 Weiwei Qian
Weiwei Qian Yuanyuan Liu
Yuanyuan Liu Xinyang Zhao
Xinyang Zhao Yuxin Dong
Yuxin Dong Jian Zhou
Jian Zhou Songtao Shou
Songtao Shou- 1Department of Emergency Medicine, Tianjin Medical University General Hospital, Tianjin, China
- 2Department of Immunology, International Cancer Center, Shenzhen University Health Science Center, Tianjin, China
Objective: Sepsis is a systemic inflammatory response syndrome triggered by infection, characterized by high clinical heterogeneity and complex immunopathological mechanisms. Immune dysregulation plays a central role in its progression. This study aims to investigate the compositional changes of immune cells, characteristics of intercellular communication, and potential regulatory mechanisms of N⁶-methyladenosine (m6A) modification in sepsis, with a particular focus on the functional remodeling of platelets.
Methods: This study integrated single-cell RNA sequencing data (GSE167363 dataset) from sepsis patients with m6A methylation sequencing data of peripheral blood mononuclear cells (PBMCs). Through systematic analysis, we compared the differences in immune cell composition, developmental trajectories, intercellular communication, and m6A modifications among healthy controls, survivors, and non-survivors, and further screened for key m6A-regulated target genes.
Results: The analysis revealed that platelets gradually accumulated during the progression of sepsis, while B cells, T cells, and regulatory T cells (Tregs) exhibited a trend toward platelet-like phenotypic remodeling. Cell–cell communication analysis showed a marked decline in communication strength among immune cells as the disease worsened, particularly a significant weakening of the APP–CD74 signaling pathway between platelets and B cells, indicating impaired immune network synergy. m⁶A methylation sequencing revealed distinct remodeling of m⁶A peaks and dysregulation of related regulatory factors in non-survivors. Further integrative analysis identified RPA1 as a key m⁶A-regulated target gene, whose expression was closely associated with APP and co-regulated by multiple m6A-modifying factors.
Conclusion: This study reveals disruptions in immune cell interactions and an m6A-dependent mechanism of platelet functional remodeling during sepsis progression. The identification of the key target gene RPA1 offers new insights into the immunopathological mechanisms of sepsis and lays a theoretical foundation for future precision interventions and therapeutic strategies.
1 Introduction
Sepsis is a life-threatening syndrome caused by a dysregulated host response to infection, often accompanied by multiple organ dysfunction (1–4). Despite advancements in antimicrobial therapy and organ support, sepsis continues to be associated with high morbidity and mortality, particularly among critically ill patients, where mortality rates can exceed 30% (5, 6). Increasing evidence suggests that the onset and progression of sepsis are closely linked to immune system dysregulation, with immune cell dysfunction considered one of the key determinants of clinical outcomes (4, 7).
Among the various immune cells, platelets have traditionally been recognized for their roles in hemostasis and thrombosis. However, recent studies have increasingly highlighted their emerging functions in immune regulation (8, 9). Platelets can actively participate in the immune response by releasing pro-inflammatory mediators, expressing immune molecules, and interacting with other immune cells (10). In the hyperinflammatory context of sepsis, platelets may not only contribute to microthrombus formation and tissue hypoperfusion but also promote inflammation and immune suppression through interactions with T cells and monocytes (11–15). Previous studies have demonstrated that sepsis can induce a spleen-derived population of protective platelets expressing high levels of CD40 ligand (1); platelet MHC class I molecules have been shown to impair CD8+ T cell function during sepsis (11); IFITM3 has been identified as a regulator of fibrinogen uptake and platelet reactivity in non-viral sepsis (16); and glycoprotein VI (GPVI) has been reported to enhance local immune defense in pneumococcal sepsis (17). These findings collectively underscore the crucial and multifaceted role of platelets in the immunoregulatory network of sepsis.
Epigenetic modifications, particularly N6-methyladenosine (m6A), the most prevalent and reversible internal modification of eukaryotic mRNA, have recently been recognized as critical regulators of immune responses and cell fate under various pathological conditions (18, 19). The m6A modification is dynamically regulated by “writers” (e.g., METTL3, METTL14), “erasers” (e.g., FTO, ALKBH5), and “readers” (e.g., YTHDF and IGF2BP families), which collectively influence mRNA stability, splicing, translation efficiency, and degradation (18, 19). Emerging evidence has revealed that m6A plays a pivotal immunoregulatory role in diverse diseases, including cancers, autoimmune disorders, and viral infections (20). In recent years, studies focusing on m6A in sepsis have gained momentum. For instance, histone protease-regulated METTL3 was shown to exacerbate sepsis-induced lung injury via m6A-mediated modification of ACSL4, promoting ferroptosis (21). Neutrophil extracellular traps (NETs) have been implicated in modulating m6A modification and regulating alveolar epithelial cell ferroptosis in sepsis-associated acute lung injury (22). METTL3 has also been reported to reprogram mitochondrial metabolism via an m6A–IGF2BP2–dependent mechanism, aggravating ferroptosis-induced myotoxicity (23). Additionally, YTHDF1 enhances WWP1 expression, promoting NLRP3 ubiquitination and thereby suppressing caspase-1–dependent pyroptosis (24). METTL3-mediated m6A modification also facilitates TLR4 pathway activation to enhance neutrophil activation during sepsis (25). Despite these advances in understanding the role of m6A in organ-specific damage during sepsis, its functional involvement in peripheral immune cells, particularly in the platelet-mediated immune regulatory network, remains poorly characterized.
Based on the aforementioned evidence, the present study integrates single-cell RNA sequencing (scRNA-seq) and m6A methylome sequencing to systematically investigate the immunoregulatory role of platelets in sepsis. We explore alterations at the levels of cellular composition, cell-cell communication, and epitranscriptomic modifications, aiming to identify key m6A regulatory factors and downstream target genes. This work provides novel mechanistic insights into the molecular underpinnings of sepsis and offers a theoretical foundation for the identification of potential therapeutic targets.
2 Materials and methods
2.1 Single-cell dataset and patient sample collection
This study utilized the publicly available scRNA-seq dataset GSE167363, which includes peripheral blood mononuclear cells (PBMCs) from 10 patients with Gram-negative sepsis (survivor group: n = 6; non-survivor group: n = 4) and 2 healthy controls (HCs) (26). Additionally, PBMCs from six sepsis patients (survivor group: n = 3; non-survivor group: n = 3), recruited from the Department of Emergency Medicine, Tianjin Medical University General Hospital, were used for m6A methylation sequencing. Informed consent was obtained from all participants, and the study protocol was approved by the Ethics Committee of Tianjin Medical University General Hospital (Approval ID: IRB2023-YX-045-01).
2.2 scRNA-seq data processing and quality control
Single-cell transcriptomic data were processed using the Seurat package (v5.0) in R. Low-quality cells were filtered out based on the following criteria: number of features < 200 or > 6000, and mitochondrial gene content > 15%. Data were normalized using the “LogNormalize” method, and the top 2000 highly variable genes were selected for downstream analysis. Principal component analysis (PCA) was performed for dimensionality reduction, with the number of components selected via elbow plot. Data integration across samples was conducted using Seurat’s canonical correlation analysis (CCA) to minimize batch effects. Louvain algorithm was employed for clustering, and visualization was performed using UMAP and t-SNE embeddings.
2.3 Cell type annotation and differential expression analysis
Cell type annotation was conducted using a combination of GPTCelltype (v1.1.1), SingleR (v1.0.0), and scCATCH (v3.1.1), with reference to established marker gene databases and published literature. Differentially expressed genes (DEGs) were identified using the wilcoxauc function from the RPresto package, with the significance threshold set at log2 fold change > 0.25 and adjusted p < 0.05. Functional enrichment analyses, including Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Set Enrichment Analysis (GSEA), were performed using clusterProfiler (v4.6.0).
2.4 Pseudotime trajectory analysis
To explore developmental dynamics, Monocle2 (v2.22.0) was applied to infer pseudotime trajectories of key immune cell subsets, including B cells, T cells, regulatory T cells (Tregs), and platelets, based on their transcriptional profiles.
