 Roberto Depascale1Anna Ghirardello1Elisabetta Zanatta1Chiara Franco1Marisol Bracalenti1Federico Pettorossi1Mariele Gatto1,2Elena Treppo3Beatrice Moccaldi1Margherita Zen1Stefano Piaserico4Christian Ciolfi4Luca Quartuccio3Andrea Doria1
Roberto Depascale1Anna Ghirardello1Elisabetta Zanatta1Chiara Franco1Marisol Bracalenti1Federico Pettorossi1Mariele Gatto1,2Elena Treppo3Beatrice Moccaldi1Margherita Zen1Stefano Piaserico4Christian Ciolfi4Luca Quartuccio3Andrea Doria1 Luca Iaccarino1*
Luca Iaccarino1*- 1Rheumatology Unit, Department of Medicine DIMED, University of Padua, Padua, Italy
- 2Academic Rheumatology Centre, Department of Clinical and Biological Sciences, University of Turin, AO Mauriziano di Torino, Turin, Italy
- 3Rheumatology Unit, Department of Medicine DAME, University of Udine, Udine, Italy
- 4Dermatology Unit, Department of Medicine DIMED, University of Padua, Padua, Italy
Aim: We aimed to describe the clinical and serological characteristics of anti-small ubiquitin-like modifier-activating enzyme (SAE)-positive cases from a multicentric cohort of patients affected with idiopathic inflammatory myopathies (IIMs).
Methods: Anti-SAE antibody-positive patients (determined by line immunoassay) from a prospective cohort of patients with IIM were retrospectively evaluated. We considered features at disease onset and during follow-up. Muscular involvement was evaluated by the Manual Muscle Test-8, creatine phosphokinase (CK) levels, and/or magnetic resonance imaging; interstitial lung disease (ILD) was evaluated by high-resolution computed tomography; and skin and joint involvement was evaluated by clinical judgment. The therapeutic approach was also reported in all patients, and a literature review was also provided.
Results: Out of 170 patients with IIM, 10 (5.9%) were anti-SAE positive, all classified as having dermatomyositis; therefore, among 80 patients with dermatomyositis, the prevalence of anti-SAE antibodies was 12.5%. The female-to-male ratio was 9:1. The median time from onset of symptoms to diagnosis was 1 year (range 0–2 years), and the mean age at onset of symptoms was 55.5 years (range 34–77 years). All patients had skin manifestations, including photosensitive rash, heliotrope rash, and Gottron’s sign and/or papules (one with ulcerations). Refractory features requiring multiple lines of immunosuppressants were observed in 60% of cases. Four patients had arthritis and/or inflammatory arthralgia; four had muscular involvement, usually mild; and none had ILD. One patient had a history of malignancy. All patients were treated with glucocorticoids and received different immunosuppressants, including cyclophosphamide.
Conclusions: All patients with anti-SAE antibody positivity were classified as having dermatomyositis, with severe and refractory skin manifestations in most cases. One case of malignancy was described; therefore, cancer screening should be warranted in all anti-SAE patients.
Introduction
Dermatomyositis (DM) is a rare and multisystemic autoimmune disorder included in the large spectrum of idiopathic inflammatory myopathies (IIMs) (1), characterized by chronic inflammation in the skin and skeletal muscles (2). DM patients can be classified into different phenotypes, according to clinical features and myositis-specific antibody (MSA) positivity (1–4). Five mutually exclusive MSAs have been associated with DM: anti-Mi2, anti-melanoma differentiation-associated protein 5 (MDA5), anti-nuclear matrix protein-2 (NXP2), anti-transcriptional intermediary factor-1-γ (TIF-1-gamma), and anti-small ubiquitin-like modifier-activating enzyme (SAE). All of them have shown diagnostic and prognostic values (5–7). Anti-SAE antibodies were first reported by Betteridge et al. in 2007 (8) and then described in 2009 in a cohort of patients with IIM from the United Kingdom (9). Anti-SAE antibodies bind the small ubiquitin-like modifier (SUMO)-activating enzyme, characterized by two subunits (SAE1 and SAE2). SUMO is involved in post-translational modification of several target proteins by “sumoylation” (9). Sumoylation is an important regulator of the normal function of many proteins, which has been hypothesized to play an important role in the pathogenesis of some human diseases (10–12). The most common technique used for the detection of anti-SAE antibodies in patient sera is radiolabeled 35S protein immunoprecipitation (IP), but enzyme-linked immunosorbent assay (ELISA) and immunoblotting can also be applied (10). The prevalence of anti-SAE autoantibodies reported in the literature ranges from 1%–3% in Asians to 6%–8% in Caucasians (8, 10, 13, 14). The clinical phenotype of anti-SAE-positive DM patients is often characterized by amyopathic dermatomyositis at disease onset. Skin manifestations can be severe and often pruritic. Muscle disease, when present, is usually mild and often develops later during the disease course. Dysphagia appears to be another common manifestation among anti-SAE-positive patients. Interstitial lung disease (ILD), usually in the form of organizing pneumonia (OP), is another possible feature, often mild and subclinical. The prevalence of cancer varies among different studies (2, 15).
This study aims to describe the clinical features of patients with anti-SAE antibody positivity in a multicenter cohort of patients affected with IIM. A narrative review of cases reported in the literature will also be provided.
Methods
Patients with anti-SAE positivity were retrospectively evaluated from a prospective multicentric cohort (Padua University Hospital and Udine University Hospital) of patients with IIM according to the EULAR/ACR and/or Bohan and Peter and/or European Neuromuscular Center (ENMC) classification criteria (16–18) since January 2011 to January 2025.
Antinuclear antibodies (ANA) were detected through screening by indirect immunofluorescence (IIF), and specific MSA/MAA (myositis-associated antibodies) were tested using multiparametric line immunoassay according to the manufacturer’s protocol (EUROLINE, Lubeck, Germany). A representative image of anti-SAE-1 positivity in case 3 by a commercial line immunoassay is visualized in Figure 1.
