 Wujianhong Liu
Wujianhong Liu Shengrong Dong
Shengrong Dong Feiran Hao†
Feiran Hao†- Department of Pathology, Shanghai Tenth People’s Hospital Affiliated to Tongji University, Shanghai, China
Colorectal cancer (CRC) is the third most common cancer worldwide, and its high incidence, mortality, and treatment resistance highlight the urgency of exploring new therapeutic targets. As research into cancer metabolic reprogramming deepens, the central role of lipid metabolism abnormalities in CRC progression has gradually become apparent. In the tumor microenvironment (TME), conditions such as hypoxia, glucose deprivation, and lactic acid accumulation alter the energy demands of tumor cells, driving metabolic reprogramming in lipid uptake, synthesis, and oxidation. This reprogramming helps maintain high energy needs and supports the malignant growth of tumor cells. This lipid metabolic reprogramming provides tumor cells with the necessary energy and enhances their proliferation, invasion, immune evasion, and resistance characteristics. Moreover, the lipid metabolic reprogramming of tumor cells is closely related to various cells within the TME, and these interactions promote, to some extent, the remodeling of the tumor microenvironment, further driving tumor development. Emerging lipid detection technologies position specific lipid molecules as promising biomarkers for auxiliary diagnosis and prognostic evaluation. Concurrently, targeting key lipid metabolic pathways offers innovative strategies to optimize existing therapies and overcome drug resistance. This review summarizes the basic and abnormal mechanisms of lipid metabolism in CRC, lipid metabolic interactions in the tumor microenvironment, the regulatory network between the gut microbiota and lipid metabolism, and the progress in therapeutic strategies targeting lipid metabolism. By exploring the interaction between CRC and lipid metabolism in depth, this review aims to provide new ideas and theoretical support for the treatment, early intervention, and prognosis evaluation of CRC.
1 Introduction
CRC is a leading cause of cancer-related mortality globally, with a 5-year survival rate of only 65% (1). In recent years, with changes in lifestyle and population aging, the incidence and mortality rates of CRC have shown an upward trend worldwide, especially in some developing countries where the growth rate is more significant (2, 3). By 2040, there will be 3.2 million new cases and 1.6 million deaths globally (4). Currently, clinical management of CRC relies on a multidisciplinary treatment approach, including surgical resection, adjuvant chemotherapy (such as the FOLFOX regimen), radiotherapy, and targeted therapy (anti-EGFR monoclonal antibodies, anti-VEGF drugs) (5–7). However, these therapeutic strategies still have numerous limitations. First, more than half of early-stage patients experience recurrence or metastasis after surgery (8); second, approximately 23% of CRC patients have metastasis at the time of diagnosis, for whom effective treatments are still lacking. The efficacy of targeted drugs is limited by molecular heterogeneity, such as only about 40% of metastatic CRC patients carrying the wild-type RAS gene benefiting from cetuximab treatment (9); additionally, about 90% of microsatellite stable (MSS) CRC patients do not meet the criteria for immune checkpoint inhibitors (such as PD-1 inhibitors), and more than half of patients with metastatic defective mismatch repair (dMMR) CRC do not respond to immune checkpoint inhibition (ICI) (10). Moreover, during treatment, tumor resistance is a significant concern. Therefore, there is an urgent need to develop new intervention strategies targeting the core biological mechanisms of CRC progression to address current therapeutic challenges.
In recent years, with the continuous advancement of metabolomics and oncology research, metabolic reprogramming has gradually become an important focus in cancer research (11, 12). Tumor cells alter their energy acquisition methods due to their high metabolic demand, preferentially utilizing glycolysis for rapid energy supply through the “Warburg effect,” while also undergoing profound remodeling of lipid and amino acid metabolic pathways (13). This metabolic adaptation not only meets the biosynthetic demands (such as membrane phospholipids and signaling molecules) but also regulates epigenetic modifications, oxidative stress responses, and the immune microenvironment through metabolic products (14). In CRC, metabolic reprogramming exhibits significant stage-dependent dynamics: early-stage tumors primarily activate glycolysis (15), while in advanced stages, tumors display various metabolic abnormalities, with lipid metabolism disorders (such as overexpression of fatty acid synthase, FASN) becoming increasingly prominent (16, 17). Especially when glucose is limited in the microenvironment, tumor cells tend to shift toward lipid metabolism (18). Lipid metabolic reprogramming plays a variety of key roles in the occurrence and development of CRC. On the one hand, excessive fatty acid synthesis provides membrane components and energy reserves for rapid proliferation (19, 20), contributing to tumor cell proliferation; on the other hand, abnormal lipid metabolism can enhance lipid-driven cell signaling (21), contributing to tumor cell proliferation; on the other hand, abnormal lipid metabolism can enhance lipid-driven cell signaling (22–24).
Although intervention strategies targeting lipid metabolism (such as the fatty acid synthase inhibitor TVB-2640) have shown potential in clinical trials, their efficacy is limited by tumor heterogeneity, metabolic pathway redundancy, and the complexity of host-microbiome interactions (25, 26). Therefore, systematically analyzing the dynamic regulatory network of lipid metabolism reprogramming in CRC and elucidating its interactions with the tumor microenvironment (TME) and gut microbiota is an essential step in overcoming the treatment challenges of CRC. In this review, we summarize the key mechanisms of lipid metabolic abnormalities in CRC in recent years, their relationship with the tumor microenvironment and gut microbiota, and discuss therapeutic strategies targeting lipid metabolism, with the aim of providing theoretical support for CRC treatment.
2 Lipid metabolism in CRC
Lipid metabolism is a core biological process that maintains cellular homeostasis, encompassing lipid uptake, lipid synthesis, and lipid oxidation (Figure 1) (27). At the same mass, lipid metabolism can provide more energy than glycolysis and amino acid metabolism. In CRC cells, lipid metabolism provides energy, builds biological membranes, and serves as secondary messengers to participate in cell activity signaling pathways (28), supporting unique functions such as cell proliferation, growth, invasion, and angiogenesis. Table 1 summarizing the key factors involved in lipid metabolism in CRC.
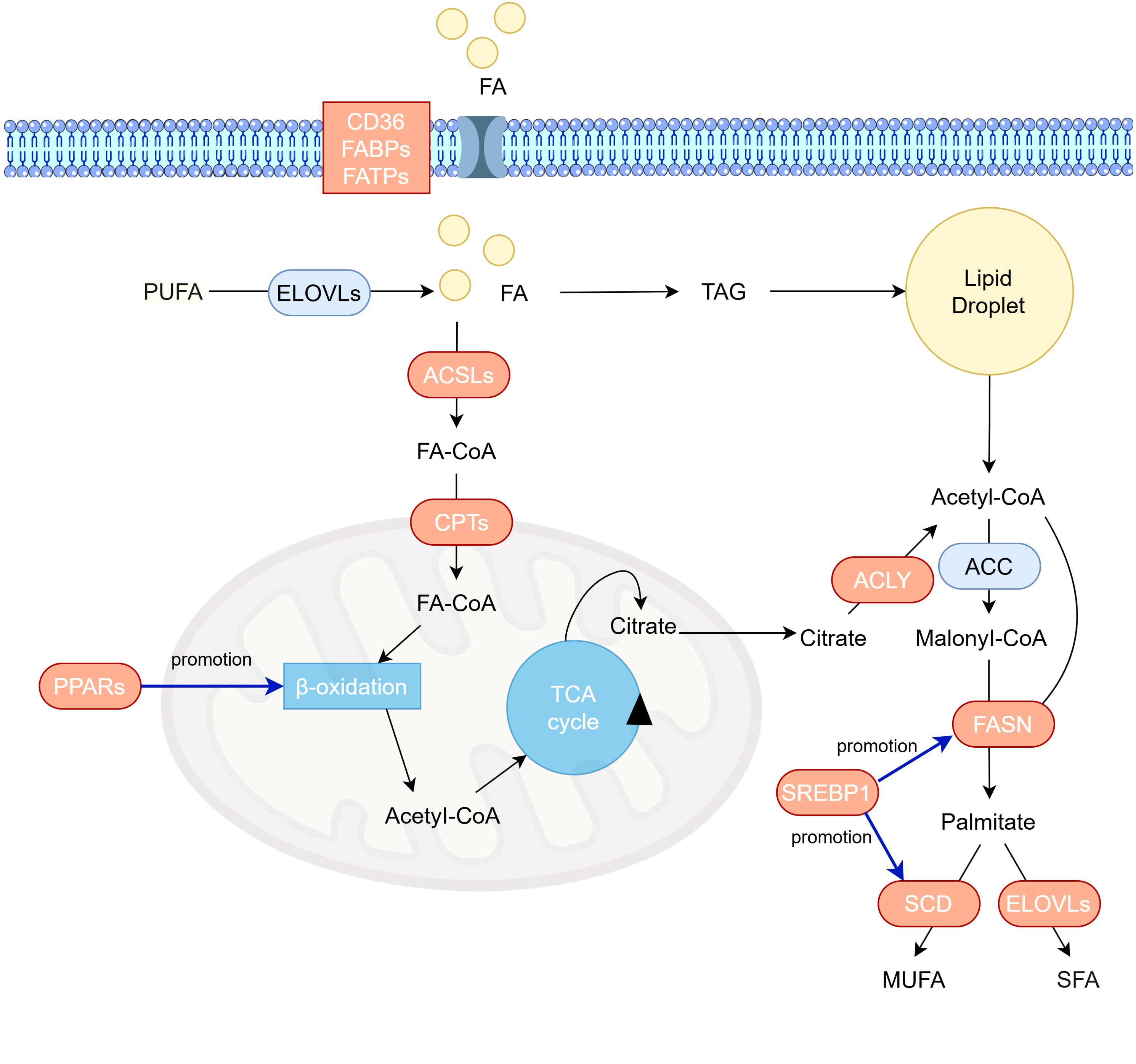
Figure 1. Overview of lipid metabolic reprogramming in colorectal cancer cells. Exogenous fatty acids enter cells via lipid transport proteins (CD36, FABPs, FATPs). Intracellularly, fatty acids are activated to acyl-CoA by ACSL. Long-chain acyl-CoA is then converted to acylcarnitine by carnitine CPT1. After translocation across the inner mitochondrial membrane, carnitine CPT2 regenerates acyl-CoA. Within the mitochondrial matrix, acyl-CoA undergoes β-oxidation to yield acetyl-CoA, which enters the tricarboxylic acid (TCA) cycle for complete oxidation. Citrate from the TCA cycle effluxes to the cytosol, where ACLY catalyzes its conversion to cytosolic acetyl-CoA.ACC subsequently carboxylates acetyl-CoA to malonyl-CoA. Acetyl-CoA and malonyl-CoA are condensed by FASN to form palmitate, which is elongated to 18–24 carbon chains by the ELOVL enzyme family in the endoplasmic reticulum. Activated palmitate can also be desaturated by SCD to generate MUFA and PUFA. Proteins with increased expression in CRC are highlighted in peach. ACC, acetyl-CoA-carboxylase; ACLY, ATP-citrate lyase; ACSL, acyl-CoA synthetase long-chain family; CPT, carnitine palmitoyl transferase; ELOVL, elongation of very-long-chain fatty acids protein; FA, fatty acid; FABP, fatty acid binding protein; FASN, fatty acid synthase; FATP, fatty acid transport protein; SCD, stearoyl-CoA desaturase; SFA, saturated fatty acid; TAG, triacylglycerols; MUFA, monosaturated fatty acid; PUFA, polyunsaturated fatty acid.
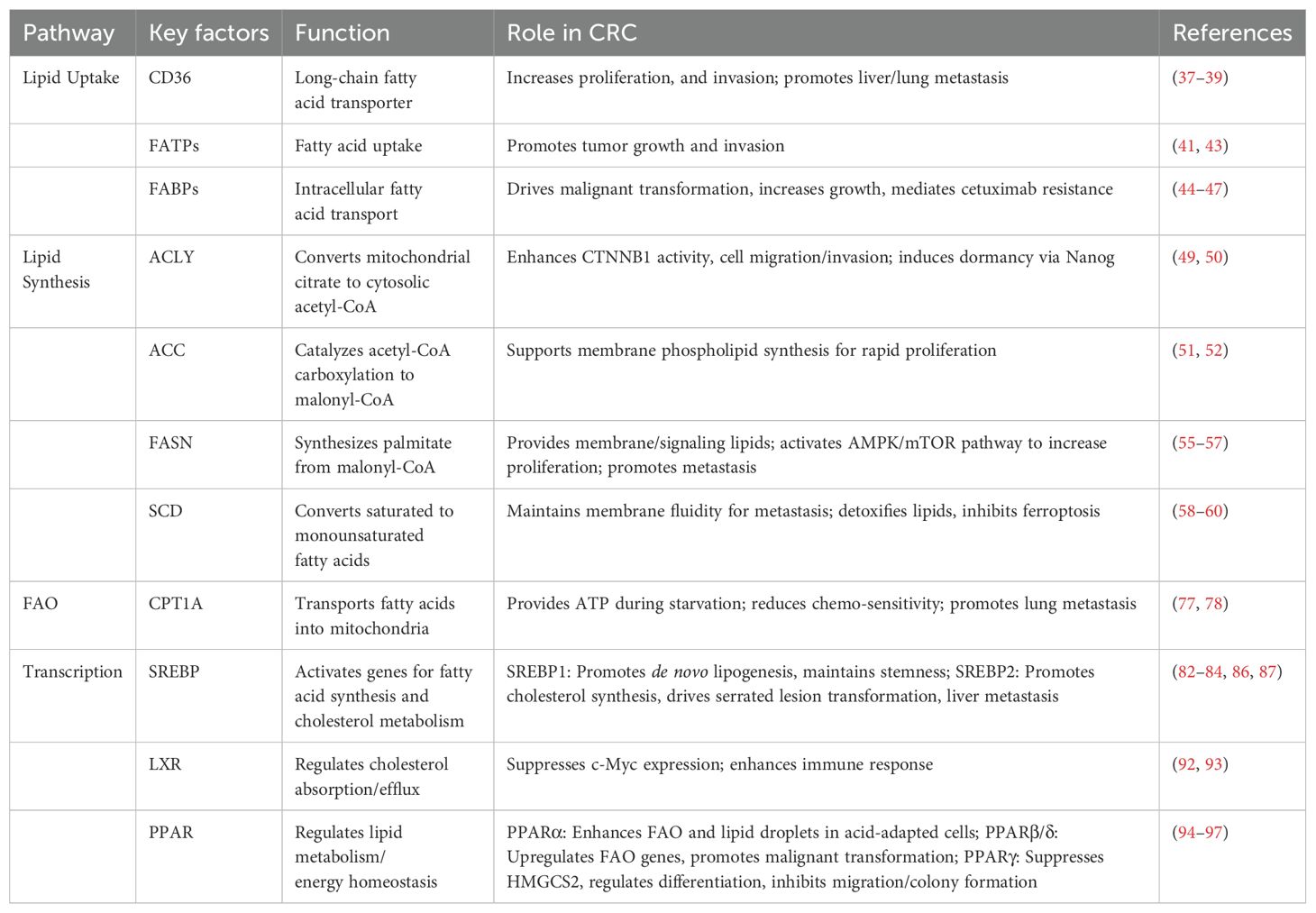
Table 1. Lipid metabolic pathways and key regulators in CRC.
