 Jiyue Xia
Jiyue Xia Youhong Jiang1
Youhong Jiang1 Wenbo Liao
Wenbo Liao Zhijun Xin
Zhijun Xin- 1Department of Orthopedic Surgery, Affiliated Hospital of Zunyi Medical University, Zunyi, Guizhou, China
- 2Department of Anesthesiology, Weifang Traditional Chinese Medicine Hospital, Weifang, Shandong, China
- 3Department of Respiratory and Critical Care Medicine, Qixingguan District People’s Hospital of Bijie, Bijie, Guizhou, China
HECT domain and ankyrin repeat-containing E3 ubiquitin-protein ligase 1 (HACE1) is a well-known tumor suppressor and is essential for embryonic development. In recent years, researchers have increasingly discovered that HACE1 plays a vital role in the pathological process of many degenerative diseases. HACE1 is regarded as a stress-responsive gene whose expression is induced by a variety of stress stimuli. The expression of HACE1 counters cell stress damage by promoting the expression of antioxidant genes and inhibiting ROS production from Rac1-dependent NADPH oxidase. Meanwhile, HACE1 serves as a crucial E3 ubiquitin ligase that activates autophagy by ubiquitinating autophagy-related receptors to clear irreversibly oxidized biomolecules within the cell. Therefore, HACE1 is essential for cellular survival by maintaining antioxidant defense mechanisms and autophagic flux. Pharmacological and genetic modulation of HACE1 expression holds potential therapeutic value in age-related diseases such as neurodegenerative disorders, cardiovascular diseases, and cancer.
Graphical Abstract.
Highlights
● HACE1 acts as a stress response factor, which activates antioxidative stress and autophagy.
● HACE1 expression is induced by physiological stimuli, while pathological stimuli deplete it.
● HACE1 is an important link between autophagy and oxidative stress.
● HACE1 plays a therapeutic role in degenerative diseases such as neurodegenerative diseases, cardiovascular diseases and cancer.
1 Introduction
Oxidative stress occurs when the production of reactive oxygen species (ROS) overwhelms the capacity of the body’s antioxidant defense systems, ultimately leading to cellular and tissue oxidative damage (1). ROS comprise both radical species, such as superoxide anions and hydroxyl radicals, and non-radical molecules like hydrogen peroxide and singlet oxygen (2). Under physiological conditions, these ROS molecules function as signaling entities, capable of activating autophagy and antioxidant defense mechanisms to maintain cellular homeostasis (3, 4). However, excessive extracellular stimulation leads to ROS accumulation and also depletes the endogenous antioxidant system, resulting in cellular redox imbalance, mitochondrial dysfunction, and DNA oxidative damage, which ultimately leads to oxidative damage and even cell death (5). Increasing evidence indicates that oxidative stress plays a crucial role in the pathogenesis of various age-related diseases, including neurodegenerative disorders, cardiovascular diseases, and cancer (6–8).
Autophagy is a catabolic process that maintains cellular homeostasis by degrading and recycling cellular components and damaged organelles (9). Research has identified autophagy as an effective alternative mechanism for antioxidant defense systems, alleviating cellular oxidative damage by removing irreversible oxidative products and damaged mitochondria within cells (10). It is well known that excessive ROS disrupts critical cellular components, including DNA, proteins, and lipids (11). Among these disruptions, DNA oxidative damage includes oxidative modifications of DNA bases and single-stranded or double-stranded DNA breaks (12). Oxidative damage to proteins causes protein carbonylation and the accumulation of unfolded proteins (13, 14). Lipid peroxidation, mainly caused by cell membrane or subcellular organelle membrane phospholipid and polyunsaturated fatty acid (PUFA), produces highly destructive carbonyl compounds and disrupts the integrity of cell membranes (15). Increasing evidence indicates that autophagy alleviates cellular oxidative damage by engulfing and degrading oxidized substances (16–18).
Recent research has found that HACE1 (HECT domain and ankyrin repeat-containing E3 ubiquitin-protein ligase 1) serves as a stress-protective gene that plays a crucial role in heart diseases, neurodegenerative diseases, and tumors. As a cross-regulator of oxidative stress and autophagy, HACE1 could mitigate the production and accumulation of oxidative damage substances by enhancing the antioxidant defense system and activating autophagy. However, previous reviews have solely focused on the role and mechanisms of HACE1 in a single disease, and a better understanding of its molecular pathogenesis is required to enable the development of targeted therapies. In this review, we first introduce the protein structure and function of HACE1 and then provide a systematic mechanism analysis of its redox signaling pathway, autophagy pathway, and tumor suppressor pathways. Therefore, the role of HACE1 in the adaptive defense system of the organism makes it a promising therapeutic target in age-related degenerative diseases. Encouragingly, researchers have discovered that certain natural antioxidants can upregulate HACE1 expression, thereby maintaining cellular homeostasis by activating autophagy and enhancing antioxidant stress responses (19–21). Undoubtedly, akin to its downstream target Nrf2, HACE1 is also destined to emerge as a novel antioxidant stress factor and attract widespread attention.
2 Basic structure and function of HACE1
HACE1, known as a renowned tumor suppressor gene, is located within the tumor suppressor region on chromosome 6q21, encoding a protein with a relative molecular mass of approximately 103 kDa (22). As shown in Figure 1, the HACE1 protein is composed of 909 amino acid residues and is generally localized in the Golgi apparatus and endoplasmic reticulum (22, 23). It consists of an N-terminal helical domain (NHD), seven ankyrin repeats (ANK), a linker middle domain (MID), and a conserved C-terminal catalytic HECT domain (24). The C-terminal HECT domain is essential for the E3 ubiquitin ligase activity of HACE1, and mutation of the conserved HECT domain cysteine residue 876 (HACE1 C876S) has been recognized to eliminate its E3 ubiquitin ligase activity (25). The NHD and MID domains influence the ubiquitination capacity of HACE1 by regulating its oligomerization state because the N-terminal helix of one monomer limits access to the C-terminal domain of the other monomer in the dimer (24, 26). Additionally, it was found that Group-I PAKs could affect the oligomerization state of HACE1 by mediating its post-translational modifications (PTMs), which consequently affects its ubiquitination activity (26). Therefore, we recommend further research on how to regulate PTMs of HACE1 to affect its activity. The ANK domain is responsible for determining the autophagic activity of HACE1 by mediating protein-protein interactions (27). In summary, every domain of HACE1 is essential for its function.

Figure 1. Basic structure and function of HACE1. (a, b) HACE1 is a tumor suppressor gene located on human chromosome 6q21 encoding a protein known as E3 ubiquitin protein ligase. (c) The ubiquitination ability of HACE1 protein is related to its oligomerization state and post-translational modifications. (d) Transcriptional activity of HACE1 is strongly correlated with CpG methylation levels in the HACE1 gene promoter.
HACE1 is a bifunctional E3 ubiquitin ligase. On one hand, HACE1 transfers ubiquitin to target substrates via its HECT domain E3 ubiquitin ligase activity, leading to subsequent proteasomal degradation. Studies have shown that the tumor suppressor function of HACE1 is closely linked to its E3 ubiquitin ligase activity. Mechanistically, HACE1 mediates the ubiquitination and degradation of Rac1 via its E3 ubiquitin ligase activity, thereby inhibiting ROS generation from Rac1-dependent NADPH oxidase and cellular hyperproliferation induced by cyclin D1 (28–30). While the activity and level of Rac1 can be modulated by regulating the ubiquitination capacity of HACE1 (24, 26, 31). On the other hand, HACE1 can activate autophagy independently of its E3 ubiquitin ligase activity. Research indicates that HACE1 activates p62-dependent selective autophagy through protein-protein interactions mediated by its ANK domain (27). Furthermore, HACE1 is capable of interacting with OPTN without relying on the E3 ubiquitin ligase activity (32). In addition, HACE1 can regulate the ubiquitination and degradation efficiency of target proteins by influencing the protein-protein interactions through the ANK domain (31). For example, HACE1 controls the level of active Rac1 and cell migration by interacting with Rac1 through the ANK domain (33). All in all, HACE1 is a critical E3 ubiquitin ligase and exerts its biological function through the activation of either the proteasome pathway or the autophagy lysosomal pathway, as shown in Figure 2.

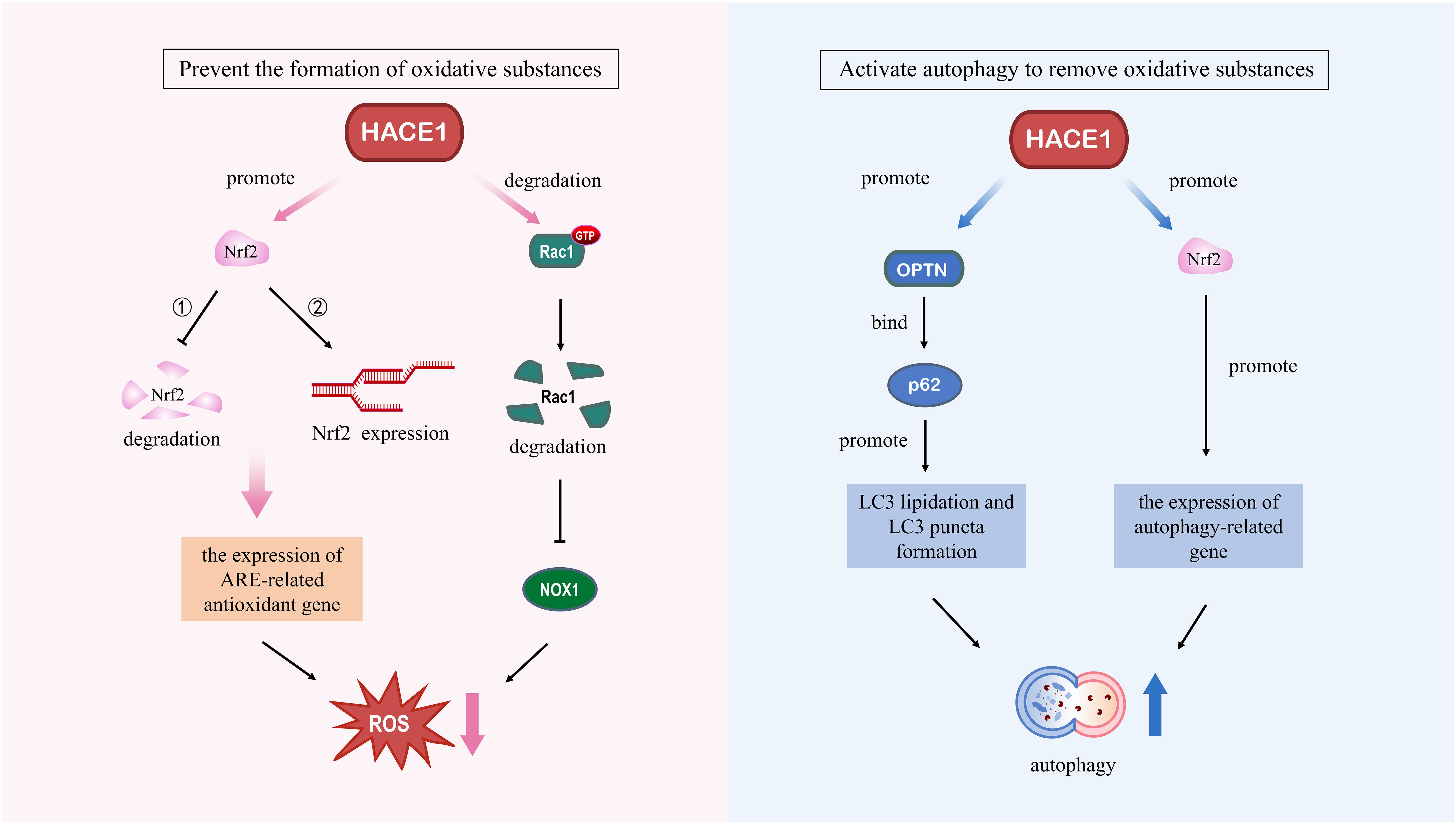
Figure 2. Molecular mechanisms of HACE1 in degenerative diseases. First, HACE1 might reduce oxidative stress by modulating Nrf2, Rac1, and CCNC. Second, HACE1 activates autophagy by ubiquitinating OPTN to promote the interaction of OPTN with p62. Moreover, HACE1 can interact directly with p62 to activate autophagy. Third, HACE1 inhibits tumor cell proliferation by suppressing the expression of CCND1.
3 HACE1 dysfunction plays a significant role in neurodegenerative diseases
Neurodegenerative diseases are a class of clinical syndromes characterized by progressive and irreversible damage to specific neurons, leading to the gradual decline of corresponding neurological functions (34). As shown in Table 1, their common pathological mechanism lies in the misfolding and aggregation of intracellular neuronal proteins (such as Aβ, tau, and α-synuclein), forming insoluble deposits that induce oxidative stress and mitochondrial dysfunction, ultimately leading to synaptic dysfunction and neuronal cell death (35). As a novel molecular target in neurodegenerative diseases, HACE1 is closely related to neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease and Huntington’s disease (36). How HACE1 deficiency leads to neurodegenerative proteinopathies is not fully understood, but two distinct mechanisms are thought to be particularly relevant. First, HACE1 deficiency causes disruption of the ubiquitin-proteasome system and autophagolysosome pathway, resulting in protein aggregates that cannot be cleared (27). Second, HACE1 deficiency causes dysfunction of mitochondria and antioxidant defense systems, resulting in increased oxidative stress and protein misfolding, which exacerbates protein aggregate formation (37, 38).
