 Min Zhang
Min Zhang Liang Lyu
Liang Lyu Liang Ge4
Liang Ge4 Dongqing Gu
Dongqing Gu- 1Department of Sleep and Psychology, Chongqing Health Center for Women and Children, Women and Children’s Hospital of Chongqing Medical University, Chongqing, China
- 2Department of College of Medical Informatics, Chongqing Medical University, Chongqing, China
- 3Department of Information, Chongqing Health Center for Women and Children (Women and Children’s Hospital of Chongqing Medical University), Chongqing, China
- 4Department of Laboratory of Infection and Immunity, West China School of Medical Sciences & Forensic Medicine, Sichuan University, Chengdu, China
- 5Department of Pathology, The Third Hospital of Mianyang, Sichuan Mental Health Center, Mianyang, Sichuan, China
- 6Department of Obstetrics and Gynecology, Chongqing Health Center for Women and Children (Women and Children’s Hospital of Chongqing Medical University), Chongqing, China
Background: Despite extensive genetic studies investigating primary biliary cholangitis (PBC), the mechanistic basis of risk-associated variants remains poorly understood. To address this gap, we performed a systematic evaluation of cumulative evidence linking genetic variants to PBC susceptibility.
Methods: A comprehensive search was conducted to identify published studies on the association between genetic variants and PBC risk. Specifically, separate analyses were conducted for genome-wide association studies (GWASs) and candidate-gene association studies to address potential heterogeneity arising from differences in study design. Meta-analyses were performed to calculate pooled odds ratio (OR) and 95% confidence interval (CI) for the candidate-gene association studies. Significant associations were further graded using Venice criteria and false-positive report probability (FPRP) tests. Functional annotation, pathway enrichment, and phenome-wide analyses were performed to elucidate biological relevance.
Results: Overall, we included 105 articles involving 71,031 cases and 140,499 controls. Meta-analyses were conducted for 70 variants across 33 genes. Among these, 44 variants were identified as significantly associated with PBC risk, comprising 30 HLA variants and 14 non-HLA variants. Separately, published GWAS have reported 115 significant variants. Nine variants (DQA1*0401, DQB1*0301, DQB1*0402, DQB1*0602, DRB1*08, DRB1*0803, DRB1*11, DRB1*1101, and rs7574865) were identified by both approaches. Additionally, meta-analyses of candidate-gene association studies provided strong evidence supporting the association of eight further variants (A*3303, B*4403, DPB1*0201, DQB1*0401, rs231725, rs231775, rs1544410, and rs9303277) with PBC at the genome-wide significance level (P < 5.0 × 10-8). Pathway analysis revealed significant enrichment of the mapped genes in immune cell regulation and immune response-regulating signaling pathways. Phenome-wide analyses further indicated that the missense variant rs231775 was significantly associated with thyroid problems and melanoma (P< 6.43×10-5).
Conclusion: This study provides the most comprehensive synopsis to date of PBC’s genetic architecture, highlighting robust HLA and non-HLA risk loci.
Systematic review registration: https:///www.crd.york.ac.uk/PROSPERO/view/CRD42021282146, identifier CRD42021282146.
1 Introduction
Primary biliary cholangitis (PBC), characterized by significant female predominance, is the most prevalent autoimmune liver disease (1). Individuals with PBC often experience symptoms that significantly impact their quality of life, including itching, fatigue, abdominal pain, and sicca complex (2). Untreated PBC is associated with an increased risk of cirrhosis and related complications, liver failure and even death (3). It is well known that genetic factors contribute to the pathogenesis of PBC. Several genome-wide association studies (GWASs) have identified variants in human leukocyte antigen (HLA) regions (e.g., DQB1*0301, DRB1*08, DRB1*1302) and outside HLA regions (non-HLA) that are associated with PBC susceptibility (4–7). Nevertheless, these loci together account for only 21% of the genetic causes of this disease (8).
Despite results from genome-wide association studies (GWASs) are prominent and increasingly available, candidate-gene association studies are still the most predominant type of research for identifying common risk alleles for PBC. Over the past decade, over 90 candidate-gene PBC association studies have been conducted, evaluating over 800 genetic loci in HLA region and non-HLA regions. While some of these genetic loci may indeed be linked to PBC risk, many others are false-positive associations that do not replicate in additional populations. The determination of whether these associations are validity typically involves a comprehensive examination of epidemiological evidence alongside biological plausibility, often through a meta-analysis which can enhance the statistical power and assess the replication and consistency of an association by consolidating data from multiple studies (9). In addition, following the guidelines developed by the Human Genome Epidemiology Network multidisciplinary workshop (10, 11), Venice criteria have been used to assess cumulative evidence of genetic associations (12–15). However, previous meta-analysis primarily focused on individual variants or those within a single gene (16–18), and no comprehensive field synopsis has been published to evaluate the cumulative evidence of associations between genetic variants and PBC risk so far.
In this study, we aimed to provide a comprehensive overview of the current understanding of the genetic architecture of PBC based on published literature. First, we conducted separate analyses for GWASs and candidate-gene association studies. For candidate-gene association studies, we performed a meta-analysis to comprehensively evaluate the association between genetic variants and PBC risk. We then evaluated the cumulative evidence for significant associations by combining Venice criteria and false-positive report probability (FPRP) tests. Finally, we conducted functional annotation, pathway analysis and phenome-wide analysis of potential pathogenic loci.
2 Materials and methods
The methodology for the meta-analysis followed the guidelines proposed by the Human Genome Epidemiology Network for a systematic review of genetic association studies and the Preferred Reporting Items for Systematic Reviews and Meta-Analyses statement (Supplementary Tables S1) (19, 20). The protocol was registered in the International Prospective Register of Systematic Reviews (CRD42021282146).
2.1 Literature search strategy and study eligibility
A comprehensive literature search of related studies was conducted using PubMed, Embase, and Web of Science (published on or before May 1, 2024), using the following keywords: “autoimmune liver disease OR primary biliary cholangitis OR primary biliary cirrhosis” AND “Genetic OR SNP OR polymorphism OR genotype OR variant OR allele OR mutation OR genome-wide association study OR GWAS.” The titles, abstracts, and full texts of the studies were reviewed as needed to identify all relevant articles. In addition, the reference lists of all included studies, reviews, and meta-analyses were manually screened for additional potential studies.
The inclusion criteria were as follows (1): original articles published in English (2); observational studies (3); investigating associations between genetic variants and risk of PBC; and (4) providing risk estimates [odds ratio (OR) and relative risk (RR]) and 95% confidence intervals (CIs) or data to calculate them. Exclusion criteria (1): participants complicated with other liver diseases (2); less than 50 cases and controls; and (3) reviews, abstracts, case reports, and letters. If several publications used the same or overlapping data, only the studies that reported results from the most recent or largest analysis was included. Two investigators (DG and YW) independently assessed the eligibility of each publication and any disagreements were discussed with the principal author (MZ).
2.2 Data extraction, preparation, and management
Two authors (DG and MZ) independently extracted data using a pre-designed collection sheet. The data included PMID, first author, publication year, study design, sample size of cases and controls, source of population, ethnicity, variants, gene, major and minor alleles, genotype and allele counts, risk estimates, and corresponding 95% CIs or P-value (for studies using multiple adjusted models, the most fully adjusted estimates were extracted).
2.3 Meta-analyses
To address potential heterogeneity arising from differences in study design, we conducted separate analyses for GWASs and candidate-gene association studies. For GWAS-derived data, we reported the SNP with the largest sample size at each locus within each ancestry group, along with its corresponding effect estimate, to avoid redundancy caused by linkage disequilibrium (LD) among SNPs at the same genomic region.
For candidate-gene association studies, we performed meta-analyses for variants with data available from at least three independent datasets. We calculated the pooled OR and 95%CIs using an additive genetic model. We meta-analyzed the associations of variants in human leukocyte antigen (HLA) gene with PBS risk and the associations of loci in non-HLA genes with PBS risk. Statistical heterogeneity among the studies was assessed using the Cochran Q statistic (P < 0.10 was considered statistically significant) and I2 statistic (I² ≤ 25% represented mild heterogeneity, 25% - 50% represented moderate heterogeneity, and ≥ 50% represented large heterogeneity) (21). A random-effects model was used if I2 ≥ 50%, while a fixed-effects model was used if I2 < 50%. For variants that showed a significant association with PBC, sensitivity analyses were performed by excluding the first published or positive study. Furthermore, we assessed potential publication bias using Begg’s test (22) and small-study bias using Egger’s test (23). In addition, we conducted subgroup meta-analyses stratified by Ethnicity (datasets ≥ 2 in either Asian or Caucasian populations). Between-subgroup heterogeneity was assessed using Cochran’s Q test, and P < 0.10 were considered indicative of significant ethnic heterogeneity. To explore potential sources of heterogeneity, we further performed meta-regression and subgroup analyses stratified by diagnostic criteria for PBC and genotyping method in meta-analyses with high heterogeneity.
