 Jinping Yin
Jinping Yin Yanli Zhu
Yanli Zhu Rongrong Liu
Rongrong Liu Weixin Wang
Weixin Wang Zhicheng Wang
Zhicheng Wang Jianfeng Wang
Jianfeng Wang- 1Department of Radiotherapy in China-Japan Union Hospital of Jilin University, Changchun, Jilin, China
- 2National Health Commission (NHC) Key Laboratory of Radiobiology, School of Public Health, Jilin University, Changchun, Jilin, China
Non-alcoholic fatty liver disease (NAFLD) is a widespread chronic liver disorder, affecting nearly a quarter of the global population. It progresses from simple steatosis to non-alcoholic steatohepatitis (NASH), fibrosis, cirrhosis, and hepatocellular carcinoma (HCC). The gut-liver axis is crucial in NAFLD progression, driven by intestinal barrier dysfunction, microbial translocation, and immune dysregulation. Neutrophil extracellular traps (NETs)—web-like structures of DNA, histones, and inflammatory proteins—promote chronic inflammation and liver injury. This review examines the role of NETs in gut-liver axis crosstalk and NAFLD progression. It explores how NETs amplify inflammation, contribute to fibrosis, and facilitate the progression from NAFLD to HCC by interacting with gut microbiota and immune signaling pathways. Therapeutic strategies targeting NETs, such as reducing their formation, enhancing degradation, and modulating the gut microbiota, offer promising approaches to mitigate disease progression. This review sheds light on the interplay between NETs and the gut-liver axis, offering new insights into NAFLD pathophysiology and potential therapeutic strategies to improve patient outcomes.
1 Introduction
Non-alcoholic fatty liver disease (NAFLD) is now one of the most common chronic liver diseases, affecting one billion people worldwide (1). It ranges from non-alcoholic fatty liver (NAFL) to the more severe non-alcoholic steatohepatitis (NASH), which can progress to fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) (2). Intestinal barrier dysfunction and bacterial translocation through the gut-liver axis drive liver disease progression (3). This bidirectional network connects the liver and intestine via the biliary tract, portal circulation, and systemic circulation, interacting closely with the immune system and gut microbiota (4, 5). Its dysfunction disrupts immune balance, worsening liver disease (6, 7).
Over the past two decades, the role of immune cells in NAFLD progression and fibrosis has gained increasing attention (8, 9). Early studies identified inflammation as a key driver of NAFLD leading to extensive research on the contributions of immune cell types and inflammatory factors (10–16).
Neutrophils, the most abundant immune cells, constitute about 70% of circulating leukocytes and serve as the first line of defense in innate immunity (17). They rapidly migrate to sites of infection or injury, but their persistent activation and excessive recruitment contribute to various inflammatory diseases (18). Neutrophils promote tissue damage by releasing proteases such as matrix metalloproteinases and neutrophil elastase and generating oxidative bursts that disrupt cell membranes (19). They also form neutrophil extracellular traps (NETs), web-like structures of DNA, histones, and inflammatory proteins that drive chronic inflammation and cancer progression (20, 21). Emerging evidence links NETs to the gut-liver axis, highlighting their role in sustaining inflammation and promoting the progression of NAFLD to advanced liver disease, including HCC (22, 23).
This review examines the role of NETs in NAFLD pathophysiology, focusing on their interactions with the gut-liver axis and how these contribute to disease onset and progression. In addition, we will explore NETs’ involvement in the transition from normal liver to NAFLD, NASH, liver fibrosis, and ultimately HCC. Additionally, it discusses potential therapeutic strategies targeting NETs and intestine, including inhibiting their formation, promoting degradation, modulating the gut microbiota, and employing multi-targeted combination therapies. By elucidating the crosstalk between NETs and the gut-liver axis, this review aims to uncover novel pathophysiological mechanisms and therapeutic opportunities for NAFLD and its complications.
2 NET formation and functional roles
2.1 Formation of NETs
NETs are highly negatively charged, web-like structures released by activated neutrophils (24). The process of their formation, termed NETosis, has been the subject of extensive research in recent years. It is now understood that NETosis occurs through three distinct mechanisms (Table 1).
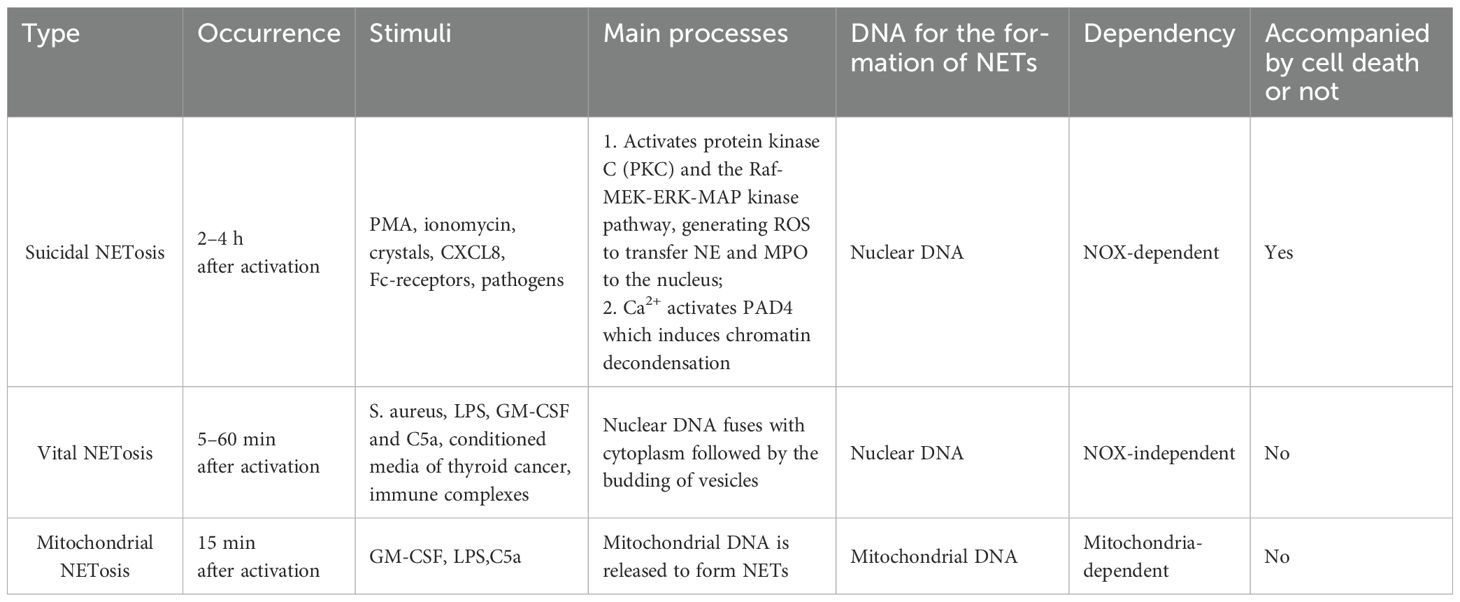
Table 1. Three mechanisms for the formation of NETs.
Suicidal NETosis, the first type of NETosis, is NADPH oxidase-dependent and results in neutrophil death, typically occurring within 2–4 hours of neutrophil activation. Stimulation by Phorbol Myristate Acetate (PMA) (25), ionomycin (26), crystals (27), CXCL8 (28), Fc receptors (29, 30) and pathogens activates the NADPH oxidase complex via the PKC-Raf-MERK-ERK pathway (31). This activation generates reactive oxygen species (ROS), which promote chromatin decondensation and facilitate the transfer of neutrophil elastase (NE) and myeloperoxidase (MPO) to the nucleus (32). Additionally, extracellular Ca2+ influx activates peptidylarginine deaminase 4 (PAD4), leading to histone citrullination, which weakens the electrostatic bond between histones and DNA, further promoting chromatin decondensation (26, 33). Ultimately, the nuclear membrane ruptures, expelling nuclear contents that fuse with cytoplasmic granules to form NETs.
The second type of NETosis, termed vital NETosis, occurs independently of NADPH oxidase and does not involve neutrophil death. It is typically induced within 5–60 minutes following neutrophil activation. This process is triggered by Toll-like receptor 2 (TLR2), Toll-like receptor 4 (TLR4), and the complement protein C3 in response to S. aureus (34, 35), as well as by lipopolysaccharides (LPS) (32, 36–40), granulocyte/macrophage colony-stimulating factor (GM-CSF) and complement factor 5a (C5a) (41, 42), conditioned media from thyroid cancer cells (43), and immune complexes (42, 44, 45). During vital NETosis, nuclear DNA merges with the cytoplasm and is subsequently released via vesicle budding. Despite nuclear extrusion, these neutrophils retain their phagocytic capacity, and their lifespan remains unaffected by DNA loss (46).
The third type, mitochondrial NETosis, typically occurs within 15 minutes of neutrophil activation. Upon stimulation by GM-CSF, LPS, or C5a, neutrophils release mitochondrial, rather than nuclear, DNA to form NETs without undergoing cell death (41).
2.2 Structure of NETs and its role in NAFLD progression
Upon activation, neutrophils undergo morphological changes, becoming flattened as nuclear lobules disappear, chromatin decondenses, and the inner and outer nuclear membranes separate. The nuclear membrane then fragments into vesicles, while the nucleoplasm and cytoplasm merge into homogeneous clumps. Eventually, the cell condenses, becoming round, and the cytoplasmic membrane ruptures, releasing intracellular components to form fibrous bundles (47). NETs display a unique ultrastructure, consisting of a chromatin filament framework, 15–17 nm in diameter, primarily composed of modified nucleosomes (24, 48). This filamentous network is interspersed with 50 nm globular structures. These globules are enriched with proteins from primary and secondary granules, including NE, MPO, cathepsin G, proteinase 3, BPI (cationic bactericidal/permeability-increasing protein), calgranulin, α-defensins, lactoferrin, LL-37 (a fragment of cathelicidin hCAP18), and PTX3. Additionally, tertiary granule components, such as matrix metalloproteinase-9 (MMP-9) and peptidoglycan recognition protein-S (PGRP-S), are incorporated (49–51).
The role of NETs in NAFLD progression can be categorized into two main effects: physiological and pathogenic (Figure 1). Regarding physiological effects, both in vivo and in vitro studies have demonstrated that DNA fibrils from NETs can adhere to gram-negative and gram-positive bacteria, as well as fungi, significantly limiting pathogen transmission (24, 34, 36, 52). However, the ability of NETs to kill pathogens remains debated. While NETs contain bactericidal proteins and enzymes, such as BPI, LL-37, α-defensins, NE, MPO, protease 3, and cathepsin G, some studies have shown pathogen death within NETs (34, 53, 54). Others have not observed such effects, suggesting that plasma protease inhibitors, which inhibit enzymes like NE, and the presence of apolipoproteins, which impair the bactericidal activity of LL-37 and α-defensins, may limit their function (55, 56) (Figure 1).
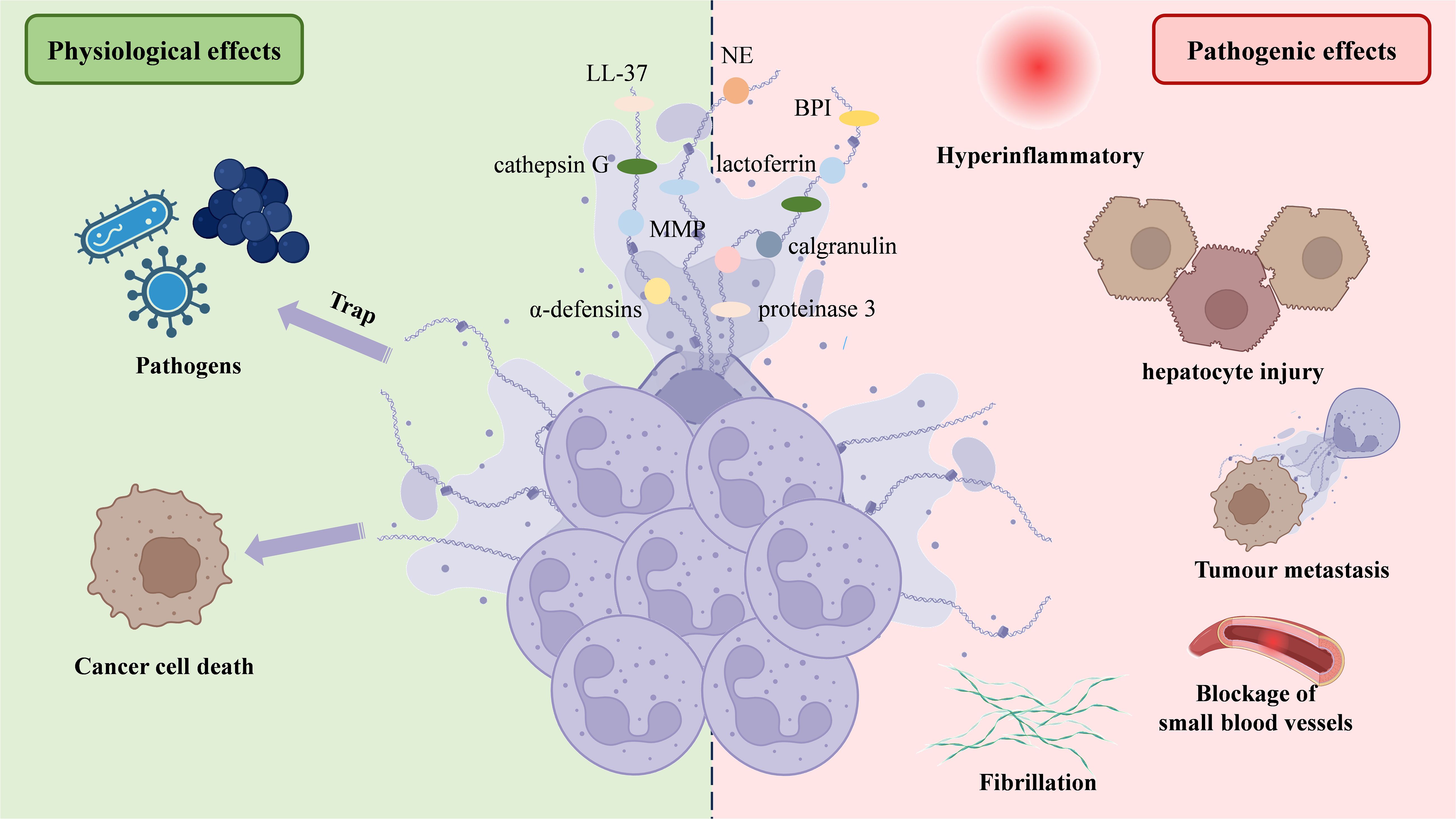
Figure 1. NETs’ structure and two-sided functionality. NETs are web-like structures formed by decondensed chromatin (DNA and histones) decorated with neutrophil-derived antimicrobial proteins and enzymes, such as NE, lactoferrin, α-defensins, and proteinase 3. NETs play a dual role in physiological and pathological processes. On the physiological side, NETs trap and kill pathogens, contributing to the immune defense, and induce cancer cell death. However, on the pathological side, excessive or dysregulated NETs can cause hyperinflammation, hepatocyte injury, promote tumor metastasis, lead to fibrillation, and obstruct small blood vessels, contributing to thrombotic and inflammatory diseases. NETs, Neutrophil extracellular traps; NE, Neutrophil elastase.
