 Hammodah R. Alfar
Hammodah R. Alfar Sidney W. Whiteheart
Sidney W. Whiteheart- Department of Molecular and Cellular Biochemistry, University of Kentucky, Lexington, KY, United States
As first responders to vascular injury and microbial invasion, platelets play a critical role in hemostasis and immunity. Previous reviews have explored how different platelet receptors can be activated by various bacterial proteins, yet strain-specific perspectives remain underexplored. In this review, we highlight eight bacterial strains that have been associated with thrombosis, each possessing unique proteins or toxins capable of activating or modulating platelets. We discuss some common themes in the molecular interactions between these bacterial components and their effects on platelet function. Some interactions influence platelet aggregation, granule secretion, pro-inflammatory cytokine release, and thrombo-inflammatory responses, while others only mediate bacterial survival. By focusing on strain-specific mechanisms, this review provides an understanding of the different strategies employed by bacteria to manipulate platelet functions. These insights may aid in developing targeted therapeutic interventions to mitigate platelet-associated complications during bacterial infections.
Introduction
As abundant, circulating, vascular guards, platelets are uniquely positioned to detect and respond to vascular damage to stop bleeding and maintain vascular homeostasis. These same qualities make platelets valuable sensors of circulating pathogens. The platelets’ abilities to bind and potentially endocytose pathogens (depending on size), become activated, and secrete a host of bioactive molecules suggest that they can be a pivotal part of the response to an infection. Interactions between platelets and viruses or bacteria have long been known, but their significance to immune responses has only recently become the focus of research.
Bacteria are well-known pathogens that can cause diseases with thrombotic complications, e.g., infective endocarditis, pneumonia, sepsis, and hemolytic uremic syndrome (HUS). They exhibit diverse abilities to interact with platelets and can induce platelet activation, adhesion, aggregation, and secretion. Some species appear to interact with platelets via multiple pathways. Previous reviews have focused on the many platelet surface receptors and how they bind pathogens. Here, we take a bacteria-centric view examining how eight different bacterial pathogens affect platelets through direct and indirect binding, secretion of bacterial proteins, and internalization. We specifically address how each species uses different mechanisms to affect platelet function and cause cardiovascular complications.
Platelets in hemostasis and immune response
Platelets are known for their roles in hemostasis and thrombosis (1–3). Upon tissue damage, they are part of the first response to vascular injury. They prevent further blood loss and limit the invasion of pathogens into circulation. Platelets also contribute to healing the injured area and limiting infection through the release of growth factors and microbicidal peptides from their granules (4). Though lacking nuclei, platelets contain most typical cellular organelles (e.g., mitochondria, endosomes, and granules), which contribute to their function in hemostasis (5, 6). Platelets have three types of granules: alpha (α), dense (δ), and lysosomal, which contain various small molecules, cytokines, chemokines, clotting factors, and enzymes, that are released upon platelet activation, are essential for function, and may contribute to pathology (4, 7–14). In the last decade, an additional secretory granule (T-granules) with tubular morphology has been proposed, which contains toll-like receptor 9 (TLR9) and protein disulfide isomerase (PDI) (15). Platelets are produced by megakaryocytes (MKs) in the bone marrow, where they are equipped with the appropriate organelles and granule contents before being released into the bloodstream (16–19). MKs in the lung have a distinct immune phenotype, but their contribution to platelet production is controversial (20–24). Recently, platelet transcriptomics data from COVID-19 and septic patients suggest that this process is altered by infection and thus may be systemically responsive to vascular health (25). Besides the proteins made by MKs, platelets also endocytose proteins from the circulation and store them in and release them from their granules (e.g., fibrinogen, IgG, albumin, and fibronectin) (26, 27). Thus, platelets are able to both sample their environment via endocytosis and alter it via secretion of their granule content.
Platelets are gaining more attention for their role in immune responses to bacteria, viruses, and parasites (28–30). These roles are not surprising, as platelets express a wide range of cell surface receptors that allow them to interact with different pathogens (31, 32). After pathogen detection, platelets release cytokines, chemokines, and microbicidal peptides that kill or trap pathogens to limit their spread (33, 34). Platelets can also alert immune cells to invading pathogens through released cytokines, chemokines, and microvesicles, which enclose different molecules (e.g., miRNA, RANTES/CCL5, P-selectin, defensins, kinocidins, and thymosins) (35). The microvesicles can alert immune cells, kill some pathogens, and influence gene expression in adjacent cells (e.g., monocytes, smooth muscle cells, and vascular cells) (5). Additionally, surface exposure of granule membrane proteins (i.e., P-selectin) drives direct interactions between platelets and circulating leukocytes (e.g., neutrophils and monocytes), further integrating platelet reactivity with immune system cells. A common facet of systemic infections is reduced platelet count (mild thrombocytopenia), but the severity and underlying mechanisms vary between pathogens (36–38). Consistently, sepsis is associated with platelet consumption and decreased platelet counts, which are prognostic of poorer patient outcomes and higher risks of recurrent infections (36).
Interactions between platelets and bacteria
The interactions between bacteria and platelets are complicated, dynamic, and evolving. Some interactions are part of the host defense system, while others affect bacterial evasion of this response. Certain bacteria directly or indirectly interact with platelets to trigger their activation or manipulate their functions, dysregulate their immune responses, or exacerbate thrombosis. While not the only mechanism by which bacteria form thrombi, bacteria can adhere to endothelial cells, disturb their permeability, and expose the procoagulant sub-endothelium, which is a normal platelet activation for hemostasis. Platelets express an array of receptors and secretory granules that enable them to recognize and respond to different bacterial species (31). Upon encountering bacteria, platelets can rapidly adhere, activate intracellular signaling pathways, and release antimicrobial substances stored within their granules to destroy the invading pathogens. The released molecules can recruit other immune cells, e.g., neutrophils and monocytes, thereby contributing to the overall host defense against bacterial infections. However, bacteria-platelet complexes can shield the bacteria from antibiotics. S. aureus generates biofilms, which shield it from immune responses and render it more resistant to antimicrobial therapies (39). These biofilms contain proteins, polysaccharides, and extracellular DNA, which ensnare and trigger platelets, promoting their aggregation (40). This platelet activation promotes the recruitment of immune cells that secrete cytokines and tissue factors, potentially leading to organ damage if the biofilm is adjacent (39, 41). Infective endocarditis (IE) is a well-known example of a biofilm-associated disease. Its pathogenesis primarily involves the development of septic vegetations—bacterial colonies embedded within fibrin and platelet aggregates that form on heart valves (42). The presence of platelets is essential for in vitro biofilm formation (40). Despite these clear interactions, the significance of platelet-bacterial interaction is unclear and challenging to modulate therapeutically. Platelet responses appear essential for the immune response; however, extensive platelet activation leads to thrombotic events that exacerbate bacterial infection, enhance bacterial survival, and ultimately are detrimental to patients.
The study of platelet-bacteria interactions has a long history. In 1901, Levaditi first reported how rabbit platelets interact with Vibrio cholera, demonstrating that platelets aggregate when incubated with the bacteria (43). However, not until the 1970’s did Clawson and White conduct specific studies of the interactions between platelets and bacteria (44–47). More recently, platelets have been shown to endocytose bacteria such as S. aureus in vivo and in vitro, and the process is enhanced by platelet activation, but the fate of bound/endocytosed bacteria was unclear (48, 49). While this remains an active area of research, some insights were clarified with recent reports showing that platelets can kill some bacterial species (e.g., S. aureus and E. coli), but not others (e.g., S. pneumoniae) (50, 51). Platelets kill E. coli in a manner enhanced by platelet factor 4 (PF4) and anti-PF4/Heparin antibodies (51), while efficient killing of S. aureus requires neither (50). Interestingly, platelets cannot kill S. pneumoniae (50). S. pneumoniae make platelets unresponsive to TRAP-6 stimulation and induce phosphatidylserine (PS) exposure on the platelet surface (50). The latter effect might indicate the conversion of platelets into a procoagulant form or the induction of platelet apoptosis. Such data emphasize the complexity of the interactions between platelets and bacteria, as such interactions depend on the bacterial species and strain. In addition to the pathophysiological complexity of platelets’ interaction with bacteria, experimental variations in the literature often result in contradictory data regarding the reactivity of specific bacterial strains with platelets. Some of the experimental variations are caused by the form of platelets used in experiments [i.e., washed platelets or platelet-rich plasma (PRP)], platelet-to-bacteria ratio, and the platelet activation assay metric (i.e., aggregation or P-selectin exposure). Hence, depending on the bacterial strain, platelets can have a positive or negative impact on bacterial spreading and survival.
Mechanisms of platelet-bacteria interaction
While platelets appear to have several ways to interact with bacteria, there are some common themes that are used by several bacterial strains. Direct interactions between bacteria and specific platelet receptors have been demonstrated. S. sanguinis binds directly to GPIb (52). Other platelet receptors, such as TLRs, FcγRIIA, complement receptors, and integrins (i.e., GPIIb/IIIa), can directly bind specific bacterial species (53). Platelets can bind bacteria indirectly via plasma proteins that are ligands for specific receptors (e.g., von Willebrand Factor (vWF)), which bridge S. aureus and GPIb (54). The amount of these plasma proteins can change during pathological infections, thus altering the potential mechanisms of the interactions (55–58). Bacteria also release specific molecules (e.g., toxins) that interact and affect platelets. E. coli and S. pneumoniae release Shiga toxin and pneumolysin, respectively, which are associated with platelet activation (5). Platelets can also internalize bacteria either directly or via opsonization of IgG-coated bacteria through FcγRIIA (59).
Interestingly, not all these bacterial interactions lead to platelet activation. Some play a supporting role by increasing platelet adhesion under shear conditions (60). Bacterial-induced platelet adhesion/aggregation exhibits distinctive features that differ from the responses to hemostatic and physiological agonists (60). First, unlike agonists such as ADP, bacterial-induced platelet aggregation is an all-or-none process (61). It depends on the concentration of bacteria introduced into the reaction (60). Secondly, platelets respond slowly to bacteria compared to hemostatic agonists (61). Once bacteria are introduced into the reaction, it may require 2–20 min for platelets to become activated and aggregate, in contrast to the <1 min needed when thrombin is added (61). This delay, a.k.a. lag time, varies based on the species, strain, and concentration of the bacteria interacting with platelets (60). Lastly, in contrast to hemostatic activation, where activation of single types of platelet receptors is sufficient, activation by bacteria often involves co-stimulation of the FcγRllA (53).
Platelet interactions with Staphylococcus aureus
S. aureus is a spherical gram-positive bacterium, commensal on the skin and mucous surfaces. Once in the bloodstream, it is a hazardous pathogen capable of inducing necrotizing infections marked by extensive inflammation and tissue damage. This is due to its ability to secrete proteins and enzymes such as proteases, lipases, nucleases, and hyaluronidase, which degrade surrounding tissues (62). S. aureus is equipped with elements that protect it from the immune response generated against tissue damage and is capable of escaping the immune system in several ways, such as the release of chemotaxis inhibitors, leukocyte toxins, complement inactivators, and other antimicrobial peptides (62).
Severe S. aureus infections have been linked to a higher risk of thrombosis, especially deep vein thrombosis (DVT) and disseminated intravascular coagulation (DIC), as the bacteria can have effects on the pro-coagulant and inflammatory pathways and on the anticoagulation factors (63, 64). S. aureus was the first bacterium shown to be endocytosed by platelets, but ADP was required (5, 44, 49). Platelets bind and extend their pseudopods to encapsulate S. aureus, thus limiting bacterial dissemination into the bloodstream (65). Platelets are involved in the eradication of systemic infections (65). Under normal conditions, platelets patrol Kupffer cells (KCs) through a “touch and go” mechanism that involves GPIb and vWF at the KC surface (66). This process is intensified during infection, where platelets are the first cells to arrive in an infected liver, even before neutrophils (66). KCs capture S. aureus and platelets switch from “touch and go” mechanism to “sustained GPIIb/IIIa-dependent adhesion”, which traps the bacteria and limits their spread (66). This process limits liver dysfunction and is essential for S. aureus eradication and host survival (66). Platelets are also involved in the generation of a more specific immune response in a process that depends on GPIb and C3. This directs some bacteria to the spleen to activate CD8α+ dendritic cells (67). S. aureus can also induce platelet aggregation and apoptosis. The aggregation response is unique compared to other bacterial species. S. aureus induces aggregation with a shorter lag time (2–5 min) than other species, such as S. sanguinis or S. gordonii (15–20 min) (68, 69). Apoptosis induction is mediated through the degradation of Bcl-xL protein (anti-apoptosis protein), which increases the exposure of PS, which supports the coagulation system (36).
S. aureus modulates thrombosis through a diverse array of surface elements, which in most cases fall into two categories: Microbial Surface Components Recognizing Adhesive Matrix Molecules (MSCRAMMs) and Secretable Expanded Repertoire Adhesive Molecules (SERAMs). MSCRAMMs are connected to the peptidoglycan through a sortase-anchoring motif, while SERAMs are affixed to the bacterial cell surface through non-covalent means (70). Using these proteins, S. aureus modulates thrombosis (induction or resolution) through the mechanisms discussed below (see Figure 1).
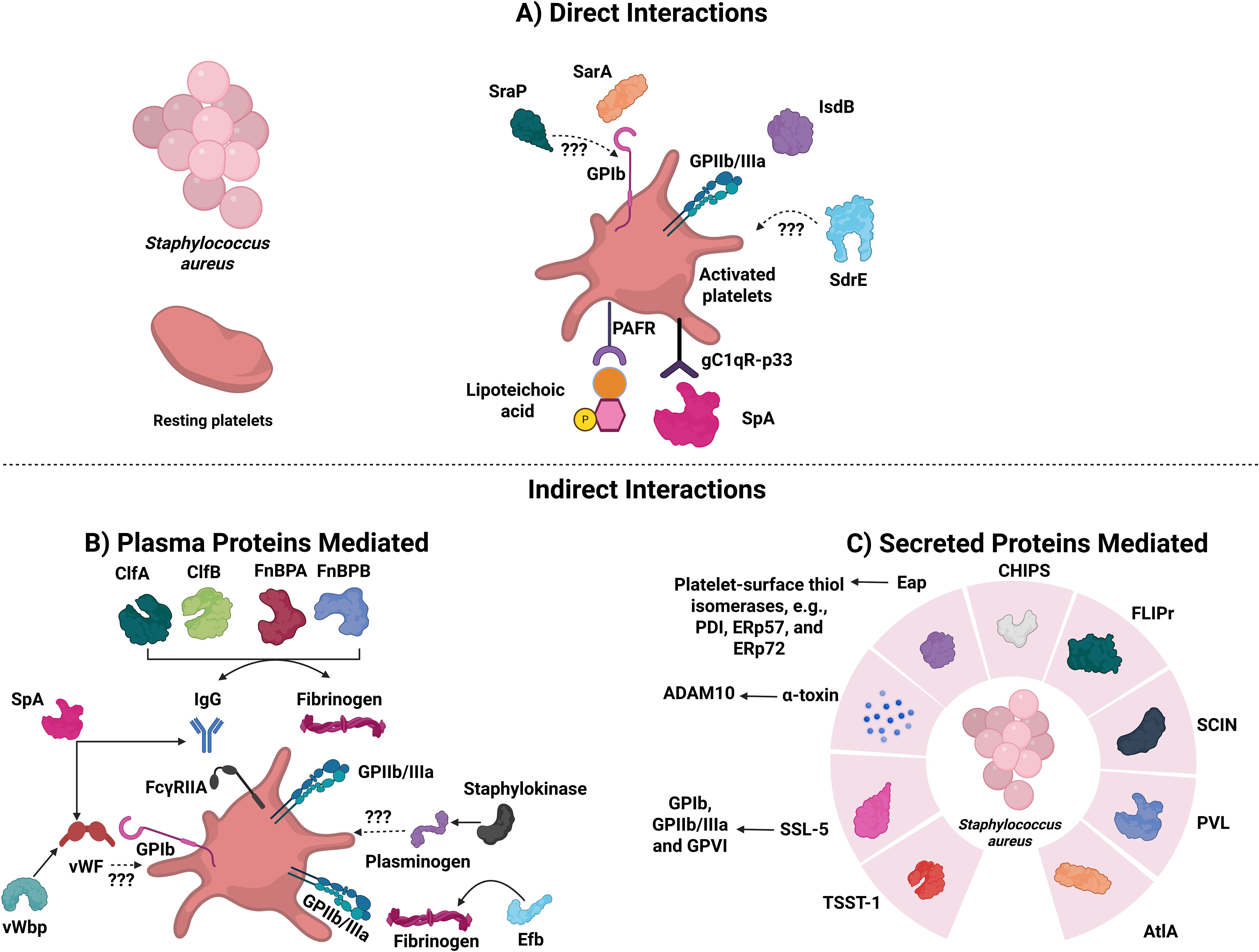
Figure 1. Staphylococcus aureus proteins that are involved in direct or indirect interactions with platelets. The diagram depicts known platelet-S. aureus direct and indirect interactors. (A) S. aureus proteins capable of activating directly. SraP, Serine-rich adhesin protein; SarA, Staphylococcal accessory regulator protein; IsdB, Iron-responsive surface determinant B; SdrE, Serine-aspartate repeat protein; SpA, Staphylococcal protein A; PAFR, Platelet-activating factor receptor. (B) S. aureus proteins capable of activating platelets indirectly (via plasma proteins). Abbreviations are: ClfA and ClfB: Clumping factors A and B; FnBPA and FnBPB: Fibronectin-binding proteins A and B; SpA: Staphylococcal protein A; vWF: von Willebrand Factor; vWbp: vWF-binding protein; and Efb: Extracellular fibrinogen binding protein. (C) S. aureus proteins capable of activating platelets indirectly (via secreted proteins). TSST-1, Toxic shock syndrome toxin-1; SSL-5, Superantigen-like-5; ADAM10, Disintegrin and metalloproteinase domain-containing protein 10; PDI, Protein disulfide isomerase; Eap, Extracellular adherence protein; CHIPS, Chemotaxis inhibitory protein of S. aureus; FLIPr, Formyl peptide receptor-like 1 inhibitory protein; SCIN, Staphylococcal complement inhibitor; PVL, Panton-valentine leucocidin; and AtlA, Major autolysin.
