 Yao Yao
Yao Yao Qiong Wu1†
Qiong Wu1† Bin Fan
Bin Fan Mingwei Sheng
Mingwei Sheng Fengmei Wang
Fengmei Wang- 1Department of Gastroenterology and Hepatology, Tianjin First Central Hospital, Nankai University, Tianjin, China
- 2Department of Urology, Tianjin First Central Hospital, Nankai University, Tianjin, China
- 3Tianjin Key Laboratory of Radiation Medicine and Molecular Nuclear Medicine, Institute of Radiation Medicine, State Key Laboratory of Advanced Medical Materials and Devices, Tianjin Institutes of Health Science, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China
- 4Department of Anesthesiology, Tianjin First Central Hospital, Nankai University, Tianjin, China
With the continuous increase of the incidence of Metabolic dysfunction-associated steatotic liver disease(MASLD), the proportion of MASLD-driven hepatocellular carcinoma(HCC) is gradually increasing, which will become a heavy burden on global public health. This article summarizes the existing literature and discusses the role of various innate immune cells in the occurrence and development of MASLD, such as cell-cell crosstalk, and uses bibliometric analysis to find current research hotspots and emerging topics, in order to provide valuable reference for scholars studying the direction of MASLD-driven HCC immunity. It also provides a solid foundation for more researchers to join the research direction. And try to inspire researchers in future research to seek breakthroughs in this regard.
1 Introduction
MASLD, formerly known as Non-Alcoholic Fatty Liver Disease (NAFLD), is defined as excess triglyceride storage in the liver in the presence of at least one cardiometabolic risk factor, including isolated metabolic dysfunction-associated steatotic liver(MASL), metabolic dysfunction-associated steatotic hepatitis (MASH), and fibrosis and cirrhosis (1). According to statistics, about 30% of the global population suffers from MASLD (2). Moreover, the incidence of MASH may increase by 56% in the next 10 years (3), and the overall burden of end-stage liver disease caused by MASLD may increase by 2 to 3 times in the next 20 years (4). Currently, approximately 2% of MASH converts to MASH-driven HCC each year (5). It is noteworthy that the progression process can also occur in the absence of cirrhosis (6). With the promotion and implementation of hepatitis B blocking therapy and the gradual improvement of clinical cure rate of hepatitis B, the proportion of MASLD-driven HCC is gradually increasing, which will be a heavy burden on global public health. Based on above, how to reduce the incidence of MASH and how to alleviate or reverse MASH-HCC are challenging point for scientific researchers.
It is well known that chronic inflammation is an important trigger for hepatocyte transformation and carcinogenesis, and that extensive liver inflammation is regulated by changes in innate immune activation. The inflammatory cascade in MASLD is caused by innate immunity, which involves myeloid innate immune cells such as macrophages (resident and recruited), monocytes, dendritic cells (DCs), neutrophils, eosinophils, basophils, and innate lymphoid cells(ILCs), and adaptive immunity, which involves B and T lymphocytes (7–9). Suppressing the excessive inflammatory response by regulating innate immune cells seems to be a feasible way forward.
With the increasing incidence of MASLD and the proportion of MASLD-driven HCC worldwide, scholars from all over the world have carried out in-depth studies on the mechanisms of inflammation and cancer transformation of MASLD, especially the role of innate immune cells. Based on this, we have summarized the role of various innate immune cells in the occurrence and development of MASLD (Figure 1), searched the research on innate immune cells in MASLD published from 2000 to 2025, used bibliometric analysis to explore the current research hotspots and emerging topics, in order to provide a solid foundation for more researchers to join the research in this direction. And we aim to inspire researchers in future studies to seek breakthroughs in this area.
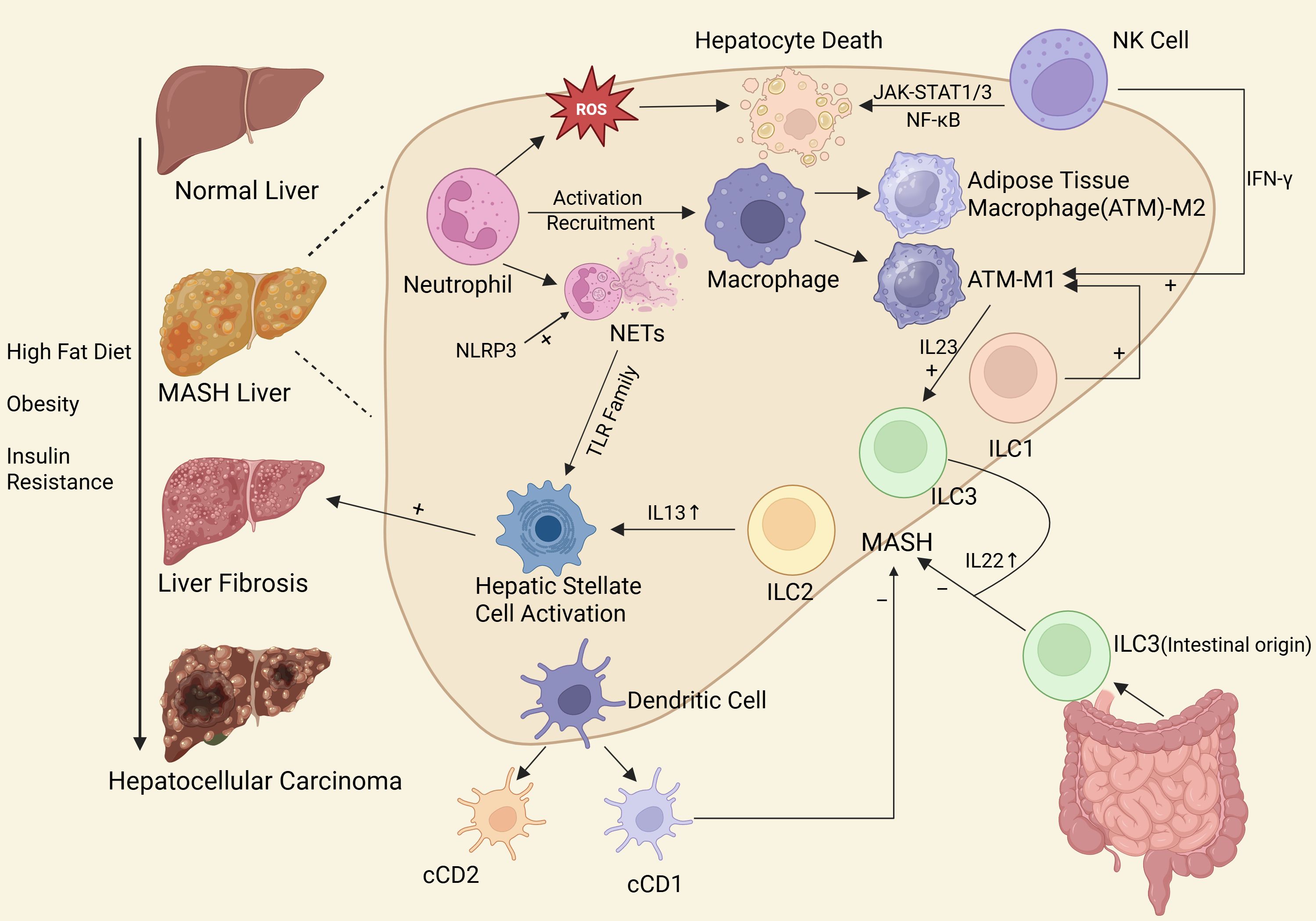
Figure 1. The role of innate immune cells in MASLD.
