 Yanggang Hong
Yanggang Hong Jiajun Li
Jiajun Li Nuo Xu1†
Nuo Xu1† Wanyi Shu
Wanyi Shu Feng Chen
Feng Chen Yuze Mi
Yuze Mi Haigang Geng
Haigang Geng- 1State Key Laboratory of Systems Medicine for Cancer, Shanghai Cancer Institute & Department of Liver Surgery, Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, China
- 3Wenzhou Medical University, Wenzhou, Zhejiang, China
- 4Department of Gastrointestinal Surgery, Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 5Oncology Department of Shanghai Sixth People’s Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, China
Background: Colorectal cancer (CRC) remains a leading cause of cancer-related mortality worldwide. Despite therapeutic advances, there is a critical need to identify novel, effective, and safe drug targets to improve precision treatment strategies.
Methods: We developed a multi-layered framework integrating Mendelian randomization (MR), colocalization analysis, genome-wide association study (GWAS) data, and expression quantitative trait loci (eQTLs) to prioritize causal and druggable genes in CRC. Single-cell and bulk RNA sequencing were used to characterize gene expression within the tumor microenvironment. Phenome-wide association studies (PheWAS) assessed off-target effects, and drug repurposing potential was evaluated using OpenTargets, DrugBank, and DGIdb. Validation of key targets was performed through RT-qPCR and immunohistochemistry (IHC) in CRC patient samples.
Results: Out of 4,479 druggable genes, MR analysis identified 47 candidates significantly associated with CRC risk. Six genes (TFRC, TNFSF14, LAMC1, PLK1, TYMS, and TSSK6) demonstrated strong colocalization signals and were further validated across replication datasets and subtype-stratified analyses. PheWAS analysis revealed minimal off-target effects for these genes. Notably, several of these genes have already been targeted by existing or investigational drugs, suggesting potential for repurposing. These genes exhibited distinct expression patterns in tumor and stromal cell types and were differentially expressed in CRC versus normal tissues. Among them, TNFSF14, an immune modulator, is particularly involved in regulating T cell activation within the tumor microenvironment.
Conclusion: This study identifies and validates six promising druggable targets for CRC, providing a strong foundation for future preclinical studies. These findings open avenues for advancing precision oncology and drug repurposing strategies in CRC treatment, contributing to the development of more effective and personalized therapeutic approaches.
1 Introduction
Colorectal cancer (CRC) is one of the most prevalent and lethal malignancies worldwide, with an increasing incidence in both developed and developing countries. According to the Global Cancer Statistics, CRC ranks as the third most common cancer and the second leading cause of cancer-related mortality globally (1). The pathogenesis of CRC is a complex interplay of genetic, environmental, and lifestyle factors, including diet, smoking, alcohol consumption, and chronic inflammation (2). The adenoma-carcinoma sequence describes the progression of normal colonic epithelium to adenomatous polyps and ultimately invasive carcinoma, driven by accumulated genetic and epigenetic alterations (3). Mutations in key genes such as APC, TP53, KRAS, and PIK3CA, alongside microsatellite instability (MSI) and CpG island methylator phenotype (CIMP), are crucial in CRC pathogenesis (4, 5). Despite advances in early detection through colonoscopy and fecal immunochemical tests, a significant proportion of CRC cases are diagnosed at an advanced stage, leading to poor survival outcomes (6).
The development of anti-cancer drugs has significantly transformed CRC treatment, yet challenges remain due to drug resistance, toxicity, and limited efficacy in certain patient subgroups. Conventional treatment strategies include surgical resection for localized disease, combined with chemotherapy and other neoadjuvant treatments such as targeted therapy or immunotherapy (7). The introduction of fluoropyrimidine-based chemotherapy (e.g., 5-fluorouracil) in combination with oxaliplatin or irinotecan has improved survival rates (8, 9). Targeted therapies against EGFR (cetuximab, panitumumab) and VEGF (bevacizumab) have further enhanced treatment options for metastatic CRC, particularly in patients with RAS wild-type tumors (10, 11). More recently, immune checkpoint inhibitors (ICIs) have revolutionized the treatment landscape for MSI/dMMR CRC, demonstrating durable responses and improved overall survival (12). However, resistance mechanisms, heterogeneity in drug response, and high treatment costs continue to impede the widespread success of these therapies, necessitating the exploration of novel drug targets.
Recent studies have highlighted the role of tumor-initiating cells (TICs), a subpopulation with stem-like properties and high tumorigenic potential, in contributing to immune evasion, drug resistance, and disease recurrence (13). TICs remodel the tumor microenvironment to suppress immune responses, facilitating tumor progression and therapeutic failure. Therefore, identifying immune-relevant and druggable targets within TIC-enriched populations represents a promising approach to overcome resistance and enhance the efficacy of immunotherapy in digestive system tumors such as CRC.
Several studies have also sought to identify novel therapeutic targets in CRC, leveraging insights from genomics, transcriptomics, and proteomics. For example, inhibition of BRAF V600E-mutant tumors using vemurafenib in combination with cetuximab has shown promising but limited clinical efficacy, highlighting the need for combination strategies. HER2-targeted therapies, such as trastuzumab and pertuzumab, have been explored in HER2-amplified CRC with encouraging results (14, 15). Other emerging drug targets include Wnt signaling inhibitors (Porcupine inhibitors), MEK inhibitors, and metabolic regulators such as IDH1/2 inhibitors (16). Additionally, RNA-based therapies, including small interfering RNA (siRNA) and antisense oligonucleotides, have demonstrated preclinical potential in targeting oncogenic pathways in CRC (17). Despite these advancements, many of these therapies remain in early development stages or face challenges in clinical translation due to toxicity, off-target effects, and tumor heterogeneity.
Given these limitations, our study employs a Mendelian randomization (MR) approach to systematically identify potential drug targets for CRC. Unlike traditional observational studies, which are often confounded by environmental and lifestyle factors, MR leverages genetic variants as instrumental variables to infer causal relationships between gene expression and disease risk (18). This method reduces biases and enhances the robustness of target discovery, offering a powerful tool for drug repurposing and biomarker identification (19, 20). In our study, we integrate genome-wide association study (GWAS) data with expression quantitative trait loci (eQTL) analysis to prioritize druggable genes associated with CRC risk. By applying rigorous statistical criteria and sensitivity analyses, we aim to identify high-confidence targets that could be exploited for therapeutic intervention. Our findings provide a foundation for future preclinical and clinical studies, potentially paving the way for precision medicine strategies in CRC treatment (Figure 1).
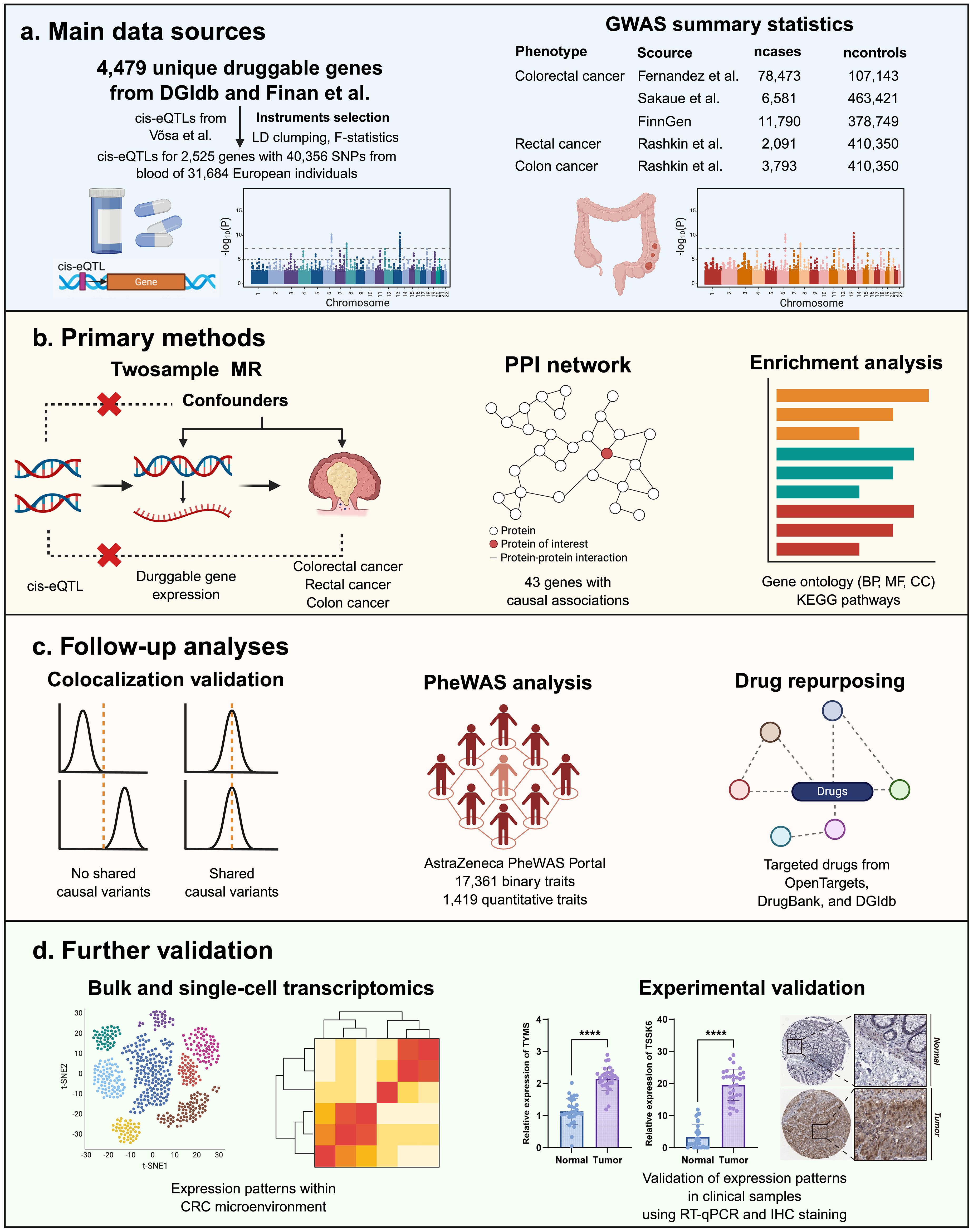
Figure 1. (A–D) Study design and workflow for identifying druggable gene targets associated with CRC.
2 Methods
2.1 Identification of druggable genes
To comprehensively identify potential therapeutic targets for CRC, we compiled a broad list of druggable genes, which encode proteins amenable to modulation by small molecules or biologics (21). Two primary resources were used, including the Drug–Gene Interaction Database (DGIdb v4.2.0) (22) and a comprehensive review of human druggable genes (21). These sources integrate data from drug-target databases, clinical studies, and chemical biology literature to define the druggable genome. From these databases, we curated a total of 4,479 unique druggable genes (Supplementary Table S1). This wide-scope inclusion strategy ensured that we captured both clinically validated targets and exploratory candidates relevant to complex diseases, including cancer. These genes were then used as input for subsequent eQTL and MR analyses to evaluate their causal relevance to CRC risk.
2.2 Acquisition of cis−eQTLs
Cis-eQTLs were defined as genetic variants located within 1 megabase (Mb) of the transcription start site of a gene and significantly associated with its expression (minor allele frequency > 0.01). We obtained cis-expression quantitative trait loci (cis-eQTLs) data from the eQTLGen Consortium (https://eqtlgen.org/), which provides large-scale eQTL information derived from blood samples of 31,684 individuals of European ancestry (23). This dataset includes expression data for 16,987 genes and over 30,000 significant cis-eQTLs. Among the 4,479 druggable genes initially identified, 2,525 had corresponding cis-eQTLs available in the eQTLGen dataset and were included in downstream MR analysis. This dataset served as the basis for selecting genetic instruments to evaluate causal relationships between gene expression and CRC risk.
2.3 Outcome data
Summary-level GWAS data for CRC were obtained from previously published meta-analyses and biobank studies, all based on individuals of European ancestry (24). The GWAS statistics could be further obtained from the GWAS catalog (https://www.ebi.ac.uk/gwas/home). The discovery analysis included 78,473 CRC cases and 107,143 controls (GCST90255675). Two replication datasets were used: one from Sakaue et al. (6,581 cases and 463,421 controls; GCST90018808) (25) and one from the FinnGen project (11,790 cases and 378,749 controls) (26). To explore site-specific effects, we also conducted stratified analyses using GWAS data for colon cancer (3,793 cases and 410,350 controls; GCST90011811) and rectal cancer (2,091 cases and 410,350 controls; GCST90011810) (27). All participants provided informed consent, and ethical approvals were obtained from the relevant institutional review boards. Detailed cohort characteristics are provided in Table 1.
