 Abire Allaoui1,2*
Abire Allaoui1,2* Abderrahmane Moundir1
Abderrahmane Moundir1 Ibtihal Benhsaien1
Ibtihal Benhsaien1 Fatima Ailal1
Fatima Ailal1 Jalila El Bakkouri2Khadija Echchilali1Abdelhamid Naitlho2Hassan El Kabli1
Jalila El Bakkouri2Khadija Echchilali1Abdelhamid Naitlho2Hassan El Kabli1 Ahmed Aziz Bousfiha1Mina Moudatir1
Ahmed Aziz Bousfiha1Mina Moudatir1- 1Clinical Immunology, Inflammation and Allergy Laboratory (LICIA), Faculty of Medicine and Pharmacy, University of Hassan II Casablanca, Casablanca, Morocco
- 2Immunopathology-Immunotherapy-Immunomonitoring Laboratory, Faculty of Medicine, Mohammed VI University of Health and Sciences, Casablanca, Morocco
Background: Common Variable Immunodeficiency (CVID) is the most prevalent symptomatic inborn errors of immunity (IEI), characterized by impaired antibody production, recurrent infections, and immune dysregulation. While extensively studied in Western populations, data from North Africa remains scarce. This study provides the first comprehensive evaluation of the clinical, immunological, and genetic landscape of CVID in a Moroccan nationwide cohort.
Methods: A multicenter, cross-sectional study was conducted across eight university hospitals in Morocco from 2019 to 2025. Sixty-one CVID patients were enrolled according to ESID (European society of immunodeficiency) and MENA (Middle-East and North Africa) guidelines. Clinical, immunological, and genetic data were analyzed. Whole-blood samples were processed for immunophenotyping, and a subset of patients underwent next-generation sequencing (NGS) targeting 474 inborn error of immunity (IEI)-associated genes.
Results: The mean age at diagnosis was 25.9 (SD 18.7) years old, with a diagnostic delay of 6.91 (SD 8.82) years. The most frequent infectious complications were pulmonary infections (88.5%) and gastrointestinal infections (63.9%). Non-infectious complications were present in 49.2% of patients, with predominant features including lymphoproliferation (50.8%), autoimmune cytopenias (39.3%), and granulomatous disease (18%). Bronchiectasis was the most common pulmonary finding (44.3%). Genetic testing (n=25) revealed 19 pathogenic variants in 13 genes, including 14 novel variants, particularly LRBA, CTLA4, PIK3CD, NFKB1, VAV1, and TCF3. The phenotype-genotype correlation, based on clinical presentation, gene function, and multidisciplinary assessment, was strong in 52.6% of cases.
Conclusion: This study provides Morocco’s first clinical and genetic landscape of CVID, highlighting a high prevalence of consanguinity-associated monogenic defects and a significant burden of infectious and immune dysregulatory complications. Our findings emphasize the need for early diagnosis, multidisciplinary management, and access to targeted therapies in non-Western settings. Further studies with functional validation of genetic variants are warranted to refine precision medicine approaches in CVID.
Introduction
Common Variable Immunodeficiency (CVID) is the most prevalent symptomatic inborn errors of immunity (IEI) (1), with a prevalence of approximately 1:50,000 to 1:25,000 (2). CVID is characterized by impaired antibody production, recurrent infections, and a broad spectrum of immune dysregulation, including autoimmunity, lymphoproliferation, and malignancies (2–4). Autoimmune cytopenias and lymphoproliferative disorders remain prominent, reinforcing the intricate interplay between immune deficiency and immune dysregulation (2, 5, 6). Despite the availability of immunoglobulin replacement therapy (IgRT) and targeted biologics, challenges persist in optimizing therapeutic strategies, particularly in resource-limited settings (7).
The global landscape of CVID has evolved with advances in immunogenetics and next-generation sequencing (NGS), which have revealed monogenic and polygenic contributions to disease pathogenesis (8–10). Studies from international cohorts have highlighted the complexity of genetic determinants and their heterogeneous correlation with clinical phenotypes (11, 12).
Additionally, the role of environmental factors and regional infectious disease burdens has become increasingly recognized in shaping disease expression and outcomes (5, 13, 14). This clinical and genetic heterogeneity requires a multidisciplinary approach for diagnosis and management.
While extensively studied in Western populations, data from North Africa, particularly Morocco, remain scarce (15–17). This study provides the first comprehensive evaluation of the clinical, immunological, and genetic landscape of CVID in a Moroccan nationwide cohort, comparing our findings with international data. By participating in elucidating the disease spectrum in a non-Western population, our research aims to bridge knowledge gaps, identify diagnostic and therapeutic challenges in non-Western countries, and contribute to the evolving understanding of CVID pathophysiology.
Methods
A multicenter, cross-sectional, nationwide study of patients diagnosed with CVID was conducted in Morocco from January 2019 to December 2025. Eight university hospitals treating PIDs have participated in the study. The Moroccan-CVID Registry was created under the aegis of the Working Group of the Moroccan Society of Primary Immunodeficiency (MSPID). All patients aged 4 years and above with CVID diagnosis according to the European Society for Immunodeficiencies (ESID) registry working definitions (18) and the Middle East and North Africa Diagnosis Guidelines for Inborn Errors of Immunity (19), with at least decreased levels of two serum immunoglobulins (low IgG and low IgA and/or low IgM, >2 standard deviations below the mean for age) and impaired antibody response or low B memory switched-class lymphocytes, with the exclusion of defined causes of hypogammaglobulinemia such as malignancies, medications, protein loss, or bone marrow failure, were considered eligible.
Data collection and variables
Data was extracted from the Moroccan-CVID Registry, which systematically compiles sociodemographic, clinical, biological, and immunological data, accompanying co-morbidities, family history, genetic data, treatments, and patient clinical courses. All data were collected by reviewing electronic medical records and included in a predesigned database on SPSS (Statistical Package for the Social Sciences) software.
Demographic data included sex, age at diagnosis, age at clinical onset, diagnostic delay, first PID- related clinical complication (infection, dysimmunity, malignancy, or other), and follow-up time.
Genetic testing, family history, and consanguinity were recorded.
Blood samples were obtained and placed in ethylenediaminetetraacetic acid (EDTA) tubes for complete blood count analysis (white blood cell count, lymphocyte count, and neutrophil count). All patients underwent HIV tests to rule out HIV infection. Fluorescently labeled monoclonal antibodies specific for T cell markers (CD3, CD4, CD8), B cell markers (CD19, CD20), and NK cell markers (CD16, CD56) were added to the samples and incubated. The enumeration was performed using flow cytometry with a FACSCanto II flow cytometer (from BD Biosciences, San Jose, CA) and analyzed using FACSDiva software. Serum immunoglobulin (Ig) levels were determined by nephelometry (BNProspec, Siemens). All patients immunoglobulin and lymphocyte subset levels were tested before the first Ig replacement therapy. Patients with transient hypogammaglobulinemia or selective IgA deficiency (normal IgG/IgM) were excluded via repeat testing.
All other diagnostic tests, such as chest X-ray, computer tomography of the chest, endoscopic interventions, sputum/blood/stool cultures, and autoimmunity tests, were evaluated according to each patients clinical course as medically required. Infectious, hematologic and malignant causes were excluded for cases of splenomegaly and lymphadenopathies.
All patients underwent Ig testing and lymphocyte subtype enumeration, but only 36 patients had B lymphocyte subset enumeration, only seven patients had a response to vaccine testing, and only 25 patients had genetic testing due to insufficient facilities and resources.
The immunological phenotype of each patient was studied and classified according to EUROClass (20) classifications.
Genetic, flow cytometry, and immunologic testing were carried out by the Immunology Laboratory, Ibn Rochd University Hospital, and Laboratoire National de Recherche Mohammed VI (LNRM6) and by a deep sequencing array (using a custom-designed Illumina SNP array) with the 474 genes involved in inborn errors of immunity (21).
Variants were sorted to find non-synonymous variants with a minor allele frequency (MAF) of less than 0.01 in the Genome Aggregation Database (gnomAD v3). This helped find variants that might be causal. Our analysis concentrated on IEI genes as categorized by the International Union of Immunological Societies (IUIS) (22). The mutations have to align with the disease’s mode of transmission: one variant for autosomal dominant or X-linked transmission in males and two variants for autosomal recessive transmission. For diseases that can be inherited in a dominant or recessive transmission, the presence of one copy of the mutated gene (heterozygous variant) was deemed adequate for analysis. To prioritize variations with a detrimental effect, in silico pathogenicity predictions were utilized, including the CADD (Combined Annotation Dependent Depletion) approach (23), the SIFT (Sorting Intolerant from Tolerant) method (24), the Polyphen2 (Polymorphism Phenotyping v2) method (25), and the Alpha Missense method (26). Candidate variants were subsequently analyzed for genotype-phenotype correlation. “Strong correlation” referred to a match between the clinical presentation and the known function of the gene based on current literature and in silico predictions, further supported by multidisciplinary team review. Defined as High (≥2 features), Moderate (1 feature), or Low (no known associations). Disease databases, including OMIM and ClinVar, were utilized to assess the genetic variant’s contribution to the disease. All discovered variants were assessed for their potential pathogenicity following the recommendations set forth by the American College of Medical Genetics and Genomics (ACMG) (27). Consequently, variants were categorized as pathogenic, likely pathogenic, of uncertain significance (VUS), likely benign, and benign. Popviz (28) was used to determine the mutation significance cutoff (MSC) with a confidence level of 95%. The significance of each mutation was evaluated during multidisciplinary meetings involving physicians, geneticists, immunologists, and other specialists, informed by clinical status and literature findings.
The follow-up of our patients adheres to a locally applied monitoring protocol: abdominal ultrasound is performed every 6 to 12 months; chest CT scans are repeated every 2 years; pulmonary function tests are conducted annually; and upper and lower gastrointestinal endoscopies are scheduled every 2 years, or earlier if clinically indicated.
