 Erik Kupschke1,2
Erik Kupschke1,2 Mirjam Schenk1,3*
Mirjam Schenk1,3*- 1Christine Kühne – Center for Allergy Research and Education, Davos, Switzerland
- 2Graduate School for Cellular and Biomedical Sciences, University of Bern, Bern, Switzerland
- 3Institute of Tissue Medicine and Pathology, Experimental Pathology, University of Bern, Bern, Switzerland
Atopic dermatitis (AD) is one of the most common chronic inflammatory skin diseases worldwide, significantly impairing patients’ quality of life. It is characterized by recurrent eczematous lesions, intense pruritus, and disruption of the epidermal barrier. The pathogenesis of AD is multifactorial and involves complex interactions between genetic predisposition, environmental triggers, skin barrier defects, microbial dysbiosis, and immune dysregulation. While much of the research in recent decades has focused on the type 2 helper T cell (Th2)-driven adaptive immune responses that dominate the acute phase of the disease, the role of innate immunity—particularly that of myeloid cells—has emerged as a crucial and underrated component in disease pathogenesis and progression. This review highlights recent findings on the role of myeloid cells in the initiation, maintenance, and amplification of inflammation in AD. Myeloid cells respond to a wide range of environmental and tissue-derived triggers, including cytokines, alarmins, and microbial products. Upon activation, they contribute to the inflammatory milieu by producing chemokines and cytokines, presenting antigens, and recruiting other immune cells to the skin. Importantly, myeloid cells not only shape the local immune landscape but also engage in crosstalk with keratinocytes and adaptive immune cells, thereby reinforcing chronic inflammation. In addition, the review outlines emerging therapeutic strategies aimed at modulating myeloid cell function or selectively targeting pro-inflammatory subsets. These approaches offer promising avenues that complement existing Th2-centered therapies, addressing disease mechanisms beyond the adaptive immune response. A deeper understanding of the diverse and dynamic roles of myeloid cells in AD may thus support the development of more comprehensive and personalized treatment strategies for long-term disease control.
Introduction to atopic dermatitis
Atopic dermatitis (AD) is one of the most common inflammatory skin diseases worldwide. It often starts in early childhood with onset before age of 5 years but can also develop in adults. AD is characterized by intense pruritus, dry skin, and recurrent eczematous lesions. The prevalence of AD has increased 2-3-fold in recent decades, currently affecting approximately 25% of children and 4-7% of adults in industrialized nations (1). This rising incidence has made AD a significant public health concern due to its substantial impact on quality of life and economic burden. The pathophysiology of AD is complex, involving genetic, immunologic, and environmental factors that lead to skin barrier dysfunction and immune dysregulation. Impaired skin barrier function increases susceptibility to irritants, allergens, and microbial colonization, particularly by Staphylococcus aureus (S. aureus) which can trigger inflammatory responses involving both innate and adaptive immunity. AD is often associated with other atopic conditions like asthma, allergic rhinitis, and food allergies as part of the “atopic march.” Management typically involves an integrated approach including skin care, anti-inflammatory treatments, and, in some cases, emerging targeted therapies (Table 1). Early diagnosis and intervention are important to control symptoms, prevent complications, and improve quality of life for patients with this challenging chronic condition. In the early stages of the disease, it is easier to break the itch-scratch cycle, as it worsens with each iteration. The inflammation causes itching, which leads to scratching. Scratching disrupts the skin barrier, allowing allergens to enter and triggering stronger inflammation. Therefore, early diagnosis and treatment are crucial to control symptoms, prevent complications, and improve the quality of life for patients with this challenging chronic condition.
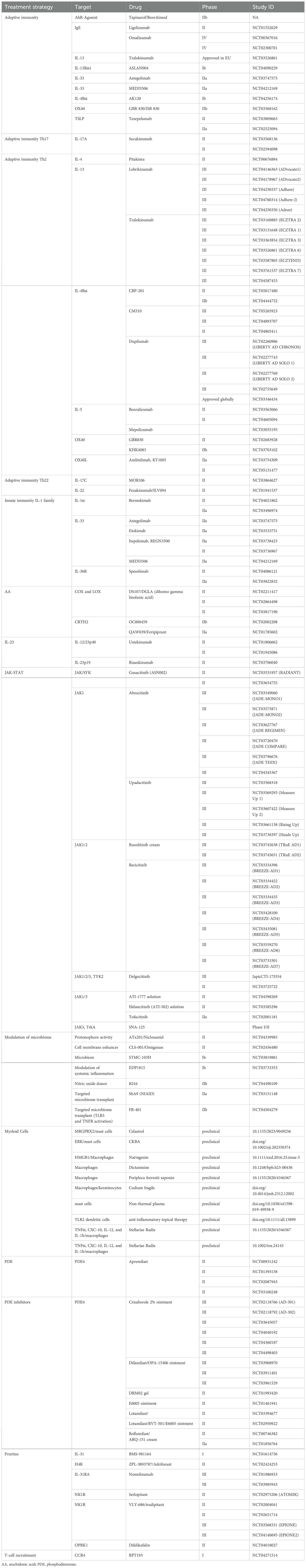
Table 1. Current therapeutical approaches to treat atopic dermatitis, sorted by treatment strategy.
Myeloid cells, including monocytes, macrophages, dendritic cells (DCs), and granulocytes, are crucial components of the innate immune system and play essential roles in skin homeostasis and inflammation. In the context of AD, these cells exhibit several intriguing characteristics that warrant further investigation, like phenotypic and functional changes in response to the skin microbiome. Furthermore, myeloid cells demonstrate dynamic roles throughout the progression of AD. For instance, myeloid-derived suppressor cells (MDSCs) show increased numbers and suppressive function in early stages of the disease but decreased presence in later stages. This dynamic behavior suggests a complex and evolving role for myeloid cells in the pathogenesis of AD.
Role of myeloid cells in AD
Monocytes and macrophages
Macrophages in the skin can originate form distinct sources. Tissue-resident macrophages which originate from yolk-sac-derived erythro-myeloid progenitors during early embryonic phase and get renewed frequently (2). The other type are macrophages originating from circulating monocytes, which are recruited to the skin in response to chemotactic signals. In addition to macrophages, monocytes can differentiate into DCs, amplifying the inflammatory response. Monocytes contribute to the inflammatory cycle in AD through production of cytokines and reactive oxygen species, and by their differentiation into active macrophages that drive skin inflammation (3). These macrophages are central players in inflammation, becoming activated at the onset of the inflammatory process. This activation triggers the expression of many genes, enhancing their ability to eliminate bacteria and regulate other cells through cytokine and chemokine secretion (4). However, excessive macrophage activation can be detrimental, contributing to conditions such as septic shock, organ dysfunction syndrome, and chronic inflammatory diseases like psoriasis and AD (3, 5). Research by Kiekens et al. demonstrated that macrophage numbers are significantly increased in the skin of both the acute and chronic inflammation phase of AD compared to non-affected and healthy skin (6).
In acute and chronic AD lesions, the majority of macrophages originate from circulating monocytes that infiltrate the dermis, where they become key players in both immune defense and inflammation (Figure 1). Whereas they are relative scarce in non-lesional skin. These macrophages, function as antigen-presenting cells (APCs), play a role in antimicrobial activity, preventing bacterial invasion, and drive inflammation by releasing pro-inflammatory cytokines (e.g., TNF-α, IL-1β) and chemokines, which recruit additional immune cells (7). In contrast, tissue resident macrophages primarily maintain skin homeostasis and have regulatory functions during inflammation. Moreover, they participate in tissue remodeling and fibrosis by secreting matrix metalloproteinases (MMPs) and other related factors. Toll-like receptor stimuli and IFN-g polarize monocyte-derived macrophages toward the classical M1 activation pathway, resulting in production of high levels of proinflammatory cytokines (including IL-1, TNF-α, IL-12, and IL-23), reactive nitrogen, and oxygen radicals, enhanced microbicidal activity, and promotion of Th1 responses. Signal transducer and activator of transcription (STAT) 1 activation is crucial in this context. Phenotypically, M1 cells express high levels of MHC class II and co-stimulatory molecules, such as CD80 and CD86, and upregulate the expression of intracellular suppressor of cytokine signaling 3 (SOCS3) and inducible nitric oxide synthase (iNOS). Therefore, M1 cells are implicated in initiating and sustaining an inflammatory response (8).