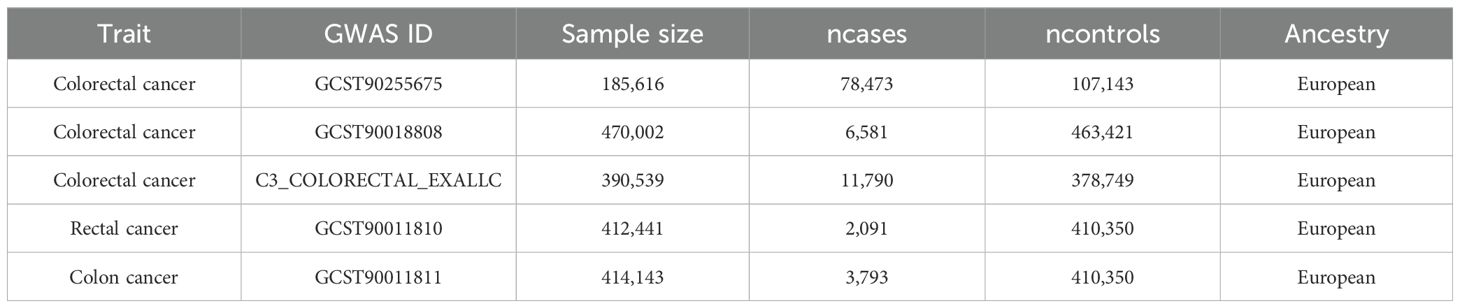
Table 1. Information on the GWAS datasets analyzed in this study.
2.4 Selection of instrumental variable
To ensure robust causal inference in MR, we applied strict criteria for selecting single nucleotide polymorphisms (SNPs) as instrumental variables (IVs) for each druggable gene. First, we extracted cis-eQTL SNPs with genome-wide significance (P < 5.0 × 10-8) (28) from the eQTLGen dataset, ensuring strong association with gene expression. To eliminate linkage disequilibrium (LD), SNPs were clumped using the 1000 Genomes Project (European population) reference panel, with an LD threshold of r2 < 0.1 and a clumping window of 10,000 kb (29). SNPs incompatible across exposure and outcome datasets were excluded. For palindromic SNPs, we inferred strand orientation based on allele frequencies or removed them if this information was unavailable. The F-statistic was calculated for each IV using the formula F = (N − k − 1)/k × [R2/(1 − R2)] to assess instrument strength (30). Only SNPs with F-statistics > 20 were retained to avoid weak instrument bias. After filtering, a total of 40,356 SNPs were selected as IVs corresponding to 2,525 druggable genes, forming the basis for downstream MR analysis (31). Details about the IVs are shown in Supplementary Table S2.
2.5 MR analysis
We conducted two sample MR analysis using the TwoSampleMR R package (version 0.6.6) to estimate the causal effects of gene expression on CRC, colon cancer, and rectal cancer risk. For genes with only a single valid instrumental SNP, we applied the Wald ratio method. When two or more independent SNPs were available, the inverse-variance weighted (IVW) method was used to derive overall causal estimates (32). To control for multiple hypothesis testing, we applied Bonferroni correction based on the total number of gene tested (33). Statistical significance thresholds were set at P < 2.02 × 10-5. This corrected threshold ensured robust identification of causal genes while minimizing the false discovery rate.
2.6 Sensitivity analysis
To assess the robustness of the MR results and detect potential violations of MR assumptions, we conducted a series of sensitivity analyses for genes with significant causal associations. For genes with multiple IVs, MR-Egger intercept tests were performed to evaluate the presence of horizontal pleiotropy, with P < 0.05 indicating potential bias (34). We also calculated Cochran’s Q statistic under both the IVW and MR-Egger models to assess heterogeneity among the SNP-specific estimates (35). Additionally, a leave-one-out analysis was carried out by iteratively excluding one SNP at a time to identify any disproportionately influential variants. Only genes that met all quality control criteria, including consistent effect directions across MR methods, non-significant MR-Egger intercepts, and stable leave-one-out results, were retained as high-confidence targets for further investigation.
2.7 Construction of protein-protein interaction network and enrichment analysis
To explore the functional interactions among genes identified by MR, we constructed a protein-protein interaction (PPI) network using the STRING database (https://string-db.org), limiting results to “Homo sapiens” and applying a minimum confidence score of 0.15. The network included only interactions supported by experimental or database evidence. To further investigate the biological relevance of these genes, we performed functional enrichment analysis using the clusterProfiler R package (36). Gene Ontology (GO) enrichment was used to categorize genes based on biological process (BP), cellular component (CC), and molecular function (MF) (37). In parallel, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was conducted to identify significantly enriched signaling pathways (38). Only terms and pathways with P < 0.05 were considered statistically significant. These analyses provided insights into the potential mechanisms through which the causal genes may influence CRC development and progression.
2.8 Colocalization analysis
Colocalization analysis was performed using the coloc R package (39) to determine whether gene expression and CRC risk share the same causal variant. SNPs were harmonized using the TwoSampleMR pipeline to ensure alignment between exposure and outcome datasets. Default prior probabilities were set to P1 = 1 × 10-4 (association with gene expression), P2 = 1 × 10-4 (association with CRC), and P12 = 1 × 10-5 (association with both). The posterior probabilities (PP) were used to evaluate five hypotheses, with strong colocalization defined as PP.H4/(PP.H3 + PP.H4) > 0.7 (40, 41), indicating a high likelihood that both traits are influenced by the same causal variant. Genes meeting this threshold were prioritized as high-confidence druggable targets.
2.9 Phenome−wide association analysis
To assess potential off-target effects and horizontal pleiotropy of the prioritized druggable genes, we conducted a phenome-wide association study (PheWAS) using the AstraZeneca PheWAS Portal (https://azphewas.com) (42). This platform includes genotype-phenotype associations derived from approximately 450,000 UK Biobank participants, covering ~15,500 binary and ~1,500 continuous phenotypes (43). For each candidate gene, we queried associated variants to identify significant relationships with traits unrelated to CRC. A significance threshold of P < 1 × 10–6 was applied, in accordance with the portal’s recommended cutoff for suggestive associations, to control for multiple testing and reduce false positives. This analysis provided insight into the broader phenotypic impact of each gene, helping to evaluate the safety and specificity of targeting these genes in CRC therapy.
2.10 Drug evaluation and repurposing
Given that most therapeutic agents target small-molecule proteins, we aimed to identify druggable targets among the 6 causal genes showing strong colocalization signals with CRC. To explore potential repurposing opportunities, we integrated our results with evidence from OpenTargets (44), DrugBank (45), and DGIdb (22). This integrative approach allowed us to prioritize candidate compounds, both approved and investigational, with favorable safety profiles that could be repositioned for CRC therapeutic applications.
2.11 Transcriptomic data analysis
The scRNA-seq data, comprising 33 samples from pre- and post-treatment tumor tissues and adjacent normal tissues of CRC patients, were retrieved from the GEO database (accession ID: GSE205506). We applied a strict quality control criteria to ensure data reliability. Cells were retained if they contained fewer than 20% mitochondrial gene content, expressed more than 200 genes, and had between 200 and 6000 detected genes, appearing in at least three cells. A total of 175,566 high-quality cells were selected for downstream analysis. To correct batch effects and improve clustering accuracy, we utilized the Harmony algorithm for data integration (46). The data was normalized using log-normalization, and the FindVariableFeatures function identified the top 2000 highly variable genes. Principal Component Analysis (PCA) was performed for dimensionality reduction, followed by soft k-means clustering using the Harmony package. Cells were then grouped into distinct clusters using the FindClusters function with a resolution of 0.6. Cell type annotation was conducted based on canonical marker genes, differential expression patterns, and known cellular profiles. Additionally, the expression patterns of potential targets were identified in bulk RNA-seq and scRNA-seq data. Bulk RNA-seq data, the TCGA-COAD cohort, was downloaded from UCSC Xena data portal (https://xena.ucsc.edu/). We first compared the expression levels of the targets between tumor and adjacent normal tissues. Subsequently, featureplots were utilized to visualize the expression patterns of target genes in CRC microenvironment. Additionally, we analyzed the association between the expression levels of target genes and key T cell exhaustion markers (PD-1, PD-L1, TIM-3) as well as the immune-suppressive cytokine IL-10 via GEPIA2 (http://gepia2.cancer-pku.cn/).
2.12 Real-time quantitative PCR
Paired tumor and adjacent normal tissues were collected from 31 patients with histologically confirmed CRC at Renji Hospital, Shanghai Jiao Tong University School of Medicine (Approval No.: KY2024-188-C). Tissue samples were immediately snap-frozen in liquid nitrogen and stored at -80°C until further processing. Total RNA was extracted using TRIzol reagent (Invitrogen) in accordance with the manufacturer’s instructions. Reverse transcription was performed using Superscript II reverse transcriptase (Invitrogen) and gene-specific primers. Real-time quantitative PCR (RT-qPCR) was conducted using the ABI Prism 7300 Sequence Detection System (Applied Biosystems) to assess the expression of LAMC1, TFRC, TNFSF14, PLK1, TYMS, TSSK6, and the reference gene GAPDH. Relative transcript levels were normalized to GAPDH expression using the relative standard curve method, following the manufacturer’s technical guidelines (Applied Biosystems). All data analysis adhered to the MIQE guidelines (47), ensuring experimental transparency and reproducibility. Detailed primer sequences were provided in Supplementary Table S3. The statistical differences were analyzed by one-way ANOVA and t-test using GraphPad Prism 10.1.2 (San Diego, CA, USA). P < 0.05 were considered significantly different.
2.13 Immunohistochemical staining
Immunohistochemical (IHC) images of both tumor and normal tissues were obtained from the Human Protein Atlas (HPA, https://www.proteinatlas.org/). However, IHC data of both normal and tumor tissues for TSSK6 were not available in the database and were therefore not included in the analysis.
3 Results
3.1 Identification of genes causally associated with CRC risk
Using a two-sample MR framework, we systematically assessed the causal relationship between the expression of 2,525 druggable genes and CRC risk. After Bonferroni correction (P < 2.02 × 10-5), 47 genes demonstrated significant causal associations in the discovery cohort, including 45 via the IVW method and 2 via the Wald ratio method (Figure 2; Supplementary Table S4). To ensure robustness, we conducted sensitivity analyses, including MR-Egger intercept, Cochran’s Q test, and leave-one-out tests. Four genes (HLA-DRB5, CTSF, TSPO, and TRPC6) were excluded due to evidence of horizontal pleiotropy (P < 0.05). The remaining 43 genes passed all quality control criteria and were retained for further validation and downstream analyses. These results provide a high-confidence set of candidate genes potentially involved in CRC pathogenesis and highlight their potential as therapeutic targets.