2 The role of various types of innate immune cells in the progression of MASLD
2.1 Macrophages
Macrophages were first identified by Elie Metchnikoff in 1884 (10). In 1876, Wilhelm von Kupffer (11) first discovered that there were a large number of tissue macrophages in the healthy liver, and these cells were named Kupffer cells (KCs). Hepatic macrophages are composed of KCs and recruited monocyte-derived macrophages. Under steady-state conditions, KCs comprise more than 90% of all hepatic macrophages, while monocytes circulate in the hepatic sinusoids but do not participate in the adult KCs repertoire. In the case of KCs depletion, bone marrow derived monocytes are rapidly recruited, colonize the liver, and produce monocyte-derived KCs that are genetically and functionally nearly identical to embryonic macrophages and acquire the ability to self-sustain in the liver without the subsequent contribution of bone marrow monocytes. Together, they constitute the largest population of innate immune cells in the liver and play a central role in maintaining liver homeostasis (12, 13).
According to the state of the microenvironment, liver macrophages can be differentiated into two different types, namely the classically activated pro-inflammatory M1 and the alternatively activated anti-inflammatory M2. In the context of MASLD, macrophage polarization has been reviewed and elucidated, which can be summarized as follows (14): In the early stage of MASH, resident KCs mainly promote triglyceride storage and exhibit an anti-inflammatory M2 phenotype. Depletion of KCs is beneficial for improve hepatocyte steatosis and insulin resistance, followed by the conversion of monocyte-derived macrophages into KCs within the hepatic sinusoids. However, monocyte-differentiated KCs can reduce lipid accumulation but exacerbate liver inflammation. Since the role of KCs in MASLD spectrum disorders has been reviewed in detail, this article focuses on the role of Adipose tissue macrophages(ATMs) in MASLD spectrum disorders, to supplement the immune role of macrophages in liver inflammation.
MASH pathology is characterized by hepatic fat accumulation and inflammatory cell infiltration, and although MASH occurs in the context of altered metabolism, there is a strong immuno-inflammatory component in the expanding adipose tissue compartment (15). ATMs, which constitute the majority of immune cells in adipose tissue, regulate tissue homeostasis in the lean state and metabolic dysregulation in obesity (16). Meanwhile, ATMs accumulate in high caloric intake environments and undergo transcriptional and phenotypic changes, from regulating metabolic dysregulation to promoting tissue inflammation and aggravating metabolic disorders (17). Adipose tissue inflammation significantly contributes to MASH progression and may even be a prerequisite for MASH progression. Boesch et al. (18) found that ablation of ATMs or removal of visceral white adipose tissue reduced insulin resistance and liver inflammation in animal models. Interestingly, Boesch et al. (18) showed that ATMs dysfunction causes loss of vascular integrity in adipose tissue characterized by albumin extravasation into perivascular tissues, which promotes the development of adipose tissue and liver inflammation. This can lead to the progression of MASLD to a more severe phenotype. This further confirms that ATMs play different roles under different conditions. When MASH-ATMs exhibit proinflammatory and lipid-related phenotypes, they secrete small extracellular vesicles (sEVs) enriched in fibrogenic miRNAs miR-155 and miR-34a to promote liver fibrosis progression (19). Growth differentiation factor-15 (GDF-15) is an effective anti-obesity target, which prevents obesity by binding and activating its receptor in the central nervous system (20, 21). Using large human cohorts of obesity, type 2 diabetes, and MASLD states, as well as several experimental mouse models of obesity, insulin resistance, and MASLD-like conditions, L’homme et al. (22) found that GDF-15 production increased first in adipose tissue at the onset of obesity and T2D and subsequently in the liver during MASLD progression. Accumulation of ATMs in adipose tissue is responsible for elevated GDF-15 in obesity and T2D patients. Notably, inactivation of GDF-15 in ATMs reduced plasma GDF-15 concentrations in mice and aggravated the development of diet-induced obesity and hepatic complications, especially progression from steatosis to MASH. In addition, Martinez-Sanchez et al. (23), a Spanish clinical trial, collected adipose tissue and liver biopsies from 42 MASLD patients with different fibrosis stages and found that the degree of liver fibrosis in MASLD patients was significantly correlated with the abundance of proinflammatory ATMs, and the modulation of ATMs improved liver fibrosis.The above studies further indicate that ATMs play a non-negligible role in the progression of MASLD. Resident ATMs can regulate metabolic disorders in obesity, and when ATMs accumulate, they can accelerate metabolic disorders. However, whether ATMs dysfunction in MASLD is a cause or a consequence of MASLD is a common question that has not yet been concluded. It is discussed in the Journal of Hepatology and suggests a possible need for longitudinal sampling and analysis of adipose tissue in large MASLD cohorts, with further clarification of links to clinical outcome data (24). This is also the direction that future researchers need to explore deeply.
2.2 Neutrophil
Inflammatory infiltration is the pathological feature of MASLD. Neutrophils are the most abundant subtype of leukocyte in the blood circulation. As the first responders of inflammation, neutrophils are the first cellular defense line of the human innate immune system, and play an indispensable role in the evolution process from simple hepatic steatosis to NASH (25). Neutrophils are primarily recognized for their role as antigen-presenting cells, which directly activate KCs and endothelial cells and promote cellular adhesion, At the same time, they plays a role in antibacterial response and inflammatory injury by producing ROS, granzymes, cytokines and neutrophil extracellular traps (NETs) (26). However, abnormal overproduction of these mediators may lead to increased inflammation. Brinkman et al. (27) first reported in 2004 that neutrophils can expel their nuclear material and establish NETs, which can trap and kill bacteria. In the following two decades, its role in various non-infectious inflammation and cancer has been gradually discovered. In 2018, Dirk et al. (28) found that neutrophils were stimulated to form NETs in a MASH mouse model, and that inhibition of NETs significantly reduced macrophage infiltration, inflammatory cytokine production, and HCC development. However, the underlying mechanisms remain unclear.
