 Evangelia Fouka
Evangelia Fouka Anders Lindén
Anders Lindén Apostolos Bossios
Apostolos Bossios- 1Division for Lung and Airway Research, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden
- 2Lung Laboratory, Center for Molecular Medicine, Karolinska University Hospital, Stockholm, Sweden
- 3National and Kapodistrian University of Athens, 2nd Department of Respiratory Medicine, University General Hospital Attikon, Athens, Greece
- 4Karolinska Severe COPD Center, Department of Respiratory Medicine and Allergy, Karolinska University Hospital, Stockholm, Sweden
- 5Karolinska Severe Asthma Center, Department of Respiratory Medicine and Allergy, Karolinska University Hospital, Stockholm, Sweden
Bronchiectasis is a chronic airway disease characterized by dysbiosis, persistent inflammation, and permanent structural airway damage. Neutrophilic inflammation is a key pathogenic feature, as indicated by enhanced neutrophil-derived proteases and formation of neutrophil extracellular traps (NETs), associated with poor prognosis. However, recent studies have identified an eosinophilic endotype in up to 30% of patients, characterized by higher levels of type 2 (T2) cytokines and fractional exhaled nitric oxide (FeNO). The role of T helper (Th) cells in the dysregulated inflammatory environment of bronchiectasis remains unclear. Evidence suggests that persistent bacterial infection can skew adaptive immunity from Th1 toward Th2 response, while the airway microbiome-IL-17 axis is also a critical regulator of chronic inflammation. T regulatory (Treg) cells have been shown to play a protective role against excessive chronic inflammation by modulating the function of several types of effector cells, including the Th17 subset. However, the capacity of this subset to delay or prevent disease progression remains to be determined Microbial dysbiosis, with loss of diversity and increased quantity of bacterial pathogens, may also be important for disease progression, and emerging evidence indicates that distinct inflammatory endotypes associate with specific microbiota alterations, especially in severe disease. In this review, we provide an overview of the immune cells and cytokine signaling that are involved in the pathogenesis of bronchiectasis. Additionally, we present the main endotypes of bronchiectasis and explore the relationships between the type of inflammation and alterations in microbiota, as well as the potential benefits of targeting specific pathophysiological mechanisms for the management of bronchiectasis. This review also examines how bacterial infection can shift adaptive immunity from Th1 toward Th2 responses, the role of the airway microbiome-IL-17 axis in chronic inflammation and the potential protective role of Treg cells against excessive inflammation. Novel therapeutic strategies are highlighted, with focus on targeting specific cytokine signaling pathways and restoring Th17/Treg balance These developments underscore a shift toward precision medicine in bronchiectasis, emphasizing the importance of identifying specific inflammatory endotypes to tailor treatment strategies effectively.
1 Introduction
Bronchiectasis is a chronic airway disease characterized by permanent and abnormal dilation of the bronchi and clinically by persistent cough and sputum production that periodically worsen due to acute respiratory infections, known as “exacerbations” (1). Typical structural airway changes include thickening of the bronchial walls, loss of normal ciliary function, mucus plugging, and the formation of sac-like or cylindrical bronchial dilations (2). Although the exact prevalence of bronchiectasis remains uncertain, a report in 2013 indicated an increase of 40% since 2003, with a prevalence of 566 per 100–000 population (3), whereas a more recent Medicare claims analysis raised the annual prevalence to 701 per 100,000 population in subjects >65 years of age (4). Notably, the term bronchiectasis represents “an umbrella term” that in reality includes a variety of associated disorders (5), with several recent studies revealing significant worldwide etiologic diversity (6–8). Several attempts have been made to define clinical phenotypes in bronchiectasis (9, 10). However, taxonomy by cause appears to be clinically relevant only in a few conditions for which specific treatments are available (11).
Prior to our understanding of modern immunology, the most accepted model of bronchiectasis pathogenesis was the “vicious cycle” hypothesis proposed by Cole in 1986 (12). This model suggested that an initial insult in susceptible individuals may trigger chronic airway inflammation, leading to structural airway damage and abnormal mucociliary clearance. Abnormal inflammatory responses stimulated by subsequent infections may result in further airway damage and remodeling, perpetuating disease progression. More recently, the “vicious cycle” hypothesis has been replaced by the “vicious vortex” concept proposed by Flume et al. (13), which suggests a complex interaction of individual elements contributing to disease pathogenesis rather than a constant sequence of events.
The pathology of bronchiectasis may relate to a range of endogenous and exogenous insults, such as bacterial resistance against antibiotics, environmental pollutants, the presence and activity of comorbidities, and inflammatory endotypes, all of which contribute to the disease’s persistence and severity (14). Antibiotic overuse and/or misuse can lead to the emergence of resistant pathogens, that may persist within biofilms complicating treatment and maintaining chronic infection, inflammation and tissue damage (15). Environmental factors, particularly air pollution, also play a significant role in the progression of bronchiectasis. Exposure to pollutants such as particulate matter (PM10) and nitrogen dioxide (NO2), even at levels below established safety thresholds, can trigger systemic inflammation, exacerbating the disease and leading to adverse health outcomes and increased healthcare utilization (16). Multimorbidity in bronchiectasis is common and, although the specific impact on bronchiectasis pathogenesis is poorly. However, it is known that such comorbidities are associated with poorer clinical outcomes and increased mortality (17, 18). Understanding this multifactorial pathology will probably be essential for developing effective management and prevention strategies for bronchiectasis.
The inflammatory milieu in bronchiectasis is typically dominated by excessive accumulation of neutrophils, seemingly regardless of etiology (19–21). The progressive imbalance between pro- and anti-inflammatory cytokines and their inhibitors results in excessive accumulation of immune effector cells in the airways, leading to direct epithelial damage and ciliary dysfunction (22, 23). However, recent studies have indicated that airway eosinophilic inflammation is present in up to 20-30% of patients, predominating over or occurring concurrently with neutrophilic inflammation (24–26). Other immune cells, such as macrophages, are believed to play essential roles under certain circumstances (27), and significant numbers of peribronchial lymphoid aggregates containing B- and CD4+ T lymphocytes and germinal centers have been found in bronchial mucosal biopsies from patients with bronchiectasis (28–30). In contrast, patients with T-cell dysfunction are at increased risk of developing bronchiectasis (31, 32). Table 1 summarizes the roles of key inflammatory cells in the pathogenesis of bronchiectasis.
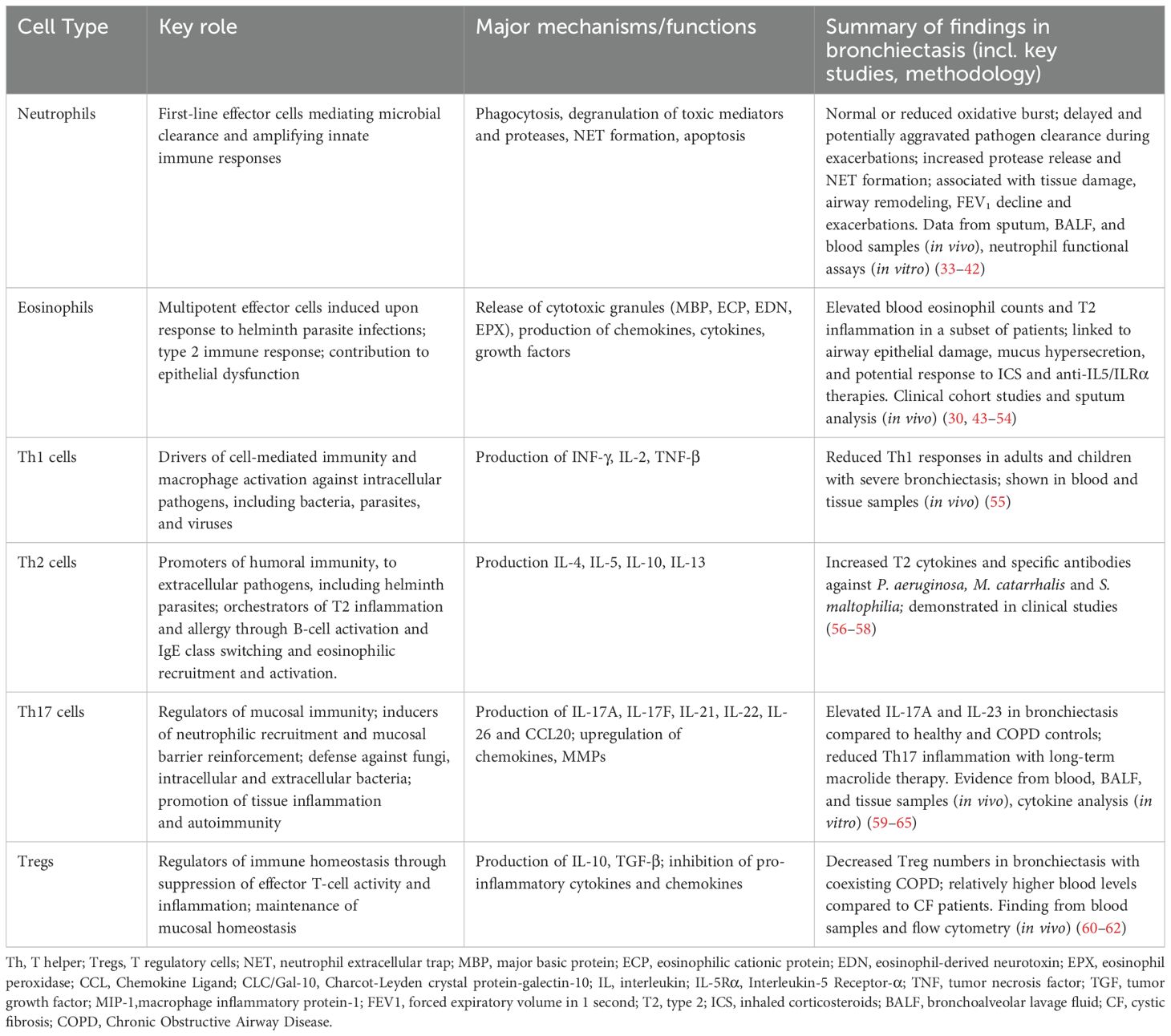
Table 1. Inflammatory cells contributing to disease pathogenesis in bronchiectasis.
There is increasing awareness of bronchiectasis as a clinical entity with new and developing treatment options. Therefore, expanding research into endotypes, which reflect the underlying pathophysiological mechanisms of the disease, may help us establish clinically adequate characterization of specific patient subpopulations. In this narrative review we provide an overview of immune cells and cytokine pathways involved in the pathogenesis of bronchiectasis not related to cystic fibrosis (CF). We also explore the role of neutrophilic inflammation as a key pathogenic feature associated with poor prognosis and the presence of an eosinophilic endotype in a significant subset of patients. The review also examines how bacterial infection can shift adaptive immunity from Th1 toward Th2 responses, the role of the airway microbiome-IL-17 axis in chronic inflammation and the potential protective role of Treg cells against excessive inflammation. Additionally, we explore the relationship between distinct inflammatory endotypes and microbiota alterations in severe bronchiectasis, as well as the potential benefits of targeting specific pathophysiological mechanisms in the management of bronchiectasis. We have performed searches in PubMed for bronchiectasis AND/OR inflammation AND/OR neutrophils AND/OR eosinophils AND/OR T helper lymphocytes AND/OR T regulatory cells between 1991 to present, with two-thirds of the articles published since 2016. Both eosinophilic and neutrophilic endotypes are covered, and data highlighting the potential protective and pathological roles of T-helper (Th) and T-regulatory (Treg) cells in this disorder are presented.
