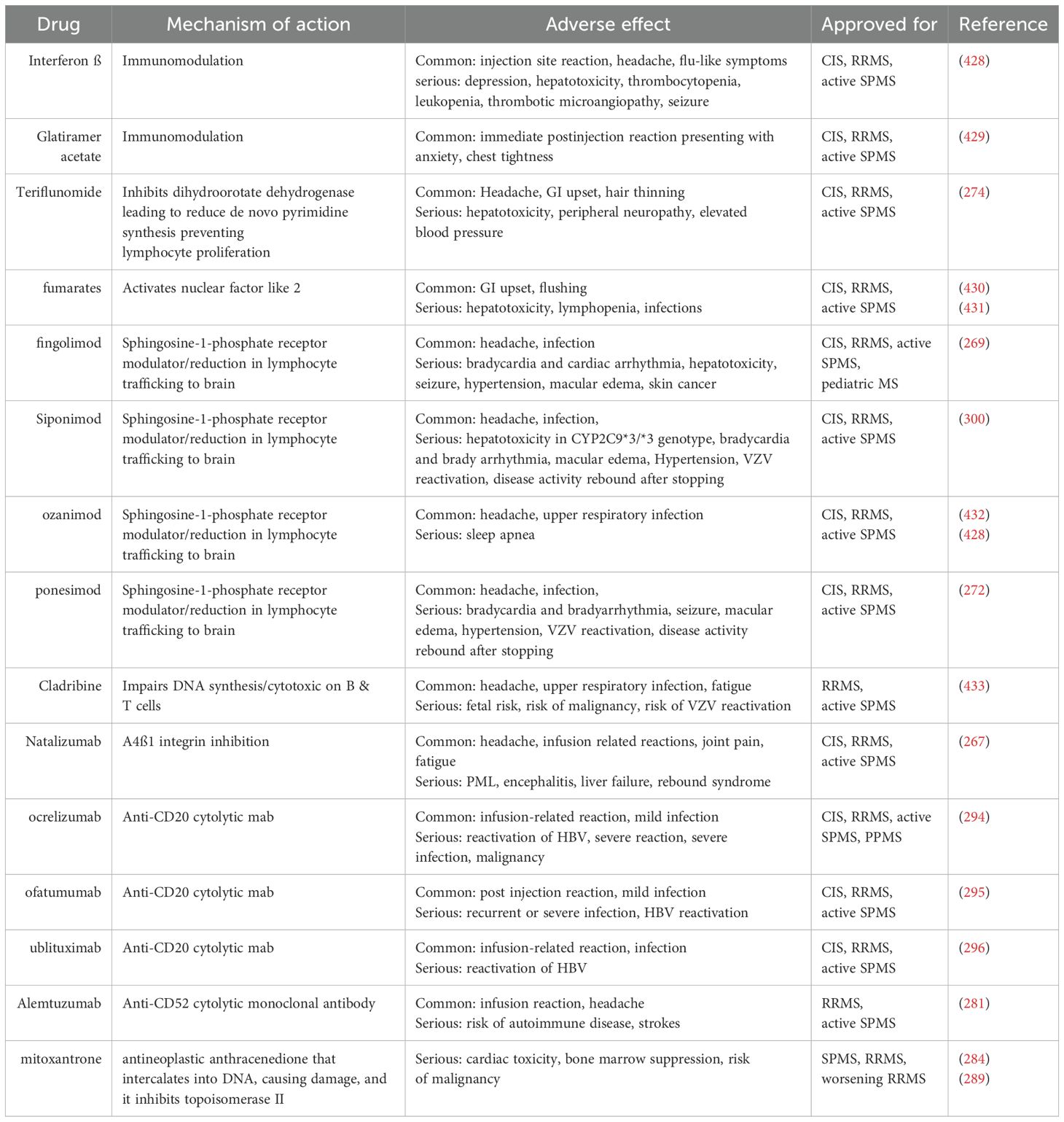
Table 1. Current therapeutics available for multiple sclerosis.
Other drugs have been used for the treatment of RRMS, such as the oral drug teriflunomide, which inhibits the proliferation of B and T-cell blasts (273–275), and oral dimethyl fumarate, which exerts its immunomodulatory function through shifting T-helper (Th) cells from proinflammatory Th1 to anti-inflammatory Th2 cells (276). Additionally, oral Cladribine is an active purine nucleoside analog prodrug that accumulates in lymphocytes due to the low activity of the 5’-nucleotidase required for their inactivation, causing the death of these cells (277, 278). Additionally, alemtuzumab is another humanized monoclonal antibody therapeutic, which is anti-CD52, a receptor expressed on lymphocytes (279–281).
The efficacy of these drugs was evaluated in clinical trials by assessing the reduction in disability via an expanded disability status scale (EDSS) and measuring the reduction in the number of relapses via the annualized relapse rate (aRR). These parameters were assessed in combination with other parameters, including the appearance of lesions on MRI and brain atrophy. The EDSS reflects the relative disability of MS patients on the basis of neurological examination of symptoms and signs of eight functional systems: vision; brain stem function; pyramidal and extrapyramidal systems; and cerebellar, cerebral, sensory, bowel, and bladder functions (282). All drugs approved for MS achieved a significant reduction in the aRR and in disability worsening when the EDSS score was equal to or greater than one third, with statistical clinical significance. Natalizumab stands out as yielding the greatest reduction in the aRR, reaching almost 70% (267).
Interestingly, despite the number of drugs approved for RRMS, there are currently very few drugs for primary progressive MS. However, some drugs have been tested for SPMS, including lamotrigine, dronabinol and dirucotide, but they do not demonstrate significant effectiveness (283–286). In a randomized trial of injectable drugs to treat SPMS and PPMS, the results were not positive (284, 287, 288). It was only later, after testing mitoxantrone, an anticancer drug, for the treatment of SPMS and PPMS that IFN-β and mitoxantrone were approved for the treatment of SPMS, not PPMS, and only in patients with worsening disease and evidence of inflammation, leaving PPMS with limited treatment options (284, 289, 290). Trials for fingolimod in PPMS (291) and natalizumab in SPMS (292) also failed to produce positive results.
With the accrual of evidence suggesting the role of B cells in the pathogenesis of MS, monoclonal antibodies such as rituximab and ocrelizumab, which target CD20 on the surface of B cells, have emerged as therapeutic candidates (72, 293, 294). In a phase III clinical trial of ocrelizumab for the treatment of RRMS, compared with subcutaneous IFN-β, the drug resulted in a 45% reduction in aRR and in the progression of disability (294). Surprisingly, phase III trials for the same drug in the PPMS have shown positive results, and ocrelizumab was recently approved by the FDA as the first drug for the treatment of PPMS (72). In 2020, ofatumumab, a fully human IgG1 kappa anti-CD20 monoclonal antibody, RRMS AND SPMS, was approved for the treatment of CIS (295). In 2022, Ublituximab, a chimeric anti-CD20 monoclonal antibody, was approved for RRMS and SPMS (296).
Despite the significant efficacy of drugs approved for RRMS, most RRMS patients still progress to SPMS, which reflects an ongoing disease process. These drugs confer symptomatic treatment of the disease and slow progressive disability but are not a cure. Furthermore, some of these drugs have serious side effects, including fulminant hepatitis reported with IFN-β; opportunistic CNS infection known as natalizumab-associated progressive multifocal leukoencephalopathy (PML) (297, 298); bradycardia or conduction defects detected with fingolimod (268, 269); and increased disease activity after the withdrawal of natalizumab, fingolimod and other drugs (299). These side effects and the persistent potential to convert to progressive disease highlight that these DMTs are immunosuppressive and immunomodulating agents that act only on the peripheral inflammatory component of the disease. These drugs are immunosuppressive, which could explain why they lack efficacy in progressive forms of MS where there is less immune cell infiltration into the CNS and where disease progression is led by innate immune mechanisms of the CNS mediated by microglia and macrophages (72, 300).
There remains a need to explore and delineate pathobiological pathways that could be targeted by therapeutics to stop the progression of MS, which might include efforts to increase myelination or change the polarization of microglia. As previously mentioned, microglia appear to be important mediators of the chronic progressive disease process in PPMS and SPMS, with research revealing that they exert their effects on the surrounding CNS parenchymal cells and the BBB. The presence of activated microglia in the NAWM of MS patients, which are believed to be prelesional areas, further makes microglia extremely attractive targets for treating MS.
In vivo disease models
Different animal models have been used to investigate MS in vivo (Table 2), especially the experimental autoimmune encephalomyelitis (EAE) model (301, 302), which includes inflammation of the CNS triggered by infiltration of autoreactive T cells and monocytes, causing demyelination (303). The EAE model can be induced in vertebrates—mostly rodents (mice, rats, and guinea pigs)—either by active immunization with a CNS antigen or by adoptive (passive) transfer of activated T cells to naive animals (304–306).
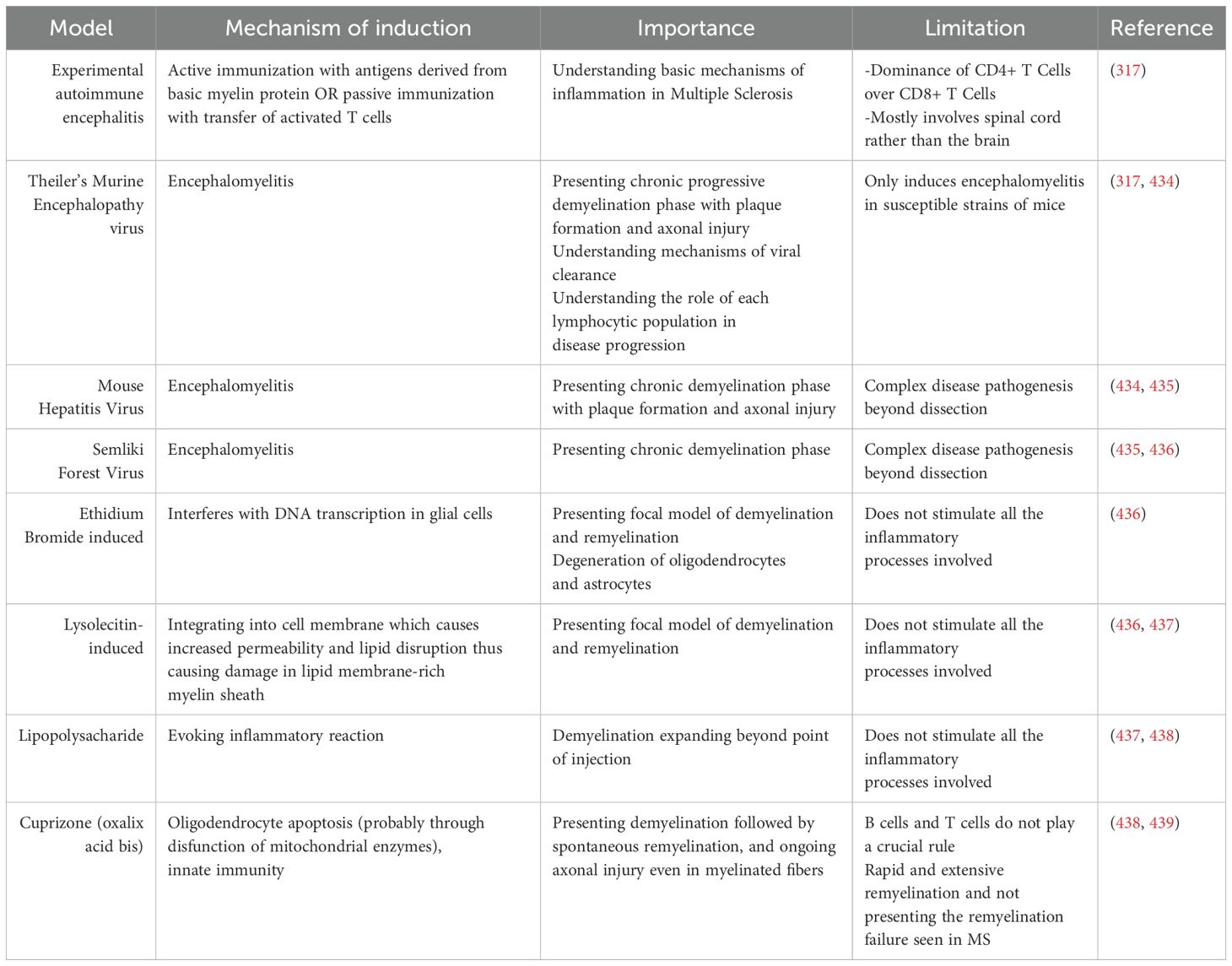
Table 2. In vivo preclinical models of multiple sclerosis.
During active immunization with a CNS antigen, signs of the disease appear within 10–17 days, whereas it takes 5–7 days in adoptive transfer (307). Adaptive immunization can be incomplete in that disease induction is dependent only on transferred CD4+ T cells and lacks the contributions of CD8+ T cells and B cells. The basic myelin proteins used for the sensitization of immune cells in active EAE models include myelin basic protein (MBP) (308, 309), myelin oligodendrocyte glycoprotein (MOG) 26959137, and proteolipid protein (PLP) (310); more recently, small antigenic peptides of these proteins, such as MBP1-37, MBP1-11, MBP1-9, MOG55-75, and PLP139-151, have been used (310, 311). Different animal strains have different potentials for developing autoreactive T cells upon immunization and hence different clinical and pathological presentations of the disease. The animals’ response is also affected by the type and dose of the antigen used as well as the animal’s age and sex (312, 313). Nevertheless, all actively immunized animals share an initial first finding of perivascular infiltration into white matter. Furthermore, most pathological changes associated with EAE are noted in the spinal cord and optic nerve but not in the brain (314–316).
The active induction of EAE recapitulates many aspects of MS, including the development of inflammation with immunoglobulin deposition, demyelination, and axonal damage, including gliosis and remyelination. In contrast, other features, including primary neurodegeneration, the involvement of CD8+ T cells, and cortical lesions, are not accurately modeled (317–319). As mentioned previously, some lesions in MS patients lack the prominent immune response features of demyelination and microglial activation. This category of lesion does not appear in EAE models, in which lesions are primarily immune mediated.
Many aspects of progressive MS are still not reflected in animal models, which contribute to the deficient development of therapeutics for progressive MS despite decades of MS research. The incidence of inflammatory demyelination in EAE patients decreases after the removal of the sensitizing brain antigen from the periphery (320, 321). Conversely, disease severity in progressive MS increases with time, which implies that a persistent stimulus must exist throughout the course of the disease, whether it is endogenous or exogenous to the CNS. Thus, EAE cannot recapitulate the progressive nature of the disease. One of the models mimicking secondary progressive MS involves repeated injections of the MOG 35-55 peptide, which causes long-term expression of the disease phenotype (322).
Owing to the potential viral etiology of MS, virus-induced in vivo models have been developed, including experimental demyelinating disease induced by Theiler’s murine encephalomyelitis virus (TMEV) (323, 324) and mouse hepatitis virus (MHV) (325). These are superior models to EAE with respect to the progressive accumulation of disability during demyelination and the longer incubation period before the appearance of symptoms, but there is a higher mortality rate in these animals in addition to the hazards associated with working with some of these viruses (326, 327).
Toxin-induced models for studying demyelination also exist, in which demyelination is induced by cytotoxic agents and does not result from immune attack (328). These models are useful for studying demyelination and remyelination mechanisms as well as potential remyelinating therapeutics (329). Examples of these focally used agents include lysolecithin (330), ethidium bromide (EtBr) (329, 331), and cuprizone. Lesions induced by these toxins differ from each other with respect to the process by which myelin is degraded as well as the degree of astrocyte loss (329).
Although naturally occurring animal models of EAE do not exist, researchers have discovered spontaneous autoimmune encephalomyelitis in transgenic mice expressing T-cell receptors specific to myelin antigen peptides (332–334). Humanized EAE mouse models have been developed in trials to overcome species-related differences in the molecular mechanisms of MS, especially the antigen presentation process, cell adhesion, and role of chemokines in disease pathogenesis (335, 336).
EAE has served as an experimental tool for the development of MS therapeutics, such as glatiramer acetate, mitoxantrone, and natalizumab (337, 338), and has been used to investigate the efficacy and safety of many other treatments, including methylprednisolone (339) for MS relapses and IFN-β, which cause disease exacerbation after treatment discontinuation (340). There are nevertheless also drugs that decrease disease activity in animal models but either fail to show any therapeutic efficacy in clinical trials or generate adverse effects. Examples of these drugs include monoclonal anti-tumor necrosis factor antibody cA2, which increases MRI activity in patients but does not improve symptoms (341); anti-CD28 Mab TGN-1412, which causes cytokine storms (342) and multiple organ failure; linomide, which causes cardiotoxicity (341) and oral tolerance (343); and sulfasalazine (344), which has no therapeutic effect. The translational failure of these therapeutics may be attributed in part to the differing genetic makeup between humans and rodents. In conclusion, there is no perfect in vivo animal model for MS, and the selection of a model should be based on the primary aim of the research and pathological mechanisms being investigated (345, 346).
