 Johanna Rausch1,2*†
Johanna Rausch1,2*† Stephanie Herold1*†
Stephanie Herold1*† Simone Liebhäuser1Yagmur Bülbül1Edite Antunes Ferreira1Till Wenz3Kevin Jan Legscha1,2
Simone Liebhäuser1Yagmur Bülbül1Edite Antunes Ferreira1Till Wenz3Kevin Jan Legscha1,2 Matthias Bros4Florian Butsch4Oliver Kriege1
Matthias Bros4Florian Butsch4Oliver Kriege1 Klaus Warnatz5,6Miriam Groß5,7
Klaus Warnatz5,6Miriam Groß5,7 Kai Lehmberg8Helena Clara Lichtenfeld8Paul La Rosée9
Kai Lehmberg8Helena Clara Lichtenfeld8Paul La Rosée9 Markus Philipp Radsak1,10
Markus Philipp Radsak1,10 Matthias Theobald1,2,11
Matthias Theobald1,2,11 Hakim Echchannaoui1,2‡
Hakim Echchannaoui1,2‡ Markus Munder1,11,12‡
Markus Munder1,11,12‡- 1Department of Hematology and Medical Oncology, University Medical Center, Johannes Gutenberg University, Mainz, Germany
- 2German Cancer Consortium (DKTK), Partner Site Frankfurt/Mainz, Mainz, Germany
- 3Department of Hematology and Medical Oncology, Johanniter Hospital Bonn, Bonn, Germany
- 4Department of Dermatology, University Medical Center, Johannes Gutenberg University, Mainz, Germany
- 5Center for Chronic Immunodeficiency, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 6Department of Rheumatology and Clinical Immunology, University Medical Center Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 7Institute of Immunodeficiency, University Medical Center Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 8Division of Pediatric Stem Cell Transplantation and Immunology, Clinic for Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 9Department of Internal Medicine II, Schwarzwald-Baar-Klinikum, Villingen-Schwenningen, Germany
- 10Department of Hematology and Oncology, Donau-Isar Hospitals, Deggendorf, Germany
- 11Research Center for Immune Therapy [Forschungszentrum für Immuntherapie (FZI)], University Medical Center (UMC) of the Johannes Gutenberg University, Mainz, Germany
- 12Department of Hematology and Medical Oncology, Diakonissen-Stiftungs-Krankenhaus Speyer, Speyer, Germany
Griscelli syndrome type 2 (GS-2) is a rare congenital immune dysfunction characterized by partial albinism and recurrent episodes of hemophagocytic lymphohistiocytosis (HLH). It is caused by a variant in the gene encoding Rab27a leading to a degranulation defect in melanocytes, natural killer (NK)- and T cells. Prognosis of patients with GS-2 is limited by repetitive episodes of life-threatening HLH with onset in early childhood. The only curative treatment is an allogeneic hematopoietic stem cell transplantation (HSCT). Here, we report on an 18 year old female patient with a homozygous missense p.Arg50Glnfs*35 variant in exon 2 of RAB27A who presented with an exceptionally late onset of severe HLH. Her phenotypically inapparent albinism complicated to correctly diagnose GS-2. Immune function assays confirmed a T- and NK cell degranulation deficiency characteristic for patients with primary HLH, while microscopic hair analysis revealed melanin clumps secondary to melanocyte functional impairment. To understand why disease onset occurred unusually late in this patient, we investigated the patient’s T cell and polymorphonuclear neutrophil (PMN) function in more detail. We could show that intracellular granzyme B storage in cytotoxic T cells was increased compared to healthy donors and that the patient’s T cells maintained some degranulation activity. Both, antigen-specific cytotoxic response and proliferation capacity of the patient’s T cells were preserved. We demonstrate for the first time that also PMN degranulation, assessed as stimulation-induced CD66b and CD11b cell membrane expression, is dysfunctional in patients with Rab27a deficiency-associated primary HLH. The patient was treated with steroids and cyclosporine A for immunosuppression to control the HLH. After two severe episodes within only a few months, she eventually received an allogeneic HSCT and has not experienced further HLH episodes for now more than 3 years after the HSCT procedure. This case should raise awareness for the possibility of initial manifestation of primary, genetically-determined HLH even in adult patients.
1 Introduction
GS-2 is an inherited autosomal recessive immune disorder characterized by primary HLH and partial albinism usually with onset in early childhood (1, 2). It results from a variant in the gene encoding Rab27a, a protein of the GTPase family involved in vesicular transport and organelle dynamics. Rab27a is highly expressed in melanocytes and leukocytes (3). In melanocytes, Rab27a is involved in melanosome transport (4) explaining the partial albinism in case of loss-of-function variants, though rare cases without apparent albinism have been described (5–8). In cytotoxic T lymphocytes (CTLs), NK- and mast cells, Rab27a plays an important role in the secretion of cytolytic granules by interacting with the priming factor Munc13-4 (2, 3, 9) which is a member of the Unc13 protein family encoded by the gene UNC13D (Unc-13 Homolog D) (10). Variants in either protein disable the release of lytic granules at the immunologic synapse causing reduced cytotoxicity (11, 12). This leads to an insufficient elimination of infectious triggers with persistent immune stimulation with secondary systemic hyperinflammation and cytokine storm presenting as HLH (13, 14).
HLH is characterized as a syndrome of fever, splenomegaly and cytopenia sustained by a dysregulated proliferation and activation of T cells and macrophages (15, 16). Diagnostic markers are severely increased levels of serum ferritin and soluble interleukin-2 receptor (sIL2R), hypertriglyceridemia, hypofibrinogenemia, hemophagocytosis in the bone marrow and low NK cell degranulation (17). Additionally, inflammatory cytokines such as interferon-gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), interleukin 1 (IL-1), IL-2, IL-6, IL-10, IL-12, IL-16 and macrophage colony stimulating factor (M-CSF) are hyper-secreted (18–22).
Primary HLH is associated with several inheritable gene defects, most prominently variants in PRF1 (perforin 1), UNC13D, STX11 (Syntaxin 11), MUNC18-2 (also called STXBP2, Syntaxin Binding Protein 2) or RAB27A (13, 15, 16). All these variants interfere with proper immunity and provoke repetitive episodes of HLH, frequently with fatal outcome. Besides primary HLH, there are many known triggers causing secondary HLH independent of known monogenetic predisposition such as infections (e.g. Epstein-Barr virus, EBV), rheumatological disorders (e.g. Still’s disease or systemic lupus erythematosus), malignant disorders (e.g. lymphomas) or immune modifying therapy (e.g. stem cell transplantation or checkpoint inhibitors) (13, 16). First line treatment for patients with HLH is guided by the HLH-1994 protocol (15, 16) with modifications or additions, however HSCT remains the only curative therapy for patients with primary HLH (15, 23).
2 Case description
An 18-year-old woman presented at our emergency unit with fever, cough, epistaxis and temporary memory loss after several weeks of fatigue. She was in a severely reduced general condition with positive shock index and thus admitted to our ward with diagnosis of sepsis for further treatment. Physical examination showed a so far undiagnosed splenomegaly without other obvious phenotypical abnormalities. Laboratory results revealed pancytopenia (leukocytes: 930/µl, hemoglobin: 8.0 g/dl, thrombocytes: 17/nl), elevated transaminases (ALT 126 U/l [normal range <35 U/l], AST 139 U/l [5–31 U/l]), CRP (104 mg/l [< 5 mg/l]) and LDH (819 U/l [< 245 U/l]) as well as an undetectable haptoglobin (<0.08g/l [0.35-2.5 g/l]) and slightly elevated bilirubin (1.3 mg/dl [0.2-1.2 mg/dl]). Additional tests showed no blasts in the differential blood count, high ferritin (5712 ng/ml [5–200 ng/ml]), elevated triglycerides (407 mg/dl [< 150 mg/dl]), hypogammaglobulinemia (IgG 3.46g/l [5.5-16.3 g/l]) and elevated sIL-2R (6,073 Mio U/ml, >2400U/ml diagnostic for HLH/94.2 ng/ml, [1.9-13.1 ng/ml]). The patient history yielded one similar prior episode of fever with meningitis-like symptoms and diarrhea at the age of 4 years. Laboratory routine revealed a mild anemia and elevated CRP (74 mg/l [< 5 mg/l]), but no elevation of transaminases. A cerebrospinal fluid diagnostic excluded a meningitis and the patient was eventually discharged with the diagnosis of a gastroenteritis. She did not report of other recurrent fevers, muscle weakness, neurologic symptoms or a diagnosed cytopenia. The family was of Middle Eastern origin, and her parents were consanguineous, though the exact degree of relationship is unknown. Her father died of a glioblastoma, her mother and her two siblings were healthy.