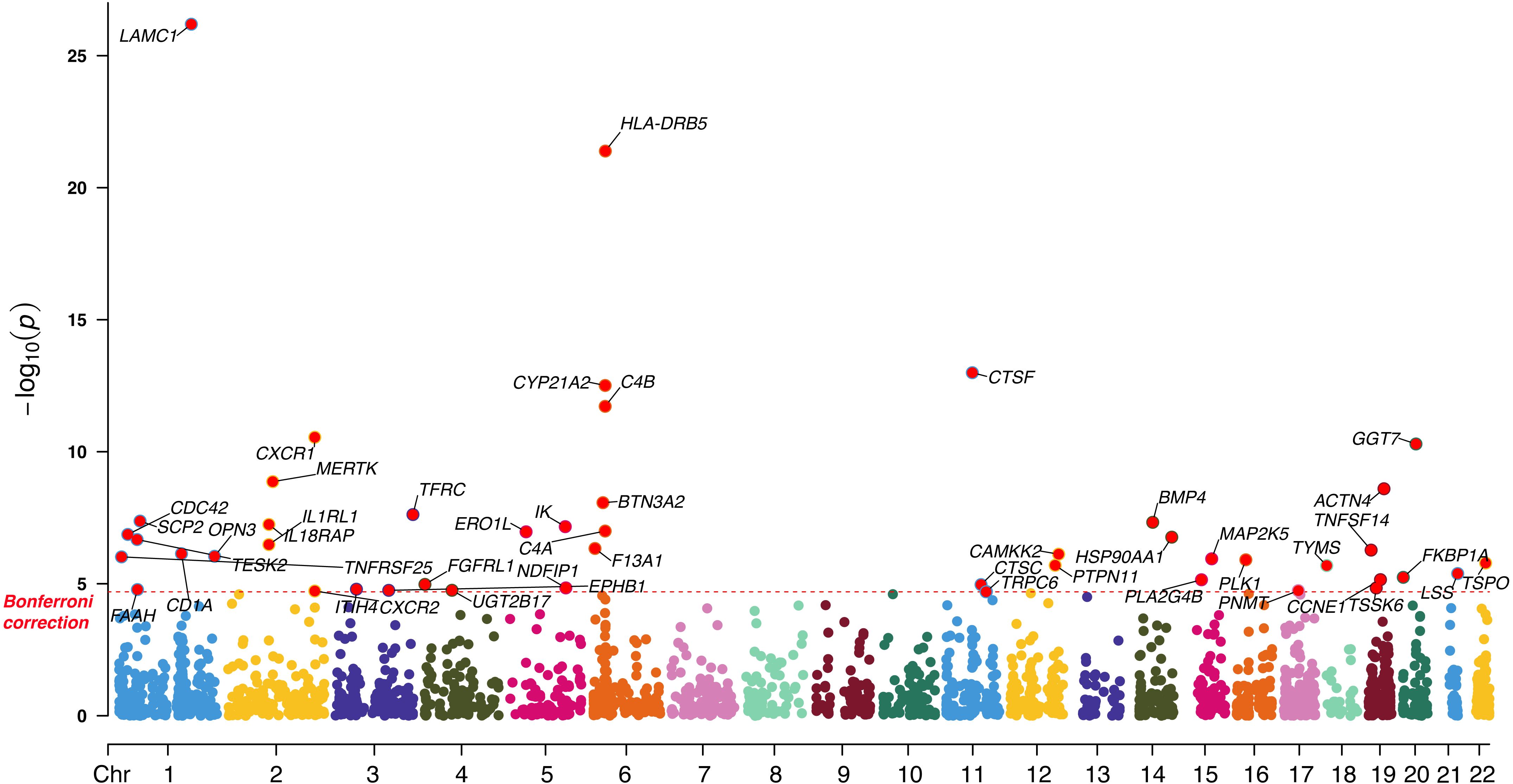
Figure 2. Manhattan plot illustrating the causal effect of significant druggable genes on CRC in the discovery cohort.
3.2 Construction of PPI network and enrichment analysis
To elucidate the functional relationships and interaction landscape among the genes causally linked to CRC, we constructed a PPI network using the STRING database. The network was restricted to interactions in “Homo sapiens” and filtered with a minimum interaction score of 0.15 to ensure confidence in the associations (Figure 3). The resulting network revealed a highly interconnected core, with genes such as PTPN11, CDC42, TFRC, HSP90AA1, and PLK1 serving as central hubs, suggesting key regulatory roles in CRC-related pathways. To further explore the biological relevance of these candidate genes, GO and KEGG pathway enrichment analyses were conducted using the R package clusterProfiler. GO analysis identified significant enrichment in terms associated with immune function and cell signaling, particularly lymphocyte-mediated immunity, dendritic cell migration, and protein localization to the nucleus (Figure 4A). Cellular component analysis highlighted localization to secretory granules, cytoplasmic vesicle lumens, and the external side of the plasma membrane, while molecular function analysis emphasized roles in protein tyrosine kinase activity and cytokine receptor activity. KEGG pathway enrichment revealed that the candidate genes were significantly associated with cancer-relevant signaling pathways (Figure 4B). The most enriched pathways included cytokine-cytokine receptor interaction, epithelial cell signaling in Helicobacter pylori infection, and leukocyte transendothelial migration. These findings underscore the potential involvement of the identified genes in inflammation, immune regulation, and tumor microenvironment remodeling-hallmarks of CRC development and progression.
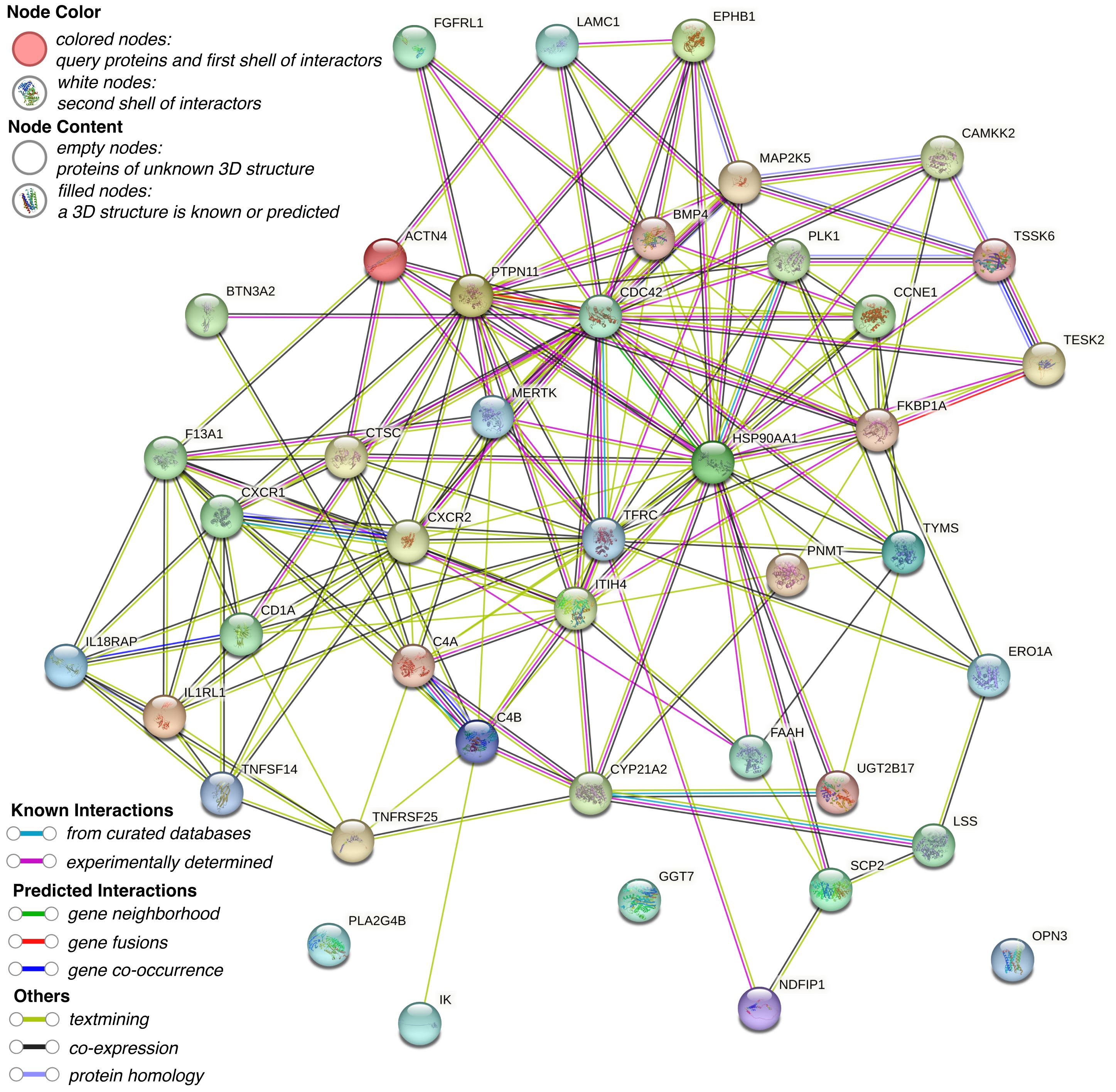
Figure 3. PPI network of CRC-associated genes constructed via STRING.
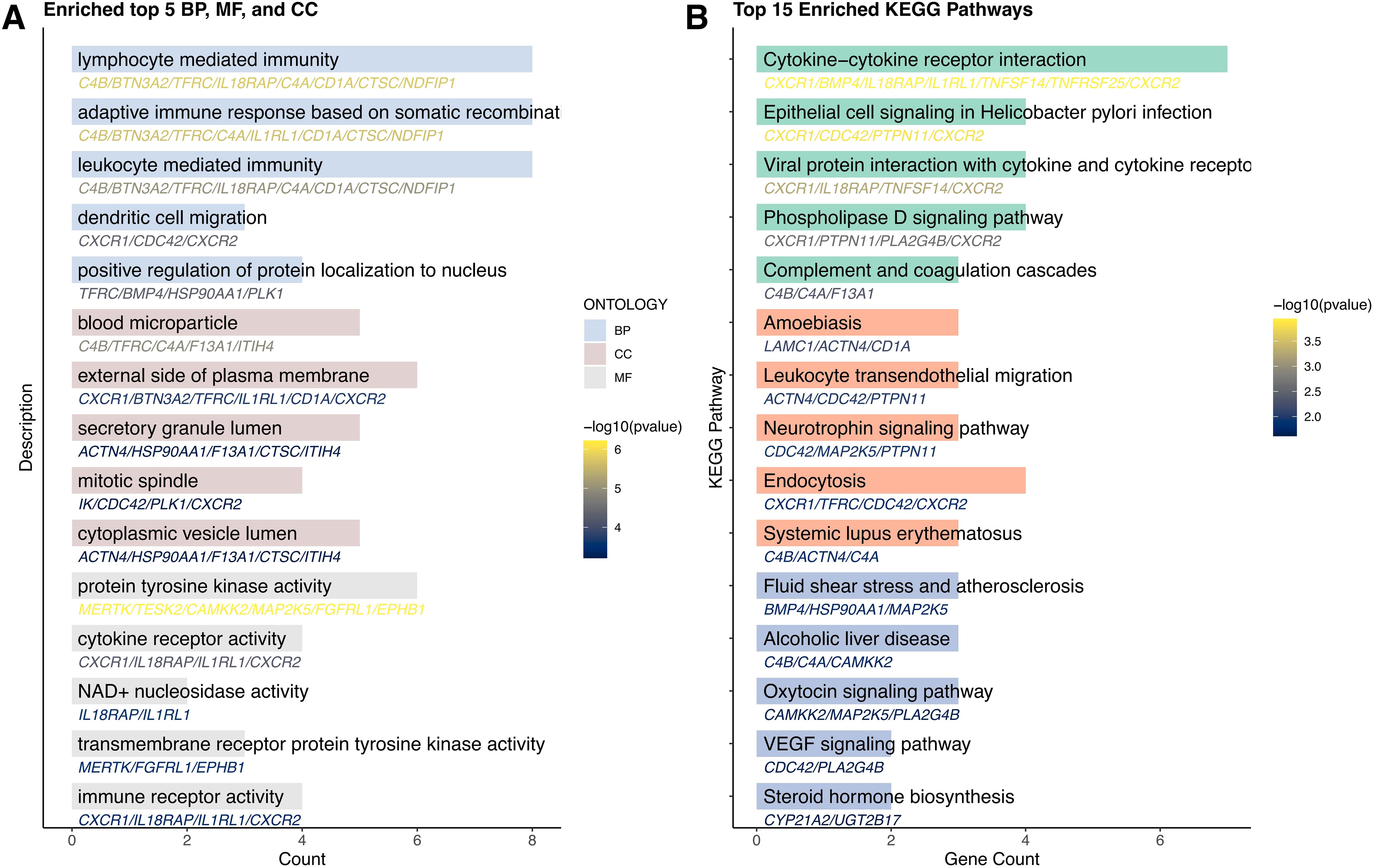
Figure 4. Results of enrichment analysis. GO (A) and KEGG (B) enrichment analyses of candidate genes.
3.3 Prioritizing 6 genes in colocalization analysis
To further refine the causal gene list and minimize potential false positives due to LD, we performed colocalization analysis on the 43 genes identified from MR. This approach assessed whether CRC-associated variants and gene expression signals shared the same causal variant. Six genes (TFRC, TNFSF14, LAMC1, PLK1, TYMS, and TSSK6) met this stringent criterion, indicating a high likelihood that gene expression and CRC risk are driven by the same genetic variants (Table 2). These genes were subsequently prioritized as high-confidence druggable targets. The remaining 37 genes, while suggestive, did not reach the colocalization threshold and were retained as secondary candidates for further evaluation. Sensitivity analyses supported the robustness of these six genes, showing no evidence of horizontal pleiotropy or significant heterogeneity (Table 3). Consistent effects across alternative MR models and leave-one-out analyses further confirmed the stability of these associations (Supplementary Figures S1, S2). These results highlight a subset of genes with both strong causal and colocalization evidence, warranting focused investigation in downstream validation and translational studies.