Statistics
To ensure efficient data management, we used a computerized database using SPSS (Statistical Package for the Social Sciences) version 25 software. Using this database facilitated the entry of each patient’s information and simplified the subsequent analyses. The data were subjected to descriptive and statistical analyses. We considered the p-value <0.05 as significant. Qualitative characteristics were presented as percentages, while quantitative characteristics were expressed as mean or median with standard deviation, using the tools available in SPSS software.
Ethical statement
The patients or parents/guardians of the young participants gave written informed consent to participate in the study and genetic testing. The study was approved by the Ethics Committee of the Ibn Rochd University Hospital in Casablanca.
Results
Demographics
A total of 61 patients diagnosed with CVID were included. In our cohort, the mean age at diagnosis was 25.9 (SD 18.7) years old, and 33 patients (54.1%) were male (Figure 1A). However, the mean diagnostic delay was 6.91 (SD 8.82) years, while the global mean age at characteristic symptom onset was 19.00 (SD 17.28) years old. Most patients initially presented with infectious complications in 80.3% of the cases (n=49), followed by non-infectious complications (19.7%, n=12) (Figure 1B). The consanguinity rate was 54.1% (n=33) (Figure 1C). A family history of immunodeficiency was noted in one case.
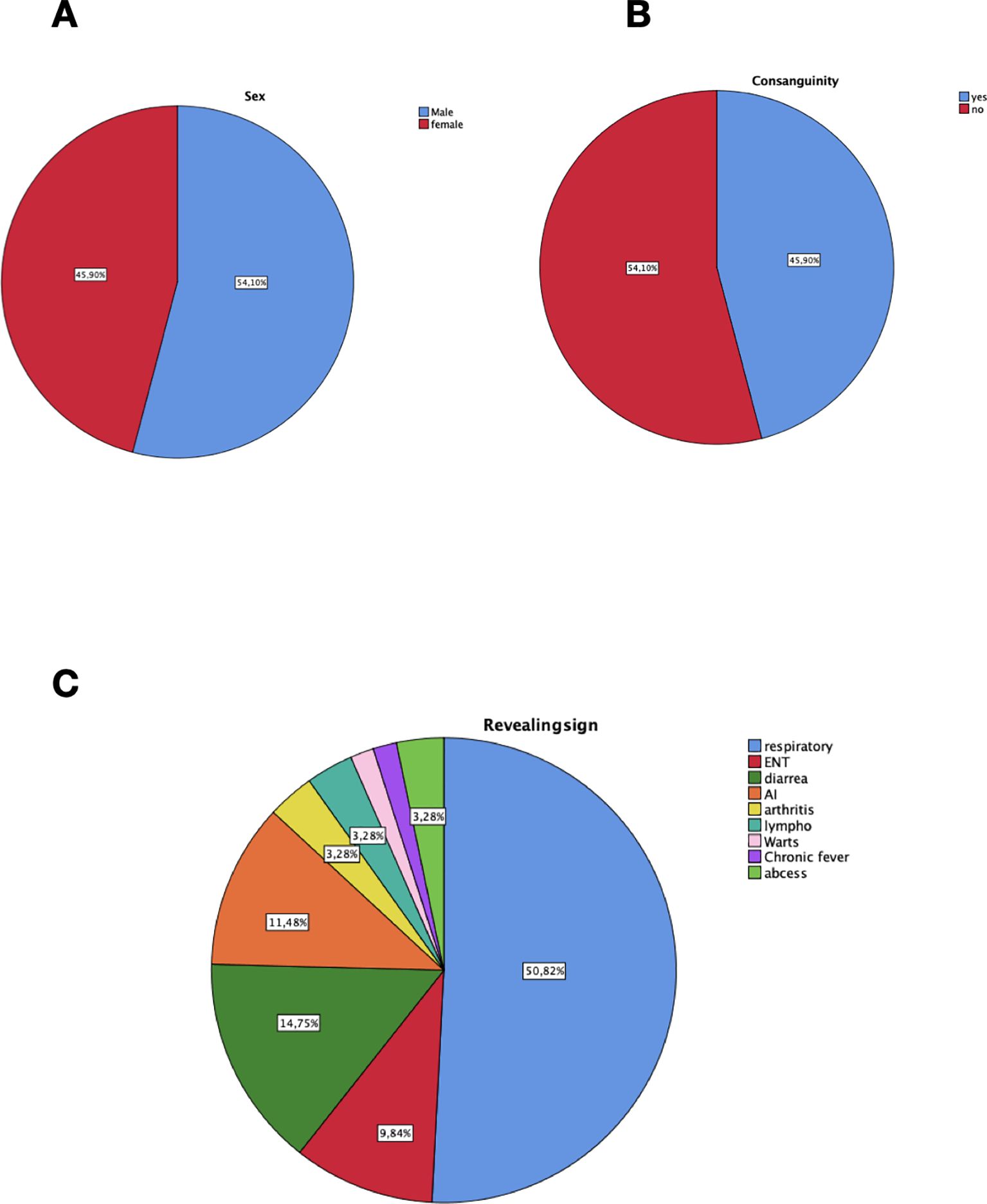
Figure 1. Demographic characteristics and major revealing signs of 61 Moroccan patients with CVID. (A) Gender distribution in our cohort. (B) Consanguinity distribution in our population. (C) Distribution of revealing signs at diagnosis. respiratory, respiratory infections; ENT, Ear nose throat infections; AI, Autoimmunity; lympho, lymphoproliferation, abcess; deep-seated infections.
Infectious complications
Pulmonary infections were the major cause of recurrent infections in 88.5% (n=54), followed by digestive tract infections in 63.9% (n=39), then ENT infections in 50.8% (n=31), and pyelonephritis was noted in one case. Skin, soft tissue, or musculoskeletal infections were noted, respectively, in 3.3% (n=2), 3.3% (n=2), and 1.6% (n=1). Almost 63.9% (n=39) had suffered at least one episode of a major bacterial infection complicated with sepsis in 8.2% (n=5), and pneumonia accounted for most episodes, 60.7% (n=37). Bacterial infections were mostly caused by streptococcus pneumonia, staphylococcus aureus, pseudomonas, salmonella, and Klebsiella pneumonia.
Gastroenteritis was caused by giardia enteritis, norovirus, and enterovirus. Helicobacter pylori was a predominant cause of gastritis. Seven cases (11.5%) of mycobacterium tuberculosis were noted (5 pulmonary infections, 2 adenitis), and two cases of herpes zoster virus infections.
Non-infectious complications
Non-infectious complications were present in 30 patients (49.2%). The most common immune dysregulation complication was lymphoproliferation with splenomegaly (50.8%, n=31), closely followed by lymphadenopathies and immune cytopenias, each one in 39.3% (n=24), autoimmune hemolytic anemia (AIHA) 11.5% (n=7), immune thrombopenia (ITP) 27.9% (n=17), Evans syndrome (AIHA+ ITP) in 8.2% (n=5), and neutropenia 9.8% (n=6), non-infectious enteropathy in 14.8% (n=9), uveitis, and inflammatory arthritis, each one in 6.6% (n=4). Granulomatosis (sarcoidosis-like disease) was noticed and confirmed histologically in 18% (n = 11) (pulmonary, hepatic, and splenic involvement). Systemic diseases were present in three patients (systemic lupus, rheumatoid arthritis, and psoriatic rheumatism). Autoimmune and inflammatory complications are regrouped in Table 1.
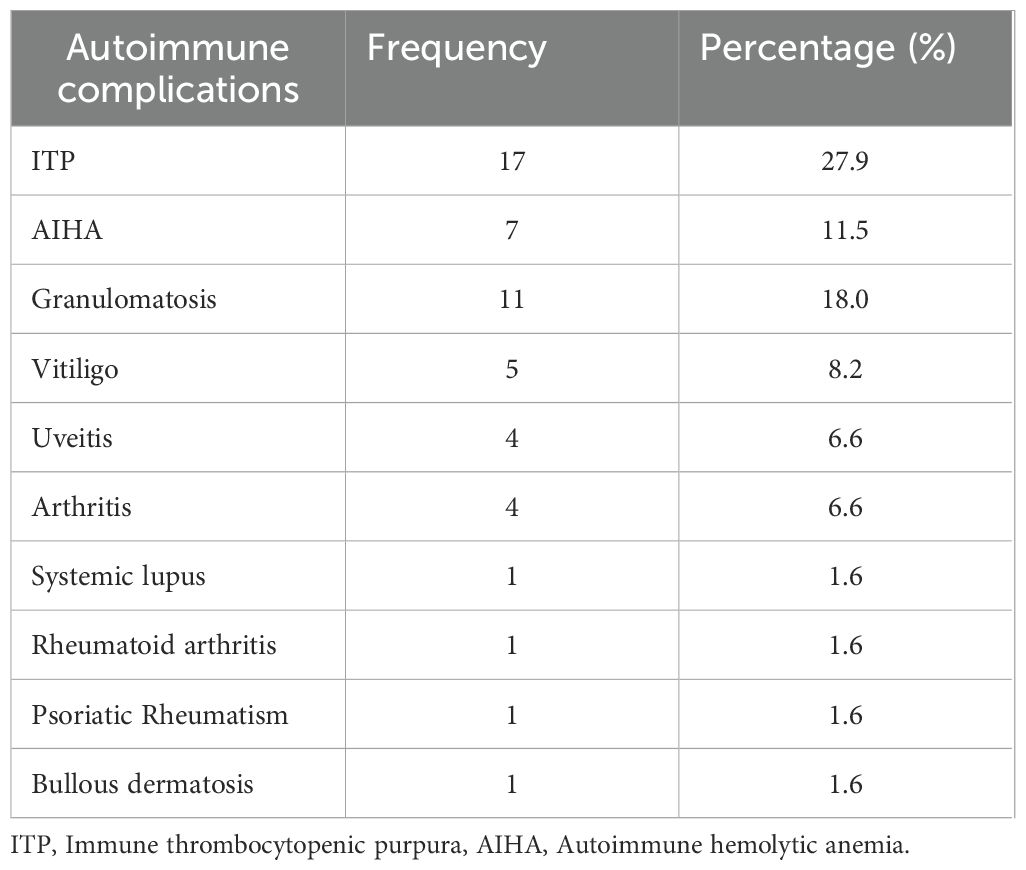
Table 1. Autoimmune complications in Moroccan patients with CVID.