Innate immunity involves signal transduction through pattern recognition receptors, such as Toll-like Receptors (TLRs), which play an indispensable role in the process of recognizing pathogen-associated molecular patterns or damage-associated molecular patterns (29). TLR3 is the most up-regulated TLR in the TLR family in hepatic stellate cells(HSCs) stimulated by NETs, and TLR3 signaling induces inflammation (increased COX-2 expression) and HSCs activation (increased α-SMA expression) and PGE2 production, which promotes liver inflammation and injury and accelerates MASH related liver fibrosis (30). In contrast, TLR3 knockout HSCs lost the ability to activate in response to NETs stimulation. It has been shown that ROS are a key factors in promoting HSC activation (31). During MASH progression, ROS are released from mitochondria into the cytoplasm, ROS accumulation promotes ferroptosis of hepatocytes, recruits neutrophils, and induces the production of NETs, while blockade of NETs can inhibit the progression of MASH (32). These findings support that recruitment of NETs promotes hepatic stellate cell activation and subsequent fibrosis development in MASH mice, that is, NETs play an important role in the pathogenesis of MASH fibrosis.
NOD-like receptor protein 3 (NLRP3) inflammasome is expressed in various cell types of the liver, including hepatocytes, HSCs, macrophages, and neutrophils, and has been identified as one of the triggers of liver inflammation in MASLD (33, 34). NLRP3 was induced and activated in the liver of MASH mice, and that NLRP3 inhibitors significantly reduced liver neutrophil infiltration and regulated MASH fibrosis progression (35). Meanwhile, NETs formation, liver injury, and fibrosis were attenuated after administration of NLRP3 inhibitors in mice fed a MASH + alcohol diet (36). How NLRP3 regulates neutrophil recruitment and NETs formation is still unclear. Breast regression protein 39 (BRP39, human homolog YKL-40) may be one of the key targets. BRP39 is a glycoprotein expressed by many cell types, including macrophages, neutrophils, fibroblasts, and epithelial cells. In NLRP3-induced MASH, BRP39 deficiency improved liver inflammation and fibrosis, and the activation of HSCs cells was significantly reduced, and the aggregation of Ly6C+ infiltrating macrophages and H3Cit+ neutrophils in the liver was significantly reduced (37). BRP39-deficient mice exhibit impaired chemotaxis and migration of circulating neutrophils and decreased expression of RNAs associated with immune activation, migration, and signaling responses in neutrophils. NLRP3 inhibitors have now been investigated in preclinical studies in metabolic and inflammatory-driven diseases (gout, cardiovascular disease, cancer, etc.), neurodegenerative diseases, and cryopyrin-associated periodic syndrome (CAPS). Although basic studies have shown the beneficial effects of NLRP3 inhibitors in MASLD spectrum disorders, there is still a gap in preclinical studies. Currently, there are no FDA-approved NLRP3-targeting inhibitors. MCC950, commonly used in basic research on MASLD spectrum disorders, is a direct NLRP3 inhibitor that shows promise but exhibits off-target effects. The clinical development of MCC950 in rheumatoid arthritis was discontinued due to hepatotoxicity. Hence the preclinical evaluation of MCC950 in the treatment of MASLD patients should be carefully evaluated (38).
NETs can also serve as a bridge between innate and adaptive immunity. While promoting innate immunity, NETs promote Treg differentiation through metabolic reprogramming of naive CD4 T cells, crosstalk with adaptive immunity, and further prevent the occurrence of MASH-HCC (39). Moreover, NETs can directly affect hepatocytes and aggravate MASH by promoting hepatocyte senescence, leading to liver fibrosis (40). In conclusion, the production of NETs by neutrophils is an important pathway to maintain the inflammatory state, exerting its effects after injury. However, excessive NETs constitute harmful factors, which can exacerbate liver injury and promote cancer progression. Probably due to the short time of NETs research (research began in the past 20 years), no NETs blockers have been used in preclinical studies.
2.3 ILCs
Innate lymphoid cells (ILCs) are a group of tissue-resident immune cells that lack T cell and B cell receptors. According to their developmental and functional trajectories, ILCs are divided into five subsets: natural killer (NK) cells, ILC1s, ILC2s, ILC3s, and lymphoid tissue-induced cells, but their grouping is still controversial (41).
2.3.1 NK cell
During the progression of MASH, liver inflammation (provided by lipotoxicity, immune-inflammatory cells, and endotoxemia) affects the mechanistic process of NK cells development and functional maturation (42). In the late stage, persistent high lipids promote programmed necrosis of NK cells, which in turn promotes the progression of MASH to fibrosis (43). Patients with obesity, simple steatosis, and progressive liver fibrosis have a greatly increased risk of developing HCC due to reduced NK cell numbers and function (44).
Hepatic NK cells have a dual role in nonalcoholic fatty liver disease. On the one hand, they can amplify the inflammatory response in the MASH stage (45). On the other hand, they can inhibit the development of fibrosis by killing activated hepatic stellate cells (46), or they can improve fibrosis by regulating the fibrotic properties of other liver-resident immune cells (47), thus playing a key role in the liver. Activated hepatic NK cells in MASH exhibit robust secretion of proinflammatory cytokines, including interferon-γ(IFN-γ), interleukin-1β(IL-1β), interleukin-12(IL-12), CCL4, CCL5, and granulocyte-macrophage colony-stimulating factor(GM-CSF). The absence of NK cells or IFN-γ preventes the accumulation of proinflammatory macrophages in visceral adipose tissue and significantly improved insulin sensitivity, corroborating the fact that NK cells can amplify the inflammatory response during the MASH phase (48). Besides, these proinflammatory cytokines secreted by NK cells activate hepatic Janus Kinase-Signal Transducers and Activators of Transcription 1/3(JAK-STAT1/3) and nuclear factor kappa-B (NF-κB) signaling pathways to induce hepatocyte injury. It is worth mentioning that NK cell-derived IFN-γ plays a key role in maintaining the balance of the inflammatory environment in NASH and promoting tissue integrity (45). NK cells produce IFN-γ to differentiate Mϕ into M1-like phenotype (non-M2-like phenotype), which plays an anti-fibrogenesis role. Loss of NKp46+ cells promotes the development of fibrosis and promotes profibrotic gene expression and a distorted M2 Mϕ phenotype (49).