2.1 Abnormal lipid uptake in CRC
Lipid uptake is an essential pathway for cells to acquire lipids, mainly involving the uptake of fatty acids (FAs) and cholesterol (29). Rapidly growing tumors require a large amount of lipids. Studies have shown that fatty acid uptake is mainly mediated by fatty acid transport proteins (FATs). Known transport proteins include fatty acid translocase (CD36), fatty acid transport proteins (FATPs), and fatty acid-binding proteins (FABPs) (30).CD36 is a cell surface scavenger receptor that participates in lipid uptake as a lipid transport protein. CD36 expression is upregulated in various tumors such as breast cancer, gastric cancer, and CRC (31–33). Targeted inhibition of CD36 can suppress the growth and metastasis of multiple cancers (34–36), indicating its crucial role in cancer development. In CRC, CD36 expression is significantly higher in tumor tissues compared to adjacent tissues, and CD36 expression is elevated in the liver and non-metastatic lesions (37). Inhibition of CD36 can, to some extent, suppress CRC metastasis (38). Bioinformatics analysis has shown that CD36 regulates cell proliferation and apoptosis in CRC via the PPAR signaling pathway (39). Mechanistic studies have demonstrated that CD36 plays an important role in CRC models. Overexpression of CD36 increases the proliferation, invasiveness, and colony-forming ability of CRC cell lines (HCT116, HT29), upregulates survivin expression, and decreases caspase-3 and PARP cleavage (38). In CD36 knockdown mouse models, the number and volume of lung metastases are significantly reduced after tail vein injection, suggesting that CD36 plays a key role in cell survival and proliferation (37). The FATPs family is another class of key transport proteins, with six homologous proteins (FATP1-6) in the human body. Previous studies have reported that FATPs are upregulated in most cancers, such as breast cancer, melanoma, renal cell carcinoma, and CRC (40–42), and promote tumor growth and invasion by regulating fatty acid uptake (42). In CRC, FATP5 overexpression is particularly significant. Research has shown that it plays a key role in regulating the cell cycle, but its impact on cancer invasiveness remains unclear and requires further investigation (43).FABPs, members of the lipid-binding protein superfamily, are widely involved in intracellular lipid transport and storage. In the APCMin mouse model, knockout of the FABP1 allele reduces the number of adenomas, suggesting the role of this protein in CRC development (44). In in vitro models, FABP5 is associated with a more active cell cycle, and knocking down FABP5 leads to cell cycle arrest at the G1 phase and apoptosis (45). FABP4 can enhance cellular lipid metabolism through the AKT pathway and induce intracellular lipid droplet formation (46). Immunohistochemistry shows that FABP4 expression is associated with stronger tumor invasiveness and poorer prognosis (47). Overall, in colorectal cancer, cancer cells can increase fatty acid uptake by upregulating the expression of FATPs, CD36, and FABPs, thereby maintaining the rapid proliferation and growth of tumors (48).
2.2 Abnormal lipid synthesis in colon cancer
CRC cells activate abnormal lipid synthesis to meet their rapid proliferation needs. Lipid synthesis is a process centered around acetyl-CoA as a metabolic hub, generating products like fatty acids and cholesterol through various enzymatic reactions. Fatty acid synthesis begins with acetyl-CoA generated in the mitochondria, which is transported to the cytoplasm through the citric acid-pyruvate cycle. In the presence of ATP-citrate lyase (ACLY), acetyl-CoA is converted into cytoplasmic acetyl-CoA. Subsequently, acetyl-CoA carboxylase (ACC) carboxylates acetyl-CoA into malonyl-CoA, which is the rate-limiting step. Fatty acid synthase (FASN) then catalyzes a cyclical process of condensation, reduction (NADPH providing electrons), dehydration, and reduction, ultimately producing palmitic acid (a 16-carbon fatty acid), which is extended to 18–24 carbon chains in the endoplasmic reticulum by the ELOVL enzyme family. Immunohistochemical analysis of CRC patients reveals ACLY overexpression correlating with metastasis. Mechanistic studies demonstrate ACLY stabilizes CTNNB1 (β-catenin) protein, enhancing its transcriptional activity to promote cancer cell migration and invasion (49). Additionally, elevated ACLY expression induces acetyl-CoA accumulation, facilitating P300-mediated H3K27 acetylation to transcriptionally activate Nanog, thereby inducing cellular dormancy in CRC (50). In humans, there are two isoenzymes of ACC, ACC1 and ACC2. ACC1 is significantly overexpressed in CRC tissues, and its activity is aberrantly activated through transcriptional and epigenetic mechanisms. For instance, circular RNA circCAPRIN1 directly binds to the signal transducer and activator of transcription 2 (STAT2), enhancing the transcriptional expression of ACC1, thereby promoting lipid synthesis and driving CRC proliferation and metastasis (51). Mechanistically, the de novo fatty acid synthesis mediated by ACC1 is crucial for maintaining tumor cell membrane structure, energy storage, and signal transduction. Knockdown of intestinal epithelial ACC1 significantly reduces the incidence of inflammation-related CRC (52). As one of the key enzymes in lipid synthesis, FASN has been widely studied in CRC (53, 54). Lipidomics research has shown that lipid markers such as mono-unsaturated/poly-unsaturated triglycerides (TG), sphingomyelins, and ceramides are significantly elevated in CRC tissues. FASN gene expression is significantly upregulated and associated with poor prognosis (55). Mechanistic studies have found that FASN can enhance CRC cell proliferation and migration via the AMPK/mTOR pathway (56). Some genes, such as HMGA1, can promote the expression of key lipid synthesizing enzymes like FASN by activating the SREBP1 signaling pathway, significantly increasing the levels of triglycerides and phosphatidylcholine in CRC cells, thus accelerating CRC proliferation and metastasis (57). The rate-limiting enzyme stearoyl-CoA desaturase (SCD) plays an indispensable role in catalyzing the synthesis of monounsaturated fatty acids (mainly oleic acid and palmitoleic acid), both of which are major components of biological membranes. SCD1 is significantly upregulated in cancer and is associated with metastasis and therapeutic resistance. Its core mechanism involves the regulation of fatty acid desaturation and balancing with ferroptosis resistance (58, 59). Inhibiting SCD1 can significantly reduce lipid metabolism and metastasis in CRC cells (60). Due to the demand for long-chain fatty acids in CRC cells, the expression of the fatty acid elongase family (ELOVLs) is usually increased in CRC to meet their demands (61). In conclusion, compared to normal cells, tumor cells exhibit a significantly higher ratio of de novo synthesized fatty acids, and this metabolic shift is positively correlated with tumor malignancy (62).
2.3 Abnormal lipid storage and oxidation in CRC
Lipid droplets are organelles that store triglycerides and cholesterol, playing a crucial role in maintaining cellular lipid and energy homeostasis (63). Tumor cells mainly store the lipids they intake and synthesize in lipid droplets (64). The increased abundance of lipid droplets is one of the symptoms of cancer aggressiveness (65). Some studies have found that in metastatic cell lines, the total triglyceride and cholesterol content in lipid droplets is elevated, while the content of saturated triglycerides is reduced, suggesting that the degree and ratio of fatty acid storage in lipid droplets are related to tumor invasiveness (66).CRC stem cells are key factors contributing to cancer initiation, drug resistance, and recurrence. The lipid droplet content in CRC stem cells is significantly increased and is directly related to the activity of CD133 and the Wnt signaling pathway, making it one of the markers of stem cells (67). Other studies have shown that Fusobacterium nucleatum can induce the acquisition of stemness characteristics in non-colorectal cancer stem cells through lipid droplet-mediated Numb degradation (68). Lipid droplets are also critical in maintaining intracellular stability. Even in the presence of sufficient oxygen, tumor cells are still in an acidic microenvironment due to the excessive production of lactic acid (69). Chronic acidic microenvironments lead to mitochondrial remodeling in tumor cells to maintain energy production, and this remodeling depends on the buffering system of lipid droplets (70). Fatty acids generated by autophagy are directed into lipid droplets to prevent lipid toxicity to mitochondria. Inhibition of lipid droplet formation disrupts mitochondrial function and impairs the transport of fatty acids to mitochondria (71, 72). Furthermore, there is a close interaction between lipid droplet formation and fatty acid oxidation (FAO), which together maintain the metabolic balance of cancer cells (72).
Fatty acids exhibit high energy density, yielding approximately 9 kcal per gram upon oxidation—more than double the energy derived from glucose. Under nutrient-replete conditions, certain malignancies preferentially utilize fatty acid oxidation for energy production (73, 74).FAO generally involves four stages: activation, transfer, β-oxidation, and complete oxidation. In the cytoplasm, fatty acids are catalyzed by acyl-CoA synthetase to form activated acyl-CoA, consuming ATP in the process. Then, long-chain acyl-CoA enters the mitochondria through the carnitine shuttle system: carnitine palmitoyltransferase I (CPT1) converts it into acylcarnitine, which is transported across the mitochondrial inner membrane, and carnitine palmitoyltransferase II (CPT2) regenerates it into acyl-CoA. This process is the rate-limiting step. In the mitochondrial matrix, acyl-CoA undergoes β-oxidation to produce acetyl-CoA, which then enters the citric acid cycle and is fully oxidized to generate large amounts of ATP (75). A study integrating CRC bulk and single-cell transcriptomic data and using GFAO_Score to represent FAO levels found that the high GFAO_Score group had higher staging and decreased sensitivity to chemotherapy drugs (76). The rate-limiting enzyme of FAO, CPT1, has three isoforms (CPT1A-C). Studies targeting CPT1A have shown that, compared to the primary site, the expression level of CPT1A in metastatic sites is significantly increased, and inhibiting CPT1A expression can reduce the lung metastatic rate of CRC. Another study targeting CPT1C found that inhibiting CPT1C can suppress CRC cell FAO, proliferation, and migration (77). Compared to normal tissues, CRC significantly increases the likelihood of peroxidation due to oxidative stress (78). Some lipid peroxidation products, such as epoxy-ketone eicosatrienoic acid (EKODE), accumulate in CRC cells and induce an inflammatory response in colonic epithelial cells by activating the JNK pathway, promoting tumor progression in the AOM/DSS-induced CRC mouse model (79).
2.4 Transcriptional regulation of lipid metabolism in CRC
The transcriptional regulation of lipid metabolism in CRC is a complex network of multi-factor collaboration, with several key transcription factors at its core. Sterol regulatory element-binding protein (SREBP) is a central transcription factor in regulating lipid metabolism. It promotes de novo lipid synthesis in CRC cells by activating the expression of genes involved in fatty acid synthesis and cholesterol metabolism, playing a crucial role in the occurrence, development, and metastasis of CRC (80). The SREBP family comprises two homologous genes, SREBP1 and SREBP2. SREBP1 primarily regulates fatty acid and triglyceride synthesis, whereas cholesterol biosynthesis is predominantly governed by SREBP2 (81). Knockdown of SREBP in CRC cell lines significantly reduces intracellular fatty acids, cholesterol, and triglycerides, inhibiting tumor cell proliferation. Further bioenergetics analysis shows that SREBP knockdown inhibits mitochondrial respiration, glycolysis, and FAO, leading to an overall metabolic shift in the cells. This metabolic remodeling results in a significant reduction in cancer cell proliferation and weakens its ability to form tumor spheroids, suggesting that SREBP-dependent lipid synthesis is critical for maintaining the stemness characteristics of CRC cells (82). Analysis of GEO tissue microarrays revealed that the expression of long non-coding RNA (lncRNA) ZFAS1 is upregulated in CRC. ZFAS1 stabilizes SREBP1 mRNA by binding to poly(A)-binding protein 2, allowing its accumulation and reprogramming lipid metabolism (83). Studies on PIK3CA mutations and tumor lipid metabolism reveal that the PIK3CA-E545K mutation promotes nuclear accumulation of SREBP1, enhancing transcription of apolipoprotein A5 (APOA5) and thereby mediating platinum-based drug resistance in CRC (84). Precursors of SREBPs (pre-SREBPs) bind to their partner SREBP cleavage-activating protein (SCAP) and reside in the endoplasmic reticulum. Upon cellular cholesterol depletion, the SREBP/SCAP complex translocates to the Golgi apparatus, where SREBP undergoes proteolytic cleavage. The N-terminal SREBP fragment then enters the nucleus and binds sterol regulatory elements (SREs) in promoter regions of target genes, upregulating key enzymes for cholesterol synthesis (e.g., HMG-CoA reductase, squalene monooxygenase) (85). This transcriptional reprogramming drives excessive de novo cholesterol synthesis and uptake, providing essential membrane components, lipid raft structures, and signaling molecule precursors for rapid cancer cell proliferation. Research on CRC metastasis demonstrates significantly elevated SREBP2 expression in liver metastases compared to primary tumors—a phenomenon not observed in brain or lymph node metastases—indicating pathway-specific activation of cholesterol synthesis in hepatic metastasis. SREBP2 knockdown markedly reduces both the number and volume of liver metastases in nude mice (86). In aPKC-deficient intestinal tumors, enhanced SREBP2 activation upregulates cholesterol biosynthesis, promoting cellular metaplasia and the formation of aggressive cellular subsets within serrated tumor lesions (87).
Liver X receptors (LXR) are another type of transcription factor that regulates lipid metabolism. There are two subtypes in humans, LXRα and LXRβ. These are encoded by different genes but have similar functions (88). In the intestinal epithelium, LXR acts as a cholesterol sensor, regulating cholesterol absorption and excretion. It negatively regulates the expression of cholesterol uptake protein Niemann-Pick C1-like 1 (NPC1L1), thereby reducing intestinal cholesterol absorption (89). At the same time, LXR induces the expression of ATP-binding cassette transporters ABCG5 and ABCG8 to promote fecal cholesterol excretion (90). This action helps maintain systemic cholesterol homeostasis and is associated with a reduced risk of CRC development. Clinical CRC cohorts further verified this role: LXR expression is decreased in CRC patients, and the tissue cholesterol content is elevated (91). LXR activation can inhibit the activity of β-catenin, a key transcription factor in the Wnt/β-catenin signaling pathway, which is frequently dysregulated in CRC. By inhibiting β-catenin, LXR reduces the expression of oncogenes such as c-Myc, thereby suppressing CRC proliferation (92). Additionally, studies have shown that in mice fed with the LXR receptor agonist GW3965, the number of tertiary lymphoid structures in tumors increases, adaptive immune responses are enhanced, and tumor formation is reduced (93).
Peroxisome proliferator-activated receptors (PPARs) play pivotal regulatory roles in the lipid metabolism of CRC. Their three subtypes (PPARα, PPARβ/δ, PPARγ) influence tumorigenesis, progression, and metabolic reprogramming through distinct mechanisms. PPARα modulates fatty acid mobilization and enhances FAO. Elevated PPARα expression in CRC exhibits dual tumor-suppressive and oncogenic roles. In PPARα-deficient murine colon cancer models, increased levels of DNMT1 and PRMT6 promote colon carcinogenesis by mediating methylation of p21 and p27, respectively; the PPAR agonist fenofibrate suppresses AOM/DSS-induced colorectal carcinogenesis (94). Conversely, within acidic tumor microenvironments, upregulated PPARα fuels cancer cell proliferation and invasion by enhancing FAO and lipid droplet (LD) accumulation, thereby supporting the energy demands of acid-adapted CRC cells (95). Under high-fat diet conditions, activated PPARβ/δ induces downstream FAO gene expression, potentiates intestinal stem cell function, and elevates tumorigenic risk (96). As a regulator of adipogenesis, PPARγ activation negatively modulates mitochondrial HMGCS2 to govern intestinal cell differentiation (97). PPARγ also serves as a receptor for linoleic acid, suppressing CRC cell migration and colony formation upon linoleic acid treatment (98).
3 Lipid metabolism interactions in the tumor microenvironment
The TME is a complex system composed of tumor cells, immune cells, stromal cells, and non-cellular components (Figure 2). Its interaction with metabolism can significantly impact tumor progression and therapeutic resistance (99, 100). Recent studies have found that lipid metabolism in the TME not only provides energy and biosynthetic precursors for tumor cells but also forms a multi-layered pro-cancer network by regulating immune cell phenotype and function, promoting angiogenesis, and maintaining cancer stem cell characteristics (68, 101, 102).