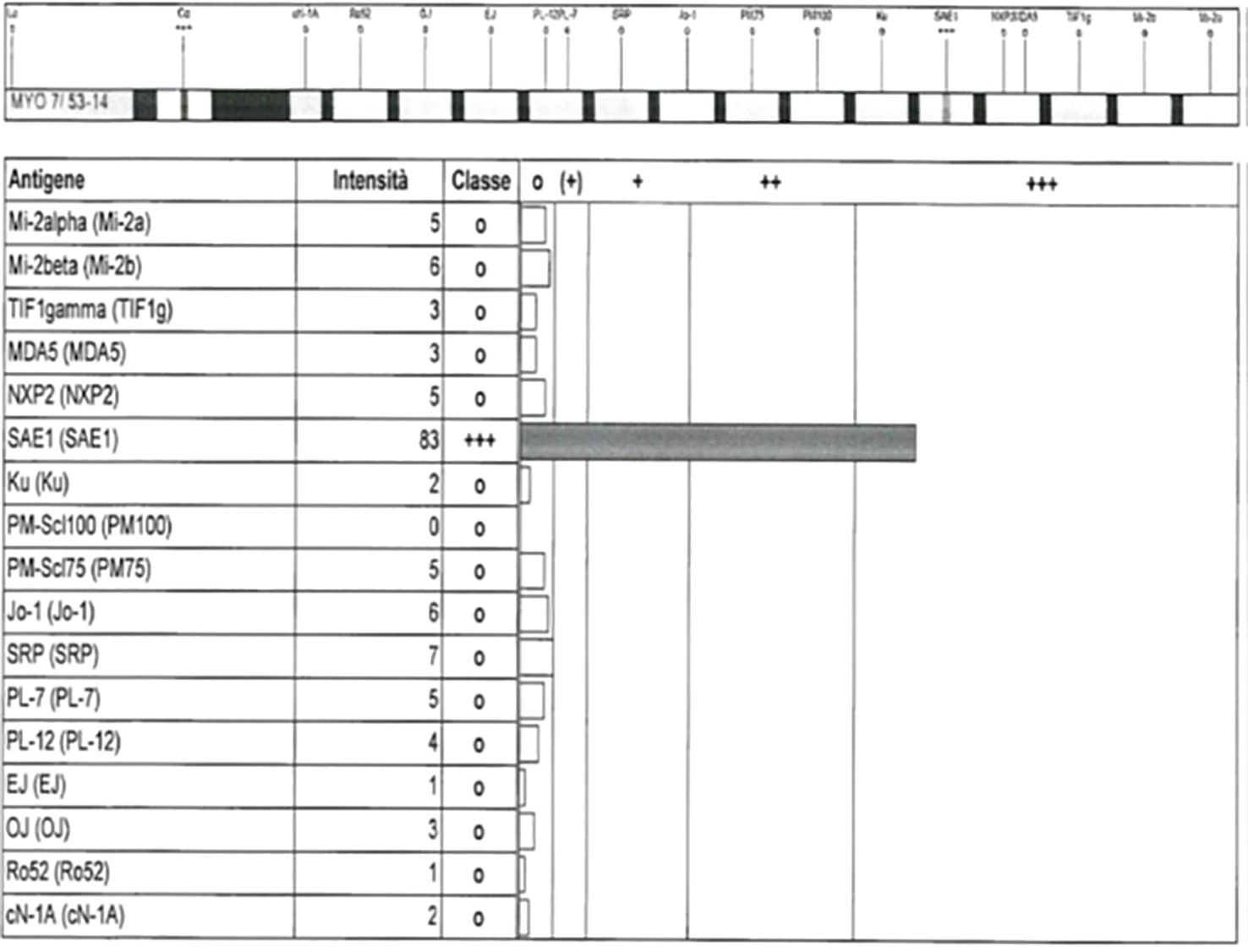
Figure 1. Representative image of anti-SAE-1 positivity in case 3 by a commercial line immunoassay (EUROLINE myositis profile 3).
In the prospective cohort, physical examination findings were obtained since the first visit. Among the laboratory tests, the closest values to the date of the visit were recorded. Muscle involvement was defined by muscular weakness assessed through the Manual Muscle Test-8 (MMT-8) and elevated creatine phosphokinase (CK) level (above the upper limit of normal) and/or muscular edema in T2-weighted images on muscle magnetic resonance imaging (MRI). All patients affected with DM underwent high-resolution computed tomography (HRCT) at baseline to assess the presence of ILD. Lung involvement was also evaluated during the follow-up according to the onset of new respiratory symptoms and/or restrictive pattern shown by pulmonary function tests (PFTs). Furthermore, all patients affected with DM were initially screened for cancer by using full-body computed tomography, esophagogastroduodenoscopy, and colonoscopy. Medications used by patients were also recorded, and response/refractoriness to treatment was defined by the physician’s judgment. Precisely, one patient was deemed refractory in case of inadequate response to glucocorticoids and at least two immunosuppressants.
The literature review was performed searching in PubMed, LiSSa, BDSP, and Cochrane Library databases for articles related to the association of DM and the anti-SAE autoantibody up until January 2025. We included all papers with anti-SAE DM case(s) description. We used the following keywords: anti-SAE, dermatomyositis, and skin in myositis.
The study was carried out in accordance with the Declaration of Helsinki and approved by our institution’s ethics committee (Azienda Ospedaliera di Padova, n. 5505/A/22).
Results
A total of 170 patients with IIM were enrolled in the study. Among them, 10 (5.9%) were anti-SAE positive, all diagnosed with DM. The prevalence of anti-SAE antibodies in patients with DM of our cohort (n=80) was 12.5%. All patients were Caucasian and 90% were women. The mean age at disease onset was 55.5 years (range 34–77 years). The median time from onset of symptoms to diagnosis was 1 year (range 0–2 years), and the median follow-up duration was 35 months (range 23–58 months).
Clinical features
The clinical and serological features of anti-SAE-positive patients are described in Table 1.