2 Inflammatory endotypes in bronchiectasis
2.1 The neutrophilic endotype
Neutrophils are the most abundant inflammatory cells in the airways of patients with bronchiectasis and are rapidly recruited from the bone marrow by several chemoattractants (such as IL-8/CXCL-8 and leukotriene B4) or proinflammatory cytokines (such as TNF-α) (66). In the lungs, neutrophils facilitate the rapid clearance of invading pathogens via a range of effector functions, including phagocytosis, degranulation of toxic mediators, and production of oxygen-free radicals (67, 68). Although phagocytosis is the primary means by which neutrophils contribute to bacterial killing, the formation of neutrophil extracellular traps (NETs) is also crucial for the trapping and elimination of pathogens through the release of extracellular neutrophil DNA webs containing large amounts of neutrophil serine proteases (SNPs) such as neutrophil elastase (NE), proteinase 3 (PR3), and cathepsin G (CatG), as well as antimicrobial peptides and platelets (69–72). Excessive levels of NETs in the airways promote an exaggerated inflammatory response, which is linked to disrupted mucociliary function, extracellular matrix degradation and hypersecretion of mucus with increased viscosity (33–35).
Neutrophilic inflammation and dysfunctional killing of pathogens are key elements in the pathogenesis of bronchiectasis (36). An early study reported that neutrophils preserve their normal phagocytic activity in the peripheral blood of patients with bronchiectasis (37). However, subsequent research has revealed that although phagocytosis does not differ between patients with bronchiectasis and healthy controls, neutrophilic oxidative burst may be reduced in the former (38). Indeed, this is compatible with the idea that the ability of airway neutrophils to kill bacteria is hampered and contributes to the pathogenesis in bronchiectasis. Prolonged neutrophil survival has been demonstrated in bronchiectasis, resulting in increased numbers of neutrophils, neither apoptotic nor necrotic, in the airways (39). Similarly, delayed apoptosis has been reported in patients with stable bronchiectasis, and this phenomenon may increase during exacerbations (40).
In bronchiectasis, airway neutrophilic inflammation, characterized by high concentrations of NETs, NE, and other neutrophil proteins, is associated with chronic airway infection, worse lung function, higher exacerbation rates, and more severe disease (20, 34, 41). Moreover, the antimicrobial peptide LL-37 may induce NET formation, and increased levels of this peptide have been associated with chronic P. pseudomonas infection, FEV₁ decline and higher exacerbation rates (42). Matrix metalloproteinases (MMP)-8 and -9 and pregnancy zone protein (PZP), a recently discovered NET-related protein, display increased levels in sputum from patients with bronchiectasis and have shown a positive correlation with clinical parameters, including radiological abnormalities, lung function, bacterial load, disease severity, and exacerbation risk (73, 74). Individuals with PI3K syndrome develop severe bronchiectasis, presumably because dysregulated PI3K activation contributes to immune dysfunction by augmenting neutrophil migration, degranulation, and ROS generation (20).
2.2 The eosinophilic endotype in bronchiectasis
Eosinophils are bone marrow-derived granulocytes that differentiate from CD34+ pluripotent progenitors and are regulated by several transcription factors, interleukins, and epithelium-derivate alarmins, including GATA-1, eotaxin-1, IL-5, IL-3, Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF), IL-33, IL-25, and thymic stromal lymphopoietin (TSLP) (75). Until recently, their role was restricted to host defense against parasitic infections (76). However, eosinophils are now recognized as key players in a range of airway disorders that can display pathological type 2 (T2) inflammation in local tissues, such as asthma, chronic rhinosinusitis with nasal polyps, eosinophilic granulomatosis with polyangiitis (EGPA) and a subset of patients with chronic obstructive pulmonary disease (COPD) (77). Degranulation of eosinophils, airway epithelium disruption, and increased mucus production may contribute to this association through the release of various mediators, such as chemokines, cytokines, and growth factors, which promote an intense airway inflammatory response (43). Moreover, their prolonged survival in inflamed tissues, mainly driven by IL-5, a cytokine produced by Th2 cells, mast cells, and type 2 innate lymphoid cells (ILC2s), is responsible for the persistence of inflammation, tissue damage, and airway remodeling in T2-related disorders (44, 45).
Recently, the eosinophilic endotype of bronchiectasis has been recognized, although the published data on these patients is limited. Along these lines, increased numbers of eosinophils have been found in the airway mucosa of patients with bronchiectasis compared with healthy controls (28). A small study has shown that eosinophilia, defined as ≥3% eosinophils in induced sputum, is associated with increased levels of T2 biomarkers, including FeNO and IL-13, although to a lesser degree compared to a severe refractory asthma reference population (25). Applying cutoff values of ≥3% and ≥300 cells/μL, a significant association between sputum and blood eosinophil counts has been demonstrated in approximately 20% of patients with bronchiectasis, despite the absence of classic eosinophil-driven comorbidities such as asthma and allergic pulmonary aspergillosis (ABPA) (24). These findings indicate that eosinophilic inflammation may play a significant role in a substantial proportion of bronchiectasis cases without coexisting asthma. This finding implies the potential for an alternative treatment strategy aligned with the current concept in COPD (46, 47). Recently, it was proposed that a T2-high endotype in bronchiectasis patients without concomitant asthma may be defined by FeNO levels of ≥25 ppb and a blood eosinophil count of ≥300 cells/μL (78). However, a recent study reported no significant differences in blood neutrophil levels between bronchiectasis patients with varying eosinophil levels (79). In addition, blood eosinophil counts of <100 cells/μL are linked to a more severe phenotype, probably reflecting a myeloid shift toward neutrophil production in response to persistent infection (80). Therefore, it remains unclear whether the presence of eosinophilia reflects predominant eosinophilic inflammation or a mixed inflammatory pattern owing to the dual nature of eosinophils (pro-inflammatory and anti-infective) (77).
Interestingly, there is evidence that certain airway bacteria promote eosinophilic inflammation in bronchiectasis. Recent studies have shown that specific airway pathogens, such as S. aureus, Streptococcus spp., and P. aeruginosa, are associated with higher blood eosinophil counts and shorter times to exacerbation (24, 81). In addition to reports that P. aeruginosa directly drives T2 inflammation in CF-related bronchiectasis (82), identification of this pathogen may explain the source of eosinophilia in the general bronchiectasis population, considering the known bactericidal capacity of eosinophils, thereby presenting a potentially treatable trait (48). There is also evidence that fungal exposure and sensitization may drive the T2 endotype in patients with bronchiectasis without evidence of ABPA, which can be identified by the presence of increased blood eosinophils, FeNO, and specific IgE levels (49).
3 T-helper and T-regulatory cells
3.1 T-helper and regulatory T (Treg) cells as immune response maestros
T lymphocytes, commonly referred to as T cells, are critical players in the adaptive immune system, involved in the initiation of cell-based immune responses that protect the host against various diseases. These cells originate from thymocyte progenitors generated in the bone marrow and are categorized into four primary types: CD4+, CD8+, γδ T cells, and natural killer T (NKT) cells (50). Upon identification of cognate antigens by T cell receptors (TCRs) through MCH molecules on the surface of antigen-presenting cells (APCs), naïve CD4+ and CD8+ T cells undergo activation, clonal expansion, and differentiation, ultimately performing effector functions, including destruction of infected cells, cytokine production, and upregulation of immune responses (51). A subset of T cells transforms into memory T cells, which exhibit rapid effector responses upon re-encountering familiar antigens, thereby providing the host with robust and enduring protection (52). In parallel, naïve CD4+ T cells, also known as Th cells, have the potential to differentiate into functionally distinct subsets (Th1, Th2, Th9, Th17, Th22, follicular helper T (Tfh), and Treg) under the regulation of different transcription factors and signaling molecules, depending on the cytokine content of a certain microenvironment (53) (Figure 1).
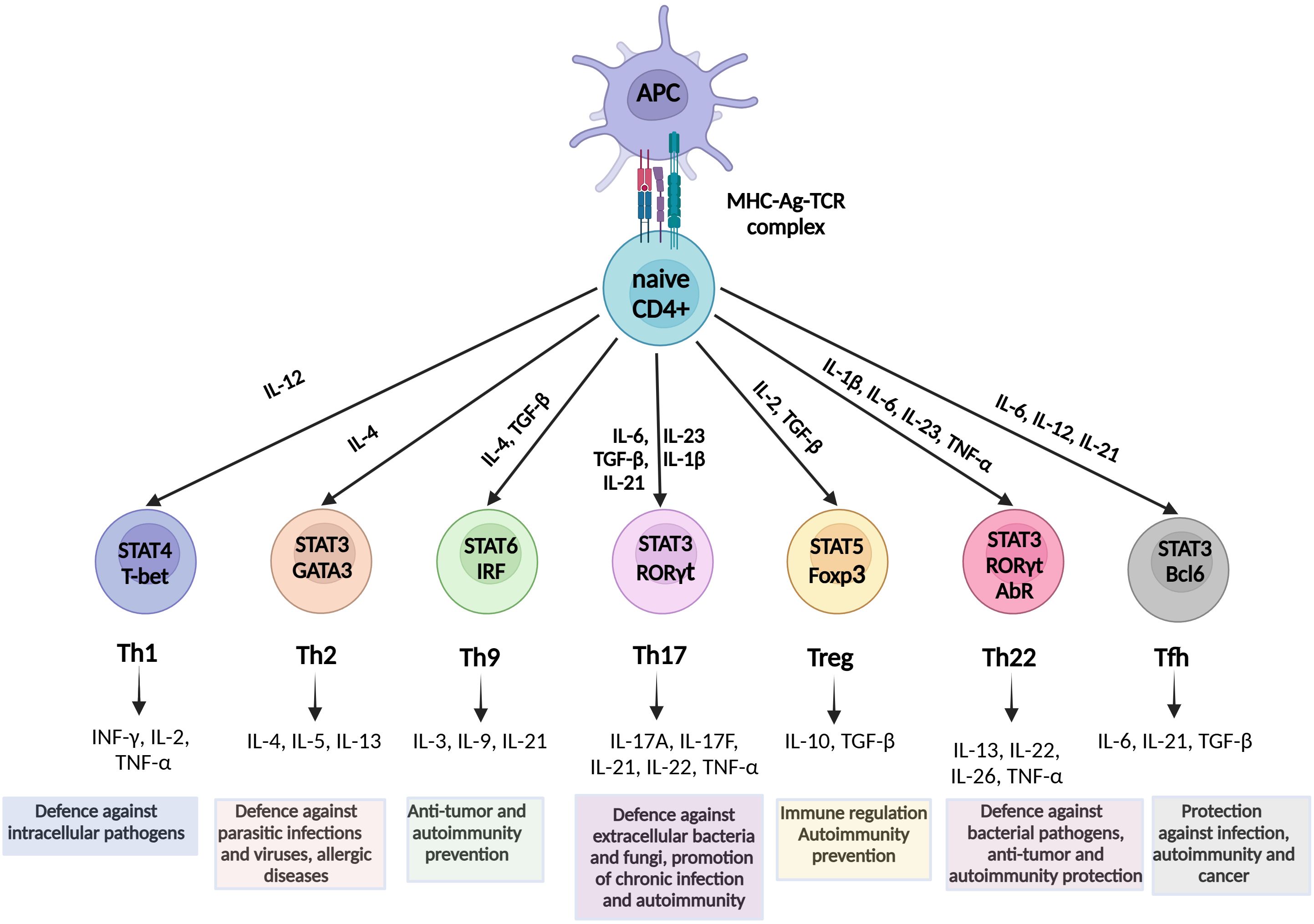
Figure 1. Schematic overview of Th cell differentiation from naive CD4+ T cells. Naïve CD4+ T cells differentiate into Th1, Th2, Th9, Th17, Th22, Treg, and Tfh subsets under the influence of specific cytokines and transcription factors. Each subset mediates distinct immune functions. Th1, intracellular pathogens and viruses defense; Th2, protection against parasitic infections, T2 immunity; Th9, anti-tumor activity, allergy; Th17, mucosal defense, chronic inflammation; Tregs, immune tolerance, autoimmunity prevention; Th22, barrier protection, autoimmunity prevention; and Tfh, B-cell activation and antibody production. Key signaling pathways and effector cytokines are indicated. STAT, Signal transducer and activator of transcription; INF, Interferon; IL, Interleukin; TNF, Tumor necrosis factor; TGF, Transforming growth factor; RORγt, related orphan receptor gamma; FOXp3, forkhead box P3; Tfh, T follicular helper.