Direct platelet interactions
Platelets express receptors that directly bind S. aureus without an adapter and induce activation. Through GPIb and GPIIb/IIIa, platelets bind to the staphylococcal accessory regulator protein (SarA) and Iron-responsive surface determinant B (IsdB), respectively (39, 71). The presence of both of these proteins is essential for bacterial adherence to platelets and aggregation (71, 72). Platelets also express the complement receptor, gC1qR-p33, which, besides binding to a plethora of plasma proteins, binds to the staphylococcal protein A (SpA) (73, 74). Under resting conditions, platelets express minimal gC1qR levels, but this increases substantially upon activation with TRAP, epinephrine, or ADP (75). This receptor plays a significant role in the pathogenesis of IE, which can be caused by S. aureus (76). Another protein that can mediate binding and activation of platelets directly is the highly glycosylated serine-rich repeat (SRR) protein called Serine-rich adhesin Protein (SraP) (73). The presence of the SRR on S. aureus suggests it could interact with GPIb, but this has not been confirmed (53, 73). Other substances released by platelets, like ADP, thromboxane A2, and PF4, can increase the impact that bacteria have on platelet activation (77). Arman et al. showed that released PF4, upon interaction with S. aureus, is essential for platelet aggregation and reduces its lag time (77). Finally, Serine-aspartate repeat protein (SdrE), associated with S. aureus cell walls, also binds platelets and can induce their aggregation (78).
Indirect platelet interactions involving plasma proteins
S. aureus binds to various extracellular matrix proteins—such as fibrinogen, fibronectin, vWF, laminin, vitronectin, complement proteins, collagen, IgG, and thrombospondin—which can act as bridges, allowing platelets to interact with the bacteria (72, 79–81). SpA, which binds directly to gC1qR-p33, binds both vWF and the Fc region of IgG and can activate both GPIb and FcγRllA, respectively (79–81). Molecules such as Clumping factors A and B (ClfA and ClfB) and fibronectin-binding proteins A and B (FnBPA and FnBPB) bind to fibrinogen and act as bridging molecules (82). These proteins are expressed and produced during various stages of the bacterial growth cycle, bind to different fibrinogen chains, and induce platelet activation and aggregation (82). ClfA binds to the C-terminus of the fibrinogen γ chain, while ClfB binds to the C-terminus of the fibrinogen α chain (82). FnBPA and FnBPB bind to the C-terminus of the γ chain of fibrinogen (73, 82). Plasma IgG can bind to ClfA, ClfB, FnBPA, and FnBPB on S. aureus, leading to platelet activation through FcγRIIA, while complement proteins provide an additional pathway for ClfA- and ClfB-mediated platelet activation, though the specific receptor involved remains unknown (73, 83).
Indirect platelet interactions: via proteins and α-toxin secretion
S. aureus secretes different toxins that can cause organ failure (e.g., leukotoxin ED, superantigens, and α-type phenol-soluble modulins (PSM)) (84), though none directly interacts with platelets (84). S. aureus secrete a small β-barrel pore-forming toxin known as α-toxin (Hla; a.k.a. α-hemolysin due to its ability to lyse red blood cells) that can activate platelets (85). α-Toxin is initially secreted in a water-soluble monomeric form, but once bound to a membrane, it oligomerizes to a heptamer with a diameter between 1–3 nm (5, 86). The formed pore allows the influx of Ca²+, K+, ATP, and smaller molecules (between 1 and 4 kDa) (87). In 1964, Siegel and Cohen showed that α-toxin induces the aggregation of human platelets and a procoagulant response when present at sub-lytic concentrations (88). Recent reports have shown that, in addition to platelet aggregation and activation, α-toxin induces platelet apoptosis, platelet-neutrophil aggregate formation, aggregated platelet deposition in the liver, and initiates platelet protein synthesis (84, 89–92).
α-Toxin binds to ADAM10 on platelet surfaces to form an active, zinc-containing, metalloprotease complex (84). Though α-toxin activates platelets, it eventually destroys them, mediating lysis and impaired thrombus stability (93). Active ADAM10 cleaves the extracellular domain of GPVI, triggers platelet secretion, and impairs the subsequent events of platelet activation, such as platelet aggregation and adhesion to fibrinogen and vWF (94, 95). The interaction between ADAM10 and α-toxin precipitates acute lung injury and hemorrhage in mice, through disruption of GPIIb/IIIa activation, also mediated by GPVI proteolysis (94). As a response to α-toxin, human platelets release β-defensin 1, a granule cargo protein that impairs the growth of S. aureus and induces neutrophil extracellular traps (NETs) formation. NET formation limits S. aureus growth (65). However, they can be degraded by α-toxin (96). S. aureus expresses many virulent factors that thwart the microbicidal activity of NETs (97, 98).
Another toxin released from S. aureus is toxic shock syndrome toxin-1 (TSST-1), which causes thrombocytopenia, platelet activation, and apoptosis in vivo, though the effects on isolated platelets are limited in vitro (99). Certain strains of S. aureus are positive for the panton-valentine leukocidin (PVL) toxin, which damages neutrophils and leads to platelet activation via neutrophil release of α-defensins and the myeloperoxidase product, hypochlorous acid (HOCl), and some HOCl-modified proteins (100). While some of these toxins directly affect platelets, it is unclear whether the damage they cause to other cells also precipitates platelet activation through the production of damage-associated molecular patterns (DAMPs).
S. aureus also activates platelets through the secretion of other proteins (39). Extracellular adherence protein (Eap; a SERAM (101)) binds to the platelet-surface thiol isomerases (e.g., PDI, ERp57, and ERp72), and promotes activation and aggregation (102). Eap also induces the binding of plasma proteins such as fibrinogen, TSP-1, vitronectin, and fibronectin in a time, concentration, and temperature-dependent manner (102). S. aureus secretes chemotaxis inhibitory protein of S. aureus (CHIPS), formyl peptide receptor-like 1 inhibitory protein (FLIPr), staphylococcal complement inhibitor (SCIN), and the major autolysin (AtlA) proteins, which all promote platelet activation and aggregation (70, 103). Superantigen-like-5 (SSL-5) is released by S. aureus and induces platelet aggregation through GPIb and GPIIb/IIIa, as well as increases the platelet adhesion to the endothelial cell matrix (103). This activation is attributed to SSL-5 binding to GPVI (104). However, SSL-5 can bind P-selectin glycoprotein ligand-1 (PSGL-1) to inhibit neutrophil rolling and migration to infected sites (105).
Direct activation of the coagulation system
S. aureus can directly trigger the coagulation cascades through its two prothrombin activators: staphylocoagulase and vWF-Binding Protein (vWbp) (106). Both activate prothrombin, leading to the formation of active staphylothrombin, which produces fibrin and can protect S. aureus against the host’s defense mechanisms (106). vWbp plays a role in the vascular adhesion of S. aureus through two distinct mechanisms: first, by binding to vWF under shear conditions, and second, by activating prothrombin, resulting in the formation of S. aureus-fibrin-platelet aggregates through the interaction with GPIIb/IIIa (79).
S. aureus can prevent thrombosis
S. aureus contains a staphylokinase that promotes the dissolution of blood clots (97). Staphylokinase binds to plasminogen with high affinity and converts the zymogen into plasmin, which cleaves fibrin (106). Additionally, S. aureus secretes an extracellular fibrinogen binding protein (Efb), which binds to fibrinogen via its N-terminus, to C3, through its C-terminus, or directly to P‐selectin on activated platelets (107, 108). These three interactions can lead to different outcomes. The binding of the N-terminus of Efb to P-selectin inhibits platelet interactions with PSGL‐1 on monocytes and neutrophils and their recruitment (107, 108). The inhibitory effect of Efb on platelet function is harmful to the host, as platelet activation is essential to eradicate S. aureus (109). However, the binding of Efb to fibrinogen and C3 is essential for bacterial survival. This enables S. aureus to shield itself from phagocytosis (107, 110). Finally, the cell wall component, lipoteichoic acid from S. aureus, inhibits platelet aggregation in response to physiological agonists and reduces thrombosis in vitro by binding to platelet-activating factor receptor (PAFR) (111, 112). Interestingly, anti-TLR2 antibodies had no effect (111).
Platelet interactions with Streptococcus pneumoniae
S. pneumoniae is a lancet-shaped, gram-positive bacterium that is a leading cause of life-threatening, community-acquired pneumonia (CAP) (5). The severity of CAP correlates with the development of thrombocytopenia (113). Besides CAP, S. pneumoniae can cause sepsis and, on rare occasions, IE (114–116). Recent studies have shown that a significant portion of CAP-associated fatalities may be linked to cardiovascular incidents occurring during infection (117, 118). Such events have various causes, including S. pneumoniae itself and its virulence factors, but there is a growing recognition that platelet activation contributes (119).
In the 1970s, the interaction between platelets and S. pneumoniae was first suggested, but this has not been without controversy (46, 120). Some reports showed platelet activation and aggregation upon the addition of S. pneumoniae, and other reports did not (46, 120). The interaction between platelets and S. pneumoniae is serotype-specific, as some serotypes induced platelet activation, while others did not (46, 120). In contrast to S. aureus, which is killed by platelets and their releasate, S. pneumoniae is not killed by platelets nor their releasates, but it affects the viability of platelets as they become unresponsive to TRAP-6 stimulation and expose PS on their surfaces (50). The latter effect might indicate the conversion into procoagulant platelets or that S. pneumoniae induces platelet apoptosis.
Schrottmaier et al. recently showed that the phosphatidylinositol 3-kinase catalytic subunit (p110β) in platelets is essential for the innate immune responses against S. pneumoniae (121). The presence of p110β in platelets is essential for neutrophil activation and to prevent S. pneumoniae propagation (121). They also found that the inhibition or genetic deletion of p110β impairs the recruitment and phagocytosis of neutrophils and monocytes, hinders their infiltration, and enhances bacterial dissemination (121). Verschoor et al. showed that platelets can bind nonencapsulated S. pneumoniae in a GPIb- and C3-dependent process (67). However, platelets did not bind or direct capsulated S. pneumoniae to the spleen to activate CD8α+ T-cells (67). The presence of the capsule prevents the deposition of complement proteins on S. pneumoniae (67). How S. pneumoniae induces platelet activation is elusive, though several mechanisms are suggested. Figure 2A summarizes how S. pneumoniae interacts/activates platelets and induces prothrombotic/pro-inflammatory conditions through one or a combination of the following mechanisms.
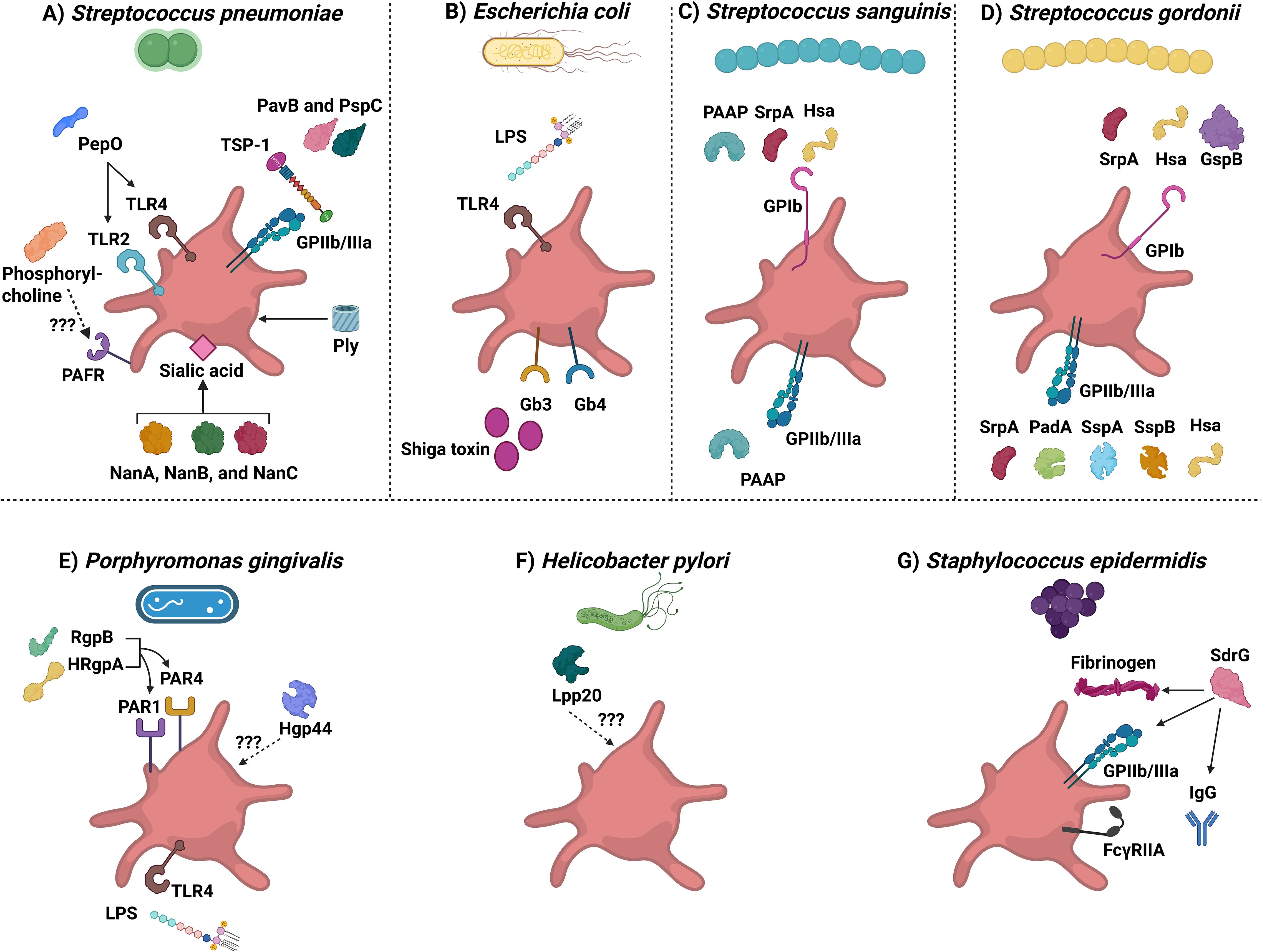
Figure 2. Bacterial strains derived molecules that interact with platelets. The diagram depicts different bacterial strains that can interact with platelets. (A) Streptococcus pneumoniae. Abbreviations are: PepO, pneumoniae endopeptidase O; TLR2 and TLR4, Toll-like receptor 2 and 4; PAFR, Platelet-activating factor receptor; NanA, NanB, and NanC, Neuraminidases A, B, and C; Ply, Pneumolysin; PavB, Pneumococcal adherence and virulence factor B; PspC, Pneumococcal surface protein C; and TSP-1: Thrombospondin-1. (B) Escherichia coli. Abbreviations are: LPS: Lipopolysaccharide; TLR4: Toll-like receptor 4; and Gb3 and 4: Globotriaosylceramide 3 and 4 receptors. (C) Streptococcus sanguinis. Abbreviations are: PAAP: Platelet-associated aggregation protein; SrpA: Serine-rich protein A; and Hsa: hemagglutinin salivary antigen. (D) Streptococcus gordonii. SrpA, Serine-rich protein A; Hsa, hemagglutinin salivary antigen; GspB, Gordonii surface proteins glycosylated streptococcal protein B; PadA, Platelet adherence protein A; SspA and SspB, Streptococcal surface protein A and B). (E) Porphyromonas gingivalis. RgpB, Arginine-specific protease B; HRgpA, high-molecular-weight arginine-specific gingipain (A) Hgp44, Hemagglutinin/adhesion domain of the Arg-gingipain A protein; PAR1 and PAR4, Protease-activated receptor 1 and 4; LPS, Lipopolysaccharide; and TLR4, Toll-like receptor 4. (F) Helicobacter pylori. Lpp20, Low molecular weight antigen. (G) Staphylococcus epidermidis. SdrG, Serine aspartate dipeptide repeat (G).