2.4 Assessment of cumulative evidence
Associations with P < 0.05 in the primary meta-analyses were evaluated using the Venice criteria to assess epidemiological credibility. The detailed methods have been described in our previous research (24). Finally, epidemiological credibility was categorized as strong, moderate, or weak, based on the grade level of A, B, or C, according to three criteria: amount of evidence, replication, and protection from bias (10, 11). In addition, FPRP was calculated for these associations (25). Specifically, FPRP values of < 0.05, 0.20 - 0.05, and > 0.20 were considered strong, moderate, and weak evidence of a true association, respectively. We up-graded the cumulative evidence if the FPRP result was strong, and down-graded the cumulative evidence if the FPRP result was weak.
2.5 Functional annotation
To provide biological insights into the significant variants identified by our meta-analysis and previous GWASs, we mapped these SNPs to genes and conducted functional annotation using the Encyclopedia of DNA Elements (ENCODE) tool HaploReg v4.1 (26). To identify the tissues most relevant to the significant genes, we conducted Genotype-Tissue Expression (GTEx) tissue enrichment analysis based on 54 tissue types available from GTEx (version 8) using the functional mapping and annotation of genome-wide association studies (FUMA) GENE2FUNC process (27). Furthermore, we evaluated the enrichment of significantly mapped genes in Gene Ontology (GO) biological processes using the WebGestalt tool (28). We utilized the Benjamin-Hochberg procedure to correct for multiple testing and considered a false discovery rate (FDR) corrected P-value of less than 0.05 as a statistical difference.
2.6 Phenome-wide analyses
In addition, phenome-wide analyses were performed to estimate associations between the newly identified functional variants and 778 phenotypes from the UK Biobank, and summary data were generated using GeneATLAS (29). P values < 6.43×10-5 (0.05/778) were considered statistically significant after adjusting for multiple comparisons of variants and 778 phenotypes.
2.7 Statistical analysis
Statistical analysis was conducted using Stata version 15 (StataCorp, College Station, TX), and a two-tailed P-value of < 0.05 was considered statistically significant unless otherwise specified.
3 Results
3.1 Characteristics of the included studies
In total, 4,224 publications were screened after duplicates were excluded from the literature search (Figure 1). Ultimately, 105 articles involving 71,031 cases and 140,499 controls were included, and these articles investigated 1,341 variants located in 419 genes or chromosomal loci associated with risk of PBC. Most of these articles were conducted in Caucasians (n=62), followed by Asians (n=48) (Figure 2). The sample size ranged from 115 to 24,510 (median, 584), and the number of cases ranged from 16 to 8,061 (median, 232). Among these articles, 15 were GWASs (4, 6, 7, 30–41) (Supplementary Table S2) and 90 were candidate-gene association studies. Sixty-nine candidate-gene association studies explored the relationship between 302 variants in 122 non-HLA genes and the risk of PBC (Supplementary Table S3, Supplementary Table S4), whereas 23 studies investigated the association between 178 variants in HLA region and the risk of PBC (Supplementary Table S5, Supplementary Table S6).
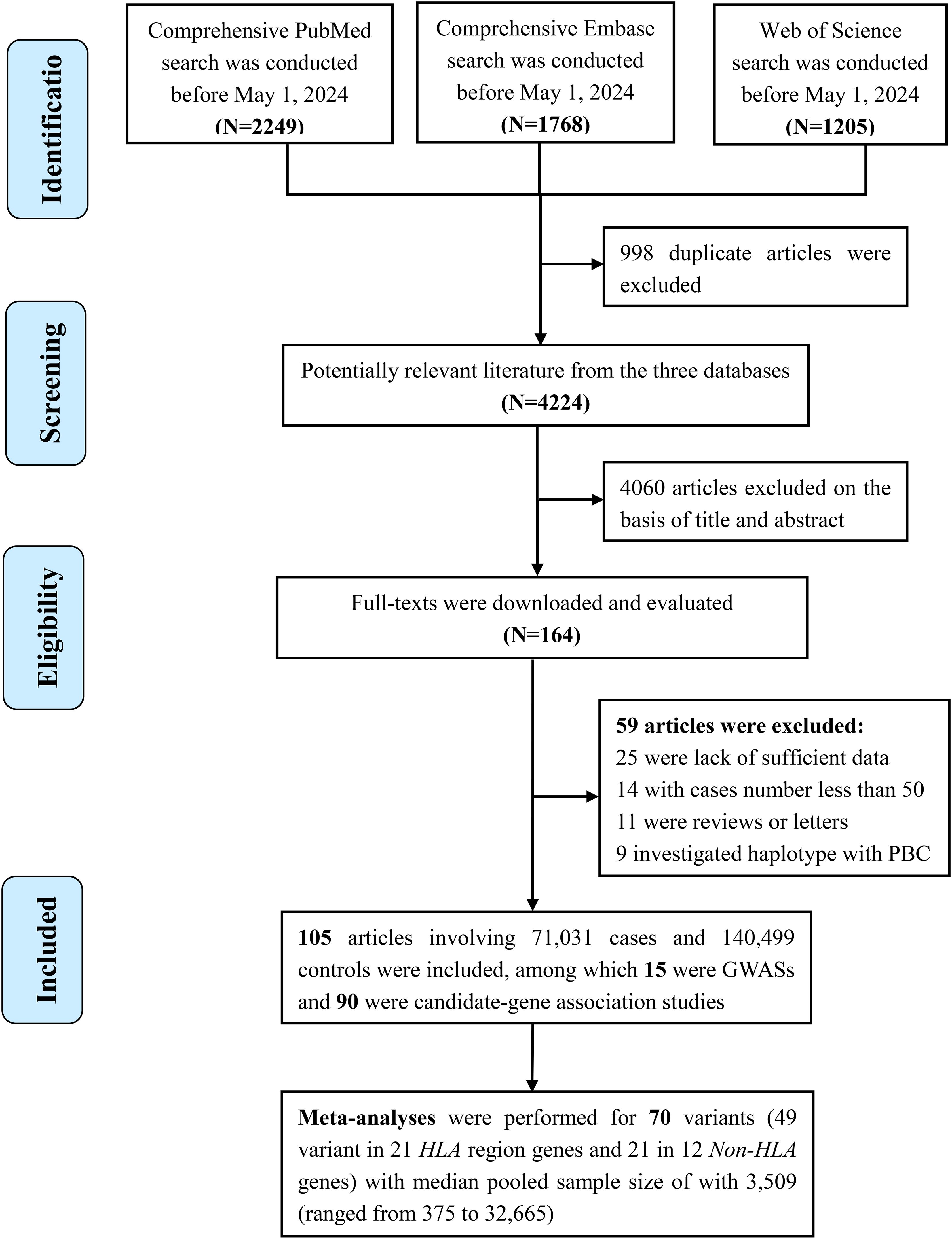
Figure 1. Flowchart of literature selection in the meta-analysis.