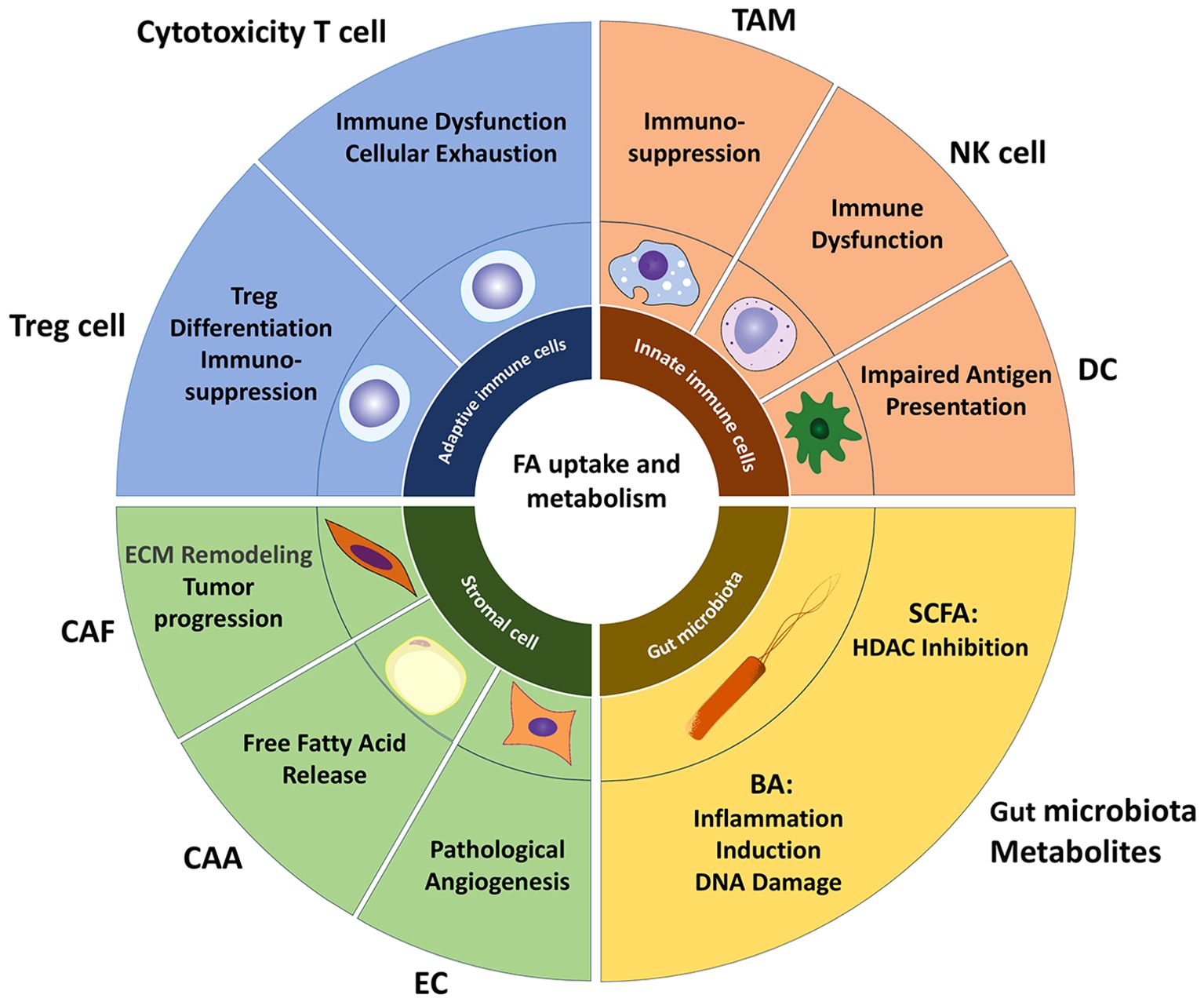
Figure 2. Lipid metabolism landscape in TME. Lipid metabolism within the TME establishes a multi-layered pro-tumorigenic network by regulating immune cell phenotypes/functions and promoting angiogenesis. Tumor-infiltrating cytotoxic T cells commonly exhibit metabolic dysfunction, where excessive lipid accumulation impairs T cell function and promotes exhaustion. Fatty acids facilitate Treg differentiation by suppressing lineage-defining transcription factors while upregulating FoxP3 expression. The majority of TAM adopt an immunosuppressive M2 phenotype, which internalizes free fatty acids (FFAs) released by tumor cells via extracellular vesicles and CD36, reinforcing their immunosuppressive activity. Intracellular lipid accumulation compromises NK cell cytotoxicity against tumors and diminishes antigen-presenting capacity in DCs. Among stromal cells, enhanced lipid metabolism supplies lipids to tumors, remodels the extracellular matrix, and induces angiogenesis, collectively driving tumor proliferation and metastasis. Microbial metabolites SCFAs and BAs—play pivotal roles in the TME: SCFAs accumulate in CRC cells, acting as HDAC inhibitors to suppress proliferation, while BAs promote CRC development by inducing inflammation and DNA damage. TAM, tumor-associated macrophage; DC, dendritic cell; CAF, cancer-associated fibroblast; CAA, cancer-associated adipocyte; EC, endothelial cell; SCFA, short-chain fatty acid; BA, Bile acid.
3.1 Immune cells and lipids
Hypoxia, acidic environments, and lipid accumulation in the tumor microenvironment can suppress the function of immune cells within it (102). Immune cells in the TME also undergo metabolic changes to maintain certain functions (103). Immune cells in the TME can be categorized into adaptive immune cells and innate immune cells. T cells are the primary adaptive immune cells in the tumor microenvironment. Within the TME, T cells are mainly divided into two categories: one consists of effector T cells and helper T cells, which inhibit tumor growth; the other consists of regulatory T cells (Tregs), which suppress immune responses (104). Studies have shown that tumor-infiltrating T cells often exhibit metabolic disorders, characterized by enhanced metabolic switching from glycolysis to lipid oxidation (105, 106). However, excessive lipid metabolism can affect T cell function and lead to T cell exhaustion (107); linoleic acid increases CPT1A in CD4+ T cells, leading to oxidative stress and inducing apoptosis (108); linoleic acid increases CPT1A in CD4+ T cells, leading to oxidative stress and inducing apoptosis (109); excessive lipids in TAM can also induce an increase in CD36 expression in CD8+T cells, leading to lipid peroxidation and functional impairment of the cells (110); at the same time, persistent antigen stimulation in the TME can induce the gradual loss of PGC1α in CD8+ T cells via the Akt signaling pathway, affecting mitochondrial replication, gradually reducing the cells’ ability to utilize lipids, and triggering CD8+ T cell exhaustion (111). Treg cells mainly rely on FAO for energy (112).In hypoxic conditions with lactate accumulation in the TME, Treg cells can upregulate CD36 expression and adjust mitochondria through the PPAR-β signaling pathway to maintain their function. Knocking out Treg cell CD3 can reduce the expression of various immune regulatory receptors (113). Using sulfonated N-succinimide oleic acid (SSO) and etomoxir to inhibit fatty acid transport and oxidation can decrease the number of Treg cells and the expression of cell markers (114).
In the tumor microenvironment, innate immune cells mainly include macrophages, NK cells, and dendritic cells. Macrophages are considered one of the main cell types leading to lipid metabolic disorders in the TME. Compared with normal samples, the expression of ALOX5 is increased and the pro-inflammatory lipid product 5-HETE is significantly up-regulated in CRC. Analysis of large Affymetrix microarray datasets shows that genes related to arachidonic acid lipid metabolism and pro-inflammatory mediators are co-expressed with relevant macrophage markers, indicating the important role of macrophages in driving pro-inflammatory responses and lipid disorders in the TME (115). Tumor-associated macrophages (TAMs) are primarily divided into the pro-inflammatory M1 type and the immunosuppressive M2 type (116). M1 macrophages rely on glycolysis, while M2 macrophages obtain energy through FAO. M1 macrophages kill tumor cells through antibody-mediated cytotoxicity, but during this process, the glucose uptake demand of the cells significantly increases. The lack of glucose and the accumulation of lactate in the TME induce the transformation of M1 macrophages into M2 type (117). Therefore, most of the macrophages in the tumor microenvironment are immunosuppressive M2 type (118). M2 macrophages have enhanced fatty acid uptake and synthesis abilities and rely on FAO for energy supply in the TME (119). Free fatty acids released by tumor cells are extensively taken up by M2 macrophages through extracellular vesicles and CD36, promoting their immunosuppressive phenotype (120). This suppressive ability is mainly reflected in the inhibition of CD8+ T cell function, and inhibition of CD36 can reduce M2 polarization and enhance CD8+ T cell function (120). Additionally, oleic acid in the environment can lead to lipid droplet accumulation in infiltrating macrophages in colon cancer and promote M2 polarization via activation of the mTOR signaling pathway. Injection of lipid droplet synthesis inhibitors can suppress tumor growth in vivo models (121). Single-cell transcriptomics reveals a terminally differentiated C5 macrophage subset characterized by robust lipid metabolic reprogramming and potent immunosuppressive function (122). A study on obesity found that the number and function of NK cells in obese patients are impaired, and adiponectin combined treatment can restore part of the NK cell function, suggesting that lipid metabolism can affect NK cells (123). Research has shown that in a high cholesterol microenvironment, CRC cells upregulate TGF-β1 secretion through ATP6V0A1-dependent cholesterol uptake, indirectly inhibiting the immune surveillance function of NK cells (124). Another study found that lipid accumulation after colorectal cancer surgery leads to increased CD36 and lipid content in NK cells, impairing their cytotoxic function against tumors (125).In the colorectal cancer tumor microenvironment, lipid metabolism reprogramming of dendritic cells (DCs) significantly affects their immune function and anti-tumor response. Single-cell transcriptomic analyses reveal significant enrichment of lipid metabolism, fatty acid metabolism, and PPARA signaling pathways in DCs. Concurrently, the transcription factor RUNX2 is markedly upregulated in tumor-infiltrating DCs, where it orchestrates downstream PPAR signaling via activation of the Wnt/β-catenin axis, thereby remodeling cellular lipid metabolism (126). The research found that lipid droplets containing electrophilic oxidized truncated (ox-tr) lipids in tumor DCs were much larger than in the control group. These lipid droplets covalently bind to heat shock protein 70, hindering MHC translocation, and resulting in a decrease in the antigen presentation ability of DCs (127). Using ACC inhibitors to reduce intracellular lipids in DCs can partially restore their function, indicating that lipid metabolism reprogramming in the TME is an important mechanism affecting DC function (128).
3.2 Adipocytes
The intestine is an organ rich in fat, and CRC establishes intimate connections with adipocytes when it invades adipose tissue (129). CRC cells induce adjacent adipocytes to dedifferentiate into cancer-associated adipocytes (CAAs) through direct contact or paracrine signaling. These CAAs are characterized by a reduction in lipid droplets, enhanced lipolysis, and increased secretion of pro-inflammatory factors (130, 131). Co-culture experiments show that CRC cells inhibit the expression of genes related to adipogenesis (such as FADS1, and SC4MOL) and activate lipolytic pathways, leading to the release of free fatty acids and cholesterol from adipocytes (130). These lipids are taken up by tumor cells via transport proteins such as CD36, providing energy and membrane synthesis materials for tumor cells (132). These fatty acids can also activate the AMPK signaling pathway, inducing autophagy and promoting the transformation of colon cancer into a mode of energy acquisition primarily based on FAO (133). Adipocytes also enhance the expression of FABP4 in surrounding tissues and mediate resistance to cetuximab (134). CAAs promote CRC cell proliferation and metastasis by secreting pro-inflammatory factors such as TNF-α and LCN2, activating the YAP/TAZ signaling pathway (131). Additionally, exosomes secreted by colon cancer cells, containing miR-146b-5p, induce the browning and lipid mobilization of white adipocytes, which is one of the causes of cachexia (135). Similarly, in colorectal cancer chemotherapy models, first-line chemotherapy drugs reduce the expression of proteins involved in ATP generation, β-oxidation, and lipid synthesis in adipocytes, leading to adipose depletion (136). This depletion of fat is associated with shortened survival and quality of life, warranting more research and attention (137).
3.3 Cancer-associated fibroblasts
Cancer-associated Fibroblasts (CAFs) are key components of the TME and interact with colorectal cancer through the secretion of cytokines, remodeling the extracellular matrix (ECM), and regulating metabolic pathways, including lipid metabolism (138, 139). CAFs upregulate the expression of genes related to fatty acid synthesis and secrete large amounts of fatty acids and phospholipids. After being taken up by CRC cells, these lipids enhance tumor cell migration ability (140). CRC cells also transfer the HSPC111 protein via exosomes to hepatic stellate cells, converting them into CAFs and preparing for liver metastasis. Furthermore, HSPC111 can phosphorylate ACLY, increasing the acetyl-CoA level in CAFs and promoting the expression and secretion of the chemokine CXCL5 through H3K27 acetylation in an epigenetic manner. The secreted CXCL5 activates the epithelial-mesenchymal transition (EMT) of CRC cells through the CXCR2 receptor, forming a positive feedback loop that further stimulates the release of HSPC111 in tumor exosomes (141). Spatial transcriptomics analysis has found that lipid metabolism-related pathways are significantly upregulated in areas enriched with inflammatory cancer-associated fibroblasts (iCAFs). In iCAF-enriched regions of patients receiving neoadjuvant chemotherapy, despite an overall decrease in metabolic activity, fatty acid metabolic activity remains high, suggesting that iCAFs may promote chemotherapy resistance by maintaining lipid metabolic activity (142).
3.4 Endothelial cells
In CRC, endothelial cells (ECs) are the central executors of tumor angiogenesis. In the tumor microenvironment, due to a lack of glucose, proliferating endothelial cells undergo metabolic reprogramming and switch to fatty acid oxidation for energy production (143). In this state, endothelial cells exhibit increased lipid uptake and enhanced expression of FABP4, and inhibition of FABP4 can reduce tumor angiogenesis (144). Studies have shown that vascular endothelial growth factor B regulates endothelial cell fatty acid uptake through the vascular fatty acid transporters, although the exact mechanism remains unclear (145). Co-culture experiments have revealed that FASN affects endothelial cell activity by regulating the secretion profile of angiogenic factors. Knocking down FASN significantly reduces microvessel density in colon cancer cell lines and induces the “normalization” of vascular structures. Overexpression of FASN enhances MMP-9 activity and VEGF secretion, promoting endothelial cell activation (146). Other studies have shown that inhibiting CRC lipid metabolism through PI3K inhibitors reduces angiogenesis, suggesting the important role of lipid metabolism in tumor vascular formation (147). Single-cell transcriptomics of obese CRC samples reveal enrichment of an ESM1+ EC subpopulation in tumors, correlating with poorer prognosis. This subpopulation exhibits upregulated pathways regulating epithelial cell migration and adhesion, indicating a pro-tumorigenic role (148).
3.5 Gut microbiota and the regulatory network of lipid metabolism
The occurrence of CRC is closely related to various factors, among which the interaction between gut microbiota and lipid metabolism is considered to play a significant role in the development of CRC (149, 150). The gut microbiota interacts with host lipid metabolism through metabolic products such as short-chain fatty acids (SCFAs) and secondary bile acids, forming a complex regulatory axis. SCFAs, such as acetate, propionate, and butyrate, are the primary metabolites produced by gut microbes through the fermentation of dietary fibers (151). The benefits of butyrate are well known, as it can regulate gut immune responses by activating G-protein-coupled receptors (such as GPR43 and GPR109A), reduce chronic inflammation, and thus decrease the risk of carcinogenesis (152). In the TME, butyrate can also enhance CD8+ T cell responses through IL-2-related signaling pathways, promoting the antitumor effects of PD-1 inhibitors (153). Butyrate also induces metabolic adaptations in activated CD8+ T cells, redirecting TCA cycle substrates toward fatty acid uptake and FAO (154). Butyrate metabolism diverges between normal intestinal epithelium and cancer cells: Normal epithelia utilize butyrate via β-oxidation, whereas CRC cells exhibit diminished catabolic capacity due to the Warburg effect, leading to intracellular accumulation (155). Once accumulated, butyrate acts as a histone deacetylase (HDAC) inhibitor, leading to CRC cell apoptosis. Metabolically, butyrate binds to pyruvate kinase M2 (PKM2), promoting its dephosphorylation and tetramerization. This activates PKM2, suppresses the Warburg effect, reduces glycolytic energy production, and inhibits CRC growth (156). Chronic butyrate exposure diminishes glycolytic capacity in CRC cells regardless of glucose availability. Butyrate increases mitochondrial pyruvate oxidation flux, thereby enhancing de novo lipid synthesis and lipid accumulation (157). Additionally, studies report upregulated SLC27A1 (FATP) expression in butyrate-treated CRC cells, augmenting fatty acid transport (158). Short-chain fatty acids (SCFAs) and their derived acyl-CoAs can also be oxidized. Under hypoxia, tumor cells convert acetate to acetyl-CoA via ACSS2, activating lipogenic genes (e.g., FASN) to fuel de novo lipogenesis and support growth (159). Conversely, CD8+ T cells utilize acetate in an ACSS2-dependent manner to rescue effector functions impaired by glucose deprivation (160), highlighting acetate’s paradoxical roles in tumor progression. Acetate is converted to butyrate in the gut by bacteria such as Roseburia spp. Via butyryl-CoA:acetyl-CoA transferase (161). Propionate, as an HDAC inhibitor, can synergize with butyrate to inhibit tumor cell proliferation, although its potency is relatively weaker (162). Furthermore, recent studies have shown that SCFAs, in addition to their HDAC inhibition effects, can induce propionylation modifications at histone H3K18 and H4K12 sites, inhibit the Wnt/β-catenin signaling pathway, upregulate key CRC oncogenes (such as MYC, FOS, and JUN), and downregulate genes related to cell proliferation (ANP32B) and the cell cycle (MKI67), leading to CRC cell death (163).
Bile acids (BA) and their derivatives are another type of metabolite produced by the gut microbiota, which plays a dual role in the development of CRC (164). Primary BAs, such as cholic acid (CA) and chenodeoxycholic acid (CDCA), are produced in the liver and can be converted into secondary BAs by the gut microbiota. Some secondary BAs, such as deoxycholic acid (DCA) and lithocholic acid (LCA), have been shown to promote CRC by inducing inflammation, DNA damage, and cell proliferation (165). However, other BAs, such as ursodeoxycholic acid (UDCA), may have protective effects by reducing inflammation and inhibiting tumor growth via the TGR5-YAP axis (166).
The gut microbiota not only exerts significant effects on the intestinal microenvironment through its metabolites but also influences the progression of CRC by regulating lipid metabolism pathways. In the Apcmin/+ mouse model, a high-fat diet affects phospholipid metabolism via the gut microbiota, leading to intestinal mucosal damage and CRC development (167).In obese CRC patients, the dysregulation of fatty acid and phospholipid metabolism is closely associated with the enrichment of pathogenic bacteria and the reduction of beneficial bacteria (168). Additionally, the intrinsic lipid metabolism reprogramming in colorectal cancer also affects the gut microbiota. The rate-limiting enzyme in cholesterol biosynthesis, squalene epoxidase (SQLE), is significantly upregulated in CRC (169). Overexpression of SQLE indirectly accelerates tumor progression through gut microbiota dysbiosis: the gut microbiota of SQLE transgenic mice shows enrichment of pathogenic bacteria (such as C. muridarum), reduced abundance of anti-inflammatory and protective bacteria (such as S. violaceusniger), elevated levels of secondary bile acids (such as DCA), reduced tight junction proteins, and impaired intestinal barrier function, which aggravates CRC progression (170).
4 Lipid metabolism-related biomarkers and analytical techniques in CRC
Lipids serve as fundamental components of cellular membranes and participate in diverse metabolic processes, playing critical physiological roles (171). During the occurrence of CRC, lipid metabolism shows significant disorders, manifested as abnormal expression and dysfunction of multiple lipid molecules and related proteins. These changes directly drive the malignant biological behavior of tumors and become potential diagnostic and prognostic biomarkers. With the rapid development of lipidomics technologies, especially the application of mass spectrometry-based high-throughput analysis platforms combined with multivariate statistical methods, complex lipid quantitative analysis in cancer has become possible, and more and more new lipid biomarkers have been discovered thereby.
4.1 Lipid metabolism-related biomarkers in CRC
Characteristic lipidomic remodeling occurs during CRC development. Profiling these distinct alterations provides valuable insights for CRC diagnosis and prognosis. CRC patients exhibit significant imbalances in fatty acid composition within serum and tumor tissues, including elevated long-chain PUFAs (e.g., arachidonic acid [AA], eicosapentaenoic acid [EPA], docosahexaenoic acid [DHA]) and reduced levels of PUFAs such as linoleic acid (LA) and α-linolenic acid (ALA) (172). Comparative analyses reveal upregulated ω-6 PUFAs in carcinomas versus adenomas, while ω-3 PUFAs are downregulated in CRC (173). Additionally, decreased monounsaturated fatty acid (MUFA) content alongside elevated saturated fatty acids (SFAs) and n-3/n-6 PUFAs has been observed in CRC tissues (174). Assessing these FA profiles may aid in early CRC detection. β-Hydroxybutyrate (BHB), a ketone body derived from FAO, demonstrates progressively increased serum levels across CRC stages, potentially serving as a biomarker for tumor metabolic reprogramming (175). Notably, when administered as part of a ketogenic diet, BHB suppresses CRC growth (176), warranting further investigation into its context-dependent functions. Associations between triglycerides (TAGs) and CRC risk remain inconsistent: A Chinese cohort study identified elevated TAG levels as positively correlated with colon cancer progression (177), whereas analysis of the UK Biobank found no statistically significant TAG-CRC association after adjusting for BMI and other factors (178). The dietary variations influence TAG measurements (55). Phosphatidylethanolamine (PE) enrichment in plasma exosomes distinguishes non-metastatic CRC patients from healthy controls, with lower PE levels observed in metastatic versus non-metastatic cases (179). Such lipidomic shifts reflect functional membrane alterations across CRC stages.
Given CRC lipid complexity, multi-lipid biomarker panels outperform single molecules for staging. A diagnostic panel comprising O-(4,8-dimethylnonanoyl)carnitine, LPC 19:0, TAG 58:1, and PC 38:7 achieves an AUC of 0.805 (95% CI: 0.684–0.922) for discriminating adenomas from CRC (180). Krishnan et al. constructed machine learning models utilizing ceramide (CE)(22:6), CE(18:3), TG(56:9), and FA combinations to identify liver metastasis in CRC (17). Liu et al. developed an integrated model (ApoA1, ApoA2, lithocholic acid [LCA], CEA) with an AUC of 0.995 (95% CI: 0.969–0.999) for CRC diagnosis (181).
4.2 Lipid analysis techniques in CRC
With the development of high-throughput technologies, lipidomics has gradually matured in CRC research and biomarker discovery. The main analytical tools currently include mass spectrometry (MS) and nuclear magnetic resonance (NMR). MS plays a core role in the analysis of lipid metabolic changes in CRC (182). MS-based lipid analysis encompasses non-imaging and imaging strategies. In non-imaging methods, gas chromatography-mass spectrometry (GC-MS) is suitable for analyzing volatile small-molecule lipids (molecular weight < 1000 Da). It has high sensitivity but limited coverage (183). Liquid chromatography-mass spectrometry/capillary electrophoresis-mass spectrometry (LC-MS/CE-MS) is widely used for complex lipids (such as phospholipids and glycerolipids), serving as the core tool of lipidomics. It can qualitatively and quantitatively analyze hundreds of lipids, especially suitable for global analysis of biological fluids (serum, urine) and tissue extracts (115, 184).
Imaging methods focus on spatial resolution analysis, providing direct information on the spatial distribution of target molecules, but compared with high-performance liquid chromatography-mass spectrometry (HPLC-MS), the analytical depth is reduced (185). Matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI) achieves lipid ionization through matrix co-crystallization, with a spatial resolution of 5–200 μm. It can be coupled with high-resolution mass spectrometry such as Fourier transform ion cyclotron resonance (FTICR) to distinguish lipid isomers, but requires optimization of matrices (such as 2,5-dihydroxybenzoic acid, α-cyano-4-hydroxycinnamic acid) and deposition techniques to suppress ion interference (186, 187); Desorption electrospray ionization mass spectrometry imaging (DESI-MSI) directly analyzes tissue sections in an open environment, suitable for rapid clinical detection (such as intraoperative diagnosis), with a resolution of approximately 50–200 μm (nano-DESI can reach < 10 μm) (188, 189); Secondary ion mass spectrometry (SIMS) must be performed under vacuum, with a resolution up to the nanometer scale (0.05–100 μm), suitable for single-cell lipid distribution research, but it is destructive to samples and can cause lipid fragmentation (190). Multimodal integrations (e.g., LIMS with infrared spectroscopy/spatial transcriptomics) and emerging single-cell LIMS (e.g., SpaceM) provide multidimensional insights into lipid metabolism across molecular, cellular, and tissue levels, advancing mechanistic and clinical applications (188). NMR spectroscopy features non-destructive analysis and simple sample preparation. Compared to MS, NMR has a weaker resolution for similar lipids and is currently mainly used for non-targeted analysis (191). For example, Mika et al. employed ¹H-NMR for global lipid profiling followed by GC-MS for major component analysis in CRC (174).
5 Targeting lipid metabolism in CRC treatment strategies
As summarized above, lipid metabolism plays a crucial role in the occurrence and development of CRC. These findings have led us to consider lipid metabolism as a potential target for intervention in order to impede the progression and drug resistance of CRC. Currently, therapeutic strategies targeting metabolic vulnerabilities have shown promising translational potential.
5.1 Targeting lipid metabolism in treatment
Inhibiting the de novo lipogenesis (DNL) pathway has become an important strategy for cancer treatment (192).FASN, the rate-limiting enzyme in DNL, is closely associated with tumor cell proliferation, metastasis, and chemotherapy resistance, making it a key target for inhibiting lipid synthesis (54, 56). Cerulenin, the first-generation FASN inhibitor, can inhibit tumor cell energy metabolism and the mTOR signaling pathway, thereby inhibiting CRC cell proliferation (193). In mice, Cerulenin inhibits CRC liver metastasis and reduces the size of liver metastatic lesions (194). Epigallocatechin gallate(EGCG), a green tea extract, exerts strong FASN inhibition, which reduces the levels of free fatty acids in tumors and decreases tumor volume in xenografted mice, without affecting the mice’s weight (195). Randomized clinical trials have found that this extract can significantly reduce the incidence of colon adenomas (RR, 0.56; 95% CI, 0.34-0.92) (196). Luteolin, a flavonoid compound, exerts anticancer effects by regulating the Wnt/β-catenin pathway and the miR-384/PTN axis (197, 198). TVB inhibitors, a new generation of potent FASN inhibitors, including TVB-2640 and TVB-3664, have shown good results in Phase I clinical trials (25, 199). In xenograft models derived from CRC patients, TVB-3664 reduced tumor lipid storage and inhibited tumor growth in 30% of the samples. Additionally, Orlistat, as a sulfatase domain inhibitor, has shown antitumor activity in breast cancer and CRC models, but its use in clinical settings is limited by its gastrointestinal carcinogenic effects (200). It is worth noting that CRC cells can counteract the effects of FASN inhibitors by upregulating CD36. In this case, the combination of CD36 inhibitors can enhance the efficacy of FASN inhibitors (38).
Apart from FASN, other lipid synthesis proteins can also be targeted. ACLY can generate cytosolic acetyl-CoA and bridge the mitochondria and non-mitochondrial processes. The ACLY inhibitor SB204990 inhibits acetyl-CoA generation, suppressing tumor cell proliferation and inducing differentiation (201). ACC allosteric inhibitors, such as TOFA, can inhibit fatty acid synthesis and induce CRC cell apoptosis in a dose-dependent manner (202). SCD1, the rate-limiting enzyme in unsaturated fatty acid synthesis, is highly expressed in CRC (58). Its inhibitor, Betulinic Acid, can impair CRC stem cell clonogenic ability and induce cell death (203). Notably, targeting the DNL pathway requires balancing therapeutic efficacy and metabolic toxicity. For example, systemic inhibition of ACC may induce thrombocytopenia, while local delivery technologies (such as nanoparticle encapsulation) are being explored to enhance selectivity (192).
Targeting lipid oxidation is also an important strategy for CRC therapy. CPT1 is the rate-limiting enzyme in mitochondrial fatty acid transport (75). Its inhibitor Etomoxir can significantly inhibit fatty acid oxidation, inducing an energy crisis in CRC cells (204). Another inhibitor, DHP-B, extracted from Piperaceae plants, can disrupt the interaction between mitochondrial CPT1A and VDAC1, leading to increased mitochondrial permeability and reduced energy synthesis, thereby inhibiting CRC cell growth and inducing apoptosis (77).
Table 2 summarizes ongoing or completed clinical studies related to lipid metabolism.
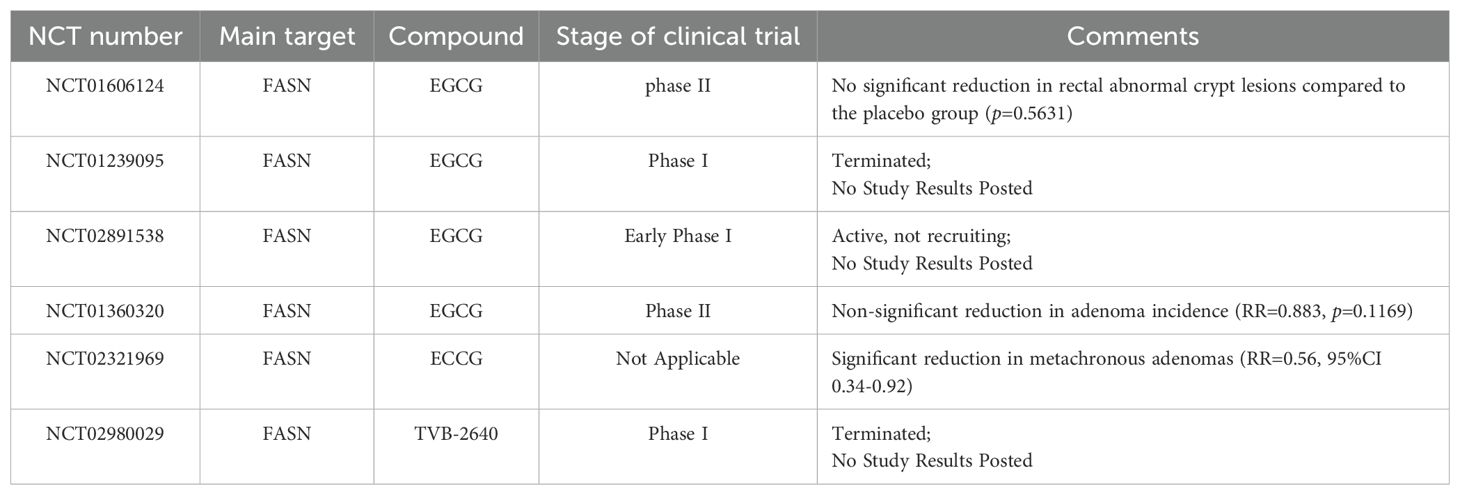
Table 2. The clinical research on targeting lipid metabolism in CRC.
Table 3 summarizes preclinical therapeutic compounds targeting lipid metabolism in CRC.
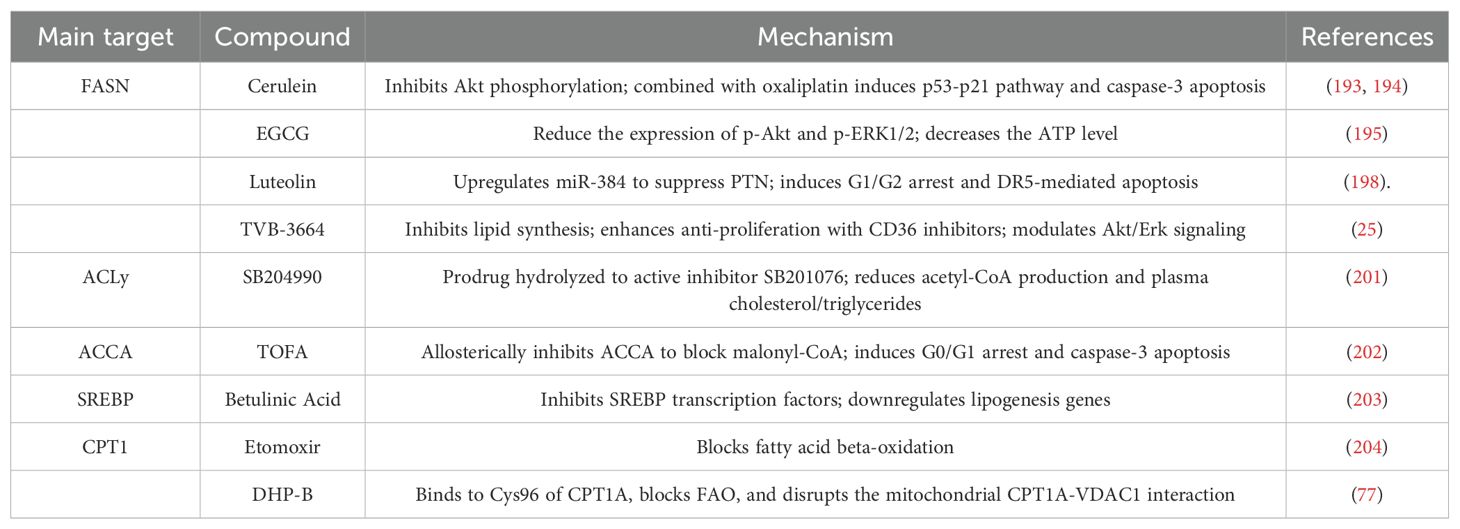
Table 3. The preclinical research on targeting lipid metabolism in CRC.
5.2 Potential of combination therapy
The combination of lipid metabolism inhibitors and chemotherapy is a promising therapeutic approach. Oxaliplatin and 5-fluorouracil are mainstream chemotherapy drugs for the treatment of CRC, and reducing resistance to them is crucial for CRC treatment (8). In xenograft mouse models, the combination of Cerulenin and oxaliplatin significantly inhibits CRC progression and reduces the dosages of both drugs, thereby extending chemotherapy duration (205); EGCG can target CRC tumor stem cells and enhance their sensitivity to 5-fluorouracil chemotherapy (206).ACLy can induce resistance in CRC cells to the active metabolite of irinotecan, SN38, but the combination of ACLy and AKT inhibitors can restore chemotherapy sensitivity in tumor cells (207). In organoids derived from CRC patients resistant to cetuximab, the FABP4 inhibitor BMS309403 can restore tumor cells’ sensitivity to cetuximab (134). In preclinical studies, the SCD1 inhibitor MF-438, when combined with 5-fluorouracil, can reduce the 5-FU dose by half while maintaining the same efficacy (16). Moreover, targeting lipid metabolism can synergize with immunotherapy: for example, SIRT is a gene involved in the conversion of glucose-lipid metabolism in cells. In CRC, SIRT1 promotes the secretion of CX3CL1, which enhances Treg cell infiltration. CX3CR1 inhibitors can suppress CRC proliferation while enhancing the inhibitory effect of PD-1 antibodies on the tumor (208). Table 4 outlines the effects of combining lipid metabolism inhibitors with chemotherapy, immunotherapy, or targeted drugs
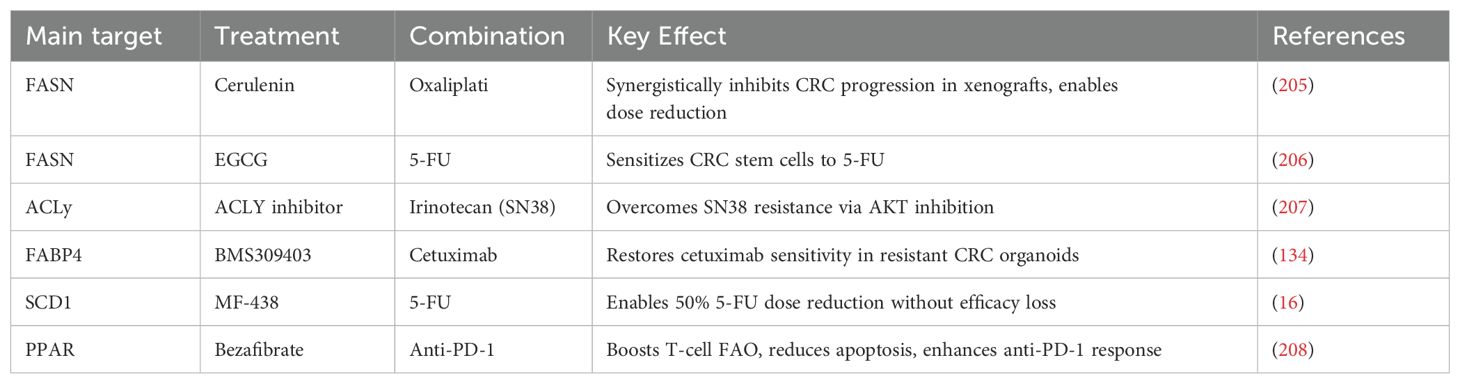
Table 4. The combination therapy in CRC.
6 Conclusions
Lipid metabolism reprogramming plays a crucial role in the progression of CRC. During the development of CRC, the expression of key enzymes involved in processes such as fatty acid synthesis, uptake, and oxidation is often increased. This process not only provides an abundant energy supply for tumor cells but also promotes malignant progression by regulating cell signaling, enhancing anti-apoptotic mechanisms, and increasing cell migration and invasion capabilities. Factors in the TME, such as nutritional status, hypoxic conditions, and immune responses, directly affect the lipid metabolism pathways of tumor cells, thereby promoting tumor invasiveness and resistance (124). Various cell types within the TME, including Tregs, CAAs, and CAFs, undergo lipid metabolism reprogramming through interactions with the tumor, fostering an immune-suppressive microenvironment that promotes metastasis (99). At the same time, the gut microbiota, an important component of the TME, can influence tumor cell metabolic reprogramming by modulating metabolic products and immune responses in the intestine. The lipid metabolism reprogramming of CRC cells also leads to further dysregulation of the gut microbiota, accelerating tumor progression.
The reprogramming of lipid metabolism in CRC leads to changes in specific lipid molecular profiles, making these molecules highly promising diagnostic and prognostic biomarkers. With the rapid development of lipidomics technologies, especially the application of MS-based high-throughput analysis platforms and multivariate statistical methods, the identification and quantification of these lipid markers have become feasible. However, although existing studies have revealed a large number of lipid metabolism-related molecules with clinical potential, their translational application in clinical practice still faces challenges such as insufficient standardization and lack of cross-cohort validation. Therefore, future research should further focus on the mechanistic analysis of lipid markers, the standardization of analysis processes, and clinical validation with multi-center large samples, so as to promote the effective application of lipidomics in the precision diagnosis and treatment of CRC.
Currently, effective treatments for advanced-stage CRC patients are still lacking. Given the central role of lipid metabolism in CRC initiation and progression, many drugs or inhibitors tested in preclinical models target lipid metabolism. Studies have shown that targeting lipid metabolism can inhibit CRC proliferation and migration, as well as reduce chemotherapy resistance (209). Some drugs, such as EGCG and TVB-2640, have entered clinical trials (25, 196). However, challenges remain: First, lipid metabolic pathways are redundant, and CRC cells compensate by activating fatty acid uptake pathways to reduce the effect of FASN inhibitors. Therefore, single-target treatments are often difficult to achieve long-term effects. Second, the spatial heterogeneity of the tumor microenvironment means that tumor cells in different regions may have distinct metabolic characteristics. Lipid metabolic capacity is enhanced in hypoxic regions (99). Furthermore, individual differences in metabolic regulation may also affect the effectiveness of targeted therapies, such as in obese patients, where enhanced lipid metabolism makes them more prone to resistance (210). Therefore, future research should integrate multi-omics technologies, such as spatial metabolomics and single-cell transcriptomics, to analyze the dynamic architecture of metabolic networks, and use artificial intelligence models to predict combination targets, ultimately achieving a leap from mechanistic exploration to precision therapy.
Author contributions
WL: Conceptualization, Writing – original draft. SD: Writing – original draft. FH: Writing – original draft. YG: Funding acquisition, Writing – review & editing. QW: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work is supported by the National Natural Science Foundation of China (82273366 and 82373243).
Acknowledgments
We appreciate the support provided by Shanghai Tenth People’s Hospital. The membrane-2d-bluelight icon by Servier is licensed under CC-BY 3.0 Unported https://creativecommons.org/licenses/by/3.0/.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CRC, Colorectal cancer; TME, tumor microenvironment; ACC, acetyl-CoA-carboxylase; ACLY, ATP-citrate lyase; ACSL, acyl-CoA synthetase long-chain family; CPT, carnitine palmitoyl transferase; ELOVL, elongation of very-long-chain fatty acids protein; FA, fatty acid; FABP, fatty acid binding protein; FASN, fatty acid synthase; FATP, fatty acid transport protein; SCD, stearoyl-CoA desaturase; SFA, saturated fatty acid; TAG, triacylglycerols; MUFA, monounsaturated fatty acid; PUFA, polyunsaturated fatty acid; SREBP, Sterol regulatory element-binding protein; LXR, Liver X receptors; PPAR, Peroxisome proliferator-activated receptor; TAM, tumor-associated macrophage; DC, dendritic cell; CAF, cancer-associated fibroblast; CAA, cancer-associated adipocyte; EC, endothelial cell; SCFA, short-chain fatty acid; BA, Bile acid; EGCG, Epigallocatechin gallate.
References
1. Siegel RL, Giaquinto AN, and Jemal A. Cancer statistics, 2024. CA Cancer J Clin. (2024) 74:12–49. doi: 10.3322/caac.21820
2. Cao W, Qin K, Li F, and Chen W. Socioeconomic inequalities in cancer incidence and mortality: An analysis of GLOBOCAN 2022. Chin Med J (Engl). (2024) 137:1407–13. doi: 10.1097/CM9.0000000000003140
3. He M, Gu R, Huang X, Zhao A, Tian S, and Zheng Y. Global burden of colorectal cancer attributable to metabolic risks from 1990 to 2021, with projections of mortality to 2035. Int J Colorectal Dis. (2025) 40:46. doi: 10.1007/s00384-025-04817-w
4. Morgan E, Arnold M, Gini A, Lorenzoni V, Cabasag CJ, Laversanne M, et al. Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN. Gut. (2023) 72:338–44. doi: 10.1136/gutjnl-2022-327736
5. Abakushina EV, Gelm YV, Pasova IA, and Bazhin AV. Immunotherapeutic approaches for the treatment of colorectal cancer. Biochem (Mosc). (2019) 84:720–8. doi: 10.1134/S0006297919070046
6. Liu R, Ji Z, Wang X, Zhu L, Xin J, Ma L, et al. Regorafenib plus sintilimab as a salvage treatment for microsatellite stable metastatic colorectal cancer: a single-arm, open-label, phase II clinical trial. Nat Commun. (2025) 16:1481. doi: 10.1038/s41467-025-56748-3
7. Gu T, Qi H, Wang J, Sun L, Su Y, and Hu H. Identification of T cell dysfunction molecular subtypes and exploration of potential immunotherapy targets in BRAF V600E-mutant colorectal cancer. Discover Oncol. (2025) 16:163. doi: 10.1007/s12672-025-01930-8
8. Abedizadeh R, Majidi F, Khorasani HR, Abedi H, and Sabour D. Colorectal cancer: a comprehensive review of carcinogenesis, diagnosis, and novel strategies for classified treatments. Cancer Metastasis Rev. (2024) 43:729–53. doi: 10.1007/s10555-023-10158-3
9. Rosati G, Aprile G, Colombo A, Cordio S, Giampaglia M, Cappetta A, et al. Colorectal cancer heterogeneity and the impact on precision medicine and therapy efficacy. Biomedicines. (2022) 10:1035. doi: 10.3390/biomedicines10051035
10. Acha-Sagredo A, Andrei P, Clayton K, Taggart E, Antoniotti C, Woodman CA, et al. A constitutive interferon-high immunophenotype defines response to immunotherapy in colorectal cancer. Cancer Cell. (2025) 43:292–307.e7. doi: 10.1016/j.ccell.2024.12.008
11. Wang D, Zhu L, Liu H, Feng X, Zhang C, Liu B, et al. Altered gut metabolites and metabolic reprogramming involved in the pathogenesis of colitis-associated colorectal cancer and the transition of colon "inflammation to cancer. J Pharm Biomed Analysis. (2025) 253:116553. doi: 10.1016/j.jpba.2024.116553
12. Schmidt DR, Patel R, Kirsch DG, Lewis CA, Vander Heiden MG, and Locasale JW. Metabolomics in cancer research and emerging applications in clinical oncology. CA Cancer J Clin. (2021) 71:333–58. doi: 10.3322/caac.21670
13. Faubert B, Solmonson A, and DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. (2020) 368:eaaw5473. doi: 10.1126/science.aaw5473
14. Sun L, Zhang H, and Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. (2022) 13:877–919. doi: 10.1007/s13238-021-00846-7
15. Marcucci F and Rumio C. On the role of glycolysis in early tumorigenesis-permissive and executioner effects. Cells. (2023) 12:1124. doi: 10.3390/cells12081124
16. Zabielska J, Stelmanska E, Szrok-Jurga S, Kobiela J, and Czumaj A. Lipids metabolism inhibition antiproliferative synergy with 5-fluorouracil in human colorectal cancer model. Int J Mol Sci. (2025) 26:1186. doi: 10.3390/ijms26031186
17. Krishnan ST, Winkler D, Creek D, Anderson D, Kirana C, Maddern GJ, et al. Staging of colorectal cancer using lipid biomarkers and machine learning. Metabolomics. (2023) 19:84. doi: 10.1007/s11306-023-02049-z
18. Ma Y, Zhang S, Jin Z, and Shi M. Lipid-mediated regulation of the cancer-immune crosstalk. Pharmacol Res. (2020) 161:105131. doi: 10.1016/j.phrs.2020.105131
19. Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, et al. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer. (2017) 16:76. doi: 10.1186/s12943-017-0646-3
20. Efeyan A, Comb WC, and Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature. (2015) 517:302–10. doi: 10.1038/nature14190
21. Gomaraschi M. Role of lipoproteins in the microenvironment of hormone-dependent cancers. Trends Endocrinol Metab. (2020) 31:256–68. doi: 10.1016/j.tem.2019.11.005
22. Snaebjornsson MT, Janaki-Raman S, and Schulze A. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. (2020) 31:62–76. doi: 10.1016/j.cmet.2019.11.010
23. Piccinin E, Cariello M, and Moschetta A. Lipid metabolism in colon cancer: Role of Liver X Receptor (LXR) and Stearoyl-CoA Desaturase 1 (SCD1). Mol Aspects Med. (2021) 78:100933. doi: 10.1016/j.mam.2020.100933
24. Wang Y, Qiu X, Li Q, Qin J, Ye L, Zhang X, et al. Single-cell and spatial-resolved profiling reveals cancer-associated fibroblast heterogeneity in colorectal cancer metabolic subtypes. J Trans Med. (2025) 23:175. doi: 10.1186/s12967-025-06103-3
25. Zaytseva YY, Rychahou PG, Le AT, Scott TL, Flight RM, Kim JT, et al. Preclinical evaluation of novel fatty acid synthase inhibitors in primary colorectal cancer cells and a patient-derived xenograft model of colorectal cancer. Oncotarget. (2018) 9:24787–800. doi: 10.18632/oncotarget.25361
26. Jog E, Jainarayanan AK, La Ferlita A, Chakraborty A, Dalwai A, Yahya S, et al. Inhibiting de novo lipogenesis identifies a therapeutic vulnerability in therapy-resistant colorectal cancer. Redox Biol. (2025) 79:103458. doi: 10.1016/j.redox.2024.103458
27. Liu H, Wang S, Wang J, Guo X, Song Y, Fu K, et al. Energy metabolism in health and diseases. Signal Transduction Targeted Ther. (2025) 10:69. doi: 10.1038/s41392-025-02141-x
28. Lam SM, Wang Z, Li B, and Shui G. High-coverage lipidomics for functional lipid and pathway analyses. Anal Chim Acta. (2021) 1147:199–210. doi: 10.1016/j.aca.2020.11.024
29. Schönfeld P and Wojtczak L. Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J Lipid Res. (2016) 57:943–54. doi: 10.1194/jlr.R067629
30. Matsushita Y, Nakagawa H, and Koike K. Lipid metabolism in oncology: why it matters, how to research, and how to treat. Cancers (Basel). (2021) 13:474. doi: 10.3390/cancers13030474
31. Feng WW, Wilkins O, Bang S, Ung M, Li J, An J, et al. CD36-mediated metabolic rewiring of breast cancer cells promotes resistance to HER2-targeted therapies. Cell Rep. (2019) 29:3405–20.e5. doi: 10.1016/j.celrep.2019.11.008
32. Wang J, Wen T, Li Z, Che X, Gong L, Jiao Z, et al. CD36 upregulates DEK transcription and promotes cell migration and invasion via GSK-3β/β-catenin-mediated epithelial-to-mesenchymal transition in gastric cancer. Aging (Albany NY). (2020) 13:1883–97. doi: 10.18632/aging.103985
33. Kuang H, Sun X, Liu Y, Tang M, Wei Y, Shi Y, et al. Palmitic acid-induced ferroptosis via CD36 activates ER stress to break calcium-iron balance in colon cancer cells. FEBS J. (2023) 290:3664–87. doi: 10.1111/febs.v290.14
34. Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. (2017) 541:41–5. doi: 10.1038/nature20791
35. Zhang F, Xia X, Chai R, Xu R, Xu Q, Liu M, et al. Inhibition of USP14 suppresses the formation of foam cell by promoting CD36 degradation. J Cell Mol Med. (2020) 24:3292–302. doi: 10.1111/jcmm.15002
36. Ruan C, Meng Y, and Song H. CD36: an emerging therapeutic target for cancer and its molecular mechanisms. J Cancer Res Clin Oncol. (2022) 148:1551–8. doi: 10.1007/s00432-022-03957-8