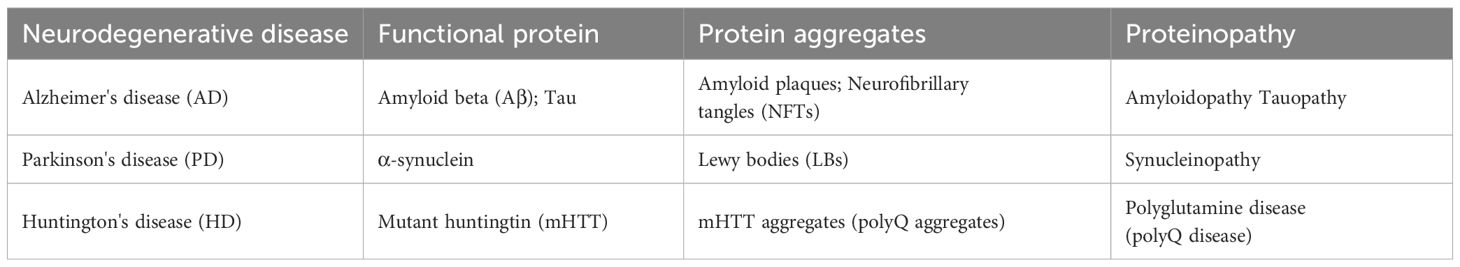
Table 1. The formation of misfolded protein aggregates is a hallmark of neurodegenerative diseases.
4 HACE1 expression promotes cellular antioxidant defense capacity
HACE1 functions as a ubiquitous early stress response gene induced by various stress stimuli, including endoplasmic reticulum stress, virus infection, nutrient deprivation, hypoxia and gamma irradiation (39, 40). The role of HACE1 in ischemia reperfusion (IR) injury has recently been reported. IR for a short period of time could upregulate HACE1 expression and activate the antioxidant stress pathway to alleviate IR-induced cardiac injury (41). However, prolonged IR stimulation resulted in depletion of HACE1 reserves, which exacerbated oxidative stress and inflammation in neurons and cardiomyocytes (42, 43). It is well known that oxidative stress is the main mechanism of tissue damage induced by IR (44). Thus, HACE1 expression induced by this hemodynamic stress is a compensatory response of the body, and lack of HACE1 results in cell sensitivity to oxidative damage and dependence on extracellular nutrients (45, 46). A growing body of research suggests that HACE1 plays an important role in regulating oxidative stress in age-related degenerative diseases.
4.1 HACE1 protects against oxidative damage by activating Nrf2
Nuclear factor erythroid-derived 2-like-2 (Nrf2), a key antioxidant transcription factor, plays an important role in enhancing cellular resistance to toxic stimuli and oxidative stress damage (47). Under resting conditions, Keap1 functions as a substrate adaptor protein for a Cul3-dependent E3 ubiquitin ligase complex. Within the E3 ubiquitin ligase complex based on the Cul3 scaffold, Keap1 forms homodimers through its BTB domain, which allows it to specifically recognize and expedite the ubiquitination and degradation of cytoplasmic Nrf2 (48). The oxidative modification of specific cysteine residues on Keap1 causes spatial conformational changes that cause Nrf2 to separate from ubiquitination (49). Furthermore, Nrf2 expression and nuclear translocation can be induced by oxidative stress, which eventually results in Nrf2 accumulation in the nucleus (50). Binding to small Maf proteins, nuclear Nrf2 activates antioxidant response elements (AREs) and triggers the expression of several genes related to detoxification and antioxidants (51).
HACE1 is an oxidative stress response factor that is essential for maintaining cellular antioxidant defense by mediating Nrf2 activation. Specifically, HACE1 prevents Nrf2 ubiquitination and proteasomal degradation by competing with Keap1, thereby stabilizing Nrf2 protein and enhancing its transcriptional activity in response to oxidative stress (52). This process requires HACE1 to have an intact domain, because the loss of either the HECT or ANK domain will lead to the disruption of Nrf2 protein homeostasis (52). Hence, the cytoprotective effect of HACE1 may be mainly attributed to the antioxidant activity of Nrf2. Rotblat et al. (45) found that HACE1 is closely associated with neurodegenerative diseases because its expression levels are reduced in the striatum of Huntington’s disease patients. Furthermore, knockout of HACE1 resulted in impaired antioxidant stress response in mice, and this sensitivity to oxidative stress could be restored by ectopic expression of Nrf2 (45). In addition to inhibiting neurodegeneration, HACE1 has been reported to activate the Nrf2/ARE signaling pathway, upregulate HO-1 and NQO1 antioxidant genes, and attenuate H/R-induced oxidative stress and inflammation, making it a promising therapeutic target and predictor of cardiac disease (42). Consistent with findings in the nervous system, the protective effect of HACE1 in the heart was also confirmed in Nrf2-mediated responses (53).
4.2 HACE1 alleviates oxidative damage through ubiquitination and degradation of active Rac1
Ras-related C3 botulinum toxin substrate 1 (Rac1) is a small GTPase belonging to the Rho GTPase family and an essential intracellular signaling molecule (54). As a signal transduction molecular switch, Rac1 alternates between inactive and active states to transduce cellular signals. The activated form of Rac1 is an indispensable subunit of NOX, which is essential for the subsequent activation of NOX and ROS generation (55). NOX is a transmembrane protein composed of two catalytic subunits (gp91phox and p22phox) and four regulatory subunits (p47phox, p40phox, p67phox and Rac1) (56). In a resting cellular state, NOX is inactive because there are no cytoplasmic subunits bound to the cell membrane (57). When cells are stimulated by internal and external factors, the cytoplasmic subunit of NOX is modified by phosphorylation to undergo a conformational change and ultimately assembles with the membrane-bound subunit to form a catalytically active oxidase (58). In addition, previous study has reported that there is a mutual activation between Rac1 and NOX, which is instrumental in sustaining physiological levels of ROS required for axonal growth of hippocampal neurons (59).
The current study has found that the E3 ubiquitin ligases HACE1 and the IAPs are responsible for the ubiquitination and proteasome degradation of activated Rac1 (60). During the further identification process, Stéphanie et al. (61) have found that HACE1 is the E3 ubiquitin ligase with the greatest influence on the regulation of Rac1 turnover and activity. As shown in the Figure 3, it has been shown that HACE1 controls the activity of the Rac1-dependent NOX complex by targeting Rac1 for degradation, which inhibits the production of superoxide and ROS (29, 62). The deficiency of HACE1 leads to the hyperactivation of Rac1, resulting in increased cellular malignancy and invasiveness, thereby promoting cancer progression (25, 55, 63). In addition to its therapeutic role in tumors, HACE1 is significant for embryonic development through the Rac1 signaling pathway (64). Lack of HACE1 causes the accumulation of Rac1 and NADPH oxidase-dependent ROS production, which ultimately results in vertebrate cardiac and neurological hypoplasia (65, 66). Zang et al. (67) have found that HACE1 expression decreased with age in mouse models of Parkinson’s disease. HACE1 knockdown exacerbates neuroinflammation and symptoms in PD in a Rac1-dependent NADPH oxidase manner (67). Therefore, targeting HACE1 to inhibit the Rac1 signaling pathway to alleviate cellular oxidative stress would be an effective therapeutic strategy in degenerative diseases.
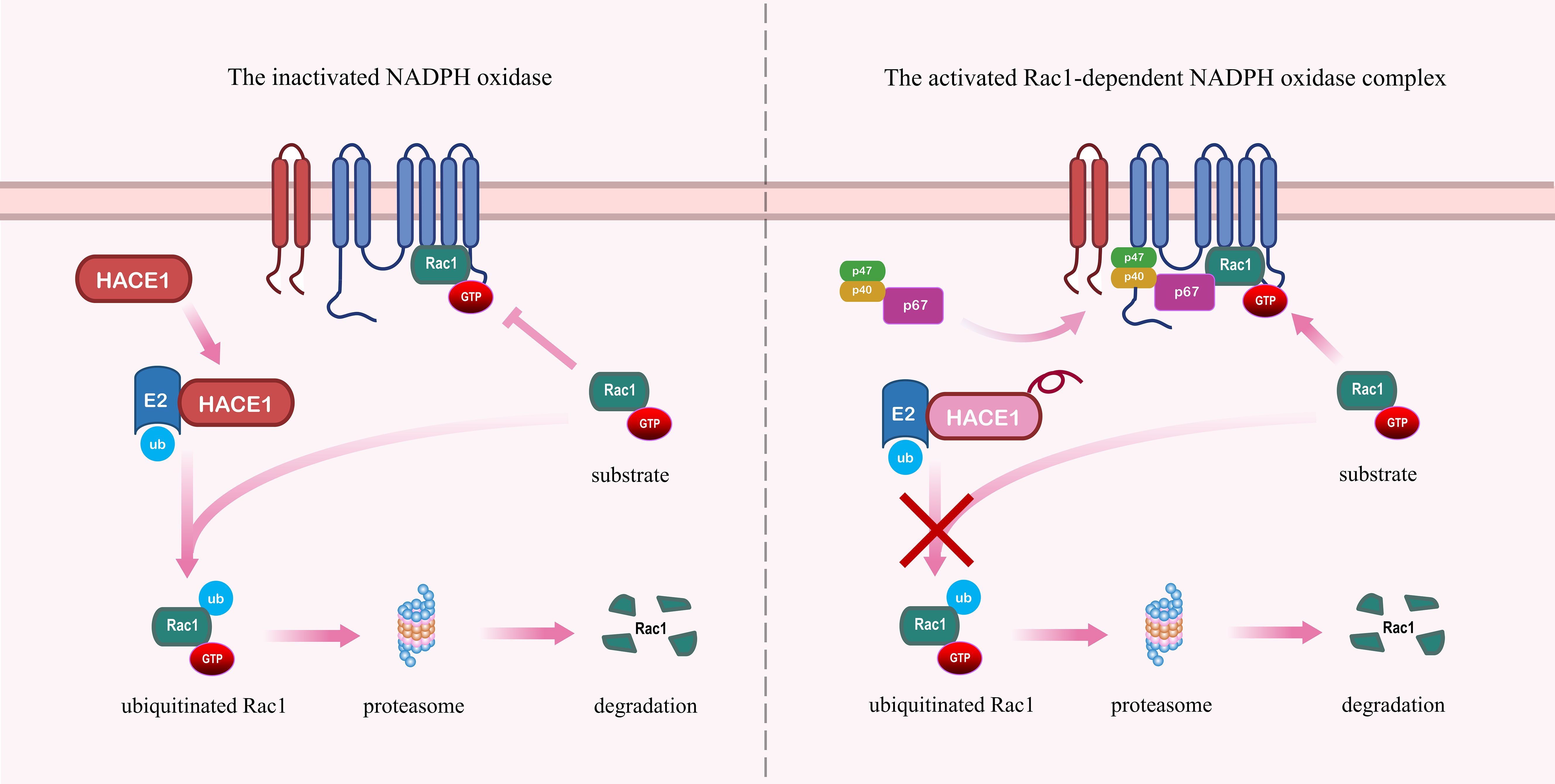
Figure 3. HACE1 prevents the formation of the NADPH oxidase complex by promoting the ubiquitination and degradation of Rac1. Left panel: HACE1 inhibits NADPH oxidase by ubiquitinating and degrading activated Rac1. Right panel: Deficiency of HACE1 impedes the ubiquitination and degradation of activated Rac1, thereby exacerbating the formation of the NADPH oxidase complex.
As research progressed, it has been discovered that activation of Rac1 is necessary for NADPH oxidase-dependent ROS production (62). Thus, one might wonder whether HACE1 regulates oxidative stress through degrading Rac1. To confirm this conjecture, we summarize the functional mechanisms and actions of HACE1 in regulating Rac1 in Table 2. The expression of HACE1 protein is influenced by various factors, including gene editing, HACE1 mutations, and the regulation of HACE1-PTMs. HIF inhibitors (FIH) have been found to inhibit the ability of HACE1 to ubiquitinate Rac1 by hydroxylating HACE1 at residue N191 of the ANK domain (68). Neuroepithelial transforming gene 1 (NET1) protects Rac1 from degradation by HACE1 and is essential for actin dynamics and meiotic spindle formation (69). Furthermore, HACE1-driven Rac1 ubiquitination degradation also requires another E3 ubiquitin ligase, TRIP12. Silencing TRIP12 blocks the inhibitory effect of HACE1 on Rac1 protein degradation and esophageal cancer (ESCA) tumor growth (70). Rac1 has been considered a central signaling hub essential for many oncogene-induced transformations, playing a crucial role in cancer cell proliferation, migration, and invasion (71). HACE1 inhibits the progression of a variety of tumors, including breast cancer, lung cancer, prostate cancer, and osteosarcoma, by regulating the ubiquitination and proteasomal degradation of Rac1. Specifically, HACE1 protects cells from oxidative stress-induced DNA damage and cycle D1-driven hyperproliferation by blocking ROS production through Rac1-dependent NADPH oxidase. In addition, HACE1 plays a crucial role in the growth and development of the organism, especially the heart and nervous system. Taken together, HACE1 regulates cellular oxidative stress in a Rac1-dependent manner and is essential in organ development and tumor suppression.