Coeliac-like and Biermer-like diseases were noted in 14.8% (n=9) and 3.3% (n=2), respectively. IBD-like disease (inflammatory bowel disease) was found and confirmed histologically in 13.1% (n=8) (Figure 2). Liver disease was represented by hepatitis, hepatomegaly, and liver nodules, found in 4.9% (n=3) and 3.3% (n=2), respectively, complicated with portal hypertension in one case.
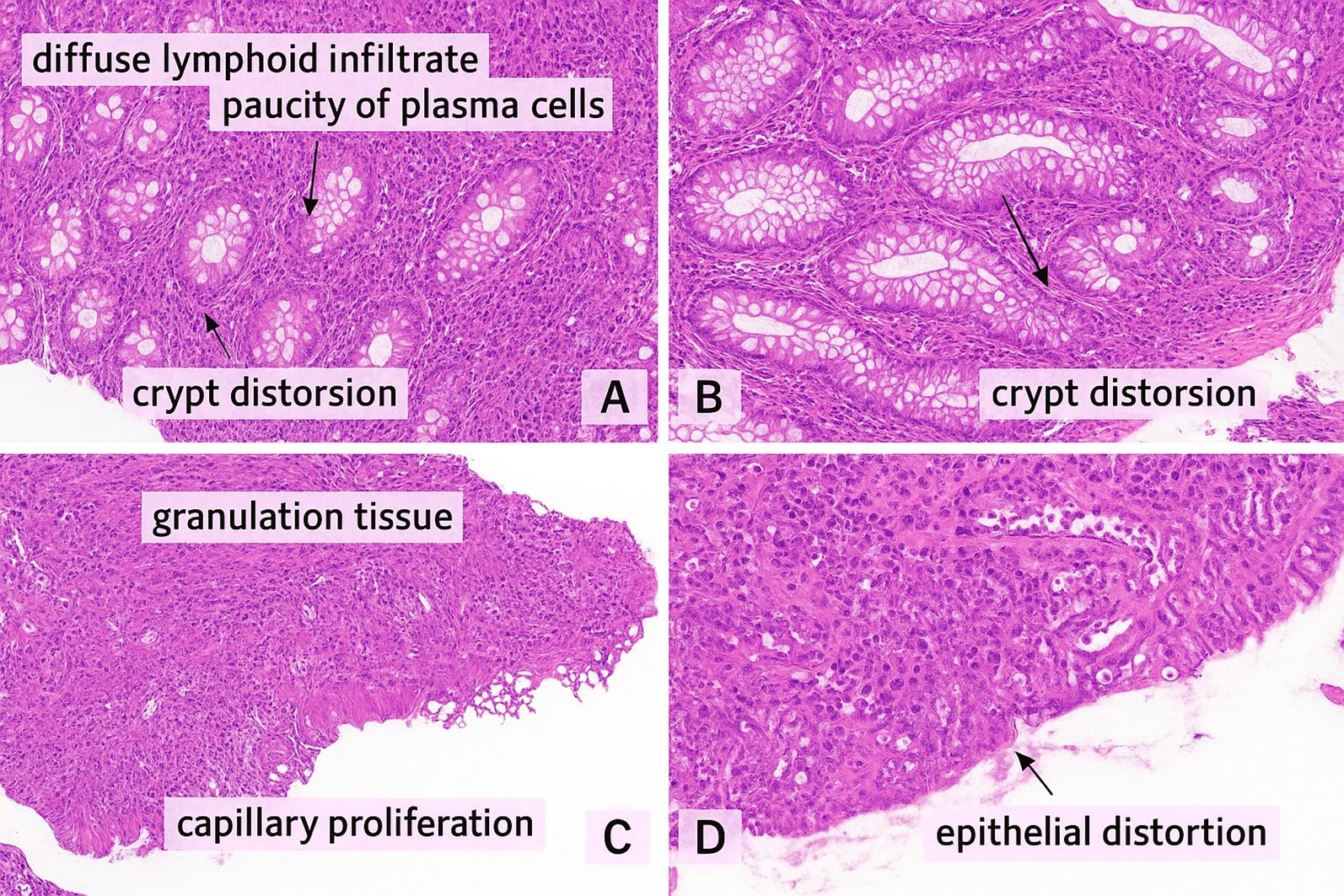
Figure 2. (A, B) Colonic mucosa with a dense, diffuse lymphoid infiltrate within the lamina propria, composed predominantly of small lymphocytes, with a striking paucity of plasma cells. No epithelioid granulomas or giant cells were observed (Hematoxylin & Eosin stain) (C, D) Colonic biopsy showing granulation tissue characterized by a mixed inflammatory infiltrate, fibrino-leukocytic debris, and capillary proliferation. The colonic epithelium exhibits glandular dedifferentiation and reduced mucin production (Hematoxylin & Eosin stain). These microscopic findings are consistent with Crohn-like enterocolitis in a 26 years old female Moroccan patient with TNFRSF13B mutation associated to CD21 deficiency.
Dyspnea and cough were the most common pulmonary symptoms. High-resolution computed tomography (HRCT) was performed in 54 patients (88.5%) (Figures 3). Bronchiectasis was the most common imaging finding (44.3%, n=27). Lung parenchymal involvement was observed in 11.5% (n=7) of patients, and the most commonly described radiological pattern was compatible with lymphocytic interstitial pneumonia in 8.2% (n=5). Combined lymphocytic interstitial pneumonia and granulomatosis (GLILD) was not confirmed histologically in any of our patients. Three patients developed chronic respiratory failure requiring oxygen therapy.
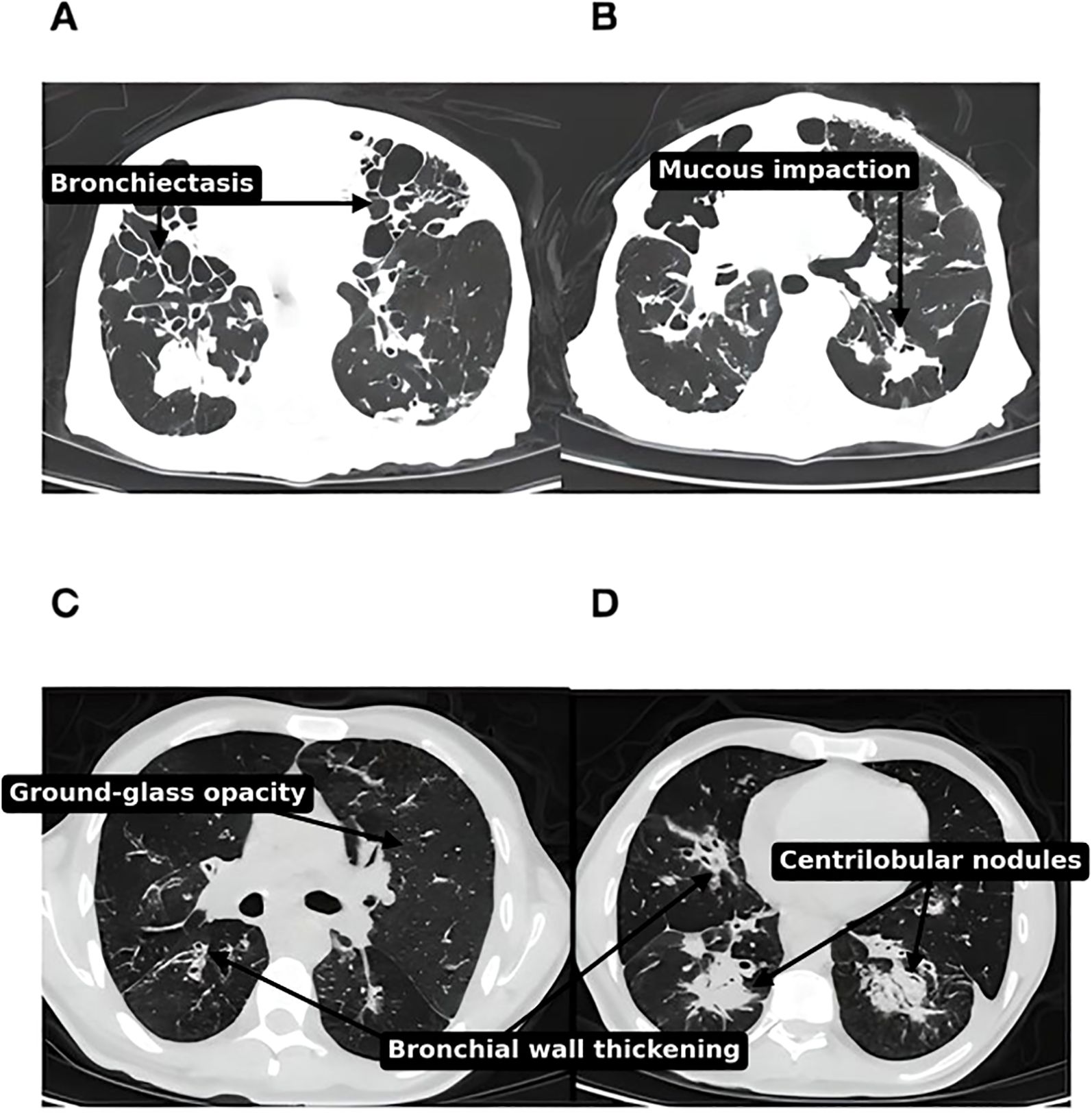
Figure 3. (A, B) Thoracic CT scan, parenchymal window, bilateral diffuse nodules and diffuse cystic bronchiectasis in a 78 years old Moroccan patient with CVID (no gene was identified). (C, D) Thoracic CT scan, parenchymal window, of diffuse nodules, reticulations and ground-glass opacities. Apart from CVID-ILD features, there are also diffuse bronchiectasis with mucoid impaction pointing to surinfection, in 55 years old male Moroccan patient with TNFRSF13B mutation.