37. Drury J, Rychahou PG, Kelson CO, Geisen ME, Wu Y, He D, et al. Upregulation of CD36, a fatty acid translocase, promotes colorectal cancer metastasis by increasing MMP28 and decreasing E-cadherin expression. Cancers (Basel). (2022) 14:252. doi: 10.3390/cancers14010252
38. Drury J, Rychahou PG, He D, Jafari N, Wang C, Lee EY, et al. Inhibition of fatty acid synthase upregulates expression of CD36 to sustain proliferation of colorectal cancer cells. Front Oncol. (2020) 10:1185. doi: 10.3389/fonc.2020.01185
39. Zhang X, Yao J, Shi H, Gao B, and Zhang L. LncRNA TINCR/microRNA-107/CD36 regulates cell proliferation and apoptosis in colorectal cancer via PPAR signaling pathway based on bioinformatics analysis. Biol Chem. (2019) 400:663–75. doi: 10.1515/hsz-2018-0236
40. Koundouros N and Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. (2020) 122:4–22. doi: 10.1038/s41416-019-0650-z
41. Binienda A, Machelak W, Zielińska M, and Fichna J. Free fatty acid receptors type 2 and 4 mediate the anticancer effects of fatty acids in colorectal cancer - in vitro and in vivo studies. Biochim Biophys Acta (BBA) - Mol Basis Disease. (2025) 1871:167708. doi: 10.1016/j.bbadis.2025.167708
42. Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov. (2018) 8:1006–25. doi: 10.1158/2159-8290.CD-17-1371
43. Geng QS, Yang MJ, Li LF, Shen ZB, Wang LH, Zheng YY, et al. Over-expression and prognostic significance of FATP5, as a new biomarker, in colorectal carcinoma. Front Mol Biosci. (2021) 8:770624. doi: 10.3389/fmolb.2021.770624
44. Dharmarajan S, Newberry EP, Montenegro G, Nalbantoglu I, Davis VR, Clanahan MJ, et al. Liver fatty acid-binding protein (L-Fabp) modifies intestinal fatty acid composition and adenoma formation in ApcMin/+ mice. Cancer Prev Res (Phila). (2013) 6:1026–37. doi: 10.1158/1940-6207.CAPR-13-0120
45. Kawaguchi K, Senga S, Kubota C, Kawamura Y, Ke Y, and Fujii H. High expression of Fatty Acid-Binding Protein 5 promotes cell growth and metastatic potential of colorectal cancer cells. FEBS Open Bio. (2016) 6:190–9. doi: 10.1002/2211-5463.12031
46. Tian W, Zhang W, Zhang Y, Zhu T, Hua Y, Li H, et al. FABP4 promotes invasion and metastasis of colon cancer by regulating fatty acid transport. Cancer Cell Int. (2020) 20:512. doi: 10.1186/s12935-020-01582-4
47. Kim SH, Pyo JS, Son BK, Oh IH, and Min KW. Clinicopathological significance and prognostic implication of nuclear fatty acid-binding protein 4 expression in colorectal cancer. Pathol Res Pract. (2023) 249:154722. doi: 10.1016/j.prp.2023.154722
48. Pakiet A, Sikora K, Kobiela J, Rostkowska O, Mika A, and Sledzinski T. Alterations in complex lipids in tumor tissue of patients with colorectal cancer. Lipids Health Dis. (2021) 20:85. doi: 10.1186/s12944-021-01512-x
49. Wen J, Min X, Shen M, Hua Q, Han Y, Zhao L, et al. ACLY facilitates colon cancer cell metastasis by CTNNB1. J Exp Clin Cancer Res. (2019) 38:401. doi: 10.1186/s13046-019-1391-9
50. Zhang M, Peng R, Wang H, Yang Z, Zhang H, Zhang Y, et al. Nanog mediated by FAO/ACLY signaling induces cellular dormancy in colorectal cancer cells. Cell Death Dis. (2022) 13:159. doi: 10.1038/s41419-022-04606-1
51. Yang Y, Luo D, Shao Y, Shan Z, Liu Q, Weng J, et al. circCAPRIN1 interacts with STAT2 to promote tumor progression and lipid synthesis via upregulating ACC1 expression in colorectal cancer. Cancer Commun (Lond). (2023) 43:100–22. doi: 10.1002/cac2.12380
52. Li S, Lu CW, Diem EC, Li W, Guderian M, Lindenberg M, et al. Acetyl-CoA-Carboxylase 1-mediated de novo fatty acid synthesis sustains Lgr5(+) intestinal stem cell function. Nat Commun. (2022) 13:3998. doi: 10.1038/s41467-022-31725-2
53. Röhrig F and Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer. (2016) 16:732–49. doi: 10.1038/nrc.2016.89
54. Fhu CW and Ali A. Fatty acid synthase: an emerging target in cancer. Molecules. (2020) 25:3935. doi: 10.3390/molecules25173935
55. Ecker J, Benedetti E, Kindt ASD, Höring M, Perl M, Machmüller AC, et al. The colorectal cancer lipidome: identification of a robust tumor-specific lipid species signature. Gastroenterology. (2021) 161:910–23.e19. doi: 10.1053/j.gastro.2021.05.009
56. Lu T, Sun L, Wang Z, Zhang Y, He Z, and Xu C. Fatty acid synthase enhances colorectal cancer cell proliferation and metastasis via regulating AMPK/mTOR pathway. Onco Targets Ther. (2019) 12:3339–47. doi: 10.2147/OTT.S199369
57. Zhao Y, Liu MJ, Zhang L, Yang Q, Sun QH, Guo JR, et al. High mobility group A1 (HMGA1) promotes the tumorigenesis of colorectal cancer by increasing lipid synthesis. Nat Commun. (2024) 15:9909. doi: 10.1038/s41467-024-54400-0
58. Sen U, Coleman C, and Sen T. Stearoyl coenzyme A desaturase-1: multitasker in cancer, metabolism, and ferroptosis. Trends Cancer. (2023) 9:480–9. doi: 10.1016/j.trecan.2023.03.003
59. Wu T, Wan J, Qu X, Xia K, Wang F, Zhang Z, et al. Nodal promotes colorectal cancer survival and metastasis through regulating SCD1-mediated ferroptosis resistance. Cell Death Dis. (2023) 14:229. doi: 10.1038/s41419-023-05756-6
60. Che G, Wang W, Wang J, He C, Yin J, Chen Z, et al. Sulfotransferase SULT2B1 facilitates colon cancer metastasis by promoting SCD1-mediated lipid metabolism. Clin Transl Med. (2024) 14:e1587. doi: 10.1002/ctm2.v14.2
61. Nagarajan SR, Butler LM, and Hoy AJ. The diversity and breadth of cancer cell fatty acid metabolism. Cancer Metab. (2021) 9:2. doi: 10.1186/s40170-020-00237-2
62. Menendez JA and Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. (2007) 7:763–77. doi: 10.1038/nrc2222
63. Olzmann JA and Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. (2019) 20:137–55. doi: 10.1038/s41580-018-0085-z
64. Petan T, Jarc E, and Jusović M. Lipid droplets in cancer: guardians of fat in a stressful world. Molecules. (2018) 23:1941. doi: 10.3390/molecules23081941
65. Butler LM, Perone Y, Dehairs J, Lupien LE, de Laat V, Talebi A, et al. Lipids and cancer: Emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv Drug Delivery Rev. (2020) 159:245–93. doi: 10.1016/j.addr.2020.07.013
66. Munir R, Lisec J, Jaeger C, and Zaidi N. Abundance, fatty acid composition and saturation index of neutral lipids in colorectal cancer cell lines. Acta Biochim Pol. (2021) 68:115–8. doi: 10.18388/abp.2020_5465
67. Tirinato L, Liberale C, Di Franco S, Candeloro P, Benfante A, La Rocca R, et al. Lipid droplets: a new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells. (2015) 33:35–44. doi: 10.1002/stem.1837
68. Liu H, Du J, Chao S, Li S, Cai H, Zhang H, et al. Fusobacterium nucleatum promotes colorectal cancer cell to acquire stem cell-like features by manipulating lipid droplet-mediated numb degradation. Adv Sci (Weinh). (2022) 9:e2105222. doi: 10.1002/advs.202105222
69. Pillai SR, Damaghi M, Marunaka Y, Spugnini EP, Fais S, and Gillies RJ. Causes, consequences, and therapy of tumors acidosis. Cancer Metastasis Rev. (2019) 38:205–22. doi: 10.1007/s10555-019-09792-7
70. Khacho M, Tarabay M, Patten D, Khacho P, MacLaurin JG, Guadagno J, et al. Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nat Commun. (2014) 5:3550. doi: 10.1038/ncomms4550
71. Mailler E, Guardia CM, Bai X, Jarnik M, Williamson CD, Li Y, et al. The autophagy protein ATG9A enables lipid mobilization from lipid droplets. Nat Commun. (2021) 12:6750. doi: 10.1038/s41467-021-26999-x
72. Liu X, Sun X, Mu W, Li Y, Bu W, Yang T, et al. Autophagic flux-lipid droplet biogenesis cascade sustains mitochondrial fitness in colorectal cancer cells adapted to acidosis. Cell Death Discovery. (2025) 11:21. doi: 10.1038/s41420-025-02301-6
73. Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, and Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. (2017) 14:11–31. doi: 10.1038/nrclinonc.2016.60
74. Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang XY, et al. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. (2018) 435:92–100. doi: 10.1016/j.canlet.2018.08.006
75. Bian X, Liu R, Meng Y, Xing D, Xu D, and Lu Z. Lipid metabolism and cancer. J Exp Med. (2021) 218:e20201606. doi: 10.1084/jem.20201606
76. Zou P, Chen C, and Wu X. Identification of fatty acid oxidation-related subtypes by integrated analysis of bulk- and single-cell transcriptome profiling in colorectal cancer. J Gastrointest Oncol. (2024) 15:147–63. doi: 10.21037/jgo-23-833
77. Hu A, Wang H, Xu Q, Pan Y, Jiang Z, Li S, et al. A novel CPT1A covalent inhibitor modulates fatty acid oxidation and CPT1A-VDAC1 axis with therapeutic potential for colorectal cancer. Redox Biol. (2023) 68:102959. doi: 10.1016/j.redox.2023.102959
78. Perše M. Oxidative stress in the pathogenesis of colorectal cancer: Cause or consequence? BioMed Res Int. (2013) 2013:725710. doi: 10.1155/2013/725710
79. Lei L, Yang J, Zhang J, and Zhang G. The lipid peroxidation product EKODE exacerbates colonic inflammation and colon tumorigenesis. Redox Biol. (2021) 42:101880. doi: 10.1016/j.redox.2021.101880
80. Yi J, Zhu J, Wu J, Thompson CB, and Jiang X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci U S A. (2020) 117:31189–97. doi: 10.1073/pnas.2017152117
81. Tang H, Zhang Y, Zhao D, Guo M, Yuan X, and Wang X. Unlocking the lipid code: SREBPs as key drivers in gastrointestinal tumour metabolism. Lipids Health Dis. (2025) 24:190. doi: 10.1186/s12944-025-02612-8
82. Wen YA, Xiong X, Zaytseva YY, Napier DL, Vallee E, Li AT, et al. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. (2018) 9:265. doi: 10.1038/s41419-018-0330-6
83. Wang H, Chen Y, Liu Y, Li Q, Luo J, Wang L, et al. The lncRNA ZFAS1 regulates lipogenesis in colorectal cancer by binding polyadenylate-binding protein 2 to stabilize SREBP1 mRNA. Mol Ther Nucleic Acids. (2022) 27:363–74. doi: 10.1016/j.omtn.2021.12.010
84. Liu YL, Xiang Z, Zhang BY, Zou YW, Chen GL, Yin L, et al. APOA5 alleviates reactive oxygen species to promote oxaliplatin resistance in PIK3CA-mutated colorectal cancer. Aging (Albany NY). (2024) 16:9410–36. doi: 10.18632/aging.205872
85. Xu M, Jiang SY, Tang S, Zhu M, Hu Y, Li J, et al. Nuclear SREBP2 condensates regulate the transcriptional activation of lipogenic genes and cholesterol homeostasis. Nat Metab. (2025) 7:1034–51. doi: 10.1038/s42255-025-01291-0
86. Zhang KL, Zhu WW, Wang SH, Gao C, Pan JJ, Du ZG, et al. Organ-specific cholesterol metabolic aberration fuels liver metastasis of colorectal cancer. Theranostics. (2021) 11:6560–72. doi: 10.7150/thno.55609
87. Muta Y, Linares JF, Martinez-Ordoñez A, Duran A, Cid-Diaz T, Kinoshita H, et al. Enhanced SREBP2-driven cholesterol biosynthesis by PKCλ/ι deficiency in intestinal epithelial cells promotes aggressive serrated tumorigenesis. Nat Commun. (2023) 14:8075. doi: 10.1038/s41467-023-43690-5
88. Buñay J, Fouache A, Trousson A, de Joussineau C, Bouchareb E, Zhu Z, et al. Screening for liver X receptor modulators: Where are we and for what use? Br J Pharmacol. (2021) 178:3277–93. doi: 10.1111/bph.15286
89. Duval C, Touche V, Tailleux A, Fruchart JC, Fievet C, Clavey V, et al. Niemann-Pick C1 like 1 gene expression is down-regulated by LXR activators in the intestine. Biochem Biophys Res Commun. (2006) 340:1259–63. doi: 10.1016/j.bbrc.2005.12.137
90. Repa JJ, Berge KE, Pomajzl C, Richardson JA, Hobbs H, and Mangelsdorf DJ. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors α and β. J Biol Chem. (2002) 277:18793–800. doi: 10.1074/jbc.M109927200
91. Sharma B, Gupta V, Dahiya D, Kumar H, Vaiphei K, and Agnihotri N. Clinical relevance of cholesterol homeostasis genes in colorectal cancer. Biochim Biophys Acta - Mol Cell Biol Lipids. (2019) 1864:1314–27. doi: 10.1016/j.bbalip.2019.06.008
92. Uno S, Endo K, Jeong Y, Kawana K, Miyachi H, Hashimoto Y, et al. Suppression of beta-catenin signaling by liver X receptor ligands. Biochem Pharmacol. (2009) 77:186–95. doi: 10.1016/j.bcp.2008.10.007
93. Das S, Parigi SM, Luo X, Fransson J, Kern BC, Okhovat A, et al. Liver X receptor unlinks intestinal regeneration and tumorigenesis. Nature. (2025) 637:1198–206. doi: 10.1038/s41586-024-08247-6
94. Luo Y, Xie C, Brocker CN, Fan J, Wu X, Feng L, et al. Intestinal PPARα Protects against colon carcinogenesis via regulation of methyltransferases DNMT1 and PRMT6. Gastroenterology. (2019) 157:744–59.e4. doi: 10.1053/j.gastro.2019.05.057
95. Rolver MG, Holland LKK, Ponniah M, Prasad NS, Yao J, Schnipper J, et al. Chronic acidosis rewires cancer cell metabolism through PPARα signaling. Int J Cancer. (2023) 152:1668–84. doi: 10.1002/ijc.v152.8
96. Mana MD, Hussey AM, Tzouanas CN, Imada S, Barrera Millan Y, Bahceci D, et al. High-fat diet-activated fatty acid oxidation mediates intestinal stemness and tumorigenicity. Cell Rep. (2021) 35:109212. doi: 10.1016/j.celrep.2021.109212
97. Kim JT, Li C, Weiss HL, Zhou Y, Liu C, Wang Q, et al. Regulation of ketogenic enzyme HMGCS2 by wnt/β-catenin/PPARγ Pathway in intestinal cells. Cells. (2019) 8:1106. doi: 10.3390/cells8091106
98. Mokhtari K, Mahdevar M, Hajipour M, Esmaeili M, Peymani M, Mirzaei S, et al. Title: Involvement of unsaturated fatty acid biosynthesis in CRC progression based on in vitro and in silico studies. BioMed Pharmacother. (2022) 153:113338. doi: 10.1016/j.biopha.2022.113338
99. Nenkov M, Ma Y, Gaßler N, and Chen Y. Metabolic reprogramming of colorectal cancer cells and the microenvironment: implication for therapy. Int J Mol Sci. (2021) 22:6262. doi: 10.3390/ijms22126262
100. Krauß D, Fari O, and Sibilia M. Lipid metabolism interplay in CRC-an update. Metabolites. (2022) 12:213. doi: 10.3390/metabo12030213
101. Long Y, Shi H, He Y, and Qi X. Analyzing the impact of metabolism on immune cells in tumor microenvironment to promote the development of immunotherapy. Front Immunol. (2023) 14:1307228. doi: 10.3389/fimmu.2023.1307228
102. Liu L, Mo M, Chen X, Chao D, Zhang Y, Chen X, et al. Targeting inhibition of prognosis-related lipid metabolism genes including CYP19A1 enhances immunotherapeutic response in colon cancer. J Exp Clin Cancer Res. (2023) 42:85. doi: 10.1186/s13046-023-02647-8
103. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature. (2017) 543:252–6. doi: 10.1038/nature21379
104. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med. (2019) 25:1251–9. doi: 10.1038/s41591-019-0522-3
105. Corn KC, Windham MA, and Rafat M. Lipids in the tumor microenvironment: From cancer progression to treatment. Prog Lipid Res. (2020) 80:101055. doi: 10.1016/j.plipres.2020.101055
106. He S, Cai T, Yuan J, Zheng X, and Yang W. Lipid Metabolism in Tumor-Infiltrating T Cells. In: Li Y, editor. Lipid Metabolism in Tumor Immunity. Springer Singapore, Singapore (2021). p. 149–67.
107. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
108. Sugiura A and Rathmell JC. Metabolic barriers to T cell function in tumors. J Immunol. (2018) 200:400–7. doi: 10.4049/jimmunol.1701041
109. Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. (2013) 342:1094–8. doi: 10.1126/science.1241908
110. Xu S, Chaudhary O, Rodríguez-Morales P, Sun X, Chen D, Zappasodi R, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity. (2021) 54:1561–77.e7. doi: 10.1016/j.immuni.2021.05.003
111. Fernandez-Marcos PJ and Auwerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr. (2011) 93:884s–90. doi: 10.3945/ajcn.110.001917
112. He S, Cai T, Yuan J, Zheng X, and Yang W. Lipid metabolism in tumor-infiltrating T cells. Adv Exp Med Biol. (2021) 1316:149–67. doi: 10.1007/978-981-33-6785-2_10
113. Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol. (2020) 21:298–308. doi: 10.1038/s41590-019-0589-5
114. Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A, et al. HIF-1α Is a metabolic switch between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression of tregs in glioblastoma. Cell Rep. (2019) 27:226–37.e4. doi: 10.1016/j.celrep.2019.03.029
115. Soundararajan R, Maurin MM, Rodriguez-Silva J, Upadhyay G, Alden AJ, Gowda SGB, et al. Integration of lipidomics with targeted, single cell, and spatial transcriptomics defines an unresolved pro-inflammatory state in colon cancer. Gut. (2025) 74:586–602. doi: 10.1136/gutjnl-2024-332535
116. Xiang Y and Miao H. Lipid metabolism in tumor-associated macrophages. Adv Exp Med Biol. (2021) 1316:87–101. doi: 10.1007/978-981-33-6785-2_6
117. Kelly B and O'Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. (2015) 25:771–84. doi: 10.1038/cr.2015.68
118. O'Neill LA and Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. (2016) 213:15–23. doi: 10.1084/jem.20151570
119. Chen D, Zhang X, Li Z, and Zhu B. Metabolic regulatory crosstalk between tumor microenvironment and tumor-associated macrophages. Theranostics. (2021) 11:1016–30. doi: 10.7150/thno.51777
120. Yang P, Qin H, Li Y, Xiao A, Zheng E, Zeng H, et al. CD36-mediated metabolic crosstalk between tumor cells and macrophages affects liver metastasis. Nat Commun. (2022) 13:5782. doi: 10.1038/s41467-022-33349-y
121. Wu H, Han Y, Rodriguez Sillke Y, Deng H, Siddiqui S, Treese C, et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol Med. (2019) 11:e10698. doi: 10.15252/emmm.201910698
122. Ji T, Fu H, Wang L, Chen J, Tian S, Wang G, et al. Single-cell RNA profiling reveals classification and characteristics of mononuclear phagocytes in colorectal cancer. PloS Genet. (2024) 20:e1011176. doi: 10.1371/journal.pgen.1011176
123. O'Shea D, Cawood TJ, O'Farrelly C, and Lynch L. Natural killer cells in obesity: impaired function and increased susceptibility to the effects of cigarette smoke. PloS One. (2010) 5:e8660. doi: 10.1371/journal.pone.0008660
124. Jin HR, Wang J, Wang ZJ, Xi MJ, Xia BH, Deng K, et al. Lipid metabolic reprogramming in tumor microenvironment: from mechanisms to therapeutics. J Hematol Oncol. (2023) 16:103. doi: 10.1186/s13045-023-01498-2
125. Niavarani SR, Lawson C, Bakos O, Boudaud M, Batenchuk C, Rouleau S, et al. Lipid accumulation impairs natural killer cell cytotoxicity and tumor control in the postoperative period. BMC Cancer. (2019) 19:823. doi: 10.1186/s12885-019-6045-y
126. Xie S, Cai Y, Chen D, Xiang Y, Cai W, Mao J, et al. Single-cell transcriptome analysis reveals heterogeneity and convergence of the tumor microenvironment in colorectal cancer. Front Immunol. (2022) 13:1003419. doi: 10.3389/fimmu.2022.1003419
127. Veglia F, Tyurin VA, Mohammadyani D, Blasi M, Duperret EK, Donthireddy L, et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun. (2017) 8:2122. doi: 10.1038/s41467-017-02186-9
128. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. (2010) 16:880–6. doi: 10.1038/nm.2172
129. Tabuso M, Homer-Vanniasinkam S, Adya R, and Arasaradnam RP. Role of tissue microenvironment resident adipocytes in colon cancer. World J Gastroenterol. (2017) 23:5829–35. doi: 10.3748/wjg.v23.i32.5829
130. Olszańska J, Pietraszek-Gremplewicz K, Domagalski M, and Nowak D. Mutual impact of adipocytes and colorectal cancer cells growing in co-culture conditions. Cell Commun Signal. (2023) 21:130. doi: 10.1186/s12964-023-01155-8
131. Song Y, Na H, Lee SE, Kim YM, Moon J, Nam TW, et al. Dysfunctional adipocytes promote tumor progression through YAP/TAZ-dependent cancer-associated adipocyte transformation. Nat Commun. (2024) 15:4052. doi: 10.1038/s41467-024-48179-3
132. Mukherjee A, Bilecz AJ, and Lengyel E. The adipocyte microenvironment and cancer. Cancer Metastasis Rev. (2022) 41:575–87. doi: 10.1007/s10555-022-10059-x
133. Wen YA, Xing X, Harris JW, Zaytseva YY, Mitov MI, Napier DL, et al. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. (2017) 8:e2593. doi: 10.1038/cddis.2017.21
134. Cheng YC, Chen MY, Yadav VK, Pikatan NW, Fong IH, Kuo KT, et al. Targeting FABP4/UCP2 axis to overcome cetuximab resistance in obesity-driven CRC with drug-tolerant persister cells. Transl Oncol. (2025) 53:102274. doi: 10.1016/j.tranon.2025.102274
135. Di W, Zhang W, Zhu B, Li X, Tang Q, and Zhou Y. Colorectal cancer prompted adipose tissue browning and cancer cachexia through transferring exosomal miR-146b-5p. J Cell Physiol. (2021) 236:5399–410. doi: 10.1002/jcp.v236.7
136. Ebadi M, Field CJ, Lehner R, and Mazurak VC. Chemotherapy diminishes lipid storage capacity of adipose tissue in a preclinical model of colon cancer. Lipids Health Dis. (2017) 16:247. doi: 10.1186/s12944-017-0638-8
137. Murphy RA, Wilke MS, Perrine M, Pawlowicz M, Mourtzakis M, Lieffers JR, et al. Loss of adipose tissue and plasma phospholipids: relationship to survival in advanced cancer patients. Clin Nutr. (2010) 29:482–7. doi: 10.1016/j.clnu.2009.11.006
138. Chen X and Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. (2019) 18:99–115. doi: 10.1038/s41573-018-0004-1
139. Kamali Zonouzi S, Pezeshki PS, Razi S, and Rezaei N. Cancer-associated fibroblasts in colorectal cancer. Clin Transl Oncol. (2022) 24:757–69. doi: 10.1007/s12094-021-02734-2
140. Gong J, Lin Y, Zhang H, Liu C, Cheng Z, Yang X, et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. (2020) 11:267. doi: 10.1038/s41419-020-2434-z
141. Zhang C, Wang XY, Zhang P, He TC, Han JH, Zhang R, et al. Cancer-derived exosomal HSPC111 promotes colorectal cancer liver metastasis by reprogramming lipid metabolism in cancer-associated fibroblasts. Cell Death Dis. (2022) 13:57. doi: 10.1038/s41419-022-04506-4
142. Peng Z, Ye M, Ding H, Feng Z, and Hu K. Spatial transcriptomics atlas reveals the crosstalk between cancer-associated fibroblasts and tumor microenvironment components in colorectal cancer. J Transl Med. (2022) 20:302. doi: 10.1186/s12967-022-03510-8
143. Dagher Z, Ruderman N, Tornheim K, and Ido Y. Acute regulation of fatty acid oxidation and amp-activated protein kinase in human umbilical vein endothelial cells. Circ Res. (2001) 88:1276–82. doi: 10.1161/hh1201.092998
144. Harjes U, Bridges E, Gharpure KM, Roxanis I, Sheldon H, Miranda F, et al. Antiangiogenic and tumour inhibitory effects of downregulating tumour endothelial FABP4. Oncogene. (2017) 36:912–21. doi: 10.1038/onc.2016.256
145. Hagberg CE, Falkevall A, Wang X, Larsson E, Huusko J, Nilsson I, et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature. (2010) 464:917–21. doi: 10.1038/nature08945
146. Zaytseva YY, Elliott VA, Rychahou P, Mustain WC, Kim JT, Valentino J, et al. Cancer cell-associated fatty acid synthase activates endothelial cells and promotes angiogenesis in colorectal cancer. Carcinogenesis. (2014) 35:1341–51. doi: 10.1093/carcin/bgu042
147. Hussain A, Qazi AK, Mupparapu N, Guru SK, Kumar A, Sharma PR, et al. Modulation of glycolysis and lipogenesis by novel PI3K selective molecule represses tumor angiogenesis and decreases colorectal cancer growth. Cancer Lett. (2016) 374:250–60. doi: 10.1016/j.canlet.2016.02.030
148. Xiao G, Zheng Y, Chen H, Luo M, Yang C, Ren D, et al. Single-cell transcriptome analysis reveals immunosuppressive landscape in overweight and obese colorectal cancer. J Transl Med. (2024) 22:134. doi: 10.1186/s12967-024-04921-5
149. Scott AJ, Alexander JL, Merrifield CA, Cunningham D, Jobin C, Brown R, et al. International Cancer Microbiome Consortium consensus statement on the role of the human microbiome in carcinogenesis. Gut. (2019) 68:1624–32. doi: 10.1136/gutjnl-2019-318556
150. Hanus M, Parada-Venegas D, Landskron G, Wielandt AM, Hurtado C, Alvarez K, et al. Immune system, microbiota, and microbial metabolites: the unresolved triad in colorectal cancer microenvironment. Front Immunol. (2021) 12:612826. doi: 10.3389/fimmu.2021.612826
151. Makki K, Deehan EC, Walter J, and Bäckhed F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe. (2018) 23:705–15. doi: 10.1016/j.chom.2018.05.012
152. Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y, et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat Commun. (2020) 11:4457. doi: 10.1038/s41467-020-18262-6
153. Zhu X, Li K, Liu G, Wu R, Zhang Y, Wang S, et al. Microbial metabolite butyrate promotes anti-PD-1 antitumor efficacy by modulating T cell receptor signaling of cytotoxic CD8 T cell. Gut Microbes. (2023) 15:2249143. doi: 10.1080/19490976.2023.2249143
154. Bachem A, Makhlouf C, Binger KJ, de Souza DP, Tull D, Hochheiser K, et al. Microbiota-derived short-chain fatty acids promote the memory potential of antigen-activated CD8(+) T cells. Immunity. (2019) 51:285–97.e5. doi: 10.1016/j.immuni.2019.06.002
155. Donohoe DR, Holley D, Collins LB, Montgomery SA, Whitmore AC, Hillhouse A, et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. (2014) 4:1387–97. doi: 10.1158/2159-8290.CD-14-0501
156. Li Q, Cao L, Tian Y, Zhang P, Ding C, Lu W, et al. Butyrate suppresses the proliferation of colorectal cancer cells via targeting pyruvate kinase M2 and metabolic reprogramming. Mol Cell Proteomics. (2018) 17:1531–45. doi: 10.1074/mcp.RA118.000752
157. Bekebrede AF, Deuren TV, Gerrits WJJ, Keijer J, and Boer VCJ. Butyrate alters pyruvate flux and induces lipid accumulation in cultured colonocytes. Int J Mol Sci. (2021) 22:10937. doi: 10.3390/ijms222010937
158. Huang C, Deng W, Xu HZ, Zhou C, Zhang F, Chen J, et al. Short-chain fatty acids reprogram metabolic profiles with the induction of reactive oxygen species production in human colorectal adenocarcinoma cells. Comput Struct Biotechnol J. (2023) 21:1606–20. doi: 10.1016/j.csbj.2023.02.022
159. Schug ZT, Vande Voorde J, and Gottlieb E. The metabolic fate of acetate in cancer. Nat Rev Cancer. (2016) 16:708–17. doi: 10.1038/nrc.2016.87
160. Qiu J, Villa M, Sanin DE, Buck MD, O'Sullivan D, Ching R, et al. Acetate promotes T cell effector function during glucose restriction. Cell Rep. (2019) 27:2063–74.e5. doi: 10.1016/j.celrep.2019.04.022
161. Depner M, Taft DH, Kirjavainen PV, Kalanetra KM, Karvonen AM, Peschel S, et al. Maturation of the gut microbiome during the first year of life contributes to the protective farm effect on childhood asthma. Nat Med. (2020) 26:1766–75. doi: 10.1038/s41591-020-1095-x
162. Ryu TY, Kim K, Han TS, Lee MO, Lee J, Choi J, et al. Human gut-microbiome-derived propionate coordinates proteasomal degradation via HECTD2 upregulation to target EHMT2 in colorectal cancer. Isme J. (2022) 16:1205–21. doi: 10.1038/s41396-021-01119-1
163. Nshanian M, Gruber JJ, Geller BS, Chleilat F, Lancaster SM, White SM, et al. Short-chain fatty acid metabolites propionate and butyrate are unique epigenetic regulatory elements linking diet, metabolism and gene expression. Nat Metab. (2025) 7:196–211. doi: 10.1038/s42255-024-01191-9
164. Sinha SR, Haileselassie Y, Nguyen LP, Tropini C, Wang M, Becker LS, et al. Dysbiosis-induced secondary bile acid deficiency promotes intestinal inflammation. Cell Host Microbe. (2020) 27:659–70.e5. doi: 10.1016/j.chom.2020.01.021
165. Ocvirk S and O'Keefe SJD. Dietary fat, bile acid metabolism and colorectal cancer. Semin Cancer Biol. (2021) 73:347–55. doi: 10.1016/j.semcancer.2020.10.003
166. Zhang H, Xu H, Zhang C, Tang Q, and Bi F. Ursodeoxycholic acid suppresses the Malignant progression of colorectal cancer through TGR5-YAP axis. Cell Death Discov. (2021) 7:207. doi: 10.1038/s41420-021-00589-8
167. Yang J, Wei H, Zhou Y, Szeto CH, Li C, Lin Y, et al. High-fat diet promotes colorectal tumorigenesis through modulating gut microbiota and metabolites. Gastroenterology. (2022) 162:135–49.e2. doi: 10.1053/j.gastro.2021.08.041
168. Li J, Chen Z, Wang Q, Du L, Yang Y, Guo F, et al. Microbial and metabolic profiles unveil mutualistic microbe-microbe interaction in obesity-related colorectal cancer. Cell Rep Med. (2024) 5:101429. doi: 10.1016/j.xcrm.2024.101429
169. Jun SY, Brown AJ, Chua NK, Yoon JY, Lee JJ, Yang JO, et al. Reduction of squalene epoxidase by cholesterol accumulation accelerates colorectal cancer progression and metastasis. Gastroenterology. (2021) 160:1194–207.e28. doi: 10.1053/j.gastro.2020.09.009
170. Li C, Wang Y, Liu D, Wong CC, Coker OO, Zhang X, et al. Squalene epoxidase drives cancer cell proliferation and promotes gut dysbiosis to accelerate colorectal carcinogenesis. Gut. (2022) 71:2253–65. doi: 10.1136/gutjnl-2021-325851
171. Pan M, Qin C, and Han X. Lipid metabolism and lipidomics applications in cancer research. Adv Exp Med Biol. (2021) 1316:1–24. doi: 10.1007/978-981-33-6785-2_1.