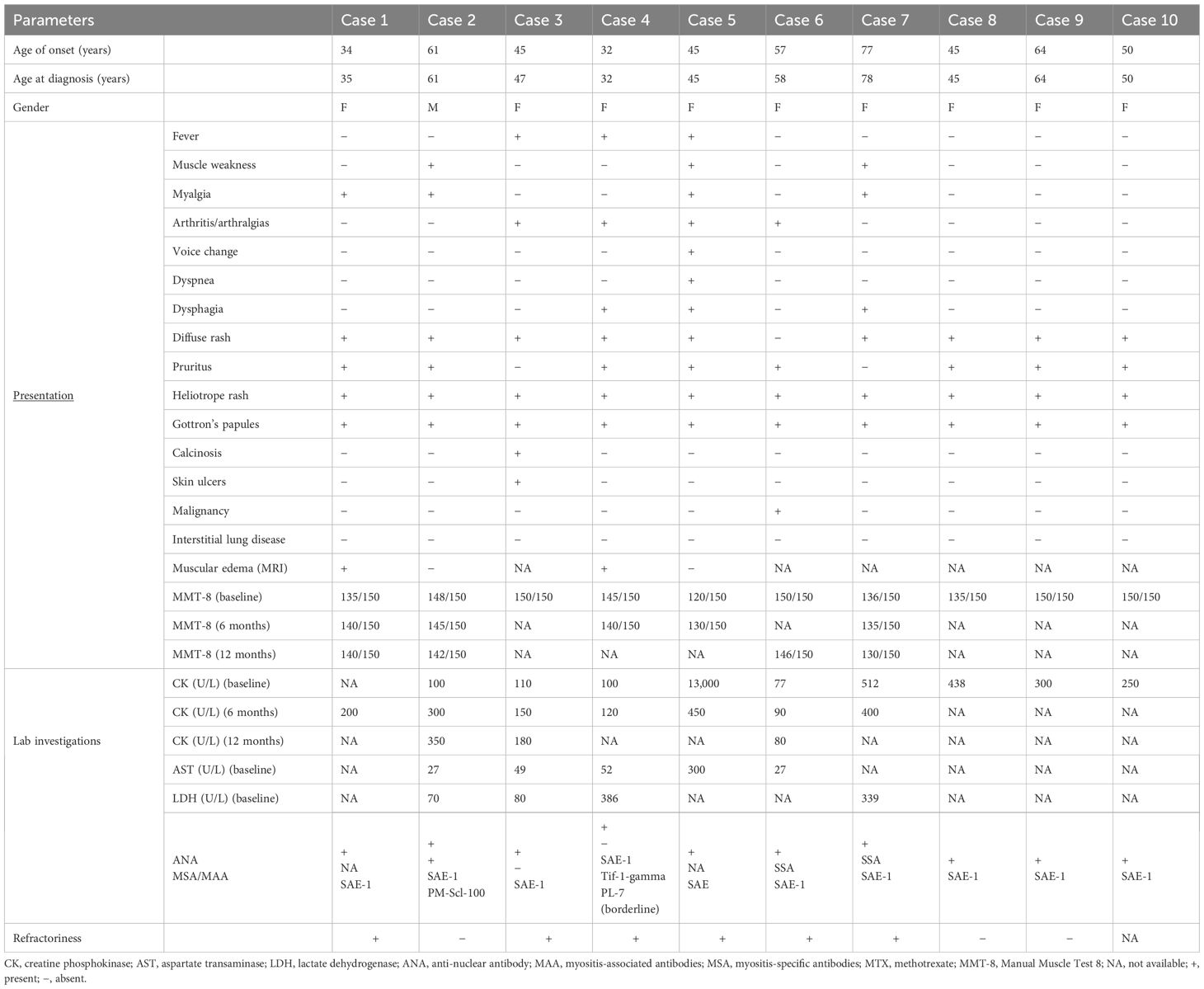
Table 1. Clinical and serological features of anti-SAE-positive patients at baseline and during follow-up.
All patients displayed skin manifestations, most of them initially presenting with a diffuse rash (Figure 2). Gottron papules and Gottron’s sign were found in 100% of patients. One patient had a diffuse skin involvement associated with panniculitis and skin ulcerations. Seven patients underwent skin biopsy showing non-specific dermatitis. Calcinosis was described in one case involving the buttocks and thighs. None had mechanic’s hands. Muscle weakness occurred in three patients (30%). In most cases, the onset of myositis occurred after skin involvement, with a mean of 8 months (range 3–8 months). In two cases, not complaining of muscular weakness but only myalgia, edema on muscular MRI was found. Three patients had elevated CK. In one case, the elevation of muscular enzymes was mild, occurring 2years after the skin manifestations. The other patient developed an acute rhabdomyolysis requiring hospitalization with very high levels of CK and acute kidney damage at disease onset. All potential causes of rhabdomyolysis, including infectious and toxic etiologies, were ruled out, and the detection of autoantibodies supported an autoimmune origin.
