 Dekai Wei
Dekai Wei Fujun Li3†
Fujun Li3† Jibing Chen
Jibing Chen- 1Graduate School, Guangxi University of Chinese Medicine, Nanning, China
- 2Ruikang Hospital affiliated to Guangxi University of Chinese Medicine, Nanning, China
- 3Department of Anesthesiology, Sichuan Clinical Research Center for Cancer, Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, Affiliated Hospital of University of Electronic Science and Technology of China, Chengdu, China
Ischemic stroke, one of the cerebrovascular diseases with the highest global disability and mortality rates, is characterized by secondary neuroinflammatory injury during its pathological progression, which remains a major challenge in clinical management. Although reperfusion therapies, including intravenous thrombolysis (IVT) and endovascular mechanical thrombectomy (EVT), have significantly improved acute-phase blood flow restoration, the neuroinflammatory cascade triggered post-reperfusion exacerbates neuronal damage. Key mechanisms include microglial overactivation and blood-brain barrier (BBB) disruption, ultimately leading to poor neurological outcomes. Recent studies have increasingly revealed the pivotal roles of exosomes and non-coding RNAs (ncRNAs) in post-ischemic stroke pathology. Specifically, exosomes, as natural nanocarriers, demonstrate targeted regulation of immune-inflammatory cascades in cerebral ischemia/reperfusion injury due to their low immunogenicity and efficient delivery capacity; complementarily, ncRNAs participate in pathophysiological processes including apoptosis, angiogenesis, inflammatory responses, and hypoxic stress through epigenetic regulatory mechanisms. This review systematically deciphers the regulatory networks of exosomes and ncRNAs in post-stroke pathological progression and neural repair, with particular focus on their molecular mechanisms in modulating specific inflammatory components. Building on current advances, we emphasize that while affirming the clinical value of reperfusion therapy, it is imperative to integrate evidence-based secondary prevention systems to address stroke management challenges. Notably, exosome-derived ncRNAs have emerged as promising diagnostic/therapeutic candidates: they not only precisely regulate inflammation-related pathways but also provide a novel strategy for developing targeted delivery systems. With deepening mechanistic understanding, exosome-based therapies are expected to revolutionize therapeutic paradigms for neuroinflammatory disorders, paving new avenues for precise intervention and functional recovery in stroke patients.
1 Introduction
Stroke, the second most common fatal disease worldwide and the leading cause of permanent disability in adults, encompasses hemorrhagic stroke (HS) and ischemic stroke (IS). IS, which accounts for 87% of cases, has its core pathological mechanism involving acute arterial occlusion-induced cerebral ischemia and hypoxia (1–4). Current clinical management of acute IS emphasizes time window-dependent vascular recanalization, predominantly relying on IVT, EVT, or their combined application. Neurons are highly sensitive to oxygen-glucose deprivation; therefore, timely restoration of blood perfusion is critical to mitigate irreversible damage. Although thrombolytic agents such as alteplase (requiring administration within 4.5 hours post-onset) and modified tenecteplase can dissolve thrombi, delayed symptom recognition and prolonged medical transfers prevent most patients from benefiting from thrombolytic therapy (5–7). In contrast, EVT extends the therapeutic window to 24 hours through physical thrombus removal, further improving outcomes in patients with large vessel occlusion (8–11). Nevertheless, fewer than 3% of global stroke patients receive EVT, with a 460-fold disparity in technical accessibility between nations, underscoring the critical imbalance in healthcare resource allocation (12).
Notably, even with successful reperfusion, nearly half of patients experience poor prognosis due to reperfusion injury, while current antiplatelet therapies merely prevent stroke recurrence without repairing existing neural damage (13–16). Three major bottlenecks constrain contemporary IS treatment: (1) Strict time windows exclude most patients from intervention opportunities; (2) The BBB blocks delivery of 95% of neurorestorative drugs; (3) Effective interventions for reperfusion injury remain elusive (17, 18). These challenges demand innovative therapeutic strategies. Exosomes, with their unique BBB-penetrating capacity, multi-target regulatory properties, and nanoscale carrier advantages, provide a groundbreaking solution to simultaneously overcome drug delivery barriers, promote neural repair, and modulate inflammatory injury.
Extracellular vesicles (EVs) constitute a heterogeneous population of membrane-bound particles released by both prokaryotic and eukaryotic cells (19). EV classification depends on biogenesis pathways: exosomes (40–100 nm) formed through multivesicular body exocytosis, ectosomes (50-1,000 nm) generated via plasma membrane budding, and apoptotic bodies (800-5,000 nm) shed during programmed cell death (20, 21). As the most therapeutically promising EV subset, exosomes inherit lipid bilayers from parental cell membranes and carry diverse functional biomolecules (proteins, lipids, nucleic acids), executing essential intercellular signaling missions (22, 23). In cerebral ischemia pathology, exosomal neuroprotection stems from bioactive cargo-microenvironment interactions. Protein and nucleic acid components directly activate neurogenesis pathways, suppress microglial hyperactivation, and promote angiogenesis with BBB and white matter stabilization. Additionally, inflammatory mediators (e.g., TNF-α, IL-6) from ischemic lesions enhance exosomal chemotaxis via receptor-mediated trans-barrier migration, increasing their accumulation at injury sites to coordinate reparative and immunomodulatory responses (24–28). Compared to synthetic nanocarriers, exosomes exhibit superior targeting precision, biocompatibility, and low immunogenicity (29, 30). These intrinsic advantages position them as ideal vectors to overcome neurorestorative drug delivery barriers, offering novel technological pathways for personalized stroke therapeutics.
ncRNAs represent a class of regulatory RNA molecules lacking protein-coding potential, primarily comprising microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) (31). Accumulating evidence demonstrates that ncRNAs directly or indirectly participate in diverse pathophysiological processes of IS, critically regulating apoptosis, vascular remodeling, inflammatory cascades, and hypoxic stress responses (32–36). The interactions among ncRNAs reveal sophisticated molecular crosstalk. Specifically, circRNAs and lncRNAs function as molecular sponges to sequester miRNAs, thereby attenuating miRNA-mRNA binding capacity and reversing miRNA-mediated negative regulation of target mRNAs. This post-transcriptional regulatory paradigm, termed the competing endogenous RNA (ceRNA) mechanism, establishes multi-layered gene expression control networks (37–39). For instance, the circRNA-miRNA-mRNA axis orchestrates fundamental cellular processes such as immune homeostasis maintenance, cell cycle progression, and stress adaptation through miRNA activity modulation (40, 41). Such networked regulatory mechanisms open new avenues for deciphering the molecular pathogenesis of IS and developing precision-targeted therapeutic strategies.
To systematically elucidate the roles of exosomes and ncRNAs in post-ischemic stroke neuroinflammation, we searched the PubMed database from January 2022 to March 2025 using the following query: “exosomes” OR “ncRNAs” OR “circRNA” OR “lncRNA” OR “miRNA” AND “stroke” AND “neuroinflammation” OR “microglia” OR “inflammation”. A three-step screening process was performed:
1. Initial search: 1,221 records identified
2. Title/abstract screening: 534 potentially eligible studies retained
3. Full-text assessment: 46 studies ultimately included based on the criteria below
1.1 Inclusion and exclusion criteria
1.1.1 Inclusion criteria
1. Disease models: Rodent ischemic stroke models or OGD/R/LPS-induced neuroinflammatory cell models
2. Study subjects: Exosomes or ncRNAs (circRNA, lncRNA, miRNA)
3. Study type: Original mechanistic/interventional studies (excluding non-empirical literature)
4. Publication requirement: Full-text articles in English
5. Data completeness: For animal studies, explicit reporting of both neuroinflammatory markers (e.g., TNF-α, IL-1β) and infarct volume data; for cell studies, explicit reporting of neuroinflammatory markers (e.g., TNF-α, IL-1β, IL-6) and cell viability/cytotoxicity data (e.g., CCK-8, LDH release, apoptosis rate)
6. Literature prioritization: For overlapping studies, the most recent or data-complete version was retained
1.1.2 Exclusion criteria
1. Methodological flaws: Absence of control groups, undisclosed animal strains or cell sources
2. Irrelevant publication types: Reviews, conference abstracts, commentaries, or studies with inaccessible full texts
3. Non-ischemic stroke models: Intracerebral hemorrhage, Alzheimer’s disease, etc.
2 Pathogenesis of IS and neuroinflammatory mechanisms
2.1 Pathophysiological basis of IS
IS occurs when cerebral blood vessels supplying the brain become obstructed. Occlusion caused by locally formed clots within cerebral vessels is termed cerebral thrombosis, which is closely associated with lipid deposits in atherosclerotic arteries. Alternatively, clots originating from extracranial sites, termed cerebral embolism, may travel through the bloodstream to cerebral vessels, with atrial fibrillation being the primary etiology (42). Due to the structural characteristics of cerebral vasculature and its hierarchical perfusion network, vascular occlusion typically manifests as focal neurological deficits confined to specific brain regions supplied by the affected vessel. The central core region experiences near-complete cessation of cerebral blood flow (CBF), resulting in rapid cellular necrosis within minutes. Surrounding this core lies a transitional zone termed the “penumbra,” where reduced perfusion permits temporary cellular survival. This salvageable tissue constitutes the primary target for neuroprotective interventions (43). Following temporary CBF deprivation in the penumbra, reperfusion restores oxygen and nutrient supply but paradoxically induces significant secondary tissue damage through ischemia-reperfusion (I/R) injury (42, 44). This dual-phase injury mechanism is not unique to cerebral ischemia but has been well-documented in other organ systems, including myocardial infarction and acute kidney injury (45–48).
2.2 Pathological characteristics of neuroinflammation and its multidimensional regulatory networks
Reperfusion injury constitutes a multifactorial process involving sustained neuroinflammatory responses, oxidative stress, excitotoxicity, calcium overload, and their intricate interconnections, collectively exacerbating post-ischemic cerebral damage (16, 49, 50). The neuroinflammatory cascade in IS strictly fulfills the four diagnostic criteria of neuroinflammation while orchestrating a sophisticated interplay between tissue destruction and repair mechanisms. As summarized in Figure 1, this interplay is intricately regulated by a network of non-coding RNAs, particularly circRNAs and lncRNAs, which function through diverse mechanisms, including sponging microRNAs and directly interacting with signaling proteins, to precisely modulate key inflammatory pathways and cell death processes such as pyroptosis. Notably, the term “neuroinflammation” suffers from conceptual overgeneralization in practical applications. Its ambiguous definitional boundaries not only risk misinterpretation of pathological mechanisms but may also induce cognitive biases among novice researchers. To address this conceptual ambiguity, previous studies have systematically defined four diagnostic hallmarks based on peripheral inflammation parallels: microglial activation, brain-specific infiltration of peripheral immune cells, significant elevation of inflammatory mediators, and secondary neurodegeneration (51–53). These pathological processes may induce either necrosis or programmed apoptosis, with the specific mode determined by injury severity and neuronal metabolic status. In post-ischemic inflammatory responses, damaged and dying cells release damage-associated molecular patterns (DAMPs) that initiate immune activation and exert critical regulatory functions (54). These DAMPs precisely engage pattern recognition receptor clusters, including Toll-like receptor (TLR) families and scavenger receptors, on microglia and cerebrovascular endothelial cells. Functioning as biological radars within the sterile microenvironment devoid of exogenous pathogens, they breach the molecular silence of the BBB to activate inflammatory responses across neurovascular unit components: microglia initiate phagocytic programs, astrocytes release chemokines, and endothelial cells deploy adhesion molecules to establish molecular pathways for subsequent leukocyte transmigration (55–57).
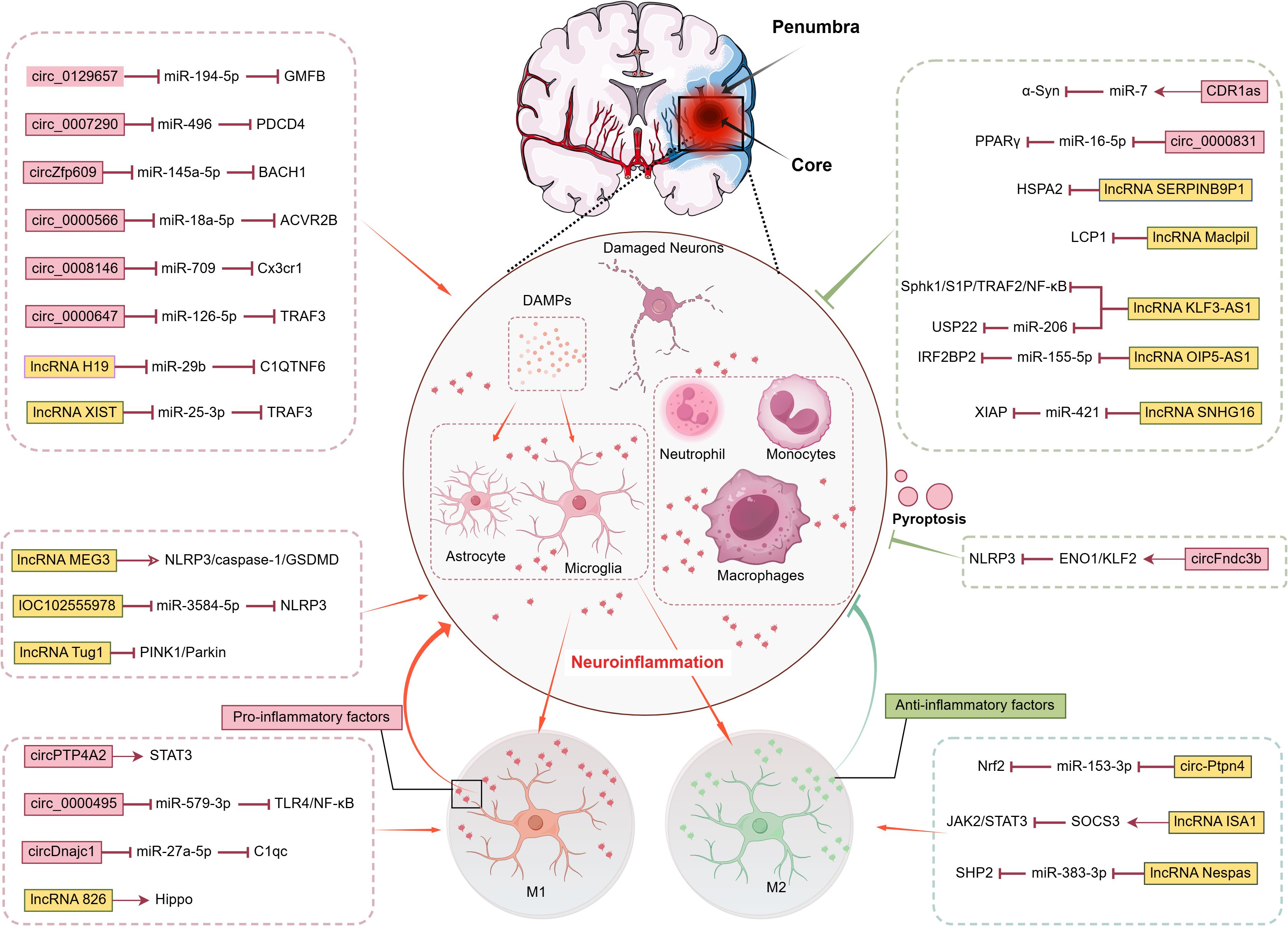
Figure 1. Post-Stroke Neuroinflammation Cascade Regulatory Network of CircRNAs and LncRNAs.
2.3 Temporal progression of neuroinflammation and damage-repair homeostasis
In summary, inflammation is a recognized core regulatory axis in stroke pathology, functioning as an integral component of I/R-induced cascades. It persists throughout the entire pathological process from onset to post-injury repair, exhibiting dual regulatory characteristics of damage-repair homeostasis across different post-stroke phases (58, 59). This process can be divided into three stages:
1. Acute phase (initial hours post-stroke): Primarily characterized by phagocytic activity toward necrotic cells and debris, mediated jointly by resident phagocytes (microglia and macrophages) and infiltrating leukocytes (mainly neutrophils).
2. Subacute phase (days post-stroke): Marks gradual inflammation resolution.
3. Late phase (days to weeks post-stroke): Dominated by collaborative effects of astrocytes and microglia, leading to glial scar formation (60).
While glial scarring is essential for reconstructing vascular networks and sealing lesion areas to restore tissue integrity, it concurrently induces axonal regeneration barriers that significantly impair central nervous system functionality. Clinical studies demonstrate that elevated inflammatory levels correlate with poorer therapeutic outcomes in IS, establishing inflammation as a critical prognostic factor. For instance, early measurements of C-reactive protein (CRP) effectively predict functional disability and mortality in acute IS patients (61). The detrimental effects of inflammation extend beyond these observations. In focal and global cerebral I/R injury, intracranial hemorrhage, or mechanical brain trauma, inflammatory responses consistently exacerbate secondary damage despite incomplete mechanistic understanding (57). Current evidence suggests that targeted modulation of neuroinflammation may provide protective effects in IS, opening new therapeutic avenues. Rational immunomodulation could suppress pathological damage while enhancing reparative functions, though potential side effects require caution. Future research should clarify molecular mechanisms and signaling pathways underlying inflammatory regulation, precisely distinguish beneficial versus harmful targets, and develop safer therapeutic strategies to improve patient prognosis.
3 Limitations of conventional therapies for IS: breakthrough directions in ncRNA-targeted neuroinflammation
3.1 Temporal-spatial dilemmas and risk limitations of reperfusion therapies
The core contradiction in acute IS treatment lies in the spatiotemporal limitations of recanalization and associated therapeutic risks. Current strategies primarily include IVT and EVT.
3.1.1 Rigid temporal window constraints
Alteplase (rt-PA), the only approved standard thrombolytic drug, must be administered within 4.5 hours of symptom onset. However, delayed hospital arrival severely limits its application. For example, only 12.43% of Chinese patients aged 65 or older arrive within 3 hours, with even lower rates in rural areas, compared to 40% to 60% in high-income countries (5, 62–66). Although next-generation thrombolytics, such as Tenecteplase and Reteplase, extend therapeutic windows through structural optimization and simplified administration, their clinical translation remains constrained by progressively increasing hemorrhagic risks, with intracranial hemorrhage risk higher than that of alteplase (64, 67, 68).
3.1.2 Spatial accessibility imbalance
While EVT extends the recanalization window to 24 hours and improves outcomes in large-vessel occlusion patients, its implementation faces three barriers: (1) strict indications excluding small-vessel disease populations; (2) insufficient equipment and technical expertise at primary healthcare institutions; and (3) procedure-related vascular injury and post-reperfusion injury risks requiring multimodal imaging-guided patient selection (12, 69–72).