Diagnostic workup (Figure 1A) demonstrated no signs of relevant viral or bacterial infection as well as no Adenovirus, Cytomegalovirus (CMV), EBV, Herpes simplex virus (HSV), Varicella-Zoster virus (VZV) as determined by PCR in serum samples. Serum PCR was weakly positive for human herpesvirus 6 (HHV-6) (5.7x102 copies/ml) and seroconversion after SARS-CoV2 infection (IgM negative/IgG positive) approximately 6 months earlier. Computed tomography (CT) scan showed hepatosplenomegaly and anasarca but no evidence of lymphoma. Anamnestic, clinical and a broad serological work-up did not reveal any signs of autoimmune disease. We performed a bone marrow biopsy to rule out hematological malignancy. Erythropoiesis constituted more than 50% of the nucleated cells, whereas granulopoiesis was reduced but with normal cellular differentiation. We excluded leishmaniosis and mycobacterial infection by PCR of the bone marrow sample. Histologically, we observed signs of hemophagocytosis. Thus, the patient fulfilled 7/8 of the initial diagnostic criteria (fever, splenomegaly, pancytopenia, hypertriglyceridemia ≥ 265mg/dl, hemophagocytosis in bone marrow and hyperferritinemia ≥500 µg/l as well as elevated sIL-2R), for the diagnosis of HLH and intravenous admission of dexamethasone was consequently started according to HLH-2004 protocol (16). On day 3 after admission, we added intravenous immunoglobulins. Cytotoxic treatment with etoposide was omitted because of the patient’s young age and missing fertility-preserving measures.
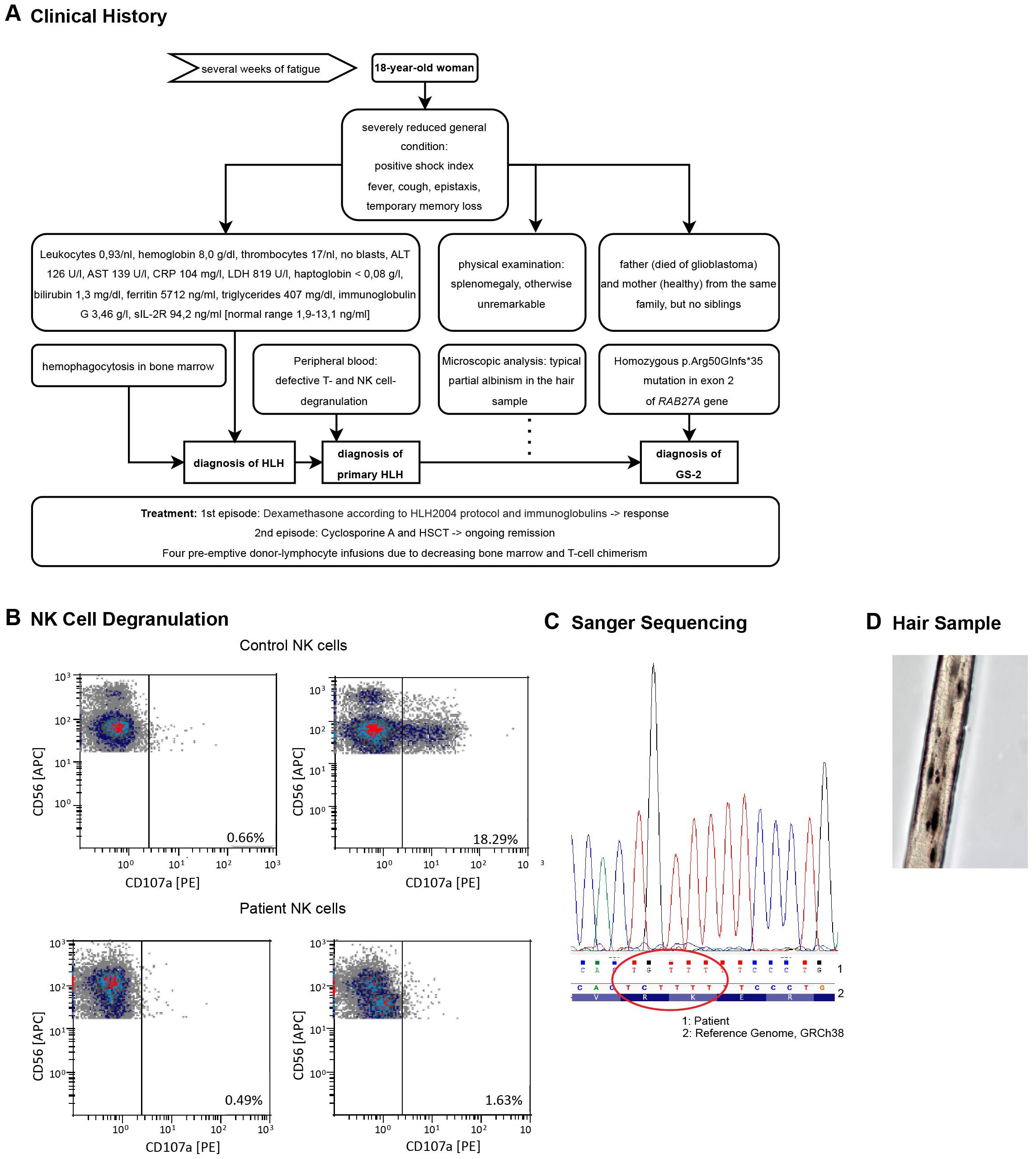
Figure 1. (A) Diagnostic workup leading to the diagnosis of GS-2. (B) Degranulation defect in patient’s NK cells. Cells were isolated from the peripheral blood during the first HLH episode. Degranulation was assessed by flow cytometry measuring the CD107a cell membrane expression. The top depicts HD samples, the bottom cells from the HLH patient. Results for NK cells are shown without (left) or after (right) in vitro stimulation with K562 cells for 2 hours. The data was kindly provided by the Center for Chronic Immunodeficiency of the Medical Center - University of Freiburg, Freiburg, Germany. (C) Sanger Sequencing results from the patient’s blood detecting the p.Arg50Glnfs*35 variant in exon 2. The results were displayed with Chromas software and aligned to the GRCh38 genome displayed by the Integrative Genomic Viewer (IGV) (Version 2.16.2). (D) Melanin clumps and partial albinism in a hair sample, light microscopy, 40X magnification.
Since no trigger factor for secondary HLH was apparent in our extensive work-up, we considered the possibility of primary HLH. NK cell function in the peripheral blood was evaluated by flow cytometry analysis of the degranulation marker CD107a (LAMP-1) and revealed defective activation-induced degranulation of NK cells in both, CD56bright and CD56dim populations (Figure 1B). This finding, complemented by the absence of infectious, rheumatological or malignant causes, led to the diagnosis of a primary HLH. Targeted gene sequencing demonstrated a homozygous p.Arg50Glnfs*35 variant (Del-Ins) in exon 2 of the RAB27A gene (Figure 1C), previously described in patients with GS-2 (24–26). Microscopic analysis of her hair revealed the typical picture of partial albinism with melanin clumping (Figure 1D), even though phenotypically no albinism was apparent. With proof of hereditary HLH, HSCT was indicated and the donor search process initiated.