In terms of pathogenic effects, excessive NET formation has been linked to increased liver inflammation, exacerbating NAFLD progression (57–59). Neutrophils can also mediate hepatocyte injury via NETs, ROS and inflammatory mediators (60). Primary tumors trigger neutrophil recruitment and NET release at pre-metastatic sites, enhancing tumor growth and spread by interacting with cytokines. This mechanism may explain liver colonization by colorectal, lung, and breast cancers (61). NET formation by vital NETosis boosts HMGB1 production in tumor cells, activating TLR9-dependent pathways (62) and the TLR4/9-COX2 pathway (63), which improve tumor cell survival and invasion. Some studies have demonstrated that tumor cells expressing advanced glycation end-product receptors bind HMGB1, activating nuclear factor kappa-B (NF-κB) signaling and inducing interleukin-8 (IL-8) release. This attracts additional neutrophils and promotes NET formation, thereby facilitating the hepatic spread of colorectal cancer (64, 65). Other research indicates that NETs contribute to thrombosis in HCC patients and may worsen liver surgery-induced distal organ damage by triggering a systemic procoagulant state and microvascular immune thrombosis (66), as confirmed in a mouse model of hepatic ischemia-reperfusion injury (IRI) (67). Zermatten et al. observed elevated NET levels in the plasma of cirrhotic patients, potentially due to impaired hepatic clearance (68). Zhao et al. found that S1PR knockdown alleviates liver inflammation and fibrosis by inhibiting NET formation (69). These findings highlight the significant role of NETs in promoting the development of NAFLD (Figure 1).
3 The gut-liver axis and NAFLD
The intestinal barrier is a crucial anatomical and functional structure that mediates interactions between the gut and liver. It restricts the spread of microbes and toxins while permitting the absorption of nutrients into circulation for delivery to the liver. The “multiple hits” hypothesis explains the pathogenesis of NAFLD, involving factors such as genetic predisposition, altered gastrointestinal hormone and adipokine secretion, insulin resistance, nutritional imbalances, gut microbiota dysbiosis, and inflammation (7, 70–72). Among these, NAFLD is particularly associated with increased intestinal permeability and shifts in gut microbiota, which further exacerbate disease progression (73). Intestinal vascular barrier dysfunction has been identified as a key factor in NAFLD development (74, 75). The mechanisms regulating gut-liver axis homeostasis are complex, involving dietary, genetic, and microbiota-related factors that collectively influence intestinal permeability and metabolite levels. The imbalance of gut-liver axis homeostasis leads to intestinal ecological disorders, and the close connection between intestinal epithelial cells is destroyed under the action of various adverse factors, permeability changes or exposure to toxic bacterial metabolites, and then the intestinal epithelium is damaged (76). Fatty liver disease, including NAFLD, NASH and the later stages of development, the pathogenesis of multiple factors interwoven into a network of different stages, different levels of interaction.
3.1 Primary or secondary changes in gut microbiota
Gut microbiota plays a critical role in maintaining gut-liver axis homeostasis. Disruptions in its composition and transport to the liver through the gut-liver axis contribute significantly to the development of NAFLD. The intestinal microbiota is predominantly composed of bacterial phyla, with Bacteroidetes and Firmicutes being the most abundant, as identified through advanced techniques like shotgun sequencing and ribosomal multi-site sequencing (77, 78). Studies have shown that gut microbiota composition is altered in NAFLD patients, with reduced relative proportions of Alistipes, Odoribacter, Rikenellaceae, Bacteroides, Oscillibacter, and lactic acid bacteria from Firmicutes, alongside an increase in Peptiophilus, Escherichia, Enterobacterium, and anaerobic bacteria (79).
These microbiota changes are influenced by factors such as mode of delivery, diet, lifestyle, drug use, and host genetics. Gut microbiota plays an essential role in immunity, digestion, endocrine functions, neurotransmission, drug metabolism, and endotoxin clearance. In NAFLD, alterations in gut microbiota are primarily driven by genetic and metabolic abnormalities (80). These disruptions exacerbate NAFLD progression by increasing intestinal permeability, releasing bacterial toxins, and causing metabolic disorders. Additionally, altered gut microbiota can reach the liver via the gut-liver axis, creating an inflammatory environment that promotes hepatic steatosis (6). Some studies have targeted the detrimental effects of gut dysbiosis in NAFLD by using microbiota-regulating drugs, such as glucagon-like peptide-1 receptor agonists (GLP-1 RAs) for type 2 diabetes (81). These treatments have shown effectiveness in reversing hepatocyte autophagy and reducing NAFLD-associated dysbiosis, further supporting the need to explore the role of gut microbiota in NAFLD (79).
There is bidirectional crosstalk between the gut microbiota and the liver through the gut-liver axis. The liver influences the gut microbiota by releasing hormones, bile, and antibodies into the intestine (7). In NAFLD, the accumulation of fat in the liver leads to lipotoxicity, disrupting metabolic processes. The regulating effect of substances secreted by the diseased liver on intestinal microbes causes changes in intestinal microbial structure, which can act on the liver again through various ways as secondary factors, causing further development or remission of NAFLD.
3.2 Increased intestinal permeability
Impairment of the intestinal barrier is a key factor in disrupting the gut-liver axis. The intestinal barrier comprises several components: the mechanical, biochemical, microbial, and immune barriers, which include intestinal epithelial cells, secretions, gut microbiota, gut-associated lymphoid tissue (GALT), and diffuse immune cells (82). The permeability of the intestinal barrier is influenced by various factors, such as the protective mucosal layer produced by goblet cells, antimicrobial peptides from Paneth cells, tight junction proteins maintaining epithelial integrity, and immune cell activation. The tight junctions, located at the apical end of epithelial cells, are critical for regulating intestinal mucosal permeability. Under pathological conditions, disruption of tight junction structure and function leads to impaired intestinal barrier function (83). Environmental factors, such as pollution (84), and dietary changes can exacerbate NAFLD by compromising the barrier, which allows the inappropriate transport of nutrients, bacteria, and toxins to the liver (85). In NAFLD patients, dysregulation of the microbiota results in a thinner mucosal layer, decreased antimicrobial peptide production, reduced tight junction protein levels, and changes in immune cell populations in the lamina propria (86). Macrophage activation triggers pro-inflammatory cytokine production and amplifies neutrophil responses, further increasing intestinal permeability and disrupting entero-hepatic axis homeostasis.
3.3 Increased metabolic endotoxin
Increased intestinal barrier permeability can induce metabolic endotoxemia, which in turn contributes to the development of steatohepatitis. Additionally, altered absorption of various metabolites can affect liver metabolism, promoting liver steatosis and fibrosis. In NAFLD patients, dysregulation of gut microbiota and heightened intestinal permeability result in greater liver exposure to bacterial products, thereby triggering metabolic endotoxemia and disrupting gut-liver axis function (87). A dysfunctional gut microbiota with increased intestinal permeability exposes the liver to bacterial compounds, including ethanol, short-chain fatty acids (SCFAs), pathogen-associated molecular patterns (PAMPs), and damage-associated molecular patterns (DAMPs) (88). These compounds, particularly DAMPs released from the compromised intestine, activate neutrophils and induce the formation of NETs through synergistic mechanisms (89–91).
3.4 Vicious cycle of NETs in intestinal diseases
The pathogenesis of various intestinal diseases involves multiple factors that promote excessive production of pro-inflammatory substances and immune responses, leading to pathological changes in the intestinal wall. Colorectal biopsies from patients with Crohn’s disease and ulcerative colitis revealed increased expression of NETs-associated proteins, such as PAD4, compared to healthy controls (92). Furthermore, treatment with infliximab, a high-affinity monoclonal antibody targeting tumor necrosis factor-α (TNF-α), resulted in elevated expression of these proteins (93). Studies demonstrated that inhibition of TNF-α reduced both NETs levels and PAD4 expression in patients with intestinal diseases, indicating elevated NETs in these conditions (93). Additionally, research using a mouse model showed that treatment with deoxyribonuclease I (DNase I) reduced NET release and alleviated colitis, as well as the development of colitis-related tumors (94). These findings suggest that NETs play a catalytic role in intestinal disease progression, forming a vicious cycle of mutual causality.
4 NETs-gut-liver axis interaction
The gut and liver engage in a dynamic relationship, influenced by both organs, the microbiome, diet, and environmental factors. This interconnection is referred to as the gut-liver axis (95). The liver receives blood from the intestine through the portal vein, which supplies most of the venous and arterial blood to the liver. From a pathophysiological perspective, the intestinal mucosal barrier and the portal vein regulate the exchange of toxins and microorganisms between the gut and liver, allowing nutrients to enter circulation and reach the liver (7, 96). This portal vein-mediated interaction facilitates the transfer of bacterial metabolites from the intestine to the liver, potentially contributing to various liver diseases (95). Numerous studies have highlighted the gut-liver axis’ pivotal role in NAFLD progression (7). NASH patients, in particular, exhibit higher levels of intestinal microbiota imbalance, along with intestinal inflammation and barrier damage. Disruption of the mucosal transport and permeability, particularly at the tight junctions between intestinal endothelial cells (e.g., occludin and claudin), is observed (97). Dysfunction of the gut-vascular barrier is considered a key factor in NAFLD development (98, 99). Several studies indicate that gut dysbiosis can impair the intestinal epithelium, weaken tight junctions, increase intestinal permeability, and expose the liver to harmful bacterial metabolites (90, 100, 101). Both NAFLD and NASH are associated with increased intestinal barrier permeability and the translocation of bacteria or their products into the bloodstream (102–104). Given the crucial role of NETs in both processes, further investigation into their involvement in the gut-liver axis and NAFLD is warranted (Figure 2).
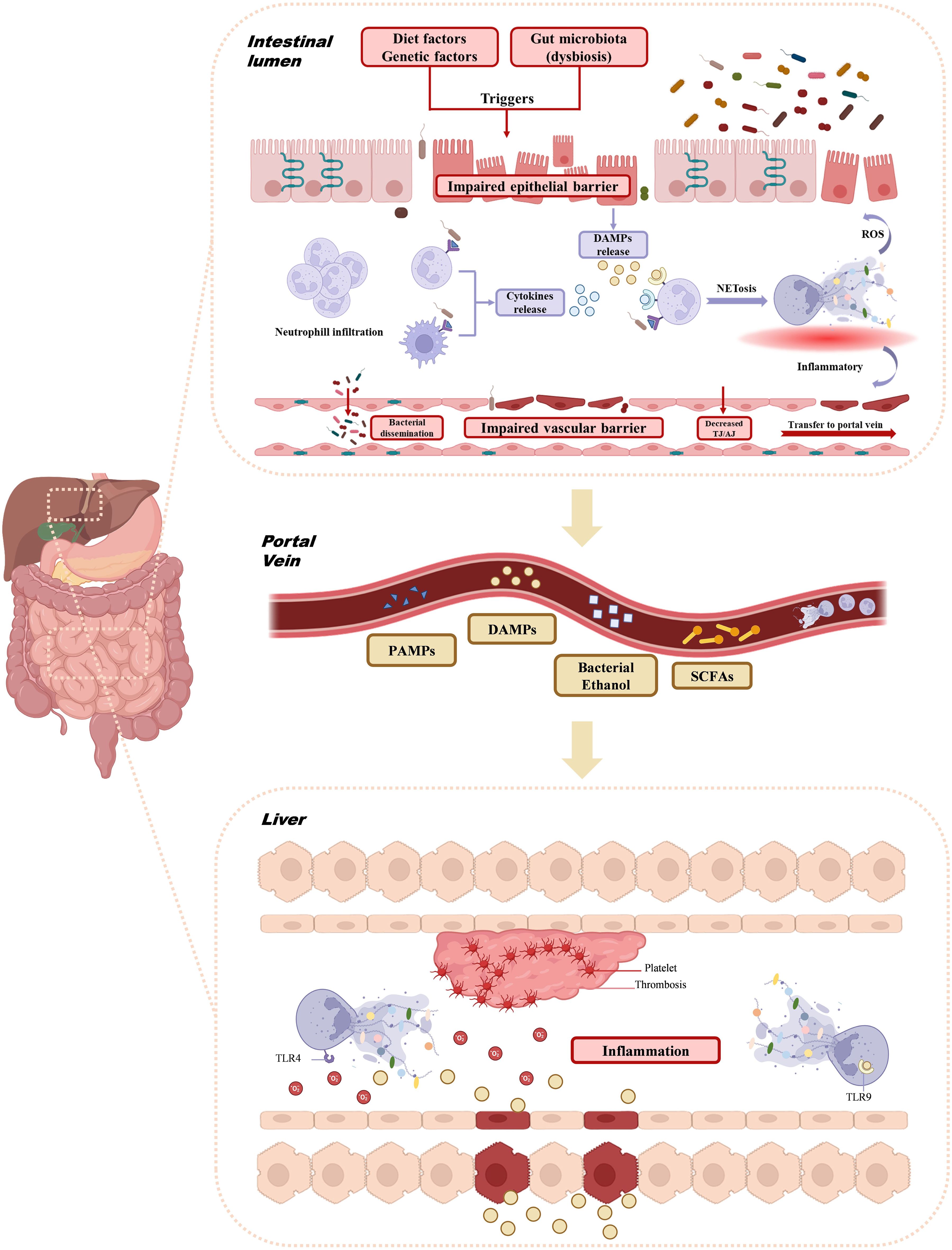
Figure 2. Interaction of NETs with gut-liver axis. Triggers such as diet, genetic factors, and gut microbiota dysbiosis impair the intestinal epithelial barrier, allowing neutrophil infiltration and promoting the formation of NETs. The resulting ROS and inflammatory environment further disrupt the intestinal barrier and portal vein. Concurrently, bacterial dysbiosis and barrier dysfunction enable bacterial products like PAMPs, DAMPs, ethanol, and SCFAs to enter the portal vein. These signals activate liver inflammation, trigger platelet aggregation via TLR4 and TLR9, and cause thrombosis and liver injury. NETs, Neutrophil extracellular traps; ROS, Reactive Oxygen Species; PAMPs, Pathogenetic associated molecular patterns; DAMPs, Damage-associated molecular patterns; SCFAs, Short-chain fatty acids; TLR, Toll-like receptor.
