 Yaroslav Kaminskiy
Yaroslav Kaminskiy Vitaly Degtyarev
Vitaly Degtyarev Alexey Stepanov3
Alexey Stepanov3 Michael Maschan
Michael Maschan- 1Department of Oncology and Pathology, Karolinska Institutet, SciLifeLab, Solna, Sweden
- 2Dmitry Rogachev National Medical Center of Pediatric Hematology, Oncology, and Immunology, Moscow, Russia
- 3Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Moscow, Russia
Central nervous system (CNS) tumors are the second most common type of cancer in children and remain the leading cause of mortality in pediatric oncology. For patients with high-risk CNS tumors, standard treatments often prove ineffective, with survival rates being less than 10%. Hence, there is an urgent need to develop alternative treatment strategies for this patient population. Globally, numerous clinical trials are actively investigating a range of novel therapeutic approaches, from pharmacological and immunological therapies to physical modalities targeting the tumor. Among these emerging therapies, CAR T cell therapy has shown great promise, with the first objective clinical responses already reported. This review aims to evaluate the current landscape of CAR T cell therapy for pediatric CNS tumors, focusing on clinical efficacy, toxicity profiles of systemic and locoregional delivery, antigen heterogeneity, and key challenges in clinical implementation. We provide a comprehensive analysis of reported clinical trials, including not only CAR T cell studies but also investigations involving tumor-infiltrating lymphocytes (TILs), NK and lymphokine-activated killer (LAK) cells, offering a broader perspective on immunotherapeutic approaches for CNS malignancies.
Introduction
CAR T cells are autologous or allogeneic lymphocytes engineered to express a chimeric receptor that targets a specific antigen on the surface of tumor cells. Upon binding of the CAR T cell to the tumor antigen, it becomes activated leading to cytotoxicity, cytokine secretion and proliferation (1). The scope of CAR T cell therapy has significantly expanded, particularly in the treatment of hematologic malignancies. In parallel, numerous clinical trials are ongoing to assess the efficacy and safety of CAR T therapy in solid tumors. However, several challenges remain, including the immunosuppressive tumor microenvironment, CAR T cell trafficking limitations, tumor antigen heterogeneity, antigen loss, T-cell exhaustion and treatment-related toxicity (2–4).
Additional challenges arise from the blood-brain barrier (BBB), which restricts the delivery of many therapeutic agents from the bloodstream into the CNS (5). The CNS is now recognized as an immunologically dynamic system, with the BBB and microglia forming the first line of immune defense. Peripheral immune cells, such as lymphocytes and monocytes, are typically absent in the CNS under normal conditions but can infiltrate through the BBB during pathological processes and be detected in the cerebrospinal fluid (CSF) (6). Indeed, systemically administered CAR T cells have also been detected in the CSF, indicating their capacity to cross the BBB (7). The toxic effects of systemic CAR T cell administration, such as cytokine release syndrome (CRS) and neurotoxicity, are well-documented, yet are less understood in the context of locoregional delivery into the CNS (8).
In neuro-oncology, implantable systems are frequently utilized for drug delivery, allowing chemotherapeutic agents to be administered directly into the brain ventricles or the tumor site (9). Consequently, CAR T cells can be infused intravenously, intraventricularly, or locally into the postoperative tumor cavity (Figure 1, Table 1). Local drug administration enables bypassing the blood-brain barrier but is associated with certain risks. According to published data, the incidence of non-infectious complications with this delivery method reaches 33%, while infectious complications occur in 27% of cases (10). The most common non-infectious complications include intracranial hemorrhage, malfunction of the implanted catheter, and neurotoxic effects of administered drugs (11). Infectious complications are predominantly bacterial in origin, though cases of fungal and viral catheter-associated encephalitis have also been reported (12). Thus, strict adherence to catheter insertion and maintenance protocols is critical to minimizing the risk of these complications (13).
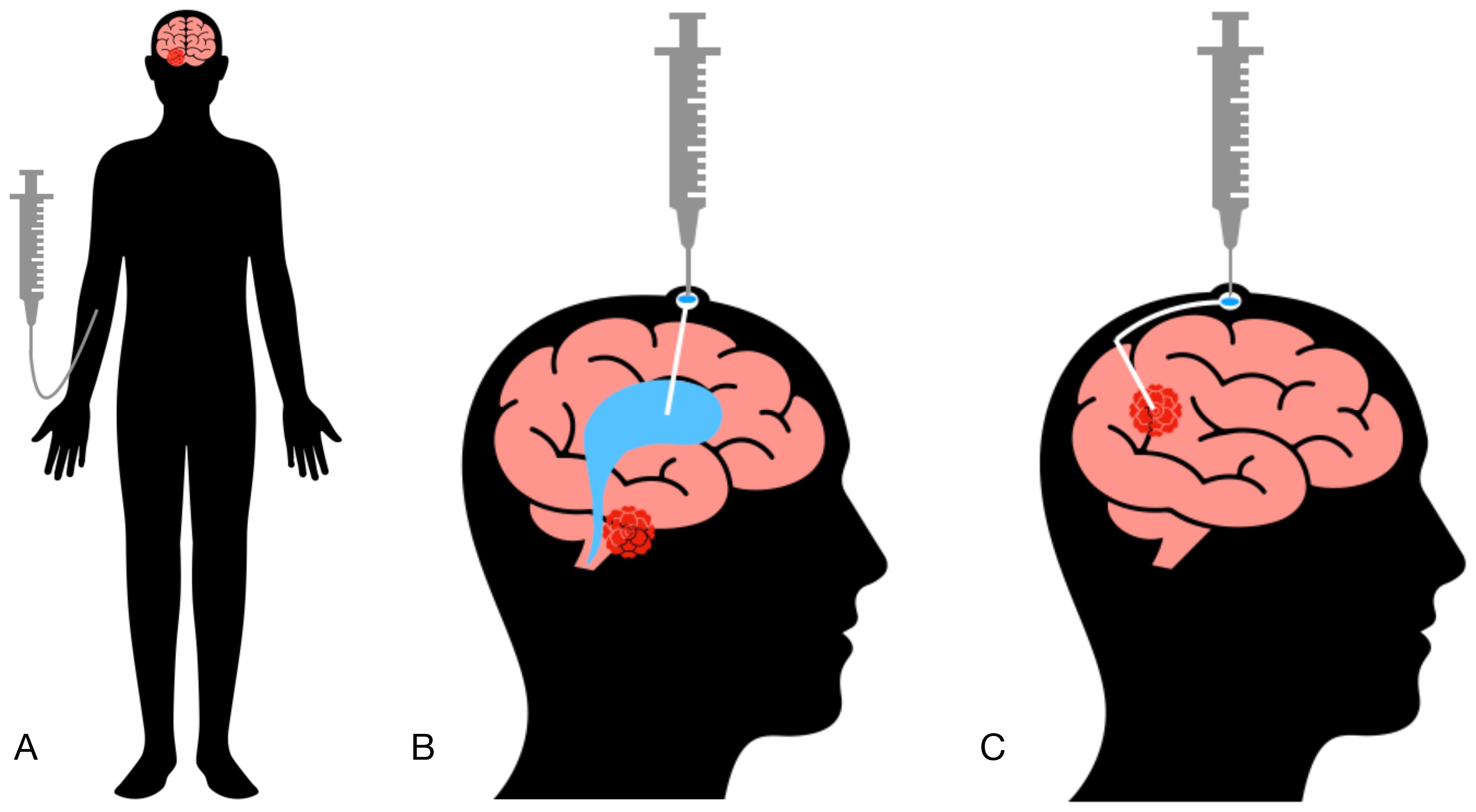
Figure 1. Methods of cellular product administration. (A) Intravenous administration (IV). (B) Intraventricular administration (ICV). (C) Intratumoral administration (IT).
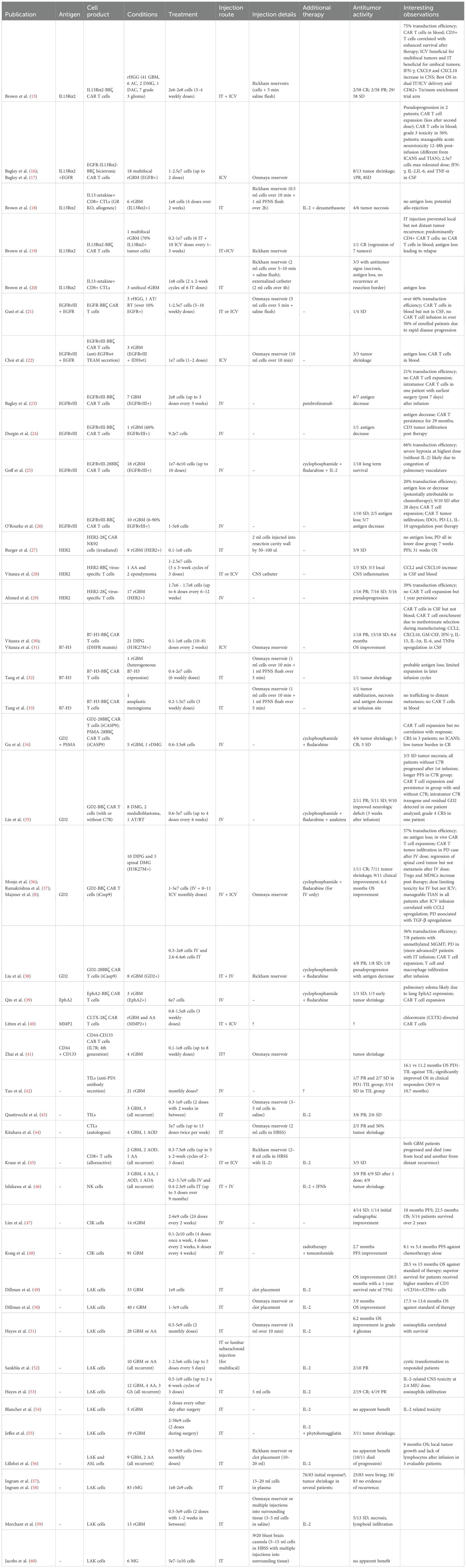
Table 1. Cellular therapy clinical trials in CNS tumors.