Pneumolysin directly mediates the activation of platelets
One virulence factor involved in platelet activation is pneumolysin (Ply). This cholesterol-dependent β-barrel cytolysin binds to target cells, assembles into the membrane, and forms pores (122). Ply plays an essential role in CAP by fostering S. pneumoniae colonization and invasion of the upper and lower respiratory tract (123). The effect of Ply on platelets has been controversial. Some reports indicate Ply induces platelet activation, as measured by flow cytometry (P-selectin expression on the surface of activated platelets) and aggregometry (platelet aggregation) (5). Other studies indicate that Ply induces platelet destruction, rendering them unresponsive to stimulation (124). Jahn et al. hypothesized that these controversial results might be due to the assays used (5). If Ply forms pores in platelets, more anti-P-selectin antibodies could get into the granules, making it appear that there was more α-granule exocytosis. Additionally, if the platelets are perforated, more light would pass through them during the aggregometry experiments, incorrectly suggesting an increase in aggregation (5). Thus, both types of assays could yield falsely positive results because the platelets were permeabilized (5, 124). This hypothesis is supported by scanning EM images that show perforated platelets after the addition of Ply (5, 124). Besides its activation, aggregation, and destruction effects on platelets, Ply induces the release of extracellular vesicles from platelets and causes neutrophils to secrete platelet-activating factor (PAF) and thromboxane A2; both are platelet activators (124–128). However, platelet aggregation is not solely due to Ply, as Ply-deficient strains of S. pneumoniae induce platelet aggregation similar to intact strains (129).
Activation of platelet-activating factor receptor on the platelet surface
PAFR is believed to contribute to the binding of S. pneumoniae to endothelial cells (130). PAFR is present on the surface of many cells (e.g., platelets, neutrophils, macrophages, and lymphocytes) and is considered to mediate inflammatory signals (131). The biological ligand of PAFR is PAF, which is released from cells such as neutrophils, macrophages, and endothelial cells (132). Phosphorylcholine, in bacterial membranes, mimics PAF and binds specifically to PAFR (133). Human platelets appear to have two binding sites for PAF (134). Nevertheless, the interactions between the platelet’s PAFR and S. pneumoniae are largely unexplored, and more work is needed to identify the effects of such interaction on the pathology associated with S. pneumoniae.
TLR2 and TLR4 interactions
TLRs are pattern recognition receptors that recognize molecules with pathogen-associated molecular patterns (PAMPs). Platelets express TLRs on their surface (e.g., TLR1, TLR2, TLR4, and TLR6) and in their endosomes (e.g., TLR7 and TLR9) (135). The most studied are TLR2 and TLR4, as they are the most abundant TLRs on the platelet surface (136). While TLR2 binds lipoteichoic acids and peptidoglycan from gram-positive bacteria, TLR4 binds lipopolysaccharide (LPS) from gram-negative bacteria (137). Earlier reports showed that encapsulated S. pneumoniae induces platelet activation and aggregation through TLR2, but unencapsulated S. pneumoniae did not (5). Other reports suggested some encapsulated S. pneumoniae failed to induce platelet activation, while some unencapsulated strains did activate platelets (5). Keane et al. showed that TLR2, but not TLR4, is essential for S. pneumoniae induction of platelet aggregation by using blocking anti-TLR2 and TLR4 monoclonal antibodies, but again this conclusion was challenged as platelet activation was still observed in the presence of TLR blocking antibodies or in single or dual platelet TLR KO mice (TLR2-/-, TLR4-/-, TLR9-/-or TLR2,4-/-) and in MyD88-/- mice (129, 138). Zhang et al. showed that recombinant S. pneumoniae endopeptidase O (PepO), induces an innate immune response in mice that is dependent on both TLR2 and TLR4 (139). Thus, the interactions between TLR2 and TLR4 on platelets and S. pneumoniae and their importance are still controversial.
FcγRIIA activation
Platelets express another receptor that can be activated by S. pneumoniae, FcγRllA. In 2014, Arman et al. showed that platelets are activated by a range of bacteria, including S. pneumoniae, and showed that FcγRllA activation is needed (77). The activation depends on IgG and GPIIb/IIIa involvement and can be potentiated by the released PF4, ADP, and thromboxane A2 from platelets (77). To activate FcγRllA, S. pneumoniae must be opsonized with IgG, and the activation of FcγRllA provides a co-stimulatory signal used by S. pneumoniae and other pathogens (53). However, which component of S. pneumoniae is bound by the opsonizing IgG is undetermined.
GPIIb/IIIa binding and activation
Recent reports indicate a direct binding of S. pneumoniae to platelets via soluble fibrin and thrombospondin-1 (TSP-1) secreted from activated platelets (140). It is suggested that the adherence and virulence factor B (PavB) and surface protein C (PspC) may potentially attach to platelet GPIIb/IIIa in the presence of TSP-1 on activated platelets (141, 142).
Neuraminidases mediate complement activation and blood hemolysis
S. pneumoniae expresses neuraminidases (e.g., NanA, NanB, and NanC) that can remove platelet surface sialic acids (143). These terminal sugars play a crucial role in factor H-mediated complement regulation on both cells and platelets, and their removal can result in uncontrolled complement activation, platelet aggregation, and destruction of red blood cells (143). Thus, S. pneumoniae could activate the complement cascade, induce platelet aggregation, and cause blood hemolysis through these enzymes (143).
Other mechanisms
Other mechanisms have been proposed for S. pneumoniae-mediated activation of platelets. The phage-derived proteins, platelet-binding locus A and platelet-binding locus B (pblA and pblB), interact with platelet membrane gangliosides (119, 144, 145). Hydrogen peroxide, generated by the pneumococcal pyruvate oxidase, has been reported to affect platelet function (119, 146, 147). Finally, endothelial cell dysfunction resulting from S. pneumoniae infection and the production of vasoactive molecules like thromboxane A2 could also activate circulating platelets (148).
Platelet interactions with Escherichia coli
The interactions between gram-negative bacteria and platelets are less studied (59). E. coli is a rod-shaped, gram-negative bacterium first identified by Theodor Escherich in 1885 (149). Most strains are human or animal commensals localized in the gastrointestinal tract (149). However, some have acquired virulent factors, which associate them with several human diseases such as sepsis and HUS (149, 150). HUS presents as a triad of microangiopathic hemolytic anemia, thrombocytopenia, and acute renal failure (137). E. coli can be divided into two main categories: intestinal and extraintestinal pathogenic E. coli (59). Each group has several strains, with enterohemorrhagic E. coli (EHEC) being the most studied strain (59). Consistent with the controversial results when studying bacteria and platelets, the interaction between E. coli and platelets has been extensively debated, specifically on the involvement of platelet TLR4 and FcγRIIA receptors and the effects of LPS on platelets (150–153). The effects are strain-dependent, which was confirmed as platelets or their relesates promote or inhibit the growth of different E. coli strains (154, 155).
Platelets can endocytose E. coli, pre-opsonized with IgG, through the FcγRII receptor to kill them (5). E. coli activates platelets through GPIIb/IIIa (150, 152) and that activation is enhanced in the presence of complement, thromboxane A2, and ADP (150, 152). The released PF4 by activated platelets binds to polyanions on E. coli to form a complex (51), which helps opsonize PF4-coated E. coli and mediate their killing in a GPIIb/IIIa- and FcγRIIa-dependent process (51). These interactions are more complicated, as the interactions between platelets and E. coli vary between individuals (150). The most common molecules that E. coli uses to affect platelet functions are LPS and Shiga toxin (Figure 2B).
Lipopolysaccharide
Gram-negative E. coli has an outer membrane containing LPS, which consists of amphipathic glycoconjugates composed of a hydrophobic lipid domain linked to a central oligosaccharide and an outer polysaccharide. In macrophages, LPS binds to TLR4 in the presence of LPS-binding protein (LBP), CD14, and MD-2 (present on the extracellular domain of TLR4) (156). Platelets express TLR4 (5) and they also express other LPS signaling complex components such as MD2 and MyD88, but not CD14 (157). In children infected with EHEC, LPS is found on the surface of platelets only in children with HUS or before developing HUS; however, it is not found in children who did not develop HUS (153). This indicates platelet activation by LPS may precede HUS development.
In 2005, Andonegui et al. showed, for the first time, that LPS injections of mice induce thrombocytopenia in a TLR4-dependent manner (158). LPS stimulation of TLR4 is essential for TNF-α production, as platelet-depleted mice failed to secrete TNFα after LPS injection (159). This effect was reversed after platelet transfusion (159). Later, it was discovered that LPS induces TLR4-dependent platelet aggregation, α-granule secretion, and dense granule secretion (157). The lipid A fragment of LPS interacts with TLR4 to initiate a pro-inflammatory condition (154). LPS can also modify the protein synthesis in platelets, triggering a pro-inflammatory response through IL-1β splicing, translation, and secretion after caspase-1 processing (160, 161). Released IL-1β was only detected in microparticles (161); but it can amplify the pro-inflammatory condition that can lead to endothelial activation and tissue damage (59, 161). Pires et al. has shown that LPS enhances human platelet activation via a TLR4–PI3K–Akt–ERK1/2–PLA2 signaling pathway (162). Interestingly, platelets possess the ability to recognize and respond to distinct LPS structures, readily differentiating those from E. coli and Salmonella minnesota (163). Specifically, platelet releasate generated in response to E. coli LPS induces a unique cytokine secretion profile in peripheral blood mononuclear cells (PBMCs) that differs from the response elicited by Salmonella minnesota LPS (163). This suggests that platelets can detect danger signals via a single receptor, TLR4, and tailor their responses to differentially modulate immune reactions depending on the specific LPS structure encountered. Recently, Burkard et al. showed direct, in vivo evidence that GPVI is a central mediator of LPS-induced pulmonary thrombo-inflammation—promoting PNC formation, neutrophil recruitment, and NETosis—while its inhibition protects mice from LPS-induced acute lung injury and respiratory failure (164).There has been extensive debate about the in vitro effects of LPS on platelets—specifically, whether it activates them, primes them, or has no effect (150–153, 160, 162, 165–167). The differences might be due to the strain of E. coli, LPS type (smooth vs. rough LPS), concentration of LPS, or technical issues such as the ratio of platelets to bacteria, washed platelets vs. PRP, incubation times, and the assay being used, aggregation or P-selectin exposure. Several reports indicate that LPS isolated from E. coli O157 was the most potent using a TLR4-dependent process to modulate the secretion of stored cytokines by human platelets (153, 168). Yet, Moriarty et al. demonstrated that LPS from E. coli O157 does not induce platelet aggregation; however, viable E. coli O157 did (150). Arbesu et al. showed that E. coli (O18:K1) activates platelets independent of TLR4, GPIIb/IIIa, or plasma proteins (169). It should be noted that other reports suggest that LPS does not activate platelets but primes platelets to respond to lower levels of classical agonists (e.g., thrombin, epinephrine, ADP, or arachidonic acid) (157, 160, 165, 166). However, these controversial results may be attributed to the presence of residual amounts of plasma CD14 in washed platelets. Platelets do not express CD14, but since soluble CD14 (sCD14) is in plasma, the presence of low quantities of plasma or serum could lead to a greater effect of LPS on platelets (170). The requirement of sCD14 might be consistent with the in vivo data regarding the effect of LPS on platelet activation (160, 170, 171). Still, other studies indicate that LPS inhibits platelet aggregation and decreases platelet adhesion to fibrinogen (172, 173).
Shiga toxin
Discovered in 1897 by Kiyoshi Shiga, the strain of E. coli, called Shiga toxin-producing E. coli (STEC), causes vascular endothelial dysfunction by releasing Shiga toxin (59). In 1977, another toxin, verotoxin was discovered and so named because it killed Vero cells in culture (174). Both Shiga toxin and verotoxin are a group of cytotoxic proteins secreted from enteric pathogens that share structures and functions (174). Shiga toxin produced by the enterohemorrhagic E. coli O157:H7 (E. coli expresses somatic (O) antigen 157 and flagella (H) antigen 7) can cause HUS, which is the most common cause of renal failure in children ≤ 3 years (175). Thrombocytopenia might result from the effects of Shiga toxin on platelets as it induces platelet aggregation (176). Shiga toxin induces microthrombi formation in the kidney’s capillaries (specifically, in the glomerular capillaries) and decreases prostacyclin production by the damaged endothelial cells, which promotes platelet aggregation (175). The formed thrombi in the renal vessels significantly affect the efficacy of glomerular filtration, leading to renal failure (175). During HUS, platelets are activated, release their granule content, and are consumed via microthrombosis (59). As such, the diagnosis of HUS can be confused with disseminated intravascular coagulopathy (DIC). The main differences between the two conditions are the prothrombin time (PT), which is within the normal range or slightly extended, and fibrinogen levels, which are also normal or elevated in HUS (177).
Shiga toxin binds to glycosphingolipid receptors on the platelet surface [Globotriaosylceramide 3 and 4 receptors (Gb3 and Gb4)] (178). The interaction of Shiga toxin with platelets has been controversial, as some reports indicate that platelets bind and internalize Shiga toxin, which leads to aggregate formation, activation, morphological changes, and increased fibrinogen binding, while others failed to confirm the interactions (176, 179–184). Using different anticoagulants and methods of platelet isolation and purification, Gosh et al. later showed that the binding of Shiga toxin occurs on the surface of activated platelets but not on resting platelets (178). The method of isolating platelets was key, as the effects of harsher isolation conditions led to their activation and subsequent binding of Shiga toxin (178).
Platelet interactions with Streptococcus sanguinis
S. sanguinis is an opportunistic bacterium that inhabits the human mouth (69). S. sanguinis is the most frequent causative microorganism of IE (185). Upon bloodstream entrance, S. sanguinis can cause several complications, such as adhering to host extracellular matrix protein and/or platelets, colonizing the heart valves and ultimately leading to IE (185, 186). S. sanguinis strains are divided into three categories based on their ability to induce platelet activation ex vivo (187, 188). Type 1: adhere and activate platelets with a short delay time, type 2: do not adhere but activate platelets with a longer delay time; and type 3: do not adhere or activate platelets (187).
The first streptococcal surface protein to bind and activate platelets to be identified was the platelet-associated aggregation protein (PAAP) (61, 189, 190). PAAP has a collagen-like epitope that can activate platelets through an uncharacterized receptor, but it is suggested to be GPIIb/IIIa or GPIb and not GPVI (61, 189–191). The interaction between platelets and S. sanguinis is shear-dependent and seems to be mediated through GPIb (60). Platelets isolated from Bernard Soulier syndrome patients (lacking GPIb on their platelets) failed to respond to S. sanguinis, and blocking antibodies against GPIb inhibited both aggregation and adhesion induced by S. sanguinis (187).
In addition to GPIb, platelet aggregation in response to S. sanguinis relies on GPIIb/IIIa and thromboxane A2. However, aggregation does not occur through direct binding to GPIIb/IIIa, as blocking this receptor with antagonists had no effect (187). Aggregation induced by S. sanguinis is mediated through both GPIIb/IIIa and GPIb, which can occur through either a vWF-independent mechanism or via glycosylated adhesions containing SRRs, such as serine-rich protein A (SrpA) and hemagglutinin salivary antigen (Hsa). Both bind to GPIb in a sialic acid-dependent manner (52, 192). The interaction with GPIb is through SrpA, which is not the only mechanism to activate platelets, as its deletion did not inhibit platelet activation but prolonged the lag time for platelet aggregation (61). In addition to GPIb, S. sanguinis can activate platelets in a complement-dependent process and through FcγRllA as well (193–195). However, certain strains of S. sanguinis stimulate the release of RANTES, PF4, sCD40L, sCD62p, and PDGF-AB from platelets, and other strains do not (196). Thus, it appears that different strains of S. sanguinis can induce platelet activation via different mechanisms, and they further differ in their requirements for thromboxane A2 or ADP for platelet activation (196). Figure 2C summarizes the main S. sanguinis proteins that can activate platelets.
Platelet interactions with Streptococcus gordonii
S. gordonii is a commensal, oral bacterium that causes several complications (i.e., IE) (197). As with other bacteria, the platelet-S. gordonii interaction is strain-specific (61). Some strains have a long lag time in aggregometry experiments, while others have shorter ones or fail to activate platelets altogether (61). S. gordonii possesses SRR adhesin proteins (e.g., gordonii surface proteins glycosylated streptococcal protein B (GspB), Hsa, and SrpA), which bind to a variety of sialylated glycoproteins or the extracellular sialoglycans on GPIbα (198, 199). These SRR adhesins trigger platelet activation by interacting with platelet GPIb in a shear-dependent process (60, 198). While Hsa binds to N-linked sialic acids on GPIb and GPIIb/IIIa, GspB binds to O-linked sialic acids on GPIb on the membrane-proximal mucin-like core of GPIb (200). S. gordonii also induces platelet activation through the platelet adherence protein A (PadA), which specifically interacts with GPIIb/IIIa to induce platelet aggregation and adhesion (201). There are multiple sites of PadA binding to GPIIb/IIIa, resulting in platelet adhesion, dense granule secretion, and spreading on immobilized S. gordonii (201). However, PadA is dispensable for S. gordonii -platelet aggregation but is essential for adhesion of bacteria to platelets (202).
PadA and Hsa are needed for S. gordonii binding to cellular fibronectin and vitronectin, and to promote the formation of biofilms (203). Platelets can bind to immobilized S. gordonii through GPIIb/IIIa and GPIbα through PadA and Hsa, respectively (204). S. gordonii expresses two cell wall-associated polypeptides, streptococcal surface protein A and B (SspA and SspB, belonging to the antigen 1/antigen 2 family) (205). Both of these proteins induce GPIIb/IIIa-dependent aggregation and their deletion extends the lag time for platelet aggregation but does not affect adhesion to platelets (61, 205).
S. gordonii-mediated platelet aggregation also involves FcγRllA (206). The activation of FcγRIIA is dependent on IgG binding and GPIIb/IIIa involvement (77). Platelet releasate is essential, with released ADP and thromboxane A2 being needed for platelet aggregation by S. gordonii (77). Conversely, released PF4 binds to bacteria and reduces the lag time for platelet aggregation by S. gordonii (77). Figure 2D summarizes the main S. gordonii proteins that can activate platelets.
Platelet interactions with Porphyromonas gingivalis
P. gingivalis is a gram-negative, anaerobic bacterium that is the major cause of periodontitis (207). P. gingivalis infection can increase thrombosis risks in patients with atherosclerosis, ischemic stroke, aneurysm, and atrial fibrillation (208–211). P. gingivalis has multiple effects on platelets. Platelets can endocytose P. gingivalis without the need for other agonists (i.e., ADP) (212). P. gingivalis secretes cysteine proteinases called gingipains, which have trypsin-like activity (213) and are essential for P. gingivalis virulence (214). Through gingipains, P. gingivalis enhances pneumococcal adhesion to alveoli by inducing PAFR expression (133). There are two types of gingipains; lysine-specific protease (Kgp) and three variants of the arginine-specific protease (Rgp): RgpAcat, RgpB, and high-molecular-weight arginine-specific gingipain A (HRgpA) (215). RgpB and HRgpA induce platelet activation and aggregation by activating PAR1 and PAR4 (213). The incubation of P. gingivalis with human whole blood increased the potential of thrombosis (216). P. gingivalis induces platelet aggregation, P-selectin expression, platelet neutrophil aggregation, and NET formation (217). This effect can or cannot be modified by the addition of ADP (217). In addition, P. gingivalis increases the free calcium concentration in platelets and induces the release of RANTES from platelets, but at the same time, it can degrade it (218). Rgp and Kgp express a gingipain-derived hemagglutinin domain (Hgp44) at the C-termini, which undergoes autoproteolytic cleavage (219). Hgp44 was shown to be essential for platelet aggregation and activation (219). Also, LPS isolated from P. gingivalis enhances platelet spreading and filopodial extensions (220). The increase of filopodial extensions is mediated by the activation of Cdc42, which is a small GTPase that is essential for filopodial formation (220). Thus, RgpB, HRgpA, Hgp44, and LPS, produced by P. gingivalis can induce platelet aggregation and activation. Figure 2E summarizes the main P. gingivalis proteins that can activate platelets.
Platelet interactions with Helicobacter pylori
H. pylori is a gram-negative bacterium known for its role in peptic ulcers, but it also contributes to cardiovascular diseases (CVDs; e.g., myocardial infarction (MI), atherosclerosis, and immune thrombocytopenic purpura (ITP)) (221). H. pylori-infected patients develop chronic ITP, which is a result of platelet destruction by autoantibodies (221). H. pylori can cause thrombocytopenia without preceding bacteremia through a mechanism mediated by autoantibodies that destroy platelets (5). Consistently, patients treated with H. pylori eradication therapy have increased platelet counts (222). The development of thrombocytopenia involves the H. pylori low-molecular-weight antigen (Lpp20), which binds to platelets and can specifically react with sera from patients with H. pylori to induce chronic ITP (Figure 2F) (223). H. pylori requires the presence of plasma proteins, such as vWF and specific IgGs, to induce platelet aggregation and activation (61, 224). Function-blocking antibodies against vWF or GPIba inhibited H. pylori-induced platelet aggregation (224). This was confirmed in Bernard Soulier Syndrome patients who failed to respond to H. pylori (61). However, as with any bacteria, some strains of H. pylori activate platelets, and some do not (221).
Platelet interactions with Staphylococcus epidermidis
S. epidermidis, is a coagulase-negative strain present on skin that can cause endocarditis and infections of medical-implemented devices (82). S. epidermidis can cause fibrin clot rupture, which leads to infected clot embolization and cause systemic infection (225). In general, coagulase-negative staphylococci are less virulent than positive bacteria such as S. aureus (226). S. epidermidis can, directly and indirectly, interact with platelets through the serine aspartate dipeptide repeat G (SdrG), which is an MSCRAMM (82). The direct interaction involves GPIIb/IIIa and indirect interaction involves fibrinogen, IgG, and FcγRII (82). S. epidermidis can crosslink GPIIb/IIIa and FcγRIIA to activate platelets (82). Figure 2G summarizes the main P. gingivalis proteins that can activate platelets.
Bacterial stimulation of platelets and its clinical significance
While platelet activation plays a key role in helping the body eliminate viral and bacterial infections, excessive platelet stimulation can worsen disease outcomes, particularly in conditions like IE and sepsis. In preclinical studies for IE and sepsis, antiplatelet therapy such as aspirin has shown promising results when used as prophylactic or adjunct therapy (227–231). In both sepsis and IE, platelets are essential in the first line to remove pathogens. However, once the infection is established, platelet activation can worsen the condition. Extensive activation leads to thrombotic events that exacerbate the infection, promote bacterial survival, and ultimately harm the patient. Therefore, in theory, inhibiting platelet activity should be beneficial. As a result, several prospective and retrospective clinical studies have investigated whether antiplatelet therapies can reduce infection-related complications. However, no clear conclusions have yet been reached regarding their effectiveness in preventing or slowing the progression of infection (232–241). The main limitations of these studies include small sample sizes, which make it challenging to achieve statistical significance, as well as significant variability in patient age, underlying health conditions, the duration and dosage of antiplatelet therapy before or after the onset of infection, and the bacterial strains responsible for the disease (242). Despite the complexity of platelet–bacteria interactions, current research provides a solid foundation for future clinical applications. For example, platelet activation markers could be used as early diagnostic or prognostic tools in sepsis or IE, while targeted modulation of platelet responses could help reduce pathological thrombosis without compromising immune defense. An ideal target would be FcγRIIA receptor on the platelet surface since it is needed for pathogen-induced platelet activation (53). Furthermore, understanding specific bacterial virulence factors that alter platelet function opens new opportunities for precision medicine, where therapies are tailored based on the infecting pathogen’s profile. Ultimately, integrating platelet-related findings into clinical practice holds promise for improving the management and outcomes of severe bacterial infections.
Conclusion
Our understanding of platelet functions as immune cells has dramatically expanded, suggesting that they are crucial to responses to microbial infections. The interactions between platelets and pathogens are dynamic, multifaceted, and complicated processes that involve host defense mechanisms and microbial evasion strategies. Bacteria have also evolved mechanisms to exploit platelet functions for their benefit. While some bacteria have surface molecules that facilitate their adhesion and activation of platelets, others do not. Instead, some bacteria use plasma proteins as adapters to connect them with platelets. The most frequently exploited plasma proteins are fibrinogen, IgG, and vWF. These bind to GPIb and GPIIb/IIIa, which are frequently involved in the direct interaction between platelets and bacteria (Figure 3). However, these interactions alone are generally not sufficient to trigger platelet activation. For most bacterial species, platelet activation relies on FcγRIIa signaling. Inhibiting FcγRIIa—either through antibody blockade or depletion of specific IgG—effectively prevents platelet activation. This demonstrates that the interaction between IgG and FcγRIIa is crucial for initiating platelet activation. This is a unique feature of the immune response of platelets. For hemostasis, one type of receptor activation is sufficient to activate platelets.
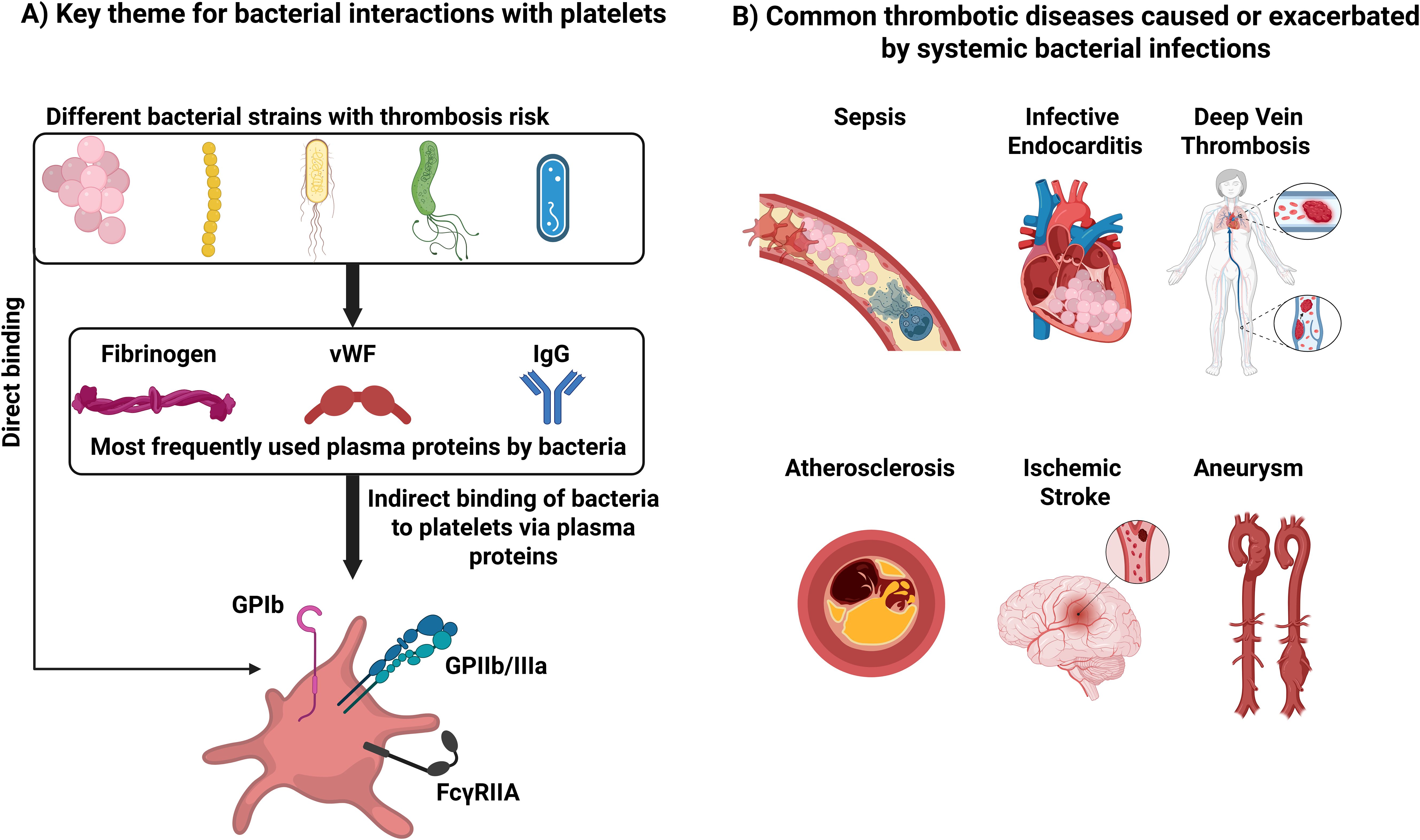
Figure 3. Key themes for bacterial interactions with platelets and cardiovascular disease. (A) Interactions that can lead to thrombosis. (B) Common thrombotic diseases caused or exacerbated by systemic bacterial infections.
Throughout this review, we have highlighted how most of these interactions lead to platelet consumption, dysregulated immune responses, or exacerbation of thrombotic events (Table 1). However, platelets are also involved in immune responses against bacteria and in their eradication. The presence of platelets is essential for TNF- α release following LPS injection in mice. Platelets are important in preventing liver injury during S. aureus infection. Platelets are key to neutrophil activation and preventing S. pneumoniae propagation. Platelets are capable of endocytosing and killing certain bacteria such as S. aureus and E. coli. Despite these insights, the importance of the platelet immune response against bacterial infection is still understudied. A major challenge in the field is the absence of FcγRIIa on mouse platelets. In mice, bacteria-driven platelet activation does not rely on FcγRIIa and is likely to follow mechanisms that might be distinct from humans. Other challenges also include contradictory findings, perhaps more reflective of the assays used or other technical issues such as the form of platelets used (washed platelets or PRP), platelets to bacteria ratio, bacterial strains, incubation times and temperature, incubation condition (static or stirring), platelet isolation methods, and platelet activation assay read out (aggregation or P-selectin exposure). It is hoped that our summary of the strategies bacteria use to affect platelets will help guide the needed research into the mechanisms underlying these effects.
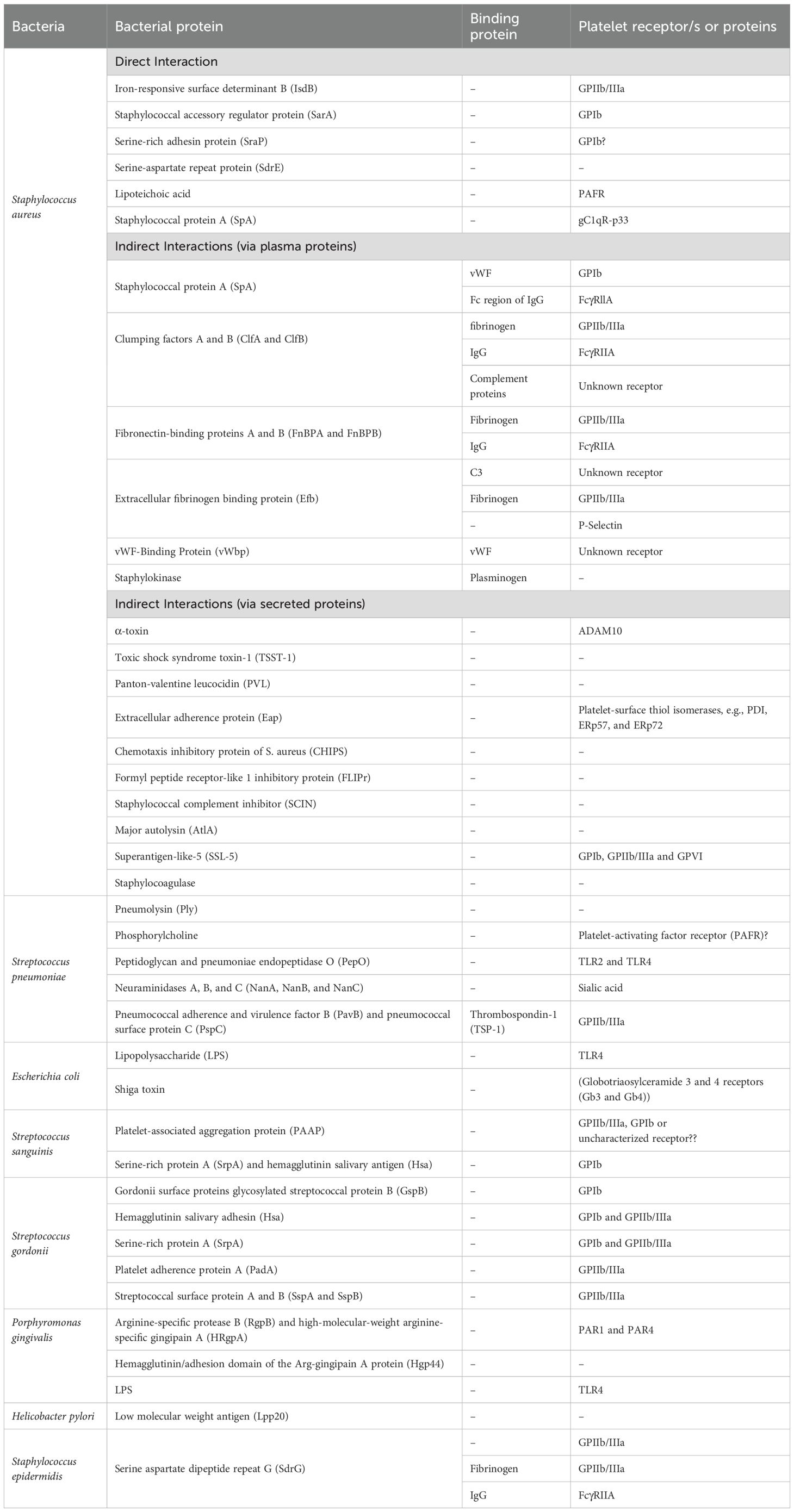
Table 1. Summary of the bacterial proteins that interact with platelets either through direct interactions, released toxins, or via bridging plasma proteins.