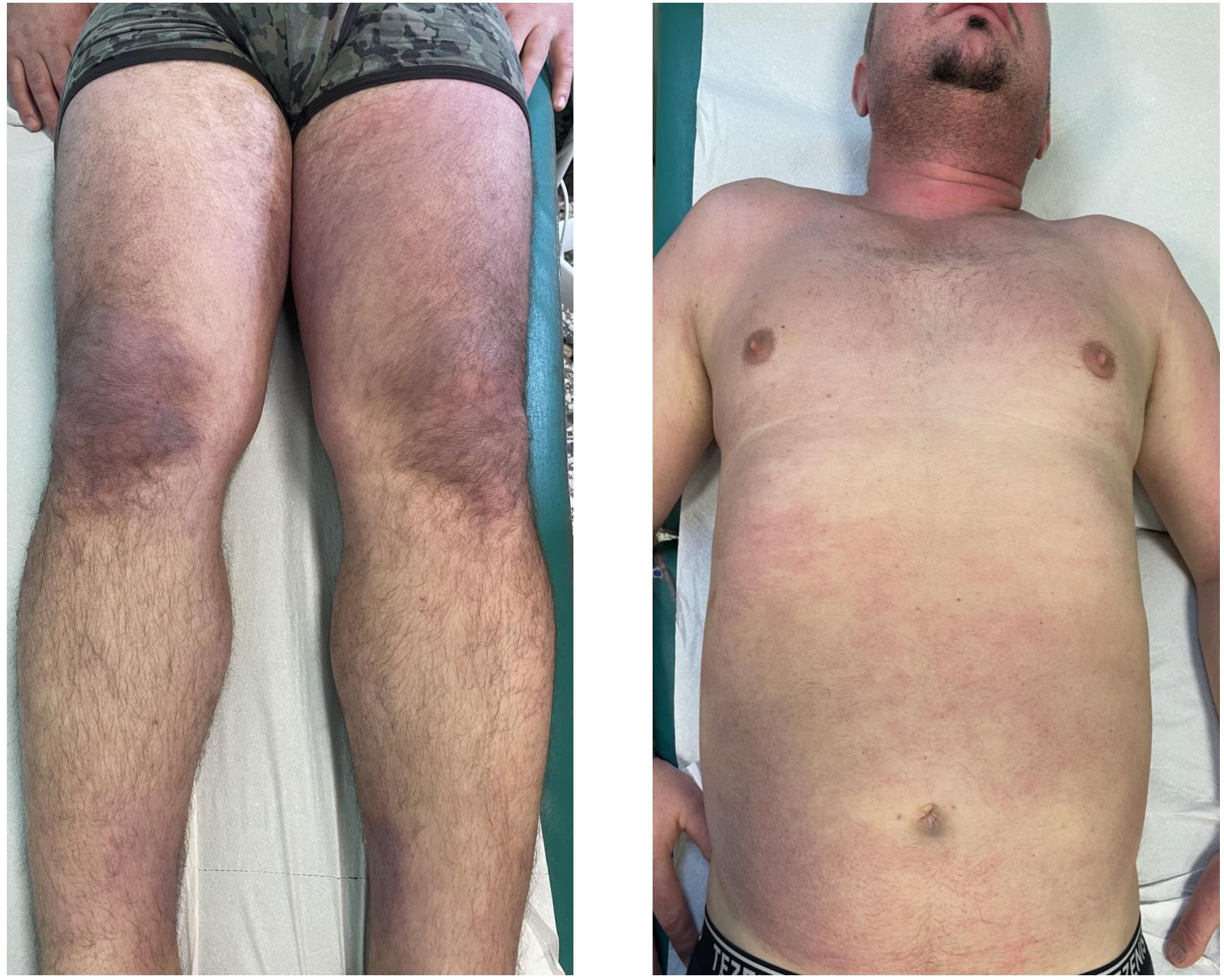
Figure 2. Case 2: A 62-year-old male patient diagnosed with anti-SAE+ DM. Wide violaceous erythematous plaques involving the knees, trunk, and hands. Consent obtained.
Muscular biopsy was performed in one patient, confirming the histological pattern of DM. Three patients reported dysphagia. Arthritis or inflammatory arthralgias were reported in four cases (40%). No patient had clinical, functional, or radiological signs of ILD at baseline, and further signs or symptoms of pulmonary involvement were found during follow-up. As reported in Table 1, in addition to anti-SAE positivity, case 4 was positive for anti-TIF1 gamma and borderline positive for anti-PL-7. In such patients, borderline anti-tRNA synthetase positivity was apparently not related to lung involvement, both at diagnosis and follow-up.
There was one case of malignancy (10%) in our cohort. The patient was a 57-year-old lady diagnosed with DM, and cancer screening found a localized and differentiated colon adenocarcinoma. She underwent colon resection in 2020, remaining cancer-free thereafter.
Treatment
Methotrexate (MTX) was the most commonly used medication in our cohort, together with glucocorticoids (70% and 100%, respectively). Six patients (60%) had a refractory cutaneous disease and required multiple medication changes, including one case with ulcers and panniculitis requiring cyclophosphamide (CYC), after failure of several immunosuppressants. Because of persistent cutaneous manifestations despite steroids and MTX, one patient was successfully treated with the JAK inhibitor baricitinib (BARI) (Table 2).
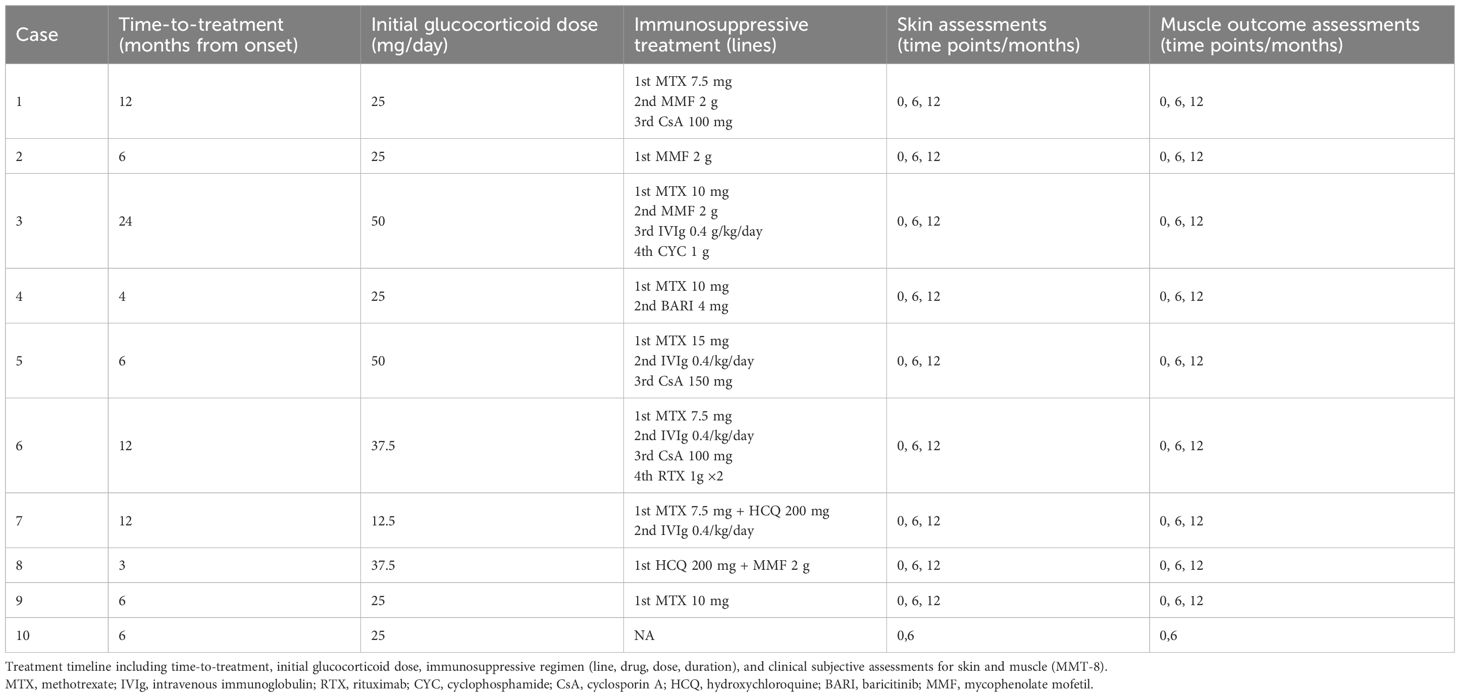
Table 2. Treatment timeline and prespecified assessments.