In vitro models
With the advent of the era of translational medicine and the rapid advancement of in vitro models, researchers are increasingly directed toward in vitro models for neurodegenerative disease and CNS disorders. In vitro models have strong potential to overcome some of the limitations mentioned in the in vivo models of MS, such as the differences between the human and rodent genomes and the resulting differences in molecular mechanisms. These limitations, at least in part, contributed to the failure of the translation of many therapeutics from animal models to clinical trials and the lack of a definitive cure for the disease process. In addition, one of the most important hurdles is the lack of models for progressive disease, as well as treatments. One of the main advantages of these in vitro models is the ability to scale them to enable high-throughput screening of drug targets and extensive studies of molecular mechanisms to reveal more therapeutic hits. In these models, interactions between different CNS cells and immune cells can be studied closely in a simple setting and manipulated with a high degree of precision.
With increasing awareness of the involvement of blood–brain barrier dysfunction in many neurological and psychological disorders, efforts have been made to model the BBB in vitro to offer a simplified, reproducible biological platform to study these disorders and translate findings into clinical practice.
BBB dysfunction has been reported in a variety of neurodegenerative disorders in addition to the previously discussed MS, including Alzheimer’s disease (AD) (347–350), amyotrophic lateral sclerosis (ALS) (351), Parkinson’s disease (PD) (352), and Huntington’s disease (HD) (353). Furthermore, BBB breakdown in epilepsy is thought to adversely affect the CNS microenvironment and neuronal physiology (354). Cognitive and neurological decline during aging is also attributed in part to BBB dysfunction (355, 356). Furthermore, posttraumatic epilepsy and neural degeneration, which are cognitive and psychological disorders that occur after traumatic brain injury, are related to BBB dysfunction (357–359). BBB dysfunction has even been implicated in neuropsychological disorders such as schizophrenia and autism. (360).
Different models have been developed to investigate BBB dysfunction in brain disorders, screen drugs for their ability to cross the BBB (361), and study neuroimmunological interactions at the BBB interface (362). These BBB models differ in the source and the type of brain microvascular endothelial cells used (Table 3). During the last few years, the design of in vitro BBB models has progressed from using 2D monolayers or transwell models to more sophisticated designs that include shear stress induced by fluid flow in microfluidic devices or 3D organoid (Table 4). The developments also included using more than one cell type in a co-culture rather than just using BMVECs alone. Coculture BBB models can provide endothelial cells with the necessary signals from other cells in the NVU when combined with astrocytes, pericytes, and neurons, which contributes meaningfully to barrier properties (363, 364). These barrier properties are not intrinsic to brain endothelial cells but rather depend on the microenvironment of the NVU (365, 366) Each of the cell types or the designs has its own advantages and disadvantages to be considered while picking up a model to answer a specific research question.
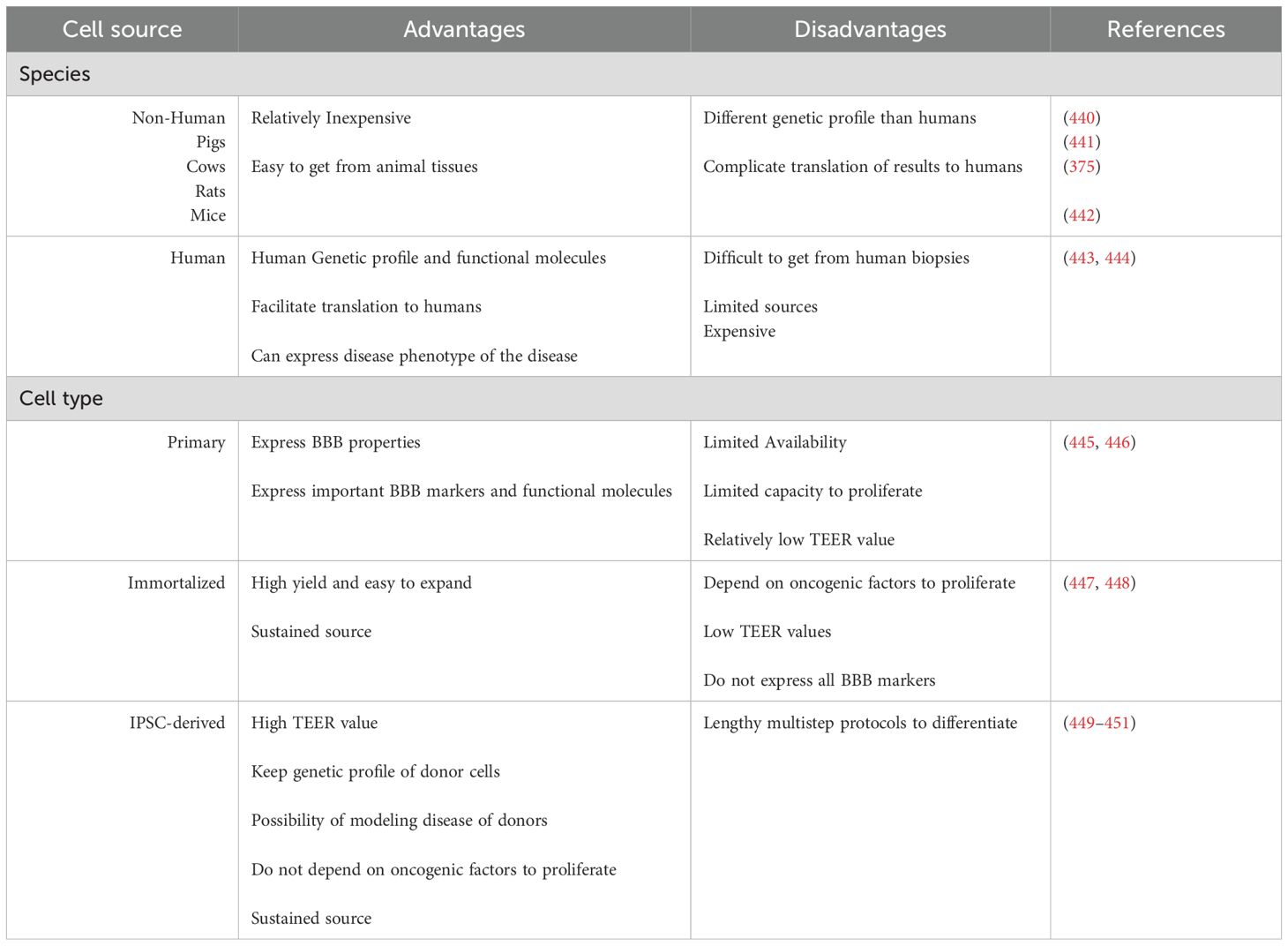
Table 3. Cell source for in-vitro BBB models.
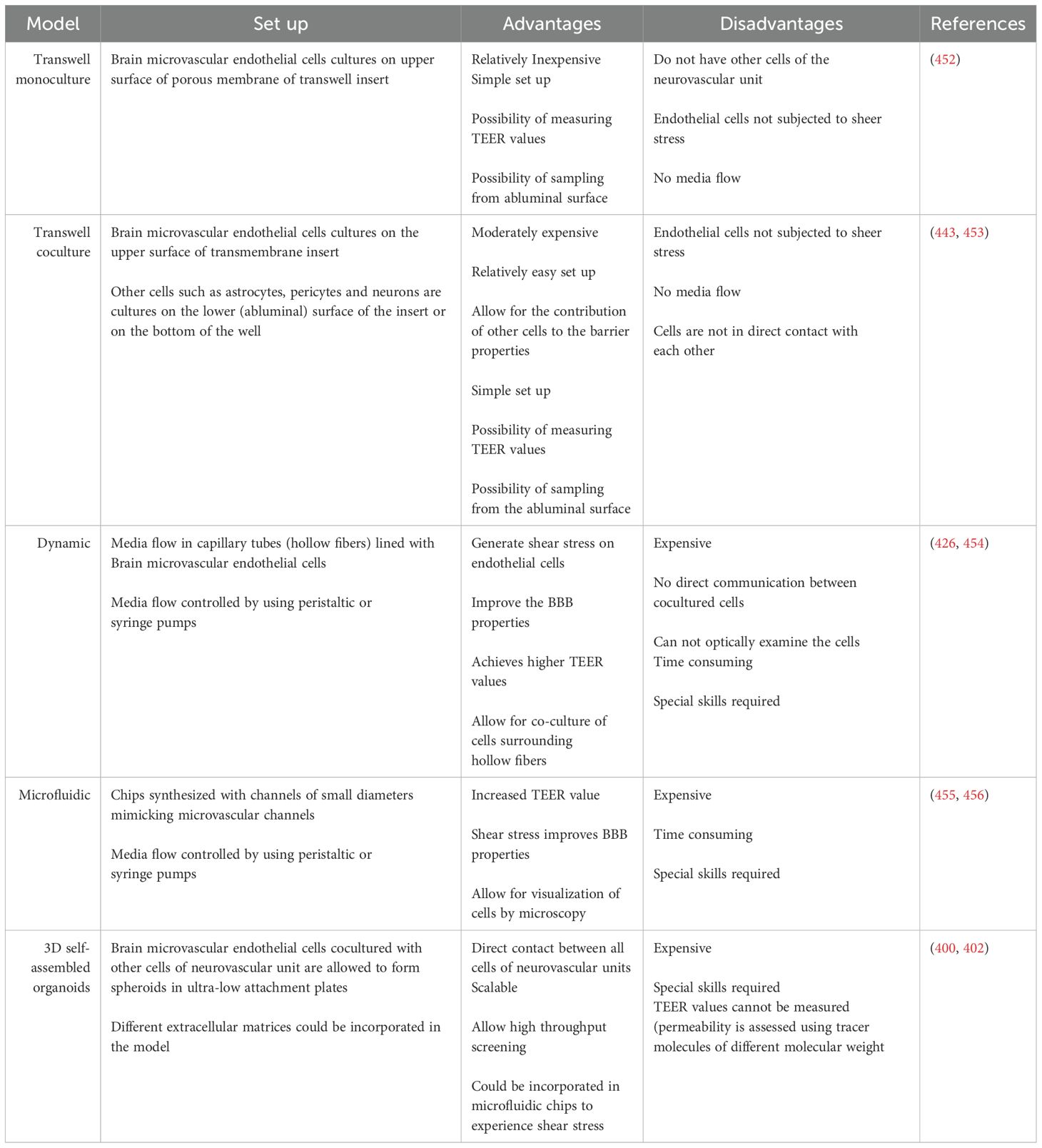
Table 4. Designs for in-vitro BBB models.
In vitro BBB models were initially developed with brain endothelial cells cultured on transwell inserts. These systems allow the measurement of transendothelial electrical resistance (TEER) values across the monolayer and direct measurement of permeability by sampling from luminal (blood facing) and abluminal (brain-facing) compartments (367–370). Transwell systems using cocultures of brain endothelial cells, astrocytes (371–375) and pericytes (376–378), either in contact or noncontact settings, presented increased TEER values, which was reflected by decreased permeability to tracer molecules such as lucifer yellow, sodium fluorescein, sucrose, and dextrans. Move to table
The transwell in vitro BBB model has been used to study immune system interactions at the BBB interface. In these models, endothelial cells can be stimulated by pathogenic factors such as LPS (379–382) or by proinflammatory cytokines such as TNF-α, IL-6, IL-1β, and IFN-δ (382–384). These systems have been extensively used to study the regulation of the transmigration of monocytes, neutrophils, and lymphocytes across the BBB (385–387) and to investigate the effects of adhesion molecules on leukocyte transmigration (148, 385, 388–392).
Transwell coculture models have also been used to study the involvement of the BBB in MS. Some studies have used 2D brain endothelial cell cultures to study the ability of patient-derived sera to modulate BBB properties, especially tight junction protein and adhesion molecule expression. Using a transwell BBB model, Shimizu et al. reported that serum from MS patients disrupts BBB function by decreasing claudin-5 expression and decreasing the TEER value, which reflects increased BBB permeability. VCAM1 expression increased in response to exposure to sera and IgG from different types of MS patients. Disruption can be prevented by the addition of MMP inhibitors. (167). Similarly, Minagar et al. demonstrated that sera from patients with exacerbated MS could decrease the expression of occludin and VE-cadherin in endothelial cells (168). Sheikh et al. demonstrated that sera from MS patients could alter the metabolic function of the brain endothelium by decreasing glycolytic activity, the oxygen consumption rate and the expression of endothelial glucose transporter 1 (GLUT-1) (393).
Additionally, other studies used in vitro transwell BBB models to study the capacity of immune cells isolated from the peripheral blood of MS patients to cross the BBB. Prat et al. showed that, compared with healthy control lymphocytes, MS patient lymphocytes exhibited enhanced migration across the in vitro transwell BBB and that transmigration could be reduced via the use of an anti-monocyte chemoattractant protein 1 monoclonal antibody (394). In another study, the authors showed that CD4+ T cells from MS patients presented increased expression of P-selectin glycoprotein ligand-1 (PSGL-1). Compared with PSGL-1-negative T cells, CD4+ T cells positive for PSGL-1 showed an increased capacity to transmigrate across the BBB (395). Despite the popularity, relative simplicity, and low cost of transwell BBB models, they may not reflect the complex interactions and contact between different cellular and acellular elements of the NVU and may lack the physiological shear stress that helps maintain several BBB properties (396).
3D BBB models have been developed to overcome this contact issue via coculture of primary brain endothelial cells, astrocytes and pericytes under low-adherence conditions, allowing the formation of BBB multicellular organoids, which exhibit BBB properties (397, 398). These organoids have the advantages of direct contact between cells, are reproducible, and can be cost-effective relative to animal models. BBB organoids could be used to study drug transport across the BBB, investigate neurological disease mechanisms, and develop therapeutics (397). 3D BBB models have been used to study general inflammatory conditions by exposing organoids to exogenous inflammatory cytokines to mimic neuroinflammatory conditions. The use of 3D BBB models to study MS-specific features has not been widely applied (399).
The absence of neurons and glial cells is a limitation of most BBB organoids, as they are critical contributors to BBB development and are necessary to study neurovascular coupling in neurological disorders in conjunction with the BBB. Nzou et al. reported the development of a human neurovascular unit organoid model that contains all six constituent human cell types found within the brain cortex: brain microvascular endothelial cells; pericytes; astrocytes; microglia; oligodendrocytes; and neurons, with endothelial cells enclosing the brain parenchymal cells (400). This 3D in vitro system contains all major cell types found in the adult human brain cortex and provides a platform to understand the fundamental principles at play with the BBB and its function and to understand the effects of substances that cross the BBB. This sophisticated human brain model system has been used to study hypoxia, inflammation, and the delivery of therapeutic agents across the BBB (401–404).
The development and incorporation of iPSC-derived brain endothelial cells (iBECs) in BBB coculture models resulted in a BBB with high TEER values (405, 406). These models have been widely used to study disease pathophysiology (407–409) and drug screening (410, 411). HiPSCs have been used to model many brain disorders, including Parkinson’s disease (PD) (412) and Alzheimer’s disease (AD) (413).Although they have not yet been applied directly to study MS, they present great potential for integrating NVU cells from MS patients to study the contribution of the genome to MS pathophysiology. In addition, these iPSC-derived endothelial cells can be manipulated by gene editing to introduce specific genetic mutations to study their effects on NVU function (414). One of the most recent advances in the field of brain organoids with relevance to multiple sclerosis is the induction of myelination in human cortical spheroids, which makes them good platforms for studying demyelination events in neurodegenerative disorders (415).
With advancements in microfluidic technology, BBB-on-a-chip models have emerged, allowing perfusion of the BBB in two-dimensional microfluidic models (416–418), hybrid systems (419, 420), or self-organizing 3-D multicellular BBB models (421, 422). 3D self-assembled BBB organoids could be incorporated into microfluidic chips. Six different human organoids, including a brain with six different cell types that form a 3D BBB, liver, heart, lung, vascular and testes, were incorporated into a single microfluidic body-on-a-chip system to study integrated functional parameters (423). The same integrated body-on-a-chip system was used to test the effect of prodrug metabolism by the liver and to prove its toxic effect on other organoids (424). The metabolism of the alkylating drug ifosfamide in liver organoids into chloroacetaldehyde results in BBB neurotoxicity downstream. Although there are no other multi-tissue organ-on-a-chip models reported to date that include the BBB with other organs, multiorgan-on-a-chip (multi-OoC) models represent strong candidates for investigating and better understanding the human body, systemic illnesses and organ communications. This system can support screening for drug efficacy and toxicity prior to translation to clinical trials and can help reduce the number of animals used for in vivo studies.
These microfluidic devices help investigate the BBB in a more physiologically relevant microenvironment, but they require special skills and equipment (425). Microfluidic BBB models have been used to study the transmigration of immune cells across the barrier. Flow has been demonstrated to enhance BBB integrity and upregulate the expression of tight junction proteins. Compared with that in static models, the transmigration of immune cells is reduced (426). Nair et al. used a microfluidic BBB model to test the effects of proinflammatory cytokines on BBB integrity and permeability. TNFα and interleukin-1 beta (IL-1β) disrupt BBB integrity and increase BBB permeability. The expression of cell adhesion molecules increases with the subsequent increase in the transmigration of human T cells and the inhibition of transmigration in the presence of natalizumab (427). Microfluidic BBB models have not yet been fully exploited in the study of the disease-specific pathophysiology of MS or immune cells in MS patients.