2.5 Cell–cell communication analysis
Cell–cell communication networks were predicted using CellphoneDB (v2.1.7) to identify significant ligand–receptor interactions, with a minimum expression threshold of 10% set for inclusion in downstream analyses. NicheNet was used to assess ligand activity and predict target gene regulatory potential. The interaction network was visualized using the CCplotR R package.
2.6 m6A RNA methylation sequencing and experimental workflow
Total RNA was extracted from PBMCs using TRIzol™, followed by DNase I treatment and enzymatic purification. For m6A profiling, 20 μg of total RNA was fragmented in Mg2+ lysis buffer at 70°C for 6 minutes, and the reaction was terminated with EDTA. RNA fragments were purified using the Zymo RNA Clean & Concentrator-5 kit. m6A immunoprecipitation (MeRIP) was performed using an anti-m6A antibody and Protein A/G magnetic beads, incubated at 4°C for 2 hours. After elution and purification, sequencing libraries were constructed and subjected to paired-end sequencing (PE150) on the Illumina NovaSeq 6000 platform.
2.7 m6A methylation data analysis
Raw sequencing reads were processed using Cutadapt (v2.5) to trim adaptors and remove low-quality reads. Clean reads were aligned to the human genome (hg38) using Hisat2. Peak calling and differential methylation analysis were performed using exomePeak2 (v2.13.2), with a significance threshold of p < 0.05. Motif discovery and annotation were conducted using HOMER (v4.10.4). Differentially methylated genes were subjected to GO and KEGG enrichment analysis. Peak distribution and m6A site visualization were completed using Integrative Genomics Viewer (IGV, v2.19.1).
2.8 Candidate gene screening and correlation analysis
To identify key candidate genes, we integrated three datasets: (1) differentially m6A-modified genes in PBMCs; (2) DEGs in platelets between the survivor and non-survivor groups; and (3) DEGs in platelets between HCs and the survivor group. Genes shared across all three datasets were defined as core candidate genes. The correlation between the expression of these candidate genes and the platelet ligand APP was further assessed. In addition, we evaluated the correlation between candidate gene expression and 10 major m6A regulatory factors.
2.9 Statistical analysis and data visualization
All data visualizations were performed using R packages, including ggplot2, ComplexHeatmap, VennDiagram, and circlize. Statistical analyses were conducted based on data distribution and type, applying the Wilcoxon rank-sum test, Student’s t-test, or ANOVA as appropriate. A p-value < 0.05 was considered statistically significant.
3 Results
3.1 Single-cell transcriptomics reveals peripheral immune heterogeneity and a potentially critical role of platelets in sepsis progression
To investigate the immunological landscape of PBMCs in Gram-negative sepsis patients with distinct clinical outcomes (survivors vs. non-survivors), we performed scRNA-seq analysis based on the publicly available dataset GSE167363, which includes PBMCs from 10 sepsis patients (survivor group: n = 6; non-survivor group: n = 4) and 2 HCs.
First, canonical immune cell marker genes were integrated to generate a bubble plot depicting marker gene expression patterns across different immune cell subsets (Figure 1A). Based on these expression profiles, unsupervised clustering was performed, followed by dimensionality reduction using t-SNE1 and UMAP1, leading to the successful identification of multiple PBMCs subpopulations, including B cells, CD14+ monocytes, FCGR3A+ monocytes, dendritic cells (DCs), T cells, regulatory T cells (Tregs), natural killer (NK) cells, platelets, erythroid progenitors, neutrophils, macrophages, plasmacytoid dendritic cells (pDCs), and hematopoietic stem cells (Figure 1B). Notably, the distribution of these cell subsets varied significantly among HCs, survivors, and non-survivors, indicating pronounced immune heterogeneity across groups (Supplementary Figures 1A–D).
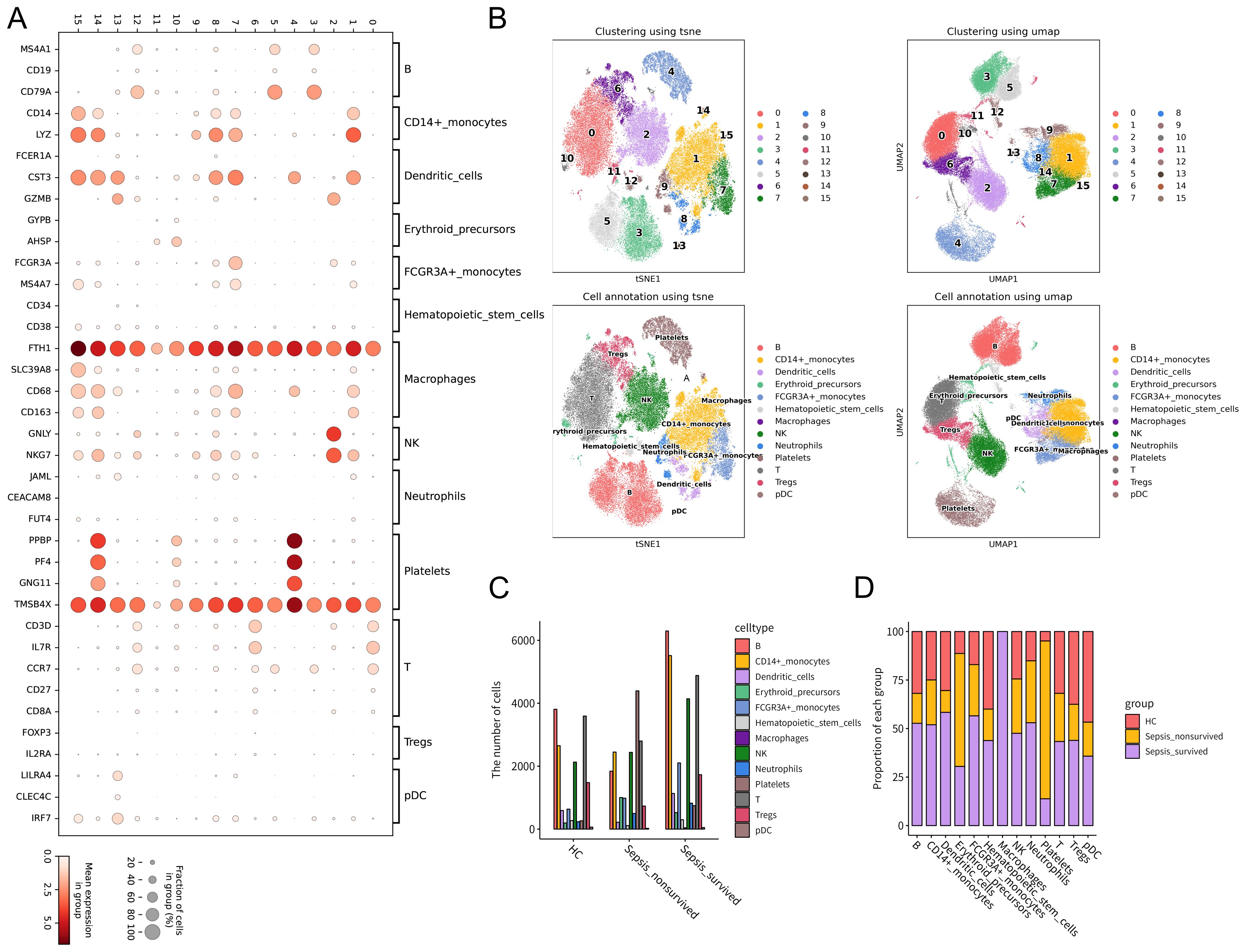
Figure 1. Single-cell transcriptomic analysis of peripheral blood mononuclear cells (PBMCs) from healthy controls (HCs) and patients with Gram-negative sepsis. Single-cell RNA sequencing was performed on PBMCs from 10 sepsis patients (survivor group: n = 6; non-survivor group: n = 4) and 2 healthy controls using the GSE167363 dataset. (A) Expression patterns of PBMCs-specific marker genes visualized in a bubble plot. Dot size represents the proportion of cells expressing a given marker, and color intensity indicates the average expression level. (B) Cell clustering was performed based on canonical marker expression, followed by dimensionality reduction and visualization using t-SNE and UMAP. (C) Comparison of total PBMCs counts across HCs, sepsis survivors, and non-survivors. (D) Relative proportions of immune cell subsets across the three groups, illustrating alterations in immune composition under different pathological conditions.