2.3.2 ILC1
ILC1 was initially identified in the liver of C57BL/6 mice and subsequently in other tissues such as salivary glands, skin, and uterus. Conventional NK cells and ILC1 can be distinguished by their T-box transcription factor properties. The T-box protein in T cells, Tbet, encoded by the Tbx21 gene is involved in IFN-γ production (50, 51). Mature NK cells are strictly dependent on Eomes(Tbet+ Eomes+) and have a Lin- CD45+ NK1.1+ NKp46+ CD49a- CD49b+ phenotype in mice, while ILC1 is independent of Eomes(Tbet+ Eomes-). The phenotype in mice is Lin− CD45+ NK1.1+ NKp46+ CD49a+ CD49b− (52).
Hepatic NKp46+ cells account for approximately 10% to 20% of the total number of intrahepatic lymphocytes in mice and 40% to 50% of the total number of intrahepatic lymphocytes in humans (53). Previous studies have shown that high-fat diet (HFD) stimulation can activate adipose tissue and increase the number of ILC1, which is the main source of IFN-γ and TNF-α. At the same time, ILC1 polarizes ATM-M1, leading to adipose tissue inflammation and insulin resistance (54). In addition, ILC1 plays at least in part the role of proinflammatory effector cells in aggravating hepatic ischemia-reperfusion injury(IRI) under steatosis conditions (55).
2.3.3 ILC2
Interleukin-33(IL-33), a cytokine of the IL-1 family, is released extracellularly in chronic hepatocyte stress and marks the accumulation and activation of ILC2 in the liver. Attenuating IL-33-dependent accumulation and activation of ILC2 can inhibit the activation of hepatic stellate cells and reduce the secretion of collagen and matrix proteins, thereby alleviating the degree of liver fibrosis (56). Liver-activated ILC2 produce IL-13, which in turn triggers HSC activation and transdifferentiation in a transcription factor-dependent manner, and participates in the progression of liver fibrosis (57). Meanwhile, Gonzalez-Polo et al. (58) studied liver tissues from patients with liver biopsy and found that ILC2 numbers are significantly increased in fibrotic tissues, activated in tissues that promote human liver fibrosis immunopathology, independent of etiology, and may be a potential new therapeutic target. However, because the interaction between the immune system and lipid metabolism is very complex, it has been shown that lipid metabolism can affect the role of ILC2 in airway inflammation and lung homeostasis. After ILC2 activation, the uptake of fatty acids (FA) is increased, and the utilization of lipid metabolism is significantly increased (59–61).
2.3.4 ILC3
The proportion of ILC3 in the liver is very small. In 2018, Wang et al. (62) showed that ILC3 can promote the formation of liver fibrosis by producing IL-22. However, subsequent studies have focused on attenuating hepatic steatosis by activation of ILC3 to produce IL-22, the major cellular source in the liver, by a high-fat diet; in other words, IL-22 secreted by ILC3 is protective against MASLD spectrum disorders. It has also been confirmed in preclinical models of MASLD that systemic treatment with IL-22 can improve hepatic steatosis, reduce human serum triglyceride levels, reduce intestinal lipid metabolism and uptake, and have liver protective effects (63–66). Hamaguchi et al. (67) found a significant increased M1 macrophages and decreased M2 macrophages in HFD-fed mice. HFD stimulates the production of IL-23 by M1 macrophages, thereby promoting the proliferation of hepatic ILC3, while IL-22 secreted by ILC3 contributes to the up-regulation of hepatic lipid metabolism and has anti-apoptotic activity. Interestingly, ILC3 deficiency leads to significant liver fibrosis even in the absence of HFD. IL-22 produced by ILC3 in the gut plays an important role in maintaining the integrity of the intestinal barrier, altering lipid metabolism and reducing metabolic imbalance (68, 69). High-fat diet can reduce the production of IL-22 by intestinal ILC3, destroy the integrity of the intestinal barrier, and aggravate liver steatosis (70). Although basic research has consistently confirmed that ILC3 proliferation promotes the increase of IL-22 secretion and can reverse MASLD, there is no such study reported in the top Journals. It may be due to the small proportion of ILC3 itself and its short discovery time, so the current research in this field has great potential.
2.4 DCs
DCs are the bridge between innate and adaptive immunity and represent the tolerant cell population of the liver in steady state. They are highly heterogeneous, including plasmacytoid dendritic cells, classical type 1 DCs (cDC1; CD103+ CD11c+ CD11b-) and classical type 2 DCs (cDC2; CD103− CD11c+ CD11b+) (71, 72). A 2022 Mexican study of 128 liver biopsy tissues showed that the frequency of CD11c+ DC(cDC) expression in liver tissues of MASLD patients was higher than that of mildly obese or non-obese patients (73). In basic research, Heier et al. demonstrated that cDC1s in mouse liver are a protective cell type that can regulate the influx of inflammatory cells and coordinate the pro-inflammatory and anti-inflammatory environment during the progression of steatohepatitis in mice (74). In parallel, the abundance of cDC1s in visceral adipose tissue is reduced in mice fed a high-fat diet and that reduced expression of cDC1-related genes is characteristic of mice prone to weight gain (75). In addition, deficiency of cDC1s reduced energy expenditure and caused adipose tissue inflammation in middle-aged mice, which was associated with impaired glucose tolerance, insulin resistance, dyslipidemia and hepatic steatosis. The mechanism may be related to the deficiency of and increased phosphorylation of liver kinase B1 (LKB1) in cDCs (76). However, the mechanism of DCs in MASLD spectrum diseases is not fully understood, and more studies are needed to reveal it.
3 Emerging therapies targeting innate immune cells in MASLD
MASLD is a systemic disease involving the crosstalk between liver, adipose tissue, muscle and intestinal tract. The key players in the intrahepatic crosstalk are hepatocytes, HSCs, liver sinusoidal endothelial cells and immune cells. At present, the focus of clinical research is mainly on macrophages and NK cells.
Future therapeutic strategies for macrophages may be aimed at changing macrophages from a pro-inflammatory phenotype to a restorative phenotype, but there are no relevant clinical studies. A recent phase 2 open-label randomized controlled trial demonstrated the safety, feasibility, and potential efficacy of using ex vivo-mature autologous monocyte-derived macrophages in patients with cirrhosis (77). In addition, CAR macrophages with antifibrotic T-cell immunity have been developed and shown to be effective in reducing experimental fibrosis (78).