In the following sections, we summarized cellular therapy clinical trials in CNS tumors and try to identify common trends on several key aspects, including: initial antitumor response, availability of clinically validated antigens, post-infusion antigen escape, delivery route, toxicity profile, dosing, and the characteristics of the tumor microenvironment before and after treatment.
Cellular therapy clinical trials in CNS tumors
CNS tumors represent the most frequently targeted solid tumors in recent and ongoing cellular therapy trials (14). Table 1 provides a comprehensive summary of published clinical trials investigating cellular therapies for CNS tumors, with the majority focusing on high-grade gliomas (HGGs), particularly primary and recurrent glioblastoma (pGBM/rGBM), diffuse intrinsic pontine glioma (DIPG), and diffuse midline glioma (DMG). Here, we analyzed a total of 42 reported clinical trials, including 23 using CAR T/NK cells, 2 using tumor-infiltrating lymphocytes (TILs), 2 using unmodified CD8+ T cells, 1 using unmodified NK cells, and the remainder involving lymphokine-activated killer (LAK) cells.
These studies employed various approaches regarding the delivery method of the cellular product, the use of lymphodepleting chemotherapy, and different CAR T cell dosing strategies. The primary focus of all trials was to evaluate the safety and tolerability of the therapy, while the secondary objective was often to assess its efficacy.
Initial antitumor response
Until recently, adoptive cellular therapy has demonstrated limited clinical benefit in CNS tumors when assessed by progression-free and overall survival (PFS/OS). One of the most promising trials showed that only 4 out of 58 patients with recurrent HGG responded, with additional 29 out of 58 experiencing stable disease following a course of CAR T cell therapy (15). However, relatively limited clinical benefit may be misleading when assessing the full potential of adoptive cell therapy in CNS tumors because adoptively transferred cells do exhibit the capacity to shrink tumor lesions, even if this is not yet enough to generate a robust clinical response. In fact, most CAR T and TIL trials involving local delivery—whether intrathecal (IT) or intraventricular (ICV)—have demonstrated at least transient radiographic improvement or tumor shrinkage in more than 50% of patients, with the exception of those reported by Gust et al. (21) and Vitanza et al. (28, 30). Indeed, in the most recent H3K27M+ DMG trial, significant tumor shrinkage was observed in 7 out of 11 patients with clinical improvement observed in 9 out of 11 patients (36). In general, tumor shrinkage was observed across different trials independent of target antigen and therapeutic dose. These findings suggest that even at low doses, CAR T cells show discernible antitumor activity. Unfortunately, this activity is almost always transient, with patients ultimately progressing and succumbing to the disease. The key challenges in CNS tumors are likely poor CAR T cell persistence and rapid antigen escape, both of which are well-known mechanisms of tumor evasion from CAR T cell therapy (61, 62). In the case of CNS tumors, antigen escape is probably secondary to poor persistence, as it tends to occur more frequently in patients who have received multiple therapeutic doses.
Overall, while current antitumor responses in CNS tumors have been limited, the ability of CAR T cells to cause tumor shrinkage—even temporarily— is promising and highlights the importance of continued research into improving cell persistence and reducing antigen escape to achieve better clinical outcomes.
Several clinically validated antigens
Unlike many other solid tumors where tumor-specific antigens are scarce, gliomas have 6 clinically validated CAR T antigens that, when targeted, demonstrated tumor regression and were found to be safe when administered locally (Table 2) These targets include IL13Rα2 (63–69), EGFRvIII (70–72), EGFR (71, 73–77), HER2 (64, 74, 78–81), B7-H3 (82–87), and GD2 (86, 88–91) (Figure 2, Table 2).
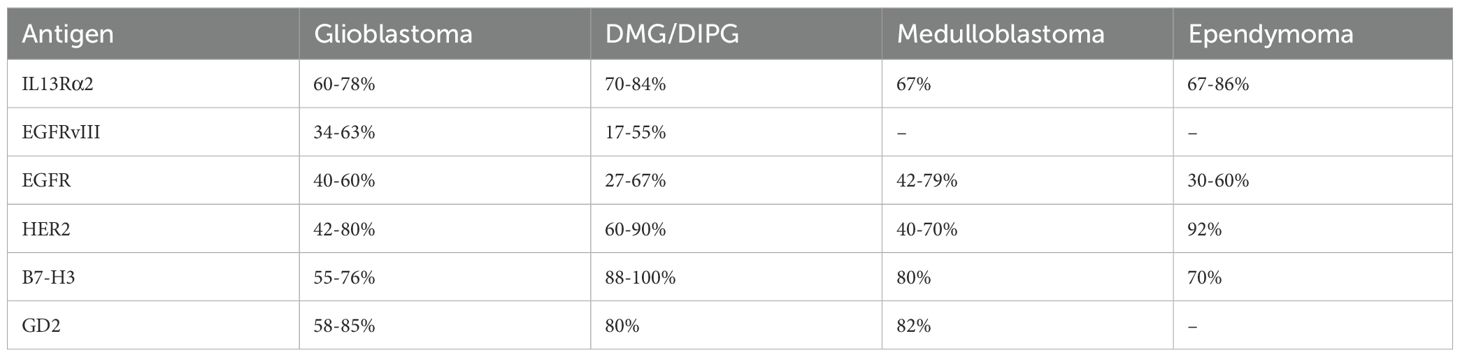
Table 2. Expression frequency of clinically validated targets for CAR T therapy in CNS tumors.
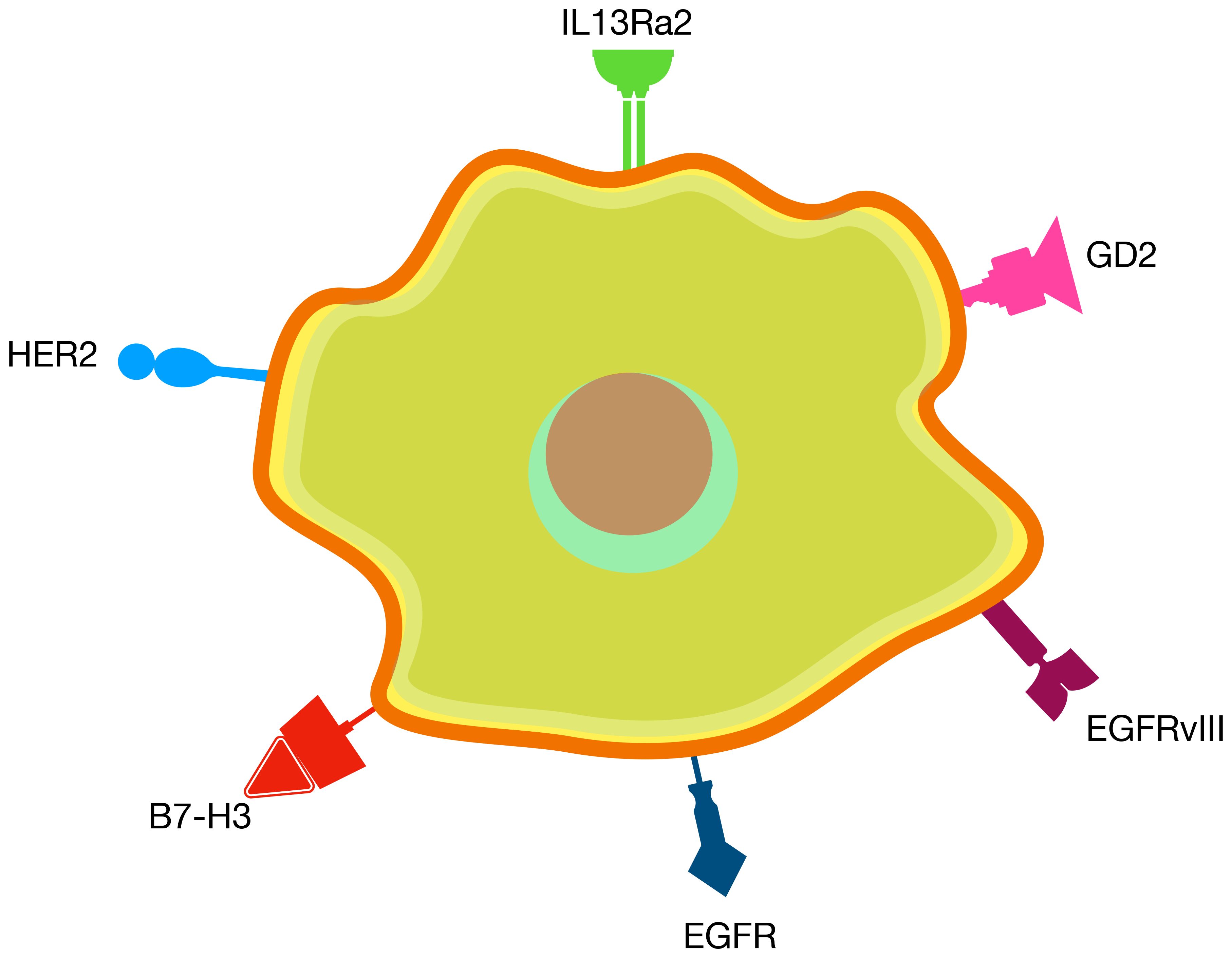
Figure 2. Clinically validated targets for CAR T therapy in CNS tumors.