After differentiation from naïve CD4+ T cells, Th1 cells provide phagocyte-dependent protective immunity against intracellular pathogens and viruses via IFN-γ, IL-2, and TNF-α production (54, 83). However, they also contribute to the resolution of acute tissue inflammation by inhibiting the powerful neutrophil chemoattractant IL-8 (84). In contrast, Th2 cells facilitate a phagocyte-independent protective mechanism associated with parasitic infections and allergic diseases via the production of IL-4, IL-5, IL-10, and IL-13 (85). Nonetheless, a reduced Th2 response reduces immune complex formation, thereby facilitating confinement of tissue damage (86). The Th9 CD4+ subgroup is believed to originate from Th2 cells, after their “reprogramming” by TGF-β and IL-4, and has been shown to promote antitumor protection and prevent autoimmunity (87). Notably, the proinflammatory Th22 subset participates in protective responses against bacterial pathogens and is associated with autoimmune pathogenesis and defense against tumors (88). Finally, Tfh cells play a critical role in protective immunity in response to various pathogenic infections, autoimmune disorders, and cancers (89, 90).
The current understanding is that Th17 cells play a vital role in preserving the integrity of mucosal epithelial barriers, while they are also crucial in protecting the host against bacterial and fungal mucosal infections through the secretion of Th17 cytokines (IL-17A, IL-17F, IL-21, IL-22, and IL-26), and potentially TNF-α and IL-6 upon certain stimulation (91, 92). These cells facilitate leukocyte tissue infiltration through upregulation of chemokines and metalloproteinases, however at the cost of promoting chronic inflammation and severe autoimmunity (93, 94).
The Treg subset is a discrete anti-inflammatory CD4+ subset that constitutes up to 10% of total CD4+ cells (95). An essential characteristic of Tregs is that they express FOXP3, a transcription factor critical for their development and function (96). According to their origin, FOXP3 Tregs can be divided into two major subsets: i) thymus-derived Tregs (tTregs), also known as natural Tregs, and ii) peripherally-induced Tregs (pTregs) (97, 98). These cells play a critical role in the maintenance of immune tolerance and have been shown to prevent the progression of several autoimmune and inflammatory diseases by suppressing numerous immune cells involved in innate and adaptive immune responses, such as B cells, CD4+ T cells, Th cells, CD8+ cells, NK cells, NKT cells, macrophages, dendritic cells, and neutrophils (99). Tregs perform their main functions through several mechanisms, such as cytokine secretion (mainly IL-10, but also TGF-β and IL-35), release of extracellular vesicles, granzyme/perforin mediated cellular cytolysis, cell-cell contact inducing modulation of dendritic cells, and metabolic perturbations (100). In addition, Tregs have been shown to suppress anticancer immune responses against autologous and tumor-expressing antigens by inhibiting the activation and differentiation of CD4+ helper and CD8+ cytotoxic T cells, thus contributing to tumor occurrence and progression (101, 102).
The balance between transcription factors Foxp3 and RORγt is critical in the Th17/Treg equilibrium, as Foxp3 activity may reduce Th17 cell differentiation through TGF-β-induced inhibition of RORγt function in the absence of other inflammatory stimuli (103). In contrast, IL-6, IL-21, and IL-23 promote Th17 versus Treg differentiation, favoring a proinflammatory environment (104).
3.2 The pathogenic potential role of T helper cells and the role of T regulatory cells
A growing body of evidence regarding the role of Th cells in CF is available, which may potentially provide valuable insights, even for patients with non-CF bronchiectasis. Preclinical studies have demonstrated that chronic infection with P. aeruginosa is characterized by a Th2-skewed response and further downregulation of the Th1 axis with decreased IFN-γ production, resulting in hampered ability to kill microbes (56, 57). In a murine model of CF, neutralization of endogenous IL-17 prior to infection with P. aeruginosa reduced bacterial load and neutrophil levels in the airway lumen (105). In contrast, transgenic mice lacking the IL-17RA receptor displayed a higher rate of infection and more significant mortality than wild-type mice after infection with two different P. aeruginosa strains, supporting that IL-17A plays a protective role in host defense against chronic pulmonary infection (106). However, higher peripheral blood Th17 signaling was strongly correlated with poorer lung function in a small study of adult CF patients compared to healthy controls (107). However, the role of Tregs in CF pathology remains to be elucidated. The results from one small study indicated a decreased peripheral blood Treg count in patients with CF colonized by P. aeruginosa. However, this decrease was not detected in subjects with intermittent P. aeruginosa infections, suggesting that chronic infection may be responsible for the Treg incapacity in CF (108).
In contrast to the case of Tregs, evidence regarding the role of specific Th cells in patients with non-CF-related bronchiectasis is scarce. A deficient Th1 response has been observed in adults and children with severe bronchiectasis (109). In contrast, a substantial Th2 response, resulting in increased levels of antibodies against P. aeruginosa, M. catarrhalis, and S. maltophilia has been reported in a controlled study on adult patients (38, 110). Notably, emerging evidence confirms the activation of T17 signaling in patients with bronchiectasis. In a small study on patients with stable bronchiectasis, investigators found significantly higher levels of several T17 cytokines in BALF than in healthy controls, including IL-17A and IL-23 (111). However, this study showed no association with clinical parameters or airway microbiology. Interestingly, while the gene expression of IL-17A in endobronchial biopsies was similar between the two groups, IL-1β levels were markedly higher in patients infected with P. aeruginosa and H. influenzae, suggesting a lesser involvement of Th17 in luminal host defense compared to innate neutrophilic activation. In contrast, higher levels of IL-17, IL-6 and Th17 cells, and lower levels of IL-10, TGF-β and Tregs in the peripheral blood were found in COPD patients with coexisting bronchiectasis and infection with P. aeruginosa, compared to patients with COPD only (112). Thus, Th17 cells may play a more prominent role in infection. Similarly, a recent study (113) demonstrated that patients with bronchiectasis unrelated to CF tend to have higher levels of Tregs in peripheral blood than CF patients, suggesting that chronic infection may impair Tregs in CF but not in non-CF bronchiectasis. Notably, treatment with a low-dose macrolide antibiotic (clarithromycin) significantly reduced both systemic and local Th17 responses in a small study of patients with bronchiectasis (114), suggesting that regulation of T17 signaling may be a beneficial, targeted treatment in these patients.
4 Interplay of inflammatory endotypes with microbiome in airway diseases
Bronchiectasis is characterized by alterations of bacterial airway microbiota, usually addressed as “dysbiosis”, including a reduced microbial diversity and increased quantities of opportunistic pathogens such as P. aeruginosa, H. influenzae, and S. pneumoniae (58). These pathogens perpetuate chronic inflammation through persistent colonization, biofilm formation, and immune evasion, thereby driving the vicious cycle of inflammation, tissue damage, and impaired mucociliary clearance (115). Thus, bacterial airway microbiota in bronchiectasis is defined by complex ecological dynamics, featuring dysbiosis and pathogen dominance, which significantly contribute to disease pathogenesis (116). Emerging evidence also underscores the potential roles of microbiota in terms of viruses and fungi as well, that may also impact bacteria and cytokine signaling, adding layers of complexity in bronchiectasis (117). Therapeutic strategies aimed at modulation of microbiota, such as inhaled antibiotics or probiotics, highlight the translational relevance of these interactions (118).
Microbial dysbiosis is a plausible factor in disease progression in patients with bronchiectasis, with genus diversity negatively correlating with disease severity, exacerbation frequency, and clinical outcomes, suggesting a bidirectional relationship between microbial community structure and host immune responses (55, 58). Bronchiectasis is characterized by frequent and diverse microbial infections and microbiota even in the stable state of the disease (59). The inflammatory response to dysbiosis may involve a complex network of cytokines that activate and recruit cells implicated in innate and adaptive immunity (60, 61). For instance, P. aeruginosa dominance is associated with increased sputum interleukin-1β, contributing to the airway injury (26). Early data from CF have indicated that dysregulated host-microbe interactions disrupt airway epithelial barrier integrity and promote a pro-inflammatory milieu, amplifying lung remodeling (62). Recent studies have highlighted the prognostic value of multi-omics data to reveal metabolic host-microbe disruptions, particularly in relation to microbial community dynamics during exacerbations (63, 64). Collectively, these studies emphasize the microbiome’s central role in bronchiectasis pathogenesis, offering insights for precision medicine approaches.
In patients with COPD who suffer from concomitant bronchiectasis, five endotypes have been described based on proteomic and microbiome profiling (65). Notably, these endotypes display distinct inflammatory, mucin, and microbiological characteristics. The first endotype was characterized by high microbial diversity and increased MUC5B, protease inhibitors, and immunoglobulin levels. The second featured Haemophilus microbiome predominance, elevated MMP8 and MMP9 levels, and a low MUC5AC/MUC5B ratio. The “infected-epithelial response” endotype, dominant by Stenotrophomonas and other Proteobacteria, was associated with increased concentrations of antimicrobial molecules, a high MUC5AC/MUC5B ratio, and low microbial diversity. The “dominant-NET” endotype was characterized by high NET proteins and Proteobacteria abundance, while the “Th2-driven” endotype featured T2 inflammation and increased dysbiosis. Similarly, in asthma-related bronchiectasis, at least two endotypes have been recognized and categorized into T2 high and T2 low subgroups, with further categorization based on specific T2 cytokines and biomarkers, allergy, and chronic bacterial infection (119).
A recent study investigated the interplay between the archetypes of inflammation signifying the key endotypes and microbiome characteristics in non-CF bronchiectasis patients from three European centers (26). Using cluster analysis of 33 sputum and serum inflammatory markers and 16S rRNA sequencing for microbiome analysis, the authors identified four distinct inflammatory endotypes associated with specific microbiome profiles. The more severe inflammatory clusters showed lower microbiome diversity and enrichment with Proteobacteria and Pseudomonas. Importantly, endotypes with severe neutrophilic and neutrophilic mixed with T2 inflammation were predictive of future exacerbation risk, suggesting their potential clinical utility in guiding targeted treatments.
Growing evidence suggests that sensitization to fungi may result in clinically relevant bronchiectasis with different endotypes. Allergic bronchopulmonary aspergillosis (ABPA) is a hypersensitivity response to Aspergillus spp., characterized by elevated blood IgE levels, peripheral eosinophilia and positive specific immunological tests for Aspergillus. The recent Cohort of Matched Asian and European Bronchiectasis (CAMEB) study identified two distinct sensitized immuno-allergic profiles in patients with bronchiectasis [68]. Patients with fungal-driven sensitization were characterized by specific responses to rAsp allergens and an airway rich in proinflammatory cytokines IL-1α, IL-1β, and TNF-α. In contrast, house dust mite sensitized patients showed increased levels of T2-related cytokines and airway eosinophilia, suggesting a potentially distinct treatable trait.
5 Discussion
An improved comprehension of the pathophysiology of bronchiectasis requires the specific understanding of three key components, impaired mucociliary function, inflammation, and infection, which together drive disease progression and clinical outcomes (13). Recent advances have significantly expanded our comprehension of bronchiectasis, moving beyond the traditional neutrophilic and eosinophilic dichotomy. These include severe neutrophilic, neutrophilic with T2 inflammation, Th2-driven, dominant-NET, infected-epithelial response, and a ‘shifted’ or mixed endotype exhibiting overlapping inflammatory features. In particular, research by Choi et al. (26) identified four inflammatory molecular endotypes in non-CF bronchiectasis, including severe neutrophilic, neutrophilic with T2 inflammation, and milder inflammatory clusters. Each of these endotypes is associated with distinct profiles of microbiota, as a result of dysbiosis, and relate to the risk of future exacerbations. This advanced endotyping approach, integrating immune profiles with microbiome features, marks a significant progression in disease understanding, with direct implications for risk assessment and personalized treatment strategies. Furthermore, the fungal sensitization endotypes reported by Mac Aogáin et al. (49), emphasize the significance of hypersensitivity responses as treatable traits, which may require specific antifungal or immunomodulatory treatments.