3.2 Precision balancing challenges in antithrombotic therapy
Antithrombotic therapy is a cornerstone of primary and secondary IS prevention, utilizing antiplatelet agents (e.g., aspirin, clopidogrel) or anticoagulants (e.g., warfarin, non-vitamin K antagonist oral anticoagulants [NOACs]) to suppress thrombogenesis (12). Treatment strategies are tailored to thrombotic etiology: antiplatelet regimens are favored for non-cardioembolic IS subtypes, such as atherosclerotic and small vessel disease (73, 74), while anticoagulation is preferred for cardioembolic origins, such as atrial fibrillation, or hypercoagulable states (75, 76). However, all antithrombotic approaches require a careful balance between ischemic protection and hemorrhagic risk, particularly gastrointestinal and intracranial hemorrhage. Effective clinical decision-making involves: Personalized bleeding risk assessment using validated scoring systems; Continuous monitoring of hemostatic parameters; and Prompt regimen adjustments based on the recurrence of thrombotic or hemorrhagic events. This delicate risk-benefit balance highlights the critical need for precision medicine in IS prophylaxis.
3.3 ncRNAs targeting neuroinflammation: molecular keys to overcoming spatiotemporal constraints
Current therapeutic strategies for IS emphasize rapid reperfusion through IVT and EVT, paired with etiology-guided antithrombotic management using antiplatelet or anticoagulant therapy. However, their clinical efficacy is limited by strict time windows, eligibility criteria, and an inability to mitigate neuroinflammation-driven secondary injury following reperfusion (77, 78). Increasing evidence positions inflammation as a central regulator in I/R cascades and a key mediator of risk factors. Notably, inflammatory processes compromise neurovascular unit (NVU) integrity prior to IS onset and continuously worsen secondary damage post-stroke (57, 79, 80). After reperfusion, aberrant neuroimmune responses initiate uncontrolled inflammatory cascades via mechanisms like inflammasome activation and mitochondrial dysfunction, resulting in BBB disruption, reactive oxygen species (ROS) bursts, and subsequent neuronal apoptosis and glial activation (81–83). Recent research identifies ncRNAs as critical regulators of post-reperfusion neuroinflammation, modulating neuroimmune interactions, suppressing inflammatory signaling pathways (e.g., NF-κB), and preserving NVU homeostasis (84). Their dynamic expression profiles, such as miR-124 and lncRNA MALAT1, not only act as early biomarkers of injury but also provide innovative therapeutic opportunities to overcome time window constraints through targeted delivery systems, such as exosomal vectors, facilitating precise anti-inflammatory interventions (32, 85–87). In this regard, stem cell-derived exosomes have emerged as highly effective ncRNA carriers, owing to their BBB permeability and biocompatibility. Experimental studies demonstrate their ability to deliver functional ncRNAs to ischemic lesions efficiently, maintaining bioactivity and coordinating molecular regulation in affected regions (88–90) (Figure 2). This advancement lays a theoretical groundwork for multidimensional strategies that integrate reperfusion, anti-inflammation, and neuroprotection, potentially overcoming the fundamental limitations of conventional IS therapies.
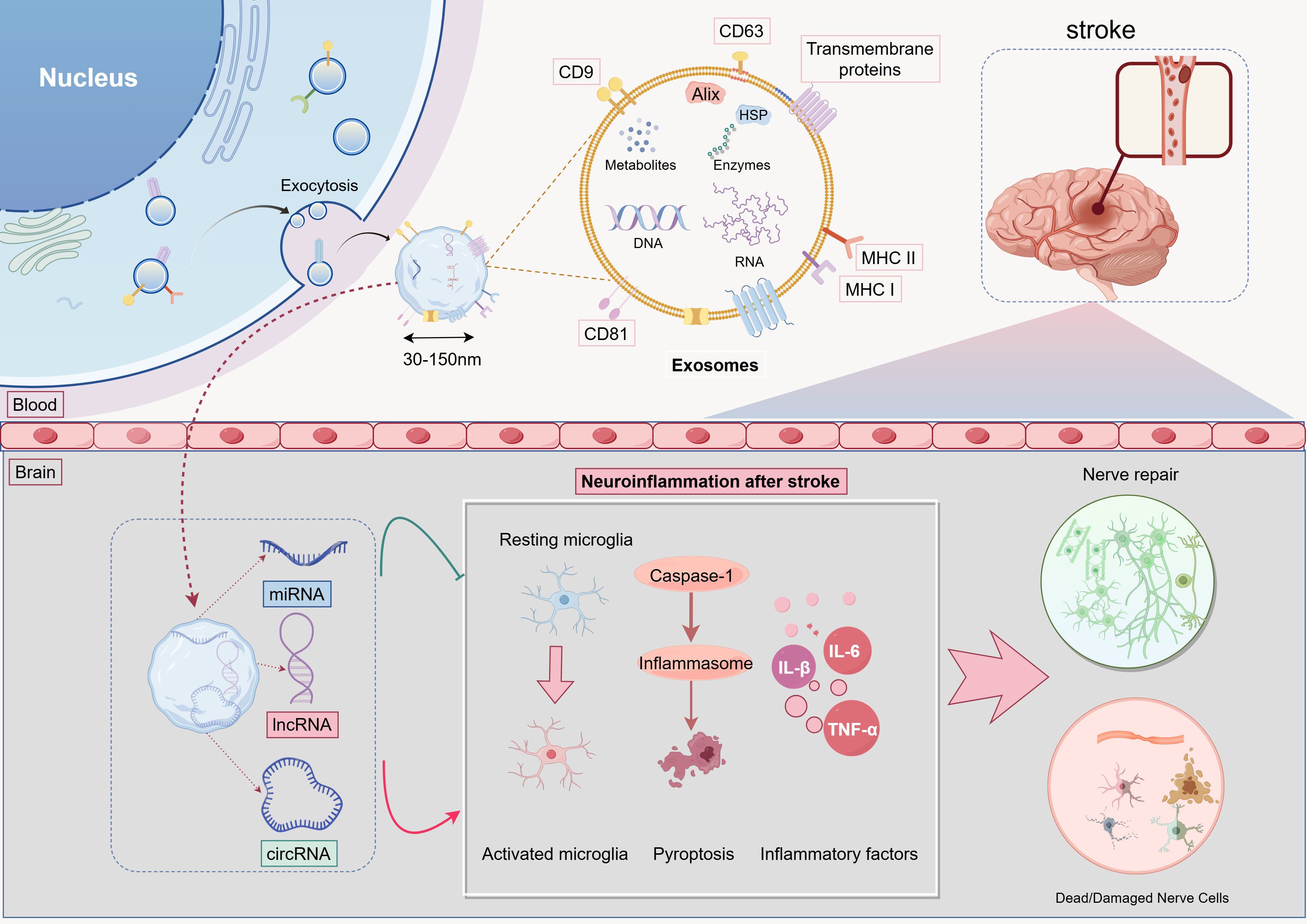
Figure 2. Exosomal ncRNAs Regulate Neuroinflammation After Stroke.
4 ncRNAs regulating neuroinflammation in IS
4.1 Regulatory roles of circRNAs in post-stroke neuroinflammatory cascades
Although members of the ncRNA family share common anti-neuroinflammatory functions, their mechanisms of action differ significantly (Table 1). As a key member of this family, circRNAs are covalently closed, single-stranded circular molecules formed through back-splicing of precursor RNAs (91). Based on their biogenesis mechanisms and structural features, circRNAs can be classified into four major subtypes: intron-derived ciRNAs (circular intronic RNAs), exon-intron chimeric EIciRNAs (exon-intron circRNAs), exon-cyclized ecircRNAs (exonic circRNAs), and tRNA-processing-derived tricRNAs (tRNA intronic circRNAs) (92, 93). Current studies reveal that circRNAs exhibit diverse functions, including acting as miRNA sponges, interacting with RNA-binding proteins (RBPs), regulating alternative splicing, transcription, and translation, generating pseudogenes, and participating in cellular transport and communication (93). In cerebral I/R injury, circRNAs primarily function as dynamic regulators of neuroinflammation. As evidenced by mechanistic studies, they not only directly modulate inflammatory responses but also critically sequester miRNAs through sponge activity to fine-tune inflammation levels, thereby steering neuroinflammatory progression (94, 95). The following representative examples illustrate this regulatory duality:
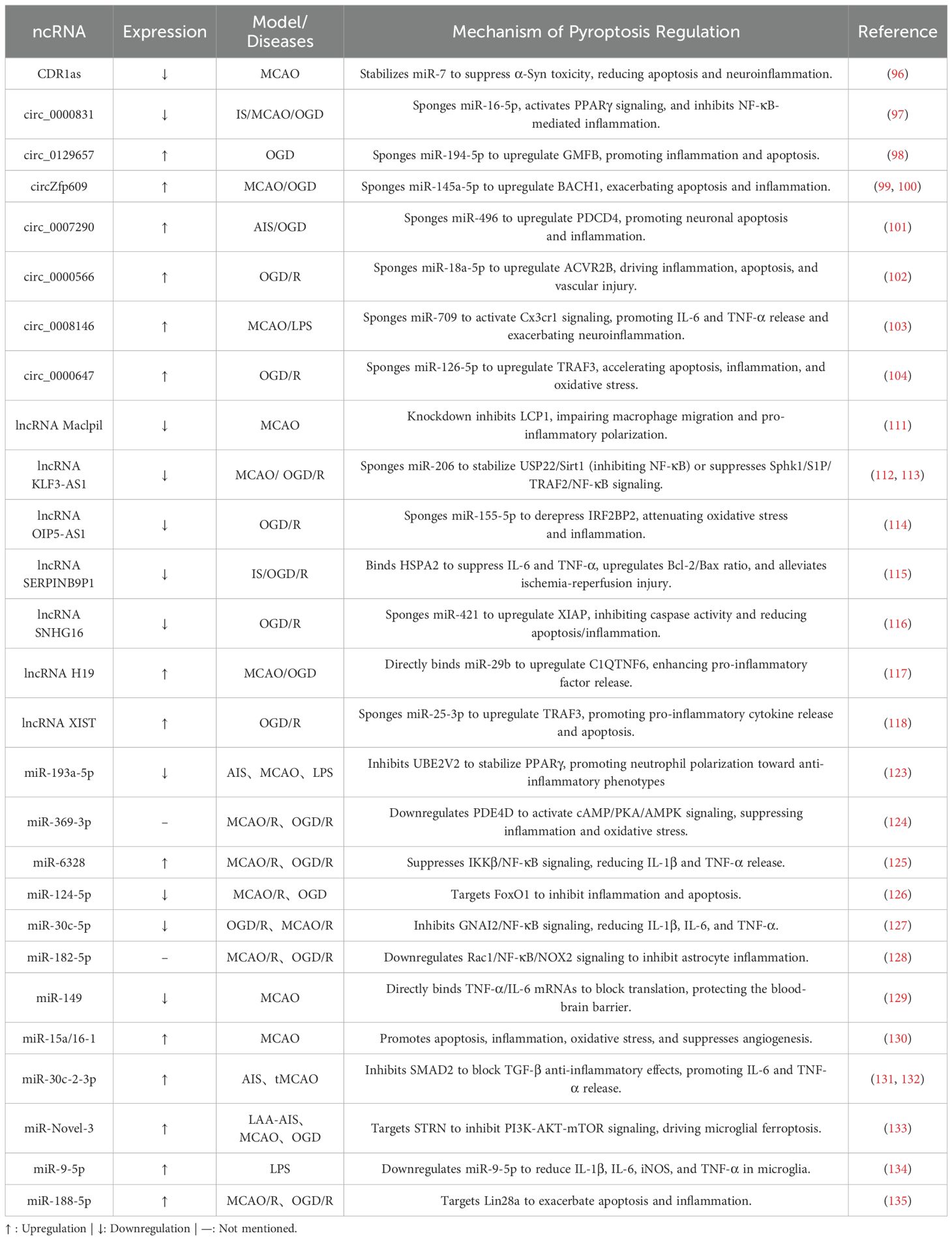
Table 1. Role of ncRNA in IS-associated neuroinflammatory responses.
CDR1as/ciRS-7 exemplifies a unique circRNA-mediated neuroprotective mechanism. Enriched with miR-7 binding sites, it stabilizes miR-7 via incomplete complementary pairing, extending its biological half-life. This interaction suppresses α-synuclein (α-Syn), a key regulator of the TLR4/NF-κB pathway, ultimately attenuating neuroinflammation, reducing neuronal apoptosis, and enhancing motor function recovery in ischemic models (96). circ_0000831 represents another pivotal anti-inflammatory circRNA, characterized by its downregulation in stroke patients. By serving as a molecular sponge for miR-16-5p, it activates adiponectin receptor 2 (AdipoR2) and the PPARγ signaling pathway, significantly alleviating neuroinflammation, vertigo, neurological deficits, and apoptosis in MCAO mice. Dual-luciferase assays confirm its direct binding to miR-16-5p, solidifying its mechanistic role (97).
Conversely, a distinct class of pro-inflammatory circRNAs exacerbates ischemic injury. For example, circ_0129657 competitively binds miR-194-5p, relieving its suppression of glia maturation factor beta (GMFB). This derepression promotes endothelial apoptosis and inflammatory cytokine secretion, while its silencing reduces cerebral infarct volume and ameliorates neurological dysfunction (98). circZfp609 mediates damage through a dual regulatory network: it serves as a sponge for miR-145a-5p both to suppress NF-κB inhibition and upregulate BACH1, thereby promoting apoptosis and pro-inflammatory factor release (e.g., IL-1β, TNF-α). Genetic knockout of circZfp609 alleviates cerebral ischemia/reperfusion injury and inhibits OGD/R-induced cellular damage by modulating astrocyte apoptosis and inflammatory responses (99, 100). Other pro-inflammatory circRNAs—including circ_0007290 (via miR-496/PDCD4), circ_0000566 (targeting miR-18a-5p), circ_0008146 (miR-709/CX3CR1 axis), and circ_0000647 (miR-126-5p/TRAF3)—likewise function as miRNA sponges to amplify ROS bursts, cytokine secretion, and endothelial injury (101–104).
In summary, circRNAs critically orchestrate neuroinflammatory responses post-stroke, primarily through sponge-mediated miRNA regulation. While anti-inflammatory members (e.g., CDR1as, circ_0000831) stabilize protective miRNAs to suppress inflammatory cascades, their pro-inflammatory counterparts (e.g., circ_0129657, circZfp609) sequester repressive miRNAs to activate apoptotic and inflammatory pathways. This functional duality positions circRNAs as promising therapeutic targets for modulating post-stroke neuroinflammation.
4.2 LncRNAs orchestrate neuroimmune crosstalk after stroke
LncRNAs are non-coding RNA molecules exceeding 200 nucleotides (nt) in length. Thousands of lncRNAs have been identified in the human genome, regulating gene expression through chromatin modification and post-transcriptional regulatory mechanisms (105–107). LncRNA dysregulation is closely associated with pathological processes in various diseases. The central nervous system harbors the most extensive repertoire of lncRNA transcripts, which participate in multidimensional regulation of neural development and homeostasis maintenance. Studies using microarray and high-throughput RNA sequencing technologies have identified hundreds of differentially expressed lncRNAs in IS patients and animal models. These expression profile abnormalities are strongly correlated with pathological processes such as BBB disruption and neuroinflammation (108–110).
At the neuroimmune interface, lncRNA Maclpil exhibits unique properties by blocking monocyte-derived macrophage (MoDM) activation and migration through inhibition of LCP1-mediated actin dynamics. Crucially, LCP1 knockout fully replicates Maclpil’s neuroprotective effects, confirming LCP1 as its key effector. Consistent with this, animal studies demonstrate that this axis reduces infarct volume and improves neurological recovery (111). Similarly, KLF3-AS1 stands out for its dual regulatory mechanisms. In cerebral I/R models, its downregulation correlates with elevated pro-inflammatory factors (TNF-α, IL-6, IL-1β). Mechanistically, KLF3-AS1 suppresses neuroinflammation via the Sphk1/NF-κB pathway (112). Furthermore, when delivered by BMSC-derived exosomes, it competitively binds miR-206 to upregulate USP22, promoting Sirt1 deubiquitination and extending its half-life to mitigate inflammation and apoptosis (113). Other protective lncRNAs (e.g., OIP5-AS1, SERPINB9P1, SNHG16) similarly modulate ceRNA networks, chaperone activity, and oxidative pathways to mitigate injury (114–116).
Conversely, some lncRNAs drive pathology. lncRNA H19 demonstrates significant clinical relevance, with its expression in neutrophils of IS patients positively correlating with leukocyte activation. Mechanistically, H19 acts as a ceRNA for miR-29b, relieving its inhibition of C1QTNF6 and driving pro-inflammatory factor release (IL-1β, TNF-α), thereby exacerbating BBB disruption and ischemic damage (117). Similarly, lncRNA XIST exhibits striking cell-type specificity in neurons. It sponges miR-25-3p to elevate TRAF3 levels, activating NF-κB-mediated inflammatory responses. Supporting its detrimental role, XIST silencing inhibits neuronal apoptosis and cytokine release, while exogenous TRAF3 overexpression reverses this protection (118).
In summary, as lncRNAs bridge epigenetic regulation and immune dynamics, they represent not just therapeutic targets but potential “molecular switches” for reprogramming neuroinflammation, offering a promising platform for developing lncRNA-mediated precision medicine strategies in stroke recovery.
4.3 miRNAs: precision modulators of post-stroke inflammatory microenvironment
MiRNAs are small non-coding RNA molecules approximately 20–24 nucleotides (nt) in length. They exert critical post-transcriptional regulatory roles by mediating target mRNA degradation or translational repression through incomplete base complementarity. Notably, a single miRNA can regulate the translation of hundreds of mRNAs, demonstrating its broad functional regulatory potential (119–122). Given their ability to directly target key inflammatory factor mRNAs, miRNAs have become a research focus in I/R injury in recent years. For example, multiple studies confirm that targeting specific miRNAs effectively modulates I/R-associated neuroinflammatory cascades, offering novel therapeutic avenues for IS.