The patient responded well to steroid therapy and was discharged on day 12 after admission to be further treated and monitored in the outpatient setting (Figure 2). One month later, she developed fever and a second episode of HLH while on 4 mg dexamethasone daily. Despite escalation of dexamethasone to 20 mg daily, she remained unresponsive with persistent pancytopenia and highly elevated ferritin levels (30464 ng/ml) (Figure 2). An immunosuppressive therapy with cyclosporine A was initiated (plasma target level of 200 ng/ml) as bridging to HSCT. After 18 days, the patient was discharged with improved general condition, decreased laboratory inflammation markers, reconstituted granulocytes and thrombocytes and reduced, but still elevated ferritin levels (4253 ng/ml). After oocyte cryopreservation, her pre-transplantation work-up disclosed normal, slightly hypocellular bone marrow (by cytology and histology), normal spleen size, but moderate hepatomegaly (midclavicular line 15 cm). The patient received an HSCT (conditioning: Fludarabine, Melphalan, Alemtuzumab) from an HLA-B-Mismatch unrelated donor 136 days after her first admission. She developed one grand-mal seizure in the context of a sepsis during neutropenia after conditioning chemotherapy. The microbiologic and pathologic workup remained without relevant findings, the cranial magnetic resonance imaging (cMRI) showed unspecific signal alterations in cortex in both hemispheres. In the follow up, radiologic findings normalized, the patient remained fully asymptomatic and anticonvulsive therapy was terminated on day 390. Three months (day 221) after HSCT she developed a late-onset acute Graft-versus-host disease (GvHD) of the skin that fully vanished after one week of local corticosteroids. Addressing a decreasing bone marrow and T cell chimerism, she received four pre-emptive donor-lymphocyte infusions (DLI) - 10, 13, 15 and 19 months after HSCT - without developing consecutive GvHD. Latest determination of chimerism in the bone marrow was 99.8% for CD15+ granulocytes, and 98.4% for CD3+ T cells. The patient has remained in remission to date with no further HLH episodes.
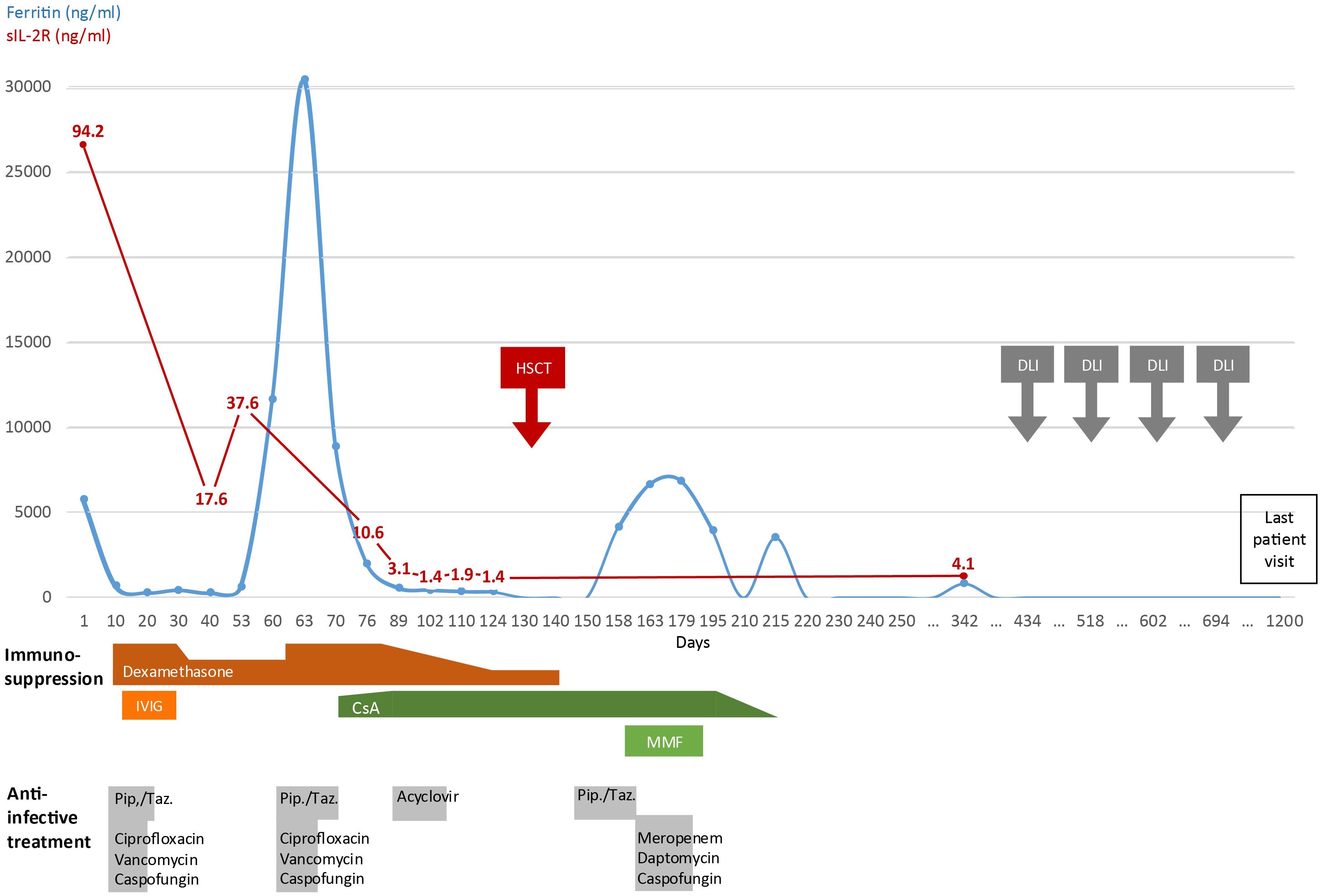
Figure 2. Timeline of clinical development. Disease activity displayed by serum ferritin and sIL-2R levels and administered therapy since first day of admission into hospital (day 1) until last patient visit. (IVIG, intravenous Immunoglobulin G; CsA, Cyclosporine A; MMF, mycophenolate mofetil; Pip./Taz., piperacillin-tazobactam; HSCT, hematopoietic stem cell transplantation; DLI, donor lymphocyte infusion).
To decipher why the first HLH episode as a sign of an underlying immune dysfunction occurred unusually late in this patient, we investigated several key cellular immune functions in vitro.
In addition to the impaired degranulation observed in NK cells, we extended the analysis to T cells. Interestingly, the patient’s CD3+ T cells showed a clear reduction rather than a complete defect in degranulation (measured as CD107a expression by flow cytometry upon stimulation (with PMA/Ionomycin) compared to healthy donors (HDs) (Figure 3A). Moreover, flow cytometry revealed an increased intracellular granzyme B (GrB) concentration compared to a HD independent of the stimulation with PMA (Figure 3B).