Skin lesions were reported in 14 patients in our cohort (22.9%). Vitiligo was the main skin lesion in 8.2% (n=5), warts in 6.6% (n=4) (Figure 4), poliosis, bullous dermatosis, and psoriasis in one case each. Atopic dermatitis and drug allergy were observed in two patients (3.3%).
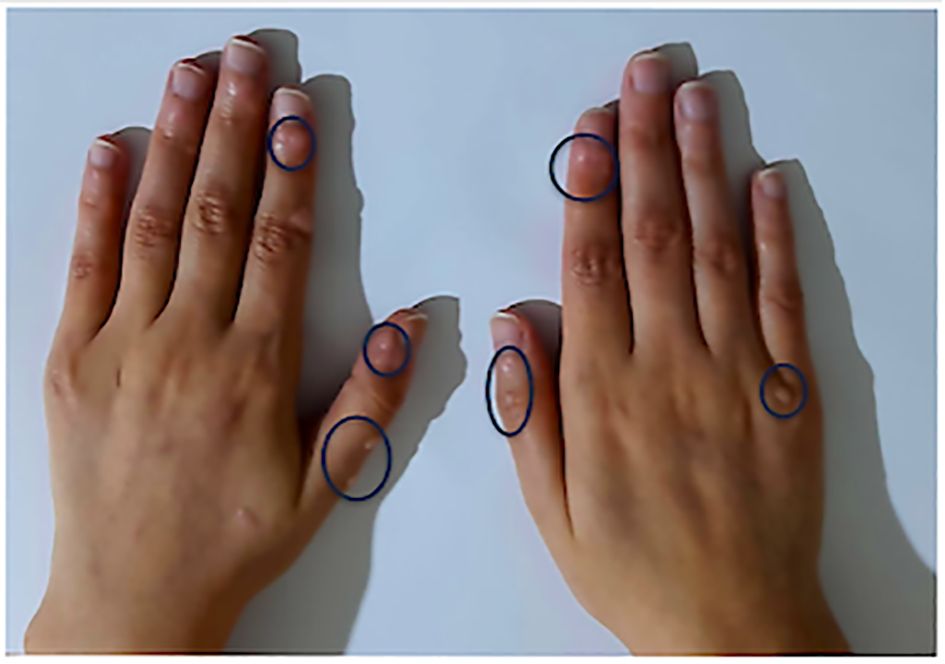
Figure 4. Warts in a patient with LRBA deficiency from our cohort. Warts are circled in blue.
Neurological immunodemyelinating lesions were found in one patient who was admitted to intensive care with impaired consciousness and died during this hospitalization following a septic choc.
Three patients presented with cancers in their clinical course: one patient with Burkitt lymphoma, one patient with marginal-zone lymphoma (gastric MALT lymphoma), and one patient with colorectal cancer.
Immunological features
There was significant variability in immunoglobulin levels and the numbers of B and T cell subpopulations between patients. Immunoglobulin levels upon diagnosis and lymphocyte subpopulation cell counts are presented in Table 2.
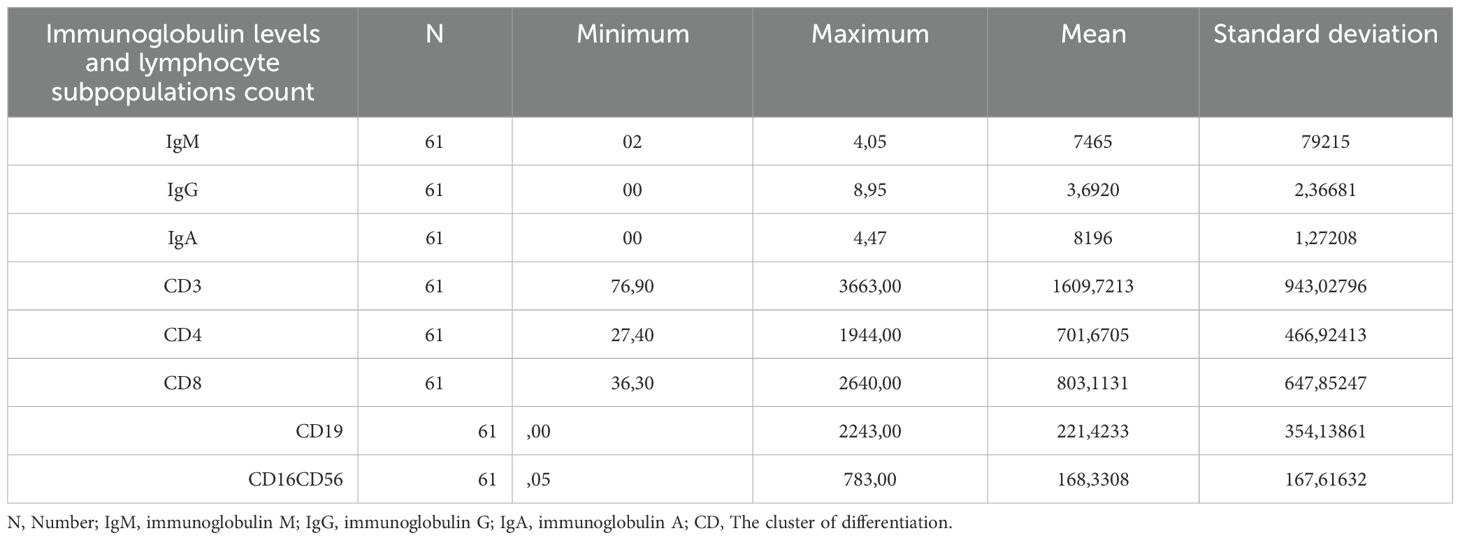
Table 2. Immunoglobulin levels and lymphocyte subpopulations count in the CVID Moroccan cohort.
Patient management and outcomes
Fifty-three patients (86.9%) were under active treatment with immunoglobulin replacement treatment (IgRT). All of the treated patients were on the intravenous route (IVIgRT).The prescribed dose was 400 mg/kg/month (ranging from 300–600 mg/kg). Good outcome with a reduced incidence of recurrent and severe infections and subsequent hospitalizations was noted in 65,6% (n=40) of the total number of patients, this percentage increases to 90% (n=36/40) among the patients who received immunoglobulines.
75.4% (n=46) of the patients were given antibiotics as a preventative measure. Prophylactic trimethoprim- sulfamethoxazole (TMP-SMX) 160/800 mg three times a week was initiated in patients with recurrent respiratory infections, independent of CD4+ counts. Patients with bronchiectasis were given azithromycin 500 mg three times a week.
Thirty patients (49.2%) received a prescription for corticosteroids. Ten patients (16.4%) were given immunosuppressants or immunomodulators to help with autoimmune and inflammatory complications of CVID, including cytopenias, granulomatosis, lymphoproliferation, and autoimmune disorders. Azathioprine had been administered to three patients. Two patients were on mycophenolate mofetil. Methotrexate was used in two patients, and one patient was put on hydroxychloroquine.
Five patients (8.2%) also used targeted therapies. Rituximab therapy was administered in three cases, one to manage immune cytopenia and the others to complete the chemotherapy regimen for Burkitt lymphoma and marginal-zone lymphoma. Two patients with LRBA and CTLA4 deficiency were put on abatacept then infliximab to manage rheumatological and intestinal inflammation. No patient benefited from bone marrow transplantation.
Three patients have died, meaning a mortality rate of 4.9% in our cohort at the time of data collection, with a mean age at death of 50 years old (SD 19.8). Dysimmune complications (severe autoimmune enteropathy) and AA amyloidosis in a patient with TCF3 mutation and septic choc in two patients without genetic confirmation, were the causes of death.
Genetics
Twenty-five patients were tested for genetics (41%). Following an in silico analysis using predictive methods (CADD, SIFT, Polyphen2), we identified 19 putative pathogenic variants in 13 genes, of which 14 (73.7%) are novel and 5 (26.3%) have been previously documented. Biallelic variants were present in 6 patients, which corresponds to 31.6% of patients with a potential genetic diagnosis. X-linked variants were present in one patient (5.26%), and monoallelic variants were present in 12 patients (63.15%). Based on ACMG guidelines, 12 (63.15%) of the variants found were labeled as VUS, 6 (31.5%) as pathogenic, and 1 (5.26%) as likely pathogenic. At the molecular level, there were 12 missense (63.15%), 3 splice donors (15.8%), 2 frameshifts (10.5%), 1 codon stop (5.26%), and 1 deletion (5.26%). All details are in Table 3.