Specific examples highlight this therapeutic potential. miR-193a-5p, for example, orchestrates neutrophil immune polarization. In acute IS patients, its significant downregulation in circulating neutrophils disrupts PPARγ-mediated anti-inflammatory signaling. Mechanistically, miR-193a-5p targets UBE2V2 to inhibit PPARγ ubiquitination, promoting N2 phenotype polarization and reducing pro-inflammatory cytokine release. Importantly, rescue experiments confirm that UBE2V2 knockdown reverses neutrophil hyperactivation in miR-193a-5p-deficient models (123). Recent advances in exosome delivery strategies have opened new avenues for miRNA-based therapies. For instance, exosomes derived from recombinant human growth differentiation factor 7 (rhGDF7)-preconditioned bone marrow mesenchymal stem cells (BMSCs) are enriched with miR-369-3p. This miRNA targets the 3’-untranslated region (3’-UTR) of the Pde4d gene, suppressing PDE4D protein expression and activating the AMPK signaling pathway, thereby significantly inhibiting I/R-induced neuronal inflammation and oxidative stress damage (124). Similarly, miR-6328 exerts critical anti-inflammatory effects in I/R injury by targeting the IKKβ/NF-κB signaling axis. Mechanistically, this miRNA binds to IKKβ to inhibit its expression, thereby blocking NF-κB pathway activation. Notably, sequencing at 72 hours post-I/R revealed elevated miR-6328 levels accompanied by reduced IKKβ, and reverse functional experiments further validated the specificity of its anti-inflammatory effects (125). In addition to these, other protective miRNAs (e.g., miR-124-5p, miR-30c-5p, miR-182-5p, miR-149) orchestrate anti-inflammatory responses primarily by suppressing neuronal apoptosis and reinforcing BBB integrity (126–129).
However, miRNA function is not context-independent. A prominent example underscoring the critical, yet often overlooked, role of age and sex factors in RNA biology is miR-15a/16-1. Significantly upregulated post-stroke and correlating with neuronal injury severity, studies in tMCAO models reveal a striking age-sex interaction: young female mice (3-month-old) exhibit the strongest neuroprotective phenotype, characterized by reduced miR-15a/16–1 induction. Combined antagonism with estradiol synergistically downregulates genes driving neurovascular death, inflammation, and oxidative stress. miR-15a/16–1 target genes further display marked age- and sex-dependent expression, solidifying their biomarker potential for tailored stroke therapy (130). Crucially, while most preclinical stroke studies utilize healthy rodent models, translating findings to human patients requires attention to human-specific factors. Recent investigations focusing on patient-derived biomarkers reveal critical mechanisms specific to human pathology. For instance, circulating miR-30c-2-3p is uniquely upregulated in plasma exosomes of acute ischemic stroke (AIS) patients. It establishes a pro-inflammatory network by targeting SMAD2, a core anti-inflammatory component of the TGF-β pathway, suggesting exosome-mediated miR-30c-2-3p delivery as a key driver of post-stroke neuroinflammation (131, 132). Notably, research delving into specific stroke subtypes uncovers further complexity. Research on the large artery atherosclerotic (LAA) stroke subtype identifies plasma exosome-derived miR-Novel-3 as a dual pathogenic factor. Specifically overexpressed in LAA-AIS patients, it concurrently drives neuroinflammation and ferroptosis by targeting STRN protein in peri-infarct microglia/macrophages, thereby suppressing PI3K-AKT-mTOR signaling to trigger inflammatory mediator release and ferroptosis-related molecular dysfunction, ultimately exacerbating ischemic neuronal injury (133).These findings collectively highlight the necessity of incorporating patient-specific complications (e.g., atherosclerosis) into stroke models to uncover clinically relevant pathological cascades. Additional detrimental miRNAs (e.g., miR-9-5p, miR-188-5p) exacerbate neuroinflammation through dysregulated TLR signaling, driving maladaptive pro-inflammatory cascades (134, 135).
5 ncRNA regulation of microglial polarization in IS
5.1 Molecular basis of microglial polarization
Microglia originate from erythromyeloid progenitors (EMPs) in the early embryonic yolk sac. Their development occurs independently of the hematopoietic stem cell-mediated system, culminating in their differentiation into tissue macrophages resident in the brain parenchyma (136–138). As the central nervous system’s primary immune effector cells, microglia constitute approximately 10% of total neural cells and dynamically maintain neuroimmune homeostasis to support brain function (139–141).
In IS, microglia display two distinct polarization states:
1. Pro-inflammatory phenotype (M1 polarization): Microglia detect DAMPs through pattern recognition receptors (PRRs), initiating neuroinflammatory cascades and releasing pro-inflammatory cytokines, such as TNF-α and IL-1β. This response disrupts the BBB and amplifies neuronal death (142–145).
2. Anti-inflammatory phenotype (M2 polarization): Microglia foster neurogenesis, angiogenesis, and synaptic remodeling by clearing apoptotic debris via phagocytosis and secreting brain-derived neurotrophic factor (BDNF), thus preserving microenvironmental homeostasis (146, 147).
In stroke, excessive DAMP stimulation skews the M1/M2 balance toward the pro-inflammatory M1 phenotype. Encouraging M2 polarization can mitigate inflammation and enhance tissue repair, improving post-stroke outcomes (53, 148). Consequently, modulating microglial polarization—particularly toward the M2 state—represents a key strategy for reducing I/R injury. Among these approaches, ncRNA-mediated regulation stands out for its precision and capacity to target multiple pathways simultaneously (Table 2).
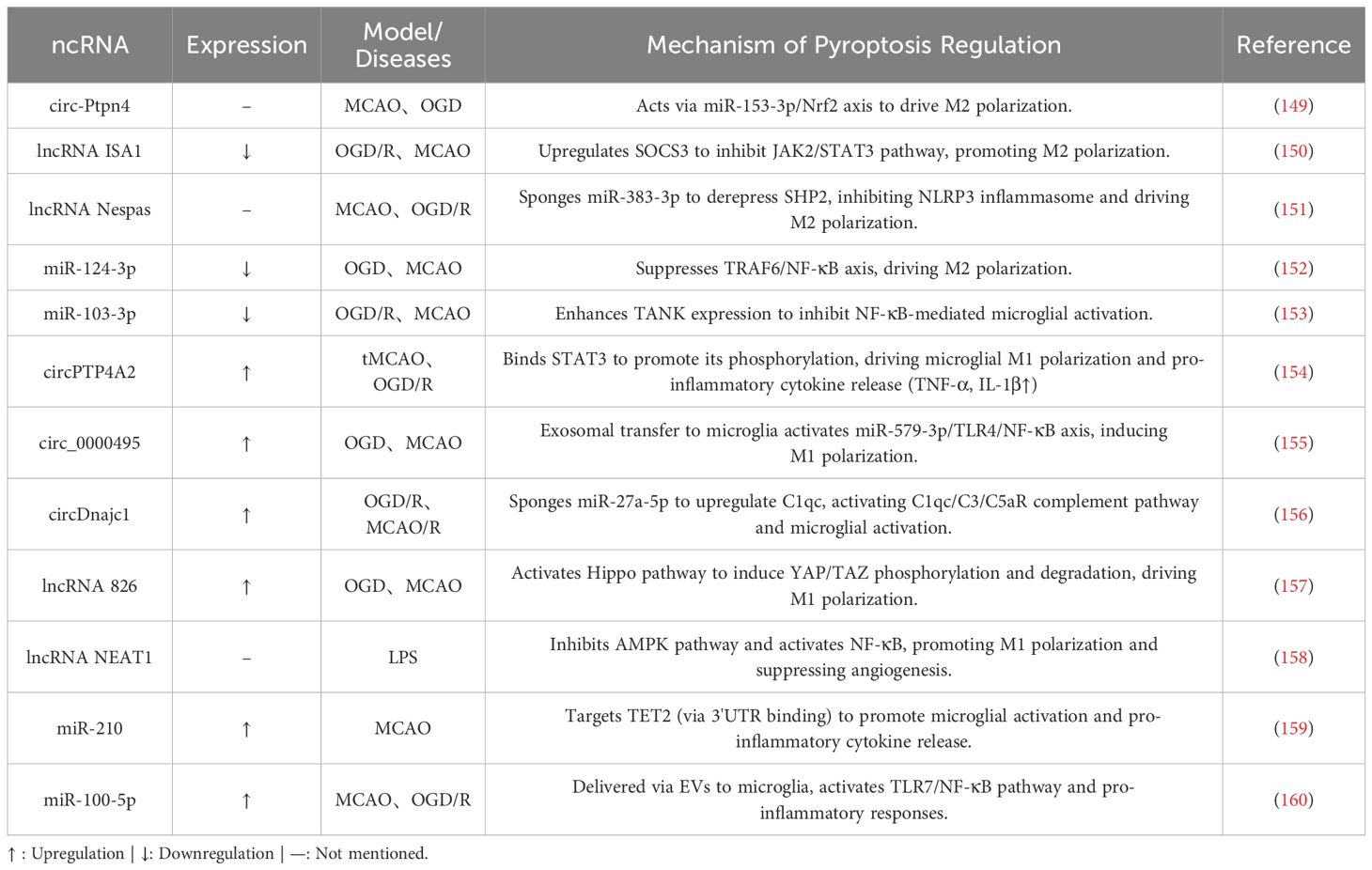
Table 2. ncRNA-mediated microglial regulation in Post-IS inflammation.
5.2 ncRNA-mediated regulation of microglial polarization
5.2.1 ncRNAs promote anti-inflammatory polarization and neural repair
Recent advancements in exosome delivery systems have introduced innovative strategies to modulate microglial polarization. For example, exosomes from hypoxia-preconditioned adipose-derived mesenchymal stem cells (ADSCs) deliver circ-Ptpn4, which significantly promotes microglial polarization toward the anti-inflammatory M2 phenotype. Mechanistic studies demonstrate that circ-Ptpn4 acts as a sponge for a specific miRNA, thereby relieving its transcriptional repression of nuclear factor erythroid 2-related factor 2 (Nrf2). This interaction activates antioxidant and anti-inflammatory signaling pathways, ultimately reducing neuronal death (149). These findings confirm the role of exosomes as effective carriers and underscore the pivotal contribution of circRNAs to phenotypic reprogramming.
The lncRNA ISA1 is a critical regulator. In MCAO mouse models, decreased ISA1 expression correlates with larger cerebral infarct volumes and greater neurological deficits. Overexpression of ISA1 exerts neuroprotective effects by activating suppressor of cytokine signaling 3 (SOCS3), which inhibits the JAK2/STAT3 signaling pathway. This suppression drives microglial polarization toward the anti-inflammatory phenotype (150). Equally important in MCAO is the role of ceRNA networks. In MCAO models, elevated miR-383-3p levels exacerbate injury by activating NLR family pyrin domain-containing 3 (NLRP3) inflammasome activity. The lncRNA Nespas mitigates this effect by sponging miR-383-3p, which relieves its inhibition of Src homology 2 domain-containing protein tyrosine phosphatase 2 (SHP2). This establishes the Nespas/miR-383-3p/SHP2 regulatory axis, offering protective effects through three mechanisms:
1. Suppressing NLRP3 inflammasome assembly;
2. Upregulating the M2 marker interleukin-10 (IL-10); and
3. Downregulating M1-associated factors, such as tumor necrosis factor-alpha (TNF-α) and inducible nitric oxide synthase (iNOS).
Together, these actions mitigate neuroinflammatory progression (151).
MiRNA regulatory networks also provide precise modulation of polarization. In OGD models, decreased miR-124-3p levels result in overactivation of the TRAF6/NF-κB signaling pathway. Supplementing with exogenous miR-124-3p inhibits TRAF6-mediated NF-κB phosphorylation, promoting microglial polarization toward the M2 phenotype and enhancing neuronal survival (152). Similarly, in MCAO rat models, miR-103-3p targets the 3’-untranslated region (3’-UTR) of TANK mRNA, thereby modulating the NF-κB signaling pathway. This regulation suppresses abnormal microglial activation, alleviating I/R injury (153).
5.2.2 ncRNAs drive pro-inflammatory polarization and neuroinflammatory injury
In contrast to their anti-inflammatory counterparts, certain ncRNAs exacerbate neuroinflammatory damage post-ischemic stroke by driving microglial polarization toward the pro-inflammatory M1 phenotype through regulation of key signaling pathways.
Within pro-inflammatory circRNA networks, circPTP4A2 serves as a representative example. In tMCAO models, its expression is markedly upregulated in plasma and peri-ischemic cortical tissues. Functional studies demonstrate that circPTP4A2 knockout significantly ameliorates cerebral injury in multiple ways: reducing infarct volume, restoring cortical blood flow homeostasis, and alleviating neurological deficits. Mechanistically, circPTP4A2 directly binds to STAT3 protein to enhance its phosphorylation levels, driving microglial M1 polarization while suppressing M2 phenotypic transition. In vitro validation confirms that circPTP4A2 silencing in OGD/R-induced BV2 microglia significantly inhibits STAT3 signaling activation, decreases pro-inflammatory factor release (TNF-α, IL-6), and increases anti-inflammatory mediator expression (154). Another key circRNA, circ_0000495, is transferred from OGD-treated human brain microvascular endothelial cells (HBMVECs) to microglia via exosomes. It competitively binds miR-579-3p to relieve its suppression of the TLR4/NF-κB pathway. This cascade significantly elevates M1 markers (TNF-α, IL-1β, IL-12) and CD86+ cell proportions while suppressing M2 markers (IL-10, CD163), ultimately exacerbating endothelial damage (155). In another pathological mechanism involving complement system activation, hypoxia-induced circDnajc1 accumulation during I/R injury sponges miR-27a-5p to alleviate its inhibition of complement component C1qc. This triggers uncontrolled activation of the complement cascade (C3/C5aR axis), driving microglial M1 polarization, promoting inflammatory mediator release, and disrupting the BBB. Targeted inhibition of circDnajc1 effectively blocks neuroinflammatory propagation (156).
Certain lncRNAs also drive pro-inflammatory activation. For example, Lnc826 is significantly upregulated in MCAO models. It selectively enhances M1 marker expression (TNF-α, IL-1β) while suppressing M2 markers (e.g., IGF-1) by modulating the Hippo signaling pathway. Notably, Lnc826 knockdown in OGD-induced cellular models reverses microglial pro-inflammatory polarization by restoring the activity of YAP, a downstream effector of the Hippo pathway (157). The pro-inflammatory effects of lncRNA NEAT1 are linked to energy metabolism dysregulation. In BV-2 microglia, NEAT1 inhibits AMPK phosphorylation, reduces SIRT1 activity, and relieves NF-κB pathway suppression, thereby driving M1 polarization. Functional experiments demonstrate that NEAT1 overexpression significantly impairs tube formation capacity in mouse cerebral arterial endothelial cells (mCAECs), while AMPK activators or NEAT1 silencing restore angiogenic activity and promote M2 polarization (158).
MiRNAs also play a critical role in exacerbating neuroinflammation. MicroRNA-210 (miR-210) exacerbates neuroinflammation through epigenetic modifications. It suppresses TET2 expression by targeting its 3’-UTR, amplifying neuroinflammatory responses post-ischemic stroke. Experimental studies demonstrate that miR-210 mimics significantly reduce TET2 levels in murine brains, whereas miR-210 knockout or antagonist interventions maintain TET2 expression. TET2 inhibits pro-inflammatory cytokines via NF-κB pathway regulation: TET2 overexpression reverses miR-210-induced elevations in IL-6/IL-1β. In stroke models, miR-210 upregulation suppresses TET2, directly driving microglial activation and pro-inflammatory polarization, as evidenced by increased CD11b+ microglia and IL-6 release. TET2 knockdown completely negates the neuroprotective effects of miR-210 inhibition, including reduced infarct volume and improved neurological function (159). Furthermore, exosome-mediated neuron-microglia communication plays a pivotal role in post-stroke neuroinflammation. Ischemia-induced neuronal hyperactivity releases sEVs enriched with miR-100-5p, which are internalized by neighboring neurons and microglia via paracrine signaling. Mechanistically, miR-100-5p directly binds Toll-like receptor 7 (TLR7) through its unique U18U19G20 motif, activating NF-κB phosphorylation cascades. This triggers explosive release of pro-inflammatory factors (IL-1β, TNF-α) and suppresses the anti-inflammatory marker arginase-1 (Arg-1). In MCAO model mice, miR-100-5p levels are markedly elevated in peri-ischemic microglia-derived sEVs. Intracerebroventricular injection of miR-100-5p antagomiR specifically silences this miRNA, effectively blocking TLR7/NF-κB signaling, reversing microglial pro-inflammatory activation, and significantly reducing infarct volume while improving motor function outcomes (160).
6 ncRNA regulation of pyroptosis in IS: linking inflammation and neuronal death
6.1 Pathological mechanisms of pyroptosis and its association with stroke
Pyroptosis, an inflammatory form of programmed cell death mediated by Gasdermin proteins, is characterized by plasma membrane rupture and the release of pro-inflammatory factors (161, 162). Unlike apoptosis, which maintains membrane integrity, pyroptosis induces the efflux of cytoplasmic inflammatory contents through membrane pores, culminating in secondary necrosis—a pathological state of altered membrane permeability following complete cell death (161, 163–165).
The Gasdermin protein family comprises six members (GSDMA, GSDMB, GSDMC, GSDMD, GSDME, and GSDMF/PJVK/DFNB59), serving as core executors of inflammatory responses and disease progression (166).
Pyroptosis is closely linked to the initiation and progression of stroke. Post-stroke, Gasdermin-mediated pyroptosis displays cascading amplification effects within the neurovascular unit (167, 168). When pyroptosis is activated in neuroglia, neurons, and brain microvascular endothelial cells (BMECs), membrane perforation leads to the explosive release of inflammatory mediators like IL-1β and IL-18. This release generates self-amplifying cytokine storms (169, 170). This pathology exacerbates brain injury through two primary mechanisms. First, pyroptosis disrupts the physical barrier by dismantling the tight junction structures of the BBB, leading to plasma protein leakage and the infiltration of toxic substances into the brain parenchyma. Second, it causes molecular signaling dysregulation, where extracellular DAMPs activate microglia via pattern recognition receptors such as TLRs. This activation drives the spread of neuroinflammation to the ischemic penumbra, accelerating the conversion of reversible injury zones into infarct cores (171–174).
Recent advances in understanding the role of pyroptosis in stroke pathophysiology have shown that ncRNAs regulate pyroptosis signaling pathways at multiple levels by targeting critical components such as NLRP3 inflammasome activation and Caspase-1 processing (Table 3). This complex epigenetic regulation provides new insights into disrupting the vicious cycle of pyroptosis, neuroinflammation, and tissue.