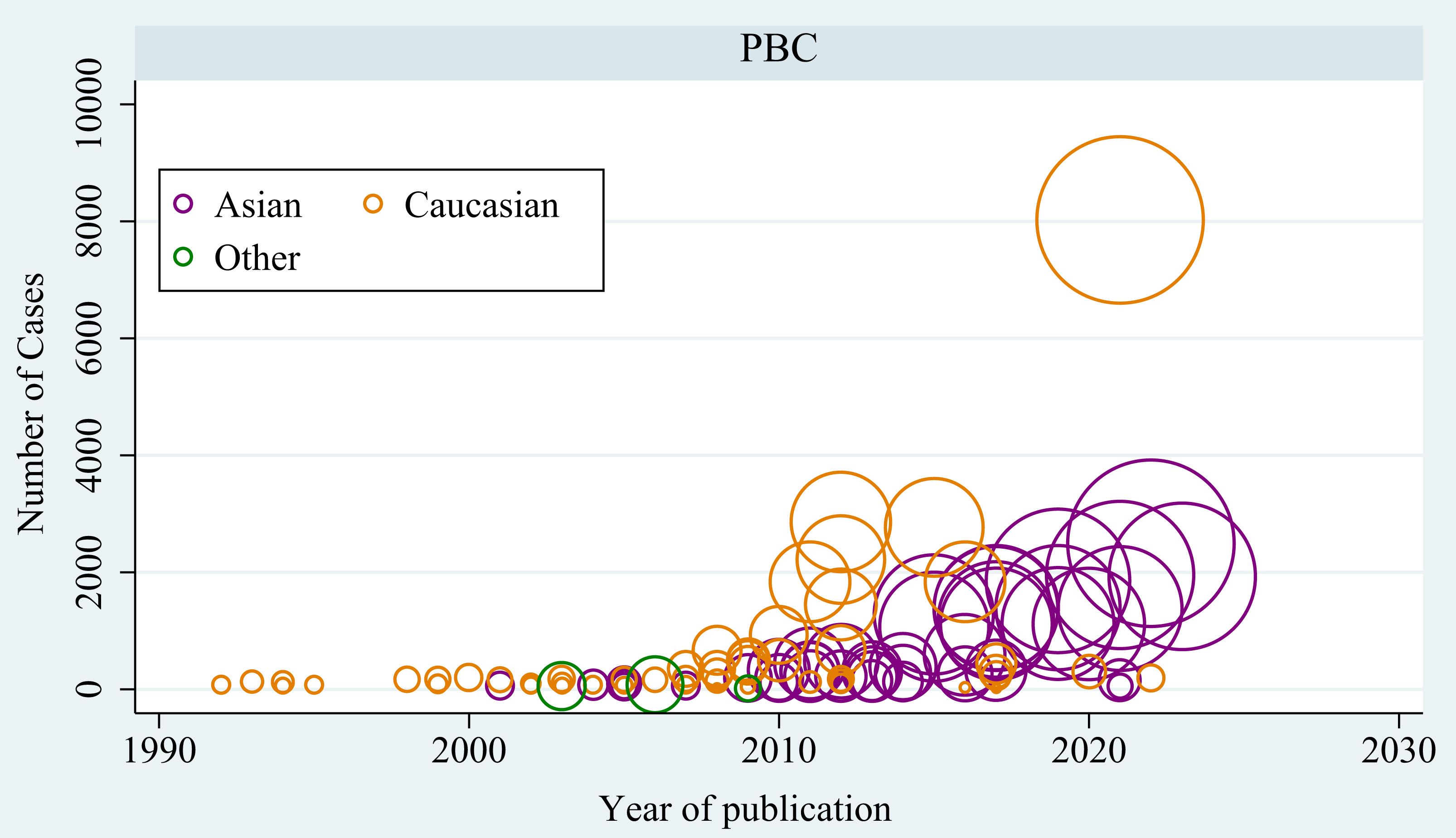
Figure 2. Characteristics of the included studies in the meta-analysis.
3.2 Genome-wide significant associations in the GWASs
Fifteen GWASs identified 111 genome-wide significant SNPs across 55 loci associated with PBC, including 71 independent SNPs in 48 loci among Europeans, 26 independent SNPs in 17 loci among Chinese populations, and 16 independent SNPs in 10 loci among Japanese populations (Table 1). Among these SNPs, 40 (or loci in linkage disequilibrium with them) were replicated in more than two GWASs.
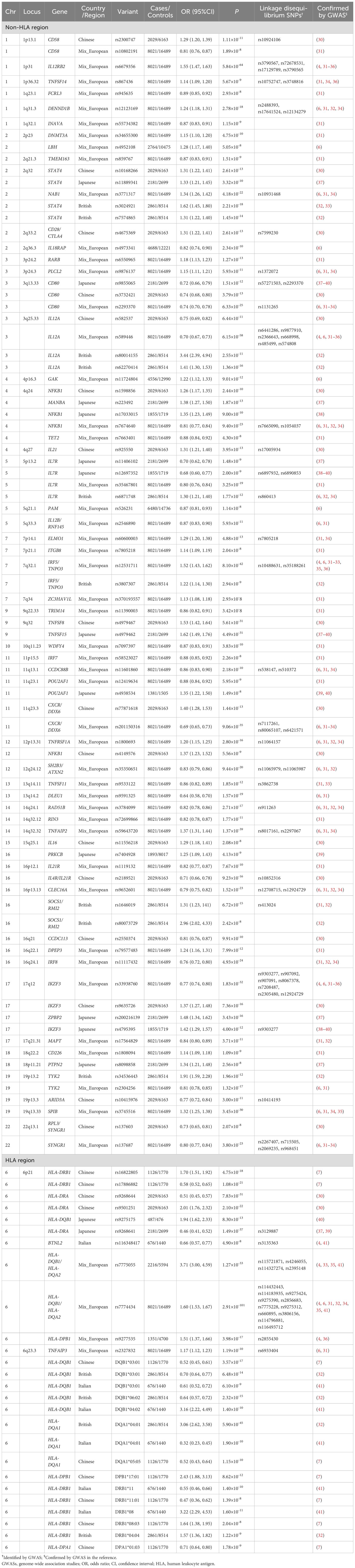
Table 1. Loci significantly associated with PBC identified by GWAS.
3.3 Results of the meta-analysis in the candidate-gene association studies
Meta-analyses were performed for 70 associations for variants (49 variant in 21 HLA region genes and 21 in 12 non-HLA genes) with available data from at least three independent sources. The median pooled sample size of the 70 meta-analyses was 3,509 (ranged from 375 to 32,665).
30 variants within six HLA genes (HLA-A, HLA-B, HLA-DQA1, HLA-DQB1, HLA-DRB1, HLA-DPB1) were found to be significantly associated with the risk of PBC (Table 2). Strong associations (OR > 2 or < 0.5) with PBC-risk were identified for 11 variants, with the strongest positive association was observed for DRB1*0801 (OR=3.11, 95% CI=1.59-6.08, P=9.16×10-4) and negative association for DQB1*0604 (OR=0.31, 95% CI=0.20-0.48, P=1.42×10-7). Ten variants (A*3303, B*4403, DPB1*0201, DPB1*0501, DQB1*0301, DQB1*0401, DQB1*0601, DRB1*08, DRB1*0803, DRB1*1101) had associations with PBC risk at genome-wide significance level (P < 5.0×10-8), among which DQB1*0301, DRB1*08, DRB1*0803 and DRB1*1101 were previously identified genome-wide significant risk loci (Table 1). No significant associations were found for another 19 variants in HLA region (Supplementary Table S7). Subgroup analyses show that among the 21 variants eligible for subgroup analysis, 11 (52.4%) displayed significant between-subgroup heterogeneity (P for subgroup heterogeneity < 0.1).
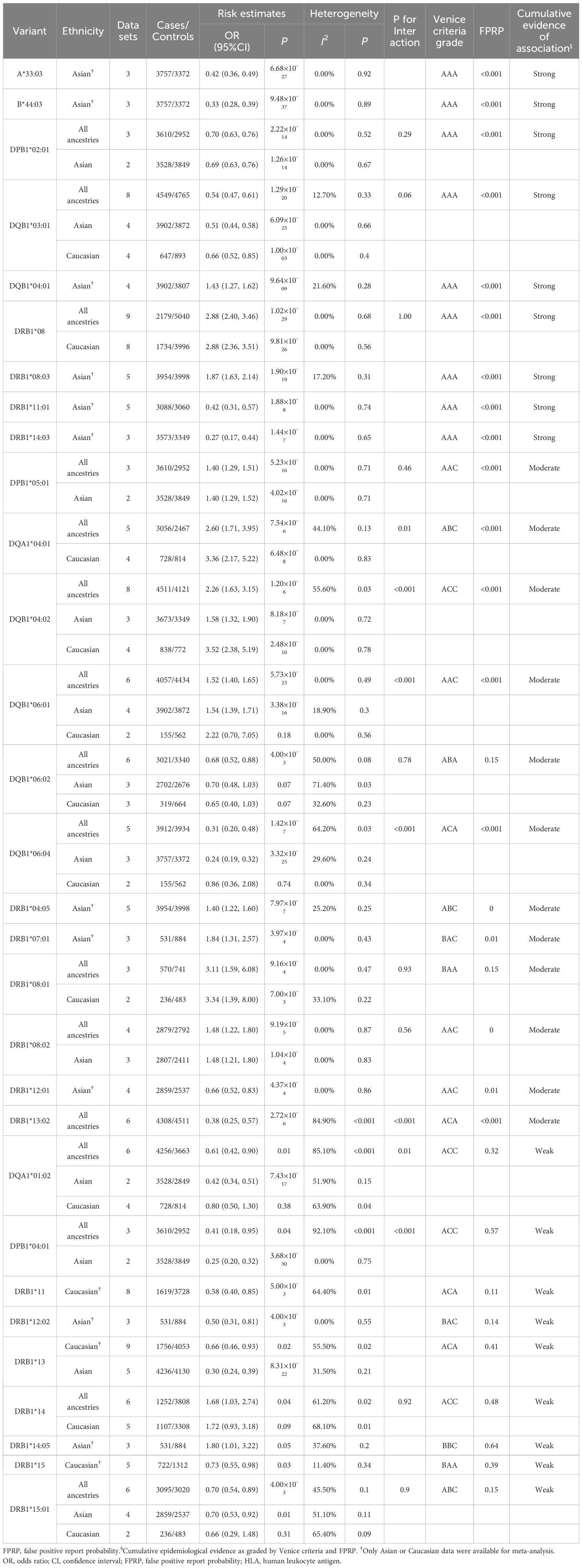
Table 2. Variants in HLA genes significantly associated with risk of primary biliary cholangitis in meta-analysis.