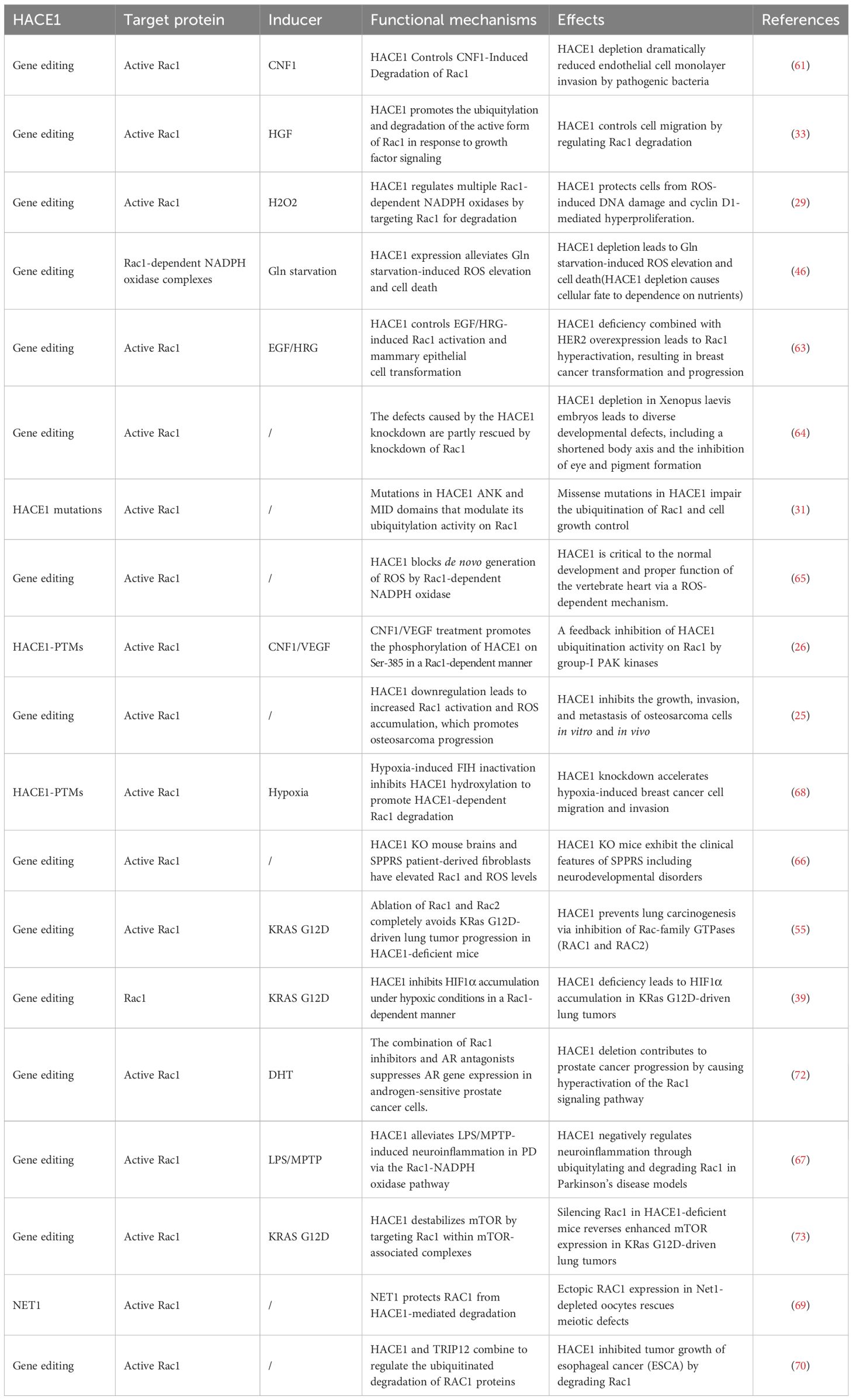
Table 2. Functional mechanisms and effects of HACE1 ubiquitination degradation activity Rac1.
5 HACE1 activates cellular autophagy through ubiquitination of autophagy-related receptor proteins
Autophagy is a catabolic degradation process necessary for cell homeostasis, which is an extremely important alternative pathway for cellular antioxidants (74). When autophagy is impaired, it leads to mitochondrial dysfunction, excessive ROS accumulation and disruption of cell homeostasis (75). Viedma-Poyatos et al. (76) discovered a vicious cycle of mutual intensification between oxidative stress and protein aggregation, resulting in abnormal intracellular protein aggregation and cytotoxicity. Accumulating evidence suggests that protein aggregation and mitochondrial dysfunction are common features associated with many age-related degenerative diseases, especially neurodegenerative diseases, cardiovascular diseases and tumors (77–79). In these diseases, autophagy efficiency decreases in various ways with age. Autophagy is essential for maintaining cell survival under oxidative stress conditions by selectively removing damaged organelles and protein aggregates (80). Therefore, the role of HACE1 gene in antioxidant stress has recently attracted increasing attention. Patients with mutations in the HACE1 gene contribute to severe neurodevelopmental deficits by causing autophagy defects and oxidative stress (37). As an autophagy adapter protein, HACE1 prevents cellular oxidative damage and tumor progression by activating autophagy to remove toxic proteins and damaged organelles through the ubiquitination of the selective autophagy receptors p62 and OPTN.
5.1 HACE1 activates selective autophagy through ubiquitination of OPTN
Optineurin (OPTN) is a multifunctional protein with multiple domains and is broadly expressed in many organs and tissues in the human body (81). OPTN consists of two coiled-coil (CC) domains, a leucine zipper (LZ) domain, an LC3-interaction region (LIR), a HACE1-interaction region (HIR), a ubiquitin-binding (UBAN) domain, and a C-terminal Npl4-type zinc finger (NZF) domain (82). The N-terminal CC domain of OPTN binds directly to the C-terminal domain of TBK1, forming stable OPTN-TBK1 heterotetrameric complexes that are recruited into ubiquitinated aggregates (83). The LZ domain of OPTN interacts with ATG9A, which facilitates the recruitment of ATG9A vesicles for autophagosome formation (84). The LZ and LIR domains of OPTN bind to the N-terminal death effector domains (DEDs) of caspase 8 and regulate TNF-α-induced apoptosis (82). In addition, Wauters et al. have discovered that OPTN binds to ubiquitinated mitochondria through its UBAN domain and recruits LC3 to wrap damaged mitochondria to form autophagosomes through its LIR domain (85). Finally, Liu et al. have found that HACE1 can interact with OPTN through its N-terminal ANK structural domain, and the region on OPTN that interacts with HACE1 is defined as the HACE1 interaction region (HIR) (32).
Previous studies have established that TANK-binding kinase 1 (TBK1) induces autophagy through phosphorylation of the autophagy receptors p62 and OPTN (86). TBK1 phosphorylates directly OPTN and promotes its interaction with LC3 to accelerate autophagy efficiency (86). In turn, OPTN enhances the phosphorylation ability of TBK1 by promoting TBK1 activation (87). This process of autophagy activation forms a positive feedback loop to increase autophagic flux. It has been shown that HACE1 is upstream of TBK1 and can inhibit virus-triggered type I IFN signaling by disrupting the formation of the MAVS-TRAF3 complex (88). Therefore, in addition to focusing on the direct interaction of HACE1 with OPTN, HACE1 may also be involved in regulating autophagy efficiency by affecting TBK1 activity.
OPTN is a recognized selective autophagy receptor that mediates interactions between ubiquitinated proteins and autophagy machinery, and lack of OPTN leads to impairment of the autophagy pathway and accumulation of oxidized proteins (32). Furthermore, the autophagy activity of OPTN is affected by its expression level as well as post-translational modifications (89). HACE1 was found to enhance the interaction between OPTN and p62 by promoting ubiquitination of OPTN, and promote the formation of autophagy receptor complex to accelerate autophagy flux (32). Here, E3 ubiquitin ligase activity of HACE1 and subsequent ubiquitination of OPTN (specifically ubiquitination at Lys193) are essential for activation of autophagy. Disruption of the polyubiquitin chain on OPTN leads to impaired formation of the p62-OPTN complex, resulting in disruption of the HACE1-OPTN-p62 autophagy axis. More and more studies show that that HACE1 activates selective autophagy mediated by the HACE1-OPTN-P62 axis through ubiquitination of OPTN, thereby inhibiting tumor cell growth and progression (20, 90, 91). Thus, HACE1 bridges the gap between the ubiquitination and autophagy pathways by ubiquitinating the selective autophagy receptor OPTN. Targeting HACE1 to induce the ubiquitination of OPTN, thereby activating cellular autophagy, holds promise for the treatment of diseases characterized by autophagy deficiency.
5.2 HACE1 alleviates cellular damage by activating autophagy dependent on p62
p62 (also known as sequestosome-1, SQSTM1) is an adapter protein with multiple functional domains, including N-terminal PB1 domain, ZZ domain, TB domain, nuclear localization signal, nuclear export signal, PEST domain, LIR domain, KIR domain and C-terminal UBA domain (92). The LIR and UBA domains are key regulatory elements of p62 as a selective autophagy adaptor protein. p62 directly binds to LC3-II via the LIR domain to mediate autophagic degradation of specifically ubiquitylated cargoes bound to the UBA domain of p62 (93). The efficiency of p62-mediated autophagy is not only influenced by its UBA domain and ubiquitinated cargo but also by oligomerization mediated by the PB1 domain (94). p62 regulates the recruitment of target cargo and the formation of autophagosomes through the oligomerization mediated by the PB1 domain (95). The KIR domain of p62 participates in the regulation of cellular oxidative stress by binding to Keap1 and promoting the autophagic degradation of it (96). Besides, the linker region between the PB1 and ZZ domains is known as the regulatory linker (RL) region, which acts as a regulatory switch for p62 activity by interacting with the ZZ domain to regulate the binding ability of the ZZ domain (97). Taken together, these findings indicate that p62 interacts with various cellular signaling molecules through its multiple domains, thereby executing diverse cellular functions.
Autophagy is a dynamic process including the formation of autophagosomes, fusion of lysosomes and autophagosomes, and degradation of autophagosomes by lysosomes (98). Prior research has demonstrated that the autophagic activity of HACE1 is dependent on p62. The ablation of p62 disrupted the HACE1-OPTN autophagy axis, which could be reactivated by reintroducing p62 (32). Furthermore, this study also found that the HACE1-OPTN-p62 axis activates autophagy by promoting LC3II lipidation and the formation of LC3 puncta, thereby clearing p62 and carbonylated proteins to reduce ROS production (32). This avoidance of the production and accumulation of oxidative damage may be the underlying mechanism of the HACE1-OPTN-p62 axis inhibiting tumor growth and tumorigenicity. When the heart experiences hemodynamic stress, HACE1 exerts cardioprotective effects by activating p62-dependent selective autophagy to clear ubiquitinated protein aggregates. In contrast, mice lacking HACE1 have impaired autophagic flow and are less able to resist pressure stress, leading to accelerated heart failure and increased mortality (27). Mechanistically, HACE1 directly interacts with p62 to affect the cellular level of p62 independently of its E3 ubiquitin ligase activity (27). These findings have identified HACE1 as a regulator of autophagy, which activates cellular autophagy by promoting the activation of p62, thereby mitigating cellular stress injury.
6 HACE1 regulates cell proliferation and oxidative stress through ubiquitinated cell cycle proteins
HACE1 is an important tumor suppressor that is depleted in many malignant tumors (63). The current study indicates that the tumor suppressor function of HACE1 may be closely related to its anti-oxidative stress capacity and autophagic activity. It has been confirmed that excessive ROS can cause oxidative damage to DNA and gene mutations, leading to cell transformation and carcinogenesis (99). HACE1 blocks the generation of ROS by Rac1-dependent NADPH oxidases, thereby alleviating the initiation and progression of tumor cells caused by DNA oxidative damage and cyclin D1-driven hyperproliferation (29). In addition, the potential therapeutic role of HACE1-activated selective autophagy in tumors has been previously discussed. Remarkably, cyclin D1 has been identified as an oncogene and is overexpressed in a variety of malignant tumors (100). Furthermore, the protein level of cyclin D1 is closely related to selective autophagy, which inhibits tumor cell proliferation through autophagy‐selective degradation of cyclin D1 (101). Therefore, these findings indicate that cyclin may be involved in HACE1 regulation of oxidative stress and autophagy that plays an important role in tumors.
6.1 HACE1 inhibits tumor cell growth, invasion, and metastasis through the degradation of cyclin D1
Cyclin D1 (CCND1) is a protein encoded by CCND1 gene located on chromosome 11q13 and involved in regulating cell cycle progression (102). During the process of cell proliferation, cyclin D1 activates and binds to cyclin-dependent kinase 4 and 6 (CDK4/6) to form the cyclin D1-CDK4/6 complex (103). This complex phosphorylates retinoblastoma (Rb) protein, leading to the release of the E2F transcription factor, which promotes gene expression associated with G1/S phase transition (104). However, overexpression of cyclin D1 leads to cell cycle checkpoint failure and CDK dysregulation, which promotes tumor initiation and progression (105). Furthermore, the current studies demonstrate that cyclin D1 can also recruit methyltransferase G9a independently of CDK kinase activity to induce H3K9 demethylation, thereby regulating gene expression and signaling (106).