NETs are recognized as a key mechanism through which the gut-liver axis influences the progression of liver diseases, including NAFLD. The impairment of the intestinal epithelial barrier, triggered by factors such as diet, genetics, and gut microbiota dysbiosis, leads to the release of cytokines from various immune cells in an inflammatory environment (105). The dysregulated gut microbiota acts as a major trigger for the overactivation of neutrophils in the intestinal wall, resulting in NET formation. Recent studies have shown that microorganisms such as adherent-invasive Escherichia coli (AIEC) and Entamoeba histolytica can stimulate NETs (106, 107). Mouries et al. observed an initial disruption of the intestinal epithelial and gut vascular barriers (GVB) in NASH (3). During diet-induced dysbiosis, the gut vascular barrier becomes compromised (3). Gao et al. demonstrated that neutrophils infiltrate and release NETs in the gut of LPS-induced endotoxemic rats, and that DNase I administration, which disrupts NETs, alleviated intestinal epithelial cell apoptosis, intestinal damage, and the systemic inflammatory response (108). The formation and clearance of NETs is a dynamic process, and if this balance is disrupted, excessive NETosis can contribute to chronic inflammation (109), further damaging the intestinal barrier. NETs generate high levels of reactive oxygen species (ROS), leading to epithelial damage, activating redox-sensitive inflammatory pathways, and promoting bacterial translocation, which can damage the vascular barrier and lead to the release of TH/AJ (110). These factors contribute to the transport of bacteria to the liver via the gut-liver axis/portal vein. On one hand, enteric-derived NETs exacerbate intestinal barrier damage, while on the other, NETs travel to the liver via the portal vein, where they synergize with NETs produced by liver neutrophils, causing liver injury. As a bridge between gut microbiota and liver inflammation, NETs can directly stimulate liver immune cells by carrying microbial components, driving further hepatic inflammation (58). Thus, the imbalance of gut microbiota, excessive NET activation, and intestinal barrier dysfunction create a vicious cycle (Figure 2).
5 NETs in NAFLD-HCC
5.1 NAFL
NAFL is characterized by hepatic fat accumulation accounting for 5–10% of liver weight and represents the early stage of NAFLD. The primary pathological feature is macrovesicular steatosis involving more than 5% of hepatocytes. Both genetic and environmental factors contribute to NAFLD development, with the gut-liver axis playing a critical role. Disruption of the intestinal barrier and gut microbiota imbalance lead to immune activation, triggering the release of inflammatory cytokines that recruit immune cells, including neutrophils, to the intestine and subsequently to the liver. Neutrophil infiltration has been observed in hepatic lobules during NAFL (111–113).
Obesity and metabolic disorders further exacerbate NAFLD through NET formation. The chronic inflammatory state associated with obesity promotes innate immune activation, enhancing NETosis, which in turn contributes to immune dysregulation, oxidative stress, and metabolic dysfunction. Studies have demonstrated increased spontaneous NET formation in mice on a high-fat diet compared to controls, with obese patients exhibiting elevated plasma NET markers, such as MPO-DNA complexes (113–115). Immunohistochemical analysis has confirmed neutrophil infiltration in the hepatic lobules of STAM mice, and DNase treatment reduced hepatic citH3 expression, suggesting that NET degradation alleviates neutrophil infiltration and hepatic injury. Chronic inflammation, neutrophil activation, NET accumulation, and ROS production form a pathological loop that exacerbates obesity-related liver damage. Increased NET in obese individuals contribute to NAFLD progression by maintaining inflammation, disrupting hepatic energy homeostasis, and promoting hepatocyte lipid accumulation and toxicity.
Metabolic disorders, driven by genetic and lifestyle factors, also modulate NETosis. Patients with type 2 diabetes exhibit increased NETs formation, primarily in response to pro-inflammatory cytokines rather than hyperglycemia itself (116). However, in vitro studies suggest hyperglycemia may impair and delay NETosis (117). Hyperlipidemia induces neutrophilia, which correlates with atherosclerosis and related cardiovascular diseases (118, 119). In atherosclerotic mouse models, cholesterol crystals directly induce NETosis or are engulfed by macrophages, triggering cytokine release—particularly interleukin-1β (IL-1β), a key NET inducer. The intracellular mechanisms of neutrophil-cholesterol crystal interaction involve ROS bursts and NE translocation to the nucleus (120). In diabetic and high-fat diet conditions, PAD4 deficiency appears to increase susceptibility to hepatic steatosis, suggesting a potential metabolic role for PAD4. In hyperglycemic patients, glucose may synergize with other stimuli, such as LPS, to enhance NETosis (121). DAMPs released during hepatic ischemia-reperfusion injury promote NET formation via TLR signaling, exacerbating liver damage and inflammation (122).
Regulatory proteins involved in lipid metabolism and inflammation further influence NET formation in NAFLD. Overexpression of Pleckstrin homology-like domain, family A, member 1 (PHLDA1) negatively regulates sterol regulatory element-binding protein 1 (SREBP-1), a key regulator of triglyceride synthesis. Reduced hepatic levels of T cell death-associated gene 51 (TDAG51) correlate with obesity, hepatic steatosis, and insulin resistance (IR), while restoring TDAG51 expression mitigates NAFLD in mice. Machine learning identified activated T cells, macrophages, and neutrophils might play roles in the progression of liver disease (123). TDAG51 also enhances FoxO1 activity in LPS-induced inflammatory responses and promotes NETs release via the TLR4-JNK axis (124). Dysregulated TLR4-mediated inflammation is implicated in various chronic inflammatory diseases, including autoimmune disorders, cancer, and metabolic syndromes (125–127).
Steatosis is linked to cytokine signaling, extracellular matrix interactions, and key inflammatory pathways, including NF-κB, MAPK, and JAK-STAT. NF-κB activation induces NLRP3 inflammasome formation, leading to IL-1β production in response to DAMPs such as cholesterol crystals, ROS, and fatty acids (128–130). These mechanisms activate TLRs, promoting inflammation and fibrosis through NF-κB and MAPK signaling (128–130). Recent studies highlight the JAK-STAT pathway’s role in inflammation, cancer, and neurodegenerative diseases, linking cytokine release and immune regulation to NAFLD pathogenesis (131). The JAK-STAT pathway has also been implicated in NASH progression. Additionally, Wohlmann et al. identified thymic stromal lymphopoietin (TSLP) as an inflammatory mediator in atopic diseases via JAK-STAT signaling (132). Given these findings, pivotal genes such as PHLDA1 and zinc finger protein 36-like 2 (ZFP36L2) may contribute to NAFLD through TLR, MAPK, and JAK-STAT pathways, representing potential therapeutic targets.
Approximately 20% of NAFLD cases progress to NASH, which carries a higher risk of advancing to fibrosis and hepatocellular carcinoma (133). However, with the rising prevalence of metabolic syndrome and its strong association with NAFLD, the global burden of NASH and its complications is increasing at an alarming rate, necessitating urgent attention (Figure 3).
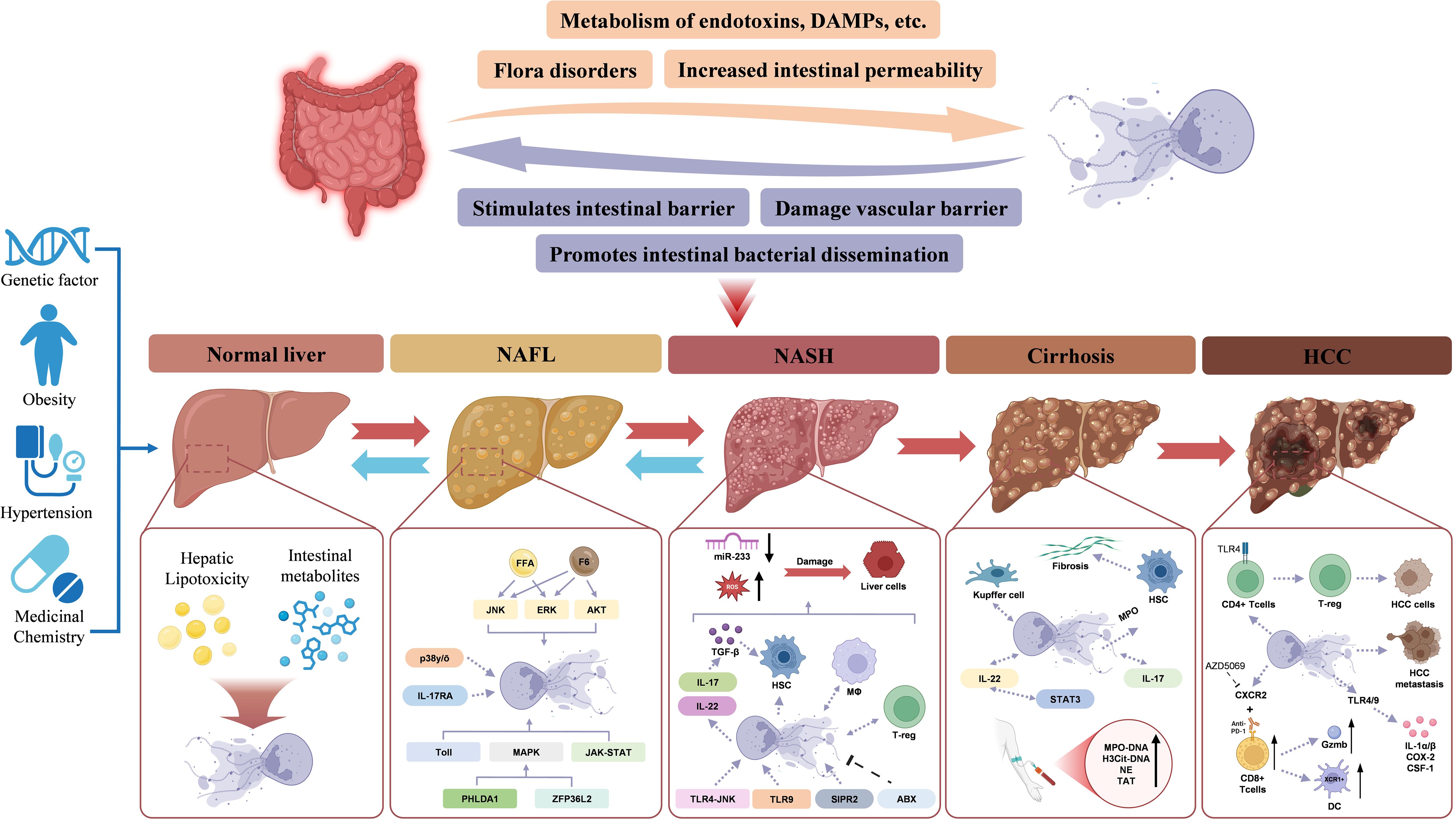
Figure 3. NETs in NAFLD-HCC. The NETs and gut-liver interactions in the progression from a health liver to NAFL, NASH, cirrhosis, and HCC. Normal Liver: Under healthy conditions, the intestinal barrier is intact, preventing endotoxin and metabolite translocation to the liver. Genetic factors, obesity, and hypertension may predispose individuals to liver dysfunction, initiating lipotoxicity and triggering NET formation. NAFLD, Dysbiosis and increased intestinal permeability allow endotoxins, DAMPs, and metabolites to reach the liver via the portal vein. NETs contribute to hepatic inflammation by amplifying lipotoxicity and activating inflammatory pathways like JNK, ERK, and AKT. NASH, With the progression to NASH, NETs exacerbate liver damage by interacting with gut-derived signals. Inflammatory cytokines (e.g., IL-17 and IL-22) activate HSCs, promoting fibrosis. MicroRNA-233 dysregulation further drives hepatocyte injury. Cirrhosis: Kupffer cells and HSCs engage in NET-mediated pathways. Fibrosis progresses due to sustained inflammation and immune activation. HCC: NETs facilitate tumor progression and contribute to HCC cells by activating CD4+ Tcells followed by T-reg. NETs, Neutrophil extracellular traps; HCC: Hepatocellular carcinoma; HSCs, Hepatic stellate cells; NAFL, Non-alcoholic fatty liver; NASH, Non-alcoholic steatohepatitis.
5.2 NASH
NASH is a more severe form of NAFLD characterized by liver inflammation and hepatocellular injury (steatohepatitis). Unlike NAFL, NASH exhibits both metabolic and inflammatory dysregulation, with pathological hallmarks including hepatocyte ballooning, chronic hepatic inflammation, and progressive fibrosis. It can lead to severe liver complications such as cirrhosis, liver failure, and HCC and may also increase the risk of extrahepatic adverse outcomes. The transition from NAFL to NASH is driven by abnormal lipid metabolism, excessive fat accumulation, and lipotoxicity, with neutrophil infiltration playing a key role in inflammation-induced liver injury. Diets rich in carbohydrates and cholesterol exacerbate neutrophil-driven liver inflammation and are linked to NASH severity.
NETosis is pivotal in the progression from NAFLD to NASH. While blocking NETs does not prevent hepatic steatosis or exsiting free fatty acid (FFA) accumulation caused by factors such as diet, it significantly reduces macrophage infiltration and shifts the inflammatory environment to a less tumor-promoting state. Elevated serum levels of NET markers in NASH patients further highlight the clinical relevance of NETs in NAFLD. Targeting NETs may be a promising strategy for reducing HCC risk in fatty liver disease (134).
NAFLD is associated with increased hepatic FFA levels (135, 136). FFAs are major activators of inflammatory pathways in NAFLD progression, and lipotoxicity-induced lipid accumulation is a key event in hepatic steatosis. Studies have shown that FFAs, such as linoleic and palmitic acid, can stimulate neutrophils to undergo NETosis, while inhibition of fatty acid synthase in human liver tissue prevents hepatic steatosis (137, 138). Experimental models indicate that blocking NET formation does not prevent fat accumulation in the liver, suggesting that NETs are a consequence rather than a cause of hepatic lipid overload. In addition to hepatocyte toxicity, FFAs have been shown to impair CD4+ T cells in NASH (139, 140). Several FFAs commonly elevated in NAFLD act as NET stimulators, promoting inflammation, recruiting immune cells such as macrophages and regulatory T cells (Tregs), and driving NASH progression toward HCC (137, 141).