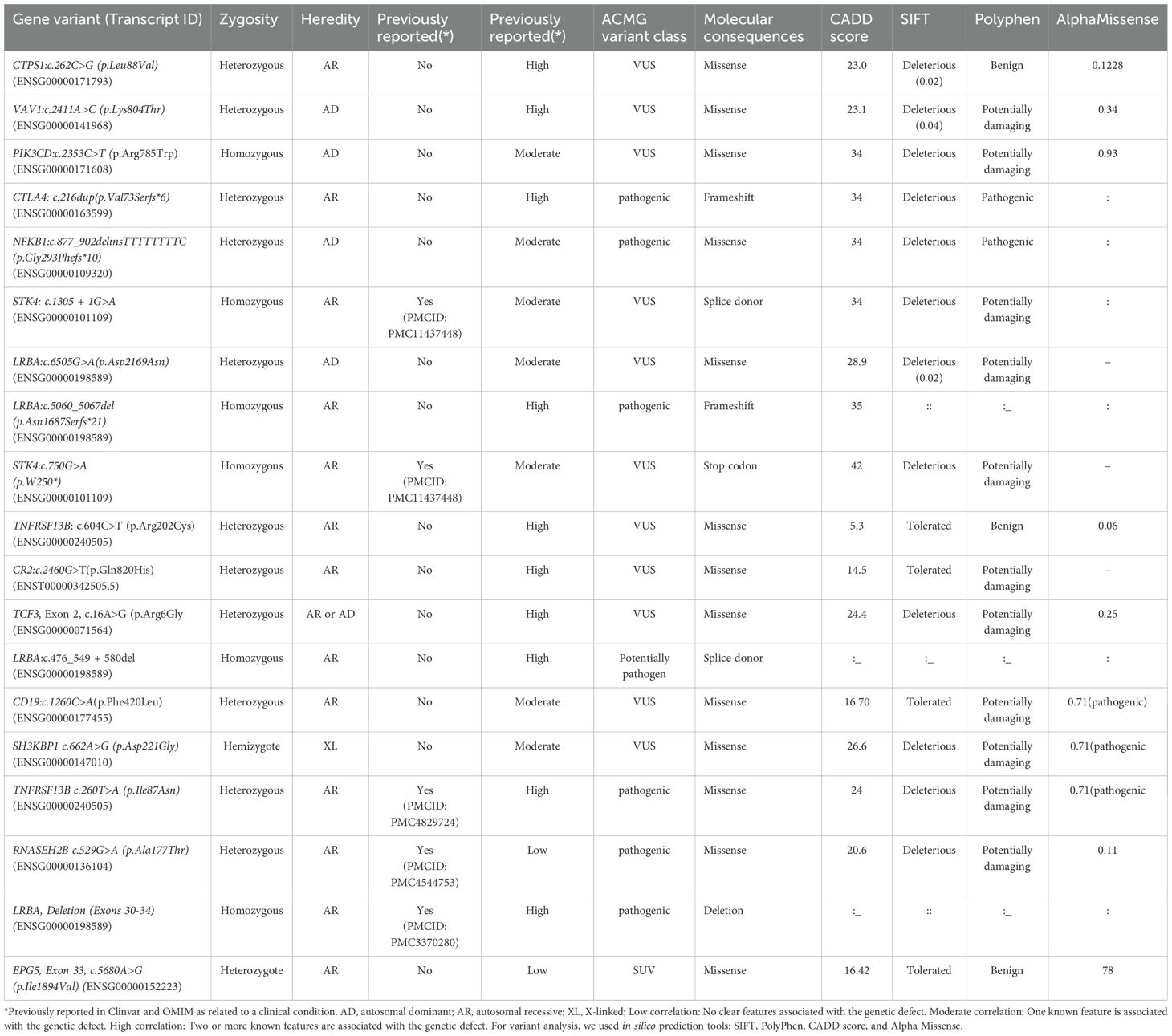
Table 3. Potential causal variant classification and in silico analysis.
We looked for all characteristics linked to the variant or gene to determine how each genotype affected the clinical phenotype. Phenotype-genotype correlation was high in 10 (52.6%) patients, moderate in 7 (36.84%), and low in 2 (10.52%). CADD vs. MAF plots showing new potential variants with high or moderate phenotype-genotype correlations are in represented in Figure 5. The identified potential causal variants were associated with CVID related-genes in 3 patients: CD19, TNFRSF13B(TACI), the association between TNRFSF13B (TACI) and CR2 (CD21 deficiency) in one patient. The variants were associated with CVID-like diseases in ten patients: LRBA in 4 cases, CTLA4 (1 case), PIK3CD (1 case), NFKB1 (1 case), VAV1 (1 case), TCF3 (1 case), SH3KBP1 (1 case). Variants associated with other immune dysregulation defects were present in six patients: STK4 deficiency in 3 cases, RNASEH2B (1 case), CTPS1 (1 case), and EPG5 (1 case). All the patients with potential causal variants are detailed in Table 4 with their clinical phenotypes, immunological features, therapeutics, and outcomes.
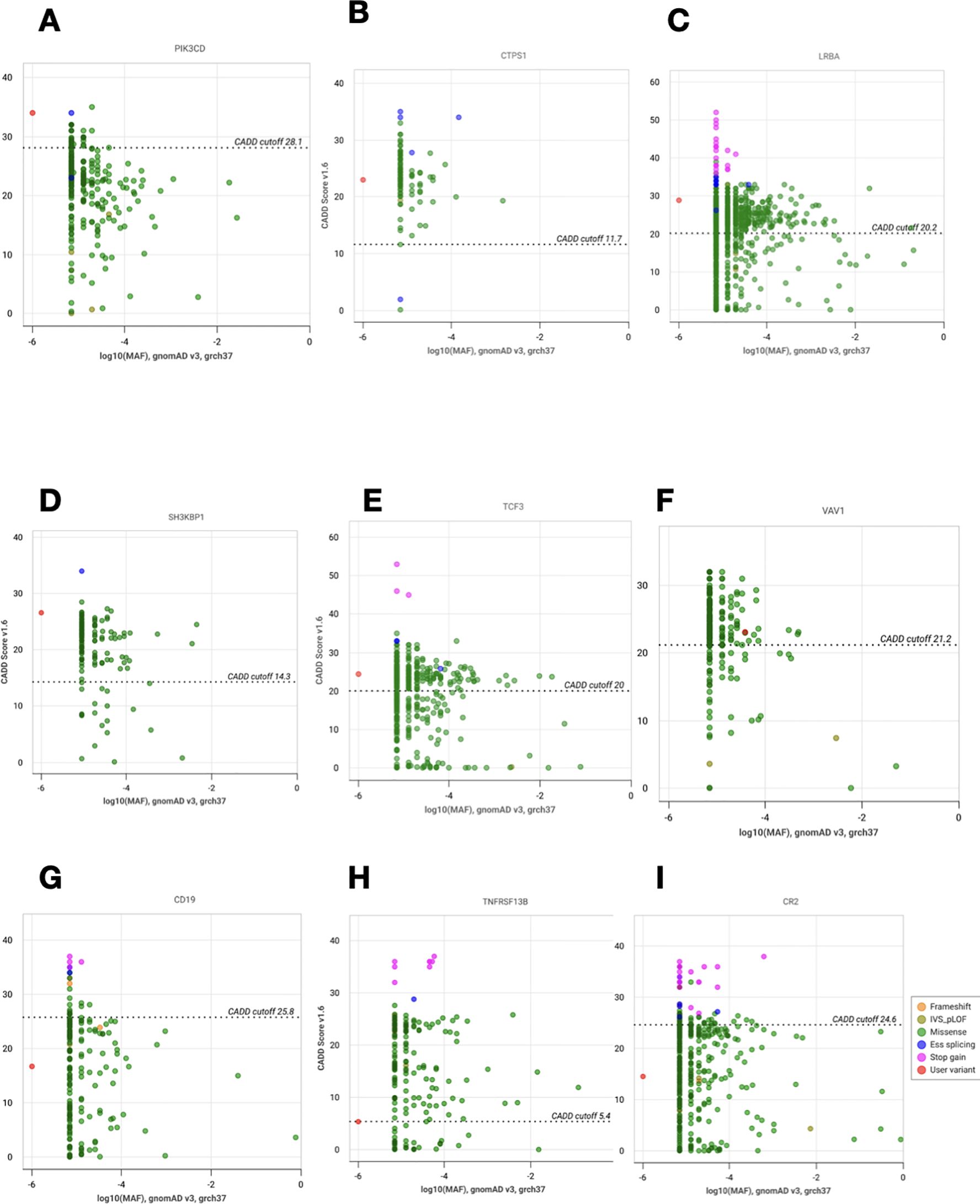
Figure 5. CADD vs. MAF plot of novel variants with high or moderate phenotype- genotype correlations. (A) Analysis of PIK3CD (p.Arg785Trp). (B) Analysis of CTPS1 (p.Leu88Val). (C) Analysis of LRBA (p.Asp2169Asn). (D) Analysis of SH3KBP1 (p.Asp221Gly). (E) Analysis of TCF3 (p.Arg6Gly). (F) Analysis of VAV1 (p.Lys804Thr). (G) Analysis of CD19 (p.Phe420Leu). (H) Analysis of TNFRSF13B (p.Arg202Cys). (I) Analysis of CR2 (p.Gln820His). The vertical and horizontal axes show combined annotation- dependent depletion scores (CADD) and minor allele frequencies (MAF), respectively. The CADD cutoff represents the MSC (mutation significance cutoff) on CADD with CI:95%. The red dot corresponds to the new variant and the other dots to the other variants reported for each gene.