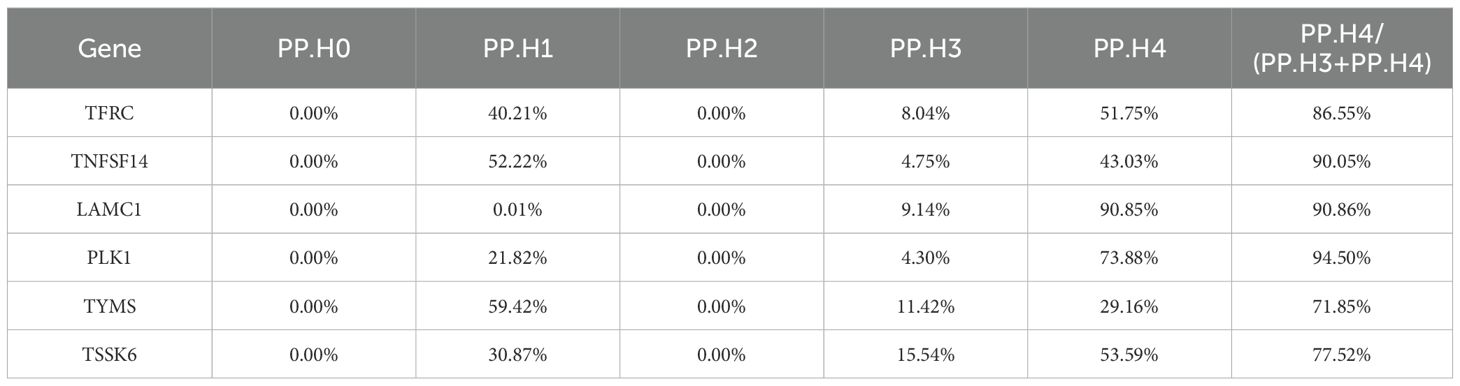
Table 2. Colocalization results of eQTLs for 6 genes with CRC-associated SNPs.
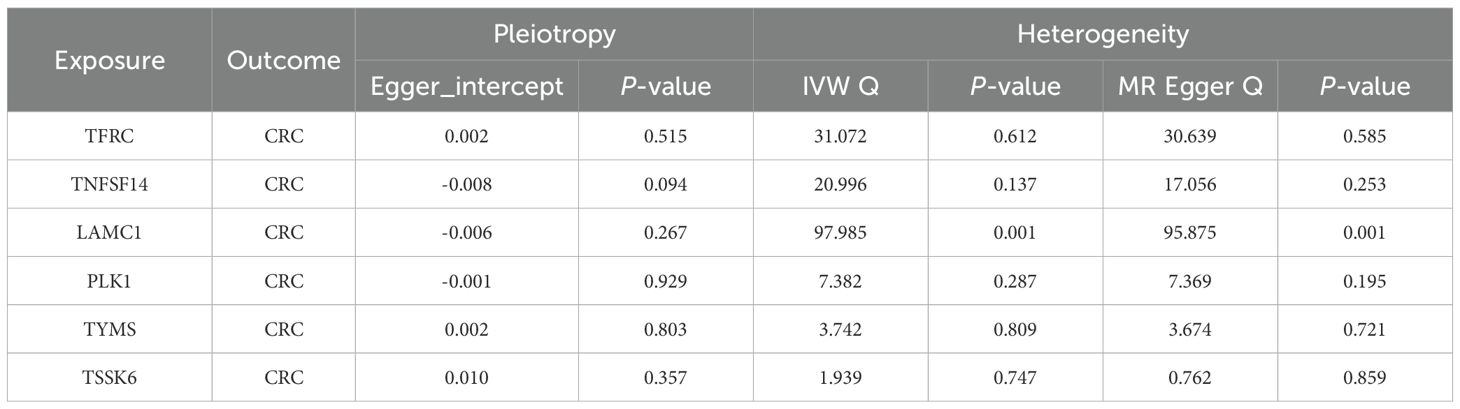
Table 3. Pleiotropy and heterogeneity test of the MR analysis.
3.4 Replication and stratified analysis
In the replication phase, among the 6 genes with significant colocalization evidence, 2 genes (LAMC1 and TNFSF14) were validated to be significant in all three replication cohorts (P < 0.05), and another 2 genes (TFRC and PLK1) were validated to be significant in at least one replication cohort (P < 0.05) (Figure 5). Additionally, the 6 genes with significant colocalization evidence were utilized for further stratified analysis in rectal cancer and colon cancer, respectively. In rectal cancer, two of the 6 genes (LAMC1 and PLK1) were identified with significant casual effect (P < 0.05) (Figure 6). In colon cancer, 2 of the 6 genes (LAMC1, and TSSK6) were identified with significant casual effect (P < 0.05) (Figure 6). Sensitivity analyses supported these findings, as no evidence of horizontal pleiotropy was detected for these genes (Supplementary Tables S5, S6). Consistent trends across four additional MR models and stability in the leave-one-out analysis confirmed the robustness of these findings (Supplementary Figures S3–S6).
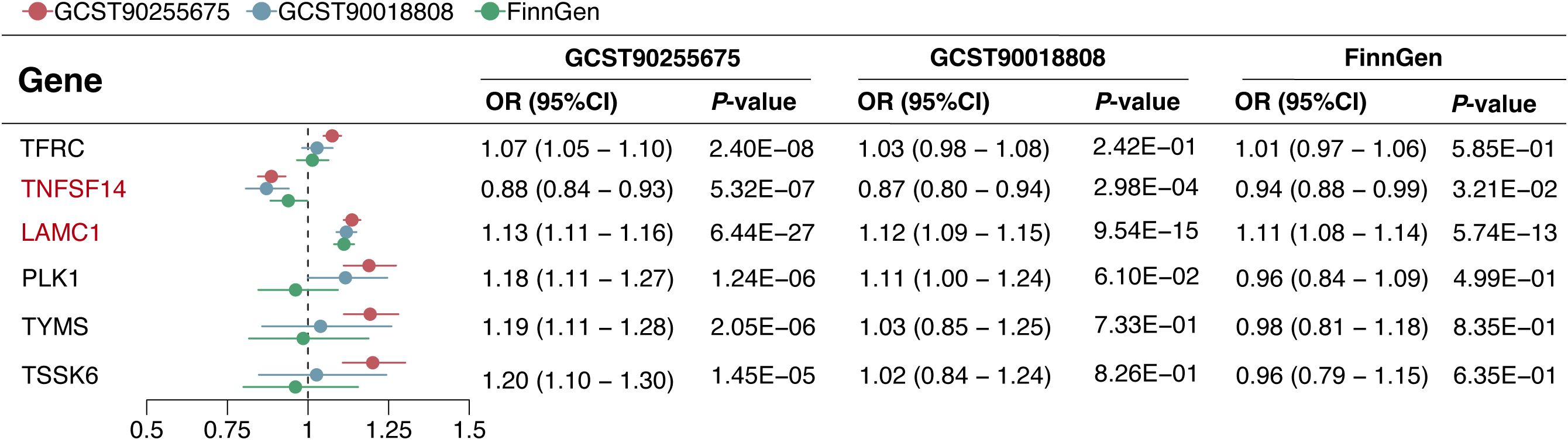
Figure 5. Causal effects of 6 candidate genes on CRC in the discovery and replication cohorts.
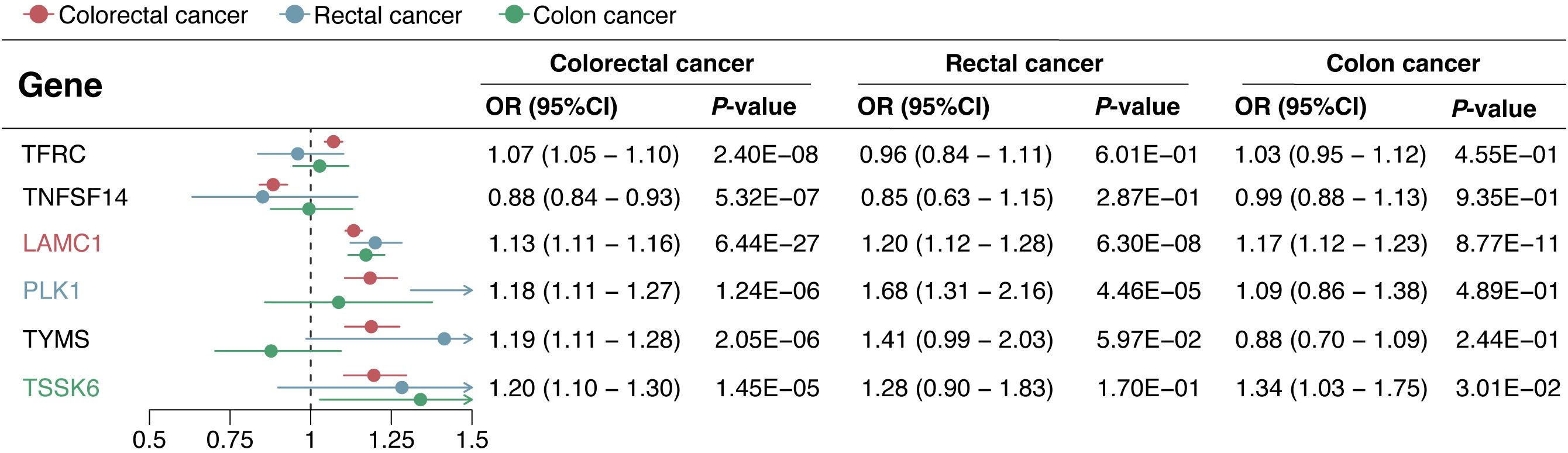
Figure 6. Causal effects of 6 candidate genes on CRC, rectal cancer, and colon cancer.
3.5 PheWAS indicated no potential side effects of drugs targeting the 6 candidate genes
The phenome-wide scan explored the relationships between potential drug targets and a variety of traits, providing valuable insights into possible side effects during drug development. This analysis revealed that none of the thirteen genes with strong colocalization evidence were linked to multiple phenotypes (P < 5 × 10-8) (Figures 7A–F).
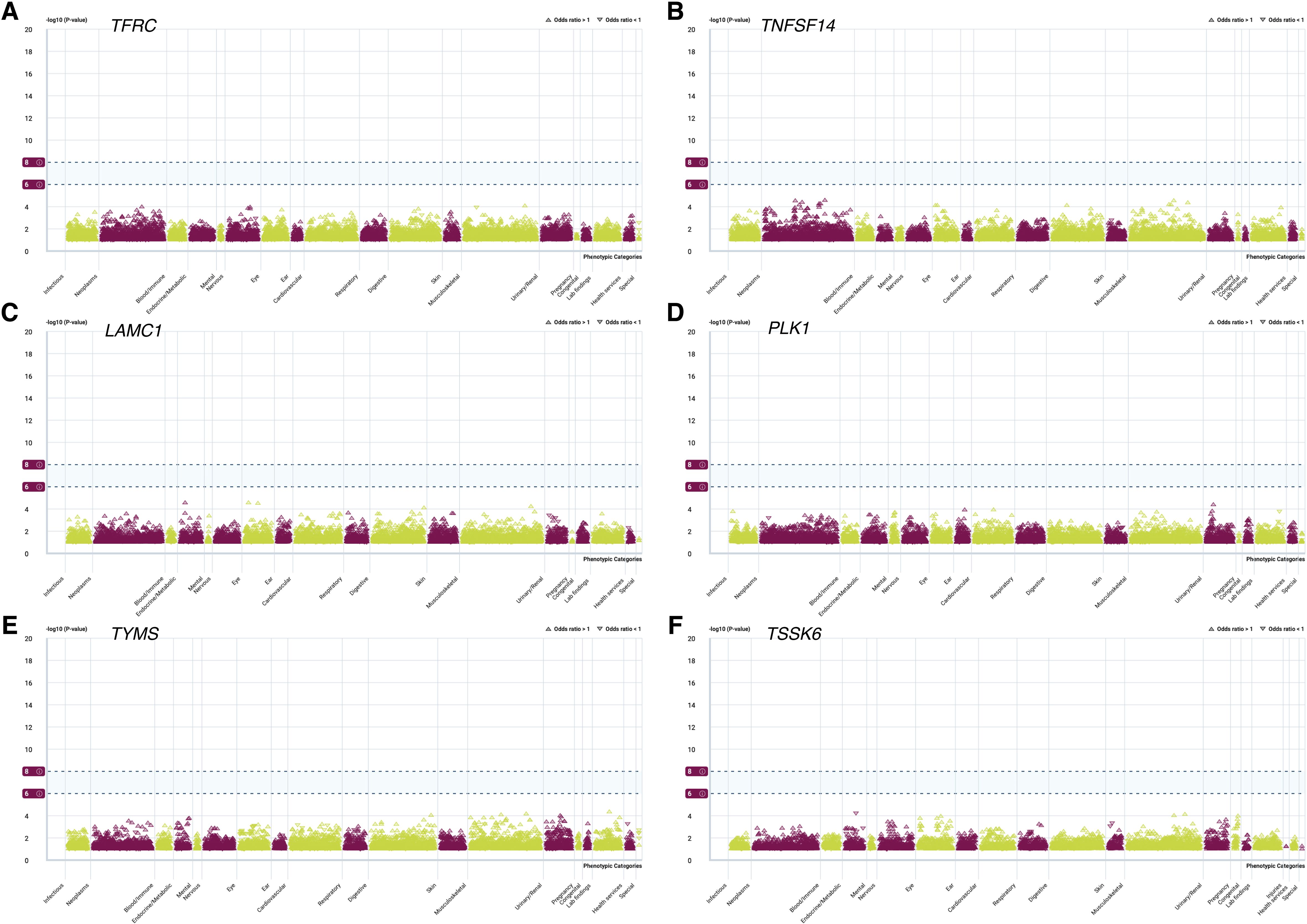
Figure 7. PheWAS analysis of associations between 6 candidate genes and various phenotypic categories. PheWAS analysis of TFRC (A), TNFSF14 (B), LAMC1 (C), PLK1 (D), TYMS (E), and TSSK6 (F).
3.6 Drug and compound prediction and existing drugs evaluation
To assess therapeutic potential, we investigated drugs targeting the six key genes (TFRC, TNFSF14, LAMC1, PLK1, TYMS, and TSSK6) using DGIdb, OpenTargets, and DrugBank. Several approved or investigational drugs were identified (Table 4). LAMC1 is targeted by agents like Ocriplasmin and Lanoteplase, mainly used for non-cancer conditions, but may be repurposed due to its role in the tumor microenvironment. TFRC, involved in iron metabolism, is already targeted by various approved iron supplements, and Pabinafusp alfa is under investigation for other diseases, showing potential for CRC applications. TNFSF14, a key immune regulator, is targeted by Baminercept, suggesting possible synergy with immunotherapies. PLK1 has multiple targeted drugs, including investigational anticancer agents like Volasertib and Onvansertib, with several approved drugs also showing potential off-target interactions. TYMS is already a standard chemotherapy target in CRC, with approved drugs such as Fluorouracil, Capecitabine, and Raltitrexed confirming its therapeutic relevance. These findings support drug repurposing strategies for CRC, potentially accelerating the development of targeted therapies.
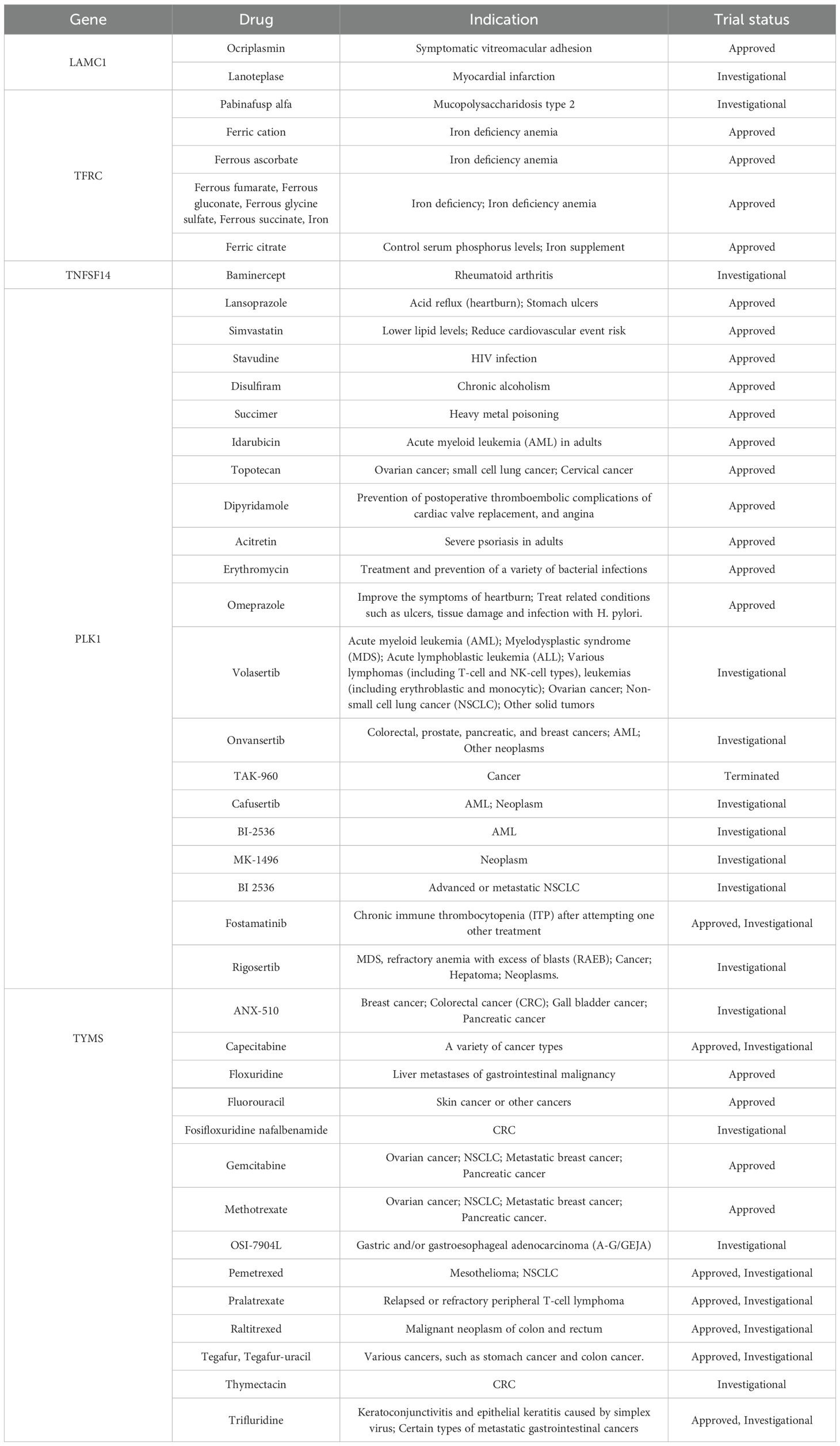
Table 4. Available drugs targeting 6 genes retrieved from DGIdb, OpenTaregts, and DrugBank.
3.7 Expression patterns of 6 targets within the CRC microenvironment
After applying the Harmony algorithm, the cellular distribution within each sample remained largely consistent, indicating the absence of significant batch effects among the samples, making them suitable for downstream analysis (Figure 8A; Supplementary Figure S7). A total of 18 distinct cell clusters were identified using a resolution parameter of 0.4 (Figure 8B). We reanalyzed 175,566 scRNA-seq cells spanning 26 samples, including primary tumor, adjacent normal tissues and PBMC samples. Based on classical marker genes, we successfully classified various cell populations, including epithelial cells, B cells, plasma cells, T/NK cells, myeloid cells, fibroblasts, myofibroblasts and endothelial cells (Figure 8C). The marker genes corresponding to each cell type displayed distinct expression patterns, reinforcing the accuracy of our cell annotation (Figures 8D, E).
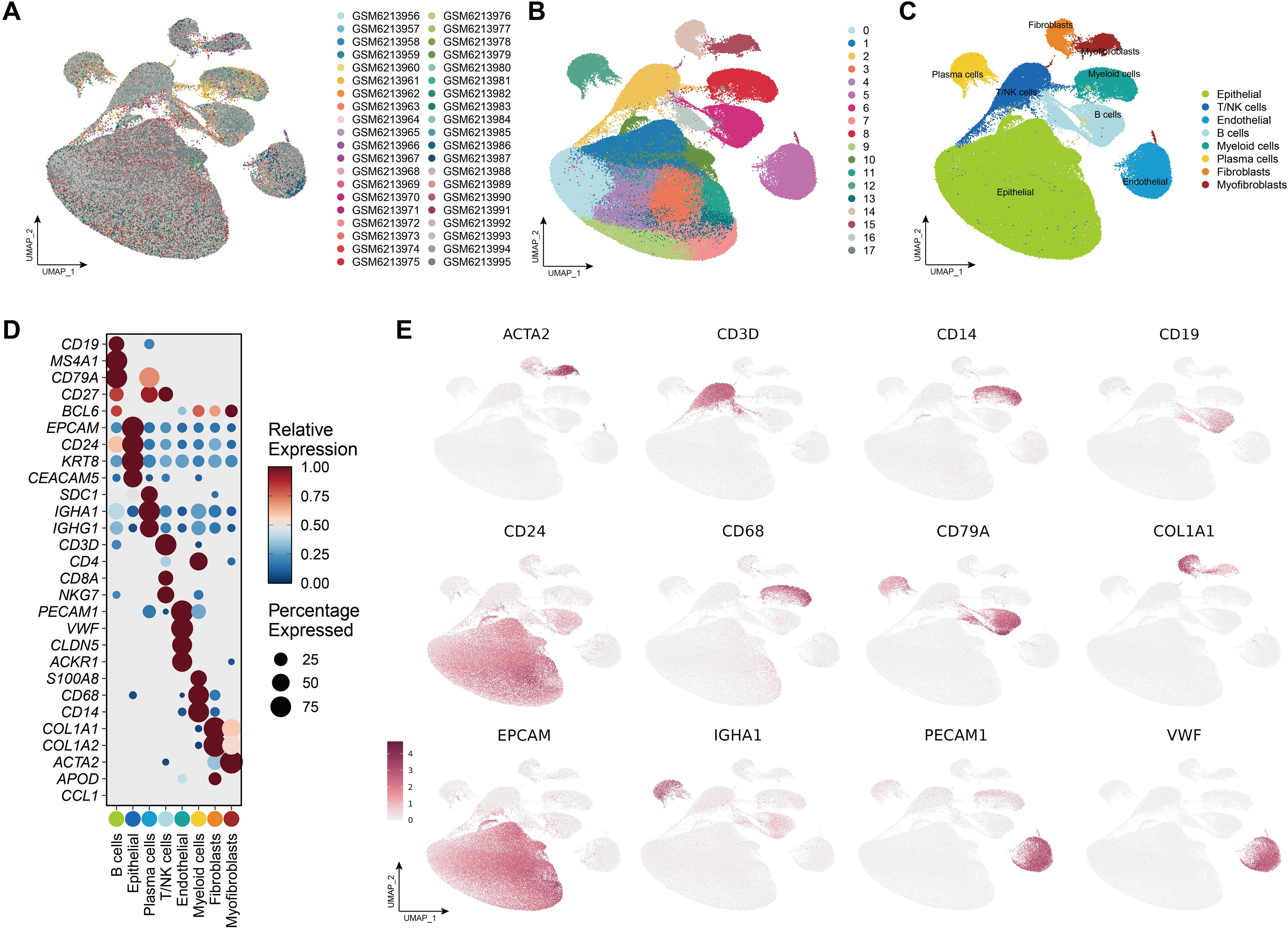
Figure 8. Single-cell landscape of the CRC tumor microenvironment. (A) UMAP plot showing the distribution of all samples included in the single-cell RNA-seq analysis. (B) Clustering of cells into distinct transcriptional states using unsupervised methods. (C) Annotation of major cell types, including Epithelial cells, T/NK cells (T cells and natural killer cells), Endothelial cells, B cells, Myeloid cells (monocytes, macrophages, dendritic cells), Plasma cells, Fibroblasts, and Myofibroblasts. (D) Dot plot showing the expression of canonical marker genes across annotated cell populations. Dot size represents the percentage of cells expressing each gene, while color reflects scaled average expression. (E) Feature plots visualizing the spatial expression patterns of selected marker genes across the UMAP space.
Detailed information on the expression levels of the 6 candidate genes is presented, along with information regarding the different expression patterns within tumor and normal samples (Figures 9A–F). The expression levels of TFRC, LAMC1, PLK1, TYMS, and TSSK6 were significantly higher in tumor tissues while TNFSF14 exhibited higher expression levels in normal tissues. Notably, LAMC1 showed high expression levels in endothelial cells, fibroblasts and myofibroblasts, and TFRC in epithelial cells. The correlation analysis of the expression levels of target genes with key T cell exhaustion markers (PD-1, PD-L1, TIM-3) and the immunosuppressive cytokine IL-10 demonstrated that TNFSF14, LAMC1, and TYMS were significantly and positively associated with these immune markers (Supplementary Figure S8).
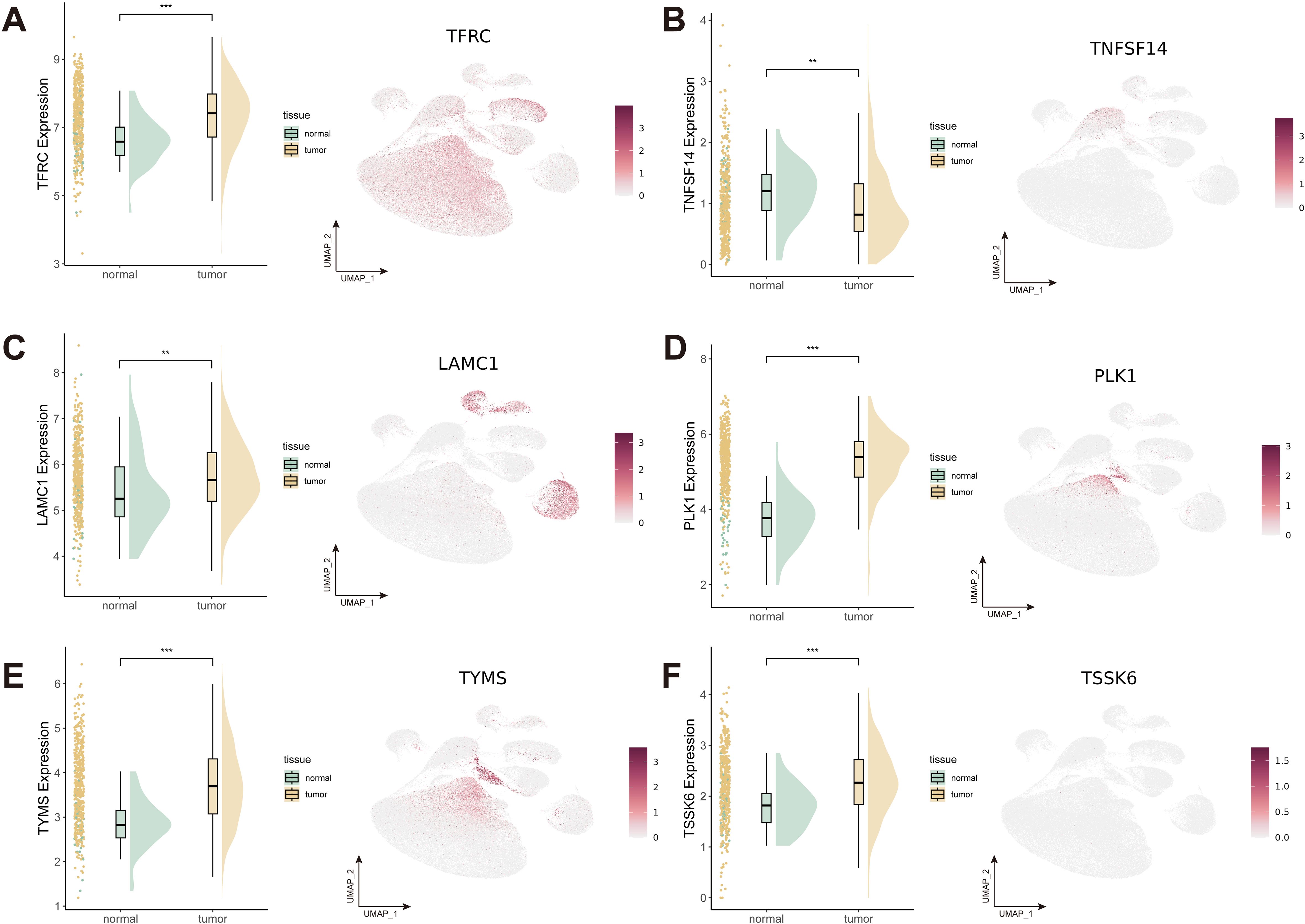
Figure 9. Expression patterns of 6 candidate genes with strong colocalization evidence. (A-F) Expression patterns of TFRC (A), TNFSF14 (B), LAMC1 (C), PLK1 (D), TYMS (E), and TSSK6 (F). **P < 0.01; ***P < 0.001.
3.8 Validation of expression patterns of 6 candidate genes
We performed RT-qPCR to validate the results of our previous analyses, where consistent expression trends were observed (Figure 10A). Specifically, TFRC, PLK1, TYMS, and TSSK6 were remarkably upregulated in tumor tissues compared to normal controls (P < 0.0001). Similarly, LAMC1 showed a significant increase (P < 0.05), while TNFSF14 was significantly downregulated in tumor tissues (P < 0.0001). Consistent with these findings, the IHC staining results of the five targets demonstrated similar trends at the protein level (Figure 10B). Strong immunoreactivity of TFRC, LAMC1, PLK1, and TYMS was observed in tumor tissues, whereas weak or no staining was observed in normal tissues. TNFSF14 showed similar staining intensities in tumor and normal tissues.

Figure 10. Experimental validation of gene expression by RT-qPCR and IHC. (A) RT-qPCR quantification of gene expression (TNFSF14, LAMC1, PLK1, TYMS, and TSSK6) in paired tumor and adjacent normal tissues from 31 CRC patients. Data presented as mean ± SD; significance determined by paired t-test (P < 0.05). (B) IHC images showing protein expression patterns. *P < 0.05; ****P < 0.0001.
4 Discussion
CRC remains a major global health challenge, with high morbidity and mortality despite advancements in screening and treatment strategies (1). Current treatment modalities, including chemotherapy, targeted therapy, and immunotherapy, have improved patient outcomes but are often limited by drug resistance and adverse effects (7). As precision oncology continues to evolve, the identification of novel drug targets is crucial for the development of more effective therapies (48, 49). Our study utilized a comprehensive approach combining MR, colocalization analysis, bulk and single-cell transcriptomics to systematically identify and validate potential druggable genes in CRC. We identified 6 candidate genes (TFRC, TNFSF14, LAMC1, PLK1, TYMS, and TSSK6) with strong evidence supporting their role in CRC pathogenesis and druggability. Additionally, through drug-gene interaction analysis using OpenTargets and DrugBank, we identified several existing drugs that may be repurposed for CRC treatment. These findings provide valuable insights into the genetic basis of CRC and highlight promising therapeutic opportunities.
LAMC1 (Laminin Subunit Gamma 1) is a key component of the extracellular matrix (ECM) and plays a crucial role in tumor progression, invasion, and metastasis by modulating cell adhesion, migration, and epithelial-to-mesenchymal transition (EMT) (50). Studies suggest that LAMC1 overexpression in CRC enhances tumor cell survival and chemoresistance by activating integrin/FAK signaling pathways. Given its role in ECM remodeling, targeting LAMC1 has the potential to disrupt malignancy-stromal interactions and prevent CRC metastasis.
TFRC (Transferrin Receptor) is essential for iron uptake and is highly expressed in rapidly proliferating tumor cells. Its overexpression in CRC promotes tumor growth by increasing intracellular iron levels, leading to enhanced DNA synthesis and oxidative metabolism (51–53). Iron metabolism dysregulation is a hallmark of cancer, and TFRC-targeting therapies, such as TfR1 antibodies or ferroptosis-inducing agents, have been explored in preclinical models.
TNFSF14, also known as LIGHT, is a member of the tumor necrosis factor (TNF) superfamily that plays a pivotal role in modulating anti-tumor immune responses. LIGHT engages two key receptors, HVEM (Herpesvirus Entry Mediator) and LTβR (Lymphotoxin Beta Receptor), to stimulate T cell proliferation, activation, and recruitment within the tumor microenvironment (TME) (54). Mechanistically, LIGHT–HVEM signaling has been shown to enhance T cell receptor–mediated activation, increase IFN-γ secretion, and support the effector function and persistence of cytotoxic CD8+ T cells (55). LIGHT–LTβR engagement promotes the formation of tertiary lymphoid structures (TLSs), improves vascular normalization, and enhances the infiltration of immune cells into the tumor core, thereby fostering a more permissive and immunologically active TME (56). In CRC, elevated LIGHT expression has been associated with enhanced infiltration of cytotoxic CD8+ T cells and a more favorable immune phenotype (57–59). Notably, TNFSF14 expression was significantly downregulated in tumor tissues compared to adjacent normal tissues, suggesting that CRC may suppress this pathway to evade immune surveillance. Given its role in activating lymphocytes, this pattern supports the immunostimulatory function of LIGHT and underscores its therapeutic potential in restoring anti-tumor immunity. However, the function of LIGHT can be context-dependent and subject to modulation by immunosuppressive signals within the TME. Specifically, TGF-β, PD-1/PD-L1, and CTLA-4 pathways may attenuate LIGHT-mediated immune activation by suppressing effector T cell responses or promoting regulatory T cell (Treg) expansion. Emerging evidence suggests that co-targeting LIGHT and immune checkpoints could synergistically overcome tumor-induced immune evasion (60). For instance, combination therapies using LIGHT agonists with anti-PD-1 or anti-CTLA-4 antibodies have shown enhanced tumor clearance in preclinical models by reinvigorating exhausted T cells and promoting durable anti-tumor immunity (61, 62). Moreover, mRNA-based approaches delivering TNFSF14 directly into dendritic cells or effector T cells may further amplify its immunostimulatory effect and help reshape an immune “cold” tumor into a “hot” one, rendering CRC more responsive to immune checkpoint blockade (62). These findings suggest that LIGHT may act as a key immunologic amplifier, whose effects are shaped by the broader signaling landscape of the TME, and that it holds promise as a combinatorial target for precision immunotherapy in CRC.
PLK1 is a serine/threonine kinase that plays a pivotal role in regulating mitotic progression (63). Dysregulation of PLK1 is highly frequent in various malignancies and is correlated with poor prognosis in many cancers (64). Previous studies have shown that PLK1 is overexpressed in CRC tumors compared with normal tissue and which is correlated with disease progression, including adverse invasion, metastasis, and prognosis (65–67). Notably, PLK1 contributes to the DNA damage response (DDR) and mitotic checkpoint activation, allowing CRC cells to survival despite of genotoxic stress. Therapeutically, PLK1 inhibitors such as Volasertib (BI 6727) and Onvansertib (NMS-P937) have demonstrated potential in preclinical and clinical studies (68–70). Onvansertib, in particular, is being investigated for its efficacy in KRAS-mutant CRC, where it enhances the response to chemotherapy (70). Additionally, CRISPR/Cas9-mediated PLK1 knockout or RNA interference (RNAi) approaches could further suppress tumor growth and increase sensitivity to DNA-damaging agents, providing a potential avenue for combination therapies.
TYMS encodes thymidylate synthase, a crucial enzyme involved in DNA synthesis and repair. It catalyzes the conversion of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP), which is essential for nucleotide biosynthesis. Overexpression of TYMS has been identified as a major mechanism of resistance to 5-fluorouracil (5-FU)-based chemotherapy in CRC, reducing treatment efficacy (71, 72). Additionally, high TYMS levels can activate the mTOR signaling pathway, further promoting tumor proliferation. Further, Martinez-Balibrea et al. found TYMS polymorphisms influence on tumor response and toxicities derived from irinotecan plus 5-fluorouracil treatment in CRC patients, and proposed a genetic-based algorithm to optimize treatment individualization (73). To overcome TYMS-related resistance, targeted TYMS inhibitors such as Raltitrexed have been developed as alternatives to 5-FU (74). Moreover, CRISPR/Cas9-mediated TYMS knockout has been proposed as a strategy to increase tumor susceptibility to fluoropyrimidine-based treatments (75). Another promising approach is the use of microRNA (miRNA) therapeutics, such as miR-192 and miR-215, which have been shown to downregulate TYMS expression and restore chemosensitivity in CRC cells (76, 77).