NETosis is an early event in NASH pathogenesis, primarily by shaping the inflammatory microenvironment through monocyte-derived macrophage recruitment. NETs also interact with immune cells to release cytokines, and NET components themselves can directly stimulate hepatocytes, exacerbating NASH. Notably, preoperative serum MPO-DNA levels in NASH patients are significantly elevated compared to individuals with normal liver function. NETs contribute to disease progression by increasing liver macrophage infiltration, as infiltrating macrophages—derived from monocytes recruited in response to inflammation—serve as key effectors in NASH, amplifying cytokine-driven inflammation (142, 143).
Neutrophil infiltration and NET formation occur early in NASH, preceding macrophage accumulation. Blocking NETs significantly alters liver inflammation by reducing monocyte-derived macrophage infiltration, although the precise mechanisms driving neutrophil recruitment into the liver remain unclear (144). NETs themselves may promote neutrophil infiltration, while excessive NET formation and its major component, ROS, contribute to hepatocyte injury. Additionally, reduced miR-223 expression enhances IL-6 production, further exacerbating liver damage and increasing susceptibility to infections in advanced liver disease (145, 146). By establishing a chronic inflammatory liver microenvironment, NETs play a crucial role in NASH progression and HCC development (Figure 3).
5.3 Fibrosis
Fibrosis represents the next stage in NAFL progression, with epidemiological studies indicating that approximately 20% of NASH patients develop fibrosis annually. As cirrhosis advances, hepatic immune function progressively declines, leading to complications such as portal hypertension, intestinal barrier dysfunction, and bacterial translocation, which can ultimately result in liver failure. During the development of NASH and liver fibrosis, the gut-liver axis, adipose-liver axis, and renin-angiotensin system (RAS) may be dysregulated and impaired (147). Myofibroblasts, pro-fibrogenic mechanisms and cell interactions in progressive NAFLD (9). Hepatic stellate cells (HSCs) and Kupffer cells are the primary mediators of liver fibrosis. MPO has been shown to activate HSCs, thereby promoting fibrotic progression (148). Kupffer cells, the liver-resident macrophages, interact with neutrophils through NETs, exhibiting a dual regulatory effect. Conversely, NETs also influence Kupffer cell function, suggesting a bidirectional interaction. This interplay indicates that NETs may drive liver fibrosis by sustaining a self-amplifying cycle following activation by a priming factor (Figure 3).
5.4 HCC
HCC represents the terminal stage of NAFLD, with strong epidemiological associations linking NAFLD to primary liver tumors, including NAFLD-related HCC, HCC of other etiologies, and liver metastases from extrahepatic malignancies. HCC accounts for 21–22% of all liver tumors, and the global proportion of HCC cases attributed to NAFLD ranges from 1% to 38%, with the highest risk observed in patients with NAFLD-related cirrhosis (149). In recent years, the increasing prevalence of NASH has contributed to a rapid rise in NAFLD-associated HCC (150). However, because NASH is often undiagnosed or misclassified as “cryptogenic cirrhosis,” and in some cases progresses directly to HCC without an intermediate cirrhotic stage, HCC is frequently detected as an initial clinical manifestation (~35–50%) with rapid disease progression (151). Notably, the stagewise progression of NASH to HCC occurs more frequently than liver cancer arising from other etiologies, suggesting the involvement of systemic or metabolic risk factors unique to NASH (152). Unlike other malignancies, HCC primarily develops in a chronic inflammatory microenvironment (153). Established risk factors for NAFLD-associated HCC include advanced age, male sex, Latino ethnicity, cirrhosis, obesity, and type 2 diabetes, all of which significantly elevate the risk of NASH-related HCC (149). Additionally, emerging evidence implicates gut microbiota dysbiosis and inflammation as key contributors to HCC development in NAFLD (140).
NASH promotes HCC by impairing immune surveillance through the suppression of CD4+ and CD8+ T cells, increasing intestinal inflammation, and disrupting gut microbiota homeostasis—processes that are pivotal in hepatocarcinogenesis (154). Several recent studies indicate that chronic steatosis induces auto-aggressive CD8+CXCR6+PD1+ T cells that eliminate parenchymal and non-parenchymal cells in an antigen-independent manner promotes chronic liver damage and a pro-tumorigenic environment (155). Under the stimulation of the tumor inflammatory microenvironment (IM), the reprogramming of Treg cells, as members of CD4+ T cells, enhances their suppression of immune responses, ultimately promoting tumor immune escape or tumor progression (156). NETs play a central role in shaping the chronic inflammatory liver microenvironment that fosters HCC development. Beyond accelerating NASH progression via the gut-liver axis, NETs are intrinsically involved in HCC pathogenesis. Elevated GM-CSF, a feature of many solid tumors, and LPS released from HCC and intestinal tumors via complement activation contribute to systemic neutrophil activation and NET formation. Increased NETosis, in turn, enhances tumor-associated thrombosis and worsens clinical outcomes. Additionally, NETs act as metastatic scaffolds, facilitating the aggregation of circulating tumor cells in peripheral tissues and “reawakening” dormant cancer cells through NET-associated proteins (157). Experimental studies suggest that DNase treatment exerts antitumor effects by disrupting NETosis.
Metabolic disorders and obesity further contribute to HCC pathogenesis through NET-mediated mechanisms, including DNA damage and oxidative stress. Obesity is a recognized risk factor for HCC in both cirrhotic and non-cirrhotic NAFLD patients, and countries with a rising prevalence of NAFLD-associated HCC typically exhibit higher obesity rates (158). Obesity-driven inflammation promotes liver tumorigenesis by increasing levels of pro-tumor cytokines such as IL-6 and TNF (159). Chronic low-grade inflammation resulting from lipid accumulation induces IL-6 and TNF release, which in turn stimulate NET formation. IL-6 activates the STAT3 pathway, promoting hepatocyte proliferation and malignant transformation (160). However, no definitive evidence links obesity to altered prognosis in NAFLD-related HCC.
Diabetes mellitus is another independent risk factor for HCC, as demonstrated in a large European cohort study involving 136,703 NAFLD patients, where diabetes emerged as the strongest predictor of HCC development (161). While hyperglycemia exacerbates NET-associated inflammation in NASH and HCC, its effects on HCC progression are not exclusive to NAFLD. Additional metabolic alterations in diabetes likely contribute to the elevated HCC risk. Moreover, NAFLD frequently coexists with other liver diseases, such as viral hepatitis and alcoholic fatty liver disease, acting as both a complicating factor and a promoter of occult liver malignancies. Compared to HCC from other etiologies, NAFLD-related HCC is characterized by an older age of onset (median: 73 years), larger tumor size, more aggressive progression, and limited eligibility for curative interventions (162). Epidemiological studies estimate a median overall survival of only 10.7 months for HCC patients (162).
Neutrophils are highly abundant within the HCC microenvironment and display significant heterogeneity. While neutrophils possess antimicrobial, immunoregulatory, and tissue-repair functions, they can also drive tissue damage, immune suppression, and tumor metastasis under specific conditions. In addition to their role in HCC progression via the gut-liver axis, metabolic syndrome, and NASH-HCC, tumor-associated neutrophils (TANs) further contribute by releasing NETs, which promote HCC progression (137). NETs exacerbate the hypercoagulable state associated with cancer by inducing tumor-associated thrombosis, thereby increasing the risk of tumor-related complications, such as organ failure (163, 164). Animal studies have demonstrated that NET depletion slows tumor growth in mice (137).
The liver is also a common site for metastases from colorectal and breast cancer. During gut-liver axis-mediated metastasis, NAFLD facilitates tumor cell dissemination, while the tumor microenvironment reciprocally promotes NAFLD progression. In vitro studies indicate that NETs facilitate the invasion and infiltration of metastatic cancer cells into the liver. Clinically, NETs are more frequently observed in colorectal and breast cancer patients with liver metastases than in those without, and elevated NET markers in patient serum serve as potential biomarkers for predicting early liver metastasis in breast cancer (165) (Figure 3).
6 NETs as a therapeutic target for NAFLD
NETs are closely associated with the conversion of NAFLD to more severe forms of NASH and have been shown to be associated with the development of liver fibrosis, cirrhosis and HCC. Targeted inhibition of the interaction between NETs and the gut-liver axis to suppress the onset and progression of NAFLD is a promising direction for the treatment of NAFLD. Potential therapeutic strategies for targeting the interaction of NETs with the gut-liver axis in NAFLD are summarized below (Table 2).
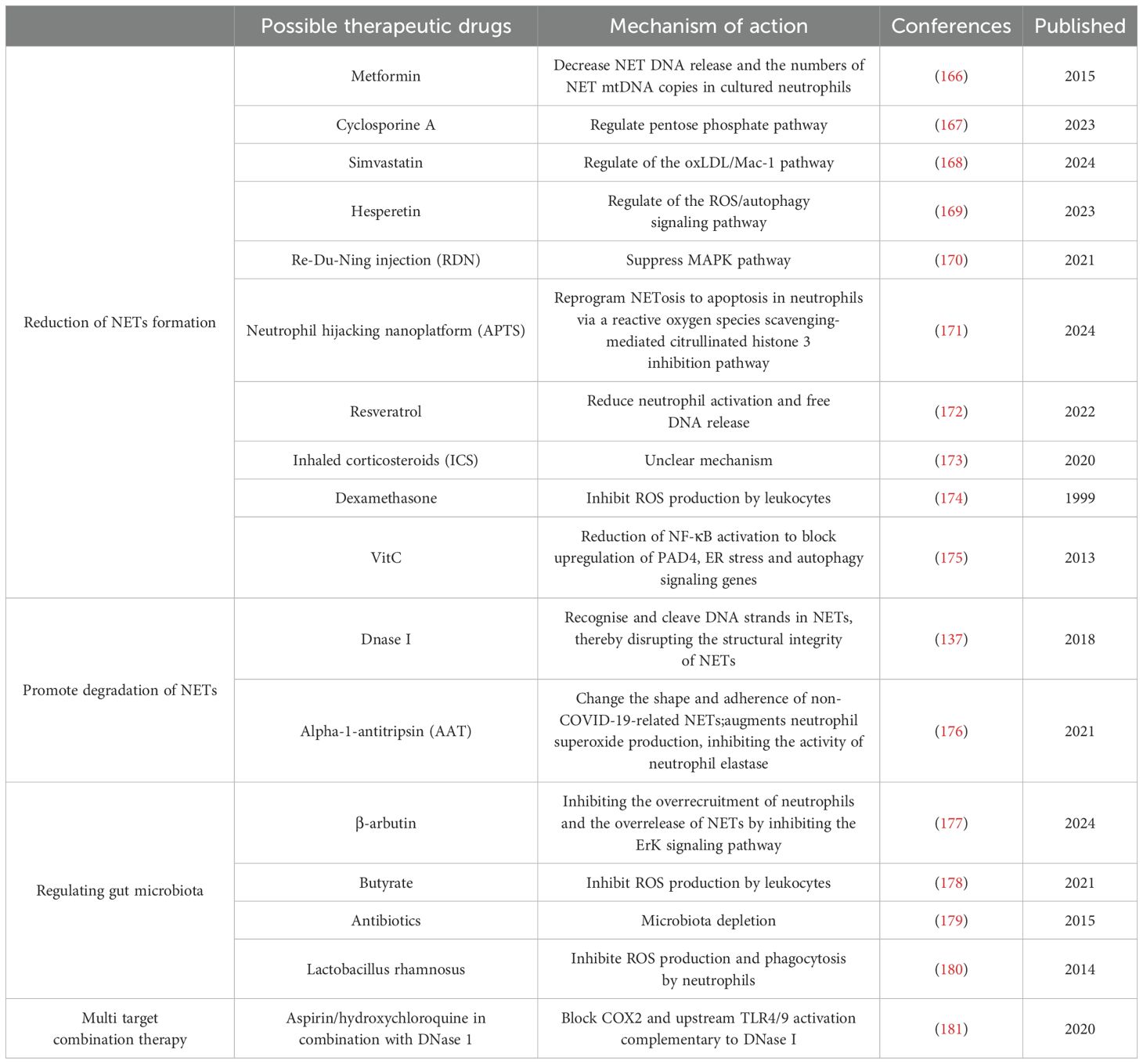
Table 2. NETs as a therapeutic target for NAFLD.
6.1 Reduction of NET formation
In recent years, several therapeutic strategies have emerged that target and inhibit the formation of NETs. In a clinical trial, metformin was found to reduce PMA-induced NETs formation (166). Tradition Chinese medicine preparations such as Re-Du-Ning (RDN) injection have also been shown to meliorate LPS-induced ALI through suppressing MAPK pathway to inhibit the formation of NETs (170). The study found that in septic mice, Hesperetin treatment reduced PMA-induced ROS production and NET formation, thereby attenuating sepsis-induced intestinal barrier damage (169). Simvastatin reduced hyperlipidemia-induced hepatic IRI by inhibiting the formation of NETs through the regulation of the oxLDL/Mac-1 pathway (168). Cyclosporine A (CsA) may reduce the severity of colitis by reducing the formation of NETs in vivo. In vitro, CsA reduces the release of ROS-dependent NETs by directly decreasing G6PD activity through activation of p53 protein, which downregulates PPP and cellular ROS levels (167). Antioxidant drugs, such as resveratrol, have been shown to be effective in reducing NETs produced by neutrophils in severely COVID-19-infected individuals by decreasing the neutrophil activation state and free DNA release (172). Inhaled corticosteroids (ICS) have also been shown to significantly reduce the formation of NETs, and in asthmatics, plasma NET levels were significantly lower in patients treated daily with ICS than in patients who used no or little ICS, but the mechanism by which they inhibit NETs formation is unknown (173). In another study, a significant reduction in reactive oxygen species production was observed in neutrophils after intravenous dexamethasone administration in human healthy subjects (174). And it’s well known that NETosis depends on the production of reactive oxygen species (182). Some studies have shown that anti-inflammatory drugs also have the ability to lower NETs (181). Significant improvement in fibrosis grading was also observed in the rat model of liver fibrosis in the aspirin and enoxaparin treatment groups (183).
Vitamin C (VitC), also known as ascorbic acid, is a water-soluble vitamin that is essential for human health. Studies have shown that in ascorbic acid deficiency, upregulation of hypoxia-inducible factor-1α (HIF-1α) blocks neutrophil apoptosis under normoxic conditions (184). Specifically, VitC may block the upregulation of PAD4, ER stress, and autophagy signaling genes by reducing NF-κB activation, thereby attenuating NETosis; in this way, VitC also significantly attenuated PMA-induced NETosis in polymorphonuclear (PMN) of healthy human volunteers (175). The antioxidant properties of VitC help to reduce oxidative stress, thereby reducing the formation of NETs (185). Interestingly, studies reporting an efficient neutrophil hijacking nanoplatform (referred to as APTS) for targeted A151 (a telomerase repeat sequence) delivery to microglia to dramatically reduce the formation of NETs by 2.2-fold via reprogramming NETosis to apoptosis in neutrophils via a reactive oxygen species scavenging-mediated citrullinated histone 3 inhibition pathway (171).
6.2 Promoting degradation of NETs
A preclinical study demonstrates the potential of recombinant human DNase I to treat cancer-related thrombosis (186). In a mouse model of necrotizing fasciitis, Group A Streptococcus (GAS) expressing DNase Sda1 has been identified as a contributor to bacterial virulence. Sda1 efficiently catabolizes NETs both in vitro and in vivo (54). DNase has shown therapeutic potential in animal models of NASH-HCC (137). However, DNase I for NETs alone has limitations. Blood concentrations of a given DNase I were found to be less stable (187). In addition, DNase I disrupts the NET structure but does not completely degrade the protein components of the NETs, suggesting that it is less effective in eliminating the inflammatory response triggered by NETs (58).
The other naturally occurring molecule, reducing pathological NET activity is alpha-1-antitripsin (AAT), a neutrophil elastase inhibitor, capable of change the shape and adherence of non-COVID-19-related NETs (176). AAT binds extracellular IL-8, reducing the neutrophils’ influx to the inflammatory site and augments neutrophil superoxide production, inhibiting the activity of neutrophil elastase (176).
6.3 Regulating gut microbiota
Numerous studies have shown that gut microbiota imbalance disrupts gut homeostasis and increases the risk of advanced NAFLD, and that this imbalance triggers hepatic inflammation and injury via the gut-liver axis, which affects bile acid metabolism and fat accumulation, and exacerbates liver fibrosis (188–193). Activation of NETs by microbiota has been reported (194). NETs were observed to be activated in a rat model of LPS-induced sepsis, and disruption of these NETs was found to attenuate intestinal damage (108). Microbiota-derived metabolite butyrate was found to inhibit neutrophil migration and NET formation in patients with Inflammatory Bowel Disease (IBD) (178). β-arbutin, a glycoside extracted from the Arctostaphylos uva-ursi leaves has also been found to contribute to the maintenance of intestinal homeostasis by inhibiting the formation of NETs, maintaining the integrity of the mucosal barrier, and shaping the composition of the intestinal flora (177).
One study focused on the effect of gut microbiota on NETs. It was found that the use of a combination of antibiotics that included ampicillin, streptomycin, metronidazole, and vancomycin reduced the number of microbes in the gut. This reduction correlated with a decrease in the formation of NETs. This means that by reducing certain types of bacteria, there may be an indirect reduction in the production of NETs in the body (179). Certain probiotics such as Lactobacillus rhamnosus strain GG have also been found to inhibit PMA and S. aureus-induced NET formation (180). These findings suggest that gut microbiota-targeted therapies hold promise as potential interventions to limit the formation of NETs during NAFLD (195, 196).
6.4 Multi target combination therapy
NETs promote HCC metastasis by activating the tumor inflammatory response, and some researchers have used two anti-inflammatory drugs, aspirin and hydroxychloroquine, to block the activation of cyclooxygenase 2 (COX2) and upstream TLR4/9 in combination with DNase I, and have demonstrated their promising effects in inhibiting HCC metastasis from multiple perspectives (181). The ability of these combination therapies to block or break down NETs and eliminate the metastatic potential of HCC cells trapped by unresolved NETs demonstrates a new use for old anti-inflammatory drugs. More strategies for combining with DNase I to combat metastasis need to be developed in the future. It is difficult to conclude that one compound works better than another in the treatment of targeted NETs, so more research is needed. Management of NETs may require the use of combination therapies that incorporate conventional treatments (e.g., fluid therapy, antibiotics, antivirals, and NET-targeting drugs) (111). Another avenue is integrating NET-targeting agents with antifibrotic compounds such as obeticholic acid or selonsertib. Since NETs can promote hepatic stellate cell activation and fibrogenesis, inhibiting NET formation while simultaneously targeting fibrogenic signaling may produce synergistic effects (197, 198).
7 Outlook
With the deepening understanding of the pathogenesis of NAFLD, it is increasingly recognized that gut microbiota dysbiosis and gut-liver axis dysfunction are among the important factors in the development of NAFLD (199, 200). Therefore, future therapeutic strategies should not only focus on the inflammatory response of the liver itself, but also consider how to intervene in the disease process by regulating the intestinal microecological balance. This provides a wide scope for the development of new therapeutic approaches. The following aspects deserve special attention:
7.1 Applications of nanomaterials
Nanoparticle delivery systems: Nanotechnology provides a new platform for drug delivery systems that can improve drug bioavailability and reduce side effects. The design of targeted nanoparticles for carrying enzymes or other active ingredients that degrading NETs promises to be an innovative therapeutic tool. For example, nanoparticles loaded with DNase I have shown significant therapeutic effects in animal models (201). Future research could further optimize the design of nanocarriers to enable more precise delivery of drugs to the liver or gut, and thus more effective against NETs-mediated inflammation and injury.
Multifunctional nanomaterials: In addition to single-function nanoparticles, multifunctional nanomaterials can be developed to combine multiple therapeutic mechanisms, such as carrying both anti-inflammatory drugs and NETs-degrading enzymes, to achieve synergistic effects.
7.2 Traditional Chinese medicine and natural products
Traditional Chinese medicine (TCM) has accumulated a wealth of experience in regulating immune responses and ameliorating chronic diseases. Some TCM such as Xuanfei Baidu Decoction (XFBD) have shown potential to regulate NETs formation via CXCL2/CXCR2 axis. Continued exploration of the active ingredients of TCM and their mechanisms of action may reveal more natural products that can be used for the treatment of NAFLD. For example, herbal components such as baicalein and tanshinone have been shown to have anti-inflammatory and antioxidant effects (202, 203).
7.3 Exosome research
Regulatory role of exosomes: exosomes, as important mediators of intercellular communication, play an important role in regulating immune responses and tissue repair processes. Studies have shown that certain types of exosomes can influence the production and clearance of NETs (204). Therefore, understanding how exosomes from different sources affect the function of NETs and how they can be used to optimize therapeutic regimens will be a key area for future research. For example, exosomes from stem cells have been shown to reduce liver inflammation by modulating immune cell function (205, 206).
Exosomes as therapeutic carriers: Exosomes can be used not only as therapeutic targets but also as drug delivery carriers. Future research could explore the use of exosomes to deliver specific drugs or enzymes to achieve more precise therapeutic effects. For example, by modifying the surface of exosomes so that they can be specifically targeted to the liver or intestines, thereby increasing the local concentration and efficacy of the drug (207, 208).
7.4 Microbiomics and personalized therapy
With the deepening of microbiomics research, there is increasing evidence of the key role of gut flora in the pathogenesis of NAFLD. Analysis of patients’ gut flora by high-throughput sequencing technology can help identify specific microbial markers and provide a basis for achieving precision medicine based on individualized characteristics (209). Future studies could further explore how to improve the metabolic status and liver health of NAFLD patients by modulating the gut microecology (e.g., using prebiotics, probiotics, or fecal bacteria transplantation).
Although the above emerging areas show great potential, it is difficult for any single approach to comprehensively address the complexities of NAFLD. Therefore, lifestyle modification, weight control, and the use of known effective medications should not be overlooked while exploring new therapies. For example, weight reduction through exercise and dietary changes, and the use of medications such as metformin to enhance insulin sensitivity are all approaches that have been shown to be effective in alleviating NAFLD symptoms (210–212). Combining these foundational measures with new strategies for targeting NETs is expected to have a synergistic effect and significantly improve the overall health of patients.
8 Conclusion
In summary, NETs play an important role in the genesis and development of NAFLD. The gut-liver axis plays an important role in the initiation of NAFL, which is mainly caused by the disruption of the intestinal barrier and the imbalance of the gut microbiota, and NETs continue to accelerate the process of hepatic fibrosis through the self-circulation of NETs activated by the stimulation of the gut-liver axis, gene induction, lipotoxicity accumulation, and the initiation of metabolic diseases. From healthy liver to NAFLD, NASH, liver fibrosis and even HCC, the formation and release of NETs is one of the key factors connecting these pathological stages. NETs are not only directly involved in hepatic inflammatory response and tissue injury, but also interact with gut microbiota through the gut-liver axis, which further promotes the disease process.
Studies have shown that an imbalance in the gut microbiota is able to exacerbate the process of liver inflammation and fibrosis by disrupting the intestinal barrier function and increasing the transfer of bacterial products to the liver (3, 213). In addition, specific metabolites in the intestinal microenvironment may regulate the production and degradation of NETs and influence the progression of NAFLD (213). Therefore, when treating NAFLD, in addition to focusing on the inflammatory state of the liver itself, the role of the gut-liver axis and gut microbiota needs to be considered comprehensively for a more comprehensive and effective management strategy.
Targeting NETs as therapeutic targets, approaches to reduce the formation of NETs, promote their degradation, modulate the gut microbiota composition, and multi-targeted combination therapies have demonstrated potential applications. In particular, modulating the gut microbiota to indirectly alleviate liver inflammation by improving gut health provides a new therapeutic perspective for NAFLD patients. Future studies should continue to explore the specific mechanisms of NETs in different stages of NAFLD-HCC and develop more new therapeutic approaches based on gut-liver axis modulation, with the aim of improving the prognosis of patients and reducing the disease burden.
Author contributions
JY: Writing – original draft, Writing – review & editing, Conceptualization. YZ: Writing – review & editing, Writing – original draft. RL: Writing – review & editing. WW: Writing – review & editing. ZW: Writing – review & editing, Project administration, Supervision. JW: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Science and Technology Development Plan of Jilin (20240101275JC and YDZJ202401200ZYTS), the Jilin Health Technology Innovation (2023JC021).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Younossi ZM. Non-alcoholic fatty liver disease – A global public health perspective. J Hepatology. (2019) 70:531–44. doi: 10.1016/j.jhep.2018.10.033
2. Samy AM, Kandeil MA, Sabry D, Abdel-Ghany AA, and Mahmoud MO. From NAFLD to NASH: Understanding the spectrum of non-alcoholic liver diseases and their consequences. Heliyon. (2024) 10:e30387. doi: 10.1016/j.heliyon.2024.e30387
3. Mouries J, Brescia P, Silvestri A, Spadoni I, Sorribas M, Wiest R, et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J Hepatol. (2019) 71:1216–28. doi: 10.1016/j.jhep.2019.08.005
4. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol. (2018) 15:397–411. doi: 10.1038/s41575-018-0011-z
5. Henao-Mejia J, Elinav E, Thaiss CA, and Flavell RA. The intestinal microbiota in chronic liver disease. Adv Immunol. (2013) 117:73–97. doi: 10.1016/B978-0-12-410524-9.00003-7
6. Hsu CL and Schnabl B. The gut-liver axis and gut microbiota in health and liver disease. Nat Rev Microbiol. (2023) 21:719–33. doi: 10.1038/s41579-023-00904-3
7. Albillos A, de Gottardi A, and Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol. (2020) 72:558–77. doi: 10.1016/j.jhep.2019.10.003
8. Huby T and Gautier EL. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat Rev Immunol. (2022) 22:429–43. doi: 10.1038/s41577-021-00639-3
9. Parola M and Pinzani M. Liver fibrosis in NAFLD/NASH: from pathophysiology towards diagnostic and therapeutic strategies. Mol Aspects Med. (2024) 95:101231. doi: 10.1016/j.mam.2023.101231
10. Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. (2001) 120:1183–92. doi: 10.1053/gast.2001.23256
11. Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, et al. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity. (2020) 52:1057–74.e7. doi: 10.1016/j.immuni.2020.04.001