The role of the Th17/Treg axis may benefit from more investigation. Although Th17-driven inflammation is increasingly recognized for enhancing neutrophilic responses and contributing to tissue damage, the regulatory functions of Tregs and the consequences of Th17/Treg imbalance remain poorly defined in bronchiectasis. We propose that this type of dysregulation of immune response represents a novel mechanistic link to airway damage and disease progression, that warrants further investigation. Moreover, the emerging field of multi-omics research, which integrates transcriptomic, proteomic, metabolomic, and microbiome data, offers unique opportunities to define bronchiectasis endotypes more precisely and to identify possible therapeutic targets. By incorporating these perspectives, our work highlights important novel directions for advancing endotype-guided and precision medicine strategies in bronchiectasis.
As pointed out above, a central pathogenic mechanism in bronchiectasis is neutrophilic inflammation combined with dysfunctional bacterial killing, which perpetuates chronic airway damage and remodeling and is associated with worse outcomes (120). Neutrophil dysfunction, with reduced oxidative burst, increased viability, and delayed apoptosis of local neutrophils, has been demonstrated in bronchiectasis, providing a rationale for prolonged airway neutrophilia and impaired bacterial killing, thus contributing to further inflammation (121). Local accumulation of neutrophils in response to microbial dysbiosis and neutrophil dysfunction is common in several other chronic airway disorders, including COPD, CF, and a subgroup of patients with non-allergic asthma, thereby resembling bronchiectasis (122, 123).
Intriguingly, evidence from patients with CF suggests that neutrophils display a pro-survival cystic fibrosis transmembrane regulator (CFTR)-dependent phenotype, which prolongs their presence in the airways and induces NETs production (124). Similarly, non-CFTR-dependent inflammatory signals, including pro-inflammatory cytokines and lipopolysaccharides, have been associated with prolonged neutrophil survival in CF patients (125). Such changes in neutrophils’ metabolic pathways, including efferocytosis failure, primary or secondary neutrophil necrosis or some level of “reprogramming” of circulating blood neutrophils impairing their functional ability, have also been demonstrated in non-CF bronchiectasis patients, regardless of disease severity (40). However, whether neutrophil dysfunction in bronchiectasis results from an intrinsic defect or is provoked by a particular inflammatory milieu remains unclear.
An emerging insight in bronchiectasis pathogenesis is the recognition of an eosinophilic endotype, though its characterization remains under investigation.Existing evidence supports the existence of a distinct eosinophilic endotype of bronchiectasis, characterized by higher levels of FeNO and T2 cytokines in the airways, which is associated with poor clinical outcomes. However, a specific eosinophilic cutoff distinguishing a pathological state has not yet been determined, as other comorbidities characterized by increased T2 inflammation, such as asthma, chronic rhinosinusitis, ABPA, and eosinophilic COPD often coexist with bronchiectasis (7, 126–129). Although differentiating these diseases can be challenging, eosinophilic bronchiectasis is typically characterized by the absence of airway hyperresponsiveness (130), more severe dyspnea and more impaired respiratory function (78), and higher severity scores on various bronchiectasis indices (131). Nevertheless, the correlation between sputum and blood eosinophilia in bronchiectasis has been found to be weak (24), and stability of blood eosinophil numbers over time remains unclear (132). Eosinophil-specific inflammatory biomarkers, such as sputum eosinophil peroxidase, which significantly increase in severe bronchiectasis and during exacerbations requiring hospitalization, may better characterize this disease endotype (133). The role of Th cells in the dysregulated inflammatory environment that signifies bronchiectasis is yet to be fully understood. Persistent bacterial infection by certain pathogens has been shown to skew adaptive immunity from Th1 toward Th2 response, in an attempt to effectively control infection with respect to antibodies, innate cytokines, and chemokines production, while at the same time preventing further expansion of neutrophilic inflammation (110). Previous studies have shown that the airway microbiome-IL-17 axis is a critical regulator of chronic inflammation in bronchiectasis (29, 134). Th17 immune responses are known to activate neutrophils, playing an important role in host defense against bacteria while simultaneously contributing to persistent airway inflammation and bronchiectasis pathogenesis (115). However, the role of specific members of the airway microbiota in the modulation of IL-17/IL-23-type immunity in bronchiectasis remains unclear (135).
Clearly, a key knowledge gap is the role of Tregs in bronchiectasis and the balance between Th17 and Treg responses, which may hold the key to understanding immune dysregulation and tissue damage. In COPD, Tregs are reduced in number and dysfunctional, both in stable state and during exacerbations (136). However, further studies are required to confirm this concept in bronchiectasis. Since Tregs and Th17 cells display counterbalancing roles, with Th17 cells promoting inflammation and pathology and Tregs maintaining self-tolerance, Th17/Treg imbalance in bronchiectasis may result in significant immune dysfunction, permitting lung tissue damage and clinical deterioration (Figure 2).
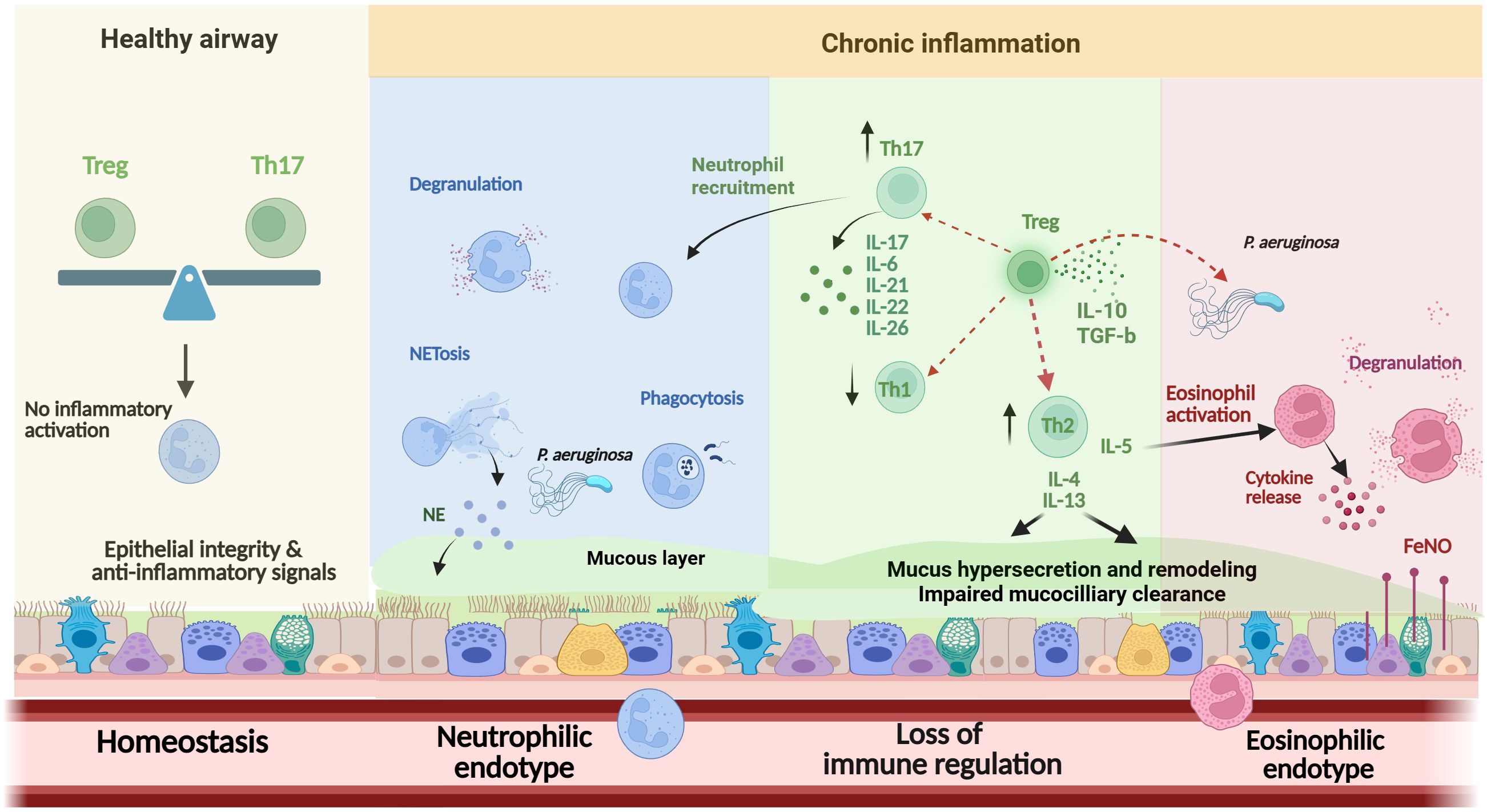
Figure 2. Overview of the main effector cells in the pathogenesis of bronchiectasis. Immune balance in healthy airways, maintained by Treg–Th17 homeostasis and epithelial-derived anti-inflammatory signals, prevents immune activation and preserves tissue integrity. In chronic dysbiosis, neutrophil dysfunction (including impaired oxidative burst, NETosis, and delayed apoptosis) and imbalances in Th1, Th2, and Th17 responses promote sustained inflammation. A distinct eosinophilic endotype, associated with T2 cytokines, elevated FeNO, and P. aeruginosa colonization, contributes to progressive epithelial damage and airway remodeling. The role of Tregs and the impact of Th17/Treg imbalance remain incompletely understood. Th, T-helper; Treg, T-regulatory; NE, Neutrophil elastase; NETs, neutrophil extracellular traps; IL, interleukin; FeNO, Fractional exhaled nitric oxide; T2, type 2 ↑, Increase; ↓, Decrease.
Certain risk factors of the host may also affect the response of T-cell and myeloid cells involved in the pathogenesis of bronchiectasis. In inflammatory bowel disease, the lung-gut axis determines a shared immune system dysfunction associated with chronic lymphocytic inflammation (137). Individuals with PCD face genetically acquired impaired mucociliary clearance, which increases their susceptibility to infections and drives local neutrophil accumulation (138). Patients with immunodeficiencies may experience persistent infections due to impaired local protective humoral immunity caused by abnormalities in B- and T-cell functions (139–141). Likewise, in Job’s syndrome, a disorder characterized by bronchiectasis, persistent lung infections, and increased IgE levels, a STAT3 gene mutation impairs IL-17 production, affecting the production of a diverse array of cytokines (142).
While most research to date has focused on CF, non-CF bronchiectasis, despite being more heterogeneous and increasingly acknowledged, has not been equally explored. The limited available data suggest that chronic infection, airway colonization, and persistent inflammation may influence T-cell responses in non-CF bronchiectasis. However, these conclusions are often drawn from small cohorts or animal models, urging the need for well-designed, disease-specific studies to understand how T-helper and Treg cell dysregulation contributes to disease progression and treatment response in this population.
The primary goal of current bronchiectasis management is to address the underlying cause of the disorder by enhancing mucociliary clearance, managing infections, and effectively preventing and treating associated complications (143). However, the emerging identification of several endotypes in bronchiectasis, especially those involving specific immune cells and their cytokine signaling, has highlighted the need for more targeted approaches that can inform treatment decisions and improve clinical outcomes in specific patient subpopulations (144). Furthermore, a recent study identified distinct inflammatory molecular endotypes in bronchiectasis patients, demonstrating that these endotypes are associated with different microbiome profiles and future exacerbation risks, emphasizing that recognizing of various inflammatory processes at an individual patient level is crucial for developing management plans that prevent exacerbations and tailor treatments effectively (26). Inflammatory mechanisms, such as neutrophil extracellular traps and eosinophilia, are instrumental in defining these endotypes, and biomarkers related to these processes may be useful in guiding therapies and enhancing the success of randomized trials (126).