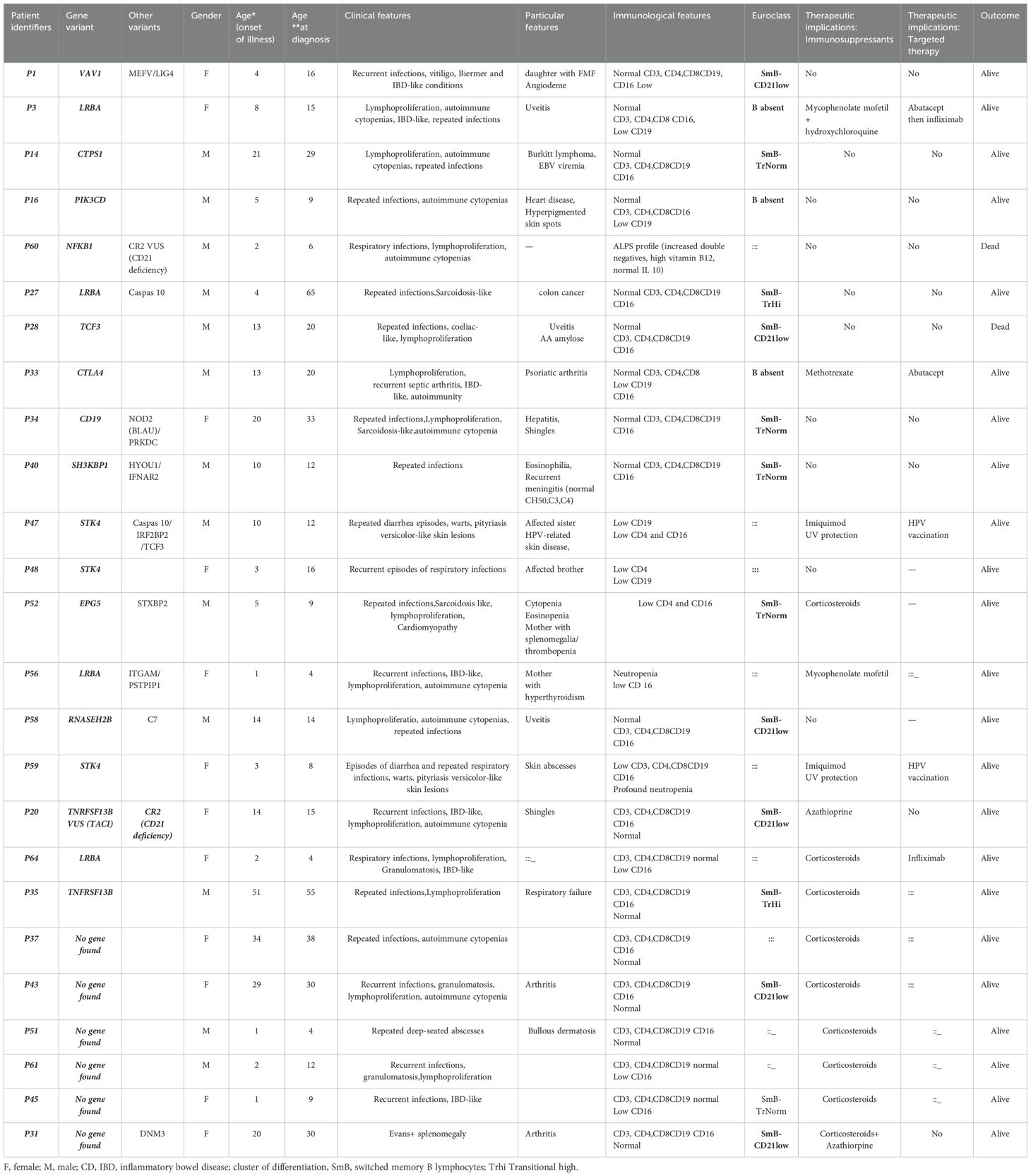
Table 4. Potential genetic variants identified in the Moroccan cohort and their clinical and immunological characteristics, and outcomes.
Family segregation study was done for STK4 gene in two siblings (P47, P48).
No functional studies were performed to evaluate the pathogenicity of identified variants.
Subgroup analysis based on consanguinity
A total of 61 CVID patients were analyzed, of whom 33 (54.1%) were born from consanguineous unions. Subgroup analysis revealed several clinically and statistically meaningful differences. Patients from consanguineous families presented with an earlier mean age at disease onset compared to non-consanguineous individuals (16.2 ± 17.1 years vs. 21.3 ± 17.3 years; p < 0.001), and experienced a shorter diagnostic delay (5.8 ± 4.6 years vs. 7.9 ± 11.2 years; p = 0.12). Complications were more frequently observed in the consanguineous group, including a significantly higher rate of severe infections (75% vs. 25%; p = 0.03) and a trend toward increased bronchiectasis (59.2% vs. 40.8%; p = 0.06). Interestingly, immune dysregulation manifestations were less prevalent among consanguineous patients, with lower rates of autoimmunity (33.3% vs. 66.7%; p = 0.05), lymphoproliferation (40% vs. 60%; p = 0.44), and granulomatous disease (18.2% vs. 81.8%; p = 0.04). A disease-causing genetic variant was identified more frequently in consanguineous individuals (60% vs. 40%; p = 0.06), reinforcing the suspected higher burden of monogenic inborn errors of immunity in this subgroup. There was no significant difference in the use of intravenous immunoglobulin therapy between the two groups (85.7% vs. 87.9%; p = 0.80). Finally, the use of immunomodulatory or immunosuppressive treatments was more common in consanguineous patients (25% vs. 12.1%), although the difference did not reach statistical significance (p = 0.19).
Discussion
Our study provides the first comprehensive and integrative overview of the clinical, immunological, and genetic spectrum of CVID patients in Morocco, a middle/low-income North African country. Our findings add to the growing body of literature on CVID in non-Western populations and highlight both similarities and specific challenges in diagnosis and management.
Epidemiology and diagnostic challenges
CVID constituted 5.7% of all IEIs in the Moroccan registry reported in 2014 (10) and currently represents 8.2% of all IEIs in the Moroccan registry (742 cases) compiled by December 2024, suggesting enhanced disease recognition and improved diagnostic capabilities in this part of the world. Our study is in line with the prevalence registered in the IEI prevalence study in the MENA region, where CVID accounted for 11,3% (n=1935) (17). Potential reasons behind the low CVID account in Morocco could be underdiagnosis, registry bias and consanguinity-driven recessive IEIs in the MENA region (17).
The mean age at diagnosis was 25.9 (SD 18.7) years, which is older than previous reports from our country (29, 30), reflecting a better recognition of CVID by practitioners caring for adult patients. However, the mean diagnostic delay is significant, 6.91 (SD 8.82) years, consistent with reported literature (17, 31, 32).
In our cohort, the consanguinity rate was notably high at 54.1%, which is significantly greater than the average of 15-25% found in the general Moroccan population (33, 34). Our findings indicate that consanguinity may be linked to earlier disease onset, more severe infections, and a higher rate of genetic diagnoses in patients with CVID. However, these results should be interpreted with caution due to several limitations. The small sample size, the retrospective nature, restricted access to genetic testing, and potential underreporting of immune dysregulation features may have impacted the subgroup comparisons. Additionally, genetic analyses were only conducted in a subset of patients, which might underestimate the true prevalence of monogenic diseases. To confirm these trends and improve tailored management strategies for consanguineous populations, larger prospective studies with comprehensive genomic screening are necessary. Nevertheless, it is essential to systematically investigate parental consanguinity in patients with CVID, as it is often linked to an increased risk of developing severe clinical complications (35).
Infectious and non-infectious complications
Pulmonary infections were the predominant infectious complication (88.5%), consistent with global data showing respiratory tract infections as the most common clinical manifestation of CVID (36, 37). Gastrointestinal infections, notably those caused by giardia enteritis, norovirus, and enterovirus, were also prevalent (63.9%), in line with previous studies indicating that up to 60% of CVID patients experience gastrointestinal infections (38). The high prevalence of Helicobacter pylori-induced gastritis and tuberculosis (11.5%) further underscores the regional burden of infectious diseases (39, 40).
Non-infectious complications were observed in 49.2% of the total cohort and were significantly more frequent in patients with a confirmed genetic defect (79%). This highlights the potential role of underlying monogenic etiologies in driving more severe phenotypes. Additionally, prolonged diagnostic delay and limited access to specialized care in our region may contribute to the higher burden of complications. Although data on atypical pathogens and IgG trough levels were not available for all patients, these factors may also have played a role and should be further explored in future studies.
Lymphoproliferation and splenomegaly being the most frequent (50.8%). These findings corroborate prior studies reporting lymphoproliferative disorders in nearly half of CVID patients (6). The prevalence of autoimmune cytopenias (39.3%), granulomatous disease (18%), and systemic autoimmune diseases (lupus, rheumatoid arthritis) aligns with previous research emphasizing the autoimmune predisposition in CVID (2). It underlies the paradoxical combination between immune deficiency and immune dysregulation in CVID and the difficulty of patients’ management to navigate all this combined complexity.
Bronchiectasis (44.3%) was the most frequent pulmonary complication, consistent with rates reported in European cohorts (41). It may be caused by repeated infections combined with local pulmonary immune dysregulation (41, 42).
The absence of histologically confirmed GLILD (granulomatous-lymphocytic interstitial lung disease) in our patients is notable, given its reported prevalence of 10-20% in Western cohorts (42, 43), and can be explained by the limited screening for chronic lung disease in our settings, especially in paucisymptomatic patients, and also by the lack of consensus on diagnostic criteria for CVID interstitial lung disease (43). This highlights the need for systematic and active screening for chronic lung disease in CVID patients, as it is associated with increased morbidity and mortality. Histologically confirmed enteropathy mimicking celiac disease (14.8%) and IBD-like disease (13.1%) reinforce the association between CVID and gastrointestinal autoimmunity and inflammation (38). Intestinal biopsies often show an increase in the number of intraepithelial lymphocytes, giving an appearance similar to lymphocytic colitis, but this intestinal inflammation in CVID is distinguished from classic IBD by the absence of plasma cells in the chorion (44).In celiac disease, the histology is also similar, except for the absence of plasma cells in the chorion in CVID, but with the constant presence of CD3+ CD8+ intraepithelial T lymphocytes (45). CVID patients have an increased risk of gastric cancer and digestive lymphoma (46). These findings further emphasize the need for early gastroenterological evaluation in CVID patients presenting with chronic diarrhea and weight loss.
Immunological and genetic landscape
The immunoglobulin profile and lymphocyte subpopulation analysis in our cohort demonstrated significant heterogeneity, as reported in the literature (34). IgG hypogammaglobulinemia was the most consistent feature, with variable reductions in IgA and IgM. Decreased memory switched B cells was also the hallmark of immune dysregulation in our cohort (52,8% of patients with <2% switched memory B cells had immune dysregulation disorders), that was also noted in the Euroclass trial by Wher et al. (20).
While most cases of CVID are sporadic, it is estimated that 5-25% of cases run in families with transmission, most often autosomal dominant (3). It has been 22 years since the first genetic etiology of CVID was described, reporting homozygous mutations of ICOS of autosomal recessive transmission in patients from the same family (47). Since then, a growing number of genetic diseases responsible for CVID have been described, affecting up to 10% or even 20% of patients, particularly those with non-infectious manifestations (8, 48, 49). However, in consanguineous cohorts such as ours, monogenic disorders can account for approximately 70% of CVID patients (50). With this increasing number of genes described, it has become clear that CVID is an umbrella disease and that many of these genetic defects cause distinct disease entities, which we currently refer to as CVID-like diseases (49).