Chimeric antigen receptor (CAR) -modified NK cells will become an effective strategy for the treatment of HCC and hepatic fibrosis in the next decade due to their specificity and low side effects (79). At present, clinical trials have focused on the efficacy of NK cells in the treatment of HCC and hepatitis virus infection(ClinicalTrials.gov, e.g., NCT05040438, NCT04162158, NCT05171309, and NCT03761875, accession date: 28 March 2023).The above studies provide hope for reversing liver fibrosis and supporting liver regeneration.
4 Conclusion and perspective
This article reviews the research progress of various innate immune cells in MASLD spectrum diseases, including macrophages, neutrophils, ILCs (ILC1, NK, ILC2, ILC3), and DCs. In the liver, the proportion of different types of innate immune cells ranges from high to low: macrophages/KCs, NK cells, DCs, neutrophils, ILC1, ILC2, and ILC3. Innate immune cells also include mast cells, eosinophils, basophils, etc. No relevant research has been reported when searching the literature, so they are not described in this paper. We used VOSviewer software to analyze all the articles published on the Web of Science from 2000 to February 25, 2025, trying to find the current research hotspots of the role of innate immune cells in MASLD. In this study, VOSviewer software was used to analyze the literature in the Web of Science Core Collection (WoSCC) database from 2000 to February 25, 2025, and try to find the current research hotspots of the role of innate immune cells in MASLD. The search formula used is as follows: (TS =((“Innate immunity cell”) OR (“Congenital immunity cell”) OR (“Nonspecific immunity cell”) OR (“Non-Specific Immunity cell”) OR (“Native Immunity cell”) OR (“Macrophage”) OR (“Dendritic cell”) OR (“Neutrophil”) OR (“Eosinophil”) OR (“Basophil”) OR (“ILCs”) OR (“Innate Lymphoid Cells”) OR (“NK cell”) OR (“ILC1”) OR (“ILC2”) OR (“ILC3”) OR (“Kupffer cell”) OR (“Kupffer cell”)) AND TS=((“non-alcoholic fatty liver disease”) or (“NAFLD”) or (“non-alcoholic fatty liver”) or (“metabolic dysfunction-associated fatty liver disease”) or (“MAFLD”) or (“non-alcoholic steatohepatitis”) or (“NASH”) or (“metabolic dysfunction-associated steatohepatitis”) or (“MASH”) or (“MASLD”))).
A total of 1578 articles focused on the relationship between innate immune cells and MASLD. Excluding conference abstracts, editorial materials, book chapters, proceeding papers, letters, and retracted publications and corrections, 1422 articles were finally included for analysis. Keyword burst detection was next performed using Vosviwer to identify emerging trends. Figure 2 is the graph obtained from VOSviewer analysis based on the average number of occurrences of all keywords in the published papers. In the figure, blue indicates that keywords appear in earlier literature, while yellow is the opposite. Specifically, yellow represents the research hotspots and trends in recent years. Figure 2 shows that KCs are still being studied by researchers since their discovery, and their recruitment to the liver, macrophage polarization and inflammatory mechanism have been relatively clear (80). Recently, neutrophils, DCs, intestinal microbes, HSCs, peroxisome proliferators-activated receptors-gamma(PPAR-γ), homeostasis have been widely studied. nnate immune cells are the core regulators of MASLD-related inflammation. At present, the mechanism of innate immune cells in the progression of MASLD is relatively unknown, and a large number of studies are still needed to clarify, which provides a reference for researchers in the future research direction.

Figure 2. Average citation years for the domains related to innate immune cells and MASLD based on keyword classification.
In addition, it is worth mentioning that the rapid progress of single-cell and spatial omics has enabled us to more precisely distinguish the metabolic characteristics and intercellular crosstalk of various immune cells in the liver of MASLD patients, thus leading to significant changes in our understanding of disease pathogenesis. In particular, the single-cell sequencing of 10 livers (n=5 healthy and n=5 cirrhotic livers) published in 2019 by Ramachandran et al. (81), is known as the benchmark. This article provides an online platform link to quickly identify the changes of each gene in each immune cell, which provides a solid foundation for subsequent research. The development of artificial intelligence such as machine learning makes it possible to use publicly available datasets for cross-merging analysis to find stable potential biomarkers in different cohorts, which is of great significance for early clinical work to identify MASH (82). In the future, we should learn to use omics data to clarify the mechanism of innate immune cells in the progression of MASLD, and work more deeply on the way of preclinical research to clinical transformation. By intervening in the immune mechanism in the process of MASLD, the MASLD patient population and the incidence of MASLD-HCC should be reduced.