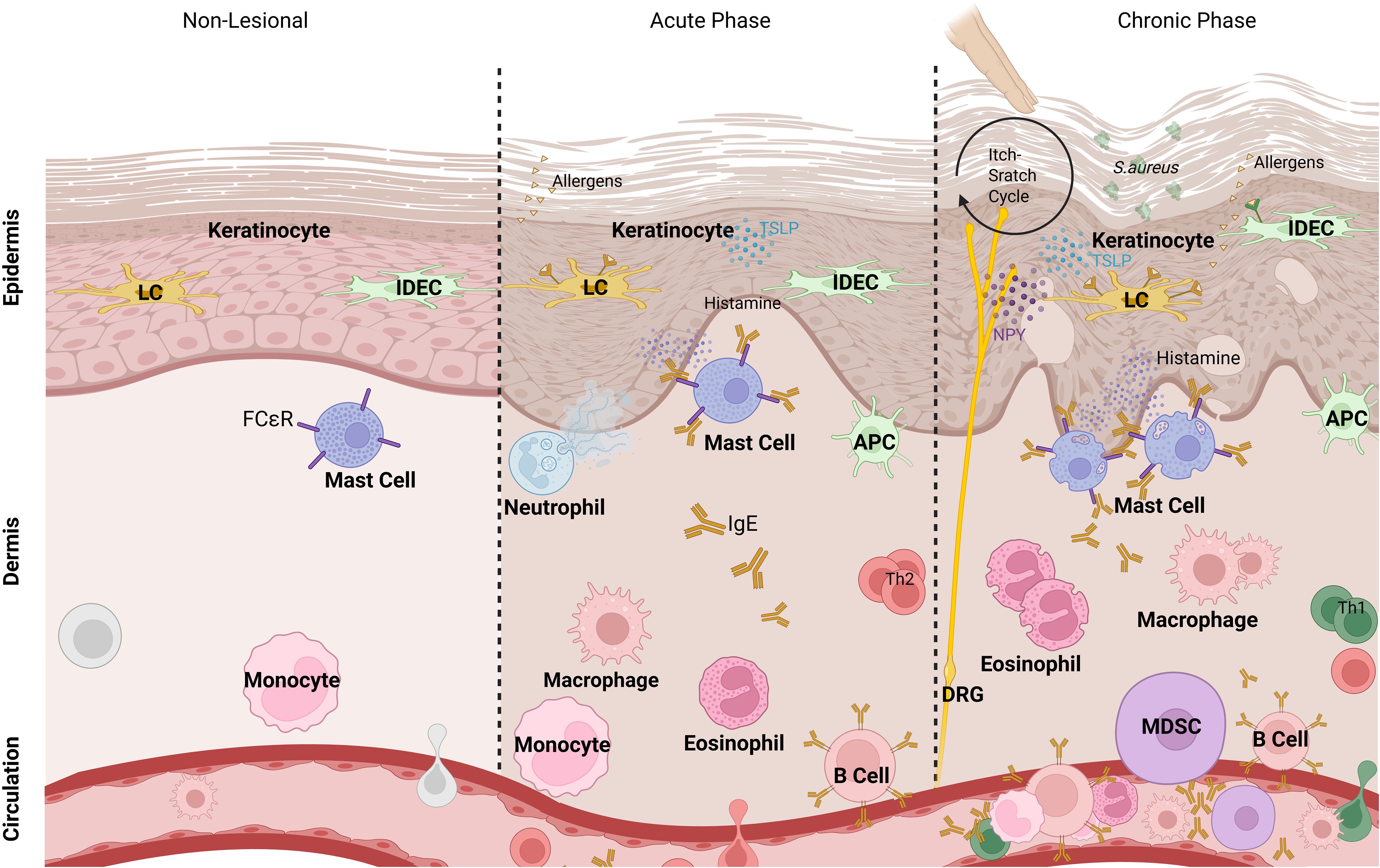
Figure 1. Myeloid cells in phases of AD. In acute and chronic phases of atopic dermatitis, keratinocytes are triggered by allergens that penetrate the disrupted skin barrier. Epidermal Langerhans cells (LCs), inflammatory dendritic epidermal cells (IDECs), and dermal macrophages capture antigens and become antigen-presenting cells (APCs), while additional immune cells like myeloid-derived suppressor cells (MDSCs) migrate to the inflamed area. Upon triggering, mast cells release histamine, while neutrophils and eosinophils release their granules. T cells, particularly Th2 cells in acute phase and Th1/Th17 cells in chronic phase, contribute to the inflammatory response. Dorsal root ganglia (DRG) interact with immune cells via neuropeptides, including neuropeptide Y (NPY), promoting itch sensation and further inflammation.
Beyond their inflammatory functions, monocyte-derived macrophages in AD can undergo alternative activation, which is marked by the increased presence of CD163+ cells in lesional AD skin (9). These CD163+ macrophages, which are indicative of alternatively activated macrophages, are more abundant in lesional AD skin compared to healthy skin, and they share a similar distribution pattern with CD68+ cells. The alternative activation indicates a distinct role of these macrophages in the chronic inflammatory environment of AD, where they may exert several functions that support the persistent nature of the disease.
While the M1/M2 framework has long served as a useful model for understanding macrophage function, it is now recognized as an oversimplification, particularly in chronic inflammatory diseases such as atopic dermatitis. Macrophages exhibit considerable phenotypic and functional plasticity, continuously adapting to their microenvironment. In the skin of AD patients, macrophages are exposed to a complex array of stimuli, including type 2 cytokines (e.g., IL-4, IL-13), microbial components (e.g., S. aureus-derived peptidoglycan), neuropeptides (e.g., neuropeptide Y), histamine, lipid mediators, and mechanical stress, which shape their activation states dynamically (10). Among microbial stimuli, S. aureus is a predominant source of PAMPs in AD (11, 12). Its colonization exacerbates barrier dysfunction and drives macrophage activation through TLR2/6 signaling, inducing proinflammatory cytokines such as IL-1β, TNF-α, and IL-6. These cytokines contribute to the inflammatory milieu and promote the differentiation of Th22s and Th17 cells. Consistently, elevated levels of Th2 and Th22 cytokines are observed in both acute and chronic AD lesions, where they further impair barrier integrity and may contribute to microbial dysbiosis (13). Prolonged exposure to S. aureus can also functionally reprogram macrophages, promoting regulatory or tolerogenic phenotypes beyond classical polarization states. Recent single-cell transcriptomic studies have identified macrophage subsets in inflamed human skin that defy traditional M1/M2 classification, instead displaying hybrid or intermediate profiles (14, 15). These include cells co-expressing pro- and anti-inflammatory markers or those with specific metabolic or tissue-remodeling programs. Such macrophage states may be further modulated by epigenetic reprogramming, tolerization due to chronic microbial exposure, or neuroimmune feedback. In AD skin, for instance, CD68+CD163+ cells co-expressing DC markers like CD1a suggest the presence of a heterogeneous pool of monocyte-derived macrophages with dual immunomodulatory and antigen-presenting functions (6). Understanding this spectrum of macrophage activation is critical for deciphering their roles in disease progression and resolution. It also holds therapeutic potential, as interventions may target distinct macrophage subtypes or reprogramming pathways, such as through TRPV4 activation or cytokine modulation, rather than broadly inhibiting or activating these cells.
Additionally, Chitinase 3-like 1 (CHI3L1), a recognized mediator in Th2-driven inflammation which is also known as breast regression protein 39 (BRP-39), has been shown to mediate the development of AD through the activation of macrophages (16). M2 macrophages, a subtype often associated with anti-inflammatory responses, have been shown to reduce disease severity in a mouse model of AD when selectively activated through the mannose receptor CD206 using the bee venom component phospholipase A2 (17). On the contrary, they have been identified as a major source of IL-31 in AD by the immunohistochemical analysis of skin biopsy samples from AD patients. Their interactions with TSLP, periostin, and basophils further contribute to AD pathogenesis and the perpetuation of the itch-scratch cycle (18). M2 macrophages are also known to produce C–C motif chemokine ligand 18 (CCL18), a chemokine strongly associated with increased morbidity in AD patients (19). IL-4 and IL-13 significantly upregulate CCL18 expression, with IL-10 also contributing, but to a lesser extent. Histamine further enhances the cytokine-induced upregulation of CCL18 mRNA expression by stimulating the histamine receptor 2 (H2R), with the strongest effect observed in IL-10-stimulated macrophages. These combined activations in macrophages drive a substantial increase in CCL18 expression, resulting in its notably high levels in lesional AD skin and in serum of affected individuals. IL-4 upregulates both H2R and H4R, while IL-13 exclusively upregulates H4R without affecting H2R. Conversely, IL-10 upregulates H2R expression but shows a trend toward downregulating H4R (20). Moreover, recent in vitro studies have identified that the transient receptor potential vanilloid 4 (TRPV4) in macrophages, exerts anti-inflammatory properties. Its activation leads to the suppression of IL-1β expression in human macrophages by inhibiting NF-κB signaling and further prevents the differentiation of monocytes into pro-inflammatory macrophages, suggesting a potential therapeutic target for modulating macrophage activity in AD (21). Such findings align with the emerging understanding that macrophage functions in AD are shaped not only by cytokine milieu but also by mechanical and metabolic cues, including ion channels, microbial ligands, and tissue stressors.
In inflamed AD lesions, macrophage markers such as RFD7, which identifies mature tissue phagocytes, and CD68 exhibit similar expression levels and distribution patterns, with CD68+ macrophages being more prevalent than RFD7+ macrophages. Macrophages with high CD36 expression are also more abundant in inflamed AD lesions. CD36, a membrane glycoprotein involved in the phagocytosis of apoptotic cells such as neutrophils, plays a crucial role in limiting tissue damage and contributing to the resolution of inflammation in AD (22–24). In addition, in inflamed AD skin macrophages and DCs share overlapping phenotypes. Both DCs and macrophages express mannose receptors (MRs) for efficient antigen uptake, with MR expression predominantly found in monocyte-derived macrophages in inflamed AD skin (25). Kiekens et al. demonstrated that some macrophage populations may express macrophage (e.g., CD68) and DC (e.g., CD1a) markers simultaneously, indicating a heterogeneous pool of macrophage/DC-like cells (6). This overlap suggests complex plasticity of macrophages and DCs in AD, contributing to disease pathology.
Dendritic cells
DCs are key antigen-presenting cells that form a crucial link between innate and adaptive immunity, playing a significant role in the pathogenesis of AD. In AD patients, the numbers of specific myeloid Dendritic Cell (mDC) subsets, including Langerhans cells (LCs) in the epidermis and inflammatory dendritic epidermal cells (IDECs), are elevated in lesional skin (Figure 1). Activation of Toll-like receptors (TLRs) triggers the maturation of DCs, marked by the upregulation of costimulatory molecules and the release of cytokines (26) (Figure 2). Research on DCs in AD skin primarily focuses on TLR2, as it recognizes PAMPs from S. aureus (Figure 1), particularly peptidoglycan, a major component of its cell wall. DCs capture antigens and migrate to lymph nodes, where they present the antigens to naive T cells, thereby driving the differentiation of Th1 and Th2 cells. Th2 cells, which dominate the immune response during acute phases of AD, produce cytokines such as IL-4, IL-5, and IL-13 upon arrival in the inflamed skin, driving the allergic inflammation characteristic of AD. DCs are uniquely specialized to detect antigens from both external and internal sources, responding to various stress signals. Upon stimulation, DCs initiate and regulate adaptive immunity, performing a highly adaptable function that allows them to adjust their behavior to the specific tissue microenvironment, whether in the skin, lung, or gut mucosa. For example, DCs in the skin, such as LCs and IDECs, exhibit distinct marker expression, cytokine production, migration patterns, and metabolic adaptations compared to DC subsets like conventional DCs (cDCs) or plasmacytoid DCs (pDCs) in the lung or gut mucosa. Despite these dynamic and specialized roles in AD, research indicates that the absolute number of DCs remains unchanged in lesional compared to non-lesional AD skin (6, 27–30). In lesional AD skin, the marked dysbiosis of the microbiome, particularly the overgrowth of S. aureus, plays a critical role in shaping DC function. S. aureus-derived components such as peptidoglycan (PGN), lipoteichoic acid (LTA), and enterotoxins activate pattern recognition receptors (especially TLR2 and TLR6) on DCs, leading to the production of IL-6, IL-1β, and IL-23 (Figure 2). This stimulation promotes Th17 and Th2 polarization in a context-dependent manner. However, chronic microbial exposure may lead to DC tolerance or functional exhaustion, characterized by diminished cytokine production and impaired antigen presentation. This paradox, enhanced stimulation coupled with reduced functional responsiveness, highlights a hallmark of the dysregulated immune environment in chronic AD.