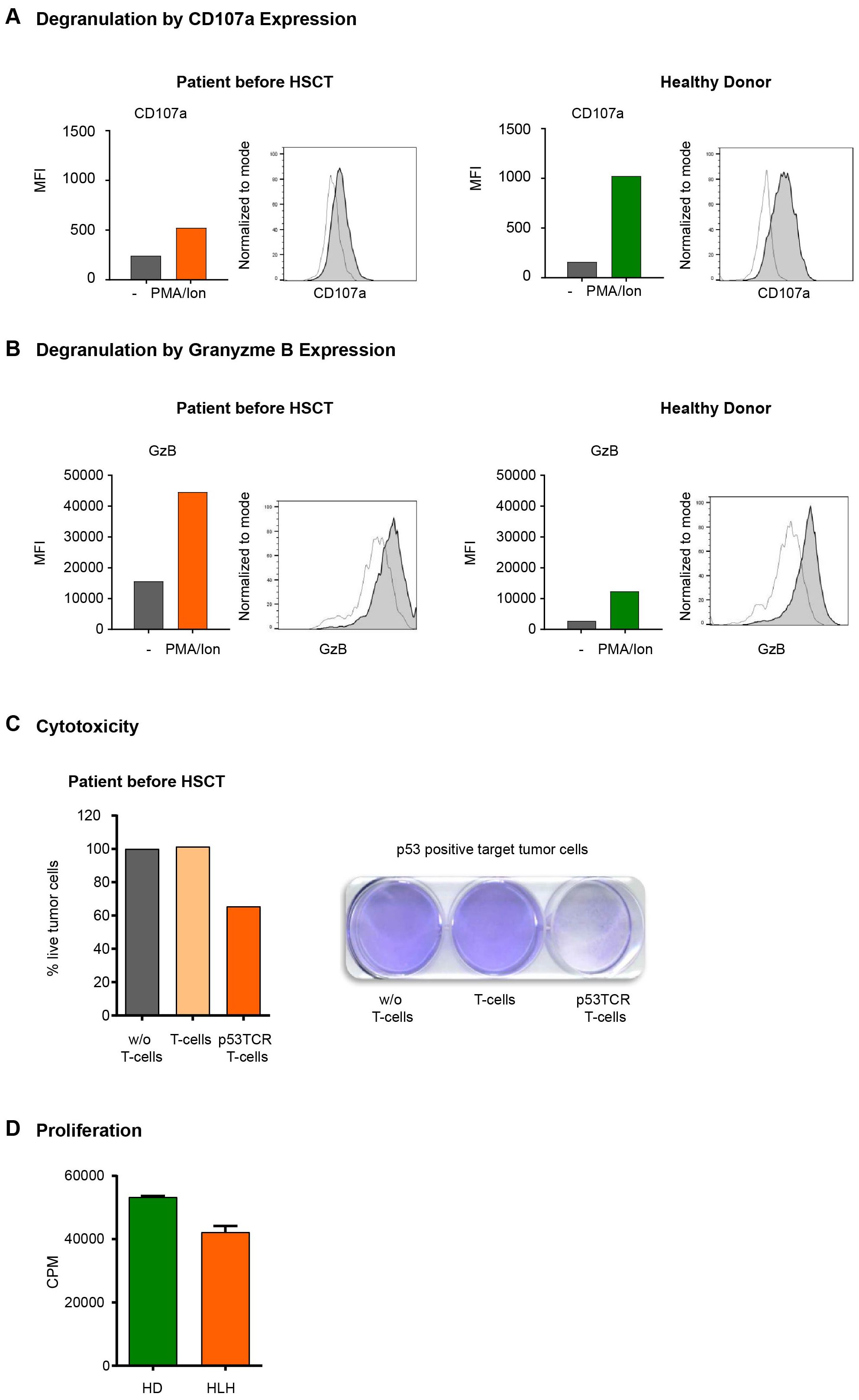
Figure 3. (A, B) Cell-membrane CD107a (A) and intracellular Granzyme B (B) expression in T cells. (A) Flow cytometry expression analysis of the degranulation marker CD107a in CD3+ T cells from HD and HLH patient detected as MFI values of CD107a expression in HD T cells and HLH patient (unstimulated vs stimulated). Medium only (transparent histogram) or stimulated with PMA/Ionomycin (gray histogram). (B) Analysis of intracellular granzyme B (GrB) expression in CD3+ T cells from healthy control donor and HLH patient. MFI values of GrB expression in HD T cells and HLH patient (unstimulated vs stimulated). Medium only (transparent histogram) or stimulated with PMA/Ionomycin (gray histogram). (C) Cytotoxic activity of CD3+ T cells. The cytolytic activity of the HLH patient’s T cells was assessed in tumor colony-forming assays after transduction with a p53(264-272) specific TCR. Tumor cells (Saos2/143) were co-cultured for 24h either without (w/o) T cells, with unmodified T cells or with p53(264-272) TCR transduced T cells. After repetitive washing steps only viable tumor cells remain attached to the cell culture plate and are visualized by staining with crystal violet dye and measured by optical density (OD) of the dye. Primary data and OD quantification with normalization (100% viability in the condition w/o T cells) of this experiment are demonstrated. (D) Proliferation of CD3+ T cells from HD and HLH patient as determined by 3H-Thymidine assay.
We next analyzed to which degree the reduced degranulation would affect T cell-mediated cytotoxicity. For this, we used our well-established model of peptide antigen specific CD3+-mediated cellular cytotoxicity (27). Briefly, peripheral blood mononuclear cells (PBMC) of the patient were retrovirally transduced with a T cell receptor (TCR) with specificity for the HLA-A2.1 restricted p53(264-272) peptide. After 4 days of peptide specific stimulation the cytolytic function of p53TCR-modified CD3+ T cells from the patient was assessed in overnight coculture with a target tumor cell line (osteosarcoma Saos2/143) expressing the p53(264-272) peptide (Figure 3C). Importantly, the patient’s TCR-expressing CD3+ T cells induced relevant antigen-specific tumor cytotoxicity compared to non-transduced T cells. Next, we measured the proliferation capacity of the patient’s T cells, following our established methodology (28). CD3+ T cells were isolated from peripheral blood and stimulated with agonistic anti-CD3/CD28-beads as described (29). Proliferation was determined by [3H]-thymidine pulsing as described before (28). Here we demonstrated that the proliferation capacity of the patient’s CD3+ T cells was in the same range as the corresponding HD controls (Figure 3D).
Expression of Rab27a in PMN has previously been demonstrated (30, 31) and its involvement in PMN tertiary and specific granule mobilization was shown (32). This latter finding was generated by sophisticated blocking strategies in normal donor PMN. We had the unique opportunity to analyze in vitro degranulation efficacy in RAB27A mutated (and therefore potential loss-of-function) PMN from our patient, before and after HSCT.
Degranulation of certain PMN granule subtypes can be quantified by cell membrane incorporation of granule-localized membranous proteins, which become externalized and are present in the cell membrane during the process of degranulation. Activation–induced upregulation of CD66b and CD11b is associated with degranulation of tertiary and specific PMN granules (32, 33). Upon lipopolysaccharide (LPS)-mediated activation we monitored cell membrane CD11b and CD66b expression by flow cytometry. In contrast to HD, our patient did not show a significant activation-induced increase in CD11b and CD66b expression, demonstrating a severe degranulation deficiency of her PMNs. Similar analysis was performed after HSCT, demonstrating a normal degranulation capacity of the allogeneic PMNs, correlating to the clinical remission of the patient (Figure 4A).