In our study, genetic testing using a targeted panel in 25 patients (41%) revealed 19 putative pathogenic variants in 13 genes, with novel variants in 73.7% of cases. The identification of mutations in LRBA, CTLA4, PIK3CD, and NFKB1 is in line with emerging literature demonstrating the genetic heterogeneity of CVID (9–11). Notably, STK4 deficiency was identified in three patients, a gene for combined immunodeficiencies; this type of entity is increasingly recognized in CVID-like phenotypes with combined immunodeficiency features (51, 52). While 52.6% of patients demonstrated a high phenotype-genotype correlation, the absence of functional validation remains a limitation.
Genetic testing in patients with CVID can potentially guide therapy, as was the case in our study, such as the use of abatacept, infliximab, and rituximab in the treatment of autoimmune manifestations of CVID in patients with CTLA-4 and LRBA mutations. mTOR inhibitors can also be considered, especially in patients with PI3K signaling defects (7).
Therapeutic management and outcomes
Immunoglobulin replacement therapy (IgRT) was administered exclusively through intravenous infusion to 86.9% of patients, leading to a reduction in the incidence of recurrent and severe infections as well as subsequent hospitalizations. However, due to the retrospective nature of our study, these results should be interpreted with caution.
The use of prophylactic antibiotics in 75.4% of cases is in line with international recommendations for infection prevention in CVID patients (41, 53), as some patients had persistent infections even after immunoglobulin replacement therapy.
The use of corticosteroids (49.2%) and immunosuppressive agents (16.4%) reflects the need for immune modulation in CVID-associated autoimmune and inflammatory complications. Targeted therapies, such as rituximab, abatacept, and infliximab, were used in 8.2% of cases, highlighting the evolving role of biologics in CVID management (54).
Mortality in our cohort was 4.9%, mainly due to infectious complications and dysimmune disorders. This rate is lower than in other large CVID studies (55, 56), which may be due to the short duration of the study and the small number of patients. Mortality is increased in CVID, 10 times higher than in the general population, as reported in some studies (55), highlighting the importance of early diagnosis and individualized therapeutic strategies.
Strengths and limitations
Our study presents several strengths. It is the first comprehensive analysis of CVID in Morocco, contributing valuable epidemiological, clinical, and genetic data from a non-Western setting. The inclusion of genetic analysis provides novel insights into the molecular basis of CVID in this population. Additionally, our study highlights specific regional challenges, including diagnostic delays and a high burden of infectious diseases, thereby informing future healthcare strategies.
Several limitations of this study should be acknowledged. First, the relatively small sample size may restrict the generalizability of our findings to broader populations. Genetic testing was performed in only 41% of patients, and functional validation of novel variants could not be conducted, limiting our ability to confirm the pathogenicity of some genetic alterations. Moreover, due to the retrospective design, potential information bias may have affected the accuracy of recorded clinical complications and therapeutic outcomes. Another important limitation concerns the diagnostic workup: full immunophenotyping was not available for all patients due to technical constraints. We followed ESID and MENA definitions, but our decisions were also guided by expert consensus within our national immunology network, relying on clinical presentation, Ig levels, and exclusion of secondary causes. Nevertheless, the absence of full immunophenotyping and standardized functional immune evaluation in all patients reflects the technical and resource constraints of our setting. Future prospective studies with larger cohorts, comprehensive immunological assessment, and long-term follow-up are warranted to better characterize the phenotypic and genotypic diversity of CVID in North African populations.
Conclusion
Our study underscores the clinical and immunological heterogeneity of CVID in a North African population, marked by a high burden of infectious and autoimmune complications and significant diagnostic delays. The substantial proportion of patients born to consanguineous unions and the high rate of monogenic diagnoses highlight the critical role of genetic factors in this setting. These findings reinforce the value of early genetic testing in consanguineous populations to enhance diagnostic accuracy, enable personalized management, and support genetic counseling. Incorporating genomic tools into national diagnostic protocols could significantly improve outcomes and reduce healthcare disparities in similar low- and middle-income contexts. Future efforts should focus on expanding access to genetic sequencing, functionally validating novel variants, and developing region-specific care strategies to optimize long-term outcomes for patients with CVID.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the ethics committee of the University Hospital Ibn Rochd in Casablanca. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
AA: Conceptualization, Data curation, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing. AM: Data curation, Formal analysis, Methodology, Software, Writing – review & editing. IB: Data curation, Supervision, Validation, Writing – review & editing. FA: Conceptualization, Investigation, Supervision, Writing – review & editing. JB: Conceptualization, Formal analysis, Methodology, Writing – review & editing. KE: Supervision, Validation, Writing – review & editing. AN: Supervision, Validation, Writing – review & editing. HK: Supervision, Validation, Writing – review & editing. AB: Project administration, Resources, Supervision, Validation, Writing – review & editing. MM: Conceptualization, Methodology, Project administration, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1602820/full#supplementary-material
References
1. Fudenberg H, Good RA, Goodman HC, Hitzig W, Kunkel HG, Roitt IM, et al. Primary immunodeficiencies. Report of a world health organization committee. Pediatrics. (1971) 47:927–46. doi: 10.1542/peds.47.5.927
2. Gathmann B, Mahlaoui N, CEREDIH, Gérard L, Oksenhendler E, Warnatz K, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. (2014) 134:116–26. doi: 10.1016/j.jaci.2013.12.1077
3. Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International consensus document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. (2016) 4:38–59. doi: 10.1016/j.jaip.2015.07.025
4. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. (2008) 112:277–86. doi: 10.1182/blood-2007-11-124545
5. Jørgensen SF, Fevang B, and Aukrust P. Autoimmunity and inflammation in CVID: a possible crosstalk between immune activation, gut microbiota, and epigenetic modifications. J Clin Immunol. (2019) 39:30–6. doi: 10.1007/s10875-018-0574-z
6. Rizvi FS, Zainaldain H, Rafiemanesh H, Jamee M, Hossein-Khannazer N, Hamedifar H, et al. Autoimmunity in common variable immunodeficiency: a systematic review and meta-analysis. Expert Rev Clin Immunol. (2020) 16:1227–35. doi: 10.1080/1744666X.2021.1850272
7. Azizi G, Ziaee V, Tavakol M, Alinia T, Yazdai R, Mohammadi H, et al. Approach to the management of autoimmunity in primary immunodeficiency. Scand J Immunol. (2017) 85:13–29. doi: 10.1111/sji.12506
8. Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova JL, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol. (2016) 7:220. doi: 10.3389/fimmu.2016.00220
9. Abolhassani H, Hammarström L, and Cunningham-Rundles C. Current genetic landscape in common variable immune deficiency. Blood. (2020) 135:656–67. doi: 10.1182/blood.2019000929
10. Ameratunga R, Lehnert K, Woon ST, Gillis D, Bryant VL, Slade CA, et al. Review: diagnosing common variable immunodeficiency disorder in the era of genome sequencing. Clin Rev Allergy Immunol. (2018) 54:261–8. doi: 10.1007/s12016-017-8645-0
11. Cunningham-Rundles C, Casanova JL, and Boisson B. Genetics and clinical phenotypes in common variable immunodeficiency. Front Genet. (2023) 14:1272912. doi: 10.3389/fgene.2023.1272912
12. Ramirez NJ, Posadas-Cantera S, Caballero-Oteyza A, Camacho-Ordonez N, and Grimbacher B. There is no gene for CVID - novel monogenetic causes for primary antibody deficiency. Curr Opin Immunol. (2021) 72:176–85. doi: 10.1016/j.coi.2021.05.010
13. Li J, Wei Z, Li YR, Maggadottir SM, Chang X, Desai A, et al. Understanding the genetic and epigenetic basis of common variable immunodeficiency disorder through omics approaches. Biochim Biophys Acta. (2016) 1860:2656–63. doi: 10.1016/j.bbagen.2016.06.014
14. Peng XP, Caballero-Oteyza A, and Grimbacher B. Common variable immunodeficiency: more pathways than roads to Rome. Annu Rev Pathol. (2023) 18:283–310. doi: 10.1146/annurev-pathmechdis-031521-024229
15. Bousfiha AA, Errami A, Jeddane L, Mellouli F, Reda SM, Adeli M, et al. Primary immunodeficiencies: epidemiology in the maghreb. Tunis Med. (2018) 96:672–7.
16. Moundir A, Ouair H, Benhsaien I, Jeddane L, Rada N, Amenzoui N, et al. Genetic diagnosis of inborn errors of immunity in an emerging country: a retrospective study of 216 moroccan patients. J Clin Immunol. (2023) 43:485–94. doi: 10.1007/s10875-022-01398-z
17. Aghamohammadi A, Rezaei N, Yazdani R, Delavari S, Kutukculer N, Topyildiz E, et al. Consensus middle east and north africa registry on inborn errors of immunity. J Clin Immunol. (2021) 41:1339–51. doi: 10.1007/s10875-021-01053-z
18. Seidel MG, Kindle G, Gathmann B, Quinti I, Buckland M, van Montfrans J, et al. The european society for immunodeficiencies (ESID) registry working definitions for the clinical diagnosis of inborn errors of immunity. J Allergy Clin Immunol Pract. (2019) 7:1763–70. doi: 10.1016/j.jaip.2019.02.004
19. Baris S, Abolhassani H, Massaad MJ, Al-Nesf M, Chavoshzadeh Z, Keles S, et al. The middle east and north africa diagnosis and management guidelines for inborn errors of immunity. J Allergy Clin Immunol Pract. (2023) 11:158–180.e11. doi: 10.1016/j.jaip.2022.10.003
20. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. (2008) 111:77–85. doi: 10.1182/blood-2007-06-091744
21. Invitae Primary Immunodeficiency Panel | Test catalog | Invitae. Available online at: https://www.invitae.com/us/providers/test-catalog/test-08100 (Accessed February 10, 2025).