At the cellular level, a comparison of total PBMCs counts among the HC, survivor, and non-survivor groups revealed distinct patterns of cellular abundance (Figure 1C, Supplementary Table S1). Further analysis of immune cell composition showed a progressive increase in the relative proportion of platelets from HCs to survivors and non-survivors (Figure 1D). This trend suggests that platelets may not only participate in the immune response during sepsis but also be closely associated with disease severity and clinical outcomes. Taken together, these findings indicate that the immune cell composition of PBMCs in Gram-negative sepsis patients undergoes significant alterations. In particular, the increasing proportion of platelets during disease progression suggests their potential key role in the pathogenesis and adverse outcomes of sepsis.
3.2 Dynamic changes in platelet differentiation trajectories and immune communication networks in sepsis
To further investigate the immunological potential of platelets in sepsis, we performed pseudotime trajectory analysis on PBMCs. The results revealed that B cells, T cells, and Tregs exhibited continuous and well-defined developmental trajectories along the pseudotime axis, ultimately converging toward a platelet-like phenotype (Figure 2A, Supplementary Figures 2A–C). These findings suggest that, under septic conditions, certain immune cells may undergo phenotypic reprogramming or functional transdifferentiation, acquiring platelet-like features. This differentiation process was accompanied by dynamic changes in gene expression profiles within these cell subsets, further supporting the presence of a platelet-directed reprogramming trajectory (Figure 2B).
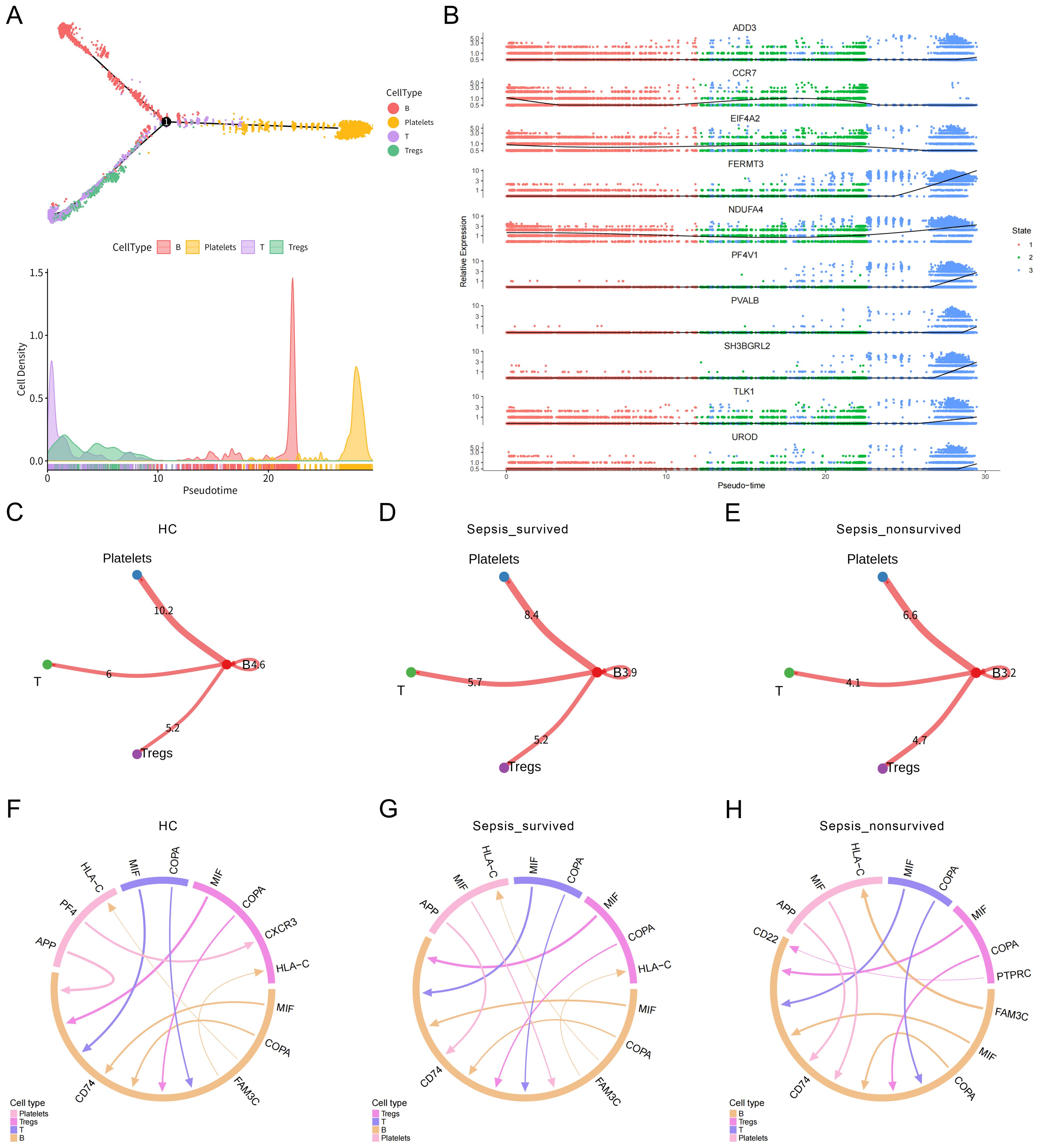
Figure 2. Pseudotime trajectory and cell–cell communication analysis of immune cell differentiation toward platelets. (A) Pseudotime trajectory analysis of peripheral blood mononuclear cells (PBMCs) illustrating immune cell developmental dynamics. Cells in different states are color-coded, revealing a potential differentiation trajectory from B cells, T cells, and Tregs toward a platelet-like phenotype. (B) Dynamic changes in gene expression profiles during the transition of B cells, T cells, and Tregs toward platelet-like states. (C–E) Heatmaps of overall cell–cell interaction strength among immune cell subsets in (C) healthy controls (HCs), (D) sepsis survivors, and (E) sepsis non-survivors. (F–H) Ligand–receptor interaction networks between B cells, Tregs, T cells, and platelets in the three groups.
To assess potential intercellular communication between immune cells and platelets, we constructed and analyzed the PBMCs cell–cell interaction network. Global interaction strength analysis indicated a progressive weakening of immune cell communication with advancing disease severity, with the non-survivor group exhibiting the sparsest interaction network (Figures 2C–E). Focusing on the ligand–receptor interactions between B cells, T cells, Tregs, and platelets, we observed that in healthy individuals, communication between these subsets and platelets was relatively active (Figure 2F). However, in the sepsis survivor group (Figure 2G) and the non-survivor group (Figure 2H), the network became increasingly impaired. Notably, the signaling intensity of the APP → CD74 axis, a key communication route between platelets and B cells, decreased progressively across groups: 10.88 in the healthy control group, 8.864 in the survivor group, and 6.636 in the non-survivor group. This trend indicates that platelet–B cell signaling is markedly suppressed during sepsis progression. Given the critical role of CD74 as both a chaperone of MHC class II molecules and a regulator of inflammatory signaling, attenuation of this axis may reflect impaired immune activation or defects in antigen presentation capacity. Taken together, these findings reveal that the immune communication network between platelets and immune cells undergoes substantial remodeling during the course of sepsis. The pronounced weakening of this network, particularly in non-survivors, suggests a functional impairment of platelets in maintaining immune homeostasis, which may contribute to immune dysregulation and poor clinical outcomes in sepsis.
3.3 Differential gene expression analysis reveals functional enrichment of platelets in sepsis
To further explore the functional alterations of PBMCs under different clinical outcomes in sepsis, we first performed differential gene expression analysis across immune cell subsets between HCs and the sepsis survivor group (Figure 3A, Supplementary Table S2). The results revealed extensive transcriptomic differences across multiple immune cell populations, suggesting that sepsis induces widespread immune activation and functional reprogramming in specific cell subsets. Subsequent comparison of PBMCs between the survivor and non-survivor groups revealed more pronounced transcriptional alterations in the non-survivors across several immune cell types (Figure 3B, Supplementary Table S3), particularly in B cells, T cells, and platelets. These findings indicate that these key immune populations may undergo functional transitions that are closely associated with disease progression.