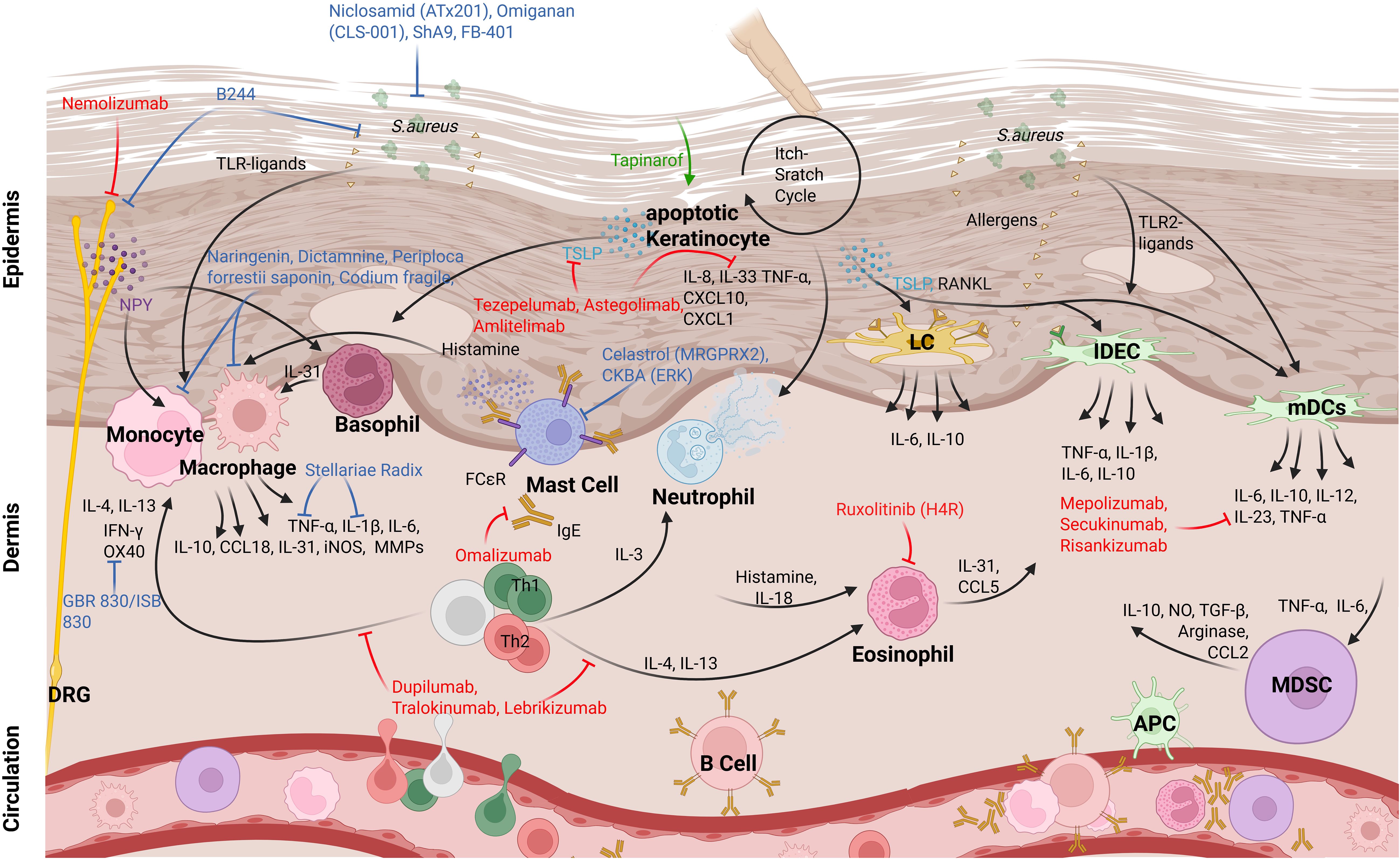
Figure 2. Myeloid cell interactions and therapeutic targets in AD skin. In AD skin, myeloid cells—including Langerhans cells (LCs), inflammatory dendritic epidermal cells (IDECs), monocyte-derived dendritic cells (mDCs), myeloid-derived suppressor cells (MDSCs), macrophages, and monocytes—interact with each other and with other immune and non-immune cells. These interactions are triggered by external stimuli (e.g., S. aureus compounds) and internal signals (e.g., TSLP from keratinocytes), leading to cytokine production (e.g., IL-6, IL-10, IL-4, IL-13). T cells are major drivers of inflammation. Current therapies (shown in red) and investigational treatments (in blue) mainly act by modulating cytokine pathways rather than directly targeting DC subsets. Some novel approaches, such as B244, target the skin microbiome to influence myeloid cell function.
Langerhans cells
LCs are the main specialized subset of DCs located in normal epidermis, acting as sentinels of the immune system (Figure 1). They play a crucial role in initiating antigen-specific immune responses by efficiently taking up antigens in the skin, processing them, and presenting antigenic peptides to T cells in the draining lymph nodes. LCs are characterized by specific surface markers, including CD1a and CD207 (31). In addition to their well-established role in triggering specific immune responses through antigen presentation, LCs also exhibit regulatory functions. This regulatory capacity is partly mediated through the production of IL-10 or the activation of the aryl hydrocarbon receptor (32, 33). These functions may help explain findings by Igyarto et al., who observed that LC-deficient mice were more prone to contact hypersensitivity reactions compared to control mice (33). Further research has validated these regulatory functions in a skin graft mouse model, demonstrating that receptor activator of nuclear factor κB (RANK) is stimulated by its ligand (RANKL), which is produced by apoptotic keratinocytes, inducing LCs to produce IL-10 (34). This IL-10, in turn, induced the development of CD4+CD25+ Tregs, which can suppress skin immune responses (35). Kaplan et al. suggested that the presence of LC-derived IL-10 during the priming phase of the immune response may skew the T cell response toward a Th2 phenotype rather than a Th1 phenotype, potentially promoting the differentiation of Tregs (36). The Th2-dominant microenvironment in AD also contributes to reduced TLR2 expression on LCs. Th2 cytokines, such as IL-4 and IL-13, downregulate TLR2 expression, impairing the cell’s ability to respond to bacterial ligands and shifting the immune response away from Th1-mediated defense (37, 38). The Dysfunctional TLR2 signaling on LCs reduces cytokine production, including IL-6, which drives Th17 immune responses via the NF-κB pathway, and IL-10. While no direct link between TLR2 and IL-10 is known, its downregulation has been observed, suggesting altered immune regulation. In acute AD, these cytokines are impaired, diminishing the skin’s ability to control inflammation and bacterial infections (38–40). In healthy skin, LCs respond to bacterial signals such as S. aureus by maturing and migrating in response to TLR2 ligation, which is essential for initiating an effective immune defense. However, in AD skin, freshly isolated LCs show significantly lower TLR2 expression compared to keratinocytes and their healthy counterparts (41, 42). This low expression prevents LCs from maturing and migrating properly when stimulated with Pam3Cys, a synthetic TLR1/2 ligand that mimics S. aureus signals by replicating the structure of its lipoprotein component (43, 44). As a result, LCs in AD fail to initiate an adequate immune response, which is crucial for fighting skin infections and regulating inflammation. This impairment is reflected by a reduction in antimicrobial peptides (AMPs) such as LL-37, HBD-2, and HBD-3 in AD skin, further compromising the skin’s ability to combat microbial colonization and infection, particularly by S.aureus (45, 46). The impaired TLR2 response in LCs is not solely due to reduced receptor expression but also involves desensitization and tolerance, likely driven by chronic exposure to microbial ligands in AD skin. Prolonged colonization by S. aureus results in persistent TLR2 stimulation, contributing to immune cell desensitization, as observed in other immune cells such as monocytes and macrophages (45, 47). Desensitized immune cells fail to respond effectively to foreign PAMPs, potentially leading to a compromised immune response against subsequent infections.
Inflammatory dendritic epidermal cells
IDECs are pivotal in the pathophysiology of AD. This FcϵRI-positive subtype of mDCs contribute significantly to the inflammatory milieu and maintenance of the inflammatory reaction in AD by producing high levels of proinflammatory cytokines (48). Since LCs are more associated with a local Th2 response in acute AD lesions, IDECs contribute to the transition from a Th2-dominated immune response to a more multifaced immune profile in chronic phases, involving Th1, Th2, and Th17, thereby shaping the overall immune landscape in the skin (Figure 1) (1, 49, 50). Unlike LCs, IDECs are highly matured in the ‘steady state’ in AD skin, yet they still exhibit a reduced TLR2 expression, similar to LCs (24). Despite this maturation, IDECs in AD skin fail to respond to TLR2 ligation, as evidenced by their inability to upregulate MHC class II, CD83 and the costimulatory molecules CD80, and CD86 after stimulation with Pam3Cys (25). This characteristics of IDECs in AD lesions further contribute to the impaired immune response and decreased production of IL-10 seen in AD.
IDECs are also involved in the sensitization process to environmental allergens, capturing these allergens that penetrate the epidermis and trigger IgE-mediated immune responses. The functional behavior of IDECs is broadly influenced by the surrounding inflammatory microenvironment, where locally released cytokines from keratinocytes and other immune cells can modulate their activity (51). In AD IDECs, much like LCs, seem to be desensitized to TLR2 signals due to chronic exposure to microbial ligands in AD skin, particularly from S. aureus. This prolonged exposure leads to tolerance, preventing proper activation and cytokine production by IDECs (42, 47). In healthy skin, IDECs, along with LCs, help control bacterial infections by recognizing and responding to microbial patterns through TLR2, but in AD, this mechanism is severely compromised. This desensitization likely stems from the altered skin microbiome in AD, where S. aureus colonization increases while Staphylococcus epidermidis fails to control its growth (52–54). This shift in microbial composition further contributes to the impaired TLR2 response, as described for LCs. Furthermore, IDECs are implicated in self-sensitization mechanisms, responding to keratinocyte-derived proteins released due to skin damage, which can induce a Th2 response to self-structures resembling environmental allergens. This Th2 dominated cytokine environment in AD downregulates TLR2 expression and skew IDEC function toward promoting allergic inflammation rather than fighting infection (37, 38, 55). Moreover, IDECs, like LCs, fail to produce sufficient IL-6 and IL-10 after TLR2 stimulation, which impairs their ability to control inflammation and regulate immune responses effectively (47, 56). This compromised function of IDECs further exacerbates the immune imbalance in AD, promoting persistent inflammation and heightened susceptibility to bacterial infections. Understanding the role of IDECs in AD not only sheds light on the mechanisms underlying this condition but also opens potential therapeutic avenues aimed at modulating their proinflammatory properties to enhance tolerance and mitigate inflammation in affected individuals.