12. Weiskirchen R and Tacke F. Immune surveillance of liver cancer in non-alcoholic fatty liver disease: excess lipids cause CD4 T-cells loss and promote hepatocellular carcinoma development. Hepatobiliary Surg Nutr. (2016) 5:433–7. doi: 10.21037/hbsn.2016.09.10
13. Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, Horsfall L, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology. (2014) 59:1393–405. doi: 10.1002/hep.26937
14. Ficht X and Iannacone M. Immune surveillance of the liver by T cells. Sci Immunol. (2020) 5:eaba2351. doi: 10.1126/sciimmunol.aba2351
15. Gomes AL, Teijeiro A, Burén S, Tummala KS, Yilmaz M, Waisman A, et al. Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell. (2016) 30:161–75. doi: 10.1016/j.ccell.2016.05.020
16. Wandrer F, Liebig S, Marhenke S, Vogel A, John K, Manns MP, et al. TNF-Receptor-1 inhibition reduces liver steatosis, hepatocellular injury and fibrosis in NAFLD mice. Cell Death Dis. (2020) 11:212. doi: 10.1038/s41419-020-2411-6
17. Malech HL, Deleo FR, and Quinn MT. The role of neutrophils in the immune system: an overview. Methods Mol Biol. (2014) 1124:3–10. doi: 10.1007/978-1-62703-845-4_1
18. Kolaczkowska E and Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
19. Wang J. Neutrophils in tissue injury and repair. Cell Tissue Res. (2018) 371:531–9. doi: 10.1007/s00441-017-2785-7
20. Guan X, Guan X, Zhao Z, and Yan H. NETs: Important players in cancer progression and therapeutic resistance. Exp Cell Res. (2024) 441:114191. doi: 10.1016/j.yexcr.2024.114191
21. Castanheira FVS and Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood. (2019) 133:2178–85. doi: 10.1182/blood-2018-11-844530
22. Hsu CL and Schnabl B. The gut–liver axis and gut microbiota in health and liver disease. Nat Rev Microbiol. (2023) 21:719–33. doi: 10.1038/s41579-023-00904-3
23. Wang H, Kim SJ, Lei Y, Wang S, Wang H, Huang H, et al. Neutrophil extracellular traps in homeostasis and disease. Signal Transduction Targeted Ther. (2024) 9:235. doi: 10.1038/s41392-024-01933-x
24. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
25. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. (2007) 176:231–41. doi: 10.1083/jcb.200606027
26. Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol. (2015) 11:189–91. doi: 10.1038/nchembio.1735
27. Schorn C, Janko C, Krenn V, Zhao Y, Munoz L, Schett G, et al. Bonding the foe – NETting neutrophils immobilize the pro-inflammatory monosodium urate crystals. Front Immunol. (2012) 3:376. doi: 10.3389/fimmu.2012.00376
28. Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Letters. (2010) 584:3193–7. doi: 10.1016/j.febslet.2010.06.006
29. Behnen M, Leschczyk C, Möller S, Batel T, Klinger M, Solbach W, et al. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcγRIIIB and Mac-1. J Immunol. (2014) 193:1954–65. doi: 10.4049/jimmunol.1400478
30. Aleyd E, Van Hout MWM, Ganzevles SH, Hoeben KA, Everts V, Bakema JE, et al. IgA enhances netosis and release of neutrophil extracellular traps by polymorphonuclear cells via FCA receptor I. J Immunol. (2014) 192:2374–83. doi: 10.4049/jimmunol.1300261
31. Rochael NC, Guimarães-Costa AB, Nascimento MTC, Desouza-Vieira TS, Oliveira MP, Garciae Souza LF, et al. Classical ROS-dependent and early/rapid ROS-independent release of Neutrophil Extracellular Traps triggered by Leishmania parasites. Sci Rep. (2015) 5:518302. doi: 10.1038/srep18302
32. Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, García-Orozco A, Alvarado-Navarro A, and Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: An overview. Front Immunol. (2017) 8. doi: 10.3389/fimmu.2017.00081
33. Sørensen OE and Borregaard N. Neutrophil extracellular traps - The dark side of neutrophils. J Clin Invest. (2016) 126:1612–20. doi: 10.1172/JCI84538
34. Pilsczek FH, Salina D, Poon KKH, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. (2010) 185:7413–25. doi: 10.4049/jimmunol.1000675
35. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
36. Yost CC, Cody MJ, Harris ES, Thornton NL, McInturff AM, Martinez ML, et al. Impaired neutrophil extracellular trap (NET) formation: A novel innate immune deficiency of human neonates. Blood. (2009) 113:6419–27. doi: 10.1182/blood-2008-07-171629
37. Tong M, Potter JA, Mor G, and Abrahams VM. Lipopolysaccharide-stimulated human fetal membranes induce neutrophil activation and release of vital neutrophil extracellular traps. J Immunol. (2019) 203:500–10. doi: 10.4049/jimmunol.1900262
38. Yipp BG, Petri B, Salina D, Jenne CN, Scott BNV, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. (2012) 18:1386–93. doi: 10.1038/nm.2847
39. Zheng X, Wu X, Wen Q, Tang H, Zhao L, Shi F, et al. Eriodictyol alleviated LPS/D-galN-induced acute liver injury by inhibiting oxidative stress and cell apoptosis via PI3K/AKT signaling pathway. Nutrients. (2023) 15:4349. doi: 10.3390/nu15204349
40. Wu X, Zheng X, Wen Q, Zhang Y, Tang H, Zhao L, et al. Swertia cincta Burkill alleviates LPS/D-GalN-induced acute liver failure by modulating apoptosis and oxidative stress signaling pathways. Aging (Albany NY). (2023) 15:5887–916. doi: 10.18632/aging.204848
41. Yousefi S, Mihalache C, Kozlowski E, Schmid I, and Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differentiation. (2009) 16:1438–44. doi: 10.1038/cdd.2009.96
42. Amini P, Stojkov D, Felser A, Jackson CB, Courage C, Schaller A, et al. Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat Commun. (2018) 9:2958. doi: 10.1038/s41467-018-05387-y
43. Cristinziano L, Modestino L, Loffredo S, Varricchi G, Braile M, Ferrara AL, et al. Anaplastic thyroid cancer cells induce the release of mitochondrial extracellular DNA traps by viable neutrophils. J Immunol. (2020) 204:1362–72. doi: 10.4049/jimmunol.1900543
44. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
45. van Dam LS, Kraaij T, Kamerling SWA, Bakker JA, Scherer UH, Rabelink TJ, et al. Intrinsically distinct role of neutrophil extracellular trap formation in antineutrophil cytoplasmic antibody–associated vasculitis compared to systemic lupus erythematosus. Arthritis Rheumatol. (2019) 71:2047–58. doi: 10.1002/art.v71.12
46. Yipp BG and Kubes P. NETosis: How vital is it? Blood. (2013) 122:2784–94. doi: 10.1182/blood-2013-04-457671
47. Pinegin B, Vorobjeva N, and Pinegin V. Neutrophil extracellular traps and their role in the development of chronic inflammation and autoimmunity. Autoimmun Rev. (2015) 14:633–40. doi: 10.1016/j.autrev.2015.03.002
48. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PloS Pathog. (2009) 5:e1000639. doi: 10.1371/journal.ppat.1000639
49. Parker H, Albrett AM, Kettle AJ, and Winterbourn CC. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J Leukocyte Biol. (2012) 91:369–76. doi: 10.1189/jlb.0711387
50. Bianchi M, Niemiec MJ, Siler U, Urban CF, and Reichenbach J. Restoration of anti-Aspergillus defense by neutrophil extracellular traps in human chronic granulomatous disease after gene therapy is calprotectin-dependent. J Allergy Clin Immunol. (2011) 127:1243–52.e7. doi: 10.1016/j.jaci.2011.01.021
51. Averhoff P, Kolbe M, Zychlinsky A, and Weinrauch Y. Single residue determines the specificity of neutrophil elastase for shigella virulence factors. J Mol Biol. (2008) 377:1053–66. doi: 10.1016/j.jmb.2007.12.034
52. Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, and Henriques-Normark B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol. (2006) 16:401–7. doi: 10.1016/j.cub.2006.01.056
53. Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. (2009) 114:2619–22. doi: 10.1182/blood-2009-05-221606
54. Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol. (2006) 16:396–400. doi: 10.1016/j.cub.2005.12.039
55. Menegazzi R, Decleva E, and Dri P. Killing by neutrophil extracellular traps: Fact or folklore? Blood. (2012) 119:1214–6. doi: 10.1182/blood-2011-07-364604
56. Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. (1989) 320:365–76. doi: 10.1056/NEJM198902093200606
57. Liu K, Wang FS, and Xu R. Neutrophils in liver diseases: pathogenesis and therapeutic targets. Cell Mol Immunol. (2021) 18:38–44. doi: 10.1038/s41423-020-00560-0
58. Honda M and Kubes P. Neutrophils and neutrophil extracellular traps in the liver and gastrointestinal system. Nat Rev Gastroenterol Hepatol. (2018) 15:206–21. doi: 10.1038/nrgastro.2017.183
59. Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J Hepatol. (2019) 71:793–801. doi: 10.1016/j.jhep.2019.06.021
60. Friedman SL, Neuschwander-Tetri BA, Rinella M, and Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
61. Hu W, Lee SML, Bazhin AV, Guba M, Werner J, and Nieß H. Neutrophil extracellular traps facilitate cancer metastasis: cellular mechanisms and therapeutic strategies. J Cancer Res Clin Oncol. (2023) 149:2191–210. doi: 10.1007/s00432-022-04310-9
62. Zhong W, Wang Q, Shen X, and Du J. The emerging role of neutrophil extracellular traps in cancer: from lab to ward. Front Oncol. (2023) 13. doi: 10.3389/fonc.2023.1163802
63. Zhang Y, Song J, Zhang Y, Li T, Peng J, Zhou H, et al. Emerging role of neutrophil extracellular traps in gastrointestinal tumors: A narrative review. Int J Mol Sci. (2023) 24:334. doi: 10.3390/ijms24010334
64. Zha C, Meng X, Li L, Mi S, Qian D, Li Z, et al. Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the HMGB1/RAGE/IL-8 axis. Cancer Biol Med. (2020) 17:154–68. doi: 10.20892/j.issn.2095-3941.2019.0353
65. Zhu B, Zhang X, Sun S, Fu Y, Xie L, and Ai P. NF-κB and neutrophil extracellular traps cooperate to promote breast cancer progression and metastasis. Exp Cell Res. (2021) 405:112707. doi: 10.1016/j.yexcr.2021.112707
66. Zhang H, Goswami J, Varley P, van der Windt DJ, Ren J, Loughran P, et al. Hepatic surgical stress promotes systemic immunothrombosis that results in distant organ injury. Front Immunol. (2020) 11:987. doi: 10.3389/fimmu.2020.00987
67. Dyer MR, Chen Q, Haldeman S, Yazdani H, Hoffman R, Loughran P, et al. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. . Sci Rep. (2018) 8:2068. doi: 10.1038/s41598-018-20479-x
68. Zermatten MG, Fraga M, Moradpour D, Bertaggia Calderara D, Aliotta A, Stirnimann G, et al. Hemostatic alterations in patients with cirrhosis: from primary hemostasis to fibrinolysis. Hepatology. (2020) 71:2135–48. doi: 10.1002/hep.31201
69. Zhao X, Yang L, Chang N, Hou L, Zhou X, Yang L, et al. Neutrophils undergo switch of apoptosis to NETosis during murine fatty liver injury via S1P receptor 2 signaling. Cell Death Dis. (2020) 11:379. doi: 10.1038/s41419-020-2582-1
70. Alisi A, Carpino G, Oliveira FL, Panera N, Nobili V, and Gaudio E. The role of tissue macrophage-mediated inflammation on NAFLD pathogenesis and its clinical implications. Mediators Inflamm. (2017) 2017:8162421. doi: 10.1155/2017/8162421
71. Alvares D, Hoffman S, Stankovic B, and Adeli K. Gut peptide and neuroendocrine regulation of hepatic lipid and lipoprotein metabolism in health and disease. Biochim Biophys Acta Mol Cell Biol Lipids. (2019) 1864:326–34. doi: 10.1016/j.bbalip.2018.12.010
72. Grabherr F, Grander C, Effenberger M, Adolph TE, and Tilg H. Gut dysfunction and non-alcoholic fatty liver disease. Front Endocrinol (Lausanne). (2019) 10:611. doi: 10.3389/fendo.2019.00611
73. Fukui H, Brauner B, Bode JC, and Bode C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J Hepatol. (1991) 12:162–9. doi: 10.1016/0168-8278(91)90933-3
74. Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. (2009) 49:1877–87. doi: 10.1002/hep.22848
75. Matsushita N, Osaka T, Haruta I, Ueshiba H, Yanagisawa N, Omori-Miyake M, et al. Effect of lipopolysaccharide on the progression of non-alcoholic fatty liver disease in high caloric diet-fed mice. Scand J Immunol. (2016) 83:109–18. doi: 10.1111/sji.2016.83.issue-2
76. Jolley KA, Bliss CM, Bennett JS, Bratcher HB, Brehony C, Colles FM, et al. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiol (Reading). (2012) 158:1005–15. doi: 10.1099/mic.0.055459-0
77. Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, et al. Structure, function and diversity of the healthy human microbiome. Nature. (2012) 486:207–14. doi: 10.1038/nature11234
78. Maestri M, Santopaolo F, Pompili M, Gasbarrini A, and Ponziani FR. Gut microbiota modulation in patients with non-alcoholic fatty liver disease: Effects of current treatments and future strategies. Front Nutr. (2023) 10:1110536. doi: 10.3389/fnut.2023.1110536
79. Zeng F, Su X, Liang X, Liao M, Zhong H, Xu J, et al. Gut microbiome features and metabolites in non-alcoholic fatty liver disease among community-dwelling middle-aged and older adults. BMC Med. (2024) 22:104. doi: 10.1186/s12916-024-03317-y
80. Khan A, Ding Z, Ishaq M, Bacha AS, Khan I, Hanif A, et al. Understanding the effects of gut microbiota dysbiosis on nonalcoholic fatty liver disease and the possible probiotics role: recent updates. Int J Biol Sci. (2021) 17:818–33. doi: 10.7150/ijbs.56214
81. Sakaguchi T, Brand S, and Reinecker HC. Mucosal barrier and immune mediators. Curr Opin Gastroenterol. (2001) 17:573–7. doi: 10.1097/00001574-200111000-00016
82. Otani T and Furuse M. Tight junction structure and function revisited. Trends Cell Biol. (2020) 30:805–17. doi: 10.1016/j.tcb.2020.08.004
83. Arciello M, Gori M, Maggio R, Barbaro B, Tarocchi M, Galli A, et al. Environmental pollution: A tangible risk for NAFLD pathogenesis. Int J Mol Sci. (2013) 14:22052–66. doi: 10.3390/ijms141122052
84. Nascimento D, Mota A, Carvalho M, Andrade EDO, Oliveira É PSF, Galvão LLP, et al. Can diet alter the intestinal barrier permeability in healthy people? A systematic review. Nutrients. (2024) 16:1871. doi: 10.3390/nu16121871