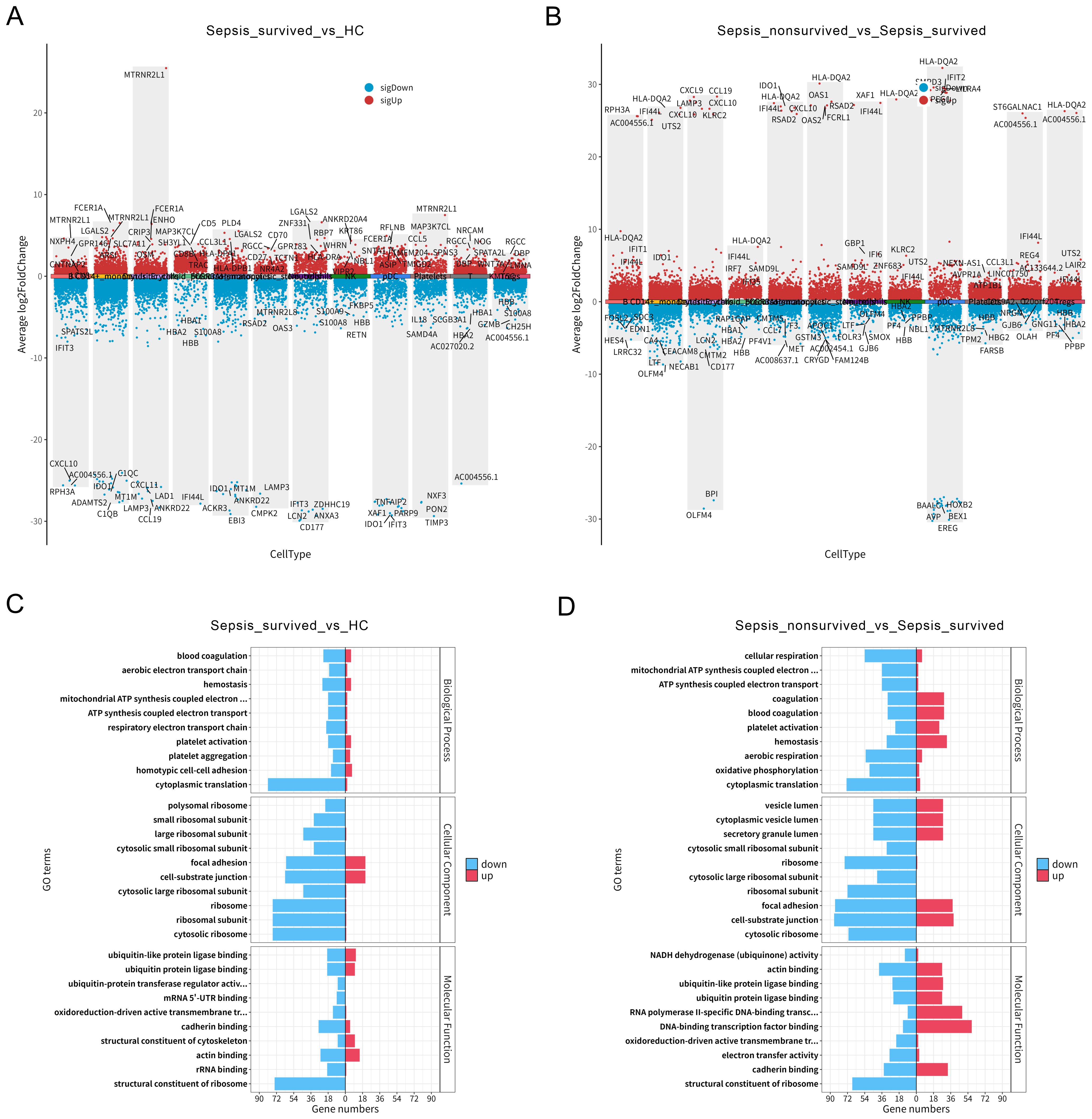
Figure 3. Differential gene expression and platelet functional enrichment in peripheral blood mononuclear cells (PBMCs) of sepsis patients. (A) Differential gene expression analysis across PBMCs subsets between healthy controls (HCs) and sepsis survivors. (B) Differential gene expression across PBMCs subsets between sepsis survivors and non-survivors. (C) Gene Ontology (GO) enrichment analysis of differentially expressed genes (DEGs) in platelets between HCs and sepsis survivors. (D) GO enrichment analysis of platelet DEGs between sepsis survivors and non-survivors.
To better understand the functional changes in platelets during sepsis, we conducted GO enrichment analysis of DEGs between HCs and survivors (Figure 3C), as well as between survivors and non-survivors (Figure 3D). The results showed that platelet DEGs in sepsis were significantly enriched in biological processes such as blood coagulation, platelet activation, and hemostasis, indicating an enhanced activation of their classical hemostatic functions. Notably, these functional enrichments were further intensified in the non-survivor group, suggesting that in addition to their conventional roles, platelets may be actively involved in inflammatory responses and immune dysregulation in advanced stages of sepsis. In summary, our data reveal substantial transcriptomic reprogramming of monocytes and platelets within the PBMCs compartment of sepsis patients. Specifically, platelets exhibit a progressively enhanced functional state across groups with worsening clinical outcomes, highlighting their potential key role in the immunopathogenesis of sepsis.
3.4 m6A methylation profiles and functional enrichment in PBMCs of sepsis patients
Previous analyses revealed a progressive weakening of immune communication between platelets and B cells during sepsis; however, the underlying molecular mechanisms remain unclear. To investigate the role of m6A in this process, we first assessed the expression patterns of canonical m6A regulatory enzymes across different PBMCs subsets in HCs, sepsis survivors, and non-survivors (Figures 4A–E, Supplementary Figures 3A–C). We observed substantial heterogeneity in the expression of demethylases (ALKBH5, FTO) and methyltransferase complex components (WTAP, METTL3, METTL14), suggesting their potential involvement in the epigenetic regulation of immune function during sepsis.