22. Poli MC, Aksentijevich I, Bousfiha A, Cunningham-Rundles C, Hambleton S, Klein C, et al. Human inborn errors of immunity: 2024 update on the classification from the International Union of Immunological Societies Expert Committee. J Hum Immun. (2025) 1:e20250003. doi: 10.70962/jhi.20250003
23. Rentzsch P, Witten D, Cooper GM, Shendure J, and Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. (2019) 47:D886–94. doi: 10.1093/nar/gky1016
24. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, and Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. (2012) 40:W452–457. doi: 10.1093/nar/gks539
25. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
26. Cheng J, Novati G, Pan J, Bycroft C, Žemgulytė A, Applebaum T, et al. Accurate proteome- wide missense variant effect prediction with AlphaMissense. Science. (2023) 381:eadg7492. doi: 10.1126/science.adg7492
27. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med Off J Am Coll Med Genet. (2015) 17:405–24. doi: 10.1038/gim.2015.30
28. Zhang P, Bigio B, Rapaport F, Zhang SY, Casanova JL, Abel L, et al. PopViz: a webserver for visualizing minor allele frequencies and damage prediction scores of human genetic variations. Bioinformatics. (2018) 34:4307–9. doi: 10.1093/bioinformatics/bty536
29. Bousfiha AA, Jeddane L, El Hafidi N, Benajiba N, Rada N, El Bakkouri J, et al. First report on the Moroccan registry of primary immunodeficiencies: 15 years of experience (1998-2012). J Clin Immunol. (2014) 34:459–68. doi: 10.1007/s10875-014-0005-8
30. Mokhantar K, Abire A, Ailal F, Bakkouri JE, Ouazahrou K, Errami A, et al. Classification of common variable immunodeficiency using measurement of B-cell subsets in moroccan patients. Turk J Immunol. (2023) 11:29–36. doi: 10.4274/tji.galenos.2023.36854
31. Bonilla FA, Khan DA, Ballas ZK, Chinen J, Frank MM, Hsu JT, et al. Practice parameter for the diagnosis and management of inborn errors of immunity (IEI). J Allergy Clin Immunol. (2015) 136:1186–1205.e1-78. doi: 10.1016/j.jaci.2015.04.049
32. Cabañero-Navalon MD, Garcia-Bustos V, Nuñez-Beltran M, Císcar Fernández P, Mateu L, Solanich X, et al. Current clinical spectrum of common variable immunodeficiency in Spain: The multicentric nationwide GTEM-SEMI-CVID registry. Front Immunol. (2022) 13:1033666. doi: 10.3389/fimmu.2022.1033666
33. El Khair A, Dahbi N, Cheffi K, Talbi J, Hilali A, and El Ossmani H. Characterization of the consanguinity in the moroccan population of doukkala. J Soc Sci. (2023) 19:15–21. doi: 10.3844/jssp.2023.15.21
34. Jaouad IC, Elalaoui SC, Sbiti A, Elkerh F, Belmahi L, and Sefiani A. Consanguineous marriages in Morocco and the consequence for the incidence of autosomal recessive disorders. J Biosoc Sci. (2009) 41:575–81. doi: 10.1017/S0021932009003393
35. Rivoisy C, Gérard L, Boutboul D, Malphettes M, Fieschi C, Durieu I, et al. Parental consanguinity is associated with a severe phenotype in common variable immunodeficiency. J Clin Immunol. (2012) 32:98–105. doi: 10.1007/s10875-011-9604-9
36. Carrabba M, Salvi M, Baselli LA, Serafino S, Zarantonello M, Trombetta E, et al. Long-term follow-up in common variable immunodeficiency: the pediatric-onset and adult-onset landscape. Front Pediatr. (2023) 11:1125994. doi: 10.3389/fped.2023.1125994
37. Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. (2007) 27:308–16. doi: 10.1007/s10875-007-9075-1
38. Velthof L, Geldof J, Truyens M, Van Dorpe J, Ferdinande L, De Vriendt C, et al. Gastrointestinal disease in common variable immunodeficiency disorder (CVID): histological patterns, diagnostic clues and pitfalls for the pathologist and gastroenterologist. J Clin Med. (2025) 14:497. doi: 10.3390/jcm14020497
39. Alsulaimany FAS, Awan ZA, Almohamady AM, Koumu MI, Yaghmoor BE, Elhady SS, et al. Prevalence of helicobacter pylori infection and diagnostic methods in the middle east and north africa region. Med Kaunas Lith. (2020) 56:169. doi: 10.3390/medicina56040169
40. Moradinazar M, Afshar ZM, Ramazani U, Shakiba M, Shirvani M, and Darvishi S. Epidemiological features of tuberculosis in the Middle East and North Africa from 1990 to 2019: results from the global burden of disease Study 2019. Afr Health Sci. (2023) 23:366–75. doi: 10.4314/ahs.v23i3.43
41. Ramzi N, Jamee M, Bakhtiyari M, Rafiemanesh H, Zainaldain H, Tavakol M, et al. Bronchiectasis in common variable immunodeficiency: A systematic review and meta-analysis. Pediatr Pulmonol. (2020) 55:292–9. doi: 10.1002/ppul.24599
42. Schussler E, Beasley MB, and Maglione PJ. Lung disease in primary antibody deficiencies. J Allergy Clin Immunol Pract. (2016) 4:1039–52. doi: 10.1016/j.jaip.2016.08.005
43. Bintalib HM, van de Ven A, Jacob J, Davidsen JR, Fevang B, Hanitsch LG, et al. Diagnostic testing for interstitial lung disease in common variable immunodeficiency: a systematic review. Front Immunol. (2023) 14:1190235. doi: 10.3389/fimmu.2023.1190235
44. Viallard JF, Lebail B, Begueret H, and Fieschi C. Les déficits immunitaires communs variables (DICV): partie 2. Mise à jour clinique et thérapeutique. Rev Med Interne. (2021) 42:473–81. doi: 10.1016/j.revmed.2020.12.015
45. Uzzan M, Ko HM, Mehandru S, and Cunningham-Rundles C. Gastrointestinal disorders associated with common variable immune deficiency (CVID) and chronic granulomatous disease (CGD). Curr Gastroenterol Rep. (2016) 18:17. doi: 10.1007/s11894-016-0491-3
46. Leone P, Vacca A, Dammacco F, and Racanelli V. Common variable immunodeficiency and gastric Malignancies. Int J Mol Sci. (2018) 19:451. doi: 10.3390/ijms19020451
47. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Dräger R, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. (2003) 4:261–8. doi: 10.1038/ni902
48. van Schouwenburg PA, Davenport EE, Kienzler AK, Marwah I, Wright B, Lucas M, et al. Application of whole genome and RNA sequencing to investigate the genomic landscape of common variable immunodeficiency disorders. Clin Immunol Orlando Fla. (2015) 160:301–14. doi: 10.1016/j.clim.2015.05.020
49. Bogaert DJA, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, and Haerynck F. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet. (2016) 53:575–90. doi: 10.1136/jmedgenet-2015-103690
50. Abolhassani H, Aghamohammadi A, Fang M, Rezaei N, Jiang C, Liu X, et al. Clinical implications of systematic phenotyping and exome sequencing in patients with primary antibody deficiency. Genet Med Off J Am Coll Med Genet. (2019) 21:243–51. doi: 10.1038/s41436-018-0012-x
51. Buchbinder D, Baker R, Lee YN, Ravell J, Zhang Y, McElwee J, et al. Identification of patients with RAG mutations previously diagnosed with common variable immunodeficiency disorders. J Clin Immunol. (2015) 35:119–24. doi: 10.1007/s10875-014-0121-5
52. Schepp J, Bulashevska A, Mannhardt-Laakmann W, Cao H, Yang F, Seidl M, et al. Deficiency of adenosine deaminase 2 causes antibody deficiency. J Clin Immunol. (2016) 36:179–86. doi: 10.1007/s10875-016-0245-x
53. Salehzadeh M, Aghamohammadi A, and Rezaei N. Evaluation of immunoglobulin levels and infection rate in patients with common variable immunodeficiency after immunoglobulin replacement therapy. J Microbiol Immunol Infect Wei Mian Yu Gan Ran Za Zhi. (2010) 43:11–7. doi: 10.1016/S1684-1182(10)60002-3
54. Fevang B. Treatment of inflammatory complications in common variable immunodeficiency (CVID): current concepts and future perspectives. Expert Rev Clin Immunol. (2023) 19:627–38. doi: 10.1080/1744666X.2023.2198208
55. Resnick ES, Moshier EL, Godbold JH, and Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. (2012) 119:1650–7. doi: 10.1182/blood-2011-09-377945
Keywords: common variable immunodeficiency, inborn errors of immunity, genetic variants, immune dysregulation, Morocco, Africa, next-generation sequencing
Citation: Allaoui A, Moundir A, Benhsaien I, Ailal F, Bakkouri JE, Echchilali K, Naitlho A, Kabli HE, Bousfiha AA and Moudatir M (2025) Clinical, immunological, and genetic landscape of common variable immunodeficiency in Morocco: a nationwide multicenter study. Front. Immunol. 16:1602820. doi: 10.3389/fimmu.2025.1602820
Received: 30 March 2025; Accepted: 23 June 2025;
Published: 09 July 2025.
Edited by:
Antonio Marzollo, University of Padua, ItalyReviewed by:
Federica Pulvirenti, Accademic Hospital Policlinico Umberto, ItalyMattia Moratti, University of Rome Tor Vergata, Italy
Copyright © 2025 Allaoui, Moundir, Benhsaien, Ailal, Bakkouri, Echchilali, Naitlho, Kabli, Bousfiha and Moudatir. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abire Allaoui, YWJpcmUuYWxsYW91aUBnbWFpbC5jb20=