As to variants located outside the HLA region (non-HLA genes), 14 variants within 11 genes were found to be significantly associated with PBC risk (Table 3). Five variants (rs231775, rs231725, rs9303277, rs1864325 and rs1544410) reached genome-wide significance (P < 5.0×10-8) across all ancestries, among which rs9303277 and rs1864325 were (or in LD with) previously identified by GWAS. Of these, rs1544410 within VDR exhibited the strongest association with PBC risk (OR=1.62, 95% CI=1.37-1.93, P=2.99×10-08). Subgroup analyses suggested only rs231775 in CTLA-4 (OR=1.31, 95% CI=1.21-1.41, P=3.28×10-12) and rs9303277 in IKZF3 were identified as genome-wide significant loci in Asian population. No significant associations were observed for another 7 variants within five non-HLA genes (Supplementary Table S8).
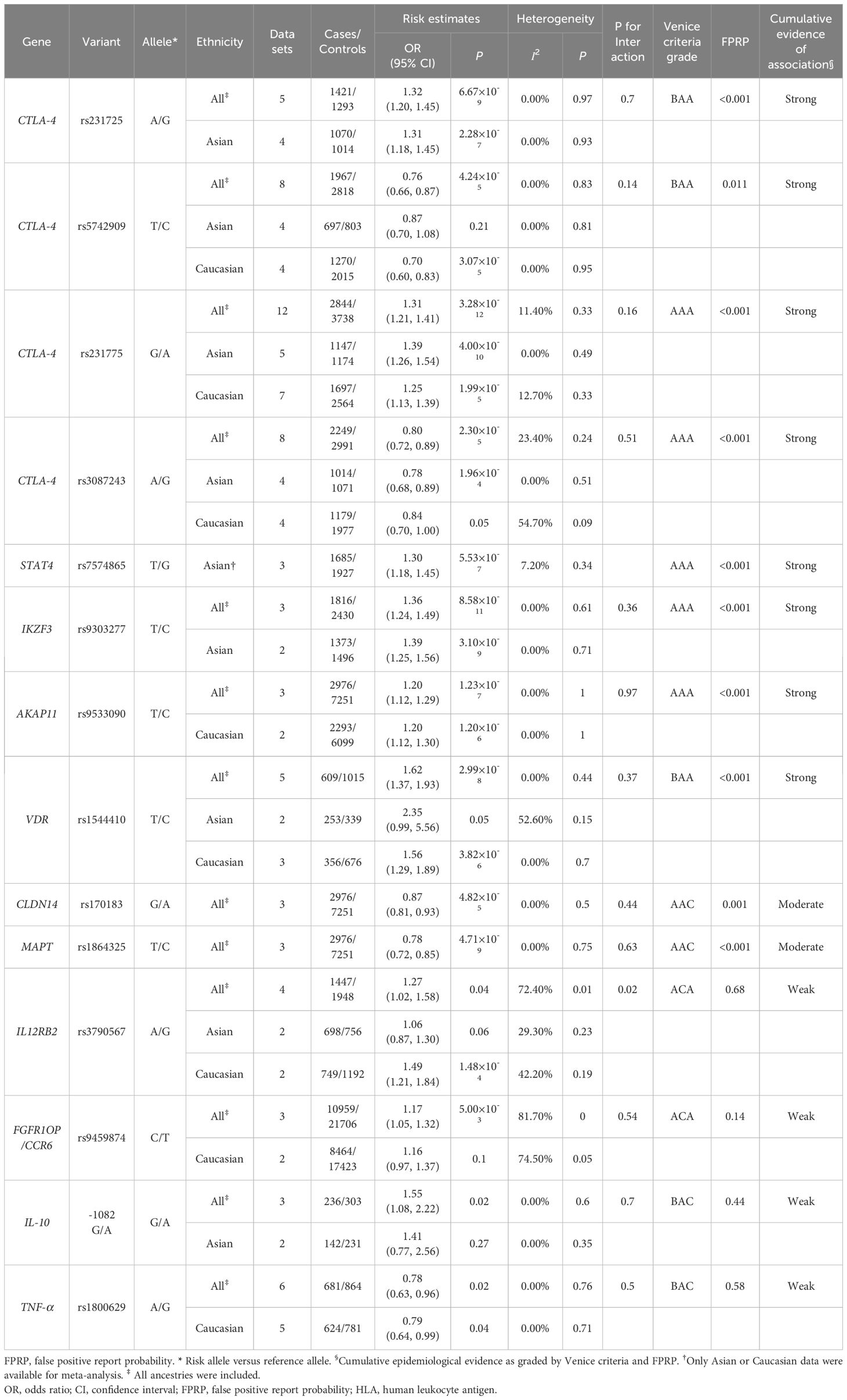
Table 3. Variants in Non-HLA genes significantly associated with risk of primary biliary cholangitis in meta-analysis.
3.4 Heterogeneity, sensitivity analysis, and bias in the meta-analysis
Of the 70 meta-analysis, 23 (32.9%) had high heterogeneity, 6 (8.6%) had moderate heterogeneity, and 41 (58.6%) had no or little heterogeneity. The proportion of high heterogeneity in the 43 significant associations was lower than that in the remaining 26 non-significant associations (22.7% vs 42.3%%). Subgroup analyses (Supplementary Table S9) showed that diagnostic criteria, genotyping methodology, and ethnicity might be the source of heterogeneity (P for interaction <0.05). Meta-regression indicated that ethnicity and diagnostic criteria might contribute to the heterogeneity of DQB10402 (P=0.011) and DQA10102 (P=0.007), respectively. Sensitivity analyses by excluding the initial published or positive study were performed for the 44 variants significantly related to PBC-risk. The results indicated that 75.0% of the significant association was robustness, and the other 25.0% was no longer significant when excluding the initial positive study (Supplementary Table S10). Publication bias was evaluated by Begg’s tests. Six variants (rs170183, DRB1*0802, DQA1*0401, DQB1*0402, DPB1*0501) indicated evidence of publication bias (P < 0.10). As to bias due to small studies (estimated by Egger tests), four variants (rs1864325, DRB1*0405, DRB1*14, DRB1*0802) showed evidence of possible small study bias (P < 0.10) (Supplementary Table S10).
3.5 Cumulative evidence assessment
In the evaluation of the cumulative evidence for the 44 significant associations (Table 2 and Table 3), grades of A were given to 34, 29, and 28 variants for the amount of evidence, replication of the association, and protection from bias, respectively by the Venice criteria. Grades of B were given to 10, five, and zero associations for each of the three criteria. Grades of C were given to 16 variants for protection from bias (Supplementary Table S10), mainly due to the loss of significance after excluding the initial report (n=10), small study bias (n=4) and significant publication bias (n=4). Significant associations with PBC-risk had a calculated FPRP < 0.05 for 29 variants, FPRP 0.05-0.20 for 6 variant, and FPRP > 0.20 for 9 variants. By integrating the Venice criteria and FPRP, cumulative epidemiological evidence of a significant relationship was graded as strong for 17 variants (9 within 5 HLA genes and 8 within 5 non-HLA genes), moderate for 14 variants (13 within 4 HLA genes and 2 within 2 non-HLA genes), and weak for 13 variants (8 within 3 HLA genes and 4 within 4 non-HLA genes).
3.6 Functional annotation and pathway analysis
Functional annotation was further conducted using HaploReg V4.1 for the variants that significant associated with PBC risk (Supplementary Table S11). The results suggested that these variants and their highly correlated SNPs might fall within a DNase I hypersensitivity site, a strong prompter, and an enhancer activity region. Of these variants, rs231775 was missense located in the CTLA4 gene. GTEx tissue enrichment analysis indicated that the significant mapped genes for PBC were significantly enriched in the small intestine, lymphocytes, lungs, spleen, brain, and blood (Supplementary Figure S1). In addition, GO pathway analysis across these significantly mapped genes revealed enrichment in 10 biological pathways (FDR < 0.05), primarily involved in immune cell regulation and immune response-regulating signaling pathways (Table 4).
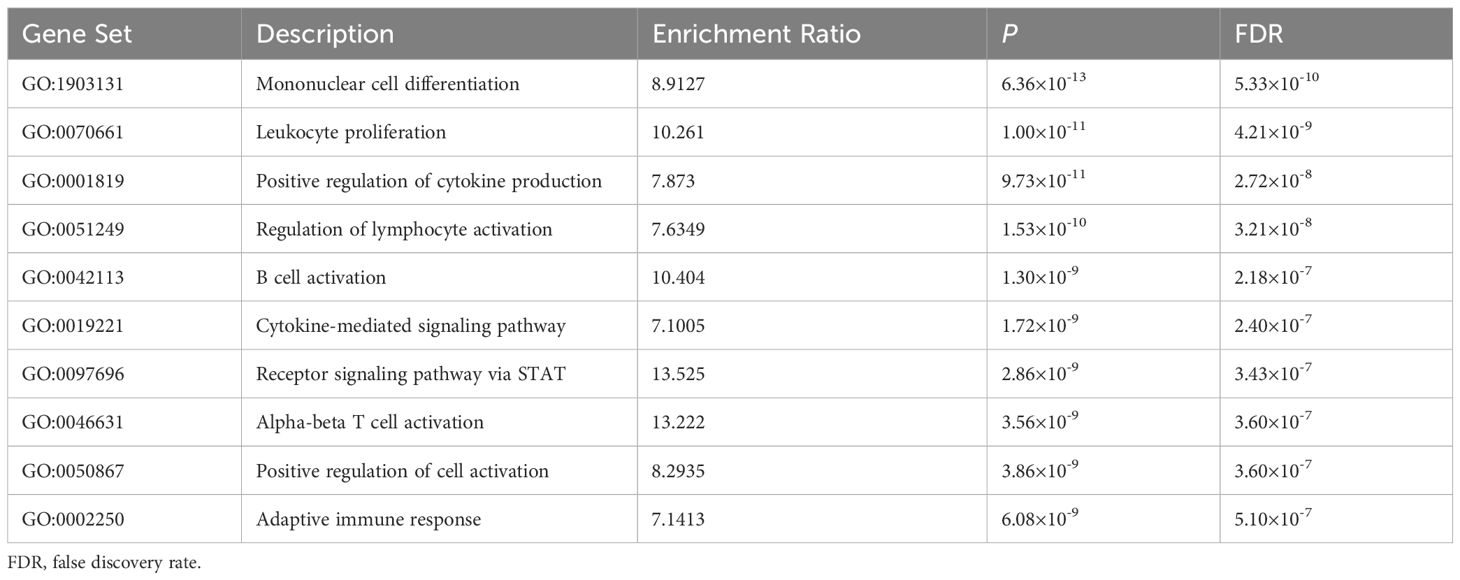
Table 4. GO pathway analysis across the significant mapped genes of primary biliary cholangitis.
3.7 Phenome-wide analysis
Finally, we performed phenome-wide analysis for the two additional genome-wide significant SNPs, rs1544410 and rs231725 (rs231775 and rs231725 were in strong LD with R2 = 0.85), identified by our meta-analysis. The results suggested that rs231725 was primarily associated with thyroid problems (such as hypothyroidism, thyroid gland disorders, and hyperthyroidism/thyrotoxicosis) and melanoma (Figure 3 and Supplementary Table S12). However, no significant association was identified for rs1544410.
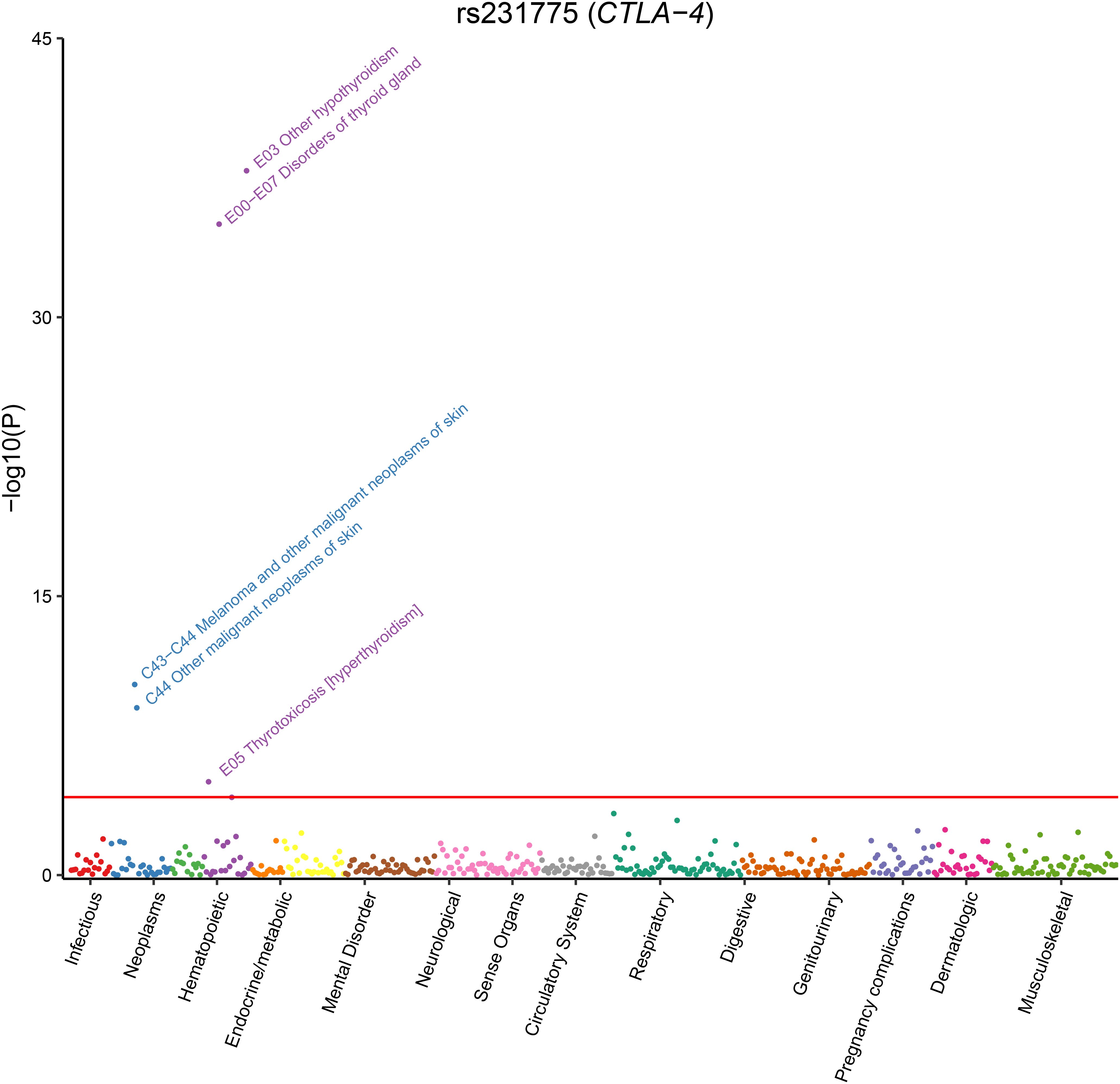
Figure 3. Phenome-wide analysis of rs231775 using data from UK Biobank.
4 Discussion
To the best of our knowledge, this is the largest and most comprehensive study to systematically assess the relationship between genetic variants (in HLA region and non-HLA region) and risk of PBC. This research incorporated data from 105 articles that involved 71,031 cases and 140,499 controls. Meta-analyses of candidate-gene association studies identified 44 variants significantly associated with PBC risk (30 variants within six HLA genes and 14 variants within 11 non-HLA genes). Separately, published GWAS reported 115 significant variants. Among these variants, nine variants (eight variants in HLA genes and rs7574865 in STAT4) were identified by both approaches. Cumulative epidemiological evidence graded 17 strong, 14 moderate, and 13 weak associations. Notably, strong evidence supports the missense variant rs231775 in CTLA4 as a genome-wide significant locus, emphasizing its potential role in PBC pathogenesis. In addition, tissue enrichment analysis and phenome-wide analysis showed that PBC may share a common genetic architecture with some autoimmune diseases. This study comprehensively evaluated published research on the relationship between genetic variants and risk of PBC. These findings improve our current understanding of the genetic architecture of this disease.
The HLA has been extensively studied in a variety of immune-mediated diseases, such as rheumatoid arthritis (42), inflammatory bowel disease (43), and autoimmune hepatitis (7, 44). Our study confirmed the importance of variations in the HLA gene in the pathogenesis of PBC. Specifically, eight variants (DQA1*0401, DQB1*0301, DQB1*0402, DQB1*0602, DRB1*08, DRB1*0803, DRB1*11, and DRB1*1101) have been shown to be associated with PBC both in published GWAS and meta-analyses of candidate-gene association studies. Among these variants, strong evidence supports four variants (DRB1*08, DRB1*1101, DRB1*0803, and DQB1*0301) were associated with PBC at the genome-wide significance level by our meta-analyses. These results are consistent with previous studies, indicating that HLA is a susceptibility gene for PBC (7, 33).
The HLA-DRB1*08 allele family has been the most extensively studied in terms of PBC susceptibility. Our meta-analysis suggests that DRB1*0803 is associated with PBC at the genome-wide significance level with strong evidence, which is also verified by a published GWAS of in the Chinese population (7). Another variant in this family that exhibits the strongest association with PBC is DRB1*0801 (OR=3.11). This variant is significantly associated with PBC in Caucasian populations, yet its association in Japanese populations has been reported as non-significant (45). However, cumulative evidence grades this association as moderate, and it has not been replicated in large-scale studies such as GWAS. Studies have indicated that DRB1*0801 plays a crucial role in disrupting hepatic self-tolerance by binding and presenting charged pyruvate dehydrogenase E2 (PDC-E2) peptides (46). Additionally, this allele family is a major susceptibility factor for autoimmune hepatitis in white European and American populations (47–49), and is also associated with a reduced risk of primary sclerosing cholangitis (50).
In addition to the HLA locus, non-HLA genes also play an important role in the pathogenesis of PBC. Strong evidence from meta-analysis indicates that rs7574865 in STAT4 is a risk variant for PBC in Asian populations. Furthermore, published GWAS have also identified its association with PBC susceptibility in the British population (32). Rs7574865 located in the third intron of the STAT4 gene. Although this variant does not disrupt any transcription factor binding sites (51), it has been suggested to affect alternative splicing and is associated with STAT4 gene upregulation (52). This allele is also linked to an increased risk of rheumatoid arthritis (53) and ulcerative colitis (54). Furthermore, strong evidence from meta-analysis supports seven additional associations, four of which reached genome-wide significance (rs231725, rs231775, rs1544410, and rs9303277). Among these, rs231775 is a missense variant located in exon 1 of CTLA4, resulting in a threonine-to-alanine amino acid change (p.Thr17Ala). Functional evidence suggests that the A (Thr) allele increases CTLA-4 surface expression, which may modulate T-cell regulation and thus contribute to pathogenesis of autoimmune diseases such as PBC, although the possibility remains that it is a tag SNP in linkage disequilibrium with an untyped causal variant (55, 56). Another significant variant, rs231725, resides in the 3′-UTR of CTLA4 and has been reported to regulate mRNA stability and translational efficiency (57). This SNP reduces CTLA-4 expression and modulates CD4+ T-cell signaling thresholds, potentially contributing to PBC pathogenesis (58). These results are consistent with previous studies showing that CTLA-4 inhibitors (such as ipilimumab) enhance T-cell activation, potentially increasing the risk of autoimmune disorders (59, 60). In contrast, abatacept—a CTLA-4 agonist that inhibits T-cell activation—is currently under evaluation in a multicenter trial for UDCA-unresponsive PBC patients (NCT02078882) (61). In addition, phenome-wide analyses have suggested that PBC may share a common genetic architecture with certain autoimmune diseases, such as hypothyroidism/myxedema, hyperthyroidism/thyrotoxicosis, and inflammatory bowel disease. These results were consistent with clinical observations that PBC could coexist with other autoimmune diseases (62, 63), or hematological disorders (64). These findings could help develop strategies for the prevention and treatment of PBC and other related diseases.
However, our study had several limitations. First, our meta-analyses were conducted for variants with at least three independent datasets, which may have resulted in other important PBC-associated variants being overlooked (354 variants with only one dataset). However, we further performed meta-analysis for variants with two datasets and identified additional 11 loci significantly associated with PBC risk (Supplementary Table S13). Second, although functional variants have been identified, it is unknown whether they are causal variants, and further research is required to address this issue. Third, despite sensitivity analyses suggested robustness for most of the associations, a large heterogeneity was found in approximately 30% of the associations. To explore the potential sources of heterogeneity, we conducted subgroup analyses and meta-regression. The results suggested that ethnicity, diagnostic criteria for PBC, and genotyping methods may all contribute to the heterogeneity. Among these, ethnicity appeared to be a major factor, which may be partially explained by differences in allele frequencies across populations. For example, the HLA allele DQB1*0601 had an allele frequency of 0.109 in East Asians but only 0.013 in Europeans (Supplementary Table S14), consistent with its significant association in Asian populations only. Such differences underscore the importance of considering population background and methodological variations in genetic meta-analyses. Finally, although the HLA region demonstrates strong association with PBC risk, the low population incidence of PBC results in poor positive/negative predictive values for clinical screening. Nonetheless, this study identified disease-associated variants within this region and provides mechanistic insights for future investigation.
This comprehensive landmark study delivers the most extensive genetic dissection of PBC to date. Meta-analyses of candidate-gene association studies identified 44 risk-associated variants, comprising 30 variants within six HLA genes and 14 variants within 11 non-HLA genes. Among these variants, 17 across 10 genes supported by strong epidemiological evidence. Published GWAS have separately reported 115 significant variants associated with PBC. Notably, nine variants were identified by both approaches: the HLA alleles DQA1*0401, DQB1*0301, DQB1*0402, DQB1*0602, DRB1*08, DRB1*0803, DRB1*11, and DRB1*1101, along with the STAT4 variant rs7574865. Our findings not only consolidate the current understanding of PBC susceptibility but also uncover previously unappreciated genetic features underlying disease pathogenesis.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
Ethical approval was not required for the study involving humans in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements.
Author contributions
DG: Data curation, Funding acquisition, Writing – original draft. LL: Formal Analysis, Validation, Writing – original draft. LG: Formal Analysis, Writing – review & editing. YW: Data curation, Writing – review & editing. MZ: Formal Analysis, Project administration, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by Senior Medical Talents Program of Chongqing for Young and Middle-aged (No. YXGD202440). The funding agency had no role in study design, data collection, data management, data analysis, data interpretation, writing of the manuscript, or submission decision.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer WH declared a shared parent affiliation with the authors LG, YW to the handling editor at the time of review.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1600364/full#supplementary-material
Abbreviations
PBC, primary biliary cholangitis; GWASs, genome-wide association studies; SNP, single nucleotide polymorphism; OR, odds ratio; CI, confidence interval; FPRP, false positive report probability; GTEx, Genotype-Tissue Expression; FDR, false discovery rate; HLA, human leukocyte antigen.