Narrative literature review and discussion
To date, we have been able to find 208 anti-SAE adult patients reported in the literature (Table 3).
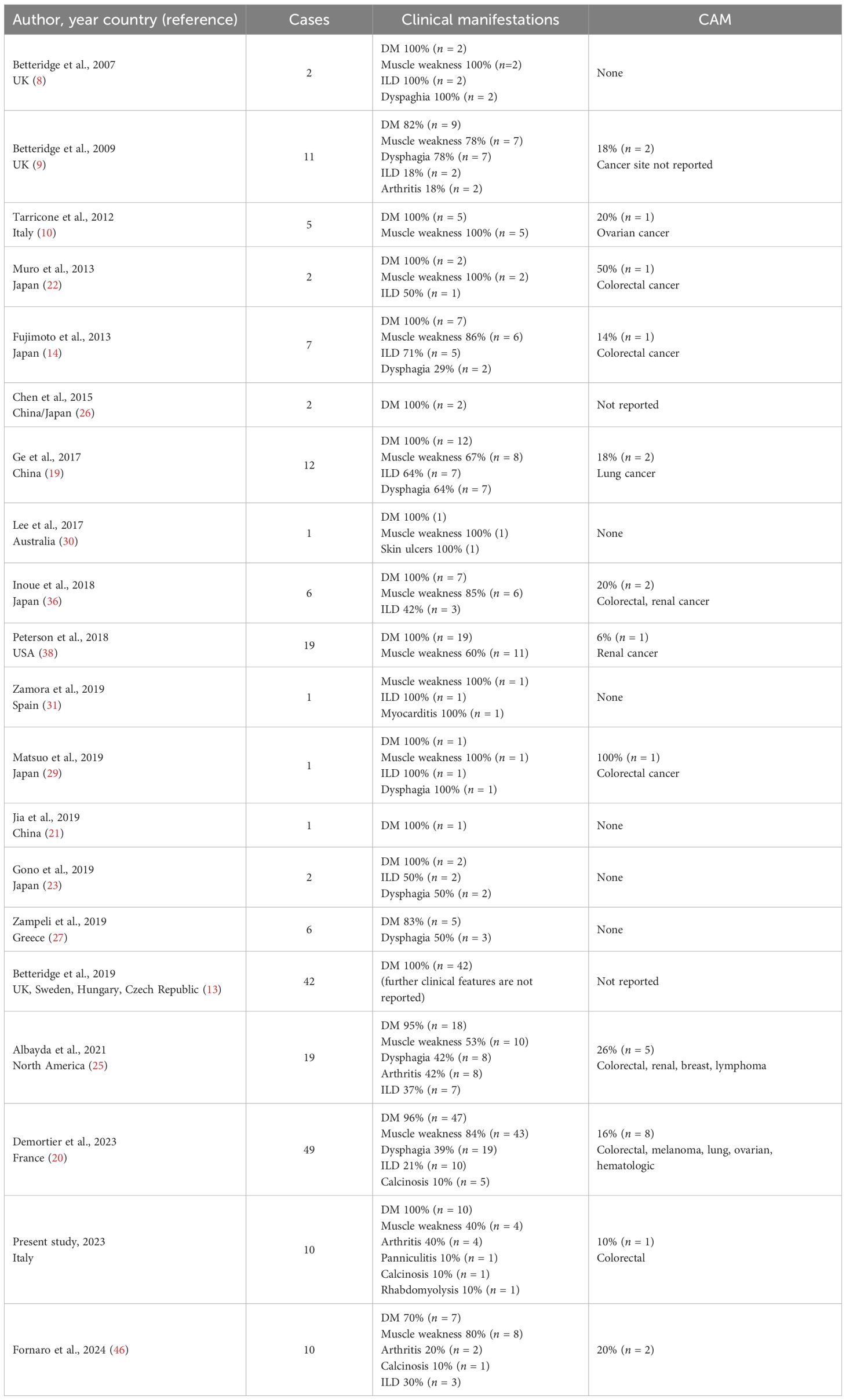
Table 3. Anti-SAE-positive DM patients reported in the literature.
According to these studies, ethnic background may influence the frequency of disease manifestations (24, 25); Middle Eastern anti-SAE patients have a higher risk of developing cancer, ILD, dysphagia, and diffuse and pruritic erythema than Caucasian patients (26). Although disease symptoms may vary among ethnicities, the prevalence of skin, muscular, and lung manifestations is similar (27, 28). In our study, anti-SAE positivity was characterized by predominant diffuse and often pruritic skin manifestations, accompanied by clinical or subclinical myopathy that typically developed after the onset of skin lesions. These findings are in line with previous studies (10, 21, 25, 29). Interestingly, we also reported one case of severe cutaneous and subcutaneous involvement with panniculitis and necrotic ulcers requiring deep immunosuppressant treatment, as rarely described in the literature (30). Although patients with anti-SAE antibodies are usually classified as having an amyopathic form of DM, in our cohort, overt muscle disease was found in three patients (30%). This finding suggests that muscle involvement should be screened in all cases, particularly during the follow-up (25). Interestingly, we also described a case of acute and potentially fatal rhabdomyolysis at disease onset. Only another single case report of a patient with severe muscle and cardiac involvement (myocarditis), leading to death, was described (31). In our cohort, no sign of ILD was found (0%). Among anti-SAE-positive patients, evidence of preserved pulmonary functions and a higher prevalence of organizing pneumonia pattern rather than other MSAs has been reported in the literature (10, 14, 32).
The coexistence of more than one MSA, as found in case 4, can be observed by multi-analytic line immunoassays, as recently reported (11). In dermatomyositis, the presence of multiple autoantibody positivities frequently does not correspond to specific clinical manifestations. It may result from analytical artifacts or antigen cross-reactivity and lack a clear consensus for interpretation in clinically discordant cases. It highlights the need for further research to elucidate this phenomenon (11).