Our findings also uncovered TSSK as a novel and promising therapeutic target in CRC, which appear to contribute to chemoresistance. TSSK6 (Testis-Specific Serine/Threonine Kinase 6) is primarily expressed in the reproductive system but has been detected in various cancers, including CRC. Although its role in CRC remains poorly understood, emerging evidence suggests that TSSK6 may contribute to tumor progression through cell cycle regulation and chemoresistance (78). Inhibiting TSSK6 could provide a novel approach to sensitizing CRC cells to chemotherapy. Future studies should explore CRISPR-mediated TSSK6 deletion to evaluate its impact on tumor growth and drug resistance.
In comparison with previous CRC biomarker discovery studies, our integrative approach offers several distinct advantages. Traditional omics-based screens have often focused on differential gene expression or somatic mutation profiling alone, which, while informative, may not establish causal relationships between gene function and CRC pathogenesis. For example, large-scale transcriptomic analyses such as The Cancer Genome Atlas (TCGA) have identified numerous dysregulated genes in CRC, but many lack functional validation or clinical translatability due to confounding factors and tumor heterogeneity. Similarly, proteomic and epigenetic studies have proposed novel targets, yet few have progressed to actionable therapies due to challenges in target prioritization. In contrast, our study leverages a MR framework combined with colocalization analysis to infer causal gene-disease relationships, reducing confounding and increasing biological plausibility. Furthermore, by integrating bulk and single-cell RNA-seq data, we not only identify gene targets but also precisely map their expression across distinct cellular compartments in the tumor microenvironment. This spatial resolution is often absent in previous screens and enables more accurate prediction of therapeutic relevance, target accessibility, and potential resistance mechanisms. Additionally, our use of PheWAS to evaluate off-target effects provides a safety-centric perspective rarely addressed in omics studies. Together, these strengths highlight the novelty and translational relevance of our prioritized targets, which exhibit strong genetic associations, therapeutic tractability, and distinct expression patterns. These features not only distinguish them from previously reported candidates but also suggest their potential utility as biomarkers for patient stratification in future clinical trials, enabling more precise selection of individuals likely to benefit from targeted or immunotherapeutic interventions.
Despite the strengths of our integrative framework, several important limitations must be acknowledged. Specifically, both the eQTL and GWAS datasets employed in this study were derived entirely from individuals of European ancestry. This lack of population diversity may restrict the transferability of our findings to non-European populations, as allele frequencies, LD structures, and gene-environment interactions can differ substantially across ethnic groups. Therefore, future studies incorporating multi-ethnic cohorts, including Asian, African, and admixed populations, are essential to validate the identified targets and ensure their generalizability and clinical relevance in a globally diverse setting. Second, we focused on cis-eQTLs, which, while minimizing pleiotropy, restricted the detection of trans-regulatory effects and precluded bidirectional MR analyses, such as those assessing CRC-related gene expression changes. Third, transcriptomic data do not fully capture protein-level dynamics, as post-transcriptional regulation and protein degradation can affect gene function (79). Further validation using proteomics and CRISPR-based functional studies is essential to confirm the biological relevance of our targets. Fourth, while we identified several promising druggable genes and matched them to known compounds, preclinical validation, such as in vivo efficacy, toxicity assessment, and combination therapy testing, is necessary to advance these findings toward clinical application. Additionally, while we observed the expression of candidate genes in broader immune and stromal compartments, a more detailed investigation into their enrichment within specific immunosuppressive populations was beyond the scope of this work. Future studies incorporating higher-resolution cell-type classification and functional validation will be important to further elucidate the roles of these genes in immune evasion. Finally, while our analysis identified druggable targets in CRC, we were unable to stratify samples by key clinical or molecular subtypes such as MSI status or consensus molecular subtypes (CMS), due to the lack of subtype annotation in the available dataset. Given the well-established molecular and immunological heterogeneity of CRC, such stratification may significantly influence the expression and therapeutic relevance of candidate genes. Future studies incorporating subtype-specific validation will be essential to refine the clinical utility of our findings and to enable more precise immunotherapeutic strategies tailored to distinct CRC subpopulations.
5 Conclusion
This integrative genomic and single-cell framework identified six high-confidence druggable genes (LAMC1, TFRC, TNFSF14, PLK1, TYMS, and TSSK6) with potential therapeutic relevance in CRC. By combining MR, colocalization, transcriptomics, and drug-gene interaction analysis, we systematically prioritized targets with strong genetic evidence, cell-specific expression, and minimal predicted off-target effects. Several of these genes are already linked to approved or investigational compounds, offering opportunities for drug repurposing and precision treatment strategies. While further functional and preclinical validation is needed, our findings provide a robust foundation for advancing targeted therapies in CRC and highlight the utility of integrative omics approaches in oncology drug discovery.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Ethics Committee of Renji Hospital, Shanghai Jiao Tong University School of Medicine (Approval No.: KY2024-188-C). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
YH: Conceptualization, Data curation, Software, Writing – original draft, Writing – review & editing. JL: Conceptualization, Data curation, Funding acquisition, Software, Writing – original draft, Writing – review & editing. NX: Data curation, Writing – original draft. WS: Data curation, Writing – original draft. FC: Data curation, Writing – original draft. YM: Writing – review & editing. HG: Conceptualization, Data curation, Project administration, Writing – review & editing. QL: Conceptualization, Data curation, Project administration, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The authors declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National College Students Innovation and Entrepreneurship Training Program, China (No. 202410343023), and the Zhejiang University Student Science and Technology Innovation Activity Plan, China (No. 2023R413025).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1604154/full#supplementary-material
References
1. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74:229–63. doi: 10.3322/caac.21834
2. Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, and Wallace MB. Colorectal cancer. Lancet. (2019) 394:1467–80. doi: 10.1016/S0140-6736(19)32319-0
3. Fearon ER and Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. (1990) 61:759–67. doi: 10.1016/0092-8674(90)90186-I
4. Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. (2015) 21:1350–6. doi: 10.1038/nm.3967
5. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. (2012) 487:330–7. doi: 10.1038/nature11252
6. Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. (2020) 70:145–64. doi: 10.3322/caac.21601
7. Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. (2016) 27:1386–422. doi: 10.1093/annonc/mdw235
8. Saltz LB, Cox JV, Blanke C, Rosen LS, Fehrenbacher L, Moore MJ, et al. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group N Engl J Med. (2000) 343:905–14. doi: 10.1056/NEJM200009283431302
9. Okines AFC, Norman AR, McCloud P, Kang YK, and Cunningham D. Meta-analysis of the REAL-2 and ML17032 trials: evaluating capecitabine-based combination chemotherapy and infused 5-fluorouracil-based combination chemotherapy for the treatment of advanced oesophago-gastric cancer. Ann Oncol. (2009) 20:1529–34. doi: 10.1093/annonc/mdp047
10. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. (2004) 350:2335–42. doi: 10.1056/NEJMoa032691
11. Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. (2009) 27:663–71. doi: 10.1200/JCO.2008.20.8397
12. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
13. Yang C, Geng H, Yang X, Ji S, Liu Z, Feng H, et al. Targeting the immune privilege of tumor-initiating cells to enhance cancer immunotherapy. Cancer Cell. (2024) 42:2064–81 e19. doi: 10.1016/j.ccell.2024.10.008
14. Siena S, Raghav K, Masuishi T, Yamaguchi K, Nishina T, Elez E, et al. HER2-related biomarkers predict clinical outcomes with trastuzumab deruxtecan treatment in patients with HER2-expressing metastatic colorectal cancer: biomarker analyses of DESTINY-CRC01. Nat Commun. (2024) 15:10213. doi: 10.1038/s41467-024-53223-3
15. Jhaveri K, Eli LD, Wildiers H, Hurvitz SA, Guerrero-Zotano A, Unni N, et al. Neratinib + fulvestrant + trastuzumab for HR-positive, HER2-negative, HER2-mutant metastatic breast cancer: outcomes and biomarker analysis from the SUMMIT trial. Ann Oncol. (2023) 34:885–98. doi: 10.1016/j.annonc.2023.08.003
16. Li Q, Geng S, Luo H, Wang W, Mo YQ, Luo Q, et al. Signaling pathways involved in colorectal cancer: pathogenesis and targeted therapy. Signal Transduct Target Ther. (2024) 9:266. doi: 10.1038/s41392-024-01953-7
17. Zhai J, Chen H, Wong CC, Peng Y, Gou H, Zhang J, et al. ALKBH5 drives immune suppression via targeting AXIN2 to promote colorectal cancer and is a target for boosting immunotherapy. Gastroenterology. (2023) 165:445–62. doi: 10.1053/j.gastro.2023.04.032
18. Smith-Byrne K, Hedman A, Dimitriou M, Desai T, Sokolov AV, Schioth HB, et al. Identifying therapeutic targets for cancer among 2074 circulating proteins and risk of nine cancers. Nat Commun. (2024) 15:3621. doi: 10.1038/s41467-024-46834-3
19. Zhang X, Yu W, Li Y, Wang A, Cao H, and Fu Y. Drug development advances in human genetics-based targets. MedComm (2020). (2024) 5:e481. doi: 10.1002/mco2.v5.2
20. Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet. (2015) 47:856–60. doi: 10.1038/ng.3314
21. Finan C, Gaulton A, Kruger FA, Lumbers RT, Shah T, Engmann J, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med. (2017) 9:eaag1166. doi: 10.1126/scitranslmed.aag1166
22. Freshour SL, Kiwala S, Cotto KC, Coffman AC, McMichael JF, Song JJ, et al. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. (2021) 49:D1144–d51. doi: 10.1093/nar/gkaa1084
23. Võsa U, Claringbould A, Westra HJ, Bonder MJ, Deelen P, Zeng B, et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet. (2021) 53:1300–10. doi: 10.1038/s41588-021-00913-z
24. Fernandez-Rozadilla C, Timofeeva M, Chen Z, Law P, Thomas M, Schmit S, et al. Deciphering colorectal cancer genetics through multi-omic analysis of 100,204 cases and 154,587 controls of European and east Asian ancestries. Nat Genet. (2023) 55:89–99. doi: 10.1038/s41588-022-01222-9
25. Sakaue S, Kanai M, Tanigawa Y, Karjalainen J, Kurki M, Koshiba S, et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet. (2021) 53:1415–24. doi: 10.1038/s41588-021-00931-x
26. Kurki MI, Karjalainen J, Palta P, Sipila TP, Kristiansson K, Donner KM, et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. (2023) 613:508–18. doi: 10.1038/s41586-022-05473-8
