 Yimin Wang1,2
Yimin Wang1,2 Fang Zhou1*
Fang Zhou1*- 1Department of Hematology, the 960th Hospital of the People’s Liberation Army Joint Logistics Support Force, Jinan, China
- 2The First Clinical Medical School, Shandong University of Traditional Chinese Medicine, Jinan, China
Chronic graft-versus-host disease (cGVHD), a severe complication of allogeneic hematopoietic stem cell transplantation (allo-HSCT), arises from donor immune cell-mediated tissue damage, chronic inflammation, and fibrosis. Current therapies fail to adequately address fibrotic progression and heighten infection risks, underscoring the need for targeted strategies. Hypoxia-inducible factor-1α (HIF-1α), a pivotal regulator, emerges as a potential therapeutic target by orchestrating immunometabolic homeostasis, suppressing fibrosis, preserving gut microbiota balance, and retaining graft-versus-leukemia (GVL) effects. However, clinical translation necessitates overcoming challenges in tissue specificity and off-target effects. Smart nanodelivery systems hold promise for enhancing precision to enable localized HIF-1α pathway modulation. This review highlights the multidimensional roles of HIF-1α in cGVHD pathogenesis and proposes nanotherapeutic approaches to reconcile immunofibrotic imbalances, advancing a paradigm shift in cGVHD management while preserving GVL efficacy.
1 Introduction
Chronic graft-versus-host disease (cGVHD), a lethal complication following allogeneic hematopoietic stem cell transplantation (allo-HSCT), is characterized by a core pathological mechanism involving persistent attack of donor-derived immune cells on host tissues, which triggers chronic inflammation, immune tolerance dysregulation, and multi-organ fibrosis. Post-transplantation, donor immune cells infiltrate host tissues and induce cellular damage, releasing damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) (1, 2). These molecules activate innate immune receptors including Toll-like receptors (TLRs), NOD-like receptors, and the NLRP3 inflammasome, thereby driving pro-inflammatory cytokine cascades (3). The chronic inflammatory microenvironment facilitates antigen-presenting cells (APCs) to activate autoreactive B and T lymphocytes. Concurrently, compromised central and peripheral immune tolerance mechanisms exacerbate the targeting of donor-recipient shared antigens by these cells, recapitulating autoimmune pathological processes (4).
In cGVHD, autoreactive CD4+ T cells differentiate into T helper 1(Th1), Th2, and Th17 subsets, with Th17 cells exacerbating chronic inflammation and tissue remodeling through aberrant interleukin-17 (IL-17) secretion (5). Concurrently, follicular helper T cells (Tfh), via IL-21 signaling, orchestrate germinal center formation to drive B cell somatic hypermutation and pathogenic antibody production (6). Sustained immune dysregulation promotes macrophage-derived profibrotic factors, including transforming growth factor-β (TGF-β) and platelet-derived growth factor-A (PDGF-A), which activate fibroblasts and enhance collagen deposition, ultimately culminating in organ fibrosis (7, 8). cGVHD manifests with heterogeneous clinical presentations ranging from lichenoid skin lesions and Sjögren-like syndrome to fibrotic involvement of joints, lungs, and fasciae. Approximately 20% of patients develop debilitating sclerotic skin lesions and bronchiolitis obliterans syndrome (BOS), which are hallmarks of progressive fibrotic lesions (9).
Although glucocorticoids combined with calcineurin inhibitors remain first-line therapy, approximately 50% of patients develop steroid-refractory or relapsed disease, while prolonged immunosuppression predisposes to life-threatening infections, metabolic disorders, and osteoporosis (10–12). Current therapeutic strategies demonstrate limited efficacy against progressive fibrotic complications (e.g., BOS and dermal sclerosis), with fibrotic organ failure constituting the predominant cause of cGVHD-related mortality. These clinical challenges underscore the urgent need for mechanistic dissection of cGVHD pathogenesis and development of novel targeted interventions.
Hypoxia-inducible factor-1α (HIF-1α), a master regulator of cellular adaptation to hypoxic microenvironments, has been shown to regulate Th17 differentiation (via RORγt activation), inhibit Tfh-B cell crosstalk (by modulating CD40/CD40L signaling), and reverse macrophage profibrotic polarization (through mTORC1/TGF-β axis suppression) (13–15). Furthermore, HIF-1α synergistically maintains immune balance via metabolic reprogramming, epigenetic regulation, and gut microbiota homeostasis. Notably, HIF-1α exerts protective effects against cGVHD while preserving graft-versus-leukemia (GVL) activity, offering a unique therapeutic advantage for balancing cGVHD suppression and leukemia relapse prevention.
This review systematically elucidates the multidimensional roles of HIF-1α in modulating immune-metabolic homeostasis, suppressing fibrosis, maintaining gut microecology, and sustaining GVL efficacy, thereby providing a mechanistic foundation for targeted cGVHD interventions with preserved antileukemic effects. However, clinical translation faces challenges: tissue-specific modulation, off-target effect risks, and synergistic strategies with conventional therapies remain imperative to explore. The development of smart nanodelivery systems may enhance targeting precision. Collectively, HIF-1α-targeted therapy holds promise for overcoming the dual immunofibrotic pathological barriers in cGVHD, establishing a breakthrough therapeutic paradigm for clinical translation.
2 Introduction to HIF-1α
2.1 Structure of HIF-1α
HIF-1α, the oxygen-sensitive subunit of the heterodimeric transcription factor HIF-1, is a master regulator of cellular adaptation to hypoxia. The human HIF-1α protein consists of 826 amino acids encoded by the HIF1A gene located on chromosome 14q21-24 (16, 17). Structurally, HIF-1α belongs to the basic helix-loop-helix (bHLH)/PER-ARNT-SIM (PAS) superfamily. Its N-terminal region contains a bHLH domain essential for DNA binding and a PAS domain mediating heterodimerization with the constitutively expressed HIF-1β/ARNT subunit (18, 19). The central region contains the oxygen-dependent degradation domain (ODDD), which is characterized by two critical proline residues (Pro402 and Pro564) targeted by prolyl hydroxylases (PHDs). Hydroxylation of these residues triggers VHL-dependent ubiquitination and proteasomal degradation under normoxia. The C-terminal region harbors two transactivation domains (TAD-N and TAD-C) and an oxygen-regulated inhibitory domain (CID). TAD-N and TAD-C synergistically activate hypoxia-responsive genes through recruitment of transcriptional coactivators such as p300/CBP (20). Notably, TAD-C activity is further regulated by asparagine hydroxylation under normoxia, which prevents coactivator binding.
2.2 Physiological functions of HIF-1α
HIF-1α enhances cellular resilience to hypoxia by suppressing apoptosis via upregulation of B-cell lymphoma-2 (BCL-2) family proteins and inhibition of pro-apoptotic factors. Simultaneously, it sustains proliferation by activating mitogenic pathways such as IGF-2/IGF-1R signaling (21, 22). HIF-1α orchestrates inflammatory responses by polarizing macrophages toward a pro-inflammatory phenotype (M1) and enhancing neutrophil survival. It amplifies cytokine production (IL-1β, TNF-α, IL-6) and regulates T cell differentiation, linking hypoxia to chronic inflammation and autoimmune pathologies (23). HIF-1α stimulates erythropoiesis by inducing erythropoietin (EPO) in renal interstitial cells, optimizing oxygen delivery during systemic hypoxia (24). The pleiotropic roles of HIF-1α extend to cancer progression, cardiovascular remodeling, and inflammatory diseases. Its dysregulation contributes to tumor angiogenesis, metabolic adaptation, and therapy resistance, positioning it as a promising therapeutic target. Emerging strategies aim to modulate HIF-1α stability or activity using small-molecule inhibitors or gene-editing approaches (25, 26).
3 HIF-1α and immune regulation
3.1 T cells
3.1.1 T cells and cGVHD
In cGVHD, the specific functions of T cells include direct cytotoxic effects, cytokine production, and regulation of other immune cells. After allo-HSCT, donor T cells may recognize recipient tissue antigens and become activated. These activated T cells can differentiate into effector T cells and memory T cells, with effector T cells capable of directly attacking the host’s tissues, leading to clinical symptoms of cGVHD. Donor CD4+ and CD8+ T cells can cause significant cGVHD symptoms, with CD8+ T cell-mediated cGVHD being thymus-dependent and preferentially damaging the recipient’s thymic medullary epithelial cells, resulting in defects in thymic negative selection (27). In cGVHD, the number and function of regulatory T cells (Tregs) may be impaired, and a reduction in Tregs is associated with the development of extensive cGVHD. For example, the imbalance in the reconstitution of CD4+ Tregs and conventional CD4+ T cells (CD4+ Tcon) after allo-HSCT promotes cGVHD progression. Additionally, Tregs can inhibit T cell function by secreting immunosuppressive factors such as IL-10 and TGF-β, as well as inducing apoptosis in effector T cells through perforin and granzyme secretion (28, 29). The interaction between T cells and B cells is also critical in cGVHD. Tfh interacting with B cells can promote B cell differentiation and antibody production. In cGVHD patients, the expression of circulating follicular helper T (cTfh) cells, extrafollicular helper T cells, and B cells is altered, and these changes correlate with cGVHD pathogenesis. For instance, cTfh cell levels are significantly reduced in cGVHD patients compared to controls, whereas extrafollicular helper T cells are markedly elevated (30, 31). Furthermore, various cytokines contribute to cGVHD by regulating T cell function and differentiation. TGF-β1 promotes Foxp3 expression in T cells and induces Th cell conversion and expansion into Tregs via the SMAD signaling pathway (32). Additionally, TGF-β1 enhances CTLA-4 expression in naïve T cells, thereby promoting Foxp3 production in Tregs (33). IL-2 therapy can restore CD4+ T cell subset homeostasis in cGVHD patients and promote immune tolerance reconstitution (34). Given the pivotal role of T cells in cGVHD, they represent potential therapeutic targets.
3.1.2 HIF-1α and T cells
HIF-1α plays a critical role in T cell differentiation and function. It is central to T cell metabolic reprogramming. Under hypoxia, HIF-1α promotes a metabolic shift from oxidative phosphorylation to glycolysis in T cells, a process essential for their activation and effector functions. For example, HIF-1α deficiency prevents metabolic reprogramming in hypoxic T cells, thereby inhibiting IFN-γ induction (35). In chronic infections and tumor microenvironments, T cells may undergo exhaustion, characterized by functional decline and upregulated inhibitory receptors. HIF-1α regulates this process. Studies show that mitochondrial dysfunction reduces α-ketoglutarate levels, thereby inhibiting PHD activity and stabilizing HIF-1α in progenitor exhausted T cells (Tpex), driving their transcriptional and metabolic reprogramming into terminally exhausted T cells (36). Moreover, HIF-1α deficiency impairs glycolytic flux, leading to reduced ATP production and compromised activation-induced cell death (AICD) in hypoxic T cells (35). HIF-1α also regulates T cell differentiation into subsets such as Th1, Th2, Th17, and Tregs. In Th17 differentiation, HIF-1α acts via retinoic acid-related orphan receptor-γt (RORγt) to drive this process (37). Specific HIF-1α knockout in Tregs enhances their suppressive capacity under hypoxia, potentially by promoting oxidative phosphorylation and upregulating inhibitory receptors such as CTLA-4 (38). HIF-1α further participates in T cell epigenetic regulation. Under normoxia, HIF-1α undergoes hydroxylation by prolyl hydroxylases (PHD1-3), triggering polyubiquitination and proteasomal degradation via the von Hippel-Lindau protein (pVHL) complex (39). HIF-1α also regulates T cell oxidative stress responses. Hypoxia stabilizes HIF-1α, which modulates glycolysis-related gene expression to influence ATP and ROS production, thereby promoting T cell metabolic adaptation and survival (40). Elucidating its specific mechanisms may aid in developing novel immunotherapies for cGVHD (Figure 1).
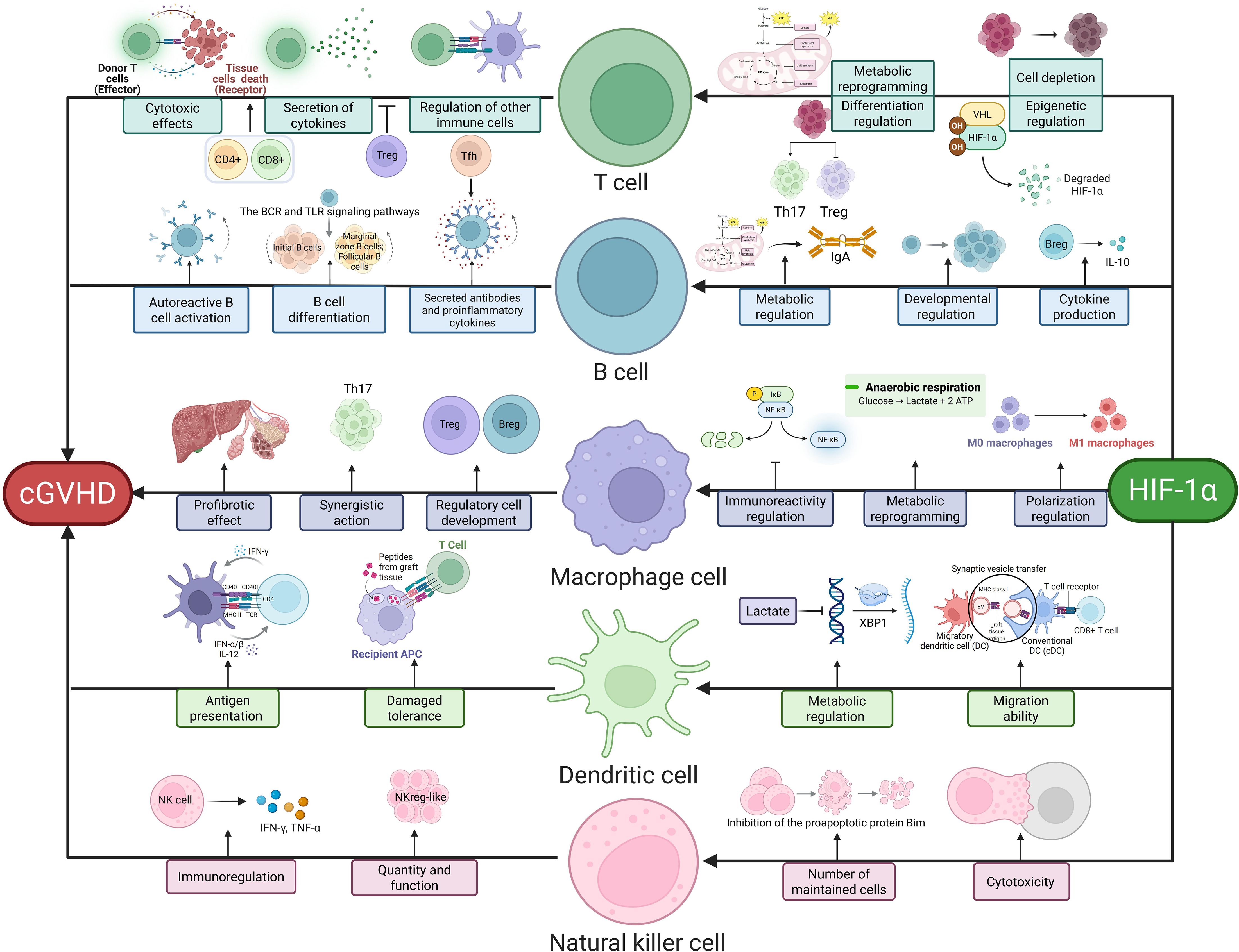
Figure 1. HIF-1α-Mediated Immune Regulation in cGVHD. This schematic delineates key immune pathways modulated by HIF-1α: T cells drive pathology via cytotoxic effects (CD8+-mediated thymic epithelial damage), cytokine secretion (Th17/Treg imbalance), and immune regulation (impaired Treg suppression via IL-10/TGF-β). B cells contribute through autoreactive activation (BCR/TLR signaling), antibody/proinflammatory cytokine secretion (e.g., anti-PDGFR antibodies), and regulatory dysfunction (Breg-derived IL-10 deficiency). Macrophages promote fibrosis via profibrotic polarization (TGF-β secretion), metabolic reprogramming, and amplified inflammation (M1/M2 imbalance). Dendritic cells facilitate antigen presentation (host antigen-driven T cell activation), impaired tolerance (reduced Treg induction), and migratory defects (HIF-1α/CCR7 axis). NK cells regulate disease through cytotoxic activity (granzyme/perforin), quantitative/functional alterations, and subset-specific modulation (NKreg-like suppression vs. CD74+-mediated inflammation). Central to these pathways, HIF-1α orchestrates metabolic reprogramming (glycolysis induction via PKM2/LDHA), epigenetic regulation (VHL/PHD axis), and cytokine signaling, thereby integrating immune dysregulation in cGVHD pathogenesis.