Even with the great advancements observed in the field of translational research and modeling platforms, a disease as complex as MS remains relatively uninvestigated. Owing to the failure of the translation of many MS drugs from animal models to humans, the lack of true therapeutic options to cure MS, the severe side effects imposed by the currently available therapeutics, and the lack of medications for the progressive forms of the disease, the exploitation of alternative modeling options with greater biological relevance to the human body has become a great opportunity for the scientific community. Improving translational and regenerative medicine approaches, such as multicellular human BBB models, create a very promising field to investigate and test therapeutics for MS. Such models could be used to study more biologically involved processes; brain organoids, which include an immune cell component, could be used to study neuroimmune interactions at the BBB interface and the crosstalk between immune cells and CNS parenchymal microglia, and myelinating brain organoids could be used to study demyelination pathogenesis and remyelination mechanisms in MS. Recent success in generating iPSCs from MS patients is a further promising step toward personalizing brain and BBB models for MS.
Author contributions
AE-T: Supervision, Writing – review & editing, Conceptualization, Writing – original draft, Validation. PH: Writing – review & editing. DP: Writing – review & editing. MS: Writing – review & editing, Supervision, Validation. AA: Validation, Supervision, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. WFIRM’s mission driven research is supported by various funding sources to include state and sponsored funding.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Simone IL, Carrara D, Tortorella C, Ceccarelli A, and Livrea P. Early onset multiple sclerosis. Neurol Sci. (2000) 21:S861–3. doi: 10.1007/s100720070027
2. Ding C, Wu Y, Chen X, Chen Y, Wu Z, Lin Z, et al. Global, regional, and national burden and attributable risk factors of neurological disorders: The Global Burden of Disease study 1990-2019. Front Public Health. (2022) 10:952161. doi: 10.3389/fpubh.2022.952161
3. Levin LI, Munger KL, Rubertone MV, Peck CA, Lennette ET, Spiegelman D, et al. Temporal relationship between elevation of epstein-barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis. JAMA. (2005) 293:2496–500. doi: 10.1001/jama.293.20.2496
4. Ascherio A, Munger KL, Lennette ET, Spiegelman D, Hernan MA, Olek MJ, et al. Epstein-Barr virus antibodies and risk of multiple sclerosis: a prospective study. JAMA. (2001) 286:3083–8. doi: 10.1001/jama.286.24.3083
5. Sundstrom P, Juto P, Wadell G, Hallmans G, Svenningsson A, Nystrom L, et al. An altered immune response to Epstein-Barr virus in multiple sclerosis: a prospective study. Neurology. (2004) 62:2277–82. doi: 10.1212/01.WNL.0000130496.51156.D7
6. Marrie RA and Wolfson C. Multiple sclerosis and Epstein-Barr virus. Can J Infect Dis. (2002) 13:111–8. doi: 10.1155/2002/745764
7. Thacker EL, Mirzaei F, and Ascherio A. Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis. Ann Neurol. (2006) 59:499–503. doi: 10.1002/ana.20820
8. Handel AE, Williamson AJ, Disanto G, Handunnetthi L, Giovannoni G, and Ramagopalan SV. An updated meta-analysis of risk of multiple sclerosis following infectious mononucleosis. PLoS One. (2010) 5. doi: 10.1371/journal.pone.0012496
9. Levin LI, Munger KL, O'Reilly EJ, Falk KI, and Ascherio A. Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann Neurol. (2010) 67:824–30. doi: 10.1002/ana.21978
10. Lunemann JD, Jelcic I, Roberts S, Lutterotti A, Tackenberg B, Martin R, et al. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J Exp Med. (2008) 205:1763–73. doi: 10.1084/jem.20072397
11. Ransohoff RM and Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. (2012) 12:623–35. doi: 10.1038/nri3265
12. Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med. (2007) 204:2899–912. doi: 10.1084/jem.20071030
13. Serafini B, Severa M, Columba-Cabezas S, Rosicarelli B, Veroni C, Chiappetta G, et al. Epstein-Barr virus latent infection and BAFF expression in B cells in the multiple sclerosis brain: implications for viral persistence and intrathecal B-cell activation. J Neuropathol Exp Neurol. (2010) 69:677–93. doi: 10.1097/NEN.0b013e3181e332ec
14. Willis SN, Stadelmann C, Rodig SJ, Caron T, Gattenloehner S, Mallozzi SS, et al. Epstein-Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain. (2009) 132:3318–28. doi: 10.1093/brain/awp200
15. Lassmann H, Niedobitek G, Aloisi F, Middeldorp JM, and NeuroProMiSe EBVWG. Epstein-Barr virus in the multiple sclerosis brain: a controversial issue–report on a focused workshop held in the Centre for Brain Research of the Medical University of Vienna, Austria. Brain. (2011) 134:2772–86. doi: 10.1093/brain/awr197
16. Bartosik-Psujek H and Psujek M. Vitamin D as an immune modulator in multiple sclerosis. Neurol Neurochir Pol. (2019) 53:113–22. doi: 10.5603/PJNNS.a2019.0015
17. Kamen DL and Tangpricha V. Vitamin D and molecular actions on the immune system: modulation of innate and autoimmunity. J Mol Med (Berl). (2010) 88:441–50. doi: 10.1007/s00109-010-0590-9
18. May E, Asadullah K, and Zugel U. Immunoregulation through 1,25-dihydroxyvitamin D3 and its analogs. Curr Drug Targets Inflammation Allergy. (2004) 3:377–93. doi: 10.2174/1568010042634596
19. Marrie RA, Rudick R, Horwitz R, Cutter G, Tyry T, Campagnolo D, et al. Vascular comorbidity is associated with more rapid disability progression in multiple sclerosis. Neurology. (2010) 74:1041–7. doi: 10.1212/WNL.0b013e3181d6b125
20. Sundstrom P, Nystrom L, and Hallmans G. Smoke exposure increases the risk for multiple sclerosis. Eur J Neurol. (2008) 15:579–83. doi: 10.1111/j.1468-1331.2008.02122.x
21. Handel AE, Williamson AJ, Disanto G, Dobson R, Giovannoni G, and Ramagopalan SV. Smoking and multiple sclerosis: an updated meta-analysis. PLoS One. (2011) 6:e16149. doi: 10.1371/journal.pone.0016149
22. Hsieh DT, Warden GI, Butler JM, Nakanishi E, and Asano Y. Multiple sclerosis exacerbation associated with high-altitude climbing exposure. Mil Med. (2020) 185:e1322–5. doi: 10.1093/milmed/usz421
23. Sabel CE, Pearson JF, Mason DF, Willoughby E, Abernethy DA, and Taylor BV. The latitude gradient for multiple sclerosis prevalence is established in the early life course. Brain. (2021) 144:2038–46. doi: 10.1093/brain/awab104
24. Wood H. Multiple sclerosis: Latitude and vitamin D influence disease course in multiple sclerosis. Nat Rev Neurol. (2017) 13:3. doi: 10.1038/nrneurol.2016.181
25. Willer CJ, Dyment DA, Risch NJ, Sadovnick AD, Ebers GC, and Canadian Collaborative Study G. Twin concordance and sibling recurrence rates in multiple sclerosis. Proc Natl Acad Sci U S A. (2003) 100:12877–82. doi: 10.1073/pnas.1932604100
26. Cotsapas C and Mitrovic M. Genome-wide association studies of multiple sclerosis. Clin Transl Immunol. (2018) 7:e1018. doi: 10.1002/cti2.1018
27. Falcao AM, van Bruggen D, Marques S, Meijer M, Jakel S, Agirre E, et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med. (2018) 24:1837–44. doi: 10.1038/s41591-018-0236-y
28. Hollenbach JA and Oksenberg JR. The immunogenetics of multiple sclerosis: A comprehensive review. J Autoimmun. (2015) 64:13–25. doi: 10.1016/j.jaut.2015.06.010
29. Ma Q, Shams H, Didonna A, Baranzini SE, Cree BAC, Hauser SL, et al. Integration of epigenetic and genetic profiles identifies multiple sclerosis disease-critical cell types and genes. Commun Biol. (2023) 6:342. doi: 10.1038/s42003-023-04713-5
30. Lin X, Yang Y, Gresle M, Cuellar-Partida G, Han X, Stankovich J, et al. Novel plasma and brain proteins that are implicated in multiple sclerosis. Brain. (2023) 146:2464–75. doi: 10.1093/brain/awac420
31. Kim W and Patsopoulos NA. Genetics and functional genomics of multiple sclerosis. Semin Immunopathol. (2022) 44:63–79. doi: 10.1007/s00281-021-00907-3
32. Goris A, Vandebergh M, McCauley JL, Saarela J, and Cotsapas C. Genetics of multiple sclerosis: lessons from polygenicity. Lancet Neurol. (2022) 21:830–42. doi: 10.1016/S1474-4422(22)00255-1
33. Desai RA, Davies AL, Tachrount M, Kasti M, Laulund F, Golay X, et al. Cause and prevention of demyelination in a model multiple sclerosis lesion. Ann Neurol. (2016) 79:591–604. doi: 10.1002/ana.24607
34. Dendrou CA, Fugger L, and Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. (2015) 15:545–58. doi: 10.1038/nri3871
35. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. (2018) 17:162–73. doi: 10.1016/S1474-4422(17)30470-2
36. Milo R and Miller A. Revised diagnostic criteria of multiple sclerosis. Autoimmun Rev. (2014) 13:518–24. doi: 10.1016/j.autrev.2014.01.012
37. Katz Sand I. Classification, diagnosis, and differential diagnosis of multiple sclerosis. Curr Opin Neurol. (2015) 28:193–205. doi: 10.1097/WCO.0000000000000206
39. Lassmann H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front Immunol. (2018) 9:3116. doi: 10.3389/fimmu.2018.03116
40. Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. (2005) 128:2705–12. doi: 10.1093/brain/awh641
41. Lassmann H, Bruck W, and Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol. (2007) 17:210–8. doi: 10.1111/j.1750-3639.2007.00064.x
42. Schmierer K and Miquel ME. Magnetic resonance imaging correlates of neuro-axonal pathology in the MS spinal cord. Brain Pathol. (2018) 28:765–72. doi: 10.1111/bpa.2018.28.issue-5
43. Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H, et al. Remyelination is extensive in a subset of multiple sclerosis patients. Brain. (2006) 129:3165–72. doi: 10.1093/brain/awl217
44. Patani R, Balaratnam M, Vora A, and Reynolds R. Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathol Appl Neurobiol. (2007) 33:277–87. doi: 10.1111/j.1365-2990.2007.00805.x
45. Kuhlmann T, Ludwin S, Prat A, Antel J, Bruck W, and Lassmann H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. (2017) 133:13–24. doi: 10.1007/s00401-016-1653-y
46. Frischer JM, Weigand SD, Guo Y, Kale N, Parisi JE, Pirko I, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol. (2015) 78:710–21. doi: 10.1002/ana.24497
47. Thompson AJ, Polman CH, Miller DH, McDonald WI, Brochet B, Filippi MMX, et al. Primary progressive multiple sclerosis. Brain. (1997) 120:1085–96. doi: 10.1093/brain/120.6.1085
48. Bramow S, Frischer JM, Lassmann H, Koch-Henriksen N, Lucchinetti CF, Sorensen PS, et al. Demyelination versus remyelination in progressive multiple sclerosis. Brain. (2010) 133:2983–98. doi: 10.1093/brain/awq250
49. Luchetti S, Fransen NL, van Eden CG, Ramaglia V, Mason M, and Huitinga I. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: a retrospective autopsy cohort analysis. Acta Neuropathol. (2018) 135:511–28. doi: 10.1007/s00401-018-1818-y
50. Revesz T, Kidd D, Thompson AJ, Barnard RO, and McDonald WI. A comparison of the pathology of primary and secondary progressive multiple sclerosis. Brain. (1994) 117:759–65. doi: 10.1093/brain/117.4.759
51. Hayashi T, Morimoto C, Burks JS, Kerr C, and Hauser SL. Dual-label immunocytochemistry of the active multiple sclerosis lesion: major histocompatibility complex and activation antigens. Ann Neurol. (1988) 24:523–31. doi: 10.1002/ana.410240408
52. Booss J, Esiri MM, Tourtellotte WW, and Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J Neurol Sci. (1983) 62:219–32. doi: 10.1016/0022-510X(83)90201-0
53. MaChado-Santos J, Saji E, Troscher AR, Paunovic M, Liblau R, Gabriely G, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain. (2018) 141:2066–82. doi: 10.1093/brain/awy151
54. International Multiple Sclerosis Genetics Consortium, Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. (2013) 45:1353–60. doi: 10.1038/ng.2770
55. Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. (2000) 192:393–404. doi: 10.1084/jem.192.3.393
56. Jacobsen M, Cepok S, Quak E, Happel M, Gaber R, Ziegler A, et al. Oligoclonal expansion of memory CD8+ T cells in cerebrospinal fluid from multiple sclerosis patients. Brain. (2002) 125, 538–50. doi: 10.1093/brain/awf059
57. Wakim LM, Woodward-Davis A, and Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci U S A. (2010) 107:17872–9. doi: 10.1073/pnas.1010201107
58. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, and Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. (2000) 100:655–69. doi: 10.1016/S0092-8674(00)80702-3
59. Thieu VT, Yu Q, Chang HC, Yeh N, Nguyen ET, Sehra S, et al. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity. (2008) 29:679–90. doi: 10.1016/j.immuni.2008.08.017
60. Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM, et al. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol. (2007) 8:145–53. doi: 10.1038/ni1424
61. Lazarevic V, Chen X, Shim JH, Hwang ES, Jang E, Bolm AN, et al. T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORgammat. Nat Immunol. (2011) 12:96–104. doi: 10.1038/ni.1969
62. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. (2007) 8:1390–7. doi: 10.1038/ni1539
63. Stockinger B and Omenetti S. The dichotomous nature of T helper 17 cells. Nat Rev Immunol. (2017) 17:535–44. doi: 10.1038/nri.2017.50
64. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. (2012) 13:991–9. doi: 10.1038/ni.2416
65. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
66. Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. (2009) 132:1175–89. doi: 10.1093/brain/awp070
67. Funaro M, Messina M, Shabbir M, Wright P, Najjar S, Tabansky I, et al. The role of B cells in multiple sclerosis: more than antibodies. Discov Med. (2016) 22:251–5.
68. Lisak RP, Benjamins JA, Nedelkoska L, Barger JL, Ragheb S, Fan B, et al. Secretory products of multiple sclerosis B cells are cytotoxic to oligodendroglia in vitro. J Neuroimmunol. (2012) 246:85–95. doi: 10.1016/j.jneuroim.2012.02.015
69. Lisak RP, Nedelkoska L, Benjamins JA, Schalk D, Bealmear B, Touil H, et al. B cells from patients with multiple sclerosis induce cell death via apoptosis in neurons in vitro. J Neuroimmunol. (2017) 309:88–99. doi: 10.1016/j.jneuroim.2017.05.004
70. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. (2008) 358:676–88. doi: 10.1056/NEJMoa0706383
71. Myhr KM, Torkildsen O, Lossius A, Bo L, and Holmoy T. B cell depletion in the treatment of multiple sclerosis. Expert Opin Biol Ther. (2019) 19:261–71. doi: 10.1080/14712598.2019.1568407
72. Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. (2017) 376:209–20. doi: 10.1056/NEJMoa1606468
73. Probstel AK, Sanderson NS, and Derfuss T. B cells and autoantibodies in multiple sclerosis. Int J Mol Sci. (2015) 16:16576–92. doi: 10.3390/ijms160716576
74. Haider L, Fischer MT, Frischer JM, Bauer J, Hoftberger R, Botond G, et al. Oxidative damage in multiple sclerosis lesions. Brain. (2011) 134:1914–24. doi: 10.1093/brain/awr128
75. Witte ME, Nijland PG, Drexhage JA, Gerritsen W, Geerts D, van Het Hof B, et al. Reduced expression of PGC-1alpha partly underlies mitochondrial changes and correlates with neuronal loss in multiple sclerosis cortex. Acta Neuropathol. (2013) 125:231–43. doi: 10.1007/s00401-012-1052-y
76. Esiri MM and Gay D. Immunological and neuropathological significance of the Virchow-Robin space. J Neurol Sci. (1990) 100:3–8. doi: 10.1016/0022-510X(90)90004-7
77. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, and Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. (2004) 14:164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x
78. Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. (2011) 134:2755–71. doi: 10.1093/brain/awr182
79. Choi SR, Howell OW, Carassiti D, Magliozzi R, Gveric D, Muraro PA, et al. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain. (2012) 135:2925–37. doi: 10.1093/brain/aws189
80. Trapp BD, Vignos M, Dudman J, Chang A, Fisher E, Staugaitis SM, et al. Cortical neuronal densities and cerebral white matter demyelination in multiple sclerosis: a retrospective study. Lancet Neurol. (2018) 17:870–84. doi: 10.1016/S1474-4422(18)30245-X
81. Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. (2011) 365:2188–97. doi: 10.1056/NEJMoa1100648
82. Haider L, Simeonidou C, Steinberger G, Hametner S, Grigoriadis N, Deretzi G, et al. Multiple sclerosis deep grey matter: the relation between demyelination, neurodegeneration, inflammation and iron. J Neurol Neurosurg Psychiatry. (2014) 85:1386–95. doi: 10.1136/jnnp-2014-307712
83. Vercellino M, Masera S, Lorenzatti M, Condello C, Merola A, Mattioda A, et al. Demyelination, inflammation, and neurodegeneration in multiple sclerosis deep gray matter. J Neuropathol Exp Neurol. (2009) 68:489–502. doi: 10.1097/NEN.0b013e3181a19a5a
84. Qiu W, Raven S, Wu JS, Bundell C, Hollingsworth P, Carroll WM, et al. Hypothalamic lesions in multiple sclerosis. J Neurol Neurosurg Psychiatry. (2011) 82:819–22. doi: 10.1136/jnnp.2009.198192
85. Gallego-Delgado P, James R, Browne E, Meng J, Umashankar S, Tan L, et al. Neuroinflammation in the normal-appearing white matter (NAWM) of the multiple sclerosis brain causes abnormalities at the nodes of Ranvier. PLoS Biol. (2020) 18:e3001008. doi: 10.1371/journal.pbio.3001008
86. Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. (2007) 130:1089–104. doi: 10.1093/brain/awm038
87. Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B, et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol. (2010) 68:477–93. doi: 10.1002/ana.22230
88. Vidaurre OG, Haines JD, Katz Sand I, Adula KP, Huynh JL, McGraw CA, et al. Cerebrospinal fluid ceramides from patients with multiple sclerosis impair neuronal bioenergetics. Brain. (2014) 137:2271–86. doi: 10.1093/brain/awu139
89. Chiou B, Lucassen E, Sather M, Kallianpur A, and Connor J. Semaphorin4A and H-ferritin utilize Tim-1 on human oligodendrocytes: A novel neuro-immune axis. Glia. (2018) 66:1317–30. doi: 10.1002/glia.23313
90. Filippi M, Paty DW, Kappos L, Barkhof F, Compston DA, Thompson AJ, et al. Correlations between changes in disability and T2-weighted brain MRI activity in multiple sclerosis: a follow-up study. Neurology. (1995) 45:255–60. doi: 10.1212/WNL.45.2.255
91. Rovaris M, Bozzali M, Santuccio G, Ghezzi A, Caputo D, Montanari E, et al. In vivo assessment of the brain and cervical cord pathology of patients with primary progressive multiple sclerosis. Brain. (2001) 124:2540–9. doi: 10.1093/brain/124.12.2540
92. Miller DH, Thompson AJ, and Filippi M. Magnetic resonance studies of abnormalities in the normal appearing white matter and grey matter in multiple sclerosis. J Neurol. (2003) 250:1407–19. doi: 10.1007/s00415-003-0243-9
93. Ciccarelli O, Werring DJ, Barker GJ, Griffin CM, Wheeler-Kingshott CA, Miller DH, et al. A study of the mechanisms of normal-appearing white matter damage in multiple sclerosis using diffusion tensor imaging–evidence of Wallerian degeneration. J Neurol. (2003) 250:287–92. doi: 10.1007/s00415-003-0992-5
94. Ge Y, Grossman RI, Babb JS, He J, and Mannon LJ. Dirty-appearing white matter in multiple sclerosis: volumetric MR imaging and magnetization transfer ratio histogram analysis. AJNR Am J Neuroradiol. (2003) 24:1935–40.
