 Xinyi Xu
Xinyi Xu Ruihan Yu
Ruihan Yu Chenyu Li
Chenyu Li Huabao Xiong
Huabao Xiong Chunxia Li
Chunxia Li- Institute of Immunology and Molecular Medicine, Jining Medical University, Jining, China
As a chronic inflammatory illness of the respiratory system, asthma occurs due to various factors and is characterized by a T helper 2 (Th2)-skewed immune response, airway hyperresponsiveness, and reversible airflow obstruction. Toll-like receptors (TLRs) perform a “double-edged sword” function in asthma-related immunological dysregulation by recognizing damage-associated molecular patterns and pathogen-associated molecular patterns. In turn, the activation of some TLRs stimulates epithelial cells to release inflammatory cytokines, exacerbating Th2-driven inflammation and contributing to airway remodeling. Certain TLR signals help inhibit allergic responses by inducing type I interferon or regulatory T cells. The TLR family comprises 10 members, each responsible for recognizing the distinct molecular structure of multiple microbial sources. Variations in environmental microbial exposure duration and host genetic background contribute to the complexity of the TLR signaling network during asthma development. In recent years, therapeutic strategies targeting TLRs have shown potential for asthma treatment. However, a comprehensive review of TLRs in asthma is lacking. Therefore, this review sought to examine the functional mechanisms of TLRs and associated signaling cascades in asthma, and explore novel prevention and treatment approaches centered on TLRs modulation.
1 Introduction
Asthma, a prevalent respiratory condition affecting over 330 million individuals worldwide (1, 2), is a fundamentally heterogeneous disease. This heterogeneity manifests as multiple distinct phenotypes, which are clinically defined by variations in presenting features, triggering factors, patterns of airway inflammation, and physiological or pathological characteristics (3). Underpinning these phenotypes is a complex and persistent inflammatory process within the airways. Key features of inflammation in asthma include airway hyperresponsiveness, eosinophilic infiltration, excessive mucus production, reversible airflow limitation, structural airway remodeling, and goblet cell hyperplasia (1). The innate immune system plays a pivotal role against this backdrop of complex airway pathology. Toll-like receptors (TLRs) represent a category of pattern recognition receptors that detect damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), triggering innate immune responses (4). TLRs function as a vital connection between adaptive and innate immunity by modulating the activation of essential cytokines and antigen-presenting cells (5). They play a crucial part in numerous diseases, including atherosclerosis, acute lymphoblastic leukemia, Parkinson’s disease, sepsis, cancer, and autoimmune disorders (6–9). Recent studies show that TLRs contribute to the onset and exacerbation of asthma. The purpose of this review is to investigate the function of TLRs in respiratory inflammation, their contribution to the pathogenesis of asthma, and new developments in targeting TLRs for asthma treatment.
2 Pathogenesis of asthma
Asthma is a heterogeneous disease with multiple phenotypes, including allergic, non-allergic, late-onset, asthma with fixed airflow limitation, and asthma with obesity, which are shaped by differences in age of onset, clinical features, triggers, and inflammatory mechanisms. The pathophysiology of asthma involves activation of both innate and adaptive immune responses, leading to chronic airway inflammation. This inflammation results in airway edema, mucus hypersecretion, and remodeling, characterized by subepithelial fibrosis, basement membrane thickening, smooth muscle hypertrophy, angiogenesis, and mucous gland hyperplasia. These structural changes are driven, in part, by Th1 (type1) and Th2 (type 2) cell-mediated immune responses, contributing to persistent airway dysfunction (10). In typical allergen-induced asthma, the pathogenesis is frequently characterized by an inflammatory environment dominated by the type 2 immune response. Under normal conditions, a balance exists between type 1 and type 2 cells. However, when harmful agents, such as allergens, viruses, and bacteria, infiltrate the respiratory tract, they initially trigger airway epithelial cells to release epithelial-derived cytokines, including interleukin-33 (IL-33), IL-25, and thymic stromal lymphopoietin. These cytokines act as early warning signals for potential epithelial and endothelial damage. Concurrently, these agents stimulate antigen-presenting cells, especially dendritic cells, to perform the following functions: uptake and processing of antigens, migration, and subsequent presentation of these antigens to naïve CD4+ T lymphocytes. This interaction activates the lymphocytes and induces their differentiation into type 2 cells, consequently disrupting the balance between type 1 and type 2 cells (11). This imbalance between type 1 and type 2 cells is considered a critical immunological factor in the etiology of asthma.
An imbalance between type 1 and type 2 immune responses is characterized by diminished type 1 cell activity and elevated production of type 2 cytokines (12). A key step within the immune system’s regulatory processes involves type 2 cells promoting the differentiation of B cells into plasma cells, which subsequently produce antibodies. Under the influence of type 2 cytokines such as IL-13 and IL-4, plasma cells preferentially synthesize immunoglobulin E (IgE). The interaction between IgE and its high-affinity FcϵRI receptor on mast cells and basophils is pivotal in the allergic cascade. When an allergen binds to two adjacent IgE molecules, cross-linking occurs, activating mast cells and triggering the release of pre-formed bioactive mediators, including histamine, tryptases, and chymase. Furthermore, mast cells synthesize substantial amounts of inflammatory mediators such as cysteinyl leukotrienes, prostaglandin D2, and type 2-associated cytokines (IL-13, IL-9, IL-5, and IL-4) (13). These mediators amplify the type 2-mediated inflammatory response, leading to increased vascular permeability, excessive mucus secretion, tracheal smooth muscle contraction, and inflammatory cell infiltration. IL-4, acting as an upstream regulatory cytokine for type 2 effector cytokines, binds to its receptor and facilitates the differentiation of naïve CD4+ T cells into the type 2 phenotype (14). Prostaglandin D2, the primary prostaglandin derived from mast cells and eosinophils, shows a positive correlation with asthma severity, attack frequency, and type 2-associated inflammatory markers. IgE-mediated allergen presentation lowers the threshold for triggering allergen-specific type 2 cell responses, thereby enhancing IgE synthesis and establishing a self-perpetuating cycle central to asthma pathogenesis (15).
Additionally, an imbalance between Th17 and regulatory T cells significantly contributes to the pathophysiology of chronic obstructive pulmonary disease. The overactivation of Th17 cells enhances the inflammatory response, while a reduction in regulatory T cells weakens immune regulation, failing to inhibit inflammation effectively and contributing to the progression of the disease (16). Three innate lymphoid cell (ILC) types are also involved in asthma development. ILC1 primarily produces interferon (IFN), while ILC3 predominantly secretes IL-17 and IL-22. ILC2 releases cytokines such as IL-13, IL-4, and IL-5, which recruit and activate inflammatory cells, including mast cells and eosinophils, causing allergic airway inflammation and exacerbating asthma symptoms (2). Additionally, factors such as airway and gut microbiota composition, allergen exposure, air pollution, oxidative stress, neuroregulation, epigenetics, sex hormones, and age are closely related to asthma (17).
3 TLRs
TLRs were first discovered in fruit flies in 1988, with the discovery of TLR4 in humans in 1997 (18, 19). To date, researchers have revealed the existence of 10 functionally active TLRs (TLR1 through TLR10) in humans and 12 in laboratory mice (20). TLR1, TLR2, TLR4, TLR5, and TLR6 are predominantly expressed on the cell surface of various immune cells, where they primarily recognize lipids and protein constituents, whereas TLR3, TLR7, TLR8, and TLR9 are expressed within endosomes and activated by specific nucleic acid ligands, including viral double-stranded RNA, dsDNA, sense single-stranded RNA, and bacterial unmethylated cytosine-phosphate-guanine (CpG) DNA respectively (21–25).
TLRs function as pattern recognition receptors that identify PAMPs and DAMPs, thereby initiating immune signaling and promoting the maturation and activation of immune cells. Upon recognizing PAMPs or DAMPs, TLRs activate intracellular signaling cascades by inducing structural alterations in their Toll/IL-1 receptor (TIR) domain, which enables the recruitment of cytoplasmic adapter proteins (26). The TLR signaling pathway is classified into two distinct categories based on the associated adaptor proteins: the myeloid differentiation factor 88 (MyD88)-dependent pathway, present in all TLRs with the exception of TLR3, and the MyD88-independent pathway, referred to as the TIR-domain-containing adapter-inducing IFN-β (TRIF)-dependent pathway (27). The MyD88-dependent pathway activates mitogen-activated protein kinase (MAPK) and nuclear factor-κB (NF-κB), leading to the production of proinflammatory cytokines. The TRIF-dependent pathway stimulates the synthesis of type I IFNs and inflammatory cytokines mediated by IFN regulatory factor 3 (IRF3) (28). Under pathological conditions, TLRs contribute to allergic reactions, inflammatory responses, and autoimmune diseases by identifying microbial components or endogenous molecules (29).
4 Role of TLRs in asthma
Multiple studies have demonstrated a correlation between TLRs and asthma as well as chronic respiratory inflammation, with genetic polymorphisms in TLRs influencing susceptibility to and severity of asthma. These variations complicate the effective management of asthma-related respiratory inflammation (30, 31). Increasing evidence indicates that TLR family members and their associated signaling pathways perform pivotal functions in the context of asthma pathobiology.
4.1 TLRs
4.1.1 TLR1
TLR1 is expressed in myeloid cells, T and B cells, natural killer cells, and microglia and astrocytes and is involved in the recognition of cell wall constituents, including viral proteins and bacterial lipoproteins (32–34). The expression of TLR1 on peripheral blood mononuclear cells is significantly higher in patients with asthma than that in healthy individuals, indicating that TLR1 plays a role in the pathogenesis of asthma (35). However, TLR1/2 mRNA treatment has demonstrated protective effects on asthma induced by house dust mites in mice (36).
Furthermore, the polymorphism of the TLR1 gene is associated with the risk of asthma. Kormann et al. confirmed that the TLR1 single-nucleotide polymorphisms (SNPs), such as rs5743594, rs5743595, and rs4833095, have a protective effect on atopic asthma (37), but the variant TLR1 rs5743618 may increase the risk of asthma at the age of 11–13 years after infant bronchiolitis (38, 39). The aforementioned studies indicate that, on the one hand, TLR1 recognizes pathogens and is highly expressed in patients with asthma, suggesting its potential role in promoting inflammation or in the pathogenesis of asthma, but on the other hand, the TLR1/2 signaling pathway has been confirmed to have protective effects on asthma, and TLR1-specific gene polymorphisms are associated with either protection against asthma or risk of asthma. These seemingly contradictory findings suggest that the specific role of TLR1 in asthma may be regulated by multiple factors, and it is worthy of further in-depth exploration.
4.1.2 TLR2
TLR2 is broadly expressed on the surface of diverse cell types, including both immune cells, such as myeloid cells and T cells (40, 41) and non-immune cells, such as epithelial, endothelial, and nerve cells (42). Typically, TLR2 forms functional heterodimers with TLR1 or TLR6 on the cell surface. This heterodimerization not only broadens the spectrum of PAMPs recognized by TLR2 but also diversifies the resulting downstream signaling cascades (43). This widespread distribution underscores the significant role of TLR2 in linking innate immunity, tissue homeostasis, and disease pathogenesis. TLR2 transduces signals from various molecules, including lipoproteins and cell wall components of Gram-positive bacteria, peptidoglycans and lipoarabinomannan from mycobacteria (44, 45), and the high mobility group protein B1 (HMGB1) (46).
In the context of asthma, the role of TLR2 is particularly evident. Pauline et al. demonstrated that specific recognition of Aspergillus fumigatus conidia PAMPs by the TLR2/MyD88 pathway elevates IL-10 levels while reducing lung eosinophilia and type 2 responses (47). Furthermore, microRNA-146a (miR-146a), a key regulatory factor in ovalbumin (OVA)-induced allergic asthma models, alleviates asthma symptoms by modulating the TLR2 signaling pathway (48). Genetic studies reveal that TLR2 deficiency enhances IgE production but suppresses IgG1 class switching following OVA sensitization (49). Specific polymorphisms, such as TLR2 rs3804100, are associated with allergic asthma (50), whereas TLR2 rs4696480 shows a significant link to asthma susceptibility (51). Interestingly, genetic variants within the TLR2-associated heterodimer network (including TLR1 and TLR6) confer strong protection against atopic asthma (37).
Collectively, these studies suggest that TLR2 predominantly exerts a protective role in asthma. Its activation can mitigate allergic inflammation through multiple mechanisms, including inhibiting type 2 responses, regulating antibody class switching, and inducing anti-inflammatory factors such as IL-10. However, the precise impact of TLR2 activation likely depends on factors such as the timing of microbial exposure and individual genetic background.
IgE and its high-affinity Fc receptor, FcϵRI, are pivotal in asthma pathogenesis. Genome-wide association studies have shown that the Fcϵ receptor Ia (FCER1A) gene, encoding the ligand-binding subunit of the high-affinity IgE receptor, is the main susceptibility locus for serum IgE levels. Genetic polymorphisms of the FCER1A gene may affect IgE levels related to asthma (52). In atopic dermatitis, an interaction between the TLR2 rs4696480 locus and the FCER1A rs2252226 locus has been linked to disease severity (53). Concurrently, TLR2 activation induces FcϵRI downregulation in human Langerhans cells (54). A suggested mechanism posits that TLR2 regulates the type 1 and type 2 balance. Nevertheless, this is complicated by the fact that TLR2 drives a type 1 deviation during the chronic phase of atopic dermatitis (55), contrasting with generally type 2-polarized immune response of asthma (56). TLR2 modulates FcϵRI expression in airway antigen-presenting cells. If TLR2 downregulates FcϵRI, it could attenuate IgE-mediated mast cell activation, thereby mitigating acute allergic responses. The relevance of the TLR2 rs4696480 polymorphism established in atopic dermatitis (57) raises the possibility of a similar effect in asthma, potentially influencing IgE levels via FCER1A expression regulation. However, further experimentation is required to confirm the direct regulatory effect of TLR2 on FcϵRI expression in the context of asthma.