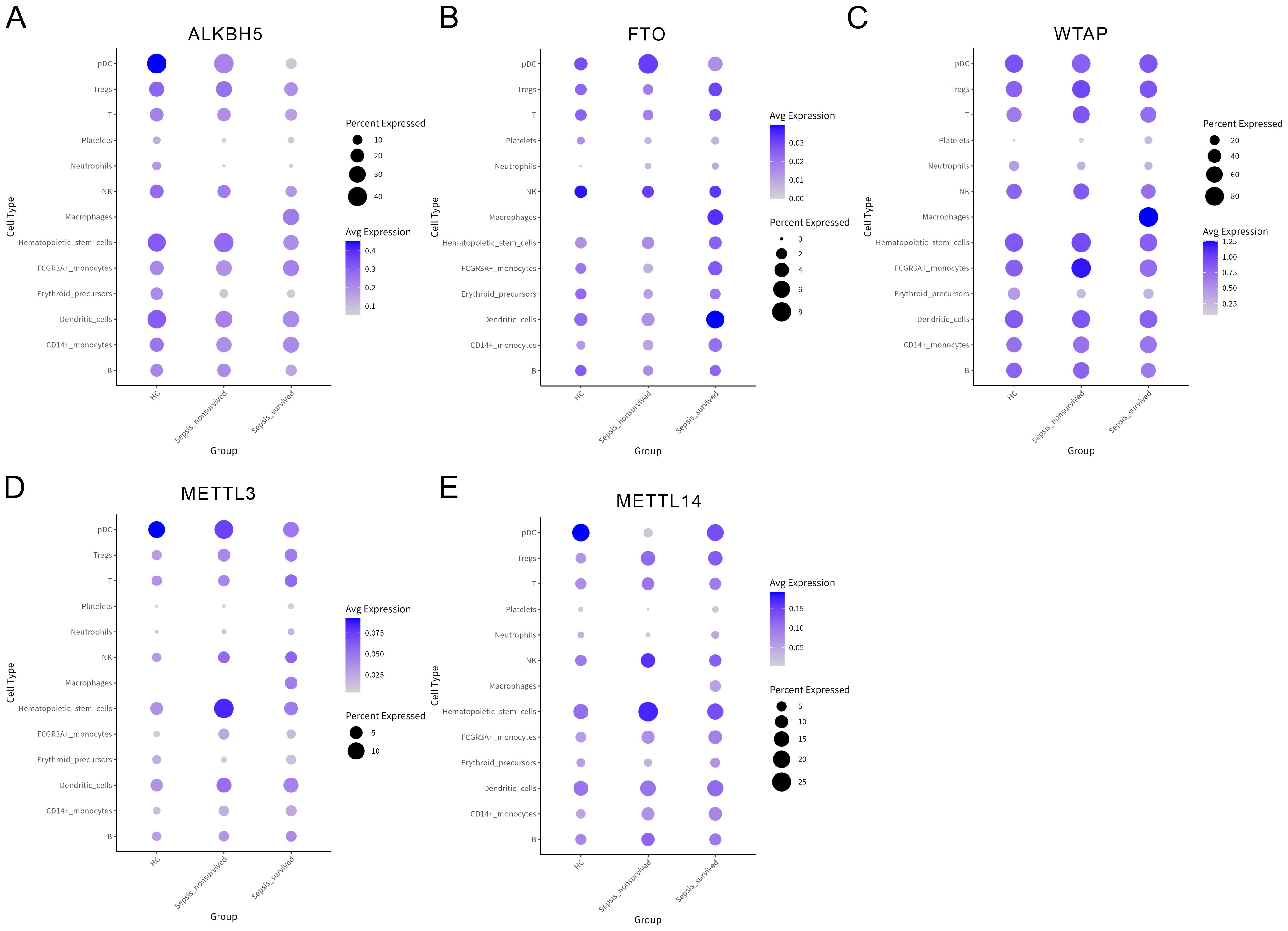
Figure 4. Expression profiles of N6-methyladenosine (m6A) regulatory enzymes across peripheral blood mononuclear cells (PBMCs) subsets. (A–E) Bubble plots showing the expression levels of key m6A regulatory factors in various PBMCs subsets, including ALKBH5 (A), FTO (B), WTAP (C), METTL3 (D), and METTL14 (E).
Next, we performed MeRIP-seq on PBMCs from survivors and non-survivors. In both groups, m6A peaks were predominantly enriched in coding sequence (CDS) regions (46.9%), followed by 3′ untranslated regions (3′UTR) (30.4%) (Figure 5A). Notably, a slight reduction in CDS-localized m6A peaks was observed in the non-survivor group compared to survivors (Figure 5B), implying that disease progression may alter m6A distribution and affect mRNA stability or translational efficiency. Differential expression analysis revealed that several m6A regulators were significantly dysregulated in the non-survivor group (Figure 5C), further supporting the dynamic involvement of m6A modification in sepsis pathogenesis. Functional enrichment analysis of differentially methylated genes revealed significant associations with pathways related to metabolic regulation, inflammatory responses, apoptosis, and immune signaling (Figures 5D, E), indicating that m6A modification may influence sepsis through multilayered regulatory mechanisms. In summary, sepsis patients exhibit dynamic remodeling of m6A methylation in PBMCs, characterized by altered regulator expression, redistribution of modification sites, and functional pathway enrichment. These results highlight the potential contribution of m6A-dependent regulation to disease progression and outcome in sepsis.

Figure 5. N6-methyladenosine (m6A) methylation landscape and functional enrichment analysis in peripheral blood mononuclear cells (PBMCs) from sepsis survivors and non-survivors. (A) Pie chart depicting the overall distribution of m6A peaks across mRNA transcripts in PBMCs from sepsis survivors (n = 3) and non-survivors (n = 3), based on MeRIP-seq analysis. (B) Average distribution of m6A peaks across transcript regions, including 5’UTR, coding sequence, and 3’UTR, in the survivor and non-survivor groups. (C) Heatmap illustrating differential expression of m6A regulatory enzymes in PBMCs between sepsis survivors and non-survivors. (D, E) Functional enrichment of differentially methylated genes based on Gene Ontology annotation (D) and Kyoto Encyclopedia of Genes and Genomes pathway analysis (E).
3.5 1.10 m6A-regulated characteristics of RPA1 and its correlation with key immune factors in sepsis
To identify key genes potentially involved in m6A-mediated immune remodeling in sepsis, we integrated three sets of differentially expressed or modified genes for intersection analysis: (1) Differentially methylated genes between sepsis survivors and non-survivors in PBMCs; (2) DEGs in platelets between survivors and non-survivors; (3) Differentially expressed platelet genes between HCs and survivors. This integrative analysis yielded 56 overlapping candidate genes (Figure 6A). To evaluate their potential role in platelet–immune cell interaction, particularly in relation to B cell communication, we examined the correlation between candidate genes and the platelet-derived ligand APP. RPA1 exhibited the strongest correlation with APP expression (Figure 6B, Supplementary Table S4), suggesting a possible role in the APP–CD74 signaling axis and platelet–B cell interaction.
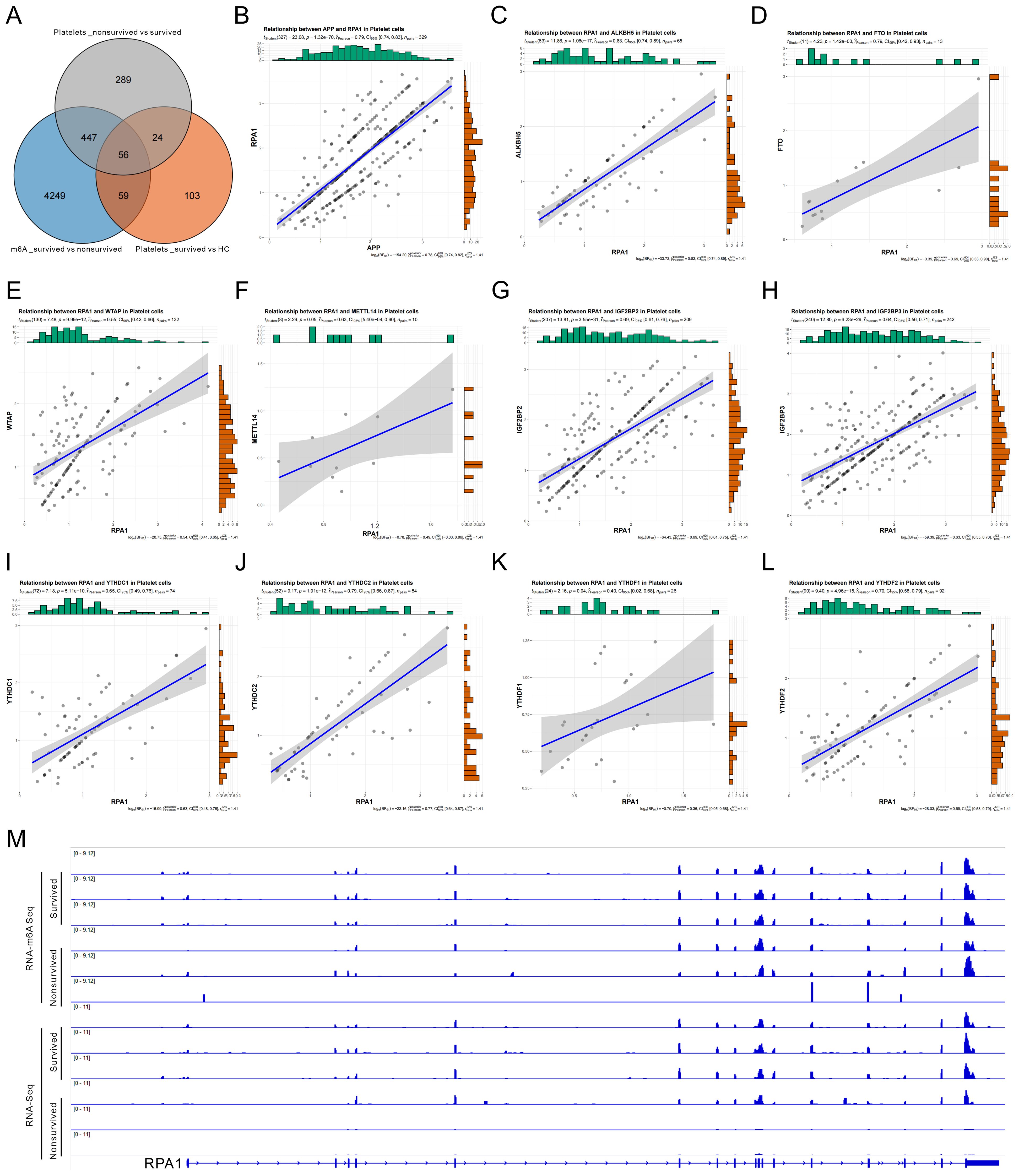
Figure 6. N6-methyladenosine (m6A)-mediated regulation of RPA1 and its correlation with key factors in sepsis. (A) Venn diagram showing the intersection of three sets of differentially regulated genes: (1) differentially m6A-methylated genes in peripheral blood mononuclear cells (PBMCs) between sepsis survivors and non-survivors; (2) differentially expressed genes (DEGs) in platelets between survivors and non-survivors; and (3) DEGs in platelets between healthy controls (HC) and survivors. (B) Correlation analysis between RPA1 and the platelet-specific ligand APP. (C–L) Correlation analysis between RPA1 and ten key m6A regulatory factors in platelets, including ALKBH5 (C), FTO (D), WTAP (E), METTL14 (F), IGF2BP2 (G), IGF2BP3 (H), YTHDC1 (I), YTHDC2 (J), YTHDF1 (K), and YTHDF2 (L). (M) Integrative Genomics Viewer tracks showing differential m6A methylation abundance and RPA1 expression levels in PBMCs from sepsis survivors versus non-survivors.