References
1. Gerussi A, Cristoferi L, Carbone M, Asselta R, and Invernizzi P. The immunobiology of female predominance in primary biliary cholangitis. J Autoimmun. (2018) 95:124–32. doi: 10.1016/j.jaut.2018.10.015
2. Levy C, Manns M, and Hirschfield G. New treatment paradigms in primary biliary cholangitis. Clin Gastroenterol Hepatol. (2023) 21:2076–87. doi: 10.1016/j.cgh.2023.02.005
3. Younossi ZM, Bernstein D, Shiffman ML, Kwo P, Kim WR, Kowdley KV, et al. Diagnosis and management of primary biliary cholangitis. Am J Gastroenterol. (2019) 114:48–63. doi: 10.1038/s41395-018-0390-3
4. Hirschfield GM, Liu X, Xu C, Lu Y, Xie G, Lu Y, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med. (2009) 360:2544–55. doi: 10.1056/NEJMoa0810440
5. Li Y, Li Z, Chen R, Lian M, Wang H, Wei Y, et al. A regulatory variant at 19p13.3 is associated with primary biliary cholangitis risk and ARID3A expression. Nat Commun. (2023) 14:1732. doi: 10.1038/s41467-023-37213-5
6. Cordell HJ, Han Y, Mells GF, Li Y, Hirschfield GM, Greene CS, et al. International genome-wide meta-analysis identifies new primary biliary cirrhosis risk loci and targetab le pathogenic pathways. Nat Commun. (2015) 6:8019. doi: 10.1038/ncomms9019
7. Wang C, Zheng X, Tang R, Han C, Jiang Y, Wu J, et al. Fine mapping of the MHC region identifies major independent variants associated with Han Chinese primary biliary cholangitis. J Autoimmun. (2020) 107:102372. doi: 10.1016/j.jaut.2019.102372
8. Gervais O, Ueno K, Kawai Y, Hitomi Y, Aiba Y, Ueta M, et al. Regional heritability mapping identifies several novel loci (STAT4, ULK4, and KCNH5) for primary biliary cholangitis in the Japanese population. Eur J Hum Genet. (2021) 29:1282–91. doi: 10.1038/s41431-021-00854-5
9. Ioannidis JP, Ntzani EE, Trikalinos TA, and Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nat Genet. (2001) 29:306–9. doi: 10.1038/ng749
10. Ioannidis JP, Boffetta P, Little J, O’Brien TR, Uitterlinden AG, Vineis P, et al. Assessment of cumulative evidence on genetic associations: interim guidelines. Int J Epidemiol. (2008) 37:120–32. doi: 10.1093/ije/dym159
11. Khoury MJ, Bertram L, Boffetta P, Butterworth AS, Chanock SJ, Dolan SM, et al. Genome-wide association studies, field synopses, and the development of the knowledge base on genetic variation and human diseases. Am J Epidemiol. (2009) 170:269–79. doi: 10.1093/aje/kwp119
12. Bertram L, McQueen MB, Mullin K, Blacker D, and Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. (2007) 39:17–23. doi: 10.1038/ng1934
13. Zhang B, Beeghly-Fadiel A, Long J, and Zheng W. Genetic variants associated with breast-cancer risk: comprehensive research synopsis, meta-analysis, and epidemiological evidence. Lancet Oncol. (2011) 12:477–88. doi: 10.1016/S1470-2045(11)70076-6
14. Chatzinasiou F, Lill CM, Kypreou K, Stefanaki I, Nicolaou V, Spyrou G, et al. Comprehensive field synopsis and systematic meta-analyses of genetic association studies in cutaneous melanoma. J Natl Cancer Inst. (2011) 103:1227–35. doi: 10.1093/jnci/djr219
15. Ma X, Zhang B, and Zheng W. Genetic variants associated with colorectal cancer risk: comprehensive research synopsis, meta-analysis, and epidemiological evidence. Gut. (2014) 63:326–36. doi: 10.1136/gutjnl-2012-304121
16. Jawed R, Zhang M, Wang C, Yang SH, Jiang P, Wu Q, et al. Replication study and meta-analysis indicate a suggestive association of RUNX3 locus with primary biliary cholangitis. Immunogenetics. (2020) 72:467–74. doi: 10.1007/s00251-020-01192-4
17. Zhang L, Gao C, Liu C, Chen J, and Xu K. Association between STAT4 polymorphisms and risk of primary biliary cholangitis: a meta-analysis. Genes Genomics. (2018) 40:1101–9. doi: 10.1007/s13258-018-0717-x
18. Yang XC, Fujino M, Cai SJ, Li SW, Liu C, and Li XK. Genetic polymorphisms of cytotoxic T-lymphocyte antigen 4 in primary biliary cholangitis: A meta-analysis. J Immunol Res. (2017) 2017:5295164. doi: 10.1155/2017/5295164
19. Sagoo GS, Little J, and Higgins JP. Systematic reviews of genetic association studies. Hum Genome Epidemiol Network. PloS Med. (2009) 6:e28. doi: 10.1371/journal.pmed.1000028
20. Moher D, Liberati A, Tetzlaff J, Altman DG, and Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. (2009) 339:b2535. doi: 10.1136/bmj.b2535
21. Lau J, Ioannidis JP, and Schmid CH. Quantitative synthesis in systematic reviews. Ann Intern Med. (1997) 127:820–6. doi: 10.7326/0003-4819-127-9-199711010-00008
22. Begg CB and Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics. (1994) 50:1088–101. doi: 10.2307/2533446
23. Egger M, Davey Smith G, Schneider M, and Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ. (1997) 315:629–34. doi: 10.1136/bmj.315.7109.629
24. Zhang M, Tang M, Fang Y, Cui H, Chen S, Li J, et al. Cumulative evidence for relationships between multiple variants in the VTI1A and TCF7L2 genes and cancer incidence. Int J Cancer. (2018) 142:498–513. doi: 10.1002/ijc.31074
25. Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, and Rothman N. Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst. (2004) 96:434–42. doi: 10.1093/jnci/djh075
26. Ward LD and Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. (2012) 40:D930–4. doi: 10.1093/nar/gkr917
27. Watanabe K, Taskesen E, van Bochoven A, and Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. (2017) 8:1826. doi: 10.1038/s41467-017-01261-5
28. Zhang B, Kirov S, and Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. (2005) 33:W741–8. doi: 10.1093/nar/gki475
29. Canela-Xandri O, Rawlik K, and Tenesa A. An atlas of genetic associations in UK Biobank. Nat Genet. (2018) 50:1593–9. doi: 10.1038/s41588-018-0248-z
30. Qiu F, Tang R, Zuo X, Shi X, Wei Y, Zheng X, et al. A genome-wide association study identifies six novel risk loci for primary biliary cholangitis. Nat Commun. (2017) 8:14828. doi: 10.1038/ncomms14828
31. Cordell HJ, Fryett JJ, Ueno K, Darlay R, Aiba Y, Hitomi Y, et al. An international genome-wide meta-analysis of primary biliary cholangitis: Novel risk loci and candidate drugs. J Hepatol. (2021) 75:572–81. doi: 10.1016/j.jhep.2021.04.055
32. Liu JZ, Almarri MA, Gaffney DJ, Mells GF, Jostins L, Cordell HJ, et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat Genet. (2012) 44:1137–41. doi: 10.1038/ng.2395
33. Juran BD, Hirschfield GM, Invernizzi P, Atkinson EJ, Li Y, Xie G, et al. Immunochip analyses identify a novel risk locus for primary biliary cirrhosis at 13q14, multiple independent associations at four established risk loci and epistasis between 1p31 and 7q32 risk variants. Hum Mol Genet. (2012) 21:5209–21. doi: 10.1093/hmg/dds359
34. Mells GF, Floyd JA, Morley KI, Cordell HJ, Franklin CS, Shin SY, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet. (2011) 43:329–32. doi: 10.1038/ng.789
35. Liu X, Invernizzi P, Lu Y, Kosoy R, Lu Y, Bianchi I, et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat Genet. (2010) 42:658–60. doi: 10.1038/ng.627
36. Hirschfield GM, Liu X, Han Y, Gorlov IP, Lu Y, Xu C, et al. Variants at IRF5-TNPO3, 17q12–21 and MMEL1 are associated with primary biliary cirrhosis. Nat Genet. (2010) 42:655–7. doi: 10.1038/ng.631
37. Hitomi Y, Ueno K, Aiba Y, Nishida N, Kono M, Sugihara M, et al. A genome-wide association study identified PTPN2 as a population-specific susceptibility gene locus for primary biliary cholangitis. Hepatology. (2024) 80:776–90. doi: 10.1097/HEP.0000000000000894