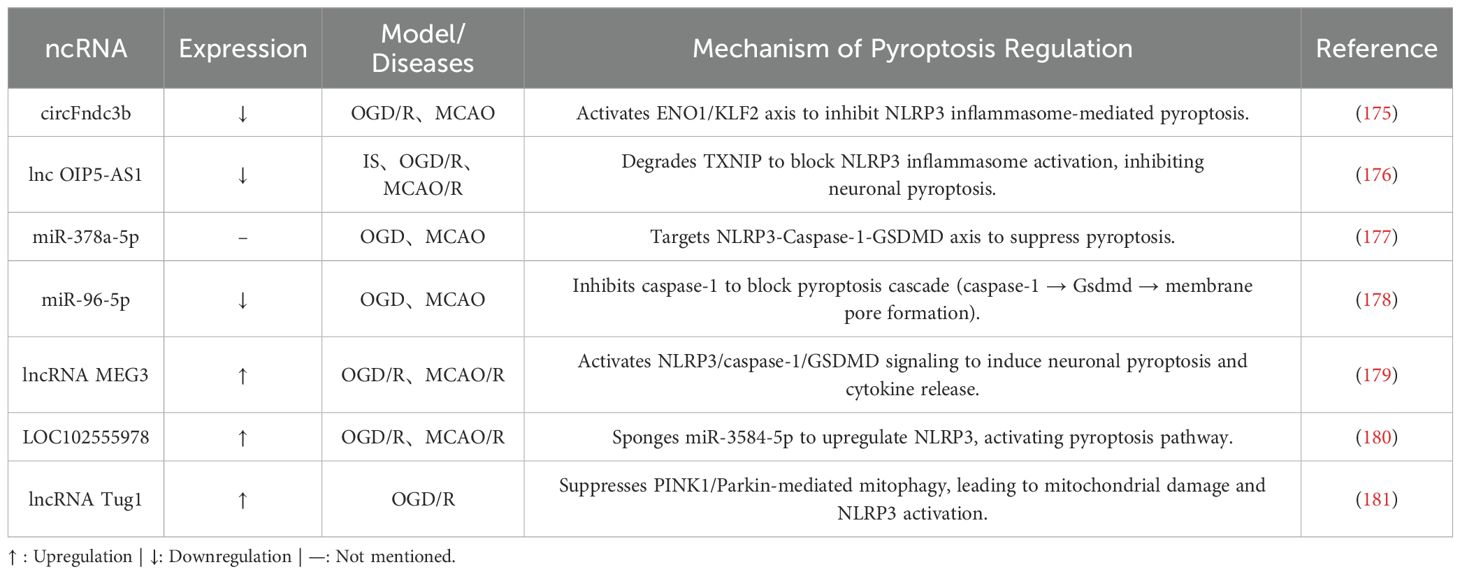
Table 3. IS-associated pyroptotic signaling via ncRNA.
6.2 ncRNA-mediated pyroptosis control
6.2.1 ncRNAs inhibiting pyroptosis
Recent studies highlight the neuroprotective potential of ncRNAs through targeted regulation of pyroptosis-related molecules.
For example, the expression of circFndc3b is inversely correlated with neuropathological severity in IS models. In MCAO models, circFndc3b levels are significantly downregulated in peri-infarct cortical regions, while exercise intervention upregulates circFndc3b to reduce infarct volume and enhance neurological recovery. Mechanistically, circFndc3b directly binds to enolase 1 (ENO1) and coordinates a dual-pathway regulation: First, it enhances ENO1-mediated stabilization of Klf2 mRNA via 3’-UTR interactions, thereby suppressing NLRP3 inflammasome-driven pyroptosis; second, it promotes ENO1-dependent stabilization of FUS mRNA to reinforce circFndc3b self-circularization, forming a self-reinforcing positive feedback loop. This mechanism not only identifies exercise intervention as a potential therapeutic target but also elucidates how circFndc3b maintains cellular homeostasis, offering critical theoretical support for post-stroke neurorestorative strategies (175).
Additionally, lncRNAs regulate pyroptosis. For example, exosome-delivered lncRNA OIP5-AS1 suppresses pyroptosis. In OGD/R-treated neurons and MCAO/R models, OIP5-AS1 levels are significantly downregulated, whereas exogenous supplementation with OIP5-AS1 reduces infarct volume and inhibits the expression of pyroptosis-related proteins. Mechanistic studies reveal that OIP5-AS1 directly binds to thioredoxin-interacting protein (TXNIP), promoting E3 ubiquitin ligase ITCH-mediated ubiquitination and degradation of TXNIP. This process blocks NLRP3 inflammasome activation and subsequent pyroptosis cascades (176).
MiRNAs also play pivotal roles in suppressing pyroptosis. Astrocyte-derived exosomes deliver miR-378a-5p to exert anti-pyroptotic effects. In OGD and MCAO models, miR-378a-5p binds to the 3’-UTR of the NLRP3 gene, inhibiting its transcription and inflammasome assembly, thereby significantly reducing pyroptosis marker levels. Functional studies confirm that miR-378a-5p expression is positively correlated with neuronal survival rates. Conversely, genetic knockout of miR-378a-5p induces dose-dependent upregulation of NLRP3 expression, exacerbating neuronal injury (177). Another anti-pyroptotic miRNA, miR-96-5p, suppresses pyroptosis by targeting the CASP1 gene rather than the broader caspase-1/GSDMD axis. In I/R models, decreased miR-96-5p expression is negatively correlated with activation of pyroptosis-related proteins (cleaved caspase-1, Gsdmd-N). Functional studies demonstrate that miR-96-5p directly binds to the CASP1 gene to inhibit its transcription. In OGD/R models, miR-96-5p mimics reduce TUNEL-positive cell ratios and improve neuronal survival. Animal experiments further validate these findings: intracerebroventricular delivery of miR-96-5p agomir downregulates the expression of pyroptosis executioner proteins in the ischemic penumbra, while also alleviating behavioral deficits and cerebral edema (178).
6.2.2 ncRNAs promoting pyroptosis
ncRNAs exacerbate post-ischemic pyroptosis through diverse regulatory mechanisms. For example, lncRNA MEG3 promotes neuronal pyroptosis in cerebral I/R injury through epigenetic regulation. Ischemia-induced downregulation of the fat mass and obesity-associated protein (FTO) increases m6A methylation of MEG3, enhancing its stability and leading to pathological accumulation. This elevated MEG3 activates the NLRP3/caspase-1/GSDMD signaling pathway, triggering pyroptosis, as demonstrated by the concurrent upregulation of NLRP3, cleaved caspase-1, and GSDMD-N in cortical tissues. Importantly, overexpressing FTO reverses the aberrant m6A modification of MEG3 and suppresses pyroptosis, highlighting the FTO-MEG3 axis as a potential therapeutic target for stroke intervention (179).
Additionally, the ceRNA mechanism contributes to pyroptosis activation. In I/R models, lncRNA LOC102555978 is significantly upregulated and acts as a molecular sponge by competitively binding miR-3584-5p. This binding relieves the miRNA’s inhibition of NLRP3, thereby activating pyroptosis cascades. Disrupting the LOC102555978/miR-3584-5p/NLRP3 axis reduces pyroptosis markers, improves cell viability, and mitigates pathological damage, as shown in experimental studies (180).
Moreover, disrupting mitochondrial homeostasis exacerbates pyroptosis. Under hypoxic conditions, lncRNA Tug1 suppresses PINK1/Parkin-mediated mitophagy, thereby promoting microglial pyroptosis. Knocking down Tug1 activates the PINK1-Parkin pathway, as evidenced by an increased LC3B-II/LC3B-I ratio and enhanced p62 degradation. This activation enhances Parkin recruitment to mitochondria, facilitating the clearance of damaged mitochondria and inhibiting NLRP3 inflammasome activation. Notably, this protective effect is abolished by PINK1 silencing or the mitophagy inhibitor mdivi-1, confirming that Tug1 promotes pyroptosis by impairing mitochondrial quality control (181).
7 Challenges and considerations for clinical translation: from technical hurdles to biological complexity
7.1 Technical bottlenecks in exosome production, storage, and delivery for ncRNA therapeutics
A comparative summary of these intricate mechanisms, detailing the functions and targets of key ncRNAs in preclinical neuroinflammation models, is provided in Table 4. Notwithstanding these promising mechanistic insights, the therapeutic promise of exosome-based ncRNA delivery hinges on overcoming substantial technical challenges in production, storage, and delivery. The therapeutic promise of exosome-based ncRNA delivery hinges on overcoming substantial technical challenges in production, storage, and delivery. Exosome isolation exemplifies these hurdles, as current techniques struggle to balance efficiency and integrity. Conventional methods, such as ultracentrifugation (UC) and ultrafiltration (UF), yield low recovery rates—typically 5–25% for UC—while risking membrane damage and vesicle deformation. Emerging alternatives, including microfluidic chips, offer rapid separation in under five minutes but demand sophisticated instrumentation, limiting scalability. Immunoaffinity purification achieves exceptional purity (>90%) yet relies on costly, specific antibodies (182). To address these limitations, multi-technique integration strategies, such as sequential centrifugation combined with UF and UC, have become essential for producing high-purity, structurally intact exosomes (183). Success in these approaches requires careful optimization of six critical parameters: recovery rate, sample compatibility, molecular integrity, separation efficiency, cost-effectiveness, and operational simplicity (184). However, unlike synthetic drugs, exosome production is fraught with industrialization risks stemming from biological variability, which complicates batch-to-batch consistency. Each phase—from cell culture to harvest—profoundly influences yield and quality, necessitating stringent oversight to meet clinical standards (185).
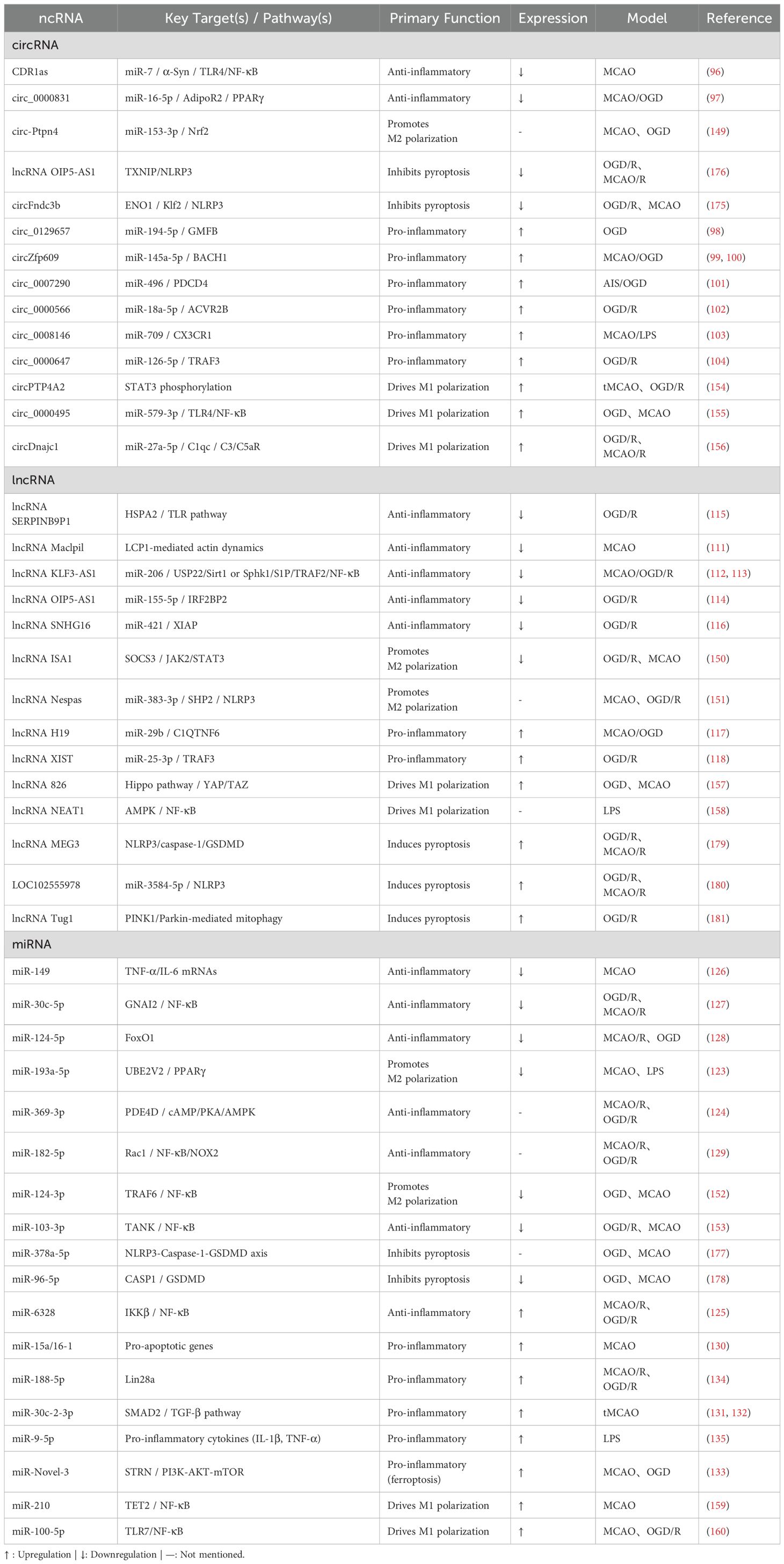
Table 4. Comparative summary of ncRNA functions and targets in IS-associated neuroinflammation across preclinical models.
Storage poses another formidable challenge, as exosomes are highly susceptible to degradation. Short-term storage at 4 °C suffices for less than 48 hours, but long-term preservation demands deep-freezing at -80 °C. Even under these conditions, exosomes suffer biofunctional decay, reduced concentration, vesicle fusion, and structural fragmentation. Prolonged storage or repeated freeze-thaw cycles exacerbate these issues, triggering aggregation that increases particle size and diminishes biological activity. Such degradation not only compromises therapeutic potential but also heightens immunogenicity risks through membrane rupture and cargo leakage (186). To mitigate these effects, protective strategies are indispensable. Storing exosomes at -80 °C in biocompatible cryoprotectants, such as 5% sucrose solution, outperforms PBS by better preserving size distribution, concentration, and membrane protein integrity. Complementing this, programmed gradient cooling at 1 °C/min suppresses ice crystal formation, reducing vesicle breakage and enhancing stability during extended storage (187).
Even with optimized production and storage, delivering ncRNA therapeutics via exosomes remains a complex endeavor. Nucleic acids face inherent vulnerabilities—nuclease degradation, poor stability, and limited transmembrane transport due to their negative charge—which impede efficient targeting and therapeutic impact (188). Achieving precise tissue- or cell-specific delivery is vital to limit off-target effects and systemic toxicity, presenting substantial challenges for delivery system design (189). Platforms like PEGylated nanoparticles improve circulation time and exploit the EPR effect (190), while focused ultrasound (FUS) enhances BBB penetration (191, 192). However, challenges such as immunogenicity persist.
Exosomes, as endogenous extracellular vesicles, offer a compelling solution, leveraging their natural nucleic acid-loading capacity, low immunogenicity, and tissue tropism. In preclinical studies of IS, intravenous injection predominates as the delivery route. Yet, natural exosomes encounter rapid clearance by the MPS, driven by plasma-derived opsonins binding to their hydrophobic surface domains. Consequently, over 90% of injected exosomes accumulate in peripheral organs, primarily the liver and spleen, leaving less than 1% to reach the ischemic brain parenchyma (193, 194). This nonspecific biodistribution undermines neuroprotective, anti-inflammatory, and pro-angiogenic effects while raising concerns about dose-dependent toxicity. Attempts to elevate brain concentrations through higher doses result in off-target accumulation in hepatic macrophages and splenic antigen-presenting cells, potentially triggering hepatotoxicity and systemic inflammation.
To overcome these delivery barriers, innovative engineering strategies have emerged. Conjugating “don’t-eat-me” signals, such as CD47 or CD55, reduces phagocytic clearance (195), while functionalizing exosomes with targeting ligands like RGD peptides enhances site-specific accumulation (196). Exploring alternative administration routes, such as intranasal delivery via the olfactory pathway, improves BBB penetration and brain enrichment. Advances in drug loading, exemplified by magnetic extrusion-derived exosome-mimetics using the ammonium sulfate gradient method, integrate enhanced payload capacity with precise targeting. Additionally, selecting specific cellular sources, such as neuron-derived exosomes bearing neural adhesion molecules, capitalizes on inherent BBB-crossing capabilities (197).These modifications collectively aim to refine delivery precision and efficacy.
Despite such progress, translating engineered exosomes into clinical practice remains hindered by persistent obstacles. Production at GMP scale incurs prohibitively high costs, compounded by suboptimal drug encapsulation efficiency and storage-related degradation. Complex pharmacokinetics—characterized by significant hepatic and splenic sequestration, low brain targeting, and a short half-life—further complicate therapeutic development. Addressing these challenges demands a comprehensive approach, where full-process GMP management plays a pivotal role. By enforcing rigorous raw material screening, terminal product quality control, strict aseptic operations, batch-to-batch consistency monitoring, and stability validation, this strategy ensures quality assurance and bridges critical logistical gaps. Ultimately, the clinical advancement of exosome-based ncRNA therapeutics hinges on systematically resolving these technical bottlenecks through integrated technological innovation and robust quality control.
7.2 The critical impact of biological heterogeneity: age, sex, and comorbidities
Biological heterogeneity, encompassing non-modifiable factors such as age, sex, and genetics, profoundly shapes the pathophysiology and progression of IS. These factors critically influence disease susceptibility, clinical presentation, and therapeutic outcomes (198, 199). Epidemiological evidence demonstrates that stroke incidence doubles every decade after age 55 (200). Sex-specific disparities are evident, with women facing a higher lifetime risk and mortality burden from stroke. Women account for 57.1% of global stroke deaths, and stroke ranks as the third leading cause of death in women over 55, compared to fifth in men (201–203). This disparity stems from female-specific risk factors, such as premature menopause and pregnancy complications, as well as amplified effects of shared comorbidities like hypertension and diabetes.
Preclinical studies reveal intricate interactions between age and sex in stroke outcomes. For instance, young female rodents show enhanced responses to neuroprotective treatments, such as reduced infarct volume with miR-15a/16–1 modulation. However, these sex differences diminish with age due to accumulated repair deficits (130). Interestingly, middle-aged female rodents exhibit greater cognitive vulnerability post-stroke but achieve optimal functional recovery with intervention (204). These findings underscore the role of estrogen-mediated neuroprotection and the age-dependent decline in neural plasticity.
Comorbidities, particularly hypertension and diabetes, further complicate the impact of biological heterogeneity on stroke. Hypertension escalates stroke risk in a dose-dependent manner (205), while diabetes independently doubles the risk and hastens stroke onset, especially in younger individuals (205, 206). Importantly, these comorbidities interact with aging to alter therapeutic efficacy. For example, the effectiveness of FosDT siRNA diminishes in aged hypertensive models and necessitates dose adjustments in diabetic contexts. This is attributed to accelerated aging of the neurovascular unit via the NF-κB-FosDT-REST inflammatory axis (207).
Diabetes significantly worsens stroke-induced neurovascular damage through mechanisms such as metabolic dysregulation, BBB disruption, white matter injury, and heightened inflammation. These factors not only impair recovery but also reduce the efficacy of standard neurorestorative therapies, such as exosomes derived from non-diabetic mesenchymal stem cells (208). However, exosomes from diabetic rat mesenchymal stem cells (T2DM-MSC-Exo) have shown unique therapeutic benefits in diabetic stroke models. They restore BBB integrity, reduce hemorrhage, attenuate neuroinflammation, and promote white matter repair through pathways involving miR-9/ABCA1 and IGF1R (209). Additionally, diabetes exacerbates post-stroke cardiac dysfunction via impaired miR-126 signaling, a deficit that exosome therapy can address by targeting multi-organ inflammatory cascades (28). These findings highlight how metabolic dysregulation, microvascular damage, and chronic inflammation compromise recovery in patients with comorbidities (208).