4.1.3 TLR3
TLR3 is located on the endosomal membranes of epithelial cells, neuroglial cells, neurons, and dendritic cells and can recognize viral double-stranded RNA, polyinosinic:polycytidylic acid. This receptor plays a crucial role in autoimmune diseases, viral infections, and the development of asthma (58–61). Abnormal activation of the TLR3 signaling pathway and excessive activation of its downstream signaling factor TRIF can trigger the recruitment of local inflammatory cells and excessive synthesis of proinflammatory mediators, thereby causing various inflammatory diseases including asthma (62). In animal models of asthma, stimulation of the TLR3/TRIF signaling pathway has been demonstrated to exacerbate airway inflammation, promote inflammatory cell infiltration, and worsen the severity of the asthma response (62). After TLR3 is activated, significant infiltration of inflammatory cells occurs in the lung tissue, which subsequently leads to epithelial damage and histamine release. Excessively released inflammatory mediators promote the migration and differentiation of lung fibroblasts and airway matrix cells, enhance extracellular matrix synthesis, and cause thickening and fibrosis of the airway wall, thereby further exacerbating the severity of asthma (62, 63). Sugiura et al. also confirmed that in asthma, activation of the TLR3/TRIF signaling pathway could affect airway remodeling by promoting the differentiation of myofibroblasts (64).
Furthermore, studies have also revealed the important role of the TLR3/NF-κB/IRF3 signaling pathway in the progression of viral-induced asthma (65, 66). For example, respiratory syncytial virus infection could upregulate the expression of TLR3, resulting in the overexpression and release of downstream inflammatory factors through the TLR3/NF-κB/IRF3 pathway in asthmatic mice; this process also enhances extracellular matrix synthesis, further aggravating asthma symptoms (67). Additionally, Ramu et al. found that the TLR3/TAK1 signaling pathway regulates the production of IL-33 in bronchial smooth muscle cells during nasal rhinovirus infection (68).
The contribution of TLR3 genetic variants to asthma pathogenesis remains an area of investigation, with evidence showing some heterogeneity. While one study in Chinese Han patients failed to detect a significant association between TLR3 SNPs and asthma susceptibility or severity (69), other investigations have reported positive links. Specifically, research on aspirin-tolerant asthma found that TLR3 polymorphisms, particularly -299698G>T and 293391G>A [Leu412Phe] were associated with various respiratory phenotypes encompassing asthma (70). Moreover, both Canadian family-based analyses and an Australian population-based case-control study associated the rs1519309 SNP within TLR3 with atopic asthma (71). Functionally, TLR3 is known to mediate viral recognition and subsequent inflammatory responses, processes that are believed to significantly influence asthma progression. Indeed, excessive activation of TLR3 has been shown to exacerbate inflammation and fibrosis. However, despite correlations observed between TLR3 genetic variants and various asthma and allergic phenotypes, the specific impact of these variants on asthma susceptibility and severity exhibits considerable heterogeneity across different populations and studies. This inconsistency underscores the complex and multifactorial nature of TLR3’s regulation within the context of asthma pathogenesis.
4.1.4 TLR4
TLR4 is primarily expressed on myeloid-derived immune cells, including macrophages, monocytes, and dendritic cells (72–74) and also present on non-immune cells such as microglia, astrocytes, neurons, and endothelial cells (75). The outer membrane component of Gram-negative bacteria, lipopolysaccharide (LPS), serves as a specific ligand for TLR4 (76). Upon LPS binding, TLR4 activates the MyD88-dependent signaling pathway, triggering a cascade that activates downstream kinases. This cascade facilitates the nuclear translocation of NF-κB-associated factors, enabling their regulation of proinflammatory gene expression, particularly for IL-6, IL-1β, and tumor necrosis factor-α genes (77). TLR4 triggers immune responses via two distinct signaling pathways: the TRIF-dependent and the MyD88-dependent pathways (78). Stimulation of the MyD88/NF-κB pathway enhances the production of proinflammatory cytokines (77, 79), whereas the TRIF pathway contributes to asthma exacerbation (80).
Research by McAlees JW et al. highlights the differential roles of TLR4 depending on the cell type involved. Their findings indicate that TLR4 expression in hematopoietic cells is crucial for neutrophilic airway inflammation, observed both following LPS exposure and in Th17-driven neutrophilic responses to house dust mite lysates and OVA. In contrast, TLR4 expression by airway epithelial cells plays a key role specifically in robust eosinophilic airway inflammation when these same allergens are used for sensitization and challenge (81). However, broader research indicates that TLR4 activation predominantly promotes eosinophilic responses and type 2 inflammation. Specifically, studies demonstrate that LPS binding to TLR4 promotes type 2 immune responses, thereby exacerbating allergic respiratory inflammation (79). The cytokines and chemokines produced during type 2 responses are pivotal in asthma development, facilitating eosinophil recruitment and survival, promoting antigen-specific antibody production, and inducing mucus cell proliferation in the bronchial epithelium (82). Beyond recognizing exogenous microbial PAMPs, TLR4 also detects endogenous DAMPs (83), including HMGB1 and cellular heat shock proteins (84). The interaction between TLR4 and HMGB1 activates the NF-κB pathway, leading to the synthesis of inflammatory mediators and exacerbation of asthma symptoms. Furthermore, suppression of HSF1 has been shown to worsen OVA-induced airway inflammation and hyperresponsiveness in mice, likely through HMGB1 upregulation and activation of the TLR4/MyD88/NF-κB pathway (85). Aberrant activation of the TLR2/TLR4 signaling pathway can result in the excessive production of inflammatory factors, such as IL-1β, IL-5, IL-8, IL-4, and IL-13, and chemotactic mediators by neutrophils, monocytes, and type 2 lymphocytes within the airway. These mediators recruit neutrophils, stimulate goblet cell hyperplasia, and amplify the type 2 immune response. Consequently, this cascade leads to the synthesis of allergen-specific IgE and IgG1, increasing airway hyperresponsiveness and potentially triggering acute asthma attacks (77, 86). Collectively, these studies demonstrate that TLR4, by bridging innate and adaptive immunity and responding to diverse stimuli (both PAMPs and DAMPs), plays a multifaceted role in asthma pathophysiology, influencing inflammation, immune deviation, inflammatory cell recruitment, airway remodeling, and hyperresponsiveness, establishing TLR4 as a critical regulatory node.
Additionally, polymorphisms in the TLR4 gene are significantly correlated with an elevated risk of asthma (87). TLR4 rs4986791 was found to be significantly associated with asthma susceptibility in a meta-analysis, especially in the Asian population (88). The findings from a comparative analysis demonstrate an association between the TLR4 rs4986791 variant and bronchial asthma risk among Chinese children (89). TLR4 rs1927911 is also associated with antibiotic exposure and bronchiolitis in childhood asthma (90). Although mice expressing cosegregating SNPs D298G and T399I in TLR4 have enhanced responses to the house dust mite allergen, their responses to OVA and LPS were not significant (91). These findings indicate that TLR4 polymorphic variants exhibit differential interactions with allergic stimuli, potentially modulating immune responses in a genotype-dependent manner. Although existing studies (including population association analyses and animal models) have provided strong evidence for the role of TLR4 in asthma, there are still some limitations. For instance, population studies are mostly correlational analyses, making it difficult to determine causal relationships.
4.1.5 TLR5
TLR5 is a receptor that recognizes bacterial flagellar proteins and is not only expressed on the surface of myeloid cells, but also on the surface of epithelial cells, microglia, and astrocytes. It is abundantly expressed in airway epithelial cells and weakly expressed in alveolar macrophages (92). Shikhagaie et al. discovered that TLR5 is widely expressed in the lungs, but its expression is reduced in severe asthma. In patients with severe asthma, a significantly negative correlation is present between intracellular TLR5 immunoreactivity and IgE (93). This might be due to the fact that patients with severe asthma have insufficient TLR signaling during viral or bacterial infections, resulting in a reduced anti-allergic type1 response and a weakened or impaired immune defense mechanism. The binding of the flagellin protein to its receptor TLR5 affects the transcriptional profile of human primary epithelial cells, including genes encoding chemokines, matrix metalloproteinases, and antimicrobial molecules, thus inducing the expression of proinflammatory cytokines and chemokine mRNAs and promoting the secretion of granulocyte-macrophage colony-stimulating factor, C-X-C motif chemokine ligand-5, C-C motif chemokine-5, and C-X-C motif chemokine ligand-10, which may contribute to airway inflammation and remodeling (94). Bacterial flagellin in the household environment initiates allergic responses to indoor allergens in a TLR5-dependent manner, thereby promoting the development of allergic asthma (92). In OVA-specific asthmatic mouse models, the activation of TLR5 by bacterial products (flagellin) exacerbates allergic airway inflammation (95).
While the recognition of flagellin generally of TLR5 promotes inflammation in asthma, genetic variations in the TLR5 gene reveal a more complex picture, sometimes offering protection. For instance, a dominant-negative polymorphism (rs5744168) is linked to alleviated asthma symptoms in patients (95). Interestingly, another TLR5 variant (rs5744174) might increase susceptibility to bronchiolitis not caused by respiratory syncytial virus (non-RSV bronchiolitis), but this variant does not appear to be associated with the subsequent development of asthma following such bronchiolitis (96).
4.1.6 TLR6
TLR6 is mostly present on the cell membrane of myeloid cells and also expressed on epithelial cells, microglia, and astrocytes, where it forms heterodimers with other TLRs, particularly TLR2 (30). TLR6 forms heterodimers with TLR2 to recognize specific pathogen-associated components such as diacyl lipopeptides, lipoteichoic acid, and zymosan. Chun et al. examined the expression of TLR6 on CD14high cells in patients with asthma and found that TLR6 expression in the asthma group was significantly lower than that in the control group, and the expression level of TLR6 was statistically significant among patients with mild, moderate, and severe asthma (35). In the asthma model of TLR6-/- mice induced by fungal or house dust mite antigens, airway hyperresponsiveness, inflammation and remodeling worsened significantly, but the levels of IL-23 and IL-17 in the whole lung decreased significantly. Exogenous IL-23 treatment restored the production of IL-17A in asthma TLR6-/- mice, significantly reducing airway hyperresponsiveness, inflammation and pulmonary fungal burden (97). This indicates that TLR6 may play a certain protective or regulatory role in asthma through the IL-23/IL-17 pathway, and its low expression may be associated with the aggravation of asthma.
In the development of asthma, TLR6 not only regulates the immune response through signaling pathways, but its genetic polymorphisms also jointly influence the susceptibility to the disease along with environmental exposure. In children exposed to a farm environment, those carrying the TLR6 rs1039559 T-allele and the TLR6 rs5743810 C-allele have a lower risk of early-onset asthma compared with healthy children (98). The polymorphism of TLR6 rs5743810 has some exploratory correlation with airway reactivity (99) and a weak correlation with childhood asthma (100). Perhaps this conclusion (such as reducing risks) requires a larger sample size and more rigorous environmental control for verification.
4.1.7 TLR7/TLR8
TLR7 is mostly expressed in human plasmacytoid dendritic cells and, to a lesser degree, T cells, B cells, neutrophils, eosinophils, and mononuclear macrophages (101). TLR8 is predominantly expressed in myeloid dendritic cells, neutrophils, and monocytes (102). TLR7 and TLR8 recognize single-stranded RNA as their natural ligand (24). Yan et al. employed a combined analysis approach based on the gene expression profiles of induced sputum derived from the comprehensive Gene Expression Omnibus databases (GSE76262 and GSE137268) and found that reduced TLR7 expression correlated with airway eosinophilic inflammation and lung function in asthma (103). In a mouse asthma model induced by dust mites, TLR7 expression was significantly downregulated. TLR7-deficient asthmatic mice exhibited substantial inflammatory cell infiltration in the lungs, accompanied by elevated levels of IL-4, IL-10, IL-12, and IFN-γ, as well as increased phosphorylation of inhibitory κB kinase-α and NF-κBp65 expression. Importantly, the administration of a TLR7 agonist reversed these detrimental effects, suggesting that TLR7 upregulation mitigates asthma inflammation by inhibiting the NF-κB signaling pathway (104). Consistent with this finding, another study demonstrated that the interaction between TLR7 and its ligand alleviates eosinophilia and allergen-induced airway hyperreactivity in asthma, a process mediated by MyD88-dependent but MK2-independent signaling pathways (105). However, the interaction of TLR8 with its ligand promotes neutrophil proliferation, induces the secretion of chemokines and neutrophil elastase, triggering an airway inflammatory response that may counteract the protective effect of TLR7 (106). In rhinovirus-induced asthma, the IFN response is defective. Administration of the TLR7/8 agonist R848 to peripheral blood mononuclear cells from preschool asthmatic children was found to decrease IFN-αR mRNA levels while inducing IFN-λR1 mRNA. These findings suggest that targeting TLR7/8-mediated modulation of type I/III IFN signaling pathways could represent a novel therapeutic strategy to enhance antiviral immunity in pediatric asthma (107).
Although the available evidence suggests a protective role of TLR7 and a proinflammatory effect of TLR8, several key issues require further exploration. Firstly, the expression and function of TLR7/8 are likely influenced by factors including cell type, the underlying inflammatory milieu (e.g., eosinophilic versus neutrophilic predominance), and disease stage. Furthermore, the observed correlations in the literature—such as the association between TLR7 expression and eosinophilic inflammation—are not yet established as causal relationships. Secondly, although evidence supports the proinflammatory properties of TLR8, whether it directly counteracts the protective effects of TLR7 within the human body, and if this interaction is truly and universally significant, requires further direct investigation.
This complexity is further highlighted by genetic association studies. For example, a study found that an increased frequency of TLR7 gene polymorphisms is correlated with a higher risk of asthma in preschool children following infant bronchiolitis (108, 109). Conversely, although case-control and case-only studies found no significant association between TLR7 and TLR8 genetic variations and overall asthma susceptibility, these same variations were linked to asthma-related phenotypes. Specifically, polymorphisms in both TLR7 and TLR8 were associated with eosinophil count, serum IgE levels, lung function, and asthma severity (110). This suggests that genetic variations in TLR7 and TLR8 may indeed contribute to asthma pathogenesis, particularly influencing disease characteristics rather than initial susceptibility.
4.1.8 TLR9
TLR9 is mainly found in monocytes, plasmacytoid dendritic cells, B cells, microglia, astrocytes, and neurons. It recognizes self-DNA within immune complexes and unmethylated CpG DNA from viral, bacterial, and parasitic sources (100). When TLR9 binds to unmethylated CpG DNA on the surface of plasmacytoid dendritic cells and B cells, it can trigger type 1 immune responses and promote the differentiation and development of regulatory T cells. This process is accompanied by the production of IFN-γ and the release of IL-10 and transforming growth factor-β, and these cytokines work together to inhibit type 2 cell responses and alleviate asthma symptoms (111). Consistent with this anti-inflammatory potential, intranasal administration of CpG can alleviate experimental fungal asthma in a TLR9-dependent manner (112).
However, the role of TLR9 in asthma is complex. Some studies suggest that the TLR9–IL-2 axis may contribute to the deterioration of allergic asthma by exacerbating excessive IL-17A production (113). Furthermore, evidence indicates that TLR9 may regulate allergic airway inflammation by activating the NLRP3 inflammasome and inducing oxidative stress (114). Despite these complexities, TLR9 agonists hold promise as potential immunomodulators or vaccine adjuvants, exhibiting promising prospects in the research of immunotherapy for allergic diseases (115).
Additionally, genetic factors play a role, as specific TLR9 polymorphisms may influence the clinical manifestations of childhood asthma. For instance, the TLR9 rs187084 polymorphism is associated with the control of childhood bronchial asthma to some extent (116), and the overall TLR9 SNP profile is significantly correlated with susceptibility to childhood asthma (109). This genetic link is further supported by studies on bronchiolitis-induced wheezing, which suggest that the TLR9 rs187084 gene polymorphism may be a potential genetic factor in its occurrence and development (117).
4.1.9 TLR10
TLR10 is a recently identified member of the human TLR family. Currently, it is regarded as the only orphan receptor in this family, and its exact function and natural ligands remain unclear (118). TLR10 is expressed as a type I transmembrane protein in various immune cells such as B cells and dendritic cells (119). Its mRNA is highly expressed in lymphoid tissues such as the spleen, lymph nodes, thymus, and tonsils.
Although there is a lack of in-depth analysis of its biological function, genetic studies have found that the polymorphisms of the human TLR10 gene are associated with various disease states, including bacterial infections, asthma, autoimmune diseases, and cancer (120–122). Lazarus et al. confirmed that TLR10 gene variations are related to asthma susceptibility (122). In particular, TLR10 is associated with the occurrence of bronchiolitis post-asthma, which may increase the risk of developing asthma in infants who have suffered from bronchiolitis before the age of 11–13 (38, 108, 116). These findings suggest that TLR10 may play a potential role in the pathogenesis of respiratory diseases, especially in the onset of childhood asthma, and warrant further in-depth research.
4.2 TLR signaling pathways involved in asthma
4.2.1 MyD88 pathway
The TLR signaling pathway typically initiates downstream signal transduction through two major adaptor proteins: MyD88 and TRIF. Among these, MyD88 plays a central role in signal transduction and is essential for the activation of most TLRs (123). Structurally, MyD88 contains three distinct functional domains: an N-terminal death domain, a central intermediate region, and a C-terminal Toll/IL-1 receptor (TIR) domain. The cytoplasmic region of TLRs shares significant homology with the TIR domain of the IL-1 receptor (IL-1R) family, which is critical for downstream signaling. TIR domain-containing adaptor proteins are essential for TIR-mediated signaling, as they are required for the activation of TLR4 and TLR2 pathways, but not for TLR5, TLR7, or TLR9 (124, 125).
Most TLRs, including TLR1/2, TLR4, TLR5, TLR6/2, TLR7/8, and TLR9, transmit signals through the MyD88-dependent pathway (126). This pathway activates the NF-κB and MAPK signaling cascades, leading to the production of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α (47, 86, 104, 115, 127). In the context of asthma, dysregulation or aberrant activation of the MyD88-dependent signaling pathway has been implicated in the development of immune imbalance, characterized by enhanced type 2 immune responses, suppression of regulatory T cell function, and exacerbated airway inflammation and remodeling. Specifically, TLR4, which recognizes both exogenous ligands such as LPS from Gram-negative bacteria and endogenous danger signals like HMGB1, activates the NF-κB and MAPK pathways via the MyD88-dependent pathway. This activation promotes Th2 cell differentiation and IgE production, which are key features of allergic asthma (85, 86). Similarly, TLR2/1 and TLR2/6, which recognize bacterial lipoproteins and peptidoglycans, also signal through the MyD88 pathway, inducing the expression of IL-12 and IFN-γ. Under certain conditions, this may suppress type 2 responses, but excessive activation can lead to chronic inflammation (47, 49). Furthermore, TLR7/8, which detects single-stranded RNA from viruses, activates the MyD88 and IRF7 pathways, resulting in the production of IFN-α/β and IL-12. This signaling is particularly relevant in viral-induced asthma, highlighting the critical role of MyD88-dependent pathways in linking microbial exposure to immune dysregulation in allergic diseases (105, 107).
4.2.2 TRIF pathway
TRIF is extensively expressed across various cell types and is located within the cytoplasm under resting conditions. The TRIF signaling pathway is shared between TLR3 and TLR4 (128). Specifically, TRIF can directly interact with TLR3 or associate with TLR4 indirectly through the TRIF-associated adaptor molecules (6). Upon binding to TLR4, activation of the TRIF pathway occurs within the endosomal compartment following TLR4/MD2 complex internalization. Activation of the TRIF pathway involves activation of tumor necrosis factor receptor-associated factor 3, induction of nuclear translocation of IRF3, and recruitment of IFN-β. These processes are mediated by TRIF-associated adaptor molecules and TIR domain-containing adaptor proteins (129). The TRIF-dependent pathway can activate IRF3 and NF-κB, inducing proinflammatory cytokine gene expression and type I IFN production. This mechanism exacerbates asthma (130).
Although the role of TLRs in asthma has been extensively investigated, several controversies remain unresolved, highlighting the complexity of TLR-mediated immune regulation in allergic diseases. First, some TLRs exhibits a dual and context-dependent role in asthma: while it can drive pro-inflammatory responses by activating NF-κB and MAPK pathways, it may also exert protective effects (95, 96). This duality underscores the need for a more nuanced understanding of functional outcomes of TLRs in different disease contexts. Second, cross-regulation among TLR signaling pathways adds another layer of complexity. TLRs can interact in both synergistic and antagonistic manners, leading to divergent immune responses. Such interactions may contribute to the heterogeneity of asthma phenotypes and complicate the interpretation of experimental findings (105, 106). Finally, individual variability and epigenetic regulation further complicate the picture. The activity of TLR signaling pathways may be influenced by epigenetic modifications which may alter gene expression and immune cell function. These factors not only contribute to inter-individual differences in disease susceptibility but also highlight the importance of integrating epigenetic perspectives in future TLR research.
4.3 The association of TLRs with microbial exposure and epigenetic regulation in allergic diseases
TLRs are pivotal in shaping immune responses within allergic diseases, acting as sentinels that recognize microbial components present in the environment. Consistent with the hygiene hypothesis, diminished early-life microbial exposure is thought to skew immune development towards type 2 imbalance, thereby increasing susceptibility to allergic conditions like asthma (131). The TLR signaling pathway directly influences the trajectory of the immune response upon encountering microbial molecules. For example, Fuchs et al. demonstrated that TLR2/6 agonists could suppress type 2 inflammation, concurrently reducing airway hyperresponsiveness and mucus secretion (132). This interplay further underscores the complexity, as the combined application of TLR agonists with miR-146a mimics was found to attenuate OVA-induced asthma inflammation in mice (48).
Crucially, the relationship between TLRs, microbial exposure, and immune outcomes is deeply intertwined with epigenetic regulation. Maternal smoking, for instance, can induce aberrant TLR signaling in newborns, a key innate mechanism for microbial recognition, potentially laying the molecular groundwork for heightened infant infection susceptibility (133). Conversely, prenatal exposure to non-pathogenic microorganisms, engaging TLR-dependent pathways, significantly lowers the asthma risk of offspring (134). This protective effect of microbe-TLR interaction appears intrinsically linked to epigenetic modifications.
The expression of miR-146a, a common regulator of both TLR4 and ST2 signaling, serves as a prime example. Its levels are modulated by the intensity of microbial encounters early in life, suggesting it may be a critical epigenetic link connecting early environmental stimuli to adult asthma susceptibility (135). Furthermore, when air allergens and respiratory viruses activate TLRs, the downstream oxidative stress can induce modifications like the oxidation of guanine bases within inflammatory gene regulatory regions. These modifications act as epigenetic marks associated with inflammation induction. Targeting the pathways that induce this epigenetic reprogramming offers a potential therapeutic strategy to reverse airway remodeling seen in various chronic airway diseases (136).
In essence, TLRs function as environmental sensing hubs, orchestrating the reprogramming of epigenetic markers, such as miR-146a, in response to microbial exposure. This dynamic regulation significantly influences the initiation and progression of asthma. However, the intricate details of these mechanisms undoubtedly warrant further investigation.
5 TLR-related asthma treatment strategies
5.1 TLR antagonists/inhibitors
The involvement of TLRs in the pathogenesis of asthma is well-documented, with excessive activation of TLRs being closely associated with the development of the disease. Therefore, blocking TLR signaling pathways has emerged as a potential therapeutic strategy (137).
Among various TLRs, TLR4 has been the most extensively studied in the context of asthma treatment. Several compounds have been investigated for their ability to modulate TLR4 signaling and alleviate asthma symptoms. For instance, saxagliptin, a dipeptidyl peptidase-4 inhibitor, has been shown to significantly reduce airway inflammation in OVA-induced asthmatic mice by inhibiting the TLR4/reactive oxygen species/NF-κB signaling pathway. It also mitigates oxidative stress in lung tissue and lowers the levels of NF-κB and TLR4, suggesting its potential as a therapeutic agent (138). Resveratrol, a natural polyphenol found in grape skins, berries, and nuts, has demonstrated therapeutic potential in various pulmonary diseases, including pulmonary fibrosis, atherosclerosis, and pulmonary hypertension (139–141). In the context of asthma, high-dose resveratrol can suppress the production of inflammatory cytokines by inhibiting the HMGB1/TLR4/NF-κB pathway, thereby reducing airway inflammation and remodeling (142). Paeonol, an active compound derived from Paeonia suffruticosa, has been shown to downregulate TLR4 expression and block the nuclear translocation of NF-κB in asthma models. It also reduces the phosphorylation of p38 and extracellular signal-regulated kinase (ERK), leading to improved symptoms in OVA-induced asthma (143).
In addition to small-molecule inhibitors, other therapeutic approaches targeting TLR signaling include the use of probiotics and prebiotics, which have been shown to suppress inflammatory cell infiltration and reduce the production of inflammatory mediators, suggesting potential benefits for asthma patients (77). Small-molecule compounds that modulate TLR activity, particularly those targeting TLR7, TLR8, and TLR9, are also being explored for the management of inflammation-related diseases, including asthma (6, 144).
5.2 TLR agonists
Currently, several studies are exploring the therapeutic potential of TLR agonists in the treatment of asthma. Among them, the TLR2 agonist Pam3CSK4 has shown promising effects. Animal studies by Liao et al. demonstrated that Pam3CSK4 reduces type 2 inflammatory cytokines and Th17 cell levels, alleviates airway inflammation, and decreases airway hyperresponsiveness, suggesting its potential in asthma management (145). In addition, TLR4 agonists are primarily used as adjuvants in allergic vaccines to promote immune tolerance to TLR4 ligands and suppress TLR4 receptor expression. Monophosphoryl lipid A, a TLR4 agonist, has shown significant therapeutic effects when used as an allergen adjuvant, particularly in patients with asthma who exhibit elevated TLR4 expression in the airways. Resiquimod, a dual agonist of TLR7 and TLR8, has also been investigated for its anti-inflammatory effects in experimental asthma models. It has been shown to reduce Th2 inflammatory cytokine release, decrease eosinophil infiltration in the bronchus, and lower serum IgE levels (146). Despite these positive findings, the use of TLR agonists in asthma treatment remains controversial due to potential adverse effects. For example, resiquimod may cause local irritation, such as erythema and itching, when applied topically. In summary, while TLR agonists show promise as potential therapeutic agents for asthma, further research is needed to fully evaluate their safety, efficacy, and optimal application in clinical settings.
5.3 Mucin 1
Mucin 1 is a transmembrane glycoprotein of the mucin family, composed of an N-terminal extracellular subunit and a C-terminal cytoplasmic subunit (147). It is expressed not only in epithelial cells but also in immune cells such as dendritic cells and macrophages (148). Mucin 1 exerts anti-inflammatory effects by inhibiting TLR activation during bacterial and viral infections (149). Importantly, it plays a key role in asthma pathogenesis by regulating the TLR4/MyD88/NF-κB signaling pathway, thereby suppressing neutrophil-mediated airway inflammation. These findings suggest that Mucin 1 may represent a promising therapeutic target for asthma treatment.
5.4 HMGB1
HMGB1 is a highly conserved nuclear protein that can be actively secreted by macrophages, monocytes, and other immune cells. It is released passively from damaged necrotic tissue cells (150, 151). As an endogenous DAMP, HMGB1 contributes to the progression of allergic respiratory disorders by interacting with TLR4 and the receptor for advanced glycation end-products. The activated TLR4/MyD88/NF-κB signaling pathway is crucial in HSF1/HMGB1-mediated asthma (85). Inhibition of the TLR4/MYD88/NF-κB pathway has demonstrated efficacy in alleviating neutrophil-induced airway inflammation in individuals with asthma (147). Experimental animal studies show that HMGB1 interacts with advanced glycation end-products, TLR2, and TLR4, leading to the activation and production of proinflammatory factors (152). Therefore, inhibiting HMGB1 can mitigate OVA-induced airway inflammation and hyper-responsiveness (84). Anti-HMGB1 antibodies not only inhibit the activation of HMGB1-mediated pathways but also reduce type 2 cytokine accumulation, inflammatory cell infiltration, and mucin production in diisononyl phthalates-induced asthma models, thereby alleviating airway hyperresponsiveness and lung tissue damage (153).
5.5 miRNAs
microRNAs (miRNAs), approximately 22 nucleotides long, are noncoding RNA molecules that regulate gene expression at the post-transcriptional level. They primarily bind to the 3′-untranslated region of target mRNAs, causing their translational inhibition or degradation (154). To date, most miRNAs have been identified within cells, exerting a considerable influence on asthma prognosis, treatment, and therapeutic drug development (155). For example, miR-21, miR-223, and let-7a have been implicated in the development and pathogenesis of various asthma forms (156). MiRNAs may contribute to alleviating asthma-related inflammation by modulating airway epithelial cells and regulating key signaling molecules, including forkhead box protein C1, PI3K, AKT, NF-κB, cyclin D1, and transforming growth factor β1. Additionally, TLR4 has been recognized as a direct target for miR-20a and miR-217.
5.6 Nanodevices
Asthma is a persistent inflammatory disorder affecting the respiratory tract, traditionally managed with inhaled corticosteroids and bronchodilators. However, these treatments have limitations, such as systemic side effects, brief retention time in the pulmonary system, and poor patient compliance. Based on the distinctive anatomical features of the lungs (encompassing a vast alveolar surface area, thin epithelial barrier, and low proteolytic activity (157), nanoparticles present a promising strategy for asthma treatment through targeted drug delivery and functional design. Among organic nanoparticles, liposomes and polymer-based nanoparticles have exhibited significant potential by encapsulating glucocorticoids (GCs) or immunomodulators. These nanoparticles enhance drug deposition rate in the airway mucosa, prolong local drug activity via controlled release mechanisms, and reduce the frequency of dosing, thereby improving treatment efficacy. Nanoparticles have emerged as a novel class of targeted and effective TLR inhibitors (158). They not only inhibit two pathways involved in TLR4 signaling (the TRIF-dependent activation of IRF and the MyD88-dependent activation of NK-κB) but also inhibit pathways associated with TLR2, TLR3, and TLR5 (6). While the application of nanoparticles in treating diseases is still in its nascent phase, their clinical efficacy requires further validation. Despite the advancements that nanotechnology has brought to the development of asthma drugs, the molecular mechanisms, performance modifications, and potential toxicological effects of nanoparticles should not be ignored.
5.7 GCs
GCs are currently the most effective drugs for asthma control (159), and one of its primary mechanisms involves regulating the expression of TLRs. GCs effectively inhibit airway inflammation by targeting multiple aspects of the inflammatory response, such as proinflammatory cytokine release inhibition, inflammatory cell infiltration decrease, and β2 receptor responsiveness enhancement in airway smooth muscle cells (160–162). For example, budesonide is an inhaled GC and relieves asthma by regulating the expression of TLR2, TLR4, and IL-10 in CD4+/CD25+ regulatory T cells, as well as the secretion of tumor necrosis factor-α and IL-6 from PBMCs in patients with asthma (163). GCs are commonly used to treat acute attacks of mild to moderate asthma. However, GCs have limited efficacy in many patients with severe asthma, primarily owing to the impairment of transcriptional inhibitors such as histone deacetylase 2, which results from oxidative stress-induced DNA damage (164). Additionally, GCs are associated with several side effects, including high blood pressure, central obesity, type 2 diabetes, insulin resistance, and osteoporosis. Managing these adverse effects remains a significant clinical challenge, highlighting the urgent need for alternative therapeutic strategies or the development of innovative pharmaceuticals to replace GCs.
6 Discussion and future prospects
TLRs are necessary for both adaptive and innate immunity, functioning as the primary defense mechanism against microbial infections in the host. While TLRs are crucial for the amplification of immune responses and pathogen recognition, their excessive activation can destabilize the immune system and facilitate the onset of inflammatory disorders. However, most reported TLRs are implicated in asthma development. Their precise role remains debated across different studies (Figure 1; Tables 1, 2). The controversy mainly revolves around three key aspects: whether TLR affects asthma through proinflammatory or regulatory mechanisms, the contrasting effects of different TLR subtypes on asthma progression, and the effect of environmental exposures (such as microorganisms or pollutants) on disease processes through TLR signaling (165). Several factors may contribute to these discrepancies, including variation in immune activation by different TLRs, differences in the timing and dose of microbial exposure, inconsistencies in animal models (e.g., OVA or house dust mite induction), and the presence of human TLR gene polymorphisms. While many TLR agonists and inhibitors have been developed for the management of inflammatory diseases, such as asthma, atherosclerosis, cancer, and autoimmune disorders, most studies have been conducted in animal models. However, owing to the significant differences between the animal model of asthma and human disease, further clinical translational studies are needed to validate these treatments for asthma. Additionally, environmental microbes can simultaneously activate multiple TLRs, making it more effective to develop antagonists or inhibitors that target multiple TLR signaling pathways rather than a single receptor. As a potential pharmacological target in asthma treatment, TLR modulation holds the potential to drive significant breakthroughs and expand therapeutic options for asthma management.
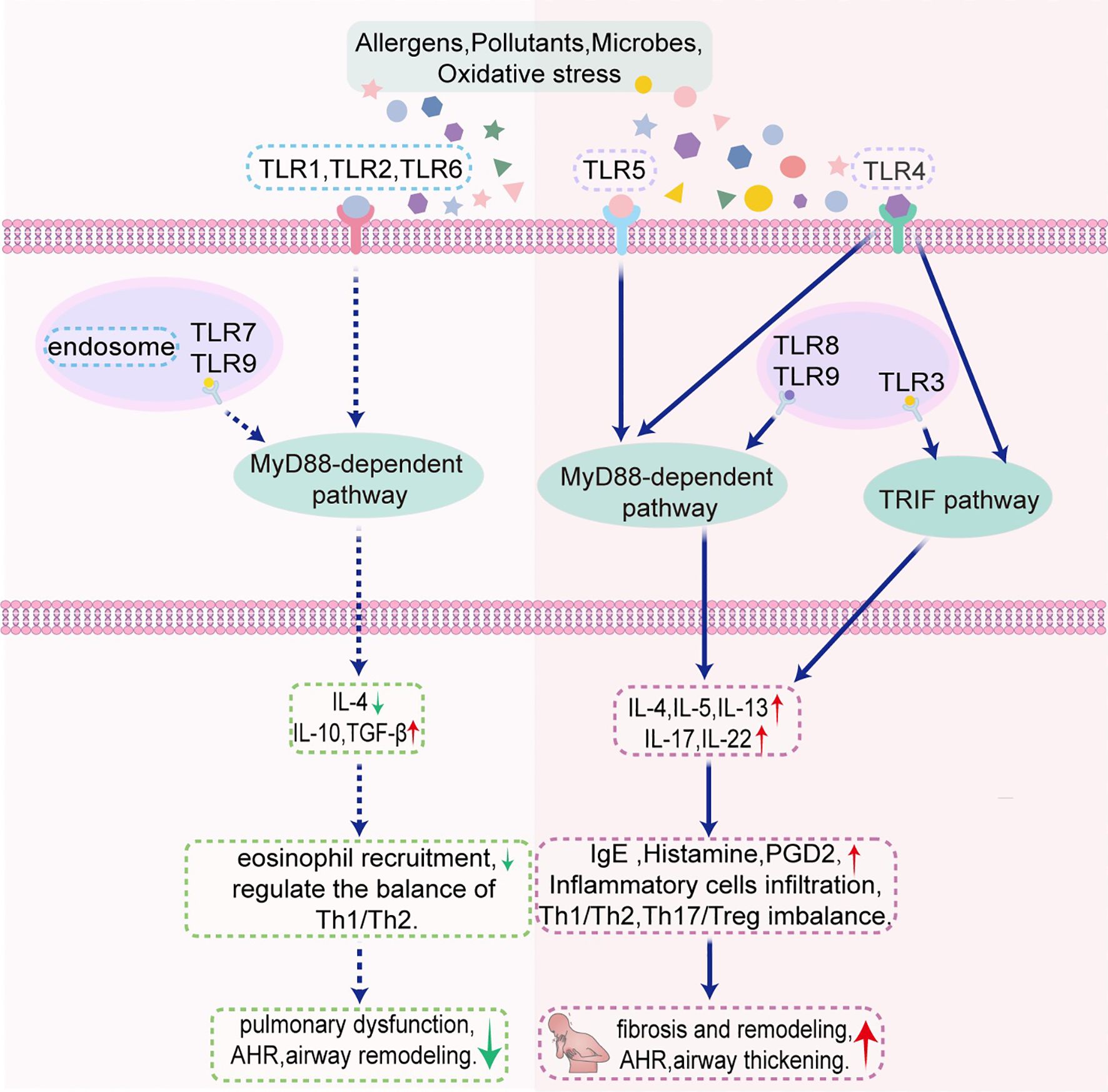
Figure 1. Schematic illustration of the mechanism by which TLRs regulate asthma in murine models through MyD88-dependent pathway and TRIF pathway. AHR, airway hyperresponsiveness; PGD2, prostaglandin D2; TLR, Toll-like receptor; TRIF, Toll/interleukin-1 receptor-domain-containing adapter-inducing interferon-β.
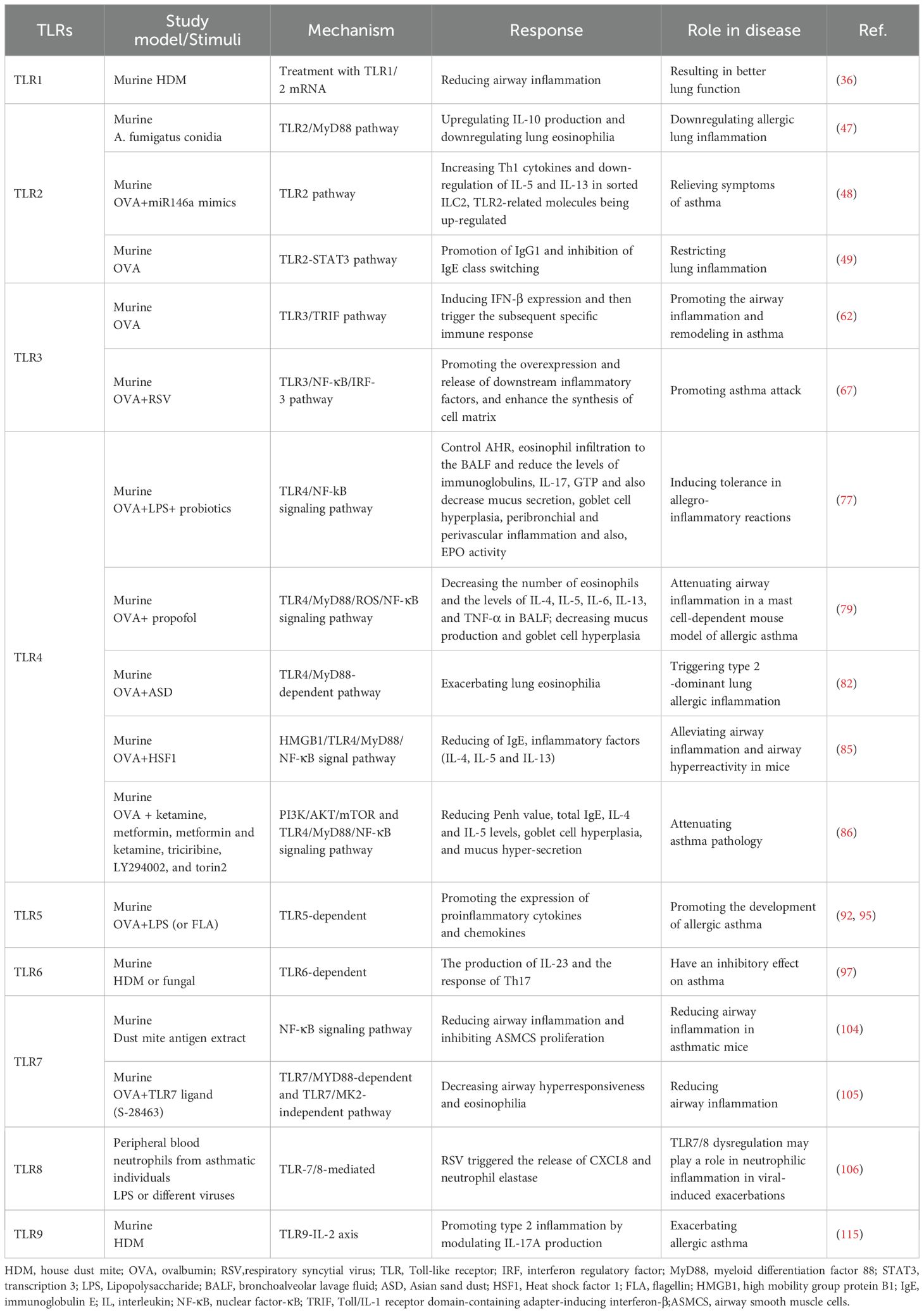
Table 1. TLRs regulate asthma through different mechanisms.
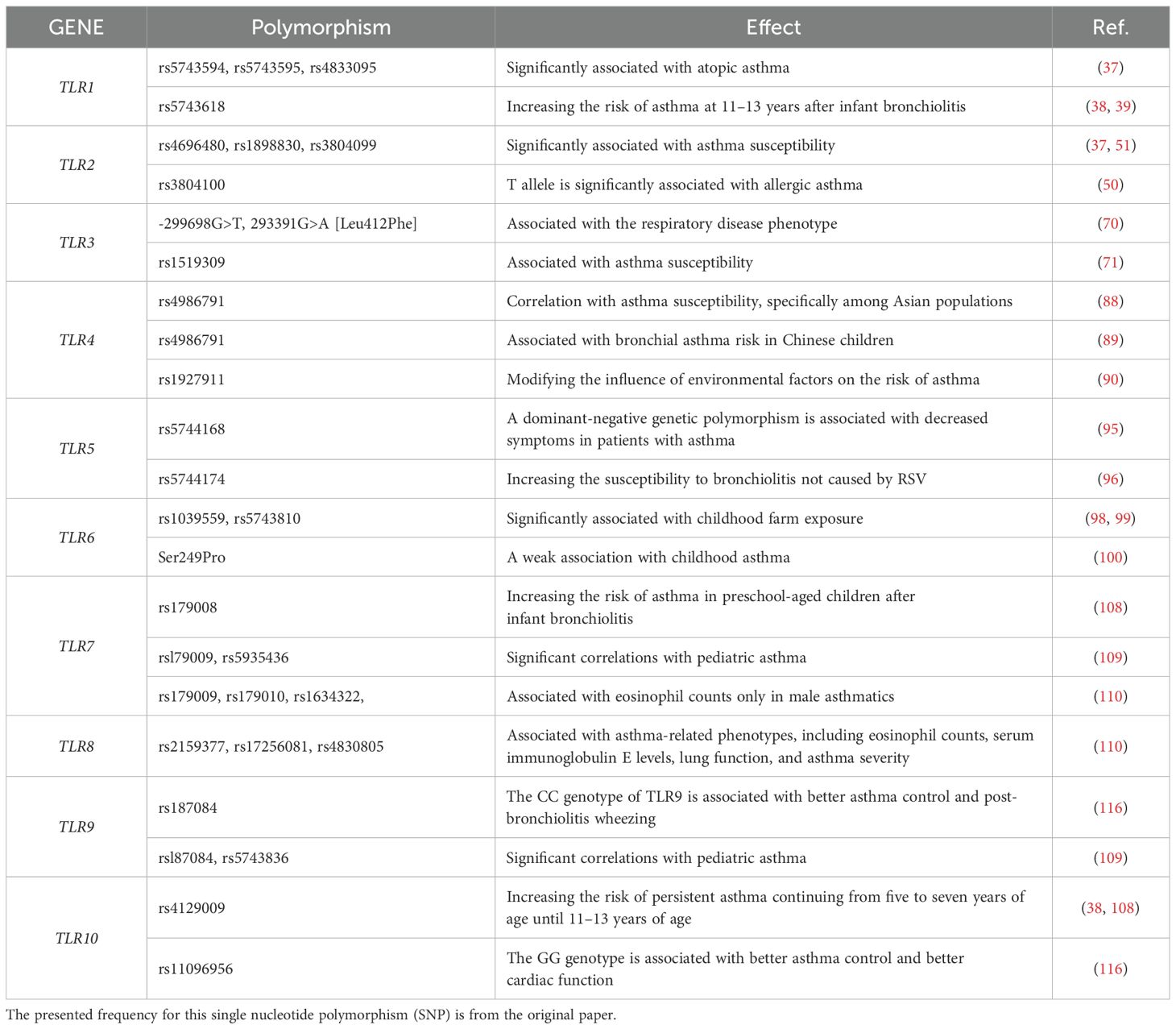
Table 2. Single nucleotide polymorphisms in TLR and association with human asthma.
Author contributions
XX: Writing – original draft. RY: Investigation, Software, Writing – original draft. ZY: Investigation, Methodology, Writing – original draft. CYL: Methodology, Writing – original draft. HX: Conceptualization, Funding acquisition, Writing – review & editing. CXL: Conceptualization, Funding acquisition, Project administration, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research received funding support from Lin He’s Academician Workstation of New Medicine and Clinical Translation in Jining Medical University, Jining, China (No. JYHL2021MS06), the Undergraduate Innovation Training Program of Jining Medical University (cx2025087), and the National Natural Science Foundation of China, Beijing, China (No. 82171810).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CpG, unmethylated cytosine-phosphate-guanine; DAMP, damage-associated molecular pattern; GC, glucocorticoid; HMGB1, high mobility group protein B1; IFN, interferon; IgE, immunoglobulin E; IL, interleukin; ILC, innate lymphoid cell; LPS, lipopolysaccharide; miRNA, microRNA; MyD88, myeloid differentiation factor 88; NF-κB, nuclear factor-κB; PAMP, pathogen-associated molecular pattern; Th cell, T helper cell; TIR, Toll/interleukin-1 receptor; TLR, Toll-like receptor; TRIF, TIR-domain-containing adapter-inducing interferon-β.
References
1. Sockrider M and Fussner L. What is asthma? Am J Respir Crit Care Med. (2020) 202:P25–p6. doi: 10.1164/rccm.2029P25
2. Papadopoulos NG, Miligkos M, and Xepapadaki P. A current perspective of allergic asthma: from mechanisms to management. Handb Exp Pharmacol. (2022) 268:69–93. doi: 10.1007/164_2021_483
3. Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. (2012) 18:716–25. doi: 10.1038/nm.2678
4. Kawai T and Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
5. Duan T, Du Y, Xing C, Wang HY, and Wang RF. Toll-like receptor signaling and its role in cell-mediated immunity. Front Immunol. (2022) 13:812774. doi: 10.3389/fimmu.2022.812774
6. Jin M, Fang J, Wang JJ, Shao X, Xu SW, Liu PQ, et al. Regulation of toll-like receptor (TLR) signaling pathways in atherosclerosis: from mechanisms to targeted therapeutics. Acta Pharmacol Sin. (2023) 44:2358–75. doi: 10.1038/s41401-023-01123-5
7. Gallardo-Zapata J, Perez-Figueroa E, Olivar-Lopez V, Medina-Sanson A, Jimenez-Hernandez E, Ortega E, et al. TLR agonists modify NK cell activation and increase its cytotoxicity in acute lymphoblastic leukemia. Int J Mol Sci. (2024) 25:7500. doi: 10.3390/ijms25137500
8. Mazzotta GM, Ceccato N, and Conte C. Synucleinopathies take their toll: are TLRs a way to go? Cells. (2023) 12:1231. doi: 10.3390/cells12091231
9. Chen YH, Wu KH, and Wu HP. Unraveling the complexities of toll-like receptors: from molecular mechanisms to clinical applications. Int J Mol Sci. (2024) 25:5037. doi: 10.3390/ijms25095037
10. Gans MD and Gavrilova T. Understanding the immunology of asthma: Pathophysiology, biomarkers, and treatments for asthma endotypes. Paediatr Respir Rev. (2020) 36:118–27. doi: 10.1016/j.prrv.2019.08.002
11. Li M, Wang C, Xu WT, and Zhong X. Sodium houttuyfonate plays a protective role in the asthmatic airway by alleviating the NLRP3-related pyroptosis and Th1/Th2 immune imbalance. Mol Immunol. (2023) 160:103–11. doi: 10.1016/j.molimm.2023.06.013
12. Qiu Q, Zhang W, Liu K, Huang F, Su J, Deng L, et al. Schisandrin A ameliorates airway inflammation in model of asthma by attenuating Th2 response. Eur J Pharmacol. (2023) 953:175850. doi: 10.1016/j.ejphar.2023.175850
13. Costanzo G, Costanzo G, Del Moro L, Nappi E, Pelaia C, Puggioni F, et al. Mast cells in upper and lower airway diseases: sentinels in the front line. Int J Mol Sci. (2023) 24:9771. doi: 10.3390/ijms24119771
14. Gandhi NA, Pirozzi G, and Graham NMH. Commonality of the IL-4/IL-13 pathway in atopic diseases. Expert Rev Clin Immunol. (2017) 13:425–37. doi: 10.1080/1744666X.2017.1298443
15. Hammad H and Lambrecht BN. The basic immunology of asthma. Cell. (2021) 184:1469–85. doi: 10.1016/j.cell.2021.02.016
16. Lopes F, Tibério I, Leme A, and Fairclough L. Editorial: The importance of Th17/Treg imbalance in asthma and COPD development and progression. Front Immunol. (2022) 13:1025215. doi: 10.3389/fimmu.2022.1025215
17. Ashley S, Eoin H, and John M. 'The microbiome and the pathophysiology of asthma. Respir Res. (2016) 17:163. doi: 10.1186/s12931-016-0479-4. M MD.
18. Medzhitov R, Preston-Hurlburt P, and Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. (1997) 388:394–7. doi: 10.1038/41131
19. Hashimoto C, Hudson KL, and Anderson KV. The Toll gene of Drosophila, required for dorsal-ventral embryonic polarity, appears to encode a transmembrane protein. Cell. (1988) 52:269–79. doi: 10.1016/0092-8674(88)90516-8
20. Arora S, Ahmad S, Irshad R, Goyal Y, Rafat S, Siddiqui N, et al. TLRs in pulmonary diseases. Life Sci. (2019) 233:116671. doi: 10.1016/j.lfs.2019.116671
21. Kawai T, Ikegawa M, Ori D, and Akira S. Decoding Toll-like receptors: Recent insights and perspectives in innate immunity. Immunity. (2024) 57:649–73. doi: 10.1016/j.immuni.2024.03.004
22. Lim CS, Jang YH, Lee GY, Han GM, Jeong HJ, Kim JW, et al. TLR3 forms a highly organized cluster when bound to a poly(I:C) RNA ligand. Nat Commun. (2022) 13:6876. doi: 10.1038/s41467-022-34602-0
23. Lee CW, Bakre A, Olivier TL, Alvarez-Narvaez S, Harrell TL, and Conrad SJ. Toll-like receptor ligands enhance vaccine efficacy against a virulent newcastle disease virus challenge in chickens. Pathogens. (2023) 12:1230. doi: 10.3390/pathogens12101230
24. Tong AJ, Leylek R, Herzner AM, Rigas D, Wichner S, Blanchette C, et al. Nucleotide modifications enable rational design of TLR7-selective ligands by blocking RNase cleavage. J Exp Med. (2024) 221:e20230341. doi: 10.1084/jem.20230341
25. Wang Y, Qiao SL, Wang J, Yu MZ, Wang NN, Mamuti M, et al. Engineered cpG-loaded nanorobots drive autophagy-mediated immunity for TLR9-positive cancer therapy. Adv Mater. (2024) 36:e2306248. doi: 10.1002/adma.202306248
26. Li D and Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther. (2021) 6:291. doi: 10.1038/s41392-021-00687-0
27. Song D, Niu J, Zhang Z, Sun Z, Wang D, Li J, et al. Purple sweet potato polysaccharide exerting an anti-inflammatory effect via a TLR-mediated pathway by regulating polarization and inhibiting the inflammasome activation. J Agric Food Chem. (2024) 72:2165–77. doi: 10.1021/acs.jafc.3c07511
28. Luo S, Lin H, Wu C, Zhu L, Hua Q, Weng Y, et al. Cholinergic macrophages promote the resolution of peritoneal inflammation. Proc Natl Acad Sci U.S.A. (2024) 121:e2402143121. doi: 10.1073/pnas.2402143121
29. Wicherska-Pawlowska K, Wrobel T, and Rybka J. Toll-like receptors (TLRs), NOD-like receptors (NLRs), and RIG-I-like receptors (RLRs) in innate immunity. TLRs, NLRs, and RLRs ligands as immunotherapeutic agents for hematopoietic diseases. Int J Mol Sci. (2021) 22:13397. doi: 10.3390/ijms222413397
30. Wang Y, Zhang S, Li H, Wang H, Zhang T, Hutchinson MR, et al. Small-molecule modulators of toll-like receptors. Acc Chem Res. (2020) 53:1046–55. doi: 10.1021/acs.accounts.9b00631
31. Stikker B, Trap L, Sedaghati-Khayat B, de Bruijn MJW, van Ijcken WFJ, de Roos E, et al. Epigenomic partitioning of a polygenic risk score for asthma reveals distinct genetically driven disease pathways. Eur Respir J. (2024) 64:2302059. doi: 10.1183/13993003.02059-2023
32. Guo S, Gao W, Zeng M, Liu F, Yang Q, Chen L, et al. Characterization of TLR1 and expression profiling of TLR signaling pathway related genes in response to Aeromonas hydrophila challenge in hybrid yellow catfish (Pelteobagrus fulvidraco female symbol x P. vachelli male symbol). Front Immunol. (2023) 14:1163781. doi: 10.3389/fimmu.2023.1163781
33. Zhou R, Liu L, and Wang Y. Viral proteins recognized by different TLRs. J Med Virol. (2021) 93:6116–23. doi: 10.1002/jmv.27265
34. Rao H, Tian H, Wang X, Huo C, Zhu L, Li Z, et al. Diversification of Toll-like receptor 1 in swamp eel (Monopterus albus). Dev Comp Immunol. (2024) 157:105190–. doi: 10.1016/j.dci.2024.105190
35. Chun E, Lee SH, Lee SY, Shim EJ, Cho SH, Min KU, et al. Toll-like receptor expression on peripheral blood mononuclear cells in asthmatics; implications for asthma management. J Clin Immunol. (2010) 30:459–64. doi: 10.1007/s10875-009-9363-z
36. Zeyer F, Mothes B, Will C, Carevic M, Rottenberger J, Nurnberg B, et al. mRNA-mediated gene supplementation of toll-like receptors as treatment strategy for asthma in vivo. PloS One. (2016) 11:e0154001. doi: 10.1371/journal.pone.0154001
37. Kormann MS, Depner M, Hartl D, Klopp N, Illig T, Adamski J, et al. Toll-like receptor heterodimer variants protect from childhood asthma. J Allergy Clin Immunol. (2008) 122:86–92,.e1-8. doi: 10.1016/j.jaci.2008.04.039
38. Törmänen S, Korppi M, Lauhkonen E, Koponen P, Teräsjärvi J, Vuononvirta J, et al. Toll-like receptor 1 and 10 gene polymorphisms are linked to postbronchiolitis asthma in adolescence. Acta Paediatr. (2018) 107:134–9. doi: 10.1111/apa.13984
39. Korppi M and Törmänen S. Toll-like receptor 1 and 10 variations increase asthma risk and review highlights further research directions. Acta Paediatr. (2019) 108:1406–10. doi: 10.1111/apa.14795
40. Radzyukevich YV, Kosyakova NI, and Prokhorenko IR. Impact of comorbidity of bronchial asthma and type 2 diabetes mellitus on the expression and functional activity of TLR2 and TLR4 receptors. Life (Basel). (2023) 13:550. doi: 10.3390/life13020550
41. Sobstyl M, Niedzwiedzka-Rystwej P, Grywalska E, Korona-Glowniak I, Sobstyl A, Bednarek W, et al. Toll-like receptor 2 expression as a new hallmark of advanced endometriosis. Cells. (2020) 9:1813. doi: 10.3390/cells9081813
42. Pergolizzi S, Fumia A, D'Angelo R, Mangano A, Lombardo GP, Giliberti A, et al. Expression and function of toll-like receptor 2 in vertebrate. Acta Histochem. (2023) 125:152028. doi: 10.1016/j.acthis.2023.152028
43. Oliveira-Nascimento L, Massari P, and Wetzler LM. The role of TLR2 in infection and immunity. Front Immunol. (2012) 3:79. doi: 10.3389/fimmu.2012.00079
44. Yarandi SS, Kulkarni S, Saha M, Sylvia KE, Sears CL, and Pasricha PJ. Intestinal bacteria maintain adult enteric nervous system and nitrergic neurons via toll-like receptor 2-induced neurogenesis in mice. Gastroenterology. (2020) 159:200–13 e8. doi: 10.1053/j.gastro.2020.03.050
45. Pattanaik KP, Sengupta S, Jit BP, Kotak R, and Sonawane A. Host-mycobacteria conflict: Immune responses of the host vs. the mycobacteria TLR2 and TLR4 ligands and concomitant host-directed therapy. Microbiol Res. (2022) 264:127153. doi: 10.1016/j.micres.2022.127153
46. Ren W, Zhao L, Sun Y, Wang X, and Shi X. HMGB1 and Toll-like receptors: potential therapeutic targets in autoimmune diseases. Mol Med. (2023) 29:117. doi: 10.1186/s10020-023-00717-3
47. Pauline P, Sofie DP, Giresse T, Rudi B, Johan G, Marta R, et al. Aspergillus fumigatus Recognition by Dendritic Cells Negatively Regulates Allergic Lung Inflammation through a TLR2/MyD88 Pathway. Am J Respir Cell Mol Biol. (2021) 64:39–49. doi: 10.1165/rcmb.2020-0083OC
48. Wang X, Lu X, Ma C, Ma L, and Han S. Combination of TLR agonist and miR146a mimics attenuates ovalbumin-induced asthma. Mol Med. (2020) 26:65. doi: 10.1186/s10020-020-00191-1
49. Li Y, Chen Q, Ji W, Fan Y, Huang L, Chu C, et al. TLR2 deficiency promotes IgE and inhibits IgG1 class-switching following ovalbumin sensitization. Ital J Pediatr. (2021) 47:162. doi: 10.1186/s13052-021-01088-3
50. Bjørnvold M, Munthe-Kaas MC, Egeland T, Joner G, Dahl-Jørgensen K, Njølstad PR, et al. A TLR2 polymorphism is associated with type 1 diabetes and allergic asthma. Genes Immun. (2009) 10:181–7. doi: 10.1038/gene.2008.100
51. Gao Y, Xiao H, Wang Y, and Xu F. Association of single-nucleotide polymorphisms in toll-like receptor 2 gene with asthma susceptibility: A meta-analysis. Med (Baltimore). (2017) 96:e6822. doi: 10.1097/md.0000000000006822
52. Potaczek DP, Michel S, Sharma V, Zeilinger S, Vogelberg C, von Berg A, et al. Different FCER1A polymorphisms influence IgE levels in asthmatics and non-asthmatics. Pediatr Allergy Immunol. (2013) 24:441–9. doi: 10.1111/pai.12083
53. Potaczek DP, Przytulska-Szczerbik A, Bazan-Socha S, Nastałek M, Wojas-Pelc A, Okumura K, et al. Interaction between functional polymorphisms in FCER1A and TLR2 and the severity of atopic dermatitis. Hum Immunol. (2020) 81:709–13. doi: 10.1016/j.humimm.2020.08.002
54. Herrmann N, Koch S, Leib N, Bedorf J, Wilms H, Schnautz S, et al. TLR2 down-regulates FcϵRI and its transcription factor PU.1 in human Langerhans cells. Allergy. (2013) 68:621–8. doi: 10.1111/all.12145
55. Vafaeian A, Rajabi F, and Rezaei N. Toll-like receptors in atopic dermatitis: pathogenesis and therapeutic implications. Heliyon. (2025) 11:e42226. doi: 10.1016/j.heliyon.2025.e42226
56. Papapostolou N and Makris M. Allergic asthma in the era of personalized medicine. J Pers Med. (2022) 12:1162. doi: 10.3390/jpm12071162
57. Zhou B, Liang S, Shang S, and Li L. Association of TLR2 and TLR9 gene polymorphisms with atopic dermatitis: a systematic review and meta-analysis with trial sequential analysis. Immunol Med. (2023) 46:32–44. doi: 10.1080/25785826.2022.2132683
58. Zhang X, Gao R, Zhang C, Teng Y, Chen H, Li Q, et al. Extracellular RNAs-TLR3 signaling contributes to cognitive impairment after chronic neuropathic pain in mice. Signal Transduct Target Ther. (2023) 8:292. doi: 10.1038/s41392-023-01543-z
59. Porsbjerg C, Nieto-Fontarigo JJ, Cerps S, Ramu S, Menzel M, Hvidtfeldt M, et al. Phenotype and severity of asthma determines bronchial epithelial immune responses to a viral mimic. Eur Respir J. (2022) 60:2102333. doi: 10.1183/13993003.02333-2021
60. Qi SY, Yang MM, Li CY, Yu K, and Deng SL. The HPV viral regulatory mechanism of TLRs and the related treatments for HPV-associated cancers. Front Immunol. (2024) 15:1407649. doi: 10.3389/fimmu.2024.1407649
61. Lincez PJ, Shanina I, and Horwitz MS. Changes in MDA5 and TLR3 sensing of the same diabetogenic virus result in different autoimmune disease outcomes. Front Immunol. (2021) 12:751341. doi: 10.3389/fimmu.2021.751341
62. Yang M, Wang HY, Chen JC, and Zhao J. Regulation of airway inflammation and remodeling in asthmatic mice by TLR3/TRIF signal pathway. Mol Immunol. (2017) 85:265–72. doi: 10.1016/j.molimm.2017.03.002
63. Jiang JJ, Chen SM, Li HY, Xie QM, and Yang YM. TLR3 inhibitor and tyrosine kinase inhibitor attenuate cigarette smoke/poly I:C-induced airway inflammation and remodeling by the EGFR/TLR3/MAPK signaling pathway. Eur J Pharmacol. (2021) 890:173654. doi: 10.1016/j.ejphar.2020.173654
64. Sugiura H, Ichikawa T, Koarai A, Yanagisawa S, Minakata Y, Matsunaga K, et al. Activation of Toll-like receptor 3 augments myofibroblast differentiation. Am J Respir Cell Mol Biol. (2009) 40:654–62. doi: 10.1165/rcmb.2008-0371OC
65. Menzel M, Ramu S, Calvén J, Olejnicka B, Sverrild A, Porsbjerg C, et al. Oxidative stress attenuates TLR3 responsiveness and impairs anti-viral mechanisms in bronchial epithelial cells from COPD and asthma patients. Front Immunol. (2019) 10:2765. doi: 10.3389/fimmu.2019.02765
66. Schaunaman N, Nichols T, Cervantes D, Hartsoe P, Ferrington DA, and Chu HW. The effect of a TLR3 agonist on airway allergic inflammation and viral infection in immunoproteasome-deficient mice. Viruses. (2024) 16:1384. doi: 10.3390/v16091384
67. Chen YQ, Zhou Y, Wang QL, Chen J, Chen H, Xie HH, et al. Conciliatory anti-allergic decoction attenuates pyroptosis in RSV-infected asthmatic mice and lipopolysaccharide (LPS)-induced 16HBE cells by inhibiting TLR3/NLRP3/NF-kappaB/IRF3 signaling pathway. J Immunol Res. (2022) 2022:1800401. doi: 10.1155/2022/1800401
68. Ramu S, Calvén J, Michaeloudes C, Menzel M, Akbarshahi H, Chung KF, et al. TLR3/TAK1 signalling regulates rhinovirus-induced interleukin-33 in bronchial smooth muscle cells. ERJ Open Res. (2020) 6:00147–2020. doi: 10.1183/23120541.00147-2020
69. Zhang Q, Fu XL, Qian FH, Cao Q, Mao ZD, Bai JL, et al. Polymorphisms in Toll-like receptor 3 are associated with asthma-related phenotypes in the Chinese Han patients. Int J Immunogenet. (2016) 43:383–90. doi: 10.1111/iji.12290
70. Palikhe NS, Kim SH, Kim JH, Losol P, Ye YM, and Park HS. Role of toll-like receptor 3 variants in aspirin-exacerbated respiratory disease. Allergy Asthma Immunol Res. (2011) 3:123–7. doi: 10.4168/aair.2011.3.2.123
71. Daley D, Park JE, He JQ, Yan J, Akhabir L, Stefanowicz D, et al. Associations and interactions of genetic polymorphisms in innate immunity genes with early viral infections and susceptibility to asthma and asthma-related phenotypes. J Allergy Clin Immunol. (2012) 130:1284–93. doi: 10.1016/j.jaci.2012.07.051
72. Rayees S, Rochford I, Joshi JC, Joshi B, Banerjee S, and Mehta D. Macrophage TLR4 and PAR2 signaling: role in regulating vascular inflammatory injury and repair. Front Immunol. (2020) 11:2091. doi: 10.3389/fimmu.2020.02091
73. Allali S, Rignault-Bricard R, de Montalembert M, Taylor M, Bouceba T, Hermine O, et al. HbS promotes TLR4-mediated monocyte activation and proinflammatory cytokine production in sickle cell disease. Blood. (2022) 140:1972–82. doi: 10.1182/blood.2021014894
74. Lim KH, Wang L, Dotse E, Wang M, Tiu CY, Wijanarko KJ, et al. TLR4 sensitizes plasmacytoid dendritic cells for antiviral response against SARS-CoV-2 coronavirus. J Leukoc Biol. (2024) 115:190–200. doi: 10.1093/jleuko/qiad111
75. Stierschneider A and Wiesner C. Shedding light on the molecular and regulatory mechanisms of TLR4 signaling in endothelial cells under physiological and inflamed conditions. Front Immunol. (2023) 14:1264889. doi: 10.3389/fimmu.2023.1264889
76. Ciesielska A, Matyjek M, and Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. (2021) 78:1233–61. doi: 10.1007/s00018-020-03656-y
77. Wu Z, Mehrabi Nasab E, Arora P, and Athari SS. Study effect of probiotics and prebiotics on treatment of OVA-LPS-induced of allergic asthma inflammation and pneumonia by regulating the TLR4/NF-kB signaling pathway. J Transl Med. (2022) 20:130. doi: 10.1186/s12967-022-03337-3
78. Pereira M, Durso DF, Bryant CE, Kurt-Jones EA, Silverman N, Golenbock DT, et al. The IRAK4 scaffold integrates TLR4-driven TRIF and MYD88 signaling pathways. Cell Rep. (2022) 40:111225. doi: 10.1016/j.celrep.2022.111225
79. Li HY, Meng JX, Liu Z, Liu XW, Huang YG, and Zhao J. Propofol attenuates airway inflammation in a mast cell-dependent mouse model of allergic asthma by inhibiting the toll-like receptor 4/reactive oxygen species/nuclear factor kappaB signaling pathway. Inflammation. (2018) 41:914–23. doi: 10.1007/s10753-018-0746-2
80. Stefan KL, Kim MV, Iwasaki A, and Kasper DL. Commensal microbiota modulation of natural resistance to virus infection. Cell. (2020) 183:1312–24 e10. doi: 10.1016/j.cell.2020.10.047
81. McAlees JW, Whitehead GS, Harley IT, Cappelletti M, Rewerts CL, Holdcroft AM, et al. Distinct Tlr4-expressing cell compartments control neutrophilic and eosinophilic airway inflammation. Mucosal Immunol. (2015) 8:863–73. doi: 10.1038/mi.2014.117
82. He M, Ichinose T, Song Y, Yoshida Y, Bekki K, Arashidani K, et al. Desert dust induces TLR signaling to trigger Th2-dominant lung allergic inflammation via a MyD88-dependent signaling pathway. Toxicol Appl Pharmacol. (2016) 296:61–72. doi: 10.1016/j.taap.2016.02.011
83. Fowler TE, Choudhary V, Melnyk S, Farsi M, Chang LY, Fortingo N, et al. Dioleoylphosphatidylglycerol inhibits heat shock protein B4 (HSPB4)-induced inflammatory pathways in vitro. Int J Mol Sci. (2023) 24:5839. doi: 10.3390/ijms24065839
84. Wang Y, Le Y, Wu J, Zhao W, Zhang Q, Xu G, et al. Inhibition of xanthine oxidase by allopurinol suppresses HMGB1 secretion and ameliorates experimental asthma. Redox Biol. (2024) 70:103021. doi: 10.1016/j.redox.2023.103021
85. Shang L, Wang L, Shi X, Wang N, Zhao L, Wang J, et al. HMGB1 was negatively regulated by HSF1 and mediated the TLR4/MyD88/NF-kappaB signal pathway in asthma. Life Sci. (2020) 241:117120. doi: 10.1016/j.lfs.2019.117120
86. Ma B, Athari SS, Mehrabi Nasab E, and Zhao L. PI3K/AKT/mTOR and TLR4/myD88/NF-κB signaling inhibitors attenuate pathological mechanisms of allergic asthma. Inflammation. (2021) 44:1895–907. doi: 10.1007/s10753-021-01466-3
87. Shukur W, Alyaqubi K, Dosh R, Al-Ameri A, Al-Aubaidy H, Al-Maliki R, et al. Association of Toll-like receptors 4 (TLR-4) gene expression and polymorphisms in patients with severe asthma. J Med Life. (2021) 14:544–8. doi: 10.25122/jml-2021-0173
88. Guo N, Tian H, Song T, and Peng Y. Association of TLR4 gene rs4986790 and rs4986791 polymorphisms with asthma susceptibility: meta-analysis and trial sequential analysis. Ann Saudi Med. (2024) 44:183–94. doi: 10.5144/0256-4947.2024.183
89. Chen X, Wang K, Yao Q, Peng L, and Wei L. The relationship between the rs4986791 variant of the TLR4 gene and the severity of bronchial asthma in children. Asian Pac J Allergy Immunol. (2024) 42:159–64. doi: 10.12932/ap-100920-0954
90. Lee E, Kwon JW, Kim HB, Yu HS, Kang MJ, Hong K, et al. Association between antibiotic exposure, bronchiolitis, and TLR4 (rs1927911) polymorphisms in childhood asthma. Allergy Asthma Immunol Res. (2015) 7:167–74. doi: 10.4168/aair.2015.7.2.167
91. Fink MY, Qi X, Shirey KA, Fanaroff R, Chapoval S, Viscardi RM, et al. Mice expressing cosegregating single nucleotide polymorphisms (D298G and N397I) in TLR4 have enhanced responses to house dust mite allergen. J Immunol. (2022) 208:2085–97. doi: 10.4049/jimmunol.2100926
92. Wilson RH, Maruoka S, Whitehead GS, Foley JF, Flake GP, Sever ML, et al. The Toll-like receptor 5 ligand flagellin promotes asthma by priming allergic responses to indoor allergens. Nat Med. (2012) 18:1705–10. doi: 10.1038/nm.2920
93. Shikhagaie MM, Andersson CK, Mori M, Kortekaas Krohn I, Bergqvist A, Dahl R, et al. Mapping of TLR5 and TLR7 in central and distal human airways and identification of reduced TLR expression in severe asthma. Clin Exp Allergy. (2014) 44:184–96. doi: 10.1111/cea.12176
94. Losol P, Ji MH, Kim JH, Choi JP, Yun JE, Seo JH, et al. Bronchial epithelial cells release inflammatory markers linked to airway inflammation and remodeling in response to TLR5 ligand flagellin. World Allergy Organ J. (2023) 16:100786. doi: 10.1016/j.waojou.2023.100786
95. Whitehead GS, Hussain S, Fannin R, Trempus CS, Innes CL, Schurman SH, et al. TLR5 activation exacerbates airway inflammation in asthma. Lung. (2020) 198:289–98. doi: 10.1007/s00408-020-00337-2
96. Törmänen S, Teräsjärvi J, Lauhkonen E, Helminen M, Koponen P, Korppi M, et al. TLR5 rs5744174 gene polymorphism is associated with the virus etiology of infant bronchiolitis but not with post-bronchiolitis asthma. Health Sci Rep. (2018) 1:e38. doi: 10.1002/hsr2.38
97. Moreira AP, Cavassani KA, Ismailoglu UB, Hullinger R, Dunleavy MP, Knight DA, et al. The protective role of TLR6 in a mouse model of asthma is mediated by IL-23 and IL-17A. J Clin Invest. (2011) 121:4420–32. doi: 10.1172/JCI44999
98. Lau MY, Dharmage SC, Burgess JA, Win AK, Lowe AJ, Lodge C, et al. The interaction between farming/rural environment and TLR2, TLR4, TLR6 and CD14 genetic polymorphisms in relation to early- and late-onset asthma. Sci Rep. (2017) 7:43681. doi: 10.1038/srep43681
99. Lauhkonen E, Koponen P, Vuononvirta J, Teräsjärvi J, Nuolivirta K, Toikka JO, et al. Gene Polymorphism of Toll-Like Receptors and Lung Function at Five to Seven Years of Age after Infant Bronchiolitis. PloS One. (2016) 11:e0146526. doi: 10.1371/journal.pone.0146526
100. Hoffjan S, Stemmler S, Parwez Q, Petrasch-Parwez E, Arinir U, Rohde G, et al. Evaluation of the toll-like receptor 6 Ser249Pro polymorphism in patients with asthma, atopic dermatitis and chronic obstructive pulmonary disease. BMC Med Genet. (2005) 6:34. doi: 10.1186/1471-2350-6-34
101. Soni C, Perez OA, Voss WN, Pucella JN, Serpas L, Mehl J, et al. Plasmacytoid dendritic cells and type I interferon promote extrafollicular B cell responses to extracellular self-DNA. Immunity. (2020) 52:1022–38 e7. doi: 10.1016/j.immuni.2020.04.015
102. Youness A, Cenac C, Faz-Lopez B, Grunenwald S, Barrat FJ, Chaumeil J, et al. TLR8 escapes X chromosome inactivation in human monocytes and CD4(+) T cells. Biol Sex Differ. (2023) 14:60. doi: 10.1186/s13293-023-00544-5
103. Yan K and Liang Y. Decreased TLR7 expression was associated with airway eosinophilic inflammation and lung function in asthma: evidence from machine learning approaches and experimental validation. Eur J Med Res. (2024) 29:116. doi: 10.1186/s40001-023-01622-5
104. Song L, Luan B, Xu Q-R, and Wang X-F. Effect of TLR7 gene expression mediating NF-κB signaling pathway on the pathogenesis of bronchial asthma in mice and the intervention role of IFN-γ. Eur Rev Med Pharmacol Sci. (2021) 25:866–79. doi: 10.26355/eurrev_202101_24655
105. Moisan J, Camateros P, Thuraisingam T, Marion D, Koohsari H, Martin P, et al. TLR7 ligand prevents allergen-induced airway hyperresponsiveness and eosinophilia in allergic asthma by a MYD88-dependent and MK2-independent pathway. Am J Physiol Lung Cell Mol Physiol. (2006) 290:L987–95. doi: 10.1152/ajplung.00440.2005
106. Tang FS, Van Ly D, Spann K, Reading PC, Burgess JK, Hartl D, et al. Differential neutrophil activation in viral infections: Enhanced TLR-7/8-mediated CXCL8 release in asthma. Respirology. (2016) 21:172–9. doi: 10.1111/resp.12657
107. Krug J, Kiefer A, Koelle J, Vuorinen T, Xepapadaki P, Stanic B, et al. TLR7/8 regulates type I and type III interferon signalling in rhinovirus 1b-induced allergic asthma. Eur Respir J. (2021) 57:2001562. doi: 10.1183/13993003.01562-2020
108. Törmänen S, Korppi M, Teräsjärvi J, Vuononvirta J, Koponen P, Helminen M, et al. Polymorphism in the gene encoding toll-like receptor 10 may be associated with asthma after bronchiolitis. Sci Rep. (2017) 7:2956. doi: 10.1038/s41598-017-03429-x
109. Shan L, Hou P, Kang X, and Shang Y. Effects of single-nucleotide polymorphisms in the TLR7 and TLR9 genes of asthmatic children. Ann Clin Lab Sci. (2018) 48:601–7.
110. Zhang Q, Qian FH, Yin XW, Cao Q, Bai JL, Du Q, et al. Associations of toll-like receptor 7 and 8 polymorphisms with asthma and asthma-related phenotypes in a chinese han population. Iran J Allergy Asthma Immunol. (2015) 14:569–80.
111. Gupta GK and Agrawal DK. CpG oligodeoxynucleotides as TLR9 agonists: therapeutic application in allergy and asthma. BioDrugs: Clin immunotherapeutics biopharmaceuticals Gene Ther. (2010) 24:225–35. doi: 10.2165/11536140-000000000-00000
112. Ramaprakash H and Hogaboam CM. Intranasal CpG therapy attenuated experimental fungal asthma in a TLR9-dependent and -independent manner. Int Arch Allergy Immunol. (2010) 152:98–112. doi: 10.1159/000265531
113. Murakami Y, Ishii T, Nunokawa H, Kurata K, Narita T, and Yamashita N. TLR9-IL-2 axis exacerbates allergic asthma by preventing IL-17A hyperproduction. Sci Rep. (2020) 10:18110. doi: 10.1038/s41598-020-75153-y
114. Zhao CC, Xie QM, Xu J, Yan XB, Fan XY, and Wu HM. TLR9 mediates the activation of NLRP3 inflammasome and oxidative stress in murine allergic airway inflammation. Mol Immunol. (2020) 125:24–31. doi: 10.1016/j.molimm.2020.06.016
115. Shokrollah F, Narjes A, Ali M, Ali KH, Masoud P, and Nima R. TLR9-based immunotherapy for the treatment of allergic diseases. Immunotherapy. (2017) 9:339–46. doi: 10.2217/imt-2016-0104
116. Rabie RA, Hussien AE, Abdelhameed HS, Shedeed SA, Almadani N, Nofal HA, et al. Role of toll-like receptors nine and ten polymorphisms in childhood bronchial asthma control and their relation to cardiac function. Diagnostics (Basel). (2025) 15:817. doi: 10.3390/diagnostics15070817
117. Nuolivirta K, Törmänen S, Teräsjärvi J, Vuononvirta J, Koponen P, Korppi M, et al. Post-bronchiolitis wheezing is associated with toll-like receptor 9 rs187084 gene polymorphism. Sci Rep. (2016) 6:31165. doi: 10.1038/srep31165
118. Chuang T-H and Ulevitch RJ. Identification of hTLR10: a novel human Toll-like receptor preferentially expressed in immune cells. Biochim Biophys Acta (BBA) - Gene Structure Expression. (2001) 1518:157–61. doi: 10.1016/s0167-4781(00)00289-x
119. Veit Hornung SR, Britsch S, Krug A, Jahrsdörfer B, Giese T, Endres S, et al. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to cpG oligodeoxynucleotides1. J Immunol. (2002) 168:4531–7. doi: 10.4049/jimmunol.168.9.4531
120. Guirado M, Gil H, Saenz-Lopez P, Reinboth J, Garrido F, Cozar JM, et al. Association between C13ORF31, NOD2, RIPK2 and TLR10 polymorphisms and urothelial bladder cancer. Hum Immunol. (2012) 73:668–72. doi: 10.1016/j.humimm.2012.03.006
121. Tenhu E, Terasjarvi J, Cruzeiro ML, Savonius O, Rugemalira E, He Q, et al. Gene polymorphisms of TLR10: effects on bacterial meningitis outcomes in Angolan children. APMIS. (2022) 130:221–9. doi: 10.1111/apm.13213
122. Lazarus R, Raby BA, Lange C, Silverman EK, Kwiatkowski DJ, Vercelli D, et al. TOLL-like receptor 10 genetic variation is associated with asthma in two independent samples. Am J Respir Crit Care Med. (2004) 170:594–600. doi: 10.1164/rccm.200404-491OC
123. Deguine J and Barton GM. MyD88: a central player in innate immune signaling. F1000Prime Rep. (2014) 6:97. doi: 10.12703/P6-97
124. Horng T, Barton GM, Flavell RA, and Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. (2002) 420:329–33. doi: 10.1038/nature01180
125. Manik MK, Pan M, Xiao L, Gu W, Kim H, Pospich S, et al. Structural basis for TIR domain-mediated innate immune signaling by Toll-like receptor adaptors TRIF and TRAM. Proc Natl Acad Sci U S A. (2025) 122:e2418988122. doi: 10.1073/pnas.2418988122
126. Lee LM, Ji M, Sinha M, Dong MB, Ren X, Wang Y, et al. Determinants of divergent adaptive immune responses after airway sensitization with ligands of toll-like receptor 5 or toll-like receptor 9. PloS One. (2016) 11:e0167693. doi: 10.1371/journal.pone.0167693
127. Takeda K and Akira S. Toll-like receptors. Curr Protoc Immunol. (2015) 109:14.12.1–14.12.10. doi: 10.1002/0471142735.im1412s109
128. Fitzpatrick JM, Minogue E, Curham L, Tyrrell H, Gavigan P, Hind W, et al. MyD88-dependent and -independent signalling via TLR3 and TLR4 are differentially modulated by Delta(9)-tetrahydrocannabinol and cannabidiol in human macrophages. J Neuroimmunol. (2020) 343:577217. doi: 10.1016/j.jneuroim.2020.577217
129. Zou PF, Shen JJ, Li Y, Zhang ZP, and Wang YL. TRAF3 enhances TRIF-mediated signaling via NF-kappaB and IRF3 activation in large yellow croaker Larimichthys crocea. Fish Shellfish Immunol. (2020) 97:114–24. doi: 10.1016/j.fsi.2019.12.024
130. Akira S, Uematsu S, and Takeuchi O. Pathogen recognition and innate immunity. Cell. (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
131. Liu AH. Revisiting the hygiene hypothesis for allergy and asthma. J Allergy Clin Immunol. (2015) 136:860–5. doi: 10.1016/j.jaci.2015.08.012
132. Fuchs B, Knothe S, Rochlitzer S, Nassimi M, Greweling M, Lauenstein HD, et al. A Toll-like receptor 2/6 agonist reduces allergic airway inflammation in chronic respiratory sensitisation to Timothy grass pollen antigens. Int Arch Allergy Immunol. (2010) 152:131–9. doi: 10.1159/000265534
133. Prescott SL. Effects of early cigarette smoke exposure on early immune development and respiratory disease. Paediatr Respir Rev. (2008) 9:3–9; quiz 10. doi: 10.1016/j.prrv.2007.11.004
134. Conrad ML, Ferstl R, Teich R, Brand S, Blümer N, Yildirim AO, et al. Maternal TLR signaling is required for prenatal asthma protection by the nonpathogenic microbe Acinetobacter lwoffii F78. J Exp Med. (2009) 206:2869–77. doi: 10.1084/jem.20090845
135. Taruselli MT, Abdul Qayum A, Abebayehu D, Caslin HL, Dailey JM, Kotha A, et al. IL-33 induces cellular and exosomal miR-146a expression as a feedback inhibitor of mast cell function. J Immunol. (2024) 212:1277–86. doi: 10.4049/jimmunol.2200916
136. Brasier AR and Boldogh I. Targeting inducible epigenetic reprogramming pathways in chronic airway remodeling. Drugs Context. (2019) 8:2019-8-3. doi: 10.7573/dic.2019-8-3
137. Gao W, Xiong Y, Li Q, and Yang H. Inhibition of toll-like receptor signaling as a promising therapy for inflammatory diseases: A journey from molecular to nano therapeutics. Front Physiol. (2017) 8:508. doi: 10.3389/fphys.2017.00508
138. Helal MG, Megahed NA, and Abd Elhameed AG. Saxagliptin mitigates airway inflammation in a mouse model of acute asthma via modulation of NF-kB and TLR4. Life Sci. (2019) 239:117017. doi: 10.1016/j.lfs.2019.117017
139. Ramli I, Cheriet T, Posadino AM, Giordo R, Zayed H, Eid AH, et al. Potential therapeutic targets of resveratrol in the prevention and treatment of pulmonary fibrosis. Front Biosci (Landmark Ed). (2023) 28:198. doi: 10.31083/j.fbl2809198
140. Beijers RJ, Gosker HR, Sanders KJ, de Theije C, Kelders M, Clarke G, et al. Resveratrol and metabolic health in COPD: A proof-of-concept randomized controlled trial. Clin Nutr. (2020) 39:2989–97. doi: 10.1016/j.clnu.2020.01.002
141. Mirhadi E, Roufogalis BD, Banach M, Barati M, and Sahebkar A. Resveratrol: Mechanistic and therapeutic perspectives in pulmonary arterial hypertension. Pharmacol Res. (2021) 163:105287. doi: 10.1016/j.phrs.2020.105287
142. Jiang H, Duan J, Xu K, and Zhang W. Resveratrol protects against asthma-induced airway inflammation and remodeling by inhibiting the HMGB1/TLR4/NF-κB pathway. Exp Ther Med. (2019) 18:459–66. doi: 10.3892/etm.2019.7594
143. Tang Y, Huang W, Song Q, Zheng X, He R, and Liu J. Paeonol ameliorates ovalbumin-induced asthma through the inhibition of TLR4/NF-κB and MAPK signaling. Evid Based Complement Alternat Med. (2018) 2018:3063145. doi: 10.1155/2018/3063145
144. Haseeb M, Choi YS, Patra MC, Jeong U, Lee WH, Qayyum N, et al. Discovery of novel small molecule dual inhibitor targeting toll-like receptors 7 and 9. J Chem Inf Model. (2024) 64:5090–107. doi: 10.1021/acs.jcim.4c00578
145. Liao X, He J, Wang R, Zhang J, Wei S, Xiao Y, et al. TLR-2 agonist Pam3CSK4 has no therapeutic effect on visceral leishmaniasis in BALB/c mice and may enhance the pathogenesis of the disease. Immunobiology. (2023) 228:152725. doi: 10.1016/j.imbio.2023.152725
146. Jha A, Thwaites RS, Tunstall T, Kon OM, Shattock RJ, Hansel TT, et al. Increased nasal mucosal interferon and CCL13 response to a TLR7/8 agonist in asthma and allergic rhinitis. J Allergy Clin Immunol. (2021) 147:694–703 e12. doi: 10.1016/j.jaci.2020.07.012
147. Liu L, Zhou L, Wang L, Mao Z, Zheng P, Zhang F, et al. MUC1 attenuates neutrophilic airway inflammation in asthma by reducing NLRP3 inflammasome-mediated pyroptosis through the inhibition of the TLR4/MyD88/NF-kappaB pathway. Respir Res. (2023) 24:255. doi: 10.1186/s12931-023-02550-y
148. Chen W, Zhang Z, Zhang S, Zhu P, Ko JK, and Yung KK. MUC1: structure, function, and clinic application in epithelial cancers. Int J Mol Sci. (2021) 22:6567. doi: 10.3390/ijms22126567
149. Milara J, Morell A, de Diego A, Artigues E, Morcillo E, and Cortijo J. Mucin 1 deficiency mediates corticosteroid insensitivity in asthma. Allergy. (2019) 74:111–21. doi: 10.1111/all.13546
150. Wang S and Zhang Y. HMGB1 in inflammation and cancer. J Hematol Oncol. (2020) 13:116. doi: 10.1186/s13045-020-00950-x
151. Cavone L, Cuppari C, Manti S, Grasso L, Arrigo T, Calamai L, et al. Increase in the level of proinflammatory cytokine HMGB1 in nasal fluids of patients with rhinitis and its sequestration by glycyrrhizin induces eosinophil cell death. Clin Exp Otorhinolaryngol. (2015) 8:123–8. doi: 10.3342/ceo.2015.8.2.123
152. Paudel YN, Angelopoulou E, Piperi C, Balasubramaniam V, Othman I, and Shaikh MF. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: Updates on receptor signalling. Eur J Pharmacol. (2019) 858:172487. doi: 10.1016/j.ejphar.2019.172487
153. Hwang YH, Lee Y, Paik MJ, and Yee ST. Inhibitions of HMGB1 and TLR4 alleviate DINP-induced asthma in mice. Toxicol Res (Camb). (2019) 8:621–9. doi: 10.1039/c9tx00048h
154. Sharma R, Tiwari A, and McGeachie MJ. Recent miRNA research in asthma. Curr Allergy Asthma Rep. (2022) 22:231–58. doi: 10.1007/s11882-022-01050-1
155. Han X, Song Y, Piao Y, Wang Z, Li Y, Cui Q, et al. Mechanism of miR-130b-3p in relieving airway inflammation in asthma through HMGB1-TLR4-DRP1 axis. Cell Mol Life Sci. (2024) 82:9. doi: 10.1007/s00018-024-05529-0
156. Soccio P, Moriondo G, Lacedonia D, Tondo P, Pescatore D, Quarato CMI, et al. MiRNA and exosomal miRNA as new biomarkers useful to phenotyping severe asthma. Biomolecules. (2023) 13:1542. doi: 10.3390/biom13101542
157. Patil JS and Sarasija S. Pulmonary drug delivery strategies: A concise, systematic review. Lung India. (2012) 29:44–9. doi: 10.4103/0970-2113.92361
158. Wang L, Zhang H, Sun L, Gao W, Xiong Y, Ma A, et al. Manipulation of macrophage polarization by peptide-coated gold nanoparticles and its protective effects on acute lung injury. J Nanobiotechnology. (2020) 18:38. doi: 10.1186/s12951-020-00593-7
159. Barnes PJ. Glucocorticosteroids: current and future directions. Br J Pharmacol. (2011) 163:29–43. doi: 10.1111/j.1476-5381.2010.01199.x
160. Reichardt SD, Amouret A, Muzzi C, Vettorazzi S, Tuckermann JP, Luhder F, et al. The role of glucocorticoids in inflammatory diseases. Cells. (2021) 10:2921. doi: 10.3390/cells10112921
161. Cain DW and Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol. (2017) 17:233–47. doi: 10.1038/nri.2017.1
162. Bansal A, Mostafa MM, Kooi C, Sasse SK, Michi AN, Shah SV, et al. Interplay between nuclear factor-kappaB, p38 MAPK, and glucocorticoid receptor signaling synergistically induces functional TLR2 in lung epithelial cells. J Biol Chem. (2022) 298:101747. doi: 10.1016/j.jbc.2022.101747
163. Pace E, Di Sano C, Ferraro M, Bruno A, Caputo V, Gallina S, et al. Budesonide increases TLR4 and TLR2 expression in Treg lymphocytes of allergic asthmatics. Pulm Pharmacol Ther. (2015) 32:93–100. doi: 10.1016/j.pupt.2015.02.003
164. Pofi R, Caratti G, Ray DW, and Tomlinson JW. Treating the side effects of exogenous glucocorticoids; can we separate the good from the bad? Endocr Rev. (2023) 44:975–1011. doi: 10.1210/endrev/bnad016
Keywords: toll-like receptors, toll-like receptors signaling, asthma, immune cells, inflammation, cytokines, therapeutic strategies
Citation: Xu X, Yu R, Yang Z, Li C, Xiong H and Li C (2025) Toll-like receptor-mediated immune imbalance in asthma: controversies, breakthroughs, and future directions. Front. Immunol. 16:1605185. doi: 10.3389/fimmu.2025.1605185
Received: 02 April 2025; Accepted: 16 June 2025;
Published: 02 July 2025.
Edited by:
Vijay Kumar, Morehouse School of Medicine, United StatesReviewed by:
Susetta Finotto, Universitätsklinikum Erlangen, GermanyDaniel P. Potaczek, University of Marburg, Germany
Copyright © 2025 Xu, Yu, Yang, Li, Xiong and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huabao Xiong, eGlvbmdoYmxAMTYzLmNvbQ==; Chunxia Li, eGlhY2h1bjExMTNAbWFpbC5qbm1jLmVkdS5jbg==