Finally, during cancer screening, one female patient from our cohort was diagnosed with non-metastatic colorectal adenocarcinoma. In the literature, 27 out of 208 patients (12.9%) with cancer-associated myositis in anti-SAE patients have been previously described (13, 19, 24, 25); however, the prevalence is underestimated because some papers did not evaluate or did not report any data regarding cancer association. In our cohort, all patients with DM, including anti-SAE-positive patients, underwent screening for neoplasms. In line with the reports in the literature, we therefore recommend screening for cancer in all patients with anti-SAE positivity (22, 33, 34).
Most patients affected with IIM respond well to glucocorticoids, although randomized clinical trials are still lacking. Nevertheless, a significant proportion of patients affected with IIM fail to respond to conventional immunosuppressants. Despite the overall good prognosis, difficult-to-treat skin disease might be an issue in the management of anti-SAE patients (35–38). As a matter of fact, in our cohort, 6 out of 10 patients (60% refractory rate) with refractory skin disease were given different immunosuppressants to control cutaneous disease activity, including CYC in a patient with severe cutaneous involvement and skin ulcers. Also, previous studies have pinpointed this feature of anti-SAE patients, reporting a percentage of difficult-to-treat and resistant skin manifestations in approximately 40% of patients (20, 25). Interestingly, a good response to BARI in the cutaneous domain was described in a young patient of our cohort. Among all IIM subtypes, growing evidence supports the role of interferon (IFN) in sustaining the pathogenesis of several manifestations in DM, particularly cutaneous disease (39). IFN signaling relies upon the Janus kinase-signal transducer and activator of transcription (JAK/STAT) cascade, which has become the target of the novel family of small-molecule JAK inhibitors in various diseases (40). Nowadays, the role of BARI in the management of IIM is under evaluation in two clinical trials (41, 42). Finally, despite the risk of infection, which should always be considered (43, 44), another option for refractory cases is the use of rituximab (RTX) (45), similar to one case from our cohort.
Our study has strengths and limitations. The main strength is that our patients were followed up prospectively with a long period of observation (mean 3 years); in addition, clinical and serological data were prospectively recorded at all visits; therefore, most of the patients had complete data for the study.
Limitations include the small number of patients described and the retrospective nature of the study. Furthermore, ethnicity can be a limitation, since all of our patients were Caucasian, and it may not reflect the heterogeneous characteristics of anti-SAE patients among different countries (20). The absence of ILD cases among anti-SAE patients in our series should be interpreted with caution. Although anti-SAE dermatomyositis typically shows lower ILD prevalence than other subsets, such as anti-MDA5, larger cohorts and longer follow-up are required to precisely estimate ILD risk (9).
Another limitation is that, in our cohort, only one patient underwent muscular biopsy for the histological confirmation of inflammatory myositis. On the other hand, current EULAR guidelines (16) recommend muscle biopsy in patients with presumed IIM, but it is not mandatory when cutaneous manifestations and serological characteristics are strongly suggestive of DM (3).
Conclusions
Amyopathic or hypomyopathic DM is the most common clinical presentation of patients with anti-SAE positivity enrolled in our cohort. Skin involvement is severe and refractory in most cases and requires multiple lines of immunosuppressive therapy. Muscular involvement is usually mild but tends to develop during follow-up. Finally, given the limited number of cases and the current gaps in knowledge, future research should aim to identify reliable biomarkers that can better define the clinical spectrum, predict disease course, and guide therapeutic decisions in anti-SAE dermatomyositis.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The study was carried out in accordance with the Declaration of Helsinki and approved by our institution’s Ethics committee (Azienda Ospedaliera di Padova, n. 5505/A/22). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
RD: Writing – original draft, Writing – review & editing. AG: Writing – original draft, Writing – review & editing. EZ: Writing – original draft, Writing – review & editing. CF: Writing – original draft, Writing – review & editing. MB: Writing – original draft, Writing – review & editing. FP: Writing – original draft, Writing – review & editing. MG: Writing – original draft, Writing – review & editing. ET: Writing – original draft, Writing – review & editing. BM: Writing – original draft, Writing – review & editing. MZ: Writing – original draft, Writing – review & editing. SP: Writing – original draft, Writing – review & editing. CC: Writing – original draft, Writing – review & editing. LQ: Writing – original draft, Writing – review & editing. AD: Writing – original draft, Writing – review & editing. LI: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Acknowledgments
We would like to thank Dr. Nicoletta Gallo (University Hospital of Padua) for image 1.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Iaccarino L, Ghirardello A, Bettio S, Zen M, Gatto M, Punzi L, et al. The clinical features, diagnosis and classification of dermatomyositis. J Autoimmun. (2014) 48–49:122–7. doi: 10.1016/j.jaut.2013.11.005
2. Franco C, Gatto M, Iaccarino L, Ghirardello A, and Doria A. Lymphocyte immunophenotyping in inflammatory myositis: a review. Curr Opin Rheumatol. (2021) 33:522–8. doi: 10.1097/BOR.0000000000000831
3. Lundberg IE, de Visser M, and Werth VP. Classification of myositis. Nat Rev Rheumatol. (2018) 14:269–78. doi: 10.1038/nrrheum.2018.41
4. Ghirardello A, Zampieri S, and Iaccarino L. Myositis specific and myositis associated autoantibodies in idiopathic inflammatory myopathies: a serologic study of 46 patients. Reumatismo. (2005) 57:22–8. doi: 10.4081/reumatismo.2005.22
5. Ghirardello A, Zampieri S, Iaccarino L, Tarricone E, Bendo R, Gambari PF, et al. Anti-mi-2 antibodies. Autoimmunity. (2005) 38:79–83. doi: 10.1080/08916930400022681
6. Ghirardello A, Zampieri S, Tarricone E, Iaccarino L, Bendo R, Briani C, et al. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity. (2006) 39:217–21. doi: 10.1080/08916930600622645
7. Zampieri S, Ghirardello A, Iaccarino L, Tarricone E, Gambari PF, and Doria A. Anti-Jo-1 antibodies. Autoimmunity. (2005) 38:73–8. doi: 10.1080/08916930400022640
8. Betteridge Z, Gunawardena H, North J, Slinn J, and McHugh N. Identification of a novel autoantibody directed against small ubiquitin-like modifier activating enzyme in dermatomyositis. Arthritis Rheum. (2007) 56:3132–7. doi: 10.1002/art.22862
9. Betteridge ZE, Gunawardena H, Chinoy H, North J, Ollier WE, Cooper RG, et al. Clinical and human leuco- cyte antigen class II haplotype associations of autoantibodies to small ubiquitin-like modifier enzyme, a dermatomyositis-specific autoantigen target, in UK Caucasian adult-onset myositis. Ann Rheum Dis. (2009) 68:1621–5. doi: 10.1136/ard.2008.097162
10. Tarricone E, Ghirardello A, Rampudda M, Bassi N, Punzi L, and Doria A. Anti-SAE antibodies in autoimmune myositis: identification by unlabelled protein immunoprecipitation in an Italian patient cohort. J Immunol Methods. (2012) 384:128–34. doi: 10.1016/j.jim.2012.07.019
11. Ghirardello A, Gatto M, Franco C, Zanatta E, Padoan R, Ienna L, et al. Detection of myositis autoantibodies by multi-analytic immunoassays in a large multicenter cohort of patients with definite idiopathic inflammatory myopathies. Diagnostics (Basel). (2023) 13:3080. doi: 10.3390/diagnostics13193080
12. Wilkinson KA and Henley JM. Mechanisms, regulation and consequences of protein SUMOylation. Biochem J. (2010) 428:133–45. doi: 10.1042/BJ20100158
13. Betteridge Z, Tansley S, Shaddick G, Chinoy H, Cooper RG, New RP, et al. Frequency, mutual exclusivity and clinical associations of myositis autoantibodies in a combined European cohort of idiopathic inflammatory myopathy patients. J Autoimmun. (2019) 101:48–55. doi: 10.1016/j.jaut.2019.04.001
14. Fujimoto M, Matsushita T, Hamaguchi Y, Kaji K, Asano Y, Ogawa F, et al. Autoantibodies to small ubiquitin-like modifier activating enzymes in Japanese patients with dermatomyositis: comparison with a UK Caucasian cohort. Ann Rheum Dis. (2013) 72:151–3. doi: 10.1136/annrheumdis-2012-201736
15. Bodoki L, Nagy-Vincze M, Griger Z, Betteridge Z, Szöllősi L, Dankó K, et al. Four dermatomyositis- specific autoantibodies—anti-TIF1c, anti-NXP2, anti-SAE and anti-MDA5-in adult and juvenile patients with idiopathic inflammatory myopathies in a Hungarian cohort. Autoimmun Rev. (2014) 13:1211–9. doi: 10.1016/j.autrev.2014.08.011
16. Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, Visser M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. (2017) 76:1955–64. doi: 10.1136/annrheumdis-2017-211468
17. Bohan A and Peter JB. Polymyositis and dermatomyositis. N Engl J Med. (1974) 292:344–7. doi: 10.1056/NEJM197502132920706
18. Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, et al. 119Th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis. Neuromuscul Disord. (2004) 14:337–45. doi: 10.1016/j.nmd.2004.02.006
19. Ge Y, Lu X, Shu X, and Wuang G. Clinical characteristics of anti-SAE antibodies in Chinese patients with dermatomyositis in comparison with different patient cohorts. Sci Rep. (2017) 7:188. doi: 10.1038/s41598-017-00240-6
20. Demortier J, Vautier M, Chosidow O, Gallay L, Bessis D, Berezne A, et al. Anti-SAE autoantibody in dermatomyositis: original comparative study and review of the literature. Rheumatol (Oxford). (2023) 62:3932–39. doi: 10.1093/rheumatology/kead154
21. Jia E, Wei J, Geng H, Qiu X, Xie J, Xiao Y, et al. Diffuse pruritic erythema as a clinical manifestation in anti-SAE antibody–associated dermatomyositis: a case report and literature review. Clin Rheumatol. (2019) 38:2189–93. doi: 10.1007/s10067-019-04562-w
22. Muro Y, Sugiura K, and Akiyama M. Low prevalence of anti-small ubiquitin-like modifier activating enzyme antibodies in dermato- myositis patients. Autoimmunity. (2013) 46:279–84. doi: 10.3109/08916934.2012.755958
23. Gono T, Tanino Y, Nishikawa A, Kawamata T, Hirai K, Okazaki Y, et al. Two cases with autoantibodies to small ubiquitin-like modifier activating enzyme: a potential unique subset of dermatomyositis-associated interstitial lung disease. Int J Rheum Dis. (2019) 22:1582–6. doi: 10.1111/1756-185X.13593
24. Muro Y, Sugiura K, Nara M, Sakamoto I, Suzuki N, and Akiyama M. High incidence of cancer in anti- small ubiquitin-like modifier activating enzyme antibody–positive dermatomyositis. Rheumatol (Oxford). (2015) 54:1745–7. doi: 10.1093/rheumatology/kev247
25. Albayda J, Mecoli C, Casciola-Rosen L, Danoff SK, Lin CT, Hines D, et al. A North American cohort of anti-SAE dermatomyositis: clinical phenotype, testing, and review of cases. ACR Open Rheumatol. (2021) 3:287–94. doi: 10.1002/acr2.11247