A major barrier to translating stroke therapeutics from bench to bedside is the reliance on preclinical models that overlook biological heterogeneity. While most studies employ young, healthy rodents, stroke primarily affects elderly individuals with multiple comorbidities (210). This mismatch is critical, as systematic reviews indicate that less than 10% of preclinical studies test interventions in comorbid models, where therapeutic efficacy is often significantly reduced (210).
Bridging this translational gap requires the adoption of more clinically relevant preclinical models. This entails using aged animals, ensuring balanced sex representation, and incorporating validated comorbidities such as hypertension and diabetes (210–212). Moreover, given species-specific physiological differences, advanced models like non-human primates (NHPs) are crucial. For instance, pharmacokinetic studies in pig-tailed macaques show that human exosomes have a circulation half-life of approximately 40 minutes—four times longer than in rodents—and display unique immune interactions, such as rapid binding to B lymphocytes (213). These insights highlight the shortcomings of rodent models for assessing biodistribution and underscore the value of NHPs in evaluating exosome pharmacodynamics to facilitate clinical translation.
To further address the translational challenges, Table 5 provides a systematic comparison of dysregulation patterns and functional consistency of key exosome/ncRNA regulators across preclinical and human contexts. This analysis identifies both promising concordant targets, such as circ_0000831 and miR-193a-5p, and critical discrepancies, like those involving lncRNA GAS5, which necessitate validation in human models.
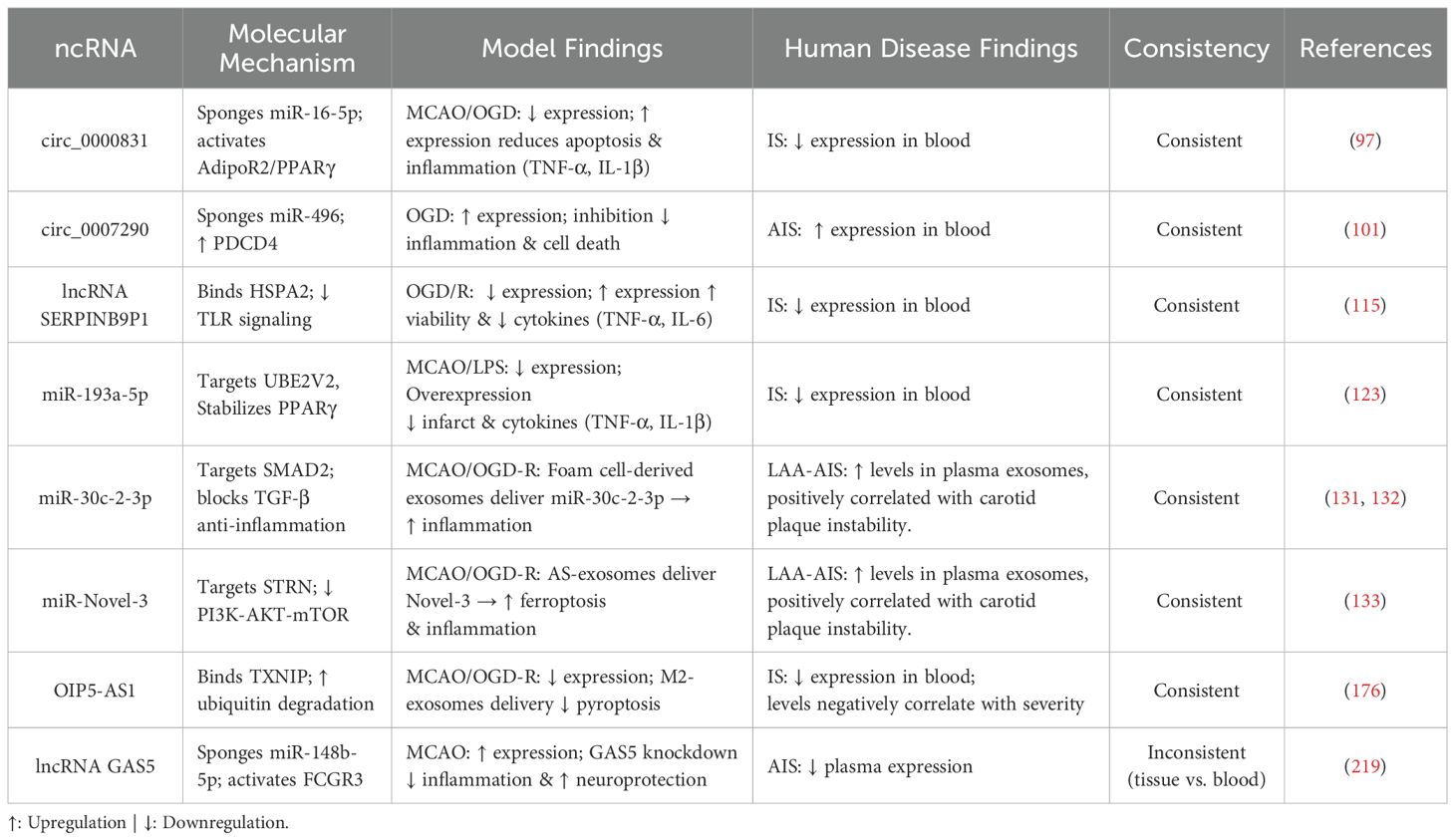
Table 5. Comparison of key findings on exosome/ncRNA regulation of neuroinflammation in preclinical models vs. human ischemic stroke.
7.3 Optimizing exosome sources for ncRNA delivery: stem cells vs. differentiated cells
The choice of exosome source is pivotal in optimizing ncRNA delivery for treating cerebral ischemia, as different cellular origins confer distinct therapeutic advantages and limitations. Both stem cell-derived and differentiated cell-derived exosomes have demonstrated potential in promoting neural plasticity and functional recovery in stroke models, largely through their cargo of ncRNAs and bioactive lipids, which mitigate inflammation and secondary neuronal damage (214).
For instance, mesenchymal stem cell (MSC)-derived and M2 microglia-derived exosomes both deliver miR-124 but exert their effects through divergent mechanisms. MSC-derived exosomes, when engineered with rabies virus glycoprotein (RVG) for enhanced targeting, penetrate the BBB more effectively and accumulate in ischemic lesions. This promotes neurogenesis and angiogenesis while indirectly ameliorating the inflammatory microenvironment (215). In contrast, M2 microglia-derived exosomes directly modulate neuroinflammation by reducing pro-inflammatory cytokines (IL-1β, TNF-α), elevating anti-inflammatory factors (IL-10, TGF-β), and inducing microglial polarization toward the M2 phenotype. However, their therapeutic impact requires repeated administration, indicating limited persistence in vivo (216).
Similarly, brain endothelial cell-derived exosomes (EC-Exo) and adipose-derived stem cell exosomes (ADSC-Exo) exhibit distinct properties in delivering miR-126. EC-Exo, naturally enriched with miR-126, efficiently crosses the damaged BBB and accumulates in endothelial cells and neurons within the ischemic border zone. By enhancing vascular integrity and promoting M2 macrophage polarization, EC-Exo reverses hyperglycemia-induced miR-126 deficiency and neurovascular damage in diabetic stroke models (28). Yet, the challenge of isolating brain endothelial cells constrains large-scale production. Conversely, ADSC-Exo requires genetic modification to boost miR-126 loading and, despite weaker BBB penetration, effectively suppresses microglial activation and pro-inflammatory cytokine secretion, indirectly fostering neurogenesis and angiogenesis (27). The ease of sourcing ADSC-Exo from adipose tissue supports clinical scalability, though repeated administration is necessary to sustain efficacy, suggesting rapid clearance.
In summary, stem cell-derived exosomes, such as those from MSCs and ADSCs, offer versatility through their amenability to engineering and their capacity for multi-dimensional repair—synergistically addressing inflammation, BBB protection, and neural/vascular regeneration. This makes them well-suited for complex, multi-target conditions like stroke. Differentiated cell-derived exosomes, such as those from M2 microglia and brain endothelial cells, provide precision by directly regulating specific pathways (e.g., microglial polarization or vascular reconstruction). However, their clinical translation is hindered by production challenges, source scarcity, and pathology-dependent risks. The optimal exosome source must therefore be selected based on a careful evaluation of targeting needs, therapeutic mechanism breadth, production feasibility, and administration logistics.
8 Discussion
IS remains a leading global cause of death and disability, characterized by brain tissue necrosis and neuronal loss. As explored in earlier sections, ncRNAs—including circRNAs, lncRNAs, and miRNAs—orchestrate the intricate inflammatory cascade central to stroke pathology and recovery. Operating through sophisticated molecular networks, these ncRNAs modulate neuroinflammatory signaling, influencing processes such as microglial polarization, cytokine production, neuronal survival, and vascular integrity. Yet, their therapeutic application hinges on overcoming the challenge of targeted delivery to the ischemic brain.
Exosomes emerge as pivotal delivery vectors in this context. These naturally occurring nanovesicles, distinguished by low immunogenicity and inherent biocompatibility, possess a unique ability to traverse the BBB. By encapsulating and protecting their ncRNA cargo, exosomes enable precise delivery to ischemic lesions and the neurovascular unit, allowing ncRNAs to reprogram detrimental immune responses—such as shifting microglial balance—while promoting reparative pathways. This exosome-ncRNA synergy marks a transformative approach to stroke therapeutics, addressing key limitations of current strategies. It extends therapeutic windows by targeting persistent secondary injury pathways, breaches the BBB for efficient CNS delivery, and finely tunes the multifaceted neuroinflammatory response. Rather than broad suppression, this modulation recalibrates the inflammatory milieu, curbing destructive pro-inflammatory cascades while supporting beneficial reparative functions, offering a nuanced strategy to mitigate ischemia/reperfusion injury and enhance neurological restoration.
The dynamic expression patterns of ncRNAs, which are fundamental to their role in regulating post-stroke neuroinflammation, combined with their stability in readily accessible biofluids such as blood and cerebrospinal fluid, render them exceptionally promising candidates for non-invasive diagnostic and prognostic biomarkers in IS (217, 218) (Table 6). Specific alterations in the levels of these regulatory molecules often correlate with the onset, severity, progression, and subtype of stroke, reflecting the underlying inflammatory state and tissue injury.
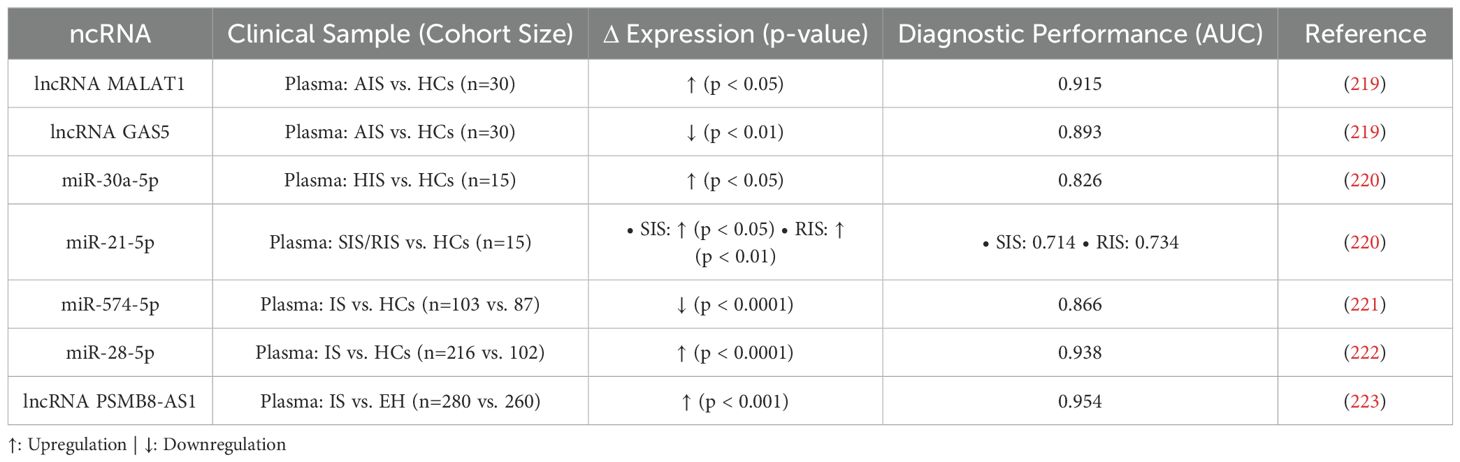
Table 6. ncRNA Biomarkers in Patient Biofluids: Diagnostic and Prognostic Potential for Ischemic Stroke-Associated Neuroinflammation.
Among promising ncRNA biomarker candidates, lncRNAs MALAT1 and GAS5 highlight both potential and complexity. In mouse MCAO models, both are upregulated in brain tissue, with MALAT1 knockout reducing neurological deficits, inflammation (e.g., IL-1β, TNF-α), and enhancing neuronal survival, indicating it worsens injury via inflammation, while GAS5 knockout curbs neuroinflammation and promotes neuroprotection (219). Yet, in AIS patient serum, MALAT1 rises while GAS5 falls, revealing a stark model-clinical divergence. This discrepancy, tied to tissue-specific regulation and species differences, complicates biomarker translation, though their opposing serum shifts, linked to disease and inflammation, affirm their diagnostic potential for AIS.
Nevertheless, their significant and opposing changes in patient serum, correlating with disease status and inflammation, underscore their strong potential as inflammation-linked diagnostic indicators for AIS Demonstrating temporal diagnostic utility, miR-30a-5p is specifically upregulated in the hyperacute stage (HIS) with excellent diagnostic efficacy (AUC=0.826), positioning it as a potential HIS-specific biomarker. Conversely, miR-21-5p shows sustained elevation during the subacute (SIS) and recovery (RIS) stages, likely linked to neuroprotective responses. Critically, combining these two miRNAs effectively distinguishes between HIS, SIS, and RIS stages, offering a high-value, non-invasive tool for stage-specific diagnosis with significant clinical translation prospects (220). Numerous other ncRNAs show significant diagnostic and/or prognostic value. For instance, serum miR-574-5p levels are significantly lower in IS patients, correlating negatively with neurological deficit severity (NIHSS) and serving as an independent risk factor for poor prognosis (221); serum HDL-C-associated miR-28-5p levels are elevated in AIS patients, negatively correlating with HDL-C (r = -0.848), demonstrating high diagnostic efficacy (AUC=0.938) and correlation with stroke severity (r=0.777) (222); and serum lncRNA PSMB8-AS1 is significantly elevated in IS versus hypertension alone (P < 0.001), showing high diagnostic accuracy (AUC=0.954), correlating with neurological deficit severity, and strongly predicting poor prognosis (mRS>2; AUC=0.888, sensitivity 97.8%) and increased 12-month recurrence risk (223).
While neuroimaging (CT/MRI) remains the diagnostic cornerstone for IS ncRNA detection emerges as a highly promising complementary approach. It offers a pathway towards earlier detection (particularly valuable in the hyperacute phase where imaging may be equivocal), enhanced sensitivity, non-invasiveness, and personalized prognosis, potentially addressing limitations like accessibility and cost. However, challenges remain: ncRNA expression can be influenced by comorbid conditions or other neurological injuries, and the precise assessment of disease progression and prognosis using specific further large-scale validation and refinement.
8.1 Limitations
While providing a systematic synthesis, this review has the following limitations: (i) It focuses primarily on neuroinflammation, not encompassing all stroke pathologies; (ii) Its conclusions rely solely on English-language studies from PubMed, potentially omitting relevant unpublished data or non-English findings, and it did not perform a meta-analysis, limiting statistical power and formal assessment of heterogeneity and publication bias; (iii) The field’s rapid evolution means new discoveries may supersede included evidence; (iv) Interpretation is constrained by inconsistent reporting of statistical parameters (e.g., power analysis, multiple testing corrections, sample size justification) across studies, variability in study quality, potential publication bias (underrepresentation of negative/null results), and low external/internal validity across diverse preclinical models; (v) Clinical biomarker data, though promising, require extensive validation.
9 Conclusions
Exosomes and ncRNAs constitute a groundbreaking frontier in understanding and managing post-IS neuroinflammation. Their capacity to precisely regulate inflammatory networks, penetrate the BBB, and serve as biomarkers offers unparalleled opportunities to address the shortcomings of existing stroke therapies. Despite persistent challenges in production, delivery, biological heterogeneity, and clinical validation, the mechanistic insights presented here lay a robust foundation. Through innovative engineering, rigorous preclinical testing in clinically relevant models, and strategic clinical translation, the full potential of exosome-ncRNA strategies can be realized, ultimately enhancing functional recovery and quality of life for stroke survivors.
Author contributions
DW: Writing – original draft, Investigation. FL: Writing – review & editing, Project administration. CG: Writing – review & editing. JC: Data curation, Writing – review & editing. YY: Methodology, Funding acquisition, Writing – review & editing, Resources, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from the Guangxi Natural Science Foundation (Grant No. 2025GXNSFAA0691007): “Protective Effects and Mechanisms of Qifu Yin in Regulating Neural Stem Cell Exosome Secretion against Cerebral Ischemia-Reperfusion Injury,” and the Guangxi Key Research and Development Program (Grant No. AB25069271): “Application Research of Large-scale Preparation of Umbilical Cord Mesenchymal Stem Cell Exosomes for the Treatment of Ischemic Stroke.”
Acknowledgments
Figure 1 was created using Figdraw (http://www.figdraw.com), with proper attribution and compliance with its terms of use.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Correction note
This article has been corrected with minor changes. These changes do not impact the scientific content of the article.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tsao CW, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, et al. Heart disease and stroke statistics-2023 update: A report from the american heart association. Circulation. (2023) 147:e93–e621. doi: 10.1161/cir.0000000000001123
3. Hilkens NA, Casolla B, Leung TW, and de Leeuw FE. Stroke. Lancet. (2024) 403:2820–36. doi: 10.1016/s0140-6736(24)00642-1
4. Kleindorfer DO, Towfighi A, Chaturvedi S, Cockroft KM, Gutierrez J, Lombardi-Hill D, et al. 2021 Guideline for the prevention of stroke in patients with stroke and transient ischemic attack: A guideline from the american heart association/American stroke association. Stroke. (2021) 52:e364–467. doi: 10.1161/str.0000000000000375
5. Tsivgoulis G, Katsanos AH, Sandset EC, Turc G, Nguyen TN, Bivard A, et al. Thrombolysis for acute ischemic stroke: current status and future perspectives. Lancet Neurol. (2023) 22:418–29. doi: 10.1016/s1474-4422(22)00519-1
6. Lee VH. Small step or giant leap? Expanding the acute stroke thrombolysis window to 24 hours. N Engl J Med. (2024) 391:273–5. doi: 10.1056/NEJMe2405846
7. Meng X, Li S, Dai H, Lu G, Wang W, Che F, et al. Tenecteplase vs alteplase for patients with acute ischemic stroke: the ORIGINAL randomized clinical trial. Jama. (2024) 332:1437–45. doi: 10.1001/jama.2024.14721
8. Langezaal LCM, van der Hoeven E, Mont’Alverne FJA, de Carvalho JJF, Lima FO, Dippel DWJ, et al. Endovascular therapy for stroke due to basilar-artery occlusion. N Engl J Med. (2021) 384:1910–20. doi: 10.1056/NEJMoa2030297
9. Nguyen TN, Abdalkader M, Fischer U, Qiu Z, Nagel S, Chen HS, et al. Endovascular management of acute stroke. Lancet. (2024) 404:1265–78. doi: 10.1016/s0140-6736(24)01410-7
10. Nogueira RG, Jadhav AP, Haussen DC, Bonafe A, Budzik RF, Bhuva P, et al. Thrombectomy 6 to 24 hours after stroke with a mismatch between deficit and infarct. N Engl J Med. (2018) 378:11–21. doi: 10.1056/NEJMoa1706442
11. Goyal M, Menon BK, van Zwam WH, Dippel DW, Mitchell PJ, Demchuk AM, et al. Endovascular thrombectomy after large-vessel ischemic stroke: a meta-analysis of individual patient data from five randomized trials. Lancet. (2016) 387:1723–31. doi: 10.1016/s0140-6736(16)00163-x
12. Asif KS, Otite FO, Desai SM, Herial N, Inoa V, Al-Mufti F, et al. Mechanical thrombectomy global access for stroke (MT-GLASS): A mission thrombectomy (MT-2020 plus) study. Circulation. (2023) 147:1208–20. doi: 10.1161/circulationaha.122.063366
14. Huang G, Zang J, He L, Zhu H, Huang J, Yuan Z, et al. Bioactive nanoenzyme reverses oxidative damage and endoplasmic reticulum stress in neurons under ischemic stroke. ACS Nano. (2022) 16:431–52. doi: 10.1021/acsnano.1c07205
15. Li C, Zhao Z, Luo Y, Ning T, Liu P, Chen Q, et al. Macrophage-disguised manganese dioxide nanoparticles for neuroprotection by reducing oxidative stress and modulating inflammatory microenvironment in acute ischemic stroke. Adv Sci (Weinh). (2021) 8:e2101526. doi: 10.1002/advs.202101526
16. Nie X, Leng X, Miao Z, Fisher M, and Liu L. Clinically ineffective reperfusion after endovascular therapy in acute ischemic stroke. Stroke. (2023) 54:873–81. doi: 10.1161/strokeaha.122.038466
17. Wassélius J, Arnberg F, von Euler M, Wester P, and Ullberg T. Endovascular thrombectomy for acute ischemic stroke. J Intern Med. (2022) 291:303–16. doi: 10.1111/joim.13425
18. Dong X. Current strategies for brain drug delivery. Theranostics. (2018) 8:1481–93. doi: 10.7150/thno.21254
19. Ma X, Peng L, Zhu X, Chu T, Yang C, Zhou B, et al. Isolation, identification, and challenges of extracellular vesicles: emerging players in clinical applications. Apoptosis. (2024) 30:422–45. doi: 10.1007/s10495-024-02036-2
20. Kalra H, Simpson RJ, Ji H, Aikawa E, Altevogt P, Askenase P, et al. Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol. (2012) 10:e1001450. doi: 10.1371/journal.pbio.1001450
21. Simpson RJ and Mathivanan S. Extracellular microvesicles: the need for internationally recognized nomenclature and stringent purification criteria. J Proteomics Bioinform. (2012) 05. doi: 10.4172/jpb.10000e10
22. van Niel G, D’Angelo G, and Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. (2018) 19:213–28. doi: 10.1038/nrm.2017.125
23. Dai J, Su Y, Zhong S, Cong L, Liu B, Yang J, et al. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct Target Ther. (2020) 5:145. doi: 10.1038/s41392-020-00261-0
24. Yokoi A and Ochiya T. Exosomes and extracellular vesicles: Rethinking the essential values in cancer biology. Semin Cancer Biol. (2021) 74:79–91. doi: 10.1016/j.semcancer.2021.03.032
25. Webb RL, Kaiser EE, Scoville SL, Thompson TA, Fatima S, Pandya C, et al. Human neural stem cell extracellular vesicles improve tissue and functional recovery in the murine thromboembolic stroke model. Transl Stroke Res. (2018) 9:530–9. doi: 10.1007/s12975-017-0599-2
26. Webb RL, Kaiser EE, Jurgielewicz BJ, Spellicy S, Scoville SL, Thompson TA, et al. Human neural stem cell extracellular vesicles improve recovery in a porcine model of ischemic stroke. Stroke. (2018) 49:1248–56. doi: 10.1161/strokeaha.117.020353
27. Geng W, Tang H, Luo S, Lv Y, Liang D, Kang X, et al. Exosomes from miRNA-126-modified ADSCs promotes functional recovery after stroke in rats by improving neurogenesis and suppressing microglia activation. Am J Transl Res. (2019) 11:780–92.
28. Venkat P, Cui C, Chopp M, Zacharek A, Wang F, Landschoot-Ward J, et al. MiR-126 mediates brain endothelial cell exosome treatment-induced neurorestorative effects after stroke in type 2 diabetes mellitus mice. Stroke. (2019) 50:2865–74. doi: 10.1161/strokeaha.119.025371
29. Liang Y, Duan L, Lu J, and Xia J. Engineering exosomes for targeted drug delivery. Theranostics. (2021) 11:3183–95. doi: 10.7150/thno.52570
30. Lai Z, Ye T, Zhang M, and Mu Y. Exosomes as vehicles for noncoding RNA in modulating inflammation: A promising regulatory approach for ischemic stroke and myocardial infarction. J Inflammation Res. (2024) 17:7485–501. doi: 10.2147/jir.S484119
31. Mercer TR, Dinger ME, and Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. (2009) 10:155–9. doi: 10.1038/nrg2521
32. Ghafouri-Fard S, Shoorei H, and Taheri M. Non-coding RNAs participate in the ischemia-reperfusion injury. BioMed Pharmacother. (2020) 129:110419. doi: 10.1016/j.biopha.2020.110419
33. Pan Y, Liu Y, Wei W, Yang X, Wang Z, and Xin W. Extracellular vesicles as delivery shippers for noncoding RNA-based modulation of angiogenesis: insights from ischemic stroke and cancer. Small. (2023) 19:e2205739. doi: 10.1002/smll.202205739
34. Yang R, Yang B, Liu W, Tan C, Chen H, and Wang X. Emerging role of non-coding RNAs in neuroinflammation mediated by microglia and astrocytes. J Neuroinflamm. (2023) 20:173. doi: 10.1186/s12974-023-02856-0
35. Kuang Y, Zheng X, Zhang L, Ai X, Venkataramani V, Kilic E, et al. Adipose-derived mesenchymal stem cells reduce autophagy in stroke mice by extracellular vesicle transfer of miR-25. J Extracell Vesicles. (2020) 10:e12024. doi: 10.1002/jev2.12024
36. Zuo ML, Wang AP, Song GL, and Yang ZB. miR-652 protects rats from cerebral ischemia/reperfusion oxidative stress injury by directly targeting NOX2. BioMed Pharmacother. (2020) 124:109860. doi: 10.1016/j.biopha.2020.109860
37. Salmena L, Poliseno L, Tay Y, Kats L, and Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. (2011) 146:353–8. doi: 10.1016/j.cell.2011.07.014
38. Zeng Z, Xia L, Fan S, Zheng J, Qin J, Fan X, et al. Circular RNA circMAP3K5 acts as a microRNA-22-3p sponge to promote resolution of intimal hyperplasia via TET2-mediated smooth muscle cell differentiation. Circulation. (2021) 143:354–71. doi: 10.1161/circulationaha.120.049715
39. Yang L, Wilusz JE, and Chen LL. Biogenesis and regulatory roles of circular RNAs. Annu Rev Cell Dev Biol. (2022) 38:263–89. doi: 10.1146/annurev-cellbio-120420-125117
40. Chen B, Yi J, Xu Y, Zheng P, Tang R, and Liu B. Construction of a circRNA-miRNA-mRNA network revealed the potential mechanism of Buyang Huanwu Decoction in the treatment of cerebral ischemia. BioMed Pharmacother. (2022) 145:112445. doi: 10.1016/j.biopha.2021.112445
41. Liu M, Sun H, Yao Q, Wang D, Zhang J, Ye X, et al. New insights into roles of IL-7R gene as a diagnostic biomarker for post-stroke depression. Front Immunol. (2024) 15:1506214. doi: 10.3389/fimmu.2024.1506214
42. Palmer SJ. Identification, care and prevention of stroke is possible. Br J Healthc. Assist. (2023) 17:236–9.
43. Xing C, Arai K, Lo EH, and Hommel M. Pathophysiologic cascades in ischemic stroke. Int J Stroke. (2012) 7:378–85. doi: 10.1111/j.1747-4949.2012.00839.x
44. Jadhav AP, Desai SM, and Jovin TG. Indications for mechanical thrombectomy for acute ischemic stroke: current guidelines and beyond. Neurology. (2021) 97:S126–s136. doi: 10.1212/wnl.0000000000012801
45. Xiang Q, Yi X, Zhu XH, Wei X, and Jiang DS. Regulated cell death in myocardial ischemia-reperfusion injury. Trends Endocrinol Metab. (2024) 35:219–34. doi: 10.1016/j.tem.2023.10.010
46. Frangogiannis NG. Pathophysiology of myocardial infarction. Compr Physiol. (2015) 5:1841–75. doi: 10.1002/cphy.c150006
47. Zhang M, Liu Q, Meng H, Duan H, Liu X, Wu J, et al. Ischemia-reperfusion injury: molecular mechanisms and therapeutic targets. Signal Transduct Target Ther. (2024) 9:12. doi: 10.1038/s41392-023-01688-x
48. Ding S, Kim YJ, Huang KY, Um D, Jung Y, and Kong H. Delivery-mediated exosomal therapeutics in ischemia-reperfusion injury: advances, mechanisms, and future directions. Nano Converg. (2024) 11:18. doi: 10.1186/s40580-024-00423-8
49. Moskowitz MA, Lo EH, and Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. (2010) 67:181–98. doi: 10.1016/j.neuron.2010.07.002
50. Salaudeen MA, Bello N, Danraka RN, and Ammani ML. Understanding the pathophysiology of ischemic stroke: the basis of current therapies and opportunity for new ones. Biomolecules. (2024) 14:305. doi: 10.3390/biom14030305
51. Estes ML and McAllister AK. Alterations in immune cells and mediators in the brain: it’s not always neuroinflammation! Brain Pathol. (2014) 24:623–30. doi: 10.1111/bpa.12198
52. O’Callaghan JP, Sriram K, and Miller DB. Defining “neuroinflammation. Ann N Y Acad Sci. (2008) 1139:318–30. doi: 10.1196/annals.1432.032
53. Woodburn SC, Bollinger JL, and Wohleb ES. The semantics of microglia activation: neuroinflammation, homeostasis, and stress. J Neuroinflamm. (2021) 18:258. doi: 10.1186/s12974-021-02309-6
54. Kono H and Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. (2008) 8:279–89. doi: 10.1038/nri2215
55. Marsh BJ, Williams-Karnesky RL, and Stenzel-Poore MP. Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience. (2009) 158:1007–20. doi: 10.1016/j.neuroscience.2008.07.067
56. Gülke E, Gelderblom M, and Magnus T. Danger signals in stroke and their role on microglia activation after ischemia. Ther Adv Neurol Disord. (2018) 11:1756286418774254. doi: 10.1177/1756286418774254
57. Przykaza Ł. Understanding the connection between common stroke comorbidities, their associated inflammation, and the course of the cerebral ischemia/reperfusion cascade. Front Immunol. (2021) 12:782569. doi: 10.3389/fimmu.2021.782569
58. Alsbrook DL, Di Napoli M, Bhatia K, Biller J, Andalib S, Hinduja A, et al. Neuroinflammation in acute ischemic and hemorrhagic stroke. Curr Neurol Neurosci Rep. (2023) 23:407–31. doi: 10.1007/s11910-023-01282-2
59. Iadecola C and Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. (2011) 17:796–808. doi: 10.1038/nm.2399
60. Drieu A, Levard D, Vivien D, and Rubio M. Anti-inflammatory treatments for stroke: from bench to bedside. Ther Adv Neurol Disord. (2018) 11:1756286418789854. doi: 10.1177/1756286418789854
61. den Hertog HM, van Rossum JA, van der Worp HB, van Gemert HM, de Jonge R, Koudstaal PJ, et al. C-reactive protein in the very early phase of acute ischemic stroke: association with poor outcome and death. J Neurol. (2009) 256:2003–8. doi: 10.1007/s00415-009-5228-x
62. Grotta JC. tPA for stroke: important progress in achieving faster treatment. Jama. (2014) 311:1615–7. doi: 10.1001/jama.2014.3322
63. Berge E, Whiteley W, Audebert H, De Marchis GM, Fonseca AC, Padiglioni C, et al. European Stroke Organization (ESO) guidelines on intravenous thrombolysis for acute ischemic stroke. Eur Stroke J. (2021) 6:I–lxii. doi: 10.1177/2396987321989865
64. Wang Y, Li S, Pan Y, Li H, Parsons MW, Campbell BCV, et al. Tenecteplase versus alteplase in acute ischemic cerebrovascular events (TRACE-2): a phase 3, multicenter, open-label, randomized controlled, non-inferiority trial. Lancet. (2023) 401:645–54. doi: 10.1016/s0140-6736(22)02600-9
65. Wolters FJ, Li L, Gutnikov SA, Mehta Z, and Rothwell PM. Medical attention seeking after transient ischemic attack and minor stroke before and after the UK face, arm, speech, time (FAST) public education campaign: results from the oxford vascular study. JAMA Neurol. (2018) 75:1225–33. doi: 10.1001/jamaneurol.2018.1603
66. Lacy CR, Suh DC, Bueno M, and Kostis JB. Delay in presentation and evaluation for acute stroke: Stroke Time Registry for Outcomes Knowledge and Epidemiology (S.T.R.O.K.E.). Stroke. (2001) 32:63–9. doi: 10.1161/01.str.32.1.63
67. Hill MD and Michel P. Tenecteplase knocking on the door. Stroke. (2018) 49:2276–7. doi: 10.1161/strokeaha.118.022318
68. Li S, Gu HQ, Li H, Wang X, Jin A, Guo S, et al. Reteplase versus alteplase for acute ischemic stroke. N Engl J Med. (2024) 390:2264–73. doi: 10.1056/NEJMoa2400314
69. Vanacker P, Lambrou D, Eskandari A, Mosimann PJ, Maghraoui A, and Michel P. Eligibility and predictors for acute revascularization procedures in a stroke center. Stroke. (2016) 47:1844–9. doi: 10.1161/strokeaha.115.012577
70. Krishnan R, Mays W, and Elijovich L. Complications of mechanical thrombectomy in acute ischemic stroke. Neurology. (2021) 97:S115–s125. doi: 10.1212/wnl.0000000000012803
71. Berkhemer OA, Fransen PS, Beumer D, van den Berg LA, Lingsma HF, Yoo AJ, et al. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. (2015) 372:11–20. doi: 10.1056/NEJMoa1411587
72. Jovin TG, Nogueira RG, Lansberg MG, Demchuk AM, Martins SO, Mocco J, et al. Thrombectomy for anterior circulation stroke beyond 6 h from time last known well (AURORA): a systematic review and individual patient data meta-analysis. Lancet. (2022) 399:249–58. doi: 10.1016/s0140-6736(21)01341-6
73. Collaborative overview of randomized trials of antiplatelet therapy–I: Prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. Antiplatelet Trialists’ Collaboration. Bmj. (1994) 308:81–106. doi: 10.1136/bmj.308.6921.81
74. Shah J, Liu S, and Yu W. Contemporary antiplatelet therapy for secondary stroke prevention: a narrative review of current literature and guidelines. Stroke Vasc Neurol. (2022) 7:406–14. doi: 10.1136/svn-2021-001166
75. Greco A, Occhipinti G, Giacoppo D, Agnello F, Laudani C, Spagnolo M, et al. Antithrombotic therapy for primary and secondary prevention of ischemic stroke: JACC state-of-the-art review. J Am Coll Cardiol. (2023) 82:1538–57. doi: 10.1016/j.jacc.2023.07.025
76. Schirmer SH, Baumhäkel M, Neuberger HR, Hohnloser SH, van Gelder IC, Lip GY, et al. Novel anticoagulants for stroke prevention in atrial fibrillation: current clinical evidence and future developments. J Am Coll Cardiol. (2010) 56:2067–76. doi: 10.1016/j.jacc.2010.09.017
77. Campbell BCV, De Silva DA, Macleod MR, Coutts SB, Schwamm LH, Davis SM, et al. Ischemic stroke. Nat Rev Dis Primers. (2019) 5:70. doi: 10.1038/s41572-019-0118-8
78. Seiffge DJ, Werring DJ, Paciaroni M, Dawson J, Warach S, Milling TJ, et al. Timing of anticoagulation after recent ischemic stroke in patients with atrial fibrillation. Lancet Neurol. (2019) 18:117–26. doi: 10.1016/s1474-4422(18)30356-9
