 Minxia Ke1,2,3
Minxia Ke1,2,3 Wenli Liu2,3Huimin Lu2Xiafei Pan1Mengyang Wu1Nianmin Qi2Zhiqiang Wang2,3
Wenli Liu2,3Huimin Lu2Xiafei Pan1Mengyang Wu1Nianmin Qi2Zhiqiang Wang2,3 Yuehong Wu1*Feng Zhang2,3*
Yuehong Wu1*Feng Zhang2,3*- 1Department of Biochemistry and Molecular Biology, College of Life Science and Medicine, Zhejiang Sci-Tech University, Hangzhou, Zhejiang, China
- 2Department of Early Research, Asia Cell & Gene Therapeutics Co., Limited, Zhejiang, China
- 3Department of Early Research, Horgos Stem Cell Therapy Co., Limited, Horgos, Xinjiang, China
Ankylosing spondylitis (AS) is a chronic autoimmune inflammatory disease primarily affecting the axial skeleton, characterized by joint erosion and ankylosis. AS significantly impacts quality of life, work capacity and mental health through chronic pain, stiffness and functional decline. Its pathogenesis is multifactorial, involving genetic predispositions, immunological dysregulation and environmental triggers. Current treatments, including nonsteroidal anti-inflammatory drugs and immunosuppressive agents, offer limited symptomatic relief and fail to improve long-term prognosis due to efficacy limitations and side effects. Recent advances in cell therapy, particularly mesenchymal stem cells (MSCs) and chimeric antigen receptor (CAR) T-cell therapy, demonstrate promise in addressing these limitations by providing immunomodulatory, anti-inflammatory and regenerative benefits. This review summarizes the pathogenesis of AS, the limitations of existing treatments and the clinical progress of MSC therapy, while exploring the potential of emerging CAR-based therapies.
1 Introduction
Ankylosing spondylitis (AS) is a chronic autoimmune inflammatory disease predominantly affecting the axial skeleton, including the spine and sacroiliac joints, resulting in progressive joint erosion and eventual ankylosis (1). The disease progresses slowly with a long duration, and its peak onset occurs in young adults aged 20–30 years (2). The global prevalence of AS varies geographically, ranging from 0.1% to 1.4%, with a male-to-female ratio averaging 3.4:1 (3). Specifically, the prevalence rates are 0.238% in Europe, 0.167% in Asia, 0.102% in Latin America, 0.319% in North America and 0.074% in Africa. In China, a comprehensive survey across 16 regions reported an overall prevalence of 0.22%, with a male prevalence of 0.36% and female prevalence of 0.09%, yielding a male-to-female ratio of 4:1 (4). According to the latest Chinese guidelines (2022), the estimated prevalence is 0.3%, which exhibits an upward trend (5).
Clinically, AS presents with significant back pain, stiffness and functional decline, ultimately leading to spinal and pelvic fusion (6). In adolescents, AS may initially manifest as non-radiographic axial spondyloarthritis (nr-axSpA), with characteristic sacroiliac joint changes emerging later (7). AS is frequently associated with other autoimmune diseases, such as acute anterior uveitis, inflammatory bowel disease and psoriasis (8). AS exerts a lifelong detrimental effect on patients, significantly impacting their quality of life, work capacity and mental health (9). Furthermore, AS is correlated with an increased risk of premature mortality (10). In the treatment of AS, nonsteroidal anti-inflammatory drugs (NSAIDs) and immunosuppressive agents have traditionally been employed. While these therapies can effectively mitigate inflammatory responses, alleviate clinical symptoms and enhance patients’ quality of life, they are still associated with suboptimal therapeutic outcomes and a range of adverse effects (11). Moreover, current treatments fail to enhance long-term prognosis, imposing a significant burden on patients and society (12). Consequently, there is an urgent requirement for more comprehensive research into the pathogenesis of AS, alongside the expedited development of innovative therapeutic strategies.
In recent years, the emergence and advancement of innovative therapies, such as cell therapy, have offered promising new avenues for the treatment of AS. Extensive research has demonstrated that mesenchymal stem cells (MSCs) possess significant immunomodulatory and regenerative properties (13). They can mitigate inflammatory responses and facilitate tissue repair through both direct cell-to-cell interactions and the secretion of bioactive soluble factors (13). Additionally, chimeric antigen receptor (CAR) T-cell therapy has emerged as a promising therapeutic strategy for autoimmune diseases, demonstrating significant potential in early clinical trials for conditions such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) and type 1 diabetes (14–16). This article reviews the pathogenesis of AS, existing treatment methods and their limitations, summarizes the clinical progress and mechanisms of MSC treatment for AS, and explores the potential of other cell therapies (such as CAR-based cell therapies) in the treatment of AS. Furthermore, we critically analyze the issues that need to be addressed before cell therapy can be routinely used to treat AS.
2 Pathogenesis of AS
The pathogenesis of AS is multifactorial, involving a complex interplay between genetic and environmental factors. Genetic factors are considered significant contributors to the development of AS, especially the human leukocyte antigen B27 (HLA-B27), which has been strongly implicated in disease susceptibility (17). The positivity rate of HLA-B27 in AS patients is over 90%, compared to only 4%-7% in the general population (17). The potential mechanisms through which HLA-B27 abnormalities contribute to the development of AS encompass: the arthritogenic peptide hypothesis, immune recognition of abnormal forms of HLA-B27, and the induction of endoplasmic reticulum stress (ERS) response due to the accumulation of misfolded HLA-B27 molecules (18).
The arthritogenic peptide hypothesis proposes that antigen-presenting cells (APCs) in AS patients present both self-antigens and microbial peptides via HLA-B27, thereby triggering a specific immune response mediated by CD8+ cytotoxic T cells (19). These T cells recognize and respond to the presented peptides, leading to the activation and clonal expansion of pathogenic T-cell clones that drive inflammation and tissue damage in the joints (19). A recent study has provided compelling evidence supporting the arthritogenic peptide hypothesis associated with HLA-B27 (20). This work identified CD8+ T cells expressing disease-related T cell receptors (TCRs) with specific TRBV9–CDR3–Jβ2.3 chains in the blood and synovial fluid of AS patients. These TRBV9 chains pair with TRAV21 chains and expand clonally within the joints. Utilizing an HLA-B27:05 yeast display peptide library, the study successfully identified microbial and self-antigen peptides capable of activating AS-associated TCRs. Structural analysis revealed that the cross-reactivity between peptide-MHC and TCRs originates from a common motif shared by self-antigens and microbial antigens, which binds specifically to the TRBV9-CDR3β TCR. These findings underscore the potential pathogenic role of both microbial and self-antigens in HLA-B27-associated diseases and highlight the arthritogenic peptide hypothesis as a key mechanism underlying the development of AS.