26. Chen Z, Hu W, Wang Y, Guo Z, Sun L, and Kuwana M. Distinct profiles of myositis-specific autoantibodies in Chinese and Japanese patients with polymyositis/dermatomyositis. Clin Rheumatol. (2015) 34:1627–31. doi: 10.1007/s10067-015-2935-9
27. Zampeli E, Venetsanopoulou A, Argyropoulou OD, Mavragani CP, Tektonidou MG, and Vlachoyiannopoulos PG. Myositis autoantibody profiles and their clinical associations in Greek patients with inflammatory myopathies. Clin Rheumatol. (2019) 38:125–32. doi: 10.1007/s10067-018-4267-z
28. Victor J, Zanardo L, He´ron-Mermin D, Poursac N, Solé G, Bordes C, et al. Retrospective analysis of anti-TIF1gamma, anti-NXP2 and anti-SAE1/2 antibodies carriers at Bordeaux University Hospital from November 2014 to February 2017. Rev Med Interne. (2019) 40:70–81. doi: 10.1016/j.revmed.2018.11.003
29. Matsuo H, Yanaba K, Umezawa Y, Nakagawa H, and Muro Y. Anti-SAE antibody-positive dermatomyositis in a Japanese patient: a case report and review of the literature. J Clin Rheumatol. (2019) 25:e115–6. doi: 10.1097/RHU.0000000000000683
30. Lee S, Findeisen J, McLean C, and Stavrakoglou A. Recalcitrant ulcers associated with anti-small ubiquitin-like modifier activating enzyme–positive dermatomyositis treated with surgery followed by intravenous immunoglobulin. Australas J Dermatol. (2018) 59:e76–8. doi: 10.1111/ajd.12659
31. Zamora E, Seder-Colomina E, Holgado S, Quirant-Sanchez B, Mate JL, Martínez-Cáceres EM, et al. Heart–lung–muscle anti-SAE syndrome: an atypical severe combination. J Clin Med. (2018) 8:20. doi: 10.3390/jcm8010020
32. Zanatta E, Cocconcelli E, Castelli G, Giraudo C, Fraia AS, De Zorzi E, et al. Interstitial lung disease with and without progressive fibrosing phenotype in patients with idiopathic inflammatory myopathies: data from a large multicentric cohort. RMD Open. (2023) 9:e003121. doi: 10.1136/rmdopen-2023-003121
33. Stockton D, Doherty VR, and Brewster DH. Risk of cancer in patients with dermatomyositis or polymyositis, and follow-up implications: a Scottish population-based cohort study. Br J Cancer. (2001) 85:41–5. doi: 10.1054/bjoc.2001.1699
34. Hill CL, Zhang Y, Sigurgeirsson B, Pukkala E, Mellemkjaer L, and Airio A. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. (2001) 357:96–100. doi: 10.1016/S0140-6736(00)03540-6
35. Aggarwal R, Charles-Schoeman C, Schessl J, Bata-Csörgő Z, Dimachkie MM, Griger Z, et al. Trial of intravenous immune globulin in dermatomyositis. N Engl J Med. (2022) 387:1264–78. doi: 10.1056/NEJMoa2117912
36. Inoue S, Okiyama N, Shobo M, Motegi S, Hirano H, Nakagawa Y, et al. Diffuse erythema with ‘angel wings’ sign in Japanese patients with anti-small ubiquitin-like modifier activating enzyme antibody–associated dermatomyositis. Br J Dermatol. (2018) 179:1414–5. doi: 10.1111/bjd.17026
37. Daly ML, Gordon PA, and Creamer D. Cutaneous features of dermato- myositis associated with myositis-specific antibodies. Br J Dermatol. (2017) 176:1662–5. doi: 10.1111/bjd.15020
38. Peterson LK, Jaskowski TD, La’ulu SL, and Tebo AE. Antibodies to small ubiquitin-like modifier activating enzyme are associated with a diagnosis of dermatomyositis: results from an unselected cohort. Immunol Res. (2018) 66:431–36. doi: 10.1007/s12026-018-9006-7
39. Gasparotto M, Franco C, Zanatta E, Ghirardello A, Zen M, Iaccarino L, et al. The interferon in idiopathic inflammatory myopathies: Different signatures and new therapeutic perspectives. A literature review. Autoimmun Rev. (2023) 22:103334. doi: 10.1016/j.autrev.2023.103334
40. Griffiths CE, Iaccarino L, Naldi L, Olivieri I, Pipitone N, Salvarani C, et al. Psoriasis and psoriatic arthritis: immunological aspects and therapeutic guidelines. Clin Exp Rheumatol. (2023) 24:S72–8.
41. Baricitinib in Patients With Relapsing or naïve Dermatomyositis (BIRD). Available online at: https://clinicaltrials.gov/ct2/show/NCT04972760 (Accessed October 2025).
42. JAK 1/2 inhibitor, baricitinib, in the treatment of adult IIM (MYOJAK). Available online at: https://clinicaltrials.gov/ct2/show/NCT04208464 (Accessed October 2025).
43. Zampieri S, Ghirardello A, Iaccarino L, Briani C, Sarzi-Puttini P, Atzeni F, et al. Polymyositis and dermatomyositis and infections. Autoimmunity. (2006) 39:191–6. doi: 10.1080/08916930600622348
44. Iaccarino L, Gatto M, Bettio S, Caso F, Rampudda M, Zen M, et al. Overlap connective tissue disease syndromes. Autoimmun Rev. (2013) 12:363–73. doi: 10.1016/j.autrev.2012.06.004
45. Nalotto L, Iaccarino L, Zen M, Gatto M, Borella E, Domenighetti M, et al. Rituximab in refractory idiopathic inflammatory myopathies and antisynthetase syndrome: Personal experience and review of the literature. Immunol Res. (2013) 56:362–70. doi: 10.1007/s12026-013-8408-9
Keywords: inflammatory myopathies, dermatomyositis, anti-SAE antibodies, refractory skin involvement, immunosuppresants
Citation: Depascale R, Ghirardello A, Zanatta E, Franco C, Bracalenti M, Pettorossi F, Gatto M, Treppo E, Moccaldi B, Zen M, Piaserico S, Ciolfi C, Quartuccio L, Doria A and Iaccarino L (2025) Patients with anti-SAE+ dermatomyositis display refractory and difficult-to-treat skin manifestations: case series from two Italian cohorts and review of literature. Front. Immunol. 16:1597282. doi: 10.3389/fimmu.2025.1597282
Received: 10 April 2025; Accepted: 30 September 2025;
Published: 16 October 2025.
Edited by:
Rosaria Talarico, ERN ReCONNET Coordination Team, ItalyReviewed by:
Antonella Notarnicola, Karolinska Institutet (KI), SwedenYu Shan, Shanghai University of Traditional Chinese Medicine, China
Copyright © 2025 Depascale, Ghirardello, Zanatta, Franco, Bracalenti, Pettorossi, Gatto, Treppo, Moccaldi, Zen, Piaserico, Ciolfi, Quartuccio, Doria and Iaccarino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luca Iaccarino, bHVjYS5pYWNjYXJpbm9AdW5pcGQuaXQ=