 Yulong Zhou
Yulong Zhou Xiyang Tang
Xiyang Tang Weiguang Du1†
Weiguang Du1† Xiaolong Yan
Xiaolong Yan Jinbo Zhao
Jinbo Zhao- 1Department of Thoracic Surgery, Tangdu Hospital, The Fourth Military Medical University, Xi’an, Shaanxi, China
- 2Department of Cardiothoracic Surgery, The 902nd Hospital of the Chinese People’s Liberation Army Joint Logistic Support Force, Bengbu, Anhui, China
- 3Department of Ophthalmology, Tangdu Hospital, The Fourth Military Medical University, Xi’an, Shaanxi, China
Therapies targeting immune checkpoints like programmed death receptor-1 and programmed death ligand-1 have demonstrated remarkable effectiveness in combating cancer. However, a subset of patients fails to respond to these therapies, underscoring the complexity of tumor immune evasion mechanisms. Exploring innovative immune regulatory targets represents a crucial research priority in this field. Signal regulatory protein α (SIRPα) is an immunosuppressive receptor expressed on myeloid cells that inhibits innate immunity through its interaction with the ligand integrin-associated protein (CD47). Blocking the SIRPα–CD47 axis can enhance myeloid cell-mediated anti-tumor responses and stimulate adaptive immunity, thereby synergizing with therapeutic antibodies and T-cell checkpoint inhibitors. Additionally, tumor-intrinsic SIRPα may facilitate tumor growth and immune evasion. This paper aims to elucidate the mechanisms of SIRPα activity in various cell types, review the advancements in SIRPα-targeted tumor therapies, and highlight the potential research value of tumor-expressed endogenous SIRPα.
1 Introduction
The clinical application of immune checkpoint inhibitors (ICIs) has profoundly transformed the landscape of cancer treatment (1). The majority of immune therapies activate adaptive immune responses that primarily target T-cell immune checkpoints (2). Programmed death receptor-1 (PD-1)/programmed death ligand-1 (PD-L1) inhibitors are currently the most widely used ICIs, with four anti-PD-1 and three anti-PD-L1 antibodies currently approved for clinical use (3, 4). The blockade of PD-1/PD-L1 can substantially slow down the progression of several solid tumors (5, 6). Despite satisfactory and lasting effects among responders, the therapeutic efficacy of these antibodies remains suboptimal for some patients. Therefore, more ICIs are yet necessary (7, 8). The immunosuppressive receptor known as signal regulatory protein α (SIRPα), expressed on myeloid cells, was developed and has received a lot of attention because of its function in mediating the immunosuppressive “don’t eat me” signal from cancer cells (9). It is widely recognized that cancer cells can upregulate integrin-associated protein (CD47) expression to exploit this “don’t eat me” signal to evade macrophage-mediated clearance and achieve immune evasion (Figure 1) (10). Studies have shown that targeting suppressive macrophages may enhance anticancer immune responses and improve the efficacy of immunotherapy combinations (11). Myeloid cells constitute a major component of the tumor microenvironment of solid cancers, whereas T-cell infiltration is often limited (12). The immunosuppressive cells within the tumor immune microenvironment inhibit T-cell activity through various mechanisms, thereby promoting cancer growth and metastasis (13, 14). Therefore, targeting myeloid cells within the tumor microenvironment, particularly through interventions aimed at their immune checkpoints, may offer novel strategies for inhibiting cancer progression. For example, blocking the CD47–SIRPα axis holds great potential as a novel immunotherapeutic approach (15). The structure and operation of SIRPα are covered in this review, along with a discussion of the molecular pathways by which SIRPα functions in various cells. We also present the research progress made toward anti-SIRPα antibody cancer therapies and discuss why a SIRPα-targeting strategy may be a valuable choice.

Figure 1. Tumor immune evasion via the “don’t eat me” signal. Cancer cells evade immune detection by exploiting the “don’t eat me” signal. The binding of SIRPα to CD47 initiates ITIM phosphorylation in the cytoplasm, recruiting SHP1 and SHP2 tyrosine phosphatases. This cascade dephosphorylates myosin IIA, preventing its accumulation at the phagocytic synapse and suppressing macrophage phagocytic signaling. SIRPα, signal regulatory protein α; ITIM, immunoreceptor tyrosine-based inhibitory motifs.
2 Structure and function of SIRPα
SIRPα is a member of the SIRP protein family, which comprises five distinct subtypes: SIRPα, SIRPβ1, SIRPβ2, SIRPγ, and SIRPδ. This protein, also referred to by various names such as CD172a, SHPS-1, p84, MFR, MYD-1, or PTPNS1, interacts exclusively with its ligand, CD47 (16, 17). SIRPα was expressed on myeloid cells, such as macrophages, neutrophils, dendritic cells, and microglial cells. It is also expressed at low levels in T-, B-, and natural killer (NK) cells (18). SIRPα is composed of three extracellular immunoglobulin superfamily domains. These domains include 1 variable and 2 constant type 1 domains. Additionally, SIRPα has one transmembrane region and an intracellular tail that can transmit inhibitory signals. Inside the intracellular tail, there are four tyrosine residues. These residues form two typical immunoreceptor tyrosine-based inhibitory motifs (ITIMs) (19, 20). Additionally, the extracellular immunoglobulin (Ig)V domain contains a ligand-binding region that allows SIRPα to interact with CD47, which consequently triggers a signaling cascade that can recruit the protein tyrosine phosphatases SHP1 and SHP2. This cascade results in the dephosphorylation of myosin IIA, which prevents its accumulation at the phagocytic synapse and ultimately leads to the suppression of phagocytic signals in macrophages, thereby protecting healthy cells from immune attacks. This inhibitory signal is known as the “don’t eat me” signal (21). Notably, the extracellular IgV domain of SIRPα is a hotspot for polymorphisms, with 10 human SIRPA alleles identified, the main variants being SIRPAV1, SIRPAV2, and SIRPAV8 (22–24). In turn, SIRPγ, which is primarily expressed on activated T-cells, has a much lower affinity for CD47 than that of SIRPα (25). Although the extracellular regions of SIRPγ and SIRPα share a high degree of homology (>70%), the intracellular domain of SIRPγ is notably shorter and fails to efficiently recruit signaling proteins, ultimately resulting in its lack of signaling potential. However, because of its binding ability to increase cell-cell adhesion, it can promote the production of synapses between T-cells and antigen-presenting cells (APCs), which increases the efficiency of antigen presentation and helps to mediate T-cell proliferation and cytokine secretion (26, 27). SIRPβ, expressed predominantly on myeloid cells, comprises 2 isoforms: SIRPβ1 and SIRPβ2. The SIRPβ2 isoform recruits the immunoreceptor tyrosine-based activation motifs-containing adaptor DAP12 via a transmembrane lysine residue to initiate immunostimulatory signaling, enhancing phagocytosis and antigen presentation by myeloid cells. Unlike SIRPα, SIRPβ2 does not interact with CD47, and its activation ligand remains unidentified. Similarly, while SIRPβ1 ligands are undefined, macrophage-specific SIRPβ1 engagement enhances phagocytic activity (28). Contrastingly, SIRPδ, a secreted isoform characterized by a single V-type Ig superfamily domain, is postulated to be expressed in spermatozoa and respiratory tissues (17).
3 Myeloid-intrinsic SIRPα regulates the tumor immune microenvironment
3.1 Functional role of SIRPα in macrophages
Blocking SIRPα can enhance antibody-dependent cellular phagocytosis (ADCP) by macrophages, features that have attracted significant attention for research (29–31) (Figure 2). Microglia play a similar functional role to macrophages in central nervous system tumors. They function as the effector cells in the disruption of the CD47-SIRPα anti-phagocytic axis (32, 33). Generally, promoting ADCP is achieved by blocking the binding of SIRPα to CD47 to abolish the “don’t eat me” signal. Furthermore, in chimeric antigen receptor macrophages, SIRPα inhibition in macrophages can activate inflammatory pathways and the cGAS–STING signaling cascade, leading to an elevated production of proinflammatory cytokines, such as interleukin-1 (IL-1), tumor necrosis factor-alpha (TNF-α), reactive oxygen species (ROS), and nitric oxide, which increase the anticancer activity (34, 35). Moreover, preventing the expression of SIRPα in macrophages induces the recruitment and migration of T-cells via increased secretion of chemokines (e.g., C-C motif chemokine ligands CCL3 and CCL4) (26). In SIRPα-knockout (SIRPα-KO) mice, SIRPα-KO macrophages were found to display robust anticancer activity and antigen-presenting capacity, which was associated with enhanced T-cell activation and proliferation. Notably, SIRPα-KO macrophages were found to promote T-cell recruitment in cancers via a Syk–Btk-dependent mechanism involving CCL8 secretion, transforming tumor-associated macrophages and granulocytic myeloid-derived suppressor cells into subsets expressing high levels of CCL8 and H2-Q10, respectively, with enhanced antigen presentation, phagocytosis, inflammatory response, and chemotaxis capacities (36). Therefore, targeting SIRPα has the potential to reprogram the tumor immune microenvironment, promoting systemic anticancer responses and preventing solid cancer progression. In brief, by blocking the expression of SIRPα in macrophages, the traditional “don’t eat me” signaling pathway can be suppressed, which will improve phagocytosis and stimulate macrophages to secrete chemokines and cytokines via additional signaling pathways. Simultaneously, it has the potential to block the SIRPα-mediated non-CD47-dependent pathway, reprogramming the suppressive tumor immune microenvironment.