79. Zhao Y, Li Q, Niu J, Guo E, Zhao C, Zhang J, et al. Neutrophil membrane-camouflaged polyprodrug nanomedicine for inflammation suppression in ischemic stroke therapy. Adv Mater. (2024) 36:e2311803. doi: 10.1002/adma.202311803
80. Chen B, Cao P, Guo X, Yin M, Li X, Jiang L, et al. Maraviroc, an inhibitor of chemokine receptor type 5, alleviates neuroinflammatory response after cerebral Ischemia/reperfusion injury via regulating MAPK/NF-κB signaling. Int Immunopharmacol. (2022) 108:108755. doi: 10.1016/j.intimp.2022.108755
81. Enzmann G, Kargaran S, and Engelhardt B. Ischemia-reperfusion injury in stroke: impact of the brain barriers and brain immune privilege on neutrophil function. Ther Adv Neurol Disord. (2018) 11:1756286418794184. doi: 10.1177/1756286418794184
82. Aronowski J, Strong R, and Grotta JC. Reperfusion injury: demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. J Cereb Blood Flow Metab. (1997) 17:1048–56. doi: 10.1097/00004647-199710000-00006
83. Stuckey SM, Ong LK, Collins-Praino LE, and Turner RJ. Neuroinflammation as a key driver of secondary neurodegeneration following stroke? Int J Mol Sci. (2021) 22. doi: 10.3390/ijms222313101
84. Chunhui G, Yanqiu Y, Jibing C, Ning L, and Fujun L. Exosomes and non-coding RNAs: bridging the gap in Alzheimer’s pathogenesis and therapeutics. Metab Brain Dis. (2025) 40:84. doi: 10.1007/s11011-024-01520-7
85. Chen J, Liu P, Dong X, Jin J, and Xu Y. The role of lncRNAs in ischemic stroke. Neurochem Int. (2021) 147:105019. doi: 10.1016/j.neuint.2021.105019
86. Tiedt S, Prestel M, Malik R, Schieferdecker N, Duering M, Kautzky V, et al. RNA-Seq Identifies Circulating miR-125a-5p, miR-125b-5p, and miR-143-3p as Potential Biomarkers for Acute Ischemic Stroke. Circ Res. (2017) 121:970–80. doi: 10.1161/circresaha.117.311572
87. Liu JJ, Long YF, Xu P, Guo HD, and Cui GH. Pathogenesis of miR-155 on nonmodifiable and modifiable risk factors in Alzheimer’s disease. Alzheimers Res Ther. (2023) 15:122. doi: 10.1186/s13195-023-01264-z
88. Ha D, Yang N, and Nadithe V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm Sin B. (2016) 6:287–96. doi: 10.1016/j.apsb.2016.02.001
89. Gao L, Qiu F, Cao H, Li H, Dai G, Ma T, et al. Therapeutic delivery of microRNA-125a-5p oligonucleotides improves recovery from myocardial ischemia/reperfusion injury in mice and swine. Theranostics. (2023) 13:685–703. doi: 10.7150/thno.73568
90. Wang Y, Niu H, Li L, Han J, Liu Z, Chu M, et al. Anti-CHAC1 exosomes for nose-to-brain delivery of miR-760-3p in cerebral ischemia/reperfusion injury mice inhibiting neuron ferroptosis. J Nanobiotechnology. (2023) 21:109. doi: 10.1186/s12951-023-01862-x
91. Chen LL and Yang L. Regulation of circRNA biogenesis. RNA Biol. (2015) 12:381–8. doi: 10.1080/15476286.2015.1020271
92. He AT, Liu J, Li F, and Yang BB. Targeting circular RNAs as a therapeutic approach: current strategies and challenges. Signal Transduct Target Ther. (2021) 6:185. doi: 10.1038/s41392-021-00569-5
93. Tang X, Ren H, Guo M, Qian J, Yang Y, and Gu C. Review on circular RNAs and new insights into their roles in cancer. Comput Struct Biotechnol J. (2021) 19:910–28. doi: 10.1016/j.csbj.2021.01.018
94. Tay Y, Rinn J, and Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. (2014) 505:344–52. doi: 10.1038/nature12986
95. Yang K, Zeng L, Ge A, Wang S, Zeng J, Yuan X, et al. A systematic review of the research progress of non-coding RNA in neuroinflammation and immune regulation in cerebral infarction/ischemia-reperfusion injury. Front Immunol. (2022) 13:930171. doi: 10.3389/fimmu.2022.930171
96. Mehta SL, Chokkalla AK, Bathula S, Arruri V, Chelluboina B, and Vemuganti R. CDR1as regulates α-synuclein-mediated ischemic brain damage by controlling miR-7 availability. Mol Ther Nucleic Acids. (2023) 31:57–67. doi: 10.1016/j.omtn.2022.11.022
97. Huang R, Zhang W, Li W, Gao Y, Zheng D, and Bi G. Overexpressing circ_0000831 is sufficient to inhibit neuroinflammation and vertigo in cerebral ischemia through a miR-16-5p-dependent mechanism. Exp Neurol. (2022) 353:114047. doi: 10.1016/j.expneurol.2022.114047
98. Qian Y, Tang B, Zhang H, and Yang H. Highly-expressed circ_0129657 inhibits proliferation as well as promotes apoptosis and inflammation in HBMECs after oxygen-glucose deprivation via miR-194-5p/GMFB axis. Autoimmunity. (2023) 56:2201405. doi: 10.1080/08916934.2023.2201405
99. Wang H, Liao S, Li H, Chen Y, and Yu J. Long non-coding RNA TUG1 sponges mir-145a-5p to regulate microglial polarization after oxygen-glucose deprivation. Front Mol Neurosci. (2019) 12:215. doi: 10.3389/fnmol.2019.00215
100. Zhou Z, Wang X, Hu Q, and Yang Z. CircZfp609 contributes to cerebral infarction via sponging miR-145a-5p to regulate BACH1. Metab Brain Dis. (2023) 38:1971–81. doi: 10.1007/s11011-023-01208-4
101. Wang F, Liu J, Wang D, Yao Y, and Jiao X. Knockdown of circ_0007290 alleviates oxygen-glucose deprivation-induced neuronal injury by regulating miR-496/PDCD4 axis. Metab Brain Dis. (2022) 37:807–18. doi: 10.1007/s11011-021-00900-7
102. Liu D, Xiao H, Liu J, Zhang Y, Li J, Zhang T, et al. Circ_0000566 contributes oxygen-glucose deprivation and reoxygenation (OGD/R)-induced human brain microvascular endothelial cell injury via regulating miR-18a-5p/ACVR2B axis. Metab Brain Dis. (2023) 38:1273–84. doi: 10.1007/s11011-023-01166-x
103. Li L, Zhang D, Yao W, Wu Z, Cheng J, Ji Y, et al. Ligustrazine exerts neuroprotective effects via circ_0008146/miR-709/Cx3cr1 axis to inhibit cell apoptosis and inflammation after cerebral ischemia/reperfusion injury. Brain Res Bull. (2022) 190:244–55. doi: 10.1016/j.brainresbull.2022.10.011
104. Dai Y, Sheng Y, Deng Y, Wang H, Zhao Z, Yu X, et al. Circ_0000647 promotes cell injury by modulating miR-126-5p/TRAF3 axis in oxygen-glucose deprivation and reperfusion-induced SK-N-SH cell model. Int Immunopharmacol. (2022) 104:108464. doi: 10.1016/j.intimp.2021.108464
105. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. (2012) 22:1775–89. doi: 10.1101/gr.132159.111
106. Bao MH, Szeto V, Yang BB, Zhu SZ, Sun HS, and Feng ZP. Long non-coding RNAs in ischemic stroke. Cell Death Dis. (2018) 9:281. doi: 10.1038/s41419-018-0282-x
107. Kopp F and Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. (2018) 172:393–407. doi: 10.1016/j.cell.2018.01.011
108. Scoyni F, Sitnikova V, Giudice L, Korhonen P, Trevisan DM, Hernandez de Sande A, et al. ciRS-7 and miR-7 regulate ischemia-induced neuronal death via glutamatergic signaling. Cell Rep. (2024) 43:113862. doi: 10.1016/j.celrep.2024.113862
109. Dykstra-Aiello C, Jickling GC, Ander BP, Shroff N, Zhan X, Liu D, et al. Altered expression of long noncoding RNAs in blood after ischemic stroke and proximity to putative stroke risk loci. Stroke. (2016) 47:2896–903. doi: 10.1161/strokeaha.116.013869
110. Srinivas T, Mathias C, Oliveira-Mateos C, and Guil S. Roles of lncRNAs in brain development and pathogenesis: Emerging therapeutic opportunities. Mol Ther. (2023) 31:1550–61. doi: 10.1016/j.ymthe.2023.02.008
111. Wang Y, Yin Q, Yang D, Jin H, Yao Y, Song J, et al. LCP1 knockdown in monocyte-derived macrophages: mitigating ischemic brain injury and shaping immune cell signaling and metabolism. Theranostics. (2024) 14:159–75. doi: 10.7150/thno.88678
112. Cao Y, Wang D, and Zhou D. MSC promotes the secretion of exosomal lncRNA KLF3-AS1 to regulate sphk1 through YY1-musashi-1 axis and improve cerebral ischemia-reperfusion injury. Mol Neurobiol. (2024) 61:10462–80. doi: 10.1007/s12035-024-04150-3
113. Xie X, Cao Y, Dai L, and Zhou D. Bone marrow mesenchymal stem cell-derived exosomal lncRNA KLF3-AS1 stabilizes Sirt1 protein to improve cerebral ischemia/reperfusion injury via miR-206/USP22 axis. Mol Med. (2023) 29:3. doi: 10.1186/s10020-022-00595-1
114. Zhang JK, Li Y, Yu ZT, Jiang JW, Tang H, Tu GL, et al. OIP5-AS1 inhibits oxidative stress and inflammation in ischemic stroke through miR-155-5p/IRF2BP2 axis. Neurochem Res. (2023) 48:1382–94. doi: 10.1007/s11064-022-03830-7
115. Lv M, Song X, Wang W, Li J, Chen J, Huang X, et al. LncRNA SERPINB9P1 mitigates cerebral injury induced by oxygen–Glucose deprivation/reoxygenation by interacting with HSPA2. Mol Neurobiol. (2025) 62:6397–409. doi: 10.1007/s12035-024-04646-y
116. Cao X, Ma J, and Li S. Mechanism of lncRNA SNHG16 in oxidative stress and inflammation in oxygen-glucose deprivation and reoxygenation-induced SK-N-SH cells. Bioengineered. (2022) 13:5021–34. doi: 10.1080/21655979.2022.2026861
117. Li G, Ma X, Zhao H, Fan J, Liu T, Luo Y, et al. Long non-coding RNA H19 promotes leukocyte inflammation in ischemic stroke by targeting the miR-29b/C1QTNF6 axis. CNS Neurosci Ther. (2022) 28:953–63. doi: 10.1111/cns.13829
118. Li Y, Zhang JK, Yu ZT, Jiang JW, Tang H, Tu GL, et al. LncRNA XIST exacerbates oxygen-glucose deprivation/reoxygenation-induced cerebral injury through the miR-25-3p/TRAF3 axis. Mol Neurobiol. (2023) 60:6109–20. doi: 10.1007/s12035-023-03450-4
119. Dong H, Lei J, Ding L, Wen Y, Ju H, and Zhang X. MicroRNA: function, detection, and bioanalysis. Chem Rev. (2013) 113:6207–33. doi: 10.1021/cr300362f
120. Carthew RW and Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. (2009) 136:642–55. doi: 10.1016/j.cell.2009.01.035
121. Ambros V. The functions of animal microRNAs. Nature. (2004) 431:350–5. doi: 10.1038/nature02871
122. Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet. (2005) 37:495–500. doi: 10.1038/ng1536
123. Han Z, Li L, Zhao H, Wang R, Yan F, Tao Z, et al. MicroRNA-193a-5p rescues ischemic cerebral injury by restoring N2-like neutrophil subsets. Transl Stroke Res. (2023) 14:589–607. doi: 10.1007/s12975-022-01071-y
124. Tao H, Li L, Dong L, Chen H, Shan X, Zhuge L, et al. Growth differentiation factor 7 pretreatment enhances the therapeutic capacity of bone marrow-derived mesenchymal stromal cells against cerebral ischemia-reperfusion injury. Chem Biol Interact. (2023) 386:110779. doi: 10.1016/j.cbi.2023.110779
125. Li Y, Jin T, Liu N, Wang J, Qin Z, Yin S, et al. A short peptide exerts neuroprotective effects on cerebral ischemia-reperfusion injury by reducing inflammation via the miR-6328/IKKβ/NF-κB axis. J Neuroinflamm. (2023) 20:53. doi: 10.1186/s12974-023-02739-4
126. Vahidi S, Bigdeli MR, Shahsavarani H, Ahmadloo S, and Roghani M. Neuroprotective Therapeutic Potential of microRNA-149-5p against Murine Ischemic Stroke. Mol Neurobiol. (2024) 61:8886–903. doi: 10.1007/s12035-024-04159-8
127. Deng X, Zeng Y, and Ding D. MiR-30c-5p-targeted regulation of GNAI2 improves neural function injury and inflammation in cerebral ischemia-reperfusion injury. Appl Biochem Biotechnol. (2024) 196:5235–48. doi: 10.1007/s12010-023-04802-5
128. Su W, Lv M, Wang D, He Y, Han H, Zhang Y, et al. Tanshinone IIA Alleviates Traumatic Brain Injury by Reducing Ischemia–Reperfusion via the miR-124-5p/FoxO1 Axis. Mediators Inflammation. (2024) 2024:7459054. doi: 10.1155/2024/7459054
129. Ding W, Gu Q, Liu M, Zou J, Sun J, and Zhu J. Astrocytes-derived exosomes pre-treated by berberine inhibit neuroinflammation after stroke via miR-182-5p/Rac1 pathway. Int Immunopharmacol. (2023) 118:110047. doi: 10.1016/j.intimp.2023.110047
130. Huang X, Li S, Qiu N, Ni A, Xiong T, Xue J, et al. Sex and age-dependent effects of miR-15a/16–1 antagomir on ischemic stroke outcomes. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms252111765
131. Fang L, Wu S, Zhu X, Cai J, Wu J, He Z, et al. MYEOV functions as an amplified competing endogenous RNA in promoting metastasis by activating TGF-β pathway in NSCLC. Oncogene. (2019) 38:896–912. doi: 10.1038/s41388-018-0484-9
132. Tang Y, Dong MH, Pang XW, Zhang H, Chu YH, Zhou LQ, et al. Macrophage exosomal miR-30c-2-3p in atherosclerotic plaques aggravates microglial neuroinflammation during large-artery atherosclerotic stroke via TGF-β/SMAD2 pathway. J Neuroinflamm. (2024) 21:292. doi: 10.1186/s12974-024-03281-7
133. Qin C, Dong MH, Tang Y, Chu YH, Zhou LQ, Zhang H, et al. The foam cell-derived exosomal miRNA Novel-3 drives neuroinflammation and ferroptosis during ischemic stroke. Nat Aging. (2024) 4:1845–61. doi: 10.1038/s43587-024-00727-8
134. Hou D, Pei C, Yu D, and Yang G. miR-188-5p silencing improves cerebral ischemia/reperfusion injury by targeting Lin28a. Metab Brain Dis. (2023) 38:2327–38. doi: 10.1007/s11011-023-01273-9
135. Wang M, Yang Y, Guo Y, Tan R, Sheng Y, Chui H, et al. Xiaoxuming decoction cutting formula reduces LPS-stimulated inflammation in BV-2 cells by regulating miR-9-5p in microglia exosomes. Front Pharmacol. (2023) 14:1183612. doi: 10.3389/fphar.2023.1183612
136. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
137. Zhao YX, Song JY, Bao XW, Zhang JL, Wu JC, Wang LY, et al. Single-cell RNA sequencing-guided fate-mapping toolkit delineates the contribution of yolk sac erythro-myeloid progenitors. Cell Rep. (2023) 42:113364. doi: 10.1016/j.celrep.2023.113364
138. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518:547–51. doi: 10.1038/nature13989
139. Kettenmann H, Hanisch UK, Noda M, and Verkhratsky A. Physiology of microglia. Physiol Rev. (2011) 91:461–553. doi: 10.1152/physrev.00011.2010
140. Prinz M, Masuda T, Wheeler MA, and Quintana FJ. Microglia and central nervous system-associated macrophages-from origin to disease modulation. Annu Rev Immunol. (2021) 39:251–77. doi: 10.1146/annurev-immunol-093019-110159
141. Borst K, Dumas AA, and Prinz M. Microglia: Immune and non-immune functions. Immunity. (2021) 54:2194–208. doi: 10.1016/j.immuni.2021.09.014
143. Yan J, Zhang Y, Wang L, Li Z, Tang S, Wang Y, et al. TREM2 activation alleviates neural damage via Akt/CREB/BDNF signaling after traumatic brain injury in mice. J Neuroinflamm. (2022) 19:289. doi: 10.1186/s12974-022-02651-3
144. Wei W, Zhang L, Xin W, Pan Y, Tatenhorst L, Hao Z, et al. TREM2 regulates microglial lipid droplet formation and represses post-ischemic brain injury. BioMed Pharmacother. (2024) 170:115962. doi: 10.1016/j.biopha.2023.115962
145. Sun R and Jiang H. Border-associated macrophages in the central nervous system. J Neuroinflamm. (2024) 21:67. doi: 10.1186/s12974-024-03059-x
146. Var SR, Shetty AV, Grande AW, Low WC, and Cheeran MC. Microglia and macrophages in neuroprotection, neurogenesis, and emerging therapies for stroke. Cells. (2021) 10. doi: 10.3390/cells10123555
147. Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, et al. Microglial and macrophage polarization—new prospects for brain repair. Nat Rev Neurol. (2015) 11:56–64. doi: 10.1038/nrneurol.2014.207
148. Jia J, Yang L, Chen Y, Zheng L, Chen Y, Xu Y, et al. The role of microglial phagocytosis in ischemic stroke. Front Immunol. (2021) 12:790201. doi: 10.3389/fimmu.2021.790201
149. Wang F, Jiang M, Chi Y, Huang G, and Jin M. Exosomes from circRNA-Ptpn4 can modify ADSC treatment and repair nerve damage caused by cerebral infarction by shifting microglial M1/M2 polarization. Mol Cell Biochem. (2024) 479:2081–92. doi: 10.1007/s11010-023-04824-x
150. Lu E, Zhou P, Li Y, Chen J, Zhang K, and Zhou K. Long noncoding RNA ISA1 protects against ischemic brain damage by promoting the transformation of microglia toward anti-inflammatory phenotype via the SOCS3/JAK2/STAT3 pathway. Neurochem Res. (2025) 50:92. doi: 10.1007/s11064-025-04343-9
151. Hong Z, Zuo Z, Zhao Y, Ai Y, Zhang L, Li L, et al. Transcranial focused ultrasound stimulation alleviates NLRP3-related neuroinflammation induced by ischemic stroke via regulation of the Nespas/miR-383-3p/SHP2 pathway. Int Immunopharmacol. (2025) 144:113680. doi: 10.1016/j.intimp.2024.113680
152. Cheng Z, Li X, Ye X, Yu R, and Deng Y. Purpurogallin Reverses Neuronal Apoptosis and Enhances “M2” Polarization of Microglia Under Ischemia via Mediating the miR-124-3p/TRAF6/NF-κB Axis. Neurochem Res. (2023) 48:375–92. doi: 10.1007/s11064-022-03752-4