Author contributions
HA: Conceptualization, Writing – original draft, Writing – review & editing. SW: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The work was supported by grants from the NIH, NHLBII (HL138179, HL56652 and HL150818), and a Department of Veterans Affairs Merit Award to SW.
Acknowledgments
The authors thank the members of the Whiteheart Laboratory and Dr. Jeremy P. Wood for their careful perusal of this manuscript. The figures were created in BioRender.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Holly SP, Gera N, Wang P, Wilson A, Guan Z, Lin L, et al. Ether lipid metabolism by AADACL1 regulates platelet function and thrombosis. Blood Adv. (2019) 3:3818–28. doi: 10.1182/bloodadvances.2018030767
2. Prakhya KS, Vekaria H, Coenen DM, Omali L, Lykins J, Joshi S, et al. Platelet glycogenolysis is important for energy production and function. Platelets. (2023) 34:2222184. doi: 10.1080/09537104.2023.2222184
3. Smith AN, Joshi S, Chanzu H, Alfar HR, Shravani Prakhya K, and Whiteheart SW. α-Synuclein is the major platelet isoform but is dispensable for activation, secretion, and thrombosis. Platelets. (2023) 34:2267147. doi: 10.1080/09537104.2023.2267147
4. Coenen DM, Alfar HR, and Whiteheart SW. Platelet endocytosis and α-granule cargo packaging are essential for normal skin wound healing. bioRxiv. (2025). doi: 10.1101/2025.02.01.636051
5. Jahn K, Kohler TP, Swiatek L-S, Wiebe S, and Hammerschmidt S. Platelets, bacterial adhesins and the pneumococcus. Cells. (2022) 11:1121. doi: 10.3390/cells11071121
6. Matharu SS, Nordmann CS, Ottman KR, Akkem R, Palumbo D, Cruz DR, et al. Deep learning, 3D ultrastructural analysis reveals quantitative differences in platelet and organelle packing in COVID-19/SARSCoV2 patient-derived platelets. Platelets. (2023) 34:2264978. doi: 10.1080/09537104.2023.2264978
7. Joshi S and Whiteheart SW. The nuts and bolts of the platelet release reaction. Platelets. (2017) 28:129–37. doi: 10.1080/09537104.2016.1240768
8. Cremer SE, Catalfamo JL, Goggs R, Seemann SE, Kristensen AT, Szklanna PB, et al. The canine activated platelet secretome (CAPS): A translational model of thrombin-evoked platelet activation response. Res Pract Thromb Haemostasis. (2021) 5:e12450. doi: 10.1002/rth2.12450
9. Joshi S, Smith A, Alfar H, Prakhya K, Pokrovskaya I, Chanzu H, et al. OC 61.4 manipulating platelet secretion to affect hemostasis. Res Pract Thromb Haemostasis. (2023) 7. doi: 10.1016/j.rpth.2023.100570
10. Joshi S, Smith A, Alfar HR, Prakhya KS, Chanzu H, Whiteheart SW, et al. Hierarchical redundancy and contextual roles of vesicle-associated membrane proteins (VAMPs) in platelet function. Arteriosclerosis Thrombosis Vasc Biol. (2024) 44:A154–4. doi: 10.1161/atvb.44.suppl_1.154
11. Mohammadmoradi S, Driehaus E, Heier K, Alfar H, Lykins J, Levitan B, et al. The protective effect of vamp8 deficiency in aortopathies: potential impact of impaired platelet cargo release. Arteriosclerosis Thrombosis Vasc Biol. (2024) 44:A149–9. doi: 10.1161/atvb.44.suppl_1.149
12. Mohammadmoradi S, Driehaus E, Alfar H, Joshi S, and Whiteheart S. Loss of the v-SNARE VAMP8 protects against aortic aneurysms: implications of impaired platelet cargo release. Circulation. (2024) 150:A4138942–A4138942. doi: 10.1161/circ.150.suppl_1.4138942
13. Mohammadmoradi S, Driehaus ER, Alfar HR, Joshi S, and Whiteheart SW. VAMP8 deficiency attenuates angII-induced abdominal aortic aneurysm formation via platelet reprogramming and enhanced extracellular matrix stability. bioRxiv. (2025). doi: 10.1101/2025.02.03.635525
14. Mohammadmoradi S, Heier K, Driehaus ER, Alfar HR, Tyagi S, McQuerry K, et al. Impact of aspirin therapy on progression of thoracic and abdominal aortic aneurysms. medRxiv. (2025) 2025. doi: 10.1016/j.atherosclerosis.2025.119224
15. Thon JN, Peters CG, Machlus KR, Aslam R, Rowley J, Macleod H, et al. T granules in human platelets function in TLR9 organization and signaling. J Cell Biol. (2012) 198:561–74. doi: 10.1083/jcb.201111136
16. Lykins J, Becker IC, Camacho V, Alfar HR, Park J, Italiano J, et al. Serglycin controls megakaryocyte retention of platelet factor 4 and influences megakaryocyte fate in bone marrow. Blood Adv. (2025) 9:15–28. doi: 10.1182/bloodadvances.2024012995
17. Machlus KR and Italiano JE Jr. The incredible journey: From megakaryocyte development to platelet formation. J Cell Biol. (2013) 201:785–96. doi: 10.1083/jcb.201304054
18. Italiano J Jr. and Shivdasani R. Megakaryocytes and beyond: the birth of platelets. J Thromb Haemostasis. (2003) 1:1174–82. doi: 10.1046/j.1538-7836.2003.00290.x
19. Patel SR, Hartwig JH, and Italiano JE. The biogenesis of platelets from megakaryocyte proplatelets. J Clin Invest. (2005) 115:3348–54. doi: 10.1172/JCI26891
20. Livada AC, Pariser DN, and Morrell CN. Megakaryocytes in the lung: History and future perspectives. Res Pract Thromb Haemostasis. (2023) 7:100053. doi: 10.1016/j.rpth.2023.100053
21. Livada AC, McGrath KE, Malloy MW, Li C, Ture SK, Kingsley PD, et al. Long-lived lung megakaryocytes contribute to platelet recovery in thrombocytopenia models. J Clin Invest. (2024) 134. doi: 10.1172/JCI181111
22. Pariser DN, Hilt ZT, Ture SK, Blick-Nitko SK, Looney MR, Cleary SJ, et al. Lung megakaryocytes are immune modulatory cells. J Clin Invest. (2021) 131. doi: 10.1172/JCI137377
23. Lefrançais E and Looney MR. Platelet biogenesis in the lung circulation. Physiology. (2019) 34:392–401. doi: 10.1152/physiol.00017.2019
24. Lefrançais E, Ortiz-Muñoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. (2017) 544:105–9. doi: 10.1038/nature21706
25. Ajanel A and Middleton EA. Alterations in the megakaryocyte transcriptome impacts platelet function in sepsis and COVID-19 infection. Thromb Res. (2023) 231:247–54. doi: 10.1016/j.thromres.2023.05.015
26. Banerjee M. Platelet endocytosis: roles in hemostasis and innate immunity. University of Kentucky (2017).
27. Banerjee M, Joshi S, Zhang J, Moncman CL, Yadav S, Bouchard BA, et al. Cellubrevin/vesicle-associated membrane protein-3–mediated endocytosis and trafficking regulate platelet functions. Blood J Am Soc Hematol. (2017) 130:2872–83. doi: 10.1182/blood-2017-02-768176
28. Banerjee M, Huang Y, Joshi S, Popa GJ, Mendenhall MD, Wang QJ, et al. Platelets endocytose viral particles and are activated via TLR (toll-like receptor) signaling. Arteriosclerosis thrombosis Vasc Biol. (2020) 40:1635–50. doi: 10.1161/ATVBAHA.120.314180
29. Alonso AL and Cox D. Platelet interactions with viruses and parasites. Platelets. (2015) 26:317–23. doi: 10.3109/09537104.2015.1025376
30. Koupenova M, Livada AC, and Morrell CN. Platelet and megakaryocyte roles in innate and adaptive immunity. Circ Res. (2022) 130:288–308. doi: 10.1161/CIRCRESAHA.121.319821
31. Assinger A. Platelets and infection–an emerging role of platelets in viral infection. Front Immunol. (2014) 5:649. doi: 10.3389/fimmu.2014.00649
32. Alfar HR, Nthenge-Ngumbau DN, Saatman KE, and Whiteheart SW. EcoHIV-infected mice show no signs of platelet activation. Viruses. (2023) 16:55. doi: 10.3390/v16010055
33. Portier I and Campbell RA. Role of platelets in detection and regulation of infection. Arteriosclerosis thrombosis Vasc Biol. (2021) 41:70–8. doi: 10.1161/ATVBAHA.120.314645
34. Ali RA, Wuescher LM, and Worth RG. Platelets: essential components of the immune system. Curr Trends Immunol. (2015) 16:65.
35. Puhm F, Boilard E, and Machlus KR. Platelet extracellular vesicles: beyond the blood. Arteriosclerosis thrombosis Vasc Biol. (2021) 41:87–96. doi: 10.1161/ATVBAHA.120.314644
36. Li C, Li J, and Ni H. Crosstalk between platelets and microbial pathogens. Front Immunol. (2020) 11:1962. doi: 10.3389/fimmu.2020.01962
37. Sim MM, Banerjee M, Hollifield M, Alfar H, Li X, Thornton A, et al. Inflammation drives coagulopathies in SARS-CoV-2 Patients. Blood. (2020) 136:34–5. doi: 10.1182/blood-2020-142848
38. Sim M, Alfar H, Hollifield M, Chung D, Fu X, Banerjee M, et al. HIV-1 and SARS-CoV2 both cause protein s, but through different mechanisms. Res Pract Thromb Haemost. (2021) 5:1509117.
39. Braï MA, Hannachi N, El Gueddari N, Baudoin J-P, Dahmani A, Lepidi H, et al. The role of platelets in infective endocarditis. Int J Mol Sci. (2023) 24:7540. doi: 10.3390/ijms24087540
40. Jung C-J, Yeh C-Y, Shun C-T, Hsu R-B, Cheng H-W, Lin C-S, et al. Platelets enhance biofilm formation and resistance of endocarditis-inducing streptococci on the injured heart valve. J Infect Dis. (2012) 205:1066–75. doi: 10.1093/infdis/jis021
41. Donlan RM. Biofilms: microbial life on surfaces. Emerging Infect Dis. (2002) 8:881. doi: 10.3201/eid0809.020063
42. Hall-Stoodley L, Costerton JW, and Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. (2004) 2:95–108. doi: 10.1038/nrmicro821
43. Levaditi C. Sur l’état de la cytase dans le plasma des animaux normaux et des organismes vaccinés contre le vibrion cholérique. (1901).
45. Clawson C, Rao G, and White JG. Platelet interaction with bacteria. IV. Stimulation of the release reaction. Am J Pathol. (1975) 81:411.
46. Clawson C and White JG. Platelet interaction with bacteria: I. Reaction phases and effects of inhibitors. Am J Pathol. (1971) 65:367.
47. Clawson C and White JG. Platelet interaction with bacteria: II. Fate of the bacteria. Am J Pathol. (1971) 65:381.
48. Hamzeh-Cognasse H, Damien P, Chabert A, Pozzetto B, Cognasse F, and Garraud O. Platelets and infections–complex interactions with bacteria. Front Immunol. (2015) 6:82. doi: 10.3389/fimmu.2015.00082
49. Youssefian T, Drouin A, Massé J-M, Guichard J, and Cramer EM. Host defense role of platelets: engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood J Am Soc Hematol. (2002) 99:4021–9. doi: 10.1182/blood-2001-12-0191
50. Wolff M, Handtke S, Palankar R, Wesche J, Kohler TP, Kohler C, et al. Activated platelets kill Staphylococcus aureus, but not Streptococcus pneumoniae—The role of FcγRIIa and platelet factor 4/heparinantibodies. J Thromb Haemostasis. (2020) 18:1459–68. doi: 10.1111/jth.14814
51. Palankar R, Kohler T, Krauel K, Wesche J, Hammerschmidt S, and Greinacher A. Platelets kill bacteria by bridging innate and adaptive immunity via platelet factor 4 and FcγRIIA. J Thromb Haemostasis. (2018) 16:1187–97. doi: 10.1111/jth.13955
52. Plummer C, Wu H, Kerrigan SW, Meade G, Cox D, and Douglas CI. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br J haematology. (2005) 129:101–9. doi: 10.1111/j.1365-2141.2005.05421.x
53. Cox D. Sepsis–it is all about the platelets. Front Immunol. (2023) 14:1210219. doi: 10.3389/fimmu.2023.1210219
54. Viljoen A, Viela F, Mathelié-Guinlet M, Missiakas D, Pietrocola G, Speziale P, et al. Staphylococcus aureus vWF-binding protein triggers a strong interaction between clumping factor A and host vWF. Commun Biol. (2021) 4:453. doi: 10.1038/s42003-021-01986-6
55. Sim M, Alfar H, Hollifield M, Chung DW, Fu X, Banerjee M, et al. Unfolded von willebrand factor interacts with protein S and limits its anticoagulant activity. Blood. (2022) 140:2710–1. doi: 10.1182/blood-2022-162612
56. Sim MM, Mollica MY, Alfar HR, Hollifield M, Chung DW, Fu X, et al. Unfolded von Willebrand factor binds protein S and reduces anticoagulant activity. Blood Vessels Thromb Hemostasis. (2025) 2:100030. doi: 10.1016/j.bvth.2024.100030
57. Steinert M, Ramming I, and Bergmann S. Impact of von willebrand factor on bacterial pathogenesis. Front Med. (2020) 7:543. doi: 10.3389/fmed.2020.00543
58. D Mahmood DF, De Simone I, Sim M, Alfar HR, Zhang Z, Dai W, et al. Elevated microclots with low D-dimer as an indicator of impaired plasmin generation in patients with viral infections. Blood. (2024) 144:3954–4. doi: 10.1182/blood-2024-198245
59. Ezzeroug Ezzraimi A, Hannachi N, Mariotti A, Rolain J-M, and Camoin-Jau L. Platelets and Escherichia coli: a complex interaction. Biomedicines. (2022) 10:1636. doi: 10.3390/biomedicines10071636
60. Cox D, Kerrigan SW, and Watson SP. Platelets and the innate immune system: mechanisms of bacterial-induced platelet activation. J Thromb Haemostasis. (2011) 9:1097–107. doi: 10.1111/j.1538-7836.2011.04264.x
61. Kerrigan SW and Cox D. Platelet–bacterial interactions. Cell Mol Life Sci. (2010) 67:513–23. doi: 10.1007/s00018-009-0207-z
62. Foster TJ. Immune evasion by staphylococci. Nat Rev Microbiol. (2005) 3:948–58. doi: 10.1038/nrmicro1289
63. Martin E, Cevik C, and Nugent K. The role of hypervirulent Staphylococcus aureus infections in the development of deep vein thrombosis. Thromb Res. (2012) 130:302–8. doi: 10.1016/j.thromres.2012.06.013
64. Franks Z, Campbell RA, de Abreu AV, Holloway JT, Marvin JE, Kraemer BF, et al. Methicillin-resistant Staphylococcus aureus-induced thrombo-inflammatory response is reduced with timely antibiotic administration. Thromb Haemost. (2013) 109:684–95. doi: 10.1160/TH12-08-0543
65. Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, et al. Novel anti-bacterial activities of β-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PloS Pathog. (2011) 7:e1002355. doi: 10.1371/journal.ppat.1002355
66. Wong CH, Jenne CN, Petri B, Chrobok NL, and Kubes P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol. (2013) 14:785–92. doi: 10.1038/ni.2631
67. Verschoor A, Neuenhahn M, Navarini AA, Graef P, Plaumann A, Seidlmeier A, et al. A platelet-mediated system for shuttling blood-borne bacteria to CD8α+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat Immunol. (2011) 12:1194–201. doi: 10.1038/ni.2140
68. Loughman A, Fitzgerald JR, Brennan MP, Higgins J, Downer R, Cox D, et al. Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor A. Mol Microbiol. (2005) 57:804–18. doi: 10.1111/j.1365-2958.2005.04731.x
69. Kerrigan SW. Platelet interactions with bacteria. the non-thrombotic role of platelets in health and Disease. (2015). doi: 10.5772/58357
70. Binsker U, Palankar R, Wesche J, Kohler TP, Prucha J, Burchhardt G, et al. Secreted immunomodulatory proteins of Staphylococcus aureus activate platelets and induce platelet aggregation. Thromb Haemostasis. (2018) 118:745–57. doi: 10.1055/s-0038-1637735
71. Miajlovic H, Zapotoczna M, Geoghegan JA, Kerrigan SW, Speziale P, and Foster TJ. Direct interaction of iron-regulated surface determinant IsdB of Staphylococcus aureus with the GPIIb/IIIa receptor on platelets. Microbiology. (2010) 156:920–8. doi: 10.1099/mic.0.036673-0
72. Shenkman B, Rubinstein E, Cheung AL, Brill GE, Dardik R, Tamarin I, et al. Adherence properties of Staphylococcus aureus under static and flow conditions: roles of agr and sar loci, platelets, and plasma ligands. Infection Immun. (2001) 69:4473–8. doi: 10.1128/IAI.69.7.4473-4478.2001
73. Kerrigan SW. The expanding field of platelet–bacterial interconnections. Platelets. (2015) 26:293–301. doi: 10.3109/09537104.2014.997690
74. Nguyen T, Ghebrehiwet B, and Peerschke EI. Staphylococcus aureus protein A recognizes platelet gC1qR/p33: a novel mechanism for staphylococcal interactions with platelets. Infection Immun. (2000) 68:2061–8. doi: 10.1128/IAI.68.4.2061-2068.2000
75. Peerschke EI, Murphy TK, and Ghebrehiwet B. Activation-dependent surface expression of gC1qR/p33 on human blood platelets. Thromb haemostasis. (2003) 89:331–9.