3.2 B cells
3.2.1 B cells and cGVHD
The roles and mechanisms of B cells in cGVHD are multifaceted, involving activation, differentiation, and specific pathological effects. In cGVHD, B cell activation primarily occurs through B cell receptor (BCR) and Toll-like receptor (TLR) signaling. Key molecules in the BCR pathway, such as B cell linker protein (BLNK) and spleen tyrosine kinase (SYK), are upregulated in cGVHD patients. This activation promotes autoreactive B cell survival and maturation via NF-κB and ERK signaling (41–43). B cell differentiation is also critical in cGVHD. Studies reveal reduced proportions and counts of naïve and immature B cells in cGVHD patients, alongside increased marginal zone and follicular B cells, which correlate with disease progression (30). Regulatory B cells (Bregs) modulate immune responses in cGVHD by maintaining immune balance, inducing transplant tolerance, and exerting anti-cGVHD effects. Bregs secrete cytokines like IL-10 to inhibit T cell and macrophage activation, thereby reducing inflammation and immune attacks (44, 45). Autoreactive B cell survival depends on B cell-activating factor (BAFF) levels: low BAFF promotes their clearance, while high BAFF enhances survival by activating AKT and ERK signaling (46, 47). In cGVHD, B cells promote autoimmunity through host-reactive antibody secretion, pro-inflammatory cytokine production, and self-antigen presentation to T cells. Anti-PDGFR stimulatory antibodies found in cGVHD patients activate ROS production and type I collagen gene expression (48). In females, post-transplant detection of allo-HY antibodies predicts human cGVHD (49). Additionally, B cells from active cGVHD patients exhibit impaired IL-10 production (50). The complex roles of B cells in cGVHD highlight the need for mechanistic studies to develop novel therapies.
3.2.2 HIF-1α and B cells
HIF-1α is critical for B cell differentiation, function, and immune regulation. Studies show that HIF-1α regulates IgA-producing B cell differentiation via metabolic reprogramming. HIF-1α-deficient B cells exhibit reduced glycolysis and impaired IgA production, whereas PHD inhibitor roxadustat stabilizes HIF-1α to enhance IgA class switching and mitigate inflammation (51). HIF-1α activity is dynamically regulated during B cell development. Bone marrow pro-B and pre-B cells exhibit high HIF activity, which declines at the immature B cell stage. Genetic HIF-1α activation in B cells reduces clonal diversity, impairs BCR editing, and arrests immature B cell development, decreasing peripheral B cell numbers. HIF-1α activation also reduces surface BCR, CD19, and BAFF-R expression while increasing pro-apoptotic Bim levels (52). B cell-specific HIF-1α knockout impairs IL-10 production, and HIF-1α cooperates with phosphorylated STAT3 to regulate IL-10 expression. HIF-1α-dependent glycolysis promotes CD1dhiCD5+ B cell expansion. Compared to wild-type mice, HIF-1α-deficient B cell mice exhibit exacerbated collagen-induced arthritis (CIA) and experimental autoimmune encephalomyelitis (EAE), which can be rescued by IL-10-expressing CD1dhiCD5+ B cell transplantation (53). In rheumatoid arthritis (RA), the CD27 IgD naïve B cell subset produces IL-6, and both HIF-1α and IL-6 are co-expressed in RA patient B cells. HIF-1α directly binds the IL6 promoter to enhance its transcription (54). By regulating metabolism, cytokine production, and immune cell development, HIF-1α maintains immune balance. Further research into its mechanisms may advance therapeutic strategies for related diseases (Figure 1).
3.3 Macrophages
3.3.1 Macrophages and cGVHD
Macrophages play a key role in the pathogenesis of cGVHD, contributing to its occurrence, progression, and pathological mechanisms. They induce fibroblast differentiation into myofibroblasts by secreting pro-fibrotic cytokines such as TGF-β, which promote collagen synthesis and deposition, ultimately leading to tissue fibrosis (55). Additionally, macrophages interact with T cells through cytokine secretion and co-stimulatory molecule expression, enhancing T cell activation and proliferation. This interaction particularly drives Th17 cell formation, thereby creating a positive feedback loop that exacerbates cGVHD pathology (56). Macrophages also influence the development and function of Tregs and Bregs; their dysfunction may reduce the number or impair the function of these regulatory subsets, disrupting immune tolerance and triggering uncontrolled immune responses in cGVHD (47). During the transition from acute graft-versus-host disease (aGVHD) to cGVHD, macrophages release inflammatory mediators and reactive oxygen species (ROS), directly damaging host tissues and organs and establishing the foundation for cGVHD onset (57). In chronic cGVHD, M2 macrophages promote tissue fibrosis via TGF-β secretion, resulting in organ dysfunction and amplified inflammatory responses (58). Given these multifaceted roles, macrophage-targeted therapies—such as anti-CSF-1R antibodies and the anti-fibrotic drug pirfenidone—hold significant clinical potential, effectively alleviating pathological damage and improving patient prognosis (59). Further research into macrophage-mediated mechanisms in cGVHD is critical for developing more effective treatments.
3.3.2 HIF-1α and macrophages
HIF-1α is central to macrophage biology, regulating immune responses, migration, polarization, and metabolism. It modulates macrophage immune activity by both inhibiting NF-κB transcriptional activity to prevent excessive inflammation and promoting NF-κB-mediated pro-inflammatory cytokine expression during LPS stimulation to bolster host defense (60, 61). HIF-1α enhances macrophage phagocytosis by suppressing apoptosis and PHD activity, thereby stabilizing its own protein levels. It also improves macrophage antibacterial capacity by regulating antimicrobial peptide release and nitric oxide (NO) production (62). In migration, HIF-1α fuels macrophages via aerobic glycolysis induction. Under severe hypoxia, HIF-1α upregulates pyruvate dehydrogenase kinase 1 (PDK1) to block pyruvate entry into the tricarboxylic acid cycle, thereby regulating glucose oxidation and enhancing migratory potential (63, 64). HIF-1α further drives macrophage polarization toward the pro-inflammatory M1 phenotype by targeting glucose metabolism. Activation of the HIF-1α/pyruvate kinase M2 (PKM2) axis promotes M1 polarization and inflammatory factor release (65, 66). In metabolic reprogramming, HIF-1α is a key regulator of macrophage glycolysis, binding to hypoxia response elements (HREs) in glycolytic enzyme gene promoters to upregulate their expression and drive aerobic glycolysis (67). Additionally, HIF-1α-induced upregulation of glycolytic enzymes and tricarboxylic acid cycle intermediates stabilizes HIF-1α itself, forming a positive feedback loop. LPS-induced disruption of the tricarboxylic acid cycle leads to metabolite accumulation (e.g., succinate, fumarate), which directly inhibits PHD activity and stabilizes HIF-1α (68, 69) (Figure 1).
3.4 Dendritic cells
3.4.1 Dendritic cells and cGVHD
Dendritic cells (DCs) exhibit a dual immunoregulatory role in cGVHD, contributing to both disease progression and immune homeostasis. As professional APC, DCs initiate pathogenic immune responses by capturing and presenting host-derived antigens to donor T cells, thereby activating alloreactive immune pathways (70). Following transplantation, donor-derived DCs rapidly infiltrate recipient tissues, where they secrete proinflammatory cytokines and express co-stimulatory molecules to drive donor T cell activation and proliferation. This process preferentially promotes Th1/Th17 cell differentiation, amplifying inflammatory cascades that exacerbate tissue damage (71). Paradoxically, DCs possess intrinsic tolerogenic potential under physiological conditions, capable of maintaining immune equilibrium through Tregs induction and functional enhancement (72). However, cGVHD pathogenesis involves critical dysfunction of these tolerogenic mechanisms. The impaired ability of DCs to generate and sustain Tregs populations results in progressive loss of immune tolerance, a hallmark of chronic disease progression (73). Post-allo-HSCT complications further compound this imbalance: conditioning regimens and acute GVHD (aGVHD)-mediated thymic damage disrupt central tolerance mechanisms, while peripheral DC dysfunction permits the escape of autoreactive T cell clones (57). Donor DCs in peripheral tissues perpetuate this cycle through indirect antigen presentation, continuously activating pathogenic T cell populations and driving cGVHD progression (74). This dual functionality positions DCs as central orchestrators of the cGVHD paradox—simultaneous hyperactivation of effector responses and failure of tolerance mechanisms. Emerging therapeutic strategies aim to recalibrate DC function through two complementary approaches: pharmacological modulation of DC activation thresholds and ex vivo generation of tolerogenic DC subsets. These interventions seek to restore the delicate balance between immunogenic and tolerogenic DC activities, offering novel pathways for clinical management of cGVHD.
3.4.2 HIF-1α and dendritic cells
HIF-1α serves as a master regulator of dendritic cell (DC) biology, coordinating metabolic adaptation, functional polarization, and migratory behavior. Lactate-mediated stabilization of HIF-1α induces NDUFA4L2 expression, which suppresses mitochondrial reactive oxygen species (ROS) production and subsequently inhibits the XBP1-dependent transcriptional program that drives pathogenic autoimmune T cell responses (75). Hypoxic conditioning studies reveal that HIF-1α-deficient DCs exhibit impaired IL-22 secretion and reduced CCR7 chemokine receptor expression, resulting in defective migratory capacity. These findings establish HIF-1α as an essential mediator of DC differentiation and tissue homing under low oxygen conditions (76). The long non-coding RNA lnc-Dpf3 directly interacts with HIF-1α to repress transcription of the glycolytic enzyme gene Ldha, effectively dampening DC glycolytic flux and migration capabilities (77). Magnesium ion signaling through the TRPM7 channel activates MAPK pathways in DCs, inducing HIF-1α expression that enhances TGF-β production while suppressing effector T cell function (78). Pathogen interaction studies demonstrate that Mycobacterium tuberculosis-infected DCs upregulate HIF-1α-driven aerobic glycolysis via TLR2-dependent mechanisms, a metabolic reprogramming event critical for DC migratory responses during infection (79) (Figure 1).
3.5 Natural killer cells
3.5.1 Natural killer cells and cGVHD
The role of natural killer (NK) cells in cGVHD remains incompletely defined, though emerging evidence suggests their significant involvement in disease pathogenesis. Clinical studies demonstrate a clinically relevant association between NK cell quantity/functionality and cGVHD outcomes. Notably, patients with extensive cGVHD exhibit reduced peripheral blood NK cell counts compared to those with limited disease. Furthermore, histological analysis reveals diminished NK cell infiltration in skin lesions of cGVHD patients who underwent HLA-matched sibling allo-HSCT relative to acute GVHD cases. This observation suggests that decreased circulating NK cell numbers may reflect enhanced tissue-specific recruitment and localization to cGVHD-affected sites (80). The therapeutic potential of NK cell modulation is highlighted by clinical outcomes in umbilical cord blood (UCB) transplantation. Pediatric UCB transplant recipients demonstrate both significantly higher NK cell reconstitution and reduced cGVHD incidence compared to other donor sources. Early detection of functionally competent NK cells post-transplantation has been identified as a critical factor contributing to this protective effect against cGVHD development (81). Mechanistically, NK cells appear to participate in complex immune regulatory networks during cGVHD pathogenesis. While they may collaborate with inhibitory CD8+ T cells to suppress autoantibody-producing B cells, activation through the MICA-NKG2D axis appears detrimental. Elevated plasma levels of soluble MICA correlate with increased cGVHD susceptibility, suggesting therapeutic potential in targeting IFN-γ production via this pathway (82). Emerging research identifies specific NK cell subsets with distinct functional roles in cGVHD modulation. A CD56bright perforin-regulatory NK cell population (NKreg-like) demonstrates potent immunosuppressive capacity, with increased numbers correlating with cGVHD suppression. This subset represents a promising candidate for cell-based therapeutic strategies (83). Conversely, CD74+ NK cells exhibit pro-inflammatory characteristics through high mitochondrial potential and cytokine secretion (IFN-γ/TNF-α), driving pulmonary cGVHD pathogenesis via CXCL10-mediated recruitment of macrophages and CD4+ T cells. Preclinical models demonstrate that targeted depletion of CD74+ NK cells using anti-CD74 antibodies effectively mitigates lung injury while preserving CD74- NK cell populations to maintain GVL effects (84). These findings collectively underscore the dual regulatory potential of NK cells in cGVHD pathophysiology. While certain subsets contribute to tissue damage through inflammatory mechanisms, others demonstrate protective immunosuppressive properties. This dichotomy emphasizes the necessity for precise subset-specific modulation strategies to optimize NK cell-mediated benefits in cGVHD management, balancing therapeutic efficacy with preservation of anti-neoplastic activity.
3.5.2 HIF-1α and natural killer cells
HIF-1α has been demonstrated to play a multifaceted regulatory role in natural killer (NK) cell metabolism, anti-tumor immunity, and infection response under hypoxic conditions. Emerging evidence reveals a complex interplay between HIF-1α and cytokine signaling pathways in the tumor microenvironment. Multiple studies indicate that HIF-1α suppresses NK cell-mediated tumor surveillance by inhibiting the IL-18-dependent NF-κB signaling axis, thereby attenuating anti-tumor responses. Notably, HIF-1α-deficient NK cells exhibit enhanced tumoricidal activity characterized by elevated IFN-γ production, increased granzyme B expression, and amplified degranulation capacity, suggesting therapeutic potential through HIF-1α inhibition (85). Contrasting findings highlight the context-dependent nature of HIF-1α function in NK cell biology. Paradoxically, other investigations demonstrate that HIF-1α deficiency impairs NK cell cytotoxicity by reducing infiltration of cells expressing soluble VEGFR-1, an anti-angiogenic factor critical for suppressing tumor-associated neovascularization. This deficiency leads to dysfunctional angiogenesis despite attenuated tumor progression (86). These opposing outcomes underscore the dual regulatory roles of HIF-1α in modulating both direct cytotoxic mechanisms and microenvironmental interactions. The cytokine milieu further modulates HIF-1α activity through distinct signaling pathways. IL-15 maintains NK cell homeostasis and effector functions through STAT3-mediated stabilization of HIF-1α, while IL-2 promotes HIF-1α expression via PI3K/mTOR signaling activation. These molecular mechanisms collectively preserve NK cell-mediated defenses against malignant transformation and pathogenic challenges (87, 88). Such cytokine-HIF-1α crosstalk appears essential for maintaining metabolic fitness and functional competence under hypoxic stress. Recent mechanistic insights reveal an essential metabolic regulatory role of HIF-1α in NK cell survival during viral infections. HIF-1α-dependent metabolic reprogramming suppresses expression of the pro-apoptotic protein Bim, thereby maintaining adequate NK cell populations required for optimal antiviral responses. This survival mechanism highlights the evolutionary conservation of HIF-1α-mediated metabolic adaptation in preserving lymphoid cell homeostasis during infection (89). Collectively, these findings position HIF-1α as a critical molecular nexus integrating environmental signals, metabolic demands, and functional outputs in NK cell biology (Figure 1).