Further correlation analysis revealed that RPA1 expression in platelets was significantly associated with multiple m6A regulators, including demethylases (ALKBH5, FTO), methyltransferase complex components (WTAP, METTL14), and reader proteins (IGF2BP2/3, YTHDC1/2, YTHDF1/2) (Figures 6C–L), indicating that RPA1 may be epigenetically regulated by m6A modification. Consistently, IGV browser visualization demonstrated reduced m6A modification levels and decreased RPA1 expression in PBMCs from the non-survivor group compared to survivors (Figure 6M), further supporting RPA1 as a putative m6A-regulated target gene in sepsis. Taken together, RPA1 emerged as a key candidate gene identified through cross-dataset integration. It is closely correlated with the platelet ligand APP and subject to regulation by multiple m6A factors. These findings suggest that RPA1 may contribute to the regulation of immune cell communication in sepsis and holds promise as a mechanistic mediator and potential therapeutic target.
4 Discussion
In this study, we systematically characterized the immunological landscape, platelet functional remodeling, and epigenetic regulatory mechanisms in PBMCs from sepsis patients with different prognostic outcomes using scRNA-seq and MeRIP-seq profiling. Our findings highlight the crucial role of platelets and m6A modification in the pathogenesis and progression of sepsis. Traditionally recognized for their roles in hemostasis and thrombosis, platelets are increasingly acknowledged as active immune regulators in sepsis (2, 27, 28). Previous studies have demonstrated that platelets can participate in immune defense by expressing immunomodulatory molecules such as P-selectin and CD40 ligand (CD40L), and by releasing inflammatory mediators, thereby contributing to pathogen recognition, immune cell recruitment, and amplification of inflammatory signaling (29–33). In our study, we observed a significant increase in the proportion of platelets as sepsis progressed, accompanied by a marked decline in platelet–B cell communication in the non-survivor group. These findings further emphasize the potential involvement of platelets in immune dysregulation and poor clinical outcomes during sepsis.
In recent years, m6A methylation, the most prevalent internal modification of eukaryotic mRNA, has been increasingly implicated in the regulation of inflammation and immune-related disorders (34–37). Although studies on m6A modification in the context of sepsis remain limited, emerging evidence suggests that m6A modulates cytokine expression, immune cell activation, and inflammatory responses (38–40). In this study, MeRIP-seq analysis revealed a reprogramming of the m6A epitranscriptome in PBMCs from non-survivors, with enhanced m6A enrichment in CDS regions, suggesting potential impacts on mRNA stability and translational efficiency. Moreover, we identified significant differential expression of several m6A regulators among different prognostic groups, including methyltransferases (METTL3, METTL14), demethylases (FTO, ALKBH5), and reader proteins such as YTHDF1 and IGF2BP2. These findings indicate a dynamic regulatory landscape of m6A modification in sepsis and highlight the potential of m6A-related enzymes as therapeutic targets for modulating immune responses in sepsis.
Of particular interest is the APP → CD74 signaling axis, which exhibited a progressively decreasing trend in our cell–cell communication analysis and has also been implicated in various immune-related diseases (41). For example, APP–CD74 interactions have been shown to mediate endothelial–macrophage communication, thereby promoting renal injury and fibrosis (41). Therapeutic modulation of this axis has been reported to enhance the phagocytic activity of tumor-associated macrophages (TAMs) in glioblastoma (GBM) (42). Single-cell transcriptomic analysis has further revealed the involvement of the APP–CD74 axis in reinforcing immune suppression and tumor progression in testicular cancer (43). Additionally, APP-CD74 interactions have been shown to inhibit β-amyloid production, suggesting a potential multifaceted role for this pathway in inflammatory and metabolic environments (44). However, investigations into the function of the APP → CD74 axis in sepsis remain limited. To our knowledge, this study is the first to report a significant attenuation of this signaling axis at the single-cell level in sepsis, correlating with poor clinical outcomes. This novel insight warrants further exploration.
Moreover, RPA1, identified as a candidate gene at the intersection of differential m6A modification and platelet-specific expression, may play a key role in platelet immune function and the epigenetic regulation of sepsis. Recent evidence has shown that RPA1, the core subunit of the single-stranded DNA-binding protein complex, plays a critical role in fundamental cellular processes, including DNA replication, damage repair, chromatin accessibility, and telomere maintenance (45). Loss of RPA1 not only compromises genomic stability but also promotes necroptotic death of T cells by activating the ZBP1-RIPK3 signaling pathway, leading to immune dysregulation and exacerbated inflammatory responses. These findings have been validated in the contexts of immune homeostasis, inflammatory diseases, and tumor immune tolerance (46–50). In addition, aberrant expression of RPA1 has been implicated in various pathological conditions, including ulcerative colitis, lipid metabolism disorders, radioresistant cancers, and neurodegenerative diseases. Its multifaceted regulatory functions have been widely reported across multiple biological systems (51, 52). However, the specific role of RPA1 within the platelet-immune regulatory network in sepsis remains largely unexplored and lacks direct experimental evidence. Our study found that RPA1 expression was strongly correlated with the platelet-derived ligand APP, and showed co-expression with several m6A regulators, including METTL3 and ALKBH5, suggesting that its expression and function may be subject to dynamic m6A-dependent modulation. Given the observed attenuation of the APP → CD74 axis in the non-survivor group, we hypothesize that RPA1 may contribute to the regulation of this ligand–receptor pair, thereby influencing immune cell activation and inflammatory signaling. This, in turn, could mediate functional reprogramming of platelets during sepsis. It is important to note that although this study is the first to propose RPA1 as a potential molecular link between platelet immune function and m6A modification, the regulatory mechanisms inferred herein are primarily based on bioinformatic correlation analyses. Direct molecular and functional validation is currently lacking. We fully acknowledge the need for future investigations employing functional experiments in cellular and animal models—such as RPA1 knockdown or overexpression—to elucidate its specific effects on platelet-immune cell interactions, cytokine secretion, and immune homeostasis. Additionally, experimental validation of the APP→CD74 signaling axis is necessary to determine whether RPA1 is involved in modulating this key ligand-receptor pathway. Furthermore, the integration of advanced technologies such as CRISPR-based gene editing, single-cell multi-omics, and high-throughput epigenomic profiling may offer deeper mechanistic insights into the role of RPA1 in platelet-mediated immune regulation and sepsis progression.