172. Juloski T, Popovic T, Martacic JD, Cuk V, Milic Perovic, Stankovic S, et al. Fatty acid in colorectal cancer in adult and aged patients of both sexes. J buon. (2021) 26:1898–907. J TJV VC, M SMP, M SS.
173. Bhatt K, Orlando T, Meuwis MA, Louis E, Stefanuto PH, and Focant JF. Comprehensive insight into colorectal cancer metabolites and lipids for human serum: A proof-of-concept study. Int J Mol Sci. (2023) 24:9614. doi: 10.3390/ijms24119614
174. Mika A, Pakiet A, Czumaj A, Kaczynski Z, Liakh I, Kobiela J, et al. Decreased triacylglycerol content and elevated contents of cell membrane lipids in colorectal cancer tissue: A lipidomic study. J Clin Med. (2020) 9:1095. doi: 10.3390/jcm9041095
175. Tan B, Qiu Y, Zou X, Chen T, Xie G, Cheng Y, et al. Metabonomics identifies serum metabolite markers of colorectal cancer. J Proteome Res. (2013) 12:3000–9. doi: 10.1021/pr400337b
176. Dmitrieva-Posocco O, Wong AC, Lundgren P, Golos AM, Descamps HC, Dohnalová L, et al. β-Hydroxybutyrate suppresses colorectal cancer. Nature. (2022) 605:160–5. doi: 10.1038/s41586-022-04649-6
177. Liu T, Peng F, Yu J, Tan Z, Rao T, Chen Y, et al. LC-MS-based lipid profile in colorectal cancer patients: TAGs are the main disturbed lipid markers of colorectal cancer progression. Anal Bioanal Chem. (2019) 411:5079–88. doi: 10.1007/s00216-019-01872-5
178. Fang Z, He M, and Song M. Serum lipid profiles and risk of colorectal cancer: a prospective cohort study in the UK Biobank. Br J Cancer. (2021) 124:663–70. doi: 10.1038/s41416-020-01143-6
179. Elmallah MIY, Ortega-Deballon P, Hermite L, Pais-De-Barros JP, Gobbo J, and Garrido C. Lipidomic profiling of exosomes from colorectal cancer cells and patients reveals potential biomarkers. Mol Oncol. (2022) 16:2710–8. doi: 10.1002/1878-0261.13223
180. Zhu Y, Zhou H, Chen H, Zhang J, Liang Y, Yang S, et al. Global serum metabolomic and lipidomic analyses reveal lipid perturbations and potential biomarkers of the colorectal cancer by adenoma-carcinoma sequence. Chin J Analytical Chem. (2023) 51:100270. doi: 10.1016/j.cjac.2023.100270
181. Xu R, Shen J, Song Y, Lu J, Liu Y, Cao Y, et al. Exploration of the application potential of serum multi-biomarker model in colorectal cancer screening. Sci Rep. (2024) 14:10127. doi: 10.1038/s41598-024-60867-0
182. Cajka T and Fiehn O. Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. Trends Analyt Chem. (2014) 61:192–206. doi: 10.1016/j.trac.2014.04.017
183. Lubes G and Goodarzi M. GC-MS based metabolomics used for the identification of cancer volatile organic compounds as biomarkers. J Pharm BioMed Anal. (2018) 147:313–22. doi: 10.1016/j.jpba.2017.07.013
184. Hu Z, Shen F, Liu Y, Zhong Z, Chen Y, Xia Z, et al. Targeted metabolomics reveals novel diagnostic biomarkers for colorectal cancer. Mol Oncol. (2025) 19:1737–50. doi: 10.1002/1878-0261.13791
185. Maimó-Barceló A, Pérez-Romero K, Rodríguez RM, Huergo C, Calvo I, Fernández JA, et al. To image or not to image: Use of imaging mass spectrometry in biomedical lipidomics. Prog Lipid Res. (2025) 97:101319. doi: 10.1016/j.plipres.2025.101319
186. Denti V, Mahajneh A, Capitoli G, Clerici F, Piga I, Pagani L, et al. Lipidomic typing of colorectal cancer tissue containing tumour-infiltrating lymphocytes by MALDI mass spectrometry imaging. Metabolites. (2021) 11:599. doi: 10.3390/metabo11090599
187. Dufresne M, Fincher JA, Patterson NH, Schey KL, Norris JL, Caprioli RM, et al. α-cyano-4-hydroxycinnamic acid and tri-potassium citrate salt pre-coated silicon nanopost array provides enhanced lipid detection for high spatial resolution MALDI imaging mass spectrometry. Anal Chem. (2021) 93:12243–9. doi: 10.1021/acs.analchem.1c01560
188. Iakab SA, Ràfols P, Correig-Blanchar X, and García-Altares M. Perspective on multimodal imaging techniques coupling mass spectrometry and vibrational spectroscopy: picturing the best of both worlds. Anal Chem. (2021) 93:6301–10. doi: 10.1021/acs.analchem.0c04986
189. Yin R, Burnum-Johnson KE, Sun X, Dey SK, and Laskin J. High spatial resolution imaging of biological tissues using nanospray desorption electrospray ionization mass spectrometry. Nat Protoc. (2019) 14:3445–70. doi: 10.1038/s41596-019-0237-4
190. Passarelli MK, Pirkl A, Moellers R, Grinfeld D, Kollmer F, Havelund R, et al. The 3D OrbiSIMS—label-free metabolic imaging with subcellular lateral resolution and high mass-resolving power. Nat Methods. (2017) 14:1175–83. doi: 10.1038/nmeth.4504
191. Li J, Vosegaard T, and Guo Z. Applications of nuclear magnetic resonance in lipid analyses: An emerging powerful tool for lipidomics studies. Prog Lipid Res. (2017) 68:37–56. doi: 10.1016/j.plipres.2017.09.003
192. Batchuluun B, Pinkosky SL, and Steinberg GR. Lipogenesis inhibitors: therapeutic opportunities and challenges. Nat Rev Drug Discovery. (2022) 21:283–305. doi: 10.1038/s41573-021-00367-2
193. Chang L, Wu P, Senthilkumar R, Tian X, Liu H, Shen X, et al. Loss of fatty acid synthase suppresses the Malignant phenotype of colorectal cancer cells by down-regulating energy metabolism and mTOR signaling pathway. J Cancer Res Clin Oncol. (2016) 142:59–72. doi: 10.1007/s00432-015-2000-8
194. Murata S, Yanagisawa K, Fukunaga K, Oda T, Kobayashi A, Sasaki R, et al. Fatty acid synthase inhibitor cerulenin suppresses liver metastasis of colon cancer in mice. Cancer Sci. (2010) 101:1861–5. doi: 10.1111/j.1349-7006.2010.01596.x
195. Khiewkamrop P, Surangkul D, Srikummool M, Richert L, Pekthong D, Parhira S, et al. Epigallocatechin gallate triggers apoptosis by suppressing de novo lipogenesis in colorectal carcinoma cells. FEBS Open Bio. (2022) 12:937–58. doi: 10.1002/2211-5463.13391
196. Shin CM, Lee DH, Seo AY, Lee HJ, Kim SB, Son WC, et al. Green tea extracts for the prevention of metachronous colorectal polyps among patients who underwent endoscopic removal of colorectal adenomas: A randomized clinical trial. Clin Nutr. (2018) 37:452–8. doi: 10.1016/j.clnu.2017.01.014
197. Nalli M, Masci D, Urbani A, La Regina G, and Silvestri R. Emerging direct targeting β-catenin agents. Molecules. (2022) 27:7735. doi: 10.3390/molecules27227735
198. Yao Y, Rao C, Zheng G, and Wang S. Luteolin suppresses colorectal cancer cell metastasis via regulation of the miR−384/pleiotrophin axis. Oncol Rep. (2019) 42:131–41. doi: 10.3892/or.2019.7136
199. Falchook G, Infante J, Arkenau HT, Patel MR, Dean E, Borazanci E, et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine. (2021) 34:100797. doi: 10.1016/j.eclinm.2021.100797
200. Cioccoloni G, Bonmassar L, Pagani E, Caporali S, Fuggetta MP, Bonmassar E, et al. Influence of fatty acid synthase inhibitor orlistat on the DNA repair enzyme O6-methylguanine-DNA methyltransferase in human normal or Malignant cells in vitro. Int J Oncol. (2015) 47:764–72. doi: 10.3892/ijo.2015.3025
201. Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. (2005) 8:311–21. doi: 10.1016/j.ccr.2005.09.008
202. Wang C, Xu C, Sun M, Luo D, Liao DF, and Cao D. Acetyl-CoA carboxylase-alpha inhibitor TOFA induces human cancer cell apoptosis. Biochem Biophys Res Commun. (2009) 385:302–6. doi: 10.1016/j.bbrc.2009.05.045
203. Potze L, di Franco S, Kessler JH, Stassi G, and Medema JP. Betulinic acid kills colon cancer stem cells. Curr Stem Cell Res Ther. (2016) 11:427–33. doi: 10.2174/1574888X11666151203223512
204. Hernlund E, Ihrlund LS, Khan O, Ates YO, Linder S, Panaretakis T, et al. Potentiation of chemotherapeutic drugs by energy metabolism inhibitors 2-deoxyglucose and etomoxir. Int J Cancer. (2008) 123:476–83. doi: 10.1002/ijc.v123:2
205. Shiragami R, Murata S, Kosugi C, Tezuka T, Yamazaki M, Hirano A, et al. Enhanced antitumor activity of cerulenin combined with oxaliplatin in human colon cancer cells. Int J Oncol. (2013) 43:431–8. doi: 10.3892/ijo.2013.1978
206. Toden S, Tran HM, Tovar-Camargo OA, Okugawa Y, and Goel A. Epigallocatechin-3-gallate targets cancer stem-like cells and enhances 5-fluorouracil chemosensitivity in colorectal cancer. Oncotarget. (2016) 7:16158–71. doi: 10.18632/oncotarget.v7i13
207. Zhou Y, Bollu LR, Tozzi F, Ye X, Bhattacharya R, Gao G, et al. ATP citrate lyase mediates resistance of colorectal cancer cells to SN38. Mol Cancer Ther. (2013) 12:2782–91. doi: 10.1158/1535-7163.MCT-13-0098
208. Zi R, Zhao X, Liu L, Wang Y, Zhang R, Bian Z, et al. Metabolic-immune suppression mediated by the SIRT1-CX3CL1 axis induces functional enhancement of regulatory T cells in colorectal carcinoma. Adv Sci (Weinh). (2025):e2404734. doi: 10.1002/advs.202404734
209. Masci D, Puxeddu M, Silvestri R, and La Regina G. Metabolic rewiring in cancer: small molecule inhibitors in colorectal cancer therapy. Molecules. (2024) 29:2110. doi: 10.3390/molecules29092110
Keywords: lipid metabolism, fatty acids, colorectal cancer, the tumor microenvironment, CRC therapy
Citation: Liu W, Dong S, Hao F, Gao Y and Wei Q (2025) Lipid metabolic reprogramming in colorectal cancer: mechanisms and therapeutic strategies. Front. Immunol. 16:1603032. doi: 10.3389/fimmu.2025.1603032
Received: 31 March 2025; Accepted: 23 June 2025;
Published: 11 July 2025.
Edited by:
Jia Jinlin, Peking Union Medical College Graduate School, ChinaReviewed by:
Guoqing Liu, Inner Mongolia University of Science and Technology, ChinaXiaoying Zhou, Guangxi Medical University, China
Yoichi Takakusagi, National Institutes for Quantum and Radiological Science and Technology, Japan
Copyright © 2025 Liu, Dong, Hao, Gao and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaohui Gao, Z2FveWFvaHVpMTg3QHRvbmdqaS5lZHUuY24=; Qing Wei, d2VpcWluZzE5NzFAdG9uZ2ppLmVkdS5jbg==
†These authors have contributed equally to this work