4 HIF-1α and gut microbiota
4.1 Gut microbiota and cGVHD
Gut microbiota dysbiosis is closely linked to the pathogenesis of cGVHD. Significant alterations in gut microbiota composition are observed in cGVHD patients, notably a reduction in beneficial bacteria and an expansion of potential pathogens. The decline of butyrate-producing bacteria (e.g., Lachnoclostridium, Clostridium, and Faecalibacterium) may impair intestinal barrier integrity, thereby exacerbating cGVHD progression (90). Gut microbiota-derived metabolites also play critical roles in cGVHD. Short-chain fatty acids (SCFAs) enhance Tregs differentiation and function by inhibiting histone deacetylase (HDAC) activity, thereby mitigating cGVHD severity (91, 92). Tryptophan metabolites such as indole and its derivatives regulate Treg/Th17 balance via aryl hydrocarbon receptor (AhR) activation, suppressing inflammation and promoting intestinal epithelial repair (93, 94). Bile acids inhibit pro-inflammatory cytokines (TNF-α, IL-12) in DCs and monocytes while stimulating IL-10 production in macrophages, thus dampening immune cell pro-inflammatory activity. Bile acids also modulate Treg/Th17 equilibrium (95, 96). Secondary bile acids like 3-oxoLCA (3-oxo lithocholic acid) and isoalloLCA directly bind the transcription factor RORγt to inhibit Th17 differentiation, whereas isoalloLCA enhances Tregs differentiation by promoting oxidative phosphorylation and CNS3 H3K27 acetylation (97). Interventions targeting gut microbiota, such as fecal microbiota transplantation (FMT), have demonstrated therapeutic potential in cGVHD (98, 99).
4.2 HIF-1α and gut microbiota
The gut microbiota modulates HIF-1α expression and stability through metabolic regulation of intestinal epithelial cells (IECs), particularly by influencing cellular oxygen consumption patterns. HIF-1α enhances intestinal barrier integrity by upregulating tight junction protein expression in IECs, creating a bidirectional regulatory axis between host physiology and microbial communities (100, 101). This transcription factor further coordinates IEC metabolic programming and immune responses, indirectly shaping gut microbiota composition and functionality. Microbial-derived SCFAs, particularly butyrate, stabilize HIF-1α through dual mechanisms: direct inhibition of PHD enzyme activity and iron chelation-mediated PHD functional impairment, both contributing to reduced HIF-1α degradation (100, 102). Post-allo-HSCT complications reveal HIF-1α’s critical protective role. Donor T cell-induced intestinal damage impairs IEC oxidative phosphorylation, paradoxically increasing luminal oxygen levels and destabilizing HIF-1α. IEC-specific HIF-1α knockout murine models demonstrate exacerbated acute graft-versus-host disease (aGVHD) severity, highlighting the protein’s essential functions in barrier maintenance and immune regulation (103). Mechanistically, HIF-1α mitigates aGVHD through three interconnected pathways: promoting IEC regeneration, modulating immune cell infiltration/activation, and maintaining beneficial microbial communities. These findings position HIF-1α as a central regulator of intestinal homeostasis under transplant-related stress conditions (Figure 2A).
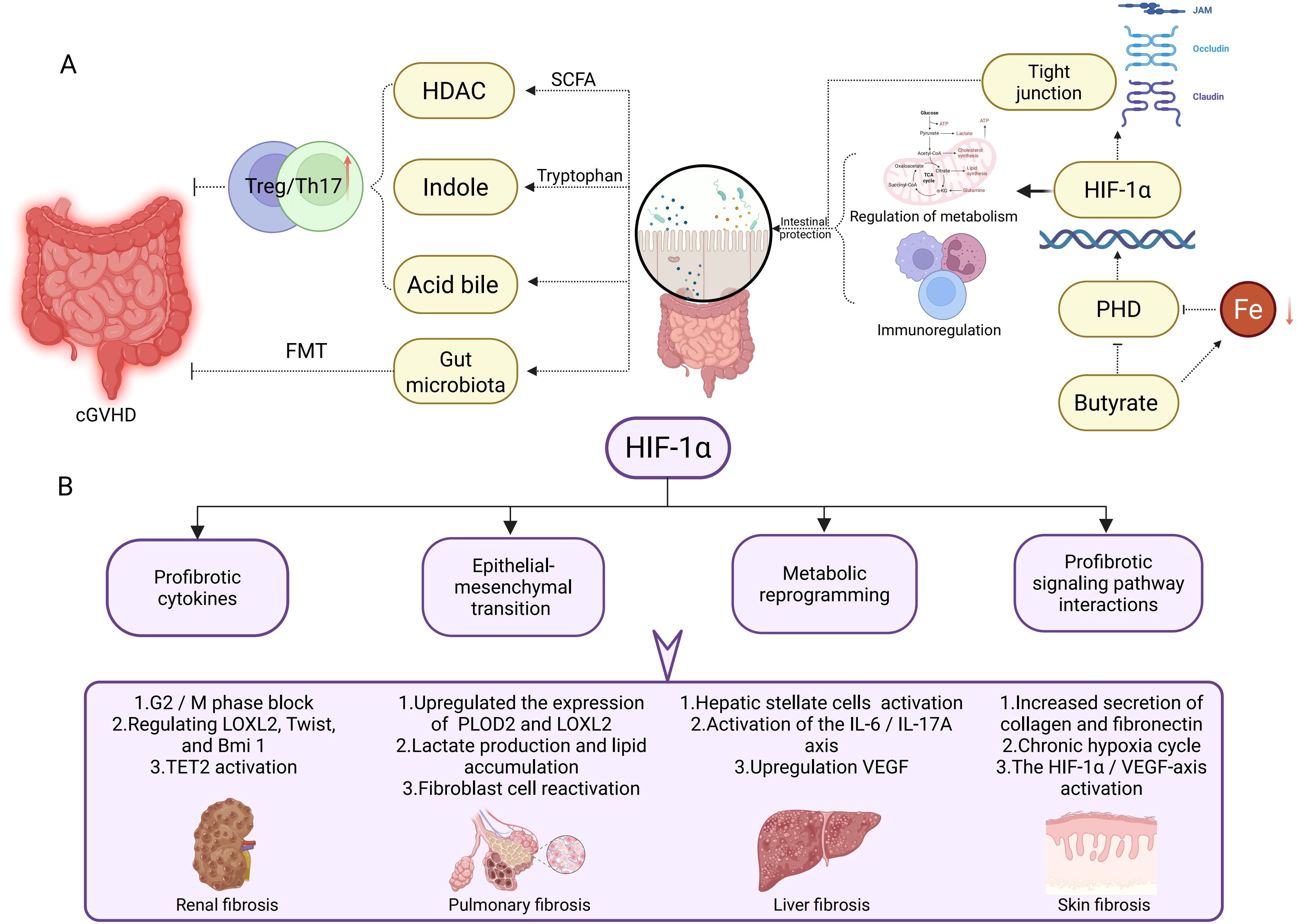
Figure 2. HIF-1α in Gut Microbiota Regulation and Tissue Fibrosis Pathogenesis. (A) This section illustrates the potential mechanisms of the gut microbiota in cGVHD via HIF-1α. Short-chain fatty acids, indoles, and bile acids modulate the Treg/Th17 cell balance, influencing intestinal barrier integrity and immune regulation. HIF-1α enhances tight junction protein expression to promote intestinal barrier protection, while concurrently regulating metabolic and immune responses to maintain gut homeostasis. (B) This section delineates the role of HIF-1α in distinct fibrotic processes. In renal fibrosis, HIF-1α induces pathogenesis via G2/M phase arrest, modulation of LOXL2, Twist, and Bmi1 expression, and TET2 activation. In pulmonary fibrosis, it mediates progression through upregulation of PLOD2 and LOXL2, lactate production, lipid accumulation, and fibroblast reactivation. Liver fibrosis is promoted by HIF-1α via hepatic stellate cell activation, the IL-6/IL-17A axis, and VEGF upregulation. In skin fibrosis, HIF-1α drives pathogenesis by enhancing collagen and fibronectin secretion, perpetuating chronic hypoxia cycles, and activating the HIF-1α/VEGF axis.
5 HIF-1α and tissue fibrosis
Fibrosis, characterized by excessive extracellular matrix (ECM) deposition, is a hallmark of cGVHD following allo-HSCT. While preclinical models of organ fibrosis (renal, pulmonary, liver, skin) provide mechanistic insights, their relevance to allo-HSCT lies in cross-pathway similarities with cGVHD-mediated tissue damage.
5.1 Renal fibrosis
In allo-HSCT, renal fibrosis associated with cGVHD or calcineurin inhibitor nephrotoxicity mirrors HIF-1α-driven mechanisms observed in idiopathic models. HIF-1α-induced G2/M arrest in tubular epithelial cells (via p53 upregulation) and EMT (mediated by LOXL2/Twist/Bmi1) promote TGF-β–driven ECM deposition (type I collagen, fibronectin) (104–107). Notably, TET2 activation—implicated in both fibrosis and immune regulation—links HIF-1α to epigenetic reprogramming in fibrotic kidneys (108). These mechanisms underscore HIF-1α as a potential target to mitigate cGVHD-induced renal injury (Figure 2B).
5.2 Pulmonary fibrosis
Activation of the HIF pathway induces structural and functional collagen dysregulation, a key contributor to mechanical dysfunction in idiopathic pulmonary fibrosis (IPF) tissues. Pulmonary complications post-allo-HSCT share pathogenic features with idiopathic pulmonary fibrosis (IPF). HIF upregulates enzymes such as PLOD2 and LOXL2 to alter collagen crosslinking, fiber nanostructure, and tissue stiffness (109). CCT6A, highly expressed in type II alveolar epithelial cells (AEC2) of fibrotic lungs, correlates with disease severity. It suppresses HIF-1α-mediated lactate production by promoting VHL-dependent ubiquitination and degradation of HIF-1α, thereby inhibiting lipid accumulation and alleviating bleomycin-induced pulmonary fibrosis in mice (110). Drp1 inhibition prevents mitochondrial fission in fibroblasts and regulates ROS/HIF-1α-dependent lipid metabolic reprogramming, suppressing fibroblast activation and mitigating fibrosis progression (111) (Figure 2B).
5.3 Liver fibrosis
HIF-1α also plays a critical role in liver fibrosis. Studies demonstrate that liver fibrosis and end-stage cirrhosis are frequently associated with hepatic hypoxia. As a key transcription factor regulating cellular hypoxia responses, HIF-1α is critically involved in hepatic stellate cell (HSC) activation and fibrosis progression. During liver fibrosis, HSCs respond to hypoxic stimuli, upregulating HIF-1α expression. HIF-1α promotes IL-6 production by directly binding to the hypoxia-response element (HRE) in the IL6 promoter of HSCs, which stimulates Th17 cells to secrete IL-17A. This interaction establishes the HIF-1α/IL-6/IL-17A axis, driving fibrotic development (112). HIF-1α further regulates elevated VEGF levels in activated HSCs. Concurrently, miR-21 overexpression in hepatocytes stabilizes HIF-1α via pVHL, amplifying the HIF-1α/VEGF signaling pathway and accelerating fibrosis (113). Additionally, HIF-1α interacts with key pathways—including PI3K/AKT/mTOR, MAPK/ERK, and NF-κB—all of which contribute to the onset and progression of liver fibrosis (61, 114–116) (Figure 2B).
5.4 Skin fibrosis
Systemic sclerosis (SSc), characterized by skin/visceral fibrosis and microvascular damage, shares pathogenic parallels with sclerodermatous cGVHD. HIF-1α hyperactivation in SSc perpetuates a cycle of chronic hypoxia, promotes excessive ECM production (collagen, fibronectin) by fibroblasts, and drives endothelial-to-mesenchymal transition via the HIF-1α/VEGF axis (117–119). This knowledge provides mechanistic insight into the development of sclerodermatous cGVHD, where similar hypoxic microenvironments and profibrotic signaling likely contribute to skin and internal organ fibrosis (Figure 2B).
6 HIF-1α and antileukemic effects
HIF-1α is highly expressed across multiple malignancies and correlates with malignant progression and poor patient prognosis. In hematologic cancers, HIF-1α plays a critical role in leukemogenesis, disease progression, and therapeutic resistance. Experimental evidence confirms that bone marrow (BM) oxygen levels are exceptionally low (~0.6%), creating a hypoxic microenvironment that protects leukemia stem cells and sustains their self-renewal capacity (120, 121).
6.1 HIF-1α and acute lymphoblastic leukemia
In acute lymphoblastic leukemia (ALL), HIF-1α functions as an oncogenic driver. Its targeted inhibition suppresses tumor proliferation, induces apoptosis, and enhances chemosensitivity. Mechanistically, deferoxamine (DFO)-mediated HIF-1α inhibition blocks tumor growth and triggers apoptosis by inactivating the ROS/HIF-1α signaling axis (122). Chemical inhibition of HIF-1α downregulates Yin-Yang 1 (YY1) expression, thereby potentiating ALL cell responsiveness to chemotherapeutic agents (123). HIF-1α knockdown in human T-ALL cells and murine models restores hypoxia-compromised mTOR activity, significantly improving chemotherapeutic sensitivity (124). Notably, Notch1 signaling is indispensable for HIF-1α-driven proliferation, invasion, and chemoresistance in T-ALL. Hypoxic HIF-1α silencing suppresses Notch1 activation, further validating HIF-1α as a promising therapeutic target for T-ALL (125). These findings collectively establish HIF-1α as a key molecular target, with therapeutic interventions against HIF-1α showing potential to optimize clinical outcomes in ALL patients.
6.2 HIF-1α and chronic lymphocytic leukemia
In chronic lymphocytic leukemia (CLL), HIF-1α serves as a pivotal oncogene that drives CLL cell survival and proliferation through multiple molecular mechanisms. Stromal cells upregulate HIF-1α expression in CLL cells via the CXCL12/CXCR4 signaling axis, particularly in TP53-deficient cells where HIF-1α accumulation confers apoptosis resistance and enhances chemoresistance (126, 127). Pharmacological HIF-1α inhibitors (e.g., BAY87-2243, EZN-2208) potentiate fludarabine-induced apoptosis and improve therapeutic response (128, 129). HIF-1α further orchestrates CLL cell interactions with tumor microenvironments by regulating chemotaxis and bone marrow/spleen niche adhesion (130). Under hypoxic conditions, HIF-1α overexpression amplifies adenosine biosynthesis and signaling through A2A receptors, establishing an immunosuppressive microenvironment that promotes tumor progression and drug resistance. Importantly, A2A receptor blockade reverses these effects and restores leukemic cell sensitivity to therapeutic agents (131). Mechanistically, HIF-1α inhibitors disrupt redox homeostasis by modulating oxidative stress pathways, effectively suppressing CLL cell proliferation (132). These findings collectively establish HIF-1α pathway targeting as a viable strategy to subvert CLL cell survival mechanisms, offering significant potential for improving clinical outcomes in CLL patients.
6.3 HIF-1α and acute myeloid leukemia
HIF-1α is an important player in the pathogenesis and progression of AML, functioning as a crucial regulator of cellular responses to hypoxia in the bone marrow microenvironment. Frequently overexpressed in AML subsets with adverse genetic abnormalities like t (8, 21) rearrangement, NPM1 mutations, and IDH1/2 or TP53 alterations, HIF-1α overexpression is strongly linked to poor clinical outcomes, including reduced relapse-free survival and resistance to chemotherapy agents such as cytarabine (133–135). The hypoxic bone marrow microenvironment stabilizes HIF-1α, which protects leukemic stem cells (LSCs) by facilitating immune evasion and chemoresistance (136). HIF-1α drives AML pathogenesis through multiple interconnected mechanisms. It promotes leukemic cell growth, proliferation, migration, and invasion. Overexpression of HIF-1α confers doxorubicin resistance in AML cells and synergizes with leukemia-derived macrophage migration inhibitory factor (MIF) to enhance cellular proliferation and survival. Targeting the HIF-1α/YAP axis enhances doxorubicin chemosensitivity by disrupting this regulatory circuit (137, 138). In acute promyelocytic leukemia (APL), HIF-1α collaborates with the PML-RARα fusion protein to maintain leukemia stem cell self-renewal and promote tumor neovascularization and migration (139). HIF-1α enhances glycolytic metabolism, promotes genomic hypermethylation and histone modifications, regulates autophagy, and inhibits apoptosis. It also mediates chemoresistance by causing cell cycle arrest at the G0/G1 phase and forming regulatory loops with proteins like YAP (137, 140). Additionally, factors such as PARP14 and hypoxia-induced CXCL2 signaling can modulate HIF-1α activity to support AML progression (141).