IL13Rα2 is a transmembrane protein subunit of the interleukin-13 receptor, playing a crucial role in immune response regulation by binding to IL-13 (92). It is significantly overexpressed in tumor cells, with minimal expression in normal tissues. EGFR and its variant EGFRvIII belong to the epidermal growth factor receptor family, promoting signaling pathways that stimulate cell proliferation (93). EGFRvIII is a tumor-specific mutation of EGFR, resulting from an in-frame deletion of exons 2 to 7. EGFRvIII is commonly expressed in gliomas and other tumors but is absent in normal tissues, whereas EGFR is found in normal tissues but exhibits elevated expression levels in cancers (94). HER2, another member of the EGFR family, is overexpressed in various tumors, including breast cancer, and has limited expression in normal tissues (95). B7-H3 is an immune checkpoint molecule with immunosuppressive properties. While its expression in normal tissues is limited, B7-H3 is highly overexpressed in many adult and pediatric tumors, making it a promising target for immunotherapy (96). GD2, a disialoganglioside from the glycosphingolipid family, enhances tumor cell adhesion, invasiveness, and immunosuppression (97). While primarily expressed in the central and peripheral nervous systems under normal conditions, GD2 is overexpressed in numerous malignant diseases across both adults and children (98).
Although there is a recognized degree of antigenic heterogeneity in gliomas, with only a subset of tumor cells expressing any given antigen, at least one of the antigens from the list is expected to be expressed in most tumors (71). For instance, Barish et al. estimated that 93% of high grade glioma cases expressed at least one of IL13Rα2, HER2 or EGFR antigens (72% at least two) (99).This makes multitargeting as a clear path forward for enhancing therapeutic efficacy, with approaches targeting multiple pathways simultaneously already being evaluated in both clinical and preclinical trials (26, 92, 100–104).
EGFRvIII, EGFR, and HER2 are more frequently expressed in adult gliomas, while IL13Rα2, B7-H3, and GD2 appear to be suitable candidates for both adult and pediatric CNS tumors. In pediatric CNS tumors, B7-H3 and GD2 are the most frequently observed antigens, followed by IL13Rα2, with HER2 and EphA2 being less common (105). GD2 is expressed in approximately 80% of DIPG and medulloblastoma cases, making it a promising target for these tumors (88, 89). EphA2 is expressed in 60 - 90% of GBM cases and so far EphA2-CAR T cells have been tested with IV administration at relatively low dose (10e7 cells) (39, 106, 107). Although the treatment was considered safe, there were reports of pulmonary edema, likely caused by EphA2 expression in lung tissue. Local administration of EphA2 CAR T cells might solve this problem by limiting systemic migration.
New CAR T targets for glioma currently tested in preclinical and clinical studies include: CD44 (41), CD70 (108), CD97 (109), CD133 (41), CD147 (110), C.TNC (DIPG) (111), CSPG4 (GBM) (112), GPC2 (DIPG, medulloblastoma) (113), EphA3 (114), MMP2 (40, 115), PD-L1 (GBM) (116), PTGFRN (117), ROBO1 (GBM) (71, 118, 119).
A recent study demonstrated that a peptide derived from the most common mutation DMGs, H3.3K27M (found in 70% of cases), can be effectively targeted by T cells in the HLA-A*02:01 context (120). Combined with evidence that most high-grade gliomas express HLA class I complexes, this supports the potential of adoptive T cell therapy as a promising strategy for targeting gliomas with the H3.3K27M mutation (105). Other, less conventional targets include CMV antigens such as pp65 or IE1-72 (expressed in 67% of adult HGG), ligands for NK receptor NKG2D (71, 107, 121) and BTN2A1/BTN3A ligands for γ9δ2 T-cell receptor (122).
Altogether, the availability of several validated antigens for gliomas provides an excellent opportunity to address antigen escape through multitargeting CAR T strategies; however, the impact of tumor heterogeneity on treatment durability remains a critical unanswered question. Future studies should investigate how initial antigen expression levels and spatial differences across metastatic sites influence long-term responses, potentially through paired biopsy analyses.
Post-infusion antigen escape
Antigen escape is a well-recognized mechanism that contributes to CAR T therapy failure (61). Several studies have documented antigen loss following CAR T cell infusion in CNS tumors, including IL13Rα2, EGFRvIII, EGFR, and B7-H3 (19, 20, 22, 23, 32). It is worth noting that concurrent chemotherapy may also contribute to decreased antigen expression (at least for EGFR) and, therefore, decreased antigen levels in trials combining CAR T cells with chemotherapy might be misleading (26). Given the availability of several clinically validated antigens, antigen escape can be mitigated through the use of bi- or tri-specific CARs, or other multitarget CAR designs (22, 100, 123). Although there is a possibility that all validated antigens could eventually be lost, to date no study has demonstrated such an occurrence, making this a promising therapeutic strategy.
In summary, implementing multitarget CAR T cell approaches offers a feasible solution to combat antigen escape in CNS tumors. The promising preclinical and clinical findings to date suggest that combining multiple antigen targets could significantly enhance the therapeutic potential of CAR T cell therapy.
Benefits of local delivery
Evidence suggests that local (IT or ICV) delivery outperforms IV administration in terms of both antitumor response and toxicity profile. With a few exceptions, IV-infused cells have shown limited CNS and tumor infiltration, as well as reduced antitumor activity (Table 1). Additionally, systemic administration restricts the range of suitable target antigens and therapeutic doses. For example, EphA2—a promising glioma target—as well as HER2 are expressed in lung epithelium, posing a risk of off-target toxicity similar to that observed in early HER2 CAR T trials (39, 124). Lowering the therapeutic dose may reduce toxicity risks but could also compromise antitumor efficacy (29). Local delivery of HER2 CAR T cells, by contrast, was well tolerated and shows signs of antitumor activity (28, 125). Furthermore, local delivery does not require lymphodepletion, which is often a prerequisite for IV administration. The superiority of local delivery has also been demonstrated in multiple preclinical brain tumor models (123, 126–128).
It appears that ICV administration is more effective for multifocal disease, as IT administration resulted in very limited distal infiltration of CAR T cells (15, 16, 19). On the other hand, ICV is also suitable for both unifocal and multifocal diseases, especially given that up to 35% of glioblastomas become multifocal (129). Although there has been no head-to-head clinical comparison between IT and ICV delivery, several trials have demonstrated this even at low therapeutic doses (8, 15, 16, 19, 22, 36, 37). It appears that a combination of IT and ICV infusions could be advantageous in combating both local and distal recurrence, as dual administration is both feasible and safe, with early indications suggesting a clinical benefit (15).
Rickham and Ommaya reservoirs are widely used devices for local delivery of therapeutic agents into the cerebrospinal fluid (CSF) or brain tissue (130). Both have shown comparable efficacy in administering adoptive cell therapy in CNS tumors (Table 1). As a general practice, between 0.5 to 8 ml of cell products were delivered through these devices (IT or ICV) over about 10 minutes, followed by a flush with saline to ensure complete delivery. In certain cases, cell products were injected directly into the surrounding brain tissue near a resected tumor cavity.
Overall, the evidence strongly supports local (IT and/or ICV) delivery as an effective method for enhancing CAR T cell therapy efficacy in CNS tumors. Implementing these strategies could obviate the need for improving CAR T cell migration and brain infiltration by delivering the cells close to the tumor site. This not only achieves higher local concentrations of CAR T cells in the tumor microenvironment but also reduces off-target effects ultimately leading to better patient outcomes.
Favorable toxicity profile
Local (IT, ICV) infusions have shown excellent tolerability in most studies, with very few instances of grade 3 or higher adverse events (Table 1). With the exception of Bagley et al. (17), neither recent CAR T trials nor older LAK studies exhibited evident dose-limiting toxicity, even at the highest doses (10e10 cells per infusion). Typically, CAR T cell doses were in the range of 10e7 to 10e8 cells per infusion, which is lower compared to LAK infusions that ranged from 10e9 to 10e10 cells. Additionally, the proportion of CAR+ T cells in infusion products rarely reached 100%, suggesting that the effective dose might be even lower. These observations imply the potential for dose escalation to 10e10 CAR T cells or more per injection. Although direct comparison between CAR T cells and LAK cells dosing may not be appropriate—considering the differences in mechanisms of action—LAK cells were often injected with high doses of IL-2 and still showed a favorable safety profile, indicating that further dose escalation for CAR T cells could be possible. Of course, CAR T cell manufacturing capabilities must be considered, as expanding sufficient cells for multiple doses at 10e10 CAR T cells could be challenging. Interestingly, early studies with LAK cells also demonstrated that local delivery of high IL-2 doses is better tolerated than IV delivery. In summary, the favorable toxicity profile of local CAR T cell infusions provides an opportunity for dose escalation to enhance antitumor efficacy while maintaining safety. Further exploration of optimized dosing regimens could maximize therapeutic benefit.