Despite uncovering a range of critical cellular dynamics, intercellular communication patterns, and epigenetic regulatory features, this study has several limitations. First, the relatively small sample size, with some analyses based on a limited number of patient samples, may reduce statistical power and limit the generalizability of the findings. Second, m6A methylation data were derived from bulk PBMCs populations, which restricts the resolution of m6A modification dynamics at the single-cell level. Third, the mechanistic inferences drawn from this study are primarily based on computational predictions, and lack direct experimental validation. Future investigations should aim to validate these findings in larger, multi-center cohorts, and leverage emerging technologies such as CRISPR-based epigenetic editing, CLIP-seq, and single-cell m6A sequencing techniques (e.g., DART-seq) to dissect the cell type-specific functions of m6A in platelets with higher precision. Additionally, the APP → CD74 signaling axis and the RPA1-centered regulatory network warrant in-depth experimental exploration to evaluate their potential as targets for therapeutic intervention in sepsis.
In summary, this study reveals significant immune cell heterogeneity, platelet immune reprogramming, and m6A-mediated gene regulation in the peripheral blood of sepsis patients. Notably, we identify RPA1 as a potential key node linking platelet immune function with m6A epitranscriptomic remodeling. These findings offer new epigenetic insights into the pathogenesis of sepsis, and lay the groundwork for future research aimed at biomarker discovery and targeted therapeutic strategies.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies on humans were approved by the Ethics Committee of Tianjin Medical University General Hospital (Approval ID: IRB2023-YX-045-01). Informed consent was obtained from all participants.
Author contributions
WQ: Conceptualization, Data curation, Formal analysis, Writing – original draft. YL: Data curation, Formal analysis, Resources, Writing – original draft. XZ: Data curation, Formal analysis, Resources, Writing – original draft. YD: Data curation, Formal analysis, Writing – original draft. JZ: Data curation, Formal analysis, Writing – original draft. SS: Data curation, Formal analysis, Funding acquisition, Resources, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by WU JIEPING MEDICAL FOUNDATION (No.2023045).
Acknowledgments
We would like to thank all authors for their contributions to this article and the corresponding author for his financial support of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1607732/full#supplementary-material
References
1. Arora J, Mendelson AA, and Fox-Robichaud A. Sepsis: network pathophysiology and implications for early diagnosis. Am J Physiol Regul Integr Comp Physiol. (2023) 324:R613–r624. doi: 10.1152/ajpregu.00003.2023
2. Hotchkiss RS, Monneret G, and Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. (2013) 13:862–74. doi: 10.1038/nri3552
3. Wang W, Ma L, Liu B, and Ouyang L. The role of trained immunity in sepsis. Front Immunol. (2024) 15:1449986. doi: 10.3389/fimmu.2024.1449986
4. Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, and Vincent J-L. Sepsis and septic shock. Nat Rev Dis Primers. (2016) 2:16045. doi: 10.1038/nrdp.2016.45
5. Chiu C and Legrand M. Epidemiology of sepsis and septic shock. Curr Opin Anaesthesiol. (2021) 34:71–6. doi: 10.1097/ACO.0000000000000958
6. La Via L, Sangiorgio G, Stefani S, Marino A, Nunnari G, Cocuzza S, et al. The global burden of sepsis and septic shock. Epidemiologia (Basel). (2024) 5:456–78. doi: 10.3390/epidemiologia5030032
7. Delano MJ and Ward PA. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol Rev. (2016) 274:330–53. doi: 10.1111/imr.2016.274.issue-1
8. Koupenova M, Livada AC, and Morrell CN. Platelet and megakaryocyte roles in innate and adaptive immunity. Circ Res. (2022) 130:288–308. doi: 10.1161/CIRCRESAHA.121.319821
9. Mandel J, Casari M, Stepanyan M, Martyanov A, and Deppermann C. Beyond hemostasis: platelet innate immune interactions and thromboinflammation. Int J Mol Sci. (2022) 23(7):3868. doi: 10.3390/ijms23073868
10. Gremmel T, Frelinger A. L., 3RD, and Michelson AD. Platelet physiology. Semin Thromb Hemost. (2024) 50:1173–86. doi: 10.1055/s-0044-1786387
11. Guo L, Shen S, Rowley JW, Tolley ND, Jia W, Manne BK, et al. Platelet MHC class I mediates CD8+ T-cell suppression during sepsis. Blood. (2021) 138:401–16. doi: 10.1182/blood.2020008958
12. Fu G, Deng M, Neal MD, Billiar TR, and Scott MJ. Platelet-monocyte aggregates: understanding mechanisms and functions in sepsis. Shock. (2021) 55:156–66. doi: 10.1097/SHK.0000000000001619
13. Guo M, Fan S, Chen Q, Jia C, Qiu M, Bu Y, et al. Platelet-derived microRNA-223 attenuates TNF-α induced monocytes adhesion to arterial endothelium by targeting ICAM-1 in Kawasaki disease. Front Immunol. (2022) 13:922868. doi: 10.3389/fimmu.2022.922868
14. Karakas D and Ni H. Unveiling platelets as immune regulatory cells. Circ Res. (2024) 134:987–9. doi: 10.1161/CIRCRESAHA.124.324167
15. Maneta E, Aivalioti E, Tual-Chalot S, Emini Veseli B, Gatsiou A, Stamatelopoulos K, et al. Endothelial dysfunction and immunothrombosis in sepsis. Front Immunol. (2023) 14:1144229. doi: 10.3389/fimmu.2023.1144229
16. Campbell RA, Manne BK, Banerjee M, Middleton EA, Ajanel A, Schwertz H, et al. IFITM3 regulates fibrinogen endocytosis and platelet reactivity in nonviral sepsis. J Clin Invest. (2022) 132(23):e153014. doi: 10.1172/JCI153014
17. Claushuis TAM, De Vos AF, Nieswandt B, Boon L, Roelofs J, De Boer OJ, et al. Platelet glycoprotein VI aids in local immunity during pneumonia-derived sepsis caused by gram-negative bacteria. Blood. (2018) 131:864–76. doi: 10.1182/blood-2017-06-788067
18. Oerum S, Meynier V, Catala M, and TISNé C. A comprehensive review of m6A/m6Am RNA methyltransferase structures. Nucleic Acids Res. (2021) 49:7239–55. doi: 10.1093/nar/gkab378
19. Zaccara S, Ries RJ, and Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. (2019) 20:608–24. doi: 10.1038/s41580-019-0168-5
20. Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. (2021) 6:74. doi: 10.1038/s41392-020-00450-x
21. Wu D, Spencer CB, Ortoga L, Zhang H, and Miao C. Histone lactylation-regulated METTL3 promotes ferroptosis via m6A-modification on ACSL4 in sepsis-associated lung injury. Redox Biol. (2024) 74:103194. doi: 10.1016/j.redox.2024.103194
22. Zhang H, Liu J, Zhou Y, Qu M, Wang Y, Guo K, et al. Neutrophil extracellular traps mediate m(6)A modification and regulates sepsis-associated acute lung injury by activating ferroptosis in alveolar epithelial cells. Int J Biol Sci. (2022) 18:3337–57. doi: 10.7150/ijbs.69141
23. Zhang H, Wu D, Wang Y, Guo K, Spencer CB, Ortoga L, et al. METTL3-mediated N6-methyladenosine exacerbates ferroptosis via m6A-IGF2BP2-dependent mitochondrial metabolic reprogramming in sepsis-induced acute lung injury. Clin Transl Med. (2023) 13:e1389. doi: 10.1002/ctm2.v13.9