Targeting HIF-1α represents a promising therapeutic strategy for AML. Inhibitors like PX-478 and echinomycin have shown potential in preclinical models by sensitizing AML cells to chemotherapy (142, 143). Hypoxia-activated prodrugs such as TH-302 have demonstrated some efficacy in clinical trials, but challenges remain in terms of toxicity and isoform-specific targeting (144). Combinatorial approaches, including HIF-1α inhibition with hypomethylating agents or BCL-2 inhibitors, may overcome resistance. Besides, elevated HIF-1α expression correlates with poor prognosis and cytarabine resistance in NPM1-mutated/FLT3-ITD-negative AML subtypes, establishing it as a predictive biomarker (133). Therapeutic HIF-1α suppression emerges as a promising strategy to restore chemosensitivity and improve clinical outcomes, positioning HIF-1α as a high-value molecular target for precision AML therapy.
6.4 Chronic myeloid leukemia
HIF-1α functions as a critical transcriptional regulator in chronic myeloid leukemia (CML), driving leukemic cell survival and proliferation through upregulation of BCR-ABL and Met oncogenic pathways (145). Clinically, elevated HIF-1α expression correlates with tyrosine kinase inhibitor (TKI) resistance in CML, significantly compromising therapeutic efficacy (146). Mechanistic studies reveal that HIF-1α pathway inhibition enhances TKI sensitivity and induces apoptosis through multiple modalities: hydroxyurea suppresses HIF-1α-mediated glycolytic reprogramming to overcome drug resistance, while 2-methoxyestradiol directly targets HIF-1α to trigger apoptotic cascades. The triple kinase inhibitor Tri-CAP further disrupts pro-survival signaling networks in CML cells (146–148). Importantly, HIF-1α overexpression serves as a biomarker for advanced disease progression in CML patients (149). These findings collectively establish HIF-1α as a high-value therapeutic target, offering novel strategies to optimize clinical outcomes in CML management (Table 1).
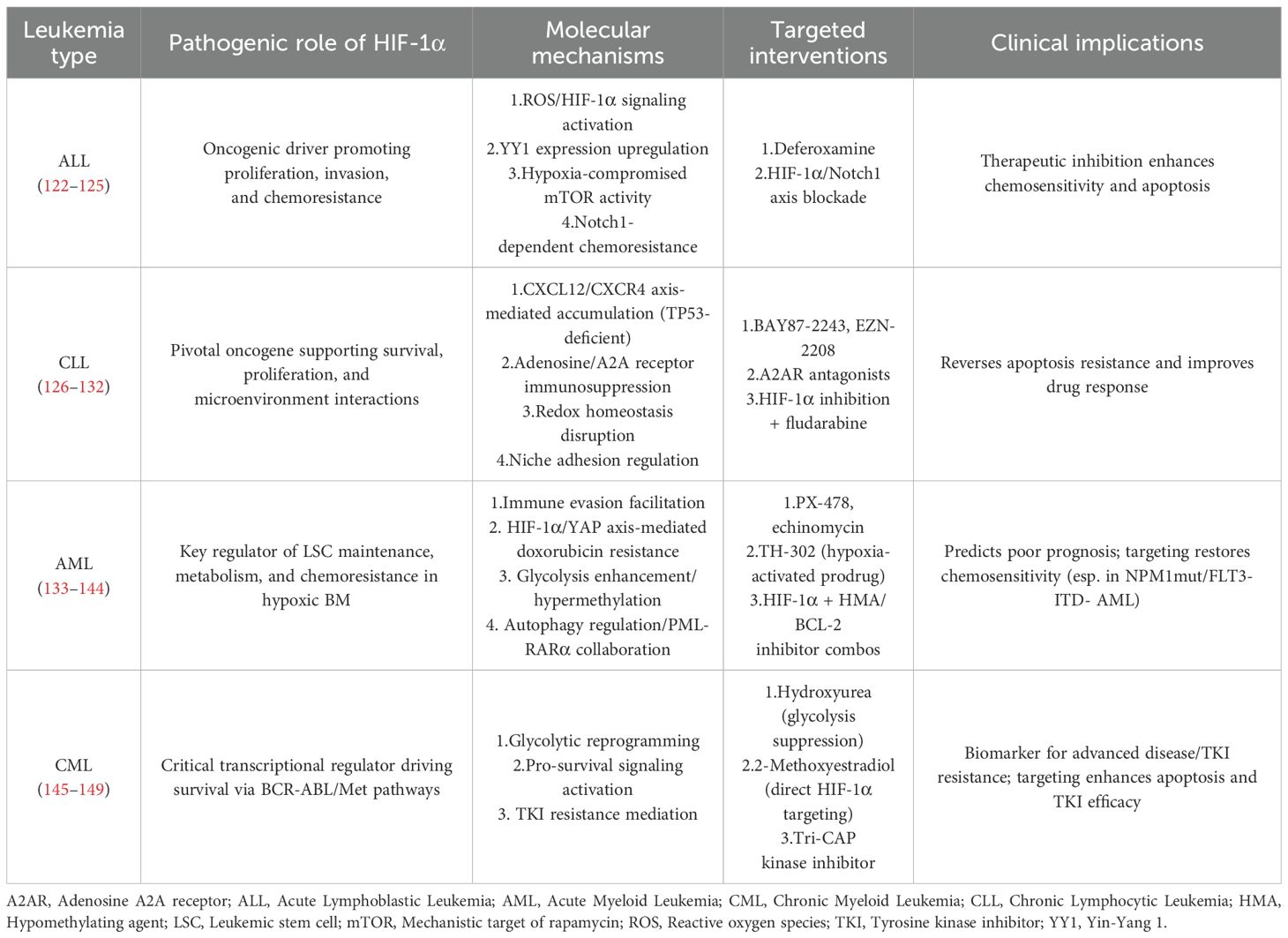
Table 1. HIF-1α and antileukemic effects.
7 Targeting HIF-1α
7.1 Advances in nanotherapeutic development
HIF-1α has emerged as a high-priority therapeutic target, driving the development of multiple inhibition strategies. Current approaches focus on four principal mechanisms: HIF1A gene silencing, suppression of HIF-1α protein translation, enhancement of proteasomal degradation, and blockade of HIF-1 transcriptional activity. These modalities collectively aim to attenuate HIF-1α signaling through distinct molecular pathways (150). Nanotechnology has revolutionized HIF-1α-targeted therapy by enabling precise drug delivery systems. Nanotherapeutics leverage unique biological properties to achieve therapeutic monitoring, diagnostic integration, and controlled release kinetics. Engineered nanocarriers with tailored surface modifications and nanoscale dimensions enhance drug delivery selectivity while improving solubility/stability, enabling multi-agent synergism, increasing bioavailability, and facilitating sustained payload release (151). These attributes position nanocarriers as powerful tools for maximizing therapeutic efficacy while minimizing systemic toxicity.
Over recent decades, researchers have engineered diverse nanocarrier architectures including lipid-based nanoparticles, extracellular vesicles (EVs), bioinspired/biohybrid systems, and emerging platforms like metal-organic frameworks (MOFs). This structural diversity not only expands nanomedicine development possibilities but also provides innovative solutions for HIF-1α-targeted interventions (151). Liposomes, as pioneering nanocarriers, have demonstrated clinical success in delivering various HIF-1α-targeted agents (152). EVs, nature-derived nanovesicles, show particular promise for bioactive molecule transport in HIF-1α modulation strategies (153). MOFs offer exceptional drug-loading capacities through their high surface-area-to-volume ratios and programmable porosity, establishing novel platforms for controlled therapeutic release (154).
7.2 Nanotherapeutic strategies targeting HIF-1α
7.2.1 HIF-1α siRNA delivery systems
RNA interference-mediated HIF-1α gene silencing represents a potent therapeutic strategy achieved through advanced nanocarrier systems designed to enhance siRNA stability and delivery precision. Cadmium telluride quantum dots functionalized with 2-deoxyglucose enable GLUT1-mediated hypoxic targeting and efficient siRNA delivery through receptor-specific endocytosis mechanisms (155). These nanosystems may target the liver in cGVHD, as the liver is highly metabolically active and glucose transporters like GLUT1 are involved in its normal function and are dysregulated in cGVHD-associated hepatic complications (156). Chimeric membrane-coated iron oxide magnetic nanoparticles demonstrate enhanced tissue penetration capabilities for deep hypoxic zone targeting, as evidenced in recent studies (157). They could potentially target the skin in cGVHD, where deep-seated hypoxic regions may develop due to inflammation and impaired microcirculation, leading to fibrotic changes. Innovative hypoxia-responsive delivery platforms utilizing 2-nitroimidazole-modified polyethyleneimine complexes (bPEI1.8k-C6) achieve oxygen tension-dependent siRNA release through controlled disassembly under low oxygen conditions (158). In cGVHD, these platforms might target the lungs, which are often affected in cGVHD, and the hypoxic microenvironment in the lungs could trigger the release of siRNA to modulate HIF-1α levels (Figure 3).
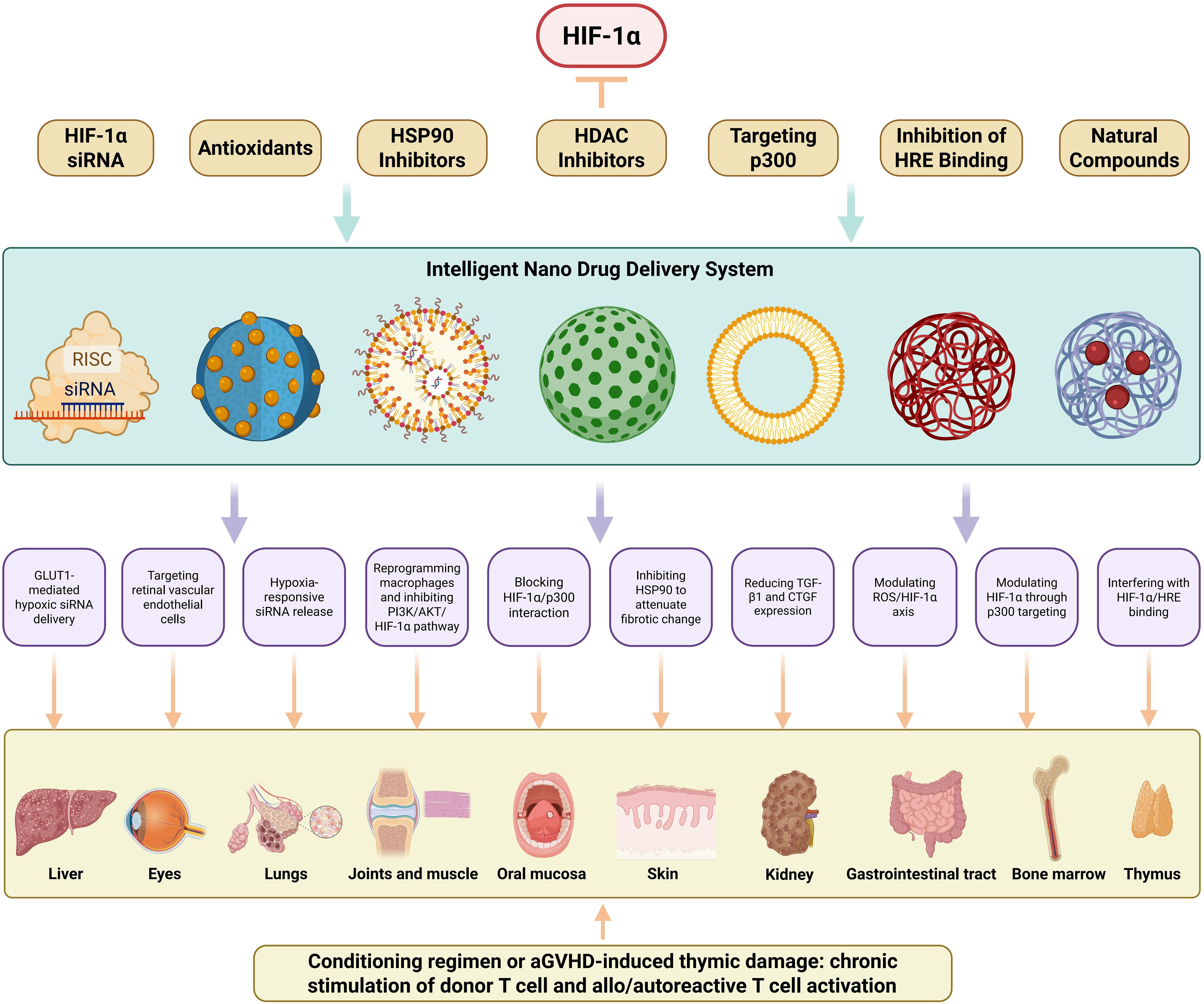
Figure 3. HIF-1α-Targeted Nanotherapeutic Strategies for Organ-Specific cGVHD Intervention. This schematic delineates a multidimensional nanotherapeutic approach targeting HIF-1α in cGVHD. Seven distinct targeting modalities are presented: HIF-1α siRNA delivery, antioxidative nanotherapy, HSP90 inhibition, HDAC inhibition, p300 interference, HRE binding blockade, and phytochemical-based therapy. Collectively, these constitute integrated nano-platforms engineered for HIF-1α gene silencing, enhanced proteasomal degradation, or transcriptional inhibition. An intelligent nanodelivery system-employing diverse nanocarriers (e.g., liposomes, extracellular vesicles, metal-organic frameworks)-facilitates hypoxia-responsive drug release, receptor-mediated targeted delivery, and enhanced biodistribution. This system enables organ-specific precision low-toxicity interventions for cGVHD-affected tissues, including liver (GLUT1-mediated hypoxic siRNA delivery), eyes (Targeting retinal vascular endothelial cells), lungs (Hypoxia-responsive siRNA release), joints/muscles (Reprogramming macrophages and inhibiting PI3K/AKT/HIF-1α pathway), oral mucosa (Blocking HIF-1α/p300 interaction), skin (Inhibiting HSP90 to attenuate fibrotic change), kidneys (Reducing TGF-β1 and CTGF expression), gastrointestinal tract (Modulating ROS/HIF-1α axis), bone marrow (Modulating HIF-1α through p300 targeting), and thymus (Interfering with HIF-1α/HRE binding).
7.2.2 Antioxidant nanotherapeutics
Antioxidant-based nanotherapeutic approaches combat HIF-1α stabilization by maintaining prolyl hydroxylase domain 2 (PHD2) activity through reactive oxygen species (ROS) scavenging. Bilobalide-containing nanoformulations significantly reduce HIF-1α expression in hypoxic cellular environments through enhanced redox homeostasis regulation (159). Ellagic acid encapsulated in nanoliposomal carriers demonstrates dual functionality, simultaneously suppressing HIF-1α activity and protecting against chemotherapy-induced hepatotoxicity in preclinical models (160). Arsenic sulfide mineral-derived nanoparticles exhibit potent ROS/HIF-1α axis modulation, effectively reducing both oxidative stress markers and hypoxia signaling pathways (161). They may target the gastrointestinal tract in cGVHD, where oxidative stress and abnormal HIF-1α activation contribute to mucosal damage and inflammation (162) (Figure 3).
7.2.3 HSP90 inhibition platforms
Heat shock protein 90 (HSP90) inhibition strategies leverage nanotechnology to induce proteasomal degradation of HIF-1α through ubiquitination pathways. Nanoporphyrin-based micellar systems co-encapsulating HSP90 inhibitors achieve synergistic HIF-1α suppression through combined photothermal therapy and photodynamic therapy modalities, with the photodynamic component generating cytotoxic singlet oxygen while photothermal effects enhance drug release kinetics (163, 164). These platforms may target the skin in cGVHD, as the skin is easily accessible for photothermal and photodynamic therapies, and modulation of HIF-1α through HSP90 inhibition can potentially reduce fibrotic and inflammatory processes in cGVHD-affected skin (Figure 3).
7.2.4 HDAC inhibition strategies
Histone deacetylase (HDAC) inhibitors demonstrate dual therapeutic benefits in both preventing graft-versus-host disease (GVHD) and preserving graft-versus-leukemia (GVL) effects, as evidenced by recent research (165). These compounds additionally promote HIF-1α degradation through epigenetic modulation mechanisms (166). The encapsulation of HDAC inhibitor SAHA within multifunctional photosensitizer-integrated nanogels significantly reduces HIF-1α levels while enhancing photodynamic therapy efficacy (167). The metallofullerenol nanomaterial Gd@C82(OH)22 exhibits multimodal inhibitory activity, functioning as both an HDAC1 inhibitor and a potent suppressor of HIF-1α and TGF-β signaling pathways (168). These may target the lymphoid organs in cGVHD, such as the spleen and lymph nodes, as they play a crucial role in the immune response in cGVHD, and modulation of HIF-1α through HDAC inhibition may help regulate the immune-mediated damage (Figure 3).