153. Bai X, Qiu Y, Wang J, Dong Y, Zhang T, and Jin H. Panax quinquefolium saponins attenuates microglia activation following acute cerebral ischemia-reperfusion injury via Nrf2/miR-103-3p/TANK pathway. Cell Biol Int. (2024) 48:201–15. doi: 10.1002/cbin.12100
154. Wang X, Zhang S, Lv B, Chen H, Zhang W, Dong L, et al. Circular RNA PTP4A2 regulates microglial polarization through STAT3 to promote neuroinflammation in ischemic stroke. CNS Neurosci Ther. (2024) 30:e14512. doi: 10.1111/cns.14512
155. Min XL, Jia WJ, Guo L, Jing R, Zhao XH, Hu JY, et al. Brain microvascular endothelial cell-derived exosomes transmitting circ_0000495 promote microglial M1-polarization and endothelial cell injury under hypoxia condition. FASEB J. (2024) 38:e23387. doi: 10.1096/fj.202301637R
156. Li Y, Ye Q, Li J, Zhang L, Yu C, Xue S, et al. Exploring the mechanism of Taohong Siwu Decoction in treating ischemic stroke injury via the circDnajc1/miR-27a-5p/C1qc signaling axis. Phytomedicine. (2025) 136:156305. doi: 10.1016/j.phymed.2024.156305
157. Chen S, Wang L, Yuan Y, Wen Y, and Shu S. Electroacupuncture regulates microglia polarization via lncRNA-mediated hippo pathway after ischemic stroke. Biotechnol Genet Eng Rev. (2023) 39:1379–95. doi: 10.1080/02648725.2023.2177046
158. Chen T, Huang X, Zhao YX, Zhou ZW, and Zhou WS. NEAT1 inhibits the angiogenic activity of cerebral arterial endothelial cells by inducing the M1 polarization of microglia through the AMPK signaling pathway. Cell Mol Biol Lett. (2024) 29:62. doi: 10.1186/s11658-024-00579-5
159. Li Y, Song R, Shen G, Huang L, Xiao D, Ma Q, et al. MicroRNA-210 downregulates TET2 (Ten-eleven translocation methylcytosine dioxygenase 2) and contributes to neuroinflammation in ischemic stroke of adult mice. Stroke. (2023) 54:857–67. doi: 10.1161/strokeaha.122.041651
160. Xin D, Li T, Zhao Y, Guo X, Gai C, Jiang Z, et al. MiR-100-5p-rich small extracellular vesicles from activated neuron to aggravate microglial activation and neuronal activity after stroke. J Nanobiotechnology. (2024) 22:534. doi: 10.1186/s12951-024-02782-0
161. Hsu SK, Li CY, Lin IL, Syue WJ, Chen YF, Cheng KC, et al. Inflammation-related pyroptosis, a novel programmed cell death pathway, and its crosstalk with immune therapy in cancer treatment. Theranostics. (2021) 11:8813–35. doi: 10.7150/thno.62521
162. Silva MT. Bacteria-induced phagocyte secondary necrosis as a pathogenicity mechanism. J Leukoc Biol. (2010) 88:885–96. doi: 10.1189/jlb.0410205
163. Newton K, Dixit VM, and Kayagaki N. Dying cells fan the flames of inflammation. Science. (2021) 374:1076–80. doi: 10.1126/science.abi5934
164. Park SY and Kim IS. Engulfment signals and the phagocytic machinery for apoptotic cell clearance. Exp Mol Med. (2017) 49:e331. doi: 10.1038/emm.2017.52
165. Sachet M, Liang YY, and Oehler R. The immune response to secondary necrotic cells. Apoptosis. (2017) 22:1189–204. doi: 10.1007/s10495-017-1413-z
166. Broz P, Pelegrín P, and Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. (2020) 20:143–57. doi: 10.1038/s41577-019-0228-2
167. Yuan X, Xia Y, Jiang P, Chen J, and Wang C. Neuroinflammation targeting pyroptosis: molecular mechanisms and therapeutic perspectives in stroke. Mol Neurobiol. (2024) 61:7448–65. doi: 10.1007/s12035-024-04050-6
168. Chen S, Mei S, Luo Y, Wu H, Zhang J, and Zhu J. Gasdermin family: a promising therapeutic target for stroke. Transl Stroke Res. (2018) 9:555–63. doi: 10.1007/s12975-018-0666-3
169. Long J, Sun Y, Liu S, Yang S, Chen C, Zhang Z, et al. Targeting pyroptosis as a preventive and therapeutic approach for stroke. Cell Death Discov. (2023) 9:155. doi: 10.1038/s41420-023-01440-y
170. Coll RC, Schroder K, and Pelegrín P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci. (2022) 43:653–68. doi: 10.1016/j.tips.2022.04.003
171. Vasudevan SO, Behl B, and Rathinam VA. Pyroptosis-induced inflammation and tissue damage. Semin Immunol. (2023) 69:101781. doi: 10.1016/j.smim.2023.101781
172. Shi Y, Zhang L, Pu H, Mao L, Hu X, Jiang X, et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischemic reperfusion brain injury. Nat Commun. (2016) 7:10523. doi: 10.1038/ncomms10523
173. Tuo QZ, Zhang ST, and Lei P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med Res Rev. (2022) 42:259–305. doi: 10.1002/med.21817
174. Li L, Shi C, Dong F, Xu G, Lei M, and Zhang F. Targeting pyroptosis to treat ischemic stroke: From molecular pathways to treatment strategy. Int Immunopharmacol. (2024) 133:112168. doi: 10.1016/j.intimp.2024.112168
175. Zhao Y, He X, Yang X, Hong Z, Xu Y, Xu J, et al. CircFndc3b mediates exercise-induced neuroprotection by mitigating microglial/macrophage pyroptosis via the ENO1/KLF2 axis in stroke mice. Adv Sci (Weinh). (2025) 12:e2403818. doi: 10.1002/advs.202403818
176. Li Z, Pang Y, Hou L, Xing X, Yu F, Gao M, et al. Exosomal OIP5-AS1 attenuates cerebral ischemia-reperfusion injury by negatively regulating TXNIP protein stability and inhibiting neuronal pyroptosis. Int Immunopharmacol. (2024) 127:111310. doi: 10.1016/j.intimp.2023.111310
177. Sun R, Liao W, Lang T, Qin K, Jiao K, Shao L, et al. Astrocyte-derived exosomal miR-378a-5p mitigates cerebral ischemic neuroinflammation by modulating NLRP3-mediated pyroptosis. Front Immunol. (2024) 15:1454116. doi: 10.3389/fimmu.2024.1454116
178. Jin F, Jin L, Wei B, Li X, Li R, Liu W, et al. miR-96-5p alleviates cerebral ischemia-reperfusion injury in mice by inhibiting pyroptosis via downregulating caspase 1. Exp Neurol. (2024) 374:114676. doi: 10.1016/j.expneurol.2024.114676
179. Yan H, Huang W, Rao J, Yan D, and Yuan J. Demethylase FTO-Mediated m6A Modification of lncRNA MEG3 Activates Neuronal Pyroptosis via NLRP3 Signaling in Cerebral Ischemic Stroke. Mol Neurobiol. (2024) 61:1023–43. doi: 10.1007/s12035-023-03622-2
180. Gao P, Shi H, Jin X, Guo S, Zhou X, and Gao W. Mechanism of astragaloside IV regulating NLRP3 through LOC102555978 to attenuate cerebral ischemia reperfusion induced microglia pyroptosis. Int Immunopharmacol. (2024) 131:111862. doi: 10.1016/j.intimp.2024.111862
181. Yao M, Wang X, Lin H, Shu H, Xu Z, Tang L, et al. LncRNA tug1 regulates post-stroke microglial pyroptosis via PINK1/parkin-mediated mitophagy. Inflammation. (2024) 48:2677–91. doi: 10.1007/s10753-024-02219-8
182. Akbar A, Malekian F, Baghban N, Kodam SP, and Ullah M. Methodologies to isolate and purify clinical grade extracellular vesicles for medical applications. Cells. (2022) 11. doi: 10.3390/cells11020186
183. Elashiry M, Elashiry MM, Elsayed R, Rajendran M, Auersvald C, Zeitoun R, et al. Dendritic cell derived exosomes loaded with immunoregulatory cargo reprogram local immune responses and inhibit degenerative bone disease. vivo. J Extracell Vesicles. (2020) 9:1795362. doi: 10.1080/20013078.2020.1795362
184. Auquière M, Muccioli GG, and des Rieux A. Methods and challenges in purifying drug-loaded extracellular vesicles. J Extracell Vesicles. (2025) 14:e70097. doi: 10.1002/jev2.70097
185. Giovannelli L, Bari E, Jommi C, Tartara F, Armocida D, Garbossa D, et al. Mesenchymal stem cell secretome and extracellular vesicles for neurodegenerative diseases: Risk-benefit profile and next steps for the market access. Bioact Mater. (2023) 29:16–35. doi: 10.1016/j.bioactmat.2023.06.013
186. Broad K, Walker SA, Davidovich I, Witwer K, Talmon Y, and Wolfram J. Unraveling multilayered extracellular vesicles: Speculation on cause. J Extracell Vesicles. (2023) 12:e12309. doi: 10.1002/jev2.12309
187. Walker SA, Davidovich I, Yang Y, Lai A, Goncalves JP, Deliwala V, et al. Sucrose-based cryoprotective storage of extracellular vesicles. Extracell Vesicle. (2022) 1. doi: 10.1016/j.vesic.2022.100016
188. Klibanov AL, Maruyama K, Torchilin VP, and Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. (1990) 268:235–7. doi: 10.1016/0014-5793(90)81016-h
189. Bian X, Zhou L, Luo Z, Liu G, Hang Z, Li H, et al. Emerging delivery systems for enabling precision nucleic acid therapeutics. ACS Nano. (2025) 19:4039–83. doi: 10.1021/acsnano.4c11858
190. Matsumura Y and Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. (1986) 46:6387–92.
191. Suk JS, Xu Q, Kim N, Hanes J, and Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Delivery Rev. (2016) 99:28–51. doi: 10.1016/j.addr.2015.09.012
192. McMahon D, O’Reilly MA, and Hynynen K. Therapeutic agent delivery across the blood-brain barrier using focused ultrasound. Annu Rev BioMed Eng. (2021) 23:89–113. doi: 10.1146/annurev-bioeng-062117-121238
193. Zhang M, Hu S, Liu L, Dang P, Liu Y, Sun Z, et al. Engineered exosomes from different sources for cancer-targeted therapy. Signal Transduct Target Ther. (2023) 8:124. doi: 10.1038/s41392-023-01382-y
194. Xia Y, Zhang J, Liu G, and Wolfram J. Immunogenicity of extracellular vesicles. Adv Mater. (2024) 36:e2403199. doi: 10.1002/adma.202403199
195. Logtenberg MEW, Scheeren FA, and Schumacher TN. The CD47-SIRPα Immune checkpoint. Immunity. (2020) 52:742–52. doi: 10.1016/j.immuni.2020.04.011
196. Fernandes Neto JM, Nadal E, Bosdriesz E, Ooft SN, Farre L, McLean C, et al. Multiple low dose therapy as an effective strategy to treat EGFR inhibitor-resistant NSCLC tumors. Nat Commun. (2020) 11:3157. doi: 10.1038/s41467-020-16952-9
197. Guo P, Busatto S, Huang J, Morad G, and Moses MA. A facile magnetic extrusion method for preparing endosome-derived vesicles for cancer drug delivery. Adv Funct Mater. (2021) 31. doi: 10.1002/adfm.202008326
198. Rexrode KM, Madsen TE, Yu AYX, Carcel C, Lichtman JH, and Miller EC. The impact of sex and gender on stroke. Circ Res. (2022) 130:512–28. doi: 10.1161/circresaha.121.319915
200. Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Executive summary: heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation. (2012) 125:188–97. doi: 10.1161/CIR.0b013e3182456d46
201. Mishra SR, Chung HF, Waller M, Dobson AJ, Greenwood DC, Cade JE, et al. Association between reproductive life span and incident nonfatal cardiovascular disease: A pooled analysis of individual patient data from 12 studies. JAMA Cardiol. (2020) 5:1410–8. doi: 10.1001/jamacardio.2020.4105
202. Parikh NI, Gonzalez JM, Anderson CAM, Judd SE, Rexrode KM, Hlatky MA, et al. Adverse pregnancy outcomes and cardiovascular disease risk: unique opportunities for cardiovascular disease prevention in women: A scientific statement from the american heart association. Circulation. (2021) 143:e902–16. doi: 10.1161/cir.0000000000000961
203. Tarnutzer AA, Lee SH, Robinson KA, Wang Z, Edlow JA, and Newman-Toker DE. ED misdiagnosis of cerebrovascular events in the era of modern neuroimaging: A meta-analysis. Neurology. (2017) 88:1468–77. doi: 10.1212/wnl.0000000000003814
204. Sampath D, Branyan TE, Markowsky KG, Gunda R, Samiya N, Obenaus A, et al. Sex differences in cognitive impairment after focal ischemia in middle-aged rats and the effect of iv miR-20a-3p treatment. Neurobiol Aging. (2023) 129:168–77. doi: 10.1016/j.neurobiolaging.2023.05.001
205. Lewington S, Clarke R, Qizilbash N, Peto R, and Collins R. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. (2002) 360:1903–13. doi: 10.1016/s0140-6736(02)11911-8
206. Stansbury JP, Jia H, Williams LS, Vogel WB, and Duncan PW. Ethnic disparities in stroke: epidemiology, acute care, and postacute outcomes. Stroke. (2005) 36:374–86. doi: 10.1161/01.STR.0000153065.39325.fd
207. Mehta SL, Chelluboina B, Morris-Blanco KC, Bathula S, Jeong S, Arruri V, et al. Post-stroke brain can be protected by modulating the lncRNA FosDT. J Cereb Blood Flow Metab. (2024) 44:239–51. doi: 10.1177/0271678x231212378
208. Venkat P and Chopp M. Exosome treatment for stroke with diabetic comorbidity. Neural Regener Res. (2022) 17:315–7. doi: 10.4103/1673-5374.319190
209. Venkat P, Zacharek A, Landschoot-Ward J, Wang F, Culmone L, Chen Z, et al. Exosomes derived from bone marrow mesenchymal stem cells harvested from type two diabetes rats promotes neurorestorative effects after stroke in type two diabetes rats. Exp Neurol. (2020) 334:113456. doi: 10.1016/j.expneurol.2020.113456
210. Sena E, van der Worp HB, Howells D, and Macleod M. How can we improve the pre-clinical development of drugs for stroke? Trends Neurosci. (2007) 30:433–9. doi: 10.1016/j.tins.2007.06.009
211. Finklestein SP, Fisher M, Furlan AJ, Goldstein LB, Gorelick PB, Kaste M, et al. Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. (1999) 30:2752–8. doi: 10.1161/01.str.30.12.2752
212. Schmidt-Pogoda A, Bonberg N, Koecke MHM, Strecker JK, Wellmann J, Bruckmann NM, et al. Why most acute stroke studies are positive in animals but not in patients: A systematic comparison of preclinical, early phase, and phase 3 clinical trials of neuroprotective agents. Ann Neurol. (2020) 87:40–51. doi: 10.1002/ana.25643
213. Driedonks T, Jiang L, Carlson B, Han Z, Liu G, Queen SE, et al. Pharmacokinetics and biodistribution of extracellular vesicles administered intravenously and intranasally to Macaca nemestrina. J Extracell Biol. (2022) 1. doi: 10.1002/jex2.59
214. Zhang ZG, Buller B, and Chopp M. Exosomes - beyond stem cells for restorative therapy in stroke and neurological injury. Nat Rev Neurol. (2019) 15:193–203. doi: 10.1038/s41582-018-0126-4
215. Yang J, Zhang X, Chen X, Wang L, and Yang G. Exosome Mediated Delivery of miR-124 Promotes Neurogenesis after Ischemia. Mol Ther Nucleic Acids. (2017) 7:278–87. doi: 10.1016/j.omtn.2017.04.010
216. Song Y, Li Z, He T, Qu M, Jiang L, Li W, et al. M2 microglia-derived exosomes protect the mouse brain from ischemia-reperfusion injury via exosomal miR-124. Theranostics. (2019) 9:2910–23. doi: 10.7150/thno.30879
217. Aderinto N, Olatunji G, Kokori E, Sanker V, Yusuf IA, Adefusi TO, et al. miR-210 in ischemic stroke: biomarker potential, challenges and future perspectives. Eur J Med Res. (2024) 29:432. doi: 10.1186/s40001-024-02029-6
218. Eyileten C, Czajka P, Domitrz I, Wierzchowska-Ciok A, Gasecka A, Mirowska-Guzel D, et al. Extracellular vesicle-derived miRNAs in ischemic stroke: roles in neuroprotection, tissue regeneration, and biomarker potential. Cell Mol Neurobiol. (2025) 45:31. doi: 10.1007/s10571-025-01551-3
219. He P, Jiang H, Zhu J, Hu M, and Song P. Identification and validation of the inflammatory response-related LncRNAs as diagnostic biomarkers for acute ischemic stroke. Sci Rep. (2025) 15:13818. doi: 10.1038/s41598-025-98101-0
220. Wang W, Li DB, Li RY, Zhou X, Yu DJ, Lan XY, et al. Diagnosis of hyperacute and acute ischemic stroke: the potential utility of exosomal microRNA-21-5p and microRNA-30a-5p. Cerebrovasc Dis. (2018) 45:204–12. doi: 10.1159/000488365
221. Yin X and Zhang C. Circulating miR-574-5p shows diagnostic and prognostic significance and regulates oxygen-glucose deprivation (OGD)-induced inflammatory activation of microglia by targeting ATP2B2. Mol Cell Probes. (2025) 79:102016. doi: 10.1016/j.mcp.2025.102016
222. Zhang X. Correlation analysis between high-density lipoprotein cholesterol-carried microRNA-28-5p and acute ischemic stroke. J Neurochem. (2025) 169:e70119. doi: 10.1111/jnc.70119
Keywords: ischemic stroke, ncRNAs, exosome, neuroinflammation, ischemia/reperfusion (I/R) injury
Citation: Wei D, Li F, Guo C, Chen J and You Y (2025) Exosomes and non-coding RNAs in the regulation of neuroinflammation after ischemic stroke: mechanisms and therapeutic perspectives. Front. Immunol. 16:1601843. doi: 10.3389/fimmu.2025.1601843
Received: 28 March 2025; Accepted: 18 August 2025;
Published: 04 September 2025; Corrected: 10 September 2025.
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Ivana Kawikova, National Institute of Mental Health, CzechiaXianshuang Liu, Henry Ford Hospital, United States
Prasad S. Koka, Biomedical Research Institute of Southern California, United States
Copyright © 2025 Wei, Li, Guo, Chen and You. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanqiu You, eW91eWFucWl1QDE2My5jb20=; Jibing Chen, amliaW5nY2hlbjM5OEAxNjMuY29t
†These authors have contributed equally to this work
‡ORCID: Chen Jibing, orcid.org/0000-0001-8596-2126
You Yanqiu, orcid.org/0009-0009-2518-9093