76. Peerschke EI, Bayer AS, Ghebrehiwet B, and Xiong YQ. gC1qR/p33 blockade reduces Staphylococcus aureus colonization of target tissues in an animal model of infective endocarditis. Infection Immun. (2006) 74:4418–23. doi: 10.1128/IAI.01794-05
77. Arman M, Krauel K, Tilley DO, Weber C, Cox D, Greinacher A, et al. Amplification of bacteria-induced platelet activation is triggered by FcγRIIA, integrin αIIbβ3, and platelet factor 4. Blood J Am Soc Hematol. (2014) 123:3166–74. doi: 10.1182/blood-2013-11-540526
78. O´ Brien L, Kerrigan SW, Kaw G, Hogan M, Penadés J, Litt D, et al. Multiple mechanisms for the activation of human platelet aggregation by Staphylococcus aureus: roles for the clumping factors ClfA and ClfB, the serine–aspartate repeat protein SdrE and protein A. Mol Microbiol. (2002) 44:1033–44. doi: 10.1046/j.1365-2958.2002.02935.x
79. Claes J, Vanassche T, Peetermans M, Liesenborghs L, Vandenbriele C, Vanhoorelbeke K, et al. Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von Willebrand factor–binding protein. Blood J Am Soc Hematol. (2014) 124:1669–76. doi: 10.1182/blood-2014-02-558890
80. Hartleib JR, Köhler N, Dickinson RB, Chhatwal GS, Sixma JJ, Hartford OM, et al. Protein A is the von Willebrand factor binding protein on Staphylococcus aureus. Blood J Am Soc Hematol. (2000) 96:2149–56.
81. O’Seaghdha M, van Schooten CJ, Kerrigan SW, Emsley J, Silverman GJ, Cox D, et al. Staphylococcus aureus protein A binding to von Willebrand factor A1 domain is mediated by conserved IgG binding regions. FEBS J. (2006) 273:4831–41. doi: 10.1111/j.1742-4658.2006.05482.x
82. Brennan MP, Loughman A, Devocelle M, Arasu S, Chubb AJ, Foster T, et al. Elucidating the role of Staphylococcus epidermidis serine–aspartate repeat protein G in platelet activation. J Thromb haemostasis. (2009) 7:1364–72. doi: 10.1111/j.1538-7836.2009.03495.x
83. Fitzgerald JR, Loughman A, Keane F, Brennan M, Knobel M, Higgins J, et al. Fibronectin-binding proteins of Staphylococcus aureus mediate activation of human platelets via fibrinogen and fibronectin bridges to integrin GPIIb/IIIa and IgG binding to the FcγRIIa receptor. Mol Microbiol. (2006) 59:212–30. doi: 10.1111/j.1365-2958.2005.04922.x
84. Surewaard BG, Thanabalasuriar A, Zeng Z, Tkaczyk C, Cohen TS, Bardoel BW, et al. α-Toxin induces platelet aggregation and liver injury during Staphylococcus aureus sepsis. Cell Host Microbe. (2018) 24:271–284.e3. doi: 10.1016/j.chom.2018.06.017
85. Berube BJ and Bubeck Wardenburg J. Staphylococcus aureus α-toxin: nearly a century of intrigue. Toxins. (2013) 5:1140–66. doi: 10.3390/toxins5061140
86. Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, and Gouaux JE. Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore. Science. (1996) 274:1859–65. doi: 10.1126/science.274.5294.1859
87. Bhakdi S and Tranum-Jensen J. Alpha-toxin of staphylococcus aureus. Microbiological Rev. (1991) 55:733–51. doi: 10.1128/mr.55.4.733-751.1991
88. Siegel I and Cohen S. Action of staphylococcal toxin on human platelets. J Infect Dis. (1964) p:488–502. doi: 10.1093/infdis/114.5.488
89. Kraemer BF, Campbell RA, Schwertz H, Franks ZG, Vieira de Abreu A, Grundler K, et al. Bacteria differentially induce degradation of Bcl-xL, a survival protein, by human platelets. Blood J Am Soc Hematol. (2012) 120:5014–20. doi: 10.1182/blood-2012-04-420661
90. Parimon T, Li Z, Bolz DD, McIndoo ER, Bayer CR, Stevens DL, et al. Staphylococcus aureus α-hemolysin promotes platelet-neutrophil aggregate formation. J Infect Dis. (2013) 208:761–70. doi: 10.1093/infdis/jit235
91. Rondina M, Schwertz H, Harris E, Kraemer B, Campbell R, Mackman N, et al. The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J Thromb Haemostasis. (2011) 9:748–58. doi: 10.1111/j.1538-7836.2011.04208.x
92. Schubert S, Schwertz H, Weyrich AS, Franks ZG, Lindemann S, Otto M, et al. Staphylococcus aureus α-toxin triggers the synthesis of B-cell lymphoma 3 by human platelets. Toxins. (2011) 3:120–33. doi: 10.3390/toxins3020120
93. Jahn K, Handtke S, Palankar R, Kohler TP, Wesche J, Wolff M, et al. α-hemolysin of Staphylococcus aureus impairs thrombus formation. J Thromb Haemostasis. (2022) 20:1464–75. doi: 10.1111/jth.15703
94. Powers ME, Becker RE, Sailer A, Turner JR, and Wardenburg JB. Synergistic action of Staphylococcus aureus α-toxin on platelets and myeloid lineage cells contributes to lethal sepsis. Cell Host Microbe. (2015) 17:775–87. doi: 10.1016/j.chom.2015.05.011
95. Bhakdi S, Muhly M, Mannhardt U, Hugo F, Klapettek K, Mueller-Eckhardt C, et al. Staphylococcal α toxin promotes blood coagulation via attack on human platelets. J Exp Med. (1988) 168:527–42. doi: 10.1084/jem.168.2.527
96. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. science. (2004) 303:1532–5. doi: 10.1126/science.1092385
97. Liesenborghs L, Verhamme P, and Vanassche T. Staphylococcus aureus, master manipulator of the human hemostatic system. J Thromb Haemostasis. (2018) 16:441–54. doi: 10.1111/jth.13928
98. Thammavongsa V, Kim HK, Missiakas D, and Schneewind O. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol. (2015) 13:529–43. doi: 10.1038/nrmicro3521
99. Guo M, Yi T, Wang Q, Wang D, Feng P, Kesheng D, et al. TSST-1 protein exerts indirect effect on platelet activation and apoptosis. Platelets. (2022) 33:998–1008. doi: 10.1080/09537104.2022.2026907
100. Niemann S, Bertling A, Brodde MF, Fender AC, Van de Vyver H, Hussain M, et al. Panton-Valentine Leukocidin associated with S. aureus osteomyelitis activates platelets via neutrophil secretion products. Sci Rep. (2018) 8:2185. doi: 10.1038/s41598-018-20582-z
101. Eisenbeis J, Peisker H, Backes CS, Bur S, Hölters S, Thewes N, et al. The extracellular adherence protein (Eap) of Staphylococcus aureus acts as a proliferation and migration repressing factor that alters the cell morphology of keratinocytes. Int J Med Microbiol. (2017) 307:116–25. doi: 10.1016/j.ijmm.2017.01.002
102. Bertling A, Niemann S, Hussain M, Holbrook L, Stanley RG, Brodde MF, et al. Staphylococcal extracellular adherence protein induces platelet activation by stimulation of thiol isomerases. Arteriosclerosis thrombosis Vasc Biol. (2012) 32:1979–90. doi: 10.1161/ATVBAHA.112.246249
103. De Haas C, Weeterings C, Vughs M, De Groot P, Van Strijp J, and Lisman T. Staphylococcal superantigen-like 5 activates platelets and supports platelet adhesion under flow conditions, which involves glycoprotein Ibα and αIIbβ3. J Thromb Haemostasis. (2009) 7:1867–74. doi: 10.1111/j.1538-7836.2009.03564.x
104. Hu H, Armstrong PC, Khalil E, Chen YC, Straub A, Li M, et al. GPVI and GPIbα mediate staphylococcal superantigen-like protein 5 (SSL5) induced platelet activation and direct toward glycans as potential inhibitors. PloS One. (2011) 6:e19190. doi: 10.1371/journal.pone.0019190
105. Bestebroer J, Poppelier MJ, Ulfman LH, Lenting PJ, Denis CV, van Kessel KP, et al. Staphylococcal superantigen-like 5 binds PSGL-1 and inhibits P-selectin–mediated neutrophil rolling. Blood. (2007) 109:2936–43. doi: 10.1182/blood-2006-06-015461
106. Peetermans M, Vanassche T, Liesenborghs L, Lijnen RH, and Verhamme P. Bacterial pathogens activate plasminogen to breach tissue barriers and escape from innate immunity. Crit Rev Microbiol. (2016) 42:866–82. doi: 10.3109/1040841X.2015.1080214
107. Posner MG, Upadhyay A, Abubaker AA, Fortunato TM, Vara D, Canobbio I, et al. Extracellular fibrinogen-binding protein (Efb) from Staphylococcus aureus inhibits the formation of platelet-leukocyte complexes. J Biol Chem. (2016) 291:2764–76. doi: 10.1074/jbc.M115.678359
108. Wallis S, Wolska N, Englert H, Posner M, Upadhyay A, Renné T, et al. A peptide from the staphylococcal protein Efb binds P-selectin and inhibits the interaction of platelets with leukocytes. J Thromb Haemostasis. (2022) 20:729–41. doi: 10.1111/jth.15613
109. Zhang X, Liu Y, Gao Y, Dong J, Mu C, Lu Q, et al. Inhibiting platelets aggregation could aggravate the acute infection caused by Staphylococcus aureus. Platelets. (2011) 22:228–36. doi: 10.3109/09537104.2010.543962
110. Ko Y-P, Kuipers A, Freitag CM, Jongerius I, Medina E, van Rooijen WJ, et al. Phagocytosis escape by a Staphylococcus aureus protein that connects complement and coagulation proteins at the bacterial surface. PloS Pathog. (2013) 9:e1003816. doi: 10.1371/journal.ppat.1003816
111. Waller AK, Sage T, Kumar C, Carr T, Gibbins JM, and Clarke SR. Staphylococcus aureus lipoteichoic acid inhibits platelet activation and thrombus formation via the Paf receptor. J Infect Dis. (2013) 208:2046–57. doi: 10.1093/infdis/jit398
112. Sheu J-R, Lee C-R, Lin C-H, Hsiao G, Ko W-C, Chen Y-C, et al, et al. Mechanisms involved in the antiplatelet activity of Staphylococcus aureus lipoteichoic acid in human platelets. Thromb Haemostasis. (2000) 83:777–84.