7.2.5 p300 targeting approaches
The transcriptional coactivator p300 enhances HIF-1α stability through acetylation of its C-terminal domain, amplifying hypoxic signaling under low oxygen conditions (169). Pharmacological intervention using lificiguat (YC-1) disrupts the HIF-1α/p300 interaction, effectively inhibiting transcriptional activation (170). Advanced liposomal systems prepared via film ultrasonic dispersion techniques demonstrate compartmentalized drug loading, with deferoxamine encapsulated in hydrophilic layers and YC-1 in hydrophobic bilayers. This dual-delivery platform maintains iron chelation benefits while preventing HIF-1α overexpression typically induced by deferoxamine (171). The liposomal system may target the bone marrow in cGVHD, as iron metabolism and HIF-1α regulation are important in the bone marrow microenvironment, and modulation of HIF-1α through p300 targeting can potentially improve hematopoiesis and reduce inflammation. Furthermore, the liposomal system encapsulated with YC-1 may also target the kidneys in cGVHD. It has been shown that YC-1 treatment reduces the protein expression levels of TGF-β1 and connective tissue growth factor (CTGF) in diabetic kidneys (172).
Photodynamic therapy challenges are addressed through innovative nanoparticle platforms combining long-lived iridium photosensitizers with YC-1, which mitigate oxygen depletion while suppressing HIF-1α upregulation (173). They could target the ocular surface in cGVHD, where photodynamic therapy can be applied topically, and HIF-1α modulation may alleviate dry eye symptoms and associated inflammation. Although chetomin shows promise in blocking HIF-1α/p300 interactions, its clinical application alongside mTOR inhibitor everolimus is limited by poor solubility-a limitation overcome through mPEG-b-PLA copolymer micelles prepared via solvent evaporation (174). These micelles may target the oral mucosa in cGVHD, as the oral cavity is often affected in cGVHD, and modulation of HIF-1α through p300 targeting can potentially reduce mucosal inflammation and ulceration (Figure 3).
7.2.6 HRE binding interference
The formation of active HIF-1 complexes through HIF-1α/β heterodimerization and subsequent binding to hypoxia response elements (HREs) represents a critical therapeutic target. Echinomycin specifically intercalates HRE sequences, effectively blocking transcriptional activation while demonstrating immunomodulatory effects through Tregs cell expansion and Th17/Th1 response reduction, potentially mediated through direct HIF-1α inhibition (175). Nanoliposome-based reformulation of echinomycin enhances therapeutic safety and efficacy through improved pharmacokinetic profiles (152). These nanoliposomal formulations of echinomycin may target the thymus in cGVHD, as the thymus is important for T-cell development and maturation, and modulation of HIF-1α through HRE binding interference can potentially regulate the immune response in cGVHD (Figure 3).
7.2.7 Phytochemical-based nanotherapeutics
Natural compounds demonstrate significant potential in HIF-1α modulation through nanotechnology-enhanced delivery systems. Berberine-iron oxide nanoparticle complexes induce proteasome-dependent HIF-1α degradation while enabling magnetic targeting capabilities (176). These complexes may target the heart in cGVHD, as the heart can be affected by cGVHD-related microvascular changes and fibrosis, and magnetic targeting can potentially deliver berberine to the affected cardiac tissues to modulate HIF-1α. Core-satellite structured upconverting nanoparticles loaded with curcumin effectively reprogram macrophages through HIF-1α-mediated photodynamic mechanisms (177). Self-assembling dihydroartemisinin dimer nanoprodrugs (SS NPs) exhibit enhanced stability and glycolytic regulation via PI3K/AKT/HIF-1α pathway inhibition, demonstrating high drug loading capacity (178). These nanoprodrugs may target the joints and muscle tissues in cGVHD and modulation of HIF-1α through glycolytic pathway regulation can potentially improve joints and muscle function (Table 2, Figure 3).
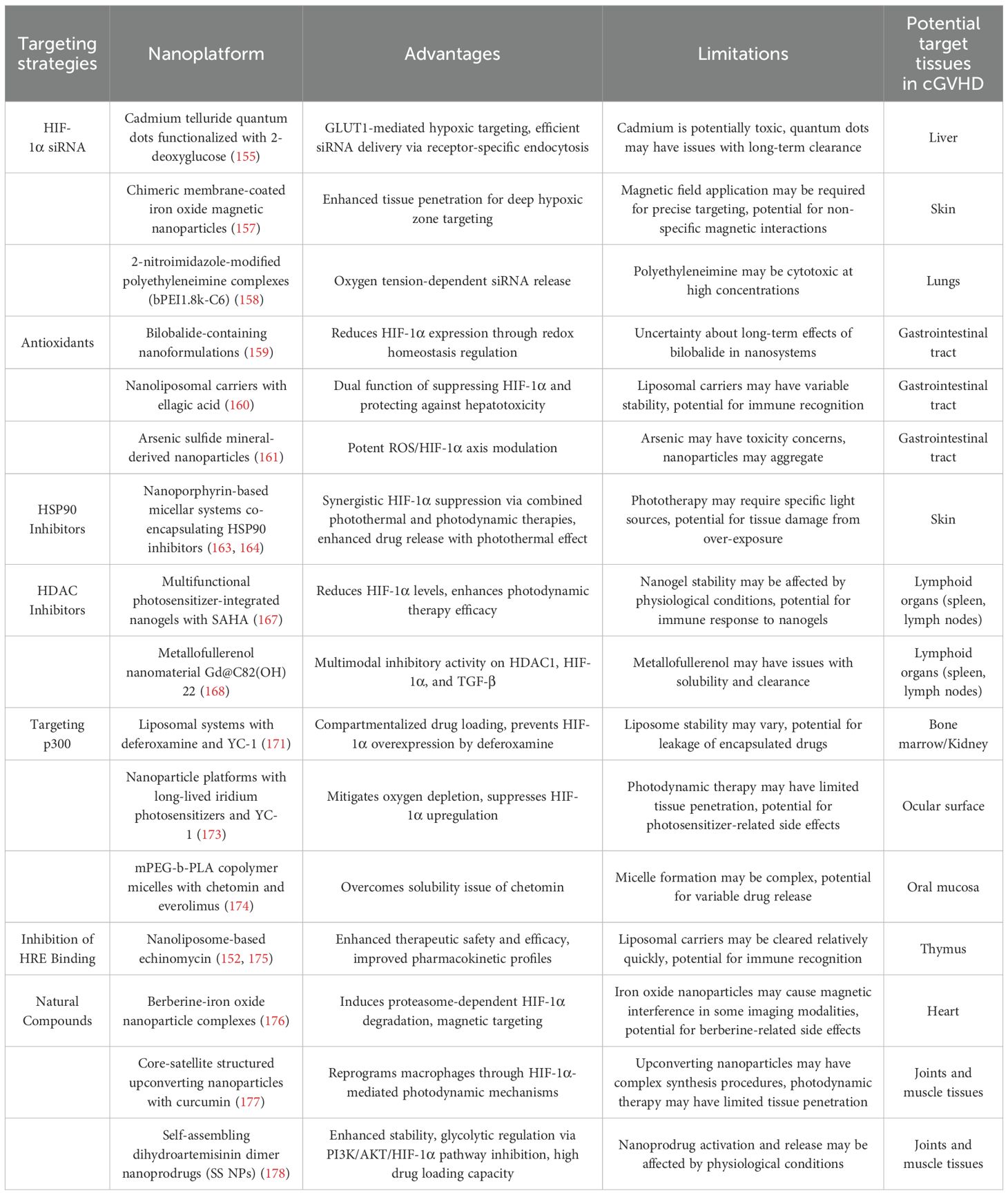
Table 2. Nanotherapeutic strategies targeting HIF-1α.
7.3 Addressing practical challenges in nanotherapeutic translation
While the preceding nanotherapeutic strategies targeting HIF-1α demonstrate considerable preclinical promise for mitigating cGVHD manifestations in specific organs, their successful clinical translation necessitates a rigorous assessment and proactive mitigation of inherent practical challenges.
7.3.1 Scalability in production
A primary translational hurdle involves achieving scalable and reproducible manufacturing of complex nanocarriers. The intricate architectures, specialized surface functionalizations, and precise drug loading requirements mandate sophisticated production processes susceptible to batch-to-batch variability, posing significant barriers to cost-effective, large-scale Good Manufacturing Practice (GMP) compliant production required for clinical trials and commercialization (179). Mitigation strategies center on adopting modular nanocarrier designs amenable to scalable production methods, implementing Quality-by-Design (QbD) principles for robust process optimization and control, and leveraging advanced analytical techniques for exhaustive physicochemical characterization to ensure batch consistency (180).
7.3.2 Regulatory hurdles and compliance strategies
Navigating the evolving regulatory landscape for nanomedicines presents distinct complexities. Regulatory agencies face the challenge of evaluating novel material interactions, complex structure-activity relationships, potential long-term biodistribution profiles, and unique safety profiles that may differ significantly from traditional small molecules or biologics. Pre-emptive mitigation involves early and continuous engagement with regulatory bodies to define appropriate non-clinical testing pathways, developing comprehensive safety assessment programs that include extensive in vitro immunogenicity screening and specialized in vivo toxicology studies evaluating organ accumulation and chronic effects, and establishing standardized characterization protocols for novel nanoplatforms (181).
7.3.3 Pharmacokinetics, toxicity, and optimization strategies
The pharmacokinetic profile and potential toxicity of nanotherapeutics remain critical considerations. The modifications that confer enhanced permeability and retention (EPR) or active targeting capabilities can also lead to unintended interactions with the mononuclear phagocyte system (MPS), resulting in accelerated blood clearance, significant liver/spleen sequestration, and potential off-target toxicities upon repeated dosing, thereby limiting bioavailability at the intended pathological site and increasing systemic burden (182). Moreover, the biodistribution and long-term biocompatibility of both the nanocarrier components and their degradation products must be thoroughly investigated (183). Mitigation approaches for pharmacokinetic and toxicity challenges focus on strategic material selection, surface engineering to minimize MPS recognition, rational dose optimization guided by detailed pharmacokinetic/pharmacodynamic (PK/PD) modelling in relevant disease models, and the development of predictive in vitro and in silico models to better estimate human safety margins.
7.3.4 Biological barriers and translational strategies
While HIF-1α-targeted nanotherapeutics show preclinical promise for cGVHD, clinical translation requires addressing disease-specific biological hurdles. Key challenges include aligning nanocarrier design with cGVHD’s multifocal nature (navigating vascular dysfunction and avoiding MPS clearance), resolving HIF-1α’s dual role (balancing fibrosis inhibition with tissue repair), and developing hypoxia-responsive release systems. Preclinical models must recapitulate cGVHD’s immunological complexity, while biomarker discovery should enable non-invasive efficacy monitoring (184). Regulatory strategies should leverage adaptive trials for organ-specific evaluation, integrating nanomedicine design with cGVHD pathophysiology to restore immune tolerance and tissue homeostasis.
8 Discussion and outlook
As a potential therapeutic target for cGVHD, HIF-1α has garnered attention due to its multifaceted roles in immune regulation, metabolic reprogramming, and fibrosis. Inhibiting HIF-1α can simultaneously intervene in inflammatory responses, abnormal T/B cell activation, and tissue fibrosis, demonstrating the advantage of “one target, multiple effects.” For example, using nanocarriers to deliver HIF-1α siRNA or small molecule inhibitors (such as Echinomycin) can precisely downregulate its expression, alleviating Th17 polarization and the fibrotic process; regulating the gut microbiota-HIF-1α axis may restore immune homeostasis, providing new ideas for combination therapy. However, there are still many challenges that cannot be ignored. The functions of HIF-1α in normal physiology (such as hypoxic adaptation) may lead to tissue damage if overly suppressed; the in vivo stability of nanomedicines and their ability to penetrate fibrotic tissues still need optimization; some inhibitors (such as HDAC inhibitors) may interfere with epigenetic regulation, leading to off-target effects; moreover, most studies are limited to animal models, requiring further validation of their safety and long-term efficacy. Future developments could include smart responsive nanocarriers (such as hypoxia-triggered drug release), combined targeting of HIF-1α with other pathways (such as PD-1/CTLA-4) (185, 186), and utilizing organoid models to screen for efficient and low-toxicity compounds. Despite the challenges, HIF-1α-targeted therapy shows great promise in cGVHD, and integrating precision medicine with nanotechnology may open new therapeutic paradigms for this refractory disease.
Author contributions
YW: Formal Analysis, Investigation, Writing – original draft, Methodology. FZ: Funding acquisition, Writing – review & editing, Conceptualization, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Shandong Society of Biomedical Engineering (SDBMEKY202502II), Shandong Province Traditional Chinese Medicine Science and Technology project (M20245202) and Shandong Provincial Key Medical and Health Discipline of Hematology (960th Hospital of the PLA Joint Logistic Support Force).
Acknowledgments
Thanks to the editors and reviewers for their hard work and important comments. Thanks to BioRender.com for the drawing material.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cooke KR, Luznik L, Sarantopoulos S, Hakim FT, Jagasia M, Fowler DH, et al. The biology of chronic graft-versus-host disease: A task force report from the national institutes of health consensus development project on criteria for clinical trials in chronic graft-versus-host disease. Biol Blood Marrow Transplant. (2017) 23:211–34. doi: 10.1016/j.bbmt.2016.09.023
2. Wilhelm K, Ganesan J, Muller T, Durr C, Grimm M, Beilhack A, et al. Graft-versus-host disease is enhanced by extracellular atp activating P2x7r. Nat Med. (2010) 16:1434–8. doi: 10.1038/nm.2242
3. Schwab L, Goroncy L, Palaniyandi S, Gautam S, Triantafyllopoulou A, Mocsai A, et al. Neutrophil granulocytes recruited upon translocation of intestinal bacteria enhance graft-versus-host disease via tissue damage. Nat Med. (2014) 20:648–54. doi: 10.1038/nm.3517
4. Hess A, Thoburn C, Bright E, and Horwitz L. Specificity of effector mechanisms in syngeneic graft-vs-host disease: recognition of the mhc class ii invariant chain peptide (Clip). Transplant Proc. (1997) 29:725–7. doi: 10.1016/s0041-1345(96)00441-1
5. Forcade E, Paz K, Flynn R, Griesenauer B, Amet T, Li W, et al. An activated Th17-prone T cell subset involved in chronic graft-versus-host disease sensitive to pharmacological inhibition. JCI Insight. (2017) 2(12):e92111. doi: 10.1172/jci.insight.92111
6. Jin H, Ni X, Deng R, Song Q, Young J, Cassady K, et al. Antibodies from donor B cells perpetuate cutaneous chronic graft-versus-host disease in mice. Blood. (2016) 127:2249–60. doi: 10.1182/blood-2015-09-668145
7. Lee SJ. Classification systems for chronic graft-versus-host disease. Blood. (2017) 129:30–7. doi: 10.1182/blood-2016-07-686642
8. MacDonald KP, Hill GR, and Blazar BR. Chronic graft-versus-host disease: biological insights from preclinical and clinical studies. Blood. (2017) 129:13–21. doi: 10.1182/blood-2016-06-686618
9. Inamoto Y, Storer BE, Petersdorf EW, Nelson JL, Lee SJ, Carpenter PA, et al. Incidence, risk factors, and outcomes of sclerosis in patients with chronic graft-versus-host disease. Blood. (2013) 121:5098–103. doi: 10.1182/blood-2012-10-464198
10. Pidala JA, Gooley TA, Luznik L, and Blazar BR. Chronic graft-versus-host disease: unresolved complication or ancient history? Blood. (2024) 144:1363–73. doi: 10.1182/blood.2023022735
11. Baumrin E, Loren AW, Falk SJ, Mays JW, and Cowen EW. Chronic graft-versus-host disease. Part ii: disease activity grading and therapeutic management. J Am Acad Dermatol. (2024) 90:19–36. doi: 10.1016/j.jaad.2022.12.023
12. Baumrin E, Loren AW, Falk SJ, Mays JW, and Cowen EW. Chronic graft-versus-host disease. Part I: epidemiology, pathogenesis, and clinical manifestations. J Am Acad Dermatol. (2024) 90:1–16. doi: 10.1016/j.jaad.2022.12.024
13. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(Reg) balance by hypoxia-inducible factor 1. Cell. (2011) 146:772–84. doi: 10.1016/j.cell.2011.07.033
14. Huang B, Phelan JD, Preite S, Gomez-Rodriguez J, Johansen KH, Shibata H, et al. In vivo crispr screens reveal a hif-1alpha-mtor-network regulates T follicular helper versus Th1 cells. Nat Commun. (2022) 13:805. doi: 10.1038/s41467-022-28378-6
15. Liu B, Xian Y, Chen X, Shi Y, Dong J, Yang L, et al. Inflammatory fibroblast-like synoviocyte-derived exosomes aggravate osteoarthritis via enhancing macrophage glycolysis. Adv Sci (Weinh). (2024) 11:e2307338. doi: 10.1002/advs.202307338
16. Kim JW, Tchernyshyov I, Semenza GL, and Dang CV. Hif-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
17. Semenza GL. Hif-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol (1985). (2000) 88:1474–80. doi: 10.1152/jappl.2000.88.4.1474
18. Gonzalez FJ, Xie C, and Jiang C. The role of hypoxia-inducible factors in metabolic diseases. Nat Rev Endocrinol. (2018) 15:21–32. doi: 10.1038/s41574-018-0096-z
19. Haddad JJ and Harb HL. Cytokines and the regulation of hypoxia-inducible factor (Hif)-1alpha. Int Immunopharmacol. (2005) 5:461–83. doi: 10.1016/j.intimp.2004.11.009
20. Jiang BH, Zheng JZ, Leung SW, Roe R, and Semenza GL. Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J Biol Chem. (1997) 272:19253–60. doi: 10.1074/jbc.272.31.19253
21. Liu W, Shen SM, Zhao XY, and Chen GQ. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int J Biochem Mol Biol. (2012) 3:165–78.
22. Loor G and Schumacker PT. Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ. (2008) 15:686–90. doi: 10.1038/cdd.2008.13
23. McGettrick AF and O’Neill LAJ. The role of hif in immunity and inflammation. Cell Metab. (2020) 32:524–36. doi: 10.1016/j.cmet.2020.08.002
24. Yeo EJ, Cho YS, Kim MS, and Park JW. Contribution of hif-1alpha or hif-2alpha to erythropoietin expression: in vivo evidence based on chromatin immunoprecipitation. Ann Hematol. (2008) 87:11–7. doi: 10.1007/s00277-007-0359-6
25. Infantino V, Santarsiero A, Convertini P, Todisco S, and Iacobazzi V. Cancer cell metabolism in hypoxia: role of hif-1 as key regulator and therapeutic target. Int J Mol Sci. (2021) 22(11):5703. doi: 10.3390/ijms22115703
26. Magar AG, Morya VK, Kwak MK, Oh JU, and Noh KC. A molecular perspective on hif-1alpha and angiogenic stimulator networks and their role in solid tumors: an update. Int J Mol Sci. (2024) 25(6):3313. doi: 10.3390/ijms25063313
27. Wu T, Young JS, Johnston H, Ni X, Deng R, Racine J, et al. Thymic damage, impaired negative selection, and development of chronic graft-versus-host disease caused by donor Cd4+ and Cd8+ T cells. J Immunol. (2013) 191:488–99. doi: 10.4049/jimmunol.1300657
28. Alho AC, Kim HT, Chammas MJ, Reynolds CG, Matos TR, Forcade E, et al. Unbalanced recovery of regulatory and effector T cells after allogeneic stem cell transplantation contributes to chronic gvhd. Blood. (2016) 127:646–57. doi: 10.1182/blood-2015-10-672345
29. Bednar KJ, Lee JH, and Ort T. Tregs in autoimmunity: insights into intrinsic brake mechanism driving pathogenesis and immune homeostasis. Front Immunol. (2022) 13:932485. doi: 10.3389/fimmu.2022.932485
30. Lyu KK, Xu MM, Du YY, He XF, Chen F, Ma X, et al. the role and mechanism of tfh and B cell in human chronic graft-versus-host-disease. Zhongguo Shi Yan Xue Ye Xue Za Zhi. (2022) 30:593–9. doi: 10.19746/j.cnki.issn.1009-2137.2022.02.045
31. Wan L, Jin Z, Hu B, Lv K, Lei L, Liu Y, et al. Il-Y aggravates murine chronic graft-versus-host disease by enhancing T and B cell responses. Front Immunol. (2020) 11:559740. doi: 10.3389/fimmu.2020.559740
32. Baron C, Somogyi R, Greller LD, Rineau V, Wilkinson P, Cho CR, et al. Prediction of graft-versus-host disease in humans by donor gene-expression profiling. PloS Med. (2007) 4:e23. doi: 10.1371/journal.pmed.0040023
33. Huai G, Markmann JF, Deng S, and Rickert CG. Tgf-beta-secreting regulatory B cells: unsung players in immune regulation. Clin Transl Immunol. (2021) 10:e1270. doi: 10.1002/cti2.1270
34. Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med. (2013) 5:179ra43. doi: 10.1126/scitranslmed.3005265
35. Shen H, Ojo OA, Ding H, Mullen LJ, Xing C, Hossain MI, et al. Hif1alpha-regulated glycolysis promotes activation-induced cell death and Ifn-gamma induction in hypoxic T cells. Nat Commun. (2024) 15:9394. doi: 10.1038/s41467-024-53593-8
36. Wu H, Zhao X, Hochrein SM, Eckstein M, Gubert GF, Knopper K, et al. Mitochondrial dysfunction promotes the transition of precursor to terminally exhausted T cells through hif-1alpha-mediated glycolytic reprogramming. Nat Commun. (2023) 14:6858. doi: 10.1038/s41467-023-42634-3
37. Corcoran SE and O’Neill LA. Hif1alpha and metabolic reprogramming in inflammation. J Clin Invest. (2016) 126:3699–707. doi: 10.1172/JCI84431
38. Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A, et al. Hif-1alpha is a metabolic switch between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression of Tregs in glioblastoma. Cell Rep. (2019) 27:226–37 e4. doi: 10.1016/j.celrep.2019.03.029
39. Corrado C and Fontana S. Hypoxia and hif signaling: one axis with divergent effects. Int J Mol Sci. (2020) 21(16):5611. doi: 10.3390/ijms21165611
40. Yu Q, Dong L, Li Y, and Liu G. Sirt1 and hif1alpha signaling in metabolism and immune responses. Cancer Lett. (2018) 418:20–6. doi: 10.1016/j.canlet.2017.12.035
41. Allen JL, Tata PV, Fore MS, Wooten J, Rudra S, Deal AM, et al. Increased bcr responsiveness in B cells from patients with chronic gvhd. Blood. (2014) 123:2108–15. doi: 10.1182/blood-2013-10-533562
42. Huang S, Cheng X, Yang G, Huang R, Feng Y, Zeng L, et al. Recent advances and research progress regarding monoclonal antibodies for chronic graft-versus-host disease. Heliyon. (2024) 10:e38460. doi: 10.1016/j.heliyon.2024.e38460
43. Poe JC, Fang J, Zhang D, Lee MR, DiCioccio RA, Su H, et al. Single-cell landscape analysis unravels molecular programming of the human B cell compartment in chronic gvhd. JCI Insight. (2023) 8(11):e169732. doi: 10.1172/jci.insight.169732
44. Perezpaya I, Garcia SG, Clos-Sansalvador M, Sanroque-Munoz M, Font-Moron M, Rodriguez-Martinez P, et al. Molecular screening of transitional B cells as a prognostic marker of improved graft outcome and reduced rejection risk in kidney transplant. Front Immunol. (2024) 15:1433832. doi: 10.3389/fimmu.2024.1433832
45. Rosser EC and Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. (2015) 42:607–12. doi: 10.1016/j.immuni.2015.04.005
46. Allen JL, Fore MS, Wooten J, Roehrs PA, Bhuiya NS, Hoffert T, et al. B cells from patients with chronic gvhd are activated and primed for survival via Baff-mediated pathways. Blood. (2012) 120:2529–36. doi: 10.1182/blood-2012-06-438911
47. Li X, Gao Q, Feng Y, and Zhang X. Developing role of B cells in the pathogenesis and treatment of chronic gvhd. Br J Haematol. (2019) 184:323–36. doi: 10.1111/bjh.15719
48. Svegliati S, Olivieri A, Campelli N, Luchetti M, Poloni A, Trappolini S, et al. Stimulatory autoantibodies to pdgf receptor in patients with extensive chronic graft-versus-host disease. Blood. (2007) 110:237–41. doi: 10.1182/blood-2007-01-071043
49. Nakasone H, Tian L, Sahaf B, Kawase T, Schoenrock K, Perloff S, et al. Allogeneic hy antibodies detected 3 months after female-to-male hct predict chronic gvhd and nonrelapse mortality in humans. Blood. (2015) 125:3193–201. doi: 10.1182/blood-2014-11-613323
50. de Masson A, Bouaziz JD, Le Buanec H, Robin M, O’Meara A, Parquet N, et al. Cd24(Hi)Cd27(+) and plasmablast-like regulatory B cells in human chronic graft-versus-host disease. Blood. (2015) 125:1830–9. doi: 10.1182/blood-2014-09-599159
51. Meng X, Asadi-Asadabad S, Cao S, Song R, Lin Z, Safhi M, et al. Metabolic rewiring controlled by hif-1alpha tunes Iga-producing B-cell differentiation and intestinal inflammation. Cell Mol Immunol. (2025) 22:54–67. doi: 10.1038/s41423-024-01233-y
52. Burrows N, Bashford-Rogers RJM, Bhute VJ, Penalver A, Ferdinand JR, Stewart BJ, et al. Dynamic regulation of hypoxia-inducible factor-1alpha activity is essential for normal B cell development. Nat Immunol. (2020) 21:1408–20. doi: 10.1038/s41590-020-0772-8
53. Meng X, Grotsch B, Luo Y, Knaup KX, Wiesener MS, Chen XX, et al. Hypoxia-inducible factor-1alpha is a critical transcription factor for il-10-producing B cells in autoimmune disease. Nat Commun. (2018) 9:251. doi: 10.1038/s41467-017-02683-x
54. Fan C, Li J, Li Y, Jin Y, Feng J, Guo R, et al. Hypoxia-inducible factor-1alpha regulates the interleukin-6 production by B cells in rheumatoid arthritis. Clin Transl Immunol. (2023) 12:e1447. doi: 10.1002/cti2.1447
55. Yamakawa T, Ohigashi H, Hashimoto D, Hayase E, Takahashi S, Miyazaki M, et al. Vitamin a-Coupled Liposomes Containing Sirna against Hsp47 Ameliorate Skin Fibrosis in Chronic Graft-Versus-Host Disease. Blood. (2018) 131:1476–85. doi: 10.1182/blood-2017-04-779934
56. Ono R, Watanabe T, Kawakami E, Iwasaki M, Tomizawa-Murasawa M, Matsuda M, et al. Co-activation of macrophages and T cells contribute to chronic gvhd in human Il-6 transgenic humanised mouse model. EBioMedicine. (2019) 41:584–96. doi: 10.1016/j.ebiom.2019.02.001
57. Olivieri A and Mancini G. Current approaches for the prevention and treatment of acute and chronic gvhd. Cells. (2024) 13(18):1524. doi: 10.3390/cells13181524
58. Stewart AG, Thomas B, and Koff J. Tgf-beta: master regulator of inflammation and fibrosis. Respirology. (2018) 23:1096–7. doi: 10.1111/resp.13415
59. Du J, Paz K, Flynn R, Vulic A, Robinson TM, Lineburg KE, et al. Pirfenidone ameliorates murine chronic gvhd through inhibition of macrophage infiltration and tgf-beta production. Blood. (2017) 129:2570–80. doi: 10.1182/blood-2017-01-758854
60. Bandarra D, Biddlestone J, Mudie S, Muller HA, and Rocha S. Hif-1alpha restricts Nf-Kappab-dependent gene expression to control innate immunity signals. Dis Model Mech. (2015) 8:169–81. doi: 10.1242/dmm.017285
61. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. Nf-kappab links innate immunity to the hypoxic response through transcriptional regulation of hif-1alpha. Nature. (2008) 453:807–11. doi: 10.1038/nature06905
62. Nizet V and Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. (2009) 9:609–17. doi: 10.1038/nri2607
63. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. (2010) 70:7465–75. doi: 10.1158/0008-5472.CAN-10-1439
64. Semba H, Takeda N, Isagawa T, Sugiura Y, Honda K, Wake M, et al. Hif-1alpha-pdk1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun. (2016) 7:11635. doi: 10.1038/ncomms11635
65. Chen X, Tang J, Shuai W, Meng J, Feng J, and Han Z. Macrophage polarization and its role in the pathogenesis of acute lung injury/acute respiratory distress syndrome. Inflammation Res. (2020) 69:883–95. doi: 10.1007/s00011-020-01378-2
66. Takeda N, O’Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, et al. Differential activation and antagonistic function of hif-alpha isoforms in macrophages are essential for no homeostasis. Genes Dev. (2010) 24:491–501. doi: 10.1101/gad.1881410
67. Basheeruddin M and Qausain S. Hypoxia-inducible factor 1-alpha (Hif-1alpha): an essential regulator in cellular metabolic control. Cureus. (2024) 16:e63852. doi: 10.7759/cureus.63852
68. Qiu B, Yuan P, Du X, Jin H, Du J, and Huang Y. Hypoxia inducible factor-1alpha is an important regulator of macrophage biology. Heliyon. (2023) 9:e17167. doi: 10.1016/j.heliyon.2023.e17167
69. Seim GL, Britt EC, John SV, Yeo FJ, Johnson AR, Eisenstein RS, et al. Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-gamma stimulation. Nat Metab. (2019) 1:731–42. doi: 10.1038/s42255-019-0083-2
70. Sesti-Costa R, de Moraes-Vieira PMM, and Cervantes-Barragan L. Dendritic cells: immune response in infectious diseases and autoimmunity. Mediators Inflammation. (2020) 2020:2948525. doi: 10.1155/2020/2948525
71. Basu A, Ramamoorthi G, Albert G, Gallen C, Beyer A, Snyder C, et al. Differentiation and regulation of T(H) cells: A balancing act for cancer immunotherapy. Front Immunol. (2021) 12:669474. doi: 10.3389/fimmu.2021.669474
72. Lewis KL and Reizis B. Dendritic cells: arbiters of immunity and immunological tolerance. Cold Spring Harb Perspect Biol. (2012) 4:a007401. doi: 10.1101/cshperspect.a007401
73. Hong C, Jin R, Dai X, and Gao X. Functional contributions of antigen presenting cells in chronic graft-versus-host disease. Front Immunol. (2021) 12:614183. doi: 10.3389/fimmu.2021.614183
74. Gail LM, Schell KJ, Lacina P, Strobl J, Bolton SJ, Steinbakk Ulriksen E, et al. Complex interactions of cellular players in chronic graft-versus-host disease. Front Immunol. (2023) 14:1199422. doi: 10.3389/fimmu.2023.1199422
75. Sanmarco LM, Rone JM, Polonio CM, Fernandez Lahore G, Giovannoni F, Ferrara K, et al. Lactate limits cns autoimmunity by stabilizing hif-1alpha in dendritic cells. Nature. (2023) 620:881–9. doi: 10.1038/s41586-023-06409-6
76. Kohler T, Reizis B, Johnson RS, Weighardt H, and Forster I. Influence of hypoxia-inducible factor 1alpha on dendritic cell differentiation and migration. Eur J Immunol. (2012) 42:1226–36. doi: 10.1002/eji.201142053
77. Liu J, Zhang X, Chen K, Cheng Y, Liu S, Xia M, et al. Ccr7 chemokine receptor-inducible lnc-dpf3 restrains dendritic cell migration by inhibiting hif-1alpha-mediated glycolysis. Immunity. (2019) 50:600–15 e15. doi: 10.1016/j.immuni.2019.01.021
78. Dai Y, Wu J, Wang J, Wang H, Guo B, Jiang T, et al. Magnesium ions promote the induction of immunosuppressive bone microenvironment and bone repair through hif-1alpha-tgf-beta axis in dendritic cells. Small. (2024) 20:e2311344. doi: 10.1002/smll.202311344
79. Maio M, Barros J, Joly M, Vahlas Z, Marin Franco JL, Genoula M, et al. Elevated glycolytic metabolism of monocytes limits the generation of hif1a-driven migratory dendritic cells in tuberculosis. Elife. (2024) 12:RP89319. doi: 10.7554/eLife.89319
80. Doglio M, Crossland RE, Alho AC, Penack O, Dickinson AM, Stary G, et al. Cell-based therapy in prophylaxis and treatment of chronic graft-versus-host disease. Front Immunol. (2022) 13:1045168. doi: 10.3389/fimmu.2022.1045168