95. Bjartmar C, Kidd G, Mork S, Rudick R, and Trapp BD. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol. (2000) 48:893–901. doi: 10.1002/1531-8249(200012)48:6<893::AID-ANA10>3.0.CO;2-B
96. Mews I, Bergmann M, Bunkowski S, Gullotta F, and Bruck W. Oligodendrocyte and axon pathology in clinically silent multiple sclerosis lesions. Mult Scler. (1998) 4:55–62. doi: 10.1177/135245859800400203
97. Allen IV, McQuaid S, Mirakhur M, and Nevin G. Pathological abnormalities in the normal-appearing white matter in multiple sclerosis. Neurol Sci. (2001) 22:141–4. doi: 10.1007/s100720170012
98. Evangelou N, Konz D, Esiri MM, Smith S, Palace J, and Matthews PM. Regional axonal loss in the corpus callosum correlates with cerebral white matter lesion volume and distribution in multiple sclerosis. Brain. (2000) 123:1845–9. doi: 10.1093/brain/123.9.1845
99. Androdias G, Reynolds R, Chanal M, Ritleng C, Confavreux C, and Nataf S. Meningeal T cells associate with diffuse axonal loss in multiple sclerosis spinal cords. Ann Neurol. (2010) 68:465–76. doi: 10.1002/ana.22054
100. Moore GR, Laule C, Mackay A, Leung E, Li DK, and Zhao G. Dirty-appearing white matter in multiple sclerosis: preliminary observations of myelin phospholipid and axonal loss. J Neurol. (2008) 255:1802–11. doi: 10.1007/s00415-008-0002-z
101. Laule C, Vavasour IM, Leung E, Li DK, Kozlowski P, Traboulsee AL, et al. Pathological basis of diffusely abnormal white matter: insights from magnetic resonance imaging and histology. Mult Scler. (2011) 17:144–50. doi: 10.1177/1352458510384008
102. Cramer SP, Simonsen H, Frederiksen JL, Rostrup E, and Larsson HB. Abnormal blood-brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI. NeuroImage Clin. (2014) 4:182–9. doi: 10.1016/j.nicl.2013.12.001
103. Henderson AP, Barnett MH, Parratt JD, and Prineas JW. Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol. (2009) 66:739–53. doi: 10.1002/ana.21800
104. van der Valk P and Amor S. Preactive lesions in multiple sclerosis. Curr Opin Neurol. (2009) 22:207–13. doi: 10.1097/WCO.0b013e32832b4c76
105. Ballabh P, Braun A, and Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. (2004) 16:1–13. doi: 10.1016/j.nbd.2003.12.016
106. Harhaj NS and Antonetti DA. Regulation of tight junctions and loss of barrier function in pathophysiology. Int J Biochem Cell Biol. (2004) 36:1206–37. doi: 10.1016/j.biocel.2003.08.007
107. Vorbrodt AW and Dobrogowska DH. Molecular anatomy of intercellular junctions in brain endothelial and epithelial barriers: electron microscopist’s view. Brain Res Brain Res Rev. (2003) 42:221–42. doi: 10.1016/S0165-0173(03)00177-2
108. Wolburg H and Lippoldt A. Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul Pharmacol. (2002) 38:323–37. doi: 10.1016/S1537-1891(02)00200-8
109. Schulze C and Firth JA. Immunohistochemical localization of adherens junction components in blood-brain barrier microvessels of the rat. J Cell Sci. (1993) 104:773–82. doi: 10.1242/jcs.104.3.773
110. Gloor SM, Wachtel M, Bolliger MF, Ishihara H, Landmann R, and Frei K. Molecular and cellular permeability control at the blood-brain barrier. Brain Res Brain Res Rev. (2001) 36:258–64. doi: 10.1016/S0165-0173(01)00102-3
111. Butt AM, Jones HC, and Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study. J Physiol. (1990) 429:47–62. doi: 10.1113/jphysiol.1990.sp018243
112. Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. (1993) 123:1777–88. doi: 10.1083/jcb.123.6.1777
113. Furuse M, Fujita K, Hiiragi T, Fujimoto K, and Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. (1998) 141:1539–50. doi: 10.1083/jcb.141.7.1539
114. Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, et al. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. (2000) 11:4131–42. doi: 10.1091/mbc.11.12.4131
115. Gow A, Southwood CM, Li JS, Pariali M, Riordan GP, Brodie SE, et al. CNS myelin and sertoli cell tight junction strands are absent in Osp/claudin-11 null mice. Cell. (1999) 99:649–59. doi: 10.1016/S0092-8674(00)81553-6
116. Huber JD, Egleton RD, and Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. (2001) 24:719–25. doi: 10.1016/S0166-2236(00)02004-X
117. Greene C, Hanley N, and Campbell M. Claudin-5: gatekeeper of neurological function. Fluids Barriers CNS. (2019) 16:3. doi: 10.1186/s12987-019-0123-z
118. Hashimoto Y, Tachibana K, Krug SM, Kunisawa J, Fromm M, and Kondoh M. Potential for tight junction protein-directed drug development using claudin binders and angubindin-1. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20164016
119. Greene C, Kealy J, Humphries MM, Gong Y, Hou J, Hudson N, et al. Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol Psychiatry. (2018) 23:2156–66. doi: 10.1038/mp.2017.156
120. Fanning AS, Jameson BJ, Jesaitis LA, and Anderson JM. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem. (1998) 273:29745–53. doi: 10.1074/jbc.273.45.29745
121. Citi S, Sabanay H, Kendrick-Jones J, and Geiger B. Cingulin: characterization and localization. J Cell Sci. (1989) 93:107–22. doi: 10.1242/jcs.93.1.107
122. Zhong Y, Saitoh T, Minase T, Sawada N, Enomoto K, and Mori M. Monoclonal antibody 7H6 reacts with a novel tight junction-associated protein distinct from ZO-1, cingulin and ZO-2. J Cell Biol. (1993) 120:477–83. doi: 10.1083/jcb.120.2.477
123. Aurrand-Lions M, Johnson-Leger C, Wong C, Du Pasquier L, and Imhof BA. Heterogeneity of endothelial junctions is reflected by differential expression and specific subcellular localization of the three JAM family members. Blood. (2001) 98:3699–707. doi: 10.1182/blood.V98.13.3699
124. Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M, Citi S, and Dejana E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem. (2000) 275:20520–6. doi: 10.1074/jbc.M905251199
125. Buckley CD, Doyonnas R, Newton JP, Blystone SD, Brown EJ, Watt SM, et al. Identification of alpha v beta 3 as a heterotypic ligand for CD31/PECAM-1. J Cell Sci. (1996) 109:437–45. doi: 10.1242/jcs.109.2.437
126. Graesser D, Solowiej A, Bruckner M, Osterweil E, Juedes A, Davis S, et al. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J Clin Invest. (2002) 109:383–92. doi: 10.1172/JCI0213595
127. Kwon EE and Prineas JW. Blood-brain barrier abnormalities in longstanding multiple sclerosis lesions. An immunohistochemical study. J Neuropathol Exp Neurol. (1994) 53:625–36. doi: 10.1097/00005072-199411000-00010
128. van Horssen J, Brink BP, de Vries HE, van der Valk P, and Bo L. The blood-brain barrier in cortical multiple sclerosis lesions. J Neuropathol Exp Neurol. (2007) 66:321–8. doi: 10.1097/nen.0b013e318040b2de
129. Larochelle C, Alvarez JI, and Prat A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. (2011) 585:3770–80. doi: 10.1016/j.febslet.2011.04.066
130. Stys PK, Zamponi GW, van Minnen J, and Geurts JJ. Will the real multiple sclerosis please stand up? Nat Rev Neurosci. (2012) 13:507–14. doi: 10.1038/nrn3275
131. Kermode AG, Thompson AJ, Tofts P, MacManus DG, Kendall BE, Kingsley DP, et al. Breakdown of the blood-brain barrier precedes symptoms and other MRI signs of new lesions in multiple sclerosis. Pathogenetic Clin implications. Brain. (1990) 113:1477–89. doi: 10.1093/brain/113.5.1477
132. Goodkin DE, Rooney WD, Sloan R, Bacchetti P, Gee L, Vermathen M, et al. A serial study of new MS lesions and the white matter from which they arise. Neurology. (1998) 51:1689–97. doi: 10.1212/WNL.51.6.1689
133. Kaech SM and Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol. (2001) 2:415–22. doi: 10.1038/87720
134. Dore-Duffy P, Washington R, and Dragovic L. Expression of endothelial cell activation antigens in microvessels from patients with multiple sclerosis. Adv Exp Med Biol. (1993) 331:243–8. doi: 10.1007/978-1-4615-2920-0_38
135. Waldner H, Collins M, and Kuchroo VK. Activation of antigen-presenting cells by microbial products breaks self tolerance and induces autoimmune disease. J Clin Invest. (2004) 113:990–7. doi: 10.1172/JCI19388
136. Korn T, Bettelli E, Oukka M, and Kuchroo VK. IL-17 and th17 cells. Annu Rev Immunol. (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
137. van Stipdonk MJ, Lemmens EE, and Schoenberger SP. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol. (2001) 2:423–9. doi: 10.1038/87730
138. Steffen BJ, Butcher EC, and Engelhardt B. Evidence for involvement of ICAM-1 and VCAM-1 in lymphocyte interaction with endothelium in experimental autoimmune encephalomyelitis in the central nervous system in the SJL/J mouse. Am J Pathol. (1994) 145:189–201.
139. Greenwood J, Wang Y, and Calder VL. Lymphocyte adhesion and transendothelial migration in the central nervous system: the role of LFA-1, ICAM-1, VLA-4 and VCAM-1. off. Immunology. (1995) 86:408–15.
140. Larochelle C, Cayrol R, Kebir H, Alvarez JI, Lecuyer MA, Ifergan I, et al. Melanoma cell adhesion molecule identifies encephalitogenic T lymphocytes and promotes their recruitment to the central nervous system. Brain. (2012) 135:2906–24. doi: 10.1093/brain/aws212
141. Larochelle C, Lecuyer MA, Alvarez JI, Charabati M, Saint-Laurent O, Ghannam S, et al. Melanoma cell adhesion molecule-positive CD8 T lymphocytes mediate central nervous system inflammation. Ann Neurol. (2015) 78:39–53. doi: 10.1002/ana.24415
142. Du F, Garg AV, Kosar K, Majumder S, Kugler DG, Mir GH, et al. Inflammatory th17 cells express integrin alphavbeta3 for pathogenic function. Cell Rep. (2016) 16:1339–51. doi: 10.1016/j.celrep.2016.06.065
143. Tsukada N, Miyagi K, Matsuda M, and Yanagisawa N. Increased levels of circulating intercellular adhesion molecule-1 in multiple sclerosis and human T-lymphotropic virus type I-associated myelopathy. Ann Neurol. (1993) 33:646–9. doi: 10.1002/ana.410330614
144. Hartung HP, Reiners K, Archelos JJ, Michels M, Seeldrayers P, Heidenreich F, et al. Circulating adhesion molecules and tumor necrosis factor receptor in multiple sclerosis: correlation with magnetic resonance imaging. Ann Neurol. (1995) 38:186–93. doi: 10.1002/ana.410380210
145. Giovannoni G, Lai M, Thorpe J, Kidd D, Chamoun V, Thompson AJ, et al. Longitudinal study of soluble adhesion molecules in multiple sclerosis: correlation with gadolinium enhanced magnetic resonance imaging. Neurology. (1997) 48:1557–65. doi: 10.1212/WNL.48.6.1557
146. Winger RC, Koblinski JE, Kanda T, Ransohoff RM, and Muller WA. Rapid remodeling of tight junctions during paracellular diapedesis in a human model of the blood-brain barrier. J Immunol. (2014) 193:2427–37. doi: 10.4049/jimmunol.1400700
147. Muller WA. Transendothelial migration: unifying principles from the endothelial perspective. Immunol Rev. (2016) 273:61–75. doi: 10.1111/imr.2016.273.issue-1
148. Abadier M, Haghayegh Jahromi N, Cardoso Alves L, Boscacci R, Vestweber D, Barnum S, et al. Cell surface levels of endothelial ICAM-1 influence the transcellular or paracellular T-cell diapedesis across the blood-brain barrier. Eur J Immunol. (2015) 45:1043–58. doi: 10.1002/eji.201445125
149. Sonar SA, Shaikh S, Joshi N, Atre AN, and Lal G. IFN-gamma promotes transendothelial migration of CD4(+) T cells across the blood-brain barrier. Immunol Cell Biol. (2017) 95:843–53. doi: 10.1038/icb.2017.56
150. Frohman EM, Racke MK, and Raine CS. Multiple sclerosis–the plaque and its pathogenesis. N Engl J Med. (2006) 354:942–55. doi: 10.1056/NEJMra052130
151. Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. (2008) 57:178–201. doi: 10.1016/j.neuron.2008.01.003
152. Oshima T, Laroux FS, Coe LL, Morise Z, Kawachi S, Bauer P, et al. Interferon-gamma and interleukin-10 reciprocally regulate endothelial junction integrity and barrier function. Microvasc Res. (2001) 61:130–43. doi: 10.1006/mvre.2000.2288
153. Mankertz J, Tavalali S, Schmitz H, Mankertz A, Riecken EO, Fromm M, et al. Expression from the human occludin promoter is affected by tumor necrosis factor alpha and interferon gamma. J Cell Sci. (2000) 113:2085–90. doi: 10.1242/jcs.113.11.2085
154. Wachtel M, Bolliger MF, Ishihara H, Frei K, Bluethmann H, and Gloor SM. Down-regulation of occludin expression in astrocytes by tumour necrosis factor (TNF) is mediated via TNF type-1 receptor and nuclear factor-kappaB activation. J Neurochem. (2001) 78:155–62. doi: 10.1046/j.1471-4159.2001.00399.x
155. Nwariaku FE, Chang J, Zhu X, Liu Z, Duffy SL, Halaihel NH, et al. The role of p38 map kinase in tumor necrosis factor-induced redistribution of vascular endothelial cadherin and increased endothelial permeability. Shock. (2002) 18:82–5. doi: 10.1097/00024382-200207000-00015
156. Nusrat A, Turner JR, and Madara JL. Molecular physiology and pathophysiology of tight junctions. IV. Regulation of tight junctions by extracellular stimuli: nutrients, cytokines, and immune cells. Am J Physiol Gastrointest Liver Physiol. (2000) 279:G851–7. doi: 10.1152/ajpgi.2000.279.5.G851
157. Unlu H and Bozcuk AN. Genetics of longevity in Drosophila–I. The effects of w, m and f mutant genes in various genotype combinations. Exp Gerontol. (1979) 14:117–24. doi: 10.1016/0531-5565(79)90026-3
158. Blum MS, Toninelli E, Anderson JM, Balda MS, Zhou J, O'Donnell L, et al. Cytoskeletal rearrangement mediates human microvascular endothelial tight junction modulation by cytokines. Am J Physiol. (1997) 273:H286–94. doi: 10.1152/ajpheart.1997.273.1.H286
159. Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. (2003) 171:6164–72. doi: 10.4049/jimmunol.171.11.6164
160. Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. (2007) 13:1173–5. doi: 10.1038/nm1651
161. Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. (2010) 24:1023–34. doi: 10.1096/fj.09-141978
162. Bolton SJ, Anthony DC, and Perry VH. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo. Neuroscience. (1998) 86:1245–57. doi: 10.1016/S0306-4522(98)00058-X
163. Sozen T, Tsuchiyama R, Hasegawa Y, Suzuki H, Jadhav V, Nishizawa S, et al. Role of interleukin-1beta in early brain injury after subarachnoid hemorrhage in mice. Stroke. (2009) 40:2519–25. doi: 10.1161/STROKEAHA.109.549592
164. Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. (2001) 21:7724–32. doi: 10.1523/JNEUROSCI.21-19-07724.2001
165. Yang Y, Estrada EY, Thompson JF, Liu W, and Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. (2007) 27:697–709. doi: 10.1038/sj.jcbfm.9600375
166. Antonetti DA, Barber AJ, Hollinger LA, Wolpert EB, and Gardner TW. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem. (1999) 274:23463–7. doi: 10.1074/jbc.274.33.23463
167. Shimizu F, Tasaki A, Sano Y, Ju M, Nishihara H, Oishi M, et al. Sera from remitting and secondary progressive multiple sclerosis patients disrupt the blood-brain barrier. PLoS One. (2014) 9:e92872. doi: 10.1371/journal.pone.0092872
168. Minagar A, Ostanin D, Long AC, Jennings M, Kelley RE, Sasaki M, et al. Serum from patients with multiple sclerosis downregulates occludin and VE-cadherin expression in cultured endothelial cells. Mult Scler. (2003) 9:235–8. doi: 10.1191/1352458503ms916oa
169. Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, et al. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med. (2006) 203:1007–19. doi: 10.1084/jem.20051342
170. Conforti R, Cirillo M, Saturnino PP, Gallo A, Sacco R, Negro A, et al. Dilated Virchow-Robin spaces and multiple sclerosis: 3 T magnetic resonance study. Radiol Med. (2014) 119:408–14. doi: 10.1007/s11547-013-0357-9
171. van Horssen J, Bo L, Dijkstra CD, and de Vries HE. Extensive extracellular matrix depositions in active multiple sclerosis lesions. Neurobiol Dis. (2006) 24:484–91. doi: 10.1016/j.nbd.2006.08.005
172. van Horssen J, Dijkstra CD, and de Vries HE. The extracellular matrix in multiple sclerosis pathology. J Neurochem. (2007) 103:1293–301. doi: 10.1111/j.1471-4159.2007.04897.x
173. Kilsdonk ID, Steenwijk MD, Pouwels PJ, Zwanenburg JJ, Visser F, Luijten PR, et al. Perivascular spaces in MS patients at 7 Tesla MRI: a marker of neurodegeneration? Mult Scler. (2015) 21:155–62. doi: 10.1177/1352458514540358
174. Korotkov SM, Glazunov VV, Rozengart EV, and Suvorov AA. The action of organic cadmium complexes of different degrees of hydrophobicity on rat liver mitochondria. Tsitologiia. (1996) 38:1075–83.
175. Montagne A, Toga AW, and Zlokovic BV. Blood-brain barrier permeability and gadolinium: benefits and potential pitfalls in research. JAMA Neurol. (2016) 73:13–4. doi: 10.1001/jamaneurol.2015.2960
176. Holman DW, Klein RS, and Ransohoff RM. The blood-brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta. (2011) 1812:220–30. doi: 10.1016/j.bbadis.2010.07.019
177. Luster AD. Chemokines–chemotactic cytokines that mediate inflammation. N Engl J Med. (1998) 338:436–45. doi: 10.1056/NEJM199802123380706
178. Ding ZK, Sumrani N, and Hong JH. Prolonged simple cryothermic immersion storage of rat heart isografts: a preliminary study. J Invest Surg. (1991) 4:171–4. doi: 10.3109/08941939109140777
179. Szczucinski A and Losy J. Chemokines and chemokine receptors in multiple sclerosis. Potential targets for new therapies. Acta Neurol Scand. (2007) 115:137–46. doi: 10.1111/j.1600-0404.2006.00749.x
180. Oliver AR, Lyon GM, and Ruddle NH. Rat and human myelin oligodendrocyte glycoproteins induce experimental autoimmune encephalomyelitis by different mechanisms in C57BL/6 mice. J Immunol. (2003) 171:462–8. doi: 10.4049/jimmunol.171.1.462
181. Rothhammer V, Heink S, Petermann F, Srivastava R, Claussen MC, Hemmer B, et al. Th17 lymphocytes traffic to the central nervous system independently of alpha4 integrin expression during EAE. J Exp Med. (2011) 208:2465–76. doi: 10.1084/jem.20110434
182. Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. (2009) 10:514–23. doi: 10.1038/ni.1716
183. Kieseier BC, Seifert T, Giovannoni G, and Hartung HP. Matrix metalloproteinases in inflammatory demyelination: targets for treatment. Neurology. (1999) 53:20–5. doi: 10.1212/WNL.53.1.20
184. Wucherpfennig KW and Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. (1995) 80:695–705. doi: 10.1016/0092-8674(95)90348-8
185. Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. (2011) 479:538–41. doi: 10.1038/nature10554
186. Odoardi F, Sie C, Streyl K, Ulaganathan VK, Schlager C, Lodygin D, et al. T cells become licensed in the lung to enter the central nervous system. Nature. (2012) 488:675–9. doi: 10.1038/nature11337
187. Westland KW, Pollard JD, Sander S, Bonner JG, Linington C, and McLeod JG. Activated non-neural specific T cells open the blood-brain barrier to circulating antibodies. Brain. (1999) 122:1283–91. doi: 10.1093/brain/122.7.1283
188. Hickey WF. Migration of hematogenous cells through the blood-brain barrier and the initiation of CNS inflammation. Brain Pathol. (1991) 1:97–105. doi: 10.1111/j.1750-3639.1991.tb00646.x
189. Sospedra M and Martin R. Immunology of multiple sclerosis. Semin Neurol. (2016) 36:115–27. doi: 10.1055/s-0036-1579739
190. Trotter JL, Pelfrey CM, Trotter AL, Selvidge JA, Gushleff KC, Mohanakumar T, et al. T cell recognition of myelin proteolipid protein and myelin proteolipid protein peptides in the peripheral blood of multiple sclerosis and control subjects. J Neuroimmunol. (1998) 84:172–8. doi: 10.1016/S0165-5728(97)00260-9
191. Pelfrey CM, Tranquill LR, Vogt AB, and McFarland HF. T cell response to two immunodominant proteolipid protein (PLP) peptides in multiple sclerosis patients and healthy controls. Mult Scler. (1996) 1:270–8. doi: 10.1177/135245859600100503
192. Bielekova B, Sung MH, Kadom N, Simon R, McFarland H, and Martin R. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J Immunol. (2004) 172:3893–904. doi: 10.4049/jimmunol.172.6.3893
193. Cao Y, Goods BA, Raddassi K, Nepom GT, Kwok WW, Love JC, et al. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci Transl Med. (2015) 7:287ra74. doi: 10.1126/scitranslmed.aaa8038
194. Viglietta V, Baecher-Allan C, Weiner HL, and Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. (2004) 199:971–9. doi: 10.1084/jem.20031579
195. Schneider A, Long SA, Cerosaletti K, Ni CT, Samuels P, Kita M, et al. In active relapsing-remitting multiple sclerosis, effector T cell resistance to adaptive T(regs) involves IL-6-mediated signaling. Sci Transl Med. (2013) 5:170ra15. doi: 10.1126/scitranslmed.3004970
196. Mohme M, Hotz C, Stevanovic S, Binder T, Lee JH, Okoniewski M, et al. HLA-DR15-derived self-peptides are involved in increased autologous T cell proliferation in multiple sclerosis. Brain. (2013) 136:1783–98. doi: 10.1093/brain/awt108
197. Jelcic I, Al Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, et al. Memory B cells activate brain-homing, autoreactive CD4(+) T cells in multiple sclerosis. Cell. (2018) 175:85–100.e23. doi: 10.1016/j.cell.2018.08.011
198. Stinissen P and Hellings N. Activation of myelin reactive T cells in multiple sclerosis: a possible role for T cell degeneracy? Eur J Immunol. (2008) 38:1190–3. doi: 10.1002/eji.200838371
199. Wu C, Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P, et al. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med. (2009) 15:519–27. doi: 10.1038/nm.1957
200. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, Lafaille JJ, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. (2013) 155:1596–609. doi: 10.1016/j.cell.2013.11.030
201. International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control C, Sawcer S, Hellenthal G, Pirinen M, Spencer CC, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. (2011) 476:214–9. doi: 10.1038/nature10251
202. Song J, Wu C, Korpos E, Zhang X, Agrawal SM, Wang Y, et al. Focal MMP-2 and MMP-9 activity at the blood-brain barrier promotes chemokine-induced leukocyte migration. Cell Rep. (2015) 10:1040–54. doi: 10.1016/j.celrep.2015.01.037
203. Losy J. Is MS an inflammatory or primary degenerative disease? J Neural Transm (Vienna). (2013) 120:1459–62. doi: 10.1007/s00702-013-1079-9
204. Bever CT Jr. and Rosenberg GA. Matrix metalloproteinases in multiple sclerosis: targets of therapy or markers of injury? Neurology. (1999) 53:1380–1. doi: 10.1212/WNL.53.7.1380
205. Cepok S, Rosche B, Grummel V, Vogel F, Zhou D, Sayn J, et al. Short-lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain. (2005) 128:1667–76. doi: 10.1093/brain/awh486
206. Schirmer L, Srivastava R, and Hemmer B. To look for a needle in a haystack: the search for autoantibodies in multiple sclerosis. Mult Scler. (2014) 20:271–9. doi: 10.1177/1352458514522104
207. Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, and Kupper TS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature. (2012) 483:227–31. doi: 10.1038/nature10851
208. Barnett MH and Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. (2004) 55:458–68. doi: 10.1002/ana.20016
209. Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, and Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. (2000) 47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
210. Pachter JS, de Vries HE, and Fabry Z. The blood-brain barrier and its role in immune privilege in the central nervous system. J Neuropathol Exp Neurol. (2003) 62:593–604. doi: 10.1093/jnen/62.6.593
211. Hatterer E, Davoust N, Didier-Bazes M, Vuaillat C, Malcus C, Belin MF, et al. How to drain without lymphatics? Dendritic cells migrate from the cerebrospinal fluid to the B-cell follicles of cervical lymph nodes. Blood. (2006) 107:806–12. doi: 10.1182/blood-2005-01-0154
212. Prodinger C, Bunse J, Kruger M, Schiefenhovel F, Brandt C, Laman JD, et al. CD11c-expressing cells reside in the juxtavascular parenchyma and extend processes into the glia limitans of the mouse nervous system. Acta Neuropathol. (2011) 121:445–58. doi: 10.1007/s00401-010-0774-y
213. Mohammad MG, Tsai VW, Ruitenberg MJ, Hassanpour M, Li H, Hart PH, et al. Immune cell trafficking from the brain maintains CNS immune tolerance. J Clin Invest. (2014) 124:1228–41. doi: 10.1172/JCI71544
214. Wheway J, Obeid S, Couraud PO, Combes V, and Grau GE. The brain microvascular endothelium supports T cell proliferation and has potential for alloantigen presentation. PLoS One. (2013) 8:e52586. doi: 10.1371/journal.pone.0052586
215. Lopes Pinheiro MA, Kamermans A, Garcia-Vallejo JJ, van Het Hof B, Wierts L, O'Toole T, et al. Internalization and presentation of myelin antigens by the brain endothelium guides antigen-specific T cell migration. Elife. (2016) 5. doi: 10.7554/eLife.13149
216. Male D and Pryce G. Kinetics of MHC gene expression and mRNA synthesis in brain endothelium. Immunology. (1988) 63:37–42.
217. Locatelli G, Wortge S, Buch T, Ingold B, Frommer F, Sobottka B, et al. Primary oligodendrocyte death does not elicit anti-CNS immunity. Nat Neurosci. (2012) 15:543–50. doi: 10.1038/nn.3062
218. Eichler F and Van Haren K. Immune response in leukodystrophies. Pediatr Neurol. (2007) 37:235–44. doi: 10.1016/j.pediatrneurol.2007.06.011
219. Templeton SP and Perlman S. Pathogenesis of acute and chronic central nervous system infection with variants of mouse hepatitis virus, strain JHM. Immunol Res. (2007) 39:160–72. doi: 10.1007/s12026-007-0079-y
220. Minagar A and Alexander JS. Blood-brain barrier disruption in multiple sclerosis. Mult Scler. (2003) 9:540–9. doi: 10.1191/1352458503ms965oa
221. Miller DH, Grossman RI, Reingold SC, and McFarland HF. The role of magnetic resonance techniques in understanding and managing multiple sclerosis. Brain. (1998) 121:3–24. doi: 10.1093/brain/121.1.3
222. Lawson LJ, Perry VH, and Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience. (1992) 48:405–15. doi: 10.1016/0306-4522(92)90500-2
223. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518:547–51. doi: 10.1038/nature13989
224. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity. (2015) 42:665–78. doi: 10.1016/j.immuni.2015.03.011
225. Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, et al. Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. (2017) 18:391–405. doi: 10.1016/j.celrep.2016.12.041
226. Perry VH, Nicoll JA, and Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. (2010) 6:193–201. doi: 10.1038/nrneurol.2010.17
227. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. (2015) 523:337–41. doi: 10.1038/nature14432
228. Li Q and Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. (2018) 18:225–42. doi: 10.1038/nri.2017.125
229. Enger E, Freden H, Haljamae H, and Ingervall B. Effects of mandibular displacement on the metabolic activity of the masseter muscle in rat. Arch Biol. (1975) 20:7–10. doi: 10.1016/0003-9969(75)90144-2
230. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
231. Pridans C, Raper A, Davis GM, Alves J, Sauter KA, Lefevre L, et al. Pleiotropic impacts of macrophage and microglial deficiency on development in rats with targeted mutation of the csf1r locus. J Immunol. (2018) 201:2683–99. doi: 10.4049/jimmunol.1701783
232. Madry C, Kyrargyri V, Arancibia-Carcamo IL, Jolivet R, Kohsaka S, Bryan RM, et al. Microglial ramification, surveillance, and interleukin-1beta release are regulated by the two-pore domain K(+) channel THIK-1. Neuron. (2018) 97:299–312.e6. doi: 10.1016/j.neuron.2017.12.002
233. Veremeyko T, Yung AWY, Dukhinova M, Kuznetsova IS, Pomytkin I, Lyundup A, et al. Cyclic AMP pathway suppress autoimmune neuroinflammation by inhibiting functions of encephalitogenic CD4 T cells and enhancing M2 macrophage polarization at the site of inflammation. Front Immunol. (2018) 9:50. doi: 10.3389/fimmu.2018.00050
234. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. (2016) 19:987–91. doi: 10.1038/nn.4338
235. Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, and Bruck W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol. (2013) 125:595–608. doi: 10.1007/s00401-013-1082-0
236. Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, and Lassmann H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain. (2017) 140:1900–13. doi: 10.1093/brain/awx113
237. Bevan RJ, Evans R, Griffiths L, Watkins LM, Rees MI, Magliozzi R, et al. Meningeal inflammation and cortical demyelination in acute multiple sclerosis. Ann Neurol. (2018) 84:829–42. doi: 10.1002/ana.25365
238. Wolf Y, Shemer A, Levy-Efrati L, Gross M, Kim JS, Engel A, et al. Microglial MHC class II is dispensable for experimental autoimmune encephalomyelitis and cuprizone-induced demyelination. Eur J Immunol. (2018) 48:1308–18. doi: 10.1002/eji.201847540
239. Blomgren K and Hagberg H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic Biol Med. (2006) 40:388–97. doi: 10.1016/j.freeradbiomed.2005.08.040
240. Rochfort KD, Collins LE, Murphy RP, and Cummins PM. Downregulation of blood-brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase-dependent ROS generation: consequences for interendothelial adherens and tight junctions. PLoS One. (2014) 9:e101815. doi: 10.1371/journal.pone.0101815
241. Weaver A, Goncalves da Silva A, Nuttall RK, Edwards DR, Shapiro SD, Rivest S, et al. An elevated matrix metalloproteinase (MMP) in an animal model of multiple sclerosis is protective by affecting Th1/Th2 polarization. FASEB J. (2005) 19:1668–70. doi: 10.1096/fj.04-2030fje
242. van Horssen J, Vos CM, Admiraal L, van Haastert ES, Montagne L, van der Valk P, et al. Matrix metalloproteinase-19 is highly expressed in active multiple sclerosis lesions. Neuropathol Appl Neurobiol. (2006) 32:585–93. doi: 10.1111/j.1365-2990.2006.00766.x
243. Fakhoury M. Role of immunity and inflammation in the pathophysiology of neurodegenerative diseases. Neurodegener Dis. (2015) 15:63–9. doi: 10.1159/000369933
244. Hirsch EC and Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. (2009) 8:382–97. doi: 10.1016/S1474-4422(09)70062-6
245. Venegas C and Heneka MT. Danger-associated molecular patterns in Alzheimer’s disease. J Leukoc Biol. (2017) 101:87–98. doi: 10.1189/jlb.3MR0416-204R
246. Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. (2005) 11:328–34. doi: 10.1038/nm1197
247. Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, and Bruck W. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain. (2002) 125:2202–12. doi: 10.1093/brain/awf235
248. Ransohoff RM and Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. (2009) 27:119–45. doi: 10.1146/annurev.immunol.021908.132528
249. Siffrin V, Brandt AU, Radbruch H, Herz J, Boldakowa N, Leuenberger T, et al. Differential immune cell dynamics in the CNS cause CD4+ T cell compartmentalization. Brain. (2009) 132:1247–58. doi: 10.1093/brain/awn354
250. Kivisakk P, Mahad DJ, Callahan MK, Trebst C, Tucky B, Wei T, et al. Human cerebrospinal fluid central memory CD4+ T cells: evidence for trafficking through choroid plexus and meninges via P-selectin. Proc Natl Acad Sci U S A. (2003) 100:8389–94. doi: 10.1073/pnas.1433000100
251. Du L, Zhang Y, Chen Y, Zhu J, Yang Y, and Zhang HL. Role of microglia in neurological disorders and their potentials as a therapeutic target. Mol Neurobiol. (2017) 54:7567–84. doi: 10.1007/s12035-016-0245-0
252. Neumann H, Kotter MR, and Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. (2009) 132:288–95. doi: 10.1093/brain/awn109
253. Lampron A, Larochelle A, Laflamme N, Prefontaine P, Plante MM, Sanchez MG, et al. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med. (2015) 212:481–95. doi: 10.1084/jem.20141656
254. Wolf SA, Steiner B, Akpinarli A, Kammertoens T, Nassenstein C, Braun A, et al. CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J Immunol. (2009) 182:3979–84. doi: 10.4049/jimmunol.0801218
255. Ratchford JN, Endres CJ, Hammoud DA, Pomper MG, Shiee N, McGready J, et al. Decreased microglial activation in MS patients treated with glatiramer acetate. J Neurol. (2012) 259:1199–205. doi: 10.1007/s00415-011-6337-x
256. Kornberg MD, Smith MD, Shirazi HA, Calabresi PA, Snyder SH, and Kim PM. Bryostatin-1 alleviates experimental multiple sclerosis. Proc Natl Acad Sci U S A. (2018) 115:2186–91. doi: 10.1073/pnas.1719902115
257. Djedovic N, Stanisavljevic S, Jevtic B, Momcilovic M, Lavrnja I, and Miljkovic D. Anti-encephalitogenic effects of ethyl pyruvate are reflected in the central nervous system and the gut. BioMed Pharmacother. (2017) 96:78–85. doi: 10.1016/j.biopha.2017.09.110
258. Li WW, Setzu A, Zhao C, and Franklin RJ. Minocycline-mediated inhibition of microglia activation impairs oligodendrocyte progenitor cell responses and remyelination in a non-immune model of demyelination. J Neuroimmunol. (2005) 158:58–66. doi: 10.1016/j.jneuroim.2004.08.011
259. Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. (2013) 4:e525. doi: 10.1038/cddis.2013.54
260. Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. (2013) 16:1211–8. doi: 10.1038/nn.3469
261. Huang CY, Chen YL, Li AH, Lu JC, Wang HL, et al. Minocycline, a microglial inhibitor, blocks spinal CCL2-induced heat hyperalgesia and augmentation of glutamatergic transmission in substantia gelatinosa neurons. J Neuroinflammation. (2014) 11:7. doi: 10.1186/1742-2094-11-7
262. Sloka S, Metz LM, Hader W, Starreveld Y, and Yong VW. Reduction of microglial activity in a model of multiple sclerosis by dipyridamole. J Neuroinflammation. (2013) 10:89. doi: 10.1186/1742-2094-10-89
263. Jin Q, Metz LM, Hader W, Starreveld Y, Yong VW, et al. Improvement of functional recovery by chronic metformin treatment is associated with enhanced alternative activation of microglia/macrophages and increased angiogenesis and neurogenesis following experimental stroke. Brain Behav Immun. (2014) 40:131–42. doi: 10.1016/j.bbi.2014.03.003
264. The IFNB Multiple Sclerosis Study GroupInterferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. The IFNB Multiple Sclerosis Study Group. Neurology. (1993) 43:655–61. doi: 10.1212/WNL.43.4.655
265. Jacobs LD, The IFNB Multiple Sclerosis Study Group, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Ann Neurol. (1996) 39:285–94. doi: 10.1002/ana.410390304
266. PRISMS Study GroupRandomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet. (1998) 352:1498–504. doi: 10.1073/pnas.1433000100
267. Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. (2006) 354:899–910. doi: 10.1056/NEJMoa044397
268. Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. (2010) 362:402–15. doi: 10.1056/NEJMoa0907839
269. Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. (2010) 362:387–401. doi: 10.1056/NEJMoa0909494
270. Cross A and Riley C. Treatment of multiple sclerosis. Continuum (Minneap Minn). (2022) 28:1025–51. doi: 10.1212/CON.0000000000001170
271. Hauser SL and Cree BAC. Treatment of multiple sclerosis: A review. Am J Med. (2020) 133:1380–1390.e2. doi: 10.1016/j.amjmed.2020.05.049
272. Kappos L, Fox RJ, Burcklen M, Freedman MS, Havrdova EK, Hennessy B, et al. Ponesimod compared with teriflunomide in patients with relapsing multiple sclerosis in the active-comparator phase 3 OPTIMUM study: A randomized clinical trial. JAMA Neurol. (2021) 78:558–67. doi: 10.1001/jamaneurol.2021.0405
273. Confavreux C, O'Connor P, Comi G, Freedman MS, Miller AE, Olsson TP, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. (2014) 13:247–56. doi: 10.1016/S1474-4422(13)70308-9
274. O’Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. (2011) 365:1293–303. doi: 10.1056/NEJMoa1014656
275. Miller AE, Wolinsky JS, Kappos L, Comi G, Freedman MS, Olsson TP, et al. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. (2014) 13:977–86. doi: 10.1016/S1474-4422(14)70191-7
276. Dubey D, Kieseier BC, Hartung HP, Hemmer B, Warnke C, Menge T, et al. Dimethyl fumarate in relapsing-remitting multiple sclerosis: rationale, mechanisms of action, pharmacokinetics, efficacy and safety. Expert Rev Neurother. (2015) 15:339–46. doi: 10.1586/14737175.2015.1025755
277. Warnke C, Kieseier BC, Hartung HP, Hemmer B, Warnke C, and Menge T. Identification of targets and new developments in the treatment of multiple sclerosis–focus on cladribine. Drug Des Devel Ther. (2010) 4:117–26. doi: 10.2147/dddt.s6627
278. Brousil JA, Roberts RJ, and Schlein AL. Cladribine: an investigational immunomodulatory agent for multiple sclerosis. Ann Pharmacother. (2006) 40:1814–21. doi: 10.1345/aph.1H037
279. Cohen JA, Wiendl H, Hartung HP, Stuve O, Kieseier BC, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. (2012) 380:1819–28. doi: 10.1016/S0140-6736(12)61769-3
280. Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. (2012) 380:1829–39. doi: 10.1016/S0140-6736(12)61768-1
281. Hartung HP, Aktas O, and Boyko AN. Alemtuzumab: a new therapy for active relapsing-remitting multiple sclerosis. Mult Scler. (2015) 21:22–34. doi: 10.1177/1352458514549398
282. van Munster CE and Uitdehaag BM. Outcome measures in clinical trials for multiple sclerosis. CNS Drugs. (2017) 31:217–36. doi: 10.1007/s40263-017-0412-5
283. Freedman MS, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, et al. A phase III study evaluating the efficacy and safety of MBP8298 in secondary progressive MS. Neurology. (2011) 77:1551–60. doi: 10.1212/WNL.0b013e318233b240
284. Hommes OR, Sorensen PS, Fazekas F, Enriquez MM, Koelmel HW, Fernandez O, et al. Intravenous immunoglobulin in secondary progressive multiple sclerosis: randomised placebo-controlled trial. Lancet. (2004) 364:1149–56. doi: 10.1016/S0140-6736(04)17101-8
285. Zajicek J, Ball S, Wright D, Vickery J, Nunn A, Miller D, et al. Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): a randomised, placebo-controlled trial. Lancet Neurol. (2013) 12:857–65. doi: 10.1016/S1474-4422(13)70159-5
286. Rice GP, Filippi M, and Comi G. Cladribine and progressive MS: clinical and MRI outcomes of a multicenter controlled trial. Cladribine MRI Study Group. Neurology. (2000) 54:1145–55. doi: 10.1212/WNL.54.5.1145
287. La Mantia L, Vacchi L, Rovaris M, Di Pietrantonj C, Ebers G, Fredrikson S, et al. Interferon beta for secondary progressive multiple sclerosis: a systematic review. J Neurol Neurosurg Psychiatry. (2013) 84:420–6. doi: 10.1136/jnnp-2012-303291
288. Miller DH and Leary SM. Primary-progressive multiple sclerosis. Lancet Neurol. (2007) 6:903–12. doi: 10.1016/S1474-4422(07)70243-0
289. Hartung HP, Gonsette R, Konig N, Kwiecinski H, Guseo A, Morrissey SP, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. (2002) 360:2018–25. doi: 10.1016/S0140-6736(02)12023-X
290. Secondary Progressive Efficacy Clinical Trial of Recombinant Interferon-Beta-1a in MS (SPECTRIMS) Study Group. Randomized controlled trial of interferon- beta-1a in secondary progressive MS: Clinical results. Neurology. (2001) 56:1496–504. doi: 10.1212/WNL.56.11.1496
291. Lublin F, Miller DH, Freedman MS, Cree BAC, Wolinsky JS, Weiner H, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet. (2016) 387:1075–84. doi: 10.1016/S0140-6736(15)01314-8
292. Kapoor R, Ho PR, Campbell N, Chang I, Deykin A, Forrestal F, et al. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): a phase 3, randomised, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurol. (2018) 17:405–15. doi: 10.1016/S1474-4422(18)30069-3
293. Akaishi T and Nakashima I. Efficiency of antibody therapy in demyelinating diseases. Int Immunol. (2017) 29:327–35. doi: 10.1093/intimm/dxx037
294. Hauser SL, Ho PR, Campbell N, Chang I, Deykin A, Forrestal F, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. (2017) 376:221–34. doi: 10.1056/NEJMoa1601277
295. Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B, et al. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med. (2020) 383:546–57. doi: 10.1056/NEJMoa1917246
296. Steinman L, Bar-Or A, Cohen JA, Comi G, Correale J, Coyle PK, et al. Ublituximab versus teriflunomide in relapsing multiple sclerosis. N Engl J Med. (2022) 387:704–14. doi: 10.1056/NEJMoa2201904
297. Clifford DB, De Luca A, Simpson DM, Arendt G, Giovannoni G, and Nath A. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol. (2010) 9:438–46. doi: 10.1016/S1474-4422(10)70028-4
298. Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. (2012) 366:1870–80. doi: 10.1056/NEJMoa1107829
299. Ghadiri M, Fitz-Gerald L, Rezk A, Li R, Nyirenda M, Haegert D, et al. Reconstitution of the peripheral immune repertoire following withdrawal of fingolimod. Mult Scler. (2017) 23:1225–32. doi: 10.1177/1352458517713147
300. Kappos L, Bar-Or A, Cree BAC, Fox RJ, Giovannoni G, Gold R, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. (2018) 391:1263–73. doi: 10.1016/S0140-6736(18)30475-6
301. Mix E, Meyer-Rienecker H, Hartung HP, and Zettl UK. Animal models of multiple sclerosis–potentials and limitations. Prog Neurobiol. (2010) 92:386–404. doi: 10.1016/j.pneurobio.2010.06.005
302. Sanabria-Castro A, Flores-Diaz M, and Alape-Giron A. Biological models in multiple sclerosis. J Neurosci Res. (2020) 98:491–508. doi: 10.1002/jnr.24528
303. Bjelobaba I, Begovic-Kupresanin V, Pekovic S, and Lavrnja I. Animal models of multiple sclerosis: Focus on experimental autoimmune encephalomyelitis. J Neurosci Res. (2018) 96:1021–42. doi: 10.1002/jnr.24224
304. Baker D and Amor S. Experimental autoimmune encephalomyelitis is a good model of multiple sclerosis if used wisely. Mult Scler Relat Disord. (2014) 3:555–64. doi: 10.1016/j.msard.2014.05.002
305. Stromnes IM and Goverman JM. Active induction of experimental allergic encephalomyelitis. Nat Protoc. (2006) 1:1810–9. doi: 10.1038/nprot.2006.285
306. Stromnes IM and Goverman JM. Passive induction of experimental allergic encephalomyelitis. Nat Protoc. (2006) 1:1952–60. doi: 10.1038/nprot.2006.284
307. Kipp M, van der Star B, Vogel DY, Puentes F, van der Valk P, Baker D, et al. Experimental in vivo and in vitro models of multiple sclerosis: EAE and beyond. Mult Scler Relat Disord. (2012) 1:15–28. doi: 10.1016/j.msard.2011.09.002
308. Einstein ER, Dalal KB, and Csejtey J. Increased protease activity and changes in basic proteins and lipids in multiple sclerosis plaques. J Neurol Sci. (1970) 11:109–21. doi: 10.1016/0022-510X(70)90121-8
309. Laatsch RH, Kies MW, Gordon S, and Alvord EC. The encephalomyelitic activity of myelin isolated by ultracentrifugation. J Exp Med. (1962) 115:777–88. doi: 10.1084/jem.115.4.777
310. Tuohy VK, Kies MW, Gordon S, and Alvord EC. A synthetic peptide from myelin proteolipid protein induces experimental allergic encephalomyelitis. J Immunol. (1988) 141:1126–30. doi: 10.4049/jimmunol.141.4.1126
311. Linthicum DS, Jones S, Horvath L, and Carnegie PR. Detection of antibodies to myelin basic protein by solid-phase radioimmunoassay with [125I]protein A. J Neuroimmunol. (1981) 1:17–26. doi: 10.1016/0165-5728(81)90004-7
312. Didonna A. Preclinical models of multiple sclerosis: advantages and limitations towards better therapies. Curr Med Chem. (2016) 23:1442–59. doi: 10.2174/0929867323666160406121218
313. Lassmann H. Comparative neuropathology of chronic experimental allergic encephalomyelitis and multiple sclerosis. Schriftenr Neurol. (1983) 25:1–135. doi: 10.1007/978-3-642-45558-2
314. Constantinescu CS, Farooqi N, O'Brien K, and Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. (2011) 164:1079–106. doi: 10.1111/j.1476-5381.2011.01302.x
315. Plant GT. Optic neuritis and multiple sclerosis. Curr Opin Neurol. (2008) 21:16–21. doi: 10.1097/WCO.0b013e3282f419ca
316. Schmitt C, Strazielle N, and Ghersi-Egea JF. Brain leukocyte infiltration initiated by peripheral inflammation or experimental autoimmune encephalomyelitis occurs through pathways connected to the CSF-filled compartments of the forebrain and midbrain. J Neuroinflammation. (2012) 9:187. doi: 10.1186/1742-2094-9-187
317. Gold R, Linington C, and Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. (2006) 129:1953–71. doi: 10.1093/brain/awl075
318. Herrero-Herranz E, Pardo LA, Gold R, and Linker RA. Pattern of axonal injury in murine myelin oligodendrocyte glycoprotein induced experimental autoimmune encephalomyelitis: implications for multiple sclerosis. Neurobiol Dis. (2008) 30:162–73. doi: 10.1016/j.nbd.2008.01.001
319. Pomeroy IM, Matthews PM, Frank JA, Jordan EK, and Esiri MM. Demyelinated neocortical lesions in marmoset autoimmune encephalomyelitis mimic those in multiple sclerosis. Brain. (2005) 128:2713–21. doi: 10.1093/brain/awh626
320. Tabira T, Itoyama Y, and Kuroiwa Y. Necessity of continuous antigenic stimulation by the locally retained antigens in chronic relapsing experimental allergic encephalomyelitis. J Neurol Sci. (1984) 66:97–106. doi: 10.1016/0022-510X(84)90145-X
321. Tabira T, Itoyama Y, and Kuroiwa Y. The role of locally retained antigens in chronic relapsing experimental allergic encephalomyelitis in Guinea pigs. Prog Clin Biol Res. (1984) 146:43–8.
322. Bannerman PG, Hahn A, Ramirez S, Morley M, Bonnemann C, Yu S, et al. Motor neuron pathology in experimental autoimmune encephalomyelitis: studies in THY1-YFP transgenic mice. Brain. (2005) 128:1877–86. doi: 10.1093/brain/awh550
323. McCarthy DP, Richards MH, and Miller SD. Mouse models of multiple sclerosis: experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. Methods Mol Biol. (2012) 900:381–401. doi: 10.1007/978-1-60761-720-4_19
324. Tsunoda I and Fujinami RS. Neuropathogenesis of Theiler’s murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J Neuroimmune Pharmacol. (2010) 5:355–69. doi: 10.1007/s11481-009-9179-x
325. Haring J and Perlman S. Mouse hepatitis virus. Curr Opin Microbiol. (2001) 4:462–6. doi: 10.1016/S1369-5274(00)00236-8
326. Matas-Rico E, Garcia-Diaz B, Llebrez-Zayas P, Lopez-Barroso D, Santin L, Pedraza C, et al. Deletion of lysophosphatidic acid receptor LPA1 reduces neurogenesis in the mouse dentate gyrus. Mol Cell Neurosci. (2008) 39:342–55. doi: 10.1016/j.mcn.2008.07.014
327. Osorio-Querejeta I, Saenz-Cuesta M, Munoz-Culla M, and Otaegui D. Models for studying myelination, demyelination and remyelination. Neuromolecular Med. (2017) 19:181–92. doi: 10.1007/s12017-017-8442-1
328. Magalon K, Cantarella C, Monti G, Cayre M, and Durbec P. Enriched environment promotes adult neural progenitor cell mobilization in mouse demyelination models. Eur J Neurosci. (2007) 25:761–71. doi: 10.1111/j.1460-9568.2007.05335.x
329. Blakemore WF and Franklin RJ. Remyelination in experimental models of toxin-induced demyelination. Curr Top Microbiol Immunol. (2008) 318:193–212. doi: 10.1007/978-3-540-73677-6_8
330. Woodruff RH and Franklin RJ. Demyelination and remyelination of the caudal cerebellar peduncle of adult rats following stereotaxic injections of lysolecithin, ethidium bromide, and complement/anti-galactocerebroside: a comparative study. Glia. (1999) 25:216–28. doi: 10.1002/(SICI)1098-1136(19990201)25:3<216::AID-GLIA2>3.0.CO;2-L
331. Levine JM and Reynolds R. Activation and proliferation of endogenous oligodendrocyte precursor cells during ethidium bromide-induced demyelination. Exp Neurol. (1999) 160:333–47. doi: 10.1006/exnr.1999.7224
332. Lafaille JJ, Nagashima K, Katsuki M, and Tonegawa S. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell. (1994) 78:399–408. doi: 10.1016/0092-8674(94)90419-7
333. Waldner H, Whitters MJ, Sobel RA, Collins M, and Kuchroo VK. Fulminant spontaneous autoimmunity of the central nervous system in mice transgenic for the myelin proteolipid protein-specific T cell receptor. Proc Natl Acad Sci U S A. (2000) 97:3412–7. doi: 10.1073/pnas.97.7.3412
334. Bettelli E, Whitters MJ, Sobel RA, Collins M, and Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med. (2003) 197:1073–81. doi: 10.1084/jem.20021603
335. Ben-Nun A, Kaushansky N, Kawakami N, Krishnamoorthy G, Berer K, Liblau R, et al. From classic to spontaneous and humanized models of multiple sclerosis: impact on understanding pathogenesis and drug development. J Autoimmun. (2014) 54:33–50. doi: 10.1016/j.jaut.2014.06.004
336. Ellmerich S, Takacs K, Mycko M, Waldner H, Wahid F, Boyton RJ, et al. Disease-related epitope spread in a humanized T cell receptor transgenic model of multiple sclerosis. Eur J Immunol. (2004) 34:1839–48. doi: 10.1002/eji.200324044
337. Kieseier BC and Hartung HP. Current disease-modifying therapies in multiple sclerosis. Semin Neurol. (2003) 23:133–46. doi: 10.1055/s-2003-41138
338. Steinman L and Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. (2006) 60:12–21. doi: 10.1002/ana.20913
339. Schmidt J, Gold R, Schonrock L, Zettl UK, Hartung HP, and Toyka KV. T-cell apoptosis in situ in experimental autoimmune encephalomyelitis following methylprednisolone pulse therapy. Brain. (2000) 123:1431–41. doi: 10.1093/brain/123.7.1431
340. van der Meide PH, de Labie MC, Ruuls SR, Groenestein RJ, Botman CA, Olsson T, et al. Discontinuation of treatment with IFN-beta leads to exacerbation of experimental autoimmune encephalomyelitis in Lewis rats. Rapid reversal of the antiproliferative activity of IFN-beta and excessive expansion of autoreactive T cells as disease promoting mechanisms. J Neuroimmunol. (1998) 84:14–23. doi: 10.1016/S0165-5728(97)00207-5
341. van Oosten BW, Barkhof F, Truyen L, Boringa JB, Bertelsmann FW, von Blomberg BM, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology. (1996) 47:1531–4. doi: 10.1212/WNL.47.6.1531
342. Hunig T. The storm has cleared: lessons from the CD28 superagonist TGN1412 trial. Nat Rev Immunol. (2012) 12:317–8. doi: 10.1038/nri3192
343. Faria AM and Weiner HL. Oral tolerance: therapeutic implications for autoimmune diseases. Clin Dev Immunol. (2006) 13:143–57. doi: 10.1080/17402520600876804
344. Noseworthy JH, O'Brien P, Erickson BJ, Lee D, Sneve D, Ebers GC, et al. The Mayo Clinic-Canadian Cooperative trial of sulfasalazine in active multiple sclerosis. Neurology. (1998) 51:1342–52. doi: 10.1212/WNL.51.5.1342
345. Dedoni S, Scherma M, Camoglio C, Siddi C, Dazzi L, Puliga R, et al. An overall view of the most common experimental models for multiple sclerosis. Neurobiol Dis. (2023) 184:106230. doi: 10.1016/j.nbd.2023.106230
346. Lassmann H and Bradl M. Multiple sclerosis: experimental models and reality. Acta Neuropathol. (2017) 133:223–44. doi: 10.1007/s00401-016-1631-4
347. Iadecola C. The pathobiology of vascular dementia. Neuron. (2013) 80:844–66. doi: 10.1016/j.neuron.2013.10.008
348. Montagne A, Scherma M, Camoglio C, Siddi C, Dazzi L, Puliga R, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. (2015) 85:296–302. doi: 10.1016/j.neuron.2014.12.032
349. Montine TJ, Koroshetz WJ, Babcock D, Dickson DW, Galpern WR, Glymour MM, et al. Recommendations of the Alzheimer’s disease-related dementias conference. Neurology. (2014) 83:851–60. doi: 10.1212/WNL.0000000000000733
350. Snyder HR, Miyake A, and Hankin BL. Advancing understanding of executive function impairments and psychopathology: bridging the gap between clinical and cognitive approaches. Front Psychol. (2015) 6:328. doi: 10.3389/fpsyg.2015.00328
351. Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, and Zlokovic BV. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. (2013) 125:111–20. doi: 10.1007/s00401-012-1039-8
352. Korczyn AD. Vascular parkinsonism–characteristics, pathogenesis and treatment. Nat Rev Neurol. (2015) 11:319–26. doi: 10.1038/nrneurol.2015.61
353. Drouin-Ouellet J, Sawiak SJ, Cisbani G, Lagace M, Kuan WL, Saint-Pierre M, et al. Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: Potential implications for its pathophysiology. Ann Neurol. (2015) 78:160–77. doi: 10.1002/ana.24406
354. Marchi N, Granata T, Ghosh C, and Janigro D. Blood-brain barrier dysfunction and epilepsy: pathophysiologic role and therapeutic approaches. Epilepsia. (2012) 53:1877–86. doi: 10.1111/j.1528-1167.2012.03637.x
355. Erdo F, Denes L, and de Lange E. Age-associated physiological and pathological changes at the blood-brain barrier: A review. J Cereb Blood Flow Metab. (2017) 37:4–24. doi: 10.1177/0271678X16679420
356. Erickson MA and Banks WA. Age-associated changes in the immune system and blood(-)Brain barrier functions. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20071632
357. Greenbaum SS and Greenbaum CH. Intraoperative tissue expansion using a Foley catheter following excision of a basal cell carcinoma. J Dermatol Surg Oncol. (1990) 16:45–8. doi: 10.1111/j.1524-4725.1990.tb00007.x
358. Friedman A and Kaufer D. Blood-brain barrier breakdown and blood-brain communication in neurological and psychiatric diseases. Cardiovasc Psychiatry Neurol 2011. (2011) p:431470. doi: 10.1155/2011/431470
359. Tomkins O, Feintuch A, Benifla M, Cohen A, Friedman A, and Shelef I. Blood-brain barrier breakdown following traumatic brain injury: a possible role in posttraumatic epilepsy. Cardiovasc Psychiatry Neurol. (2011) 2011:765923. doi: 10.1155/2011/765923
360. Majumdar R. Neutron hole strength distribution of the 2f7/2 state of 207Pb. Phys Rev C Nucl Phys. (1990) 42:631–4. doi: 10.1103/PhysRevC.42.631
361. Kaisar MA, Sajja RK, Prasad S, Abhyankar VV, Liles T, and Cucullo L. New experimental models of the blood-brain barrier for CNS drug discovery. Expert Opin Drug Discov. (2017) 12:89–103. doi: 10.1080/17460441.2017.1253676
362. Erickson MA, Wilson ML, and Banks WA. In vitro modeling of blood-brain barrier and interface functions in neuroimmune communication. Fluids Barriers CNS. (2020) 17:26. doi: 10.1186/s12987-020-00187-3
363. Gastfriend BD, Palecek SP, and Shusta EV. Modeling the blood-brain barrier: Beyond the endothelial cells. Curr Opin BioMed Eng. (2018) 5:6–12. doi: 10.1016/j.cobme.2017.11.002
364. Hatherell K, Couraud PO, Romero IA, Weksler B, and Pilkington GJ. Development of a three-dimensional, all-human in vitro model of the blood-brain barrier using mono-, co-, and tri-cultivation Transwell models. J Neurosci Methods. (2011) 199:223–9. doi: 10.1016/j.jneumeth.2011.05.012
365. Stewart PA and Wiley MJ. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: a study using quail–chick transplantation chimeras. Dev Biol. (1981) 84:183–92. doi: 10.1016/0012-1606(81)90382-1
366. Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, and McMahon AP. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. (2008) 322:1247–50. doi: 10.1126/science.1164594
367. Abbott NJ, Hughes CC, Revest PA, and Greenwood J. Development and characterisation of a rat brain capillary endothelial culture: towards an in vitro blood-brain barrier. J Cell Sci. (1992) 103:23–37. doi: 10.1242/jcs.103.1.23
368. Annunziata P, Cioni C, Toneatto S, and Paccagnini E. HIV-1 gp120 increases the permeability of rat brain endothelium cultures by a mechanism involving substance P. AIDS. (1998) 12:2377–85. doi: 10.1097/00002030-199818000-00006
369. Franke H, Galla H, and Beuckmann CT. Primary cultures of brain microvessel endothelial cells: a valid and flexible model to study drug transport through the blood-brain barrier in vitro. Brain Res Brain Res Protoc. (2000) 5:248–56. doi: 10.1016/S1385-299X(00)00020-9
370. Helms HC, Abbott NJ, Burek M, Cecchelli R, Couraud PO, Deli MA, et al. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J Cereb Blood Flow Metab. (2016) 36:862–90. doi: 10.1177/0271678X16630991
371. Gaillard PJ, Voorwinden LH, Nielsen JL, Ivanov A, Atsumi R, Engman H, et al. Establishment and functional characterization of an in vitro model of the blood-brain barrier, comprising a co-culture of brain capillary endothelial cells and astrocytes. Eur J Pharm Sci. (2001) 12:215–22. doi: 10.1016/S0928-0987(00)00123-8
372. Cohen-Kashi Malina K, Cooper I, and Teichberg VI. Closing the gap between the in-vivo and in-vitro blood-brain barrier tightness. Brain Res. (2009) 1284:12–21. doi: 10.1016/j.brainres.2009.05.072
373. Abbott NJ, Voorwinden LH, Nielsen JL, Ivanov A, Atsumi R, and Engman H. An improved in vitro blood-brain barrier model: rat brain endothelial cells co-cultured with astrocytes. Methods Mol Biol. (2012) 814:415–30. doi: 10.1007/978-1-61779-452-0_28
374. Culot M, Dolman DE, Drndarski S, Fredriksson SM, et al. An in vitro blood-brain barrier model for high throughput (HTS) toxicological screening. Toxicol In Vitro. (2008) 22:799–811. doi: 10.1016/j.tiv.2007.12.016
375. Perriere N, Lundquist S, Vanuxeem D, Nion S, Landry C, Delplace Y, et al. A functional in vitro model of rat blood-brain barrier for molecular analysis of efflux transporters. Brain Res. (2007) 1150:1–13. doi: 10.1016/j.brainres.2007.02.091
376. Wilhelm I, Fazakas C, and Krizbai IA. In vitro models of the blood-brain barrier. Acta Neurobiol Exp (Wars). (2011) 71:113–28. doi: 10.55782/ane-2011-1828
377. Patabendige A, Skinner RA, and Abbott NJ. Establishment of a simplified in vitro porcine blood-brain barrier model with high transendothelial electrical resistance. Brain Res. (2013) 1521:1–15. doi: 10.1016/j.brainres.2012.06.057
378. Helms HC, Waagepetersen HS, Nielsen CU, and Brodin B. Paracellular tightness and claudin-5 expression is increased in the BCEC/astrocyte blood-brain barrier model by increasing media buffer capacity during growth. AAPS J. (2010) 12:759–70. doi: 10.1208/s12248-010-9237-6
379. Banks WA, Waagepetersen HS, Nielsen CU, Brodin B, et al. Lipopolysaccharide-induced blood-brain barrier disruption: roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J Neuroinflammation. (2015) 12:223. doi: 10.1186/s12974-015-0434-1
380. Descamps L, Gray AM, Erickson MA, Salameh TS, Damodarasamy M, and Sheibani N. Protective effect of glial cells against lipopolysaccharide-mediated blood-brain barrier injury. Glia. (2003) 42:46–58. doi: 10.1002/glia.10205
381. Veszelka S, Pasztoi M, Farkas AE, Krizbai I, Ngo TK, Niwa M, et al. Pentosan polysulfate protects brain endothelial cells against bacterial lipopolysaccharide-induced damages. Neurochem Int. (2007) 50:219–28. doi: 10.1016/j.neuint.2006.08.006
382. Wong D, Dorovini-Zis K, and Vincent SR. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp Neurol. (2004) 190:446–55. doi: 10.1016/j.expneurol.2004.08.008
383. de Vries HE, Blom-Roosemalen MC, van Oosten M, de Boer AG, van Berkel TJ, Breimer DD, et al. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J Neuroimmunol. (1996) 64:37–43. doi: 10.1016/0165-5728(95)00148-4
384. Deli MA, Descamps L, Dehouck MP, Cecchelli R, Joo F, Abraham CS, et al. Exposure of tumor necrosis factor-alpha to luminal membrane of bovine brain capillary endothelial cells cocultured with astrocytes induces a delayed increase of permeability and cytoplasmic stress fiber formation of actin. J Neurosci Res. (1995) 41:717–26. doi: 10.1002/jnr.490410602
385. Greenwood J, Heasman SJ, Alvarez JI, Prat A, Lyck R, and Engelhardt B. Review: leucocyte-endothelial cell crosstalk at the blood-brain barrier: a prerequisite for successful immune cell entry to the brain. Neuropathol Appl Neurobiol. (2011) 37:24–39. doi: 10.1111/j.1365-2990.2010.01140.x
386. Biernacki K, Prat A, Blain M, and Antel JP. Regulation of Th1 and Th2 lymphocyte migration by human adult brain endothelial cells. J Neuropathol Exp Neurol. (2001) 60:1127–36. doi: 10.1093/jnen/60.12.1127
387. von Wedel-Parlow M, Schrot S, Lemmen J, Treeratanapiboon L, Wegener J, and Galla HJ. Neutrophils cross the BBB primarily on transcellular pathways: an in vitro study. Brain Res. (2011) 1367:62–76. doi: 10.1016/j.brainres.2010.09.076
388. Coisne C, Faveeuw C, Delplace Y, Dehouck L, Miller F, Cecchelli R, et al. Differential expression of selectins by mouse brain capillary endothelial cells in vitro in response to distinct inflammatory stimuli. Neurosci Lett. (2006) 392:216–20. doi: 10.1016/j.neulet.2005.09.028
389. Coisne C, Faveeuw C, Delplace Y, Dehouck L, Miller F, Cecchelli R, et al. Mouse syngenic in vitro blood-brain barrier model: a new tool to examine inflammatory events in cerebral endothelium. Lab Invest. (2005) 85:734–46. doi: 10.1038/labinvest.3700281
390. DeStefano JG, Xu ZS, Williams AJ, Yimam N, and Searson PC. Effect of shear stress on iPSC-derived human brain microvascular endothelial cells (dhBMECs). Fluids Barriers CNS. (2017) 14:20. doi: 10.1186/s12987-017-0068-z
391. Engelhardt B and Coisne C. Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two-walled castle moat surrounding the CNS castle. Fluids Barriers CNS. (2011) 8:4. doi: 10.1186/2045-8118-8-4
392. Wilson EH, Weninger W, and Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest. (2010) 120:1368–79. doi: 10.1172/JCI41911
393. Sheikh MH, Henson SM, Loiola RA, Mercurio S, Colamatteo A, Maniscalco GT, et al. Immuno-metabolic impact of the multiple sclerosis patients’ sera on endothelial cells of the blood-brain barrier. J Neuroinflammation. (2020) 17:153. doi: 10.1186/s12974-020-01810-8
394. Prat A, Henson SM, Loiola RA, Mercurio S, Colamatteo A, and Maniscalco GT. Migration of multiple sclerosis lymphocytes through brain endothelium. Arch Neurol. (2002) 59:391–7. doi: 10.1001/archneur.59.3.391
395. Bahbouhi B, Berthelot L, Pettre S, Michel L, Wiertlewski S, Weksler B, et al. Peripheral blood CD4+ T lymphocytes from multiple sclerosis patients are characterized by higher PSGL-1 expression and transmigration capacity across a human blood-brain barrier-derived endothelial cell line. J Leukoc Biol. (2009) 86:1049–63. doi: 10.1189/jlb.1008666
396. Cucullo L, Hossain M, Puvenna V, Marchi N, and Janigro D. The role of shear stress in Blood-Brain Barrier endothelial physiology. BMC Neurosci. (2011) 12:40. doi: 10.1186/1471-2202-12-40
397. Cho CF, Hossain M, Puvenna V, Marchi N, Janigro D, et al. Blood-brain-barrier spheroids as an in vitro screening platform for brain-penetrating agents. Nat Commun. (2017) 8:15623. doi: 10.1038/ncomms15623
398. Urich E, Patsch C, Aigner S, Graf M, Iacone R, and Freskgard PO. Multicellular self-assembled spheroidal model of the blood brain barrier. Sci Rep. (2013) 3:1500. doi: 10.1038/srep01500
399. Cho H, Seo JH, Wong KHK, Terasaki Y, Park J, Bong K, et al. Three-dimensional blood-brain barrier model for in vitro studies of neurovascular pathology. Sci Rep. (2015) 5:15222. doi: 10.1038/srep15222
400. Nzou G, Wicks RT, Wicks EE, Seale SA, Sane CH, Chen A, et al. Human cortex spheroid with a functional blood brain barrier for high-throughput neurotoxicity screening and disease modeling. Sci Rep. (2018) 8:7413. doi: 10.1038/s41598-018-25603-5
401. Kostka K, Sokolova V, El-Taibany A, Kruse B, Porada D, Wolff N, et al. The application of ultrasmall gold nanoparticles (2 nm) functionalized with doxorubicin in three-dimensional normal and glioblastoma organoid models of the blood-brain barrier. Molecules. (2024) 29. doi: 10.3390/molecules29112469
402. Nzou G, Wicks RT, VanOstrand NR, Mekky GA, Seale SA, El-Taibany A, et al. Multicellular 3D neurovascular unit model for assessing hypoxia and neuroinflammation induced blood-brain barrier dysfunction. Sci Rep. (2020) 10:9766. doi: 10.1038/s41598-020-66487-8
403. Sokolova V, Mekky G, van der Meer SB, Seeds MC, Atala AJ, and Epple M. Transport of ultrasmall gold nanoparticles (2 nm) across the blood-brain barrier in a six-cell brain spheroid model. Sci Rep. (2020) 10:18033. doi: 10.1038/s41598-020-75125-2
404. Sokolova V, Nzou G, van der Meer SB, Ruks T, Heggen M, Loza K, et al. Ultrasmall gold nanoparticles (2 nm) can penetrate and enter cell nuclei in an in vitro 3D brain spheroid model. Acta Biomater. (2020) 111:349–62. doi: 10.1016/j.actbio.2020.04.023
405. Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, and Shusta EV. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci Rep. (2014) 4:4160. doi: 10.1038/srep04160
406. Neal EH, Marinelli NA, Shi Y, McClatchey PM, Balotin KM, Gullett DR, et al. A simplified, fully defined differentiation scheme for producing blood-brain barrier endothelial cells from human iPSCs. Stem Cell Rep. (2019) 12:1380–8. doi: 10.1016/j.stemcr.2019.05.008
407. Lim RG, Quan C, Reyes-Ortiz AM, Lutz SE, Kedaigle AJ, Gipson TA, et al. Huntington’s disease iPSC-derived brain microvascular endothelial cells reveal WNT-mediated angiogenic and blood-brain barrier deficits. Cell Rep. (2017) 19:1365–77. doi: 10.1016/j.celrep.2017.04.021
408. Patel R, Page S, and Al-Ahmad AJ. Isogenic blood-brain barrier models based on patient-derived stem cells display inter-individual differences in cell maturation and functionality. J Neurochem. (2017) 142:74–88. doi: 10.1111/jnc.2017.142.issue-1
409. Vatine GD, Al-Ahmad A, Barriga BK, Svendsen S, Salim A, Garcia L, et al. Modeling Psychomotor Retardation using iPSCs from MCT8-Deficient Patients Indicates a Prominent Role for the Blood-Brain Barrier. Cell Stem Cell. (2017) 20:831–843.e5. doi: 10.1016/j.stem.2017.04.002
410. Mantle JL, Min L, and Lee KH. Minimum transendothelial electrical resistance thresholds for the study of small and large molecule drug transport in a human in vitro blood-brain barrier model. Mol Pharm. (2016) 13:4191–8. doi: 10.1021/acs.molpharmaceut.6b00818
411. Clark PA, Al-Ahmad AJ, Qian T, Zhang RR, Wilson HK, Weichert JP, et al. Analysis of cancer-targeting alkylphosphocholine analogue permeability characteristics using a human induced pluripotent stem cell blood-brain barrier model. Mol Pharm. (2016) 13:3341–9. doi: 10.1021/acs.molpharmaceut.6b00441
412. Schondorf DC, Aureli M, McAllister FE, Hindley CJ, Mayer F, Schmid B, et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun. (2014) 5:4028. doi: 10.1038/ncomms5028
413. Kondo T, Asai M, Tsukita K, Kutoku Y, Ohsawa Y, Sunada Y, et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell. (2013) 12:487–96. doi: 10.1016/j.stem.2013.01.009
414. Bosworth AM, Faley SL, Bellan LM, and Lippmann ES. Modeling neurovascular disorders and therapeutic outcomes with human-induced pluripotent stem cells. Front Bioeng Biotechnol. (2017) 5:87. doi: 10.3389/fbioe.2017.00087
415. Madhavan M, Nevin ZS, Shick HE, Garrison E, Clarkson-Paredes C, Karl M, et al. Induction of myelinating oligodendrocytes in human cortical spheroids. Nat Methods. (2018) 15:700–6. doi: 10.1038/s41592-018-0081-4
416. Brown JA, Pensabene V, Markov DA, Allwardt V, Neely MD, Shi M, et al. Recreating blood-brain barrier physiology and structure on chip: A novel neurovascular microfluidic bioreactor. Biomicrofluidics. (2015) 9:054124. doi: 10.1063/1.4934713
417. Yeon JH, Na D, Choi K, Ryu SW, Choi C, and Park JK. Reliable permeability assay system in a microfluidic device mimicking cerebral vasculatures. BioMed Microdevices. (2012) 14:1141–8. doi: 10.1007/s10544-012-9680-5
418. Prabhakarpandian B, Shen MC, Nichols JB, Mills IR, Sidoryk-Wegrzynowicz M, Aschner M, et al. SyM-BBB: a microfluidic Blood Brain Barrier model. Lab Chip. (2013) 13:1093–101. doi: 10.1039/c2lc41208j
419. Katt ME, Linville RM, Mayo LN, Xu ZS, and Searson PC. Functional brain-specific microvessels from iPSC-derived human brain microvascular endothelial cells: the role of matrix composition on monolayer formation. Fluids Barriers CNS. (2018) 15:7. doi: 10.1186/s12987-018-0092-7
420. Linville RM, DeStefano JG, Sklar MB, Xu Z, Farrell AM, Bogorad MI, et al. Human iPSC-derived blood-brain barrier microvessels: validation of barrier function and endothelial cell behavior. Biomaterials. (2019) 190-191:24–37. doi: 10.1016/j.biomaterials.2018.10.023
421. Campisi M, Shin Y, Osaki T, Hajal C, Chiono V, and Kamm RD. 3D self-organized microvascular model of the human blood-brain barrier with endothelial cells, pericytes and astrocytes. Biomaterials. (2018) 180:117–29. doi: 10.1016/j.biomaterials.2018.07.014
422. Bang S, Shin Y, Osaki T, Hajal C, Chiono V, Kamm RD, et al. A low permeability microfluidic blood-brain barrier platform with direct contact between perfusable vascular network and astrocytes. Sci Rep. (2017) 7:8083. doi: 10.1038/s41598-017-07416-0
423. Skardal A, Aleman J, Forsythe S, Rajan S, Murphy S, Devarasetty M, et al. Drug compound screening in single and integrated multi-organoid body-on-a-chip systems. Biofabrication. (2020) 12:025017. doi: 10.1088/1758-5090/ab6d36
424. Rajan SAP, Aleman J, Wan M, Pourhabibi Zarandi N, Nzou G, Murphy S, et al. Probing prodrug metabolism and reciprocal toxicity with an integrated and humanized multi-tissue organ-on-a-chip platform. Acta Biomater. (2020) 106:124–35. doi: 10.1016/j.actbio.2020.02.015
425. Sivandzade F and Cucullo L. In-vitro blood-brain barrier modeling: A review of modern and fast-advancing technologies. J Cereb Blood Flow Metab. (2018) 38:1667–81. doi: 10.1177/0271678X18788769
426. Meena M, Vandormael R, De Laere M, Pintelon I, Berneman Z, Watts R, et al. A microfluidic in vitro three-dimensional dynamic model of the blood-brain barrier to study the transmigration of immune cells. Brain Sci. (2022) 12. doi: 10.3390/brainsci12101293
427. Nair AL, Groenendijk L, Overdevest R, Fowke TM, Annida R, Mocellin O, et al. Human BBB-on-a-chip reveals barrier disruption, endothelial inflammation, and T cell migration under neuroinflammatory conditions. Front Mol Neurosci. (2023) 16:1250123. doi: 10.3389/fnmol.2023.1250123
428. Cohen JA, Comi G, Selmaj KW, Bar-Or A, Arnold DL, Steinman L, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24-month, phase 3 trial. Lancet Neurol. (2019) 18:1021–33. doi: 10.1016/S1474-4422(19)30238-8
429. Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind, placebo-controlled trial. 1995. Neurol. (2001) 57:S16–24. doi: 10.1056/NEJMoa1114287
430. Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. (2012) 367:1098–107. doi: 10.1056/NEJMoa1114287
431. Naismith RT, Wundes A, Ziemssen T, Jasinska E, Freedman MS, Lembo AJ, et al. Diroximel fumarate demonstrates an improved gastrointestinal tolerability profile compared with dimethyl fumarate in patients with relapsing-remitting multiple sclerosis: results from the randomized, double-blind, phase III EVOLVE-MS-2 study. CNS Drugs. (2020) 34:185–96. doi: 10.1007/s40263-020-00700-0
432. Comi G, Wundes A, Ziemssen T, Jasinska E, Freedman MS, Lembo AJ, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): a multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. (2019) 18:1009–20. doi: 10.1016/S1474-4422(19)30239-X
433. Giovannoni G, Kappos L, Selmaj KW, Bar-Or A, Arnold DL, Steinman L, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. (2010) 362:416–26. doi: 10.1056/NEJMoa0902533
434. Oleszak EL, Comi G, Cook S, Rammohan K, Rieckmann P, and Soelberg Sorensen P. Theiler’s virus infection: a model for multiple sclerosis. Clin Microbiol Rev. (2004) 17:174–207. doi: 10.1128/CMR.17.1.174-207.2004
435. Bender SJ and Weiss SR. Pathogenesis of murine coronavirus in the central nervous system. J Neuroimmune Pharmacol. (2010) 5:336–54. doi: 10.1007/s11481-010-9202-2
436. Amor S, Chang JR, Friedman H, Katsetos CD, and Platsoucas CD. Role of immune responses in protection and pathogenesis during Semliki Forest virus encephalitis. J Gen Virol. (1996) 77:281–91. doi: 10.1099/0022-1317-77-2-281
437. Plemel JR, Michaels NJ, Weishaupt N, Caprariello AV, Keough MB, Rogers JA, et al. Mechanisms of lysophosphatidylcholine-induced demyelination: A primary lipid disrupting myelinopathy. Glia. (2018) 66:327–47. doi: 10.1002/glia.23245
438. Felts PA, Woolston AM, Fernando HB, Asquith S, Gregson NA, Mizzi OJ, et al. Inflammation and primary demyelination induced by the intraspinal injection of lipopolysaccharide. Brain. (2005) 128:1649–66. doi: 10.1093/brain/awh516
439. Kipp M, Clarner T, Dang J, Copray S, and Beyer C. The cuprizone animal model: new insights into an old story. Acta Neuropathol. (2009) 118:723–36. doi: 10.1007/s00401-009-0591-3
440. Franke H, Galla HJ, and Beuckmann CT. An improved low-permeability in vitro-model of the blood-brain barrier: transport studies on retinoids, sucrose, haloperidol, caffeine and mannitol. Brain Res. (1999) 818:65–71. doi: 10.1016/S0006-8993(98)01282-7
441. Dehouck MP, Meresse S, Delorme P, Fruchart JC, and Cecchelli R. An easier, reproducible, and mass-production method to study the blood-brain barrier in vitro. J Neurochem. (1990) 54:1798–801. doi: 10.1111/j.1471-4159.1990.tb01236.x
442. Burek M, Salvador E, and Forster CY. Generation of an immortalized murine brain microvascular endothelial cell line as an in vitro blood brain barrier model. J Vis Exp. (2012):e4022. doi: 10.3791/4022
443. Stone NL, England TJ, and O’Sullivan SE. A novel transwell blood brain barrier model using primary human cells. Front Cell Neurosci. (2019) 13:230. doi: 10.3389/fncel.2019.00230
444. Barberio C, Withers A, Mishra Y, Couraud PO, Romero IA, Weksler B, et al. A human-derived neurovascular unit in vitro model to study the effects of cellular cross-talk and soluble factors on barrier integrity. Front Cell Neurosci. (2022) 16:1065193. doi: 10.3389/fncel.2022.1065193
445. Wang Y, Withers A, Mishra Y, Couraud PO, Romero IA, and Weksler B. In vitro model of the blood-brain barrier established by co-culture of primary cerebral microvascular endothelial and astrocyte cells. Neural Regener Res. (2015) 10:2011–7. doi: 10.4103/1673-5374.172320
446. Bernas MJ, Cardoso FL, Daley SK, Weinand ME, Campos AR, Ferreira AJ, et al. Establishment of primary cultures of human brain microvascular endothelial cells to provide an in vitro cellular model of the blood-brain barrier. Nat Protoc. (2010) 5:1265–72. doi: 10.1038/nprot.2010.76
447. Weksler BB, Cardoso FL, Daley SK, Weinand ME, Campos AR, Ferreira AJ, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. (2005) 19:1872–4. doi: 10.1096/fj.04-3458fje
448. Stins MF, Badger J, and Sik Kim K. Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells. Microb Pathog. (2001) 30:19–28. doi: 10.1006/mpat.2000.0406
449. Boyer-Di Ponio J, Subileau EA, Perriere N, Charneau P, Holloway K, Leveque M, et al. Instruction of circulating endothelial progenitors in vitro towards specialized blood-brain barrier and arterial phenotypes. PLoS One. (2014) 9:e84179. doi: 10.1371/journal.pone.0084179
450. Cecchelli R, El-Ayoubi F, Glacial F, Ganeshamoorthy K, Driancourt C, Godet M, et al. A stable and reproducible human blood-brain barrier model derived from hematopoietic stem cells. PLoS One. (2014) 9:e99733. doi: 10.1371/journal.pone.0099733
451. Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, et al. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol. (2012) 30:783–91. doi: 10.1038/nbt.2247
452. Kim J, Shin SA, Lee CS, and Chung HJ. An improved in vitro blood-brain barrier model for the evaluation of drug permeability using transwell with shear stress. Pharmaceutics. (2023) 16. doi: 10.3390/pharmaceutics16010048
453. Kim W, Kim J, Lee SY, Kim HM, Joo KM, and Nam DH. Simplified in vitro 3D co-culture-based blood-brain barrier model using transwell. Biochem Biophys Res Commun. (2022) 620:63–8. doi: 10.1016/j.bbrc.2022.06.083
454. Elbakary B and Badhan RKS. A dynamic perfusion based blood-brain barrier model for cytotoxicity testing and drug permeation. Sci Rep. (2020) 10:3788. doi: 10.1038/s41598-020-60689-w
455. Ahn SI, Sei YJ, Park HJ, Kim J, Ryu Y, Choi JJ, et al. Microengineered human blood-brain barrier platform for understanding nanoparticle transport mechanisms. Nat Commun. (2020) 11:175. doi: 10.1038/s41467-019-13896-7
Keywords: multiple sclerosis, autoimme disease, neuroinf lammation, blood brain barrier, neurovascular unit, in-vitro model
Citation: El-Taibany AA, Heydarian P, Porada DA, Seeds MC and Atala A (2025) Multiple sclerosis: etiology in the context of neurovascular unit and immune system involvement and advancements with in vitro blood–brain barrier models. Front. Immunol. 16:1595276. doi: 10.3389/fimmu.2025.1595276
Received: 17 March 2025; Accepted: 16 May 2025;
Published: 10 June 2025.
Edited by:
Valentina Tomassini, University of Studies G. d’Annunzio Chieti and Pescara, ItalyReviewed by:
Margaret A Jordan, James Cook University, AustraliaTianmeng Chen, University of Pittsburgh, United States
Copyright © 2025 El-Taibany, Heydarian, Porada, Seeds and Atala. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aya A. El-Taibany, YWVsdGFpYmFAd2FrZWhlYWx0aC5lZHU=