85. Wu M-Y and Fan J-G. Gut microbiome and nonalcoholic fatty liver disease. Hepatobiliary Pancreatic Dis Int. (2023) 22:444–51. doi: 10.1016/j.hbpd.2023.06.006
86. Wiest R, Albillos A, Trauner M, Bajaj JS, and Jalan R. Targeting the gut-liver axis in liver disease. J Hepatol. (2017) 67:1084–103. doi: 10.1016/j.jhep.2017.05.007
87. de Vos WM, Tilg H, Van Hul M, and Cani PD. Gut microbiome and health: mechanistic insights. Gut. (2022) 71:1020. doi: 10.1136/gutjnl-2021-326789
88. Block H, Rossaint J, and Zarbock A. The fatal circle of NETs and NET-associated DAMPs contributing to organ dysfunction. . Cells. (2022) 11:1919. doi: 10.3390/cells11121919
89. Di Vincenzo F, Del Gaudio A, Petito V, Lopetuso LR, and Scaldaferri F. Gut microbiota, intestinal permeability, and systemic inflammation: a narrative review. Intern Emerg Med. (2024) 19:275–93. doi: 10.1007/s11739-023-03374-w
90. Denning NL, Aziz M, Gurien SD, and Wang P. DAMPs and NETs in sepsis. Front Immunol. (2019) 10:2536. doi: 10.3389/fimmu.2019.02536
91. Haboubi N. Reporting colonic biopsies in patients with inflammatory bowel disease; a practical approach. Inflammation Bowel Dis. (2019) 25:679–84. doi: 10.1093/ibd/izy288
92. Dinallo V, Marafini I, Di Fusco D, Laudisi F, Franzè E, Di Grazia A, et al. Neutrophil extracellular traps sustain inflammatory signals in ulcerative colitis. J Crohn’s Colitis. (2019) 13:772–84. doi: 10.1093/ecco-jcc/jjy215
93. Wang C-PJ, Ko GR, Lee YY, Park J, Park W, Park T-E, et al. Polymeric DNase-I nanozymes targeting neutrophil extracellular traps for the treatment of bowel inflammation. Nano Convergence. (2024) 11:6. doi: 10.1186/s40580-024-00414-9
94. Pabst O, Hornef MW, Schaap FG, Cerovic V, Clavel T, and Bruns T. Gut-liver axis: barriers and functional circuits. Nat Rev Gastroenterol Hepatol. (2023) 20:447–61. doi: 10.1038/s41575-023-00771-6
95. Ponziani FR, Zocco MA, Cerrito L, Gasbarrini A, and Pompili M. Bacterial translocation in patients with liver cirrhosis: physiology, clinical consequences, and practical implications. Expert Rev Gastroenterol Hepatology. (2018) 12:641–56. doi: 10.1080/17474124.2018.1481747
96. Chelakkot C, Ghim J, and Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med. (2018) 50:1–9. doi: 10.1038/s12276-018-0126-x
97. Cheng C, Tan J, Qian W, Zhang L, and Hou X. Gut inflammation exacerbates hepatic injury in the high-fat diet induced NAFLD mouse: Attention to the gut-vascular barrier dysfunction. Life Sci. (2018) 209:157–66. doi: 10.1016/j.lfs.2018.08.017
98. Ke Z, Huang Y, Xu J, Liu Y, Zhang Y, Wang Y, et al. Escherichia coli NF73-1 disrupts the gut-vascular barrier and aggravates high-fat diet-induced fatty liver disease via inhibiting Wnt/β-catenin signalling pathway. Liver Int. (2024) 44:776–90. doi: 10.1111/liv.15823
99. Rahman K, Desai C, Iyer SS, Thorn NE, Kumar P, Liu Y, et al. Loss of junctional adhesion molecule A promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology. (2016) 151:733–46.e12. doi: 10.1053/j.gastro.2016.06.022
100. Nair B, Kamath AJ, Tergaonkar V, Sethi G, and Nath LR. Mast cells and the gut-liver Axis: Implications for liver disease progression and therapy. Life Sci. (2024) 351:122818. doi: 10.1016/j.lfs.2024.122818
101. Cohen JC, Horton JD, and Hobbs HH. Human fatty liver disease: old questions and new insights. Science. (2011) 332:1519–23. doi: 10.1126/science.1204265
102. Tilg H and Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology. (2010) 52:1836–46. doi: 10.1002/hep.24001
103. Suzuki A and Diehl AM. Nonalcoholic steatohepatitis. Annu Rev Med. (2017) 68:85–98. doi: 10.1146/annurev-med-051215-031109
104. Abraham C, Abreu MT, and Turner JR. Pattern recognition receptor signaling and cytokine networks in microbial defenses and regulation of intestinal barriers: implications for inflammatory bowel disease. Gastroenterology. (2022) 162:1602–16.e6. doi: 10.1053/j.gastro.2021.12.288
105. Vong L, Yeung CW, Pinnell LJ, and Sherman PM. Adherent-invasive escherichia coli exacerbates antibiotic-associated intestinal dysbiosis and neutrophil extracellular trap activation. Inflammation Bowel Dis. (2016) 22:42–54. doi: 10.1097/MIB.0000000000000591
106. Fonseca Z, Díaz-Godínez C, Mora N, Alemán OR, Uribe-Querol E, Carrero JC, et al. Entamoeba histolytica Induce Signaling via Raf/MEK/ERK for Neutrophil Extracellular Trap (NET) Formation. Front Cell Infect Microbiol. (2018) 8:226. doi: 10.3389/fcimb.2018.00226
107. Gao X, Hao S, Yan H, Ding W, Li K, and Li J. Neutrophil extracellular traps contribute to the intestine damage in endotoxemic rats. J Surg Res. (2015) 195:211–8. doi: 10.1016/j.jss.2014.12.019
108. Knopf J, Mahajan A, Muñoz LE, and Herrmann M. Formation and clearance of NETs in health and disease. Cells. (2022) 11:4022. doi: 10.3390/cells11244022
109. Biasi F, Leonarduzzi G, Oteiza PI, and Poli G. Inflammatory bowel disease: Mechanisms, redox considerations, and therapeutic targets. Antioxidants Redox Signaling. (2013) 19:1711–47. doi: 10.1089/ars.2012.4530
110. Mutua V and Gershwin LJ. A review of neutrophil extracellular traps (NETs) in disease: potential anti-NETs therapeutics. Clin Rev Allergy Immunol. (2021) 61:194–211. doi: 10.1007/s12016-020-08804-7
111. Wärnberg J and Marcos A. Low-grade inflammation and the metabolic syndrome in children and adolescents. Curr Opin Lipidol. (2008) 19:11–5. doi: 10.1097/MOL.0b013e3282f4096b
112. Bonaventura A, Vecchié A, Abbate A, and Montecucco F. Neutrophil extracellular traps and cardiovascular diseases: an update. Cells. (2020) 9:231. doi: 10.3390/cells9010231
113. D’Abbondanza M, Martorelli EE, Ricci MA, De Vuono S, Migliola EN, Godino C, et al. Increased plasmatic NETs by-products in patients in severe obesity. Sci Rep. (2019) 9:14678. doi: 10.1038/s41598-019-51220-x
114. Wang H, Wang Q, Venugopal J, Wang J, Kleiman K, Guo C, et al. Obesity-induced endothelial dysfunction is prevented by neutrophil extracellular trap inhibition. Sci Rep. (2018) 8:4881. doi: 10.1038/s41598-018-23256-y
115. Carestia A, Frechtel G, Cerrone G, Linari MA, Gonzalez CD, Casais P, et al. NETosis before and after Hyperglycemic Control in Type 2 Diabetes Mellitus Patients. PloS One. (2016) 11:e0168647. doi: 10.1371/journal.pone.0168647
116. Joshi MB, Lad A, Bharath Prasad AS, Balakrishnan A, Ramachandra L, and Satyamoorthy K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett. (2013) 587:2241–6. doi: 10.1016/j.febslet.2013.05.053
117. Friedman GD, Klatsky AL, and Siegelaub AB. The leukocyte count as a predictor of myocardial infarction. N Engl J Med. (1974) 290:1275–8. doi: 10.1056/NEJM197406062902302
118. Giugliano G, Brevetti G, Lanero S, Schiano V, Laurenzano E, and Chiariello M. Leukocyte count in peripheral arterial disease: A simple, reliable, inexpensive approach to cardiovascular risk prediction. Atherosclerosis. (2010) 210:288–93. doi: 10.1016/j.atherosclerosis.2009.11.009
119. Demers M, Wong SL, Martinod K, Gallant M, Cabral JE, Wang Y, et al. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology. (2016) 5:e1134073. doi: 10.1080/2162402X.2015.1134073
120. Demers M, Wong SL, Martinod K, Gallant M, Cabral JE, Wang Y, et al. Priming neutrophils toward NETosis promotes tumor growth. Oncoimmunology. (2016) 5(5):e1134073. doi: 10.1080/2162402X.2015.1134073
121. Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. (2015) 62:600–14. doi: 10.1002/hep.27841
122. Wu X, Zheng X, and Ye G. WGCNA combined with machine learning to explore potential biomarkers and treatment strategies for acute liver failure, with experimental validation. iLIVER. (2024) 3:100133. doi: 10.1016/j.iliver.2024.100133
123. Yousof TR, Bouchard CC, Alb M, Lynn EG, Lhoták S, Jiang H, et al. Restoration of the ER stress response protein TDAG51 in hepatocytes mitigates NAFLD in mice. J Biol Chem. (2024) 300:105655. doi: 10.1016/j.jbc.2024.105655
124. Takeuchi O and Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
125. Duffy L and O’Reilly SC. Toll-like receptors in the pathogenesis of autoimmune diseases: recent and emerging translational developments. Immunotargets Ther. (2016) 5:69–80. doi: 10.2147/ITT.S89795
126. Kashani B, Zandi Z, Pourbagheri-Sigaroodi A, Bashash D, and Ghaffari SH. The role of toll-like receptor 4 (TLR4) in cancer progression: A possible therapeutic target? J Cell Physiol. (2021) 236:4121–37. doi: 10.1002/jcp.v236.6
127. Alegre F, Pelegrin P, and Feldstein AE. Inflammasomes in liver fibrosis. Semin Liver Dis. (2017) 37:119–27. doi: 10.1055/s-0037-1601350
128. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
129. Wu X, Dong L, Lin X, and Li J. Relevance of the NLRP3 inflammasome in the pathogenesis of chronic liver disease. Front Immunol. (2017) 8:1728. doi: 10.3389/fimmu.2017.01728
130. Cai B, Cai JP, Luo YL, Chen C, and Zhang S. The specific roles of JAK/STAT signaling pathway in sepsis. Inflammation. (2015) 38:1599–608. doi: 10.1007/s10753-015-0135-z
131. Wohlmann A, Sebastian K, Borowski A, Krause S, and Friedrich K. Signal transduction by the atopy-associated human thymic stromal lymphopoietin (TSLP) receptor depends on Janus kinase function. Biol Chem. (2010) 391:181–6. doi: 10.1515/bc.2010.029
132. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, and Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol. (2015) 13:643–54.e1-9. doi: 10.1016/j.cgh.2014.04.014
133. Ryerson AB, Eheman CR, Altekruse SF, Ward JW, Jemal A, Sherman RL, et al. Annual Report to the Nation on the Status of Cancer, 1975-2012, featuring the increasing incidence of liver cancer. Cancer. (2016) 122:1312–37. doi: 10.1002/cncr.v122.9
134. Lambert JE, Ramos-Roman MA, Browning JD, and Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. (2014) 146:726–35. doi: 10.1053/j.gastro.2013.11.049
135. Zhang J, Zhao Y, Xu C, Hong Y, Lu H, Wu J, et al. Association between serum free fatty acid levels and nonalcoholic fatty liver disease: a cross-sectional study. Sci Rep. (2014) 4:5832. doi: 10.1038/srep05832
136. van der Windt DJ, Sud V, Zhang H, Varley PR, Goswami J, Yazdani HO, et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology. (2018) 68:1347–60. doi: 10.1002/hep.29914
137. O’Farrell M, Duke G, Crowley R, Buckley D, Martins EB, Bhattacharya D, et al. FASN inhibition targets multiple drivers of NASH by reducing steatosis, inflammation and fibrosis in preclinical models. Sci Rep. (2022) 12:15661. doi: 10.1038/s41598-022-19459-z
138. Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. (2004) 40:185–94. doi: 10.1002/hep.20283
139. Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. (2016) 531:253–7. doi: 10.1038/nature16969
140. Wang H, Zhang H, Wang Y, Brown ZJ, Xia Y, Huang Z, et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol. (2021) 75:1271–83. doi: 10.1016/j.jhep.2021.07.032
141. Krenkel O and Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. (2017) 17:306–21. doi: 10.1038/nri.2017.11
142. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. (2012) 61:416–26. doi: 10.1136/gutjnl-2011-300304
143. Warnatsch A, Ioannou M, Wang Q, and Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. (2015) 349:316–20. doi: 10.1126/science.aaa8064
144. Németh T and Mócsai A. Feedback amplification of neutrophil function. Trends Immunol. (2016) 37:412–24. doi: 10.1016/j.it.2016.04.002
145. Boussif A, Rolas L, Weiss E, Bouriche H, Moreau R, and Périanin A. Impaired intracellular signaling, myeloperoxidase release and bactericidal activity of neutrophils from patients with alcoholic cirrhosis. J Hepatol. (2016) 64:1041–8. doi: 10.1016/j.jhep.2015.12.005
146. Lee KC, Wu PS, and Lin HC. Pathogenesis and treatment of non-alcoholic steatohepatitis and its fibrosis. Clin Mol Hepatol. (2023) 29:77–98. doi: 10.3350/cmh.2022.0237
147. Pulli B, Ali M, Iwamoto Y, Zeller MW, Schob S, Linnoila JJ, et al. Myeloperoxidase-hepatocyte-stellate cell cross talk promotes hepatocyte injury and fibrosis in experimental nonalcoholic steatohepatitis. Antioxid Redox Signal. (2015) 23:1255–69. doi: 10.1089/ars.2014.6108
148. Huang DQ, El-Serag HB, and Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. (2021) 18:223–38. doi: 10.1038/s41575-020-00381-6
149. Mittal S, El-Serag HB, Sada YH, Kanwal F, Duan Z, Temple S, et al. Hepatocellular carcinoma in the absence of cirrhosis in United States veterans is associated with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. (2016) 14:124–31.e1. doi: 10.1016/j.cgh.2015.07.019
150. Dyson J, Jaques B, Chattopadyhay D, Lochan R, Graham J, Das D, et al. Hepatocellular cancer: the impact of obesity, type 2 diabetes and a multidisciplinary team. J Hepatol. (2014) 60:110–7. doi: 10.1016/j.jhep.2013.08.011
151. Ringelhan M, Pfister D, O’Connor T, Pikarsky E, and Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol. (2018) 19:222–32. doi: 10.1038/s41590-018-0044-z
152. Stine JG, Wentworth BJ, Zimmet A, Rinella ME, Loomba R, Caldwell SH, et al. Systematic review with meta-analysis: risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment Pharmacol Ther. (2018) 48:696–703. doi: 10.1111/apt.2018.48.issue-7
153. Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, Aksenov AA, et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature. (2017) 551:340–5. doi: 10.1038/nature24302