Abnormal forms of HLA-B27, such as homodimers, are suggested to bind to specific killer cell immunoglobulin-like receptors (KIRs) expressed on natural killer (NK) cells and CD4+ T cells (21–23). This interaction triggers the release of inflammatory cytokines and chemokines, thereby enhancing T cell activation and stimulating other immune cells to initiate an inflammatory response (21–23). The unfolded protein response (UPR) hypothesis suggests that the accumulation of misfolded HLA-B27 in the ER during protein biosynthesis leads to an inflammatory response (24). HLA-B27 misfolding is associated with specific polymorphisms that characterize this allele, leading to inefficient folding and peptide loading of the heavy chain (24). This misfolding can trigger ER-associated degradation (ERAD) of the heavy chains, primarily mediated by the E3 ubiquitin ligase HRD1 (SYVN1) and the ubiquitin-conjugating enzyme UBE2JL (25). Activation of the UPR has been associated with cytokine dysregulation, leading to increased production of IL-23, IFNβ, and IL-1α (26, 27). In addition to the above hypotheses, there is also evidence that HLA-B27 can disrupt the composition of the gut microbiota, leading to microbial dysbiosis, metabolic dysfunction, and loss of mucosal tolerance. This disruption can result in the release of pro-inflammatory cytokines such as IFN-γ, TNF, and IL-17, as well as the activation of regulatory T cells (Tregs) and helper T cells (Th1, Th2, and Th17 cells) (28–31). These changes contribute to chronic inflammation in the joints, skin, or gut, further complicating the pathogenesis of AS (28–31).
In addition to HLA-B27, more than 100 genes have been identified as contributing to the susceptibility of AS (32). ER aminopeptidase 1 (ERAP1) stands out as the second most significant gene associated with AS pathogenesis (33, 34). ERAP1 polymorphisms directly influence the generation of the peptide repertoire, thereby modulating the formation of pathogenic peptides that contribute to AS development (33, 34). The IL-23 receptor and the Th17/IL-23 axis are critical factors in the inflammatory cascade of AS (35). Genetic polymorphisms within these pathways have been robustly associated with disease pathogenesis, emphasizing their role in the inflammatory process. Additionally, IFNs, as key early inflammatory mediators, can induce the production of pro-inflammatory cytokines TNFα and IL-1 and activate the NF-κB signaling pathway, thereby participating in the pathogenesis of AS (36). Toll-like receptor 7 (TLR7) has also been implicated in AS susceptibility, although its role varies by sex. TLR7 acts as a protective factor in females with AS but serves as a risk factor in males, suggesting sex-specific mechanisms in disease pathogenesis (37). Additionally, the janus kinase-signal transducer and activators of transcription (JAK-STAT) pathway, a canonical signaling pathway in the inflammatory network, plays a pivotal role in AS pathogenesis (6). This pathway integrates signals from various cytokines and growth factors, driving the transcriptional response that perpetuates inflammation and tissue damage in AS (6).
Collectively, these genetic and molecular pathways underscore the complex multifactorial nature of AS, emphasizing the intricate interplay between genetic predisposition, immune signaling, and inflammatory mediators in disease development. Future research should aim to elucidate the precise mechanisms by which these genetic variants contribute to AS pathogenesis and investigate potential therapeutic targets within these pathways.
3 Current AS treatment options and their limitations
The treatment drugs for AS recommended jointly by the Assessment of Spondylo Arthritis International Society (ASAS), the European League Against Rheumatism (EULAR), and the Chinese Society of Rheumatology (CSR) encompass NSAIDs, biologic disease-modifying antirheumatic drugs (bDMARDs), sulfasalazine (SSZ), methotrexate (MTX), and corticosteroids (38). The efficacy of these medications varies significantly, with each class of drugs presenting distinct advantages and limitations (Figure 1).

Figure 1. Current treatments for AS. (A) Nonsteroidal anti-inflammatory drugs (NSAIDs): NSAIDs are the first-line treatment for AS, providing rapid relief of back pain, morning stiffness, and joint swelling. Commonly used NSAIDs include ibuprofen, naproxen, diclofenac and indomethacin. (B) Biological agents: Biological agents, including TNF-α inhibitors (TNFi), interleukin inhibitors, and JAK inhibitors (JAKi), constitute a targeted and efficacious therapeutic strategy for the management of AS. These agents modulate specific inflammatory pathways, offering a more precise treatment option for patients, particularly those who exhibit an inadequate response to NSAIDs. (C) Conventional synthetic disease-modifying antirheumatic drugs (csDMARDs): Drugs like sulfasalazine and methotrexate are used for patients with peripheral joint involvement or those with contraindications to biologics. (D) Physical therapy: Physical therapy and surgical interventions are both essential components in the comprehensive management of AS. Physical therapy aims to enhance mobility and strength through personalized exercise regimens, while surgery is considered for severe cases to correct deformities or alleviate symptoms that have not responded to conservative treatments.
3.1 NSAIDs
NSAIDs are the first-line treatment for AS, exerting their anti-inflammatory effects by inhibiting cyclooxygenase (COX), also known as prostaglandin endoperoxide synthase (PGHS-1 and PGHS-2) (39). These enzymes play an essential role in the biosynthesis of prostaglandins, which are key mediators of inflammation, pain, and fever. Nevertheless, despite their extensive clinical application, NSAIDs exhibit notable limitations. A recent report in Germany revealed that only 19.1% of AS patients achieved complete remission with NSAIDs (40). 30% of patients responded to NSAIDs, but many of them experienced side effects (8, 41). Long-term use of NSAIDs can induce adverse reactions in the cardiovascular, gastrointestinal, and renal systems (8, 41). Additionally, approximately one-third of patients are completely unresponsive or intolerant to NSAIDs, necessitating alternative treatment approaches (42). Consequently, bDMARDs such as TNF-α inhibitors and IL-17 inhibitors, along with Janus kinase inhibitors (JAKi), have been adopted as second-line therapies following NSAIDs failure (43).