Author contributions
YY: Writing – review & editing, Writing – original draft, Software. QW: Writing – review & editing, Visualization, Formal analysis. BF: Writing – original draft, Data curation, Software. XP: Writing – review & editing, Funding acquisition, Validation. MS: Writing – review & editing, Funding acquisition, Resources, Supervision. FW: Funding acquisition, Writing – review & editing, Supervision.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. Incubation Fund of Tianjin First Central Hospital (No.2025FYMS07); National Natural Science Foundation of China (82370670); Tianjin Key Medical Construction Project (No.TJYXZDXK-3-022C); The Natural Science Foundation of Tianjin (23JCQNJC01230).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1599748/full#supplementary-material
References
1. Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F, et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology. (2023) 78:1966–86. doi: 10.1097/HEP.0000000000000520
2. Huang DQ, El-Serag HB, and Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. (2021) 18:223–38. doi: 10.1038/s41575-020-00381-6
3. Stine JG, Wentworth BJ, Zimmet A, Rinella ME, Loomba R, Caldwell SH, et al. Systematic review with meta-analysis: risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment Pharmacol Ther. (2018) 48:696–703. doi: 10.1111/apt.14937
4. Estes C, Razavi H, Loomba R, Younossi Z, and Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. (2018) 67:123–33. doi: 10.1002/hep.29466
5. Llovet JM, Willoughby CE, Singal AG, Greten TF, Heikenwälder M, El-Serag HB, et al. Nonalcoholic steatohepatitis-related hepatocellular carcinoma: pathogenesis and treatment. Nat Rev Gastroenterol Hepatol. (2023) 20:487–503. doi: 10.1038/s41575-023-00754-7
6. Younossi ZM, Golabi P, Paik JM, Henry A, Van Dongen C, and Henry L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review. Hepatology. (2023) 77:1335–47. doi: 10.1097/HEP.0000000000000004
7. Huby T and Gautier EL. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat Rev Immunol. (2022) 22:429–43. doi: 10.1038/s41577-021-00639-3
8. Carpenter S and O’Neill LAJ. From periphery to center stage: 50 years of advancements in innate immunity. Cell. (2024) 187:2030–51. doi: 10.1016/j.cell.2024.03.036
9. Sutti S and Albano E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat Rev Gastroenterol Hepatol. (2020) 17:81–92. doi: 10.1038/s41575-019-0210-2
10. Wynn TA, Chawla A, and Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. (2013) 496:445–55. doi: 10.1038/nature12034
11. Kupffer C. Ueber sternzellen der leber: briefliche mittheilung an prof. Waldeyer Archiv Für Mikroskopische Anatomie. (1876) 12:353–8. doi: 10.1007/bf02933897
12. van der Heide D, Weiskirchen R, and Bansal R. Therapeutic targeting of hepatic macrophages for the treatment of liver diseases. Front Immunol. (2019) 10:2852. doi: 10.3389/fimmu.2019.02852
13. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun. (2016) 7:10321. doi: 10.1038/ncomms10321
14. Wu J, Pan J, Zhou W, Ji G, and Dang Y. The role of N6-methyladenosine in macrophage polarization: A novel treatment strategy for non-alcoholic steatohepatitis. BioMed Pharmacother. (2024) 171:116145. doi: 10.1016/j.biopha.2024.116145
15. du Plessis J, van Pelt J, Korf H, Mathieu C, van der Schueren B, Lannoo M, et al. Association of adipose tissue inflammation with histologic severity of nonalcoholic fatty liver disease. Gastroenterology. (2015) 149:635–48.e14. doi: 10.1053/j.gastro.2015.05.044
16. Rohm TV, Meier DT, Olefsky JM, and Donath MY. Inflammation in obesity, diabetes, and related disorders. Immunity. (2022) 55:31–55. doi: 10.1016/j.immuni.2021.12.013
17. Osborn O and Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. (2012) 18:363–74. doi: 10.1038/nm.2627
18. Boesch M, Lindhorst A, Feio-Azevedo R, Brescia P, Silvestri A, Lannoo M, et al. Adipose tissue macrophage dysfunction is associated with a breach of vascular integrity in NASH. J Hepatol. (2024) 80:397–408. doi: 10.1016/j.jhep.2023.10.039
19. Rohm TV, Dos Reis FCG, Cunha eRocha K, Isaac R, Strayer S, Murphy C, et al. Adipose tissue macrophages in metabolic dysfunction-associated steatohepatitis secrete extracellular vesicles that activate liver fibrosis in obese male mice. Gastroenterology. (2025) 169(4):691–704. doi: 10.1053/j.gastro.2025.03.033
20. Hsu JY, Crawley S, Chen M, Ayupova DA, Lindhout DA, Higbee J, et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature. (2017) 550:255–9. doi: 10.1038/nature24042
21. Mullican SE, Lin-Schmidt X, Chin CN, Chavez JA, Furman JL, Armstrong AA, et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat Med. (2017) 23:1150–7. doi: 10.1038/nm.4392
22. L'homme L, Sermikli BP, Haas JT, Fleury S, Quemener S, Guinot V, et al. Adipose tissue macrophage infiltration and hepatocyte stress increase GDF-15 throughout development of obesity to MASH. Nat Commun. (2024) 15:7173. doi: 10.1038/s41467-024-51078-2
23. Martínez-Sánchez C, Bassegoda O, Deng H, Almodóvar X, Ibarzabal A, de Hollanda A, et al. Therapeutic targeting of adipose tissue macrophages ameliorates liver fibrosis in non-alcoholic fatty liver disease. JHEP Rep. (2023) 5:100830. doi: 10.1016/j.jhepr.2023.100830
24. Colella F and Ramachandran P. Adipose tissue macrophage dysfunction in human MASLD - Cause or consequence? J Hepatol. (2024) 80:390–3. doi: 10.1016/j.jhep.2023.12.007
25. Amulic B, Cazalet C, Hayes GL, Metzler KD, and Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. (2012) 30:459–89. doi: 10.1146/annurev-immunol-020711-074942
26. Burn GL, Foti A, Marsman G, Patel DF, and Zychlinsky A. The neutrophil. Immunity. (2021) 54:1377–91. doi: 10.1016/j.immuni.2021.06.00
27. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
28. van der Windt DJ, Sud V, Zhang H, Varley PR, Goswami J, Yazdani HO, et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology. (2018) 68:1347–60. doi: 10.1002/hep.29914