Multiple dosing
CAR T cell expansion, commonly observed 1–3 weeks post-infusion and associated with antitumor responses in hematologic malignancies, was noted in several CNS trials but did not clearly correlate with clinical benefit (16, 26, 32, 36–38). Similarly, long-term persistence of CAR T cells (up to 29 months) was observed in two studies, but this was achieved with IV administration and without any obvious clinical response (24, 29). Hostile tumor microenvironment and limited tumor trafficking are likely barriers to achieving sustained CAR T cell expansion and persistence in CNS tumors. To overcome this, it is now recognized that multiple-dose infusion regimens are needed to achieve meaningful clinical responses. Recent CAR T trials have administered up to 18 doses, with weekly infusions being the most common strategy.
Implementing multiple dosing strategies appears essential for achieving clinical responses in CNS tumors. Given the challenges with CAR T cell persistence and expansion in the tumor microenvironment, frequent and repeated dosing could increase the likelihood of achieving durable tumor control and maximizing the therapeutic potential of adoptive cellular therapies.
Post-treatment tumor microenvironment
Following CAR T cell infusion, several soluble factors were observed to be upregulated in CSF, including IFN-γ (8, 15, 30, 31, 38), IL-6 (8, 21, 30, 38), CCL2 (28, 30), CXCL10 (15, 28, 30, 31), TGFβ (8, 26, 36), GM-CSF (31) and TARC (31). IL-6 and TGFβ are known to inhibit T cell immunity, and CCL2 can facilitate the recruitment of myeloid cells and Tregs potentially contributing to acquired resistance against CAR T therapy (131–133). Indeed, myeloid cell infiltration post-infusion was reported in two studies, while IDO1, PD-L1, and IL-10 upregulation in the TME was documented in one study, further supporting the emergence of compensatory inhibitory pathways post-CAR T therapy (26, 38).
Hence, post-infusion TME inhibitory mechanisms, such as upregulation of inhibitory cytokines and increased myeloid cell infiltration, emphasize the need to engineer CAR T cells resistant to these challenges.
Novel strategies for CAR T cell engineering
In general, tumor microenvironment of high-grade gliomas is considered immunologically ‘cold’ due to its limited effector T cell infiltration (134). These tumors are characterized by a significant presence of suppressive myeloid cells and Tregs, as well as harsh conditions such as hypoxia, nutrient deprivation, and the accumulation of immunosuppressive factors like extracellular adenosine, galectin-1, FGF, IDO, IL-6, IL-10, PD-L1, PGE2, and TGFβ (64, 71, 103, 107, 115).
Several strategies have been proposed to counteract the inhibitory effects of the tumor microenvironment and enhance CAR T cell survival and persistence in glioma. These include constitutively active IL-7 receptor (C7R) (35, 135), constitutively active IL-18 receptor (Zip18R) (111), transgenic IL-12 (136), IL-15 (71, 137), locally released anti-CD73 antibody (103), and PD-1 knock-out (128). Overexpression of CCR2, CCR4, CXCR1/2, and CXCR4 reportedly improved CAR T cell trafficking to brain tumors in preclinical studies (118, 138–140).CAR T cell pretreatment with metformin/rapamycin also enhanced mitochondrial respiration, leading to improved CAR T cell efficacy in glioma (141).
C7R is particularly promising strategy, as a clinical trial has demonstrated that CAR T cells expressing C7R maintained stable blood levels from 3 hours to 4 weeks post-infusion unlike typical CAR T cells, whose blood levels decline rapidly (35). Co-expression of C7R in CAR T cells boosts STAT5 signaling, enhancing cell survival and proliferation during repeated antigen challenges while avoiding antigen-independent expansion (135). Preclinical models demonstrated that CAR T cells with C7R eradicate glioblastoma even at doses insufficient for efficacy in standard CAR T cells and exhibit increased resistance to apoptotic signals via BCL2 upregulation and FAS/CASP8 downregulation.
Zip18R, a synthetic IL-18-based receptor, improves CAR T cell expansion and cytokine production by activating MyD88-dependent signaling pathways (111). In glioma and sarcoma models, CAR T cells engineered with Zip18R displayed significantly greater antitumor efficacy than unmodified CAR T cells, with enhanced persistence and elevated secretion of effector cytokines such as IFN-γ, IL-2, and TNF-α. Importantly, Zip18R did not induce autonomous growth in the absence of antigen, supporting its use in solid tumors where chronic antigen exposure induces T cell dysfunction.
Similarly, intratumoral delivery of IL-12 has been shown to potentiate CAR T cell activity in preclinical glioblastoma models (136). A single local dose of IL-12 reshaped the tumor microenvironment by reducing regulatory T cells and inducing proinflammatory macrophage and CD4+ T cell infiltration. Combined with EGFRvIII-specific CAR T cells, IL-12 treatment led to durable tumor control and long-term survival, while avoiding systemic toxicity, demonstrating the power of localized cytokine modulation to rescue CAR T cell function in glioma.
Transgenic expression of IL-15 in CAR T cells targeting GD2 significantly enhanced tumor control in an orthotopic glioblastoma model (137). These IL-15-expressing CAR T cells showed improved persistence, greater tumor infiltration, and higher cytokine production, including IL-2, IFN-γ, and TNF-α, compared to conventional CAR T cells. The therapy yielded a complete response rate of 50% in preclinical studies, highlighting IL-15 as a potent enhancer of CAR T cell function and durability in the glioma setting.
Another promising strategy involves local release of an anti-CD73 antibody fragment by CAR-engineered NK cells (103). This approach enables tumor-specific suppression of adenosine-mediated immunosuppression by inhibiting CD73 activity in the glioblastoma microenvironment. In preclinical models, CD73-targeting constructs reduced adenosine accumulation, restored NKG2D expression on NK cells, enhanced cytotoxicity, and improved NK cell infiltration. These effects were associated with reorganization of the tumor immune landscape and suppression of glioma progression, establishing CD73 as a critical immunometabolic checkpoint in glioma therapy.
Checkpoint blockade via PD-1 knock-out is another method to restore CAR T cell function in glioblastoma (128). Using CRISPR-Cas9, PD-1 was deleted in EGFRvIII-specific CAR T cells alongside TCR and B2M to generate universal donor CAR T cells resistant to PD-L1-mediated immunosuppression. In preclinical glioma models, these triple-edited CAR T cells exhibited enhanced antitumor activity, elevated Th1 cytokine secretion (IFN-γ, TNF-α), and improved persistence following repeated antigen stimulation. In vivo, intraventricular administration of PD-1-deficient CAR T cells significantly prolonged survival and produced durable cures in some mice, demonstrating the importance of PD-1 pathway disruption in overcoming the glioma immune barrier.
CAR T cell trafficking to the brain tumor site can be enhanced through chemokine receptor engineering. CXCR1- and CXCR2-expressing CAR T cells exploited tumor-secreted IL-8 for selective homing (140). These modified cells showed superior intratumoral infiltration and persistence, which correlated with improved tumor clearance and induction of memory responses across preclinical models of glioblastoma and other solid tumors (91). CAR NK cells engineered with CXCR4 demonstrated chemotaxis toward CXCL12-producing gliomas, resulting in enhanced trafficking, tumor infiltration, and antitumor effects (139). CXCR4-overexpressing cells achieved complete remission in several treated animals and prolonged survival compared to controls, highlighting this receptor’s critical role in navigating the glioma chemokine landscape (111). Similarly, expression of CCR2 and CCR4 on CAR T cells could increase chemotaxis to CCL2/4/5/17/22-secreting gliomas (118).
Finally, metabolic reprogramming with metformin and rapamycin (Met+Rap) pretreatment significantly improved CAR T cell function under glioma-specific hypoxic conditions (141). This combination activated AMPK and suppressed mTOR signaling, inducing mitochondrial biogenesis via PGC-1α upregulation. As a result, CAR T cells exhibited enhanced oxidative metabolism, greater spare respiratory capacity, and resistance to exhaustion. In vivo, Met+Rap–pretreated CAR T cells achieved better tumor infiltration, persistent cytotoxicity, and extended survival in intracerebral glioma models.
Together, these multifaceted engineering strategies illustrate the growing sophistication of CAR T cell design, emphasizing the importance of combinatorial approaches to overcome the distinct immunological barriers posed by glioma.
Conclusion
High-risk CNS tumors are resistant to standard treatments such as radiation and chemotherapy, and they remain the leading cause of mortality in pediatric oncology. As a result, there is a critical need for new therapeutic approaches to improve patients’ quality of life and achieve disease remission. Recent clinical studies of CAR T-cell therapy in CNS tumors have shown promising results, though there is still room for improvement.
A detailed analysis of these clinical studies has highlighted the initial antitumor activity of CAR T-cell therapy and its low toxicity. The presence of several clinically validated antigens for CNS tumor therapy opens opportunities for multi-target strategies to prevent antigen escape. Local delivery methods, such as intraventricular and intratumoral administration, show advantages over systemic delivery, ensuring higher concentrations of cells in the tumor while minimizing toxicity. Increasing the dosage and improving the persistence of CAR T-cells could also significantly enhance treatment efficacy. Therefore, with proper modifications, CAR T-cell therapy has the potential to substantially improve the survival rates of patients with high-risk CNS tumors.