Targeting neutrophilic inflammation in bronchiectasis has consistently posed a challenge as many approaches have aimed to prevent neutrophil recruitment to the lungs, with evident risks in a chronic disorder where bacteria operate. CXCR2 antagonists have effectively achieved this outcome but have been associated with increased infections and exacerbations (145). In contrast, targeting metabolic reprogramming, such as with 5’ adenosine monophosphate-activated protein kinase (AMPK) activators, which modulate multiple intracellular metabolic pathways, including glycolysis, and reverse phagocytic dysfunction and NET formation, could potentially reverse neutrophil dysfunction and dysregulated inflammation (36). Brensocatib, a reversible inhibitor of CatC that blocks the activation of NSPs in the bone marrow during neutrophil maturation, has shown broad anti-inflammatory effects beyond its known impact on serine proteases, resulting in significant improvements in clinical outcomes, particularly reductions in rate of pulmonary exacerbations and FEV1 decline (146–149). In two recent phase II randomized, placebo-controlled, dose-finding studies, the novel selective inhibitors of CatC, BI 1291583 and HSK31858, were found to be well-tolerated and effective in reducing the risk of exacerbation in adults with bronchiectasis (150, 151). Focusing on T2 inflammation in individuals with bronchiectasis offers promising potential for enhancing clinical outcomes, aligning with the move toward precision medicine in managing bronchiectasis (152). Although recent guidelines do not recommend the routine administration of ICS in patients with bronchiectasis, emerging evidence suggests that their use may be associated with a reduced frequency of exacerbations in patients with elevated blood eosinophil levels (153). Of particular significance is the potential for eosinophils and FeNO to serve as biomarkers of IL-5, IL-4Ra, or TSLP-driven inflammation, as these cytokines may constitute targets for future biological therapies, regardless of the co-existence of asthma or COPD. So far, the effectiveness of anti-IL-5 or anti-IL-5 receptor monoclonal antibodies in severe eosinophilic bronchiectasis has been documented in a case series, which demonstrated marked improvements in exacerbation rates and lung function after six months of treatment (154). A phase III, randomized, placebo-controlled trial (NCT05006573) was conducted to evaluate the efficacy and safety of benralizumab in adults with bronchiectasis and eosinophilic inflammation. However, due to its early termination, there is no available published data or outcomes, highlighting the need for further research in this area (155).
Furthermore, recent evidence from CF has demonstrated that CFTR-targeted medications can reduce eosinophils and mucus plugs and reverse exaggerated airway enlargement, suggesting a potential novel therapeutic approach for patients with non-CF bronchiectasis with evidence of underlying CFTR dysfunction (156).
Finally, elucidating the contribution of the Th17/Treg axis in the pathogenesis of bronchiectasis may reveal novel disease-modulatory therapies. In this context, it is interesting that macrolides, which are part of validated and standardized therapy, do reduce both neutrophilic and Th17-driven inflammation (20, 143). In fact, there is evidence that anti-IL-17 treatment reduces LPS-induced inflammation in murine asthma models (157, 158) and in an elastase-induced murine emphysema model (159). Future research on targeting Th17/Treg effector cells and related cytokines and therapeutic strategies that focus on restoring the Th17/Treg axis balance may serve as promising future targets for modulating lung inflammation and preventing or delaying tissue damage in bronchiectasis (160). This is especially true considering the increasing interest and potential therapeutic role of Tregs in autoimmune and inflammatory diseases, including cancer (161). Taken together, these insights highlight the complexity of bronchiectasis pathogenesis and the urgent need for endotype-guided therapeutic strategies.
6 Conclusions
Patients with bronchiectasis exhibit several endotypes with specific inflammatory characteristics, reflecting the complex interplay between dysfunctional host immunity, pathogens, and environmental factors. Identified endotypes include not only the classic neutrophilic and eosinophilic types, but also mixed or ‘shifted’ endotypes, Th2-driven and dominant-NET profiles, each with distinct immunopathological features. The key conclusions of this review are summarized below:
● Precise determination of endotype is critical for characterizing and subtyping bronchiectasis, given that certain inflammatory profiles are strongly associated with clinical phenotypes, disease severity, and clinical outcomes.
● Recognition of inflammatory endotypes provides a promising tool for developing targeted therapies, necessitating the integration of basic mechanistic investigations, translational studies, and clinical validation of candidate biomarkers.
● Recent advances in multi-omics and immunophenotyping offer unique prospects to define bronchiectasis endotypes with greater precision and to identify feasible therapeutic targets.
● The role of T-helper and T-regulatory cells remains underexplored in non-CF bronchiectasis; addressing these gaps through well-designed, disease-specific studies is essential for developing tailored immunomodulatory treatments.
Author contributions
EF: Data curation, Conceptualization, Writing – original draft, Visualization, Investigation, Validation, Writing – review & editing, Methodology, Supervision. AL: Supervision, Methodology, Writing – original draft, Writing – review & editing, Data curation, Visualization, Investigation, Conceptualization, Validation. AB: Writing – original draft, Data curation, Supervision, Visualization, Conceptualization, Investigation, Writing – review & editing, Validation, Methodology.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by project grants from the Swedish Heart-Lung Foundation (AL: 2021-0286, AB: 2021-0434, 2022-0478, 20241637) and the Swedish Research Council (AL: 2021-01527).
Conflict of interest
EF has received payment or honoraria for lectures from AstraZeneca, Boehringer Ingelheim, Chiesi, ELPEN, Specialty Therapeutics, GlaxoSmithKline and Menarini, support for attending meetings and/or travel from GSK, Menarini, AstraZeneca and Chiesi and consulting fees from GlaxoSmithKline. AL has received project grants from the Swedish Heart-Lung Foundation #2024-0343, the Swedish Research Council #2021-01527, unrestricted research grants from AstraZeneca AB and Chiesi AB, and payment for tasks as chairman at scientific meetings or consultant at advisory boards, from AstraZeneca AB, Chiesi AB, and GlaxoSmithKline AB in Sweden, and expert consultancies for patent attorneys of different companies. AB has received grants from the Swedish Heart-Lung Foundation 2021-0434, 2022-0478 and 20241637, lecture fees from Chiesi, AstraZeneca, GlaxoSmithKline, a grant from AstraZeneca paid to the KI institution, and is Head of Assembly 5 Airway diseases, asthma, COPD, and chronic cough of the European Respiratory Society.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. O’Donnell AE. Bronchiectasis - A clinical review. N Engl J Med. (2022) 387:533–45. doi: 10.1056/NEJMra2202819
2. Whitwell F. A study of the pathology and pathogenesis of bronchiectasis. Thorax. (1952) 7:213–39. doi: 10.1136/thx.7.3.213
3. Quint JK, Millett ER, Joshi M, Navaratnam V, Thomas SL, Hurst JR, et al. Changes in the incidence, prevalence and mortality of bronchiectasis in the UK from 2004 to 2013: a population-based cohort study. Eur Respir J. (2016) 47:186–93. doi: 10.1183/13993003.01033-2015
4. Henkle E, Chan B, Curtis JR, Aksamit TR, Daley CL, and Winthrop KL. Characteristics and health-care utilization history of patients with bronchiectasis in US medicare enrollees with prescription drug plans, 2006 to 2014. Chest. (2018) 154:1311–20. doi: 10.1016/j.chest.2018.07.014
5. Chalmers JD and Elborn JS. Reclaiming the name ‘bronchiectasis’. Thorax. (2015) 70:399–400. doi: 10.1136/thoraxjnl-2015-206956
6. Polverino E, De Soyza A, Dimakou K, Traversi L, Bossios A, Crichton ML, et al. The association between bronchiectasis and chronic obstructive pulmonary disease: data from the european bronchiectasis registry (EMBARC). Am J Respir Crit Care Med. (2024) 210:119–27. doi: 10.1164/rccm.202309-1614OC
7. Polverino E, Dimakou K, Traversi L, Bossios A, Haworth CS, Loebinger MR, et al. Bronchiectasis and asthma: data from the european bronchiectasis registry (EMBARC). J Allergy Clin Immunol. (2024) 153:1553–62. doi: 10.1016/j.jaci.2024.01.027
8. Gómez-Olivas JD, Oscullo G, and Martínez-García M. Etiology of bronchiectasis in the world: data from the published national and international registries. J Clin Med. (2023) 12:5782. doi: 10.3390/jcm12185782
9. Aliberti S, Lonni S, Dore S, McDonnell MJ, Goeminne PC, Dimakou K, et al. Clinical phenotypes in adult patients with bronchiectasis. Eur Respir J. (2016) 47:1113–22. doi: 10.1183/13993003.01899-2015
10. Chalmers JD, Aliberti S, Filonenko A, Shteinberg M, Goeminne PC, Hill AT, et al. Characterization of the “Frequent exacerbator phenotype” in bronchiectasis. Am J Respir Crit Care Med. (2018) 197:1410–20. doi: 10.1164/rccm.201711-2202OC
11. Chang-Macchiu P, Traversi L, and Polverino E. Bronchiectasis phenotypes. Curr Opin Pulm Med. (2019) 25:281–8. doi: 10.1097/MCP.0000000000000569