3.2 Conventional synthetic DMARDs
csDMARDs are a class of drugs that can alleviate and improve symptoms in AS, including MTX, SSZ, and hydroxychloroquine (44). However, it typically takes several months to achieve therapeutic effects (44). MTX is an anti-metabolite that competitively inhibits dihydrofolate reductase, thereby interfering with DNA synthesis and modulating the expression of various cytokines (45). Patients receiving MTX should be regularly monitored for side effects through detailed questioning and frequent blood tests (46). SSZ exerts its effects by inhibiting the synthesis of prostaglandins (47). However, a recently published guideline recommends SSZ only for patients with persistent peripheral arthritis who are intolerant to or contraindicated for TNF inhibitors (48). In addition, the administration of csDMARDs at higher doses is associated with an increased risk of various adverse events, including gastrointestinal perforations, thromboembolism, and serious infections (49).
3.3 Targeted biological agents
3.3.1 TNF inhibitors
TNF-α plays a crucial role in spondylitis and sacroiliitis, as well as in extra-articular manifestations such as uveitis (50, 51). TNF-α inhibitors (TNFi) are the most widely used and studied therapeutic agents in the treatment of AS (50, 51). Since their introduction in the early 21st century, TNFi agents have significantly improved the management of AS. Five TNF-α inhibitors are available for the treatment of AS (52, 53). Infliximab (IFX) was the first TNFi approved for treating AS. IFX is a chimeric monoclonal antibody (75% human, 25% mouse) that blocks TNF-α from activating the cellular receptor complex and is administered intravenously (IV) (54). Adalimumab (ADA), a fully humanized monoclonal antibody (IgG1), inhibits TNF-α from binding to its receptor sites and is administered subcutaneously (SC) (55). Etanercept (ETN) is a dimeric chimeric protein that combines the extracellular binding domain of human TNF receptor-2 with the Fc region of human IgG1 (56). This fusion blocks TNF from binding to cell surface receptors, inhibiting the inflammatory cascade. ETN is administered SC. Golimumab (GLM) is a fully human monoclonal antibody that specifically binds to both soluble and transmembrane TNFs, thereby inhibiting their interaction with TNF receptors (57). Administration of GLM can be performed via IV or SC routes. Lastly, Certolizumab pegol (CZP) is a PEGylated antigen-binding fragment of a recombinant human monoclonal antibody that selectively binds to and neutralizes both soluble and membrane-bound TNF-α, and is administered SC (58).
TNFi agents have demonstrated efficacy and tolerability in the treatment of AS; however, a significant number of cases have reported treatment failure. Studies have shown that approximately 35% of AS patients are primary non-responders to TNFi therapy, a condition referred to as primary clinical failure (2). Additionally, 30% of AS patients experience TNFi treatment failure within the first year of therapy (1). Notably, the rate of TNFi treatment failure is twice as high in female AS patients compared to males (59). This disparity may be attributed to differences in sex hormone balance and gene-specific expression (59). The primary cause of clinical non-response to infliximab or adalimumab is believed to be the development of antidrug antibodies (ADAs), which can affect drug bioavailability and reduce efficacy (60). The immunogenicity of biologics is unpredictable, but it can be mitigated by selecting humanized or fully human antibodies (61, 62). Beyond immunogenicity, variations in patient genetic background, disease activity, drug dosage and schedule, route of administration, concomitant medications (including immunosuppressants), and other factors all contribute to the differing sustained efficacy of each drug (60, 63).
Furthermore, TNFi treatment also brings certain side effects, limiting its applicability. AS patients with heart failure (HF) have been observed to experience worsening of their HF condition after using TNFi (64, 65). Therefore, the American College of Rheumatology (ACR) guidelines tend to recommend non-TNFi bDMARDs for treating AS patients with HF (64, 65). Infections are the most common serious adverse events associated with TNF inhibitors (66). An analysis of 71 clinical trials revealed that 40% of serious infections were attributed to the use of TNF inhibitors (67). The most common infections in IFX treatment were upper respiratory infections (24%) and skin symptoms (24%), such as itching, rash, or fungal infections (68). Other common adverse reactions included bronchitis (28%) and infusion-related symptoms (24%) (68). Moreover, the incidence of malignancies was found to be threefold higher in patients treated with IFX and ADA for rheumatoid spondylitis (69). The use of immunosuppressive drugs, including TNFi, can increase cancer risk through various pathways, with the risk varying depending on the type of cancer (69). Additionally, a significant increase in tuberculosis risk has been observed with TNFi use (70).
3.3.2 IL-17/23 inhibitors
Bone marrow cells within the spine can produce IL-23 in response to mechanical stress and various other factors (71). IL-23 promotes the differentiation of Th17 cells and stimulates multiple cell types to produce IL-17 (72). Elevated levels of IL-17 and IL-23 have been observed in the peripheral blood of patients with AS compared to healthy individuals (73). IL-17A and IL-17F can amplify inflammatory responses in vitro when combined with TNF inflammatory regulatory factors (74). Consequently, IL-17 inhibitors, such as secukinumab, have emerged as effective second-line treatments for AS, offering significant relief of spinal pain and improved sleep quality (75, 76). However, some patients still experience treatment failure or severe side effects (74).
In a clinical study of secukinumab for AS, the most common adverse event was nasopharyngitis (11.2%), followed by mild or moderate oral candidiasis (5.3%) and serious adverse events (4.3%) (76, 77). Additionally, 6.6% of patients discontinued treatment due to adverse events (76, 77). The incidence of inflammatory bowel disease (IBD) was comparable to that observed with TNF inhibitors (76, 77). Other adverse reactions included acute uveitis, cardiovascular diseases, neutropenia, leukopenia, and staphylococcus aureus subcutaneous abscesses (76, 77). Notably, two Phase II clinical trials of IL-17 blockers for Crohn’s disease were terminated early due to worsening disease activity or a high incidence of serious adverse events (66). Therefore, AS patients with IBD or uveitis symptoms are advised to avoid IL-17 inhibitors (78).