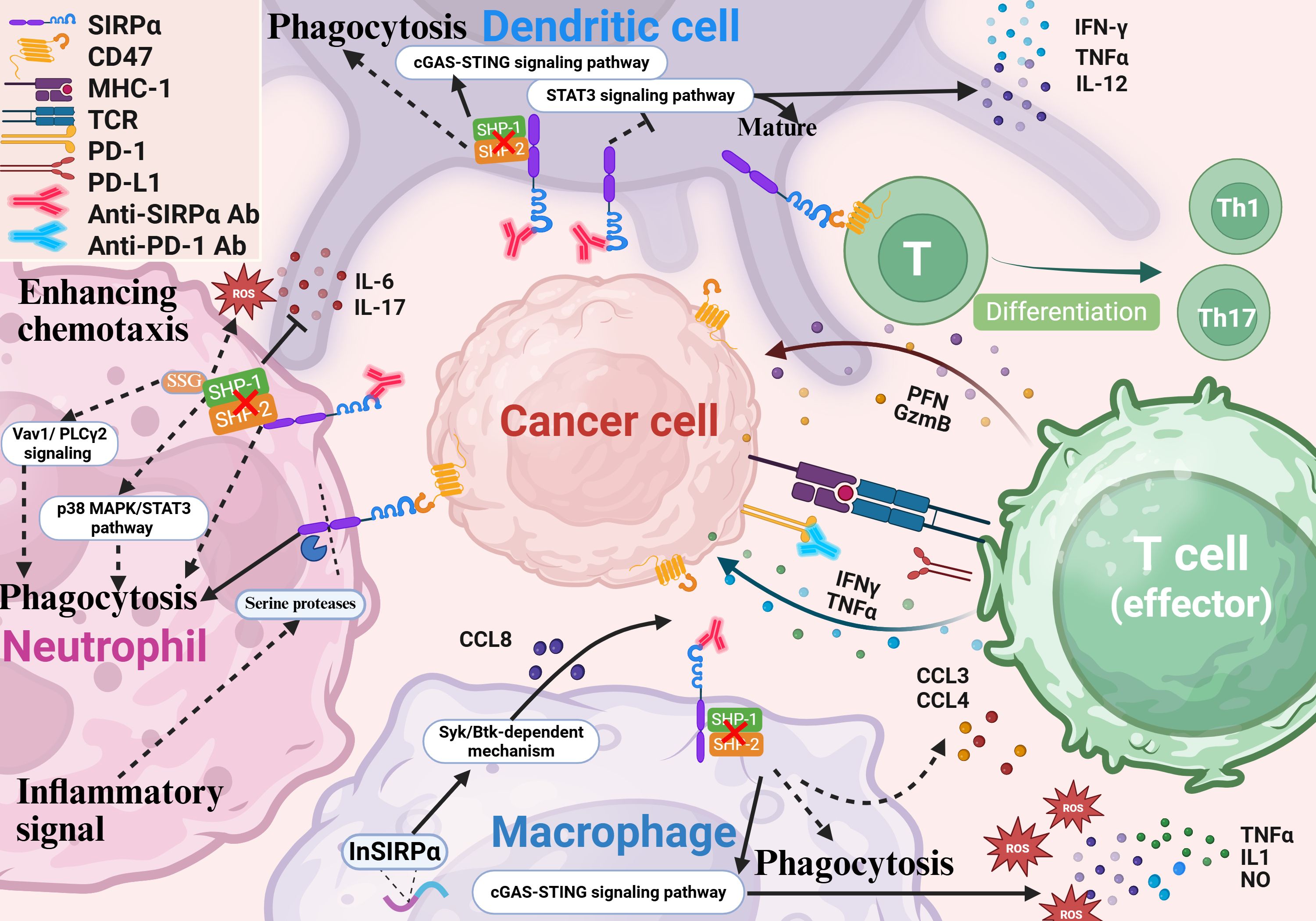
Figure 2. SIRPα blockade enhances innate and adaptive immunity. Inhibition of SIRPα boosts the phagocytic and antigen-presenting capabilities of myeloid cells. In macrophages, SIRPα blockade activates inflammatory pathways and the cGAS-STING cascade in CAR macrophages, increasing the secretion of IL-1, TNF-α, ROS, nitric oxide, and chemokines like CCL3 and CCL4. It also promotes T-cell recruitment in tumors through a Syk-Btk-dependent mechanism. In neutrophils, SIRPα inhibition enhances chemotaxis, infiltration, and cytotoxicity. During inflammation, neutrophil ITIM cleavage generates a truncated receptor that binds CD47 without transmitting inhibitory signals, further enhancing chemotaxis, ROS release, and phagocytosis. In DCs, SIRPα blockade suppresses STAT3 signaling, increases cytokine secretion (e.g., IL-12, TNF-α, and IFN-γ), and promotes DC maturation. It also activates cGAS-STING signaling, improving tumor antigen cross-presentation. Additionally, SIRPα on DCs modulates naive T-cell differentiation into helper T-cells. SIRPα, signal regulatory protein α; CAR, chimeric antigen receptor; DC, dendritic cell; IL, interleukin; TNF, tumor necrosis factor; INF, interferon.
3.2 Functional role of SIRPα in neutrophils
In cancer therapeutics, anti-SIRPα antibodies exert their antitumor effects by disrupting the CD47-SIRPα interaction and relieving inhibitory signaling on neutrophils.) (37). When combined with tumor-targeting antibodies, the Fc region of these therapeutic antibodies engages activating Fcγ receptors (e.g., FcγRIIIa) on neutrophils, triggering antibody dependent cell-mediated cytotoxicity (ADCC) and subsequently enhancing neutrophil-mediated tumor cell killing (3, 38). Sodium stibogluconate (SSG), a selective SHP-1 inhibitor, enhances neutrophil cytotoxicity by blocking phosphatase-mediated suppression of Vav1 and PLCγ2 signaling. Co-administration of SSG with CD47-SIRPα blockade amplifies ADCC efficacy through dual inhibition of immunosuppressive pathways (39). SIRPα signaling suppresses neutrophil phagocytic activity and cytotoxicity through the SHP-1/p38 MAPK/STAT3 pathway while promoting IL-6 and IL-17 secretion. After SIRPα-KO, neutrophils polarize toward the anti-tumor N1 phenotype, with enhanced phagocytic function and reduced inflammatory cytokine secretion, thereby inhibiting the growth of lung cancer (40). However, compared with IgA, IgG-mediated ADCC exhibits relatively low efficiency (41–45). Blocking SIRPα on neutrophils with anti-SIRPα antibodies significantly enhances ADCC mediated by IgA2 variants of cetuximab and trastuzumab against HER2-positive breast cancer cells and EGFR-positive epidermoid carcinoma cells (46). Paradoxically, SIRPα overexpression in autoimmune lesions (e.g., rheumatoid arthritis and inflammatory bowel disease) exacerbates inflammation through dysregulated innate immunity (47). During chronic inflammation, neutrophil-derived serine proteases cleave the SIRPα ITIM domain in an IL-17-dependent manner. The resultant truncated SIRPα retains CD47-binding capacity but loses inhibitory signaling, unleashing neutrophil chemotaxis, ROS production, and phagocytic activity (48).
3.3 Functional role of SIRPα in dendritic cells
As specialized APCs, dendritic cells (DCs) are crucial in facilitating T-cell activation and maintaining immune tolerance (49, 50). When DCs come into contact with cancer cells, they send a “don’t eat me” signal through the classic ITIM–SHP1 complex that mediates anti-phagocytic effects but also through SIRPα, which detects cancer mitochondrial DNA for cross-priming or activate the STAT3 signaling pathway to suppress the production of cytokines (such as IL-12, TNF-α, and interferon-γ) and consequently inhibit DC maturation. Additionally, the PI3K–AKT signaling pathway also plays a pivotal role in regulating the activation and maturation of DCs through SIRPα (51, 52). Combined therapy with radiotherapy/anti-SIRPα/anti-PD-1 for colorectal cancer was shown to effectively induce cGAS–STING signaling in DCs both in vitro and in vivo, facilitating efficient cross-presentation of tumor-associated antigens (53, 54). Moreover, when SIRPα was silenced in DCs, increased secretion of cytokines (e.g., TNF-α, IL-12, and IL-6), enhanced the secretion of interferon-γ by CD8+ T lymphocytes, and effectively killed cervical cancer cells in vitro (55). Of note, the interaction between SIRPα on DCs and CD47 on T-cells modulates the differentiation of naïve T-cells into T-helper (Th) cells. Mice lacking SIRPα exhibit enhanced resistance to autoimmune diseases caused by Th1 or Th17 cells, such as encephalomyelitis and colitis (56–59). Besides regulating T-cells by presenting tumor antigens, SIRPα can further influence the differentiation and function of T-cells by regulating their own maturation. Thus, blocking SIRPα can promote DC maturation and enhance their antigen-presenting function, thereby facilitating the function of cytotoxic T-cells.
4 Role of tumor-intrinsic SIRPα in tumor progression
In summary, targeting the immune checkpoint receptor SIRPα can boost both innate and adaptive immune responses, offering novel strategies for cancer immunotherapy. Surprisingly, some solid cancers (such as renal cell carcinoma, colorectal cancer, and osteosarcoma) exhibit high levels of SIRPα expression. Despite the limited research on endogenous SIRPα in cancer cells, multiple pivotal studies have shed light on the role of endogenous SIRPα in the malignant progression of cancers (Figure 3) (60, 61). Specifically, in osteosarcoma cells, the upregulation of SIRPα activates the extracellular signal-regulated kinase (ERK) pathway, leading to the phosphorylation of specificity protein 1 (Sp1) at the threonine 278 site. This phosphorylated protein then binds to the promoter region of solute carrier family 7 member 3 (SLC7A3), resulting in increased SLC7A3 expression and enhanced cellular arginine uptake capacity. These processes collectively promote the metastasis of osteosarcoma (62). Contrastingly, in acute promyelocytic leukemia (APL) cells, overexpression of SIRPα exhibits distinct effects, potentially inhibiting the β-catenin signaling pathway and upregulating Foxo3a expression, which in turn induces apoptosis and inhibits tumor cell proliferation (63). In hepatocellular carcinoma cells, SIRPα has been shown to negatively regulate tumor initiation, primarily through the inhibition of the ERK and NF-κB pathways (64). Similarly, SIRPα is used by non-small cell lung cancer as a critical regulator of the EGFR pathway. Knockdown of SIRPα induces the upregulation of p27, subsequently inhibiting cell cycle progression and reducing tumor growth. However, increased p27 expression leads to its mislocalization to the cytoplasm, paradoxically promoting cancer cell invasiveness. Conversely, the enhanced expression of SIRPα boosts the cell’s migratory and proliferative capabilities. These findings suggest that SIRPα may exert dual oncogenic or tumor-suppressive properties, depending on its regulation of multiple signaling pathways within cancer cells (65). Additionally, Z. Zhou and his research team uncovered a unique function of SIRPα in melanoma cells: as a marker for melanoma cells, the expression level of SIRPα diminishes progressively as melanoma progresses. SIRPα interacts with CD47, modulating the function of CD8+ T-cells. Studies have shown that cytotoxic T-cells exert stronger anti-melanoma effects on cells overexpressing SIRPα, and the addition of anti-PD-L1 antibodies significantly enhances this killing effect. This suggests that endogenous SIRPα in melanoma cells plays a positive role in PD-1/PD-L1-induced T-cell-mediated anticancer immunity, while the absence of SIRPα may lead to increased resistance to PD-L1 therapy (66). Microglia critically shape developing neural circuits by eliminating redundant synapses via phagocytic activity. Genetic ablation of neuronal SIRPα suppressed microglial synaptic engulfment, resulting in elevated retinal synapse density. Conversely, sustained neuronal SIRPα expression prolonged phagocytic activity and decreased synaptic numbers. Mechanistically, neuronal SIRPα serves as a decoy receptor that sequesters inhibitory CD47 signals from microglial SIRPα, thereby enabling synapse clearance. This SIRPα-CD47 regulatory axis elucidates a molecular basis for pathological synapse loss in neurological conditions (67). SIRPα is not expressed in normal astrocytes but exhibits functional expression in astrocytomas, potentially participating in cell adhesion and signaling through CD47-dependent phosphorylation and SHP-2 recruitment, thereby influencing tumor invasiveness. Furthermore, SIRPα may regulate tumor proliferation and survival by either suppressing growth factor signaling or modulating the PI3K/AKT pathway. Its potential as a therapeutic target or prognostic biomarker in astrocytomas warrants further investigation (68).
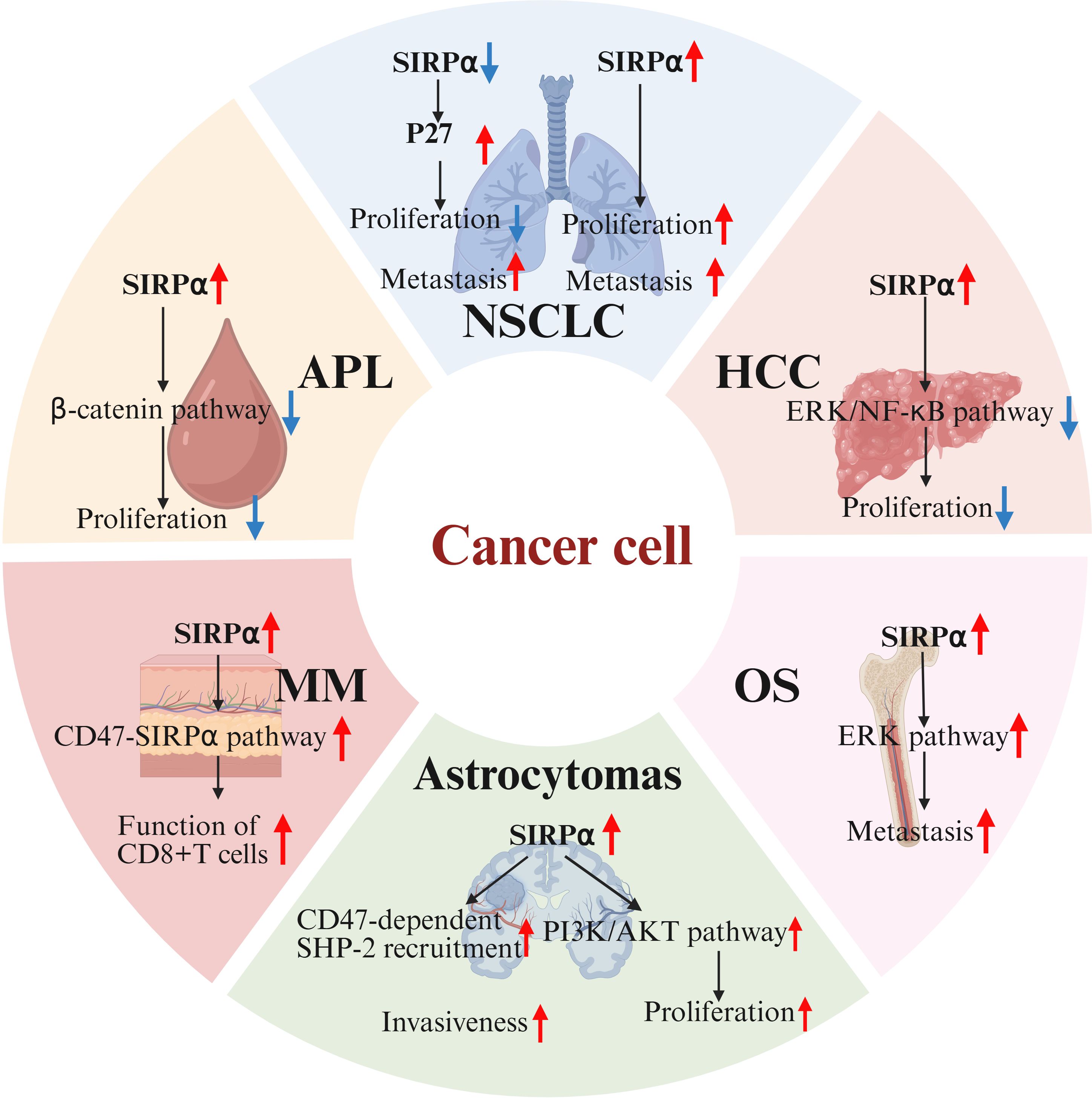
Figure 3. Endogenous SIRPα in tumor malignancy. In osteosarcoma(OS), SIRPα overexpression activates the ERK pathway, driving metastasis. In acute promyelocytic leukemia(APL), SIRPα upregulation may inhibit β-catenin signaling, suppressing tumor proliferation. In hepatocellular carcinoma(HCC), SIRPα negatively regulates tumor initiation by inhibiting ERK and NF-κB pathways. In non-small cell lung cancer(NSCLC), SIRPα knockdown upregulates p27, reducing tumor growth, but p27 mislocalization to the cytoplasm paradoxically increases invasiveness. Conversely, SIRPα overexpression enhances cell proliferation and migration. In melanoma(MM), SIRPα overexpression augments CD8+ T-cell function. In astrocytomas, SIRPα may be involved in cell adhesion and signal transduction through CD47-dependent phosphorylation and SHP-2 recruitment, thereby affecting tumor invasiveness. Additionally, SIRPα may regulate tumor proliferation and survival by inhibiting growth factor signaling or by modulating the PI3K/AKT pathway. SIRPα, signal regulatory protein α; ERK, extracellular signal-regulated kinase.
5 Advances in therapeutic targeting of SIRPα in solid tumors
5.1 Preclinical studies on anti-SIRPα therapy in solid tumors
Multiple anti-SIRPα antibodies developed for solid tumor treatment in preclinical studies have demonstrated significant efficacy in suppressing tumor progression (38, 69) (Table 1). Yanagita et al. validated the tumor-inhibitory effect of the mouse-derived anti-SIRPα monoclonal antibody MY-1, which showed enhanced cytotoxicity against HER2-positive breast cancer cells in vitro. It significantly inhibited the growth of SIRPα-expressing renal cell carcinoma and melanoma cells, but not of non-SIRPα-expressing cells. Combination with rituximab or anti-PD-1 antibody further enhanced the ability of MY-1 to suppress the growth of Burkitt lymphoma and colorectal cancer cells. Moreover, when used as a monotherapy, MY-1’s anticancer activity against renal cell carcinoma and melanoma was mediated by macrophages, but also NK and CD8+ T -cells (60). In SIRPα-deficient mice, MY-1 monotherapy showed inhibition of cancer growth by binding to SIRPβ and promoting ADCP (70). The effects of MY-1 differ between tumors with and without SIRPα expression, indicating that endogenous SIRPα in cancer cells is involved in certain regulatory mechanisms.
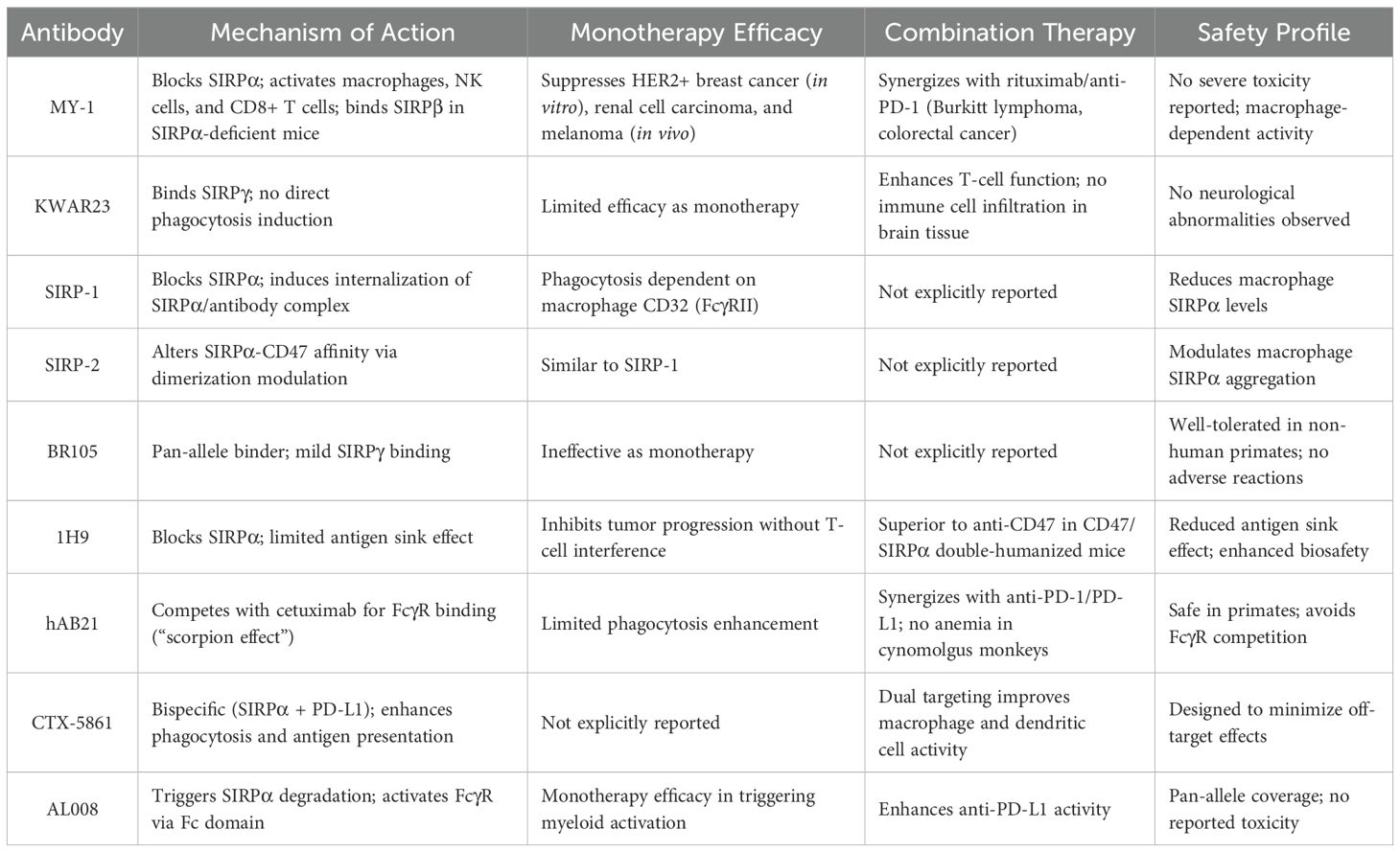
Table 1. Preclinical characteristics of anti-SIRPα antibodies in solid tumors.
Humanized SIRPα antibodies can effectively block various human SIRPα variants. Several antibody monotherapies each have their own characteristics. KWAR23 alone fails to induce macrophage phagocytosis. Moreover, no immune cell infiltration or obvious neurological abnormalities were observed in the brains of mice treated with KWAR23; however, it binds to SIRPγ and affects T-cell function (38). The phagocytic activity of SIRP-1 and -2 is important as monotherapy depends on the “eat me” receptor CD32 (FcγRII) in macrophages. SIRP-1 functions by directly blocking SIRPα and inducing internalization of the SIRPα/antibody complex, thereby reducing the levels of SIRPα in macrophages, while SIRP-2 alters the affinity of SIRPα for CD47 by affecting its dimerization/aggregation in macrophages (69). BR105 is ineffective when used alone; although it can mildly bind to SIRPγ, it does not inhibit T-cell activation. Toxicity studies in non-human primates showed that BR105 is well-tolerated, with no treatment-related adverse reactions observed (71). 1H9 exhibits a similar effect in inhibiting cancer progression without affecting T-cell function. When comparing anti-SIRPα and anti-CD47 antibodies using CD47/SIRPα double-humanized mice, it was found that 1H9 exhibits significantly reduced antigen sink effect and enhanced biosafety owing to the limited tissue distribution of SIRPα expression (72).
When combined with therapeutic antibodies, such as rituximab, all antibodies demonstrate significant inhibitory effects on the growth of hematological malignancies and solid cancers both in vitro and in vivo. Additionally, several antibodies when used in combination with ICIs exhibit good safety and therapeutic effects. Competition between hAB21 and cetuximab for macrophage FcγR limits the ability of anti-SIRPα antibodies to enhance macrophage phagocytosis. Alternatively, hAB21 with an active Fc structure can co-bind to SIRPα and FcγR in macrophages, leading to heterotrimeric interactions that restrict the binding of cetuximab to macrophage FcγR, thereby reducing phagocytic signaling. This phenomenon is known as the “scorpion effect.” When combined with anti-PD-1 or anti-PD-L1 antibody blockade therapy, hAB21 significantly inhibits the growth of tumor cells and does not cause anemia or other adverse outcomes when used in cynomolgus monkeys (11). CTX-5861 is a bispecific antibody targeting both SIRPα and PD-L1, designed to enhance macrophage phagocytosis and improve the efficiency of antigen presentation by DCs (73). AL008, a specific antibody targeting pan-alleles of SIRPα, demonstrates monotherapy efficacy by triggering SIRPα degradation and stimulating the activation of FcγR on bone marrow cells via its Fc domain. Additionally, the antitumor activity of anti-PD-L1 drugs has also been enhanced (74).
5.2 Clinical studies on anti-SIRPα therapy in solid tumors
Although the aforementioned anti-SIRPα antibodies have not entered the clinical trial stage, some anti-SIRPα antibodies have demonstrated good biosafety and cancer treatment efficacy in preclinical studies and have thus entered clinical trials (Table 2). ADU-1805, a humanized IgG2 anti-SIRPα antibody, does not affect T-cell activation or bind to red blood cells/platelets. In non-human primates, ADU-1805 exhibited no toxicity. Furthermore, ADU-1805 does not bind to macrophage FcγRIIA to trigger the “scorpion effect,” nor does it induce NK cell-mediated ADCC, lacks activity in mediating complement-dependent cytotoxicity, and does not stimulate cytokine secretion in human whole blood, further substantiating its clinical viability. ADU-1805 is undergoing clinical trials (NCT05856981), and the results are yet to be announced (27, 75). BI 765063 is a humanized IgG4 monoclonal antibody antagonist of SIRPα that binds with high affinity to SIRPαV1 but not to SIRPγ, thereby preserving T-cell function. Ongoing research (NCT05249426) to test whether different combinations of BI 765063, Ezabenlimab, chemotherapy, cetuximab and BI 836880 are helpful for patients with head and neck or liver cancer. Another clinical trial (NCT03990233) is currently evaluating the safety and efficacy of BI 765063 as monotherapy or in combination with ezabenlimab in patients with advanced solid tumors. BI 765063 monotherapy was found to be well-tolerated and showed activity, with treatment biopsies from responders demonstrating increased CD8+ T-cell infiltration and activation (76). Additionally, a clinical trial in Japan (NCT04653142) assessed the safe dose of BI 765063 in Japanese patients and found that its safety and pharmacokinetic parameters were consistent with those observed in Caucasian patients (77). A study (NCT05446129) aimed at evaluating the safety, feasibility, efficacy, and biological activity of the neoadjuvant treatment with Ezabenlimab combined with BI 765063 and pembrolizumab combined with BI 765063 in newly diagnosed patients with locally regional colorectal cancer has been dropped by the pharmaceutical company. BI 770371 is a pan-specific monoclonal antibody against SIRPα currently being evaluated the tolerability of different doses of BI 770371 when used alone or in combination with ezabbenlimab (NCT05327946). It is considered that the toxicity profile of BI 770371, both as a monotherapy and in combination therapy, is manageable. Another study (NCT05068102) aimed at finding out how the two drugs, BI 765063 and BI 770371, are absorbed in tumors and how they are distributed in the body is underway (78). CC-95251(BMS-986351) is a fully human monoclonal antibody targeting SIRPα, with preclinical studies showing its ability to enhance macrophage phagocytic activity when combined with the therapeutic antibody rituximab (79). A clinical trial (NCT03783403) is evaluating CC-95251 as a monotherapy and in combination with cetuximab and rituximab for safety, tolerability, and preliminary clinical activity in participants with advanced solid and hematological malignancies. Unfortunately, the clinical trial has been terminated owing to changes in business objectives (80). DS-1103a, a recombinant humanized IgG4 antibody targeting SIRPα, is currently being assessed in combination with T-DXd for its efficacy, recommended dosage, and pharmacokinetic properties in patients with advanced solid tumors (NCT05765851). IBI397, a pan-allelic antibody against SIRPα, underwent clinical trials for advanced malignant tumors but the trial (NCT05245916) was withdrawn owing to changes in the company’s development strategy.
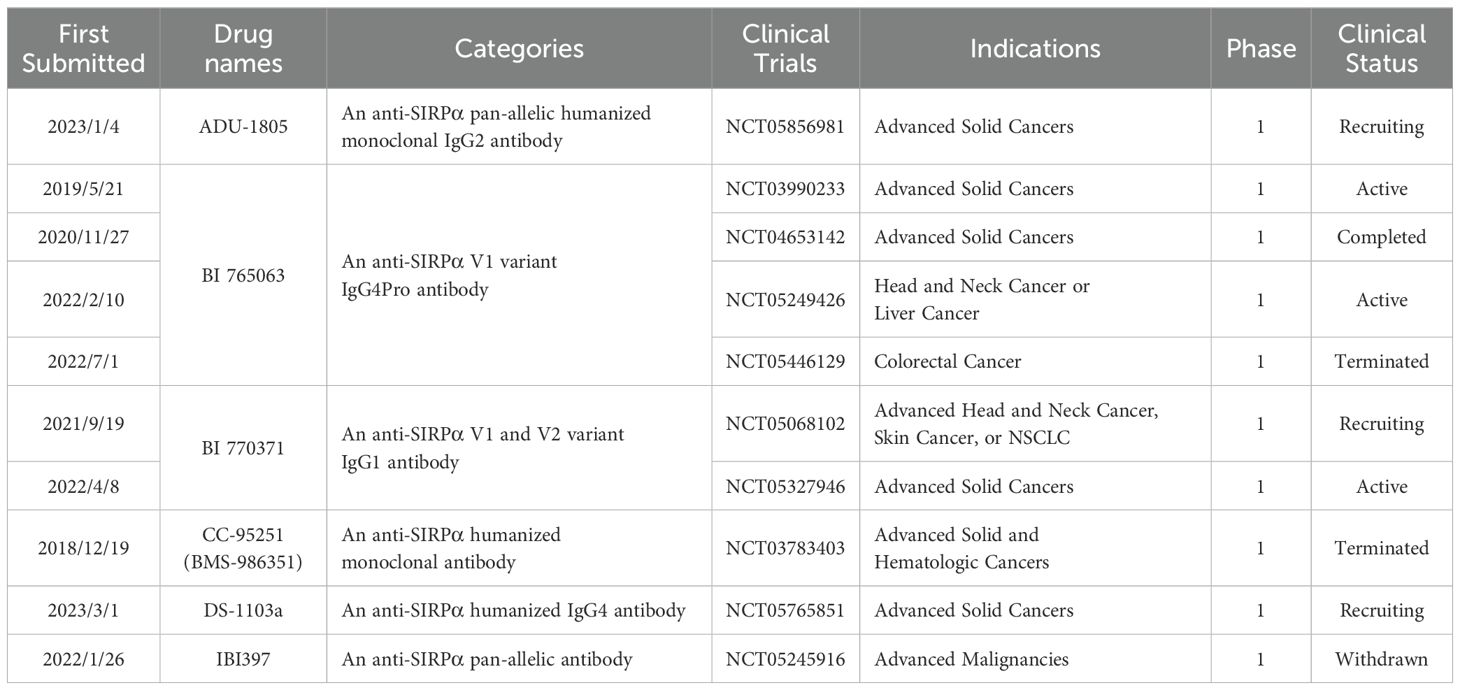
Table 2. Various anti-SIRPα antibodies are involved in multiple clinical trials.
6 Why select an anti-SIRPα antibody therapeutic strategy?
6.1 Limitations of CD47-targeted therapy in solid tumors
Current developments of CD47–SIRPα signaling pathway inhibitors can be roughly categorized into three types: (i) blockers of CD47 molecules in target cells, which includes anti-CD47 antibodies and SIRPα-Fc fusion antibodies, (ii) blockers of SIRPα molecules in immune effector cells, and (iii) inhibitors of glutaminase-like proteases (81). Anti-CD47 antibodies have been shown to achieve objective (total or partial) remission in 50% of patients by showing considerable anticancer activity in hematological malignancies. However, treatment of solid cancers has led to adverse effects, including anemia (57% of patients) and lymphocytopenia (34% of patients) (82–84). Although strong effects in preclinical studies were observed, especially those that retain large Fc receptor (FcR) inactivation potential in human IgG1 molecules, their clinical value may be limited by non-tumor toxicity (18). The primary reason is that CD47 lacks cancer specificity and is widely distributed in healthy tissues, leading to a substantial “antigen sink”; thus, high doses of anti-CD47 drugs are required to attain anticancer efficacy. Moreover, many anti-CD47 antibodies retain effector functions via their immunoglobulin Fc domains, which may trigger macrophages to engage in ADCP against healthy cells (85–87). Indeed, anemia and thrombocytopenia are common side effects of such anti-CD47 antibodies, often requiring red blood cell transfusions and low-dose initiation strategies to mitigate the adverse situation (82, 88). To manage these risks, current research on anti-CD47 antibodies is focused on molecules that reduce FcγR binding ability, such as IgG4 antibodies. Most of these molecules can still induce severe anemia in non-human primates and cancer patients. Moreover, anti-CD47 antibodies may affect how much CD47 interacts with other receptors, such as integrins, vascular endothelial growth factor receptor-2 (89), thrombospondin-1 (90), and SIRPγ (91). Notably, blocking the interaction between CD47 and SIRPγ can inhibit T-cell extravasation and activation, thereby diminishing the anticancer response. Hence, CD47 signals appear to have a more complex biological functions and its blockade may elicit unexpected cellular responses (3, 27). Additionally, the anticancer activity of anti-CD47 antibodies depends on CS1 glycoprotein antigen (SLAMF7) phagocytic signaling, which is generally absent in solid cancers but is expressed in hematological malignancies (86, 92). Since 2022, multiple Phase III clinical trials of magrolimab were terminated or suspended owing to a lack of survival benefits or adverse reactions, with the regulatory agency also pausing some clinical studies of magrolimab in solid cancers (93). The side effects caused by non-targeted cancer cells and the negative impact on the interaction between CD47 and other receptors have become major obstacles limiting the widespread application of first-type antibodies in the treatment of solid cancers (94).
6.2 Prospects of therapeutic targeting of SIRPα in solid tumors
SIRPα is predominantly expressed in myeloid cells, including monocytes, granulocytes, DCs, macrophages, and microglia, which demonstrates a more limited histological distribution than CD47. SIRPα blocking agents are less likely to be influenced by constraints on antigen expression. Therefore, therapies targeting SIRPα have the potential to avoid side effects associated with targeting CD47 (95, 96) (Table 3). Research on anti-SIRPα antibodies indicates that, similar to CD47 blocking antibodies devoid of Fc, SIRPα blocking agents lacking Fc can effectively induce anticancer immune responses when used along with T-cell-targeted therapies (11). Moreover, monotherapy with anti-SIRPα can alter the composition of the immune cell population in the tumor microenvironment, as evidenced by a significant increase in the proportion of M1 macrophages and a decrease in M2 macrophages (60, 97). SIRPα can negatively regulate DC activation and maturation, thus inhibiting SIRPα can enhance DC responses (52). Anti-SIRPα antibody therapy can stimulate an influx of tumor-infiltrating NK cells and CD8+ T-cells, as well as induce DC activation and promote T-cell effector function when used in combination with anti-PD-1 antibodies (36). The blockade of the SIRPα–CD47 signaling pathway combined with T-cell ICIs can enhance adaptive immune responses. Although the strategy of inhibiting SIRPα has advantages, such as increased antitumor responses and lack of red blood cell toxicity, the high polymorphism rate of the distal IgV domain in the extracellular region of SIRPα raises a risk of cross-reactivity with other members of the SIRP family. This makes the development of clinically beneficial SIRPα inhibitors particularly challenging (27).When glutaminase-like proteases are inhibited, newly synthesized CD47 molecules are unable to effectively bind to their natural binding partners owing to the lack of pyroglutamate modification. Unlike antagonistic molecules targeting CD47 or SIRPα directly, small-molecule inhibitors for this pathway do not compete with natural binding partners in the tumor microenvironment. Moreover, small-molecule inhibitors have high tissue penetration and potential oral bioavailability, which makes them an attractive option. However, the risk of blocking other functions of CD47 persists with small-molecule inhibitors (81). Collectively, anti-SIRPα antibodies capable of blocking all SIRPα alleles hold promise as competitive candidates for achieving the clinical goal of halting the progression of solid cancers.
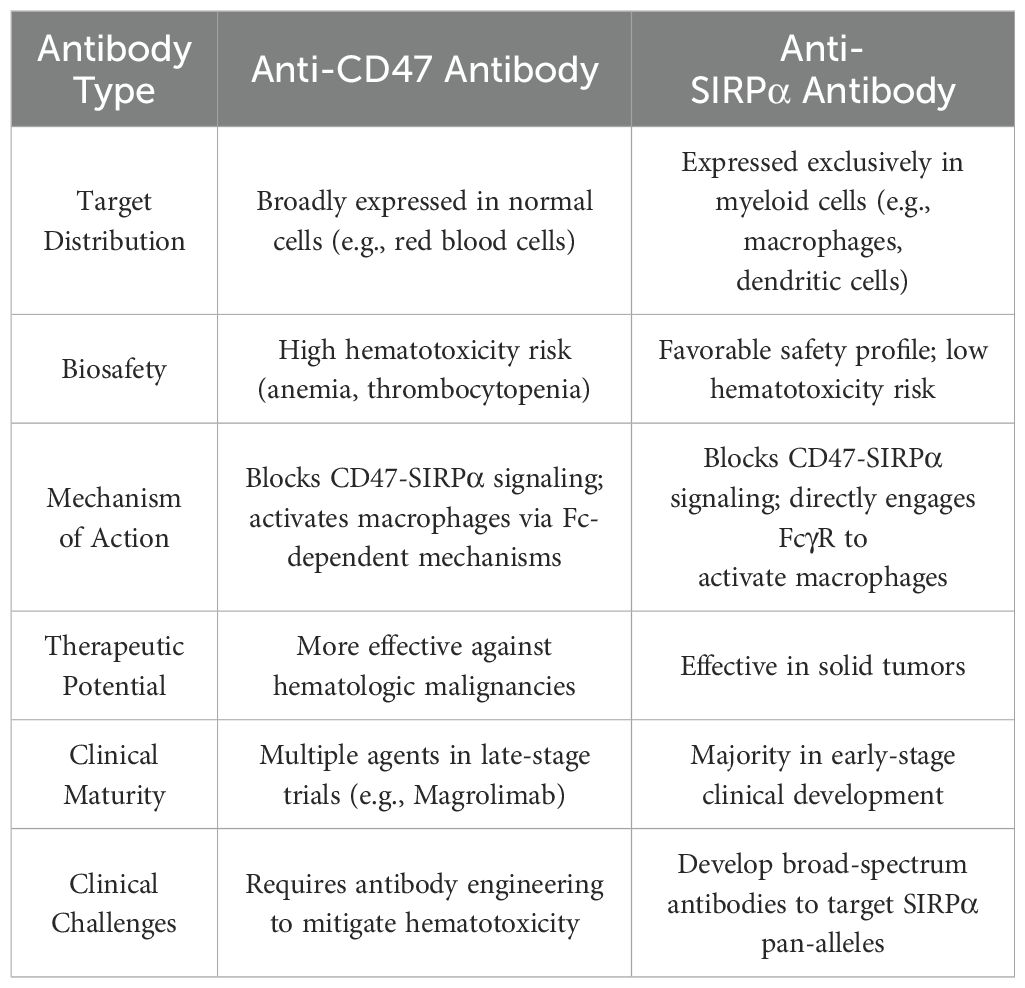
Table 3. The pros and cons of anti-CD47 antibodies versus anti-SIRPα antibodies.
7 Discussion
The CD47–SIRPα interaction plays a key regulatory role in numerous biological processes that influence cellular fate. It is not only viewed as a highly promising target in the field of cancer immunotherapy, but also holds significant importance for maintaining physiological tissue homeostasis (25, 98–100). Collectively, myeloid-intrinsic SIRPα modulates the tumor immune microenvironment by regulating the immunomodulatory functions of macrophages, neutrophils, and DCs. Contrastingly, tumor-intrinsic SIRPα primarily influences malignant phenotypes—such as proliferation, migration, and invasion—via direct intracellular signaling pathways(Figure 4). Notably, although T-cells do not express SIRPα, macrophages and DCs exert multifaceted regulation over T-cell functionality through SIRPα-dependent mechanisms. Blockade of SIRPα enhances antigen presentation in macrophages, promotes the release of pro-inflammatory cytokines, and recruits T-cells to remodel the immunosuppressive tumor microenvironment. Similarly, SIRPα inhibition in DCs alleviates its suppressive effects on antigen presentation, activates cGAS-STING signaling, and stimulates cytokine secretion, directly augmenting CD8+ T-cell cytotoxicity while balancing Th cell differentiation to optimize immune responses. Intriguingly, tumor-intrinsic SIRPα expression may also regulate T cell function. For instance, melanoma cells with low SIRPα expression exhibit suppressed CD8+ T-cell cytotoxicity. However, the molecular mechanisms by which tumor-intrinsic SIRPα modulates T-cell activity remain poorly characterized. These insights underscore the multifaceted role of SIRPα in the tumor ecosystem. Researchers have developed various humanized anti-SIRPα antibodies that have shown excellent anticancer effects in preclinical studies, and some of these antibodies have entered clinical trials. Although the unique pharmacokinetics and biosafety of anti-SIRPα antibodies are highly anticipated, the high polymorphism rate of the human SIRPα V domain poses a challenge for the development of SIRPα-targeting drugs. Fortunately, three allelic combinations (V1/V1, V1/V2, and V2/V2) cover almost the entire human population. In addition to the development of humanized pan-allele-targeting antibodies, future research should focus on designing drug delivery strategies that specifically target the tumor immune microenvironment, developing novel SIRPα-targeting therapeutics, and elucidating the molecular mechanisms of other SIRP family members. These efforts are crucial for advancing the clinical translation of SIRPα-targeted therapies for solid tumors. Further studies are warranted to dissect the cell type-specific functions of SIRPα across immune subsets and tumor cells, which will inform the development of precision immunotherapies tailored to distinct immunological and oncogenic contexts. Consequently, when designing therapeutic strategies targeting SIRPα-overexpressing cancers, it is critical to consider not only the immunostimulatory effects of SIRPα inhibition on myeloid cell-mediated immunity within the tumor microenvironment but also its direct impact on tumor cells and whether such effects may counteract potential immunotherapeutic benefits. In brief, targeting SIRPα may constitute a prospective path for future research in cancer immunotherapy, and studying the role of endogenous SIRPα in cancer cells and progression has significant scientific value.
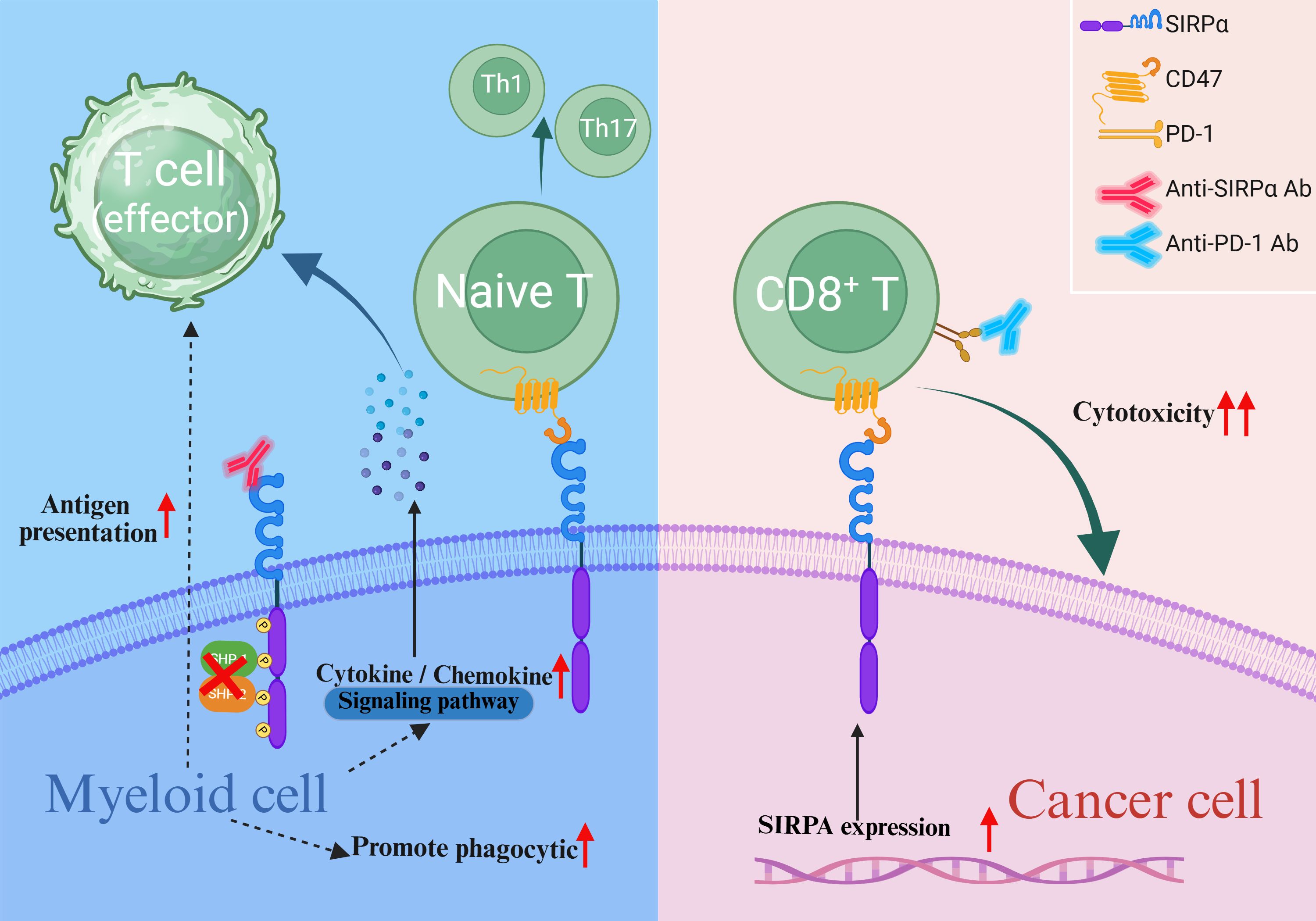
Figure 4. Endogenous SIRPα in tumor malignancy. Blockade of myeloid-intrinsic SIRPα enhances the antigen-presenting capacity of myeloid cells, promotes the release of pro-inflammatory cytokines and chemokines, augments the cytotoxic activity of CD8+ T cells, and modulates the differentiation of T helper cells. Elevated tumor-intrinsic SIRPα expression may also potentiate CD8+ T cell cytotoxicity.
Author contributions
YZ: Writing – original draft. XT: Writing – review & editing. WD: Writing – review & editing, Data curation. CS: Investigation, Writing – review & editing. XY: Supervision, Writing – review & editing. NM: Writing – review & editing. JZ: Supervision, Conceptualization, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Outstanding Youth Foundation of Shaanxi (2024-JC-JCQN-79).
Acknowledgments
The authors would like to appreciate Editage for English language editing and biorender for picture drawing (https://app.biorender.com/).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Johnson PC, Gainor JF, Sullivan RJ, Longo DL, and Chabner B. Immune checkpoint inhibitors - the need for innovation. New Engl J Med. (2023) 388:1529–32. doi: 10.1056/NEJMsb2300232
2. Liu Q, Guan Y, and Li S. Programmed death receptor (PD-)1/PD-ligand (L)1 in urological cancers: the “all-around warrior” in immunotherapy. Mol Cancer. (2024) 23:183. doi: 10.1186/s12943-024-02095-8
3. van Helden MJ, Zwarthoff SA, Arends RJ, Reinieren-Beeren IMJ, Paradé M, Driessen-Engels L, et al. BYON4228 is a pan-allelic antagonistic SIRPα antibody that potentiates destruction of antibody-opsonized tumor cells and lacks binding to SIRPγ on T cells. J Immunother Cancer. (2023) 11:e0065–e0067. doi: 10.1136/jitc-2022-006567
4. Lee D, Cho M, Kim E, Seo Y, and Cha JH. PD-L1: From cancer immunotherapy to therapeutic implications in multiple disorders. Mol Ther. (2024) 32:4235–55. doi: 10.1016/j.ymthe.2024.09.026
5. Sun Q, Hong Z, Zhang C, Wang L, Han Z, and Ma D. Immune checkpoint therapy for solid tumours: clinical dilemmas and future trends. Signal Transduct Target Ther. (2023) 8:320. doi: 10.1038/s41392-023-01522-4
6. Nadal R, Valderrama BP, and Bellmunt J. Progress in systemic therapy for advanced-stage urothelial carcinoma. Nat Rev Clin Oncol. (2024) 21:8–27. doi: 10.1038/s41571-023-00826-2
7. Friedman CF, Manning-Geist BL, Zhou Q, Soumerai T, Holland A, Da Cruz Paula A, et al. Nivolumab for mismatch-repair-deficient or hypermutated gynecologic cancers: a phase 2 trial with biomarker analyses. Nat Med. (2024) 30:1330–8. doi: 10.1038/s41591-024-02942-7
8. Agarwal N, Brugarolas J, Ghatalia P, George S, Haanen JB, Gurney H, et al. Randomized phase II dose comparison LITESPARK-013 study of belzutifan in patients with advanced clear cell renal cell carcinoma. Ann Oncol. (2024) 35:1148–56. doi: 10.1016/j.annonc.2024.08.2338
9. Sun W, Hu S, and Wang X. Advances and clinical applications of immune checkpoint inhibitors in hematological Malignancies. Cancer Commun (London England). (2024) 44:1071–97. doi: 10.1002/cac2.12587
10. Yamada-Hunter SA, Theruvath J, McIntosh BJ, Freitas KA, Lin F, Radosevich MT, et al. Engineered CD47 protects T cells for enhanced antitumour immunity. Nature. (2024) 630:457–65. doi: 10.1038/s41586-024-07443-8
11. Kuo TC, Chen A, Harrabi O, Sockolosky JT, Zhang A, Sangalang E, et al. Targeting the myeloid checkpoint receptor SIRPα potentiates innate and adaptive immune responses to promote anti-tumor activity. J Hematol Oncol. (2020) 13:160. doi: 10.1186/s13045-020-00989-w
12. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. (2015) 21:938–45. doi: 10.1038/nm.3909
13. Joyce JA and Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Sci (New York NY). (2015) 348:74–80. doi: 10.1126/science.aaa6204
14. Vaccaro K, Allen J, Whitfield TW, Maoz A, Reeves S, Velarde J, et al. Targeted therapies prime oncogene-driven lung cancers for macrophage-mediated destruction. J Clin Invest. (2024) 134:e169315. doi: 10.1172/JCI169315
15. Bouwstra R, van Meerten T, and Bremer E. CD47-SIRPα blocking-based immunotherapy: Current and prospective therapeutic strategies. Clin Trans Med. (2022) 12:e943. doi: 10.1002/ctm2.v12.8
16. Barclay AN and Brown MH. The SIRP family of receptors and immune regulation. Nat Rev Immunol. (2006) 6:457–64. doi: 10.1038/nri1859
17. van Beek EM, Cochrane F, Barclay AN, and van den Berg TK. Signal regulatory proteins in the immune system. J Immunol (Baltimore Md: 1950). (2005) 175:7781–7. doi: 10.4049/jimmunol.175.12.7781
18. Logtenberg MEW, Scheeren FA, and Schumacher TN. The CD47-SIRPα Immune checkpoint. Immunity. (2020) 52:742–52. doi: 10.1016/j.immuni.2020.04.011
19. Behrens LM, van den Berg TK, and van Egmond M. Targeting the CD47-SIRPα Innate immune checkpoint to potentiate antibody therapy in cancer by neutrophils. Cancers. (2022) 14:3366. doi: 10.3390/cancers14143366
20. Wang H, Chu F, Zhang XF, Zhang P, Li LX, Zhuang YL, et al. TPX2 enhances the transcription factor activation of PXR and enhances the resistance of hepatocellular carcinoma cells to antitumor drugs. Cell Death Dis. (2023) 14:64. doi: 10.1038/s41419-022-05537-7
21. Eladl E, Tremblay-LeMay R, Rastgoo N, Musani R, Chen W, Liu A, et al. Role of CD47 in hematological Malignancies. J Hematol Oncol. (2020) 13:96. doi: 10.1186/s13045-020-00930-1
22. Treffers LW, Zhao XW, van der Heijden J, Nagelkerke SQ, van Rees DJ, Gonzalez P, et al. Genetic variation of human neutrophil Fcγ receptors and SIRPα in antibody-dependent cellular cytotoxicity towards cancer cells. Eur J Immunol. (2018) 48:344–54. doi: 10.1002/eji.201747215
23. Takenaka K, Prasolava TK, Wang JC, Mortin-Toth SM, Khalouei S, Gan OI, et al. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat Immunol. (2007) 8:1313–23. doi: 10.1038/ni1527
24. Sim J, Sockolosky JT, Sangalang E, Izquierdo S, Pedersen D, Harriman W, et al. Discovery of high affinity, pan-allelic, and pan-mammalian reactive antibodies against the myeloid checkpoint receptor SIRPα. mAbs. (2019) 11:1036–52. doi: 10.1080/19420862.2019.1624123
25. Feng Y, Huang C, Wang Y, and Chen J. SIRPα: A key player in innate immunity. Eur J Immunol. (2023) 53:e2350375. doi: 10.1002/eji.202350375
26. Gauttier V, Pengam S, Durand J, Biteau K, Mary C, Morello A, et al. Selective SIRPα blockade reverses tumor T cell exclusion and overcomes cancer immunotherapy resistance. J Clin Invest. (2020) 130:6109–23. doi: 10.1172/JCI135528
27. Voets E, Paradé M, Lutje Hulsik D, Spijkers S, Janssen W, Rens J, et al. Functional characterization of the selective pan-allele anti-SIRPα antibody ADU-1805 that blocks the SIRPα-CD47 innate immune checkpoint. J Immunother Cancer. (2019) 7:340. doi: 10.1186/s40425-019-0772-0
28. Visser N, Nelemans LC, He Y, Lourens HJ, Corrales MG, Huls G, et al. Signal regulatory protein beta 2 is a novel positive regulator of innate anticancer immunity. Front Immunol. (2023) 14:1287256. doi: 10.3389/fimmu.2023.1287256
29. Morrissey MA, Kern N, and Vale RD. CD47 ligation repositions the inhibitory receptor SIRPA to suppress integrin activation and phagocytosis. Immunity. (2020) 53:290–302.e6. doi: 10.1016/j.immuni.2020.07.008
30. Catalán R, Orozco-Morales M, Hernández-Pedro NY, Guijosa A, Colín-González AL, Ávila-Moreno F, et al. CD47-SIRPα Axis as a biomarker and therapeutic target in cancer: current perspectives and future challenges in nonsmall cell lung cancer. J Immunol Res. (2020) 2020:9435030. doi: 10.1155/2020/9435030
31. Jia X, Yan B, Tian X, Liu Q, Jin J, Shi J, et al. CD47/SIRPα pathway mediates cancer immune escape and immunotherapy. Int J Biol Sci. (2021) 17:3281–7. doi: 10.7150/ijbs.60782
32. Martins TA, Kaymak D, Tatari N, Gerster F, Hogan S, Ritz MF, et al. Enhancing anti-EGFRvIII CAR T cell therapy against glioblastoma with a paracrine SIRPγ-derived CD47 blocker. Nat Commun. (2024) 15:9718. doi: 10.1038/s41467-024-54129-w
33. Chen J, Dai Q, Yang Q, Bao X, Zhou Y, Zhong H, et al. Therapeutic nucleus-access BNCT drug combined CD47-targeting gene editing in glioblastoma. J Nanobiotechnol. (2022) 20:102. doi: 10.1186/s12951-022-01304-0
34. Zhang H, Huo Y, Zheng W, Li P, Li H, Zhang L, et al. Silencing of SIRPα enhances the antitumor efficacy of CAR-M in solid tumors. Cell Mol Immunol. (2024) 21:1335–49. doi: 10.1038/s41423-024-01220-3
35. Li Y, Yi J, Ma R, Wang Y, Lou X, Dong Y, et al. A polymeric nanoplatform enhances the cGAS-STING pathway in macrophages to potentiate phagocytosis for cancer immunotherapy. J Controlled Release. (2024) 373:447–62. doi: 10.1016/j.jconrel.2024.07.039
36. Huang C, Wang X, Wang Y, Feng Y, Wang X, Chen S, et al. Sirpα on tumor-associated myeloid cells restrains antitumor immunity in colorectal cancer independent of its interaction with CD47. Nat Cancer. (2024) 5:500–16. doi: 10.1038/s43018-023-00691-z
37. Shaul ME and Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol. (2019) 16:601–20. doi: 10.1038/s41571-019-0222-4
38. Ring NG, Herndler-Brandstetter D, Weiskopf K, Shan L, Volkmer JP, George BM, et al. Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc Natl Acad Sci United States Am. (2017) 114:E10578–e85. doi: 10.1073/pnas.1710877114
39. van Rees DJ, Brinkhaus M, Klein B, Verkuijlen P, Tool ATJ, Schornagel K, et al. Sodium stibogluconate and CD47-SIRPα blockade overcome resistance of anti-CD20-opsonized B cells to neutrophil killing. Blood Adv. (2022) 6:2156–66. doi: 10.1182/bloodadvances.2021005367
40. Pan L, Wang B, Chen M, Ma Y, Cui B, Chen Z, et al. Lack of SIRP-alpha reduces lung cancer growth in mice by promoting anti-tumour ability of macrophages and neutrophils. Cell Prolif. (2023) 56:e13361. doi: 10.1111/cpr.13361
41. Boross P, Lohse S, Nederend M, Jansen JH, van Tetering G, Dechant M, et al. IgA EGFR antibodies mediate tumour killing in vivo. EMBO Mol Med. (2013) 5:1213–26. doi: 10.1002/emmm.201201929
42. Borrok MJ, Luheshi NM, Beyaz N, Davies GC, Legg JW, Wu H, et al. Enhancement of antibody-dependent cell-mediated cytotoxicity by endowing IgG with FcαRI (CD89) binding. mAbs. (2015) 7:743–51. doi: 10.1080/19420862.2015.1047570
43. Lohse S, Loew S, Kretschmer A, Jansen JHM, Meyer S, Ten Broeke T, et al. Effector mechanisms of IgA antibodies against CD20 include recruitment of myeloid cells for antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity. Br J Haematol. (2018) 181:413–7. doi: 10.1111/bjh.2018.181.issue-3
44. Dechant M, Beyer T, Schneider-Merck T, Weisner W, Peipp M, van de Winkel JG, et al. Effector mechanisms of recombinant IgA antibodies against epidermal growth factor receptor. J Immunol (Baltimore Md: 1950). (2007) 179:2936–43. doi: 10.4049/jimmunol.179.5.2936
45. Chan C, Stip M, Nederend M, Jansen M, Passchier E, van den Ham F, et al. Enhancing IgA-mediated neutrophil cytotoxicity against neuroblastoma by CD47 blockade. J Immunother Cancer. (2024) 12:e008478. doi: 10.1136/jitc-2023-008478
46. Treffers LW, Ten Broeke T, Rösner T, Jansen JHM, van Houdt M, Kahle S, et al. IgA-mediated killing of tumor cells by neutrophils is enhanced by CD47-SIRPα Checkpoint inhibition. Cancer Immunol Res. (2020) 8:120–30. doi: 10.1158/2326-6066.CIR-19-0144
47. Xie MM, Dai B, Hackney JA, Sun T, Zhang J, Jackman JK, et al. An agonistic anti-signal regulatory protein α antibody for chronic inflammatory diseases. Cell Rep Med. (2023) 4:101130. doi: 10.1016/j.xcrm.2023.101130
48. Zen K, Guo Y, Bian Z, Lv Z, Zhu D, Ohnishi H, et al. Inflammation-induced proteolytic processing of the SIRPα cytoplasmic ITIM in neutrophils propagates a proinflammatory state. Nat Commun. (2013) 4:2436. doi: 10.1038/ncomms3436
49. van Kooyk Y and Geijtenbeek TB. DC-SIGN: escape mechanism for pathogens. Nat Rev Immunol. (2003) 3:697–709. doi: 10.1038/nri1182
50. Hargadon KM. Tumor microenvironmental influences on dendritic cell and T cell function: A focus on clinically relevant immunologic and metabolic checkpoints. Clin Trans Med. (2020) 10:374–411. doi: 10.1002/ctm2.v10.1
51. Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, et al. Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein α Signaling. Immunity. (2017) 47:363–73.e5. doi: 10.1016/j.immuni.2017.07.016
52. Liu Q, Wen W, Tang L, Qin CJ, Lin Y, Zhang HL, et al. Inhibition of SIRPα in dendritic cells potentiates potent antitumor immunity. Oncoimmunology. (2016) 5:e1183850. doi: 10.1080/2162402X.2016.1183850
53. Hsieh RC, Krishnan S, Wu RC, Boda AR, Liu A, Winkler M, et al. ATR-mediated CD47 and PD-L1 up-regulation restricts radiotherapy-induced immune priming and abscopal responses in colorectal cancer. Sci Immunol. (2022) 7:eabl9330. doi: 10.1126/sciimmunol.abl9330
54. Ji K, Zhang Y, Jiang S, Sun L, Zhang B, Hu D, et al. SIRPα blockade improves the antitumor immunity of radiotherapy in colorectal cancer. Cell Death Discov. (2023) 9:180. doi: 10.1038/s41420-023-01472-4
55. Li X, Zhou W, Liang Y, Xu C, Xie Z, Liang J, et al. The immunotherapeutic effect of SIRPα-silenced DCs against cervical cancer. J Immunol Res. (2020) 2020:1705187. doi: 10.1155/2020/1705187
56. Saito Y, Iwamura H, Kaneko T, Ohnishi H, Murata Y, Okazawa H, et al. Regulation by SIRPα of dendritic cell homeostasis in lymphoid tissues. Blood. (2010) 116:3517–25. doi: 10.1182/blood-2010-03-277244
57. Fukunaga A, Nagai H, Yu X, Oniki S, Okazawa H, Motegi S, et al. Src homology 2 domain-containing protein tyrosine phosphatase substrate 1 regulates the induction of Langerhans cell maturation. Eur J Immunol. (2006) 36:3216–26. doi: 10.1002/eji.200635864
58. Tomizawa T, Kaneko Y, Kaneko Y, Saito Y, Ohnishi H, Okajo J, et al. Resistance to experimental autoimmune encephalomyelitis and impaired T cell priming by dendritic cells in Src homology 2 domain-containing protein tyrosine phosphatase substrate-1 mutant mice. J Immunol (Baltimore Md: 1950). (2007) 179:869–77. doi: 10.4049/jimmunol.179.2.869
59. Fortin G, Raymond M, Van VQ, Rubio M, Gautier P, Sarfati M, et al. A role for CD47 in the development of experimental colitis mediated by SIRPalpha+CD103- dendritic cells. J Exp Med. (2009) 206:1995–2011. doi: 10.1084/jem.20082805
60. Yanagita T, Murata Y, Tanaka D, Motegi SI, Arai E, Daniwijaya EW, et al. Anti-SIRPα antibodies as a potential new tool for cancer immunotherapy. JCI Insight. (2017) 2:e89140. doi: 10.1172/jci.insight.89140
61. Oyoshi H, Du J, Sakai SA, Yamashita R, Okumura M, Motegi A, et al. Comprehensive single-cell analysis demonstrates radiotherapy-induced infiltration of macrophages expressing immunosuppressive genes into tumor in esophageal squamous cell carcinoma. Sci Adv. (2023) 9:eadh9069. doi: 10.1126/sciadv.adh9069
62. Wang P, Song Y, Li H, Zhuang J, Shen X, Yang W, et al. SIRPA enhances osteosarcoma metastasis by stabilizing SP1 and promoting SLC7A3-mediated arginine uptake. Cancer Lett. (2023) 576:216412. doi: 10.1016/j.canlet.2023.216412
63. Pan C, Zhu D, Zhuo J, Li L, Wang D, Zhang CY, et al. Role of signal regulatory protein α in arsenic trioxide-induced promyelocytic leukemia cell apoptosis. Sci Rep. (2016) 6:23710. doi: 10.1038/srep23710
64. Yan HX, Wang HY, Zhang R, Chen L, Li BA, Liu SQ, et al. Negative regulation of hepatocellular carcinoma cell growth by signal regulatory protein alpha1. Hepatol (Baltimore Md). (2004) 40:618–28. doi: 10.1002/hep.20360
65. Marshall E, Pikor L, Chari R, Kennett J, Lam S, and Lam W. P1.02–052 signal regulatory protein a (SIRPA): A&xa0;Key regulator of the EGFR pathway demonstrates both tumor suppressive and oncogenic properties: topic: driver genes in NSCLC, resistance, and other. J Thorac Oncol. (2017) 12:S519. doi: 10.1016/j.jtho.2016.11.636
66. Zhou Z, Chen MM, Luo Y, Mojumdar K, Peng X, Chen H, et al. Tumor-intrinsic SIRPA promotes sensitivity to checkpoint inhibition immunotherapy in melanoma. Cancer Cell. (2022) 40:1324–40.e8. doi: 10.1016/j.ccell.2022.10.012
67. Jiang D, Burger CA, Akhanov V, Liang JH, Mackin RD, Albrecht NE, et al. Neuronal signal-regulatory protein alpha drives microglial phagocytosis by limiting microglial interaction with CD47 in the retina. Immunity. (2022) 55:2318–35.e7. doi: 10.1016/j.immuni.2022.10.018
68. Chen TT, Brown EJ, Huang EJ, and Seaman WE. Expression and activation of signal regulatory protein alpha on astrocytomas. Cancer Res. (2004) 64:117–27. doi: 10.1158/0008-5472.CAN-3455-2
69. Andrejeva G, Capoccia BJ, Hiebsch RR, Donio MJ, Darwech IM, Puro RJ, et al. Novel SIRPα Antibodies that induce single-agent phagocytosis of tumor cells while preserving T cells. J Immunol (Baltimore Md: 1950). (2021) 206:712–21. doi: 10.4049/jimmunol.2001019