81. Bejanyan N, Brunstein CG, Cao Q, Lazaryan A, Luo X, Curtsinger J, et al. Delayed immune reconstitution after allogeneic transplantation increases the risks of mortality and chronic gvhd. Blood Adv. (2018) 2:909–22. doi: 10.1182/bloodadvances.2017014464
82. Boukouaci W, Busson M, Peffault de Latour R, Rocha V, Suberbielle C, Bengoufa D, et al. Mica-129 genotype, soluble mica, and anti-mica antibodies as biomarkers of chronic graft-versus-host disease. Blood. (2009) 114:5216–24. doi: 10.1182/blood-2009-04-217430
83. Lauener MP, Tanaka E, Mei A, Abdossamadi S, Ostroumov E, Geltink RIK, et al. Expansion and characterization of immune suppressive cd56(Bright)Perforin(-) regulatory-like natural killer cells in chronic graft-versus-host disease. Cytotherapy. (2024) 26:1472–83. doi: 10.1016/j.jcyt.2024.07.013
84. Dou Y, Nian Z, Wang D, Sun G, Zhou L, Hu Z, et al. Reconstituted cd74(+) nk cells trigger chronic graft versus host disease after allogeneic bone marrow transplantation. J Autoimmun. (2024) 147:103274. doi: 10.1016/j.jaut.2024.103274
85. Ni J, Wang X, Stojanovic A, Zhang Q, Wincher M, Buhler L, et al. Single-cell rna sequencing of tumor-infiltrating nk cells reveals that inhibition of transcription factor hif-1alpha unleashes nk cell activity. Immunity. (2020) 52:1075–87 e8. doi: 10.1016/j.immuni.2020.05.001
86. Krzywinska E, Kantari-Mimoun C, Kerdiles Y, Sobecki M, Isagawa T, Gotthardt D, et al. Loss of hif-1alpha in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat Commun. (2017) 8:1597. doi: 10.1038/s41467-017-01599-w
87. Cluff E, Magdaleno CC, Fernandez E, House T, Swaminathan S, Varadaraj A, et al. Hypoxia-inducible factor-1 alpha expression is induced by Il-2 via the Pi3k/mtor pathway in hypoxic nk cells and supports effector functions in nkl cells and ex vivo expanded nk cells. Cancer Immunol Immunother. (2022) 71:1989–2005. doi: 10.1007/s00262-021-03126-9
88. Coulibaly A, Velasquez SY, Kassner N, Schulte J, Barbarossa MV, and Lindner HA. Stat3 governs the hif-1alpha response in Il-15 primed human nk cells. Sci Rep. (2021) 11:7023. doi: 10.1038/s41598-021-84916-0
89. Victorino F, Bigley TM, Park E, Yao CH, Benoit J, Yang LP, et al. Hif1alpha is required for nk cell metabolic adaptation during virus infection. Elife. (2021) 10:e68484. doi: 10.7554/eLife.68484
90. Markey KA, Schluter J, Gomes ALC, Littmann ER, Pickard AJ, Taylor BP, et al. The microbe-derived short-chain fatty acids butyrate and propionate are associated with protection from chronic gvhd. Blood. (2020) 136:130–6. doi: 10.1182/blood.2019003369
91. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. (2013) 341:569–73. doi: 10.1126/science.1241165
92. Yue X, Zhou H, Wang S, Chen X, and Xiao H. Gut microbiota, microbiota-derived metabolites, and graft-versus-host disease. Cancer Med. (2024) 13:e6799. doi: 10.1002/cam4.6799
93. Dant TA, Lin KL, Bruce DW, Montgomery SA, Kolupaev OV, Bommiasamy H, et al. T-cell expression of ahr inhibits the maintenance of pt(Reg) cells in the gastrointestinal tract in acute gvhd. Blood. (2017) 130:348–59. doi: 10.1182/blood-2016-08-734244
94. Lamas B, Natividad JM, and Sokol H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol. (2018) 11:1024–38. doi: 10.1038/s41385-018-0019-2
95. Fiorucci S, Biagioli M, Zampella A, and Distrutti E. Bile acids activated receptors regulate innate immunity. Front Immunol. (2018) 9:1853. doi: 10.3389/fimmu.2018.01853
96. Li T and Chiang JYL. Bile acid signaling in metabolic and inflammatory diseases and drug development. Pharmacol Rev. (2024) 76:1221–53. doi: 10.1124/pharmrev.124.000978
97. Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile acid metabolites control T(H)17 and T(Reg) cell differentiation. Nature. (2019) 576:143–8. doi: 10.1038/s41586-019-1785-z
98. DeFilipp Z, Damania AV, Kim HT, Chang CC, El-Jawahri A, McAfee SL, et al. Third-party fecal microbiota transplantation for high-risk treatment-naive acute gvhd of the lower gi tract. Blood Adv. (2024) 8:2074–84. doi: 10.1182/bloodadvances.2024012556
99. Liu Y, Zhao Y, Qi J, Ma X, Qi X, Wu D, et al. Fecal microbiota transplantation combined with ruxolitinib as a salvage treatment for intestinal steroid-refractory acute gvhd. Exp Hematol Oncol. (2022) 11:96. doi: 10.1186/s40164-022-00350-6
100. Di Mattia M, Sallese M, Neri M, and Lopetuso LR. Hypoxic functional regulation pathways in the gi tract: focus on the hif-1alpha and microbiota’s crosstalk. Inflammation Bowel Dis. (2024) 30:1406–18. doi: 10.1093/ibd/izae046
101. Pral LP, Fachi JL, Correa RO, Colonna M, and Vinolo MAR. Hypoxia and hif-1 as key regulators of gut microbiota and host interactions. Trends Immunol. (2021) 42:604–21. doi: 10.1016/j.it.2021.05.004
102. Goggins BJ, Minahan K, Sherwin S, Soh WS, Pryor J, Bruce J, et al. Pharmacological hif-1 stabilization promotes intestinal epithelial healing through regulation of alpha-integrin expression and function. Am J Physiol Gastrointest Liver Physiol. (2021) 320:G420–G38. doi: 10.1152/ajpgi.00192.2020
103. Seike K, Kiledal A, Fujiwara H, Henig I, Burgos da Silva M, van den Brink MRM, et al. Ambient oxygen levels regulate intestinal dysbiosis and gvhd severity after allogeneic stem cell transplantation. Immunity. (2023) 56:353–68 e6. doi: 10.1016/j.immuni.2023.01.007
104. Ning X, Zhang K, Wu Q, Liu M, and Sun S. Emerging role of twist1 in fibrotic diseases. J Cell Mol Med. (2018) 22:1383–91. doi: 10.1111/jcmm.13465
105. Liu L, Zhang P, Bai M, He L, Zhang L, Liu T, et al. P53 upregulated by hif-1alpha promotes hypoxia-induced G2/M arrest and renal fibrosis in vitro and in vivo. J Mol Cell Biol. (2019) 11:371–82. doi: 10.1093/jmcb/mjy042
106. Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, et al. Hypoxia promotes fibrogenesis in vivo via hif-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. (2007) 117:3810–20. doi: 10.1172/JCI30487
107. Du R, Xia L, Ning X, Liu L, Sun W, Huang C, et al. Hypoxia-induced bmi1 promotes renal tubular epithelial cell-mesenchymal transition and renal fibrosis via Pi3k/Akt signal. Mol Biol Cell. (2014) 25:2650–9. doi: 10.1091/mbc.E14-01-0044
108. Liu D, Sun H, Li K, Zhao Z, Liu Z, Zhang G, et al. Hif-1alpha mediates renal fibrosis by regulating metabolic remodeling of renal tubule epithelial cells. Biochem Biophys Res Commun. (2022) 618:15–23. doi: 10.1016/j.bbrc.2022.06.008
109. Brereton CJ, Yao L, Davies ER, Zhou Y, Vukmirovic M, Bell JA, et al. Pseudohypoxic hif pathway activation dysregulates collagen structure-function in human lung fibrosis. Elife. (2022) 11:e69348. doi: 10.7554/eLife.69348
110. Yan P, Yang K, Xu M, Zhu M, Duan Y, Li W, et al. Cct6a alleviates pulmonary fibrosis by inhibiting hif-1alpha-mediated lactate production. J Mol Cell Biol. (2024) 16(5):mjae021. doi: 10.1093/jmcb/mjae021
111. Tong Z, Du X, Zhou Y, Jing F, Ma J, Feng Y, et al. Drp1-mediated mitochondrial fission promotes pulmonary fibrosis progression through the regulation of lipid metabolic reprogramming by ros/hif-1alpha. Cell Signal. (2024) 117:111075. doi: 10.1016/j.cellsig.2024.111075
112. Kou K, Li S, Qiu W, Fan Z, Li M, and Lv G. Hypoxia-inducible factor 1alpha/il-6 axis in activated hepatic stellate cells aggravates liver fibrosis. Biochem Biophys Res Commun. (2023) 653:21–30. doi: 10.1016/j.bbrc.2023.02.032
113. Sun J, Shi L, Xiao T, Xue J, Li J, Wang P, et al. Microrna-21, via the hif-1alpha/vegf signaling pathway, is involved in arsenite-induced hepatic fibrosis through aberrant cross-talk of hepatocytes and hepatic stellate cells. Chemosphere. (2021) 266:129177. doi: 10.1016/j.chemosphere.2020.129177
114. Yang J, Li S, Liu S, Zhang Y, Shen D, Wang P, et al. Metformin ameliorates liver fibrosis induced by congestive hepatopathy via the mtor/hif-1alpha signaling pathway. Ann Hepatol. (2023) 28:101135. doi: 10.1016/j.aohep.2023.101135
115. Sutton KM, Hayat S, Chau NM, Cook S, Pouyssegur J, Ahmed A, et al. Selective inhibition of mek1/2 reveals a differential requirement for erk1/2 signalling in the regulation of hif-1 in response to hypoxia and igf-1. Oncogene. (2007) 26:3920–9. doi: 10.1038/sj.onc.1210168
116. Jiang H, Huang X, Wang J, Zhou Y, Ren C, Zhou T, et al. Hepatoprotective effect of medicine food homology flower saffron against ccl(4)-induced liver fibrosis in mice via the akt/hif-1alpha/vegf signaling pathway. Molecules. (2023) 28(21):7238. doi: 10.3390/molecules28217238
117. Beyer C, Schett G, Gay S, Distler O, and Distler JH. Hypoxia. Hypoxia in the pathogenesis of systemic sclerosis. Arthritis Res Ther. (2009) 11:220. doi: 10.1186/ar2598
118. Maciejewska M, Sikora M, Stec A, Zaremba M, Maciejewski C, Pawlik K, et al. Hypoxia-inducible factor-1alpha (Hif-1alpha) as a biomarker for changes in microcirculation in individuals with systemic sclerosis. Dermatol Ther (Heidelb). (2023) 13:1549–60. doi: 10.1007/s13555-023-00952-w
119. Mao J, Liu J, Zhou M, Wang G, Xiong X, and Deng Y. Hypoxia-induced interstitial transformation of microvascular endothelial cells by mediating hif-1alpha/vegf signaling in systemic sclerosis. PloS One. (2022) 17:e0263369. doi: 10.1371/journal.pone.0263369
120. Zhao Y, Xing C, Deng Y, Ye C, and Peng H. Hif-1alpha signaling: essential roles in tumorigenesis and implications in targeted therapies. Genes Dis. (2024) 11:234–51. doi: 10.1016/j.gendis.2023.02.039
121. Gaudichon J, Jakobczyk H, Debaize L, Cousin E, Galibert MD, Troadec MB, et al. Mechanisms of extramedullary relapse in acute lymphoblastic leukemia: reconciling biological concepts and clinical issues. Blood Rev. (2019) 36:40–56. doi: 10.1016/j.blre.2019.04.003
122. You H, Wang D, Wei L, Chen J, Li H, and Liu Y. Deferoxamine inhibits acute lymphoblastic leukemia progression through repression of ros/hif-1alpha, wnt/beta-catenin, and P38mapk/erk pathways. J Oncol. (2022) 2022:8281267. doi: 10.1155/2022/8281267
123. Antonio-Andres G, Martinez-Ruiz GU, Morales-Martinez M, Jimenez-Hernandez E, Martinez-Torres E, Lopez-Perez TV, et al. Transcriptional regulation of yin-yang 1 expression through the hypoxia inducible factor-1 in pediatric acute lymphoblastic leukemia. Int J Mol Sci. (2022) 23(3):1728. doi: 10.3390/ijms23031728
124. Fahy L, Calvo J, Chabi S, Renou L, Le Maout C, Poglio S, et al. Hypoxia favors chemoresistance in T-all through an hif1alpha-mediated mtorc1 inhibition loop. Blood Adv. (2021) 5:513–26. doi: 10.1182/bloodadvances.2020002832
125. Zou J, Li P, Lu F, Liu N, Dai J, Ye J, et al. Notch1 is required for hypoxia-induced proliferation, invasion and chemoresistance of T-cell acute lymphoblastic leukemia cells. J Hematol Oncol. (2013) 6:3. doi: 10.1186/1756-8722-6-3
126. Vitale C, Griggio V, Riganti C, Todaro M, Kopecka J, Jones R, et al. Targeting hif-1alpha regulatory pathways as a strategy to hamper tumor-microenvironment interactions in cll. Cancers (Basel). (2021) 13(12):2883. doi: 10.3390/cancers13122883
127. Griggio V, Vitale C, Todaro M, Riganti C, Kopecka J, Salvetti C, et al. Hif-1alpha is over-expressed in leukemic cells from tp53-disrupted patients and is a promising therapeutic target in chronic lymphocytic leukemia. Haematologica. (2020) 105:1042–54. doi: 10.3324/haematol.2019.217430
128. Valsecchi R, Coltella N, Magliulo D, Bongiovanni L, Scarfo L, Ghia P, et al. Ezn-2208 treatment suppresses chronic lymphocytic leukaemia by interfering with environmental protection and increases response to fludarabine. Open Biol. (2020) 10:190262. doi: 10.1098/rsob.190262
129. Seiffert M. Hif-1alpha: A potential treatment target in chronic lymphocytic leukemia. Haematologica. (2020) 105:856–8. doi: 10.3324/haematol.2019.246330
130. Valsecchi R, Coltella N, Belloni D, Ponente M, Ten Hacken E, Scielzo C, et al. Hif-1alpha regulates the interaction of chronic lymphocytic leukemia cells with the tumor microenvironment. Blood. (2016) 127:1987–97. doi: 10.1182/blood-2015-07-657056
131. Serra S, Vaisitti T, Audrito V, Bologna C, Buonincontri R, Chen SS, et al. Adenosine signaling mediates hypoxic responses in the chronic lymphocytic leukemia microenvironment. Blood Adv. (2016) 1:47–61. doi: 10.1182/bloodadvances.2016000984
132. Yosifov DY, Idler I, Bhattacharya N, Reichenzeller M, Close V, Ezerina D, et al. Oxidative stress as candidate therapeutic target to overcome microenvironmental protection of cll. Leukemia. (2020) 34:115–27. doi: 10.1038/s41375-019-0513-x
133. Chanswangphuwana C, Sukswai N, Tangnuntachai N, and Rojnuckarin P. Survival impact of hypoxia-inducible factor-1 alpha (Hif-1alpha) in nucleophosmin1 mutated acute myeloid leukemia. Ann Hematol. (2024) 103:5417–23. doi: 10.1007/s00277-024-06124-w