IL-23 inhibitors initially showed promise in early studies but failed to demonstrate efficacy in Phase III clinical trials in Germany (72). Furthermore, in the treatment of AS patients with ustekinumab, an IL-23 inhibitor, it was observed that individuals at high risk for cardiovascular disease exhibited a significantly elevated risk of acute coronary syndrome and stroke (79).
3.3.3 JAK inhibitors
JAK inhibitors (JAKi) interfere with the JAK-STAT signaling pathway by inhibiting one or more JAK enzymes (JAK1, JAK2, JAK3, TYK2), thereby regulating the expression of numerous inflammatory cytokines involved in autoimmune and inflammatory diseases (80). Since the approval of tofacitinib in 2012 for rheumatoid arthritis (RA), several other JAKi, including baricitinib, upadacitinib, filgotinib, and peficitinib, have been introduced into clinical practice (69). These agents have demonstrated robust efficacy in controlling disease activity, often outperforming traditional TNF inhibitors (81). However, the broad impact of JAKi on the JAK-STAT pathway, which is involved in multiple signaling cascades, raises concerns about potential off-target effects and associated safety risks.
Recent real-world clinical data and randomized trials have highlighted significant safety concerns associated with the use of Janus kinase inhibitors (JAKi). Potential serious adverse events (AEs) linked to JAKi include major adverse cardiovascular events (MACE), venous thromboembolic events (VTEs), herpes zoster, serious infections (including tuberculosis), and malignancies (82). For instance, the ORAL Surveillance trial revealed that tofacitinib was associated with a higher incidence of MACE and malignancies compared to TNFi in patients with RA (83). Additionally, tofacitinib exhibited a twofold higher risk of herpes zoster relative to bDMARDs, and this elevated risk was also observed with other JAKi (69).
These findings have prompted regulatory agencies, including the FDA and the European Medicines Agency (EMA), to issue warnings and impose restrictions on the use of JAKi, particularly in patients with cardiovascular risk factors or a history of malignancies (43). The FDA has extended boxed warnings for increased risks of MACE, VTE, infection, malignancy, and mortality to the entire class of JAKi (43). This regulatory stance underscores the critical importance of careful patient selection and individualized risk-benefit assessment when considering JAKi therapy.
Despite the availability of various treatment options, challenges persist in the management of AS. While biologics and JAK inhibitors provide substantial therapeutic benefits, they are associated with significant safety concerns, especially in patients with comorbidities such as cardiovascular disease or a history of infections. Additionally, the high costs of biologics may restrict their accessibility for certain patient populations.
3.4 Physical therapy
Surgical intervention may be considered for patients with AS in cases of severe spinal deformity, spinal fractures, or other significant complications when non-surgical treatments have failed. In AS, multi-level ankylosis compromises spinal stability, leading to fractures that are 3–4 times more prevalent than in the general population and predominantly affect the cervical spine or cervical-thoracic junction (84). Given the complexity, surgery is preferred over conservative treatment for better outcomes. However, it carries high risks of complications both peri-operatively and post-operatively (85). Other conventional physical therapies include cryotherapy, ultrasound therapy, electrotherapy, kinesiotherapy, and massage (86). Systematic physical activity is essential as it effectively mitigates the progression of AS. Nevertheless, physical therapy may have certain limitations, including the requirement for consistent effort and time commitment, varying effectiveness depending on individual conditions, and potentially high costs. There is an increasing emphasis on adopting a personalized and multidimensional approach to AS treatment, which integrates diverse therapeutic modalities. In light of these limitations, there is increasing interest in investigating alternative therapeutic approaches, such as cell therapy.
4 Mechanisms and therapeutic effects of MSCs in the treatment of AS
4.1 Overview of MSCs
MSCs are multipotent adult stem cells derived from the mesoderm during early embryonic development, characterized by their self-renewal capacity and potential for multilineage differentiation (Figure 2). Initially identified in bone marrow by Friedenstein et al., MSCs have since been isolated from various tissues, including umbilical cord, dental pulp, and adipose tissue (87, 88). The International Society for Cellular Therapy (ISCT) has established standardized criteria for the identification of MSCs, which include: (1) adherence to plastic in vitro; (2) expression of specific surface markers, such as CD105, CD90, and CD73, while lacking expression of CD45, CD34, CD14 or CD11a, CD79a or CD19, and HLA II molecules; and (3) the ability to differentiate into osteoblasts, chondrocytes, and adipocytes in vitro (88).

Figure 2. Characterization of MSCs. MSCs are derived from diverse tissue sources, including bone marrow, dental pulp, umbilical cord, and iPSCs. These cells exhibit specific surface marker expression profiles, such as CD105, CD90, and CD73, while lacking the expression of hematopoietic markers CD45, CD34, CD14 or CD11b, B cell markers CD79a or CD19, and HLA-DR. Notably, MSCs possess multipotent differentiation potential, enabling them to differentiate into various lineages, including adipocytes, osteoblasts, and chondrocytes.
Beyond their differentiation potential, MSCs exhibit robust immunomodulatory functions, capable of modulating both innate and adaptive immune responses. They reduce the pro-inflammatory phenotype by directly or indirectly interacting with dendritic cells, macrophages, NK cells, B cells, and T cells (89). Notably, MSCs can adapt their polarization phenotypes in response to the local microenvironment, shifting between anti-inflammatory and pro-inflammatory states according to disease conditions. This adaptability makes MSCs a promising therapeutic candidate for autoimmune diseases, including AS, where the inflammatory milieu can be dynamically targeted.
4.2 Immunomodulatory effects and mechanisms of MSCs in the treatment of AS
MSCs are multipotent progenitor cells with the capacity to modulate immune responses and promote tissue repair through the secretion of soluble factors and direct cell-to-cell interactions. These cells exhibit potent immunosuppressive properties by secreting a variety of molecules, including indoleamine 2,3-dioxygenase (IDO), prostaglandin E2 (PGE2), hepatocyte growth factor (HGF), transforming growth factor-beta1 (TGF-β1), insulin-like growth factor-1 (IGF-1), nitric oxide (NO), heme oxygenase-1 (HO-1), cyclooxygenase-2 (COX-2), and IL-10 (90–92).
HLA-B27 is a well-established immunogenetic marker for AS, with the arthritogenic peptide hypothesis suggesting that abnormal antigen presentation to CD8+ T cells by HLA class I molecules triggers a specific immune response. MSCs have the ability to regulate T cell proliferation, differentiation, and activity, and can reduce the production of pro-inflammatory cytokines. MSCs can upregulate IDO expression in response to inflammatory cytokines, notably IFN-γ. IDO catalyzes the conversion of tryptophan to kynurenine, thereby inhibiting T cell proliferation through disruption of cellular protein synthesis (13, 93). Additionally, MSCs produce inducible nitric oxide synthase (iNOS), which induces macrophages to release NO, thereby suppressing T cell function (94). MSCs also inhibit the differentiation of Th17 cells, a subset of T cells implicated in the pathogenesis of AS. Huang et al. described the inhibitory effect of human umbilical cord-derived MSCs on T cells in patients with SpA (95). In co-culture with peripheral blood mononuclear cells (PBMCs), umbilical cord-derived MSCs significantly reduced the production of IL-17, showing potential for the treatment of SpA. Regulatory T cells (Tregs) are a subset of T cells with potent immunosuppressive functions, acting by suppressing effector T cells and mitigating inflammation-induced tissue damage. Both peripheral blood and synovial fluid examinations in AS patients have shown a reduced number of Tregs, which is positively correlated with lower FOXP3 expression levels (96, 97). Multiple studies have shown that MSCs induce Treg proliferation, a key mechanism by which they limit inflammation. For instance, bone marrow-derived MSCs promote the differentiation of CD4+ T cells into Tregs in co-culture with PBMCs, expressing high levels of CD25 and FOXP3 (98). Moreover, bone marrow-derived MSCs induce Treg proliferation through the secretion of TGF-β1 and interaction with macrophages (99). IDO is also implicated in MSC-induced Treg generation (100). MSCs can directly interact with T cells, exhibiting the most potent inhibitory effects on activated T cells through direct cell-to-cell contact (101). This interaction is further enhanced by the upregulation of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) in MSCs, which strengthens their engagement with T cells (101).
Monocytes and macrophages in AS can polarize into pro-inflammatory (M1) or anti-inflammatory (M2) phenotypes, a process closely related to active inflammation, tissue damage, and regenerative reconstruction. In late-stage AS patients, monocytes were significantly polarized into M2 macrophages, with the M2/M1 ratio positively correlated with structural lesion damage (mSASSS) and negatively correlated with inflammatory markers (ESR, CRP) and the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) (102). MSCs influence the polarization of macrophages, which may be caused by cell-to-cell contact mechanisms and soluble factors (such as IDO, PGE2, IL-10, and COX-2) (13). For example, MSCs inhibit the proliferation of M1 macrophages and activate the production of M2 macrophages through the activation of TNF-mediated COX-2 and TNF-stimulated gene 6 (TSG-6) (13). Our previous work also demonstrated that in a mouse spondylitis model, the injection of umbilical cord-derived MSCs reduced the levels of inflammatory cytokines (TNF-α and CCL-2) in the spleen and serum of mice (103).
NK cells are a critical component of the innate immune system. HLA-B27 is specifically recognized by the inhibitory receptor KIR3DL1 on NK cells, with a correlation between KIR receptor expression and AS activity (104). This suggests that NK cells play a significant role in AS pathogenesis. MSCs can regulate NK cell phenotype through cell-to-cell interactions or secretion of factors such as TGF-β1 and PGE2, inhibiting their proliferation, cytokine secretion, and cytotoxicity (105). MSCs also suppress IL-2-stimulated NK cell proliferation (106). Interestingly, MSCs secrete HLA-G5 and IFNγ, which inhibit NK cell cytotoxicity and innate immune responses while promoting Treg proliferation (90).
Dendritic cells (DCs) are key antigen-presenting cells that synthesize IL-23, a major pro-inflammatory cytokine in AS (26, 107). IL-23 induces the differentiation of lymph node T cells into pro-inflammatory Th17 cells and stimulates IL-23R+ lymphocytes in the sacroiliac joints to secrete IL-22, which in turn activates osteoblasts and leads to local bone formation (108, 109). MSCs inhibit the upregulation of antigen-presenting and co-stimulatory signals (CD1a, CD40, CD80, CD86, and HLA-DR) during DC differentiation and prevent the increase in CD40, CD86, and CD83 expression during DC maturation (110). Moreover, MSCs and their supernatants interfere with DC endocytosis, reducing their ability to secrete IL-12 and activate allogeneic T cells (110). Jiang et al. also proposed a similar view that MSCs can reduce the expression of CD83 on mature DCs, indicating that DCs have lost their mature characteristics (111). MSCs can also inhibit DC maturation stimulated by CSF and IL-4 through the secretion of PGE2 (111). Additionally, MSCs inhibit DC differentiation through the production of IL-10 and cell-to-cell contact (112).
4.3 Heterotopic ossification (HO): a potential mechanism of MSCs in the treatment of AS
HO represents a pathological condition defined by the ectopic formation of new bone tissue in soft tissues beyond the normal skeletal system, typically evidenced by the presence of osteoblasts and chondrocytes. HO is one of most significant pathological features of AS (113). In AS, HO is predominantly manifested in soft tissues such as spinal ligaments and tendons, where the appearance of chondrocytes leads to the development of new bone (113). This process commonly occurs in conjunction with the progression of inflammation and bone erosion observed in AS patients. It can lead to joint stiffness, spinal ankylosis, and spinal deformity, and may even result in the “folded person” phenomenon. Although inflammation has long been considered a trigger for HO in AS, existing AS treatments such as NSAIDs and TNFi can rapidly alleviate inflammation and pain, but they do not significantly prevent the progression of bone lesions in AS patients.
4.3.1 Stages of HO in AS
The formation of bone tissue primarily happens through two distinct processes: intramembranous ossification and endochondral ossification (114). Intramembranous ossification is directly mediated by osteoblasts, which facilitate the local deposition of calcium phosphate crystals and subsequently contribute to bone formation (115). Endochondral ossification, which is initially mediated by chondrocytes and subsequently replaced by osteoblasts for the formation of bone tissue, plays a pivotal role in the progression of HO in AS (116).
HO in AS can be divided into four stages: inflammation, chondrogenesis, osteogenic activity, and pathological bone formation (117, 118). The initial inflammatory stage, mediated by both innate and adaptive immune cells, is a crucial trigger for HO in AS. Neutrophils from AS patients exhibited enhanced formation of neutrophil extracellular traps that carry bioactive IL-17A and IL-1β, which promote the differentiation of MSCs toward bone-forming cells (119). This inflammatory microenvironment sets the stage for subsequent pathological alterations. During the chondrogenesis stage, chondrocyte differentiation and cartilage formation occur, particularly in the ligaments of patients with early-stage AS (118). This cartilage formation serves as an intermediate phase before the onset of calcification. As the disease progresses, calcified cartilage is resorbed by osteoclasts, which are numerous in areas of ligament inflammation and on the surfaces of calcified cartilage. This osteoclast-mediated resorption of calcified cartilage initiates ossification, representing a pathologic process similar to acquired HO (118). In the osteogenic activity stage, osteoblasts replace the resorbed cartilage with bone tissue, leading to the formation of mature bone (120). As the disease progresses, approximately 60% to 70% of AS patients exhibit radiographic evidence of sacroiliac joint ankylosis, bridging ligament bone spurs in the axial skeleton, and enthesophytes or peripheral joint osteophytes (121). HO in AS is a complex and multifaceted pathological process, and understanding its stages and mechanisms is essential for developing targeted therapeutic strategies to manage HO in AS patients.
4.3.2 The molecular mechanisms of endogenous MSCs in HO in AS
During bone formation, chondrocytes differentiate from MSCs and promote the recruitment and proliferation of MSCs. These MSCs subsequently differentiate into chondrocytes and osteoblasts, eventually forming a mature bone tissue structure (117). MSCs derived from AS patients exhibit enhanced osteogenic differentiation capacity, and MSCs migrating into cartilaginous tissues can promote pathological ossification by differentiating into osteoblasts. HLA-B27 promotes pathological ossification caused by AS-MSCs through the sXBP1/RARB/TNAP pathway (122). In addition to inducing ER stress, HLA-B27 accelerates bone formation by interacting with the activin receptor-like kinase-2 (ALK2) subunit of the BMP signaling pathway, thereby enhancing the sensitivity of the BMP-TGF signaling pathway to TGF-β and upregulating the expression of tissue nonspecific alkaline phosphatase (TNAP) (123). Mutations in TNAP haplotypes, including rs3767155 (G), rs3738099 (G), and rs1780329 (T), are primarily associated with ankylosis in AS (124). The ossification of AS-MSCs requires the synergistic action of HLA-B27 and TNAP, which may explain why not all HLA-B27-positive individuals develop ankylosis.
Furthermore, the reduction of DKK-1 in AS-MSCs mediated by inflammatory cytokines is a key factor in pathological bone formation. Compared with controls, MSCs from AS patients exhibit insufficient DKK-1 expression, mainly due to IL-17-mediated inhibition of DKK-1 and stimulation of osteoblast function (125). Additionally, the imbalance of BMP-2 and Noggin secretion may lead to abnormal osteogenic differentiation of AS-MSCs (126). Osteoprogenitor cells secrete chemokine ligand CXCL12 and stem cell factors, stimulating the proliferation of myeloid MSCs. Osteocytes secrete sclerostin and granulocyte colony-stimulating factor, regulating the differentiation of lymphocytes and myeloid cells (127). In summary, these studies reveal the intricate interplay between the immune and skeletal systems, with numerous common cytokines implicated in both.
4.3.3 Therapeutic potential of transplanted MSCs for HO in AS
In the preceding section, numerous studies have demonstrated the immunomodulatory role of MSCs in the inflammatory process of AS. MSCs suppress inflammatory signals that are essential for osteogenesis, such as IL-17, thereby potentially inhibiting HO (128). Moreover, our previous preclinical animal experiments have shown the therapeutic effects of MSC transplantation on AS, with MSC treatment inhibiting HO, maintaining clear facet joint spaces, and slowing down structural lesions in the intervertebral disc, nucleus pulposus, annulus fibrosus, and cartilage (103). However, further in-depth exploration is still needed regarding the effects and mechanisms of MSC transplantation on AS, especially in terms of HO.
4.4 Clinical application of MSCs in the treatment of AS
In recent years, the immunomodulatory and regenerative properties of MSCs have garnered significant attention, prompting the initiation of several clinical trials to explore their therapeutic potential for AS (Figure 3). The earliest reported use of stem cells for AS was serendipitous: a patient with acute myeloid leukemia and AS experienced marked relief of AS symptoms and improved clinical indicators following peripheral blood stem cell transplantation (129). This patient remained symptom-free from AS for approximately 3 years post-transplantation, without the need for anti-TNF or NSAID therapy (129).

Figure 3. Immunomodulatory mechanisms of MSCs and current clinical trials in AS. MSCs inhibit the proliferation of T cells, promote the differentiation of regulatory T cells (Tregs), suppress dendritic cell (DC) maturation, and induce macrophages to adopt an immunosuppressive phenotype. Additionally, several clinical trials are currently underway to validate the safety and efficacy of MSCs in AS, including the ongoing trial in our research group.