27. Rashkin SR, Graff RE, Kachuri L, Thai KK, Alexeeff SE, Blatchins MA, et al. Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat Commun. (2020) 11:4423. doi: 10.1038/s41467-020-18246-6
28. Risch N and Merikangas K. The future of genetic studies of complex human diseases. Science. (1996) 273:1516–7. doi: 10.1126/science.273.5281.1516
29. Gkatzionis A, Burgess S, and Newcombe PJ. Statistical methods for cis-Mendelian randomization with two-sample summary-level data. Genet Epidemiol. (2023) 47:3–25. doi: 10.1002/gepi.22506
30. Pierce BL, Ahsan H, and Vanderweele TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol. (2011) 40:740–52. doi: 10.1093/ije/dyq151
31. Hong Y, Wang Y, and Shu W. Deciphering the genetic underpinnings of neuroticism: A Mendelian randomization study of druggable gene targets. J Affect Disord. (2025) 370:147–58. doi: 10.1016/j.jad.2024.11.002
32. Burgess S, Davey Smith G, Davies NM, Dudbridge F, Gill D, Glymour MM, et al. Guidelines for performing Mendelian randomization investigations: update for summer 2023. Wellcome Open Res. (2019) 4:186. doi: 10.12688/wellcomeopenres
33. Hong Y, Shu W, Jiang X, Wang Y, Chen R, Yang Q, et al. Prioritizing endocrine-disrupting chemicals targeting systemic lupus erythematosus genes via Mendelian randomization and colocalization analyses. Ecotoxicol Environ Saf. (2025) 295:118126. doi: 10.1016/j.ecoenv.2025.118126
34. Bowden J, Davey Smith G, and Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. (2015) 44:512–25. doi: 10.1093/ije/dyv080
35. Greco MF, Minelli C, Sheehan NA, and Thompson JR. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat Med. (2015) 34:2926–40. doi: 10.1002/sim.v34.21
36. Yu G, Wang LG, Han Y, and He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. (2012) 16:284–7. doi: 10.1089/omi.2011.0118
37. Gene Ontology Consortium: going forward. Nucleic Acids Res. (2015) 43:D1049–56. doi: 10.1093/nar/gku1179
38. Kanehisa M and Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. (2000) 28:27–30. doi: 10.1093/nar/28.1.27
39. Giambartolomei C, Vukcevic D, SChadt EE, Franke L, Hingorani AD, Wallace C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PloS Genet. (2014) 10:e1004383. doi: 10.1371/journal.pgen.1004383
40. Ying H, Wu X, Jia X, Yang Q, Liu H, Zhao H, et al. Single-cell transcriptome-wide Mendelian randomization and colocalization reveals immune-mediated regulatory mechanisms and drug targets for COVID-19. EBioMedicine. (2025) 113:105596. doi: 10.1016/j.ebiom.2025.105596
41. Wu X, Ying H, Yang Q, Yang Q, Liu H, Ding Y, et al. Transcriptome-wide Mendelian randomization during CD4(+) T cell activation reveals immune-related drug targets for cardiometabolic diseases. Nat Commun. (2024) 15:9302. doi: 10.1038/s41467-024-53621-7
42. Codd V, Wang Q, Allara E, Musicha C, Kaptoge S, Stoma S, et al. Polygenic basis and biomedical consequences of telomere length variation. Nat Genet. (2021) 53:1425–33. doi: 10.1038/s41588-021-00944-6
43. Wang Q, Dhindsa RS, Carss K, Harper AR, Nag A, Tachmazidou I, et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature. (2021) 597:527–32. doi: 10.1038/s41586-021-03855-y
44. Ochoa D, Hercules A, Carmona M, Suveges D, Gonzalez-Uriarte A, Malangone C, et al. Open Targets Platform: supporting systematic drug-target identification and prioritisation. Nucleic Acids Res. (2021) 49:D1302–D10. doi: 10.1093/nar/gkaa1027
45. Knox C, Wilson M, Klinger CM, Franklin M, Oler E, Wilson A, et al. DrugBank 6.0: the drugBank knowledgebase for 2024. Nucleic Acids Res. (2024) 52:D1265–D75. doi: 10.1093/nar/gkad976
46. Satija R, Farrell JA, Gennert D, Schier AF, and Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. (2015) 33:495–502. doi: 10.1038/nbt.3192
47. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. (2009) 55:611–22. doi: 10.1373/clinchem.2008.112797
48. Yang C, Guo Y, Qian R, Huang Y, Zhang L, Wang J, et al. Mapping the landscape of synthetic lethal interactions in liver cancer. Theranostics. (2021) 11:9038–53. doi: 10.7150/thno.63416
49. Yang C, Zhang S, Cheng Z, Liu Z, Zhang L, Jiang K, et al. Multi-region sequencing with spatial information enables accurate heterogeneity estimation and risk stratification in liver cancer. Genome Med. (2022) 14:142. doi: 10.1186/s13073-022-01143-6
50. Whiffin N, Hosking FJ, Farrington SM, Palles C, Dobbins SE, Zgaga L, et al. Identification of susceptibility loci for colorectal cancer in a genome-wide meta-analysis. Hum Mol Genet. (2014) 23:4729–37. doi: 10.1093/hmg/ddu177
51. Wang X, Zhou Y, Ning L, Chen J, Chen H, and Li X. Knockdown of ANXA10 induces ferroptosis by inhibiting autophagy-mediated TFRC degradation in colorectal cancer. Cell Death Dis. (2023) 14:588. doi: 10.1038/s41419-023-06114-2
52. Liu TY, Hu CC, Han CY, Mao SY, Zhang WX, Xu YM, et al. IGF2BP2 promotes colorectal cancer progression by upregulating the expression of TFRC and enhancing iron metabolism. Biol Direct. (2023) 18:19. doi: 10.1186/s13062-023-00373-x
53. Hwang J, Park A, Kim C, Kim CG, Kwak J, Kim B, et al. Inhibition of IRP2-dependent reprogramming of iron metabolism suppresses tumor growth in colorectal cancer. Cell Commun Signal. (2024) 22:412. doi: 10.1186/s12964-024-01769-6
54. Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, et al. Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol. (2004) 5:141–9. doi: 10.1038/ni1029
55. Seo GY, Shui JW, Takahashi D, Song C, Wang Q, Kim K, et al. LIGHT-HVEM signaling in innate lymphoid cell subsets protects against enteric bacterial infection. Cell Host Microbe. (2018) 24:249–60 e4. doi: 10.1016/j.chom.2018.07.008
56. Johansson-Percival A and Ganss R. Therapeutic induction of tertiary lymphoid structures in cancer through stromal remodeling. Front Immunol. (2021) 12:674375. doi: 10.3389/fimmu.2021.674375
57. Qiao G, Qin J, Kunda N, Calata JF, Mahmud DL, Gann P, et al. LIGHT elevation enhances immune eradication of colon cancer metastases. Cancer Res. (2017) 77:1880–91. doi: 10.1158/0008-5472.CAN-16-1655
58. Qiao G, Kone LB, Phillips EH, Lee SS, Brown GE, Khetani SR, et al. LIGHT enhanced bispecific antibody armed T-cells to treat immunotherapy resistant colon cancer. Oncogene. (2022) 41:2054–68. doi: 10.1038/s41388-022-02209-w
59. Maker AV, Ito H, Mo Q, Weisenberg E, Qin LX, Turcotte S, et al. Genetic evidence that intratumoral T-cell proliferation and activation are associated with recurrence and survival in patients with resected colorectal liver metastases. Cancer Immunol Res. (2015) 3:380–8. doi: 10.1158/2326-6066.CIR-14-0212
60. Shuptrine CW, Perez VM, Selitsky SR, Schreiber TH, and Fromm G. Shining a LIGHT on myeloid cell targeted immunotherapy. Eur J Cancer. (2023) 187:147–60. doi: 10.1016/j.ejca.2023.03.040
61. Yoo KJ, Johannes K, Gonzalez LE, Patel A, Shuptrine CW, Opheim Z, et al. LIGHT (TNFSF14) costimulation enhances myeloid cell activation and antitumor immunity in the setting of PD-1/PD-L1 and TIGIT checkpoint blockade. J Immunol. (2022) 209:510–25. doi: 10.4049/jimmunol.2101175
62. Khosravi GR, Mostafavi S, Bastan S, Ebrahimi N, Gharibvand RS, and Eskandari N. Immunologic tumor microenvironment modulators for turning cold tumors hot. Cancer Commun (Lond). (2024) 44:521–53. doi: 10.1002/cac2.12539
63. Combes G, Alharbi I, Braga LG, and Elowe S. Playing polo during mitosis: PLK1 takes the lead. Oncogene. (2017) 36:4819–27. doi: 10.1038/onc.2017.113
64. Knecht R, Elez R, Oechler M, Solbach C, von Ilberg C, and Strebhardt K. Prognostic significance of polo-like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Res. (1999) 59:2794–7.
65. Takahashi T, Sano B, Nagata T, Kato H, Sugiyama Y, Kunieda K, et al. Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Sci. (2003) 94:148–52. doi: 10.1111/j.1349-7006.2003.tb01411.x
66. Ran Z, Chen W, Shang J, Li X, Nie Z, Yang J, et al. Clinicopathological and prognostic implications of polo-like kinase 1 expression in colorectal cancer: A systematic review and meta-analysis. Gene. (2019) 721:144097. doi: 10.1016/j.gene.2019.144097
67. Han DP, Zhu QL, Cui JT, Wang PX, Qu S, Cao QF, et al. Polo-like kinase 1 is overexpressed in colorectal cancer and participates in the migration and invasion of colorectal cancer cells. Med Sci Monit. (2012) 18:BR237–46. doi: 10.12659/MSM.882900
68. Van den Bossche J, Lardon F, Deschoolmeester V, De Pauw I, Vermorken JB, Specenier P, et al. Spotlight on volasertib: preclinical and clinical evaluation of a promising plk1 inhibitor. Med Res Rev. (2016) 36:749–86. doi: 10.1002/med.2016.36.issue-4
69. Gjertsen BT and Schoffski P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia. (2015) 29:11–9. doi: 10.1038/leu.2014.222
70. Ahn DH, Ridinger M, Cannon TL, Mendelsohn L, Starr JS, Hubbard JM, et al. Onvansertib in combination with chemotherapy and bevacizumab in second-line treatment of KRAS-mutant metastatic colorectal cancer: A single-arm, phase II trial. J Clin Oncol. (2025) 43:840–51. doi: 10.1200/JCO-24-01266
71. Johnston PG, Lenz HJ, Leichman CG, Danenberg KD, Allegra CJ, Danenberg PV, et al. Thymidylate synthase gene and protein expression correlate and are associated with response to 5-fluorouracil in human colorectal and gastric tumors. Cancer Res. (1995) 55:1407–12.
72. Gonen M, Hummer A, Zervoudakis A, Sullivan D, Fong Y, Banerjee D, et al. Thymidylate synthase expression in hepatic tumors is a predictor of survival and progression in patients with resectable metastatic colorectal cancer. J Clin Oncol. (2003) 21:406–12. doi: 10.1200/JCO.2003.06.060
73. Martinez-Balibrea E, Abad A, Martinez-Cardus A, Gines A, Valladares M, Navarro M, et al. UGT1A and TYMS genetic variants predict toxicity and response of colorectal cancer patients treated with first-line irinotecan and fluorouracil combination therapy. Br J Cancer. (2010) 103:581–9. doi: 10.1038/sj.bjc.6605776
74. Van Cutsem E, Cunningham D, Maroun J, Cervantes A, and Glimelius B. Raltitrexed: current clinical status and future directions. Ann Oncol. (2002) 13:513–22. doi: 10.1093/annonc/mdf054
75. Liu T, Han Y, Yu C, Ji Y, Wang C, Chen X, et al. MYC predetermines the sensitivity of gastrointestinal cancer to antifolate drugs through regulating TYMS transcription. EBioMedicine. (2019) 48:289–300. doi: 10.1016/j.ebiom.2019.10.003
76. Valenzuela G, Contreras HR, Marcelain K, Burotto M, and Gonzalez-Montero J. Understanding microRNA-mediated chemoresistance in colorectal cancer treatment. Int J Mol Sci. (2025) 26:1168. doi: 10.3390/ijms26031168
77. Elrebehy MA, Al-Saeed S, Gamal S, El-Sayed A, Ahmed AA, Waheed O, et al. miRNAs as cornerstones in colorectal cancer pathogenesis and resistance to therapy: A spotlight on signaling pathways interplay - A review. Int J Biol Macromol. (2022) 214:583–600. doi: 10.1016/j.ijbiomac.2022.06.134
78. Delgado M, Gallegos Z, Stippec S, McGlynn K, Cobb MH, and Whitehurst AW. Testis-specific serine kinase 6 (TSSK6) is abnormally expressed in colorectal cancer and promotes oncogenic behaviors. J Biol Chem. (2024) 300:107380. doi: 10.1016/j.jbc.2024.107380
Keywords: colorectal cancer, genetics, druggable targets, single-cell transcriptomics, precision oncology
Citation: Hong Y, Li J, Xu N, Shu W, Chen F, Mi Y, Geng H and Li Q (2025) Integrative genomic and single-cell framework identifies druggable targets for colorectal cancer precision therapy. Front. Immunol. 16:1604154. doi: 10.3389/fimmu.2025.1604154
Received: 01 April 2025; Accepted: 28 April 2025;
Published: 27 May 2025.
Edited by:
Chen Yang, Southern Medical University, ChinaReviewed by:
Zhicheng Liu, Huazhong University of Science and Technology, ChinaShaozhuo Huang, Erasmus Medical Center, Netherlands
Yan Li, Peter Doherty Institute for Infection and Immunity, Australia
Copyright © 2025 Hong, Li, Xu, Shu, Chen, Mi, Geng and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanggang Hong, c3VuMTYwNDE0QGljbG91ZC5jb20=; Haigang Geng, R2VuZ2hhaWdhbmdAc2p0dS5lZHUuY24=; Qian Li, bGlxaWFuOTkwNzI3QDE2My5jb20=
†These authors have contributed equally to this work