154. Yahoo N, Dudek M, Knolle P, and Heikenwälder M. Role of immune responses in the development of NAFLD-associated liver cancer and prospects for therapeutic modulation. J Hepatol. (2023) 79:538–51. doi: 10.1016/j.jhep.2023.02.033
155. Wu X, Zhou Z, Cao Q, Chen Y, Gong J, Zhang Q, et al. Reprogramming of Treg cells in the inflammatory microenvironment during immunotherapy: a literature review. Front Immunol. (2023) 14:1268188. doi: 10.3389/fimmu.2023.1268188
156. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. (2013) 123:3446–58. doi: 10.1172/jci67484
157. Nair S, Mason A, Eason J, Loss G, and Perrillo RP. Is obesity an independent risk factor for hepatocellular carcinoma in cirrhosis? Hepatology. (2002) 36:150–5. doi: 10.1053/jhep.2002.33713
158. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. (2010) 140:197–208. doi: 10.1016/j.cell.2009.12.052
159. Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. (2007) 317:121–4. doi: 10.1126/science.1140485
160. Kawamura Y, Arase Y, Ikeda K, Seko Y, Imai N, Hosaka T, et al. Large-scale long-term follow-up study of Japanese patients with non-alcoholic Fatty liver disease for the onset of hepatocellular carcinoma. Am J Gastroenterol. (2012) 107:253–61. doi: 10.1038/ajg.2011.327
161. Eslam M, Sanyal AJ, and George J. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. (2020) 158:1999–2014.e1. doi: 10.1053/j.gastro.2019.11.312
162. Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. (2018) 10:eaan8292. doi: 10.1126/scitranslmed.aan8292
163. Cedervall J, Zhang Y, and Olsson AK. Tumor-induced NETosis as a risk factor for metastasis and organ failure. Cancer Res. (2016) 76:4311–5. doi: 10.1158/0008-5472.CAN-15-3051
164. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. (2020) 583:133–8. doi: 10.1038/s41586-020-2394-6
165. Wang H, Li T, Chen S, Gu Y, and Ye S. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol. (2015) 67:3190–200. doi: 10.1002/art.v67.12
166. Xu C, Ye Z, Jiang W, Wang S, and Zhang H. Cyclosporine A alleviates colitis by inhibiting the formation of neutrophil extracellular traps via the regulating pentose phosphate pathway. Mol Med. (2023) 29:169. doi: 10.1186/s10020-023-00758-8
167. Wang S, Li Z, Chang Y, Dong K, He D, and Cheng X. Simvastatin inhibits the formation of NETs by the mac-1 pathway to reduce hepatic ischemia-reperfusion injury under high-fat conditions. Discov Med. (2024) 36:1721–31. doi: 10.24976/Discov.Med.202436187.158
168. Chen F, Chu C, Wang X, Yang C, Deng Y, Duan Z, et al. Hesperetin attenuates sepsis-induced intestinal barrier injury by regulating neutrophil extracellular trap formation via the ROS/autophagy signaling pathway. Food Funct. (2023) 14:4213–27. doi: 10.1039/D2FO02707K
169. Yang C, Song C, Liu Y, Qu J, Li H, Xiao W, et al. Re-Du-Ning injection ameliorates LPS-induced lung injury through inhibiting neutrophil extracellular traps formation. Phytomedicine. (2021) 90:153635. doi: 10.1016/j.phymed.2021.153635
170. Yin N, Wang W, Pei F, Zhao Y, Liu C, Guo M, et al. A neutrophil hijacking nanoplatform reprograming NETosis for targeted microglia polarizing mediated ischemic stroke treatment. Adv Sci (Weinh). (2024) 11:e2305877. doi: 10.1002/advs.202305877
171. de Souza Andrade MM, Leal VNC, Fernandes IG, Gozzi-Silva SC, Beserra DR, Oliveira EA, et al. Resveratrol downmodulates neutrophil extracellular trap (NET) generation by neutrophils in patients with severe COVID-19. Antioxidants. (2022) 11:1690. doi: 10.3390/antiox11091690
172. Gál Z, Gézsi A, Pállinger É, Visnovitz T, Nagy A, Kiss A, et al. Plasma neutrophil extracellular trap level is modified by disease severity and inhaled corticosteroids in chronic inflammatory lung diseases. Sci Rep. (2020) 10:4320. doi: 10.1038/s41598-020-61253-2
173. Dandona P, Mohanty P, Hamouda W, Aljada A, Kumbkarni Y, and Garg R. Effect of dexamethasone on reactive oxygen species generation by leukocytes and plasma interleukin-10 concentrations: A pharmacodynamic study. Clin Pharmacol Ther. (1999) 66:58–65. doi: 10.1016/S0009-9236(99)70054-8
174. Mohammed BM, Fisher BJ, Kraskauskas D, Farkas D, Brophy DF, Fowler AA 3rd, et al. Vitamin C: a novel regulator of neutrophil extracellular trap formation. Nutrients. (2013) 5:3131–51. doi: 10.3390/nu5083131
175. Bai X, Hippensteel J, Leavitt A, Maloney JP, Beckham D, Garcia C, et al. Hypothesis: Alpha-1-antitrypsin is a promising treatment option for COVID-19. Med Hypotheses. (2021) 146:110394. doi: 10.1016/j.mehy.2020.110394
176. Qin D, Liu J, Guo W, Ju T, Fu S, Liu D, et al. Arbutin alleviates intestinal colitis by regulating neutrophil extracellular traps formation and microbiota composition. Phytomedicine. (2024) 130:155741. doi: 10.1016/j.phymed.2024.155741
177. Li G, Lin J, Zhang C, Gao H, Lu H, Gao X, et al. Microbiota metabolite butyrate constrains neutrophil functions and ameliorates mucosal inflammation in inflammatory bowel disease. Gut Microbes. (2021) 13:1968257. doi: 10.1080/19490976.2021.1968257
178. Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, et al. Neutrophil ageing is regulated by the microbiome. Nature. (2015) 525:528–32. doi: 10.1038/nature15367
179. Vong L, Lorentz RJ, Assa A, Glogauer M, and Sherman PM. Probiotic Lactobacillus rhamnosus inhibits the formation of neutrophil extracellular traps. J Immunol. (2014) 192:1870–7. doi: 10.4049/jimmunol.1302286
180. Yang LY, Luo Q, Lu L, Zhu WW, Sun HT, Wei R, et al. Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma via provoking tumorous inflammatory response. J Hematol Oncol. (2020) 13:3. doi: 10.1186/s13045-019-0836-0
181. Nishinaka Y, Arai T, Adachi S, Takaori-Kondo A, and Yamashita K. Singlet oxygen is essential for neutrophil extracellular trap formation. Biochem Biophys Res Commun. (2011) 413:75–9. doi: 10.1016/j.bbrc.2011.08.052
182. Li CJ, Yang ZH, Shi XL, and Liu DL. Effects of aspirin and enoxaparin in a rat model of liver fibrosis. World J Gastroenterol. (2017) 23:6412–9. doi: 10.3748/wjg.v23.i35.6412
183. Vissers MCM and Wilkie RP. Ascorbate deficiency results in impaired neutrophil apoptosis and clearance and is associated with up-regulation of hypoxia-inducible factor 1α. J Leukocyte Biol. (2007) 81:1236–44. doi: 10.1189/jlb.0806541
184. Wang Z, Liu L, and Liu L. Vitamin C as a treatment for organ failure in sepsis. Eur J Med Res. (2023) 28:222. doi: 10.1186/s40001-023-01183-7
185. Várady CBS, Oliveira AC, Monteiro RQ, and Gomes T. Recombinant human DNase I for the treatment of cancer-associated thrombosis: A pre-clinical study. Thromb Res. (2021) 203:131–7. doi: 10.1016/j.thromres.2021.04.028
186. Prince WS, Baker DL, Dodge AH, Ahmed AE, Chestnut RW, and Sinicropi DV. Pharmacodynamics of recombinant human DNase I in serum. Clin Exp Immunol. (1998) 113:289–96. doi: 10.1046/j.1365-2249.1998.00647.x
187. Boursier J, Mueller O, Barret M, MaChado M, Fizanne L, Araujo-Perez F, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. (2016) 63:764–75. doi: 10.1002/hep.28356
188. Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut. (2013) 62:1787–94. doi: 10.1136/gutjnl-2012-303816
189. Cheng R, Wang L, Le S, Yang Y, Zhao C, Zhang X, et al. A randomized controlled trial for response of microbiome network to exercise and diet intervention in patients with nonalcoholic fatty liver disease. Nat Commun. (2022) 13:2555. doi: 10.1038/s41467-022-29968-0
190. Caussy C, Hsu C, Lo MT, Liu A, Bettencourt R, Ajmera VH, et al. Link between gut-microbiome derived metabolite and shared gene-effects with hepatic steatosis and fibrosis in NAFLD. Hepatology. (2018) 68:918–32.
191. Ji Y, Yin Y, Sun L, and Zhang W. The molecular and mechanistic insights based on gut-liver axis: nutritional target for non-alcoholic fatty liver disease (NAFLD) improvement. Int J Mol Sci. (2020) 21:3066. doi: 10.3390/ijms21093066
192. Vallianou N, Christodoulatos GS, Karampela I, Tsilingiris D, Magkos F, Stratigou T, et al. Understanding the role of the gut microbiome and microbial metabolites in non-alcoholic fatty liver disease: current evidence and perspectives. Biomolecules. (2021) 12:56. doi: 10.3390/biom12010056
193. Chen K, Shao LH, Wang F, Shen XF, Xia XF, Kang X, et al. Netting gut disease: neutrophil extracellular trap in intestinal pathology. Oxid Med Cell Longev. (2021) 2021:5541222. doi: 10.1155/2021/5541222
194. Carpi RZ, Barbalho SM, Sloan KP, Laurindo LF, Gonzaga HF, Grippa PC, et al. The effects of probiotics, prebiotics and synbiotics in non-alcoholic fat liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH): A systematic review. Int J Mol Sci. (2022) 23:633–46. doi: 10.3390/ijms23158805
195. Chen J and Vitetta L. Gut microbiota metabolites in NAFLD pathogenesis and therapeutic implications. Int J Mol Sci. (2020) 21:5214. doi: 10.3390/ijms21155214
196. Liu J, Sun J, Yu J, Chen H, Zhang D, Zhang T, et al. Gut microbiome determines therapeutic effects of OCA on NAFLD by modulating bile acid metabolism. NPJ Biofilms Microbiomes. (2023) 9:29. doi: 10.1038/s41522-023-00399-z
197. Harrison SA, Wong VW, Okanoue T, Bzowej N, Vuppalanchi R, Younes Z, et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J Hepatol. (2020) 73:26–39. doi: 10.1016/j.jhep.2020.02.027
198. Kuang J, Wang J, Li Y, Li M, Zhao M, Ge K, et al. Hyodeoxycholic acid alleviates non-alcoholic fatty liver disease through modulating the gut-liver axis. Cell Metab. (2023) 35:1752–66.e8. doi: 10.1016/j.cmet.2023.07.011
199. Li HY, Huang SY, Zhou DD, Xiong RG, Luo M, Saimaiti A, et al. Theabrownin inhibits obesity and non-alcoholic fatty liver disease in mice via serotonin-related signaling pathways and gut-liver axis. J Adv Res. (2023) 52:59–72. doi: 10.1016/j.jare.2023.01.008
200. Lee YY, Park HH, Park W, Kim H, Jang JG, Hong KS, et al. Long-acting nanoparticulate DNase-1 for effective suppression of SARS-CoV-2-mediated neutrophil activities and cytokine storm. Biomaterials. (2021) 267:120389. doi: 10.1016/j.biomaterials.2020.120389
201. Li YY, Wang XJ, Su YL, Wang Q, Huang SW, Pan ZF, et al. Baicalein ameliorates ulcerative colitis by improving intestinal epithelial barrier via AhR/IL-22 pathway in ILC3s. Acta Pharmacol Sin. (2022) 43:1495–507. doi: 10.1038/s41401-021-00781-7
202. Sadeghi Z, Cerulli A, Marzocco S, Moridi Farimani M, Masullo M, and Piacente S. Anti-inflammatory activity of tanshinone-related diterpenes from perovskia artemisioides roots. J Nat Prod. (2023) 86:812–21. doi: 10.1021/acs.jnatprod.2c01004
203. Zhang L, Zheng B, Bai Y, Zhou J, Zhang XH, Yang YQ, et al. Exosomes-transferred LINC00668 contributes to thrombosis by promoting NETs formation in inflammatory bowel disease. Adv Sci (Weinh). (2023) 10:e2300560. doi: 10.1002/advs.202300560
204. Zhang Z, Shang J, Yang Q, Dai Z, Liang Y, Lai C, et al. Exosomes derived from human adipose mesenchymal stem cells ameliorate hepatic fibrosis by inhibiting PI3K/Akt/mTOR pathway and remodeling choline metabolism. J Nanobiotechnology. (2023) 21:29. doi: 10.1186/s12951-023-01788-4
205. Zhao H, Shang Q, Pan Z, Bai Y, Li Z, Zhang H, et al. Exosomes from adipose-derived stem cells attenuate adipose inflammation and obesity through polarizing M2 macrophages and beiging in white adipose tissue. Diabetes. (2018) 67:235–47. doi: 10.2337/db17-0356
206. Lin Y, Yan M, Bai Z, Xie Y, Ren L, Wei J, et al. Huc-MSC-derived exosomes modified with the targeting peptide of aHSCs for liver fibrosis therapy. J Nanobiotechnology. (2022) 20:432. doi: 10.1186/s12951-022-01636-x
207. Tamura R, Uemoto S, and Tabata Y. Augmented liver targeting of exosomes by surface modification with cationized pullulan. Acta Biomater. (2017) 57:274–84. doi: 10.1016/j.actbio.2017.05.013
208. Zheng W, Zhao S, Yin Y, Zhang H, Needham DM, Evans ED, et al. High-throughput, single-microbe genomics with strain resolution, applied to a human gut microbiome. Science. (2022) 376:eabm1483. doi: 10.1126/science.abm1483
209. Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. (2015) 149:367–78.e5. doi: 10.1053/j.gastro.2015.04.005
210. Romero-Gómez M, Zelber-Sagi S, and Trenell M. Treatment of NAFLD with diet, physical activity and exercise. J Hepatol. (2017) 67:829–46. doi: 10.1016/j.jhep.2017.05.016
211. de Oliveira S, Houseright RA, Graves AL, Golenberg N, Korte BG, Miskolci V, et al. Metformin modulates innate immune-mediated inflammation and early progression of NAFLD-associated hepatocellular carcinoma in zebrafish. J Hepatol. (2019) 70:710–21. doi: 10.1016/j.jhep.2018.11.034
212. Pabst O, Hornef MW, Schaap FG, Cerovic V, Clavel T, and Bruns T. Gut–liver axis: barriers and functional circuits. Nat Rev Gastroenterol Hepatology. (2023) 20:447–61. doi: 10.1038/s41575-023-00771-6
Keywords: inflammation, liver disease, gut microbiota, pathophysiological progression, immune dysregulation
Citation: Yin J, Zhu Y, Liu R, Wang W, Wang Z and Wang J (2025) A new insight: crosstalk between neutrophil extracellular traps and the gut-liver axis for nonalcoholic fatty liver disease. Front. Immunol. 16:1599956. doi: 10.3389/fimmu.2025.1599956
Received: 25 March 2025; Accepted: 27 May 2025;
Published: 27 June 2025.
Edited by:
Hongji Zhang, University of Virginia, United StatesReviewed by:
Lexing Yu, National Center for Liver Cancer, ChinaYantong Wan, Southern Medical University, China
Gang Ye, Sichuan Agricultural University, China
Copyright © 2025 Yin, Zhu, Liu, Wang, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhicheng Wang, emhpY2hlbmdAamx1LmVkdS5jbg==; Jianfeng Wang, amZ3YW5nQGpsdS5lZHUuY24=
†These authors have contributed equally to this work