In 2013, the Wang group conducted a comprehensive study to evaluate the feasibility, safety, and efficacy of bone marrow-derived MSC therapy in 31 AS patients who were intolerant to NSAIDs (130). AS patients participating in this study received four intravenous infusions of MSCs on days 0, 7, 14, and 21, with each infusion containing 1×10^6 cells/kg. The results showed that the proportion of patients achieving ASAS20 response was 77.4% at week 4, 54.8% at week 12, and 32.3% at week 16, with a mean response duration of 7.1 weeks following the fourth infusion. The mean ASDAS-CRP score decreased from 3.6 ± 0.6 at baseline to 2.4 ± 0.5 at week 4, but increased to 3.2 ± 0.8 at week 20. MRI assessments revealed a mean total inflammatory extent (TIE) of 533,482.5 at baseline, which decreased to 480,692.3 at week 4 (p > 0.05) and further to 400,547.2 at week 20 (p < 0.05). No adverse reactions were reported. In 2017, the Li group explored the therapeutic effect of umbilical cord-derived MSCs on AS (131). In this study, umbilical cord-derived MSCs were administered via intravenous infusion to five patients with AS. The cell doses ranged from 1.2 to 3.5×10^6 cells/kg, and each patient received between 1 to 3 infusions. The study revealed that following treatment, both the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) and the Bath Ankylosing Spondylitis Functional Index (BASFI) demonstrated significant reductions. Specifically, BASDAI decreased from a baseline of 4.686 ± 0.999 to 1.880 ± 1.499 at the 3-month follow-up (P=0.014), while BASFI declined from 42.000 ± 21.213 at baseline to 10.900 ± 13.585 at the 3-month follow-up (P=0.062). However, the Bath Ankylosing Spondylitis Metrological Index (BASMI) increased nonsignificantly (P=0.676). The erythrocyte sedimentation rate decreased in 3 patients, and the C-reactive protein level was significantly reduced in 1 patient. Overall, symptoms of AS improved in all patients. No serious adverse reactions were noted; however, mild transient fever occurred in three patients within 2–6 hours post intravenous administration. More recently, we conducted a clinical study (NCT05962762) further confirmed the safety and efficacy of umbilical cord-derived MSCs for AS treatment. Other ongoing trials (NCT01420432, NCT01709656, NCT02809781) continue to evaluate the therapeutic potential of MSC infusion for AS (Table 1).

Table 1. Clinical trials for AS treatment with MSCs.
MSC therapy has demonstrated significant potential in improving clinical symptoms and alleviating pain in patients with AS, with a favorable safety profile. This emerging therapeutic strategy offers a promising alternative to current treatments, such as biologics and JAK inhibitors, which are often associated with notable safety concerns and high costs. The immunomodulatory and regenerative properties of MSCs, which include the secretion of soluble factors and direct interactions with immune cells, may address the underlying pathogenesis of AS more effectively, with fewer adverse effects. However, several challenges remain to be addressed. Future research should focus on optimizing MSC sourcing, dosing, and administration routes, as well as conducting well-designed clinical trials to further validate their efficacy and safety in AS. Continued research and larger-scale clinical trials are anticipated to provide valuable insights and drive the development of this innovative treatment strategy, ultimately offering new hope for patients with AS.
5 CAR-based cell therapies in autoimmune diseases and their potential in AS treatment
A CAR is a chimeric antigen receptor molecule constructed through gene engineering technology, designed to confer specificity to immune effector cells, such as T lymphocytes, for a particular target antigen epitope (132). This modification enhances the ability of T cells to recognize and respond to antigen signals, thereby facilitating their activation and cytotoxic activity (132). Initially developed for cancer treatment, CAR T-cell therapy has demonstrated remarkable efficacy in managing hematologic malignancies and solid tumors. Building on these successes, CAR T-cell therapy is now being explored for its potential applications in autoimmune diseases (Figure 4). The rationale behind this expansion lies in the ability of CAR T cells to selectively deplete pathogenic immune cells, such as autoreactive B cells, T cells, and antigen-presenting cells (APCs), which drive the pathogenesis of autoimmune disorders. This approach aims to reset the immune system by eliminating the cells responsible for aberrant immune responses, thereby offering a novel therapeutic strategy for diseases characterized by high levels of autoantibodies or overactive lymphocytes.
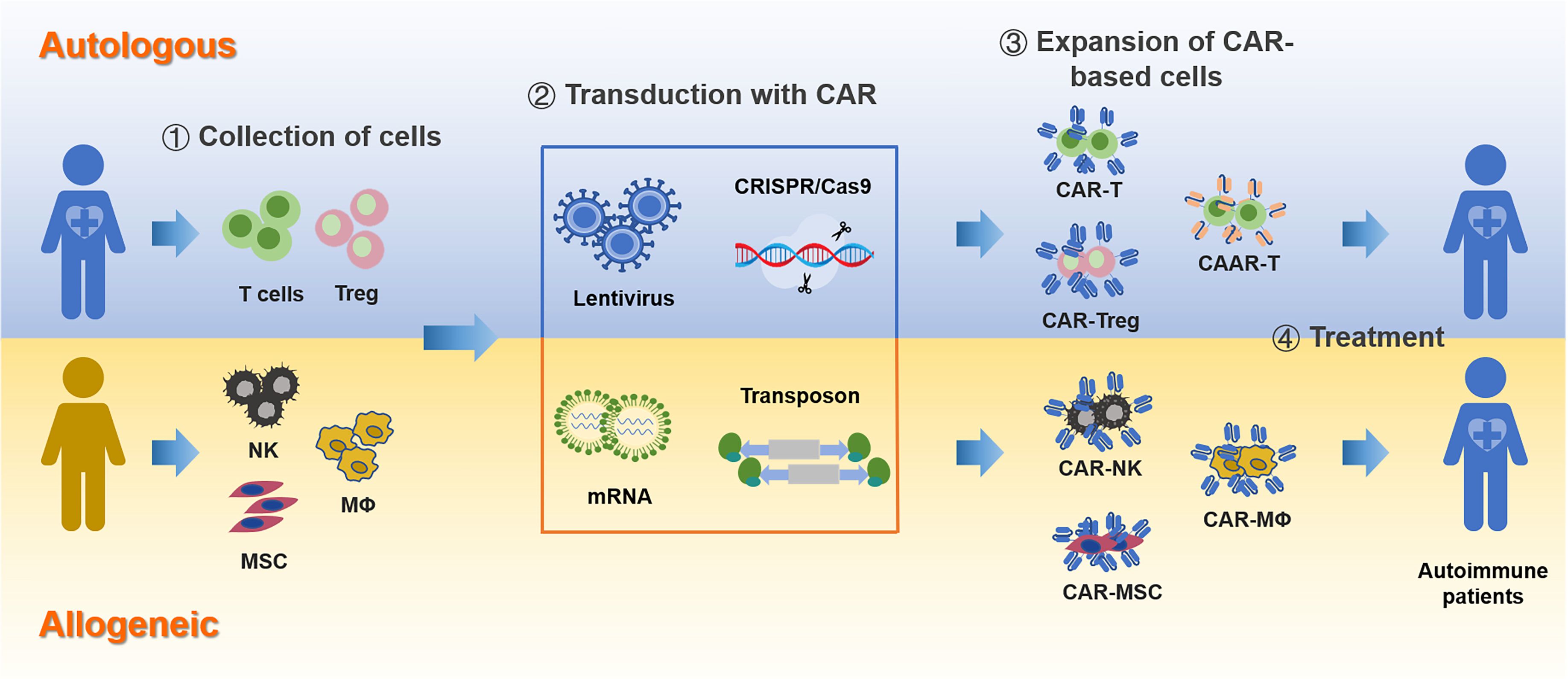
Figure 4. CAR-based immunotherapy for autoimmune diseases. The process of developing CAR-based therapies involves several key steps, starting from the selection of the cell source to the final deployment of engineered CAR cells.