134. Velasco-Hernandez T, Trincado JL, Vinyoles M, Closa A, Martinez-Moreno A, Gutierrez-Aguera F, et al. Integrative single-cell expression and functional studies unravels a sensitization to cytarabine-based chemotherapy through hif pathway inhibition in aml leukemia stem cells. Hemasphere. (2024) 8:e45. doi: 10.1002/hem3.45
135. Yang L, Chen Y, and Wu Y. The hypoxia signaling pathway in the development of acute myeloid leukemia. BioMed Pharmacother. (2025) 186:117999. doi: 10.1016/j.biopha.2025.117999
136. Wu Q, You L, Nepovimova E, Heger Z, Wu W, Kuca K, et al. Hypoxia-inducible factors: master regulators of hypoxic tumor immune escape. J Hematol Oncol. (2022) 15:77. doi: 10.1186/s13045-022-01292-6
137. Zhu B, Pan S, Liu J, Wang S, Ni Y, Xiao L, et al. Hif-1alpha forms regulatory loop with yap to coordinate hypoxia-induced adriamycin resistance in acute myeloid leukemia cells. Cell Biol Int. (2020) 44:456–66. doi: 10.1002/cbin.11246
138. Abdul-Aziz AM, Shafat MS, Sun Y, Marlein CR, Piddock RE, Robinson SD, et al. Hif1alpha drives chemokine factor pro-tumoral signaling pathways in acute myeloid leukemia. Oncogene. (2018) 37:2676–86. doi: 10.1038/s41388-018-0151-1
139. Coltella N, Percio S, Valsecchi R, Cuttano R, Guarnerio J, Ponzoni M, et al. Hif factors cooperate with pml-raralpha to promote acute promyelocytic leukemia progression and relapse. EMBO Mol Med. (2014) 6:640–50. doi: 10.1002/emmm.201303065
140. Drolle H, Wagner M, Vasold J, Kutt A, Deniffel C, Sotlar K, et al. Hypoxia regulates proliferation of acute myeloid leukemia and sensitivity against chemotherapy. Leuk Res. (2015) 39:779–85. doi: 10.1016/j.leukres.2015.04.019
141. Li L, Zhao L, Man J, and Liu B. Cxcl2 benefits acute myeloid leukemia cells in hypoxia. Int J Lab Hematol. (2021) 43:1085–92. doi: 10.1111/ijlh.13512
142. Liu W, Dou C, Zhang C, Chen P, Zhang S, Wang R, et al. Px-478 Induces Apoptosis in Acute Myeloid Leukemia under Hypoxia by Inhibiting the Pi3k/Akt/Mtor Pathway through Downregulation of Gbe1. Biochem Pharmacol. (2024) 230:116620. doi: 10.1016/j.bcp.2024.116620
143. Wang Y, Liu Y, Bailey C, Zhang H, He M, Sun D, et al. Therapeutic targeting of tp53-mutated acute myeloid leukemia by inhibiting hif-1alpha with echinomycin. Oncogene. (2020) 39:3015–27. doi: 10.1038/s41388-020-1201-z
144. Benito J, Ramirez MS, Millward NZ, Velez J, Harutyunyan KG, Lu H, et al. Hypoxia-activated prodrug th-302 targets hypoxic bone marrow niches in preclinical leukemia models. Clin Cancer Res. (2016) 22:1687–98. doi: 10.1158/1078-0432.CCR-14-3378
145. Tsubaki M, Takeda T, Matsuda T, Kimura A, Tanaka R, Nagayoshi S, et al. Hypoxia-inducible factor 1alpha inhibitor induces cell death via suppression of bcr-abl1 and met expression in bcr-abl1 tyrosine kinase inhibitor sensitive and resistant chronic myeloid leukemia cells. BMB Rep. (2023) 56:78–83. doi: 10.5483/BMBRep.2022-0095
146. Zhao F, Mancuso A, Bui TV, Tong X, Gruber JJ, Swider CR, et al. Imatinib resistance associated with bcr-abl upregulation is dependent on hif-1alpha-induced metabolic reprograming. Oncogene. (2010) 29:2962–72. doi: 10.1038/onc.2010.67
147. Guo B, Pomicter AD, Li F, Bhatt S, Chen C, Li W, et al. Trident cold atmospheric plasma blocks three cancer survival pathways to overcome therapy resistance. Proc Natl Acad Sci U.S.A. (2021) 118(51):e2107220118. doi: 10.1073/pnas.2107220118
148. Zhang Y, Chen H, Shen Y, and Zhou X. Combined effects of 2-methoxyestradiol (Hypoxia-inducible factor 1alpha inhibitor) and dasatinib (a second-generation tyrosine kinase inhibitor) on chronic myelocytic leukemia cells. J Immunol Res. (2022) 2022:6324326. doi: 10.1155/2022/6324326
149. Chen H, Shen Y, Gong F, Jiang Y, and Zhang R. Hif-alpha promotes chronic myelogenous leukemia cell proliferation by upregulating P21 expression. Cell Biochem Biophys. (2015) 72:179–83. doi: 10.1007/s12013-014-0434-2
150. Zhang X, He C, and Xiang G. Engineering nanomedicines to inhibit hypoxia-inducible factor-1 for cancer therapy. Cancer Lett. (2022) 530:110–27. doi: 10.1016/j.canlet.2022.01.012
151. Liu Q, Zou J, Chen Z, He W, and Wu W. Current research trends of nanomedicines. Acta Pharm Sin B. (2023) 13:4391–416. doi: 10.1016/j.apsb.2023.05.018
152. Bailey CM, Liu Y, Peng G, Zhang H, He M, Sun D, et al. Liposomal formulation of hif-1alpha inhibitor echinomycin eliminates established metastases of triple-negative breast cancer. Nanomedicine. (2020) 29:102278. doi: 10.1016/j.nano.2020.102278
153. Rana R, Devi SN, Bhardwaj AK, Yashavarddhan MH, Bohra D, and Ganguly NK. Exosomes as nature’s nano carriers: promising drug delivery tools and targeted therapy for glioma. BioMed Pharmacother. (2025) 182:117754. doi: 10.1016/j.biopha.2024.117754
154. Faheem A, Lawrence MC, Bushra GA, Meli MV, and Blight BA. Metal-organic frameworks as anchors for giant unilamellar vesicle immobilization. J Mater Chem B. (2025) 13:2317–26. doi: 10.1039/d4tb02055c
155. Zhu H, Zhang S, Ling Y, Meng G, Yang Y, and Zhang W. Ph-responsive hybrid quantum dots for targeting hypoxic tumor sirna delivery. J Control Release. (2015) 220:529–44. doi: 10.1016/j.jconrel.2015.11.017
156. Huang Y, Zou Y, Jiao Y, Shi P, Nie X, Huang W, et al. Targeting glycolysis in alloreactive T cells to prevent acute graft-versus-host disease while preserving graft-versus-leukemia effect. Front Immunol. (2022) 13:751296. doi: 10.3389/fimmu.2022.751296
157. Liu S, Yu B, Wang S, Shen Y, and Cong H. Preparation, surface functionalization and application of fe(3)O(4) magnetic nanoparticles. Adv Colloid Interface Sci. (2020) 281:102165. doi: 10.1016/j.cis.2020.102165
158. Kang L, Fan B, Sun P, Huang W, Jin M, Wang Q, et al. An effective tumor-targeting strategy utilizing hypoxia-sensitive sirna delivery system for improved anti-tumor outcome. Acta Biomater. (2016) 44:341–54. doi: 10.1016/j.actbio.2016.08.029
159. Priyanka A, Nisha VM, Anusree SS, and Raghu KG. Bilobalide attenuates hypoxia induced oxidative stress, inflammation, and mitochondrial dysfunctions in 3t3-L1 adipocytes via its antioxidant potential. Free Radic Res. (2014) 48:1206–17. doi: 10.3109/10715762.2014.945442
160. Stojiljkovic N, Ilic S, Stojanovic N, Jankovic-Velickovic L, Stojnev S, Kocic G, et al. Nanoliposome-encapsulated ellagic acid prevents cyclophosphamide-induced rat liver damage. Mol Cell Biochem. (2019) 458:185–95. doi: 10.1007/s11010-019-03541-8
161. Wang T, Wen T, Li H, Han B, Hao S, Wang C, et al. Arsenic sulfide nanoformulation induces erythroid differentiation in chronic myeloid leukemia cells through degradation of bcr-abl. Int J Nanomedicine. (2019) 14:5581–94. doi: 10.2147/IJN.S207298
162. Han K, Wang F, Ma X, Wu Y, Zhang H, Zhao Y, et al. Human placental mesenchymal stromal cells alleviate intestinal oxidative damage in mice with graft-versus-host disease via Cd73/adenosine/Pi3k/Akt/Gsk-3beta axis. Cell Signal. (2024) 123:111372. doi: 10.1016/j.cellsig.2024.111372
163. Long Q, Lin TY, Huang Y, Li X, Ma AH, Zhang H, et al. Image-guided photo-therapeutic nanoporphyrin synergized hsp90 inhibitor in patient-derived xenograft bladder cancer model. Nanomedicine. (2018) 14:789–99. doi: 10.1016/j.nano.2017.12.014
164. Lin TY, Guo W, Long Q, Ma A, Liu Q, Zhang H, et al. Hsp90 inhibitor encapsulated photo-theranostic nanoparticles for synergistic combination cancer therapy. Theranostics. (2016) 6:1324–35. doi: 10.7150/thno.14882
165. Kim S, Santhanam S, Lim S, and Choi J. Targeting histone deacetylases to modulate graft-versus-host disease and graft-versus-leukemia. Int J Mol Sci. (2020) 21(12):4281. doi: 10.3390/ijms21124281
166. Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. (2001) 7:437–43. doi: 10.1038/86507
167. Liu N, Liu H, Chen H, Wang G, Teng H, and Chang Y. Polyphotosensitizer nanogels for gsh-responsive histone deacetylase inhibitors delivery and enhanced cancer photodynamic therapy. Colloids Surf B Biointerf. (2020) 188:110753. doi: 10.1016/j.colsurfb.2019.110753
168. Pan Y, Wang L, Kang SG, Lu Y, Yang Z, Huynh T, et al. Gd-metallofullerenol nanomaterial suppresses pancreatic cancer metastasis by inhibiting the interaction of histone deacetylase 1 and metastasis-associated protein 1. ACS Nano. (2015) 9:6826–36. doi: 10.1021/nn506782f
169. Shi Y and Gilkes DM. Hif-1 and hif-2 in cancer: structure, regulation, and therapeutic prospects. Cell Mol Life Sci. (2025) 82:44. doi: 10.1007/s00018-024-05537-0
170. Li SH, Shin DH, Chun YS, Lee MK, Kim MS, and Park JW. A novel mode of action of yc-1 in hif inhibition: stimulation of fih-dependent P300 dissociation from hif-1alpha. Mol Cancer Ther. (2008) 7:3729–38. doi: 10.1158/1535-7163.MCT-08-0074
171. Lang J, Zhao X, Wang X, Zhao Y, Li Y, Zhao R, et al. Targeted co-delivery of the iron chelator deferoxamine and a hif1alpha inhibitor impairs pancreatic tumor growth. ACS Nano. (2019) 13:2176–89. doi: 10.1021/acsnano.8b08823
172. Hu Z, Zhu Q, Wang Y, Deng X, Yang H, Zhou M, et al. Lipid nephrotoxicity mediated by hif-1alpha activation accelerates tubular injury in diabetic nephropathy. Ren Fail. (2024) 46:2347446. doi: 10.1080/0886022X.2024.2347446
173. Zhao J, Sun S, Li X, Zhang W, and Gou S. Enhancing photodynamic therapy efficacy of upconversion-based nanoparticles conjugated with a long-lived triplet excited state iridium(Iii)-naphthalimide complex: toward highly enhanced hypoxia-inducible factor-1. ACS Appl Bio Mater. (2020) 3:252–62. doi: 10.1021/acsabm.9b00774
174. Doddapaneni BS, Al-Fatease AM, Rao DA, and Alani AWG. Dual-drug loaded micelle for combinatorial therapy targeting hif and mtor signaling pathways for ovarian cancer treatment. J Control Release. (2019) 307:272–81. doi: 10.1016/j.jconrel.2019.06.036
175. Yao Y, Wang L, Zhou J, and Zhang X. Hif-1alpha inhibitor echinomycin reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. J Transl Med. (2017) 15:28. doi: 10.1186/s12967-017-1132-9
176. Sreeja S and Krishnan Nair CK. Tumor control by hypoxia-specific chemotargeting of iron-oxide nanoparticle - berberine complexes in a mouse model. Life Sci. (2018) 195:71–80. doi: 10.1016/j.lfs.2017.12.036
177. Zhang LJ, Huang R, Shen YW, Liu J, Wu Y, Jin JM, et al. Enhanced anti-tumor efficacy by inhibiting hif-1alpha to reprogram tams via core-satellite upconverting nanoparticles with curcumin mediated photodynamic therapy. Biomater Sci. (2021) 9:6403–15. doi: 10.1039/d1bm00675d
178. Li Y, Pei Q, Cui B, Zhang H, Han L, Li W, et al. A redox-responsive dihydroartemisinin dimeric nanoprodrug for enhanced antitumor activity. J Nanobiotechnol. (2021) 19:441. doi: 10.1186/s12951-021-01200-z
179. Yang M, Chen Y, Zhu L, You L, Tong H, Meng H, et al. Harnessing nanotechnology: emerging strategies for multiple myeloma therapy. Biomolecules. (2024) 14(1):83. doi: 10.3390/biom14010083
180. Han SH, Park SY, Cha HM, Lee KB, Lim JH, and Lee DY. A robust scale-down model development and process characterization for monoclonal antibody biomanufacturing using multivariate data analysis. J Biotechnol. (2025) 401:11–20. doi: 10.1016/j.jbiotec.2025.02.007
181. Patne AY, Mohapatra S, and Mohapatra SS. Role of nanomedicine in transforming pharmacotherapy for substance use disorder (Sud). Wiley Interdiscip Rev Nanomed Nanobiotechnol. (2025) 17:e70008. doi: 10.1002/wnan.70008
182. Zelepukin IV, Shevchenko KG, and Deyev SM. Rediscovery of mononuclear phagocyte system blockade for nanoparticle drug delivery. Nat Commun. (2024) 15:4366. doi: 10.1038/s41467-024-48838-5
183. Liu K, Yao Y, Xue S, Zhang M, Li D, Xu T, et al. Recent advances of tumor microenvironment-responsive nanomedicines-energized combined phototherapy of cancers. Pharmaceutics. (2023) 15(10):2480. doi: 10.3390/pharmaceutics15102480
184. Hartl D, de Luca V, Kostikova A, Laramie J, Kennedy S, Ferrero E, et al. Translational precision medicine: an industry perspective. J Transl Med. (2021) 19:245. doi: 10.1186/s12967-021-02910-6
185. Wu Y and Yu XZ. Tipping the gvh/gvl balance by targeting hif1alpha. Cell Rep Med. (2023) 4:101295. doi: 10.1016/j.xcrm.2023.101295
Keywords: HIF-1α, chronic graft-versus-host disease, immunomodulation, fibrosis, graft-versus-leukemia, nanomedicine
Citation: Wang Y and Zhou F (2025) Targeting hypoxia-inducible factor 1α in chronic graft-versus-host disease to enhance graft-versus-leukemia responses: implications for nanotherapeutics. Front. Immunol. 16:1605669. doi: 10.3389/fimmu.2025.1605669
Received: 03 April 2025; Accepted: 10 July 2025;
Published: 28 July 2025.
Edited by:
Lonnie Shea, University of Michigan, United StatesReviewed by:
Pradeep Shrestha, University of Texas MD Anderson Cancer Center, StatesSenthilnathan Palaniyandi, University of Missouri, United States
Darshan Badal, University of Missouri, United States
Copyright © 2025 Wang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Zhou, emhvdWZhbmcxQG1lZG1haWwuY29tLmNu