113. Luquero-Bueno S, Galván-Román JM, Curbelo J, Lancho-Sánchez A, Roy-Vallejo E, Ortega-Gómez M, et al. Platelet count as an evolution marker of late mortality and cardiovascular events after an episode of community-acquired pneumonia. Eur Respir Soc. (2019) 54:PA4532. doi: 10.1183/13993003.congress-2019.PA4532
114. Yamazaki K, Miura T, Sunohara D, Komatsu T, Mochidome T, Kasai T, et al. Infective endocarditis caused by Streptococcus pneumoniae from sinusitis: A case report. J Cardiol cases. (2022) 25:279–81. doi: 10.1016/j.jccase.2021.11.003
115. de Egea V, Muñoz P, Valerio M, de Alarcón A, Lepe JA, Miró JM, et al. Characteristics and outcome of Streptococcus pneumoniae endocarditis in the XXI century: a systematic review of 111 cases (2000–2013). Medicine. (2015) 94. doi: 10.1097/MD.0000000000001562
116. Ahl J, Littorin N, Forsgren A, Odenholt I, Resman F, and Riesbeck K. High incidence of septic shock caused by Streptococcus pneumoniae serotype 3-a retrospective epidemiological study. BMC Infect Dis. (2013) 13:1–7. doi: 10.1186/1471-2334-13-492
117. Krüger S and Frechen D. Cardiovascular complications in community-acquired pneumonia. Clin Pulmonary Med. (2015) 22:62–7. doi: 10.1097/CPM.0000000000000080
118. Desai A, Aliberti S, Amati F, Stainer A, and Voza A. Cardiovascular complications in community-acquired pneumonia. Microorganisms. (2022) 10:2177. doi: 10.3390/microorganisms10112177
119. Feldman C and Anderson R. Platelets and their role in the pathogenesis of cardiovascular events in patients with community-acquired pneumonia. Front Immunol. (2020) 11:577303. doi: 10.3389/fimmu.2020.577303
120. Guckian JC. Effect of pneumococci on blood clotting, platelets, and polymorphonuclear leukocytes. Infection Immun. (1975) 12:910–8. doi: 10.1128/iai.12.4.910-918.1975
121. Schrottmaier WC, Kral-Pointner JB, Salzmann M, Mussbacher M, Schmuckenschlager A, Pirabe A, et al. Platelet p110β mediates platelet-leukocyte interaction and curtails bacterial dissemination in pneumococcal pneumonia. Cell Rep. (2022) 41. doi: 10.1016/j.celrep.2022.111614
122. Marshall JE, Faraj BH, Gingras AR, Lonnen R, Sheikh MA, El-Mezgueldi M, et al. The crystal structure of pneumolysin at 2.0 Å resolution reveals the molecular packing of the pre-pore complex. Sci Rep. (2015) 5:13293. doi: 10.1038/srep13293
123. Anderson R, Nel JG, and Feldman C. Multifaceted role of pneumolysin in the pathogenesis of myocardial injury in community-acquired pneumonia. Int J Mol Sci. (2018) 19:1147. doi: 10.3390/ijms19041147
124. Jahn K, Handtke S, Palankar R, Weissmueller S, Nouailles G, Kohler TP, et al. Pneumolysin induces platelet destruction, not platelet activation, which can be prevented by immunoglobulin preparations in vitro. Blood Adv. (2020) 4:6315–26. doi: 10.1182/bloodadvances.2020002372
125. Ohkuni H, Nagamune H, Ozaki N, Tabata A, Todome Y, Watanabe Y, et al. Characterization of recombinant Streptococcus mitis-derived human platelet aggregation factor. Apmis. (2012) 120:56–71. doi: 10.1111/j.1600-0463.2011.02813.x
126. Nel JG, Durandt C, Mitchell TJ, Feldman C, Anderson R, and Tintinger GR. Pneumolysin mediates platelet activation in vitro. Lung. (2016) 194:589–93. doi: 10.1007/s00408-016-9900-5
127. Letsiou E, Teixeira Alves LG, Felten M, Mitchell TJ, Müller-Redetzky HC, Dudek SM, et al. Neutrophil-derived extracellular vesicles activate platelets after pneumolysin exposure. Cells. (2021) 10:3581. doi: 10.3390/cells10123581
128. Nel JG, Durandt C, Theron AJ, Tintinger GR, Pool R, Richards GA, et al. Pneumolysin mediates heterotypic aggregation of neutrophils and platelets in vitro. J Infection. (2017) 74:599–608. doi: 10.1016/j.jinf.2017.02.010
129. Keane C, Tilley D, Cunningham A, Smolenski A, Kadioglu A, Cox D, et al. Invasive Streptococcus pneumoniae trigger platelet activation via Toll-like receptor 2. J Thromb Haemostasis. (2010) 8:2757–65. doi: 10.1111/j.1538-7836.2010.04093.x
130. Iovino F, Brouwer MC, van de Beek D, Molema G, and Bijlsma JJ. Signalling or binding: The role of the platelet-activating factor receptor in invasive pneumococcal disease. Cell Microbiol. (2013) 15:870–81. doi: 10.1111/cmi.12129
131. Harishkumar R, Hans S, Stanton JE, Grabrucker AM, Lordan R, and Zabetakis I. Targeting the platelet-activating factor receptor (PAF-R): antithrombotic and anti-atherosclerotic nutrients. Nutrients. (2022) 14:4414. doi: 10.3390/nu14204414
132. Rijneveld AW, Weijer S, Florquin S, Speelman P, Shimizu T, Ishii S, et al. Improved host defense against pneumococcal pneumonia in platelet-activating factor receptor-deficient mice. J Infect Dis. (2004) 189:711–6. doi: 10.1086/381392
133. Kamio N, Hayata M, Tamura M, Tanaka H, and Imai K. Porphyromonas gingivalis enhances pneumococcal adhesion to human alveolar epithelial cells by increasing expression of host platelet-activating factor receptor. FEBS Lett. (2021) 595:1604–12. doi: 10.1002/1873-3468.14084
134. Chao W and Olson MS. Platelet-activating factor: receptors and signal transduction. Biochem J. (1993) 292:617–29. doi: 10.1042/bj2920617
135. Mancuso ME and Santagostino E. Platelets: much more than bricks in a breached wall. Br J haematology. (2017) 178:209–19. doi: 10.1111/bjh.14653
136. Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, and Garraud O. Evidence of Toll-like receptor molecules on human platelets. Immunol Cell Biol. (2005) 83:196–8. doi: 10.1111/j.1440-1711.2005.01314.x
137. Nagai Y and Takatsu K. Role of the immune system in obesity-associated inflammation and insulin resistance. In: Nutrition in the Prevention and Treatment of Abdominal Obesity, vol. p. Academic Press, Cambridge, MA, USA (2014). p. 281–93.
138. de Stoppelaar SF, Claushuis TA, Schaap MC, Hou B, van der Poll T, Nieuwland R, et al. Toll-like receptor signalling is not involved in platelet response to Streptococcus pneumoniae in vitro or in vivo. PloS One. (2016) 11:e0156977. doi: 10.1371/journal.pone.0156977
139. Zhang H, Kang L, Yao H, He Y, Wang X, Xu W, et al. Streptococcus pneumoniae endopeptidase O (PepO) elicits a strong innate immune response in mice via TLR2 and TLR4 signaling pathways. Front Cell infection Microbiol. (2016) 6:23. doi: 10.3389/fcimb.2016.00023
140. Niemann S, Kehrel BE, Heilmann C, Rennemeier C, Peters G, and Hammerschmidt S. Pneumococcal association to platelets is mediated by soluble fibrin and supported by thrombospondin-1. Thromb haemostasis. (2009) 102:735–42. doi: 10.1182/blood.2020005382
141. Binsker U, Kohler TP, Krauel K, Kohler S, Schwertz H, and Hammerschmidt S. Pneumococcal adhesins PavB and PspC are important for the interplay with human thrombospondin-1. J Biol Chem. (2015) 290:14542–55. doi: 10.1074/jbc.M114.623876
142. Binsker U, Kohler TP, Krauel K, Kohler S, Habermeyer J, Schwertz H, et al. Serotype 3 pneumococci sequester platelet-derived human thrombospondin-1 via the adhesin and immune evasion protein Hic. J Biol Chem. (2017) 292:5770–83. doi: 10.1074/jbc.M116.760504
143. Syed S, Hakala P, Singh AK, Lapatto HA, King SJ, Meri S, et al. Role of pneumococcal NanA neuraminidase activity in peripheral blood. Front Cell Infection Microbiol. (2019) p:218. doi: 10.3389/fcimb.2019.00218
144. Mitchell J and Sullam PM. Streptococcus mitis phage-encoded adhesins mediate attachment to α2-8-linked sialic acid residues on platelet membrane gangliosides. Infection Immun. (2009) 77:3485–90. doi: 10.1128/IAI.01573-08
145. Tunjungputri RN, Mobegi FM, Cremers AJ, van der Gaast-de Jongh CE, Ferwerda G, Meis JF, et al. Phage-derived protein induces increased platelet activation and is associated with mortality in patients with invasive pneumococcal disease. MBio. (2017) 8. doi: 10.1128/mbio.01984-16
146. Anderson R and Feldman C. Review manuscript: Mechanisms of platelet activation by the pneumococcus and the role of platelets in community-acquired pneumonia. J Infection. (2017) 75:473–85. doi: 10.1016/j.jinf.2017.09.013
147. Brissac T, Shenoy AT, Patterson LA, and Orihuela CJ. Cell invasion and pyruvate oxidase-derived H2O2 are critical for Streptococcus pneumoniae-mediated cardiomyocyte killing. Infection Immun. (2018) 86. doi: 10.1128/iai.00569-17
148. Rae N, Finch S, and Chalmers JD. Cardiovascular disease as a complication of community-acquired pneumonia. Curr Opin pulmonary Med. (2016) 22:212–8. doi: 10.1097/MCP.0000000000000261
149. Lim JY, Yoon JW, and Hovde CJ. A brief overview of Escherichia coli O157: H7 and its plasmid O157. J Microbiol Biotechnol. (2010) 20:5. doi: 10.4014/jmb.0908.08007
150. Moriarty R, Cox A, McCall M, Smith S, and Cox D. Escherichia coli induces platelet aggregation in an FcγRIIa-dependent manner. J Thromb Haemostasis. (2016) 14:797–806. doi: 10.1111/jth.13226
151. Matus V, Valenzuela JG, Hidalgo P, Pozo LM, Panes O, Wozniak A, et al. Human platelet interaction with E. coli O111 promotes tissue-factor-dependent procoagulant activity, involving Toll like receptor 4. PloS One. (2017) 12:e0185431. doi: 10.1371/journal.pone.0185431
152. Watson CN, Kerrigan SW, Cox D, Henderson IR, Watson SP, and Arman M. Human platelet activation by Escherichia coli: roles for FcγRIIA and integrin αIIbβ3. Platelets. (2016) 27:535–40. doi: 10.3109/09537104.2016.1148129
153. Staåhl A-l, Svensson M, Mörgelin M, Svanborg C, Tarr PI, Mooney JC, et al. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood. (2006) 108:167–76. doi: 10.1182/blood-2005-08-3219
154. Ezzeroug Ezzraimi A, Hannachi N, Mariotti A, Rolland C, Levasseur A, Baron SA, et al. The antibacterial effect of platelets on Escherichia coli strains. Biomedicines. (2022) 10:1533. doi: 10.3390/biomedicines10071533
155. Tohidnezhad M, Varoga D, Wruck CJ, Podschun R, Sachweh BH, Bornemann J, et al. Platelets display potent antimicrobial activity and release human beta-defensin 2. Platelets. (2012) 23:217–23. doi: 10.3109/09537104.2011.610908
156. Ciesielska A, Matyjek M, and Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. (2021) 78:1233–61. doi: 10.1007/s00018-020-03656-y
157. Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, et al. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J Immunol. (2009) 182:7997–8004. doi: 10.4049/jimmunol.0802884
158. Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, and Kubes P. Platelets express functional Toll-like receptor-4. Blood. (2005) 106:2417–23. doi: 10.1182/blood-2005-03-0916
159. Aslam R, Speck ER, Kim M, Crow AR, Bang KA, Nestel FP, et al. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-α production in vivo. Blood. (2006) 107:637–41. doi: 10.1182/blood-2005-06-2202
160. Shashkin PN, Brown GT, Ghosh A, Marathe GK, and McIntyre TM. Lipopolysaccharide is a direct agonist for platelet RNA splicing. J Immunol. (2008) 181:3495–502. doi: 10.4049/jimmunol.181.5.3495
161. Brown GT and McIntyre TM. Lipopolysaccharide signaling without a nucleus: kinase cascades stimulate platelet shedding of proinflammatory IL-1β–rich microparticles. J Immunol. (2011) 186:5489–96. doi: 10.4049/jimmunol.1001623
162. Lopes Pires ME, Clarke SR, Marcondes S, and Gibbins JM. Lipopolysaccharide potentiates platelet responses via toll-like receptor 4-stimulated Akt-Erk-PLA2 signalling. PloS One. (2017) 12:e0186981. doi: 10.1371/journal.pone.0186981
163. Berthet J, Damien P, Hamzeh-Cognasse H, Arthaud C-A, Eyraud M-A, Zéni F, et al. Human platelets can discriminate between various bacterial LPS isoforms via TLR4 signaling and differential cytokine secretion. Clin Immunol. (2012) 145:189–200. doi: 10.1016/j.clim.2012.09.004
164. Burkard P, Schonhart C, Vögtle T, Köhler D, Tang L, Johnson D, et al. A key role for platelet GPVI in neutrophil recruitment, migration, and NETosis in the early stages of acute lung injury. Blood. (2023) 142:1463–77. doi: 10.1182/blood.2023019940
165. Montrucchio G, Bosco O, Del Sorbo L, Pecetto PF, Lupia E, Goffi A, et al. Mechanisms of the priming effect of low doses of lipopolysaccharides on leukocyte-dependent platelet aggregation in whole blood. Thromb haemostasis. (2003) 90:872–81. doi: 10.1160/TH03-02-0085
166. Rivadeneyra L, Carestia A, Etulain J, Pozner RG, Fondevila C, Negrotto S, et al. Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB. Thromb Res. (2014) 133:235–43. doi: 10.1016/j.thromres.2013.11.028
167. Claushuis TA, Van Der Veen AI, Horn J, Schultz MJ, Houtkooper RH, Van’T Veer C, et al. Platelet Toll-like receptor expression and activation induced by lipopolysaccharide and sepsis. Platelets. (2019) 30:296–304. doi: 10.1080/09537104.2018.1445841
168. Cognasse F, Hamzeh‐Cognasse H, Lafarge S, Delezay O, Pozzetto B, McNicol A, et al. Toll-like receptor 4 ligand can differentially modulate the release of cytokines by human platelets. Br J haematology. (2008) 141:84–91. doi: 10.1111/j.1365-2141.2008.06999.x
169. Arbesu I, Bucsaiova M, Fischer M, and Mannhalter C. Platelet-borne complement proteins and their role in platelet–bacteria interactions. J Thromb Haemostasis. (2016) 14:2241–52. doi: 10.1111/jth.13495
170. D’Atri LP and Schattner MA. Platelet toll-like receptors in thromboinflammation. Front Biosci (Landmark Ed). (2017) 22:1867–83. doi: 10.2741/4576
171. Damien P, Cognasse F, Eyraud M-A, Arthaud C-A, Pozzetto B, Garraud O, et al. LPS stimulation of purified human platelets is partly dependent on plasma soluble CD14 to secrete their main secreted product, soluble-CD40-Ligand. BMC Immunol. (2015) 16:1–7. doi: 10.1186/s12865-015-0067-2
172. Hashimoto K, Jayachandran M, Owen WG, and Miller VM. Aggregation and microparticle production through toll-like receptor 4 activation in platelets from recently menopausal women. J Cardiovasc Pharmacol. (2009) 54:57. doi: 10.1097/FJC.0b013e3181ab373d
173. Morganti RP, Cardoso MH, Pereira FG, Lorand-Metze I, Nucci GD, Marcondes S, et al. Mechanisms underlying the inhibitory effects of lipopolysaccharide on human platelet adhesion. Platelets. (2010) 21:260–9. doi: 10.3109/09537101003637240
174. Konowalchuk J, Speirs J, and Stavric S. Vero response to a cytotoxin of Escherichia coli. Infection Immun. (1977) 18:775–9. doi: 10.1128/iai.18.3.775-779.1977
175. Amirlak I and Amirlak B. Haemolytic uraemic syndrome: an overview. Nephrology. (2006) 11:213–8. doi: 10.1111/j.1440-1797.2006.00556.x
176. Rose P, Armour J, Williams C, and Hill F. Verotoxin and neuraminidase induced platelet aggregating activity in plasma: their possible role in the pathogenesis of the haemolytic uraemic syndrome. J Clin Pathol. (1985) 38:438–41. doi: 10.1136/jcp.38.4.438
177. Tarr PI. Shiga toxin-associated hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: distinct mechanisms of pathogenesis. Kidney Int. (2009) 75:S29–32. doi: 10.1038/ki.2008.615
178. Ghosh S, Polanowska‐Grabowska R, Fujii J, Obrig T, and Gear A. Shiga toxin binds to activated platelets. J Thromb Haemostasis. (2004) 2:499–506. doi: 10.1111/j.1538-7933.2004.00638.x
179. Karpman D, Papadopoulou D, Nilsson K, Sjögren A-C, Mikaelsson C, and Lethagen S. Platelet activation by Shiga toxin and circulatory factors as a pathogenetic mechanism in the hemolytic uremic syndrome. Blood J Am Soc Hematol. (2001) 97:3100–8. doi: 10.1182/blood.V97.10.3100
180. Cooling LL, Walker KE, Gille T, and Koerner TA. Shiga toxin binds human platelets via globotriaosylceramide (Pk antigen) and a novel platelet glycosphingolipid. Infection Immun. (1998) 66:4355–66. doi: 10.1128/IAI.66.9.4355-4366.1998
181. Yagi H, Narita N, Matsumoto M, Sakurai Y, Ikari H, Yoshioka A, et al. Enhanced low shear stress induced platelet aggregation by Shiga-like toxin 1 purified from Escherichia coli O157. Am J Hematol. (2001) 66:105–15. doi: 10.1002/1096-8652(200102)66:2<105::AID-AJH1025>3.0.CO;2-1
182. Thorpe C, Flaumenhaft R, Hurley B, Jacewicz M, Acheson D, and Keusch G. Shiga toxins do not directly stimulate alpha-granule secretion or enhance aggregation of human platelets. Acta haematologica. (1999) 102:51–5. doi: 10.1159/000040968
183. Viisoreanu D, Polanowska-Grabowska R, Suttitanamongkol S, Obrig TG, and Gear AR. Human platelet aggregation is not altered by Shiga toxins 1 or 2. Thromb Res. (2000) 98:403–10. doi: 10.1016/S0049-3848(00)00191-2
184. Yoshimura K, Fujii J, Yutsudo T, Kikuchi R, Soejima T, Shirahata S, et al. No direct effects of Shiga toxin 1 and 2 on the aggregation of human platelets in vitro. Thromb haemostasis. (1998) 80:529–30. doi: 10.1055/s-0037-1615248
185. Martini AM, Moricz BS, Ripperger AK, Tran PM, Sharp ME, Forsythe AN, et al. Association of novel Streptococcus sanguinis virulence factors with pathogenesis in a native valve infective endocarditis model. Front Microbiol. (2020) 11:10. doi: 10.3389/fmicb.2020.00010
186. Martini AM, Moricz BS, Woods LJ, and Jones BD. Type IV pili of Streptococcus sanguinis contribute to pathogenesis in experimental infective endocarditis. Microbiol Spectr. (2021) 9:e01752–21. doi: 10.1128/Spectrum.01752-21
187. Kerrigan SW, Douglas I, Wray A, Heath J, Byrne MF, Fitzgerald D, et al. A role for glycoprotein Ib in Streptococcus sanguis–induced platelet aggregation. Blood J Am Soc Hematol. (2002) 100:509–16. doi: 10.1182/blood.V100.2.509
188. Erickson PR and Herzberg M. Purification and partial characterization of a 65-kDa platelet aggregation-associated protein antigen from the surface of Streptococcus sanguis. J Biol Chem. (1990) 265:14080–7. doi: 10.1016/S0021-9258(18)77270-0
189. Erickson P and Herzberg M. A collagen-like immunodeterminant on the surface of Streptococcus sanguis induces platelet aggregation. J Immunol (Baltimore Md.: 1950). (1987) 138:3360–6.