Cyclin D1 acts as a signal transduction factor in response to various intracellular and extracellular signaling factors. For example, extracellular growth factors induce Cyclin D1 expression by activating the Ras/Raf/MAPK signaling pathway (107). Recent studies have shown that the degradation of cyclin D1 can be regulated by proteasomal and autophagic lysosomes, and GSK3β-induced T286 phosphorylation of cyclin D1 is considered to be the main mechanism of cyclin D1 proteasomal degradation (108). Activation of the PI3K/AKT signaling pathway promotes Ser9 phosphorylation of GSK3β, which leads to GSK3β inactivation and cyclin D1 stabilization (109). Additionally, cytoplasmic membrane-associated cyclin D1 augmented the phosphorylation of AKT1 at Ser473 (110). This creates the positive feedback loop required to maintain intracellular cyclin D1 levels. AMBRA1 (autophagy and beclin 1 regulator 1) is a major regulator of cyclin D1 and mediates ubiquitination and proteasomal degradation of cyclin D1 (111). It has been reported that ubiquitinated cyclin D1 can be degraded via p62-mediated selective autophagy (101).
As a well-known tumor suppressor, HACE1 can inhibit the growth, invasion, and metastasis of multiple cancer types. Penninger and colleagues have found that the tumor suppressor effect of HACE1 may be attributed to cyclin D1, and HACE1 prevents cellular stress-induced tumor cell proliferation and adhesion-dependent growth through the degradation of cyclin D1 (30). Meanwhile, they have also shown that HACE1 inhibits ROS de novo synthesis and cyclin D1 expression by blocking Rac1-dependent NOX activity (29). Although there are no studies to show that HACE1 directly targets cyclin D1 for degradation, cyclin D1 transcription requires Rac1 and NOX activities to complete (112). Lack of HACE1 results in increased Rac1-dependent cyclin D1 transcription (29). Moreover, the expression level of cyclin D1 is inhibited by OPTN, a substrate protein of HACE1, which did this by limiting Rac1 activation (113). Taken together, these studies support the notion that HACE1 exerts a tumor suppressor function by regulating cyclin D1.
6.2 HACE1 regulates mitochondrial oxidative stress and apoptosis through ubiquitination of cyclin C
Cyclin C (CCNC) is an important cell cycle protein that regulates transcription and forms the kinase module of the mediator complex with the partner kinase CDK8 and the two auxiliary subunits Med12 and Med13 (114). The mediator complex stimulates the assembly of a pre-initiation complex (PIC) and recruitment of RNA Polymerase II, which is responsible for the transcription of all protein-coding genes and most non-coding RNA genes (115, 116). Apart from transcriptional function, cyclin C is involved in the regulation of oxidative stress by activating the mitochondria-dependent cell death pathway (117). Loss of cyclin C blocks the stress-induced mitochondrial apoptosis pathway, rendering malignant tumor cells insensitive to chemotherapy (118). Recent studies report that subcellular localization of cyclin C is critical for regulating cell fate (119). Cyclin C is primarily located in the nucleus during the resting state, interacting with the Med13 to regulate transcription (120). Under oxidative stress, cyclin C translocates from the nucleus to the cytoplasm, leading to activation of the mitochondrial apoptotic pathway (121). These findings suggest that subcellular localization of cyclin C is essential for oxidative stress-induced apoptosis.
The subcellular localization of cyclin C is closely related to its protein post-translational modification. Research suggests that the phosphorylation of cyclin C is essential for its translocation from the cytoplasm to the nucleus. Oxidative stress activates Slt2p to phosphorylate cyclin C protein at Ser266 triggering the mitochondrial intrinsic pathway of apoptosis (122). HACE1 mediates the non-proteolytic ubiquitination of cyclin C in the cytoplasm, thereby promoting nuclear–mitochondrial translocation of cyclin C and maintaining the chemosensitivity of gastric cancer cells to cisplatin (118). However, it has also been shown that cyclin C relocalizes for cytoplasmic degradation in response to oxidative stress, thereby inducing normal stress-responsive gene expression (123). This apparent contradiction could be explained by the differential stages of oxidative stress processes in different cell types. However, further research is needed to explore these speculative mechanisms. In conclusion, these studies suggest that cyclin C is an important regulator of cell fate determination by regulating oxidative stress-induced mitochondrial apoptosis.
7 Modifications influencing HACE1 expression
As an important factor in the modulation of cellular stability and survival, HACE1 plays a significant role in regulating oxidative stress, autophagy, and tumor suppression. Researches have found that HACE1 is widely expressed in a variety of normal human tissues, with particularly abundant expression in the heart, kidney, and brain (22). Recently, an increasing number of studies have indicated that HACE1 inactivation is strongly associated with poor prognosis in neurodegenerative diseases, cardiovascular diseases, and tumors. Thus, targeting HACE1 is a promising strategy for the treatment of age-related diseases, and the specific agonists that promote HACE1 expression and the precise molecular mechanisms will be the focus of future research.
7.1 Methylation of HACE1
HACE1 is an indispensable tumor suppressor gene, and hypermethylation of its gene region has been found in a variety of human malignancies, including gastric cancer, colorectal cancer, liver cancer, Wilms tumor, B-cell lymphomagenesis (124). DNA methylation of CpG islands in the promoter is an important mechanism for silencing gene expression. Under normal physiological circumstances, most of the genome is methylated, and CpG islands commonly near promoters remain typically unmethylated (125). During the process of tumorigenesis, human cancer cells undergo CpG island promoter hypermethylation and loss of non-CpG island promoter CpG methylation, resulting in cancer-specific methylation patterns (126). This epigenetic alteration affects tumor suppressor genes and oncogene expression, which may lead to the malignant progression of tumors. Previous research has shown that inactivation of HACE1 renders mice more susceptible to spontaneous and carcinogen-induced tumors (30). The inactivation of HACE1 may be attributed to gene silencing due to hypermethylation of CpG islands on its promoter region (124). Analysis of the TCGA database data further has confirmed that hypermethylation of CpG islands in the HACE1 promoter region in tumors is associated with low HACE1 expression (124). Taken together, the epigenetic phenomenon of CpG island hypermethylation in HACE1 gene promoters, leading to repression and silencing of expression, is an important contributor to oncogenesis.
There are three CpG islands in HACE1 gene regions, including CpG-88, CpG-177, and CpG-29. The hypermethylation of CpG29 and CpG177 islands upstream of the HACE1 transcription start site (TSS) is associated with its low expression level was reported in human Wilms tumor (22). Increased methylation of CpG177 is associated with decreased HACE1 expression in tumor tissue compared to normal tissue (30). This has also been shown in other tumors, such as aggressive natural killer cell leukemia (ANKL) (127) and natural killer-cell neoplasms (128). These findings indicate that the expression of HACE1 through demethylation may be a promising therapeutic strategy for malignant tumors.
Previous studies have provided evidence that HACE1 inactivation in multiple cancers is due to promoter methylation (63). Enhancing HACE1 expression by regulating methylation leads to extensive research on its regulatory mechanism and its tumor-suppressive effect. For example, effective restoration of HACE1 expression in nephroblastoma by the addition of the specific DNA methylation inhibitor 5-azacytidine (5-Aza) (22). NKX6.3 attenuates Helicobacter pylori CagA-induced cellular oxidative stress by inhibiting DNA methyltransferase 1 (DNMT1) and promoting HACE1 expression. The antioxidant activity of NKX6.3 could be eliminated by HACE1 silencing (129). The histone methyltransferase inhibitor 3-deazaneplanocin A (DZNep) inhibits lymphoma cell growth by promoting HACE1 expression by reducing histone methylation modifications (130). Demethylation of the HACE1 gene promoter has increased HACE1 expression, which inhibited liver cancer cell proliferation by activating OPTN-dependent selective autophagy (90). LINC00161 is a long non-coding RNA closely related to the occurrence and development of hepatocellular carcinoma (HCC), and its expression is upregulated in HCC cells. Inhibition of HACE1 expression by inducing HACE1 promoter methylation can promote the growth and migration of HCC cells. Knockdown of LINC00161 can reduce the methylation level and increase the expression of HACE1, thereby inhibiting the progression of HCC (131). The expression of MBD3, a demethylation molecule, induced by propofol in a dose-dependent manner, promotes HACE1 protein expression and inhibits lung cancer cell proliferation by activating HACE1-OPTN axis autophagy. However, these therapeutic effects of propofol are eliminated by MBD3 knockout (91). DNA methylation is an epigenetic modification that regulates gene expression that regulates gene expression. It is an important direction for future development and has research value and exploratory significance.
7.2 Antioxidants serve as a potential HACE1 activator
Gene expression is regulated by multiple pathways, including epigenetic regulation through DNA methylation and histone acetylation (132). We have also discussed in the previous section that HACE1 expression is regulated by the DNA methylation level of the promoter regions. However, HACE1 protein expression is regulated at multiple levels, including protein degradation and gene expression. Previous studies have reported that dimethyl fumarate (DMF), an Nrf2 activator, induces the protein expression of both Nrf2 and HACE1 in a dose-dependent manner, thereby inhibiting the proliferation of T-cell acute lymphoblastic leukemia cells (19). Additionally, Huang and colleagues have found that Magnolol, extracted from the renowned traditional Chinese medicine Magnolia officinalis, exerts its antitumor effects by activating HACE1-OPTN axis-mediated autophagy (20). Finally, the supplementation of the blueberry anthocyanin malvidin 3-glucoside (MG) in colon tissues has been found to increase HACE1 expression and improve the gut microbiota composition (21). The prevalence of degenerative diseases in recent years has triggered extensive research into their pathomechanisms and treatment. At the same time, growing evidence suggests that HACE1 also plays a crucial role in degenerative diseases. HACE1 is an important regulator of cell survival, it maintains cellular homeostasis by activating cellular autophagy and the antioxidant defense system, and its biological mechanism is shown in Figure 4. Although the mechanism of action of HACE1 has been well studied, unfortunately, there are no recognized activators of targeted therapy. Therefore, a promising research focus to develop specific and potent activators of HACE1 is an urgent research problem that needs to be addressed.
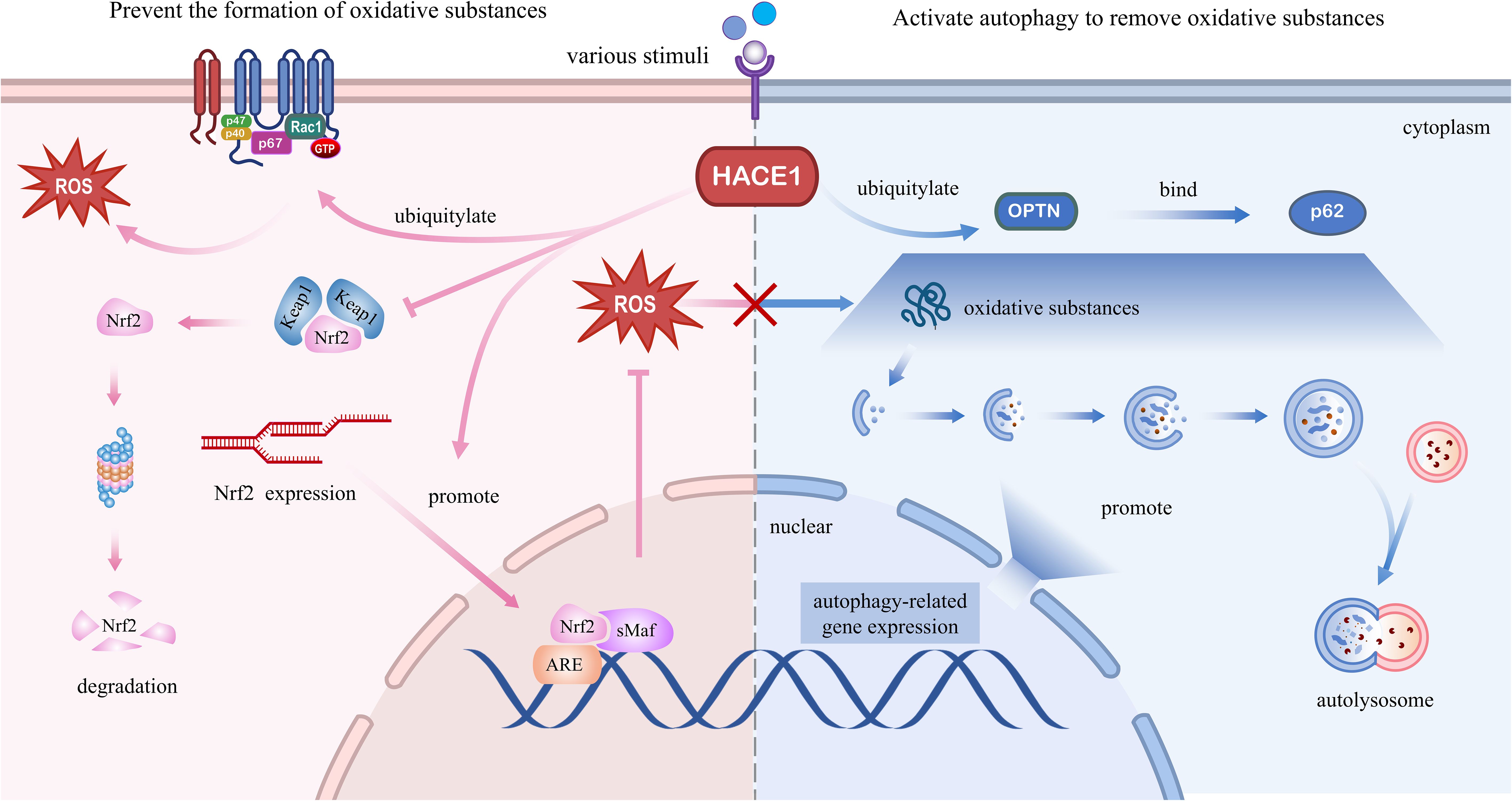
Figure 4. Molecular regulatory mechanisms of HACE1 in degenerative diseases. Left panel: HACE1 prevents the formation of oxidative substances. HACE1 inhibits NADPH oxidase 1-dependent ROS generation by ubiquitinating and degrading Rac1. HACE1 activates the Nrf2/ARE signaling pathway by competitively binding to Nrf2 with Keap1, enhancing the stability of the Nrf2 protein, and promoting the de novo synthesis of Nrf2. Right panel: HACE1 activates autophagy to remove oxidative substances. HACE1 activates autophagy by promoting the expression of autophagy-related genes and interacting with the autophagy receptors OPTN and p62.
8 Conclusions and perspectives
With the intensification of global aging, the incidence of various age-related degenerative neurological and cardiovascular diseases continues to increase (133). Although the pathogenesis of aging diseases is complex and still unclear, oxidative stress and impaired autophagy have been shown to be key pathogenic mechanisms in these diseases (134). In this review, we provide an overview of oxidative stress and autophagy and summarize the literature related to HACE1, discuss the mechanisms by which HACE1 regulates oxidative stress and autophagy, and highlight its potential therapeutic role in degenerative diseases (Figure 5). HACE1 has been identified as an oxidative stress response gene that upregulates cellular antioxidant defenses by targeting Nrf2 and Rac1, reducing the source of oxidative substances. In addition, HACE1 activates autophagy by ubiquitinating selective autophagy receptors OPTN and p62, increasing the degradation of oxidative species. It is well known that the continuous accumulation of non-degradable highly oxidized proteins leading to protein aggregate formation is an important hallmark of aging (135). In addition, the changes of HACE1 content in the serum of patients with heart failure imply its prognostic significance (53).
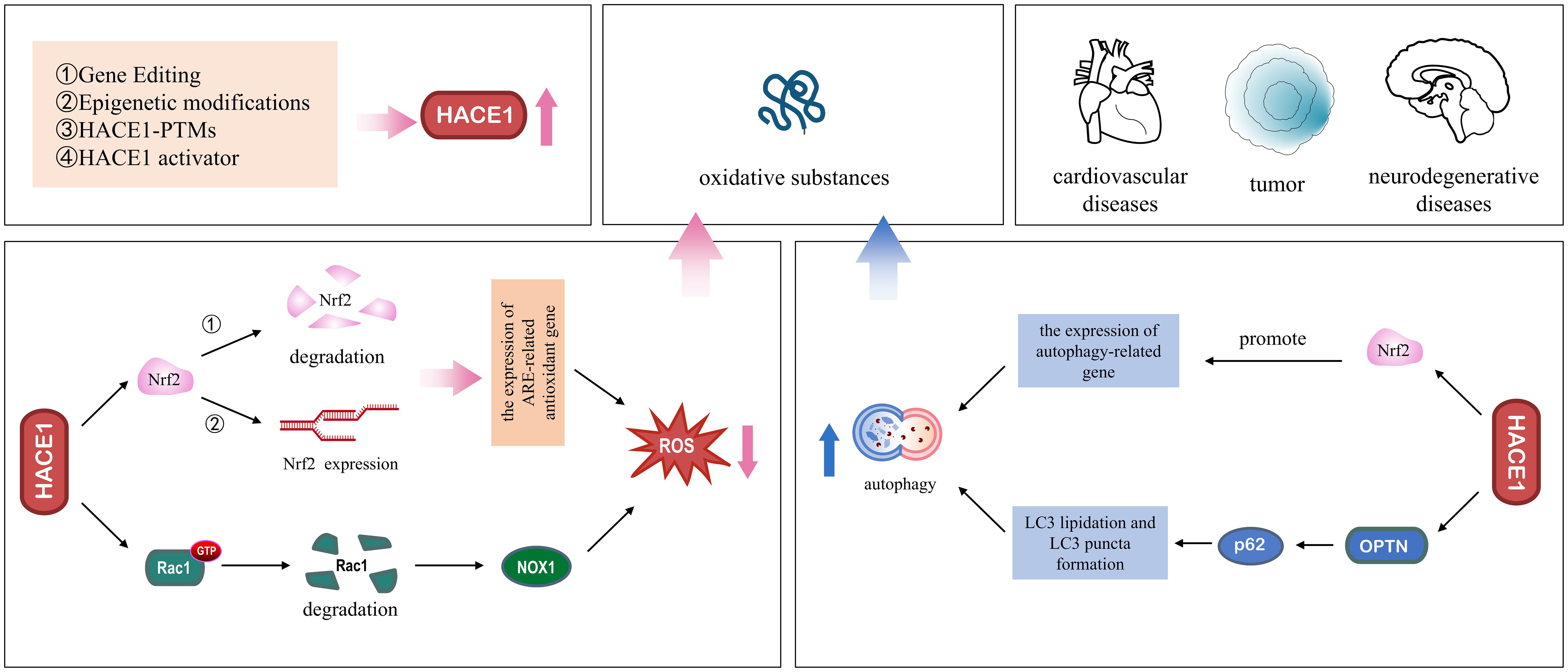
Figure 5. Schematic diagram depicting the role of HACE1 in degenerative diseases.
The two main systems for protein degradation in mammalian cells are ubiquitination-dependent proteasomal degradation and autophagy. HACE1 is an important E3 ubiquitin ligase that acts as a link between the ubiquitination and autophagy machineries. The most common consequence of protein ubiquitination is the subsequent proteasomal degradation of target proteins. When the proteasome is overloaded or its function is inhibited, misfolded proteins can accumulate and form toxic aggregates. Autophagy is an important alternative mechanism for clearing damaged organelles and toxic proteins, especially when oxidative stress causes excessive production of irreversibly oxidized biomolecules. It has already been found that HACE1 is crucial for cell survival, embryonic development, and organ formation. HACE1 maintains cell viability by activating selective autophagy to remove the damaged cell components. Mechanistically, HACE1 scavenges oxidative stress-induced misfolded protein accumulation by activating selective autophagy via ubiquitination of autophagy-associated substrates.
Although Rotblat et al. previously measured HACE1 protein levels in human striatum tissue. Unfortunately, they did not establish a statistical correlation between HACE1 expression and age (45). It seems fortunate that the relationship between HACE1 expression and age has been found in PD mice (67). Based on these reports, we speculate that HACE1 expression would decline with age and exacerbate the onset and progression of degenerative diseases. HACE1, an E3 ubiquitin ligase, is frequently inactivated and has been shown to be a putative tumor suppressor for a variety of malignancies. Therefore, restoring ubiquitination function by adding intracellular free HACE1 protein seems to be a good therapeutic option. However, there are many challenges to this treatment strategy. Specifically, because specific target proteins are numerous and incompletely defined, upregulation of HACE1 E3 ligase activity may lead to degradation or functional changes in other substrate proteins. Additionally, HACE1 ubiquitin ligase activity is disturbed by many factors, resulting in poor stability and difficult delivery.
Ischemia/reperfusion injury is defined as the restoration of blood flow to tissues or organs after transient ischemia, resulting in increased cellular dysfunction and structural damage, which is more severe than ischemia alone (136). The core pathological mechanism is the sudden influx of a large number of oxygen molecules during reperfusion, which triggers the explosive generation of ROS, leading to lipid peroxidation, protein oxidation, and DNA damage (137). In the ischemia-reperfusion injury model, HACE1 has been demonstrated to stabilize mitochondrial function and reduce oxidative stress by activating the Nrf2 signaling pathway, thereby alleviating cellular oxidative damage. It has been evidenced that Nrf2 could regulate the expression of ferroptosis-related proteins, thereby inhibiting the occurrence and development of ferroptosis (138). The study found that HACE1 prevented mitochondrial damage and ferroptosis by targeting Nrf2 in an Ang II-induced mouse model of heart failure, ultimately alleviating cardiac fibrosis and cardiac dysfunction (53). Therefore, the regulatory capacity of HACE1 in ferroptosis has begun to attract attention.
Herein, we sought to use HACE1 as a therapeutic target to explore its protective role and mechanism in aging diseases. However, it is important to note the paradoxical phenomenon of oxidative stress and autophagy in tumors. Nrf2 plays a role in suppressing cancer by protecting normal cells from oxidative stress by activating downstream antioxidant genes. However, the abnormal expression of Nrf2 in tumor cells can also help cancer cells resist oxidative damage and hinder the effect of radiotherapy and chemotherapy (139). It has been found that Nrf2 activation by HACE1 in glioma cells leads to an enhanced malignant phenotype and decreased radiosensitivity (52). Autophagy maintains normal cellular homeostasis in early tumor stages by removing damaged proteins and organelles. However, during tumor development, autophagy provides energy and material support for tumor cells by decomposing intracellular substances and helps tumor cells survive in the unfavorable microenvironment (140). In addition, contrary to that activating autophagy, it was recently reported that overexpression of HACE1 in HT29 cells with HMBOX1 knockdown promotes ubiquitination and degradation of ATG5 K63 to inhibit autophagy and reduce 5-FU resistance in colorectal cancer (141).
Recent research has shown that HACE1 expression is influenced by pharmacological and genetic strategies and plays a potential therapeutic role in age-related diseases such as neurodegeneration, cardiovascular disease, and cancer. Gene editing or vector-mediated approaches can compensate for HACE1 protein, restore its missing protein function, regulate the level of intracellular oxidative substances and affect the development of diseases. Furthermore, epigenetic regulation, including histone modification and DNA methylation, has been shown to regulate HACE1 expression, thereby affecting tumor cell growth. In conclusion, gene therapy and epigenetic regulation to regulate HACE1 protein expression is a feasible therapeutic strategy. In addition, Certain natural antioxidants have been reported to promote the expression of HACE1, which may be involved in cell survival mechanisms including oxidative stress and autophagy regulation through the ubiquitination pathway. Therefore, understanding the fine molecular regulation of oxidative stress and autophagy by HACE1, as well as the close relationship between HACE1 and various cytokines, could provide valuable information. This information may be useful in the future to improve the treatment of age-related diseases and develop new selective therapies.
Author contributions
JX: Conceptualization, Writing – original draft. YJ: Methodology, Writing – original draft. XX: Methodology, Writing – original draft. TL: Investigation, Writing – original draft. WL: Writing – review & editing. ZX: Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Graduate Education Innovation Project of Guizhou Province (No. 2024YJSKYJJ342) and the Graduate Research Fund of Zunyi Medical University (No. ZYK242).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AREs, antioxidant response elements; CCNC, Cyclin C; CCND1, Cyclin D1; DNMT1, DNA methyltransferase 1; HACE1, HECT domain and ankyrin repeat-containing E3 ubiquitin-protein ligase 1; HIF1-α, hypoxia-inducible factor 1-α; H/R, hypoxia/reoxygenation; Keap1, Kelch-like ECH-associated protein 1; LC3, microtubule-associated protein 1 light chain 3; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, NADPH oxidase; Nrf2, Nuclear factor erythroid-derived 2-like-2; OPTN, optineurin; p62, sequestosome-1; Rac1, Ras-related C3 botulinum toxin substrate 1; ROS, reactive oxygen species; TBK1, TANK-binding kinase 1.
References
1. Sanford SL, Welfer GA, Freudenthal BD, and Opresko PL. Mechanisms of telomerase inhibition by oxidized and therapeutic dNTPs. Nat Commun. (2020) 11:5288. doi: 10.1038/s41467-020-19115-y
2. Lin FC, Lee SS, Li YC, Ho YC, Chen WY, Chen CJ, et al. Protective Effects of Kirenol against Lipopolysaccharide-Induced Acute Lung Injury through the Modulation of the Proinflammatory NFkappaB Pathway and the AMPK2-/Nrf2-Mediated HO-1/AOE Pathway. Antioxidants. (2021) 10:204. doi: 10.3390/antiox10020204
3. Guo R, Wang SS, Jiang XY, Zhang Y, Guo Y, Cui HY, et al. CHK2 promotes metabolic stress-induced autophagy through ULK1 phosphorylation. Antioxidants. (2022) 11:1166. doi: 10.3390/antiox11061166
4. Ryu YS, Fernando P, Kang KA, Piao MJ, Zhen AX, Kang HK, et al. Marine Compound 3-bromo-4,5-dihydroxybenzaldehyde Protects Skin Cells against Oxidative Damage via the Nrf2/HO-1 Pathway. Mar Drugs. (2019) 17:234. doi: 10.3390/md17040234
5. Mugoni V, Panella R, Cheloni G, Chen M, Pozdnyakova O, Stroopinsky D, et al. Vulnerabilities in mIDH2 AML confer sensitivity to APL-like targeted combination therapy. Cell Res. (2019) 29:446–59. doi: 10.1038/s41422-019-0162-7
6. Tie F, Fu Y, Hu N, and Wang H. Silibinin protects against H(2)O(2)-induced oxidative damage in SH-SY5Y cells by improving mitochondrial function. Antioxidants. (2022) 11:1101. doi: 10.3390/antiox11061101
7. Liu C, Chen L, Ma Y, Hu K, Wu P, Pan L, et al. Pulmonary circulation-mediated heart targeting for the prevention of heart failure by inhalation of intrinsically bioactive nanoparticles. Theranostics. (2021) 11:8550–69. doi: 10.7150/thno.61875
8. Degtyareva NP, Saini N, Sterling JF, Placentra VC, Klimczak LJ, Gordenin DA, et al. Mutational signatures of redox stress in yeast single-strand DNA and of aging in human mitochondrial DNA share a common feature. PloS Biol. (2019) 17:e3000263. doi: 10.1371/journal.pbio.3000263
9. Lv XF, Zhang YJ, Liu X, Zheng HQ, Liu CZ, Zeng XL, et al. TMEM16A ameliorates vascular remodeling by suppressing autophagy via inhibiting Bcl-2-p62 complex formation. Theranostics. (2020) 10:3980–93. doi: 10.7150/thno.41028
10. Launay N, Aguado C, Fourcade S, Ruiz M, Grau L, Riera J, et al. Autophagy induction halts axonal degeneration in a mouse model of X-adrenoleukodystrophy. Acta Neuropathol. (2015) 129:399–415. doi: 10.1007/s00401-014-1378-8
11. Liu Y, Zhong D, He Y, Jiang J, Xie W, Tang Z, et al. Photoresponsive hydrogel-coated upconversion cyanobacteria nanocapsules for myocardial infarction prevention and treatment. Adv Sci. (2022) 9:e2202920. doi: 10.1002/advs.202202920
12. Gurunathan S, Jeyaraj M, Kang MH, and Kim JH. Melatonin enhances palladium-nanoparticle-induced cytotoxicity and apoptosis in human lung epithelial adenocarcinoma cells A549 and H1229. Antioxidants. (2020) 9:357. doi: 10.3390/antiox9040357
13. Yu C, Li D, Wang C, Xia K, Wang J, Zhou X, et al. Injectable kartogenin and apocynin loaded micelle enhances the alleviation of intervertebral disc degeneration by adipose-derived stem cell. Bioact Mater. (2021) 6:3568–79. doi: 10.1016/j.bioactmat.2021.03.018
14. Cores A, Carmona-Zafra N, Martin-Camara O, Sanchez JD, Duarte P, Villacampa M, et al. Curcumin-piperlongumine hybrids with a multitarget profile elicit neuroprotection in in vitro models of oxidative stress and hyperphosphorylation. Antioxidants. (2021) 11:28. doi: 10.3390/antiox11010028
15. Shimoyoshi S, Takemoto D, Ono Y, Kitagawa Y, Shibata H, Tomono S, et al. Sesame lignans suppress age-related cognitive decline in senescence-accelerated mice. Nutrients. (2019) 11:1582. doi: 10.3390/nu11071582
16. Kong L, Deng J, Zhou X, Cai B, Zhang B, Chen X, et al. Sitagliptin activates the p62-Keap1-Nrf2 signaling pathway to alleviate oxidative stress and excessive autophagy in severe acute pancreatitis-related acute lung injury. Cell Death Dis. (2021) 12:928. doi: 10.1038/s41419-021-04227-0
17. Chu Q, Yu X, Jia R, Wang Y, Zhang Y, Zhang S, et al. Flavonoids from Apios americana Medikus Leaves Protect RAW264.7 Cells against Inflammation via Inhibition of MAPKs, Akt-mTOR Pathways, and Nfr2 Activation. Oxid Med Cell Longev. (2019) 2019:1563024. doi: 10.1155/2019/1563024
18. Cai Y, Feng Z, Jia Q, Guo J, Zhang P, Zhao Q, et al. Cordyceps cicadae ameliorates renal hypertensive injury and fibrosis through the regulation of SIRT1-mediated autophagy. Front Pharmacol. (2021) 12:801094. doi: 10.3389/fphar.2021.801094
19. Xu JG, Cheng Q, Zhang GH, Kong LP, Li L, Liu KG, et al. Effect of dimethyl fumarate (DMF) on T-cell acute lymphoblastic leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi. (2022) 30:1–5. doi: 10.19746/j.cnki.issn.1009-2137.2022.01.01
20. Huang K, Zhang B, Feng Y, and Ma H. Magnolol promotes the autophagy of esophageal carcinoma cells by upregulating HACE1 gene expression. Acta Biochim Biophys Sinica. (2024) 56:1044–54. doi: 10.3724/abbs.2024044
21. Liu F, Smith AD, Wang TTY, Pham Q, Cheung L, Yang H, et al. Biological pathways via which the anthocyanin malvidin alleviated the murine colitis induced by Citrobacter rodentium. Food Funct. (2023) 14:1048–61. doi: 10.1039/d2fo02873e
22. Anglesio MS, Evdokimova V, Melnyk N, Zhang L, Fernandez CV, Grundy PE, et al. Differential expression of a novel ankyrin containing E3 ubiquitin-protein ligase, Hace1, in sporadic Wilms’ tumor versus normal kidney. Hum Mol Genet. (2004) 13:2061–74. doi: 10.1093/hmg/ddh215
23. Tang D, Xiang Y, De Renzis S, Rink J, Zheng G, Zerial M, et al. The ubiquitin ligase HACE1 regulates Golgi membrane dynamics during the cell cycle. Nat Commun. (2011) 2:501. doi: 10.1038/ncomms1509
24. Singh S, Machida S, Tulsian NK, Choong YK, Ng J, Shankar S, et al. Structural basis for the enzymatic activity of the HACE1 HECT-type E3 ligase through N-terminal helix dimerization. Adv Sci. (2023) 10:e2207672. doi: 10.1002/advs.202207672
25. El-Naggar AM, Clarkson PW, Negri GL, Turgu B, Zhang F, Anglesio MS, et al. HACE1 is a potential tumor suppressor in osteosarcoma. Cell Death Dis. (2019) 10:21. doi: 10.1038/s41419-018-1276-4
26. Acosta MI, Urbach S, Doye A, Ng YW, Boudeau J, Mettouchi A, et al. Group-I PAKs-mediated phosphorylation of HACE1 at serine 385 regulates its oligomerization state and Rac1 ubiquitination. Sci Rep. (2018) 8:1410. doi: 10.1038/s41598-018-19471-2
27. Zhang L, Chen X, Sharma P, Moon M, Sheftel AD, Dawood F, et al. HACE1-dependent protein degradation provides cardiac protection in response to haemodynamic stress. Nat Commun. (2014) 5:3430. doi: 10.1038/ncomms4430
28. Ferenczi S, Kuti D, Cserhati M, Krifaton C, Szoboszlay S, Kukolya J, et al. Effects of single and repeated oral doses of ochratoxin A on the lipid peroxidation and antioxidant defense systems in mouse kidneys. Toxins. (2020) 12:732. doi: 10.3390/toxins12110732
29. Daugaard M, Nitsch R, Razaghi B, McDonald L, Jarrar A, Torrino S, et al. Hace1 controls ROS generation of vertebrate Rac1-dependent NADPH oxidase complexes. Nat Commun. (2013) 4:2180. doi: 10.1038/ncomms3180
30. Zhang L, Anglesio MS, O’Sullivan M, Zhang F, Yang G, Sarao R, et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat Med. (2007) 13:1060–9. doi: 10.1038/nm1621
31. Andrio E, Lotte R, Hamaoui D, Cherfils J, Doye A, Daugaard M, et al. Identification of cancer-associated missense mutations in hace1 that impair cell growth control and Rac1 ubiquitylation. Sci Rep. (2017) 7:44779. doi: 10.1038/srep44779
32. Liu Z, Chen P, Gao H, Gu Y, Yang J, Peng H, et al. Ubiquitylation of autophagy receptor Optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell. (2014) 26:106–20. doi: 10.1016/j.ccr.2014.05.015
33. Castillo-Lluva S, Tan CT, Daugaard M, Sorensen PH, and Malliri A. The tumour suppressor HACE1 controls cell migration by regulating Rac1 degradation. Oncogene. (2013) 32:1735–42. doi: 10.1038/onc.2012.189
34. Venkatachalam N, Bakavayev S, Engel D, Barak Z, and Engel S. Primate differential redoxome (PDR) - A paradigm for understanding neurodegenerative diseases. Redox Biol. (2020) 36:101683. doi: 10.1016/j.redox.2020.101683
35. Alov P, Stoimenov H, Lessigiarska I, Pencheva T, Tzvetkov NT, Pajeva I, et al. In silico identification of multi-target ligands as promising hit compounds for neurodegenerative diseases drug development. Int J Mol Sci. (2022) 23:13650. doi: 10.3390/ijms232113650
36. Zang C, Liu H, Ning J, Chen Q, Jiang Y, Shang M, et al. Emerging role and mechanism of HACE1 in the pathogenesis of neurodegenerative diseases: A promising target. BioMed Pharmacother. (2024) 172:116204. doi: 10.1016/j.biopha.2024.116204
37. Ugarteburu O, Sanchez-Viles M, Ramos J, Barcos-Rodriguez T, Garrabou G, Garcia-Villoria J, et al. Physiopathological bases of the disease caused by HACE1 mutations: alterations in autophagy, mitophagy and oxidative stress response. J Clin Med. (2020) 9:913. doi: 10.3390/jcm9040913
38. Ehrnhoefer DE, Southwell AL, Sivasubramanian M, Qiu X, Villanueva EB, Xie Y, et al. HACE1 is essential for astrocyte mitochondrial function and influences Huntington disease phenotypes in vivo. Hum Mol Genet. (2018) 27:239–53. doi: 10.1093/hmg/ddx394
39. Turgu B, Zhang F, El-Naggar A, Negri GL, Kogler M, Tortola L, et al. HACE1 blocks HIF1alpha accumulation under hypoxia in a RAC1 dependent manner. Oncogene. (2021) 40:1988–2001. doi: 10.1038/s41388-021-01680-1
40. Kumar B, Roy A, Asha K, Sharma-Walia N, Ansari MA, and Chandran B. HACE1, an E3 ubiquitin protein ligase, mitigates Kaposi’s sarcoma-associated herpesvirus infection-induced oxidative stress by promoting Nrf2 activity. J Virol. (2019) 93:e01812–18. doi: 10.1128/JVI.01812-18
41. Liu BX, Zheng J, Tang ZW, Gao L, Wang M, Sun Y, et al. HACE1 protects against myocardial ischemia-reperfusion injury via inhibition of mitochondrial fission in mice. BMC Cardiovasc Disord. (2025) 25:77. doi: 10.1186/s12872-024-04445-2
42. Chen TY and Zheng SK. Hace1 overexpression mitigates myocardial hypoxia/reoxygenation injury via the effects on Keap1/Nrf2 pathway. In Vitro Cell Dev Biol Anim. (2022) 58:830–9. doi: 10.1007/s11626-022-00725-3
43. Zhang X, Wang X, Yin L, Wang D, Jiao H, Liu X, et al. HACE1 exerts a neuroprotective role against oxidative stress in cerebral ischemia-reperfusion injury by activating the PI3K/AKT/Nrf2 pathway. Neuroscience. (2024) 559:249–62. doi: 10.1016/j.neuroscience.2024.09.002
44. Sun L, Jin Y, Dong L, Sumi R, Jahan R, and Li Z. The neuroprotective effects of Coccomyxa gloeobotrydiformis on the ischemic stroke in a rat model. Int J Biol Sci. (2013) 9:811–7. doi: 10.7150/ijbs.6734
45. Rotblat B, Southwell AL, Ehrnhoefer DE, Skotte NH, Metzler M, Franciosi S, et al. HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc Natl Acad Sci U S A. (2014) 111:3032–7. doi: 10.1073/pnas.1314421111
46. Cetinbas N, Daugaard M, Mullen AR, Hajee S, Rotblat B, Lopez A, et al. Loss of the tumor suppressor Hace1 leads to ROS-dependent glutamine addiction. Oncogene. (2015) 34:4005–10. doi: 10.1038/onc.2014.316
47. Zhou Y, Zhou Y, Yang M, Wang K, Liu Y, Zhang M, et al. Digoxin sensitizes gemcitabine-resistant pancreatic cancer cells to gemcitabine via inhibiting Nrf2 signaling pathway. Redox Biol. (2019) 22:101131. doi: 10.1016/j.redox.2019.101131
48. Payne NC, Kalyakina AS, Singh K, Tye MA, and Mazitschek R. Bright and stable luminescent probes for target engagement profiling in live cells. Nat Chem Biol. (2021) 17:1168–77. doi: 10.1038/s41589-021-00877-5
49. Zhang Y, Xu Y, Lu W, Li J, Yu S, Brown EJ, et al. G6PD-mediated increase in de novo NADP(+) biosynthesis promotes antioxidant defense and tumor metastasis. Sci Adv. (2022) 8:eabo0404. doi: 10.1126/sciadv.abo0404
50. Iosub-Amir A, Bai F, Sohn YS, Song L, Tamir S, Marjault HB, et al. The anti-apoptotic proteins NAF-1 and iASPP interact to drive apoptosis in cancer cells. Chem Sci. (2019) 10:665–73. doi: 10.1039/c8sc03390k
51. Shen B, Feng H, Cheng J, Li Z, Jin M, Zhao L, et al. Geniposide alleviates non-alcohol fatty liver disease via regulating Nrf2/AMPK/mTOR signaling pathways. J Cell Mol Med. (2020) 24:5097–108. doi: 10.1111/jcmm.15139
52. Da C, Pu J, Liu Z, Wei J, Qu Y, Wu Y, et al. HACE1-mediated NRF2 activation causes enhanced Malignant phenotypes and decreased radiosensitivity of glioma cells. Signal Transduct Target Ther. (2021) 6:399. doi: 10.1038/s41392-021-00793-z
53. Yin P, Wu Y, Long X, Zhu S, Chen S, Lu F, et al. HACE1 expression in heart failure patients might promote mitochondrial oxidative stress and ferroptosis by targeting NRF2. Aging-US. (2023) 15:13888–900. doi: 10.18632/aging.205272
54. Toyama Y, Kontani K, Katada T, and Shimada I. Conformational landscape alternations promote oncogenic activities of Ras-related C3 botulinum toxin substrate 1 as revealed by NMR. Sci Adv. (2019) 5:eaav8945. doi: 10.1126/sciadv.aav8945
55. Kogler M, Tortola L, Negri GL, Leopoldi A, El-Naggar AM, Mereiter S, et al. HACE1 prevents lung carcinogenesis via inhibition of RAC-family GTPases. Cancer Res. (2020) 80:3009–22. doi: 10.1158/0008-5472.CAN-19-2270
56. Lacerda DD, Türck P, de Lima-Seolin BG, Colombo R, Ortiz VD, Bonetto JHP, et al. Pterostilbene reduces oxidative stress, prevents hypertrophy and preserves systolic function of right ventricle in model. Brit J Pharmacol. (2017) 174:3302–14. doi: 10.1111/bph.13948
57. Paolillo R, Boulanger M, Gâtel P, Gabellier L, De Toledo M, Tempé D, et al. The NADPH oxidase NOX2 is a marker of adverse prognosis involved in chemoresistance of acute myeloid leukemias. Haematologica. (2022) 107:2562–75. doi: 10.3324/haematol.2021.279889
58. Boussetta T, Gougerot-Pocidalo MA, Hayem G, Ciappelloni S, Raad H, Arabi Derkawi R, et al. The prolyl isomerase Pin1 acts as a novel molecular switch for TNF-alpha-induced priming of the NADPH oxidase in human neutrophils. Blood. (2010) 116:5795–802. doi: 10.1182/blood-2010-03-273094
59. Wilson C, Munoz-Palma E, Henriquez DR, Palmisano I, Nunez MT, Di Giovanni S, et al. A feed-forward mechanism involving the NOX complex and RyR-mediated Ca2+ Release during axonal specification. J Neurosci. (2016) 36:11107–19. doi: 10.1523/JNEUROSCI.1455-16.2016
60. Orme M, Bianchi K, and Meier P. Ubiquitin-mediated regulation of RhoGTPase signaling: IAPs and HACE1 enter the fray. EMBO J. (2012) 31:1–2. doi: 10.1038/emboj.2011.452
61. Torrino S, Visvikis O, Doye A, Boyer L, Stefani C, Munro P, et al. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev Cell. (2011) 21:959–65. doi: 10.1016/j.devcel.2011.08.015
62. Lu QB, Du Q, Wang HP, Tang ZH, Wang YB, and Sun HJ. Salusin-beta mediates tubular cell apoptosis in acute kidney injury: Involvement of the PKC/ROS signaling pathway. Redox Biol. (2020) 30:101411. doi: 10.1016/j.redox.2019.101411
63. Goka ET and Lippman ME. Loss of the E3 ubiquitin ligase HACE1 results in enhanced Rac1 signaling contributing to breast cancer progression. Oncogene. (2015) 34:5395–405. doi: 10.1038/onc.2014.468
64. Iimura A, Yamazaki F, Suzuki T, Endo T, Nishida E, and Kusakabe M. The E3 ubiquitin ligase Hace1 is required for early embryonic development in Xenopus laevis. BMC Dev Biol. (2016) 16:31. doi: 10.1186/s12861-016-0132-y
65. Razaghi B, Steele SL, Prykhozhij SV, Stoyek MR, Hill JA, Cooper MD, et al. hace1 Influences zebrafish cardiac development via ROS-dependent mechanisms. Dev Dyn. (2018) 247:289–303. doi: 10.1002/dvdy.24600
66. Nagy V, Hollstein R, Pai TP, Herde MK, Buphamalai P, Moeseneder P, et al. HACE1 deficiency leads to structural and functional neurodevelopmental defects. Neurol Genet. (2019) 5:e330. doi: 10.1212/NXG.0000000000000330
67. Zang CX, Wang L, Yang HY, Shang JM, Liu H, Zhang ZH, et al. HACE1 negatively regulates neuroinflammation through ubiquitylating and degrading Rac1 in Parkinson’s disease models. Acta Pharmacol Sin. (2022) 43:285–94. doi: 10.1038/s41401-021-00778-2
68. Kim I, Shin SH, Lee JE, and Park JW. Oxygen sensor FIH inhibits HACE1-dependent ubiquitination of Rac1 to enhance metastatic potential in breast cancer cells. Oncogene. (2019) 38:3651–66. doi: 10.1038/s41388-019-0676-y
69. Wang S, Wu X, Zhang M, Chang S, Guo Y, Song S, et al. NET1 is a critical regulator of spindle assembly and actin dynamics in mouse oocytes. Reprod Biol Endocrin. (2024) 22:5. doi: 10.1186/s12958-023-01177-4
70. Hu Y, Zhu Z, Xu Y, Zaman MF, Ge Y, Hu J, et al. Inhibition of esophageal cancer progression through HACE1-TRIP12 interaction and associated RAC1 ubiquitination and degradation. J Cancer. (2024) 15:3114–27. doi: 10.7150/jca.93833
71. Ma X, Ma X, Zhu L, Zhao Y, Chen M, Li T, et al. The E3 ubiquitin ligase MG53 inhibits hepatocellular carcinoma by targeting RAC1 signaling. Oncogenesis. (2022) 11:40. doi: 10.1038/s41389-022-00414-6
72. Goka ET, Mesa Lopez DT, and Lippman ME. Hormone-dependent prostate cancers are dependent on rac signaling for growth and survival. Mol Cancer Ther. (2021) 20:1052–61. doi: 10.1158/1535-7163.MCT-20-0695
73. Turgu B, El-Naggar A, Kogler M, Tortola L, Zhang HF, Hassan M, et al. The HACE1 E3 ligase mediates RAC1-dependent control of mTOR signaling complexes. EMBO Rep. (2023) 24:e56815. doi: 10.15252/embr.202356815
74. Wang Q, Guo W, Hao B, Shi X, Lu Y, Wong CW, et al. Mechanistic study of TRPM2-Ca(2+)-CAMK2-BECN1 signaling in oxidative stress-induced autophagy inhibition. Autophagy. (2016) 12:1340–54. doi: 10.1080/15548627.2016.1187365
75. Duan X, Kong Z, Mai X, Lan Y, Liu Y, Yang Z, et al. Autophagy inhibition attenuates hyperoxaluria-induced renal tubular oxidative injury and calcium oxalate crystal depositions in the rat kidney. Redox Biol. (2018) 16:414–25. doi: 10.1016/j.redox.2018.03.019
76. Viedma-Poyatos A, Gonzalez-Jimenez P, Pajares MA, and Perez-Sala D. Alexander disease GFAP R239C mutant shows increased susceptibility to lipoxidation and elicits mitochondrial dysfunction and oxidative stress. Redox Biol. (2022) 55:102415. doi: 10.1016/j.redox.2022.102415
77. Heo H, Park H, Lee MS, Kim J, Kim J, Jung SY, et al. TRIM22 facilitates autophagosome-lysosome fusion by mediating the association of GABARAPs and PLEKHM1. Autophagy. (2024) 20:1098–113. doi: 10.1080/15548627.2023.2287925
78. Gouveia M, Xia K, Colon W, Vieira SI, and Ribeiro F. Protein aggregation, cardiovascular diseases, and exercise training: Where do we stand? Ageing Res Rev. (2017) 40:1–10. doi: 10.1016/j.arr.2017.07.005
79. Manfrini N, Mancino M, Miluzio A, Oliveto S, Balestra M, Calamita P, et al. FAM46C and FNDC3A are multiple myeloma tumor suppressors that act in concert to impair clearing of protein aggregates and autophagy. Cancer Res. (2020) 80:4693–706. doi: 10.1158/0008-5472.CAN-20-1357
80. Qu L, Wu J, Tang Y, Yun X, Lo HH, Yu L, et al. a natural autophagic agent for alleviating oxidative stress-induced cell death in neuronal cells and caenorhabditis elegans models. Pharm (Basel). (2022) 15:1052. doi: 10.3390/ph15091052
81. Shi XC, Xia B, Zhang JF, Zhang RX, Zhang DY, Liu H, et al. Optineurin promotes myogenesis during muscle regeneration in mice by autophagic degradation of GSK3beta. PloS Biol. (2022) 20:e3001619. doi: 10.1371/journal.pbio.3001619
82. Nakazawa S, Oikawa D, Ishii R, Ayaki T, Takahashi H, Takeda H, et al. Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat Commun. (2016) 7:12547. doi: 10.1038/ncomms12547
83. Li F, Xu D, Wang Y, Zhou Z, Liu J, Hu S, et al. Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy. (2018) 14:66–79. doi: 10.1080/15548627.2017.1391970
84. O’Loughlin T, Kruppa AJ, Ribeiro ALR, Edgar JR, Ghannam A, Smith AM, et al. OPTN recruitment to a Golgi-proximal compartment regulates immune signalling and cytokine secretion. J Cell Sci. (2020) 133:jcs239822. doi: 10.1242/jcs.239822
85. Wauters F, Cornelissen T, Imberechts D, Martin S, Koentjoro B, Sue C, et al. LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy. (2020) 16:203–22. doi: 10.1080/15548627.2019.1603548
86. Wang X, Jiang L, Wang G, Shi W, Hu Y, Wang B, et al. Influenza A virus use of BinCARD1 to facilitate the binding of viral NP to importin alpha7 is counteracted by TBK1-p62 axis-mediated autophagy. Cell Mol Immunol. (2022) 19:1168–84. doi: 10.1038/s41423-022-00906-w
87. Heo JM, Ordureau A, Paulo JA, Rinehart J, and Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. (2015) 60:7–20. doi: 10.1016/j.molcel.2015.08.016
88. Mao HT, Wang Y, Cai J, Meng JL, Zhou Y, Pan Y, et al. HACE1 negatively regulates virus-triggered type I IFN signaling by impeding the formation of the MAVS-TRAF3 complex. Viruses. (2016) 8:146. doi: 10.3390/v8050146
89. Zhang Y, Li T, Cai X, Long D, Wang X, Liu C, et al. Sirt5-mediated desuccinylation of OPTN protects retinal ganglion cells from autophagic flux blockade in diabetic retinopathy. Cell Death Dis. (2022) 8:63. doi: 10.1038/s41420-022-00861-5
90. Yu Z, Li Y, Han T, and Liu Z. Demethylation of the HACE1 gene promoter inhibits the proliferation of human liver cancer cells. Oncol Lett. (2019) 17:4361–8. doi: 10.3892/ol.2019.10139
91. Li S, Yang H, Zhao M, Gong L, Wang Y, Lv Z, et al. Demethylation of HACE1 gene promoter by propofol promotes autophagy of human A549 cells. Oncol Lett. (2020) 20:280. doi: 10.3892/ol.2020.12143
92. Valionyte E, Yang Y, Griffiths SA, Bone AT, Barrow ER, Sharma V, et al. The caspase-6-p62 axis modulates p62 droplets based autophagy in a dominant-negative manner. Cell Death Differ. (2022) 29:1211–27. doi: 10.1038/s41418-021-00912-x
93. Feng L, Li J, Yang L, Zhu L, Huang X, Zhang S, et al. Tamoxifen activates Nrf2-dependent SQSTM1 transcription to promote endometrial hyperplasia. Theranostics. (2017) 7:1890–900. doi: 10.7150/thno.19135
94. Overa KS, Garcia-Garcia J, Bhujabal Z, Jain A, Overvatn A, Larsen KB, et al. TRIM32, but not its muscular dystrophy-associated mutant, positively regulates and is targeted to autophagic degradation by p62/SQSTM1. J Cell Sci. (2019) 132:jcs236596. doi: 10.1242/jcs.236596
95. Wurzer B, Zaffagnini G, Fracchiolla D, Turco E, Abert C, Romanov J, et al. Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. Elife. (2015) 4:e08941. doi: 10.7554/eLife.08941
96. Zhao Y, Zhu Q, Bu X, Zhou Y, Bai D, Guo Q, et al. Triggering apoptosis by oroxylin A through caspase-8 activation and p62/SQSTM1 proteolysis. Redox Biol. (2020) 29:101392. doi: 10.1016/j.redox.2019.101392
97. Zhang Y, Mun SR, Linares JF, Ahn J, Towers CG, Ji CH, et al. ZZ-dependent regulation of p62/SQSTM1 in autophagy. Nat Commun. (2018) 9:4373. doi: 10.1038/s41467-018-06878-8
98. Zhang S, Xie F, Li K, Zhang H, Yin Y, Yu Y, et al. Gold nanoparticle-directed autophagy intervention for antitumor immunotherapy via inhibiting tumor-associated macrophage M2 polarization. Acta Pharm Sin B. (2022) 12:3124–38. doi: 10.1016/j.apsb.2022.02.008
99. Ahn JH, Cho MG, Sohn S, and Lee JH. Inhibition of PP2A activity by H(2)O(2) during mitosis disrupts nuclear envelope reassembly and alters nuclear shape. Exp Mol Med. (2019) 51:1–18. doi: 10.1038/s12276-019-0260-0
100. Park JH, Kim H, Moon HR, Park BW, Park JH, Sim WS, et al. Human cardiac stem cells rejuvenated by modulating autophagy with MHY-1685 enhance the therapeutic potential for cardiac repair. Exp Mol Med. (2021) 53:1423–36. doi: 10.1038/s12276-021-00676-x
101. Wu SY, Lan SH, Wu SR, Chiu YC, Lin XZ, Su IJ, et al. Hepatocellular carcinoma-related cyclin D1 is selectively regulated by autophagy degradation system. Hepatology. (2018) 68:141–54. doi: 10.1002/hep.29781
102. Xu C, Ge S, Cheng J, Gao H, Zhang F, and Han A. Pathological and prognostic characterization of craniopharyngioma based on the expression of TrkA, beta-catenin, cell cycle markers, and BRAF V600E mutation. Front Endocrinol (Lausanne). (2022) 13:859381. doi: 10.3389/fendo.2022.859381
103. Huo J, Li J, Liu Y, Yang L, Cao X, Zhao C, et al. Amphiphilic aminated derivatives of [60]Fullerene as potent inhibitors of tumor growth and metastasis. Adv Sci. (2022) 9:e2201541. doi: 10.1002/advs.202201541
104. Osaki Y, Manolopoulou M, Ivanova AV, Vartanian N, Mignemi MP, Kern J, et al. Blocking cell cycle progression through CDK4/6 protects against chronic kidney disease. JCI Insight. (2022) 7:e158754. doi: 10.1172/jci.insight.158754
105. Takuma K, Fujihara S, Fujita K, Iwama H, Nakahara M, Oura K, et al. Antitumor effect of regorafenib on microRNA expression in hepatocellular carcinoma cell lines. Int J Mol Sci. (2022) 23:1667. doi: 10.3390/ijms23031667
106. Li Z, Jiao X, Di Sante G, Ertel A, Casimiro MC, Wang M, et al. Cyclin D1 integrates G9a-mediated histone methylation. Oncogene. (2019) 38:4232–49. doi: 10.1038/s41388-019-0723-8
107. Sudhan DR, Schwarz LJ, Guerrero-Zotano A, Formisano L, Nixon MJ, Croessmann S, et al. Extended adjuvant therapy with neratinib plus fulvestrant blocks ER/HER2 crosstalk and maintains complete responses of ER(+)/HER2(+) breast cancers: implications to the ExteNET trial. Clin Cancer Res. (2019) 25:771–83. doi: 10.1158/1078-0432.CCR-18-1131
108. Shimura T, Kobayashi J, Komatsu K, and Kunugita N. DNA damage signaling guards against perturbation of cyclin D1 expression triggered by low-dose long-term fractionated radiation. Oncogenesis. (2014) 3:e132. doi: 10.1038/oncsis.2014.48
109. Chan KKL, Siu MKY, Jiang YX, Wang JJ, Leung THY, and Ngan HYS. Estrogen receptor modulators genistein, daidzein and ERB-041 inhibit cell migration, invasion, proliferation and sphere formation via modulation of FAK and PI3K/AKT signaling in ovarian cancer. Cancer Cell Int. (2018) 18:65. doi: 10.1186/s12935-018-0559-2
110. Chen K, Jiao X, Ashton A, Di Rocco A, Pestell TG, Sun Y, et al. The membrane-associated form of cyclin D1 enhances cellular invasion. Oncogenesis. (2020) 9:83. doi: 10.1038/s41389-020-00266-y
111. Chaikovsky AC, Li C, Jeng EE, Loebell S, Lee MC, Murray CW, et al. The AMBRA1 E3 ligase adaptor regulates the stability of cyclin D. Nature. (2021) 592:794–8. doi: 10.1038/s41586-021-03474-7
112. Maya-Mendoza A, Ostrakova J, Kosar M, Hall A, Duskova P, Mistrik M, et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol Oncol. (2015) 9:601–16. doi: 10.1016/j.molonc.2014.11.001
113. Petracchini S, Hamaoui D, Doye A, Asnacios A, Fage F, Vitiello E, et al. Optineurin links Hace1-dependent Rac ubiquitylation to integrin-mediated mechanotransduction to control bacterial invasion and cell division. Nat Commun. (2022) 13:6059. doi: 10.1038/s41467-022-33803-x
114. Liu Q, Bischof S, Harris CJ, Zhong Z, Zhan L, Nguyen C, et al. The characterization of Mediator 12 and 13 as conditional positive gene regulators in Arabidopsis. Nat Commun. (2020) 11:2798. doi: 10.1038/s41467-020-16651-5
115. Abdella R, Talyzina A, Chen S, Inouye CJ, Tjian R, and He Y. Structure of the human Mediator-bound transcription preinitiation complex. Science. (2021) 372:52–6. doi: 10.1126/science.abg3074
116. Cao L, Luo Y, Guo X, Liu S, Li S, Li J, et al. SAFA facilitates chromatin opening of immune genes through interacting with anti-viral host RNAs. PloS Pathog. (2022) 18:e1010599. doi: 10.1371/journal.ppat.1010599
117. Stieg DC, Chang KT, Cooper KF, and Strich R. Cyclin C regulated oxidative stress responsive transcriptome in mus musculus embryonic fibroblasts. G3 Genes Genomes Genet. (2019) 9:1901–8. doi: 10.1534/g3.119.400077
118. Jiang HY, Chen YL, Xu XX, Li CY, Chen Y, Li DP, et al. Ubiquitylation of cyclin C by HACE1 regulates cisplatin-associated sensitivity in gastric cancer. Clin Transl Med. (2022) 12:e770. doi: 10.1002/ctm2.770
119. Willis SD, Hanley SE, Doyle SJ, Beluch K, Strich R, and Cooper KF. Cyclin C-cdk8 kinase phosphorylation of rim15 prevents the aberrant activation of stress response genes. Front Cell Dev Biol. (2022) 10:867257. doi: 10.3389/fcell.2022.867257
120. Chang KT, Jezek J, Campbell AN, Stieg DC, Kiss ZA, Kemper K, et al. Aberrant cyclin C nuclear release induces mitochondrial fragmentation and dysfunction in MED13L syndrome fibroblasts. iScience. (2022) 25:103823. doi: 10.1016/j.isci.2022.103823
121. Bharathi V, Girdhar A, and Patel BK. Role of CNC1 gene in TDP-43 aggregation-induced oxidative stress-mediated cell death in S. cerevisiae model of ALS. Biochim Biophys Acta Mol Cell Res. (2021) 1868:118993. doi: 10.1016/j.bbamcr.2021.118993
122. Jin C, Strich R, and Cooper KF. Slt2p phosphorylation induces cyclin C nuclear-to-cytoplasmic translocation in response to oxidative stress. Mol Biol Cell. (2014) 25:1396–407. doi: 10.1091/mbc.E13-09-0550
123. Cooper KF, Scarnati MS, Krasley E, Mallory MJ, Jin C, Law MJ, et al. Oxidative-stress-induced nuclear to cytoplasmic relocalization is required for Not4-dependent cyclin C destruction. J Cell Sci. (2012) 125:1015–26. doi: 10.1242/jcs.096479
124. Li JC, Chang X, Chen Y, Li XZ, Zhang XL, Yang SM, et al. Loss of the tumor suppressor HACE1 contributes to cancer progression. Curr Drug Targets. (2019) 20:1018–28. doi: 10.2174/1389450120666190227184654
125. Avrahami D, Li C, Zhang J, Schug J, Avrahami R, Rao S, et al. Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved beta cell function. Cell Metab. (2015) 22:619–32. doi: 10.1016/j.cmet.2015.07.025
126. Hartung T, Rhein M, Kalmbach N, Thau-Habermann N, Naujock M, Muschen L, et al. Methylation and expression of mutant FUS in motor neurons differentiated from induced pluripotent stem cells from ALS patients. Front Cell Dev Biol. (2021) 9:774751. doi: 10.3389/fcell.2021.774751
127. Gao LM, Zhao S, Liu WP, Zhang WY, Li GD, Kucuk C, et al. Clinicopathologic characterization of aggressive natural killer cell leukemia involving different tissue sites. Am J Surg Pathol. (2016) 40:836–46. doi: 10.1097/PAS.0000000000000634
128. Kucuk C, Hu X, Iqbal J, Gaulard P, Klinkebiel D, Cornish A, et al. HACE1 is a tumor suppressor gene candidate in natural killer cell neoplasms. Am J Pathol. (2013) 182:49–55. doi: 10.1016/j.ajpath.2012.09.012
129. Yoon JH, Kim O, Nam SW, Lee JY, and Park WS. NKX6.3 regulates reactive oxygen species production by suppressing NF-kB and DNMT1 activities in gastric epithelial cells. Sci Rep. (2017) 7:2807. doi: 10.1038/s41598-017-02901-y
130. Bouzelfen A, Kora H, Alcantara M, Bertrand P, Latouche JB, and Jardin F. Heterogeneous epigenetic regulation of HACE1 in Burkitt- Lymphoma-derived cells. Leuk Res. (2017) 60:53–7. doi: 10.1016/j.leukres.2017.06.006
131. Zhang Y, Chen S, You L, He Z, Xu P, and Huang W. LINC00161 upregulated by M2-like tumor-associated macrophages promotes hepatocellular carcinoma progression by methylating HACE1 promoters. Cytotechnology. (2024) 76:777–93. doi: 10.1007/s10616-024-00653-y
132. Alam H, Tang M, Maitituoheti M, Dhar SS, Kumar M, Han CY, et al. KMT2D deficiency impairs super-enhancers to confer a glycolytic vulnerability in lung cancer. Cancer Cell. (2020) 37:599–617 e7. doi: 10.1016/j.ccell.2020.03.005
133. Lee KH, Bae IY, Park SI, Park JD, and Lee HG. Antihypertensive effect of Korean Red Ginseng by enrichment of ginsenoside Rg3 and arginine-fructose. J Ginseng Res. (2016) 40:237–44. doi: 10.1016/j.jgr.2015.08.002
134. Miceli C, Santin Y, Manzella N, Coppini R, Berti A, Stefani M, et al. Oleuropein Aglycone Protects against MAO-A-Induced Autophagy Impairment and Cardiomyocyte Death through Activation of TFEB. Oxid Med Cell Longev. (2018) 2018:8067592. doi: 10.1155/2018/8067592
135. Konig J, Ott C, Hugo M, Jung T, Bulteau AL, Grune T, et al. Mitochondrial contribution to lipofuscin formation. Redox Biol. (2017) 11:673–81. doi: 10.1016/j.redox.2017.01.017
136. Son M, Oh S, Choi CH, Park KY, Son KH, and Byun K. Pyrogallol-phloroglucinol-6,6-bieckol from ecklonia cava attenuates tubular epithelial cell (TCMK-1) death in hypoxia/reoxygenation injury. Mar Drugs. (2019) 17:602. doi: 10.3390/md17110602
137. Parra-Flores P, Riquelme JA, Valenzuela-Bustamante P, Leiva-Navarrete S, Vivar R, Cayupi-Vivanco J, et al. The association of ascorbic acid, deferoxamine and N-acetylcysteine improves cardiac fibroblast viability and cellular function associated with tissue repair damaged by simulated ischemia/reperfusion. Antioxidants. (2019) 8:614. doi: 10.3390/antiox8120614
138. Lin ZH, Liu Y, Xue NJ, Zheng R, Yan YQ, Wang ZX, et al. Quercetin protects against MPP(+)/MPTP-induced dopaminergic neuron death in Parkinson’s disease by inhibiting ferroptosis. Oxid Med Cell Longev. (2022) 2022:7769355. doi: 10.1155/2022/7769355
139. Lin L, Wu Q, Lu F, Lei J, Zhou Y, Liu Y, et al. Nrf2 signaling pathway: current status and potential therapeutic targetable role in human cancers. Front Oncol. (2023) 13:1184079. doi: 10.3389/fonc.2023.1184079
140. Sun K, Deng W, Zhang S, Cai N, Jiao S, Song J, et al. Paradoxical roles of autophagy in different stages of tumorigenesis: protector for normal or cancer cells. Cell Biosci. (2013) 3:35. doi: 10.1186/2045-3701-3-35
Keywords: autophagy, cellular stress, HACE1, oxidative stress, ubiquitylate
Citation: Xia J, Jiang Y, Xin X, Li T, Liao W and Xin Z (2025) HACE1 as a bridge between oxidative stress and autophagy. Front. Immunol. 16:1595601. doi: 10.3389/fimmu.2025.1595601
Received: 19 March 2025; Accepted: 15 August 2025;
Published: 29 August 2025.
Edited by:
Geeta Rai, Banaras Hindu University, IndiaReviewed by:
Guan-Jun Yang, Ningbo University, ChinaEcem Kalemoglu, Jersey City Medical Center, United States
Copyright © 2025 Xia, Jiang, Xin, Li, Liao and Xin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhijun Xin, eGluemhpanVuMjAwOEAxNjMuY29t