Myeloid-derived suppressor cells
MDSCs have long been studied for their crucial role in malignancy and tumor maintenance through immunosuppression. However, they are now increasingly recognized as significant players in inflammation. Originating from immature myeloid cells, MDSCs possess immunoregulatory functions and have the potential to differentiate into mature DCs, macrophages, or granulocytes. These cells are categorized into two subtypes: monocytic MDSCs, characterized by a CD14+, CD11b+, Ly6C+, and Ly6G- phenotype, and polymorphonuclear MDSCs, identified by a CD14-, CD11b+, Ly6Clow, and Ly6G+ phenotype (57, 58). MDSCs exert their immunosuppressive effects through direct cell-to-cell contact and by secreting interleukins and chemokines. Their suppression of T-cell activity is mediated by arginase and iNOS, both of which deplete L-arginine, a molecule essential for T-cell differentiation and proliferation (59) (Figure 2). The depletion of L-arginine results in the downregulation of CD3ζ and MHC class II, along with the inhibition of Janus kinase 3 (JAK3) and STAT5 in T cells, ultimately reducing T-cell proliferation (60). Additionally, iNOS produces nitric oxide (NO), which induces dose-dependent apoptosis in T cells by modulating Bcl-2 expression (61). The role of NO in cutaneous inflammation is closely related to its surrounding conditions and concentration, with both pro-inflammatory and anti-inflammatory effects reported (61). In a mouse model of AD, exposure to S. aureus activated TLR 2–6 in the skin, leading to IL-6 production by skin cells. This IL-6 subsequently stimulated the recruitment of suppressive CD11b+Gr1+ MDSCs, which inhibited T cell-mediated responses through the iNOS-dependent pathway (62). Ligation of TLR1–2 was observed to not stimulate MDSCs recruitment. MDSCs also induce Tregs response through CTLA4 and membrane-bound TGF-β, which suppresses natural killer cell cytotoxicity, as demonstrated in recent mouse studies (63, 64). Furthermore, MDSCs produce IL-10, which promotes the differentiation of macrophages into predominantly anti-inflammatory M2 cells and fosters a Th2-mediated immune response (65, 66).
The therapeutic potential of MDSCs has been demonstrated in several mouse models, where these cells were attracted to inflamed sites through chemotactic stimuli. MDSCs were observed migrating to the spleen, lungs, lymph nodes, and inflamed skin, suggesting they selectively infiltrate inflamed tissues (67–73). However, infiltration into the skin was observed only in AD mice, reinforcing the idea that MDSCs selectively target sites of inflammation (72, 73). In one study, this was achieved by inducing CCR5 expression, while another study utilized the injection of CXCL17 (74, 75). Furthermore, MDSCs generated from human umbilical cord blood (hUCB) increased IFN-γ expression in the spleen and lymph nodes. IFN-γ is known for its therapeutic effects in AD (76, 77) and its potential to enhance the immunomodulatory capabilities of MDSCs (78, 79). However, it is important to note that mouse-derived IFN-γ cannot bind to the human IFN-γ receptor, rendering it incapable of affecting human MDSCs (80). Therefore, further studies are necessary to elucidate the role of IFN-γ induced by MDSCs in AD mouse models.
It is generally accepted that the numbers of immature granulocytic and monocytic MDSCs are elevated in patients with inflammatory conditions. This observation has been consistently reported in patients with inflammatory skin diseases and inflammatory bowel disease (58, 81–83). hUCB-MDSCs injected into mice with AD-like symptoms induced by Dermatophagoides farinae (Df) alleviated skin lesions in a dose-dependent manner, with higher doses (1 × 105; and 1 × 106 cells) proving more effective than lower doses (1 × 104 cells). The MDSCs also reduced epidermal thickness and decreased inflammatory cell infiltration (11, 64). MDSCs (1 × 105 and/or 1 × 106 cells) restored skin barrier function and improved skin fibrosis, suggesting that the anti-inflammatory effects and wound-healing capacities of MDSC therapy contribute to recovery from skin barrier impairment, dysfunction, and skin fibrosis in Df-induced AD-NC/Nga mice. This likely results from abnormal repair in response to skin damage. Additionally, MDSC treatment reduced IgE production and lowered Th2- and Th17-mediated cytokine levels (64, 84).
Contrasting with findings in AD, MDSCs from psoriasis patients may recruit Tregs, which are less capable of exerting their regulatory functions (82, 85). They also may exhibit reduced expression of surface PD-1 and lower production of the anti-inflammatory cytokine IL-10 (58, 85). To becoming functionally deficient, MDSCs can acquire proinflammatory functions when exposed to an inflammatory environment, especially in psoriasis patients. These proinflammatory functions include the overexpression of IL-1β, IL-6, IL-8, and TNF-α (86, 87); the production of MMPs such as MMP1 and MMP9, which facilitate the transmigration and accumulation of MDSCs in tissues (88); and the secretion of monocyte chemoattractant protein 1 (MCP1), which acts as a chemoattractant for proinflammatory cells (82). Additionally, GRO and IL-8 secreted by MDSCs recruit neutrophils to the skin and inflammatory cells like neutrophils and monocytes to organs beyond the skin (89).
Granulocytes
Granulocytes including neutrophils, eosinophils, and basophils, play critical roles in the pathogenesis of AD. Basophils contribute to the initiation of AD by increasing IL-4 expression and interacting with keratinocytes and dermal macrophages, leading to epidermal hyperplasia and skin barrier dysfunction, but play a minor role in chronic lesions (90, 91). It was observed that by stimulation with TSLP basophils interact with neutrophils, sensory neurons and T cells enhancing the inflammation and itch in AD skin (90). The interaction of basophils and neutrophils contributes to the severity of AD whereas the reduction of basophils leads to decreased infiltration of eosinophils and neutrophils, as well as skin thickness (92).
Eosinophils
Eosinophils in AD patients show increased and upregulated expression of histamine receptor 4 (H4R), driven by IL-4 and IL-13 through the JAK/STAT pathway, which results in elevated IL-31 production (93). The use of a direct H4R antagonist has been shown to improve disease severity, primarily by targeting pruritus (94). Moreover, the IL-18 receptor (IL-18Rα) is upregulated in eosinophils of AD patients, with histamine enhancing IL-18 expression through H2R and H4R, highlighting the roles of IL-18 and histamine in eosinophil-mediated inflammation in AD (95). Notably, the JAK-inhibitor Ruxolitinib significantly reduces H4R expression in eosinophils (Figure 2), presenting a treatment for AD (93).
Neutrophils
Neutrophils are involved in the inflammatory processes of AD particularly during acute phases or when secondary infections occur (Figure 1). They infiltrate the skin and contribute to inflammation, although their presence is not as prominent as in other skin conditions like psoriasis (96). But when they are increased in the lesional skin, the number is comparable to the number of neutrophils in psoriasis skin (97). In irritated skin the rapid infiltration of neutrophils was observed recently especially in Asian forms of AD (98, 99). This mobilization of neutrophils is affected by the Th2 cytokine mediated Th2-STAT6-C3 complement-NETs (neutrophil extracellular traps) cascade (100). The neutrophil-to-lymphocyte ratio (NLR), a serum inflammatory parameter, is a prognostic factor in several diseases (101–103). The NLR level in AD patients was observed to be higher than in healthy individuals and correlates with the severity of the inflamed and lesional skin (104–107). Therefore, elevated circulating neutrophils are more and more associated with AD severity, and a high NLR may serve as a parameter for AD severity.
TNFα released by mast cells can directly prime circulating neutrophils and enables them to migrate to surrounding tissue (108). Together with the dominant colonization of S. aureus, mast cells are the main trigger for neutrophil host response against microbial infections like NETs (109). Thus, NET formation is regulated by mast cell tryptase in vivo (110). Next to TNFα several cytokines like IL-3, IL-8, and IL-33 affect neutrophils on a genetic or functional level (111–113) (Figure 2). In recent studies it was found that the elevated serum level of high-mobility group box 1 protein (HMGB1) correlates with AD severity in patients (114). HMGB1 promotes the attraction of neutrophils to skin wounds and supports the development of NETs. Additionally, extracellular HMGB1 can contribute to tissue damage by inducing NET formation under inflammatory conditions (115, 116).
As mentioned before, neutrophils infiltrate the tissue in early stages during the development of AD lesions. The recruitment is mediated through CXCR3 signaling, which initially gets activated by inflammatory cytokines such as CXCL1 and CXCL10 (117). CXCR3 activated neutrophils additionally start producing cytokines which amplify the inflammatory response. The chemokine IL-8 (CXCL8), detected in the lesional stratum corneum of AD patients, is linked to skin-barrier dysfunction. By binding to CXCR1 and CXCR2 receptors on human neutrophils, IL-8 activates various signaling pathways (118).
AD is characterized by a complex interplay between myeloid cells and other immune cells, with significant contributions from various cytokines and receptors that drive inflammation and pruritus. The Th2 cytokines, IL-4, IL-13, and IL-31, are key players in this process. These cytokines activate sensory fibers by engaging the TRP channel ankyrin transmembrane protein 1 (TRPA1) and the transient receptor potential (TRP) channel vallinoid 1 (TRPV1), which facilitate calcium influx into these fibers, and amplifies the sensation of itch (119). TRP channels, which respond to a variety of signals including chemical compounds, mechanical stimuli, temperature changes, and osmotic stress, are crucial not only in sensory perception but also in the progression of AD. Notably, the activation of TRPV3 on T cells can further increase TSLP production and the sensation of itch (120). TSLP, secreted by keratinocytes, interacts directly with sensory neurons through its receptor, further contributing to pruritus. The release of TSLP by epidermal cells is enhanced by activated T cells and through crosstalk with other immune cells, highlighting its central role in AD pathogenesis (121). The clinical efficacy of targeting these pathways is underscored by the use of anti-TSLP monoclonal antibody, tezepelumab, which showed a substantial, though not statistically significant, improvement in key clinical characteristics of AD, such as the pruritus rating scale (NRS), Investigator’s Global Assessment (IGA), and reductions in both the Eczema Area and Severity Index (EASI) and the SCORing AD index (SCORAD) (122).