Author contributions
YK: Conceptualization, Writing – original draft, Writing – review & editing. MM: Funding acquisition, Writing – review & editing. VD: Visualization, Writing – original draft, Writing – review & editing. AS: Conceptualization, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fimmu.2025.1689398.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. ChatGPT to proofread the language and grammar.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AA, Anaplastic astrocytoma; AC, astrocytoma; AOA, anaplastic oligoastrocytoma; AOD, anaplastic oligodendroglioma; ASL, autologous stimulated lymphocyte; AT/RT, Atypical teratoid/rhabdoid tumor; BBB, blood-brain barrier; DIPG, diffuse intrinsic pontine glioma; DMG, diffuse midline glioma; CSF, cerebrospinal fluid; CR, complete response; CRS, cytokine release syndrome; GS, gliosarcoma; KO, knock-out; (p/r)GBM, (primary/recurrent) glioblastoma multiforme; (r)HGG, (recurrent) high-grade glioma; ICV, intraventricular; IT, intratumoral; IV, intravenous; LAK, lymphokine-activated killer; (r)MG, (recurrent) malignant glioma; OS, overall survival; PFS, progression-free survival; PD, progressive disease; PR, partial response; SD, stable disease; TAM, tumor-associated macrophage; TIL, tumor-infiltrating lymphocyte; wt, wild type.
References
1. Sharma A, De Leon G, Porter AB, Grill MF, Rosenthal AC, Brown CE, et al. CAR-T cell therapy in neuro-oncology: applications and toxicity. Neuroimmunol Neuroinflamm. (2018). doi: 10.20517/2347-8659.2018.51
2. Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, et al. Current progress in CAR-T cell therapy for solid tumors. Int J Biol Sci. (2019) 15:2548–60. doi: 10.7150/ijbs.34213
3. Titov A, Kaminskiy Y, Ganeeva I, Zmievskaya E, Valiullina A, Rakhmatullina A, et al. Knowns and unknowns about CAR-T cell dysfunction. Cancers (Basel). (2022) 14. doi: 10.3390/cancers14041078
4. Ma J, Chen CC, and Li M. Macrophages/microglia in the glioblastoma tumor microenvironment. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22115775
5. Abbott NJ. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J Inherit Metab Dis. (2013) 36:437–49. doi: 10.1007/s10545-013-9608-0
6. Jackson CM, Choi J, and Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol. (2019) 20:1100–9. doi: 10.1038/s41590-019-0433-y
7. Berger SC, Fehse B, Akyüz N, Geffken M, Wolschke C, Janson D, et al. Molecular monitoring of T-cell kinetics and migration in severe neurotoxicity after real-world CD19-specific chimeric antigen receptor T cell therapy. Haematologica. (2023) 108:444–56.
8. Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. (2022) 603:934–41.
9. Kramer K, Smith M, and Souweidane MM. Safety profile of long-term intraventricular access devices in pediatric patients receiving radioimmunotherapy for central nervous system Malignancies. Pediatr Blood Cancer. (2014) 61:1590–2. doi: 10.1002/pbc.25080
10. Cohen-Pfeffer JL, Gururangan S, Lester T, Lim DA, Shaywitz AJ, Westphal M, et al. Intracerebroventricular delivery as a safe, long-term route of drug administration. Pediatr Neurol. (2017) 67:23–35. doi: 10.1016/j.pediatrneurol.2016.10.022
11. Peyrl A, Chocholous M, Azizi AA, Czech T, Dorfer C, Mitteregger D, et al. Safety of Ommaya reservoirs in children with brain tumors: A 20-year experience with 5472 intraventricular drug administrations in 98 patients. J Neurooncol. (2014) :120:139–45. doi: 10.1007/s11060-014-1531-1
12. Brouwer AJ, Groenendaal F, Van Den Hoogen A, Verboon-Maciolek M, Hanlo P, Rademaker KJ, et al. Incidence of infections of ventricular reservoirs in the treatment of post-haemorrhagic ventricular dilatation: A retrospective study (1992-2003). Arch Dis Child Fetal Neonatal Ed. (2007) 92. doi: 10.1136/adc.2006.096339
13. Slavc I, Cohen-Pfeffer JL, Gururangan S, Krauser J, Lim DA, Maldaun M, et al. Best practices for the use of intracerebroventricular drug delivery devices. Mol Genet Metab. (2018) 124:184–8. doi: 10.1016/j.ymgme.2018.05.003
14. Ivica NA and Young CM. Tracking the car-t revolution: Analysis of clinical trials of car-t and tcr-t therapies for the treatment of cancer (1997–2020). Healthc (Switzerland). (2021) 9. doi: 10.20944/preprints202107.0198.v1
15. Brown CE, Hibbard JC, Alizadeh D, Blanchard MS, Natri HM, Wang D, et al. Locoregional delivery of IL-13Rα2-targeting CAR-T cells in recurrent high-grade glioma: a phase 1 trial. Nat Med. (2024) 30:1001–12. doi: 10.1038/s41591-024-02875-1
16. Bagley SJ, Logun M, Fraietta JA, Wang X, Desai AS, Bagley LJ, et al. Intrathecal bivalent CAR T cells targeting EGFR and IL13Rα2 in recurrent glioblastoma: phase 1 trial interim results. Nat Med. (2024) 30:1320–9. doi: 10.1038/s41591-024-02893-z
17. Bagley SJ, Desai AS, Fraietta JA, Silverbush D, Chafamo D, Freeburg NF, et al. Intracerebroventricular bivalent CAR T cells targeting EGFR and IL-13Rα2 in recurrent glioblastoma: a phase 1 trial. Nat Med. (2025). doi: 10.1038/s41591-025-03745-0
18. Brown CE, Rodriguez A, Palmer J, Ostberg JR, Naranjo A, Wagner JR, et al. Off-the-shelf, steroid-resistant, IL13Rα2-specific CAR T cells for treatment of glioblastoma. Neuro Oncol. (2022) 24:1318–30. doi: 10.1093/neuonc/noac024
19. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
20. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. (2015) 21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428
21. Gust J, Cole BL, Ronsley R, Wilson AL, Seidel K, Wendler J, et al. Locoregional infusion of EGFR806-CAR T cells for recurrent or refractory pediatric CNS tumors: results of the completed brainChild02 phase 1 clinical trial. Neuro Oncol. (2025). doi: 10.1093/neuonc/noaf064
22. Choi BD, Gerstner ER, Frigault MJ, Leick MB, Mount CW, Balaj L, et al. Intraventricular CARv3-TEAM-E T cells in recurrent glioblastoma. N Engl J Med. (2024) 390:1290–8. doi: 10.1056/NEJMoa2314390
23. Bagley SJ, Binder ZA, Lamrani L, Marinari E, Desai AS, Nasrallah MP, et al. Repeated peripheral infusions of anti-EGFRvIII CAR T cells in combination with pembrolizumab show no efficacy in glioblastoma: a phase 1 trial. Nat Cancer. (2024) 5:517–31. doi: 10.1038/s43018-023-00709-6
24. Durgin JS, Henderson FJ, Nasrallah MP, Mohan S, Wang S, Lacey SF, et al. Case report: prolonged survival following EGFRvIII CAR T cell treatment for recurrent glioblastoma. Front Oncol. (2021) 11:669071. doi: 10.3389/fonc.2021.669071
25. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor–transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. (2019) 42. doi: 10.1097/CJI.0000000000000260
26. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9. doi: 10.1126/scitranslmed.aaa0984
27. Burger MC, Forster MT, Romanski A, Straßheimer F, Macas J, Zeiner PS, et al. Intracranial injection of natural killer cells engineered with a HER2-targeted chimeric antigen receptor in patients with recurrent glioblastoma. Neuro Oncol. (2023) 25:2058–71. doi: 10.1093/neuonc/noad087
28. Vitanza NA, Johnson AJ, Wilson AL, Brown C, Yokoyama JK, Künkele A, et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: an interim analysis. Nat Med. (2021) 27:1544–52. doi: 10.1038/s41591-021-01404-8
29. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol. (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
30. Vitanza NA, Wilson AL, Huang W, Seidel K, Brown C, Gustafson JA, et al. Intraventricular B7-H3 CAR T cells for diffuse intrinsic pontine glioma: preliminary first-in-human bioactivity and safety. Cancer Discov. (2023) 13:114–31. doi: 10.1158/2159-8290.CD-22-0750
31. Vitanza NA, Ronsley R, Choe M, Seidel K, Huang W, Rawlings-Rhea SD, et al. Intracerebroventricular B7-H3-targeting CAR T cells for diffuse intrinsic pontine glioma: a phase 1 trial. Nat Med. (2025). doi: 10.1038/s41591-024-03451-3
32. Tang X, Wang Y, Huang J, Zhang Z, Liu F, Xu J, et al. Administration of B7-H3 targeted chimeric antigen receptor-T cells induce regression of glioblastoma. Signal Transduct Target Ther. (2021) 6:125. doi: 10.1038/s41392-021-00505-7
33. Tang X, Liu F, Liu Z, Cao Y, Zhang Z, Wang Y, et al. Bioactivity and safety of B7-H3-targeted chimeric antigen receptor T cells against anaplastic meningioma. Clin Trans Immunol Australia. (2020) 9:e1137. doi: 10.1002/cti2.1137
34. Gu J, Li J, Xu Y, Zhang G, Xie J, Jia R, et al. Preliminary exploration of PSMA CAR-T combined with GD2 CAR-T for the treatment of refractory/relapsed gliomas. J Transl Med. (2025) 23:591. doi: 10.1186/s12967-025-06523-1
35. Lin FY, Stuckert A, Tat C, White M, Ruggieri L, Zhang H, et al. Phase I trial of GD2.CART cells augmented with constitutive interleukin-7 receptor for treatment of high-grade pediatric CNS tumors. J Clin Oncol. (2024) 42:2769–79. doi: 10.1200/JCO.23.02019
36. Monje M, Mahdi J, Majzner R, Yeom KW, Schultz LM, Richards RM, et al. Intravenous and intracranial GD2-CAR T cells for H3K27M+ diffuse midline gliomas. Nature. (2024). doi: 10.1038/s41586-024-08171-9
37. Ramakrishna S, Good Z, Desai M, Zamler D, Mancusi R, Mahdi J, et al. Abstract 959: Immune signatures of GD2 CAR T cell activity in H3K27M+ diffuse midline glioma patients. Cancer Res. (2023) 83:959. doi: 10.1158/1538-7445.AM2023-959
38. Liu Z, Zhou J, Yang X, Liu Y, Zou C, Lv W, et al. Safety and antitumor activity of GD2-Specific 4SCAR-T cells in patients with glioblastoma. Mol Cancer. (2023) 22:3. doi: 10.1186/s12943-022-01711-9
39. Lin Q, Ba T, Ho J, Chen D, Cheng Y, Wang L, et al. First-in-human trial of ephA2-redirected CAR T-cells in patients with recurrent glioblastoma: A preliminary report of three cases at the starting dose. Front Oncol. (2021) 11:694941. doi: 10.3389/fonc.2021.694941
40. Litten JB, Ramakrishnan A, Astrow SH, Harrison C, Aliki A, and Badie B. Phase 1b multicenter study to evaluate CHM 1101 in patients with recurrent or progressive glioblastoma. J Clin Oncol. (2023) 41:TPS2086–TPS2086. doi: 10.1200/JCO.2023.41.16_suppl.TPS2086
41. Zhai Y, Li G, Jiang T, and Zhang W. P12.13.B Locoregional Bi-Specific Car-T cells targeting Cd44 And Cd133 in recurrent glioblastoma: The first in-human clinical trial results. Neuro Oncol. (2024) 26:v69–9.