190. Erickson PR and Herzberg M. The Streptococcus sanguis platelet aggregation-associated protein. Identification and characterization of the minimal platelet-interactive domain. J Biol Chem. (1993) 268:1646–9. doi: 10.1016/S0021-9258(18)53901-6
191. von Hundelshausen P and Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. (2007) 100:27–40. doi: 10.1161/01.RES.0000252802.25497.b7
192. Ford I, Douglas C, Preston F, Lawless A, and Hampton K. Mechanisms of platelet aggregation by Streptococcus sanguis, a causative organism in infective endocarditis. Br J haematology. (1993) 84:95–100. doi: 10.1111/j.1365-2141.1993.tb03030.x
193. Ford I, Douglas C, Heath J, Rees C, and Preston F. Evidence for the involvement of complement proteins in platelet aggregation by Streptococcus sanguis NCTC 7863. Br J haematology. (1996) 94:729–39. doi: 10.1046/j.1365-2141.1996.d01-1857.x
194. Ford I, Douglas C, Cox D, Rees D, Heath J, and Preston F. The role of immunoglobulin G and fibrinogen in platelet aggregation by Streptococcus sanguis. Br J haematology. (1997) 97:737–7446. doi: 10.1046/j.1365-2141.1997.1342950.x
195. Pampolina C and McNicol A. Streptococcus sanguis-induced platelet activation involves two waves of tyrosine phosphorylation mediated by FcγRIIA and αIIbβ3. Thromb haemostasis. (2005) 93:932–9. doi: 10.1046/j.1365-2141.1997.1342950.x
196. McNicol A, Agpalza A, Jackson E, Hamzeh‐Cognasse H, Garraud O, and Cognasse F. Streptococcus sanguinis-induced cytokine release from platelets. J Thromb Haemostasis. (2011) 9:2038–49. doi: 10.1111/j.1538-7836.2011.04462.x
197. Dadon Z, Cohen A, Szterenlicht YM, Assous MV, Barzilay Y, Raveh-Brawer D, et al. Spondylodiskitis and endocarditis due to Streptococcus gordonii. Ann Clin Microbiol Antimicrobials. (2017) 16:1–4. doi: 10.1186/s12941-017-0243-8
198. Yakovenko O, Nunez J, Bensing B, Yu H, Mount J, Zeng J, et al. Serine-rich repeat adhesins mediate shear-enhanced streptococcal binding to platelets. Infection Immun. (2018) 86. doi: 10.1128/iai.00160-18
199. Bensing BA, López JA, and Sullam PM. The Streptococcus gordonii surface proteins GspB and Hsa mediate binding to sialylated carbohydrate epitopes on the platelet membrane glycoprotein Ibα. Infection Immun. (2004) 72:6528–37. doi: 10.1128/IAI.72.11.6528-6537.2004
200. Takamatsu D, Bensing BA, Cheng H, Jarvis GA, Siboo IR, López JA, et al. Binding of the Streptococcus gordonii surface glycoproteins GspB and Hsa to specific carbohydrate structures on platelet membrane glycoprotein Ibα. Mol Microbiol. (2005) 58:380–92. doi: 10.1111/j.1365-2958.2005.04830.x
201. Keane C, Petersen HJ, Tilley DO, Haworth J, Cox D, Jenkinson HF, et al. Multiple sites on Streptococcus gordonii surface protein PadA bind to platelet GPIIbIIIa. Thromb haemostasis. (2013) 110:1278–87. doi: 10.1160/TH13-07-0580
202. Petersen HJ, Keane C, Jenkinson HF, Vickerman MM, Jesionowski A, Waterhouse JC, et al. Human platelets recognize a novel surface protein, PadA, on Streptococcus gordonii through a unique interaction involving fibrinogen receptor GPIIbIIIa. Infection Immun. (2010) 78:413–22. doi: 10.1128/IAI.00664-09
203. Haworth JA, Jenkinson HF, Petersen HJ, Back CR, Brittan JL, Kerrigan SW, et al. Concerted functions of Streptococcus gordonii surface proteins PadA and Hsa mediate activation of human platelets and interactions with extracellular matrix. Cell Microbiol. (2017) 19:e12667. doi: 10.1128/IAI.00664-09
204. Keane C, Petersen H, Reynolds K, Newman DK, Cox D, Jenkinson HF, et al. Mechanism of outside-in αIIbβ3-mediated activation of human platelets by the colonizing Bacterium, Streptococcus gordonii. Arteriosclerosis thrombosis Vasc Biol. (2010) 30:2408–15. doi: 10.1161/ATVBAHA.110.216515
205. Kerrigan SW, Jakubovics NS, Keane C, Maguire P, Wynne K, Jenkinson HF, et al. Role of Streptococcus gordonii surface proteins SspA/SspB and Hsa in platelet function. Infection Immun. (2007) 75:5740–7. doi: 10.1128/IAI.00909-07
206. McNicol A, Zhu R, Pesun R, Pampolina C, Jackson EC, Bowden GH, et al. A role for immunoglobulin G in donor-specific Streptococcus sanguis-induced platelet aggregation. Thromb haemostasis. (2006) 95:288–93. doi: 10.1128/IAI.00909-07
207. Chopra A, Bhat SG, and Sivaraman K. Porphyromonas gingivalis adopts intricate and unique molecular mechanisms to survive and persist within the host: a critical update. J Oral Microbiol. (2020) 12:1801090. doi: 10.1080/20002297.2020.1801090
208. Zhang J, Xie M, Huang X, Chen G, Yin Y, Lu X, et al. The effects of porphyromonas gingivalis on atherosclerosis-related cells. Front Immunol. (2021) 12:766560. doi: 10.3389/fimmu.2021.766560
209. Von Seckendorff AF, Nomenjanahary M-S, Labreuche J, Ollivier V, Di Meglio L, Dupont S, et al. Periodontitis in ischemic stroke: impact of Porphyromonas gingivalis on thrombus composition and ischemic stroke outcomes. Res Pract Thromb Haemostasis. (2024) 8:102313. doi: 10.1016/j.rpth.2023.102313
210. Mei F, Xie M, Huang X, Long Y, Lu X, Wang X, et al. Porphyromonas gingivalis and its systemic impact: current status. Pathogens. (2020) 9:944. doi: 10.3390/pathogens9110944
211. Ge J, Zhu X, Weng C, Yuan D, Zhao J, Zhao L, et al. Periodontitis impacts on thrombotic diseases: from clinical aspect to future therapeutic approaches. Int J Oral Sci. (2024) 16:58. doi: 10.1038/s41368-024-00325-9
212. Li X, Iwai T, Nakamura H, Inoue Y, Chen Y, Umeda M, et al. An ultrastructural study of Porphyromonas gingivalis-induced platelet aggregation. Thromb Res. (2008) 122:810–9. doi: 10.1016/j.thromres.2008.03.011
213. Lourbakos A, Yuan Y, Jenkins AL, Travis J, Andrade-Gordon P, Santulli R, et al. Activation of protease-activated receptors by gingipains from Porphyromonas gingivalis leads to platelet aggregation: a new trait in microbial pathogenicity. Blood J Am Soc Hematol. (2001) 97:3790–7. doi: 10.1016/j.thromres.2008.03.011
214. Guevara T, Rodríguez-Banqueri A, Lasica AM, Ksiazek M, Potempa BA, Potempa J, et al. Structural determinants of inhibition of Porphyromonas gingivalis gingipain K by KYT-36, a potent, selective, and bioavailable peptidase inhibitor. Sci Rep. (2019) 9:4935. doi: 10.1038/s41598-019-41354-3
215. Li N and Collyer CA. Gingipains from Porphyromonas gingivalis—complex domain structures confer diverse functions. Eur J Microbiol Immunol. (2011) 1:41–58. doi: 10.1556/EuJMI.1.2011.1.7
216. Chen WA, Fletcher HM, Gheorghe JD, Oyoyo U, and Boskovic DS. Platelet plug formation in whole blood is enhanced in the presence of Porphyromonas gingivalis. Mol Oral Microbiol. (2020) 35:251–9. doi: 10.1111/omi.12314
217. Chen WA, Fletcher HM, Payne KJ, Aka S, Thornburg MB, Gheorghe JD, et al. Platelet and neutrophil responses to Porphyromonas gingivalis in human whole blood. Mol Oral Microbiol. (2021) 36:202–13. doi: 10.1111/omi.12336
218. Klarström Engström K, Khalaf H, Kälvegren H, and Bengtsson T. The role of Porphyromonas gingivalis gingipains in platelet activation and innate immune modulation. Mol Oral Microbiol. (2015) 30:62–73. doi: 10.1111/omi.12067
219. Naito M, Sakai E, Shi Y, Ideguchi H, Shoji M, Ohara N, et al. Porphyromonas gingivalis-induced platelet aggregation in plasma depends on Hgp44 adhesin but not Rgp proteinase. Mol Microbiol. (2006) 59:152–67. doi: 10.1111/j.1365-2958.2005.04942.x
220. Senini V, Amara U, Paul M, and Kim H. Porphyromonas gingivalis lipopolysaccharide activates platelet Cdc42 and promotes platelet spreading and thrombosis. J Periodontology. (2019) 90:1336–45. doi: 10.1002/JPER.18-0596
221. Fitzgerald JR, Foster TJ, and Cox D. The interaction of bacterial pathogens with platelets. Nat Rev Microbiol. (2006) 4:445–57. doi: 10.1038/nrmicro1425
222. Takeuchi H and Okamoto A. Helicobacter pylori infection and chronic immune thrombocytopenia. J Clin Med. (2022) 11:4822. doi: 10.3390/jcm11164822
223. Takeuchi H, Islam JM, Kaneko A, Kimura A, Shida T, Oboshi W, et al. Helicobacter pylori protein that binds to and activates platelet specifically reacts with sera of H. pylori-associated chronic immune thrombocytopenia. Platelets. (2021) 32:1120–3. doi: 10.1080/09537104.2021.1945570
224. Byrne MF, Kerrigan SW, Corcoran PA, Atherton JC, Murray FE, Fitzgerald DJ, et al. Helicobacter pylori binds von Willebrand factor and interacts with GPIb to induce platelet aggregation. Gastroenterology. (2003) 124:1846–54. doi: 10.1016/S0016-5085(03)00397-4
225. Ma TM, VanEpps JS, and Solomon MJ. Structure, mechanics, and instability of fibrin clot infected with Staphylococcus epidermidis. Biophys J. (2017) 113:2100–9. doi: 10.1016/j.bpj.2017.09.001
226. Becker K, Heilmann C, and Peters G. Coagulase-negative staphylococci. Clin Microbiol Rev. (2014) 27:870–926. doi: 10.1128/CMR.00109-13
227. Veloso TR, Que Y-A, Chaouch A, Giddey M, Vouillamoz J, Rousson V, et al. Prophylaxis of experimental endocarditis with antiplatelet and antithrombin agents: a role for long-term prevention of infective endocarditis in humans? J Infect Dis. (2015) 211:72–9. doi: 10.1093/infdis/jiu426
228. Kupferwasser LI, Yeaman MR, Nast CC, Kupferwasser D, Xiong Y-Q, Palma M, et al. Salicylic acid attenuates virulence in endovascular infections by targeting global regulatory pathways in Staphylococcus aureus. J Clin Invest. (2003) 112:222–33. doi: 10.1172/JCI200316876
229. Kupferwasser LI, Yeaman MR, Shapiro SM, Nast CC, Sullam PM, Filler SG, et al. Acetylsalicylic acid reduces vegetation bacterial density, hematogenous bacterial dissemination, and frequency of embolic events in experimental Staphylococcus aureus endocarditis through antiplatelet and antibacterial effects. Circulation. (1999) 99:2791–7. doi: 10.1161/01.CIR.99.21.2791
230. Oury C, Meyers S, Jacques N, Leeten K, Jiang Z, Musumeci L, et al. Protective effect of ticagrelor against infective endocarditis induced by virulent Staphylococcus aureus in mice. Basic to Trans Sci. (2023) 8:1439–53. doi: 10.1016/j.jacbts.2023.02.003
231. Veloso TR, Oechslin F, Que Y-A, Moreillon P, Entenza JM, and Mancini S. Aspirin plus ticlopidine prevented experimental endocarditis due to Enterococcus faecalis and Streptococcus gallolyticus. Pathog Dis. (2015) 73:ftv060. doi: 10.1016/j.jacbts.2023.02.003
232. Vanassche T, Peetermans WE, Herregods M-C, Herijgers P, and Verhamme P. Anti-thrombotic therapy in infective endocarditis. Expert Rev Cardiovasc Ther. (2011) 9:1203–19. doi: 10.1586/erc.11.100
233. Habib A, Irfan M, Baddour LM, Le KY, Anavekar NS, Lohse CM, et al. Impact of prior aspirin therapy on clinical manifestations of cardiovascular implantable electronic device infections. Europace. (2013) 15:227–35. doi: 10.1093/europace/eus292
234. Chan K-L, Tam J, Dumesnil JG, Cujec B, Sanfilippo AJ, Jue J, et al. Effect of long-term aspirin use on embolic events in infective endocarditis. Clin Infect Dis. (2008) 46:37–41. doi: 10.1086/524021
235. Chan K-L, Dumesnil JG, Cujec B, Sanfilippo AJ, Jue J, Turek MA, et al. A randomized trial of aspirin on the risk of embolic events in patients with infective endocarditis. J Am Coll Cardiol. (2003) 42:775–80. doi: 10.1016/S0735-1097(03)00829-5
236. Anavekar NS, Tleyjeh IM, Anavekar NS, Mirzoyev Z, Steckelberg JM, Haddad C, et al. Impact of prior antiplatelet therapy on risk of embolism in infective endocarditis. Clin Infect Dis. (2007) 44:1180–6. doi: 10.1086/513197
237. Pepin J, Tremblay V, Bechard D, Rodier F, Walker C, Dufresne D, et al. Chronic antiplatelet therapy and mortality among patients with infective endocarditis. Clin Microbiol infection. (2009) 15:193–9. doi: 10.1111/j.1469-0691.2008.02665.x
238. Snygg-Martin U, Rasmussen RV, Hassager C, Bruun NE, Andersson R, and Olaison L. The relationship between cerebrovascular complications and previously established use of antiplatelet therapy in left-sided infective endocarditis. Scandinavian J Infect Dis. (2011) 43:899–904. doi: 10.3109/00365548.2011.603742
239. Wang X and Zhou H. Impact of antiplatelet therapy on outcomes of sepsis: A systematic review and meta-analysis. PloS One. (2025) 20:e0322293. doi: 10.1371/journal.pone.0322293
240. Hsu J, Donnelly JP, Chaudhary NS, Moore JX, Safford MM, Kim J, et al. Aspirin use and long-term rates of sepsis: A population-based cohort study. PloS One. (2018) 13:e0194829. doi: 10.1371/journal.pone.0194829
241. Eisen DP, Leder K, Woods RL, Lockery JE, McGuinness SL, Wolfe R, et al. Effect of aspirin on deaths associated with sepsis in healthy older people (ANTISEPSIS): a randomised, double-blind, placebo-controlled primary prevention trial. Lancet Respir Med. (2021) 9:186–95. doi: 10.1016/S2213-2600(20)30411-2
Keywords: GPVI, FcRγIIA, GPIB, and GPIIb/IIIa, immune responses, platelet activation
Citation: Alfar HR and Whiteheart SW (2025) Bacterial interactions with platelets: defining key themes. Front. Immunol. 16:1610289. doi: 10.3389/fimmu.2025.1610289
Received: 11 April 2025; Accepted: 10 June 2025;
Published: 03 July 2025.
Edited by:
David Stegner, University of Würzburg, GermanyReviewed by:
Sven Hammerschmidt, University of Greifswald, GermanyIsabelle Salles-Crawley, University of London, United Kingdom
Copyright © 2025 Alfar and Whiteheart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sidney W. Whiteheart, d2hpdGVoZUB1a3kuZWR1