70. Sakamoto M, Murata Y, Tanaka D, Kakuchi Y, Okamoto T, Hazama D, et al. Anticancer efficacy of monotherapy with antibodies to SIRPα/SIRPβ1 mediated by induction of antitumorigenic macrophages. Proc Natl Acad Sci United States America. (2022) 119:e2109923118. doi: 10.1073/pnas.2109923118
71. Wu ZH, Li N, Mei XF, Chen J, Wang XZ, Guo TT, et al. Preclinical characterization of the novel anti-SIRPα antibody BR105 that targets the myeloid immune checkpoint. J Immunother Cancer. (2022) 10:e004054. doi: 10.1136/jitc-2021-004054
72. Liu J, Xavy S, Mihardja S, Chen S, Sompalli K, Feng D, et al. Targeting macrophage checkpoint inhibitor SIRPα for anticancer therapy. JCI Insight. (2020) 5:e134728. doi: 10.1172/jci.insight.134728
73. Eskiocak U, Guzman W, Daly T, Nelson A, Bakhru P, Lajoie JM, et al. Abstract 3239: CTX-5861 mediated SIRPα blockade combines with tumor targeting antibodies, checkpoint blockade and/or CD137 agonism to elicit curative anti-tumor activity in syngeneic mouse models. Cancer Res. (2019) 79:3239. doi: 10.1158/1538-7445.AM2019-3239
74. Yang J, Deresa I, Ho WH, Long H, Maslyar D, Rosenthal A, et al. AL008 enhances myeloid antitumor function by inhibiting SIRPα Signaling and activating fc receptors. J Immunol (Baltimore Md: 1950). (2023) 210:204–15. doi: 10.4049/jimmunol.2200157
75. Voets E, Kreijtz J, Vink P, Hulsik DL, Parade M, Spijkers S, et al. Abstract 1203: Preclinical development of ADU-1805, a highly selective pan-allele anti-SIRPα antibody that blocks the SIRPα-CD47 innate immune checkpoint. Cancer Res. (2019) 79:1203–. doi: 10.1158/1538-7445.AM2019-1203
76. Champiat S, Cassier PA, Kotecki N, Korakis I, Vinceneux A, Jungels C, et al. Safety, pharmacokinetics, efficacy, and preliminary biomarker data of first-in-class BI 765063, a selective SIRPα inhibitor: Results of monotherapy dose escalation in phase 1 study in patients with advanced solid tumors. J Clin Oncol. (2021) 39:2623–. doi: 10.1200/jco.2021.39.15_suppl.2623
77. Kotecki N, Champiat S, Delord JP, Vinceneux A, Jungels C, Marabelle A, et al. 983P Phase I dose escalation study in patients (pts) with advanced solid tumours receiving first-in-class BI 765063, a selective signal-regulatory protein &x3b1; (SIRP&x3b1); inhibitor, in combination with ezabenlimab (BI 754091), a programmed cell death protein 1 (PD-1) inhibitor. Ann Oncol. (2021) 32:S841–S2. doi: 10.1016/j.annonc.2021.08.1367
78. Gutierrez M, Jamal R, Yamamoto N, Doi T, Elgadi M, Ferrada JL, et al. 697P Open-label, phase I, dose escalation/expansion trial of the anti-SIRP&x3b1; monoclonal antibody BI 770371 in patients with advanced solid tumours, alone or in combination with the anti-PD-1 monoclonal antibody ezabenlimab. Ann Oncol. (2023) 34:S485. doi: 10.1093/annonc/mdy282.080
79. Chan H, Trout C, Mikolon D, Adams P, Guzman R, Fenalti G, et al. Discovery and preclinical characterization of CC-95251, an anti-SIRPα Antibody that enhances macrophage-mediated phagocytosis of non-hodgkin lymphoma (NHL) cells when combined with rituximab. Blood. (2021) 138:2271. doi: 10.1182/blood-2021-147262
80. Chan H, Trout CV, Mikolon D, Adams P, Guzman R, Mavrommatis K, et al. Discovery and preclinical activity of BMS-986351, an antibody to SIRPα That enhances macrophage-mediated tumor phagocytosis when combined with opsonizing antibodies. Cancer Res Commun. (2024) 4:505–15. doi: 10.1158/2767-9764.CRC-23-0634
81. Logtenberg MEW, Jansen JHM, Raaben M, Toebes M, Franke K, Brandsma AM, et al. Glutaminyl cyclase is an enzymatic modifier of the CD47- SIRPα axis and a target for cancer immunotherapy. Nat Med. (2019) 25:612–9. doi: 10.1038/s41591-019-0356-z
82. Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, et al. CD47 blockade by hu5F9-G4 and rituximab in non-hodgkin’s lymphoma. New Engl J Med. (2018) 379:1711–21. doi: 10.1056/NEJMoa1807315
83. Sikic BI, Lakhani N, Patnaik A, Shah SA, Chandana SR, Rasco D, et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody hu5F9-G4 in patients with advanced cancers. J Clin Oncol. (2019) 37:946–53. doi: 10.1200/JCO.18.02018
84. Son J, Hsieh RC, Lin HY, Krause KJ, Yuan Y, Biter AB, et al. Inhibition of the CD47-SIRPα axis for cancer therapy: A systematic review and meta-analysis of emerging clinical data. Front Immunol. (2022) 13:1027235. doi: 10.3389/fimmu.2022.1027235
85. Tahk S, Vick B, Hiller B, Schmitt S, Marcinek A, Perini ED, et al. SIRPα-αCD123 fusion antibodies targeting CD123 in conjunction with CD47 blockade enhance the clearance of AML-initiating cells. J Hematol Oncol. (2021) 14:155. doi: 10.1186/s13045-021-01163-6
86. Jain S, Van Scoyk A, Morgan EA, Matthews A, Stevenson K, Newton G, et al. Targeted inhibition of CD47-SIRPα requires Fc-FcγR interactions to maximize activity in T-cell lymphomas. Blood. (2019) 134:1430–40. doi: 10.1182/blood.2019001744
87. Osorio JC, Smith P, Knorr DA, and Ravetch JV. The antitumor activities of anti-CD47 antibodies require Fc-FcγR interactions. Cancer Cell. (2023) 41:2051–65.e6. doi: 10.1016/j.ccell.2023.10.007
88. Ansell SM, Maris MB, Lesokhin AM, Chen RW, Flinn IW, Sawas A, et al. Phase I study of the CD47 blocker TTI-621 in patients with relapsed or refractory hematologic Malignancies. Clin Cancer Res. (2021) 27:2190–9. doi: 10.1158/1078-0432.CCR-20-3706
89. Kaur S, Martin-Manso G, Pendrak ML, Garfield SH, Isenberg JS, and Roberts DD. Thrombospondin-1 inhibits VEGF receptor-2 signaling by disrupting its association with CD47. J Biol Chem. (2010) 285:38923–32. doi: 10.1074/jbc.M110.172304
90. Soto-Pantoja DR, Stein EV, Rogers NM, Sharifi-Sanjani M, Isenberg JS, and Roberts DD. Therapeutic opportunities for targeting the ubiquitous cell surface receptor CD47. Expert Opin Ther Targets. (2013) 17:89–103. doi: 10.1517/14728222.2013.733699
91. Brooke G, Holbrook JD, Brown MH, and Barclay AN. Human lymphocytes interact directly with CD47 through a novel member of the signal regulatory protein (SIRP) family. J Immunol (Baltimore Md: 1950). (2004) 173:2562–70. doi: 10.4049/jimmunol.173.4.2562
92. Chen J, Zhong MC, Guo H, Davidson D, Mishel S, Lu Y, et al. SLAMF7 is critical for phagocytosis of haematopoietic tumour cells via Mac-1 integrin. Nature. (2017) 544:493–7. doi: 10.1038/nature22076
93. Stefanidis E, Semilietof A, Pujol J, Seijo B, Scholten K, Zoete V, et al. Combining SiRPα decoy-coengineered T cells and antibodies augments macrophage-mediated phagocytosis of tumor cells. J Clin Invest. (2024) 134:e161660. doi: 10.1172/JCI161660
94. Chen Q, Guo X, and Ma W. Opportunities and challenges of CD47-targeted therapy in cancer immunotherapy. Oncol Res. (2023) 32:49–60. doi: 10.32604/or.2023.042383
95. Singla B, Lin HP, Ahn W, Xu J, Ma Q, Sghayyer M, et al. Loss of myeloid cell-specific SIRPα, but not CD47, attenuates inflammation and suppresses atherosclerosis. Cardiovasc Res. (2022) 118:3097–111. doi: 10.1093/cvr/cvab369
96. Shi H, Wang X, Li F, Gerlach BD, Yurdagul A Jr., Moore MP, et al. CD47-SIRPα axis blockade in NASH promotes necroptotic hepatocyte clearance by liver macrophages and decreases hepatic fibrosis. Sci Trans Med. (2022) 14:eabp8309. doi: 10.1126/scitranslmed.abp8309
97. Brown JM, Recht L, and Strober S. The promise of targeting macrophages in cancer therapy. Clin Cancer Res. (2017) 23:3241–50. doi: 10.1158/1078-0432.CCR-16-3122
98. Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. (2009) 138:271–85. doi: 10.1016/j.cell.2009.05.046
99. Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. (2016) 536:86–90. doi: 10.1038/nature18935
Keywords: CD47, immunotherapy, immune checkpoint inhibitor, SIRPα, solid tumor
Citation: Zhou Y, Tang X, Du W, Shu C, Yan X, Ma N and Zhao J (2025) Deciphering the role of signal regulatory protein α in immunotherapy for solid tumors. Front. Immunol. 16:1612234. doi: 10.3389/fimmu.2025.1612234
Received: 15 April 2025; Accepted: 27 May 2025;
Published: 16 June 2025.
Edited by:
Wenxue Ma, University of California, San Diego, United StatesReviewed by:
Kamalika Mojumdar, University of Texas MD Anderson Cancer Center, United StatesHao Sun, Dana–Farber Cancer Institute, United States
Copyright © 2025 Zhou, Tang, Du, Shu, Yan, Ma and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinbo Zhao, emhhb2ppbmJvQGFsaXl1bi5jb20=; Nan Ma, bWFuYW44NDA4MDhAMTYzLmNvbQ==; Xiaolong Yan, eWFueGlhb2xvbmdAZm1tdS5lZHUuY24=
†These authors share first authorship