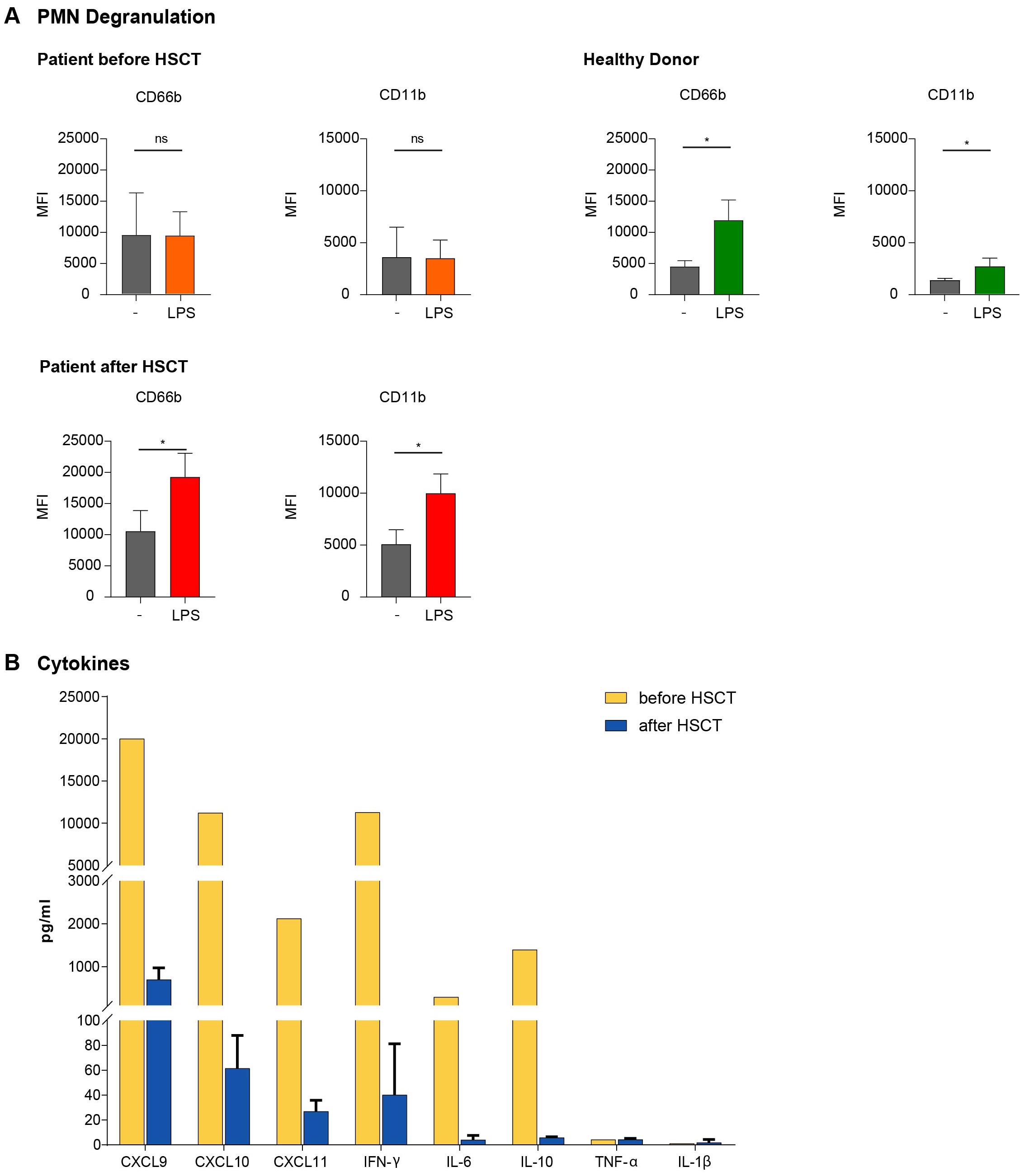
Figure 4. (A) Defective PMN Degranulation activity in GS-2. PMN of HD and the GS-2 patient were stimulated with LPS for 90 minutes. Expression of the surface markers CD66b and CD11b was assessed by flow cytometry (MFI with mean ± SEM) as correlate for the extent of degranulation/activation. n=5 samples from different HD, n=3 samples from the GS-2 patient acquired at different time points prior to and after HSCT (month 5, 6, 7), respectively. Statistical calculations were performed with Welch´s t test (*p <0.05; ns, not significant) with unstimulated control as reference. (B) HLH-associated cytokine elevations in the peripheral blood of the GS-2 patient Cytokine levels were assessed during the second HLH episode and at three different time points post-transplantation (months 5, 6, 7). All samples were taken without concurrent infection, fever or immunosuppresive medication. We compared pre-transplant HLH-associated (yellow) with the mean of post-transplant (blue) cytokine levels.
Finally, we determined a broad array of inflammatory cytokines and chemokines by Bead Array technology (27) in the serum of the patient. The serum samples were collected during the second hyperinflammatory HLH episode as well as after HSCT in the absence of clinically apparent inflammatory problems or immunosuppressive medication. Cytokine levels were assessed prior to stem cell transplantation during the second HLH episode and post transplantation. As expected, IFN-γ (11290 vs. 28pg/ml), IL-6 (293 vs. 4 pg/ml) and IL-10 (1395 vs. 6 pg/ml) were significantly increased during the HLH episode. We did not detect changes in TNF-α or IL-1β levels, but observed an increase of several chemokines, especially of CXCL9 (>20000 [above highest standard] vs. 374 pg/ml), CXCL10 (11235 vs. 61 pg/ml) and CXCL11 (2125 vs 27 pg/ml), all ligands of the chemokine receptor CXCR3 (Figure 4B).
3 Discussion
We report a case of exceptionally late onset of GS-2 in a patient with a homozygous missense p.Arg50Glnfs*35 variant in exon 2 of RAB27A. Phenotypically, partial albinism was inapparent, however a hair sample confirmed partial albinism with melanin clumps. Several reports of patients with GS-2 sine albinism exist (6–8). One retrospective study based on data of the International HLH Registry of the Familial hemophagocytic lymphohistiocytosis (FHL) study group from 1989 to 2013 identified six cases of GS-2 without albinism (8). These patients had biallelic variants in RAB27A (localization A76, R141, Y159, and S163) that altered the binding site with Munc13–4 but not melanophilin (MLPH). The disruption of Rab27a/Munc13-4-binding disabled the secretion of cytolytic granules in leukocytes while normal skin pigmentation was maintained as reflected by intact melanophilin binding (8). In a different report, a novel RAB27A gene variant (Val143Ala) was discovered in a patient without formation of typical melanin clumps in his hair. Here again, Rab27a/MLPH binding was preserved (5). The homozygous p.Arg50Glnfs*35 Rab27a variant in our patient is usually associated to albinism (34–36). Albeit our patient showed no phenotypical signs of albinism, her hair analyses revealed partial melanin clumping. Consequently, physicians should not be misled by phenotypic appearance, since subtle melanin abnormalities might not be apparent macroscopically.
Exceptional about this case was the unusually late onset of the first life-threatening HLH episode at 18 years of age. Late manifestations of primary HLH are rare but genetic alterations should still be considered as a potential underlying cause (37). A different case report with first manifestation at the age of 29 years reported a novel GATA2 variant and, like in our case, a HSCT was conducted for curative treatment (38). In patients with RAB27A variant, a similar finding was reported in a case report of a 24-year-old female with a heterozygous variant presenting with neurologic symptoms and albinism (24). The patient carried a c.259G > C; p.Ala87Pro missense variant in exon 4 and importantly also a c.149delG; p.Arg50Glnfs*35 variant in exon 2 identical to the homozygous variant in our patient. A recent case report described a novel homozygous RAB27A c.551G > A p.(R184Q) mutated patient without albinism and an onset of disease at 35 years and concurrent EBV infection (39). Possible reasons why symptoms arise after adolescence in some patients even with homozygous variants remain incompletely understood. The timing of symptom onset might be influenced by external or internal triggering events, such as infections which may exacerbate the underlying molecular defect (40–42). This is supported by a murine model with a MUNC13–4 deficiency (responsible for FHL 3), in which an infectious trigger (lymphocytic choriomeningitis virus) was necessary for the development of an HLH (43). Our patient had no concurrent infection but rather a history of infection with SARS-CoV six months ago which has been associated with the development of secondary HLH (44–46). The negative IgG-levels for EBV, CMV, HSV and VZV suggest an infection naïve patient history during childhood and adolescence. The residual protein expression from the gene variant might have delayed disease manifestation as previously discussed (47). Unfortunately, Rab27a protein expression levels were not tested and these data can therefore not be provided. Alternatively, we speculate that a compensation by other functional molecules or activation of alternative signaling pathways might mitigate the impact of the variant Rab27a protein for a time, postponing clinical symptoms. The analysis of our patient’s T cell function suggests a preserved low-level degranulation capacity (Figures 3A, B), an intact proliferation capacity (Figure 3D) and a functioning antigen-specific cytotoxic response at least in vitro (Figure 3C), possibly contributing to a late occurrence of HLH episodes. Impaired T cell cytotoxicity had been described earlier in a murine model of Rab27 deficiency (48). However, this deficient cytotoxicity was apparent towards FAS-negative target cells, while in our assay system, the Fas-FasL cytotoxic pathway is present and has likely contributed (besides the partial T cell degranulation capacity) to the preserved T cell cytotoxicity. The intracellular storage of GrB was higher than in the healthy donor sample as described before (49), either as a coping strategy or as reaction to a prolonged antigen exposure due to the insufficient antigen clearance (12, 39). Whether the remaining activity of CTLs had been sufficient to fight pathogens and prevent hyperinflammation remains unclear.