The interaction between myeloid immune cells and the neural system is called immune-neuro crosstalk and involves more than just TSLP (123). Sensory nerve fibers originating from the dorsal root ganglia and trigeminal ganglia innervate the skin and transmit excitatory signals (124). In healthy skin the nervous system and the immune system work together to maintain the homeostasis (125–128). Whereas in AD patients a higher density of nerve fibers around blood vessels and in the epidermis was detected in lesional skin. A change in one of the systems or an imbalance of their interaction can therefore affect both. Thus, neuropeptides and transmitters are able to initiate degranulation of mast cells resulting in an itch-scratch cycle, which is a fundamental aspect of AD pathogenesis (124, 129). A neurotransmitter that plays a crucial role in AD is the neuropeptide Y (NPY), which affects several myeloid cells like mast cells, LCs, monocytes, macrophages, and neutrophils (130, 131). Studies revealed higher levels of NPY in lesional compared to healthy skin (132, 133). Next to NPY several other neuromediators (acetylcholine, substance P, etc.) and cytokines (TSLP, Il-4, Il13, IL-33, etc.) affect the immune system (131, 134).
Although certain immune mechanisms in atopic dermatitis (AD)—such as the Th1/Th2 imbalance and the associated downregulation of type 1 immunity—are generally recognized, the detailed pathogenesis remains complex and heterogeneous (135, 136). The immune landscape of AD becomes even more complex with the identification of distinct endotypes through blood transcriptome analysis. This analysis has proposed two primary AD endotypes based on eosinophil-related expression signatures. The eosinophil-high cluster is characterized by greater dysregulation and a strong correlation between disease activity and IL-5 signaling pathways, while the eosinophil-low endotype shows minimal transcriptomic dysregulation and no significant association with disease activity (137). Expanding on this, recent research has identified four serum biomarker-based clusters (138). The first is marked by high levels of C–C chemokines and dominance of IL-1R1, the second by a mix of TH1, TH2, TH17, and epithelial-related chemokines, the third by a TH2, TH22, and pulmonary and activation-regulated chemokine (PARC) dominance, associated with more severe disease, and the fourth by a TH2 and eosinophil-low profile, which corresponds to a milder form of AD (138). In adults with moderate to severe AD, serum biomarker analysis further distinguishes between a low-inflammatory and a high-inflammatory group, with the latter showing elevated levels of TNFβ, monocyte chemoattractant protein 3 (MCP-3/CCL7), and IL-13 (139).
Ethnic differences in AD endotypes have also been identified, reflecting variations in immune responses. For instance, AD lesions in African Americans display an increased infiltration of DCs expressing the high-affinity IgE receptor (FcER1+), along with a skewed immune response towards TH2/TH22 and reduced TH1 and TH17 responses compared to European-American patients (140). In contrast, AD in Asian populations is characterized by a combined upregulation of TH2/TH17 responses, along with features resembling psoriasis, and a lower expression of TH1 compared to European Americans (99, 141). These findings underline the importance of considering racial differences in the management of more severe forms of AD, as these differences may influence disease presentation and treatment responses.
Perspectives and future directions
Myeloid cells are crucial contributors to the pathophysiology of AD. Depending on their activation state, they participate in a range of functions including the initiation and maintenance of inflammation, regulation of the skin barrier, antigen presentation, and the modulation of neuroimmune interactions. The complexity and context-dependence of these responses are reflected in the growing number of therapeutic approaches targeting distinct myeloid pathways. From classical cytokine inhibition to novel small molecules and microbiome-based therapies, many of these strategies aim to interrupt key signaling pathways involving myeloid cells. The relevance of these cells as both effectors and modulators of skin inflammation makes them attractive targets for future interventions.
However, despite increasing insight into myeloid cell function in AD, several challenges remain. Current classification models often fail to capture the full phenotypic and functional heterogeneity of these cells in inflamed skin. Emerging technologies such as single-cell RNA sequencing and spatial transcriptomics have started to reveal diverse and dynamic myeloid subsets that cannot be adequately described using conventional markers. Future studies should address how these cells change over time during flares, chronic inflammation, or treatment, and how their plasticity contributes to tissue remodeling, barrier dysfunction, and itch. Translationally, strategies that reprogram rather than deplete dysfunctional myeloid populations, for example through the inhibition of key amplifiers of inflammation, may provide more targeted and sustainable therapeutic effects. In parallel, the development of cell-type-specific biomarkers could support precision medicine approaches and facilitate therapeutic monitoring.
Author contributions
EK: Writing – original draft, Writing – review & editing. MS: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Facheris P, Jeffery J, Del Duca E, and Guttman-Yassky E. The translational revolution in atopic dermatitis: the paradigm shift from pathogenesis to treatment. Cell Mol Immunol. (2023) 20(5):448–74. doi: 10.1038/s41423-023-00992-4
2. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518:547–51. doi: 10.1038/nature13989
3. Austermann J, Roth J, and Barczyk-Kahlert K. The good and the bad: monocytes’ and macrophages’ Diverse functions in inflammation. Cells. (2022) 11. doi: 10.3390/cells11121979
4. Lee SB, Park H, Lee JE, Kim KS, and Jeon YH. In vivo optical reporter-gene-based imaging of macrophage infiltration of DNCB-induced atopic dermatitis. Int J Mol Sci. (2020) 21:1–12. doi: 10.3390/ijms21176205
5. Valledor AF, Comalada M, Santamaría-Babi LF, Lloberas J, and Celada A. Macrophage proinflammatory activation and deactivation: a question of balance. Adv Immunol. (2010) 108:1–20. doi: 10.1016/B978-0-12-380995-7.00001-X
6. Kiekens RCM, Thepen T, Oosting AJ, Bihari IC, Van De Winkel JGJ, Bruijnzeel-Koomen CAFM, et al. Heterogeneity within tissue-specific macrophage and dendritic cell populations during cutaneous inflammation in atopic dermatitis. Br J Dermatol. (2001) 145:957–65. doi: 10.1046/j.1365-2133.2001.04508.x
7. Ovsiy I, Riabov V, Manousaridis I, Michel J, Moganti K, Yin S, et al. IL-4 driven transcription factor FoxQ1 is expressed by monocytes in atopic dermatitis and stimulates monocyte migration. Sci Rep. (2017) 7:1–9. doi: 10.1038/s41598-017-17307-z
8. Murray PJ. Macrophage polarization. Annu Rev Physiol. (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
9. Sugaya M, Miyagaki T, Ohmatsu H, Suga H, Kai H, Kamata M, et al. Association of the numbers of CD163(+) cells in lesional skin and serum levels of soluble CD163 with disease progression of cutaneous T cell lymphoma. J Dermatol Sci. (2012) 68:45–51. doi: 10.1016/j.jdermsci.2012.07.007
10. O’Neill LAJ and Artyomov MN. Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol. (2019) 19:273–81. doi: 10.1038/s41577-019-0128-5
11. Kim J, Kim BE, and Leung DYM. Pathophysiology of atopic dermatitis: Clinical implications. Allergy Asthma Proc. (2019) 40:84–92. doi: 10.2500/aap.2019.40.4202
12. Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci Transl Med. (2017) 9. doi: 10.1126/scitranslmed.aah4680
13. Gittler JK, Shemer A, Suárez-Fariñas M, Fuentes-Duculan J, Gulewicz KJ, Wang CQF, et al. Progressive activation of TH2/TH22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. (2012) 130:1344–54. doi: 10.1016/j.jaci.2012.07.012
14. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. (2014) 40:274–88. doi: 10.1016/j.immuni.2014.01.006
15. Reynolds G, Vegh P, Fletcher J, Poyner EFM, Stephenson E, Goh I, et al. Developmental cell programs are co-opted in inflammatory skin disease. Sci (1979). (2021) 371. doi: 10.1126/science.aba6500
16. Kwak EJ, Hong JY, Kim MN, Kim SY, Kim SH, Park CO, et al. Chitinase 3-like 1 drives allergic skin inflammation via Th2 immunity and M2 macrophage activation. Clin Exp Allergy. (2019) 49:1464–74. doi: 10.1111/cea.v49.11
17. Shin D, Choi W, and Bae H. Bee venom phospholipase A2 alleviate house dust mite-induced atopic dermatitis-like skin lesions by the CD206 mannose receptor. Toxins. (2018) 10:146. doi: 10.3390/toxins10040146
18. Hashimoto T, Yokozeki H, Karasuyama H, and Satoh T. IL-31-generating network in atopic dermatitis comprising macrophages, basophils, thymic stromal lymphopoietin, and periostin. J Allergy Clin Immunol. (2023) 151:737–746.e6. doi: 10.1016/j.jaci.2022.11.009
19. Basu MN, Mortz CG, Jensen TK, Barington T, Lambertsen KL, and Halken S. Biomarkers in asthma in the context of atopic dermatitis in young children. Pediatr Allergy Immunol. (2022) 33. doi: 10.1111/pai.13823
20. Mommert S, Schaper JT, Schaper-Gerhardt K, Gutzmer R, and Werfel T. Histamine increases th2 cytokine-induced CCL18 expression in human M2 macrophages. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms222111648
21. Atsumi Y, Toriyama M, Kato H, Nakamura M, Morita A, Takaishi M, et al. Anti-inflammatory role of TRPV4 in human macrophages. Immunohorizons. (2023) 7:81–96. doi: 10.4049/immunohorizons.2200100