29. Fitzgerald KA and Kagan JC. Toll-like receptors and the control of immunity. Cell. (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
30. Xia Y, Wang Y, Xiong Q, He J, Wang H, Islam M, et al. Neutrophil extracellular traps promote MASH fibrosis by metabolic reprogramming of HSC. Hepatology. (2025) 81:947–61. doi: 10.1097/HEP.0000000000000762
31. Weiskirchen R and Tacke F. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg Nutr. (2014) 3:344–63. doi: 10.3978/j.issn.2304-3881.2014.11.03
32. Lv T, Xiong X, Yan W, Liu M, Xu H, and He Q. Mitochondrial general control of amino acid synthesis 5 like 1 promotes nonalcoholic steatohepatitis development through ferroptosis-induced formation of neutrophil extracellular traps. Clin Transl Med. (2023) 13:e1325. doi: 10.1002/ctm2.1325
33. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. (2014) 59:898–910. doi: 10.1002/hep.26592
34. de Carvalho Ribeiro M and Szabo G. Role of the inflammasome in liver disease. Annu Rev Pathol. (2022) 17:345–65. doi: 10.1146/annurev-pathmechdis-032521-102529
35. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
36. Babuta M, Morel C, de Carvalho Ribeiro M, Calenda C, Ortega-Ribera M, Thevkar Nagesh P, et al. Neutrophil extracellular traps activate hepatic stellate cells and monocytes via NLRP3 sensing in alcohol-induced acceleration of MASH fibrosis. Gut. (2024) 73:1854–69. doi: 10.1136/gutjnl-2023-331447
37. Kui L, Kim AD, Onyuru J, Hoffman HM, and Feldstein AE. BRP39 regulates neutrophil recruitment in NLRP3 inflammasome-induced liver inflammation. Cell Mol Gastroenterol Hepatol. (2024) 17:481–97. doi: 10.1016/j.jcmgh.2023.12.002
38. Cabral JE, Wu A, Zhou H, Pham MA, Lin S, and McNulty R. Targeting the NLRP3 inflammasome for inflammatory disease therapy. Trends Pharmacol Sci. (2025) 46:503–19. doi: 10.1016/j.tips.2025.04.007
39. Wang H, Zhang H, Wang Y, Brown ZJ, Xia Y, Huang Z, et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol. (2021) 75:1271–83. doi: 10.1016/j.jhep.2021.07.032
40. Xu M, Xu H, Ling YW, Liu JJ, Song P, Fang ZQ, et al. Neutrophil extracellular traps-triggered hepatocellular senescence exacerbates lipotoxicity in non-alcoholic steatohepatitis. J Adv Res. (2025) S2090-1232(25)00175-4. doi: 10.1016/j.jare.2025.03.015
41. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.01
42. Bourayou E, Perchet T, Meunier S, Bouvier H, Mailhe MP, Melanitou E, et al. Bone marrow monocytes sustain NK cell-poiesis during non-alcoholic steatohepatitis. Cell Rep. (2024) 43:113676. doi: 10.1016/j.celrep.2024.113676
43. Gu M, Zhang Y, Lin Z, Hu X, Zhu Y, Xiao W, et al. Decrease in UCP1 by sustained high lipid promotes NK cell necroptosis to exacerbate nonalcoholic liver fibrosis. Cell Death Dis. (2024) 15:518. doi: 10.1038/s41419-024-06910-4
44. Jenne CN and Kubes P. Immune surveillance by the liver. Nat Immunol. (2013) 14:996–1006. doi: 10.1038/ni.2691
45. Wang F, Zhang X, Liu W, Zhou Y, Wei W, Liu D, et al. Activated natural killer cell promotes nonalcoholic steatohepatitis through mediating JAK/STAT pathway. Cell Mol Gastroenterol Hepatol. (2022) 13:257–74. doi: 10.1016/j.jcmgh.2021.08.019
46. Tian Z, Chen Y, and Gao B. Natural killer cells in liver disease. Hepatology. (2013) 57:1654–62. doi: 10.1002/hep.26115
47. Shi FD, Ljunggren HG, La Cava A, and Van Kaer L. Organ-specific features of natural killer cells. Nat Rev Immunol. (2011) 11:658–71. doi: 10.1038/nri3065
48. Wensveen FM, Jelenc ̌ic ́ V, Valentic ́ S, Šestan M, Wensveen TT, Theurich S, et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol. (2015) 16:376–85. doi: 10.1038/ni.3120
49. Tosello-Trampont AC, Krueger P, Narayanan S, Landes SG, Leitinger N, Hahn YS, et al. NKp46(+) natural killer cells attenuate metabolism-induced hepatic fibrosis by regulating macrophage activation in mice. Hepatology. (2016) 63:799–812. doi: 10.1002/hep.28389
50. Luci C, Vieira E, Perchet T, Gual P, and Golub R. Natural killer cells and type 1 innate lymphoid cells are new actors in non-alcoholic fatty liver disease. Front Immunol. (2019) 10:1192. doi: 10.3389/fimmu.2019.01192
51. Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. (2012) 36:55–67. doi: 10.1016/j.immuni.2011.11.016
52. Zhou J, Tian Z, and Peng H. Tissue-resident NK cells and other innate lymphoid cells. Adv Immunol. (2020) 145:37–53. doi: 10.1016/bs.ai.2019.11.002
53. Zhang J, Marotel M, Fauteux-Daniel S, Mathieu AL, Viel S, Marçais A, et al. T-bet and Eomes govern differentiation and function of mouse and human NK cells and ILC1. Eur J Immunol. (2018) 48:738–50. doi: 10.1002/eji.201747299
54. O'Sullivan TE, Rapp M, Fan X, Weizman OE, Bhardwaj P, Adams NM, et al. Adipose-resident group 1 innate lymphoid cells promote obesity-associated insulin resistance. Immunity. (2016) 45:428–41. doi: 10.1016/j.immuni.2016.06.016
55. Kang J, Liggett JR, Patil D, Ranjit S, Loh K, Duttargi A, et al. Type 1 innate lymphoid cells are proinflammatory effector cells in ischemia-reperfusion injury of steatotic livers. Front Immunol. (2022) 13:899525. doi: 10.3389/fimmu.2022.899525
56. Loh Z, Fitzsimmons RL, Reid RC, Ramnath D, Clouston A, Gupta PK, et al. Inhibitors of class I histone deacetylases attenuate thioacetamide-induced liver fibrosis in mice by suppressing hepatic type 2 inflammation. Br J Pharmacol. (2019) 176:3775–90. doi: 10.1111/bph.14768
57. McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. (2013) 39:357–71. doi: 10.1016/j.immuni.2013.07.018
58. Gonzalez-Polo V, Pucci-Molineris M, Cervera V, Gambaro S, Yantorno SE, Descalzi V, et al. Group 2 innate lymphoid cells exhibit progressively higher levels of activation during worsening of liver fibrosis. Ann Hepatol. (2019) 18:366–72. doi: 10.1016/j.aohep.2018.12.001
59. Karagiannis F, Masouleh SK, Wunderling K, Surendar J, Schmitt V, Kazakov A, et al. Lipid-droplet formation drives pathogenic group 2 innate lymphoid cells in airway inflammation. Immunity. (2020) 52:620–34. doi: 10.1016/j.immuni.2020.03.003