12. Cole PJ. Inflammation: a two-edged sword–the model of bronchiectasis. Eur J Respir Dis Suppl. (1986) 147:6–15.
13. Flume PA, Chalmers JD, and Olivier KN. Advances in bronchiectasis: endotyping, genetics, microbiome, and disease heterogeneity. Lancet. (2018) 392:880–90. doi: 10.1016/S0140-6736(18)31767-7
14. De Angelis A, Johnson ED, Sutharsan S, and Aliberti S. Exacerbations of bronchiectasis. Eur Respir Rev. (2024) 33:240085. doi: 10.1183/16000617.0085-2024
15. Inchingolo R, Pierandrei C, Montemurro G, Smargiassi A, Lohmeyer FM, and Rizzi A. Antimicrobial resistance in common respiratory pathogens of chronic bronchiectasis patients: A literature review. Antibiotics (Basel). (2021) 10:326. doi: 10.3390/antibiotics10030326
16. Goeminne PC, Cox B, Finch S, Loebinger MR, Bedi P, Hill AT, et al. The impact of acute air pollution fluctuations on bronchiectasis pulmonary exacerbation: a case-crossover analysis. Eur Respir J. (2018) 52:1702557. doi: 10.1183/13993003.02557-2017
17. Marsland I, Sobala R, De Soyza A, and Witham M. Multimorbidity in bronchiectasis: a systematic scoping review. ERJ Open Res. (2023) 9:00296–2022. doi: 10.1183/23120541.00296-2022
18. Mateus SP, Ribeiro-Alves M, Salles REB, Costa W, Costa CHD, Lopes AJ, et al. Mortality and comorbidities in patients with bronchiectasis over a 3-year follow-up. Med (Baltimore). (2022) 101:e32537. doi: 10.1097/MD.0000000000032537
19. King PT. The pathophysiology of bronchiectasis. Int J Chron Obstruct Pulmon Dis. (2009) 4:411–9. doi: 10.2147/COPD.S6133
20. Keir HR, Shoemark A, Dicker AJ, Perea L, Pollock J, Giam YH, et al. Neutrophil extracellular traps, disease severity, and antibiotic response in bronchiectasis: an international, observational, multicohort study. Lancet Respir Med. (2021) 9:873–84. doi: 10.1016/S2213-2600(20)30504-X
21. Jasper AE, McIver WJ, Sapey E, and Walton GM. Understanding the role of neutrophils in chronic inflammatory airway disease. F1000Res. (2019) 8:F1000 Faculty Rev-557. doi: 10.12688/f1000research
22. Oriano M, Gramegna A, Terranova L, Sotgiu G, Sulaiman I, Ruggiero L, et al. Sputum neutrophil elastase associates with microbiota and Pseudomonas aeruginosa in bronchiectasis. Eur Respir J. (2020) 56:2000769. doi: 10.1183/13993003.00769-2020
23. Parr DG, Guest PG, Reynolds JH, Dowson LJ, and Stockley RA. Prevalence and impact of bronchiectasis in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. (2007) 176:1215–21. doi: 10.1164/rccm.200703-489OC
24. Shoemark A, Shteinberg M, De Soyza A, Haworth CS, Richardson H, Gao Y, et al. Characterization of eosinophilic bronchiectasis: A european multicohort study. Am J Respir Crit Care Med. (2022) 205:894–902. doi: 10.1164/rccm.202108-1889OC
25. Tsikrika S, Dimakou K, Papaioannou AI, Hillas G, Thanos L, Kostikas K, et al. The role of non-invasive modalities for assessing inflammation in patients with non-cystic fibrosis bronchiectasis. Cytokine. (2017) 99:281–6. doi: 10.1016/j.cyto.2017.08.005
26. Choi H, Ryu S, Keir HR, Giam YH, Dicker AJ, Perea L, et al. Inflammatory molecular endotypes in bronchiectasis: A european multicenter cohort study. Am J Respir Crit Care Med. (2023) 208:1166–76. doi: 10.1164/rccm.202303-0499OC
27. Boyton RJ, Reynolds CJ, Quigley KJ, and Altmann DM. Immune mechanisms and the impact of the disrupted lung microbiome in chronic bacterial lung infection and bronchiectasis. Clin Exp Immunol. (2013) 171:117–23. doi: 10.1111/cei.12003
28. Gaga M, Bentley AM, Humbert M, Barkans J, O’Brien F, Wathen CG, et al. Increases in CD4+ T lymphocytes, macrophages, neutrophils and interleukin 8 positive cells in the airways of patients with bronchiectasis. Thorax. (1998) 53:685–91. doi: 10.1136/thx.53.8.685
29. Frija-Masson J, Martin C, Regard L, Lothe MN, Touqui L, Durand A, et al. Bacteria-driven peribronchial lymphoid neogenesis in bronchiectasis and cystic fibrosis. Eur Respir J. (2017) 49:1601873. doi: 10.1183/13993003.01873-2016
30. Silva JR, Jones JA, Cole PJ, and Poulter LW. The immunological component of the cellular inflammatory infiltrate in bronchiectasis. Thorax. (1989) 44:668–73. doi: 10.1136/thx.44.8.668
31. Attia EF, Miller RF, and Ferrand RA. Bronchiectasis and other chronic lung diseases in adolescents living with HIV. Curr Opin Infect Dis. (2017) 30:21–30. doi: 10.1097/QCO.0000000000000325
32. Yogi S, Yamashiro T, Kamiya H, Kamiya A, Miyara T, Moromizato H, et al. Thoracic manifestations of adult T-cell leukemia/lymphoma on chest CT: difference between clinical subtypes. Diagn Interv Radiol. (2019) 25:55–61. doi: 10.5152/dir.2018.18038
33. Oriano M, Amati F, Gramegna A, De Soyza A, Mantero M, Sibila O, et al. Protease-antiprotease imbalance in bronchiectasis. Int J Mol Sci. (2021) 22:309–33. doi: 10.3390/ijms22115996
34. Chalmers JD, Moffitt KL, Suarez-Cuartin G, Sibila O, Finch S, Furrie E, et al. Neutrophil elastase activity is associated with exacerbations and lung function decline in bronchiectasis. Am J Respir Crit Care Med. (2017) 195:1384–93. doi: 10.1164/rccm.201605-1027OC
35. Twigg MS, Brockbank S, Lowry P, FitzGerald SP, Taggart C, and Weldon S. The role of serine proteases and antiproteases in the cystic fibrosis lung. Mediators Inflamm. (2015) 2015:293053. doi: 10.1155/2015/293053
36. Giam YH, Shoemark A, and Chalmers JD. Neutrophil dysfunction in bronchiectasis: an emerging role for immunometabolism. Eur Respir J. (2021) 58:210241. doi: 10.1183/13993003.03157-2020
37. Pasteur MC, Helliwell SM, Houghton SJ, Webb SC, Foweraker JE, Coulden RA, et al. An investigation into causative factors in patients with bronchiectasis. Am J Respir Crit Care Med. (2000) 162:1277–84. doi: 10.1164/ajrccm.162.4.9906120
38. King PT, Hutchinson P, Holmes PW, Freezer NJ, Bennett-Wood V, Robins-Browne R, et al. Assessing immune function in adult bronchiectasis. Clin Exp Immunol. (2006) 144:440–6. doi: 10.1111/j.1365-2249.2006.03091.x
39. Watt AP, Brown V, Courtney J, Kelly M, Garske L, Elborn JS, et al. Neutrophil apoptosis, proinflammatory mediators and cell counts in bronchiectasis. Thorax. (2004) 59:231–6. doi: 10.1136/thx.2003.008037
40. Bedi P, Davidson DJ, McHugh BJ, Rossi AG, and Hill AT. Blood neutrophils are reprogrammed in bronchiectasis. Am J Respir Crit Care Med. (2018) 198:880–90. doi: 10.1164/rccm.201712-2423OC
41. Shoemark A, Cant E, Carreto L, Smith A, Oriano M, Keir HR, et al. A point-of-care neutrophil elastase activity assay identifies bronchiectasis severity, airway infection and risk of exacerbation. Eur Respir J. (2019) 53:1384–93. doi: 10.1183/13993003.congress-2019.OA4947
42. Sibila O, Perea L, Cantó E, Shoemark A, Cassidy D, Smith AH, et al. Antimicrobial peptides, disease severity and exacerbations in bronchiectasis. Thorax. (2019) 74:835–42. doi: 10.1136/thoraxjnl-2018-212895
43. Mormile M, Mormile I, Fuschillo S, Rossi FW, Lamagna L, Ambrosino P, et al. Eosinophilic airway diseases: from pathophysiological mechanisms to clinical practice. Int J Mol Sci. (2023) 24:2003157. doi: 10.3390/ijms24087254
44. Zimmermann N and Rothenberg ME. Mechanism of enhanced eosinophil survival in inflammation. Blood. (2015) 125:3831–2. doi: 10.1182/blood-2015-04-640623
45. Frøssing L, Von Bülow A, and Porsbjerg C. Bronchiectasis in severe asthma is associated with eosinophilic airway inflammation and activation. J Allergy Clin Immunol Glob. (2023) 2:36–42. doi: 10.1016/j.jacig.2022.10.001
46. Singh D. Blood eosinophil counts in chronic obstructive pulmonary disease: A biomarker of inhaled corticosteroid effects. Tuberc Respir Dis (Seoul). (2020) 83:185–94. doi: 10.4046/trd.2020.0026
47. Martínez-García M, Oscullo G, and Gomez-Olivas JD. Peripheral cellular biomarkers in bronchiectasis. Respir Med Res. (2023) 84:101063. doi: 10.1016/j.resmer.2023.101063
48. Martínez-García M, Méndez R, Olveira C, Girón R, García-Clemente M, Máiz L, et al. The U-shaped relationship between eosinophil count and bronchiectasis severity: the effect of inhaled corticosteroids. Chest. (2023) 164:606–13. doi: 10.1016/j.chest.2023.04.029
49. Mac Aogáin M, Tiew PY, Lim AYH, Low TB, Tan GL, Hassan T, et al. Distinct “Immunoallertypes” of disease and high frequencies of sensitization in non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med. (2019) 199:842–53. doi: 10.1136/thoraxjnl-2018-212895
50. Sun L, Su Y, Jiao A, Wang X, and Zhang B. T cells in health and disease. Signal Transduct Target Ther. (2023) 8:235. doi: 10.1038/s41392-023-01471-y
51. Hwang JR, Byeon Y, Kim D, and Park SG. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp Mol Med. (2020) 52:750–61. doi: 10.1038/s12276-020-0435-8
52. Farber DL, Yudanin NA, and Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. (2014) 14:24–35. doi: 10.1038/nri3567
53. Zhu J. T helper cell differentiation, heterogeneity, and plasticity. Cold Spring Harb Perspect Biol. (2018) 10:9–22. doi: 10.1101/cshperspect.a030338
54. Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, et al. T-bet is a STAT1-induced regulator of IL-12R expression in naïve CD4+ T cells. Nat Immunol. (2002) 3:549–57. doi: 10.1038/ni794
55. Rogers GB, Zain NM, Bruce KD, Burr LD, Chen AC, Rivett DW, et al. A novel microbiota stratification system predicts future exacerbations in bronchiectasis. Ann Am Thorac Soc. (2014) 11:496–503. doi: 10.1513/AnnalsATS.201310-335OC
56. Mauch RM, Jensen P, Moser C, Levy CE, and Høiby N. Mechanisms of humoral immune response against Pseudomonas aeruginosa biofilm infection in cystic fibrosis. J Cyst Fibros. (2018) 17:143–52. doi: 10.1016/j.jcf.2017.08.012
57. Tiringer K, Treis A, Fucik P, Gona M, Gruber S, Renner S, et al. A Th17- and Th2-skewed cytokine profile in cystic fibrosis lungs represents a potential risk factor for Pseudomonas aeruginosa infection. Am J Respir Crit Care Med. (2013) 187:621–9. doi: 10.1164/rccm.201206-1150OC
58. Richardson H, Dicker AJ, Barclay H, and Chalmers JD. The microbiome in bronchiectasis. Eur Respir Rev. (2019) 28:185–94. doi: 10.1183/16000617.0048-2019
59. Wang Y, Xiao J, Yang X, Liu Y, Du J, Bossios A, et al. Pulmonary microbiology and microbiota in adults with non-cystic fibrosis bronchiectasis: a systematic review and meta-analysis. Respir Res. (2025) 26:77. doi: 10.1186/s12931-025-03140-w
60. Solarat B, Perea L, Faner R, de la Rosa D, Martínez-García M, and Sibila O. Pathophysiology of chronic bronchial infection in bronchiectasis. Arch Bronconeumol. (2023) 59:101–8. doi: 10.1016/j.arbres.2022.09.004
61. King PT. The role of the immune response in the pathogenesis of bronchiectasis. BioMed Res Int. (2018) 2018:6802637. doi: 10.1155/2018/6802637
62. Aldallal N, McNaughton EE, Manzel LJ, Richards AM, Zabner J, Ferkol TW, et al. Inflammatory response in airway epithelial cells isolated from patients with cystic fibrosis. Am J Respir Crit Care Med. (2002) 166:1248–56. doi: 10.1164/rccm.200206-627OC
63. Mac Aogáin M, Narayana JK, Tiew PY, Ali N, Yong VFL, Jaggi TK, et al. Integrative microbiomics in bronchiectasis exacerbations. Nat Med. (2021) 27:688–99. doi: 10.1038/s41591-021-01289-7
64. Taylor SL, Woodman RJ, Chen AC, Burr LD, Gordon DL, McGuckin MA, et al. FUT2 genotype influences lung function, exacerbation frequency and airway microbiota in non-CF bronchiectasis. Thorax. (2017) 72:304–10. doi: 10.1136/thoraxjnl-2016-208775
65. Huang JT, Cant E, Keir HR, Barton AK, Kuzmanova E, Shuttleworth M, et al. Endotyping chronic obstructive pulmonary disease, bronchiectasis, and the “Chronic obstructive pulmonary disease-bronchiectasis association. Am J Respir Crit Care Med. (2022) 206:417–26. doi: 10.1164/rccm.202108-1943OC
66. Kobayashi SD and DeLeo FR. Role of neutrophils in innate immunity: a systems biology-level approach. Wiley Interdiscip Rev Syst Biol Med. (2009) 1:309–33. doi: 10.1002/wsbm.v1:3
67. Borregaard N, Sørensen OE, and Theilgaard-Mönch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol. (2007) 28:340–5. doi: 10.1016/j.it.2007.06.002
68. Malech HL, Deleo FR, and Quinn MT. The role of neutrophils in the immune system: an overview. Methods Mol Biol. (2014) 1124:3–10. doi: 10.1038/s12276-020-0435-8
69. Keir HR and Chalmers JD. Neutrophil extracellular traps in chronic lung disease: implications for pathogenesis and therapy. Eur Respir Rev. (2022) 31:24–35. doi: 10.1183/16000617.0241-2021
70. Voynow JA and Shinbashi M. Neutrophil elastase and chronic lung disease. Biomolecules. (2021) 11. doi: 10.3390/biom11081065
71. Chalmers JD, Mall MA, Chotirmall SH, O’Donnell AE, Flume PA, Hasegawa N, et al. Targeting neutrophil serine proteases in bronchiectasis. Eur Respir J. (2025) 65:549–57. doi: 10.1183/13993003.01050-2024
72. Mutua V and Gershwin LJ. A review of neutrophil extracellular traps (NETs) in disease: potential anti-NETs therapeutics. Clin Rev Allergy Immunol. (2021) 61:194–211. doi: 10.1007/s12016-020-08804-7
73. Guan WJ, Gao YH, Xu G, Lin ZY, Tang Y, Gu YY, et al. Sputum matrix metalloproteinase-8 and -9 and tissue inhibitor of metalloproteinase-1 in bronchiectasis: clinical correlates and prognostic implications. Respirology. (2015) 20:1073–81. doi: 10.1111/resp.2015.20.issue-7
74. Finch S, Shoemark A, Dicker AJ, Keir HR, Smith A, Ong S, et al. Pregnancy zone protein is associated with airway infection, neutrophil extracellular trap formation, and disease severity in bronchiectasis. Am J Respir Crit Care Med. (2019) 200:992–1001. doi: 10.1164/rccm.201812-2351OC
75. Janson C, Bjermer L, Lehtimäki L, Kankaanranta H, Karjalainen J, Altraja A, et al. Eosinophilic airway diseases: basic science, clinical manifestations and future challenges. Eur Clin Respir J. (2022) 9:2040707. doi: 10.1080/20018525.2022.2040707
76. Rosenberg HF, Dyer KD, and Foster PS. Eosinophils: changing perspectives in health and disease. Nat Rev Immunol. (2013) 13:9–22. doi: 10.1038/nri3341
77. Wechsler ME, Munitz A, Ackerman SJ, Drake MG, Jackson DJ, Wardlaw AJ, et al. Eosinophils in health and disease: A state-of-the-art review. Mayo Clin Proc. (2021) 96:2694–707. doi: 10.1016/j.mayocp.2021.04.025
78. Oriano M, Gramegna A, Amati F, D’Adda A, Gaffuri M, Contoli M, et al. T2-high endotype and response to biological treatments in patients with bronchiectasis. Biomedicines. (2021) 9:529–42. doi: 10.3390/biomedicines9070772
79. Martinez-García M, Olveira C, Girón R, García-Clemente M, Máiz-Carro L, Sibila O, et al. Peripheral neutrophil-to-lymphocyte ratio in bronchiectasis: A marker of disease severity. Biomolecules. (2022) 12:981–9. doi: 10.3390/biom12101399
80. Wang X, Villa C, Dobarganes Y, Olveira C, Girón R, García-Clemente M, et al. Phenotypic clustering in non-cystic fibrosis bronchiectasis patients: the role of eosinophils in disease severity. Int J Environ Res Public Health. (2021) 18:1121–33. doi: 10.3390/ijerph18168431
81. Morimoto C, Matsumoto H, Ito I, Nagaski T, Oguma T, and Hirai T. Roles of Staphylococcus aureus and sensitization to staphylococcal enterotoxin in bronchiectasis. Respir Investig. (2022) 23:247–57. doi: 10.1016/j.resinv.2022.09.006
82. Moser C, Kjaergaard S, Pressler T, Kharazmi A, Koch C, and Høiby N. The immune response to chronic Pseudomonas aeruginosa lung infection in cystic fibrosis patients is predominantly of the Th2 type. Apmis. (2000) 108:329–35. doi: 10.1034/j.1600-0463.2000.d01-64.x
83. Butcher MJ and Zhu J. Recent advances in understanding the Th1/Th2 effector choice. Fac Rev. (2021) 10:30. doi: 10.12703/r/10-30
84. Basu A, Ramamoorthi G, Albert G, Gallen C, Beyer A, Snyder C, et al. Differentiation and regulation of T(H) cells: A balancing act for cancer immunotherapy. Front Immunol. (2021) 12:669474. doi: 10.3389/fimmu.2021.669474
85. Ruterbusch M, Pruner KB, Shehata L, and Pepper M. In vivo CD4(+) T cell differentiation and function: revisiting the th1/th2 paradigm. Annu Rev Immunol. (2020) 38:705–25. doi: 10.1146/annurev-immunol-103019-085803
86. Walker JA and McKenzie ANJ. T(H)2 cell development and function. Nat Rev Immunol. (2018) 18:121–33. doi: 10.1038/nri.2017.118
87. Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. (2008) 9:1341–6. doi: 10.1038/ni.1659
88. Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest. (2009) 119:3573–85. doi: 10.1172/JCI40202
89. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. (2014) 41:529–42. doi: 10.1016/j.immuni.2014.10.004
90. Feng H, Zhao Z, and Dong C. Adapting to the world: The determination and plasticity of T follicular helper cells. J Allergy Clin Immunol. (2022) 150:981–9. doi: 10.1016/j.jaci.2022.09.018
91. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
92. Che KF, Kaarteenaho R, Lappi-Blanco E, Levänen B, Sun J, Wheelock Å, et al. Interleukin-26 production in human primary bronchial epithelial cells in response to viral stimulation: modulation by th17 cytokines. Mol Med. (2017) 23:247–57. doi: 10.2119/molmed.2016.00064
93. Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol. (2023) 23:38–54. doi: 10.1038/s41577-022-00746-9
94. Zambrano-Zaragoza JF, Romo-Martínez EJ, Durán-Avelar Mde J, García-Magallanes N, and Vibanco-Pérez N. Th17 cells in autoimmune and infectious diseases. Int J Inflam. (2014) 2014:651503. doi: 10.1155/2014/651503
95. Bayati F, Mohammadi M, Valadi M, Jamshidi S, Foma AM, and Sharif-Paghaleh E. The therapeutic potential of regulatory T cells: challenges and opportunities. Front Immunol. (2020) 11:585819. doi: 10.3389/fimmu.2020.585819
96. Golzari-Sorkheh M and Zúñiga-Pflücker JC. Development and function of FOXP3+ regulators of immune responses. Clin Exp Immunol. (2023) 213:13–22. doi: 10.1093/cei/uxad048
97. Plitas G and Rudensky AY. Regulatory T cells: differentiation and function. Cancer Immunol Res. (2016) 4:721–5. doi: 10.1158/2326-6066.CIR-16-0193
98. Chen Z, Lin F, Gao Y, Li Z, Zhang J, Xing Y, et al. FOXP3 and RORγt: transcriptional regulation of Treg and Th17. Int Immunopharmacol. (2011) 11:536–42. doi: 10.1016/j.intimp.2010.11.008
99. Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-McCrann JM, et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol Immunol. (2018) 15:458–69. doi: 10.1038/s41423-018-0004-4
100. Grover P, Goel PN, and Greene MI. Regulatory T cells: regulation of identity and function. Front Immunol. (2021) 12:750542. doi: 10.3389/fimmu.2021.750542
101. Li C, Jiang P, Wei S, Xu X, and Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer. (2020) 19:116. doi: 10.1186/s12943-020-01234-1
102. Cinier J, Hubert M, Besson L, Di Roio A, Rodriguez C, Lombardi V, et al. Recruitment and expansion of tregs cells in the tumor environment-how to target them? Cancers (Basel). (2021) 13:e0119325. doi: 10.1371/journal.pone.0119325
103. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. (2008) 453:236–40. doi: 10.1038/nature06878
104. Kimura A and Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. (2010) 40:1830–5. doi: 10.1002/eji.201040391
105. Hsu D, Taylor P, Fletcher D, van Heeckeren R, Eastman J, van Heeckeren A, et al. Interleukin-17 pathophysiology and therapeutic intervention in cystic fibrosis lung infection and inflammation. Infect Immun. (2016) 84:2410–21. doi: 10.1128/IAI.00284-16
106. Bayes HK, Ritchie ND, and Evans TJ. Interleukin-17 is required for control of chronic lung infection caused by pseudomonas aeruginosa. Infect Immun. (2016) 84:3507–16. doi: 10.1128/IAI.00717-16
107. Mulcahy EM, Hudson JB, Beggs SA, Reid DW, Roddam LF, and Cooley MA. High peripheral blood th17 percent associated with poor lung function in cystic fibrosis. PloS One. (2015) 10:e0120912. doi: 10.1371/journal.pone.0120912
108. Hector A, Schäfer H, Pöschel S, Fischer A, Fritzsching B, Ralhan A, et al. Regulatory T-cell impairment in cystic fibrosis patients with chronic pseudomonas infection. Am J Respir Crit Care Med. (2015) 191:914–23. doi: 10.1164/rccm.201407-1381OC
109. Pizzutto SJ, Yerkovich ST, Upham JW, Hales BJ, Thomas WR, and Chang AB. Children with chronic suppurative lung disease have a reduced capacity to synthesize interferon-gamma in vitro in response to non-typeable Haemophilus influenzae. PloS One. (2014) 9:e104236. doi: 10.1371/journal.pone.0104236
110. Jaat FG, Hasan SF, Perry A, Cookson S, Murali S, Perry JD, et al. Anti-bacterial antibody and T cell responses in bronchiectasis are differentially associated with lung colonization and disease. Respir Res. (2018) 19:106. doi: 10.1186/s12931-018-0811-2
111. Chen AC, Martin ML, Lourie R, Rogers GB, Burr LD, Hasnain SZ, et al. Adult non-cystic fibrosis bronchiectasis is characterised by airway luminal Th17 pathway activation. PloS One. (2015) 10:e0119325. doi: 10.1371/journal.pone.0119325
112. Xie C, Wen Y, Zhao Y, Zeng S, Guo Q, Liang Q, et al. Clinical features of patients with bronchiectasis with comorbid chronic obstructive pulmonary disease in China. Med Sci Monit. (2019) 25:6805–11. doi: 10.12659/MSM.917034
113. Westhölter D, Beckert H, Straßburg S, Welsner M, Sutharsan S, Taube C, et al. Pseudomonas aeruginosa infection, but not mono or dual-combination CFTR modulator therapy affects circulating regulatory T cells in an adult population with cystic fibrosis. J Cyst Fibros. (2021) 20:1072–9. doi: 10.1016/j.jcf.2021.05.001
114. Fouka E, Lamprianidou E, Arvanitidis K, Filidou E, Kolios G, Miltiades P, et al. Low-dose clarithromycin therapy modulates Th17 response in non-cystic fibrosis bronchiectasis patients. Lung. (2014) 192:849–55. doi: 10.1007/s00408-014-9619-0
115. Boyton RJ and Altmann DM. Bronchiectasis: current concepts in pathogenesis, immunology, and microbiology. Annu Rev Pathol. (2016) 11:523–54. doi: 10.1146/annurev-pathol-012615-044344
116. Narayana JK, Mac Aogáin M, Hansbro PM, and Chotirmall SH. The bronchiectasis microbiome: current understanding and treatment implications. Curr Opin Pulm Med. (2025) 31:135–44. doi: 10.1097/MCP.0000000000001131
117. Mac Aogáin M and Chotirmall SH. Microbiology and the microbiome in bronchiectasis. Clin Chest Med. (2022) 43:23–34. doi: 10.1016/j.ccm.2021.11.002
118. Mac Aogáin M, Tiew PY, Jaggi TK, Narayana JK, Singh S, Hansbro PM, et al. Targeting respiratory microbiomes in COPD and bronchiectasis. Expert Rev Respir Med. (2024) 18:111–25. doi: 10.1080/17476348.2024.2355155
119. Matsumoto H. Bronchiectasis in severe asthma and asthmatic components in bronchiectasis. Respir Investig. (2022) 60:187–96. doi: 10.1016/j.resinv.2021.11.004
120. Keir HR and Chalmers JD. Pathophysiology of bronchiectasis. Semin Respir Crit Care Med. (2021) 42:499–512. doi: 10.1055/s-0041-1730891
121. Chalmers JD, Elborn S, and Greene CM. Basic, translational and clinical aspects of bronchiectasis in adults. Eur Respir Rev. (2023) 32:230015. doi: 10.1183/16000617.0015-2023
122. Lindén A and Dahlén B. Interleukin-17 cytokine signalling in patients with asthma. Eur Respir J. (2014) 44:1319–31. doi: 10.1183/09031936.00002314
123. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2016) 138:16–27. doi: 10.1016/j.jaci.2016.05.011
124. Gray RD, Hardisty G, Regan KH, Smith M, Robb CT, Duffin R, et al. Delayed neutrophil apoptosis enhances NET formation in cystic fibrosis. Thorax. (2018) 73:134–44. doi: 10.1136/thoraxjnl-2017-210134
125. Khan MA, Ali ZS, Sweezey N, Grasemann H, and Palaniyar N. Progression of cystic fibrosis lung disease from childhood to adulthood: neutrophils, neutrophil extracellular trap (NET) formation, and NET degradation. Genes (Basel). (2019) 10:183. doi: 10.3390/genes10030183
126. Martins M, Keir HR, and Chalmers JD. Endotypes in bronchiectasis: moving towards precision medicine. A narrative review. Pulmonology. (2023) 29:505–17. doi: 10.1016/j.pulmoe.2023.03.004
127. Sheng H, Yao X, Wang X, Wang Y, Liu X, and Zhang L. Prevalence and clinical implications of bronchiectasis in patients with overlapping asthma and chronic rhinosinusitis: a single-center prospective study. BMC Pulm Med. (2021) 21:211. doi: 10.1186/s12890-021-01575-7
128. Wang S, Zhang J, Zhang C, and Shao C. Clinical characteristics of allergic bronchopulmonary aspergillosis in patients with and without bronchiectasis. J Asthma. (2022) 59:1162–8. doi: 10.1080/02770903.2021.1904979
129. Sunata K, Miyata J, Kawashima Y, Konno R, Ishikawa M, Hasegawa Y, et al. Inflammatory profile of eosinophils in asthma-COPD overlap and eosinophilic COPD: a multi-omics study. Front Immunol. (2024) 15:1445769. doi: 10.3389/fimmu.2024.1445769
130. Shteinberg M, Chalmers JD, Narayana JK, Dicker AJ, Rahat MA, Simanovitch E, et al. Bronchiectasis with chronic rhinosinusitis is associated with eosinophilic airway inflammation and is distinct from asthma. Ann Am Thorac Soc. (2024) 21:748–58. doi: 10.1513/AnnalsATS.202306-551OC
131. Campisi R, Nolasco S, Mancuso M, Spinella M, Vignera F, Crimi N, et al. Eosinophilic bronchiectasis: prevalence, severity, and associated features-A cohort study. J Clin Med. (2024) 13:748–58. doi: 10.3390/jcm13164932
132. Martínez-García MA, Olveira C, Girón R, García-Clemente M, Máiz L, Sibila O, et al. Reliability of blood eosinophil count in steady-state bronchiectasis. Pulmonology. (2025) 31:2416836. doi: 10.1016/j.pulmoe.2023.11.006
133. Pollock J, Keir HR, Shoemark A, Dicker AJ, Giam YH, Crichton ML, et al. (2022). Proteomic markers of eosinophilic inflammation and disease severity in bronchiectasis. A62 CYSTIC FIBROSIS AND NON-CF BRONCHIECTASIS: MECHANISTIC INSIGHTS, in: American Thoracic Society International Conference Abstracts: American Thoracic Society. pp. A2004–A.
134. Tan HL, Regamey N, Brown S, Bush A, Lloyd CM, and Davies JC. The Th17 pathway in cystic fibrosis lung disease. Am J Respir Crit Care Med. (2011) 184:252–8. doi: 10.1164/rccm.201102-0236OC
135. Mannion JM, McLoughlin RM, and Lalor SJ. The airway microbiome-IL-17 axis: a critical regulator of chronic inflammatory disease. Clin Rev Allergy Immunol. (2023) 64:161–78. doi: 10.1007/s12016-022-08928-y
136. Zheng X, Zhang L, Chen J, Gu Y, Xu J, and Ouyang Y. Dendritic cells and Th17/Treg ratio play critical roles in pathogenic process of chronic obstructive pulmonary disease. BioMed Pharmacother. (2018) 108:1141–51. doi: 10.1016/j.biopha.2018.09.113
137. Tulic MK, Piche T, and Verhasselt V. Lung-gut cross-talk: evidence, mechanisms and implications for the mucosal inflammatory diseases. Clin Exp Allergy. (2016) 46:519–28. doi: 10.1111/cea.2016.46.issue-4
138. Goutaki M and Shoemark A. Diagnosis of primary ciliary dyskinesia. Clin Chest Med. (2022) 43:127–40. doi: 10.1016/j.ccm.2021.11.008
139. Carmier D, Dartigeas C, Mankikian J, Rousselot-Denis C, Lissandre S, Diot P, et al. Serious bronchopulmonary involvement due to chronic lymphocytic leukaemia. Eur Respir Rev. (2013) 22:416–9. doi: 10.1183/09059180.00008812
140. Patrawala M, Cui Y, Peng L, Fuleihan RL, Garabedian EK, Patel K, et al. Pulmonary disease burden in primary immune deficiency disorders: data from USIDNET registry. J Clin Immunol. (2020) 40:340–9. doi: 10.1007/s10875-019-00738-w
141. Holmes AH, Pelton S, Steinbach S, and Luzzi GA. HIV related bronchiectasis. Thorax.V. (1995) 50:1227. doi: 10.1007/s10875-019-00738-w
142. Tsilifis C, Freeman AF, and Gennery AR. STAT3 hyper-igE syndrome-an update and unanswered questions. J Clin Immunol. (2021) 41:864–80. doi: 10.1007/s10875-021-01051-1
143. Polverino E, Goeminne PC, McDonnell MJ, Aliberti S, Marshall SE, Loebinger MR, et al. European Respiratory Society guidelines for the management of adult bronchiectasis. Eur Respir J. (2017) 50:864–80. doi: 10.1183/13993003.00629-2017
144. Boaventura R, Sibila O, Agusti A, and Chalmers JD. Treatable traits in bronchiectasis. Eur Respir J. (2018) 52:1700629. doi: 10.1183/13993003.01269-2018
145. De Soyza A, Pavord I, Elborn JS, Smith D, Wray H, Puu M, et al. A randomised, placebo-controlled study of the CXCR2 antagonist AZD5069 in bronchiectasis. Eur Respir J. (2015) 46:1021–32. doi: 10.1183/13993003.00148-2015
146. Cipolla D, Zhang J, Korkmaz B, Chalmers JD, Basso J, Lasala D, et al. Dipeptidyl peptidase-1 inhibition with brensocatib reduces the activity of all major neutrophil serine proteases in patients with bronchiectasis: results from the WILLOW trial. Respir Res. (2023) 24:133. doi: 10.1186/s12931-023-02444-z
147. Johnson ED, Long MB, Perea L, Shih VH, Fernandez C, Teper A, et al. Broad immunomodulatory effects of the dipeptidyl-peptidase-1 inhibitor brensocatib in bronchiectasis: data from the phase 2, double-blind, placebo-controlled WILLOW trial. Am J Respir Crit Care Med. (2025) 46:1021–32. doi: 10.1164/rccm.202408-1545OC
148. Chalmers JD, Loebinger MR, Teper A, McShane PJ, Fernandez C, Fucile S, et al. Brensocatib in patients with bronchiectasis: subgroup analyses from the WILLOW trial. ERJ Open Res. (2025) 11:133. doi: 10.1183/23120541.00505-2024
149. Chalmers JD, Burgel PR, Daley CL, De Soyza A, Haworth CS, Mauger D, et al. Phase 3 trial of the DPP-1 inhibitor brensocatib in bronchiectasis. N Engl J Med. (2025) 392:1569–81. doi: 10.1056/NEJMoa2411664
150. Chalmers JD, Shteinberg M, Mall MA, O’Donnell AE, Watz H, Gupta A, et al. Cathepsin C (dipeptidyl peptidase 1) inhibition in adults with bronchiectasis: AIRLEAF, a phase II randomised, double-blind, placebo-controlled, dose-finding study. Eur Respir J. (2025) 65:00505–2024. doi: 10.1183/13993003.01551-2024
151. Zhong NS, Qiu R, Cao J, Huang YM, Zhou H, Xu XX, et al. Effects of the DPP-1 inhibitor HSK31858 in adults with bronchiectasis in China (SAVE-BE): a phase 2, multicentre, double-blind, randomised, placebo-controlled trial. Lancet Respir Med. (2025) 392:1569–81. doi: 10.1016/S2213-2600(25)00019-0
152. Long MB, Chotirmall SH, Shteinberg M, and Chalmers JD. Rethinking bronchiectasis as an inflammatory disease. Lancet Respir Med. (2024) 12:901–14. doi: 10.1016/S2213-2600(24)00176-0
153. Pollock J, Polverino E, Dhar R, Dimakou K, Traversi L, Bossios A, et al. Use of inhaled corticosteroids in bronchiectasis: data from the European Bronchiectasis Registry (EMBARC). Thorax. (2025) 13:414–24. doi: 10.1136/thorax-2024-221825
154. Rademacher J, Konwert S, Fuge J, Dettmer S, Welte T, and Ringshausen FC. Anti-IL5 and anti-IL5Rα therapy for clinically significant bronchiectasis with eosinophilic endotype: a case series. Eur Respir J. (2020) 55:901–14. doi: 10.1183/13993003.01333-2019
155. Choi H, McShane PJ, Aliberti S, and Chalmers JD. Bronchiectasis management in adults: state of the art and future directions. Eur Respir J. (2024) 63:358–68. doi: 10.1183/13993003.00518-2024
156. Patel SD, Bono TR, Rowe SM, and Solomon GM. CFTR targeted therapies: recent advances in cystic fibrosis and possibilities in other diseases of the airways. Eur Respir Rev. (2020) 29:1901333. doi: 10.1183/16000617.0068-2019
157. Camargo LDN, Righetti RF, Aristóteles L, Dos Santos TM, de Souza FCR, Fukuzaki S, et al. Effects of anti-IL-17 on inflammation, remodeling, and oxidative stress in an experimental model of asthma exacerbated by LPS. Front Immunol. (2017) 8:1835. doi: 10.3389/fimmu.2017.01835
158. Camargo LDN, Dos Santos TM, de Andrade FCP, Fukuzaki S, Dos Santos Lopes F, de Arruda Martins M, et al. Bronchial vascular remodeling is attenuated by anti-IL-17 in asthmatic responses exacerbated by LPS. Front Pharmacol. (2020) 11:1269. doi: 10.3389/fphar.2020.01269
159. Fukuzaki S, Righetti RF, Santos TMD, Camargo LDN, Aristóteles L, Souza FCR, et al. Preventive and therapeutic effect of anti-IL-17 in an experimental model of elastase-induced lung injury in C57Bl6 mice. Am J Physiol Cell Physiol. (2021) 320:C341–c54. doi: 10.1152/ajpcell.00017.2020
160. Zhao J, Lloyd CM, and Noble A. Th17 responses in chronic allergic airway inflammation abrogate regulatory T-cell-mediated tolerance and contribute to airway remodeling. Mucosal Immunol. (2013) 6:335–46. doi: 10.1038/mi.2012.76
Keywords: bronchiectasis, neutrophilic inflammation, eosinophilic inflammation, T helper cells, T regulatory cells, microbiome, endotypes
Citation: Fouka E, Lindén A and Bossios A (2025) The role of T-helper and T regulatory cells in driving neutrophilic and eosinophilic inflammation in bronchiectasis. Front. Immunol. 16:1598257. doi: 10.3389/fimmu.2025.1598257
Received: 22 March 2025; Accepted: 26 May 2025;
Published: 16 June 2025.
Edited by:
Ramcés Falfán-Valencia, National Institute of Respiratory Diseases-Mexico (INER), MexicoReviewed by:
Hayoung Choi, Hallym University, Republic of KoreaVincent Laiman, Gadjah Mada University, Indonesia
Copyright © 2025 Fouka, Lindén and Bossios. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Apostolos Bossios, YXBvc3RvbG9zLmJvc3Npb3NAa2kuc2U=