Defective NK- and T cell degranulation is a recognized key factor of GS-2. Novel about our case is the analysis of PMN degranulation in primary patient material as part of the diagnostic workup. Previous studies suggested that RAB27A deficient PMN exhibit an impaired myeloperoxidase (MPO) or matrix metalloproteinase 9 (MMP-9) exocytosis upon granulocyte macrophage colony-stimulating factor (GM-CSF) or LPS stimulation in murine models (31, 50). Similarly, RAB27A-downregulation (31) or Rab27a inhibition (32) lead to reduced tertiary and specific granule mobilization in human PMNs. In line with these results, we demonstrate for the first time that mobilization of gelatinase granules (CD11b) and azurophilic granules (CD66b) upon LPS stimulation is indeed defective in PMN of patients with GS-2 (Figure 4A). Interestingly, these results in primary human PMN partially are in contrast to data from the murine RAB27A knockout model, which largely suggest Rab27a-independent up-regulation of CD11b in neutrophils, yet upon GM-CSF stimulation (50).
In line with previous reports, we observed high levels of IFN-γ, IL-10, IL-6 and of the CXCR3 ligands CXCL9, CXCL10 and CXCL11 in the peripheral blood during active HLH (51–54). Especially elevated IFN-γ and IL-10 levels have been shown to be characteristic of HLH with high sensitivity and specificity (20, 51, 54). Emapalumab, an IFN-γ-blocking antibody, was successfully tested in a phase 2–3 study in children with primary HLH and is now available as specific cytokine-directed therapy for future therapeutic approaches in patients with primary HLH (18, 52). CXCL9, CXCL10 and CXCL11 are IFN-γ-inducible ligands of CXCR3, which functions as an inflammatory chemokine receptor on CD4+ Th1, CD8+ CTL, NK and dendritic cells (DC) (53) and is upregulated after DC-mediated T cell activation. Together, the high levels of these cytokines in our patient are consistent within the setting of HLH.
In summary, we present a case of GS-2 with unusually late onset and clinical phenotype without remarkable or suggestive features. We demonstrate completely absent NK- and PMN-degranulation, but partially preserved T cell degranulation. The T cells remain functional with regard to their capacity to proliferate and to mount antigen-specific cytotoxicity. The reason why the patient remained asymptomatic throughout adolescence is unexplained. Our case report emphasizes the importance to consider genetic testing for primary HLH in adult patients with causally unclear HLH.
4 Methods
4.1 Isolation of T cells and intracellular granzyme B/surface CD107a expression
Peripheral blood mononuclear cells (PBMCs) from healthy control donor (HD) and HLH patient were isolated by Ficoll density gradient centrifugation. CD3 positive T cells were isolated with the EasySep™ Human T cell Enrichment Kit (Stemcell Technologies, Vancouver, Canada) and kept overnight in RPMI 1640 + 10% human AB Serum, 1% P/S and 1% L-Glutamine. To detect the surface membrane expression of the lysosomal-associated membrane protein 1 (LAMP1/CD107a) (as a surrogate marker for degranulation) and the intracellular granzyme B (GrB), CD3+ T cells were stimulated with Phorbol 12-myristate 13-acetate (PMA) + ionomycin (Sigma-Aldrich) for 5h in the presence of monensin (eBioscience) as described earlier (27, 29). CD107a surface expression and intracellular GrB expression were analyzed by flow cytometry and quantitated by showing mean fluorescence intensity (MFI).
4.2 NK cell degranulation
NK degranulation assays were performed as described in Bryceson et al. (2012) (55).
4.3 Proliferation assay
CD3 positive T cells isolated from the PBMCs of HD and the HLH patient were stimulated with agonistic anti-CD3/CD28-beads in the presence of IL-2 for 5–6 days (27) and T cell proliferation was assessed by the incorporation of [3H]thymidine as described before (28).
4.4 Cytotoxic assay
The cytolytic activity of the HLH patient’s CD3+ T cells was assessed in tumor colony-forming assay (CFA) after retroviral transduction with a p53(264-272) specific T cell receptor, as described (27). Briefly, effector T cells were co-cultured with antigen+ (Saos2/143) target tumor cells in 6-well plates in 37°C with 5% CO2 at an effector-to-target (E:T) ratio of 2:1. After 24h, T cells as well as non-adherent lysed tumor cells were washed out and the remaining adherent viable tumor cells were fixed (4% PFA) and stained with 0.5% crystal violet dye (Merck KGaA, Germany). Crystal violet was washed off by adding PBS and the plate scanned for visual evaluation of colony counts. For quantitative analysis, the dye was dissolved by adding 5% SDS and the corresponding optical density (absorbance) measured at 570nm using a microplate reader (Dynex MRX, Magellan BioScience), and values expressed as percent of tumor viability (27, 29).
4.5 Degranulation of PMN
Degranulation of PMN granule subtypes can be quantified by cell membrane incorporation of granule-localized membranous proteins, e.g. by activation–induced upregulation of CD66b and CD11b (32, 33). PMN of healthy donors and the HLH patient were stimulated with lipopolysaccharide (LPS) for 90 minutes. The surface expression of the markers CD11b and CD66b was then assessed by flow cytometry measuring the MFI (mean ± SEM). Statistical calculations were performed with Welch´s t test (*p <0.05) with unstimulated control as reference, n=5 from different healthy donors, n=3 from the HLH patient acquired at different time points prior and after HSCT.
4.6 Cytokines/chemokines
Secreted cytokines/chemokines in vitro culture and in serum were determined by Cytometric Bead Array (BD Biosciences, Franklin Lakes, NJ) according to the manufacturer protocol. Cytokine levels were assessed at one time point during the second HLH episode and at three different time points (months 5, 6, 7 post-transplantation). All samples were taken without concurrent infection or fever.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Rhineland-Palatinate Medical Association Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
JR: Conceptualization, Investigation, Writing – original draft, Writing – review & editing, Project administration, Visualization. SH: Conceptualization, Investigation, Writing – original draft, Writing – review & editing, Project administration, Visualization. SL: Investigation, Writing – review & editing. YB: Investigation, Writing – review & editing. EAF: Investigation, Writing – review & editing. TW: Investigation, Writing – review & editing. KJL: Methodology, Writing – review & editing. MB: Investigation, Writing – review & editing, Resources. FB: Investigation, Writing – review & editing, Resources. OK: Writing – review & editing, Project administration. KW: Writing – review & editing, Supervision. MG: Investigation, Writing – review & editing. KL: Writing – review & editing, Supervision. HL: Investigation, Writing – review & editing. PL: Writing – review & editing. MR: Writing – review & editing. MT: Resources, Writing – review & editing. HE: Conceptualization, Methodology, Project administration, Resources, Writing – original draft, Writing – review & editing. MM: Conceptualization, Project administration, Resources, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was partly supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) -Project-ID 318346496 -SFB1292 TP06 (to SL, YB, EF, KJL, MM, MT and HE), SFB1292 TP21N (to MR).
Acknowledgments
We thank the patient for her consent to publish her clinical case and to donate primary cellular material for this study. We thank the Advanced Diagnostics Unit of the Center for Chronic Immunodeficiency (CCI) Freiburg for excellent services.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Griscelli C, Durandy A, Guy-Grand D, Daguillard F, Herzog C, and Prunieras M. A syndrome associating partial albinism and immunodeficiency. Am J Med. (1978) 65:691–702. doi: 10.1016/0002-9343(78)90858-6
2. Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. (2000) 25:173–6. doi: 10.1038/76024
3. Fukuda M. Versatile role of Rab27 in membrane trafficking: focus on the Rab27 effector families. J Biochem. (2005) 137:9–16. doi: 10.1093/jb/mvi002
4. Bahadoran P, Aberdam E, Mantoux F, Buscà R, Bille K, Yalman N, et al. Rab27a: A key to melanosome transport in human melanocytes. J Cell Biol. (2001) 152:843–9. doi: 10.1083/jcb.152.4.843
5. Ohishi Y, Ammann S, Ziaee V, Strege K, Groß M, Amos CV, et al. Griscelli syndrome type 2 sine albinism: unraveling differential RAB27A effector engagement. Front Immunol. (2020) 11:612977. doi: 10.3389/fimmu.2020.612977
6. Netter P, Chan SK, Banerjee PP, Monaco-Shawver L, Noroski LM, Hanson IC, et al. A novel Rab27a mutation binds melanophilin, but not Munc13-4, causing immunodeficiency without albinism. J Allergy Clin Immunol. (2016) 138:599–601.e3. doi: 10.1016/j.jaci.2015.12.1337
7. Tesi B, Rascon J, Chiang SCC, Burnyte B, Löfstedt A, Fasth A, et al. A RAB27A 5′ untranslated region structural variant associated with late-onset hemophagocytic lymphohistiocytosis and normal pigmentation. J Allergy Clin Immunol. (2018) 142:317–321.e8. doi: 10.1016/j.jaci.2018.02.031
8. Cetica V, Hackmann Y, Grieve S, Sieni E, Ciambotti B, Coniglio ML, et al. Patients with Griscelli syndrome and normal pigmentation identify RAB27A mutations that selectively disrupt MUNC13–4 binding. J Allergy Clin Immunol. (2015) 135:1310–1318.e1. doi: 10.1016/j.jaci.2014.08.039
9. Neeft M, Wieffer M, De Jong AS, Negroiu G, Metz CHG, Van Loon A, et al. Munc13–4 is an effector of rab27a and controls secretion of lysosomes in hematopoietic cells. MBoC. (2005) 16:731–41. doi: 10.1091/mbc.e04-10-0923
10. Galgano D, Soheili T, Voss M, Torralba-Raga L, Tesi B, Cichocki F, et al. Alternative UNC13D promoter encodes a functional munc13–4 isoform predominantly expressed in lymphocytes and platelets. Front Immunol. (2020) 11:1154. doi: 10.3389/fimmu.2020.01154
11. Jenkins MR, Rudd-Schmidt JA, Lopez JA, Ramsbottom KM, Mannering SI, Andrews DM, et al. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J Exp Med. (2015) 212:307–17. doi: 10.1084/jem.20140964
12. Stinchcombe JC, Barral DC, Mules EH, Booth S, Hume AN, Machesky LM, et al. Rab27a is required for regulated secretion in cytotoxic T lymphocytes. J Cell Biol. (2001) 152:825–34. doi: 10.1083/jcb.152.4.825
13. Allen CE and McClain KL. Pathophysiology and epidemiology of hemophagocytic lymphohistiocytosis. Hematology. (2015) 2015:177–82. doi: 10.1182/asheducation-2015.1.177
14. Janka GE. Hemophagocytic syndromes. Blood Rev. (2007) 21:245–53. doi: 10.1016/j.blre.2007.05.001
15. La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. (2019) 133:2465–77. doi: 10.1182/blood.2018894618
16. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2007) 48:124–31. doi: 10.1002/pbc.21039
17. Ishii E. Hemophagocytic lymphohistiocytosis in children: pathogenesis and treatment. Front Pediatr. (2016) 4:47/abstract. doi: 10.3389/fped.2016.00047/abstract
18. Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med. (2020) 382:1811–22. doi: 10.1056/NEJMoa1911326
19. Fisman DN. Hemophagocytic syndromes and infection. Emerging Infect Dis. (2000) 6:601–8. doi: 10.3201/eid0606.000608
20. Henter J, Elinder G, Soder O, Hansson M, Andersson B, and Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. (1991) 78:2918–22. doi: 10.1182/blood.V78.11.2918.2918
21. Fujiwara F, Hibi S, and Imashuku S. Hypercytokinemia in hemophagocytic syndrome. J Pediatr Hematology/Oncology. (1993) 15. Available online at: https://journals.lww.com/jpho-online/fulltext/1993/02000/hypercytokinemia_in_hemophagocytic_syndrome.12.aspx (Accessed August 22, 2023).
22. Jin Z, Suolitiken D, Wang Y, and Wang Z. The diagnostic importance of multiple cytokines in adult hemophagocytic lymphohistiocytosis. J Clin Lab Anal. (2022) 37:e24669. doi: 10.1002/jcla.24669
23. Janka GE and Lehmberg K. Hemophagocytic lymphohistiocytosis: pathogenesis and treatment. Hematol Am Soc Hematol Educ Program. (2013) 2013:605–11. doi: 10.1182/asheducation-2013.1.605
24. Henkes M, Finke J, Warnatz K, Ammann S, Stadt UZ, Janka G, et al. Late-onset hemophagocytic lymphohistiocytosis (HLH) in an adult female with Griscelli syndrome type 2 (GS2). Ann Hematol. (2015) 94:1057–60. doi: 10.1007/s00277-014-2284-9
25. Sanal O, Ersoy FG, Tezcan I, Metin AE, Yel L, Nasche GLM, et al. Griscelli disease: genotype–phenotype correlation in an array of clinical heterogeneity. J Clin Immunol. (2002) 22:237–43. doi: 10.1023/A:1016045026204
26. Stadt UZ, Beutel K, Kolberg S, Schneppenheim R, Kabisch H, Janka G, et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses ofPRF1, UNC13D, STX11, andRAB27A. Hum Mutat. (2006) 27:62–8. doi: 10.1002/humu.20274
27. Legscha KJ, Antunes Ferreira E, Chamoun A, Lang A, Awwad MHS, Ton GNHQ, et al. Δ133p53α enhances metabolic and cellular fitness of TCR-engineered T cells and promotes superior antitumor immunity. J Immunother Cancer. (2021) 9:e001846. doi: 10.1136/jitc-2020-001846
28. Vonwirth V, Bülbül Y, Werner A, Echchannaoui H, Windschmitt J, Habermeier A, et al. Inhibition of arginase 1 liberates potent T cell immunostimulatory activity of human neutrophil granulocytes. Front Immunol. (2021) 11:617699. doi: 10.3389/fimmu.2020.617699
29. Echchannaoui H, Petschenka J, Ferreira EA, Hauptrock B, Lotz-Jenne C, Voss RH, et al. A Potent Tumor-Reactive p53-Specific Single-Chain TCR without On- or Off-Target Autoimmunity In Vivo. Mol Ther. (2019) 27:261–71. doi: 10.1016/j.ymthe.2018.11.006
30. Lominadze G, Powell DW, Luerman GC, Link AJ, Ward RA, and McLeish KR. Proteomic analysis of human neutrophil granules*S. Mol Cell Proteomics. (2005) 4:1503–21. doi: 10.1074/mcp.M500143-MCP200
31. Munafó DB, Johnson JL, Ellis BA, Rutschmann S, Beutler B, and Catz SD. Rab27a is a key component of the secretory machinery of azurophilic granules in granulocytes. Biochem J. (2007) 402:229–39. doi: 10.1042/BJ20060950
32. Herrero-Turrión MJ, Calafat J, Janssen H, Fukuda M, and Mollinedo F. Rab27a regulates exocytosis of tertiary and specific granules in human neutrophils. J Immunol. (2008) 181:3793–803. doi: 10.4049/jimmunol.181.6.3793
33. Ries F, Alflen A, Aranda Lopez P, Beckert H, Theobald M, Schild H, et al. Antifungal drugs influence neutrophil effector functions. Antimicrob Agents Chemother. (2019) 63:e02409–18. doi: 10.1128/AAC.02409-18
34. Sefsafi Z, Hasbaoui BE, Kili A, Agadr A, and Khattab M. Macrophage activation syndrome associated with griscelli syndrome type 2: case report and review of literature. Pan Afr Med J. (2018) 29:75. doi: 10.11604/pamj.2018.29.75.12353
35. Mehdizadeh M and Zamani G. Griscelli Syndrome: A case report. Pediatr Hematol Oncol. (2007) 24:525–9. doi: 10.1080/08880010701533793
36. Gailson T, Pandit S, and Chandrasekaran S. Griscelli syndrome type 2. QJM: Int J Med. (2020) 113:137. doi: 10.1093/qjmed/hcz144
37. Rohr J, Beutel K, Maul-Pavicic A, Vraetz T, Thiel J, Warnatz K, et al. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica. (2010) 95:2080–7. doi: 10.3324/haematol.2010.029389
38. Mika T, Vangala D, Eckhardt M, La Rosée P, Lange C, Lehmberg K, et al. Case report: hemophagocytic lymphohistiocytosis and non-tuberculous mycobacteriosis caused by a novel GATA2 variant. Front Immunol. (2021) 12:682934. doi: 10.3389/fimmu.2021.682934
39. Zondag TCE, Torralba-Raga L, Van Laar JAM, Hermans MAW, Bouman A, Hollink IHIM, et al. Novel RAB27A variant associated with late-onset hemophagocytic lymphohistiocytosis alters effector protein binding. J Clin Immunol. (2022) 42:1685–95. doi: 10.1007/s10875-022-01315-4
40. Strippoli R, Caiello I, and Benedetti FD. Reaching the threshold: A multilayer pathogenesis of macrophage activation syndrome. J Rheumatol. (2013) 40:761–7. doi: 10.3899/jrheum.121233
41. Nodehi H, Faranoush M, Arshi S, Nabavi M, Bemanian MH, Shokri S, et al. Neonatal onset of hemophagocytic lymphohistiocytosis due to prenatal varicella-zoster infection in a neonate with griscelli syndrome type 2. Iranian J Allergy Asthma Immunol. (2022) 21:488–93. doi: 10.18502/ijaai.v21i4.10297
42. Al-Sulaiman R, Othman A, El-Akouri K, Fareed S, AlMulla H, Sukik A, et al. A founder RAB27A variant causes Griscelli syndrome type 2 with phenotypic heterogeneity in Qatari families. Am J Med Genet Part A. (2020) 182:2570–80. doi: 10.1002/ajmg.a.61829
43. Crozat K, Hoebe K, Ugolini S, Hong NA, Janssen E, Rutschmann S, et al. Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: a mouse model of type 3 familial hemophagocytic lymphohistiocytosis. J Exp Med. (2007) 204:853–63. doi: 10.1084/jem.20062447
44. Kalita P, Laishram D, Dey B, Mishra J, Barman B, and Barman H. Secondary hemophagocytic lymphohistiocytosis in post-COVID-19 patients: A report of two cases. Cureus. (2021) 13(8):1–6. Available online at: https://www.cureus.com/articles/67827-secondary-hemophagocytic-lymphohistiocytosis-in-post-covid-19-patients-a-report-of-two-cases.
45. Naous E, Nassani BM, Yaghi C, Nasr F, and Medlej R. Hemophagocytic lymphohistiocytosis, a new cause of death during ‘post-acute COVID-19 syndrome?’ A case report. J Hematopathol. (2021) 14:229–33. doi: 10.1007/s12308-021-00452-w
46. Bandaru SS, Capace A, Busa V, and Williams A. Secondary hemophagocytic lymphohistiocytosis in a post-COVID-19 patient. Cureus. (2022) 14(2):1–4. Available online at: https://www.cureus.com/articles/87992-secondary-hemophagocytic-lymphohistiocytosis-in-a-post-covid-19-patient.
47. Maimaris J, Roa-Bautista A, Sohail M, Booth C, Cugno C, Chenchara L, et al. Griscelli syndrome type 2: comprehensive analysis of 149 new and previously described patients with RAB27A deficiency. J Clin Immunol. (2024) 45:50. doi: 10.1007/s10875-024-01842-2
48. Haddad EK, Wu X, Hammer JA, and Henkart PA. Defective granule exocytosis in Rab27a-deficient lymphocytes from Ashen mice. J Cell Biol. (2001) 152:835–42. doi: 10.1083/jcb.152.4.835
49. Mellor-Heineke S, Villanueva J, Jordan MB, Marsh R, Zhang K, Bleesing JJ, et al. Elevated granzyme B in cytotoxic lymphocytes is a signature of immune activation in hemophagocytic lymphohistiocytosis. Front Immunol. (2013) 4:72. doi: 10.3389/fimmu.2013.00072
50. Ramadass M, Johnson JL, and Catz SD. Rab27a regulates GM-CSF-dependent priming of neutrophil exocytosis. J Leukoc Biol. (2017) 101:693–702. doi: 10.1189/jlb.3AB0416-189RR
51. Xu XJ, Tang YM, Song H, Yang SL, Xu WQ, Zhao N, et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatrics. (2012) 160:984–990.e1. doi: 10.1016/j.jpeds.2011.11.046
52. Zinter MS and Hermiston ML. Calming the storm in HLH. Blood. (2019) 134:103–4. doi: 10.1182/blood.2019001333
53. Groom JR and Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. (2011) 89:207–15. doi: 10.1038/icb.2010.158
54. Xu XJ, Luo ZB, Song H, Xu WQ, Henter JI, Zhao N, et al. Simple evaluation of clinical situation and subtypes of pediatric hemophagocytic lymphohistiocytosis by cytokine patterns. Front Immunol. (2022) 13:850443. doi: 10.3389/fimmu.2022.850443
Keywords: Griscelli syndrome type 2, hemophagocytic lymphohistiocytosis, RAB27a variant, polymorphonuclear neutrophils, degranulation defect, hyperinflammation, case report
Citation: Rausch J, Herold S, Liebhäuser S, Bülbül Y, Antunes Ferreira E, Wenz T, Legscha KJ, Bros M, Butsch F, Kriege O, Warnatz K, Groß M, Lehmberg K, Lichtenfeld HC, La Rosée P, Radsak MP, Theobald M, Echchannaoui H and Munder M (2025) Case Report: Late-onset primary hemophagocytic lymphohistiocytosis leading to the diagnosis of Griscelli syndrome type 2 in a young woman with phenotypically inapparent partial albinism. Front. Immunol. 16:1604460. doi: 10.3389/fimmu.2025.1604460
Received: 01 April 2025; Accepted: 23 July 2025;
Published: 07 August 2025.
Edited by:
Hirokazu Kanegane, Tokyo Medical and Dental University, JapanCopyright © 2025 Rausch, Herold, Liebhäuser, Bülbül, Antunes Ferreira, Wenz, Legscha, Bros, Butsch, Kriege, Warnatz, Groß, Lehmberg, Lichtenfeld, La Rosée, Radsak, Theobald, Echchannaoui and Munder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johanna Rausch, Sm9oYW5uYS5SYXVzY2hAdW5pbWVkaXppbi1tYWluei5kZQ==; Stephanie Herold, U3RlcGhhbmllLkhlcm9sZEB1bmltZWRpemluLW1haW56LmRl
†These authors share first authorship
‡These authors share last authorship