 Daoran Xu
Daoran Xu Liyu Hu†
Liyu Hu† Chao Liu
Chao Liu- Department of Spinal Surgery, Affiliated Jiangyin Clinical College of Xuzhou Medical University, Jiangyin, China
Copper is a vital trace element integral to numerous biological processes, including iron metabolism, neurotransmitter synthesis, mitochondrial respiration, oxidative stress regulation, and energy production. However, disturbances in copper metabolism can result in pathological conditions, including cuproptosis—a newly recognized form of programmed cell death (PCD) marked by copper accumulation and the disruption of copper-dependent metabolic pathways. Cuproptosis has been associated with various diseases, such as cancer, metabolic disorders and neurodegenerative disorders. In the context of spinal cord injury (SCI), multiple pathological mechanisms, including oxidative stress, inflammation, and PCD could impact the patient’s prognosis with SCI. This review seeks to elucidate the pathophysiological underpinnings of SCI, the mechanisms and biological significance of copper homeostasis and the role of cuproptosis in SCI.
1 Introduction
Copper, an essential transition metal, serves as a critical cofactor for fundamental biological processes ranging from mitochondrial electron transport to neurotransmitter biosynthesis (1–3). While the human body maintains copper concentrations within a narrow physiological range (~100–200 mg total), this delicate homeostasis represents a metabolic tightrope walk. Both copper deficiency and overload trigger severe pathologies—from Menkes disease (MD)/Wilson disease (WD) to cancer progression—highlighting the metal’s Janus-faced role in human health (4, 5). The recent discovery of cuproptosis, a copper-dependent cell death modality driven by mitochondrial copper accumulation, has reshaped our understanding of copper’s pathophysiological roles. Unlike apoptosis or ferroptosis, cuproptosis uniquely involves Ferredoxin 1 (FDX1)-mediated lipoylated protein aggregation and iron-sulfur (Fe-S) cluster disassembly, establishing it as a mechanistically distinct player in disease pathogenesis (6).
Emerging evidence implicates cuproptosis in neurodegenerative disorders and malignancies (7–9), with newly published studies elucidating its potential role in spinal cord injury (SCI) progression (10, 11). SCI is a prevalent and catastrophic form of central nervous system (CNS) injury that results in motor, sensory, and autonomic dysfunction. As of 2016, there were approximately 27 million cases globally (12). Furthermore, SCI imposes a substantial medical burden, with annual medical expenditures for SCI patients in the United States reaching $3 billion in 2021 (13). In the context of SCI, cuproptosis may contribute to proteotoxic stress, mitochondrial dysfunction, and inflammatory responses, thereby exacerbating tissue damage (14–16). SCI is a complex condition characterized by both primary and secondary injury mechanisms (17). The primary injury is generally induced by a mechanical force that disrupts spinal cord tissue, resulting in immediate cell death and the loss of axonal connectivity (18). Subsequently, a series of secondary injury processes ensue, including oxidative stress, inflammation, excitotoxicity, and programmed cell death (PCD), which further exacerbate the initial damage and promote additional tissue degeneration (13, 18–20) (Figure 1).
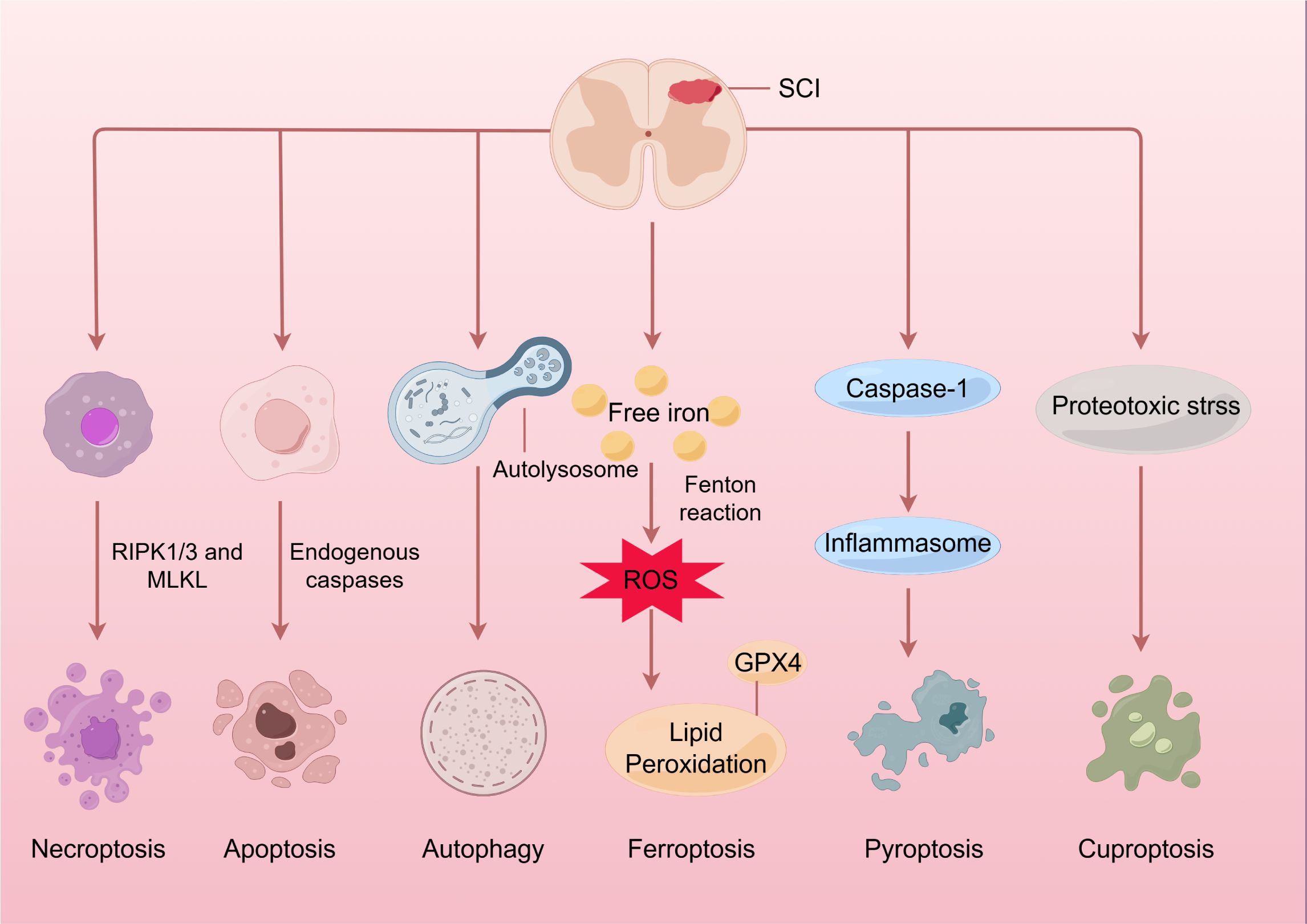
Figure 1. Spinal cord injury and programmed cell death. Initially, the application of mechanical force damages the cell membrane and releases intracellular components, further triggering autophagic processes as a defense mechanism. Following this, a range of pathophysiological processes occur, including oxidative stress, inflammation, and immune response, which eventually result in the apoptosis, necroptosis, and pyroptosis of neuronal and glial cells through specific mechanisms. Moreover, an overabundance of ROS may lead to lipid peroxidation and trigger ferroptosis. It’s intriguing that an excess of copper at the injury site might induce mitochondrial proteotoxic stress and cause cuproptosis. SCI, Spinal cord injury; RIPK1/3, receptor-interacting protein kinases; MLKL, mixed lineage kinase domain-like protein; ROS, reactive oxygen species; GPX4, glutathione peroxidase 4.
Research by Salsabili et al. has demonstrated that serum copper concentrations are significantly elevated in patients with spinal cord injuries compared to healthy individuals (21). The accumulation of copper in SCI initiates an inflammatory response, marked by the activation of microglia, which subsequently leads to the release of pro-inflammatory cytokines and chemokines. This inflammatory cascade further exacerbates tissue damage and impedes recovery (22, 23). Elucidating the role of cuproptosis in SCI is crucial for the development of targeted therapeutic interventions. Potential therapeutic strategies may involve the use of copper chelators to reduce copper accumulation and agents that modulate cuproptosis and mitochondrial function (12, 14, 24). Comprehensive therapeutic strategies that address multiple cellular pathways are imperative.
This review seeks to elucidate the mechanisms and biological functions of cuproptosis, the pathophysiological processes underlying SCI, the role of cell death in SCI, and the emerging significance of cuproptosis in the context of SCI. By comprehending the interplay between cuproptosis and SCI, novel therapeutic strategies can be developed to mitigate cell death and promote spinal cord repair. Further research is imperative to clarify the precise mechanisms and therapeutic potential of targeting cuproptosis in SCI.
2 Pathophysiological mechanisms of spinal cord injury
SCI represents a devastating traumatic insult to the CNS, inducing sensorimotor deficits and autonomic dysregulation that irrevocably compromise patients’ quality of life while imposing staggering socioeconomic burdens (12, 25). The prevailing pathophysiological paradigms underlying SCI are intricate and multidimensional, encompassing both primary and secondary injury processes (17). A comprehensive understanding of the interactions among these mechanisms is crucial for the development of effective therapeutic interventions.
2.1 Primary injury mechanisms
SCI predominantly results from acute mechanical forces that damage the integrity of spinal cord tissue, inducing structural compromise through compression, distraction, or shearing mechanisms (26). The biomechanical insult triggers a series of pathological processes: on the one hand, the mechanical force causes significant disruption to neuronal and glial cells, resulting in immediate cell death and the loss of axonal connectivity (27, 28). On the other hand, the Blood-Spinal Cord Barrier (BSCB), which ordinarily serves to protect the spinal cord from potentially harmful substances in the bloodstream, becomes compromised, allowing the infiltration of these substances (29). Additionally, the injury triggers the release of pro-inflammatory cytokines and chemokines, setting the stage for the secondary injury cascade (30, 31).
2.2 Secondary injury mechanisms
2.2.1 Oxidative stress
Oxidative stress constitutes a principal secondary injury mechanism in SCI. Subsequent to the initial mechanical trauma, a cascade of progressive processes is initiated, culminating in additional neuronal damage and functional impairment (12, 18). Oxidative stress response is mainly characterized by high levels reactive oxygen species (ROS), including superoxide anion radical (O2-), hydrogen peroxide (H2O2), hydroxyl radical (·OH), and peroxynitrite (32). It can be seen as a double-edged sword. Initially, low oxidative stress levels might help protect damaged tissues and cells (33). Yet, prolonged high oxidative stress and excessive ROS production can worsen further the inflammatory response and trigger various PCD pathways (34, 35). Mitochondria, which are not only essential for cellular energy metabolism and mitochondrial homeostasis, but also are the main organelles that generate ROS after SCI, play a significant role. This secondary injury is marked by an elevated production of ROS, which can surpass the cellular antioxidant defenses, thereby inducing mitochondrial dysfunction and apoptosis (36, 37).
Within the pathomechanical cascade of SCI, the disruption of mitochondrial homeostasis is of paramount importance, since mitochondria are central to energy production and cell death regulation (14, 38). The accumulation of oxidative stress not only facilitates neuronal apoptosis but also intensifies inflammation and tissue damage (39). Empirical studies have demonstrated that interventions designed to mitigate oxidative stress can substantially enhance outcomes in SCI models, underscoring the significance of targeting this mechanism in therapeutic strategies (34, 40, 41).
Furthermore, the interaction between oxidative stress and inflammatory responses adds complexity to the injury process. Consequently, comprehending the role of oxidative stress in SCI is essential for devising effective strategies to alleviate secondary injury and facilitate recovery. Therapeutic strategies aimed at mitigating oxidative stress and its downstream effects may offer potential for enhancing functional recovery and outcomes in patients with spinal cord injuries (12, 18).
2.2.2 Inflammation
Inflammation constitutes a pivotal element of the secondary injury cascade. Subsequent to the initial mechanical trauma, a multifaceted series of inflammatory responses is activated, potentially exacerbating damage and impeding recovery (42). While the primary injury results in immediate cellular damage, it is the ensuing inflammatory processes that substantially contribute to the magnitude of secondary injury (23). This cascade is initiated within hours post-trauma and may persist for several months, engaging various immune cells and signaling molecules that can either facilitate healing or precipitate further degeneration (18, 42, 43).
The inflammatory response is typified by the infiltration of immune cells, including neutrophils and macrophages, which secrete pro-inflammatory cytokines and chemokines (40, 44). These signaling molecules, such as IL-1β, TNF-α, and IL-6, are crucial in orchestrating the inflammatory response and may directly result in heightened oxidative stress and apoptosis of functional cells, such as neurons (45, 46). Besides, the chemokine CCL3, which is instrumental in the recruitment of inflammatory cells, has been demonstrated to exacerbate secondary damage following SCI by promoting inflammation and enlarging the lesion area (47). Similarly, C-C motif chemokine ligand 20 (CCL20) has been implicated in regulating neuroinflammation through the recruitment of Th17 cells, further complicating the inflammatory landscape following SCI (48).
In summary, inflammation should not be regarded solely as a byproduct of SCI; rather, it plays a pivotal role in the secondary injury cascade. The regulation of inflammation is essential for mitigating damage and facilitating recovery, underscoring the necessity for targeted therapeutic interventions that can effectively modulate the inflammatory response (23, 49).
2.2.3 Excitotoxicity
Excitotoxicity is a pathological process resulting from the excessive activation of excitatory neurotransmitters, notably glutamate (50). In the pathomechanistic framework of SCI, the initial trauma triggers a series of biochemical events that intensify neuronal damage, predominantly through the excessive release of glutamate (51). This surplus of glutamate can induce neuronal death and additional complications by activating various receptors, including NMDA receptors. While NMDA receptors play a vital role in synaptic plasticity, their overstimulation can lead to excitotoxicity (52, 53).
Within the process of SCI, neurotrauma causes a rapid increase in extracellular glutamate, which results in excitotoxicity, inflammation, glial scar development, and ultimately neuronal death. This elevation is contingent upon the severity of the injury, with more severe injuries correlating with higher levels of excitatory amino acids (51, 54). Furthermore, excitotoxicity not only facilitates immediate neuronal death but also predisposes the tissue to secondary injury mechanisms, including mitochondrial dysfunction and persistent neuroinflammation (55, 56).
Research indicates that interventions designed to modulate glutamate levels can effectively mitigate the effects of excitotoxicity. For example, the administration of pharmacological agents such as levetiracetam has shown potential in facilitating functional recovery following SCI by stabilizing astrocytes, which are essential for glutamate uptake and homeostasis (57). The removal of CNS glutamate into the systemic blood circulation through intravenous (IV) administration of blood glutamate scavengers (BGS) such as the enzyme recombinant glutamate-oxaloacetate transaminase 1 (rGOT1) and its co-substrate. The availability of the methods to remove excess glutamate from CNS without crossing the blood-brain barrier and with little to no negative effects could offer a substantial therapeutic benefit (54). Interestingly, mitochondria both aggravate glutamate toxicity due to impairments in the tricarboxylic acid (TCA) cycle and become a victims of glutamate toxicity, which disrupts oxidative phosphorylation (58).
2.2.4 Types of cell death in spinal cord injury
2.2.4.1 Apoptosis
Apoptosis is a key pathological feature of secondary cord injury, resulting in spinal cord dysfunction and permanent irreversible changes in neuronal cells, including neurons, astrocytes, oligodendrocytes, and microglia (59, 60). Caspases plays a crucial role in mediating apoptosis and is key to controlling this process (61). To date, there are three principal signaling pathways implicated in apoptosis encompassing the intrinsic (mitochondrial), extrinsic (death receptor), and endoplasmic reticulum (ER) pathways (61, 62). Within the context of SCI, apoptotic pathways of these damaged cells is predominantly initiated by factors such as oxidative stress, inflammation, and excitotoxicity (63). Oxidative stress, resulting from mitochondrial dysfunction and the generation of ROS, culminates in cellular damage and the activation of the intrinsic apoptotic pathway (64). Inflammation, marked by the secretion of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β), initiates the extrinsic pathway of apoptosis (45, 65). Excitotoxicity, induced by the excessive activation of excitatory neurotransmitters such as glutamate, leads to calcium influx and subsequent cellular damage, thereby further promoting apoptotic processes (14, 51).
The intrinsic apoptotic pathway is initiated by cellular stress, which prompts the release of pro-apoptotic proteins (cytochrome c) into the cytoplasm by increasing the permeability of mitochondrial membrane. This event facilitates the formation of caspase-3 and caspase-7 by activating caspase-9 and Cyt-c, subsequently activating apoptosis and regulating other endogenous apoptotic cascades leading to cell death (66). In contrast, the extrinsic apoptotic pathway is triggered by the interaction of death ligands, such as Fas ligand (FasL) with their corresponding death Fas receptors. This interaction results in the recruitment of adaptor proteins, such as Fas-associated death domain protein (FADD), consequently activating pro-caspase-8 and forms a death-inducing signaling complex (DISC), thereby mediating the executive stage of apoptosis (61, 65, 67). ER stress arises from the accumulation of misfolded proteins within the ER, which activates the unfolded protein response (UPR), increasing the expression of apoptotic proteins, and leading to apoptosis in spinal cord neurons (62).
In the pursuit of effective therapeutic interventions to mitigate apoptosis in SCI, several promising strategies have emerged. Caspase inhibitors, such as zVAD-fmk, function by obstructing the activation of caspases, thereby preventing apoptosis (68). The administration of ligustilide (LIG) post-SCI enhances mitophagy through the BNIP3-LC3 interaction, reduces oxidative stress, restores normal mitochondrial function, and ultimately weakens the intrinsic pathway of apoptosis (69). Zhou et al. have confirmed that stem cells and exosomes prevent apoptosis following SCI by modulating Fas/FasL-mediated extrinsic pathways (70). Furthermore, in SCI model rats, nerve growth factor (NGF) has been demonstrated to reduce neuronal apoptosis and suppress the ER stress-induced apoptotic response proteins CHOP, GRP78, and caspase-12 (71). A comprehensive understanding of the initiating factors, key signaling pathways, and potential therapeutic strategies to attenuate apoptosis is crucial for advancing treatment approaches. However, further research is essential to substantiate these strategies and to formulate comprehensive treatment protocols for SCI.
2.2.4.2 Autophagy
Autophagy is a crucial cellular process that relies on lysosomes to degrade damaged organelles, misfolded proteins and other cellular materials for recycling and clearing, thereby mitigating cellular toxicity (72). There are currently three well-known types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Generally, the term autophagy refers to macroautophagy (73). Autophagy in SCI is predominantly initiated by a variety of factors, including oxidative stress, nutrient deprivation, and mechanical injury. Oxidative stress, which arises from mitochondrial dysfunction and the subsequent generation of ROS, results in cellular damage and triggers autophagy as a protective mechanism (12). Similarly, nutrient deprivation, frequently observed following injury, activates autophagy to preserve cellular homeostasis (74). Excessive autophagy, however, can lead to the autophagic death of spinal cord neurons, exacerbating tissue damage. Hence, tight regulation of autophagy is essential to maintain optimal levels (13, 75). Besides, mechanical injury associated with SCI can lead to damage of the cell membrane and the release of intracellular components, thereby further stimulating autophagic processes (76).
Key signaling pathways involved in the regulation of autophagy encompass the mTOR pathway, the AMPK pathway, and the ULK1 complex (76). The mTOR pathway serves as a principal regulator of autophagy; specifically, the inhibition of mTOR is associated with the promotion of autophagic processes (77). Conversely, the AMPK pathway is activated in response to cellular energy stress, resulting in the inhibition of mTOR and the subsequent activation of autophagy (78). The ULK1 complex, which comprises ULK1, ATG13, FIP200, and ATG101, plays a critical role in the initiation of autophagy (79). Recent studies have further elucidated mechanisms of autophagy following SCI, particularly emphasizing the importance of mitophagy (12, 80). Mitophagy, a specialized form of autophagy targeting damaged mitochondria, is activated in response to mitochondrial damage, oxidative stress, and ROS (81). This process is crucial for the clearance of damaged mitochondria and the maintenance of cellular homeostasis.
Potential therapeutic strategies for modulating autophagy in SCI include the use of mTOR inhibitors, AMPK activators, and autophagy agonists. mTOR inhibitors, such as rapamycin and KU0063794, have been demonstrated to enhance autophagy, thereby reducing neural tissue damage and locomotor impairment following SCI (82, 83). Similarly, AMPK activators, including Netrin-1, promote autophagic processes by inhibiting mTOR, leading to improved functional recovery in rat models of SCI (84). Additionally, autophagy agonists like resveratrol have also been shown to facilitate functional recovery and reduce neuroinflammation through the activation of autophagy via the AMPK/mTOR pathway post-SCI (78).
In conclusion, autophagy plays a crucial role in the pathophysiology of SCI, supporting cellular protection and recovery processes (13). Targeting autophagy may provide a viable approach to alleviating secondary injury and enhancing functional recovery in individuals with SCI (76). Continued research is imperative to substantiate these approaches and to formulate comprehensive treatment protocols for SCI.
2.2.4.3 Pyroptosis
Pyroptosis, identified as a PCD in 2001, primarily takes place in macrophages, dendritic cells, and neutrophils, but can also be seen in neuron cells (85, 86). It is a lytic, pro−inflammatory PCD program and characterized by its reliance on gasdermin proteins as the executioners of cell death (86). Pyroptosis in SCI is predominantly initiated by various factors, including infection, immune activation, and mechanical trauma. Infections, especially those caused by pathogens that stimulate the inflammasome, result in the production of pro-inflammatory cytokines and the subsequent induction of pyroptosis (87). Immune activation, marked by the release of danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), also triggers inflammasome activation and facilitates pyroptosis (88, 89). Mechanical trauma, exemplified by SCI, can result in damage to the cell membrane and the subsequent release of intracellular components, thereby triggering inflammasome activation (87).
Critical signaling pathways implicated in pyroptosis encompass the NLRP3 inflammasome, AIM2 inflammasome, and caspase-4/5/11 pathways (86). The NLRP3 inflammasome is triggered by diverse stimuli, such as ROS formation, lysosomal rupture, and ion channel gating (90).This activation triggers the formation of the inflammasome complex and then activates caspase-1. Once activated, caspase-1 cleaves pro-interleukin-1β (pro-IL-1β) and pro-interleukin-18 (pro-IL-18) into their active forms, which are subsequently released into the extracellular space, thereby promoting inflammatory responses (91, 92). Moreover, caspase-1 cleaves gasdermin D, resulting in the formation of pores in the cell membrane and subsequent cell lysis (93). The AIM2 inflammasome is induced by the presence of cytosolic DNA, which triggers the assembly of the inflammasome complex and the activation of caspase-1 (87, 94). The caspase-4/5/11 pathways are initiated by bacterial lipopolysaccharide (LPS) and directly cleave gasdermin D, resulting in pyroptosis (95).
Potential therapeutic strategies for mitigating pyroptosis in SCI include the use of inflammasome inhibitors, anti-inflammatory agents, and autophagy modulators. Inflammasome inhibitors such as MCC950 and Topotecan (TPT) have demonstrated efficacy in inhibiting the activation of the NLRP3 inflammasome, thereby reducing pyroptosis and neuronal death, and enhancing functional recovery post-SCI (96, 97). Anti-inflammatory agents, such as inhibitors targeting IL-1β and IL-18, have been shown to decrease the release of pro-inflammatory cytokines and attenuate pyroptosis (98, 99). Notably, Betulinic acid (BA), an autophagy modulator, has been shown to regulate autophagy and mitophagy through the AMPK-mTOR-TFEB signaling pathway, significantly inhibiting pyroptosis and facilitating functional recovery following SCI (100). By focusing on the modulation of pyroptosis, it may be possible to mitigate secondary injury and enhance functional recovery in patients with SCI.
2.2.4.4 Necroptosis
Necroptosis refers to one form of PCD mediated by genetic programming and regulatory processes (101). Necroptosis is triggered by interactions involving death receptor family ligands with agonists such as tumor necrosis factor (TNF), FasL, and TRAIL (102). Current studies have shown that necroptosis plays a crucial role in the pathogenesis of SCI. This regulated necrotic process, orchestrated by receptor-interacting protein kinases (RIPK1 and RIPK3) and the mixed lineage kinase domain-like protein (MLKL) family, significantly influences the inflammatory response and neuronal and glial cell death subsequent to SCI (103–105).
After SCI, the accumulation of RIPK1 and RIPK3 proteins is associated with lysosomal dysfunction and the suppression of autophagy, both of which are vital for maintaining cellular homeostasis and survival. Following rapamycin treatment, lysosomal function is restored in cells, resulting in decreased levels of RIPK1, necroptosis, and cell death (103). The interaction between necroptosis and neuroinflammation is especially significant, as the inflammatory milieu resulting from necroptotic cell death can exacerbate neuronal damage and impede recovery (106, 107).
Inhibitors of RIPK1, such as necrostatins, have demonstrated potential in preclinical models by decreasing cell death and enhancing functional outcomes (108). MLKL is a pivotal regulator of necroptosis. MLKL levels increased significantly post-SCI, and inhibition of MLKL improved recovery of neurological function in SCI (105). Elucidating the mechanisms underlying necroptosis in SCI not only advances our understanding of the injury process but also facilitates the development of novel interventions aimed at promoting recovery and regeneration in the injured spinal cord (13, 102).
2.2.4.5 Ferroptosis
Ferroptosis, a well-known PCD and first formally defined in 2012, is characterized by disturbances in iron metabolism and the accumulation of lipid reactive oxygen species (109). In SCI, ferroptosis is predominantly initiated by factors such as oxidative stress, iron overload, and the dysregulation of lipid metabolism. Oxidative stress, resulting from impaired mitochondrial function and the generation of ROS, contributes to lipid peroxidation and the subsequent activation of ferroptosis (12). Additionally, iron overload could further intensify lipid peroxidation and the ferroptotic process (110). The dysregulation of lipid metabolism, characterized by the accumulation of polyunsaturated fatty acids (PUFAs) and the deficiency in various elements of the antioxidant glutathione (GSH) pathway, significantly contributes to the process of ferroptosis (111, 112).
Central to the signaling pathways implicated in ferroptosis are the GPX4-dependent and ACSL4-dependent pathways. The enzyme glutathione-dependent antioxidant enzyme glutathione peroxidase 4 (GPX4) is instrumental in safeguarding cells against ferroptosis by reducing lipid peroxides. Inhibition or deficiency of GPX4 results in the accumulation of lipid peroxides, thereby triggering ferroptosis (111, 113). Conversely, the enzyme Acyl-CoA synthetase long-chain family member4 (ACSL4) facilitates the synthesis of lipid substrates necessary for lipid peroxidation, thereby promoting ferroptosis (114). Recent studies have elucidated further mechanisms involved in ferroptosis within SCI, highlighting the significance of mitochondrial quality control (MQC). Mitochondria play a crucial role in modulating ferroptosis by influencing iron homeostasis, energy metabolism, and lipid synthesis (115, 116).
Several studies have demonstrated that ferroptosis inhibitors, such as ferrostatin-1 and Celastrol, can reduce ROS accumulation, thereby inhibiting ferroptosis in oligodendrocytes and ultimately promoting functional recovery following SCI (117, 118). Alpha-tocopherol (Vitamin E, Vit E), an effective antioxidant, has been shown to mitigate ROS accumulation, iron overload, lipid peroxidation, and mitochondrial dysfunction, thereby alleviating SCI-induced ferroptosis by downregulating Alox15 in rats with SCI (119). Interestingly, there is a notable rise in iron accumulation in the motor cortex of both SCI patients and rats, triggering the accumulation of lipid ROS and causing motor neuron ferroptosis (120). Iron deposition is a critical pathological event in ferroptosis; deferoxamine (DFO), a potent iron chelator, has been shown to increase the expression levels of GPX4, xCT, and GSH, thereby improving survival of neuronal survival and inhibiting gliosis (121). To conclude, targeting ferroptosis may offer a promising way to reduce secondary injury and improve functional recovery by these potential therapeutic strategies in patients with SCI.
3 Molecular mechanisms underlying cuproptosis
3.1 Copper in cellular processes
3.1.1 Copper homeostasis
Copper, an indispensable trace element, is crucial for a broad spectrum of physiological processes across nearly all cell types. Copper is integral to various biological functions, including iron metabolism, neurotransmitter synthesis, mitochondrial respiration, oxidative stress regulation, and energy production (122, 123). The current recommended daily intake of copper for healthy adults is 0.8–2.4 mg, while the total copper concentration in the human body ranges from 100 to 200 mg (124). To maintain systemic copper homeostasis within physiological limits, cells employ a complex regulatory network of copper enzymes, molecular chaperones, and membrane transport proteins, which collaboratively regulate copper uptake, efflux, and utilization (15, 24).
The absorption of dietary copper predominantly occurs in the small intestine and duodenum, primarily mediated by the copper transport protein 1 (CTR1), also known as solute carrier family 31 member 1 (SLC31A1), located on the apical side of intestinal epithelial cells (125). Dietary copper is mainly present in the form of divalent copper ions, upon transport to the cell surface in the blood, divalent copper ions are converted to monovalent copper ions by six-transmembrane epithelial antigen of the prostate (STEAP) proteins for further uptake (126). When intracellular copper levels are deficient, the expression of CTR1 is upregulated to facilitate the absorption of copper ions. However, under conditions of oxidative stress, the functionality of CTR1 may be compromised, thereby disrupting copper ion homeostasis (127). In addition, copper can also enter cells via divalent metal transporter 1 (DMT1) and through passive diffusion (128, 129).
Once inside the cell, copper ions are directly delivered to their target proteins by copper chaperones, which are a group of proteins responsible for transporting copper to specific intracellular sites and enzymes (130). The copper chaperone for superoxide dismutase (CCS) is specifically involved in the insertion of copper and the formation of disulfide bonds in Cu-Zn superoxide dismutase 1 (SOD1), and it regulates the localization of Cu-Zn SOD1 between the cytoplasm and mitochondria (131, 132). SOD1 is an enzyme ubiquitously present in cells, playing a crucial role in the elimination of superoxide radicals and protecting cells from oxidative damage; its mutations can lead to familial amyotrophic lateral sclerosis (ALS) (132, 133). The cytochrome c oxidase copper chaperone 17 (COX17), a copper metallochaperone, primarily responsible for transferring copper to synthesis of cytochrome oxidase 1/2 (SCO1/2) and cytochrome c oxidase copper chaperone 11 (COX11). SCO1/2 and COX11 are essential for the formation of cytochrome c oxidase 1 (COX1) and cytochrome c oxidase 2 (COX2) by delivering copper ions, respectively (122, 134). Notably, cytochrome c oxidase (CCO), the terminal complex in the electron transport chain (ETC) composed of two main components: COX1 and COX2, plays a crucial role in regulating intracellular biochemical processes (123, 135). Research indicates that the binding of copper to transcription factors via the antioxidant 1 copper chaperone (ATOX1) in the nucleus can further modulate gene expression (123). Additionally, ATOX1 facilitates the transport of copper to the trans-Golgi network proteins ATPase copper transporting alpha (ATP7A) and ATPase copper transporting beta (ATP7B). Both proteins function as P-type Cu-transporting ATPases, harnessing ATP hydrolysis energy to transfer copper ions across cellular membranes or to copper-dependent enzymes, thereby ensuring the maintenance of intracellular copper homeostasis (136, 137). Copper-transporting ATPases exhibit tissue-specific expression patterns. ATP7A is present in nearly all cells of the body, with the exception of hepatocytes. Copper is introduced into the bloodstream from enterocytes in the small intestine via the copper ion transporter ATP7A, which facilitates its systemic distribution (136, 138).
Copper predominantly attaches to ceruloplasmin (CP) and albumin in the bloodstream, and is then carried to the liver, which is the main site for copper storage and elimination. Surplus copper is expelled from liver cells into bile in vesicular form through ATP7B (139–141). Metallothioneins (MT1/2) and GSH serve as natural intracellular copper ion chelators, binding copper to store excess ions and prevent copper toxicity and cellular damage (142, 143) (Figure 2). The maintenance of intracellular copper homeostasis primarily depends on copper chaperones and transport proteins; an imbalance in copper homeostasis can result in cellular metabolic disorders and even cell death (144). This intricate equilibrium is essential for normal physiological processes and the prevention of disorders associated with copper dysregulation, including Alzheimer’s diseases (AD) and Parkinson’s diseases (PD) (145, 146).
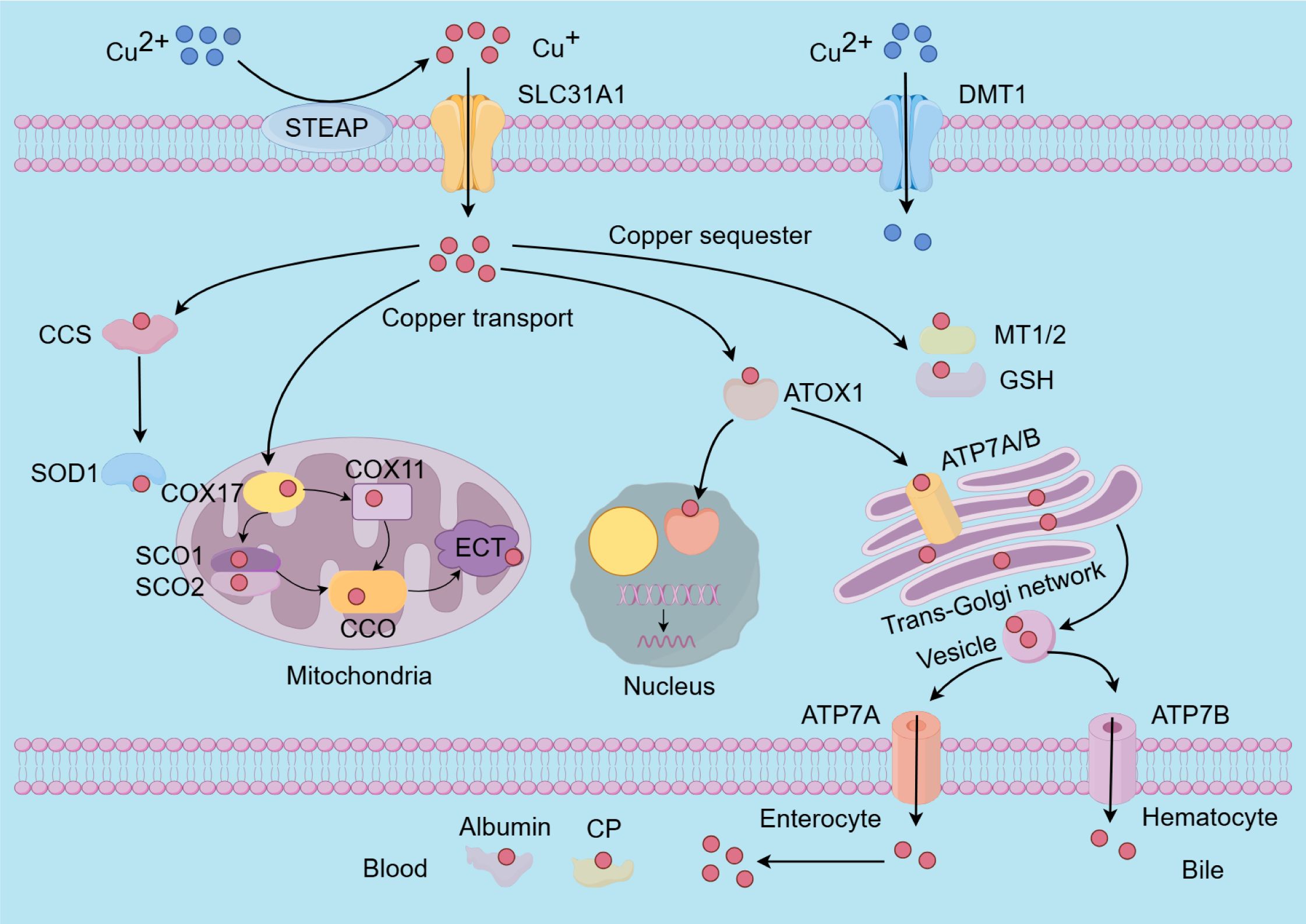
Figure 2. The molecular mechanisms underlying copper metabolism involve several key processes. The absorption of copper ions is primarily facilitated by the transporters SLC31A1 and DMT1. Notably, SLC31A1 is specific to the uptake of monovalent copper ions, necessitating the reduction of divalent copper ions to monovalent copper ions by STEAP before uptake. Within cells, copper is transported to and interacts with specific cytoplasmic copper chaperones, including COX17, CCS, and ATOX1, which direct copper to distinct cellular compartments such as the mitochondrial electron transport chain, the trans-Golgi network, and the nucleus. Copper insertion and disulfide bond formation in SOD1 are specifically facilitated by the CCS. To mitigate the cytotoxic effects of excess intracellular copper, copper ions can bind to MT1/2 and GSH for storage. Additionally, the maintenance of intracellular copper homeostasis is achieved through the export of surplus copper ions into the bloodstream via ATP7A and ATP7B. Subsequently, copper is transported throughout the body by CP and albumin. SLC31A1, Solute carrier family 31 member 1; DMT1, divalent metal transporter 1; STEAP, six-transmembrane epithelial antigen of the prostate; COX17/11, cytochrome c oxidase copper chaperone 17/11; CCS, cytoplasmic-mitochondrial metallochaperones; ATOX1, antioxidant protein 1; MT1/2, metallothioneins; GSH, glutathione; Cu-Zn SOD1, Cu-Zn superoxide dismutase 1; SCO1/2, sythesis of cytochrome oxidase 1/2; CCO, cytochrome oxidase; ATP7A/B, ATPase 7A/B; CP, cuproenzymes.
In the CNS, ATP7A and ATP7B are crucial for regulating copper levels, with disruptions in this regulatory mechanism associated to MD and WD, respectively (147, 148). Severe copper deficiency can lead to impaired energy production due to mitochondrial cytochrome c oxidase dysfunction, while excessive intracellular copper levels may contribute to the onset of various diseases (149). Importantly, the redox properties of copper pose potential risks, as an excess can lead to the generation of harmful ROS, which may damage cellular components and contribute to the pathogenesis of cancer and CNS disorders (24, 131, 150).
3.2 Molecular components involved in cuproptosis
3.2.1 Molecular mechanisms of cuproptosis
Although copper-induced cell death was recognized in the 1980s, the precise mechanism remained unclear (151). Over the past few decades, significant interest has emerged in exploring the relationship between copper and PCD, leading to extensive research on the mechanisms of copper-triggered cell death (8). The precise form of cell death induced by excessive copper ions has been a subject of debate, with researchers historically categorizing it as apoptosis, autophagy, or ferroptosis, all of which are closely associated with ROS and inflammatory responses (150).
It wasn’t until 2022 that the precise mechanism of copper-induced cell death was elucidated by Tsvetkov et al., who termed this process “cuproptosis” (6). This research challenges the traditional understanding of cuproptosis and initiates a new era of investigation into copper-induced cell death. Cuproptosis, a recently discovered type of cell death, is mediated by the ancient mechanism of protein lipoylation, a highly conserved post-translational modification that targets lysine residues (6, 150). This modification is exclusively observed in four specific enzymes: dihydrolipoamide S-succinyltransferase, glycine cleavage system H protein, dihydrolipoamide branched-chain transacylase E2 (DBT), and dihydrolipoamide S-acetyltransferase (DLAT). Notably, DLAT plays an essential role in the pyruvate dehydrogenase complex (PDC), facilitating the transformation of pyruvate into acetyl coenzyme A within the TCA cycle (123, 152).