24. Zhang S, Guan X, Liu W, Zhu Z, Jin H, Zhu Y, et al. YTHDF1 alleviates sepsis by upregulating WWP1 to induce NLRP3 ubiquitination and inhibit caspase-1-dependent pyroptosis. Cell Death Discov. (2022) 8:244. doi: 10.1038/s41420-022-00872-2
25. Luo S, Liao C, Zhang L, Ling C, Zhang X, Xie P, et al. METTL3-mediated m6A mRNA methylation regulates neutrophil activation through targeting TLR4 signaling. Cell Rep. (2023) 42:112259. doi: 10.1016/j.celrep.2023.112259
26. Qiu X, Li J, Bonenfant J, Jaroszewski L, Mittal A, Klein W, et al. Dynamic changes in human single-cell transcriptional signatures during fatal sepsis. J Leukoc Biol. (2021) 110:1253–68. doi: 10.1002/JLB.5MA0721-825R
27. Rawish E, Nording H, MüNTE T, and Langer HF. Platelets as mediators of neuroinflammation and thrombosis. Front Immunol. (2020) 11:548631. doi: 10.3389/fimmu.2020.548631
28. Rubio I, Osuchowski MF, Shankar-Hari M, Skirecki T, Winkler MS, Lachmann G, et al. Current gaps in sepsis immunology: new opportunities for translational research. Lancet Infect Dis. (2019) 19:e422–36. doi: 10.1016/S1473-3099(19)30567-5
29. Hwaiz R, Rahman M, Zhang E, and Thorlacius H. Rac1 regulates platelet shedding of CD40L in abdominal sepsis. Lab Invest. (2014) 94:1054–63. doi: 10.1038/labinvest.2014.92
30. Inwald DP, Faust SN, Lister P, Peters MJ, Levin M, Heyderman R, et al. Platelet and soluble CD40L in meningococcal sepsis. Intensive Care Med. (2006) 32:1432–7. doi: 10.1007/s00134-006-0250-2
31. Rahman M, Zhang S, Chew M, Syk I, Jeppsson B, and Thorlacius H. Platelet shedding of CD40L is regulated by matrix metalloproteinase-9 in abdominal sepsis. J Thromb Haemost. (2013) 11:1385–98. doi: 10.1111/jth.12273
32. Valet C, Magnen M, Qiu L, Cleary SJ, Wang KM, Ranucci S, et al. Sepsis promotes splenic production of a protective platelet pool with high CD40 ligand expression. J Clin Invest. (2022) 132(32):e153920. doi: 10.1172/JCI153920
33. Yu W, Yang M, Lv B, Yu Y, and Zhu W. CD40L-Activated DC Promotes Th17 Differentiation and Inhibits Th2 Differentiation in Sepsis-Induced Lung Injury via cGAS-STING Signaling. Biochem Genet. (2024) 63(3):2455–69. doi: 10.1007/s10528-024-10835-0
34. Chen J, Wei X, Wang X, Liu T, Zhao Y, Chen L, et al. TBK1-METTL3 axis facilitates antiviral immunity. Cell Rep. (2022) 38:110373. doi: 10.1016/j.celrep.2022.110373
35. Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L, et al. N6-methyladenosine modification controls circular RNA immunity. Mol Cell. (2019) 76:96–109.e9. doi: 10.1016/j.molcel.2019.07.016
36. Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature. (2019) 566:270–4. doi: 10.1038/s41586-019-0916-x
37. Yin H, Zhang X, Yang P, Zhang X, Peng Y, Li D, et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat Commun. (2021) 12:1394. doi: 10.1038/s41467-021-21514-8
38. Chen H, Zhang X, Su H, Zeng J, Chan H, Li Q, et al. Immune dysregulation and RNA N6-methyladenosine modification in sepsis. Wiley Interdiscip Rev RNA. (2023), 14(3):e1764. doi: 10.1002/wrna.v14.3
39. Li F, Zhang Y, Peng Z, Wang Y, Zeng Z, and Tang Z. Diagnostic, clustering, and immune cell infiltration analysis of m6A regulators in patients with sepsis. Sci Rep. (2023) 13:2532. doi: 10.1038/s41598-022-27039-4
40. Liu Y, Song R, Lu Z, Zhao L, Zhan X, Li Y, et al. The RNA m(6)A demethylase ALKBH5 drives emergency granulopoiesis and neutrophil mobilization by upregulating G-CSFR expression. Cell Mol Immunol. (2024) 21:6–18. doi: 10.1038/s41423-023-01115-9
41. Liu B, Li F, Wang Y, Gao X, Li Y, Wang Y, et al. APP-CD74 axis mediates endothelial cell-macrophage communication to promote kidney injury and fibrosis. Front Pharmacol. (2024) 15:1437113. doi: 10.3389/fphar.2024.1437113
42. Ma C, Chen J, Ji J, Zheng Y, Liu Y, Wang J, et al. Therapeutic modulation of APP-CD74 axis can activate phagocytosis of TAMs in GBM. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:167449. doi: 10.1016/j.bbadis.2024.167449
43. Chen G, Wang W, Wei X, Chen Y, Peng L, Qu R, et al. Single-cell transcriptomic analysis reveals that the APP-CD74 axis promotes immunosuppression and progression of testicular tumors. J Pathol. (2024) 264:250–69. doi: 10.1002/path.v264.3
44. Matsuda S, Matsuda Y, and D’adamio L. CD74 interacts with APP and suppresses the production of Abeta. Mol Neurodegener. (2009) 4:41. doi: 10.1186/1750-1326-4-41
45. Haring SJ, Mason AC, Binz SK, and Wold MS. Cellular functions of human RPA1: multiple roles of domains in replication, repair, and checkpoints. J Biol Chem. (2008) 283:19095–111. doi: 10.1074/jbc.M800881200
46. Liu P, ROJO de la VEGA M, Sammani S, Mascarenhas JB, Kerins M, Dodson M, et al. RPA1 binding to NRF2 switches ARE-dependent transcriptional activation to ARE-NRE–dependent repression. Proc Natl Acad Sci. (2018) 115:E10352–61. doi: 10.1073/pnas.1812125115
47. Sharma R, Sahoo SS, Honda M, Granger SL, Goodings C, Sanchez L, et al. Gain-of-function mutations in RPA1 cause a syndrome with short telomeres and somatic genetic rescue. Blood J Am Soc Hematol. (2022) 139:1039–51. doi: 10.1182/blood.2021011980
48. Yin Q, Li Y, Zhou Z, Li X, Li M, Liu C, et al. RPA1 controls chromatin architecture and maintains lipid metabolic homeostasis. Cell Rep. (2022) 40(2):11107. doi: 10.1016/j.celrep.2022.111071
49. Song J, Zhang X, Yin Y, Guo M, Zhao X, Wang L, et al. Loss of RPA1 impairs peripheral T cell homeostasis and exacerbates inflammatory damage through triggering T cell necroptosis. Advanced Sci. (2023) 10:2206344. doi: 10.1002/advs.202206344
50. Yu H, Bu C, Liu Y, Gong T, Liu X, Liu S, et al. Global crotonylome reveals CDYL-regulated RPA1 crotonylation in homologous recombination–mediated DNA repair. Sci Adv. (2020) 6:eaay4697. doi: 10.1126/sciadv.aay4697
51. Gall-Duncan T, Luo J, Jurkovic C-M, Fischer LA, Fujita K, Deshmukh AL, et al. Antagonistic roles of canonical and Alternative-RPA in disease-associated tandem CAG repeat instability. Cell. (2023) 186:4898–919. doi: 10.1016/j.cell.2023.09.008
Keywords: sepsis, single-cell transcriptomics, m6A methylation, platelets, RPA1, APP-CD74 signaling axis
Citation: Qian W, Liu Y, Zhao X, Dong Y, Zhou J and Shou S (2025) Single-cell transcriptomic and m6A methylation analyses reveal platelet-mediated immune regulatory mechanisms in sepsis. Front. Immunol. 16:1607732. doi: 10.3389/fimmu.2025.1607732
Received: 09 April 2025; Accepted: 02 June 2025;
Published: 23 June 2025.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Cuncai Guo, Washington University in St. Louis, United StatesBoyang Liu, The First Affiliated Hospital of Chongqinge Medical University, China
Copyright © 2025 Qian, Liu, Zhao, Dong, Zhou and Shou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Songtao Shou, enl5c2hvdUB0bXUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship