 Jonas Aakre Wik
Jonas Aakre Wik Emma Riiser Berge
Emma Riiser Berge Kristine Stromsnes
Kristine Stromsnes Bjørn Steen Skålhegg
Bjørn Steen Skålhegg- 1Hybrid Technology Hub - Centre of Excellence, Institute of Basic Medical Sciences, University of Oslo, Oslo, Norway
- 2Department of Immunology and Transfusion Medicine, Oslo University Hospital, Oslo, Norway
- 3Division for Molecular Nutrition, Institute of Basic Medical Sciences, University of Oslo, Oslo, Norway
The immune system protects the body against dangers that include pathogens, damage and cancer. Modern cancer therapies have sought to bolster immune responses against cancer using immunotherapy, which may include various forms of immune checkpoint therapy (ICT) in addition to methods of adoptive cell transfer (ACT), which is often associated with transfer of chimeric antigen receptor (CAR) T cells. Despite favorable outcomes in some patients and some cancers, as many as 60-80% of patients fail to benefit from ICT due to primary or adaptive resistance. This highlights the need for deeper understanding of how cancers suppress the immune system. Solid tumors, which make up approximately 90% of all cancers, are characterized by an immunosuppressive tumor microenvironment (TME). A hallmark of the TME is dysfunctional vascularization and impaired perfusion, which hinder effective drug delivery and promote hypoxia-induced metabolic reprograming in both cancer and immune cells. As the TME imposes intense metabolic stress through nutrient competition and lactate-driven acidification – both of which activates immunosuppressive pathways, targeting the TME itself may be beneficial in enhancing the efficacy of immunotherapy. Here we will briefly discuss the potential of targeting the metabolism of the TME as a means to promote normalized tumor vascularization and/or enhance anti-tumor immune responses.
1 Introduction
Cancer is a group of more than 100 diseases acquired by cellular defects, resulting in several hallmark features, such as uncontrolled cell growth, resistance to apoptosis, immune evasion, as well as metabolic dysregulation and metastasis (1). Approximately 90% of all cancers form tumors, which are composed of cancer cells, stromal cells and tissue-resident and infiltrating immune cells (2). The composition of the tumor, which includes all cellular and acellular factors is referred to as the tumor microenvironment (TME) (3–5). Cancers can arise in virtually all tissues of the body and is driven by a wide range of different inherited and acquired mutations, resulting in immense heterogeneity (6–8). At their core cancers are characterized by the loss of proliferative control, typically caused by the inactivation of tumor suppressor genes or the hyperactivation of oncogenes. Due to their role in regulating growth and cell cycle progression loss-of-function mutations in tumor suppressor genes results in loss of proliferative control, whereas activation of oncogenes promote uncontrolled growth (9–11). Most commonly, cancer is caused by mutations in genes encoding p53, PIK3CA, FAT4 and KRAS, with p53 mutations being observed in more than 50% of cancers (12, 13). Moreover, single-cell sequencing and spatial transcriptomics have further revealed that heterogeneity exists within the tumor itself (14–16).
The rapid rate of proliferation of cancer cells has been exploited therapeutically for decades. Chemotherapeutic drugs and radiotherapy preferentially target rapidly proliferating cells by either inducing DNA damage or by blocking central pathways involved in DNA replication (17). The rapid growth coupled with dysfunctional DNA repair boosts the accumulation of mutations, and thus accelerates the rate of cancer evolution. This makes the treatment of cancer especially challenging, as cancer cells not only develop resistance to chemotherapeutic drugs but can also adapt to harsh environmental conditions in the TME, such as hypoxia and nutrient deprivation, further enhancing their survival (18, 19). Although sequencing of cancer cells is increasingly used to predict treatment efficacy, primary and acquired therapy resistance still prevents efficient treatment of cancer in many patients (20). Moreover, as most cancer drugs work by targeting rapid proliferation, this also affects healthy cells with a high proliferation rate, often resulting in hair loss, gastrointestinal distress and immune suppression, among others (17). Together, this highlights the need for additional cancer targeting strategies. The ability to acquire resistance to treatment indicates that a single target is most likely insufficient to efficiently to adequately treat cancer patients. The identification of non-redundant pathways may result in a synergistic effect, thus requiring lower doses and potentially reducing adverse effects. This has been proven safe and effective in treatment of hypertension, and in vivo models suggest this also has the potential in cancer immunotherapies (21–23).
2 The immune system
The immune system is a complex, interactive network of defense and surveillance mechanisms, comprising physical barriers such as the skin and mucosal surfaces, as well as specialized lymphoid organs, immune cells and molecules. Together, these components work in concert to protect the host from non-self, including pathogens. The active immune system is divided into two core responses, namely the innate and the adaptive immune systems, which are fundamentally differentiated by their speed, precision, and capacity to resolve infections in addition to differ in their ability to develop immunological memory (24, 25). The innate immune system serves as the first line of defense, continuously surveilling the body for general signs of infection or damage by recognizing conserved molecular patterns - pathogen association molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) (26). Upon detection of foreign or damaged self, cells of the innate immune system are rapidly activated and recruited from the circulation within minutes to hours, eliciting a response that can be sustained for several days (25). Although the innate immune system lacks immunological memory, professional antigen-presenting cells (APCs) can also induce activation of the adaptive immune system, composed of B and T lymphocytes that can maintain memory to specific pathogens lasting decades (25, 27–29). This task is mainly performed by type 1 macrophages (M1), dendritic cells (DCs), and B lymphocytes. APCs capture antigens, process them, and present resulting antigenic peptide fragments via their major histocompatibility complex class II (MHC II) molecules, expressed on their surface to activate adaptive immune cells in lymphoid tissues (25, 30, 31). While MHC II molecules are exclusively expressed by APCs, all nucleated cells express MHC class I (MHC I) molecules, which present endogenous antigenic peptides to enable immune surveillance and thereby the elimination of infected or abnormal cells (30, 31). For T cells, antigenic peptide-MHC complexes are recognized by the T cell antigen receptor (TCR), which can discriminate between self- and non-self-molecules with remarkable specificity (31–33). Forming an essential part of the TCR is the CD3 complex, functioning as an intracellular signaling hub that translates extracellular antigen recognition into downstream signaling events that drive T cell activation to differentiation, effector function, and clonal expansion (33).
T cells are broadly classified into two helper and cytotoxic T cells, designated CD4+ helper (Th) and CD8+ cytotoxic T cells (CTLs), respectively (24). CD4+ T cells recognize antigens presented on MHC II molecules and play a central role in regulating and coordinating immune responses. The CD4+ T cells can be broadly categorized into CD4+ effector cells, orTh cells, and regulatory T cells (Tregs). While CD4+ effector T cells are crucial for clearing infection and repairing tissue damage, Tregs are responsible for preventing excessive tissue damage and autoimmunity, striking a balance to maintain a functional and healthy immune environment (34–37). In contrast to CD4+ cells, CD8+ CTLs recognize antigens presented on MHC I molecules, and are responsible for directly eliminating target cells through the release of effector cytokines or cytotoxic granules (38). Although most effector T cells, including both CD4+ and CD8+ subsets, have a transient lifespan, a small fraction differentiates into memory T cells, thereby ensuring long-term immune surveillance against the same antigen (39).
3 Cancer immunotherapy
The immune system’s intrinsic role in defending against non-self has fueled the longstanding hypothesis that immune cells can recognize cancer cells as foreign and thereby be weaponized to eliminate them. Indeed, the antigenic composition of tumors differs significantly from that of their non-transformed tissue counterparts, a distinction largely driven by their genetic instability – a core hallmark of cancer (40–42). The concept of leveraging the immune system to combat cancer, now known as immunotherapy, was first systematically introduced in the 1890s by William B. Coley, who documented several cases of spontaneous remission after administering a cocktail of killed bacteria and their products to stimulate the immune system in patients with inoperable cancer (43). What began as a foundational discovery led to decades of rigorous research in cancer immunology, ultimately positioning immunotherapy as the fourth cornerstone of cancer treatment alongside surgery, radiotherapy and chemotherapy (44). Immunotherapy now encompasses a wide variety of treatments, including immune checkpoint therapy (ICT), which will be the focus of this review, adoptive cell transfer (ACT), such as chimeric antigen receptor (CAR) T cells, as well as engineered antibodies (reviewed extensively in (45)).
The concept of immunotherapy builds on the ability of controlling aberrant antigen-induced immune cell activation (46). T cell stimulation through the TCR/CD3 complex requires concurrent perturbation of co-receptors. Whereas the initial interaction of the TCR with the MHC molecule secure antigen-specificity, co-receptor signaling is essential for tuning the activation process. This tuning depends on a balance between activating and inhibitory signals induced by perturbation of cell surface receptors with distinct functions. This is exemplified by the interplay between CD28 and the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) molecules, which compete for binding of CD80 and CD86 expressed on APCs (33). Whereas stimulation of the CD28 molecule provides a positive signal, CTLA-4 engagement delivers an inhibitory signal, thereby modulating the magnitude of initial T cell activation (33, 47). Additionally, programmed cell death receptor 1 (PD-1), also referred to as CD279, is activated by PD ligand-1 or 2 (PD-L1/2) and downregulates T cell activity. In line with this, PD-1 stimulation has an important role in regulating immunological tolerance and is vital in preventing autoimmunity and collateral tissue damage (48–51). Additionally, inflammatory cytokines elicited by the inflammatory process are essential signaling molecules that drive a productive T cell response and facilitate memory formation by shaping the activation of specific differentiation pathways within the cell (52–54).
It is now well established that cancer cells can trigger immune responses. This phenomenon is demonstrated by the utilization of tumor-infiltrating lymphocyte (TIL) therapy, where T cells are isolated from resected tumors,expanded ex vivo, then transferred back to the patient (55, 56). However, they also evolve mechanisms enabling them to evade immune detection and destruction, making immune evasion a defining hallmark of cancer (57–59). During cancer evolution, these mechanisms are continuously sculpted under the selective pressure of immunosurveillance, a phenomenon known as immunoediting (59). In this process, as patrolling immune cells selectively eliminate highly immunogenic cancer cells, they simultaneously impose a selection pressure that favors the survival and expansion of rare subclones with immune-evasive traits. Over time, these subclones can adapt, proliferate, and ultimately become immune-resistant (59). Consequently, from the earliest stages of tumor development, the immune system edits tumor immunogenicity, leading to the emergence of an immunoedited tumor dominated by cancer cell variants that have successfully evaded immune control.
Immune evasion can occur through multiple, non-mutually exclusive mechanisms (reviewed in (59) and (60)), with loss of tumor antigen presentation being one of the most well-characterized, allowing cancer cells to effectively hide in plain sight. This can result from defects in the machinery responsible for antigen processing and presentation, such as downregulation or loss of MHC I molecules on the cell surface, a phenomenon observed in 40-90% of cancers which shields cancer cells from recognition and elimination by CTLs (61, 62). In blood cancers, this “invisibility cloak” has been successfully targeted through the development of monoclonal antibodies or CAR T cells, another form of ACT, that recognize and bind tumor-specific surface antigens independently of the MHC I receptor (63). However, these approaches have shown limited efficacy against solid tumors, which, as mentioned, account for approximately 90% of all cancers (2).
In addition to avoiding detection from the immune system, cancers can also suppress the effector functions of anti-tumorigenic immune cells. A key example is the co-option of the PD-1 pathway by cancer cells (48, 49, 51). In addition to activated T cells, PD-1 expression is also detected on B cells and natural killer (NK) cells. In all three cell types, PD-1 expression is normally decreased when an inflammatory response is resolved during acute antigen clearance (50, 64). However, in cases of persistent antigen exposure, such as cancer and chronic infections, PD-1 expression remains elevated, which contributes to T cell exhaustion (48, 51, 65, 66). The two PD-1 ligands, PD-L1 and PD-L2, exhibit distinct expression patterns. While PD-L2 is predominantly expressed on APCs in lymphoid tissues, PD-L1 is broadly expressed across hematopoietic (e.g., T cells, B cells, macrophages) and non-hematopoietic cells (e.g., endothelial cells) (51, 67). PD-L1 is upregulated by pro-inflammatory cytokines, particularly interferon-γ (IFN-γ), as a feedback mechanism to tune down immune activity (50, 68). However, in many solid tumors, PD-L1 expression is aberrantly elevated within the TME due to constitutive oncogenic signaling or as an adaptive response to inflammatory cues (51, 65).
To restore T cell activity in anti-tumor immune responses, antibodies targeting co-inhibitory receptors and their ligands have been developed (48, 51, 69, 70). These include, but are not limited to, immune checkpoint inhibitors targeting the CTLA-4 and PD-1 pathways, which have demonstrated clinical efficacy in certain cancers and patient subsets (70). However, as many as 60-80% of patients with solid tumors either fail to respond or experience only transient benefits from ICT, highlighting the diverse mechanisms tumors employ to evade the immune system (71, 72). In solid tumors, many of these mechanisms are driven by the TME, a complex and dynamic ecosystem encompassing cellular, physical and chemical components that are continuously restructured and manipulated throughout tumor progression. In addition to cancer cells, the cellular constituents of the TME include diverse non-malignant stromal cells along with tissue-resident and infiltrating immune cell populations that can be reprogrammed to support tumor growth (5, 73–75). In addition to cellular components, non-cellular components – such as the extracellular matrix (ECM), metabolites, soluble signaling molecules, and the surrounding hypoxic and acidic milieu – play important roles in tumor progression and therapy resistance (5, 73–75). Targeting the TME and its diverse components to enhance immunotherapy holds significant therapeutic promise, as many of its defining features are conserved across a range of tumor types (5, 75). In this review, we will provide a brief overview of key immunosuppressive hallmarks of the TME and discuss their potential as therapeutic targets to enhance immunotherapy in cancer, with a particular focus on ICT.
4 Cellular metabolism and metabolic reprogramming
Over the last decades it has become clear that extracellular metabolites are crucial for optimal T cell activation. Nutrients and their metabolites exhibit significant interplay with the three core activation signals while also independently influencing the functional polarization of T cells. Consequently, metabolic inputs have been proposed as a novel dimension necessary for licensing the T cell immune response (reviewed in (76) and (77)). The connection between cellular function and metabolic phenotype in health and disease is therefore becoming increasingly evident across multiple fields, including immunology, cancer, and tumor angiogenesis (41, 52, 78–83). While energy and biomass production remain critical, recent insights explain how metabolism affects diverse processes such as activation, proliferation, migration and differentiation.
Although metabolic reprograming has only recently emerged as important in these processes, it has been known in cancer cells for close to a century. As early as in the 1920’s, Otto Warburg discovered that cancer cells, despite the presence of oxygen, preferentially rely on glycolysis and lactate production rather than mitochondrial respiration, a metabolic reprogramming now known as “The Warburg effect” (Figure 1) (84, 85). Cancer cells frequently upregulate glucose transporters and key enzymes in the glycolytic pathway, including Glucose transporter type 1 (GLUT1), Hexokinase 2 (HK2), 6-phosphofructokinase 2/fructose 2,6-bisphosphatase 3 (PFKFB3), pyruvate kinase muscle form 2 (PKM2) and lactate dehydrogenase A (LDHA) (86). Together, these proteins facilitate a rapid turnover of glucose and a subsequent increase in lactate production. Although this metabolic phenotype was originally believed to be caused by a mitochondrial defect in cancer cells, it is now recognized that the mitochondria remain functional and possess remarkable metabolic plasticity. This flexibility enables them to dynamically utilize a wide range of substrates, including glutamine and fatty acids to fuel diverse cellular processes (Figure 1) (87–89).
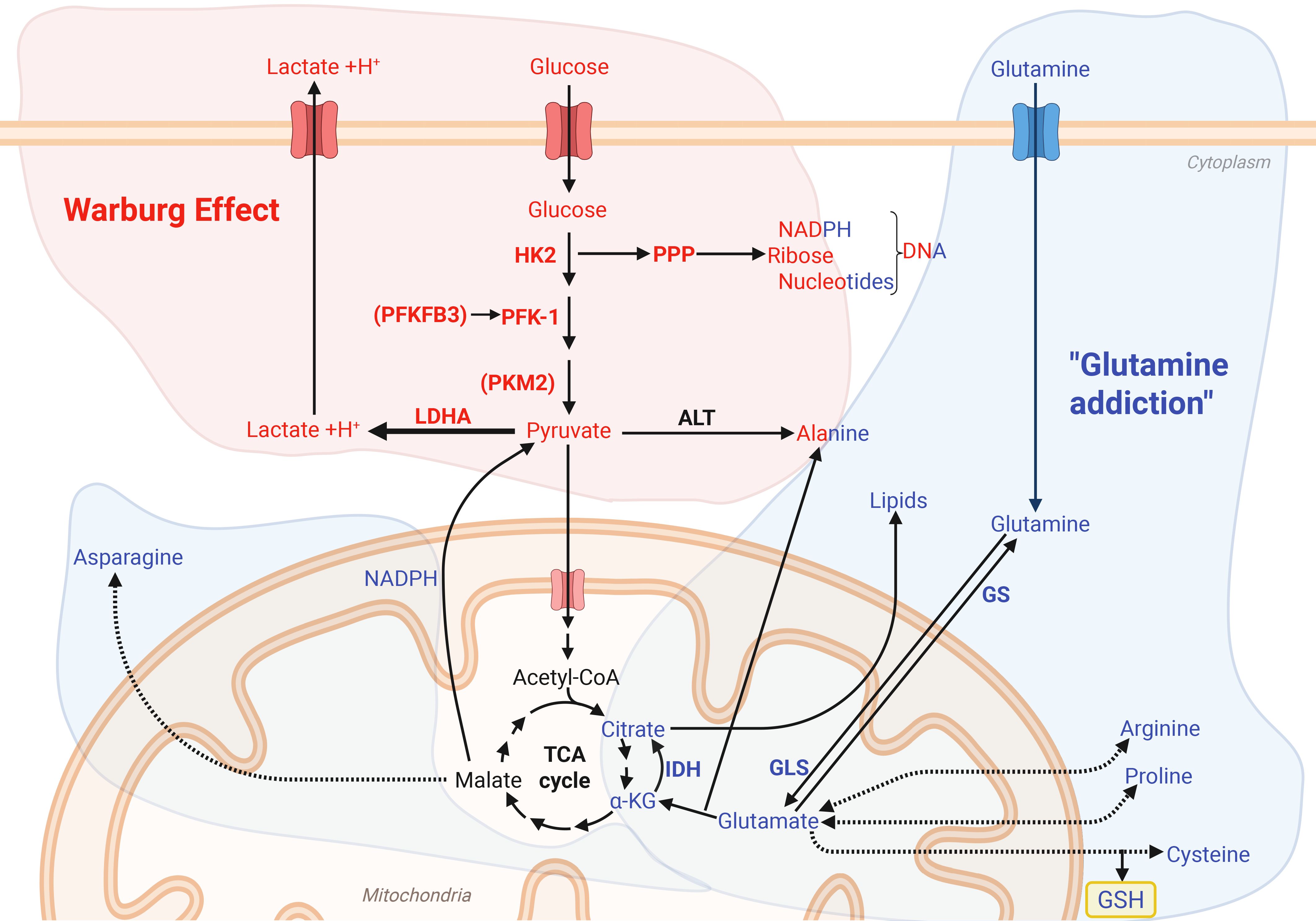
Figure 1. The Warburg effect and glutamine metabolism drives energetic and biosynthetic pathways. The Warburg effect (84, 85) metabolizes glucose into lactate despite the presence of oxygen, as well as providing important glycolytic metabolites which support redox homeostasis through the pentose phosphate pathway (PPP) and the carbohydrate backbones for DNA and RNA synthesis. Glutamine addiction (blue) describes the reliance on glutamine to provide precursor for amino acid synthesis, fueling the TCA cycle for regeneration of NADPH and generation of reduced glutathione (GSH). Figure was made using BioRender.
This metabolic plasticity may also provide resistance to metabolic inhibitors, as demonstrated by Boudreau et al. (90), where the glycolytic pancreatic cancer cell line MIA PaCa-2 adopted an oxidative phenotype after long-term exposure to an inhibitor of lactate production. In line with this, glutamine reliance, a phenomenon sometimes referred to as “glutamine addiction”, is observed in several cancer cell lines (83, 87, 88, 91, 92). Although most cells are capable of synthesizing glutamine, rapidly proliferating cells rely on additional extracellular sources, so it is sometimes referred to as a conditionally essential amino acid (88, 91, 93). Interestingly, glutamine deprivation has been shown to reduce the rate of glycolysis by regulating both the expression and activity of key glycolytic enzymes (94, 95). Furthermore, the expression of the enzyme glutaminase 1 (GLS1), which is responsible for the deamidation of glutamine to glutamate, is upregulated in many cancer cell lines and correlates with decreased survival in patients (96). Indeed, Reinfeld et al. (97) showed that myeloid cells and T cells have a higher capacity for glucose uptake than cancer cells, while cancer cells have a higher capacity for glutamine uptake. The enzymes isocitrate dehydrogenase (IDH) 1 and 2 are also important in the TCA cycle, as they catalyze the formation of α-ketoglutarate from isocitrate (98). This supports redox homeostasis and lipid synthesis, along with providing α-ketoglutarate which can regulate epigenetics or be used as a backbone for glutamate and glutamine synthesis (98–100). In cancers, mutations in IDH1 or IDH2 can result in the formation of D-2-hydroxyglutarate (2-HG) (98). Some cancer cells also rely on fatty acid oxidation, a trait which is associated with upregulated expression of the mitochondrial fatty acid transporter carnitine palmitoyl transferase 1 (CPT1) in certain tumors (89).
Metabolic plasticity and reprogramming are important for both cancer and immune cells. An increasing body of evidence demonstrates that, in some contexts, healthy cells - including immune cells and endothelial cells - can adopt similar metabolic phenotypes (101–103). Indeed, immune cells, including macrophages and T cells, undergo metabolic reprogramming upon activation (81). Macrophages, which are traditionally classified into pro-inflammatory M1 or anti-inflammatory, wound-healing M2 subtypes, adapt to a glycolytic and oxidative metabolic program, respectively (Figure 2) (104–109). Similar to cancer cells, M1 macrophages upregulate PFKFB3 to boost the rate of glycolysis (104). In line with this, deletion or inhibition of PFKFB3 in macrophages is shown to result in reduced secretion of pro-inflammatory cytokines, including IL-1β, IL-6 and tumor necrosis factor following stimulation with lipopolysaccharide. Moreover, mice with a myeloid-specific PFKFB3 deficiency exhibit increased survival in a murine sepsis model, as well as increased lymphangiogenesis following myocardial infarction (110, 111). Additionally, the tricarboxylic acid (TCA) cycle is used by M1 macrophages to produce succinate, which stabilizes hypoxia-inducible factor 1 α (HIF-1α), and citrate, which serves as a precursor for fatty acid synthesis and the antimicrobial metabolite itaconate (112, 113). In contrast, M2 macrophages utilize glutamine and fatty acids as substrates for the TCA cycle, fueling adenosine triphosphate (ATP) production through oxidative phosphorylation (114). Additionally, they metabolize tryptophane via the enzyme Indoleamine 2,3-dioxygenase (IDO) to generate the anti-inflammatory metabolite kynurenine (115, 116). IDO1 activity suppresses T-cell activity by depleting tryptophan, which is required for Th1 and CD8+ T-cell-mediated immunity, while kynurenine binds to the aryl hydrocarbon receptor (AHR), which directly activates Treg differentiation and activity, resulting in reduced anti-tumor responses (116–118).
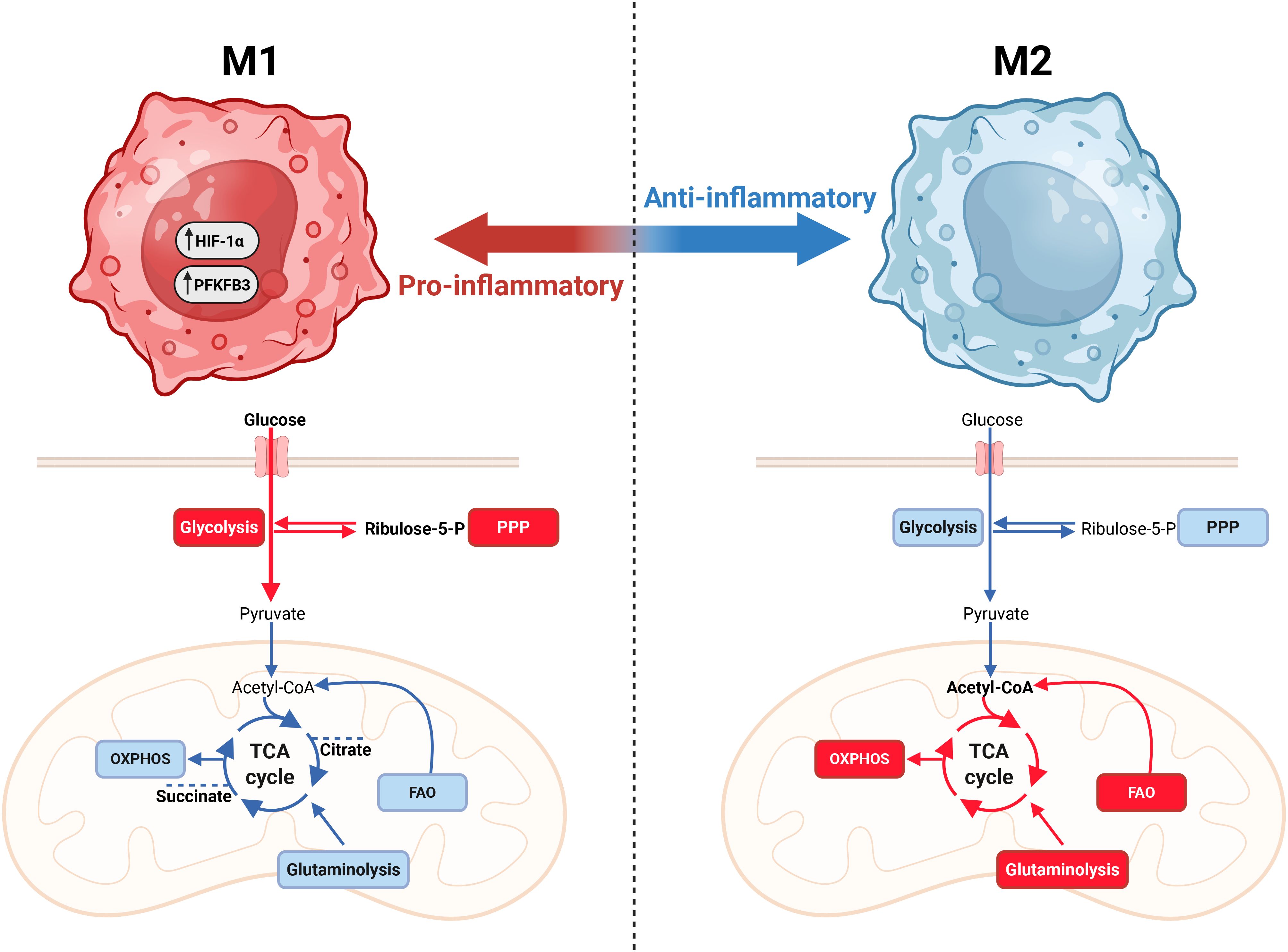
Figure 2. Macrophage metabolism is closely linked with effector function. Macrophages can polarize into pro-inflammatory (M1-like) and anti-inflammatory (M2-like) phenotypes. M1 macrophages are characterized by a high reliance on glycolysis and a less oxidative phenotype. Additionally, M1 macrophages utilize the TCA cycle to produce citrate and succinate to drive stabilization of HIF-1α to aid pro-inflammatory effector functions. The anti-inflammatory, wound-healing M2-like macrophages are less glycolytic and more oxidative, utilizing glutamine and fatty acid oxidation (FAO). Figure was made using BioRender.
Metabolic reprogramming is also important for T cells, with different T cell subsets adopting distinct metabolic adaptations (Figure 3) (103, 119, 120). In their naïve state, T cells primarily rely on oxidative phosphorylation to produce ATP, maintaining a relatively low metabolic rate prior to activation. However, upon activation through perturbations of the TCR/CD3 complex in conjunction with CD28, their metabolic rate rapidly increases (121, 122). This process is aided by the presence of abundant mRNA encoding key glycolytic enzymes, particularly HK2, along with the availability of idle ribosomes, enabling the rapid production of proteins upon upregulation (121). Additionally, activation of Akt, also known as protein kinase B, quickly upregulates the cytosolic localization of GLUT1 required to increase glucose uptake (123). Chang et al. (124) demonstrated the metabolic plasticity of T cells by replacing glucose with galactose. Despite their inability to metabolize galactose through glycolysis, T-cell proliferation is reportedly unaffected by replacing glucose with galactose, however glucose deprivation resulted in decreased production of the cytokine interferon-γ (IFN-γ), as idle glyceraldehyde 3-phosphate dehydrogenase (GAPDH) bound the IFN-γ mRNA and prevented its translation (124) In addition to glucose, T cells also require glutamine to become fully activated, and glutamine deprivation or inhibition of GLS1 reduces both proliferation and cytokine secretion in CD4+ T cells (125–128). In fact, we recently demonstrated that, similar to cancer cells, glutamine deprivation in CD4+ T cells also regulate glycolysis (129). Furthermore, CD4+ T cell subsets also adapt to distinct metabolic profiles (Figure 3), as previously reviewed in detail (130)), highlighting the close relationship between metabolism and functionality in T cells.
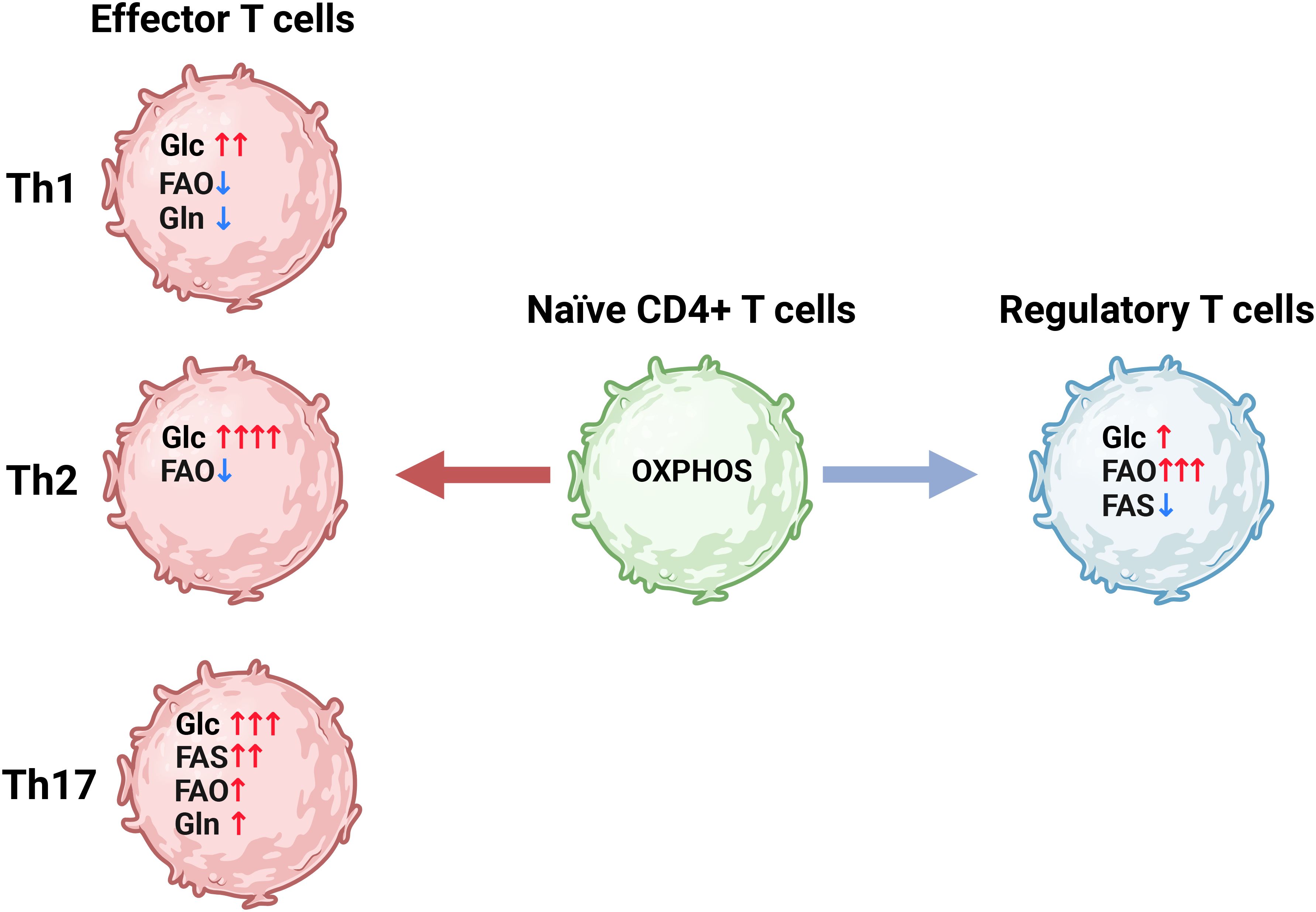
Figure 3. CD4+ T cell differentiation is correlated with distinct metabolic programs. Naïve CD4+ T cells (green) have a relatively low metabolic rate driven mainly by oxidative phosphorylation (OXPHOS). Upon activation the naïve CD4+ T cells differentiate into various subsets of effector CD4+ T cells (Th) (red) which utilize distinct metabolic programs characterized by differential reliance on glycolysis (Glc) glutamine metabolism (Gln) fatty acid oxidation (FAO) and fatty acid synthesis (FAS). Alternatively, CD4+ T cells can differentiate into regulatory T cells (Tregs), which are less glycolytic and rely more on FAO to fuel ATP production compared to the effector CD4+ T cells. Figure was made using BioRender.
Metabolism in endothelial cells has also been extensively studied due to their critical role in angiogenesis. Importantly, deciphering metabolism in these cells has led to the identification of therapeutic targets with the potential to regulate angiogenesis (reviewed in (131)). It is known that glycolysis may account for up to 85% of the ATP production in endothelial cells despite sufficient levels of circulating oxygen (102). Notably, the rate of glycolysis can be upregulated in response to pro-angiogenic and inflammatory factors (102, 132–135). In fact, upregulation of glycolysis through increased expression of PFKFB3 is a strong driver of tip cell formation in vessel sprouting (102, 135). However, the proliferating tip and stalk cells, which are the building blocks of sprouting angiogenesis, rely not only on glycolysis, but also on fatty acid oxidation to produce dNTPs for DNA synthesis, and on glutamine metabolism to fuel the TCA cycle, thereby driving vessel propagation (Figure 4) (93, 101, 136).
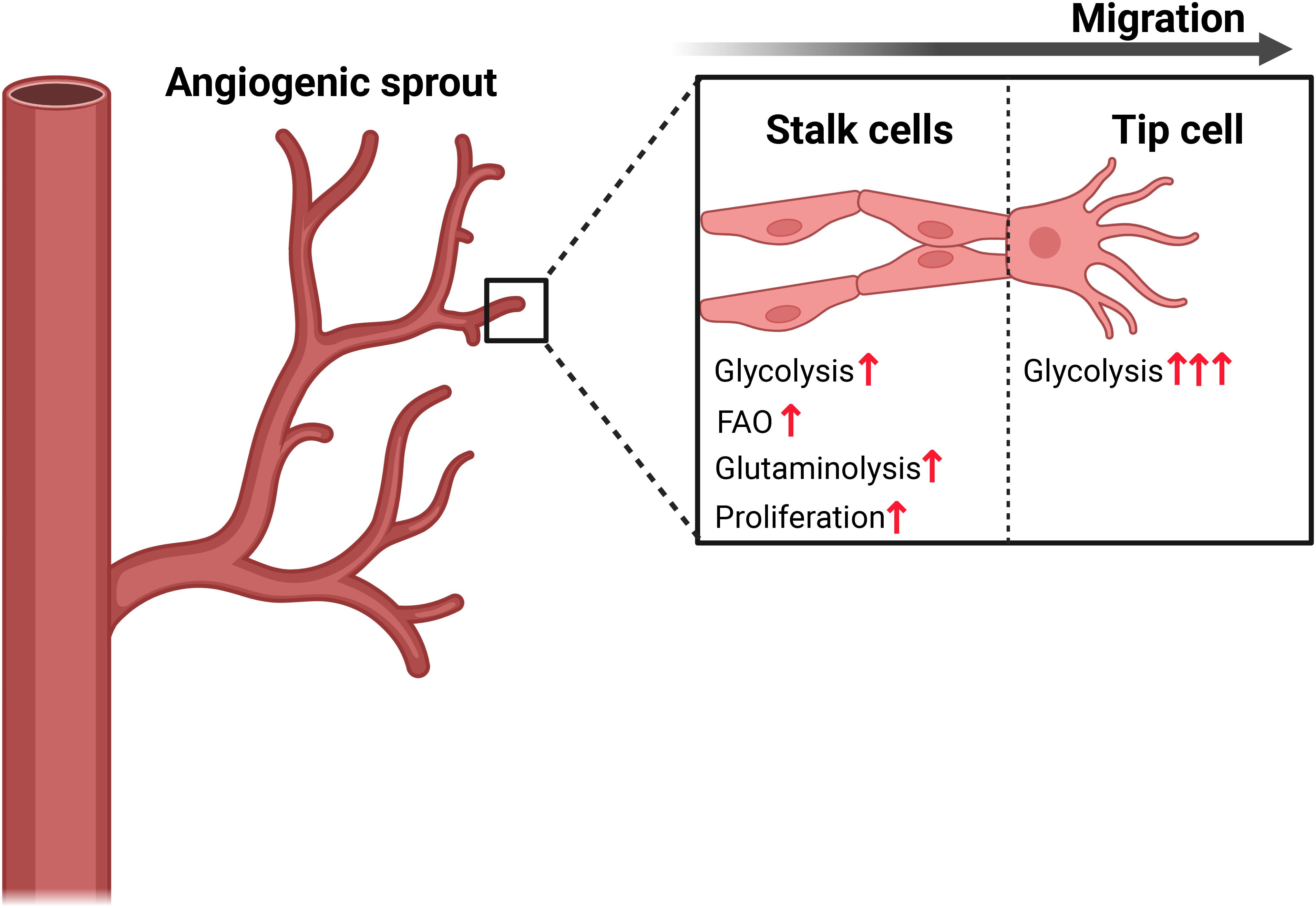
Figure 4. Endothelial cell metabolism drives migration and proliferation in vessel sprouting. The highly migratory endothelial tip cell is characterized by a higher rate of glycolysis driven by expression of PFKFB3, while the stalk cells rely on a combination of glycolysis, fatty acid oxidation (FAO) and glutaminolysis to provide building blocks required for rapid biomass proliferation. Figure was made using BioRender.
Despite the differences in metabolic profiles, most of the regulatory mechanisms are shared and conserved across cell types. Among these are mammalian target of rapamycin complex 1 and 2 (mTORC 1 and 2), HIF-1α and c-MYC (137–140). mTOR is a serine/threonine kinase that functions as part of two distinct complexes: mTORC1 and mTORC2. mTORC1 is an important regulator of anabolic metabolism, including protein and lipid synthesis, while also supporting catabolic processes by enhancing glycolysis through stabilization of HIF-1α and inducing enzymes responsible for glutaminolysis via the transcriptional activity of c-MYC (141). mTORC2 is associated with cell survival and fine-tuning of metabolic activity. It promotes fatty acid oxidation by regulating the transcription factor Forkhead box 01 (FOXO1) and enhance glycolysis through activation of Akt (142).
Stabilization of HIF-1α induces glycolytic metabolism through upregulating expression of GLUT1, HK, PFKFB3, PKM2, LDHA and Monocarboxylate transporter 4 (MCT4) (104, 143–146). Although HIF-1α is primarily stabilized by the absence of oxygen, several mechanisms can also promote its stabilization in the presence of oxygen, including the regulation by mTORC1 (141, 142). In cancer, HIF-1α stabilization is associated with a dismal prognosis for patients (147, 148). However, it also plays an important role in the functional activation of immune cells, highlighting its dual role in both promoting cancer cell growth and supporting anti-tumor immunity [reviewed in detail (149)].
5 The tumor microenvironment drives immune suppression
The TME is generally poorly vascularized, with dysfunctional and leaky blood vessels resulting in decreased availability of nutrients, hypoxia and acidification that collectively contribute to immune suppression through several mechanisms (Figure 5). Whereas hypoxia is associated with increasing tumor mass, acidification is associated with reprogrammed cancer cells producing lactate. Tumor hypoxia induces stabilization of HIF-1α which, as mentioned, drives glycolysis and is hence responsible for the lactate production (4, 5, 74, 75, 97, 150). Lactate accumulation is known to restrict T-cell proliferation by disrupting the redox homeostasis and inhibiting GAPDH activity (151). Interestingly, T cells produce acidic niches within lymph nodes to restrict their own effector functions to avoid hyperactivation, demonstrating the physiological importance of lactate (152). In addition to acidification, lactate is known to induce histone modification referred to as lactylation, which is associated with enhanced polarization of M2 cells, suppressed T cell effector functions and increased Treg differentiation, thereby supporting an anti-immune and pro-tumorigenic phenotype (153–155). In line with this, reduced lactylation has been associated with a favorable outcome in patients with solid tumors (153, 156). In addition to regulating glycolysis, HIF-1α stabilization can also act immunosuppressive by inducing the expression of PD-L1 in DCs, macrophages and myeloid derived suppressor cells (149). The lactate-induced acidification has also been shown to reduce the efficacy of immune checkpoint inhibition by influencing the binding properties of antibodies targeting PD-L1 (157). The hypoxia-lactate axis also contributes to the formation of dysfunctional blood vessels, together favoring tumor metastasis and repression of immune cell infiltration (158, 159).
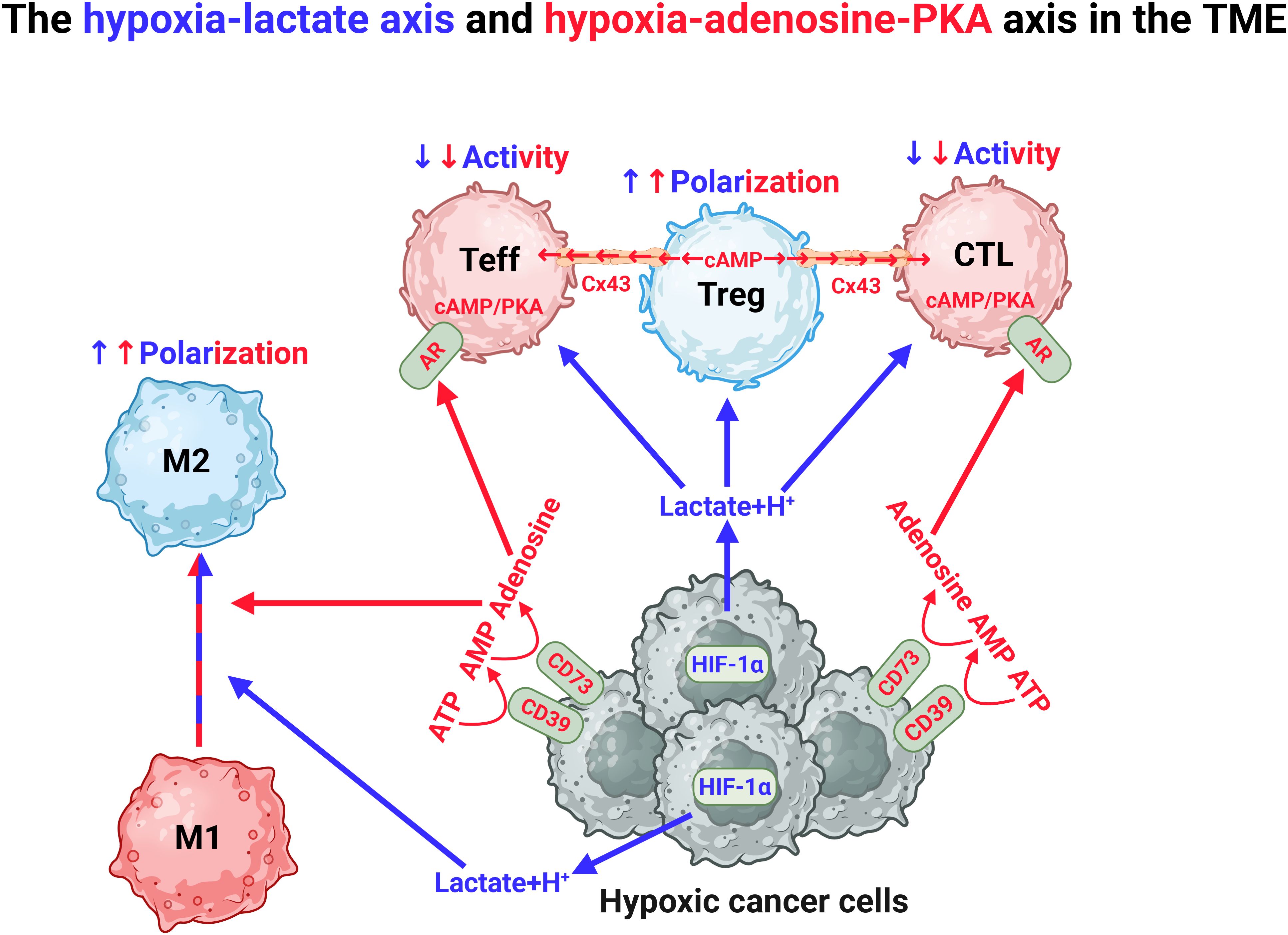
Figure 5. Hypoxia in the TME drives immunosuppression through the hypoxia-lactate axis and the hypoxia-adenosine-PKA axis. The TME is characterized by the presence of hypoxia. This drives the production of lactate, which induces polarization of anti-inflammatory M2 macrophages and Tregs. Hypoxia also induces expression of CD39 and CD73 and stepwise production of adenosine, which stimulates adenosine receptors (AR) that produce endogenous cAMP in CD4+ effector cells (Teff) and cytotoxic lymphocytes (CTL). Cyclic AMP is also produced by Tregs which transfer this to Teffs and CTLs through the junction protein connexin 43 (Cx43). In both situations, endogenous cAMP will induce PKA activation leading to inhibition of proinflammatory function of Teff and CTL. Figure was made using BioRender.
Tumor hypoxia also induces expression of CD39 and CD73, which together catalyzes the formation of adenosine from adenosine monophosphate (AMP) and ATP. Adenosine exerts its effects through ligation of the 4 subtypes of purinergic adenosine receptors (A1, A2A, A2B, and A3) (160, 161). The ARs differ in their affinity for adenosine, but are all coupled to various cellular signaling pathways through G-protein coupled receptors (GPCRs). To this end, A2AR and A2BR are upregulated in response to hypoxia and anergic signaling in T cells and induce immune suppression by activating adenylate cyclases (ACs), that initiate the synthesis of intracellular cyclic AMP (cAMP), which in turn activates the cAMP-dependent protein kinase A (PKA) (161). PKA is a holoenzyme consisting of a regulatory (R) subunit dimer and two catalytic (C) subunits. While the R subunits are encoded by four separate genes (PRKAR1A and -B, PRKAR2A and -B), the catalytic subunits are primarily encoded by two major genes, PRKACA and PRKACB, which give rise to several tissue- and cell-specific splice variants, including immune cell-specific Cβ2 (162, 163). In addition to Cβ2, the splice variants Cα1 and Cβ1 are expressed in immune cells. The prevailing dogma is that PKA activation suppresses both early and late phases of T cell activation, including proliferation and clonal expansion, the latter mediated by downregulation of IL-2 production (164, 165). In line with its inhibitory role, PKA also promotes differentiation into Tregs and Th2, which favors the tumor, while repressing effector functions of Th1, Th17 and CD8+ T cells (166, 167). Additionally, Tregs can directly induce PKA activation by transporting cAMP into target T cells through the gap-junction protein connexin 43 (Cx43) (168). As a result, in the TME, PKA activation drives T cell exhaustion, in conjunction with upregulation of PD-1 and CTLA-4 expression (72). Recently, we showed that deletion of the immune-specific PKA Cβ2 significantly suppressed tumor growth and enhanced survival in a murine metastatic cancer model (169). This was associated with increased infiltration of pro-inflammatory Th1, Th9 and Th17 cells into the tumors (169). Moreover, in 2020, Na et al. demonstrated that knockout of PKA Cβ in macrophages prevents M2 polarization and that liposomal delivery of PKA inhibitors to tumor-infiltrating macrophages enhances the therapeutic efficacy of anti-CTLA-4 antibodies, effectively counteracting breast cancer tumor growth and metastatic potential in mice (170). Together this suggest that Cβ may convey signals supporting a proinflammatory phenotype. In support of this, mice that are ablated for Cβ2 are prone to develop autoimmunity, a phenotype also reflected in upregulation of proinflammatory immune cells (171).
In addition to the hypoxia-lactate-adenosine axis, some cancer mutations result in metabolic phenotypes that contribute to an immunosuppressive TME. Cancer cells with IDH1 or IDH2 mutations cause accumulation of 2-HG, which supports the cancer cells by maintaining a stem-like phenotype, while suppressing T-cell- and macrophage-mediated immune activity (172). 2-HG is taken up by CD8+ T cells, where it destabilizes HIF-1α and acts as an inhibitor of LDH, resulting in reduced chemotaxis, cytotoxic activity and production of IFN-γ (172–174).
6 Targeting the tumor microenvironment
As the TME exerts broad immunosuppressive characteristics, targeting the TME also offers the potential for new therapies. As mentioned, the hypoxia-lactate axis and the hypoxia-adenosine-PKA axis are known to inhibit the immune system. Thus, targeting hypoxia directly by hyperbaric oxygen (HBO) treatment has been proposed (175). Although this approach is reported to enhance immune activity and reduce growth of pulmonary tumors in a mouse model (176) a recent meta-analysis found overall weak evidence that HBO treatment alone improves long-term survival (176) and its use in humans remains limited due to the lack of high-quality studies (177). Although HBO treatment might not be effective as a cancer treatment, targeting the downstream effects of hypoxia on metabolism, PKA activity and angiogenesis may still have therapeutic benefits. Moreover, targeting IDH1 and IDH2 have been explored. This is due to the fact that the cancer-specific mutation of IDH has potential to be targeted without affecting the non-mutated IDH isoforms in healthy cells (178).
6.1 Targeting tumor vascularization
The hypoxia-angiogenesis-axis has also been explored as a therapeutic strategy, which has led to the approval of monoclonal antibodies targeting VEGF for use in some cancers, including renal and colorectal cancer (179, 180). When used in combination with chemotherapy, VEGF blockade has been shown to increase progression free survival in many types of cancer (181). The combination of anti-angiogenic drugs with ICT has been proposed as a strategy to increase immune cell infiltration and improve therapeutic efficacy (182). It is also hypothesized that rather than blocking angiogenesis, it might be more beneficial to normalize the tumor vasculature to enhance vascular integrity and improve tumor perfusion (3). However, although VEGF is the main driver of angiogenesis, it is well established that additional, not yet fully understood mechanisms can also contribute to this process (131). These VEGF-independent pathways may aid resistance to VEGF-targeted therapies, highlighting the need to identify and target alternative pro-angiogenic signals.
The Notch pathway, which is known for regulating angiogenic and inflammatory pathways in endothelial cells, is of interest in targeting pathological angiogenesis [reviewed in ref (183)]. Notch signaling is induced when one of the four Notch receptors (1–4) is activated by binding to a ligand from the Delta-like (DLL1, DLL4) or Jagged (Jag1, Jag2) families (183). The Notch pathway is traditionally viewed as anti-angiogenic, and its inhibition leads to increased angiogenesis (102). However, we recently demonstrated that blocking Jag1 resulted in an upregulation of DLL4, which we and others, have reported reduces expression of VEGFR2 (184–186). Jag1 blockade was further associated with a reduction in M2-like macrophages, which might be also enhance the immune function in the TME as well (185).
Targeting endothelial cell metabolism has been proposed as a strategy to bypass resistance mechanisms, an approach that Treps et al. (131, 187) describe as “targeting the engine of angiogenesis”. In accordance with this, targeting endothelial glycolysis, glutamine metabolism and fatty acid metabolism have been explored as therapeutic strategies for pathological angiogenesis (187).
Inhibition of endothelial glycolysis through PFKFB3 blockade has been shown to reduce pathological angiogenesis in several disease models, including tumor neovascularization (132, 135, 188). Both pharmacological inhibition and partial deletion of endothelial PFKFB3 reduced tumor vascularization and metastasis, while also increasing vessel stability, which aided drug delivery and enhanced the effect of chemotherapy in a murine liver cancer model (132). Additionally, it was demonstrated that endothelial PFKFB3 and lactate enhanced polarization of M2-like macrophages in a murine ischemia model, suggesting a potential role in modulating immune responses in cancer as well (189). Lactate accumulation also directly influences angiogenesis by stabilizing HIF-1α and promoting vascularization, while simultaneously reducing vessel integrity through the activation of inflammatory pathways. Moreover, prolonged exposure to lactate drives endothelial dysfunction in the TME (74, 190, 191). Although inhibition of PFKFB3 has also been demonstrated to reduce cancer cell proliferation, the concentrations of the PFKFB3 inhibitor 3-(pyridin-3-yl)-1-(pyridin-4-yl)prop-2-en-1-one (3PO) required to achieve this effect were shown to simultaneously reduce vessel integrity and facilitate metastasis (191–193). Moreover, as there are numerous reports of off-target effects associated with 3PO, further studies are needed to determine the safety and feasibility of targeting PFKFB3 in the context of tumor vascularization (133, 194–196).
Glutamine metabolism also presents a promising target in tumor vascularization, as its importance is shared between the endothelium and the tumor (83, 93, 96, 136, 197). GLS1 inhibition has been shown to be highly effective in reducing endothelial cell proliferation as it provides an important precursor for α-ketoglutarate and the amino acid asparagine (93, 136). This approach holds potential for a synergistic effect because, as mentioned, cancer cells rely on glutamine to fuel the TCA cycle (96). Although GLS1 inhibition also reduces proliferation and cytokine secretion from CD4+ T cells, this may be circumvented by using the GLS1 inhibitor telaglenalstat (CB839). Although CB839 has been shown to have an inhibitory capacity (IC50) in the nanomolar range in sensitive cancers, it appears to inhibit proliferation without inducing apoptosis in endothelial cells in the micromolar range, while we have showed that proliferation of CD4+ T cells is not significantly reduced by concentrations up to 5 micromolar (125, 126). Moreover, CB839 also appears to induce an M1-like phenotype in macrophages, which may further enhance the anti-cancer response of the immune system (198). Additionally, glutamine deprivation or GLS1 inhibition represses glycolysis and lactate production in several cancer cells through upregulation of thioredoxin interacting protein (TXNIP) and phosphorylation of PFKFB3, potentially reducing lactate-induced differentiation of M2 macrophages (94, 95). Further studies will be needed to assess the potential synergistic effects of GLS1 inhibition and immunotherapy.
Fatty acid oxidation is another important pathway in proliferating endothelial cells. As demonstrated by Schoors et al. (101), endothelial cells use fatty acids to produce nucleotides for DNA synthesis, and inhibition of the mitochondrial fatty acid transporter CPT1A reduces proliferation. CPT1A is upregulated in certain cancers and is associated with resistance to induction of apoptosis (199, 200). Moreover, CPT1A is important for driving Treg differentiation (199, 200). CPT1A also seems to drive an anti-inflammatory phenotype, as deletion of CPT1A resulted in increased lung damage in a murine LPS-induced sepsis model (201) while inducing CPT1A expression in the RAW264.7 macrophage cell line reduced expression of iNOS and impaired phagocytotic capacity (202). In line with this, targeting CPT1A has been shown to enhance the effect of PD-1 blockade in a murine lung cancer model (199).
Adenosine, produced in response to tumor hypoxia, is another key driver of angiogenesis, promoting HIF-1α stabilization and VEGF production through ligation of the A2AR (203, 204). Stimulation of A2AR enhances glycolysis in endothelial cells, making the hypoxia-adenosine axis a promising target for modulating both the immunosuppressive and the pro-angiogenic features of the TME (204). The PKA pathway also plays a crucial role in angiogenesis by regulating endothelial cell proliferation, migration and modulating VEGF signaling (203, 205). Another cAMP effector, exchange protein directly activated by cAMP (Epac), contributes to the regulation of angiogenesis by inhibiting γ-secretase, an enzyme required for the intracellular cleavage Notch and thereby activation of the Notch pathway. In line with this, inhibition of Epac has been shown to reduce pathological angiogenesis by enhancing Notch activation and suppressing VEGF signaling (206).
6.2 Targeting lactate production in the TME
Due to the central role of lactate in the TME, targeting its production and transport have been proposed as potential therapeutic strategies (207). Given the extensive study of glycolysis and lactate production (74, 82, 190, 208), a myriad of inhibitors has been developed against key glycolytic enzymes, including LDH, HK, PFKFB3, and PKM2 in addition to the lactate transporters MCT1 and MCT4 (82, 195, 209–211).
When the glucose analog 2-deoxy-D-glucose (2-DG) inhibits HK, glycolysis is completely blocked. Although 2-DG has been shown to reduce proliferation in various cancer cells, its therapeutic potential is limited due to low specificity and hence off-target effects and toxicity – including immune suppression and gastrointestinal distress – together highlighting the need for more precise approaches (212–214).
PFKFB3, which is upregulated in many cancers, has been proposed as a more cancer-specific therapeutic target. This has resulted in extensive research into developing PFKFB3 inhibitors [reviewed in detail in (215)]. Although PFKFB3 is not directly involved in glycolysis, PFKFB3 inhibition reduces lactate production and proliferation in several cancer cell lines (209, 216). However, PFKFB3 expression is also important in immune cells, including M1 macrophages and effector T cells (109, 217). In line with this, treatment with the PFKFB3 inhibitor 3PO alleviated inflammation and reduced mortality in murine sepsis models (110). Paradoxically, PFKFB3 expression is reported to correlate with pro-invasive and pro-inflammatory activity in rheumatoid arthritis patients (218, 219), while selective inhibition of endothelial PFKFB3 reduces polarization of M2 macrophages (189). These contradictory findings stress the need for more research to explore how PFKFB3 inhibition may affect the cancer-immune interaction.
Direct targeting of LDH prevents the production of lactate and results in accumulation of pyruvate and NADH (208). This has been shown to induce oxidative stress and inhibit tumor progression in glycolytic cancers. However, cancer cells can circumvent this effect by rewiring their metabolism towards an oxidative phenotype, indicating that targeting LDH alone is insufficient (90, 220). Still, as reducing lactate levels in the TME might enhance the anti-tumor activity of the immune system, targeting LDHA still holds therapeutic potential. Notably, Renner et al. and Babl et al. (210, 221) showed that blocking lactate secretion enhanced the efficacy of anti-PD-L1 treatment by alleviating the immunosuppressive effects of lactate on T cells. Moreover, it was recently shown that the lactate-induced acidification of the TME also negatively affects the interaction between PD-L1 and anti-PD-L1 antibodies (157). Pilon-Thomas et al. (222) demonstrated that buffering the pH within the TME using sodium bicarbonate enhanced the anti-tumor activity of TILs, indicating that targeting the hypoxia-lactate axis may enhance the efficacy of several forms of immunotherapy.
6.3 Targeting the hypoxia-adenosine-PKA axis
As mentioned, the hypoxia in the TME induces expression of the ectonucleotidases CD39 and CD73, resulting in conversion of ATP to adenosine, which facilitates immune suppression through binding to the adenosine receptors A2AR and A2BR (160, 161). In turn, activation of A2AR and A2BR leads to AC-induced cAMP production and activation of PKA and Epac (223). Endogenous cAMP production is also induced by numerous other receptors, including β-adrenergic receptors, dopamine receptors, and prostaglandin receptors (PGER) such as PGE2R (223). Activation of PKA is known to suppress the nuclear factor of κ-light chain of activated B cells (NFκB) and STAT1 pathways in macrophages, resulting in a shift towards M2 macrophage polarization (72, 170, 224). In T cells, PKA phosphorylates C-terminal Src kinase (Csk), that phosphorylates lymphocyte-specific protein tyrosine kinase (Lck) on tyrosine 505, preventing downstream activation of T-cell signaling, including the NFκB and NFAT pathways (164, 225). This further results in decreased differentiation of CD4+ T cells to Th1 and Th17 cells, while increasing differentiation of Tregs (166, 167). The PKA pathway thus serves as an immune checkpoint, which offers potential as a therapeutic target.
Targeting the adenosine axis through the CD73-CD39-AR pathway has been proposed as a novel approach to immunotherapy, either alone or in combination with existing treatments such as PD-L1 blockade (160, 161). In mouse models, blocking CD39 has been demonstrated to reduce the tumor burden and increase infiltration of immune cells, including DCs and NK cells (161, 226). It is further reported that CD73 blockade, which has also been shown to increase the efficacy of both anti-CTLA-4 and anti-PD-L1 therapies, enhances the efficacy of radiotherapy by promoting the anti-tumor activity of the immune system (227) (228). In line with this, blocking A2AR also boosts anti-tumor activity (229). As these targets are bound to the extracellular side of the cell membrane, they can be targeted not only by small-molecule inhibitors, but also by using therapeutic antibodies, and multiple drug candidates have already entered phase I clinical trials (161). The expression of both CD39 and CD73 is enhanced by tumor-derived lactate, indicating that targeting lactate may potentiate immunotherapy through the adenosine-PKA axis (230). Indeed, Sun et al. (231) demonstrated that LDH blockade using oxamate enhanced the efficacy of CAR T cell therapy in a murine model for glioblastoma by suppressing the expression of both CD39 and CD73, highlighting the fact that targeting the TME can enhance several forms of immunotherapy.
Although PKA can also be directly targeted using small-molecule inhibitors, its widespread expression across most tissues poses a substantial challenge due to the high risk of off-target effects. This can be addressed by selectively targeting specific PKA subunits. As previously mentioned, Na et al. (170) demonstrated that the PKA subunit Cβ drives pro-tumoral function in macrophages. Notably, immune cells express a unique subunit, Cβ2, which may serve as a target in the PKA axis that will not induce systemic toxicity (171). In support of this, we recently demonstrated that tumor growth and metastasis was reduced in a murine model for metastatic breast cancer ablated for Cβ2, which was further associated with increased overall survival (169). Given that current PKA inhibitors broadly suppress all PKA activity, there is a clear need to develop novel, isoform-specific inhibitors that selectively target the Cβ variants.
7 The complexity of using metabolic inhibitors in therapeutic applications
Developing a drug is a lengthy and complex process typically involving several stages that include early drug identification and optimization followed by preclinical development and application for regulatory approval to initiate clinical trials (232). Once regulatory approval is granted, the compound enters clinical testing in humans, which is conducted in at least three phases. Phase I focuses on evaluating safety and determining appropriate dosage; Phase II assesses efficacy and monitors for adverse side effects; and Phase III involves large-scale trials to confirm the efficacy and safety in a broader patient population and compares the new treatment to current standard-of-care therapies (232). During this process more than 90% of preclinical drug candidates are disqualified for further development (233). Due to the complexity of metabolism, redundancies in metabolic pathways and the fact that various cells share vital metabolic features, most drugs developed to target metabolic enzymes show low efficacy or will have side effects. An example is the glutamine antagonist 6−diazo−5−oxo−L−norleucine (DON). DON, which is a non-proteogenic amino acid that blocks glutamine metabolism in all cells, resulting in severe adverse effects in patients due to distinct roles of glutamine in different cells and tissues (234, 235). Because of this, research on novel glutaminase inhibitors have led to the identification of several compounds including inhibitors BPTES which is an allosteric inhibitor with a low µM IC50 (236) and CB839, which is effective in the low nM range (237). Both BPTES and CB839 are glutaminase isoform type 1 specific with different mechanisms of action where CB839 appears to be well-tolerated with few side effects by patients and it is currently in several clinical trials for the treatment of various diseases including different cancers (238). (Table 1, https://clinicaltrials.gov/).
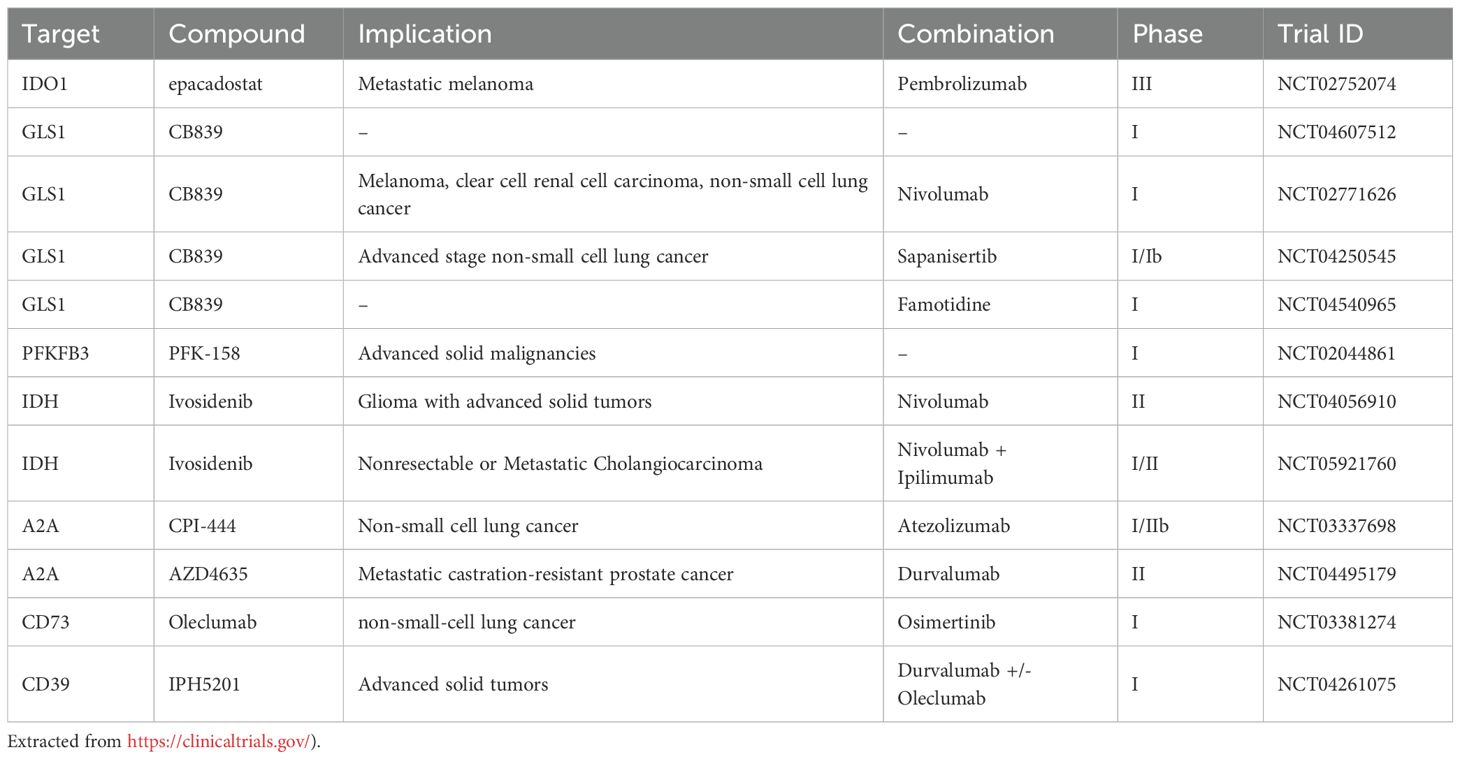
Table 1. Brief selection of inhibitors in clinical trials.
Furthermore, it has become clear that efficacy of drugs targeting metabolism may be limited due to the inherent flexibility, compensation and redundancies in metabolic pathways (90). As a result, even inhibitors with nanomolar affinity for their target may show limited therapeutic effect when used as monotherapies. Several examples illustrate this challenge. For instance, drugs targeting glycolytic enzymes downstream of hexokinase can be bypassed through the pentose phosphate pathway (PPP) where glucose 6-phosphate is shunted into the PPP and re-enters glycolysis as intermediates, effectively circumventing vital metabolic steps in the glycolytic pathway (239). Similarly, pyruvate from glycolysis can enter the TCA cycle via different routes depending on oxygen levels, allowing cells to maintain energy production under both aerobic and anaerobic conditions. Another example comes from the fact that pyruvate can enter the TCA cycle via different routes depending on oxygen levels in both an oxidative and energy-dependent fashion (240). In addition to this, drugs targeting LDH may simply shift metabolism towards a more oxidative phenotype and in that way be inefficient in inhibiting energy extraction in rapidly proliferating cells (90). Moreover, inhibiting glutaminase in glutamine consuming cells, may be compensated for by increased combustion of glucose, during which lack of carbons from glutamine is compensated for by glucose (88, 241). Finally, drug sensitive cancer cell lines can also develop resistance to metabolic inhibitors by rewiring their metabolic programs (90, 242–246). Together, these examples highlights the limitations of metabolic inhibitors when used alone and underscore the need for combination therapies. this stresses the complexity of metabolic inhibitors used as mono therapeutics. Due to this the use of metabolic inhibitors in conjunction with other treatments is emerging.
The rationale behind combinatorial strategies in cancer treatment includes targeting more than one metabolic process/pathway simultaneously reducing the required dose of individual agents and minimizing the risk of tumor immune evasion. Using the combination of drugs targeting metabolic pathways that support an immunosuppressive TME with ICT has been shown to further boost the anti-tumor responses of the immune system in preclinical models. In line with this, the IDH inhibitor ivosidenib is currently being tested as a candidate for combination therapy across several clinical trials in combination with PD-1 blockade (Table 1). There are also several non-metabolic drug candidates targeting cell surface receptors such as A2A and A2B, which have entered clinical trials in combination with PD-1 blockade, and while early reports indicate some adverse effects, including autoimmunity, these are considered manageable (247). The evidence supporting the use of inhibitors targeting glycolysis or glutamine metabolism in combination with ICT in patients is currently lacking, and hence need further exploration. At present, LDH inhibitors such as FX11, GNE-140, NCI-737, Galloflavin have also been postulated to be used to prevent tumor immune evasion when used in conjunction with PD-1 blockade (248–251). The same, but less convincing, is the case with CB839 in conjunction with PD-1 (96). In a mouse melanoma models, CB839 on its own has little effect, but when combined with anti−PD−1 but also anti−CTLA−4, it significantly suppressed tumor growth and increased infiltration of CD4+ and CD8+ T−cells (252). However, a phase I/II study of the safety and efficacy of CB839 in combination with the PD-1 inhibitor nivolumab in patients with metastatic melanoma, renal cell carcinoma, and non-small-cell lung cancer was well tolerated, but did not show increased efficacy (253). The reason for this is not known. However, the patients were not stratified based on metabolic phenotyping. As the metabolic landscape is highly variable across patients and tumors (254–256), future clinical trials should attempt to incorporate metabolic profiling to determine whether specific metabolic phenotypes correlate with improved outcomes of combination therapies.
Another obstacle in developing drugs targeting metabolism is that drugs may fail to reproduce the beneficial effect seen in preclinical models, and thus, be screened out in early clinical trials (232, 233). A striking example of this is the ECHO-301 trial, a phase III clinical trial where the IDO1 inhibitor epacadostat in combination with anti-PD1 treatment failed to provide a significantly improved patient outcome (257). However, Muller et al. (258) argues that there are several points that were inadequately discussed, which may explain the outcome, including uncertainty of whether IDO1 activity was sufficiently inhibited within the tumor, pathways bypassing IDO1 were not considered and the choice of immunotherapy over DNA damaging therapy, highlighting the need for increased understanding of metabolism within the TME. The lack of effect of a metabolic drug targeting the TME in clinical trials may be attributed to the fact that most inhibitors are screened in single cell cultures and homogenous tumor models. The latter may encompass human tumors in patient derived xenografts (PDX) animal models that may not encapsulate the complexity of tumors in the individual patient. In line with these tumors are frequently sequenced to determine patient-specific features to determine prognosis and treatment strategy. However, downstream of genetic mutations patient-specific metabolic profiles may require differential treatments despite that patients may harbor related tumors and identical oncogenic mutations. Because of this, it may be necessary to also determine metabolic phenotypes to better utilize metabolic inhibitors. Metabolic phenotypes in e.g. the TME have until now been difficult to determine. However, with extracellular flux analysis using Seahorse technology coupled with Flow cytometry has opened for more opportunities and more accurately in profiling tumor metabolic phenotypes from biopsies (259). Seahorse technology has been used to determine the metabolic phenotype of a wide array of cell types, mitochondria, 3D cell culture spheroids and now recently intact tissue biopsies (260). Seahorse profile analysis when combined with bioinformatics and artificial intelligence may in the future be useful and potential instrumental in determining combinations of drugs and treatment regimens for patient-specific targeting.
10 Concluding remarks
ICT is considered a game-changer in modern cancer treatment. However, favorable responses are observed in only 20-40% of patients with solid tumors (71, 72). Moreover, even when effective, current treatment strategies are often associated with a wide range of adverse effects, including liver, kidney and cardiovascular toxicity, and ICT may trigger autoimmune responses (261). These limitations highlight the need for additional therapeutic targets that can enhance anti-tumor efficacy while minimizing the side effects. Identifying targets capable of inducing synergistic or multifaceted responses might reduce the required treatment doses, thereby limit off-target toxicity while enhancing tumor clearance.
We have briefly summarized how hypoxia-driven metabolic processes in the TME contribute to the reprogramming of infiltrating immune cells and the development of a dysfunctional tumor vasculature- both of which aids cancer immune evasion and hinder effective drug delivery to the tumor. Although these factors currently pose a barrier to efficient cancer therapy, advancing our understanding of these mechanisms may enable the development of new treatment strategies for solid tumors. Given the central role of the tumor vasculature in the TME, anti-angiogenic drugs are being used in cancer therapy, and it is hypothesized that their combination with ICT may further enhance anti-tumor immune activity (262–264). However, although angiogenesis can be blocked by targeting the VEGF pathway, resistance to VEGF blockade is common (185, 187). Moreover, the combined blockade of VEGF and ICT is also correlated with adverse effects, including an increased risk of cardiovascular disease, highlighting the need for alternative therapeutic targets (265, 266). In this context, targeting metabolism in the TME might offer an alternative strategy for combination therapy. However, this approach requires the identification of metabolic targets - such as enzymes and pathways - that can be safely targeted, ideally offering synergistic effects when combined with existing therapies. Notably, the PFKFB3 inhibitor 3PO was shown to increase vessel integrity and enhance tumor perfusion, resulting in decreased hypoxia and increased drug delivery (132). As hypoxia results in upregulation of CD39 and CD73, increased tumor perfusion may also abrogate PKA-mediated immune suppression through reducing adenosine production in the TME (161). However, as PFKFB3 and glycolysis are also important in immune cell activation, it remains unclear if this would ultimately enhance or impair the efficacy of ICT (52, 104, 110, 111, 218). However, inhibition of LDH has been shown to reduce tumor growth in immunocompetent mice, but not in RAG knockout mice, indicating that inhibition of glycolysis may be beneficial in combination with ICT (249). Moreover, blocking lactate transport by targeting MCT1 and MCT4 has been associated with enhanced efficacy of PD-L1 blockade (210, 221). While this may partly stem from effects on reducing lactate-induced inhibition of T cells, reducing tumor acidification may also increase antibody affinity within the TME (210, 267). Glycolysis can further be targeted indirectly by disrupting glutamine metabolism via inhibition of the transcription factor MondoA (94). Although glutaminolysis is required for adequate T cell proliferation and cytokine secretion (126, 129), the GLS1 inhibitor CB839 - which is currently approved for phase 1B clinical trials - has shown minimal effects on CD4+ T cells at higher doses than those required for growth inhibition in cancer cells (125). Moreover, CB839 reduces endothelial cell proliferation without cytotoxic effects, as well as promoting M1-like macrophage polarization, suggesting its potential for combination with ICT to further boost anti-tumor immune responses (198). Additionally, these strategies may also be combined with drugs targeting cancer-specific mutations, including mutated IDH1 and IDH2, which are also known contributors of TME-induced immunosuppression.
This body of evidence suggests that targeting the metabolism of the TME might have synergistic effects by alleviating multiple aspects of TME-induced vascular dysfunction and immune suppression. Although this review has focused on ICT, there is evidence that these concepts are applicable to ACT as well. Future studies are needed to elucidate the synergistic potential for combining metabolic inhibition with ICT.
Author contributions
JW: Conceptualization, Writing – review & editing, Writing – original draft. EB: Writing – review & editing, Supervision, Visualization, Writing – original draft. KS: Writing – original draft, Writing – review & editing. BS: Conceptualization, Writing – review & editing, Funding acquisition, Writing – original draft, Supervision.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was funded by the UiO-MED and UiO-IMB, Grants to EB and BSS, Throne Holst Foundation, grant #BSS2019/2024 awarded to BSS. JAW was funded by the Norwegian Cancer Society awarded to Stefan Krauss. KS was funded by Instituto de Salud Carlos III CB16/10/00435 1034 (CIBERFES); PID2022-142470OB-I00, MICIU/AEI/10.13039/501100011033 and “ERDF 1035 A way of making Europe”; PROMETEO (CIPROM/2022/56)-”Consellería de Educación, Universi- 1036 dades, y Empleo de la Generalitat Valenciana”; Red EXERNET-RED DE EJERCICIO FISICO Y SA- 1037 LUD (RED2022-134800-T) Agencia Estatal de Investigación (Ministerio de Ciencias e Innovación).The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
3. Choi Y and Jung K. Normalization of the tumor microenvironment by harnessing vascular and immune modulation to achieve enhanced cancer therapy. Exp Mol Med. (2023) 55:2308–19. doi: 10.1038/s12276-023-01114-w
4. de Visser KE and Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. (2023) 41:374–403. doi: 10.1016/j.ccell.2023.02.016
5. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
6. Bianchi JJ, Zhao X, Mays JC, and Davoli T. Not all cancers are created equal: Tissue specificity in cancer genes and pathways. Curr Opin Cell Biol. (2020) 63:135–43. doi: 10.1016/j.ceb.2020.01.005
7. Elia I, Schmieder R, Christen S, and Fendt S-M. Organ-specific cancer metabolism and its potential for therapy. In: Herzig S, editor. Metabolic Control. Springer International Publishing, Cham (2016). p. 321–53.
8. Zhang S, Xiao X, Yi Y, Wang X, Zhu L, Shen Y, et al. Tumor initiation and early tumorigenesis: molecular mechanisms and interventional targets. Signal Transduction Targeted Ther. (2024) 9:149. doi: 10.1038/s41392-024-01848-7
9. Zhu K, Liu Q, Zhou Y, Tao C, Zhao Z, Sun J, et al. Oncogenes and tumor suppressor genes: comparative genomics and network perspectives. BMC Genomics. (2015) 16:S8. doi: 10.1186/1471-2164-16-S7-S8
10. Macleod K. Tumor suppressor genes. Curr Opin Genet Dev. (2000) 10:81–93. doi: 10.1016/S0959-437X(99)00041-6
11. Weinberg RA. Oncogenes and tumor suppressor genes. CA: A Cancer J Clin. (1994) 44:160–70. doi: 10.3322/canjclin.44.3.160
12. Wang H, Guo M, Wei H, and Chen Y. Targeting p53 pathways: mechanisms, structures and advances in therapy. Signal Transduction Targeted Ther. (2023) 8:92. doi: 10.1038/s41392-023-01347-1
13. Hall DCN and Benndorf RA. Aspirin sensitivity of PIK3CA-mutated Colorectal Cancer: potential mechanisms revisited. Cell Mol Life Sci. (2022) 79:393. doi: 10.1007/s00018-022-04430-y
14. Chen S, Zhou Z, Li Y, Du Y, and Chen G. Application of single-cell sequencing to the research of tumor microenvironment. Front Immunol. (2023) 14:1285540. doi: 10.3389/fimmu.2023.1285540
15. Lomakin A, Svedlund J, Strell C, Gataric M, Shmatko A, Rukhovich G, et al. Spatial genomics maps the structure, nature and evolution of cancer clones. Nature. (2022) 611:594–602. doi: 10.1038/s41586-022-05425-2
16. Hu J, Wang S-G, Hou Y, Chen Z, Liu L, Li R, et al. Multi-omic profiling of clear cell renal cell carcinoma identifies metabolic reprogramming associated with disease progression. Nat Genet. (2024) 56:442–57. doi: 10.1038/s41588-024-01662-5
17. Dickens E and Ahmed S. Principles of cancer treatment by chemotherapy. Surg - Oxford Int Edition. (2021) 39:215–20. doi: 10.1016/j.mpsur.2021.01.009
18. Ciriello G, Magnani L, Aitken SJ, Akkari L, Behjati S, Hanahan D, et al. Cancer evolution: A multifaceted affair. Cancer Discov. (2024) 14:36–48. doi: 10.1158/2159-8290.CD-23-0530
19. Vendramin R, Litchfield K, and Swanton C. Cancer evolution: Darwin and beyond. EMBO J. (2021) 40:e108389. doi: 10.15252/embj.2021108389
20. Damodaran S, Berger MF, and Roychowdhury S. Clinical tumor sequencing: opportunities and challenges for precision cancer medicine. Am Soc Clin Oncol Educ Book. (2015) 35:e175–82. doi: 10.14694/EdBook_AM.2015.35.e175
21. Calzetta L and Koziol-White C. Pharmacological interactions: Synergism, or not synergism, that is the question. Curr Res Pharmacol Drug Discov. (2021) 2:100046. doi: 10.1016/j.crphar.2021.100046
22. Kovoor JG, Chow CK, Salam A, Webster R, Shiel L, Nelson MR, et al. Participants’ views of ultra-low dose combination therapy for high blood pressure: a mixed-methods study from the QUARTET trial. J Hum Hypertension. (2024) 38:516–22. doi: 10.1038/s41371-024-00915-4
23. Calzetta L, Page C, Matera MG, Cazzola M, and Rogliani P. Drug-drug interactions and synergy: from pharmacological models to clinical application. Pharmacol Rev. (2024) 76:1159–220. doi: 10.1124/pharmrev.124.000951
24. Murphy K, Weaver C, and Janeway C. Janeway’s immunobiology. New York: Garland Publishing Inc (2017).
25. Marshall JS, Warrington R, Watson W, and Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol. (2018) 14:49. doi: 10.1186/s13223-018-0278-1
26. Tang D, Kang R, Coyne CB, Zeh HJ, and Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. (2012) 249:158–75. doi: 10.1111/j.1600-065X.2012.01146.x
27. Wang R, Lan C, Benlagha K, Camara NOS, Miller H, Kubo M, et al. The interaction of innate immune and adaptive immune system. MedComm. (2024) 5:e714. doi: 10.1002/mco2.714
28. Landsverk OJ, Snir O, Casado RB, Richter L, Mold JE, Réu P, et al. Antibody-secreting plasma cells persist for decades in human intestine. J Exp Med. (2017) 214:309–17. doi: 10.1084/jem.20161590
29. Kumar BV, Connors TJ, and Farber DL. Human T cell development, localization, and function throughout life. Immunity. (2018) 48:202–13. doi: 10.1016/j.immuni.2018.01.007
30. Mantegazza AR, Magalhaes JG, Amigorena S, and Marks MS. Presentation of phagocytosed antigens by MHC class I and II. Traffic. (2013) 14:135–52. doi: 10.1111/tra.12026
31. Gaudino SJ and Kumar P. Cross-talk between antigen presenting cells and T cells impacts intestinal homeostasis, bacterial infections, and tumorigenesis. Front Immunol. (2019) 10. doi: 10.3389/fimmu.2019.00360
32. Mariuzza RA, Agnihotri P, and Orban J. The structural basis of T-cell receptor (TCR) activation: An enduring enigma. J Biol Chem. (2020) 295:914–25. doi: 10.1016/S0021-9258(17)49904-2
33. Shah K, Al-Haidari A, Sun J, and Kazi JU. T cell receptor (TCR) signaling in health and disease. Signal Transduction Targeted Ther. (2021) 6:412. doi: 10.1038/s41392-021-00823-w
34. Taams LS, Palmer DB, Akbar AN, Robinson DS, Brown Z, and Hawrylowicz CM. Regulatory T cells in human disease and their potential for therapeutic manipulation. Immunology. (2006) 118:1–9. doi: 10.1111/j.1365-2567.2006.02348.x
35. Capone A and Volpe E. Transcriptional regulators of T helper 17 cell differentiation in health and autoimmune diseases. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.00348
36. Chandwaskar R and Awasthi A. Emerging roles of Th9 cells as an anti-tumor helper T cells. Int Rev Immunol. (2019) 38:204–11. doi: 10.1080/08830185.2019.1648453
37. Geginat J, Paroni M, Maglie S, Alfen JS, Kastirr I, Gruarin P, et al. Plasticity of human CD4 T cell subsets. Front Immunol. (2014) 5. doi: 10.3389/fimmu.2014.00630
38. Schmidt ME and Varga SM. The CD8 T cell response to respiratory virus infections. Front Immunol. (2018) 9. doi: 10.3389/fimmu.2018.00678
39. Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, and Kedl RM. T cell responses: naïve to memory and everything in between. Adv Physiol Education. (2013) 37:273–83. doi: 10.1152/advan.00066.2013
40. Hanahan D and Weinberg Robert A. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
41. Li X, Sun X, and Carmeliet P. Hallmarks of endothelial cell metabolism in health and disease. Cell Metab. (2019) 30:414–33. doi: 10.1016/j.cmet.2019.08.011
42. Ribatti D. The concept of immune surveillance against tumors. The first theories. Oncotarget. (2017) 8:7175–80. doi: 10.18632/oncotarget.12739
43. McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. (2006) 26:154–8.
44. Ullenhag G. Cancer treatment of today in view of the Nobel Prize. Ups J Med Sci. (2018) 123:205–6. doi: 10.1080/03009734.2018.1548528
45. Zhang M, Liu C, Tu J, Tang M, Ashrafizadeh M, Nabavi N, et al. Advances in cancer immunotherapy: historical perspectives, current developments, and future directions. Mol Cancer. (2025) 24:136. doi: 10.1186/s12943-025-02305-x
46. Naran K, Nundalall T, Chetty S, and Barth S. Principles of immunotherapy: implications for treatment strategies in cancer and infectious diseases. Front Microbiol. (2018) 9:3158. doi: 10.3389/fmicb.2018.03158
47. Noel PJ, Boise LH, Green JM, and Thompson CB. CD28 costimulation prevents cell death during primary T cell activation. J Immunol. (1996) 157:636–42. doi: 10.4049/jimmunol.157.2.636
48. Brunner-Weinzierl MC and Rudd CE. CTLA-4 and PD-1 control of T-cell motility and migration: implications for tumor immunotherapy. Front Immunol. (2018) 9. doi: 10.3389/fimmu.2018.02737
49. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
50. Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, et al. The diverse function of PD-1/PD-L pathway beyond cancer. Front Immunol. (2019) 10:2298. doi: 10.3389/fimmu.2019.02298
51. Han Y, Liu D, and Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. (2020) 10:727–42.
52. Kolan SS, Li G, Wik JA, Malachin G, Guo S, Kolan P, et al. Cellular metabolism dictates T cell effector function in health and disease. Scandinavian J Immunol. (2020) 92:e12956. doi: 10.1111/sji.12956
53. Curtsinger JM and Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol. (2010) 22:333–40. doi: 10.1016/j.coi.2010.02.013
54. Martinez-Sanchez ME, Huerta L, Alvarez-Buylla ER, and Villarreal Luján C. Role of cytokine combinations on CD4+ T cell differentiation, partial polarization, and plasticity: continuous network modeling approach. Front Physiol. (2018) 9. doi: 10.3389/fphys.2018.00877
55. Rohaan MW, Borch TH, Berg J, Met Ö, Kessels R, Foppen MHG, et al. Tumor-infiltrating lymphocyte therapy or ipilimumab in advanced melanoma. New Engl J Med. (2022) 387:2113–25. doi: 10.1056/NEJMoa2210233
56. Rohaan MW, Wilgenhof S, and Haanen J. Adoptive cellular therapies: the current landscape. Virchows Arch. (2019) 474:449–61. doi: 10.1007/s00428-018-2484-0
57. Gupta I, Hussein O, Sastry KS, Bougarn S, Gopinath N, Chin-Smith E, et al. Deciphering the complexities of cancer cell immune evasion: Mechanisms and therapeutic implications. Adv Cancer Biol - Metastasis. (2023) 8:100107. doi: 10.1016/j.adcanc.2023.100107
58. Bohn T, Rapp S, Luther N, Klein M, Bruehl TJ, Kojima N, et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol. (2018) 19:1319–29. doi: 10.1038/s41590-018-0226-8
59. Roerden M and Spranger S. Cancer immune evasion, immunoediting and intratumour heterogeneity. Nat Rev Immunol. (2025) 18:674–85. doi: 10.1038/s41577-024-01111-8
60. Wei Q and Taskén K. Immunoregulatory signal networks and tumor immune evasion mechanisms: insights into therapeutic targets and agents in clinical development. Biochem J. (2022) 479:2219–60. doi: 10.1042/BCJ20210233
61. Cornel AM, Mimpen IL, and Nierkens S. MHC class I downregulation in cancer: underlying mechanisms and potential targets for cancer immunotherapy. Cancers. (2020) 12:1760. doi: 10.3390/cancers12071760
62. DhatChinamoorthy K, Colbert JD, and Rock KL. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.636568
63. Sterner RC and Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:69. doi: 10.1038/s41408-021-00459-7
64. Brown KE, Freeman GJ, Wherry EJ, and Sharpe AH. Role of PD-1 in regulating acute infections. Curr Opin Immunol. (2010) 22:397–401. doi: 10.1016/j.coi.2010.03.007
65. Porichis F and Kaufmann DE. Role of PD-1 in HIV pathogenesis and as target for therapy. Curr HIV/AIDS Rep. (2012) 9:81–90. doi: 10.1007/s11904-011-0106-4
66. Wherry EJ and Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
67. Keir ME, Butte MJ, Freeman GJ, and Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
68. Lin X, Kang K, Chen P, Zeng Z, Li G, Xiong W, et al. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol Cancer. (2024) 23:108. doi: 10.1186/s12943-024-02023-w
69. Camacho LH. CTLA-4 blockade with ipilimumab: biology, safety, efficacy, and future considerations. Cancer Med. (2015) 4:661–72. doi: 10.1002/cam4.371
70. Sharma P, Goswami S, Raychaudhuri D, Siddiqui BA, Singh P, Nagarajan A, et al. Immune checkpoint therapy—current perspectives and future directions. Cell. (2023) 186:1652–69. doi: 10.1016/j.cell.2023.03.006
71. Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
72. Wu VH, Yung BS, Faraji F, Saddawi-Konefka R, Wang Z, Wenzel AT, et al. The GPCR-Gα(s)-PKA signaling axis promotes T cell dysfunction and cancer immunotherapy failure. Nat Immunol. (2023) 24:1318–30. doi: 10.1038/s41590-023-01529-7
73. McKeown SR. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br J Radiol. (2014) 87:20130676–. doi: 10.1259/bjr.20130676
74. de la Cruz-López KG, Castro-Muñoz LJ, Reyes-Hernández DO, García-Carrancá A, and Manzo-Merino J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front Oncol. (2019) 9. doi: 10.3389/fonc.2019.01143
75. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
76. Wilfahrt D and Delgoffe GM. Metabolic waypoints during T cell differentiation. Nat Immunol. (2024) 25:206–17. doi: 10.1038/s41590-023-01733-5
77. Raynor JL and Chi H. Nutrients: Signal 4 in T cell immunity. J Exp Med. (2024) 221. doi: 10.1084/jem.20221839
78. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U.S.A. (2007) 104:19345–50.
79. Reina-Campos M, Scharping NE, and Goldrath AW. CD8+ T cell metabolism in infection and cancer. Nat Rev Immunol. (2021) 21:718–38. doi: 10.1038/s41577-021-00537-8
80. Rhoads JP, Major AS, and Rathmell JC. Fine tuning of immunometabolism for the treatment of rheumatic diseases. Nat Rev Rheumatol. (2017) 13:313–20. doi: 10.1038/nrrheum.2017.54
81. O’Neill LA, Kishton RJ, and Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
82. Stine ZE, Schug ZT, Salvino JM, and Dang CV. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov. (2022) 21:141–62. doi: 10.1038/s41573-021-00339-6
83. Choi Y-K and Park K-G. Targeting glutamine metabolism for cancer treatment. Biomol Ther (Seoul). (2018) 26:19–28. doi: 10.4062/biomolther.2017.178
84. Vander Heiden MG, Cantley LC, and Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
85. Potter M, Newport E, and Morten KJ. The Warburg effect: 80 years on. Biochem Soc Trans. (2016) 44:1499–505. doi: 10.1042/BST20160094
86. Chandel NS. Glycolysis. Cold Spring Harb Perspect Biol. (2021) 13. doi: 10.1101/cshperspect.a040535
87. Ni R, Li Z, Li L, Peng D, Ming Y, Li L, et al. Rethinking glutamine metabolism and the regulation of glutamine addiction by oncogenes in cancer. Front Oncol. (2023) 13:1143798. doi: 10.3389/fonc.2023.1143798
88. Smith B, Schafer Xenia L, Ambeskovic A, Spencer Cody M, Land H, and Munger J. Addiction to coupling of the Warburg effect with glutamine catabolism in cancer cells. Cell Rep. (2016) 17:821–36. doi: 10.1016/j.celrep.2016.09.045
89. Koundouros N and Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. (2020) 122:4–22. doi: 10.1038/s41416-019-0650-z
90. Boudreau A, Purkey HE, Hitz A, Robarge K, Peterson D, Labadie S, et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat Chem Biol. (2016) 12:779–86. doi: 10.1038/nchembio.2143
91. Pavlova NN, Hui S, Ghergurovich JM, Fan J, Intlekofer AM, White RM, et al. As extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab. (2018) 27:428–38.e5. doi: 10.1016/j.cmet.2017.12.006
92. Luo M, Brooks M, and Wicha MS. Asparagine and glutamine: co-conspirators fueling metastasis. Cell Metab. (2018) 27:947–9. doi: 10.1016/j.cmet.2018.04.012
93. Kim B, Li J, Jang C, and Arany Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. (2017) 36:2321–33. doi: 10.15252/embj.201796436
94. Kaadige MR, Looper RE, Kamalanaadhan S, and Ayer DE. Glutamine-dependent anapleurosis dictates glucose uptake and cell growth by regulating MondoA transcriptional activity. Proc Natl Acad Sci U.S.A. (2009) 106:14878–83. doi: 10.1073/pnas.0901221106
95. Reid MA, Lowman XH, Pan M, Tran TQ, Warmoes MO, Ishak Gabra MB, et al. IKKβ promotes metabolic adaptation to glutamine deprivation via phosphorylation and inhibition of PFKFB3. Genes Dev. (2016) 30:1837–51. doi: 10.1101/gad.287235.116
96. Jin J, Byun J-K, Choi Y-K, and Park K-G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp Mol Med. (2023) 55:706–15. doi: 10.1038/s12276-023-00971-9
97. Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature. (2021) 593:282–8. doi: 10.1038/s41586-021-03442-1
98. Liu Y, Xu W, Li M, Yang Y, Sun D, Chen L, et al. The regulatory mechanisms and inhibitors of isocitrate dehydrogenase 1 in cancer. Acta Pharm Sin B. (2023) 13:1438–66. doi: 10.1016/j.apsb.2022.12.019
99. Thomas D, Wu M, Nakauchi Y, Zheng M, Thompson-Peach CAL, Lim K, et al. Dysregulated lipid synthesis by oncogenic IDH1 mutation is a targetable synthetic lethal vulnerability. Cancer Discov. (2023) 13:496–515. doi: 10.1158/2159-8290.CD-21-0218
100. McBrayer SK, Mayers JR, DiNatale GJ, Shi DD, Khanal J, Chakraborty AA, et al. Transaminase inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell. (2018) 175:101–16.e25. doi: 10.1016/j.cell.2018.08.038
101. Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I, et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature. (2015) 520:192–7. doi: 10.1038/nature14362
102. De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. (2013) 154:651–63. doi: 10.1016/j.cell.2013.06.037
103. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
104. Tawakol A, Singh P, Mojena M, Pimentel-Santillana M, Emami H, MacNabb M, et al. HIF-1alpha and PFKFB3 mediate a tight relationship between proinflammatory activation and anerobic metabolism in atherosclerotic macrophages. Arteriosclerosis thrombosis Vasc Biol. (2015) 35:1463–71. doi: 10.1161/ATVBAHA.115.305551
105. Ip WKE, Hoshi N, Shouval DS, Snapper S, and Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. (2017) 356:513–9. doi: 10.1126/science.aal3535
106. Newsholme P, Curi R, Pithon Curi TC, Murphy CJ, Garcia C, and Pires de Melo M. Glutamine metabolism by lymphocytes, macrophages, and neutrophils: its importance in health and disease. J Nutr Biochem. (1999) 10:316–24. doi: 10.1016/S0955-2863(99)00022-4
107. Karshovska E, Wei Y, Subramanian P, Mohibullah R, Geißler C, Baatsch I, et al. HIF-1α (Hypoxia-Inducible Factor-1α) Promotes Macrophage Necroptosis by Regulating miR-210 and miR-383. Arteriosclerosis thrombosis Vasc Biol. (2020) 40:583–96. doi: 10.1161/ATVBAHA.119.313290
108. Langston PK, Shibata M, and Horng T. Metabolism supports macrophage activation. Front Immunol. (2017) 8. doi: 10.3389/fimmu.2017.00061
109. Jiang H, Shi H, Sun M, Wang Y, Meng Q, Guo P, et al. PFKFB3-driven macrophage glycolytic metabolism is a crucial component of innate antiviral defense. J Immunol. (2016) 197:2880–90. doi: 10.4049/jimmunol.1600474
110. Xu J, Wang L, Yang Q, Ma Q, Zhou Y, Cai Y, et al. Deficiency of myeloid pfkfb3 protects mice from lung edema and cardiac dysfunction in LPS-induced endotoxemia. Front Cardiovasc Med. (2021) 8. doi: 10.3389/fcvm.2021.745810
111. Gong Y, Lan H, Yu Z, Wang M, Wang S, Chen Y, et al. Blockage of glycolysis by targeting PFKFB3 alleviates sepsis-related acute lung injury via suppressing inflammation and apoptosis of alveolar epithelial cells. Biochem Biophys Res Commun. (2017) 491:522–9. doi: 10.1016/j.bbrc.2017.05.173
112. Williams NC and O’Neill LAJ. A role for the Krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation. Front Immunol. (2018) 9. doi: 10.3389/fimmu.2018.00141
113. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
114. Wculek SK, Dunphy G, Heras-Murillo I, Mastrangelo A, and Sancho D. Metabolism of tissue macrophages in homeostasis and pathology. Cell Mol Immunol. (2022) 19:384–408. doi: 10.1038/s41423-021-00791-9
115. Wang XF, Wang HS, Wang H, Zhang F, Wang KF, Guo Q, et al. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: focus on macrophage polarization of THP-1 cells. Cell Immunol. (2014) 289:42–8. doi: 10.1016/j.cellimm.2014.02.005
116. Salminen A. Role of indoleamine 2,3-dioxygenase 1 (IDO1) and kynurenine pathway in the regulation of the aging process. Ageing Res Rev. (2022) 75:101573. doi: 10.1016/j.arr.2022.101573
117. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, and Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. (2010) 185:3190–8. doi: 10.4049/jimmunol.0903670
118. Zhou Y, Yao L, Ma T, Wang Z, Yin Y, Yang J, et al. Indoleamine 2,3-dioxygenase-1 involves in CD8(+)T cell exhaustion in glioblastoma via regulating tryptophan levels. Int Immunopharmacol. (2024) 142:113062. doi: 10.1016/j.intimp.2024.113062
119. Bishop EL, Gudgeon N, and Dimeloe S. Control of T cell metabolism by cytokines and hormones. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.653605
120. Palmer CS, Ostrowski M, Balderson B, Christian N, and Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol. (2015) 6. doi: 10.3389/fimmu.2015.00001
121. Wolf T, Jin W, Zoppi G, Vogel IA, Akhmedov M, Bleck CKE, et al. Dynamics in protein translation sustaining T cell preparedness. Nat Immunol. (2020) 21:927–37. doi: 10.1038/s41590-020-0714-5
122. Menk AV, Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S, et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep. (2018) 22:1509–21. doi: 10.1016/j.celrep.2018.01.040
123. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol (Baltimore Md: 1950). (2008) 180:4476–86. doi: 10.4049/jimmunol.180.7.4476
124. Chang C-H, Curtis Jonathan D, Maggi Leonard B, Faubert B, Villarino Alejandro V, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
125. Cederkvist H, Kolan SS, Wik JA, Sener Z, and Skålhegg BS. Identification and characterization of a novel glutaminase inhibitor. FEBS Open Bio. (2021). doi: 10.1002/2211-5463.13319
126. Sener Z, Cederkvist FH, Volchenkov R, Holen HL, and Skålhegg BS. T helper cell activation and expansion is sensitive to glutaminase inhibition under both hypoxic and normoxic conditions. PloS One. (2016) 11:e0160291. doi: 10.1371/journal.pone.0160291
127. Chang W-K, Yang KD, Chuang H, Jan J-T, and Shaio M-F. Glutamine protects activated human T cells from apoptosis by up-regulating glutathione and Bcl-2 levels. Clin Immunol. (2002) 104:151–60. doi: 10.1006/clim.2002.5257
128. Chang W-K, Yang KD, and Shaio M-F. Effect of glutamine on Th1 and Th2 cytokine responses of human peripheral blood mononuclear cells. Clin Immunol. (1999) 93:294–301. doi: 10.1006/clim.1999.4788
129. Wik JA, Chowdhury A, Kolan S, Bastani NE, Li G, Alam K, et al. Endogenous glutamine is rate-limiting for anti-CD3 and anti-CD28 induced CD4+ T-cell proliferation and glycolytic activity under hypoxia and normoxia. Biochem J. (2022) 479:1221–35. doi: 10.1042/BCJ20220144
130. Wik JA and Skålhegg BS. T cell metabolism in infection. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.840610
131. Falkenberg KD, Rohlenova K, Luo Y, and Carmeliet P. The metabolic engine of endothelial cells. Nat Metab. (2019) 1:937–46. doi: 10.1038/s42255-019-0117-9
132. Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A, et al. Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell. (2016) 30:968–85. doi: 10.1016/j.ccell.2016.10.006
133. Wik JA, Lundback P, la Cour Poulsen L, Haraldsen G, Skalhegg BS, and Hol J. 3PO inhibits inflammatory NFkappaB and stress-activated kinase signaling in primary human endothelial cells independently of its target PFKFB3. PloS One. (2020) 15:e0229395. doi: 10.1371/journal.pone.0229395
134. Wik JA, Phung D, Kolan S, Haraldsen G, Skålhegg BS, and Hol Fosse J. Inflammatory activation of endothelial cells increases glycolysis and oxygen consumption despite inhibiting cell proliferation. FEBS Open Bio. (2021) 11:1719–30. doi: 10.1002/2211-5463.13174
135. Xu Y, An X, Guo X, Habtetsion TG, Wang Y, Xu X, et al. Endothelial PFKFB3 plays a critical role in angiogenesis. Arteriosclerosis thrombosis Vasc Biol. (2014) 34:1231–9. doi: 10.1161/ATVBAHA.113.303041
136. Huang H, Vandekeere S, Kalucka J, Bierhansl L, Zecchin A, Brüning U, et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. (2017) 36:2334–52. doi: 10.15252/embj.201695518
137. Simcox J and Lamming DW. The central moTOR of metabolism. Dev Cell. (2022) 57:691–706. doi: 10.1016/j.devcel.2022.02.024
138. Tarrado-Castellarnau M, Atauri Pd, and Cascante M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget. (2016) 7. doi: 10.18632/oncotarget.10911
139. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
140. Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-α. Nat Med. (2015) 21:638–46. doi: 10.1038/nm.3868
141. Valvezan AJ and Manning BD. Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat Metab. (2019) 1:321–33. doi: 10.1038/s42255-019-0038-7
142. Linke M, Fritsch SD, Sukhbaatar N, Hengstschläger M, and Weichhart T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. (2017) 591:3089–103. doi: 10.1002/1873-3468.12711
143. Kim J-w, Tchernyshyov I, Semenza GL, and Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
144. Kierans SJ and Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol. (2021) 599:23–37. doi: 10.1113/JP280572
145. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. (2011) 208:1367–76. doi: 10.1084/jem.20110278
146. Ullah MS, Davies AJ, and Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem. (2006) 281:9030–7. doi: 10.1074/jbc.M511397200
147. Masoud GN and Li W. HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B. (2015) 5:378–89. doi: 10.1016/j.apsb.2015.05.007
148. Brian MO. Hypoxia-inducible factor in cancer: from pathway regulation to therapeutic opportunity. BMJ Oncol. (2024) 3:e000154. doi: 10.1136/bmjonc-2023-000154
149. McGettrick AF and O’Neill LAJ. The role of HIF in immunity and inflammation. Cell Metab. (2020) 32:524–36. doi: 10.1016/j.cmet.2020.08.002
150. Allison KE, Coomber BL, and Bridle BW. Metabolic reprogramming in the tumour microenvironment: a hallmark shared by cancer cells and T lymphocytes. Immunology. (2017) 152:175–84. doi: 10.1111/imm.12777
151. Quinn WJ 3rd, Jiao J, TeSlaa T, Stadanlick J, Wang Z, Wang L, et al. Lactate limits T cell proliferation via the NAD(H) redox state. Cell Rep. (2020) 33:108500. doi: 10.1016/j.celrep.2020.108500
152. Wu H, Estrella V, Beatty M, Abrahams D, El-Kenawi A, Russell S, et al. T-cells produce acidic niches in lymph nodes to suppress their own effector functions. Nat Commun. (2020) 11:4113. doi: 10.1038/s41467-020-17756-7
153. Xu B, Liu Y, Li N, and Geng Q. Lactate and lactylation in macrophage metabolic reprogramming: current progress and outstanding issues. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1395786
154. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
155. Gu J, Zhou J, Chen Q, Xu X, Gao J, Li X, et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-β signaling in regulatory T cells. Cell Rep. (2022) 39:110986. doi: 10.1016/j.celrep.2022.110986
156. Sun X, Dong H, Su R, Chen J, Li W, Yin S, et al. Lactylation-related gene signature accurately predicts prognosis and immunotherapy response in gastric cancer. Front Oncol. (2024) 14:1485580. doi: 10.3389/fonc.2024.1485580
157. Oh W, Kim AMJ, Dhawan D, Knapp DW, and Lim S-O. Lactic acid inhibits the interaction between PD-L1 protein and PD-L1 antibody in the PD-1/PD-L1 blockade therapy-resistant tumor. Mol Ther. (2025) 33:723–33. doi: 10.1016/j.ymthe.2024.12.044
158. Azoitei N, Becher A, Steinestel K, Rouhi A, Diepold K, Genze F, et al. PKM2 promotes tumor angiogenesis by regulating HIF-1α through NF-κB activation. Mol cancer. (2016) 15:3–. doi: 10.1186/s12943-015-0490-2
159. Lugano R, Ramachandran M, and Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci. (2020) 77:1745–70. doi: 10.1007/s00018-019-03351-7
160. Allard B, Longhi MS, Robson SC, and Stagg J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
161. Xia C, Yin S, To KKW, and Fu L. CD39/CD73/A2AR pathway and cancer immunotherapy. Mol Cancer. (2023) 22:44. doi: 10.1186/s12943-023-01733-x
162. Lu TW, Wu J, Aoto PC, Weng JH, Ahuja LG, Sun N, et al. Two PKA RIα holoenzyme states define ATP as an isoform-specific orthosteric inhibitor that competes with the allosteric activator, cAMP. Proc Natl Acad Sci U S A. (2019) 116:16347–56. doi: 10.1073/pnas.1906036116
163. Funderud A, Aas-Hanssen K, Aksaas AK, Hafte TT, Corthay A, Munthe LA, et al. Isoform-specific regulation of immune cell reactivity by the catalytic subunit of protein kinase A (PKA). Cell Signalling. (2009) 21:274–81. doi: 10.1016/j.cellsig.2008.10.013
164. Vang T, Torgersen KM, Sundvold V, Saxena M, Levy FO, Skålhegg BS, et al. Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J Exp Med. (2001) 193:497–507. doi: 10.1084/jem.193.4.497
165. Ramstad C, Sundvold V, Johansen HK, and Lea T. cAMP-dependent protein kinase (PKA) inhibits T cell activation by phosphorylating ser-43 of raf-1 in the MAPK/ERK pathway. Cell Signal. (2000) 12:557–63. doi: 10.1016/S0898-6568(00)00097-8
166. Postler TS. Chapter Seven - A most versatile kinase: The catalytic subunit of PKA in T-cell biology. In: Postler TS and Galluzzi L, editors. International Review of Cell and Molecular Biology, vol. 361. Amsterdam, Netherlands: Academic Press (2021). p. 301–18.
167. Klein-Hessling S, Jha MK, Santner-Nanan B, Berberich-Siebelt F, Baumruker T, Schimpl A, et al. Protein kinase A regulates GATA-3-dependent activation of IL-5 gene expression in Th2 cells1. J Immunol. (2003) 170:2956–61. doi: 10.4049/jimmunol.170.6.2956
168. Kuczma M, Wang CY, Ignatowicz L, Gourdie R, and Kraj P. Altered connexin 43 expression underlies age-dependent decrease of regulatory T cell suppressor function in nonobese diabetic mice. J Immunol (Baltimore Md: 1950). (2015) 194:5261–71. doi: 10.4049/jimmunol.1400887
169. Guo S, Kolan S, Li G, Hammarström CL, Grimolizzi F, Stuhr LEB, et al. Reduced EO771-induced tumour growth and increased overall-survival of mice ablated for immune cell-specific catalytic subunit Cβ2 of protein kinase A. Immunol Letters. (2024) 268:106884. doi: 10.1016/j.imlet.2024.106884
170. Na YR, Kwon JW, Kim DY, Chung H, Song J, Jung D, et al. Protein kinase A catalytic subunit is a molecular switch that promotes the pro-tumoral function of macrophages. Cell Rep. (2020) 31:107643. doi: 10.1016/j.celrep.2020.107643
171. Moen LV, Sener Z, Volchenkov R, Svarstad AC, Eriksen AM, Holen HL, et al. Ablation of the Cβ2 subunit of PKA in immune cells leads to increased susceptibility to systemic inflammation in mice. Eur J Immunol. (2017) 47:1880–9. doi: 10.1002/eji.201646809
172. Du X and Hu H. The roles of 2-hydroxyglutarate. Front Cell Dev Biol. (2021) 9. doi: 10.3389/fcell.2021.651317
173. Notarangelo G, Spinelli JB, Perez EM, Baker GJ, Kurmi K, Elia I, et al. Oncometabolite D-2HG alters T cell metabolism to impair CD8+ T cell function. Science. (2022) 377:1519–29. doi: 10.1126/science.abj5104
174. de Goede KE, Harber KJ, Gorki FS, Verberk SGS, Groh LA, Keuning ED, et al. d-2-Hydroxyglutarate is an anti-inflammatory immunometabolite that accumulates in macrophages after TLR4 activation. Biochim Biophys Acta (BBA) - Mol Basis Disease. (2022) 1868:166427. doi: 10.1016/j.bbadis.2022.166427
175. Moen I and Stuhr LE. Hyperbaric oxygen therapy and cancer–a review. Target Oncol. (2012) 7:233–42. doi: 10.1007/s11523-012-0233-x
176. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med. (2015) 7:277ra30. doi: 10.1126/scitranslmed.aaa1260
177. Bennett MH, Feldmeier J, Smee R, and Milross C. Hyperbaric oxygenation for tumour sensitisation to radiotherapy. Cochrane Database Systematic Rev. (2018) 2018. doi: 10.1073/pnas.0709747104
178. Dang L, Yen K, and Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. (2016) 27:599–608. doi: 10.1093/annonc/mdw013
179. Elebiyo TC, Rotimi D, Evbuomwan IO, Maimako RF, Iyobhebhe M, Ojo OA, et al. Reassessing vascular endothelial growth factor (VEGF) in anti-angiogenic cancer therapy. Cancer Treat Res Commun. (2022) 32:100620. doi: 10.1016/j.ctarc.2022.100620
180. Cao Y, Langer R, and Ferrara N. Targeting angiogenesis in oncology, ophthalmology and beyond. Nat Rev Drug Discov. (2023) 22:476–95. doi: 10.1038/s41573-023-00671-z
181. Meadows KL and Hurwitz HI. Anti-VEGF therapies in the clinic. Cold Spring Harb Perspect Med. (2012) 2. doi: 10.1101/cshperspect.a006577
182. Ciciola P, Cascetta P, Bianco C, Formisano L, and Bianco R. Combining immune checkpoint inhibitors with anti-angiogenic agents. J Clin Med. (2020) 9. doi: 10.3390/jcm9030675
183. Akil A, Gutiérrez-García AK, Guenter R, Rose JB, Beck AW, Chen H, et al. Notch signaling in vascular endothelial cells, angiogenesis, and tumor progression: an update and prospective. Front Cell Dev Biol. (2021) 9. doi: 10.3389/fcell.2021.642352
184. Benedito R, Roca C, Sörensen I, Adams S, Gossler A, Fruttiger M, et al. The notch ligands dll4 and jagged1 have opposing effects on angiogenesis. Cell. (2009) 137:1124–35. doi: 10.1016/j.cell.2009.03.025
185. Gjølberg TT, Wik JA, Johannessen H, Krüger S, Bassi N, Christopoulos PF, et al. Antibody blockade of Jagged1 attenuates choroidal neovascularization. Nat Commun. (2023) 14:3109. doi: 10.1038/s41467-023-38563-w
186. Williams CK, Li JL, Murga M, Harris AL, and Tosato G. Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood. (2006) 107:931–9. doi: 10.1182/blood-2005-03-1000
187. Treps L, Conradi LC, Harjes U, and Carmeliet P. Manipulating angiogenesis by targeting endothelial metabolism: hitting the engine rather than the drivers-A new perspective? Pharmacol Rev. (2016) 68:872–87. doi: 10.1124/pr.116.012492
188. Schoors S, De Bock K, Cantelmo AR, Georgiadou M, Ghesquiere B, Cauwenberghs S, et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. (2014) 19:37–48. doi: 10.1016/j.cmet.2013.11.008
189. Zhang J, Muri J, Fitzgerald G, Gorski T, Gianni-Barrera R, Masschelein E, et al. Endothelial lactate controls muscle regeneration from ischemia by inducing M2-like macrophage polarization. Cell Metab. (2020) 31:1136–53.e7. doi: 10.1016/j.cmet.2020.05.004
190. Chen J, Huang Z, Chen Y, Tian H, Chai P, Shen Y, et al. Lactate and lactylation in cancer. Signal Transduction Targeted Ther. (2025) 10:38. doi: 10.1038/s41392-024-02082-x
191. Fan M, Yang K, Wang X, Chen L, Gill PS, Ha T, et al. Lactate promotes endothelial-to-mesenchymal transition via Snail1 lactylation after myocardial infarction. Sci Adv. (2023) 9:eadc9465. doi: 10.1126/sciadv.adc9465
192. Conradi LC, Brajic A, Cantelmo AR, Bouche A, Kalucka J, Pircher A, et al. Tumor vessel disintegration by maximum tolerable PFKFB3 blockade. Angiogenesis. (2017) 20:599–613. doi: 10.1007/s10456-017-9573-6
193. Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther. (2008) 7:110–20. doi: 10.1158/1535-7163.MCT-07-0482
194. Emini Veseli B, Perrotta P, Van Wielendaele P, Lambeir A-M, Abdali A, Bellosta S, et al. Small molecule 3PO inhibits glycolysis but does not bind to 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3). FEBS Lett. (2020) 594:3067–75. doi: 10.1002/1873-3468.13878
195. Brooke DG, van Dam EM, Watts CK, Khoury A, Dziadek MA, Brooks H, et al. Targeting the Warburg Effect in cancer; relationships for 2-arylpyridazinones as inhibitors of the key glycolytic enzyme 6-phosphofructo-2-kinase/2,6-bisphosphatase 3 (PFKFB3). Bioorg Med Chem. (2014) 22:1029–39. doi: 10.1016/j.bmc.2013.12.041
196. Burmistrova O, Olias-Arjona A, Lapresa R, Jimenez-Blasco D, Eremeeva T, Shishov D, et al. Targeting PFKFB3 alleviates cerebral ischemia-reperfusion injury in mice. Sci Rep. (2019) 9:11670–. doi: 10.1038/s41598-019-48196-z
197. Eelen G, Dubois C, Cantelmo AR, Goveia J, Brüning U, DeRan M, et al. Role of glutamine synthetase in angiogenesis beyond glutamine synthesis. Nature. (2018) 561:63–9. doi: 10.1038/s41586-018-0466-7
198. Vizcaino-Castro A, Chen S, Hoogeboom BN, Boerma A, Daemen T, and Oyarce C. Effect of repurposed metabolic drugs on human macrophage polarization and antitumoral activity. Clin Immunol. (2025) 272:110440. doi: 10.1016/j.clim.2025.110440
199. Ma L, Chen C, Zhao C, Li T, Ma L, Jiang J, et al. Targeting carnitine palmitoyl transferase 1A (CPT1A) induces ferroptosis and synergizes with immunotherapy in lung cancer. Signal Transduction Targeted Ther. (2024) 9:64. doi: 10.1038/s41392-024-01772-w
200. Sun L, Wang X, Chen L, Gao Z, Xu S, Hu C, et al. CPT1A mediates chemoresistance in human hypopharyngeal squamous cell carcinoma via ATG16L1-dependent cellular autophagy. Cell Insight. (2023) 2:100127. doi: 10.1016/j.cellin.2023.100127
201. Wang M, Wu D, Liao X, Hu H, Gao J, Meng L, et al. CPT1A-IL-10-mediated macrophage metabolic and phenotypic alterations ameliorate acute lung injury. Clin Trans Med. (2024) 14:e1785. doi: 10.1002/ctm2.1785
202. Calle P, Muñoz A, Sola A, and Hotter G. CPT1a gene expression reverses the inflammatory and anti-phagocytic effect of 7-ketocholesterol in RAW264.7 macrophages. Lipids Health Dis. (2019) 18:215. doi: 10.1186/s12944-019-1156-7
203. Adair TH. An emerging role for adenosine in angiogenesis. Hypertension. (2004) 44:618–20. doi: 10.1161/01.HYP.0000144802.18301.2f
204. Liu Z, Yan S, Wang J, Xu Y, Wang Y, Zhang S, et al. Endothelial adenosine A2a receptor-mediated glycolysis is essential for pathological retinal angiogenesis. Nat Commun. (2017) 8:584. doi: 10.1038/s41467-017-00551-2
205. Aslam M. cAMP/PKA-mediated in vitro angiogenesis: A novel interplay between PKA, RhoA/ROCK, and VEGFR2 signalling. Eur Heart J. (2024) 45. doi: 10.1093/eurheartj/ehae666.3836
206. Liu H, Mei FC, Yang W, Wang H, Wong E, Cai J, et al. Epac1 inhibition ameliorates pathological angiogenesis through coordinated activation of Notch and suppression of VEGF signaling. Sci Adv. (2020) 6:eaay3566. doi: 10.1126/sciadv.aay3566
207. Claps G, Faouzi S, Quidville V, Chehade F, Shen S, Vagner S, et al. The multiple roles of LDH in cancer. Nat Rev Clin Oncol. (2022) 19:749–62. doi: 10.1038/s41571-022-00686-2
208. Du M, Yu T, Zhan Q, Li H, Zou Y, Geng M, et al. Development of a novel lactate dehydrogenase A inhibitor with potent antitumor activity and immune activation. Cancer Sci. (2022) 113:2974–85. doi: 10.1111/cas.15468
209. Clem BF, O’Neal J, Tapolsky G, Clem AL, Imbert-Fernandez Y, Kerr DA 2nd, et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther. (2013) 12:1461–70. doi: 10.1158/1535-7163.MCT-13-0097
210. Babl N, Decking SM, Voll F, Althammer M, Sala-Hojman A, Ferretti R, et al. MCT4 blockade increases the efficacy of immune checkpoint blockade. J Immunother Cancer. (2023) 11. doi: 10.1136/jitc-2023-007349
211. Todenhöfer T, Seiler R, Stewart C, Moskalev I, Gao J, Ladhar S, et al. Selective inhibition of the lactate transporter MCT4 reduces growth of invasive bladder cancer. Mol Cancer Ther. (2018) 17:2746–55. doi: 10.1158/1535-7163.MCT-18-0107
212. Pajak B, Siwiak E, Sołtyka M, Priebe A, Zieliński R, Fokt I, et al. 2-deoxy-d-glucose and its analogs: from diagnostic to therapeutic agents. Int J Mol Sci. (2020) 21:234. doi: 10.3390/ijms21010234
213. Huang Z, Chavda VP, Vora LK, Gajjar N, Apostolopoulos V, Shah N, et al. 2-deoxy-D-glucose and its derivatives for the COVID-19 treatment: an update. Front Pharmacol. (2022) 13. doi: 10.3389/fphar.2022.899633
214. Dey S, Murmu N, Mondal T, Saha I, Chatterjee S, Manna R, et al. Multifaceted entrancing role of glucose and its analogue, 2-deoxy-D-glucose in cancer cell proliferation, inflammation, and virus infection. Biomedicine Pharmacother. (2022) 156:113801. doi: 10.1016/j.biopha.2022.113801
215. Wang Y, Qu C, Liu T, and Wang C. PFKFB3 inhibitors as potential anticancer agents: Mechanisms of action, current developments, and structure-activity relationships. Eur J Med Chem. (2020) 203:112612. doi: 10.1016/j.ejmech.2020.112612
216. Seo M, Kim JD, Neau D, Sehgal I, and Lee YH. Structure-based development of small molecule PFKFB3 inhibitors: a framework for potential cancer therapeutic agents targeting the Warburg effect. PLoS One. (2011) 6:e24179. doi: 10.1371/journal.pone.0024179
217. Telang S, Clem BF, Klarer AC, Clem AL, Trent JO, Bucala R, et al. Small molecule inhibition of 6-phosphofructo-2-kinase suppresses t cell activation. J Transl Med. (2012) 10:95–. doi: 10.1186/1479-5876-10-95
218. Yang Z, Fujii H, Mohan SV, Goronzy JJ, and Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med. (2013) 210:2119–34. doi: 10.1084/jem.20130252
219. Shen Y, Wen Z, Li Y, Matteson EL, Hong J, Goronzy JJ, et al. Metabolic control of the scaffold protein TKS5 in tissue-invasive, proinflammatory T cells. Nat Immunol. (2017) 18:1025–34. doi: 10.1038/ni.3808
220. Oshima N, Ishida R, Kishimoto S, Beebe K, Brender JR, Yamamoto K, et al. Dynamic imaging of LDH inhibition in tumors reveals rapid in vivo metabolic rewiring and vulnerability to combination therapy. Cell Rep. (2020) 30:1798–810.e4. doi: 10.1016/j.celrep.2020.01.039
221. Renner K, Bruss C, Schnell A, Koehl G, Becker HM, Fante M, et al. Restricting glycolysis preserves T cell effector functions and augments checkpoint therapy. Cell Rep. (2019) 29:135–50.e9. doi: 10.1016/j.celrep.2019.08.068
222. Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. (2016) 76:1381–90. doi: 10.1158/0008-5472.CAN-15-1743
223. Zhang H, Liu Y, Liu J, Chen J, Wang J, Hua H, et al. cAMP-PKA/EPAC signaling and cancer: the interplay in tumor microenvironment. J Hematol Oncol. (2024) 17:5. doi: 10.1186/s13045-024-01524-x
224. Skålhegg BS, Funderud A, Henanger HH, Hafte TT, Larsen AC, Kvissel AK, et al. (PKA)–a potential target for therapeutic intervention of dysfunctional immune cells. Curr Drug Targets. (2005) 6:655–64. doi: 10.2174/1389450054863644
225. Vang T, Liu WH, Delacroix L, Wu S, Vasile S, Dahl R, et al. LYP inhibits T-cell activation when dissociated from CSK. Nat Chem Biol. (2012) 8:437–46. doi: 10.1038/nchembio.916
226. Brauneck F, Seubert E, Wellbrock J, Schulze Zur Wiesch J, Duan Y, Magnus T, et al. Combined blockade of TIGIT and CD39 or A2AR enhances NK-92 cell-mediated cytotoxicity in AML. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms222312919
227. An R, Wu C, Tang C, Zhang C, Han F, Xu Z, et al. Blockade of CD73 potentiates radiotherapy antitumor immunity and abscopal effects via STING pathway. Cell Death Discov. (2024) 10:404. doi: 10.1038/s41420-024-02171-4
228. Allard B, Pommey S, Smyth MJ, and Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res. (2013) 19:5626–35. doi: 10.1158/1078-0432.CCR-13-0545
229. Borodovsky A, Barbon CM, Wang Y, Ye M, Prickett L, Chandra D, et al. AZD4635 inhibitor of A2AR signaling rescues immune cell function including CD103+ dendritic cells enhancing anti-tumor immunity. J ImmunoTher Cancer. (2020) 8:e000417. doi: 10.1136/jitc-2019-000417
230. Alvarado-Ortiz E and Sarabia-Sá NM. Hypoxic link between cancer cells and the immune system: The role of adenosine and lactate. Oncol Res. (2025) 33:1803–18. doi: 10.32604/or.2025.065953
231. Sun T, Liu B, Li Y, Wu J, Cao Y, Yang S, et al. Oxamate enhances the efficacy of CAR-T therapy against glioblastoma via suppressing ectonucleotidases and CCR8 lactylation. J Exp Clin Cancer Res. (2023) 42:253. doi: 10.1186/s13046-023-02815-w
232. Singh N, Vayer P, Tanwar S, Poyet J-L, Tsaioun K, and Villoutreix BO. Drug discovery and development: introduction to the general public and patient groups. Front Drug Discov. (2023) 3. doi: 10.3389/fddsv.2023.1201419
233. Sun D, Gao W, Hu H, and Zhou S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm Sin B. (2022) 12:3049–62. doi: 10.1016/j.apsb.2022.02.002
234. Rahman A, Smith FP, Luc P-VT, and Woolley PV. Phase I study and clinical pharmacology of 6-diazo-5-oxo-L-norleucine (DON). Investigational New Drugs. (1985) 3:369–74. doi: 10.1007/BF00170760
235. Earhart RH, Amato DJ, Chang AY, Borden EC, Shiraki M, Dowd ME, et al. Phase II trial of 6-diazo-5-oxo-L-norleucine versus aclacinomycin-A in advanced sarcomas and mesotheliomas. Invest New Drugs. (1990) 8:113–9. doi: 10.1007/bf00216936
236. DeLaBarre B, Gross S, Fang C, Gao Y, Jha A, Jiang F, et al. Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry. (2011) 50:10764–70. doi: 10.1021/bi201613d
237. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. (2014) 13:890–901. doi: 10.1158/1535-7163.MCT-13-0870
238. Vogl DT, Younes A, Stewart K, Orford KW, Bennett M, Siegel D, et al. Phase 1 study of CB-839, a first-in-class, glutaminase inhibitor in patients with multiple myeloma and lymphoma. Blood. (2015) 126:3059. doi: 10.1182/blood.V126.23.3059.3059
239. Stincone A, Prigione A, Cramer T, Wamelink MMC, Campbell K, Cheung E, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. (2015) 90:927–63. doi: 10.1111/brv.12140
240. Diers AR, Broniowska KA, Chang C-F, and Hogg N. Pyruvate fuels mitochondrial respiration and proliferation of breast cancer cells: effect of monocarboxylate transporter inhibition. Biochem J. (2012) 444:561–71. doi: 10.1042/BJ20120294
241. Mazat J-P and Ransac S. The fate of glutamine in human metabolism. The interplay with glucose in proliferating cells. Metabolites. (2019) 9:81. doi: 10.3390/metabo9050081
242. Sumi C, Okamoto A, Tanaka H, Kusunoki M, Shoji T, Uba T, et al. Suppression of mitochondrial oxygen metabolism mediated by the transcription factor HIF-1 alleviates propofol-induced cell toxicity. Sci Rep. (2018) 8:8987–. doi: 10.1038/s41598-018-27220-8
243. Hossain F, Sorrentino C, Ucar DA, Peng Y, Matossian M, Wyczechowska D, et al. Notch signaling regulates mitochondrial metabolism and NF-κB activity in triple-negative breast cancer cells via IKKα-dependent non-canonical pathways. Front Oncol. (2018) 8. doi: 10.3389/fonc.2018.00575
244. Jung K-H, Lee EJ, Park JW, Lee JH, Moon SH, Cho YS, et al. EGF receptor stimulation shifts breast cancer cell glucose metabolism toward glycolytic flux through PI3 kinase signaling. PloS One. (2019) 14:e0221294–e. doi: 10.1371/journal.pone.0221294
245. Pérez-Escuredo J, Dadhich RK, Dhup S, Cacace A, Van Hée VF, De Saedeleer CJ, et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle. (2016) 15:72–83. doi: 10.1080/15384101.2015.1120930
246. Chiu CF, Guerrero JJG, Regalado RRH, Zhou J, Notarte KI, Lu YW, et al. Insights into metabolic reprogramming in tumor evolution and therapy. Cancers (Basel). (2024) 16. doi: 10.3390/cancers16203513
247. Leone RD and Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. (2018) 6:57. doi: 10.1186/s40425-018-0360-8
248. Gubser PM, Wijesinghe S, Heyden L, Gabriel SS, de Souza DP, Hess C, et al. Aerobic glycolysis but not GLS1-dependent glutamine metabolism is critical for anti-tumor immunity and response to checkpoint inhibition. Cell Rep. (2024) 43. doi: 10.1016/j.celrep.2024.114632
249. Verma S, Budhu S, Serganova I, Dong L, Mangarin LM, Khan JF, et al. Pharmacologic LDH inhibition redirects intratumoral glucose uptake and improves antitumor immunity in solid tumor models. J Clin Invest. (2024) 134. doi: 10.1172/JCI177606
250. Gong J, Hendifar A, Tuli R, Chuang J, Cho M, Chung V, et al. Combination systemic therapies with immune checkpoint inhibitors in pancreatic cancer: overcoming resistance to single-agent checkpoint blockade. Clin Trans Med. (2018) 7:e32. doi: 10.1186/s40169-018-0210-9
251. Hermans D, Gautam S, García-Cañaveras JC, Gromer D, Mitra S, Spolski R, et al. Lactate dehydrogenase inhibition synergizes with IL-21 to promote CD8+ T cell stemness and antitumor immunity. Proc Natl Acad Sci. (2020) 117:6047–55. doi: 10.1073/pnas.1920413117
252. Varghese S, Pramanik S, Williams LJ, Hodges HR, Hudgens CW, Fischer GM, et al. The glutaminase inhibitor CB-839 (Telaglenastat) enhances the antimelanoma activity of T-cell–mediated immunotherapies. Mol Cancer Ther. (2021) 20:500–11. doi: 10.1158/1535-7163.MCT-20-0430
253. Gouda MA, Voss MH, Tawbi H, Gordon M, Tykodi SS, Lam ET, et al. A phase I/II study of the safety and efficacy of telaglenastat (CB-839) in combination with nivolumab in patients with metastatic melanoma, renal cell carcinoma, and non-small-cell lung cancer. ESMO Open. (2025) 10:104536. doi: 10.1016/j.esmoop.2025.104536
254. Xue W, Wu K, Guo X, Chen C, Huang T, Li L, et al. The pan-cancer landscape of glutamate and glutamine metabolism: A comprehensive bioinformatic analysis across 32 solid cancer types. Biochim Biophys Acta (BBA) - Mol Basis Disease. (2024) 1870:166982. doi: 10.1016/j.bbadis.2023.166982
255. Benedetti E, Liu EM, Tang C, Kuo F, Buyukozkan M, Park T, et al. A multimodal atlas of tumour metabolism reveals the architecture of gene–metabolite covariation. Nat Metab. (2023) 5:1029–44. doi: 10.1038/s42255-023-00817-8
256. Xiang L, Mou J, Shao B, Wei Y, Liang H, Takano N, et al. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. (2019) 10:40. doi: 10.1038/s41419-018-1291-5
257. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. (2019) 20:1083–97. doi: 10.1016/S1470-2045(19)30274-8
258. Muller AJ, Manfredi MG, Zakharia Y, and Prendergast GC. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin Immunopathol. (2019) 41:41–8. doi: 10.1007/s00281-018-0702-0
259. Goedhart NB and Simon-Molas H. Metabolic profiling of tumor and immune cells integrating seahorse and flow cytometry. In: López-Soto A and Folgueras AR, editors. Cancer Immunosurveillance: Methods and Protocols. Springer US, New York, NY (2025). p. 103–26.
260. Yoo I, Ahn I, Lee J, and Lee N. Extracellular flux assay (Seahorse assay): Diverse applications in metabolic research across biological disciplines. Molecules Cells. (2024) 47:100095. doi: 10.1016/j.mocell.2024.100095
261. Kichloo A, Albosta M, Dahiya D, Guidi JC, Aljadah M, Singh J, et al. Systemic adverse effects and toxicities associated with immunotherapy: A review. World J Clin Oncol. (2021) 12:150–63. doi: 10.5306/wjco.v12.i3.150
262. Vanneman M and Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. (2012) 12:237–51. doi: 10.1038/nrc3237
263. Ren S, Xiong X, You H, Shen J, and Zhou P. The combination of immune checkpoint blockade and angiogenesis inhibitors in the treatment of advanced non-small cell lung cancer. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.689132
264. Lee WS, Yang H, Chon HJ, and Kim C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp Mol Med. (2020) 52:1475–85. doi: 10.1038/s12276-020-00500-y
265. Crocetto F, Ferro M, Buonerba C, Bardi L, Dolce P, Scafuri L, et al. Comparing cardiovascular adverse events in cancer patients: A meta-analysis of combination therapy with angiogenesis inhibitors and immune checkpoint inhibitors versus angiogenesis inhibitors alone. Crit Rev Oncol Hematol. (2023) 188:104059. doi: 10.1016/j.critrevonc.2023.104059
266. Ren X, Deng L, Dong X, Bai Y, Li G, and Wang Y. Adverse reactions of immune checkpoint inhibitors combined with angiogenesis inhibitors: A pharmacovigilance analysis of drug-drug interactions. Int J Immunopathol Pharmacol. (2024) 38:3946320241305390. doi: 10.1177/03946320241305390
Keywords: metabolism, cancer, T cells, macrophages, immunotherapy, tumor microenvironment, angiogenesis
Citation: Wik JA, Berge ER, Stromsnes K and Skålhegg BS (2025) Metabolism in the tumor microenvironment: implications for pathogenesis and therapeutics. Front. Immunol. 16:1610255. doi: 10.3389/fimmu.2025.1610255
Received: 11 April 2025; Accepted: 06 October 2025;
Published: 29 October 2025.
Edited by:
Adil Rasheed, Augusta University, United StatesReviewed by:
Khan M. Imran, University of North Carolina at Chapel Hill, United StatesJesús Jareb Benito-Lopez, National Institute of Respiratory Diseases-Mexico (INER), Mexico
Copyright © 2025 Wik, Berge, Stromsnes and Skålhegg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bjørn Steen Skålhegg, Yi5zLnNrYWxoZWdnQG1lZGlzaW4udWlvLm5v