The mechanism of cuproptosis involves a copper ionophore, elesclomol, which binds to divalent copper ions in the extracellular environment and facilitates their transport into cells. Within the mitochondria, elesclomol interacts with the direct target FDX1, subsequently releasing it. FDX1, a critical enzyme in the cuproptosis pathway, possesses a strong reducing capability, enabling it to lipoylate the aforementioned enzymes, and reduce divalent copper ions to monovalent copper ions (24, 153). Subsequently, monovalent copper ions directly interact with lipoylated mitochondrial protein fractions involved in the TCA cycle and affect Fe–S clusters, which are critical components of the ETC. This interaction induces the aggregation of lipoylated proteins, the loss of Fe-S cluster-containing proteins, and the upregulation of heat shock protein70 (HSP70). Consequently, the dysfunction of the TCA cycle and ETC occurs, ultimately leading to cell death mediated by proteotoxic stress (6, 153). Moreover, previous studies have demonstrated that HSP70 is significantly upregulated under conditions of stress, such as oxidative stress, hypoxia, and metal overload, thereby alleviating protein misfolding and aggregation to mitigate proteotoxic stress and prevent cellular damage (154, 155).
In their study, Tsvetkov et al. have also demonstrated that the ROS inhibitor N-acetylcysteine (NAC), along with inhibitors targeting other established cell death pathways—such as ferroptosis inhibitors (ferrostatin-1), and necroptosis inhibitors (necrostatin-1)—were ineffective in mitigating cell death mediated by copper ionophores. Unprecedentedly, GSH, a cellular copper chelator, successfully prevented this process by binding with copper ions, highlighting the distinct nature of cuproptosis (6, 156) (Figure 3). Furthermore, the abrogation of cuproptosis was achieved through the knockout of seven genes, including FDX1 and six genes associated with the lipoic acid pathway (LIPT1, LIAS, and DLD) or protein targets of lipoylation (DLAT, PDHA1, and PDHB) (6). In contrast, the cuproptosis was up-regulated by inhibiting three genes, MTF1, GLS and CDKN2A (157, 158) (Table 1).
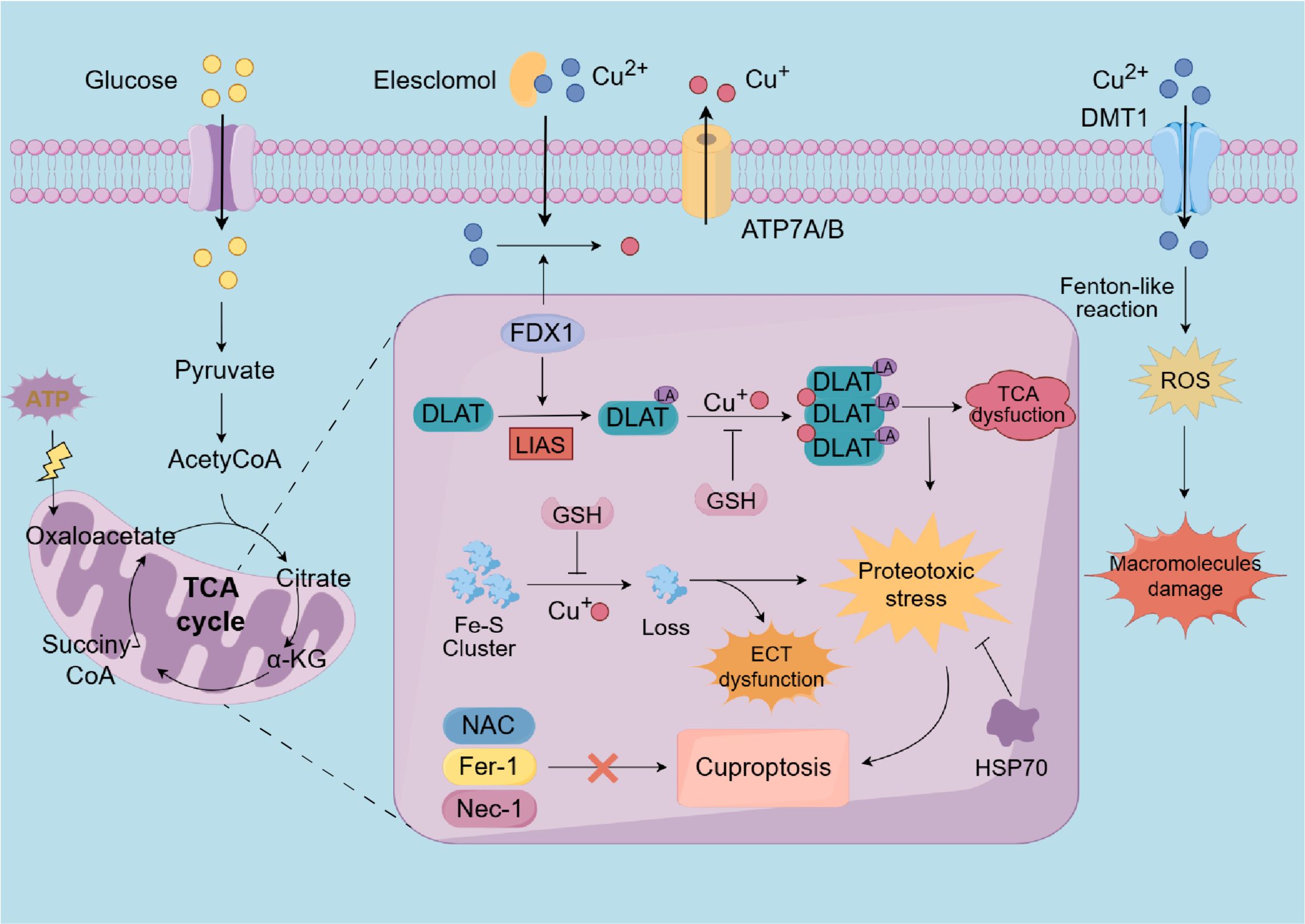
Figure 3. A diagrammatic representation of the cuproptosis process. Extracellular copper is sequestered by the copper ionophore elesclomol and transported into intracellular compartments. As an upstream regulator of protein lipoylation, FDX1/LIAS may enhance the lipoylation of TCA cycle enzymes, such as DLAT. Furthermore, within the mitochondria, FDX1 facilitates the reduction of divalent copper to its monovalent state. Subsequently, the monovalent copper could interact with DLAT and Fe-S cluster proteins results in DLAT aggregation and the depletion of Fe-S cluster proteins. These aberran interactions result in the TCA cycle and ETC to malfunction, triggering cuproptosis through proteotoxic stress, which eventually disrupts ATP synthesis. Notably, HSP70 and copper chelators, such as GSH, can inhibit cuproptosis. However, NAC, ferrostatin-1, and necrostatin-1 do not mitigate copper-induced cell death. Additionally, excessive copper ions can also induce oxidative stress and elevate ROS levels, thereby damaging intracellular macromolecules. FDX1, Ferredoxin 1; LIAS, lipoic acid synthetase; TCA, tricarboxylic acid; Fe-S, iron-sulfur cluster; DLAT, dihydrolipoamide S-acetyltransferase; ETC, electron transport chain; HSP70, heat shock protein 70; GSH, glutathione; NAC, N-acetylcysteine; ferrostatin-1, ferroptosis inhibitors; necrostatin-1, necroptosis inhibitors; ROS, reactive oxygen species.
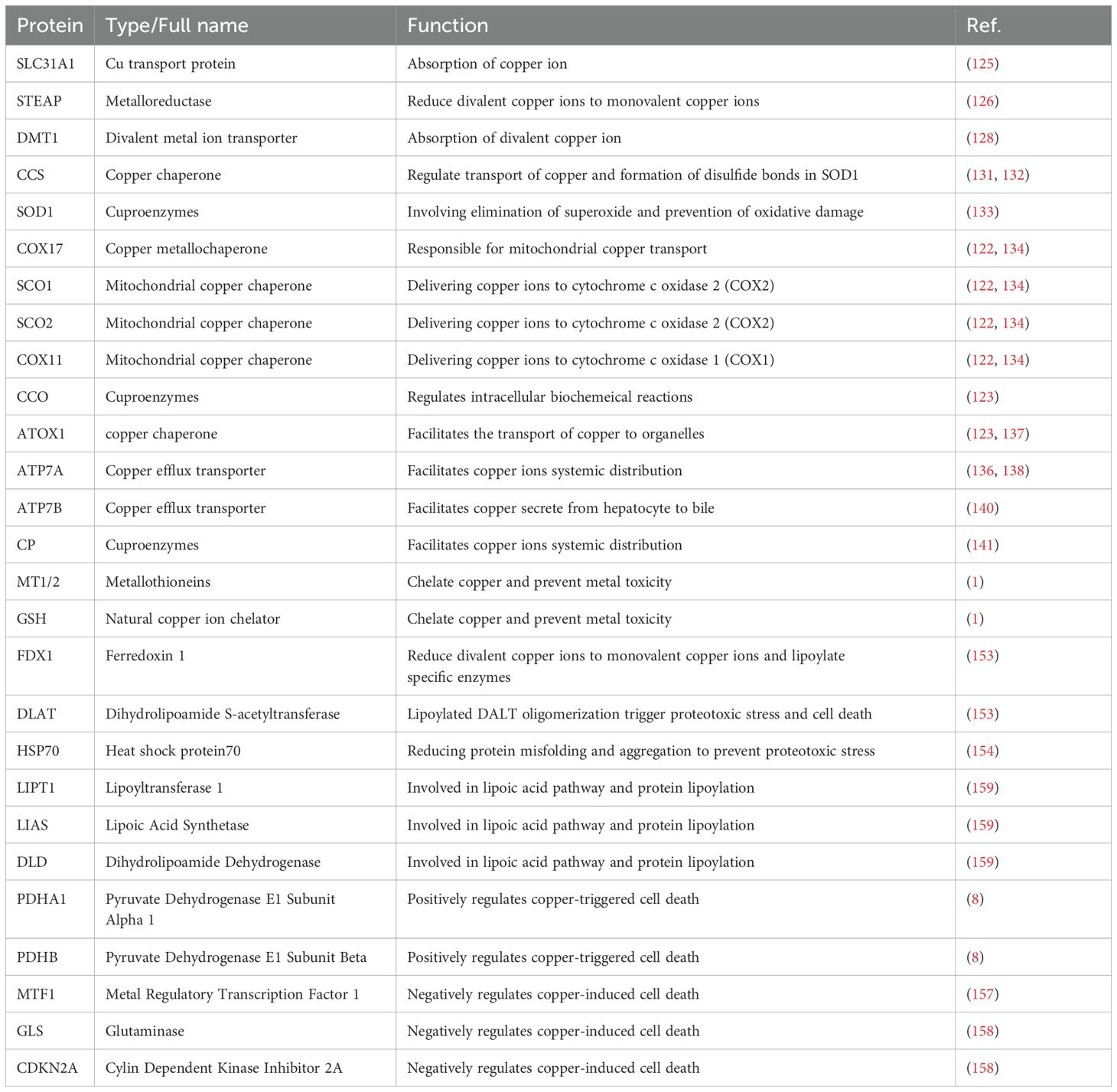
Table 1. Copper homeostasis and cuproptosis related regulatory proteins.
3.2.2 ROS generation and mitochondrial dysfunction
Every cell in the human body endeavors to maintain a balance between oxidant and antioxidant species, both of which are essential for cellular metabolism, signal transduction, and various biological functions (160). As an indispensable micronutrient, copper plays a crucial role in numerous biological processes to sustain cellular homeostasis. However, excessive copper disturbs the equilibrium between oxidants and antioxidants, resulting in ROS production and inducing oxidative stress, which then damages biological macromolecules like lipids, proteins, and DNA (24, 161). Additionally, elevated copper concentrations significantly impair the activity of Cu-Zn SOD1, catalase, and glutathione peroxidase, thereby compromising the antioxidant defense system and exacerbating the cytotoxic effects of ROS (162, 163). In patients with SCI, certain substances trigger reactions that damage the subcellular structures of neurons and glial cells, ultimately resulting in cell death. This phenomenon may be a primary factor contributing to the poor prognosis observed in these patients (12, 164).
Mitochondria, which are of great significance for cellular energy metabolism and maintaining mitochondrial homeostasis, also serve as the primary organelles responsible for generating ROS following SCI. Notably, ROS exacerbate mitochondrial damage, establishing a detrimental cycle that culminates in cell death (12). Copper-caused mitochondrial dysfunction and the promotion of mitochondrial fission are linked to the activation of mitophagy, which is closely associated with the upregulation of PINK1 and Parkin expression (165). Furthermore, copper has the potential to influence mitochondrial dynamics, including the processes of fission and fusion, which are vital for maintaining mitochondrial integrity and functionality, thereby contributing to cellular toxicity (166). Specifically, it has been suggested that the reduction of mitochondrial membrane potential is critical for copper-triggered astrocyte death, whereas neuronal damage is more closely associated with oxidative and nitrosative stress (167).
Historically, the specific mechanism of cell death induced by copper ions has been a subject of debate, and numerous studies have indicated a strong association with ROS (150). Evidence has demonstrated that copper directly targets complex IV (cytochrome c oxidase, CCO) of the ETC, upregulating its protein expression under copper overload conditions, without affecting other respiratory complexes (168, 169). Interestingly, Tsvetkov et al. identified a link between copper-induced cell death (cuproptosis) and mitochondrial metabolism, suggesting that copper impacts components of the TCA cycle rather than directly targeting the ETC (6).
In summary, while there is no direct correlation between ROS and the cuproptosis mechanism, ROS remain a critical factor in other pathways of copper-triggered cell death and should not be overlooked. The TCA cycle is intricately associated with cuproptosis due to its involvement in energy metabolism, redox homeostasis, and cellular signaling pathways. Further investigation is required to elucidate the specific mechanisms by which copper modulates TCA cycle dynamics and to understand how these interactions influence cell fate decisions in both physiological and pathological conditions.
3.2.3 Ferredoxin1: the regulator of protein lipoylation and diseases
FDX1, an evolutionarily conserved protein characterized by the presence of Fe–S clusters, serves as an upstream regulator of protein lipoylation and is integral to the metabolism of steroids, cholesterol, and bile acids (170). FDX1’s involvement in cellular redox reactions and energy metabolism highlights its essential role in maintaining cellular homeostasis and its necessity for elesclomol-induced cuproptosis (24). Recent studies have emphasized the pivotal function of FDX1 across various cancer types, including clear cell renal cell carcinoma (ccRCC), hepatocellular carcinoma (HCC), and gliomas, where its expression levels have been associated with patient survival outcomes (171–173). In the molecular pathogenesis of HCC, increased FDX1 expression has been associated with enhanced immune cell infiltration, particularly involving natural killer cells and macrophages, potentially contributing to improved patient prognoses (172). Conversely, FDX1 deficiency may result in metabolic reprogramming and oxidative stress, which activate PINK1/Parkin-mediated mitophagy and the PI3K/AKT pathway, thereby facilitating HCC proliferation, invasion, and metastasis (174).
Furthermore, the expression of FDX1 is significantly diminished in aged ovaries compared to younger ones, indicating its potential as a biomarker and therapeutic target for age-related conditions (175). Modulating the expression of FDX1 may represent a promising strategy to enhance the efficacy of treatments designed to induce cuproptosis in cancer cells, potentially leading to novel therapeutic approaches in oncology (176). To summarize, FDX1 functions as a crucial regulator of cuproptosis, impacting cancer tumor progression, prognosis, immune infiltration, and therapeutic responses (174). Further investigation into the mechanisms and interactions of FDX1 could reveal new pathways for targeted therapies in both cancer and age-related diseases.
3.3 Cuproptosis in immune response
Copper is recognized for its immunomodulatory properties, affecting various components of the immune system. Recent research has underscored the significance of copper in modulating the innate immune response, especially through its interactions with immune cells and signaling pathways (177, 178). For example, copper has been demonstrated to augment the activity of alpha-kinase 1 (ALPK1), a cytosolic pattern-recognition receptor integral to host defense mechanisms against bacterial infections. This activation results in an elevated production of proinflammatory cytokines and an augmented recruitment of immune cells, thereby facilitating a robust immune response (179).
Additionally, over copper could induce cuproptosis—a newly explored PCD form mediated by elevated copper concentrations. Recently, numerous findings have shown that it is closely linked with the immune microenvironment in various tumors (178, 180). The regulation of cuproptosis in immune cells may impact their survival and functionality, thereby potentially influencing the overall immune response to pathogens and tumors (181). For instance, within acute myeloid leukemia mechanisms, specific patterns of cuproptosis regulation have been linked to varying immune profiles, indicating that modulating cuproptosis could serve as a strategy to enhance immunotherapy outcomes (182). The accumulation of copper at infection sites has been identified as a mechanism through which the host exerts antimicrobial effects, utilizing copper toxicity as a tool employed by phagocytic cell (183, 184). Copper plays a dual role as both an essential cofactor for immune function and a potential mediator of cell death, totally underscoring its intricate involvement in the immune response is vital for us. Consequently, the interaction between copper homeostasis, cuproptosis, and immune regulation represents a promising research avenue that may yield innovative therapeutic strategies for augmenting immune responses against infections and cancer.
3.4 Cuproptosis in disease states
3.4.1 Cancer
The relationship between copper and tumorigenesis in cancer is complex, with dysregulated copper metabolism playing a dual role in both tumorigenesis and cancer therapy (185). Cuproptosis, a distinct form of cell death triggered by accumulated copper levels, has been shown to significantly impact the regulation of cancer cell survival and proliferation (8). This copper-dependent mechanism is unique compared to other types of PCD, such as apoptosis and ferroptosis, and involves specific TCA cycle pathways that can lead to proteotoxic stress under conditions of copper overload (6, 186). Interestingly, the tumor suppressor protein p53 may regulate the biogenesis of Fe-S clusters and GSH, both of which are critical components of the cuproptotic pathway, suggesting that p53 might play a significant role in mitigating copper toxicity and cuproptosis (187).
Emerging research has further elucidated the role of copper in cancer biology, highlighting its ability to promote tumor growth through cuproplasia—copper-dependent cellular proliferation—while also inducing cell death via the mechanism of cuproptosis (185). This complex interplay underscores the significance of copper metabolism as a pivotal factor in tumorigenesis, impacting both the initiation and progression of diverse cancer types (185). Copper ionophores, such as elesclomol and disulfiram, increase intracellular copper levels and induce cell death, which is mediated by oxidative stress, mitochondrial respiration disruption and protein lipoylation in cancer therapy (153, 188). Furthermore, the molecular mechanisms underlying cuproptosis remain under investigation, with current research efforts directed towards elucidating the interactions between this form of cell death and other cellular pathways and death mechanisms (186, 189). As the field of cancer research continues to evolve, the exploration of cuproptosis and its implications for tumorigenesis remains a promising area of investigation, with the potential to uncover novel insights into cancer biology and therapy (185, 190, 191).
3.4.2 Metabolic disorders
In metabolic disorders, including diabetes, the regulation of copper homeostasis may be compromised, potentially resulting in aberrant cuproptosis within cells engaged in diverse pathophysiological processes (192). Copper is integral to enzymatic activity and cellular signaling. The dysregulation of copper levels can induce oxidative stress, mitochondrial dysfunction and even cuproptosis, a critical contributor to the pathogenesis of diabetic complications (161, 193).
Novel insights have underscored the significance of copper in the pathophysiology of diabetic retinopathy, indicating that disruptions in copper metabolism may play a role in vascular dysfunction and oxidative damage within retinal cells (194). Additionally, the newly defined concept of cuproptosis, closely associated with copper accumulation, implies that imbalances in copper homeostasis may adversely affect cellular viability and function in diabetic tissues (195, 196). Elucidating the mechanisms through which copper dysregulation impacts cuproptosis could yield valuable insights into prospective therapeutic strategies for addressing diabetes-related complications (193, 197). Specifically, interventions targeting copper metabolism, such as chelation therapy or the application of copper coordination complexes, may present novel treatment pathways (198).
These approaches have the potential to alleviate the detrimental effects of oxidative stress and enhance cellular health in individuals with diabetes (192, 196). In addition, the interaction between copper homeostasis and various metabolic pathways highlights the intricate nature of managing metabolic disorders. Ongoing research into the relationships among copper, cuproptosis, and metabolic health holds the potential to inform novel strategies aimed at improving treatment outcomes for diabetes and related conditions (196, 199, 200).
3.4.3 Neurodegenerative diseases
In neurodegenerative disorders such as AD and PD, copper imbalance has been identified as a contributing factor (201, 202). The phenomenon of cuproptosis may be involved in the pathogenesis of these conditions, given that the homeostatic balance of copper within the brain is essential for sustaining neuronal health (202). Copper plays a critical role in numerous biological processes, including neurotransmission and antioxidant defense mechanisms. The dysregulation of copper levels can result in elevated oxidative stress, a major contributor to neurodegenerative processes (201, 203).
Copper ion imbalance has been observed in AD brains, where bivalent copper could attach to amyloid β (Aβ) protein, leading to the production of ROS and subsequent neuronal damage (204, 205). Copper consumption and environmental copper levels are significant risk factors for PD, whereas the copper chelator ATH434 therapy lowers copper levels to ease motor and olfactory issues in PD patients (202, 206). Moreover, numerous studies have demonstrated that in AD, copper dyshomeostasis is correlated with the accumulation of Aβ plaques and neurofibrillary tangles, which are hallmark features of the disorder (145, 207). In a similar vein, in PD, dysregulated copper levels have been associated with the aggregation of α-synuclein, a protein that constitutes Lewy bodies within affected neurons (146). Furthermore, the interaction between copper and other metals, such as iron and zinc, further complicates the neurodegenerative milieu, as imbalances can intensify oxidative damage and neuronal death (208, 209).
Notably, within the context of SCI, the BSCB integrity is compromised, leading to an immediate increase in its permeability, allowing a large influx of copper ions in the bloodstream pour into the spinal cord tissue and causing a copper overload situation (210, 211). Subsequently, the oxidative stress, inflammatory, and immune responses induced by SCI further disrupt the normal functioning of intracellular copper ion transport proteins, result in an imbalance in copper homeostasis and possibly initiating cuproptosis (131, 211). Nevertheless, the precise moments for the accumulation of copper ions and the onset of cuproptosis post-injury are not yet determined.
Recent research has underscored the potential of targeting copper-related pathways for therapeutic interventions. Specifically, copper-modulating therapies are currently under investigation in clinical trials for AD, with the anticipation that these therapies may also prove beneficial for patients with PD (212, 213). Collectively, the dysregulation of copper within the CNS constitutes a pivotal factor in the onset and advancement of neurodegenerative disorders and SCI. Additional research is imperative to elucidate the specific mechanisms through which copper modulates relevant processes and to investigate potential therapeutic strategies aimed at targeting copper homeostasis and cuproptosis (202, 213).
4 Role of cuproptosis in spinal cord injury
4.1 Cuproptosis and other programmed cell death in SCI
While copper ionophores interacting with copper can induce cuproptosis, previous studies have also found that excessive copper in cells could trigger other cell death pathways, including apoptosis, autophagy, pyroptosis, necroptosis and ferroptosis (Figure 4). This suggests a notable interaction of molecular mechanisms between cuproptosis and other forms of cell death. Moreover, we further elaborated on and compared the key differences in mechanisms for these PCD (Table 2). Comprehensively understanding the role of copper in other cell death pathways may help develop more effective SCI treatment strategies targeting cuproptosis.
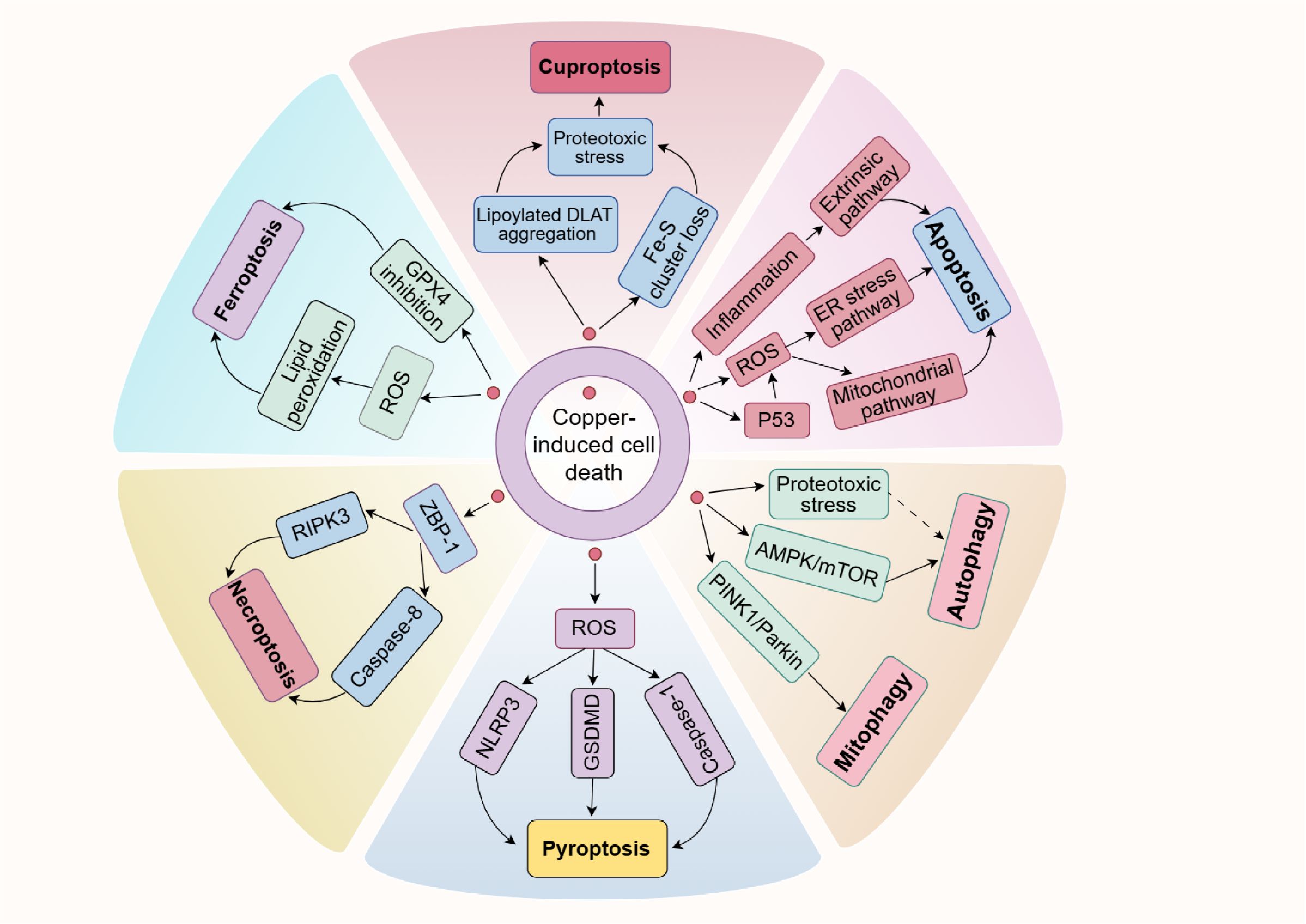
Figure 4. Copper is closely associated with various forms of PCD. On the one hand, an excess of copper can enhance p53 expression and ROS levels or initiate inflammation, causing cell apoptosis. On the other hand, copper exposure increases ROS levels or reduces GPX4 expression, which subsequently leads to ferroptosis or pyroptosis via specific pathways. It has been shown that copper induces necroptosis mediated by ZBP1. Furthermore, copper can also trigger autophagy and mitophagy by inhibiting the AMPK/mTOR pathway or activating the PINK1/Parkin signaling pathway, respectively. It is noteworthy that recent research has shown copper could induce cuproptosis, a newly discovered cell death type, characterized by the accumulation of lipoylated proteins and the depletion of Fe-S clusters. Possible connections are shown with dashed arrows.
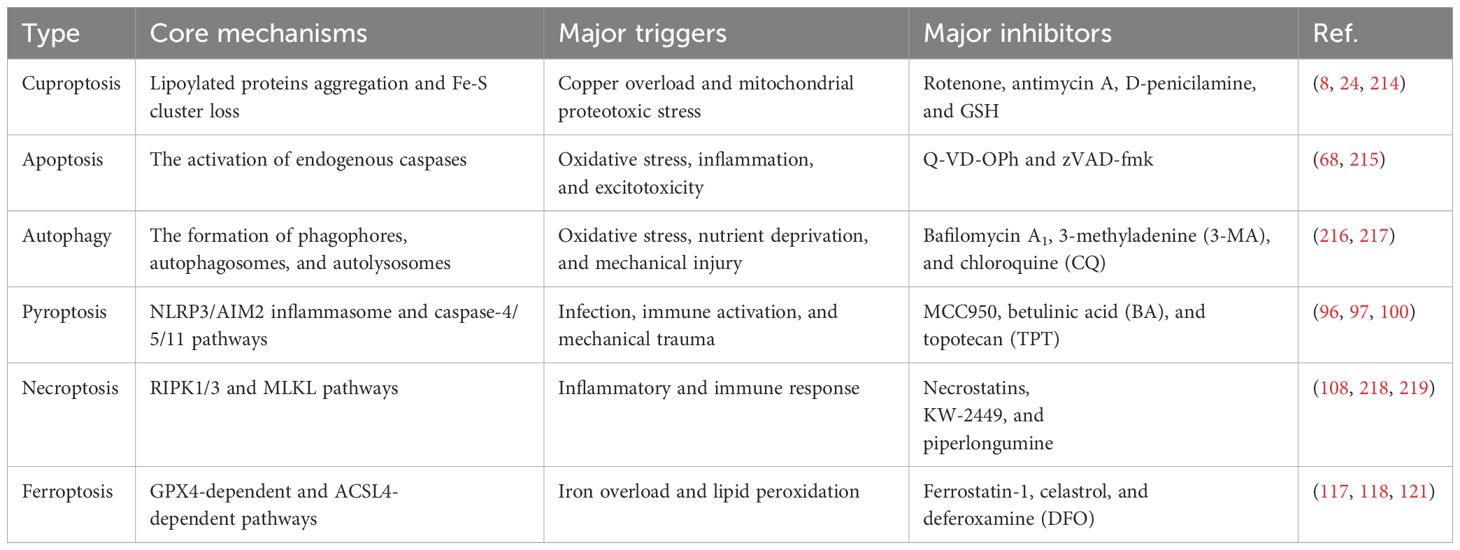
Table 2. The differences of cuproptosis and other PCD.
4.1.1 Link with apoptosis
The crosstalk between cuproptosis and apoptosis constitutes an emerging area of research, particularly in the pathomechanistic continuum of SCI. Previous studies have demonstrated that excessive copper exposure can induce oxidative stress through the overproduction of ROS, subsequently promoting hepatic apoptosis via the mitochondrial pathway or ER stress pathway (64, 220, 221). Specifically, in MCF7 human breast cancer cells, copper ions have been observed to elevate the expression of p53, subsequently inducing the production of ROS and cell apoptosis (222). In this case, p53 may mitigate cuproptosis by regulating the expression of crucial signaling molecules, including Fe-S clusters and the copper chelator GSH (187).
Additionally, excess copper is also implicated in various inflammatory vascular diseases, whereas copper chelators have been shown to inhibit inflammation (223). Inflammation is characterized by the secretion of various pro-inflammatory cytokines, which initiate the extrinsic pathway of apoptosis following SCI (45, 70). Nuclear factor kappa B (NF-κB) is a key activator of inflammatory processes; inhibiting its expression may thus be a promising strategy for treating SCI and other inflammatory diseases (99, 131, 224).
These studies indicate that ROS, p53, and inflammation may mediate the crosstalk between cuproptosis and apoptosis. Within the process of SCI, the pathological milieu is marked by oxidative stress and inflammation, both of which can intensify neuronal damage and impede recovery. By elucidating the mechanisms of this crosstalk, researchers may uncover novel therapeutic targets to enhance recovery and improve the quality of life for individuals affected by SCI.
4.1.2 Link with autophagy
Copper, recognized for its involvement in numerous biological processes, can induce oxidative stress and the ROS levels, potentially eliciting autophagic responses (225, 226). In SCI pathology, autophagy exhibits dual roles, functioning as both protective and detrimental processes. When overactivated, the autophagic machinery can enhance cell death by selectively degrading proteins that protect against damage or oxidative stress (227). However, some studies have shown that autophagy is an essential cytoprotective pathway and blocking autophagy in neurons and oligodendrocytes will limit functional recovery after SCI (74, 228, 229). Furthermore, the interaction between copper concentrations and autophagy may hold substantial significance in the progression of diseases such as neurodegeneration, wherein both cuproptosis and autophagy are integral to cellular survival and function (24, 202, 230). As mentioned above, excessive copper could lead to the TCA and ECT dysfunction, ultimately inducing cellular death mediated by proteotoxic stress (6, 153). Meanwhile, this process may induce autophagy respone to clear or recycle misfolded proteins, thereby mitigating cellular toxicity (72).
The regulation of autophagy in relation to copper is complex and involves multiple signaling pathways. Research has demonstrated that exposure to copper results in liver metabolic issues and suppresses the AMPK/mTOR signaling pathway, which in turn triggers liver autophagy (231). Moreover, copper exposure can induce mitophagy via the PINK1/Parkin pathway in chicken livers, potentially mitigating copper-induced mitochondrial apoptosis (232). Collectively, these studies enhance our understanding of copper-triggered cell death, suggesting that the initiation of cuproptosis may also influence autophagic activity.
4.1.3 Link with pyroptosis
Copper exposure has been implicated in the induction of NLRP3-dependent cellular pyroptosis, which serves as a mediator of inflammation and neurological toxicity (233). In chicken hepatocytes, copper-induced ROS have been shown to activate the NLRP3 inflammasome and promote the expression of caspase-1 protein, subsequently leading to pyroptosis. The application of NAC, a ROS scavenger, and Z-YVAD-FMK, a caspase inhibitor, has been found to mitigate the excessive pyroptosis induced by copper (234). This study suggests that ROS generated by copper accumulation may play a critical role as mediators of pyroptosis. Similarly, Zhou et al. demonstrated that copper exposure elevates oxidative stress and ROS levels in MN9D cells, which subsequently result in the upregulation of GSDMD protein and inflammasome-mediated pyroptosis (235). Additionally, copper ions can interfere with mitochondrial copper balance, leading to Aβ aggregation and increased levels of NLRP3, caspase-1, and GSDMD, which in turn activate pyroptosis in AD cell models (236). The prospective involvement of copper disturbance and cuproptosis in this context introduces novel research opportunities aimed at devising effective therapeutic strategies for managing SCI-associated cell death and facilitating recovery.
4.1.4 Link with necroptosis
Exposure to nano-copper has been shown to induce Z-DNA combined protein-1 (ZBP-1)-mediated necroptosis in the immune organs of chickens, while hesperidin appears to mitigate the toxic effects associated with nano-copper exposure (237). Jiao and colleagues have demonstrated that the activation of ZBP1 results in the recruitment of RIPK3 and caspase-8, which subsequently activate the NLRP3 inflammasome, thereby triggering necroptosis (238). A bioinformatics analysis further suggests a link between ZBP1 and both cuproptosis and necroptosis (239). Moreover, a recent study indicates that copper can activate autophagy via ULK1 and ATG16L1, which in turn may attenuate RIPK3- and MLKL-mediated necroptosis (240). Although there is currently no evidence addressing the interaction between cuproptosis and necroptosis in patients with SCI, these findings imply a potential correlation and crosstalk between copper-exposure-induced cell death and necroptosis signaling pathways.
4.1.5 Link with ferroptosis
Cuproptosis and ferroptosis are metal-associated forms of cell death, characterized by both similarities and differences in their underlying mechanisms, as previously discussed. While earlier research suggested that ferroptosis is exclusively triggered by metal ions, emerging evidence indicates that copper can also promote ferroptosis under certain conditions (24, 241). Gai and colleagues have demonstrated that copper can initiate the ferroptosis process by accumulating lipid peroxides and inhibiting the activity of GPX4 (242). GSH, an essential cofactor for GPX4, directly influences the enzyme’s activity and stability, leading to lipid peroxidation and ferroptosis in cells (113). The accumulation of copper or iron within cells results in increased GSH consumption, thereby initiating cuproptosis and creating a favorable environment for ferroptosis (226). Intriguingly, buthionine sulfoximine (BSO), known as a ferroptosis inhibitor due to its role in reducing GSH synthesis, has also been observed to induce cuproptosis (6).
In the oxidative phosphorylation process, iron plays a critical role in mitochondrial ATP synthase, with numerous Fe-S clusters present in Complex I and Complex II (243). Recent studies has indicated that the depletion of Fe-S cluster proteins can lead to ferroptosis, while the reduction of Fe-S clusters is also a key mechanism in cuproptosis (244). The TCA cycle is fundamental to cellular metabolism, not only facilitating the oxidation of nutrients to generate energy and providing essential metabolites for biosynthetic processes, but also being intricately linked to the ETC in ATP production (245). Blocking the mitochondrial TCA cycle or ETC can reduce cystine-deprivation or erastin-induced ferroptosis, suggesting that the TCA cycle is crucial for ferroptosis (246). Furthermore, the TCA cycle is crucial for cuproptosis, with the aggregation of lipoylated DLAT and the loss of Fe-S clusters proteins being a primary inducer of copper-induced cell death (6). These findings imply that there may be an interaction between cuproptosis and ferroptosis through the TCA cycle and ETC.
An analysis of genomic data across 33 cancer types reveals intricate molecular interactions between the regulators of cuproptosis and ferroptosis at the multiomic level (247). Additionally, both iron and copper have the capacity to elevate ROS levels by promoting GSH depletion or facilitating the Fenton reaction, which is pivotal in elucidating the interplay between ferroptosis and cuproptosis (226). Although cuproptosis is primarily attributed to the aggregation of lipoylated proteins in the TCA cycle rather than ROS generation, it continues to play a crucial role in other forms of copper-triggered cell death, such as ferroptosis and apoptosis (6, 222, 226, 248). To conclude, while cuproptosis and ferroptosis have unique molecular mechanisms and traits, they might influence each other through critical signaling pathways and interactions of protein molecules within the context of SCI.
4.2 Impact of cuproptosis on the spinal cord microenvironment after injury
4.2.1 Inflammatory response
Following SCI, an immediate inflammatory response is initiated, which significantly influences subsequent pathophysiological processes. This response is marked by the recruitment of immune cells, including neutrophils, monocytes, and macrophages, to the site of injury, which plays a crucial role in wound healing and tissue regeneration (40, 249). However, excessive inflammation can exacerbate tissue damage and hinder recovery, thereby contributing to secondary injury (23). Current studies have elucidated the involvement of various cellular mechanisms in the regulation of the inflammatory response. Notably, the chemokine CCL2 has been shown to significantly impact inflammation through the PI3K/Akt signaling pathway, suggesting that interventions targeting this pathway may offer therapeutic benefits in managing inflammation following SCI (250). Additionally, microRNAs, such as miR-182, have been identified as critical regulators of inflammation and apoptosis in neurons post-SCI, indicating that their modulation could enhance recovery outcomes (251).
Li et al. suggest that copper overload may cause heightened polarization of peripheral M2 macrophages and significantly decrease T cell numbers, with cuproptosis possibly affecting inflammatory pathway activation and worsening the immune microenvironment within the injured spinal cord, thereby impacting prognosis (10). The interaction between cuproptosis and traditional inflammatory responses warrants further investigation, as it may uncover novel therapeutic targets for mitigating inflammation and enhancing recovery post-SCI. Moreover, copper exposure could induce the dysfunction of multiple inflammatory-related pathways, such as NF-κB signaling pathway, a crucial regulator of inflammation in SCI, and the inhibition of NF-κB pathway has been associated with improved clinical outcomes (251–253). Investigating the interaction between cuproptosis and these well-established pathways may offer valuable insights into novel strategies for managing inflammation and promoting recovery in patients with SCI.
4.2.2 Glial cell response
Glial cells, encompassing microglia, astrocytes and oligodendrocytes, are integral to the functioning of the spinal cord. Microglia play a critical role in the development of the glial scar post-SCI, and the reduction of microglia disrupts glial scar formation, subsequently increasing parenchymal immune infiltrates, decreasing neuronal and oligodendrocyte survival, and adversely effecting on motor function and prognosis (254). Astrocytes, which are pivotal for maintaining the BSCB and providing neurotrophic support, can undergo reactive gliosis following injury. Although this process initially serves a protective function, it can result in the formation of a glial scar that may ultimately impede axonal regeneration and contribute to secondary damage (255, 256). Studies have shown that astrocytes and microglia are crucial for maintaining copper metabolism. Disruptions in this balance, such as copper accumulation or disease states, can impair neuronal health and promote neurodegenerative processes (230). Furthermore, oligodendrocytes are tasked with the myelination of axons within the CNS. Following SCI, oligodendrocyte progenitor cells (OPCs) undergo proliferation and differentiation to facilitate the remyelination of damaged axons, a process critical for the restoration of neuronal function (257).
Mao and colleagues have shown that Mpeg1 is significantly expressed in microglia after SCI. It may decrease cuproptosis, lessen the inflammatory response during SCI, and potentially protect spinal cord tissue from harm (22). Moreover, the interactions among astrocytes, oligodendrocytes, and microglia are pivotal in the context of immunity. In patients with SCI, DLD, a regulator of cuproptosis, can positively regulate cuproptosis, induce the polarization of M2 macrophages, and ultimately affect the immune microenvironment and prognosis (10).
Although there is scarce experimental evidence directly linking copper overexposure to cuproptosis in neural cells, numerous studies in other cell types and tissues underscore the crucial role of cuproptosis as a mechanism of copper-induced cytotoxicity. Regrettably, it remains unclear whether cuproptosis specifically impacts spinal cord neurons, glial cells, or immune cells due to a lack of cell-type-specific studies after SCI. The dysregulation of these glial responses, due to copper accumulation and cuproptosis, may impair their ability to support neuronal survival and repair, thereby exacerbating neuronal loss and functional impairments (201, 230). Elucidating the mechanisms through which cuproptosis influences the function of neurons, glial cells, and immune cells in SCI is pivotal for the development of targeted therapeutic strategies that enhance neuroprotection and facilitate recovery.
4.3 Therapeutic implications
4.3.1 Modulating cuproptosis as a therapeutic strategy
Given the involvement of cuproptosis in SCI, modulating this process may represent a promising therapeutic strategy. This could involve either inhibiting cuproptosis to prevent excessive cell death or promoting it to eliminate damaged cells that contribute to the pathology of the injury. Recent research highlights the significance of cuproptosis in various neurological conditions, such as stroke, cancer, and neurodegenerative diseases, suggesting that understanding its mechanisms could aid in developing novel treatment strategies for SCI (185, 258, 259).
Within the pathomechanical cascade of SCI, the dysregulation of copper metabolism and cuproptosis may exacerbate neuronal damage and hinder recovery processes. A recent investigation identified that several crucial cuproptosis-related genes (CRGs), such as ATP7A, CP, copper-dependent lysyl oxidase-like 2 (Loxl2), and Phosphodiesterase 3B (PDE3B), exhibit upregulation in neurons following SCI, thereby initiating cuproptosis and neuroinflammation at the central lesion sites. Interestingly, treatment with neurotrophin-3 (NT3)-loaded chitosan markedly suppressed the expression of these genes and facilitated functional recovery in animal models of SCI (260). In addition, among individuals with SCI who do not receive timely intervention, serum copper levels increase within 24 hours, indicating a strong correlation with clinical outcomes (11). Zhou et al. revealed that seven CRGs exhibited distinct expression patterns in SCI patients, with ATP7B, DLD, and SLC31A1 showing increased expression, while DLST, DBT, LIAS, and LIPT1 showed decreased expression (211). These alterations might affect mitochondrial function, oxidative stress, and immune response, potentially hindering the recovery of neurological function in SCI patients. Therefore, targeting cuproptosis offers a potential strategy to restore copper homeostasis and mitigate the detrimental effects of copper overload on neuronal and glial cells. This approach aligns with emerging research emphasizing the importance of cellular and molecular therapies in augmenting neuroprotection and facilitating functional recovery following SCI (10, 22).
Moreover, the investigation into copper ionophores and nanoparticle delivery systems, which have demonstrated potential in cancer therapy, may be repurposed for the treatment of SCI. These approaches may enable the precise modulation of cuproptosis, thereby improving therapeutic efficacy while minimizing potential adverse effects (24, 202). Beyond this, emerging evidence suggests that p53, HSP70 and autophagy-related proteins may cooperatively mitigate cuproptosis by enhancing Fe-S cluster biogenesis, alleviating associated proteotoxic stress, respectively, thereby restoring mitochondrial function. As research advances, it will be crucial to clarify the exact pathways of cuproptosis and how they interact with other cell death processes like apoptosis, autophagy, and ferroptosis to develop comprehensive treatment protocols for SCI (226, 261). To summarize, the modulation of cuproptosis represents a promising new approach for therapeutic intervention in SCI, necessitating further exploration of its underlying mechanisms and potential clinical applications.
4.3.2 Copper - chelating agents
Originally developed for the treatment of WD, copper chelators have recently been investigated as inhibitors of cuproptosis in recent years (24, 262). These agents can decrease the bioavailability of copper, potentially reducing oxidative stress and cellular toxicity linked to high copper levels, which are involved in numerous pathological processes such as neurodegeneration and cell death (8, 263).
Tetrathiomolybdate (TTM) is one of the earliest drugs developed for the treatment of WD, primarily by modulating copper homeostasis (264). Furthermore, research by Kim et al. has demonstrated that in ovarian and endometrial tumors, TTM can downregulate HIF-1α through a mechanism dependent on HIF-prolyl hydroxylase and inhibit the function of the copper-dependent mitochondrial complex IV (265). D-penicillamine, another prominent copper chelator used in the treatment of WD, could also be used as a supplementary treatment in conventional cancer therapies. The potential mechanism involves metal ion-catalyzed H2O2-mediated oxidative stress, which selectively induces cancer cell death and enhances radio- and chemo-sensitization (8, 266). Interestingly, GSH, a natural copper ion chelator, can directly bind to excess intracellular copper, thereby reducing proteotoxic stress and preventing cellular cuproptosis (6).
During SCI progression, the application of copper chelators and specific proteins may restore copper homeostasis, thereby alleviating the detrimental effects associated with copper overload. However, there is no evidence showing that these chelators are applied after SCI at present. Additionally, the potential synergistic effects of combining copper chelators with antioxidants or anti-inflammatory drugs are still speculative and require further experimental validation. Future researches should comprehensively elucidating the molecular mechanisms underlying cuproptosis and utilize specific inhibitors that against it, which might offer valuable insights for the development of targeted therapies for SCI, with the potential to enhance recovery outcomes and mitigate secondary damage.
The interaction between copper metabolism and cellular processes underscores the critical importance of sustaining copper homeostasis and cuproptosis, particularly in conditions such as SCI, where oxidative stress, inflammatory responses and mitochondrial dysfunction are prominent. Future research should aim to elucidate the precise pathways implicated in copper-triggered cell death and explore the potential of chelation therapy to modulate multiple pathways, thereby providing novel therapeutic strategies for neurotrauma.
5 Conclusion
Cuproptosis represents a newly identified type of cell death characterized by distinct pathophysiological mechanisms. Although significant progress has been made in comprehending the intricacies of cuproptosis in recent years, numerous aspects of this process remain incompletely understood. Current research actively explores the role of copper in cellular processes, the molecular components involved in cuproptosis, and the implications of this form of cell death in numerous pathological states. Further research into cuproptosis will not only deepen our comprehension of fundamental biological processes but also hold significant implications for the development of novel therapeutic strategies for a range of diseases. Additionally, cuproptosis may exhibit a complex and multifaceted role in SCI. Elucidating its molecular mechanisms, its interactions with other cell death pathways, its effects on the spinal cord microenvironment, and its therapeutic potential is essential for the advancement of effective treatments for SCI. Future research should aim to further elucidate these aspects of cuproptosis in SCI, with the ultimate objective of improving prognosis and quality of life for individuals affected by SCI.
Author contributions
DX: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. LH: Writing – original draft. JZ: Writing – original draft. XD: Methodology, Validation, Writing – review & editing. YZ: Supervision, Writing – original draft, Writing – review & editing. CL: Conceptualization, Methodology, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The schematic diagrams of this review were drawn by Figdraw.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chen J, Jiang Y, Shi H, Peng Y, Fan X, and Li C. The molecular mechanisms of copper metabolism and its roles in human diseases. Pflugers Archiv: Eur J Physiol. (2020) 472:1415–29. doi: 10.1007/s00424-020-02412-2
2. Harris ED. Copper as a cofactor and regulator of copper,Zinc superoxide dismutase. J Nutr. (1992) 122:636–40. doi: 10.1093/jn/122.suppl_3.636
3. Gale J and Aizenman E. The physiological and pathophysiological roles of copper in the nervous system. Eur J Neurosci. (2024) 60:3505–43. doi: 10.1111/ejn.16370
4. Vetlényi E and Rácz G. the physiological function of copper, the etiological role of copper excess and deficiency. Orvosi hetilap. (2020) 161:1488–96. doi: 10.1556/650.2020.31854
5. Wang Q, Sun J, Chen T, Song S, Hou Y, Feng L, et al. Ferroptosis, pyroptosis, and cuproptosis in alzheimer’s disease. ACS Chem Neurosci. (2023) 14:3564–87. doi: 10.1021/acschemneuro.3c00343
6. Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated tca cycle proteins. Sci (New York NY). (2022) 375:1254–61. doi: 10.1126/science.abf0529
7. Manchia M, Paribello P, Pinna M, and Faa G. The role of copper overload in modulating neuropsychiatric symptoms. Int J Mol Sci. (2024) 25:6487. doi: 10.3390/ijms25126487
8. Bian C, Zheng Z, Su J, Chang S, Yu H, Bao J, et al. Copper homeostasis and cuproptosis in tumor pathogenesis and therapeutic strategies. Front Pharmacol. (2023) 14:1271613. doi: 10.3389/fphar.2023.1271613
9. Fontes A, Pierson H, Bierła JB, Eberhagen C, Kinschel J, Akdogan B, et al. Copper impairs the intestinal barrier integrity in wilson disease. Metabolism: Clin Exp. (2024) 158:155973. doi: 10.1016/j.metabol.2024.155973
10. Li C, Wu C, Ji C, Xu G, Chen J, Zhang J, et al. The pathogenesis of dld-mediated cuproptosis induced spinal cord injury and its regulation on immune microenvironment. Front Cell Neurosci. (2023) 17:1132015. doi: 10.3389/fncel.2023.1132015
11. Seelig J, Heller RA, Hackler J, Haubruck P, Moghaddam A, Biglari B, et al. Selenium and copper status - potential signposts for neurological remission after traumatic spinal cord injury. J Trace elements Med biology: Organ Soc Minerals Trace Elements (GMS). (2020) 57:126415. doi: 10.1016/j.jtemb.2019.126415
12. Yin Z, Wan B, Gong G, and Yin J. Ros: executioner of regulating cell death in spinal cord injury. Front Immunol. (2024) 15:1330678. doi: 10.3389/fimmu.2024.1330678
13. Song Q, Cui Q, Sun S, Wang Y, Yuan Y, and Zhang L. Crosstalk between cell death and spinal cord injury: neurology and therapy. Mol Neurobiol. (2024) 61:10271–87. doi: 10.1007/s12035-024-04188-3
14. Slater PG, Domínguez-Romero ME, Villarreal M, Eisner V, and Larraín J. Mitochondrial function in spinal cord injury and regeneration. Cell Mol Life sciences: CMLS. (2022) 79:239. doi: 10.1007/s00018-022-04261-x
15. Tian Z, Jiang S, Zhou J, and Zhang W. Copper homeostasis and cuproptosis in mitochondria. Life Sci. (2023) 334:122223. doi: 10.1016/j.lfs.2023.122223
16. Wang M, Zheng L, Ma S, Lin R, Li J, and Yang S. Cuproptosis: emerging biomarkers and potential therapeutics in cancers. Front Oncol. (2023) 13:1288504. doi: 10.3389/fonc.2023.1288504
17. Yu T, Yang LL, Zhou Y, Wu MF, and Jiao JH. Exosome-mediated repair of spinal cord injury: A promising therapeutic strategy. Stem Cell Res Ther. (2024) 15:6. doi: 10.1186/s13287-023-03614-y
18. Anjum A, Yazid MD, Fauzi Daud M, Idris J, Ng AMH, Selvi Naicker A, et al. Spinal cord injury: pathophysiology, multimolecular interactions, and underlying recovery mechanisms. Int J Mol Sci. (2020) 21:7533. doi: 10.3390/ijms21207533
19. Xiao CL, Lai HT, Zhou JJ, Liu WY, Zhao M, and Zhao K. Nrf2 signaling pathway: focus on oxidative stress in spinal cord injury. Mol Neurobiol. (2025) 62:2230–49. doi: 10.1007/s12035-024-04394-z
20. McEwen ML, Sullivan PG, Rabchevsky AG, and Springer JE. Targeting mitochondrial function for the treatment of acute spinal cord injury. Neurotherapeutics: J Am Soc Exp Neurother. (2011) 8:168–79. doi: 10.1007/s13311-011-0031-7
21. Salsabili N, Mehrsai AR, and Jalaie S. Concentration of blood and seminal plasma elements and their relationships with semen parameters in men with spinal cord injury. Andrologia. (2009) 41:24–8. doi: 10.1111/j.1439-0272.2008.00885.x
22. Mao D, Chen Q, Tong S, Xu Z, Yu G, Chang C, et al. Integrated bioinformatics analysis identified cuproptosis-related hub gene mpeg1 as potential biomarker in spinal cord injury. Sci Rep. (2025) 15:1993. doi: 10.1038/s41598-025-86170-0
23. Jin Y, Song Y, Lin J, Liu T, Li G, Lai B, et al. Role of inflammation in neurological damage and regeneration following spinal cord injury and its therapeutic implications. Burns Trauma. (2023) 11:tkac054. doi: 10.1093/burnst/tkac054
24. Xue Q, Kang R, Klionsky DJ, Tang D, Liu J, and Chen X. Copper metabolism in cell death and autophagy. Autophagy. (2023) 19:2175–95. doi: 10.1080/15548627.2023.2200554
25. Tian T, Zhang S, and Yang M. Recent progress and challenges in the treatment of spinal cord injury. Protein Cell. (2023) 14:635–52. doi: 10.1093/procel/pwad003
26. Alcántar-Garibay OV, Incontri-Abraham D, and Ibarra A. Spinal cord injury-induced cognitive impairment: A narrative review. Neural regeneration Res. (2022) 17:2649–54. doi: 10.4103/1673-5374.339475
27. Grossman SD, Rosenberg LJ, and Wrathall JR. Temporal-spatial pattern of acute neuronal and glial loss after spinal cord contusion. Exp Neurol. (2001) 168:273–82. doi: 10.1006/exnr.2001.7628
28. Muradov JM, Ewan EE, and Hagg T. Dorsal column sensory axons degenerate due to impaired microvascular perfusion after spinal cord injury in rats. Exp Neurol. (2013) 249:59–73. doi: 10.1016/j.expneurol.2013.08.009
29. Mautes AE, Weinzierl MR, Donovan F, and Noble LJ. Vascular events after spinal cord injury: contribution to secondary pathogenesis. Phys Ther. (2000) 80:673–87. doi: 10.1093/ptj/80.7.673
30. Pang QM, Chen SY, Fu SP, Zhou H, Zhang Q, Ao J, et al. Regulatory role of mesenchymal stem cells on secondary inflammation in spinal cord injury. J Inflammation Res. (2022) 15:573–93. doi: 10.2147/jir.S349572
31. Fan H, Tang HB, Chen Z, Wang HQ, Zhang L, Jiang Y, et al. Inhibiting hmgb1-rage axis prevents pro-inflammatory macrophages/microglia polarization and affords neuroprotection after spinal cord injury. J Neuroinflamm. (2020) 17:295. doi: 10.1186/s12974-020-01973-4
32. Fatima G, Sharma VP, Das SK, and Mahdi AA. Oxidative stress and antioxidative parameters in patients with spinal cord injury: implications in the pathogenesis of disease. Spinal cord. (2015) 53:3–6. doi: 10.1038/sc.2014.178
33. Visavadiya NP, Patel SP, VanRooyen JL, Sullivan PG, and Rabchevsky AG. Cellular and subcellular oxidative stress parameters following severe spinal cord injury. Redox Biol. (2016) 8:59–67. doi: 10.1016/j.redox.2015.12.011
34. Sohn HM, Hwang JY, Ryu JH, Kim J, Park S, Park JW, et al. Simvastatin Protects Ischemic Spinal Cord Injury from Cell Death and Cytotoxicity through Decreasing Oxidative Stress: In Vitro Primary Cultured Rat Spinal Cord Model under Oxygen and Glucose Deprivation-Reoxygenation Conditions. J orthopaedic Surg Res. (2017) 12:36. doi: 10.1186/s13018-017-0536-9
35. Dimitrijevic MR, Danner SM, and Mayr W. Neurocontrol of movement in humans with spinal cord injury. Artif organs. (2015) 39:823–33. doi: 10.1111/aor.12614
36. Bock FJ and Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. (2020) 21:85–100. doi: 10.1038/s41580-019-0173-8
37. Li Q, Gao S, Kang Z, Zhang M, Zhao X, Zhai Y, et al. Rapamycin enhances mitophagy and attenuates apoptosis after spinal ischemia-reperfusion injury. Front Neurosci. (2018) 12:865. doi: 10.3389/fnins.2018.00865
38. Yao S, Pang M, Wang Y, Wang X, Lin Y, Lv Y, et al. Mesenchymal stem cell attenuates spinal cord injury by inhibiting mitochondrial quality control-associated neuronal ferroptosis. Redox Biol. (2023) 67:102871. doi: 10.1016/j.redox.2023.102871
39. Wang JL, Luo X, and Liu L. Targeting card6 attenuates spinal cord injury (Sci) in mice through inhibiting apoptosis, inflammation and oxidative stress associated ros production. Aging. (2019) 11:12213–35. doi: 10.18632/aging.102561
40. Anwar MA, Al Shehabi TS, and Eid AH. Inflammogenesis of secondary spinal cord injury. Front Cell Neurosci. (2016) 10:98. doi: 10.3389/fncel.2016.00098
41. Jia Z, Zhu H, Li J, Wang X, Misra H, and Li Y. Oxidative stress in spinal cord injury and antioxidant-based intervention. Spinal cord. (2012) 50:264–74. doi: 10.1038/sc.2011.111
42. Hellenbrand DJ, Quinn CM, Piper ZJ, Morehouse CN, Fixel JA, and Hanna AS. Inflammation after spinal cord injury: A review of the critical timeline of signaling cues and cellular infiltration. J Neuroinflamm. (2021) 18:284. doi: 10.1186/s12974-021-02337-2
43. Zhang Y, Yang S, Liu C, Han X, Gu X, and Zhou S. Deciphering glial scar after spinal cord injury. Burns Trauma. (2021) 9:tkab035. doi: 10.1093/burnst/tkab035
44. Mishra RR, Nielsen BE, Trudrung MA, Lee S, Bolstad LJ, Hellenbrand DJ, et al. The effect of tissue inhibitor of metalloproteinases on scar formation after spinal cord injury. Cells. (2024) 13:1547. doi: 10.3390/cells13181547
45. Bai Y, Guo N, Xu Z, Chen Y, Zhang W, Chen Q, et al. S100a1 expression is increased in spinal cord injury and promotes inflammation, oxidative stress and apoptosis of pc12 cells induced by lps via erk signaling. Mol Med Rep. (2023) 27:30. doi: 10.3892/mmr.2022.12917
46. Donnelly DJ and Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol. (2008) 209:378–88. doi: 10.1016/j.expneurol.2007.06.009
47. Pelisch N, Rosas Almanza J, Stehlik KE, Aperi BV, and Kroner A. Ccl3 contributes to secondary damage after spinal cord injury. J Neuroinflamm. (2020) 17:362. doi: 10.1186/s12974-020-02037-3
48. Hu J, Yang Z, Li X, and Lu H. C-C motif chemokine ligand 20 regulates neuroinflammation following spinal cord injury via th17 cell recruitment. J Neuroinflamm. (2016) 13:162. doi: 10.1186/s12974-016-0630-7
49. Lu ZJ, Pan QL, and Lin FX. Epigenetic modifications of inflammation in spinal cord injury. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2024) 179:117306. doi: 10.1016/j.biopha.2024.117306
50. Arnold FJ, Putka AF, Raychaudhuri U, Hsu S, Bedlack RS, Bennett CL, et al. Revisiting glutamate excitotoxicity in amyotrophic lateral sclerosis and age-related neurodegeneration. Int J Mol Sci. (2024) 25:5587. doi: 10.3390/ijms25115587
51. Park E, Velumian AA, and Fehlings MG. The role of excitotoxicity in secondary mechanisms of spinal cord injury: A review with an emphasis on the implications for white matter degeneration. J neurotrauma. (2004) 21:754–74. doi: 10.1089/0897715041269641
52. Han P and Whelan PJ. Tumor necrosis factor alpha enhances glutamatergic transmission onto spinal motoneurons. J neurotrauma. (2010) 27:287–92. doi: 10.1089/neu.2009.1016
53. Springer JE, Rao RR, Lim HR, Cho SI, Moon GJ, Lee HY, et al. The functional and neuroprotective actions of neu2000, a dual-acting pharmacological agent, in the treatment of acute spinal cord injury. J neurotrauma. (2010) 27:139–49. doi: 10.1089/neu.2009.0952
54. Goldshmit Y, Jona G, Schmukler E, Solomon S, Pinkas-Kramarski R, and Ruban A. Blood glutamate scavenger as a novel neuroprotective treatment in spinal cord injury. J neurotrauma. (2018) 35:2581–90. doi: 10.1089/neu.2017.5524
55. Xu GY, Hughes MG, Ye Z, Hulsebosch CE, and McAdoo DJ. Concentrations of glutamate released following spinal cord injury kill oligodendrocytes in the spinal cord. Exp Neurol. (2004) 187:329–36. doi: 10.1016/j.expneurol.2004.01.029
56. Yonutas HM, Hubbard WB, Pandya JD, Vekaria HJ, Geldenhuys WJ, and Sullivan PG. Bioenergetic restoration and neuroprotection after therapeutic targeting of mitoneet: new mechanism of pioglitazone following traumatic brain injury. Exp Neurol. (2020) 327:113243. doi: 10.1016/j.expneurol.2020.113243
57. Lima R, Gomes ED, Cibrão JR, Rocha LA, Assunção-Silva RC, Rodrigues CS, et al. Levetiracetam treatment leads to functional recovery after thoracic or cervical injuries of the spinal cord. NPJ Regenerative Med. (2021) 6:11. doi: 10.1038/s41536-021-00121-7
58. Vaglio-Garro A, Kozlov AV, Smirnova YD, and Weidinger A. Pathological interplay between inflammation and mitochondria aggravates glutamate toxicity. Int J Mol Sci. (2024) 25:2276. doi: 10.3390/ijms25042276
59. Shi Z, Yuan S, Shi L, Li J, Ning G, Kong X, et al. Programmed cell death in spinal cord injury pathogenesis and therapy. Cell proliferation. (2021) 54:e12992. doi: 10.1111/cpr.12992
60. Lin FX, Pan QL, Gu HY, Zeng FJ, and Lu ZJ. The role of resveratrol on spinal cord injury: from bench to bedside. Mol Neurobiol. (2024) 61:104–19. doi: 10.1007/s12035-023-03558-7
61. He W, Li ZQ, Gu HY, Pan QL, and Lin FX. Targeted therapy of spinal cord injury: inhibition of apoptosis is a promising therapeutic strategy. Mol Neurobiol. (2024) 61:4222–39. doi: 10.1007/s12035-023-03814-w
62. Bi Y, Chen X, Cao Y, Yu D, Zhao J, Jing Y, et al. Nuclear heme oxidase-1 inhibits endoplasmic reticulum stress-mediated apoptosis after spinal cord injury. BioMed Res Int. (2020) 2020:7576063. doi: 10.1155/2020/7576063
63. Lu J, Ashwell KW, and Waite P. Advances in secondary spinal cord injury: role of apoptosis. Spine. (2000) 25:1859–66. doi: 10.1097/00007632-200007150-00022
64. Andrabi SS, Yang J, Gao Y, Kuang Y, and Labhasetwar V. Nanoparticles with antioxidant enzymes protect injured spinal cord from neuronal cell apoptosis by attenuating mitochondrial dysfunction. J Controlled release: Off J Controlled Release Soc. (2020) 317:300–11. doi: 10.1016/j.jconrel.2019.12.001
65. Sobrido-Cameán D and Barreiro-Iglesias A. Role of caspase-8 and fas in cell death after spinal cord injury. Front Mol Neurosci. (2018) 11:101. doi: 10.3389/fnmol.2018.00101
66. Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, and Kroemer G. Mechanisms of cytochrome C release from mitochondria. Cell Death differentiation. (2006) 13:1423–33. doi: 10.1038/sj.cdd.4401950
67. Martin DA and Elkon KB. Mechanisms of apoptosis. Rheumatic Dis Clinics North America. (2004) 30:441–54. doi: 10.1016/j.rdc.2004.04.008
68. Austin JW and Fehlings MG. Molecular mechanisms of fas-mediated cell death in oligodendrocytes. J neurotrauma. (2008) 25:411–26. doi: 10.1089/neu.2007.0436
69. Yao H, Cai C, Huang W, Zhong C, Zhao T, Di J, et al. Enhancing mitophagy by ligustilide through bnip3-lc3 interaction attenuates oxidative stress-induced neuronal apoptosis in spinal cord injury. Int J Biol Sci. (2024) 20:4382–406. doi: 10.7150/ijbs.98051
70. Zhou X, Chu X, Yuan H, Qiu J, Zhao C, Xin D, et al. Mesenchymal stem cell derived evs mediate neuroprotection after spinal cord injury in rats via the microrna-21-5p/fasl gene axis. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2019) 115:108818. doi: 10.1016/j.biopha.2019.108818
71. Zhang H, Wu F, Kong X, Yang J, Chen H, Deng L, et al. Nerve growth factor improves functional recovery by inhibiting endoplasmic reticulum stress-induced neuronal apoptosis in rats with spinal cord injury. J Trans Med. (2014) 12:130. doi: 10.1186/1479-5876-12-130
72. Ray SK. Modulation of autophagy for neuroprotection and functional recovery in traumatic spinal cord injury. Neural regeneration Res. (2020) 15:1601–12. doi: 10.4103/1673-5374.276322
73. Li W, Yang Q, and Mao Z. Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mol Life sciences: CMLS. (2011) 68:749–63. doi: 10.1007/s00018-010-0565-6
74. Saraswat Ohri S, Bankston AN, Mullins SA, Liu Y, Andres KR, Beare JE, et al. Blocking autophagy in oligodendrocytes limits functional recovery after spinal cord injury. J neuroscience: Off J Soc Neurosci. (2018) 38:5900–12. doi: 10.1523/jneurosci.0679-17.2018
75. Yin J, Yin Z, Wang B, Zhu C, Sun C, Liu X, et al. Angiopoietin-1 protects spinal cord ischemia and reperfusion injury by inhibiting autophagy in rats. Neurochemical Res. (2019) 44:2746–54. doi: 10.1007/s11064-019-02893-3
76. Liao HY, Wang ZQ, Ran R, Zhou KS, Ma CW, and Zhang HH. Biological functions and therapeutic potential of autophagy in spinal cord injury. Front Cell Dev Biol. (2021) 9:761273. doi: 10.3389/fcell.2021.761273
77. Du Y and Cai X. Therapeutic potential of natural compounds from herbs and nutraceuticals in spinal cord injury: regulation of the mtor signaling pathway. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2023) 163:114905. doi: 10.1016/j.biopha.2023.114905
78. Meng HY, Shao DC, Li H, Huang XD, Yang G, Xu B, et al. Resveratrol improves neurological outcome and neuroinflammation following spinal cord injury through enhancing autophagy involving the ampk/mtor pathway. Mol Med Rep. (2018) 18:2237–44. doi: 10.3892/mmr.2018.9194
79. Popelka H and Klionsky DJ. One step closer to understanding mammalian macroautophagy initiation: interplay of 2 horma architectures in the ulk1 complex. Autophagy. (2015) 11:1953–5. doi: 10.1080/15548627.2015.1087635
80. Wu C, Chen Y, Chen X, Zhang Y, Zhao X, Deng Y, et al. 20-Deoxyingenol Activates Mitophagy through Tfeb and Promotes Functional Recovery after Spinal Cord Injury. Mol Neurobiol. (2025) 62:445–60. doi: 10.1007/s12035-024-04283-5
81. Shefa U, Jeong NY, Song IO, Chung HJ, Kim D, Jung J, et al. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural regeneration Res. (2019) 14:749–56. doi: 10.4103/1673-5374.249218
82. Sekiguchi A, Kanno H, Ozawa H, Yamaya S, and Itoi E. Rapamycin promotes autophagy and reduces neural tissue damage and locomotor impairment after spinal cord injury in mice. J neurotrauma. (2012) 29:946–56. doi: 10.1089/neu.2011.1919
83. Cordaro M, Paterniti I, Siracusa R, Impellizzeri D, Esposito E, and Cuzzocrea S. Ku0063794, a dual mtorc1 and mtorc2 inhibitor, reduces neural tissue damage and locomotor impairment after spinal cord injury in mice. Mol Neurobiol. (2017) 54:2415–27. doi: 10.1007/s12035-016-9827-0
84. Bai L, Mei X, Shen Z, Bi Y, Yuan Y, Guo Z, et al. Netrin-1 improves functional recovery through autophagy regulation by activating the ampk/mtor signaling pathway in rats with spinal cord injury. Sci Rep. (2017) 7:42288. doi: 10.1038/srep42288
85. D’Souza CA and Heitman J. Dismantling the cryptococcus coat. Trends Microbiol. (2001) 9:112–3. doi: 10.1016/s0966-842x(00)01945-4
86. Al Mamun A, Wu Y, Monalisa I, Jia C, Zhou K, Munir F, et al. Role of pyroptosis in spinal cord injury and its therapeutic implications. J advanced Res. (2021) 28:97–109. doi: 10.1016/j.jare.2020.08.004
87. Yin J, Gong G, Wan W, and Liu X. Pyroptosis in spinal cord injury. Front Cell Neurosci. (2022) 16:949939. doi: 10.3389/fncel.2022.949939
88. Yin TC, Li YC, Sung PH, Chiang JY, Shao PL, Yip HK, et al. Adipose-derived mesenchymal stem cells overexpressing prion improve outcomes via the nlrp3 inflammasome/damp signalling after spinal cord injury in rat. J Cell Mol Med. (2023) 27:482–95. doi: 10.1111/jcmm.17620
89. Broz P and Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
90. Kelley N, Jeltema D, Duan Y, and He Y. The nlrp3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20:3328. doi: 10.3390/ijms20133328
91. Gu HY and Liu N. Mechanism of effect and therapeutic potential of nlrp3 inflammasome in spinal cord injury. Exp Neurol. (2025) 384:115059. doi: 10.1016/j.expneurol.2024.115059
92. Mortezaee K, Khanlarkhani N, Beyer C, and Zendedel A. Inflammasome: its role in traumatic brain and spinal cord injury. J Cell Physiol. (2018) 233:5160–9. doi: 10.1002/jcp.26287
93. Burdette BE, Esparza AN, Zhu H, and Wang S. Gasdermin D in pyroptosis. Acta Pharm Sin B. (2021) 11:2768–82. doi: 10.1016/j.apsb.2021.02.006
94. Wang B and Yin Q. Aim2 inflammasome activation and regulation: A structural perspective. J Struct Biol. (2017) 200:279–82. doi: 10.1016/j.jsb.2017.08.001
95. Hu X, Chen H, Xu H, Wu Y, Wu C, Jia C, et al. Role of pyroptosis in traumatic brain and spinal cord injuries. Int J Biol Sci. (2020) 16:2042–50. doi: 10.7150/ijbs.45467
96. Jiao J, Zhao G, Wang Y, Ren P, and Wu M. Mcc950, a selective inhibitor of nlrp3 inflammasome, reduces the inflammatory response and improves neurological outcomes in mice model of spinal cord injury. Front Mol Biosci. (2020) 7:37. doi: 10.3389/fmolb.2020.00037
97. Jiang W, He F, Ding G, and Wu J. Topotecan reduces neuron death after spinal cord injury by suppressing caspase-1-dependent pyroptosis. Mol Neurobiol. (2022) 59:6033–48. doi: 10.1007/s12035-022-02960-x
98. Dai W, Wang X, Teng H, Li C, Wang B, and Wang J. Celastrol inhibits microglial pyroptosis and attenuates inflammatory reaction in acute spinal cord injury rats. Int Immunopharmacol. (2019) 66:215–23. doi: 10.1016/j.intimp.2018.11.029
99. Liu Z, Yao X, Sun B, Jiang W, Liao C, Dai X, et al. Pretreatment with kaempferol attenuates microglia-mediate neuroinflammation by inhibiting mapks-nf-Kb signaling pathway and pyroptosis after secondary spinal cord injury. Free Radical Biol Med. (2021) 168:142–54. doi: 10.1016/j.freeradbiomed.2021.03.037
100. Wu C, Chen H, Zhuang R, Zhang H, Wang Y, Hu X, et al. Betulinic acid inhibits pyroptosis in spinal cord injury by augmenting autophagy via the ampk-mtor-tfeb signaling pathway. Int J Biol Sci. (2021) 17:1138–52. doi: 10.7150/ijbs.57825
101. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. (2005) 1:112–9. doi: 10.1038/nchembio711
102. Hu X, Xu Y, Zhang H, Li Y, Wang X, Xu C, et al. Role of necroptosis in traumatic brain and spinal cord injuries. J advanced Res. (2022) 40:125–34. doi: 10.1016/j.jare.2021.12.002
103. Liu S, Li Y, Choi HMC, Sarkar C, Koh EY, Wu J, et al. Lysosomal damage after spinal cord injury causes accumulation of ripk1 and ripk3 proteins and potentiation of necroptosis. Cell Death Dis. (2018) 9:476. doi: 10.1038/s41419-018-0469-1
104. Newton K. Ripk1 and ripk3: critical regulators of inflammation and cell death. Trends Cell Biol. (2015) 25:347–53. doi: 10.1016/j.tcb.2015.01.001
105. Jiao J, Wang Y, Ren P, Sun S, and Wu M. Necrosulfonamide ameliorates neurological impairment in spinal cord injury by improving antioxidative capacity. Front Pharmacol. (2019) 10:1538. doi: 10.3389/fphar.2019.01538
106. Shao L, Liu X, Zhu S, Liu C, Gao Y, and Xu X. The role of smurf1 in neuronal necroptosis after lipopolysaccharide-induced neuroinflammation. Cell Mol Neurobiol. (2018) 38:809–16. doi: 10.1007/s10571-017-0553-6
107. Zhao H, Zong X, Li L, Li N, Liu C, Zhang W, et al. Electroacupuncture inhibits neuroinflammation induced by astrocytic necroptosis through rip1/mlkl/tlr4 pathway in a mouse model of spinal cord injury. Mol Neurobiol. (2024) 61:3258–71. doi: 10.1007/s12035-023-03650-y
108. Fan H, Zhang K, Shan L, Kuang F, Chen K, Zhu K, et al. Reactive astrocytes undergo M1 microglia/macrohpages-induced necroptosis in spinal cord injury. Mol neurodegeneration. (2016) 11:14. doi: 10.1186/s13024-016-0081-8
109. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
110. Hu X, Xu Y, Xu H, Jin C, Zhang H, Su H, et al. Progress in understanding ferroptosis and its targeting for therapeutic benefits in traumatic brain and spinal cord injuries. Front Cell Dev Biol. (2021) 9:705786. doi: 10.3389/fcell.2021.705786
111. Cao JY and Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life sciences: CMLS. (2016) 73:2195–209. doi: 10.1007/s00018-016-2194-1
112. Ryan F, Blex C, Ngo TD, Kopp MA, Michalke B, Venkataramani V, et al. Ferroptosis inhibitor improves outcome after early and delayed treatment in mild spinal cord injury. Acta neuropathologica. (2024) 147:106. doi: 10.1007/s00401-024-02758-2
113. Forcina GC and Dixon SJ. Gpx4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics. (2019) 19:e1800311. doi: 10.1002/pmic.201800311
114. Jia B, Li J, Song Y, and Luo C. Acsl4-mediated ferroptosis and its potential role in central nervous system diseases and injuries. Int J Mol Sci. (2023) 24:10021. doi: 10.3390/ijms241210021
115. Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol. (2021) 220:e202105043. doi: 10.1083/jcb.202105043
116. Wang H, Liu C, Zhao Y, and Gao G. Mitochondria regulation in ferroptosis. Eur J Cell Biol. (2020) 99:151058. doi: 10.1016/j.ejcb.2019.151058
117. Ge H, Xue X, Xian J, Yuan L, Wang L, Zou Y, et al. Ferrostatin-1 alleviates white matter injury via decreasing ferroptosis following spinal cord injury. Mol Neurobiol. (2022) 59:161–76. doi: 10.1007/s12035-021-02571-y
118. Shen W, Li C, Liu Q, Cai J, Wang Z, Pang Y, et al. Celastrol inhibits oligodendrocyte and neuron ferroptosis to promote spinal cord injury recovery. Phytomedicine: Int J phytotherapy phytopharmacology. (2024) 128:155380. doi: 10.1016/j.phymed.2024.155380
119. Zhu R, Kang Y, Li Q, Peng K, Shi X, Yin Z, et al. Alpha-tocopherol inhibits ferroptosis and promotes neural function recovery in rats with spinal cord injury via downregulating alox15. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2024) 175:116734. doi: 10.1016/j.biopha.2024.116734
120. Feng Z, Min L, Chen H, Deng W, Tan M, Liu H, et al. Iron overload in the motor cortex induces neuronal ferroptosis following spinal cord injury. Redox Biol. (2021) 43:101984. doi: 10.1016/j.redox.2021.101984
121. Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX, Sun C, et al. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural regeneration Res. (2019) 14:532–41. doi: 10.4103/1673-5374.245480
122. Pan C, Ji Z, Wang Q, Zhang Z, Wang Z, Li C, et al. Cuproptosis: mechanisms, biological significance, and advances in disease treatment-a systematic review. CNS Neurosci Ther. (2024) 30:e70039. doi: 10.1111/cns.70039
123. Zhu Z, Song M, Ren J, Liang L, Mao G, and Chen M. Copper homeostasis and cuproptosis in central nervous system diseases. Cell Death Dis. (2024) 15:850. doi: 10.1038/s41419-024-07206-3
124. Bost M, Houdart S, Oberli M, Kalonji E, Huneau JF, and Margaritis I. Dietary copper and human health: current evidence and unresolved issues. J Trace elements Med biology: Organ Soc Minerals Trace Elements (GMS). (2016) 35:107–15. doi: 10.1016/j.jtemb.2016.02.006
125. Lönnerdal B. Intestinal regulation of copper homeostasis: A developmental perspective. Am J Clin Nutr. (2008) 88:846s–50s. doi: 10.1093/ajcn/88.3.846S
126. Ohgami RS, Campagna DR, McDonald A, and Fleming MD. The steap proteins are metalloreductases. Blood. (2006) 108:1388–94. doi: 10.1182/blood-2006-02-003681
127. Liang ZD, Tsai WB, Lee MY, Savaraj N, and Kuo MT. Specificity protein 1 (Sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol Pharmacol. (2012) 81:455–64. doi: 10.1124/mol.111.076422
128. Lv X, Zhao L, Song Y, Chen W, and Tuo Q. Deciphering the role of copper homeostasis in atherosclerosis: from molecular mechanisms to therapeutic targets. Int J Mol Sci. (2024) 25:11462. doi: 10.3390/ijms252111462
129. Schoeberl A, Gutmann M, Theiner S, Corte-Rodríguez M, Braun G, Vician P, et al. The copper transporter ctr1 and cisplatin accumulation at the single-cell level by la-icp-tofms. Front Mol Biosci. (2022) 9:1055356. doi: 10.3389/fmolb.2022.1055356
130. Lutsenko S. Human copper homeostasis: A network of interconnected pathways. Curr Opin Chem Biol. (2010) 14:211–7. doi: 10.1016/j.cbpa.2010.01.003
131. An Y, Li S, Huang X, Chen X, Shan H, and Zhang M. The role of copper homeostasis in brain disease. Int J Mol Sci. (2022) 23:13850. doi: 10.3390/ijms232213850
132. Kawamata H and Manfredi G. Import, maturation, and function of sod1 and its copper chaperone ccs in the mitochondrial intermembrane space. Antioxidants Redox Signaling. (2010) 13:1375–84. doi: 10.1089/ars.2010.3212
133. Fujii J, Homma T, and Osaki T. Superoxide radicals in the execution of cell death. Antioxidants (Basel Switzerland). (2022) 11:501. doi: 10.3390/antiox11030501
134. Ren X, Zhao L, Hao Y, Huang X, Lv G, and Zhou X. Copper-instigated modulatory cell mortality mechanisms and progress in kidney diseases. Renal failure. (2025) 47:2431142. doi: 10.1080/0886022x.2024.2431142
135. Nývltová E, Dietz JV, Seravalli J, Khalimonchuk O, and Barrientos A. Coordination of metal center biogenesis in human cytochrome C oxidase. Nat Commun. (2022) 13:3615. doi: 10.1038/s41467-022-31413-1
136. Lenartowicz M and Krzeptowski W. Structure and function of atp7a and atp7b proteins–cu-transporting atpases. Postepy biochemii. (2010) 56:317–27.
137. Han J, Luo J, Wang C, Kapilevich L, and Zhang XA. Roles and mechanisms of copper homeostasis and cuproptosis in osteoarticular diseases. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2024) 174:116570. doi: 10.1016/j.biopha.2024.116570
138. Morrell A, Tallino S, Yu L, and Burkhead JL. The role of insufficient copper in lipid synthesis and fatty-liver disease. IUBMB Life. (2017) 69:263–70. doi: 10.1002/iub.1613
139. Zhou Y and Zhang L. The interplay between copper metabolism and microbes: in perspective of host copper-dependent atpases atp7a/B. Front Cell infection Microbiol. (2023) 13:1267931. doi: 10.3389/fcimb.2023.1267931
140. Zhu Z, Zhu K, Zhang J, Zhou Y, and Zhang Q. Elucidating the evolving role of cuproptosis in breast cancer progression. Int J Biol Sci. (2024) 20:4872–87. doi: 10.7150/ijbs.98806
141. Culbertson EM and Culotta VC. Copper in infectious disease: using both sides of the penny. Semin Cell Dev Biol. (2021) 115:19–26. doi: 10.1016/j.semcdb.2020.12.003
142. Wang L, ChenLiu Z, Wang D, and Tang D. Cross-talks of gsh, mitochondria, rna M6a modification, nrf2, and P53 between ferroptosis and cuproptosis in hcc: A review. Int J Biol macromolecules. (2025) 302:140523. doi: 10.1016/j.ijbiomac.2025.140523
143. Liu Y, Shen M, Zhu S, Du Z, Lin L, Ma J, et al. Metallothionein rescues doxorubicin cardiomyopathy via mitigation of cuproptosis. Life Sci. (2025) 363:123379. doi: 10.1016/j.lfs.2025.123379
144. Asthana A, Bollapalli M, Tangirala R, Bakthisaran R, and Mohan Rao C. Hsp27 suppresses the cu(2+)-induced amyloidogenicity, redox activity, and cytotoxicity of A-synuclein by metal ion stripping. Free Radical Biol Med. (2014) 72:176–90. doi: 10.1016/j.freeradbiomed.2014.04.012
145. Sabalic A, Mei V, Solinas G, and Madeddu R. The role of copper in alzheimer’s disease etiopathogenesis: an updated systematic review. Toxics. (2024) 12:755. doi: 10.3390/toxics12100755
146. Choi TS, Lee J, Han JY, Jung BC, Wongkongkathep P, Loo JA, et al. Supramolecular modulation of structural polymorphism in pathogenic A-synuclein fibrils using copper(Ii) coordination. Angewandte Chemie (International ed English). (2018) 57:3099–103. doi: 10.1002/anie.201712286
147. Lutsenko S, Roy S, and Tsvetkov P. Mammalian copper homeostasis: physiological roles and molecular mechanisms. Physiol Rev. (2025) 105:441–91. doi: 10.1152/physrev.00011.2024
148. Członkowska A, Litwin T, Dusek P, Ferenci P, Lutsenko S, Medici V, et al. Wilson disease. Nat Rev Dis Primers. (2018) 4:21. doi: 10.1038/s41572-018-0018-3
149. Tsui KH, Hsiao JH, Lin LT, Tsang YL, Shao AN, Kuo CH, et al. The cross-communication of cuproptosis and regulated cell death in human pathophysiology. Int J Biol Sci. (2024) 20:218–30. doi: 10.7150/ijbs.84733
150. Xie J, Yang Y, Gao Y, and He J. Cuproptosis: mechanisms and links with cancers. Mol Cancer. (2023) 22:46. doi: 10.1186/s12943-023-01732-y
151. Halliwell B and Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. (1984) 219:1–14. doi: 10.1042/bj2190001
152. Rowland EA, Snowden CK, and Cristea IM. Protein lipoylation: an evolutionarily conserved metabolic regulator of health and disease. Curr Opin Chem Biol. (2018) 42:76–85. doi: 10.1016/j.cbpa.2017.11.003
153. Wang W, Mo W, Hang Z, Huang Y, Yi H, Sun Z, et al. Cuproptosis: harnessing transition metal for cancer therapy. ACS nano. (2023) 17:19581–99. doi: 10.1021/acsnano.3c07775
154. Zhang L, Deng R, Guo R, Jiang Y, Guan Y, Chen C, et al. Recent progress of methods for cuproptosis detection. Front Mol Biosci. (2024) 11:1460987. doi: 10.3389/fmolb.2024.1460987
155. Santiago AM, Gonçalves DL, and Morano KA. Mechanisms of sensing and response to proteotoxic stress. Exp Cell Res. (2020) 395:112240. doi: 10.1016/j.yexcr.2020.112240
156. Li Q, Wang F, Shi L, Tang Q, Li B, Wang X, et al. Nanotrains of DNA copper nanoclusters that triggered a cascade fenton-like reaction and glutathione depletion to doubly enhance chemodynamic therapy. ACS Appl materials interfaces. (2022) 14:37280–90. doi: 10.1021/acsami.2c05944
157. Song L, Zeng R, Yang K, Liu W, Xu Z, and Kang F. The biological significance of cuproptosis-key gene mtf1 in pan-cancer and its inhibitory effects on ros-mediated cell death of liver hepatocellular carcinoma. Discover Oncol. (2023) 14:113. doi: 10.1007/s12672-023-00738-8
158. Che J, Yang X, Zhao X, Li Y, Jin Z, and Xu C. Risk factor prediction and immune correlation analysis of cuproptosis-related gene in osteoarthritis. J Cell Mol Med. (2024) 28:e18574. doi: 10.1111/jcmm.18574
159. Zhao R, Sukocheva O, Tse E, Neganova M, Aleksandrova Y, Zheng Y, et al. Cuproptosis, the novel type of oxidation-induced cell death in thoracic cancers: can it enhance the success of immunotherapy? Cell communication signaling: CCS. (2024) 22:379. doi: 10.1186/s12964-024-01743-2
160. Vo TTT, Peng TY, Nguyen TH, Bui TNH, Wang CS, Lee WJ, et al. The crosstalk between copper-induced oxidative stress and cuproptosis: A novel potential anticancer paradigm. Cell communication signaling: CCS. (2024) 22:353. doi: 10.1186/s12964-024-01726-3
161. Lou QM, Lai FF, Li JW, Mao KJ, Wan HT, and He Y. Mechanisms of cuproptosis and its relevance to distinct diseases. Apoptosis: an Int J programmed Cell Death. (2024) 29:981–1006. doi: 10.1007/s10495-024-01983-0
162. Ahuja A, Dev K, Tanwar RS, Selwal KK, and Tyagi PK. Copper mediated neurological disorder: visions into amyotrophic lateral sclerosis, alzheimer and menkes disease. J Trace elements Med biology: Organ Soc Minerals Trace Elements (GMS). (2015) 29:11–23. doi: 10.1016/j.jtemb.2014.05.003
163. Yang S, Li Y, Zhou L, Wang X, Liu L, and Wu M. Copper homeostasis and cuproptosis in atherosclerosis: metabolism, mechanisms and potential therapeutic strategies. Cell Death Discov. (2024) 10:25. doi: 10.1038/s41420-023-01796-1
164. Li J, Peng C, Huang C, Wan L, Wang K, Wu P, et al. Metal ruthenium complexes treat spinal cord injury by alleviating oxidative stress through interaction with antioxidant 1 copper chaperone protein. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2024) 11:e2407225. doi: 10.1002/advs.202407225
165. Aschner M, Skalny AV, Lu R, Martins AC, Tizabi Y, Nekhoroshev SV, et al. Mitochondrial pathways of copper neurotoxicity: focus on mitochondrial dynamics and mitophagy. Front Mol Neurosci. (2024) 17:1504802. doi: 10.3389/fnmol.2024.1504802
166. Meyer JN, Leuthner TC, and Luz AL. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology. (2017) 391:42–53. doi: 10.1016/j.tox.2017.07.019
167. Reddy PV, Rao KV, and Norenberg MD. The mitochondrial permeability transition, and oxidative and nitrosative stress in the mechanism of copper toxicity in cultured neurons and astrocytes. Lab investigation; J Tech Methods Pathol. (2008) 88:816–30. doi: 10.1038/labinvest.2008.49
168. Ruiz LM, Jensen EL, Rossel Y, Puas GI, Gonzalez-Ibanez AM, Bustos RI, et al. Non-cytotoxic copper overload boosts mitochondrial energy metabolism to modulate cell proliferation and differentiation in the human erythroleukemic cell line K562. Mitochondrion. (2016) 29:18–30. doi: 10.1016/j.mito.2016.04.005
169. Tsui KH, Li CJ, and Lin LT. Melatonin supplementation attenuates cuproptosis and ferroptosis in aging cumulus and granulosa cells: potential for improving ivf outcomes in advanced maternal age. Reprod Biol endocrinology: RB&E. (2024) 22:138. doi: 10.1186/s12958-024-01311-w
170. Sheftel AD, Stehling O, Pierik AJ, Elsässer HP, Mühlenhoff U, Webert H, et al. Humans possess two mitochondrial ferredoxins, fdx1 and fdx2, with distinct roles in steroidogenesis, heme, and fe/S cluster biosynthesis. Proc Natl Acad Sci United States America. (2010) 107:11775–80. doi: 10.1073/pnas.1004250107
171. Jiang A, Ye J, Zhou Y, Zhu B, Lu J, Ge S, et al. Copper death inducer, fdx1, as a prognostic biomarker reshaping tumor immunity in clear cell renal cell carcinoma. Cells. (2023) 12:349. doi: 10.3390/cells12030349
172. Quan Y, Li W, Yan R, Cheng J, Xu H, and Chen L. Tumor cuproptosis and immune infiltration improve survival of patients with hepatocellular carcinoma with a high expression of ferredoxin 1. Front Oncol. (2023) 13:1168769. doi: 10.3389/fonc.2023.1168769
173. Zhang G, Shen L, Li Z, and Zhao Y. Fdx1 serves as a prognostic biomarker and promotes glioma progression by regulating the immune response. Aging. (2023) 15:4963–85. doi: 10.18632/aging.204772
174. Sun B, Ding P, Song Y, Zhou J, Chen X, Peng C, et al. Fdx1 downregulation activates mitophagy and the pi3k/akt signaling pathway to promote hepatocellular carcinoma progression by inducing ros production. Redox Biol. (2024) 75:103302. doi: 10.1016/j.redox.2024.103302
175. Wu CC, Li CJ, Lin LT, Lin PH, Wen ZH, Cheng JT, et al. Cuproptosis-related gene fdx1 identified as a potential target for human ovarian aging. Reprod Sci (Thousand Oaks Calif). (2024) 32(3):867–75. doi: 10.1007/s43032-024-01573-0
176. Zhao Q and Qi T. The implications and prospect of cuproptosis-related genes and copper transporters in cancer progression. Front Oncol. (2023) 13:1117164. doi: 10.3389/fonc.2023.1117164
177. Liu Y, Zhu J, Xu L, Wang B, Lin W, and Luo Y. Copper regulation of immune response and potential implications for treating orthopedic disorders. Front Mol Biosci. (2022) 9:1065265. doi: 10.3389/fmolb.2022.1065265
178. Liu WQ, Lin WR, Yan L, Xu WH, and Yang J. Copper homeostasis and cuproptosis in cancer immunity and therapy. Immunol Rev. (2024) 321:211–27. doi: 10.1111/imr.13276
179. Lu J, Liu X, Li X, Li H, Shi L, Xia X, et al. Copper regulates the host innate immune response against bacterial infection via activation of alpk1 kinase. Proc Natl Acad Sci United States America. (2024) 121:e2311630121. doi: 10.1073/pnas.2311630121
180. Zhang X, Tang B, Luo J, Yang Y, Weng Q, Fang S, et al. Cuproptosis, ferroptosis and panoptosis in tumor immune microenvironment remodeling and immunotherapy: culprits or new hope. Mol Cancer. (2024) 23:255. doi: 10.1186/s12943-024-02130-8
181. Zhang S, Huang Q, Ji T, Li Q, and Hu C. Copper homeostasis and copper-induced cell death in tumor immunity: implications for therapeutic strategies in cancer immunotherapy. biomark Res. (2024) 12:130. doi: 10.1186/s40364-024-00677-8
182. Luo D, Liu S, Luo J, Chen H, He Z, Gao Z, et al. Characterization of cuproptosis identified immune microenvironment and prognosis in acute myeloid leukemia. Clin Trans oncology: Off Publ Fed Spanish Oncol Societies Natl Cancer Institute Mexico. (2023) 25:2393–407. doi: 10.1007/s12094-023-03118-4
183. Huang Z, Cao L, and Yan D. Inflammatory immunity and bacteriological perspectives: A new direction for copper treatment of sepsis. J Trace elements Med biology: Organ Soc Minerals Trace Elements (GMS). (2024) 84:127456. doi: 10.1016/j.jtemb.2024.127456
184. Arendsen LP, Thakar R, and Sultan AH. The use of copper as an antimicrobial agent in health care, including obstetrics and gynecology. Clin Microbiol Rev. (2019) 32:e00125-18. doi: 10.1128/cmr.00125-18
185. Tang D, Kroemer G, and Kang R. Targeting cuproplasia and cuproptosis in cancer. Nat Rev Clin Oncol. (2024) 21:370–88. doi: 10.1038/s41571-024-00876-0
186. Lin CH, Chin Y, Zhou M, Sobol RW, Hung MC, and Tan M. Protein lipoylation: mitochondria, cuproptosis, and beyond. Trends Biochem Sci. (2024) 49:729–44. doi: 10.1016/j.tibs.2024.04.002
187. Xiong C, Ling H, Hao Q, and Zhou X. Cuproptosis: P53-regulated metabolic cell death? Cell Death differentiation. (2023) 30:876–84. doi: 10.1038/s41418-023-01125-0
188. Li H, Wang J, Wu C, Wang L, Chen ZS, and Cui W. The combination of disulfiram and copper for cancer treatment. Drug Discov Today. (2020) 25:1099–108. doi: 10.1016/j.drudis.2020.04.003
189. Feng Y, Yang Z, Wang J, and Zhao H. Cuproptosis: unveiling a new frontier in cancer biology and therapeutics. Cell communication signaling: CCS. (2024) 22:249. doi: 10.1186/s12964-024-01625-7
190. Li SR, Tao SY, Li Q, Hu CY, and Sun ZJ. Harnessing nanomaterials for copper-induced cell death. Biomaterials. (2025) 313:122805. doi: 10.1016/j.biomaterials.2024.122805
191. Wang X, Zhou M, Liu Y, and Si Z. Cope with copper: from copper linked mechanisms to copper-based clinical cancer therapies. Cancer Lett. (2023) 561:216157. doi: 10.1016/j.canlet.2023.216157
192. Chang W and Li P. Copper and diabetes: current research and prospect. Mol Nutr Food Res. (2023) 67:e2300468. doi: 10.1002/mnfr.202300468
193. Qu J, Wang Y, and Wang Q. Cuproptosis: potential new direction in diabetes research and treatment. Front Endocrinol. (2024) 15:1344729. doi: 10.3389/fendo.2024.1344729
194. Hu Q, Zhang X, Huang J, Peng H, Sun Y, Sang W, et al. The stat1-slc31a1 axis: potential regulation of cuproptosis in diabetic retinopathy. Gene. (2024) 930:148861. doi: 10.1016/j.gene.2024.148861
195. Shen J, San W, Zheng Y, Zhang S, Cao D, Chen Y, et al. Different types of cell death in diabetic endothelial dysfunction. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2023) 168:115802. doi: 10.1016/j.biopha.2023.115802
196. Zhong S, Wang N, and Zhang C. Podocyte death in diabetic kidney disease: potential molecular mechanisms and therapeutic targets. Int J Mol Sci. (2024) 25:9035. doi: 10.3390/ijms25169035
197. Pan Z, Huang L, Gan Y, Xia Y, and Yu W. The molecular mechanisms of cuproptosis and small-molecule drug design in diabetes mellitus. Molecules (Basel Switzerland). (2024) 29:2852. doi: 10.3390/molecules29122852
198. Subramaniam J, Aditi A, Arumugam K, Sri S, Bharathidevi SR, and Ramkumar KM. Copper dyshomeostasis and diabetic complications: chelation strategies for management. Mini Rev medicinal Chem. (2024) 25(4):277–92. doi: 10.2174/0113895575308206240911104945
199. Cai L, Tan Y, Holland B, and Wintergerst K. Diabetic cardiomyopathy and cell death: focus on metal-mediated cell death. Cardiovasc Toxicol. (2024) 24:71–84. doi: 10.1007/s12012-024-09836-7
200. Song J, Zhu K, Wang H, Wu M, Wu Y, and Zhang Q. Deciphering the emerging role of programmed cell death in diabetic wound healing. Int J Biol Sci. (2023) 19:4989–5003. doi: 10.7150/ijbs.88461
201. Li X, Chen X, and Gao X. Copper and cuproptosis: new therapeutic approaches for alzheimer’s disease. Front Aging Neurosci. (2023) 15:1300405. doi: 10.3389/fnagi.2023.1300405
202. Huang M, Zhang Y, and Liu X. The mechanism of cuproptosis in parkinson’s disease. Ageing Res Rev. (2024) 95:102214. doi: 10.1016/j.arr.2024.102214
203. Lamtai M, Zghari O, Ouakki S, Marmouzi I, Mesfioui A, El Hessni A, et al. Chronic copper exposure leads to hippocampus oxidative stress and impaired learning and memory in male and female rats. Toxicological Res. (2020) 36:359–66. doi: 10.1007/s43188-020-00043-4
204. Kepp KP. Bioinorganic chemistry of alzheimer’s disease. Chem Rev. (2012) 112:5193–239. doi: 10.1021/cr300009x
205. Bush AI and Tanzi RE. Therapeutics for alzheimer’s disease based on the metal hypothesis. Neurotherapeutics: J Am Soc Exp Neurother. (2008) 5:421–32. doi: 10.1016/j.nurt.2008.05.001
206. Cole-Hunter T, Zhang J, So R, Samoli E, Liu S, Chen J, et al. Long-term air pollution exposure and parkinson’s disease mortality in a large pooled european cohort: an elapse study. Environ Int. (2023) 171:107667. doi: 10.1016/j.envint.2022.107667
207. van der Kant R, Goldstein LSB, and Ossenkoppele R. Amyloid-B;-independent regulators of tau pathology in alzheimer disease. Nat Rev Neurosci. (2020) 21:21–35. doi: 10.1038/s41583-019-0240-3
208. Doroszkiewicz J, Farhan JA, Mroczko J, Winkel I, Perkowski M, and Mroczko B. Common and trace metals in alzheimer’s and parkinson’s diseases. Int J Mol Sci. (2023) 24:15721. doi: 10.3390/ijms242115721
209. Lippi SLP, Neely CLC, and Amaya AL. Trace concentrations, heavy implications: influences of biometals on major brain pathologies of alzheimer’s disease. Int J Biochem Cell Biol. (2022) 143:106136. doi: 10.1016/j.biocel.2021.106136
210. Figley SA, Khosravi R, Legasto JM, Tseng YF, and Fehlings MG. Characterization of vascular disruption and blood-spinal cord barrier permeability following traumatic spinal cord injury. J neurotrauma. (2014) 31:541–52. doi: 10.1089/neu.2013.3034
211. Zhou Y, Li X, Wang Z, Ng L, He R, Liu C, et al. Machine learning-driven prediction model for cuproptosis-related genes in spinal cord injury: construction and experimental validation. Front Neurol. (2025) 16:1525416. doi: 10.3389/fneur.2025.1525416
212. Davies KM, Mercer JF, Chen N, and Double KL. Copper dyshomoeostasis in parkinson’s disease: implications for pathogenesis and indications for novel therapeutics. Clin Sci (London England: 1979). (2016) 130:565–74. doi: 10.1042/cs20150153
213. Chen LL, Fan YG, Zhao LX, Zhang Q, and Wang ZY. The metal ion hypothesis of alzheimer’s disease and the anti-neuroinflammatory effect of metal chelators. Bioorganic Chem. (2023) 131:106301. doi: 10.1016/j.bioorg.2022.106301
214. Huo S, Wang Q, Shi W, Peng L, Jiang Y, Zhu M, et al. Atf3/spi1/slc31a1 signaling promotes cuproptosis induced by advanced glycosylation end products in diabetic myocardial injury. Int J Mol Sci. (2023) 24:1667. doi: 10.3390/ijms24021667
215. Moujalled D, Strasser A, and Liddell JR. Molecular mechanisms of cell death in neurological diseases. Cell Death differentiation. (2021) 28:2029–44. doi: 10.1038/s41418-021-00814-y
216. Seglen PO and Gordon PB. 3-methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci United States America. (1982) 79:1889–92. doi: 10.1073/pnas.79.6.1889
217. Hama Y, Ogasawara Y, and Noda NN. Autophagy and cancer: basic mechanisms and inhibitor development. Cancer Sci. (2023) 114:2699–708. doi: 10.1111/cas.15803
218. Zhao Y, Wang Q, Zhu J, Cai J, Feng X, Song Q, et al. Identification of kw-2449 as a dual inhibitor of ferroptosis and necroptosis reveals that autophagy is a targetable pathway for necroptosis inhibitors to prevent ferroptosis. Cell Death Dis. (2024) 15:764. doi: 10.1038/s41419-024-07157-9
219. He X, Li M, Ye Z, You X, Wang J, Xiao X, et al. Identification of piperlongumine as potent inhibitor of necroptosis. Drug design Dev Ther. (2023) 17:1387–94. doi: 10.2147/dddt.S397971
220. Liu H, Guo H, Jian Z, Cui H, Fang J, Zuo Z, et al. Copper induces oxidative stress and apoptosis in the mouse liver. Oxid Med Cell Longevity. (2020) 2020:1359164. doi: 10.1155/2020/1359164
221. Liu H, Lai W, Liu X, Yang H, Fang Y, Tian L, et al. Exposure to copper oxide nanoparticles triggers oxidative stress and endoplasmic reticulum (Er)-stress induced toxicology and apoptosis in male rat liver and brl-3a cell. J hazardous materials. (2021) 401:123349. doi: 10.1016/j.jhazmat.2020.123349
222. Ostrakhovitch EA and Cherian MG. Role of P53 and reactive oxygen species in apoptotic response to copper and zinc in epithelial breast cancer cells. Apoptosis: an Int J programmed Cell Death. (2005) 10:111–21. doi: 10.1007/s10495-005-6066-7
223. Wei H, Zhang WJ, McMillen TS, Leboeuf RC, and Frei B. Copper chelation by tetrathiomolybdate inhibits vascular inflammation and atherosclerotic lesion development in apolipoprotein E-deficient mice. Atherosclerosis. (2012) 223:306–13. doi: 10.1016/j.atherosclerosis.2012.06.013
224. Liu J, Tang J, Zuo Y, Yu Y, Luo P, Yao X, et al. Stauntoside B inhibits macrophage activation by inhibiting nf-Kb and erk mapk signalling. Pharmacol Res. (2016) 111:303–15. doi: 10.1016/j.phrs.2016.06.022
225. Wan F, Zhong G, Ning Z, Liao J, Yu W, Wang C, et al. Long-term exposure to copper induces autophagy and apoptosis through oxidative stress in rat kidneys. Ecotoxicology Environ Saf. (2020) 190:110158. doi: 10.1016/j.ecoenv.2019.110158
226. Zhang C, Huang T, and Li L. Targeting cuproptosis for cancer therapy: mechanistic insights and clinical perspectives. J Hematol Oncol. (2024) 17:68. doi: 10.1186/s13045-024-01589-8
227. Denton D and Kumar S. Autophagy-dependent cell death. Cell Death differentiation. (2019) 26:605–16. doi: 10.1038/s41418-018-0252-y
228. Li Y, Guo Y, Fan Y, Tian H, Li K, and Mei X. Melatonin enhances autophagy and reduces apoptosis to promote locomotor recovery in spinal cord injury via the pi3k/akt/mtor signaling pathway. Neurochemical Res. (2019) 44:2007–19. doi: 10.1007/s11064-019-02838-w
229. Wu J and Lipinski MM. Autophagy in neurotrauma: good, bad, or dysregulated. Cells. (2019) 8:693. doi: 10.3390/cells8070693
230. Gromadzka G, Wilkaniec A, Tarnacka B, Hadrian K, Bendykowska M, Przybyłkowski A, et al. The role of glia in wilson’s disease: clinical, neuroimaging, neuropathological and molecular perspectives. Int J Mol Sci. (2024) 25:7545. doi: 10.3390/ijms25147545
231. Chen J, Liao J, Yu W, Cao H, Hu G, Tang Z, et al. Copper toxicity in the liver of broiler chicken: insights from metabolomics and ampk-mtor mediated autophagy perspective. Poultry Sci. (2024) 103:104011. doi: 10.1016/j.psj.2024.104011
232. Yang F, Liao J, Yu W, Qiao N, Guo J, Han Q, et al. Exposure to copper induces mitochondria-mediated apoptosis by inhibiting mitophagy and the pink1/parkin pathway in chicken (Gallus gallus) livers. J hazardous materials. (2021) 408:124888. doi: 10.1016/j.jhazmat.2020.124888
233. Dong J, Wang X, Xu C, Gao M, Wang S, Zhang J, et al. Inhibiting nlrp3 inflammasome activation prevents copper-induced neuropathology in a murine model of wilson’s disease. Cell Death Dis. (2021) 12:87. doi: 10.1038/s41419-021-03397-1
234. Liao J, Yang F, Tang Z, Yu W, Han Q, Hu L, et al. Inhibition of caspase-1-dependent pyroptosis attenuates copper-induced apoptosis in chicken hepatocytes. Ecotoxicology Environ Saf. (2019) 174:110–9. doi: 10.1016/j.ecoenv.2019.02.069
235. Zhou Q, Zhang Y, Lu L, Shi W, Zhang H, Qin W, et al. Upregulation of postsynaptic camp/pka/creb signaling alleviates copper(II)-induced oxidative stress and pyroptosis in mn9d cells. Toxicology. (2023) 494:153582. doi: 10.1016/j.tox.2023.153582
236. Zhu MJ, Zhang L, and Wang CP. Copper overload promotes B-amyloid induced nlrp3/caspase-1/gsdmd-mediated pyroptosis in alzheimer’s disease. J Integr Neurosci. (2024) 23:194. doi: 10.31083/j.jin2310194
237. Su L, Wang S, Li Q, Guo P, Wu Y, Zhao L, et al. Hesperidin alleviates zbp1-drived panoptosis induced by copper nanoparticles in immune organs of gallus. J Trace elements Med biology: Organ Soc Minerals Trace Elements (GMS). (2025) 87:127575. doi: 10.1016/j.jtemb.2024.127575
238. Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Publisher correction: Z-nucleic-acid sensing triggers zbp1-dependent necroptosis and inflammation. Nature. (2020) 580:E10. doi: 10.1038/s41586-020-2207-y
239. Miao Y, Liu J, Liu X, Yuan Q, Li H, Zhang Y, et al. Machine learning identification of cuproptosis and necroptosis-associated molecular subtypes to aid in prognosis assessment and immunotherapy response prediction in low-grade glioma. Front Genet. (2022) 13:951239. doi: 10.3389/fgene.2022.951239
240. Tang S, Liang C, Hou W, Hu Z, Chen X, Zhao J, et al. Atp7b R778l mutant hepatocytes resist copper toxicity by activating autophagy and inhibiting necroptosis. Cell Death Discov. (2023) 9:344. doi: 10.1038/s41420-023-01641-5
241. Li Y, Du Y, Zhou Y, Chen Q, Luo Z, Ren Y, et al. Iron and copper: critical executioners of ferroptosis, cuproptosis and other forms of cell death. Cell communication signaling: CCS. (2023) 21:327. doi: 10.1186/s12964-023-01267-1
242. Cai DH, Liang BF, Chen BH, Liu QY, Pan ZY, Le XY, et al. A novel water-soluble cu(Ii) gluconate complex inhibits cancer cell growth by triggering apoptosis and ferroptosis related mechanisms. J inorganic Biochem. (2023) 246:112299. doi: 10.1016/j.jinorgbio.2023.112299
243. Prasad Panda S and Kesharwani A. Micronutrients/mirs/atp networking in mitochondria: clinical intervention with ferroptosis, cuproptosis, and calcium burden. Mitochondrion. (2023) 71:1–16. doi: 10.1016/j.mito.2023.05.003
244. Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. Nfs1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. (2017) 551:639–43. doi: 10.1038/nature24637
245. Sun K, Zhi Y, Ren W, Li S, Zhou X, Gao L, et al. The mitochondrial regulation in ferroptosis signaling pathway and its potential strategies for cancer. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2023) 169:115892. doi: 10.1016/j.biopha.2023.115892
246. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. (2019) 73:354–63.e3. doi: 10.1016/j.molcel.2018.10.042
247. Shen Y, Li D, Liang Q, Yang M, Pan Y, and Li H. Cross-talk between cuproptosis and ferroptosis regulators defines the tumor microenvironment for the prediction of prognosis and therapies in lung adenocarcinoma. Front Immunol. (2022) 13:1029092. doi: 10.3389/fimmu.2022.1029092
248. Liu N and Chen M. Crosstalk between ferroptosis and cuproptosis: from mechanism to potential clinical application. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2024) 171:116115. doi: 10.1016/j.biopha.2023.116115
249. Freyermuth-Trujillo X, Segura-Uribe JJ, Salgado-Ceballos H, Orozco-Barrios CE, and Coyoy-Salgado A. Inflammation: A target for treatment in spinal cord injury. Cells. (2022) 11:269. doi: 10.3390/cells11172692
250. Fang S, Tang H, Li HL, Han TC, Li ZJ, Yin ZS, et al. Ccl2 Knockdown Attenuates Inflammatory Response after Spinal Cord Injury through the Pi3k/Akt Signaling Pathway: Bioinformatics Analysis and Experimental Validation. Mol Neurobiol. (2024) 61:1433–47. doi: 10.1007/s12035-023-03641-z
251. Fei M, Li Z, Cao Y, Jiang C, Lin H, and Chen Z. Microrna-182 improves spinal cord injury in mice by modulating apoptosis and the inflammatory response via ikkβ/nf-Kb. Lab investigation; J Tech Methods Pathol. (2021) 101:1238–53. doi: 10.1038/s41374-021-00606-5
252. Ding Y and Chen Q. The nf-Kb pathway: A focus on inflammatory responses in spinal cord injury. Mol Neurobiol. (2023) 60:5292–308. doi: 10.1007/s12035-023-03411-x
253. Deng H, Zhu S, Yang H, Cui H, Guo H, Deng J, et al. The dysregulation of inflammatory pathways triggered by copper exposure. Biol Trace element Res. (2023) 201:539–48. doi: 10.1007/s12011-022-03171-0
254. Bellver-Landete V, Bretheau F, Mailhot B, Vallières N, Lessard M, Janelle ME, et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat Commun. (2019) 10:518. doi: 10.1038/s41467-019-08446-0
255. Liu Y, Cai X, Shi B, Mo Y, Zhang J, Luo W, et al. Mechanisms and therapeutic prospects of microglia-astrocyte interactions in neuropathic pain following spinal cord injury. Mol Neurobiol. (2024) 62(4):4654–76. doi: 10.1007/s12035-024-04562-1
256. Li G, Cao Y, Shen F, Wang Y, Bai L, Guo W, et al. Mdivi-1 inhibits astrocyte activation and astroglial scar formation and enhances axonal regeneration after spinal cord injury in rats. Front Cell Neurosci. (2016) 10:241. doi: 10.3389/fncel.2016.00241
257. Mekhail M, Almazan G, and Tabrizian M. Oligodendrocyte-protection and remyelination post-spinal cord injuries: A review. Prog Neurobiol. (2012) 96:322–39. doi: 10.1016/j.pneurobio.2012.01.008
258. Xing L, Wang Z, Hao Z, Pan P, Yang A, and Wang J. Cuproptosis in stroke: focusing on pathogenesis and treatment. Front Mol Neurosci. (2024) 17:1349123. doi: 10.3389/fnmol.2024.1349123
259. Yang Y, Wu J, Wang L, Ji G, and Dang Y. Copper homeostasis and cuproptosis in health and disease. MedComm. (2024) 5:e724. doi: 10.1002/mco2.724
260. Wang S, Li C, Fan W, Chen T, Xu W, Hu X, et al. Neurotrophin-3/chitosan inhibits cuproptosis-related genes to enable functional recovery after spinal cord injury. Int J Biol macromolecules. (2025) 310:143403. doi: 10.1016/j.ijbiomac.2025.143403
261. Zha J, Chen F, Dong H, Shi P, Yao Y, Zhang Y, et al. Disulfiram targeting lymphoid Malignant cell lines via ros-jnk activation as well as nrf2 and nf-kb pathway inhibition. J Trans Med. (2014) 12:163. doi: 10.1186/1479-5876-12-163
262. Zhang L, Tsai IC, Ni Z, Chen B, Zhang S, Cai L, et al. Copper chelation therapy attenuates periodontitis inflammation through the cuproptosis/autophagy/lysosome axis. Int J Mol Sci. (2024) 25:5890. doi: 10.3390/ijms25115890
263. Brewer GJ. The use of copper-lowering therapy with tetrathiomolybdate in medicine. Expert Opin investigational Drugs. (2009) 18:89–97. doi: 10.1517/13543780802621859
264. Brewer GJ, Askari F, Dick RB, Sitterly J, Fink JK, Carlson M, et al. Treatment of wilson’s disease with tetrathiomolybdate: V. Control of free copper by tetrathiomolybdate and a comparison with trientine. Trans research: J Lab Clin Med. (2009) 154:70–7. doi: 10.1016/j.trsl.2009.05.002
265. Kim KK, Abelman S, Yano N, Ribeiro JR, Singh RK, Tipping M, et al. Tetrathiomolybdate inhibits mitochondrial complex iv and mediates degradation of hypoxia-inducible factor-1α in cancer cells. Sci Rep. (2015) 5:14296. doi: 10.1038/srep14296
266. Sciegienka SJ, Solst SR, Falls KC, Schoenfeld JD, Klinger AR, Ross NL, et al. D-penicillamine combined with inhibitors of hydroperoxide metabolism enhances lung and breast cancer cell responses to radiation and carboplatin via H(2)O(2)-mediated oxidative stress. Free Radical Biol Med. (2017) 108:354–61. doi: 10.1016/j.freeradbiomed.2017.04.001
Keywords: spinal cord injury, reactive oxygen species, copper homeostasis, cuproptosis, programmed cell death
Citation: Xu D, Hu L, Zhou J, Deng X, Zhu Y and Liu C (2025) The emerging role of cuproptosis in spinal cord injury. Front. Immunol. 16:1595852. doi: 10.3389/fimmu.2025.1595852
Received: 18 March 2025; Accepted: 26 May 2025;
Published: 16 June 2025.
Edited by:
SongOu Zhang, Ningbo University, ChinaReviewed by:
Arun Kumar Yadawa, Texas A and M University, United StatesQin Hu, Shanghai Jiao Tong University, China
Yu Zhou, Orthopedic Hospital affiliated to Chongqing University of Traditional Chinese Medicine, China
Copyright © 2025 Xu, Hu, Zhou, Deng, Zhu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiongwei Deng, ZGVuZ3NwaW5lQDE2My5jb20=; Yunrong Zhu, enlyMTM1NzlAaG90bWFpbC5jb20=; Chao Liu, bGl1Y2hhb3RvdWdhb0AxNjMuY29t
†These authors have contributed equally and share first authorship