42. Yao Y, Chen D, Tang C, Ji C, Li Z, and Qian Q. Safety, efficacy, and biomarker analysis of response to engineered tumor-infiltrating lymphocytes secreting anti-PD-1 antibody in recurrent glioblastoma: An open-label, two-arms, phase 1 study. J Clin Oncol. (2023) 41:2042. doi: 10.1200/JCO.2023.41.16_suppl.2042
43. Quattrocchi KB, Miller CH, Cush S, Bernard SA, Dull ST, Smith M, et al. Pilot study of local autologous tumor infiltrating lymphocytes for the treatment of recurrent Malignant gliomas. J Neurooncol. (1999) 45:141–57. doi: 10.1023/A:1006293606710
44. Kitahara T, Watanabe O, Yamaura A, Makino H, Watanabe T, Suzuki G, et al. Establishment of interleukin 2 dependent cytotoxic T lymphocyte cell line specific for autologous brain tumor and its intracranial administration for therapy of the tumor. J Neurooncol. (1987) 4:329–36. doi: 10.1007/BF00195603
45. Kruse CA, Cepeda L, Owens B, Johnson SD, Stears J, and Lillehei KO. Treatment of recurrent glioma with intracavitary alloreactive cytotoxic T lymphocytes and interleukin-2. Cancer Immunol Immunother. (1997) 45:77–87. doi: 10.1007/s002620050405
46. Ishikawa E, Tsuboi K, Saijo K, Harada H, Takano S, Nose T, et al. Autologous natural killer cell therapy for human recurrent Malignant glioma. Anticancer Res. (2004) 24:1861–71.
47. Lim J, Park Y, Ahn JW, Sim J, Kang SJ, Hwang S, et al. Autologous adoptive immune-cell therapy elicited a durable response with enhanced immune reaction signatures in patients with recurrent glioblastoma: An open label, phase I/IIa trial. PLoS One. (2021) 16:e0247293. doi: 10.1371/journal.pone.0247293
48. Kong DS, Nam DH, Kang SH, Lee JW, Chang JH, Kim JH, et al. Phase III randomized trial of autologous cytokine-induced killer cell immunotherapy for newly diagnosed glioblastoma in Korea. Oncotarget. (2017) 8:7003–13.
49. Dillman RO, Duma CM, Ellis RA, Cornforth AN, Schiltz PM, Sharp SL, et al. Intralesional lymphokine-activated killer cells as adjuvant therapy for primary glioblastoma. J Immunother. (2009) 32. doi: 10.1097/CJI.0b013e3181b2910f
50. Dillman RO, Duma CM, Schiltz PM, DePriest C, Ellis RA, Okamoto K, et al. Intracavitary placement of autologous lymphokine-activated killer (LAK) cells after resection of recurrent glioblastoma. J Immunother. (2004) 27:398–404. doi: 10.1097/00002371-200409000-00009
51. Hayes RL, Arbit E, Odaimi M, Pannullo S, Scheff R, Kravchinskiy D, et al. Adoptive cellular immunotherapy for the treatment of Malignant gliomas. Crit Rev Oncol Hematol. (2001) 39:31–42. doi: 10.1016/S1040-8428(01)00122-6
52. Sankhla SK, Nadkarni JS, and Bhagwati SN. Adoptive immunotherapy using lymphokine-activated killer (LAK) cells and interleukin-2 for recurrent Malignant primary brain tumors. J Neurooncol. (1996) 27:133–40. doi: 10.1007/BF00177476
53. Hayes RL, Koslow M, Hiesiger EM, Hymes KB, Hochster HS, Moore EJ, et al. Improved long term survival after intracavitary interleukin-2 and lymphokine-activated killer cells for adults with recurrent Malignant glioma. Cancer. (1995) 76:840–52.
54. Blancher A, Roubinet F, Grancher AS, Tremoulet M, Bonaté A, Delisle MB, et al. Local immunotherapy of recurrent glioblastoma multiforme by intracerebral perfusion of interleukin-2 and LAK cells. Eur Cytokine Netw. (1993) 4:331–41.
55. Jeffes EW 3rd, Beamer YB, Jacques S, Coss JS, Nep RL, Beckman M, et al. Therapy of recurrent high-grade gliomas with surgery, autologous mitogen-activated IL-2-stimulated (MAK) killer lymphocytes, and rIL-2: II. Correlation of survival with MAK cell tumor necrosis factor production in vitro. Lymphokine Cytokine Res. (1991) 10:89–94.
56. Lillehei KO, Mitchell DH, Johnson SD, McCleary EL, and Kruse CA. Long-term follow-up of patients with recurrent Malignant gliomas treated with adjuvant adoptive immunotherapy. Neurosurgery. (1991) 28:16–23.
57. Ingram M, Shelden CH, Jacques S, Skillen RG, Bradley WG, Techy GB, et al. Preliminary clinical trial of immunotherapy for Malignant glioma. J Biol Response Mod. (1987) 6:489–98.
58. Ingram M, Buckwalter JG, Jacques DB, Freshwater DB, Abts RM, Techy GB, et al. Immunotherapy for recurrent Malignant glioma: an interim report on survival. Neurol Res. (1990) 12:265–73. doi: 10.1080/01616412.1990.11739955
59. Merchant RE, Grant AJ, Merchant LH, and Young HF. Adoptive immunotherapy for recurrent glioblastoma multiforme using lymphokine activated killer cells and recombinant interleukin-2. Cancer. (1988) 62:665–71.
60. Jacobs SK, Wilson DJ, Melin G, Parham CW, Holcomb B, Kornblith PL, et al. Interleukin-2 and lymphokine activated killer (LAK) cells in the treatment of Malignant glioma: clinical and experimental studies. Neurol Res. (1986) 8:81–7. Available online at: https://www.tandfonline.com/doi/abs/10.1080/01616412.1986.11739735.
61. Majzner RG and Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. (2018) 8:1219–26. doi: 10.1158/2159-8290.CD-18-0442
62. Albelda SM. CAR T cell therapy for patients with solid tumours: key lessons to learn and unlearn. Nat Rev Clin Oncol. (2024) 21:47–66. doi: 10.1038/s41571-023-00832-4
63. Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, and Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol. (2014) 16:1304–12. doi: 10.1093/neuonc/nou045
64. Salvato I and Marchini A. Immunotherapeutic strategies for the treatment of glioblastoma: current challenges and future perspectives. Cancers. (2024) 16. doi: 10.3390/cancers16071276
65. Bhardwaj R, Suzuki A, Leland P, Joshi BH, and Puri RK. Identification of a novel role of IL-13Rα2 in human Glioblastoma multiforme: Interleukin-13 mediates signal transduction through AP-1 pathway. J Transl Med. (2018).
66. Knudson KM, Hwang SJ, McCann MS, Joshi BH, Husain SR, and Puri RK. Recent advances in IL-13Rα2-directed cancer immunotherapy. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.878365
67. Berlow NE, Svalina MN, Quist MJ, Settelmeyer TP, Zherebitskiy V, Kogiso M, et al. IL-13 receptors as possible therapeutic targets in diffuse intrinsic pontine glioma. PLoS One. (2018). doi: 10.1371/journal.pone.0193565
68. Zeng J, Zhang J, Yang YZ, Wang F, Jiang H, Chen HD, et al. IL13RA2 is overexpressed in Malignant gliomas and related to clinical outcome of patients. Am J Transl Res. (2020) 12:4702–14.
69. Donovan LK, Delaidelli A, Joseph SK, Bielamowicz K, Fousek K, Holgado BL, et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat Med. (2020) 26:720–31. doi: 10.1038/s41591-020-0827-2
70. Tang OY, Tian L, Yoder T, Xu R, Kulikovskaya I, Gupta M, et al. PD1 expression in EGFRvIII-directed CAR T cell infusion product for glioblastoma is associated with clinical response. Front Immunol. (2022) 13:872756. doi: 10.3389/fimmu.2022.872756
71. Aggarwal P, Luo W, Pehlivan KC, Hoang H, Rajappa P, Cripe TP, et al. Pediatric versus adult high grade glioma: Immunotherapeutic and genomic considerations. Front Immunol. (2022) 13:1038096. doi: 10.3389/fimmu.2022.1038096
72. Li G, Mitra SS, Monje M, Henrich KN, Bangs CD, Nitta RT, et al. Expression of epidermal growth factor variant III (EGFRvIII) in pediatric diffuse intrinsic pontine gliomas. J Neurooncol. (2012) 108:395–402. doi: 10.1007/s11060-012-0842-3
73. Oprita A, Baloi SC, Staicu GA, Alexandru O, Tache DE, Danoiu S, et al. Updated insights on EGFR signaling pathways in glioma. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22020587
74. Korshunov A, Golanov A, Ozerov S, and Sycheva R. Prognostic value of tumor-associated antigens immunoreactivity and apoptosis in medulloblastomas. An analysis of 73 cases. Brain Tumor Pathol. (1999) 16:37–44. doi: 10.1007/BF02478900
75. Krüger C, Paplomatas P, Kreuter N, Hellwig M, Eckhardt A, Conze C, et al. Epidermal growth factor receptor specific immunotherapy for SHH medulloblastoma tested in an in vitro blood-brain barrier-model. Cancer Treat Res Commun. (2025). doi: 10.1016/j.ctarc.2025.100950
76. Friedrich C, von Bueren AO, Kolevatova L, Bernreuther C, Grob T, Sepulveda-Falla D, et al. Epidermal growth factor receptor overexpression is common and not correlated to gene copy number in ependymoma. Child’s Nerv Syst. (2016) 32:281–90. doi: 10.1007/s00381-015-2981-2
77. Sievers P, Sill M, Schrimpf D, Stichel D, Reuss DE, Sturm D, et al. A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro Oncol. (2021) 23:34–43. doi: 10.1093/neuonc/noaa251
78. Ramezani M, Siami S, Rezaei M, Khazaei S, and Sadeghi M. An immunohistochemical study of HER2 expression in primary brain tumors. Biomed (Taipei). (2020) 10:21–7. doi: 10.37796/2211-8039.1001
79. Donovan LK, Delaidelli A, Joseph SK, Bielamowicz K, Fousek K, Holgado BL, et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat Med. (2020) 26:720–31. doi: 10.1038/s41591-020-0827-2
80. Nellan A, Rota C, Majzner R, Lester-McCully CM, Griesinger AM, Mulcahy Levy JM, et al. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J Immunother Cancer. (2018) 6:30. Available online at: http://jitc.bmj.com/content/6/1/30.abstract.
81. Wang SS, Davenport AJ, Iliopoulos M, Hughes-Parry HE, Watson KA, Arcucci V, et al. HER2 chimeric antigen receptor T cell immunotherapy is an effective treatment for diffuse intrinsic pontine glioma. Neurooncol Adv. (2023) 5:vdad024. doi: 10.1093/noajnl/vdad024
82. Tang X, Zhao S, Zhang Y, Wang Y, Zhang Z, Yang M, et al. B7-H3 as a novel CAR-T therapeutic target for glioblastoma. Mol Ther Oncol. (2019) :14:279–87. doi: 10.1016/j.omto.2019.07.002
83. Tang X, Zhao S, Zhang Y, Wang Y, Zhang Z, Yang M, et al. B7-H3 as a novel CAR-T therapeutic target for glioblastoma. Mol Ther Oncol. (2019) :14:279–87. doi: 10.1016/j.omto.2019.07.002
84. Nehama D, Di Ianni N, Musio S, Du H, Patané M, Pollo B, et al. B7-H3-redirected chimeric antigen receptor T cells target glioblastoma and neurospheres. EBioMedicine. (2019) :47:33–43. doi: 10.1016/j.ebiom.2019.08.030
85. Zhou Z, Luther N, Ibrahim GM, Hawkins C, Vibhakar R, Handler MH, et al. B7-H3, a potential therapeutic target, is expressed in diffuse intrinsic pontine glioma. J Neurooncol. (2013) 111:257–64. doi: 10.1007/s11060-012-1021-2
86. Haydar D, Houke H, Chiang J, Yi Z, Odé Z, Caldwell K, et al. Cell-surface antigen profiling of pediatric brain tumors: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro Oncol. (2021) 23:999–1011. doi: 10.1093/neuonc/noaa278
87. Kranendonk MEG, Hoogendijk R, Lammers JAS, van der Lugt J, Tolboom N, van Mastrigt E, et al. Do we need B7-H3 immunohistochemistry for the inclusion of children with high-grade central nervous system tumors in clinical trials targeting B7-H3? Neuro Oncol. (2025). doi: 10.1093/neuonc/noaf095
88. Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, Haile S, et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat Med. (2018) 24:572–9. doi: 10.1038/s41591-018-0006-x
89. Ciccone R, Quintarelli C, Camera A, Pezzella M, Caruso S, Manni S, et al. GD2-targeting CAR T-cell therapy for patients with GD2+ Medulloblastoma. Clin Cancer Res. (2024) 30:2545–57. doi: 10.1158/1078-0432.CCR-23-1880
90. Gargett T, Ebert LM, Truong NTH, Kollis PM, Sedivakova K, Yu W, et al. GD2-targeting CAR-T cells enhanced by transgenic IL-15 expression are an effective and clinically feasible therapy for glioblastoma. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2022-005187
91. Prapa M, Chiavelli C, Golinelli G, Grisendi G, Bestagno M, Di Tinco R, et al. GD2 CAR T cells against human glioblastoma. NPJ Precis Oncol. (2021) 5. doi: 10.1038/s41698-021-00233-9
92. Jaén M, Martín-Regalado Á, Bartolomé RA, Robles J, and Casal JI. Interleukin 13 receptor alpha 2 (IL13Rα2): Expression, signaling pathways and therapeutic applications in cancer. Biochim Biophys Acta Rev Cancer. (2022) 1877:188802.
93. Faulkner C, Palmer A, Williams H, Wragg C, Haynes HR, White P, et al. EGFR and EGFRvIII analysis in glioblastoma as therapeutic biomarkers. Br J Neurosurg. (2015) 29:23–9. doi: 10.3109/02688697.2014.950631
95. Iqbal N and Iqbal N. Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol Biol Int. (2014) 2014:852748. doi: 10.1155/2014/852748
96. Zhou WT and Jin WL. B7-H3/CD276: an emerging cancer immunotherapy. Front Immunol. (2021) 12:701006. doi: 10.3389/fimmu.2021.701006
97. Machy P, Mortier E, and Birklé S. Biology of GD2 ganglioside: implications for cancer immunotherapy. Front Pharmacol. (2023) 14:1249929. doi: 10.3389/fphar.2023.1249929
98. Nazha B, Inal C, and Owonikoko TK. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front Oncol. (2020) 10:1000. doi: 10.3389/fonc.2020.01000
99. Barish ME, Weng L, Awabdeh D, Zhai Y, Starr R, D’Apuzzo M, et al. Spatial organization of heterogeneous immunotherapy target antigen expression in high-grade glioma. Neoplasia. (2022) 30:100801.
100. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. (2018) 20:506–18. doi: 10.1093/neuonc/nox182
101. Okada H, Low KL, Kohanbash G, McDonald HA, Hamilton RL, and Pollack IF. Expression of glioma-associated antigens in pediatric brain stem and non-brain stem gliomas. J Neurooncol. (2008) 88:245–50. doi: 10.1007/s11060-008-9566-9
102. Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. (2021) 13. doi: 10.1126/scitranslmed.abe7378
103. Wang J, Toregrosa-Allen S, Elzey BD, Utturkar S, Lanman NA, Bernal-Crespo V, et al. Multispecific targeting of glioblastoma with tumor microenvironment-responsive multifunctional engineered NK cells. Proc Natl Acad Sci U S A. (2021) 118. doi: 10.1073/pnas.2107507118
104. Hegde M, Corder A, Chow KKH, Mukherjee M, Ashoori A, Kew Y, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. (2013) 21:2087–101. doi: 10.1038/mt.2013.185
105. Haydar D, Houke H, Chiang J, Yi Z, Odé Z, Caldwell K, et al. Cell-surface antigen profiling of pediatric brain tumors: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro Oncol. (2021) 23:999–1011. doi: 10.1093/neuonc/noaa278
106. Wykosky J, Gibo DM, Stanton C, and Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. (2005) 3:541–51. doi: 10.1158/1541-7786.MCR-05-0056
107. Montoya M, Gallus M, Phyu S, Haegelin J, de Groot J, and Okada H. A roadmap of CAR-T-cell therapy in glioblastoma: challenges and future perspectives. Cells. (2024) 13. doi: 10.3390/cells13090726
108. Seyfrid M, Maich WT, Shaikh VM, Tatari N, Upreti D, Piyasena D, et al. CD70 as an actionable immunotherapeutic target in recurrent glioblastoma and its microenvironment. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2021-003289
109. Zhou S, Lin W, Jin X, Niu R, Yuan Z, Chai T, et al. CD97 maintains tumorigenicity of glioblastoma stem cells via mTORC2 signaling and is targeted by CAR Th9 cells. Cell Rep Med. (2024), 101844. doi: 10.1016/j.xcrm.2024.101844
110. Park S, Maus MV, and Choi BD. CAR-T cell therapy for the treatment of adult high-grade gliomas. NPJ Precis Oncol. (2024) 8:279. doi: 10.1038/s41698-024-00753-0
111. Wickman E, Lange S, Wagner J, Ibanez J, Tian L, Lu M, et al. IL-18R supported CAR T cells targeting oncofetal tenascin C for the immunotherapy of pediatric sarcoma and brain tumors. J Immunother Cancer. (2024) 12. doi: 10.1136/jitc-2024-009743
112. Pellegatta S, Savoldo B, Di Ianni N, Corbetta C, Chen Y, Patané M, et al. Constitutive and TNFα-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci Transl Med. (2018) 10:eaao2731. doi: 10.1126/scitranslmed.aao2731
113. Day ZI, Roberts-Thomson S, Nouri YJ, Dalton NS, Wang SS, Davenport A, et al. Defining the extracellular matrix for targeted immunotherapy in adult and pediatric brain cancer. NPJ Precis Oncol. (2025) 9. doi: 10.1038/s41698-025-00956-z
114. Lertsumitkul L, Iliopoulos M, Wang SS, McArthur SJ, Ebert LM, Davenport AJ, et al. EphA3-targeted chimeric antigen receptor T cells are effective in glioma and generate curative memory T cell responses. J Immunother Cancer. (2024) 12:e009486. doi: 10.1136/jitc-2024-009486
115. Wang D, Starr R, Chang WC, Aguilar B, Alizadeh D, Wright SL, et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci Transl Med. (2020) 12. doi: 10.1126/scitranslmed.aaw2672
116. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wöhrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. (2015) 17:1064–75. doi: 10.1093/neuonc/nou307
117. Kuroda H, Kijima N, Tachi T, Ikeda S, Murakami K, Nakagawa T, et al. Prostaglandin F2 receptor negative regulator as a potential target for chimeric antigen receptor-T cell therapy for glioblastoma. Cancer Immunol Immunother. (2025) 74:136. doi: 10.1007/s00262-025-03979-4
118. Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, and Brown CE. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol Rev. (2019) 290:60–84. doi: 10.1111/imr.12773
119. Chokshi CR, Shaikh MV, Brakel B, Rossotti MA, Tieu D, Maich W, et al. Targeting axonal guidance dependencies in glioblastoma with ROBO1 CAR T cells. Nat Med. (2024) 30:2936–46. doi: 10.1038/s41591-024-03138-9
120. Chheda ZS, Kohanbash G, Okada K, Jahan N, Sidney J, Pecoraro M, et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med. (2018) 215:141–57. doi: 10.1084/jem.20171046
121. Jiu DH, Sun B, Yang D, Xu H, Zhu J, Wei J, et al. Eradication of medulloblastoma by NKG2D-specific CAR T-cells. J Clin Oncol. (2024) 38:2522.
122. Vazaios K, Hernández López P, Aarts-Riemens T, Daudeij A, Kemp V, Hoeben RC, et al. Unusual partners: γδ-TCR-based T cell therapy in combination with oncolytic virus treatment for diffuse midline gliomas. Int J Mol Sci. (2025) 26. doi: 10.3390/ijms26052167
123. Donovan LK, Delaidelli A, Joseph SK, Bielamowicz K, Fousek K, Holgado BL, et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat Med. (2020) 26:720–31. doi: 10.1038/s41591-020-0827-2
124. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, and Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
125. Burger MC, Forster MT, Romanski A, Straßheimer F, Macas J, Zeiner PS, et al. Intracranial injection of natural killer cells engineered with a HER2-targeted chimeric antigen receptor in patients with recurrent glioblastoma. Neuro Oncol. (2023) 25:2058–71. doi: 10.1093/neuonc/noad087
126. Brown CE, Aguilar B, Starr R, Yang X, Chang WC, Weng L, et al. Optimization of IL13R&x3b1;2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol Ther. (2018) 26:31–44. doi: 10.1016/j.ymthe.2017.10.002
127. Nellan A, Rota C, Majzner R, Lester-McCully CM, Griesinger AM, Mulcahy Levy JM, et al. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J Immunother Cancer. (2018) 6:30. doi: 10.1186/s40425-018-0340-z
128. Choi BD, Yu X, Castano AP, Darr H, Henderson DB, Bouffard AA, et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J Immunother Cancer. (2019) 7:304. doi: 10.1186/s40425-019-0806-7
129. Baro V, Cerretti G, Todoverto M, Della Puppa A, Chioffi F, Volpin F, et al. Newly diagnosed multifocal GBM: A monocentric experience and literature review. Curr Oncol. (2022) 29:3472–88. doi: 10.3390/curroncol29050280
130. Buonerba C, Di Lorenzo G, Marinelli A, Federico P, Palmieri G, Imbimbo M, et al. A comprehensive outlook on intracerebral therapy of Malignant gliomas. Crit Rev Oncol Hematol. (2011) 80:54–68. doi: 10.1016/j.critrevonc.2010.09.001
131. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. (2016) 76:5671–82. doi: 10.1158/0008-5472.CAN-16-0144
133. Lamano JB, Lamano JB, Li YD, DiDomenico JD, Choy W, Veliceasa D, et al. Glioblastoma-derived IL6 induces immunosuppressive peripheral myeloid cell PD-L1 and promotes tumor growth. Clin Cancer Res. (2019) 25:3643–57. doi: 10.1158/1078-0432.CCR-18-2402
134. Frederico SC, Hancock JC, Brettschneider EES, Ratnam NM, Gilbert MR, and Terabe M. Making a cold tumor hot: the role of vaccines in the treatment of glioblastoma. Front Oncol. (2021) 11:672508. doi: 10.3389/fonc.2021.672508
135. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. (2017) 7:1238–47. doi: 10.1158/2159-8290.CD-17-0538
136. Agliardi G, Liuzzi AR, Hotblack A, De Feo D, Núñez N, Stowe CL, et al. Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nat Commun. (2021) 12:444. doi: 10.1038/s41467-020-20599-x
137. Gargett T, Ebert LM, Truong NTH, Kollis PM, Sedivakova K, Yu W, et al. GD2-targeting CAR-T cells enhanced by transgenic IL-15 expression are an effective and clinically feasible therapy for glioblastoma. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2022-005187
138. Goutnik M, Iakovidis A, Still MEH, Moor RSF, Melnick K, Yan S, et al. Advancements in chimeric antigen receptor-expressing T-cell therapy for glioblastoma multiforme: Literature review and future directions. Neurooncol Adv. (2024) 6:vdae025. doi: 10.1093/noajnl/vdae025
139. Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1α-secreting glioblastoma HHS Public Access. J Immunother. (2015) 38:197–210. Available online at: http://clinicaltrials.gov/ct2/showNCT01454596.
140. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. (2019) 10(1):4016.
141. Hatae R, Kyewalabye K, Yamamichi A, Chen T, Phyu S, Chuntova P, et al. Enhancing CAR-T cell metabolism to overcome hypoxic conditions in the brain tumor microenvironment. JCI Insight. (2024) 9:e177141. Available online at: https://pmc.ncbi.nlm.nih.gov/articles/PMC11128202/.
Keywords: CNS tumors, glioma, DMG, DIPG, glioblastoma, CAR T, CAR T solid tumors, brain cancer
Citation: Kaminskiy Y, Degtyarev V, Stepanov A and Maschan M (2025) CAR T cell therapy for central nervous system solid tumors: current progress and future directions. Front. Immunol. 16:1600403. doi: 10.3389/fimmu.2025.1600403
Received: 26 March 2025; Accepted: 23 July 2025;
Published: 15 August 2025; Corrected: 23 September 2025.
Edited by:
Philipp C. Rommel, University of Pennsylvania, United StatesReviewed by:
David Akhavan, University of Kansas Medical Center, United StatesDipendra Khadka, NADIANBIO Co. Ltd., Republic of Korea
Copyright © 2025 Kaminskiy, Degtyarev, Stepanov and Maschan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Maschan, bW1hc2NoYW5AeWFuZGV4LnJ1