38. Hitomi Y, Ueno K, Kawai Y, Nishida N, Kojima K, Kawashima M, et al. POGLUT1, the putative effector gene driven by rs2293370 in primary biliary cholangitis susceptibility locus chromosome 3q13.33. Sci Rep. (2019) 9:102. doi: 10.1038/s41598-018-36490-1
39. Kawashima M, Hitomi Y, Aiba Y, Nishida N, Kojima K, Kawai Y, et al. Genome-wide association studies identify PRKCB as a novel genetic susceptibility locus for primary biliary cholangitis in the Japanese population. Hum Mol Genet. (2017) 26:650–9. doi: 10.1093/hmg/ddw406
40. Nakamura M, Nishida N, Kawashima M, Aiba Y, Tanaka A, Yasunami M, et al. Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. Am J Hum Genet. (2012) 91:721–8. doi: 10.1016/j.ajhg.2012.08.010
41. Invernizzi P, Ransom M, Raychaudhuri S, Kosoy R, Lleo A, Shigeta R, et al. Classical HLA-DRB1 and DPB1 alleles account for HLA associations with primary biliary cirrhosis. Genes Immun. (2012) 13:461–8. doi: 10.1038/gene.2012.17
42. Chia R, Saez-Atienzar S, Murphy N, Chio A, Blauwendraat C, International Myasthenia Gravis Genomics C, et al. Identification of genetic risk loci and prioritization of genes and pathways for myasthenia gravis: a genome-wide association study. Proc Natl Acad Sci U.S.A. (2022) 119:e2108672119. doi: 10.1073/pnas.2108672119
43. Sazonovs A, Kennedy NA, Moutsianas L, Heap GA, Rice DL, Reppell M, et al. HLA-DQA1*05 carriage associated with development of anti-drug antibodies to infliximab and adalimumab in patients with crohn’s disease. Gastroenterology. (2020) 158:189–99. doi: 10.1053/j.gastro.2019.09.041
44. Li Y, Sun Y, Liu Y, Wang B, Li J, Wang H, et al. Genome-wide meta-analysis identifies susceptibility loci for autoimmune hepatitis type 1. Hepatology. (2022) 76:564–75. doi: 10.1002/hep.32417
45. Nakamura M, Yasunami M, Kondo H, Horie H, Aiba Y, Komori A, et al. Analysis of HLA-DRB1 polymorphisms in Japanese patients with primary biliary cirrhosis (PBC): The HLA-DRB1polymorphism determines the relative risk of antinuclear antibodies for disease progression in PBC. Hepatol Res. (2010) 40:494–504. doi: 10.1111/j.1872-034X.2010.00631.x
46. Chow IT, James EA, Gates TJ, Tan V, Moustakas AK, Papadopoulos GK, et al. Differential binding of pyruvate dehydrogenase complex-E2 epitopes by DRB1*08:01 and DRB1*11:01 Is predicted by their structural motifs and correlates with disease risk. J Immunol. (2013) 190:4516–24. doi: 10.4049/jimmunol.1202445
47. Littera R, Perra A, Miglianti M, Piras IS, Mocci S, Lai S, et al. The double-sided of human leukocyte antigen-G molecules in type 1 autoimmune hepatitis. Front Immunol. (2022) 13:1007647. doi: 10.3389/fimmu.2022.1007647
48. Lammert C, McKinnon EJ, Chalasani N, and Phillips EJ. Novel HLA class I alleles outside the extended DR3 haplotype are protective against autoimmune hepatitis. Clin Transl Gastroenterol. (2019) 10:e00032. doi: 10.14309/ctg.0000000000000032
49. Czaja AJ, Kruger M, Santrach PJ, Moore SB, and Manns MP. Genetic distinctions between types 1 and 2 autoimmune hepatitis. Am J Gastroenterol. (1997) 92:2197–200.
50. Liu JZ, Hov JR, Folseraas T, Ellinghaus E, Rushbrook SM, Doncheva NT, et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet. (2013) 45:670–5. doi: 10.1038/ng.2616
51. Zervou MI, Sidiropoulos P, Petraki E, Vazgiourakis V, Krasoudaki E, Raptopoulou A, et al. Association of a TRAF1 and a STAT4 gene polymorphism with increased risk for rheumatoid arthritis in a genetically homogeneous population. Hum Immunol. (2008) 69:567–71. doi: 10.1016/j.humimm.2008.06.006
52. Lamana A, Lopez-Santalla M, Castillo-Gonzalez R, Ortiz AM, Martin J, Garcia-Vicuna R, et al. The Minor Allele of rs7574865 in the STAT4 Gene Is Associated with Increased mRNA and Protein Expression. PloS One. (2015) 10:e0142683. doi: 10.1371/journal.pone.0142683
53. Gu E, Lu J, Xing D, Chen X, Xie H, Liang J, et al. Rs7574865 polymorphism in signal transducers and activators of transcription 4 gene and rheumatoid arthritis: an updated meta-analysis of 28 case-control comparisons. Int J Rheum Dis. (2015) 18:3–16. doi: 10.1111/1756-185X.12363
54. Liu QF, Li Y, Zhao QH, Wang ZY, Hu S, Yang CQ, et al. Association of STAT4 rs7574865 polymorphism with susceptibility to inflammatory bowel disease: A systematic review and meta-analysis. Clin Res Hepatol Gastroenterol. (2015) 39:627–36. doi: 10.1016/j.clinre.2015.04.002
55. Breunis WB, Tarazona-Santos E, Chen R, Kiley M, Rosenberg SA, and Chanock SJ. Influence of cytotoxic T lymphocyte-associated antigen 4 (CTLA4) common polymorphisms on outcome in treatment of melanoma patients with CTLA-4 blockade. J Immunother. (2008) 31:586–90. doi: 10.1097/CJI.0b013e31817fd8f3
56. Anjos S, Nguyen A, Ounissi-Benkalha H, Tessier MC, and Polychronakos C. A common autoimmunity predisposing signal peptide variant of the cytotoxic T-lymphocyte antigen 4 results in inefficient glycosylation of the susceptibility allele. J Biol Chem. (2002) 277:46478–86. doi: 10.1074/jbc.M206894200
57. Ligers A, Teleshova N, Masterman T, Huang WX, and Hillert J. CTLA-4 gene expression is influenced by promoter and exon 1 polymorphisms. Genes Immun. (2001) 2:145–52. doi: 10.1038/sj.gene.6363752
58. Juran BD, Atkinson EJ, Schlicht EM, Fridley BL, and Lazaridis KN. Primary biliary cirrhosis is associated with a genetic variant in the 3’ flanking region of the CTLA4 gene. Gastroenterology. (2008) 135:1200–6. doi: 10.1053/j.gastro.2008.06.077
59. Kong YC and Flynn JC. Opportunistic autoimmune disorders potentiated by immune-checkpoint inhibitors anti-CTLA-4 and anti-PD-1. Front Immunol. (2014) 5:206. doi: 10.3389/fimmu.2014.00206
60. Weber JS, Kahler KC, and Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. (2012) 30:2691–7. doi: 10.1200/JCO.2012.41.6750
61. Webb GJ, Siminovitch KA, and Hirschfield GM. The immunogenetics of primary biliary cirrhosis: A comprehensive review. J Autoimmun. (2015) 64:42–52. doi: 10.1016/j.jaut.2015.07.004
62. Liu Y, Han K, Liu C, Duan F, Cheng J, and Yang S. Clinical characteristics and prognosis of concomitant primary biliary cholangitis and autoimmune diseases: A retrospective study. Can J Gastroenterol Hepatol. (2021) 2021:5557814. doi: 10.1155/2021/5557814
63. Huang P, Hou Y, Zou Y, Ye X, Yu R, and Yang S. The causal effects of primary biliary cholangitis on thyroid dysfunction: A two-sample mendelian randomization study. Front Genet. (2021) 12:791778. doi: 10.3389/fgene.2021.791778
Keywords: primary biliary cholangitis, variants, meta-analysis, genetic architecture, cumulative evidence, functional annotation, phenome-wide analysis
Citation: Zhang M, Lyu L, Ge L, Wang Y and Gu D (2025) Genetic architecture of primary biliary cholangitis: strong evidence for HLA and non-HLA risk loci. Front. Immunol. 16:1600364. doi: 10.3389/fimmu.2025.1600364
Received: 26 March 2025; Accepted: 12 August 2025;
Published: 29 August 2025.
Edited by:
Seik-Soon Khor, Nanyang Technological University, SingaporeReviewed by:
Wei Gong, Shanghai Jiao Tong University, ChinaWentao Huang, Sichuan University, China
Brian Juran, Mayo Clinic, United States
Copyright © 2025 Zhang, Lyu, Ge, Wang and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongqing Gu, Z3Vkb25ncWluZzExQDEyNi5jb20=; YiZhou Wang, eWl6aG91X3dhbmcyMDIxQDE2My5jb20=
†These authors have contributed equally to this work