22. Lonati A, Mommaas MA, Pasolini G, Lavazza A, Rowden G, and De Panfilis G. Macrophages, but not Langerhans cell-like cells of dendritic lineage, express the CD36 molecule in normal human dermis: relevance to downregulatory cutaneous immune responses? J Invest Dermatol. (1996) 106:96–101. doi: 10.1111/1523-1747.ep12328158
23. Kasraie S and Werfel T. Role of macrophages in the pathogenesis of atopic dermatitis. Mediators Inflammation. (2013) 2013:942375. doi: 10.1155/2013/942375
24. Castleman MJ, Febbraio M, and Hall PR. CD36 is essential for regulation of the host innate response to Staphylococcus aureus alpha-toxin-mediated dermonecrosis. J Immunol. (2015) 195:2294. doi: 10.4049/jimmunol.1500500
25. Tan MCAA, Mommaas AM, Drijfhout JW, Jordens R, Onderwater JJM, Verwoerd D, et al. Mannose receptor-mediated uptake of antigens strongly enhances HLA class II-restricted antigen presentation by cultured dendritic cells. Eur J Immunol. (1997) 27:2426–35. doi: 10.1002/eji.1830270942
26. Herrmann N, Koch S, Leib N, Bedorf J, Wilms H, Schnautz S, et al. TLR2 down-regulates FcϵRI and its transcription factor PU.1 in human Langerhans cells. Allergy. (2013) 68:621–8. doi: 10.1111/all.2013.68.issue-5
27. Bieber T, Novak N, Herrman N, and Koch S. Role of dendritic cells in atopic dermatitis: An update. Clin Rev Allergy Immunol. (2011) 41:254–8. doi: 10.1007/s12016-010-8224-0
28. Liu J, Zhang X, Cheng Y, and Cao X. Dendritic cell migration in inflammation and immunity. Cell Mol Immunol. (2021) 18:2461–71. doi: 10.1038/s41423-021-00726-4
29. Patente TA, Pinho MP, Oliveira AA, Evangelista GCM, Bergami-Santos PC, and Barbuto JAM. Human dendritic cells: Their heterogeneity and clinical application potential in cancer immunotherapy. Front Immunol. (2019) 10:422571. doi: 10.3389/fimmu.2018.03176
30. Alvarez D, Vollmann EH, and von Andrian UH. Mechanisms and consequences of dendritic cell migration. Immunity. (2008) 29:325. doi: 10.1016/j.immuni.2008.08.006
31. Atmatzidis DH, Lambert WC, and Lambert MW. Langerhans cell: exciting developments in health and disease. J Eur Acad Dermatol Venereology. (2017) 31:1817–24. doi: 10.1111/jdv.2017.31.issue-11
32. Koch S, Stroisch TJ, Vorac J, Herrmann N, Leib N, Schnautz S, et al. AhR mediates an anti-inflammatory feedback mechanism in human Langerhans cells involving FcϵRI and IDO. Allergy. (2017) 72:1686–93. doi: 10.1111/all.2017.72.issue-11
33. Igyarto BZ, Jenison MC, Dudda JC, Roers A, Müller W, Koni PA, et al. Langerhans cells suppress contact hypersensitivity responses via cognate CD4 interaction and langerhans cell-derived IL-10. J Immunol. (2009) 183:5085–93. doi: 10.4049/jimmunol.0901884
34. Clayton K, Vallejo AF, Davies J, Sirvent S, and Polak ME. Langerhans cells-programmed by the epidermis. Front Immunol. (2017) 8:316131. doi: 10.3389/fimmu.2017.01676
35. Yoshiki R, Kabashima K, Sugita K, Atarashi K, Shimauchi T, and Tokura Y. IL-10-producing Langerhans cells and regulatory T cells are responsible for depressed contact hypersensitivity in grafted skin. J Invest Dermatol. (2009) 129:705–13. doi: 10.1038/jid.2008.304
36. Kaplan DH, Jenison MC, Saeland S, Shlomchik WD, and Shlomchik MJ. Epidermal langerhans cell-deficient mice develop enhanced contact hypersensitivity. Immunity. (2005) 23:611–20. doi: 10.1016/j.immuni.2005.10.008
38. Wu Q, Martin RJ, LaFasto S, Efaw BJ, Rino JG, Harbeck RJ, et al. Toll-like receptor 2 down-regulation in established mouse allergic lungs contributes to decreased mycoplasma clearance. Am J Respir Crit Care Med. (2008) 177:720–9. doi: 10.1164/rccm.200709-1387OC
39. Zihad SMNK, Sifat N, Islam MA, Monjur-Al-Hossain ASMM, Sikdar KMYK, Sarker MMR, et al. Role of pattern recognition receptors in sensing Mycobacterium tuberculosis. Heliyon. (2023) 9:2405–8440. doi: 10.1016/j.heliyon.2023.e20636
40. Grebenciucova E and VanHaerents S. Interleukin 6: at the interface of human health and disease. Front Immunol. (2023) 14:1255533. doi: 10.3389/fimmu.2023.1255533
41. Li D, Lei H, Li Z, Li H, Wang Y, Lai Y, et al. A novel lipopeptide from skin commensal activates TLR2/CD36-p38 MAPK signaling to increase antibacterial defense against bacterial infection. PloS One. (2013) 8. doi: 10.1371/journal.pone.0058288
42. Grice EA and Segre JA. The skin microbiome. Nat Rev Microbiol. (2011) 9:244–53. doi: 10.1038/nrmicro2537
43. Iwamoto K, Nümm TJ, Koch S, Herrmann N, Leib N, and Bieber T. Langerhans and inflammatory dendritic epidermal cells in atopic dermatitis are tolerized toward TLR2 activation. Allergy. (2018) 73:2205–13. doi: 10.1111/all.2018.73.issue-11
44. Kennerknecht K, Noschka R, Löffler F, Wehrstedt S, Pedersen GK, Mayer D, et al. Toll like-receptor agonist Pam3Cys modulates the immunogenicity of liposomes containing the tuberculosis vaccine candidate H56. Med Microbiol Immunol. (2020) 209:163. doi: 10.1007/s00430-020-00657-3
45. Tsybikov NN, Petrisheva I, Fefelova EV, Kuznik BI, and Magen E. Expression of TLR2 and TLR4 on peripheral blood monocytes during exacerbation of atopic dermatitis. Allergy Asthma Proc. (2015) 36:e140–5. doi: 10.2500/aap.2015.36.3901
46. Wang V, Boguniewicz J, Boguniewicz M, and Ong PY. The infectious complications of atopic dermatitis. Ann Allergy Asthma Immunol. (2020) 126:3. doi: 10.1016/j.anai.2020.08.002
47. Niebuhr M, Lutat C, Sigel S, and Werfel T. Impaired TLR-2 expression and TLR-2-mediated cytokine secretion in macrophages from patients with atopic dermatitis. Allergy. (2009) 64:1580–7. doi: 10.1111/j.1398-9995.2009.02050.x
48. Wollenberg A, Kraft S, Hanau D, and Bieber T. Immunomorphological and ultrastructural characterization of langerhans cells and a novel, inflammatory dendritic epidermal cell (IDEC) population in lesional skin of atopic eczema. J Invest Dermatol. (1996) 106:446–53. doi: 10.1111/1523-1747.ep12343596
49. Novak N, Gros E, Bieber T, and Allam JP. Human skin and oral mucosal dendritic cells as ‘good guys’ and ‘bad guys’ in allergic immune responses. Clin Exp Immunol. (2010) 161:28. doi: 10.1111/j.1365-2249.2010.04162.x
50. Fania L, Moretta G, Antonelli F, Scala E, Abeni D, Albanesi C, et al. Multiple roles for cytokines in atopic dermatitis: from pathogenic mediators to endotype-specific biomarkers to therapeutic targets. Int J Mol Sci. (2022) 23:2684. doi: 10.3390/ijms23052684
51. Altrichter S, Kriehuber E, Moser J, Valenta R, Kopp T, and Stingl G. Serum igE autoantibodies target keratinocytes in patients with atopic dermatitis. J Invest Dermatol. (2008) 128:2232–9. doi: 10.1038/jid.2008.80
52. Lai Y, Cogen AL, Radek KA, Park HJ, MacLeod DT, Leichtle A, et al. Activation of TLR2 by a small molecule produced by Staphylococcus epidermidis increases antimicrobial defense against bacterial skin infections. J Invest Dermatol. (2010) 130:2211–21. doi: 10.1038/jid.2010.123
53. Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. (2012) 22:850–9. doi: 10.1101/gr.131029.111
54. Liu H, Archer NK, Dillen CA, Wang Y, Ashbaugh AG, Ortines RV, et al. Staphylococcus aureus Epicutaneous Exposure Drives Skin Inflammation via IL-36-Mediated T Cell Responses. Cell Host Microbe. (2017) 22:653–666.e5. doi: 10.1016/j.chom.2017.10.006
55. Flo TH, Halaas Ø, Torp S, Ryan L, Lien E, Dybdahl B, et al. Differential expression of Toll-like receptor 2 in human cells. J Leukoc Biol. (2001) 69:474–81. doi: 10.1189/jlb.69.3.474
56. Niebuhr M, Heratizadeh A, Wichmann K, Satzger I, and Werfel T. Intrinsic alterations of pro-inflammatory mediators in unstimulated and TLR-2 stimulated keratinocytes from atopic dermatitis patients. Exp Dermatol. (2011) 20:468–72. doi: 10.1111/j.1600-0625.2011.01277.x
57. Smith AR and Reynolds JM. Editorial: The contribution of myeloid-derived suppression to inflammatory disease. J Leukoc Biol. (2014) 96:361–4. doi: 10.1189/jlb.3CE0414-205R
58. Soler DC and McCormick TS. Expanding the list of dysregulated immunosuppressive cells in psoriasis. J Invest Dermatol. (2016) 136:1749–51. doi: 10.1016/j.jid.2016.04.029
59. Kusmartsev SA, Li Y, and Chen S-H. Gr-1+ Myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J Immunol. (2000) 165:779–85. doi: 10.4049/jimmunol.165.2.779
60. Gabrilovich DI and Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
61. Man MQ, Wakefield JS, Mauro TM, and Elias PM. Regulatory role of nitric oxide in cutaneous inflammation. Inflammation. (2022) 45(3):949–64. doi: 10.1007/s10753-021-01615-8
62. Skabytska Y, Wölbing F, Günther C, Köberle M, Kaesler S, Chen KM, et al. Cutaneous innate immune sensing of Toll-like receptor 2–6 ligands suppresses T cell immunity by inducing myeloid-derived suppressor cells. Immunity. (2014) 41:762–75. doi: 10.1016/j.immuni.2014.10.009
63. Li H, Han Y, Guo Q, Zhang M, and Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-β1. J Immunol. (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
64. Kim CH, Hong SM, Kim S, Yu JI, Jung SH, Bang CH, et al. Skin repair and immunoregulatory effects of myeloid suppressor cells from human cord blood in atopic dermatitis. Front Immunol. (2023) 14. doi: 10.3389/fimmu.2023.1263646
65. Sinha P, Clements VK, Bunt SK, Albelda SM, and Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. (2007) 179:977–83. doi: 10.4049/jimmunol.179.2.977
66. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. (2006) 66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299
67. Kurtz A. Mesenchymal stem cell delivery routes and fate. Int J Stem Cells. (2008) 1:1–7. doi: 10.15283/ijsc.2008.1.1.1
68. Pendharkar AV, Chua JY, Andres RH, Wang N, Gaeta X, Wang H, et al. Biodistribution of neural stem cells after intravascular therapy for hypoxic-ischemia. Stroke. (2010) 41:2064–70. doi: 10.1161/STROKEAHA.109.575993
69. Rosado-De-Castro PH, Schmidt FR, Battistella V, Lopes De Souza SA, Gutfilen B, Goldenberg RC, et al. Biodistribution of bone marrow mononuclear cells after intra-arterial or intravenous transplantation in subacute stroke patients. Regenerative Med. (2013) 8:145–55. doi: 10.2217/rme.13.2
70. Fabian C, Naaldijk Y, Leovsky C, Johnson AA, Rudolph L, Jaeger C, et al. Distribution pattern following systemic mesenchymal stem cell injection depends on the age of the recipient and neuronal health. Stem Cell Res Ther. (2017) 8. doi: 10.1186/s13287-017-0533-2
71. Casiraghi F, Azzollini N, Cassis P, Imberti B, Morigi M, Cugini D, et al. Pretransplant infusion of mesenchymal stem cells prolongs the survival of a semiallogeneic heart transplant through the generation of regulatory T cells. J Immunol. (2008) 181:3933–46. doi: 10.4049/jimmunol.181.6.3933
72. Cripps JG and Gorham JD. MDSC in autoimmunity. Int Immunopharmacol. (2011) 11:789–93. doi: 10.1016/j.intimp.2011.01.026
73. Schwacha MG, Scroggins SR, Montgomery RK, Nicholson SE, and Cap AP. Burn injury is associated with an infiltration of the wound site with myeloid-derived suppressor cells. Cell Immunol. (2019) 338:21–6. doi: 10.1016/j.cellimm.2019.03.001
74. Soler DC, Sugiyama H, Young AB, Massari JV, McCormick TS, and Cooper KD. Psoriasis patients exhibit impairment of the high potency CCR5(+) T regulatory cell subset. Clin Immunol. (2013) 149:111–8. doi: 10.1016/j.clim.2013.06.007
75. Oka T, Sugaya M, Takahashi N, Takahashi T, Shibata S, Miyagaki T, et al. CXCL17 attenuates imiquimod-induced psoriasis-like skin inflammation by recruiting myeloid-derived suppressor cells and regulatory T cells. J Immunol. (2017) 198:3897–908. doi: 10.4049/jimmunol.1601607
76. Brar K and Leung DYM. Recent considerations in the use of recombinant interferon gamma for biological therapy of atopic dermatitis. Expert Opin Biol Ther. (2016) 16:507. doi: 10.1517/14712598.2016.1135898
77. Jang IG, Yang JK, Lee HJ, Yi JY, Kim HO, Kim CW, et al. Clinical improvement and immunohistochemical findings in severe atopic dermatitis treated with interferon gamma. J Am Acad Dermatol. (2000) 42:1033–40. doi: 10.1067/mjd.2000.104793
78. Mojic M, Takeda K, and Hayakawa Y. The dark side of IFN-γ: its role in promoting cancer immunoevasion. Int J Mol Sci. (2017) 19. doi: 10.3390/ijms19010089
79. Yang F, Li Y, Zou W, Xu Y, Wang H, Wang W, et al. Adoptive transfer of IFN-γ-induced M-MDSCs promotes immune tolerance to allografts through iNOS pathway. Inflammation Res. (2019) 68:545–55. doi: 10.1007/s00011-019-01237-9
80. Hess NJ, Brown ME, and Capitini CM. GVHD pathogenesis, prevention and treatment: lessons from humanized mouse transplant models. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.723544
81. Haile LA, von Wasielewski R, Gamrekelashvili J, Krüger C, Bachmann O, Westendorf AM, et al. Myeloid-derived suppressor cells in inflammatory bowel disease: A new immunoregulatory pathway. Gastroenterology. (2008) 135:871–881.e5. doi: 10.1053/j.gastro.2008.06.032
82. Myeloid-derived suppressor cells are elevated in patients with psoriasis and produce various molecules. Mol Med Rep. (2016) 14(4):3935–40. doi: 10.3892/mmr.2016.5685
83. Cao LY, Chung JS, Teshima T, Feigenbaum L, Cruz PD, Jacobe HT, et al. Myeloid-derived suppressor cells in psoriasis are an expanded population exhibiting diverse T-cell–suppressor mechanisms. J Invest Dermatol. (2016) 136:1801–10. doi: 10.1016/j.jid.2016.02.816
84. Nakajima S, Tie D, Nomura T, and Kabashima K. Novel pathogenesis of atopic dermatitis from the view of cytokines in mice and humans. Cytokine. (2021) 148. doi: 10.1016/j.cyto.2021.155664
85. Soler DC, Young AB, Fiessinger L, Galimberti F, Debanne S, Groft S, et al. Increased, but functionally impaired, CD14+ HLA-DR–/low myeloid-derived suppressor cells in psoriasis: A mechanism of dysregulated T cells. J Invest Dermatol. (2016) 136:798–808. doi: 10.1016/j.jid.2015.12.036
86. Okubo Y and Koga M. Peripheral blood monocytes in psoriatic patients overproduce cytokines. J Dermatol Sci. (1998) 17:223–32. doi: 10.1016/S0923-1811(98)00019-X
87. Mizutani H, Ohmoto Y, Mizutani T, Murata M, and Shimizu M. Role of increased production of monocytes TNF-α, IL-1β and IL-6 in psoriasis: Relation to focal infection, disease activity and responses to treatments. J Dermatol Sci. (1997) 14:145–53. doi: 10.1016/S0923-1811(96)00562-2
88. Melani C, Sangaletti S, Barazzetta FM, Werb Z, and Colombo MP. Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor-bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Res. (2007) 67:11438–46. doi: 10.1158/0008-5472.CAN-07-1882
89. Ilkovitch D and Lopez DM. Immune modulation by melanoma-derived factors. Exp Dermatol. (2008) 17:977–85. doi: 10.1111/j.1600-0625.2008.00779.x
90. Mali SS and Bautista DM. Basophils add fuel to the flame of eczema itch. Cell. (2021) 184:294–6. doi: 10.1016/j.cell.2020.12.035
91. Pellefigues C, Naidoo K, Mehta P, Schmidt AJ, Jagot F, Roussel E, et al. Basophils promote barrier dysfunction and resolution in the atopic skin. J Allergy Clin Immunol. (2021) 148:799–812.e10. doi: 10.1016/j.jaci.2021.02.018
92. Hou T, Tsang MSM, Kan LLY, Li P, Chu IMT, Lam CWK, et al. Il-37 targets tslp-primed basophils to alleviate atopic dermatitis. Int J Mol Sci. (2021) 22:7393. doi: 10.3390/ijms22147393
93. Schaper-Gerhardt K, Köther B, Wolff L, Kabatas A, Gehring M, Nikolouli E, et al. The H4 R is highly expressed on eosinophils from AD patients and IL-4 upregulates expression and function via the JAK/STAT pathway. Allergy. (2021) 76:1261–4. doi: 10.1111/all.14599
94. Werfel T, Layton G, Yeadon M, Whitlock L, Osterloh I, Jimenez P, et al. Efficacy and safety of the histamine H4 receptor antagonist ZPL-3893787 in patients with atopic dermatitis. J Allergy Clin Immunol. (2019) 143:1830–1837.e4. doi: 10.1016/j.jaci.2018.07.047
95. Beyer L, Kabatas AS, Mommert S, Stark H, Werfel T, Gutzmer R, et al. Histamine activates human eosinophils via H2R and H4R predominantly in atopic dermatitis patients. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms231810294
96. Akhtar S, Alsayed RKME, Ahmad F, AlHammadi A, Al-Khawaga S, AlHarami SMAM, et al. Epigenetic control of inflammation in Atopic Dermatitis. Semin Cell Dev Biol. (2024) 154:199–207. doi: 10.1016/j.semcdb.2023.04.005. Preprint.
97. Choy DF, Hsu DK, Seshasayee D, Fung MA, Modrusan Z, Martin F, et al. Comparative transcriptomic analyses of atopic dermatitis and psoriasis reveal shared neutrophilic inflammation. J Allergy Clin Immunol. (2012) 130:1335–1343.e5. doi: 10.1016/j.jaci.2012.06.044
98. Zheng C, Cao T, Ye C, and Zou Y. Neutrophil recruitment by CD4 tissue-resident memory T cells induces chronic recurrent inflammation in atopic dermatitis. Clin Immunol. (2023) 256:109805. doi: 10.1016/j.clim.2023.109805
99. Chan TC, Sanyal RD, Pavel AB, Glickman J, Zheng X, Xu H, et al. Atopic dermatitis in Chinese patients shows TH2/TH17 skewing with psoriasiform features. J Allergy Clin Immunol. (2018) 142:1013–7. doi: 10.1016/j.jaci.2018.06.016
100. Zheng Z, Li Y, Jia S, Zhu M, Cao L, Tao M, et al. Lung mesenchymal stromal cells influenced by Th2 cytokines mobilize neutrophils and facilitate metastasis by producing complement C3. Nat Commun. (2021) 12:1–15. doi: 10.1038/s41467-021-26460-z
101. Li CH, Chiou HYC, Lin MH, Kuo CH, Lin YC, Lin YC, et al. Immunological map in COVID-19. J Microbiology Immunol Infection. (2021) 54:547–56. doi: 10.1016/j.jmii.2021.04.006
102. Huang Z, Fu Z, Huang W, and Huang K. Prognostic value of neutrophil-to-lymphocyte ratio in sepsis: A meta-analysis. Am J Emerg Med. (2020) 38:641–7. doi: 10.1016/j.ajem.2019.10.023
103. El-Gazzar AG, Kamel MH, Elbahnasy OKM, and El-Naggar MES. Prognostic value of platelet and neutrophil to lymphocyte ratio in COPD patients. Expert Rev Respir Med. (2020) 14:111–6. doi: 10.1080/17476348.2019.1675517
104. Batmaz SB. Simple markers for systemic inflammation in pediatric atopic dermatitis patients. Indian J Dermatol. (2018) 63:305–10. doi: 10.4103/ijd.IJD_427_17
105. Inokuchi-Sakata S, Ishiuji Y, Katsuta M, Kharma B, Yasuda KI, Tominaga M, et al. Role of eosinophil relative count and neutrophil-to-lymphocyte ratio in the assessment of severity of atopic dermatitis. Acta Derm Venereol. (2021) 101:adv00491–adv00491. doi: 10.2340/00015555-3838
106. Dogru M and Citli R. The neutrophil-lymphocyte ratio in children with atopic dermatitis: A case-control study. Clinica Terapeutica. (2017) 168:e262–5. doi: 10.7417/T.2017.2017
107. Jiang Y and Ma W. Assessment of neutrophil-to-lymphocyte ratio and platelet-to-lymphocyte ratio in atopic dermatitis patients. Med Sci Monitor. (2017) 23:1340–6. doi: 10.12659/MSM.900212
108. Dudeck J, Kotrba J, Immler R, Hoffmann A, Voss M, Alexaki VI, et al. Directional mast cell degranulation of tumor necrosis factor into blood vessels primes neutrophil extravasation. Immunity. (2021) 54:468–483.e5. doi: 10.1016/j.immuni.2020.12.017
109. Schramm R and Thorlacius H. Neutrophil recruitment in mast cell-dependent inflammation: Inhibitory mechanisms of glucocorticoids. Inflammation Res. (2004) 53:644–52. doi: 10.1007/s00011-004-1307-8
110. Pejler G, Alanazi S, Grujic M, Adler J, Olsson AK, Sommerhoff CP, et al. Mast cell tryptase potentiates neutrophil extracellular trap formation. J Innate Immun. (2022) 14:433–46. doi: 10.1159/000520972
111. Kimata H and Lindley I. Detection of plasma interleukin-8 in atopic dermatitis. Arch Dis Child. (1994) 70:119–22. doi: 10.1136/adc.70.2.119
112. Neuber K, Hilger RA, and Konig W. Interleukin-3, interleukin-8, FMLP and C5a enhance the release of leukotrienes from neutrophils of patients with atopic dermatitis. Immunology. (1991) 73:83.
113. Chen YL, Gutowska-Owsiak D, Hardman CS, Westmoreland M, MacKenzie T, Cifuentes L, et al. Proof-of-concept clinical trial of etokimab shows a key role for IL-33 in atopic dermatitis pathogenesis. Sci Transl Med. (2019) 11. doi: 10.1126/scitranslmed.aax2945
114. Cuppari C, Manti S, Salpietro A, Valenti S, Capizzi A, Arrigo T, et al. HMGB1 levels in children with atopic eczema/dermatitis syndrome (AEDS). Pediatr Allergy Immunol. (2016) 27:99–102. doi: 10.1111/pai.2016.27.issue-1
115. Hoste E, Maueröder C, van Hove L, Catrysse L, Vikkula HK, Sze M, et al. Epithelial HMGB1 delays skin wound healing and drives tumor initiation by priming neutrophils for NET formation. Cell Rep. (2019) 29:2689–2701.e4. doi: 10.1016/j.celrep.2019.10.104
116. Mu S, Li Z, Lin L, Wang D, Yang F, Chen L, et al. SIRT1-mediated HMGB1 deacetylation suppresses neutrophil extracellular traps related to blood–brain barrier impairment after cerebral venous thrombosis. Mol Neurobiol. (2024) 61:6060–76. doi: 10.1007/s12035-024-03959-2
117. Walsh CM, Hill RZ, Schwendinger-Schreck J, Deguine J, Brock EC, Kucirek N, et al. Neutrophils promote CXCR3-dependent itch in the development of atopic dermatitis. Elife. (2019) 8. doi: 10.7554/eLife.48448
118. Amarbayasgalan T, Takahashi H, Dekio I, and Morita E. Interleukin-8 content in the stratum corneum as an indicator of the severity of inflammation in the lesions of atopic dermatitis. Int Arch Allergy Immunol. (2012) 160:63–74. doi: 10.1159/000339666
119. Oetjen LK, Mack MR, Feng J, Whelan TM, Niu H, Guo CJ, et al. Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell. (2017) 171:217–28.e13. doi: 10.1016/j.cell.2017.08.006
120. Zhao J, Munanairi A, Liu XY, Zhang J, Hu L, Hu M, et al. PAR2 mediates itch via TRPV3 signaling in keratinocytes. J Invest Dermatol. (2020) 140:1524–32. doi: 10.1016/j.jid.2020.01.012
121. Marschall P, Wei R, Segaud J, Yao W, Hener P, German BF, et al. Dual function of Langerhans cells in skin TSLP-promoted TFH differentiation in mouse atopic dermatitis. J Allergy Clin Immunol. (2021) 147:1778–94. doi: 10.1016/j.jaci.2020.10.006
122. Simpson EL, Parnes JR, She D, Crouch S, Rees W, Mo M, et al. Tezepelumab, an anti-thymic stromal lymphopoietin monoclonal antibody, in the treatment of moderate to severe atopic dermatitis: A randomized phase 2a clinical trial. J Am Acad Dermatol. (2019) 80:1013–21. doi: 10.1016/j.jaad.2018.11.059
123. Huang X, Li F, and Wang F. Neural regulation of innate immunity in inflammatory skin diseases. Pharm (Basel). (2023) 16. doi: 10.3390/ph16020246
124. Roger A, Reynders A, Hoeffel G, and Ugolini S. Neuroimmune crosstalk in the skin: a delicate balance governing inflammatory processes. Curr Opin Immunol. (2022) 77:102212. doi: 10.1016/j.coi.2022.102212
125. Sugiura H, Omoto M, Hirota Y, Danno K, and Uehara M. Density and fine structure of peripheral nerves in various skin lesions of atopic dermatitis. Arch Dermatol Res. (1997) 289:125–31. doi: 10.1007/s004030050167
126. Dou YC, Hagströmer L, Emtestam L, and Johansson O. Increased nerve growth factor and its receptors in atopic dermatitis: an immunohistochemical study. Arch Dermatol Res. (2006) 298:31–7. doi: 10.1007/s00403-006-0657-1
127. Deng J, Parthasarathy V, Marani M, Bordeaux Z, Lee K, Trinh C, et al. Extracellular matrix and dermal nerve growth factor dysregulation in prurigo nodularis compared to atopic dermatitis. Front Med (Lausanne). (2022) 9:1022889. doi: 10.3389/fmed.2022.1022889
128. Tominaga M and Takamori K. Itch and nerve fibers with special reference to atopic dermatitis: therapeutic implications. J Dermatol. (2014) 41:205–12. doi: 10.1111/jde.2014.41.issue-3
129. Zhang Y, Zhang H, Jiang B, Tong X, Yan S, and Lu J. Current views on neuropeptides in atopic dermatitis. Exp Dermatol. (2021) 30:1588–97. doi: 10.1111/exd.v30.11
130. Chen WC, Liu YBin, Liu WF, Zhou YY, He HF, and Lin S. Neuropeptide Y is an immunomodulatory factor: direct and indirect. Front Immunol. (2020) 11:580378. doi: 10.3389/fimmu.2020.580378
131. Steinhoff M, Ahmad F, Pandey A, Datsi A, AlHammadi A, Al-Khawaga S, et al. Neuroimmune communication regulating pruritus in atopic dermatitis. J Allergy Clin Immunol. (2022) 149:1875–98. doi: 10.1016/j.jaci.2022.03.010
132. Oh SH, Bae BG, Park CO, Noh JY, Park IH, Wu WH, et al. Association of stress with symptoms of atopic dermatitis. Acta Derm Venereol. (2010) 90:582–8. doi: 10.2340/00015555-0933
133. Anderson ZT, Dawson AD, Slominski AT, and Harris ML. Current insights into the role of neuropeptide Y in skin physiology and pathology. Front Endocrinol (Lausanne). (2022) 13. doi: 10.3389/fendo.2022.838434
134. Paramita DA, Nasution K, and Lubis NZ. Relationship of substance P with the degree of atopic dermatitis severity. Clin Cosmet Investig Dermatol. (2021) 14:551–5. doi: 10.2147/CCID.S306557
135. Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol. (2015) 15:271–82. doi: 10.1038/nri3831
136. Böhm I and Bauer R. Th1-Zellen, Th2-Zellen und atopische Dermatitis. Hautarzt. (1997) 48:223–7. doi: 10.1007/s001050050573
137. Möbus L, Rodriguez E, Harder I, Boraczynski N, Szymczak S, Hübenthal M, et al. Blood transcriptome profiling identifies 2 candidate endotypes of atopic dermatitis. J Allergy Clin Immunol. (2022) 150:385–95. doi: 10.1016/j.jaci.2022.02.001
138. Bakker DS, Nierkens S, Knol EF, Giovannone B, Delemarre EM, van der Schaft J, et al. Confirmation of multiple endotypes in atopic dermatitis based on serum biomarkers. J Allergy Clin Immunol. (2021) 147:189–98. doi: 10.1016/j.jaci.2020.04.062
139. Sims JT, Chang CY, Higgs RE, Engle SM, Liu Y, Sissons SE, et al. Insights into adult atopic dermatitis heterogeneity derived from circulating biomarker profiling in patients with moderate-to-severe disease. Exp Dermatol. (2021) 30:1650–61. doi: 10.1111/exd.v30.11
140. Sanyal RD, Pavel AB, Glickman J, Chan TC, Zheng X, Zhang N, et al. Atopic dermatitis in African American patients is TH2/TH22-skewed with TH1/TH17 attenuation. Ann Allergy Asthma Immunol. (2019) 122:99–110.e6. doi: 10.1016/j.anai.2018.08.024
141. Wen HC, Czarnowicki T, Noda S, Malik K, Pavel AB, Nakajima S, et al. Serum from Asian patients with atopic dermatitis is characterized by TH2/TH22 activation, which is highly correlated with nonlesional skin measures. J Allergy Clin Immunol. (2018) 142:324–328.e11. doi: 10.1016/j.jaci.2018.02.047
Keywords: atopic dermatitis, myeloid cells, dendritic cells, immunology, inflammatory disease, therapeutic targets, immune drivers
Citation: Kupschke E and Schenk M (2025) The myeloid switch: immune drivers in atopic dermatitis — roles in pathogenesis and emerging therapeutic targeting. Front. Immunol. 16:1608338. doi: 10.3389/fimmu.2025.1608338
Received: 08 April 2025; Accepted: 04 June 2025;
Published: 30 June 2025.
Edited by:
Renato C. Monteiro, Université de Paris, FranceReviewed by:
Pablo C. Ortiz-Lazareno, Centro de Investigación Biomédica de Occidente (CIBO), MexicoMasato Tamari, National Center for Child Health and Development (NCCHD), Japan
Copyright © 2025 Kupschke and Schenk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mirjam Schenk, bWlyamFtLnNjaGVua0Bjay1jYXJlLmNo; bWlyamFtLnNjaGVua0B1bmliZS5jaA==