60. Wilhelm C, Harrison OJ, Schmitt V, Pelletier M, Spencer SP, Urban Jr JF, et al. Critical role of fatty acid metabolism in ILC2-mediated barrier protection during malnutrition and helminth infection. J Exp Med. (2016) 213:1409–18. doi: 10.1084/jem.20151448
61. Roy-Dorval A, Deagle RC, Roth F, Raybaud M, Ismailova N, Krisna SS, et al. Analysis of lipid uptake, storage, and fatty acid oxidation by group 2 innate lymphoid cells. Front Immunol. (2024) 15:1493848. doi: 10.3389/fimmu.2024.1493848
62. Wang S, Li J, Wu S, Cheng L, Shen Y, Ma W, et al. Type 3 innate lymphoid cell: a new player in liver fibrosis progression. Clin Sci (Lond). (2018) 132:2565–82. doi: 10.1042/CS20180482
63. Tang KY, Lickliter J, Huang ZH, Xian ZS, Chen HY, Huang C, et al. Safety, pharmacokinetics, and biomarkers of F-652, a recombinant human interleukin-22 dimer, in healthy subjects. Cell Mol Immunol. (2019) 16:473–82. doi: 10.1038/s41423-018-0029-8
64. Yang L, Zhang Y, Wang L, Fan F, Zhu L, Li Z, et al. Amelioration of high fat diet induced liver lipogenesis and hepatic steatosis by interleukin-22. J Hepatol. (2010) 53:339–47. doi: 10.1016/j.jhep.2010.03.004
65. Talbot J, Hahn P, Kroehling L, Nguyen H, Li D, and Littman DR. Feeding-dependent VIP neuron-ILC3 circuit regulates the intestinal barrier. Nature. (2020) 579:575–80. doi: 10.1038/s41586-020-2039-9
66. Guendel F, Kofoed-Branzk M, Gronke K, Tizian C, Witkowski M, Cheng HW, et al. Group 3 innate lymphoid cells program a distinct subset of IL-22BP-producing dendritic cells demarcating solitary intestinal lymphoid tissues. Immunity. (2020) 53:1015–32.e8. doi: 10.1016/j.immuni.2020.10.012
67. Hamaguchi M, Okamura T, Fukuda T, Nishida K, Yoshimura Y, Hashimoto Y, et al. Group 3 innate lymphoid cells protect steatohepatitis from high-fat diet induced toxicity. Front Immunol. (2021) 12:648754. doi: 10.3389/fimmu.2021.648754
68. Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature. (2014) 514:237–41. doi: 10.1038/nature13564
69. Mao K, Baptista AP, Tamoutounour S, Zhuang L, Bouladoux N, Martins AJ, et al. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature. (2018) 554:255–9. doi: 10.1038/nature25437
70. Nguyen HH, Talbot J, Li D, Raghavan V, and Littman DR. Modulating intestinal neuroimmune VIPergic signaling attenuates the reduction in ILC3-derived IL-22 and hepatic steatosis in MASLD. Hepatol Commun. (2024) 8:e0528. doi: 10.1097/HC9.0000000000000528
71. Lukacs-Kornek V and Schuppan D. Dendritic cells in liver injury and fibrosis: shortcomings and promises. J Hepatol. (2013) 59:1124–6. doi: 10.1016/j.jhep.2013.05.033
72. Lukacs-Kornek V and Turley SJ. Self-antigen presentation by dendritic cells and lymphoid stroma and its implications for autoimmunity. Curr Opin Immunol. (2011) 23:138–45. doi: 10.1016/j.coi.2010.11.012
73. Barranco-Fragoso B, Pal SC, Díaz-Orozco LE, Dorantes-Heredia R, Qi X, and Méndez-Sánchez N. Identification of hepatic dendritic cells in liver biopsies showing steatosis in patients with metabolic dysfunction-associated fatty liver disease (MAFLD) associated with obesity. Med Sci Monit. (2022) 28:e937528. doi: 10.12659/MSM.937528
74. Heier EC, Meier A, Julich-Haertel H, Djudjaj S, Rau M, Tschernig T, et al. Murine CD103+ dendritic cells protect against steatosis progression towards steatohepatitis. J Hepatol. (2017) 66:1241–50. doi: 10.1016/j.jhep.2017.01.008
75. Herna E, Cueto FJ, Cook ECL, Redondo-Urzainqui A, Charro-Zanca S, Robles-Vera I, et al. Conventional type 1 dendritic cells protect against age-related adipose tissue dysfunction and obesity. Cell Mol Immunol. (2022) 19:260–75. doi: 10.1038/s41423-021-00812-7
76. van der Zande HJ, Brombacher EC, Lambooij JM, Pelgrom LR, Zawistowska-Deniziak A, Patente TA, et al. Dendritic cell-intrinsic LKB1-AMPK/SIK signaling controls metabolic homeostasis by limiting the hepatic Th17 response during obesity. JCI Insight. (2023) 8:e15794. doi: 10.1172/jci.insight.157948
77. Brennan PN, MacMillan M, Manship T, Moroni F, Glover A, Troland D, et al. Autologous macrophage therapy for liver cirrhosis: a phase 2 open-label randomized controlled trial. Nat Med. (2025) 31:979–87. doi: 10.1038/s41591-024-03406-8
78. Dai H, Zhu C, Huai Q, Xu W, Zhu J, Zhang X, et al. Chimeric antigen receptor-modified macrophages ameliorate liver fibrosis in preclinical models. J Hepatol. (2024) 80:913–27. doi: 10.1016/j.jhep.2024.01.034
79. Thangaraj JL, Coffey M, Lopez E, and Kaufman DS. Disruption of TGF-β signaling pathway is required to mediate effective killing of hepatocellular carcinoma by human iPSC-derived NK cells. Cell Stem Cell. (2024) 31:1327–43.e5. doi: 10.1016/j.stem.2024.06.009
80. Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. (2019) 16:145–59. doi: 10.1038/s41575-018-0082-x
81. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. (2019) 575:512–8. doi: 10.1038/s41586-019-1631-3
82. Rusu EC, Clavero-Mestres H, Sa AM, Veciana-Molins M, Bertran L, Monfort-Lanzas P, et al. Uncovering hepatic transcriptomic and circulating proteomic signatures in MASH: A meta-analysis and machine learning-based biomarker discovery. Comput Biol Med. (2025) 191:110170. doi: 10.1016/j.compbiomed.2025.110170
Keywords: MASLD, MASH, HCC, innate immune cells, ATMS
Citation: Yao Y, Wu Q, Fan B, Peng X, Sheng M and Wang F (2025) Mechanisms of innate immune cells in Metabolic dysfunction-associated steatotic liver disease. Front. Immunol. 16:1599748. doi: 10.3389/fimmu.2025.1599748
Received: 25 March 2025; Accepted: 05 September 2025;
Published: 25 September 2025.
Edited by:
Mohammad Amjad Kamal, Princess Nourah bint Abdulrahman University, Saudi ArabiaReviewed by:
Rui Qiang, The Second Affiliated Hospital of Soochow University, ChinaErnest Saenz, Heidelberg University, Germany
Copyright © 2025 Yao, Wu, Fan, Peng, Sheng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingwei Sheng, c2hlbmdtaW5nd2VpQHRtdS5lZHUuY24=; Fengmei Wang, d2FuZ2ZlbmdtZWl0akAxMjYuY29t
†These authors have contributed equally to this work