5.1 Emerging CAR targets in autoimmune diseases
CD19 and B cell maturation antigen (BCMA) have emerged as key B-cell surface targets, demonstrating significant therapeutic potential in conditions such as systemic lupus erythematosus (SLE), idiopathic inflammatory myopathies, and systemic sclerosis (133). CD19 is expressed throughout multiple stages of B cell development, from pro-B cells to plasmablasts, but not in plasma cells (134). This widespread expression, coupled with CD19’s multifunctional role in B cell activation, maturation, and signaling, makes it an attractive target for B cell-directed therapies in autoimmune diseases such as SLE. Molecules like BCMA, CD38, and CD138 are predominantly expressed on plasma cells, with BCMA and CD38 also present on plasmablasts (134). This differential expression pattern allows therapeutic strategies to selectively target specific subsets or broader spectra of the B cell lineage, depending on the disease context and desired therapeutic effect.
Beyond B-cell targets, CAR T-cell therapies are being developed to directly target specific autoantibodies involved in autoimmune diseases. For instance, in pemphigus vulgaris, a skin disease characterized by autoantibodies against desmoglein 3 (Dsg3), anti-Dsg3 CAR T-cell therapy is currently undergoing clinical trials (135). Additionally, CAR T-cell therapies targeting cytokines are also in development, with a focus on modulating the inflammatory milieu in autoimmune diseases. Key targets include IL-23, which plays a critical role in mediating inflammatory responses (136). By targeting these cytokines, CAR T cells may potentially disrupt the pro-inflammatory signaling pathways, leading to reduced disease activity and improved clinical outcomes.
An innovative therapeutic strategy focuses on the precise elimination of pathogenic T-cell subsets that proliferate abnormally in specific autoimmune diseases. For example, targeting TRBV9+ T cells in AS aims to selectively eliminate pathogenic T cells while preserving normal immune cell populations (137). This strategy enhances treatment precision and reduces adverse effects on healthy cells, potentially improving the safety and efficacy of CAR T-cell therapy in autoimmune diseases. These advancements reflect the ongoing evolution of CAR T-cell therapy, moving beyond traditional cancer applications to address the complex immunopathology of autoimmune diseases. Future research is expected to identify additional targets and refine current strategies, thereby significantly broadening the therapeutic potential of CAR T-cell therapy in this field.
5.2 Clinical application of CAR-based cell therapies in the treatment of autoimmune diseases
Preclinical and clinical studies have demonstrated the promising therapeutic potential of CAR T-cell therapy in various autoimmune diseases, including multiple sclerosis, type 1 diabetes, inflammatory bowel disease, SLE, and pemphigus vulgaris (138). A notable case reported by the Mougiakakos group involved a woman with severe refractory SLE (SELENA score: 16) and Class III/IV lupus nephritis who received anti-CD19 CAR T-cell therapy (139). Following fludarabine lymphodepletion and CAR T-cell infusion, significant clinical improvement was observed within five weeks, characterized by normalization of dsDNA autoantibody titers and complement levels (C3 and C4). The SLE disease activity index score decreased from 16 at baseline to 0 at follow-up, and no significant adverse reactions were reported. The research team subsequently administered CAR T-cell therapy to four additional patients with refractory SLE, all of whom achieved a low lupus disease activity state (LLDAS) and successfully discontinued all SLE-specific medications (https://doi.org/10.1136/annrheumdis-2022-eular.1120). In another clinical study conducted by the Zhang group, patients with SLE and stage IV diffuse large B-cell lymphoma (DLBCL) exhibited continuous relief from disease activity following the infusion of CAR T cells targeting CD19 and BCMA (139). Follow-up examinations confirmed effective B-cell depletion, with stable disease remission lasting up to 23 months. These findings are encouraging and suggest that CAR T-cell therapy may offer a novel treatment option for patients with autoimmune diseases. However, the potential risks associated with CAR T-cell therapy, such as cytokine release syndrome (CRS) and neurotoxicity, necessitate further investigation (140). Additionally, the high cost of CAR T-cell therapy limits its widespread application.
To address these challenges, advancements in preparation techniques and diversification of cell types are being explored. Recent studies have investigated the expression of CARs in alternative cell types, such as NK cells, macrophages, regulatory T cells (Tregs), and MSCs (138). NK cells, known for their MHC-independent cytotoxicity and high safety profile, present a promising avenue for developing allogeneic therapies aimed at targeting pathogenic immune cells (141). Macrophages can phagocytose specific antigens and promote inflammatory responses, while also cross-presenting antigens to activate T cells. In contrast to the direct cytotoxic mechanisms, the activation of Tregs or MSCs through CAR-mediated antigen stimulation leverages their immunomodulatory properties to regulate immune responses. Tregs can secrete immunosuppressive molecules such as TGF-β, IL-10, and IL-35, making them suitable candidates for treating autoimmune diseases and preventing organ transplant rejection by inhibiting excessive T-cell activation (15). The Fransson group utilized CAR technology to target myelin oligodendrocyte glycoprotein (MOG) and co-express FoxP3, resulting in the generation of antigen-specific CAR Tregs (142). These MOG-CAR Tregs demonstrated the ability to inhibit effector T-cell proliferation in vitro and alleviate symptoms in experimental autoimmune encephalomyelitis (EAE) mouse models by reducing pro-inflammatory cytokine levels. Moreover, the MacDonald group reported that allogeneic HLA-A2 antigen-specific CAR Tregs (A2-CAR Tregs) maintained high expression levels of FoxP3, CD25, and CTLA-4 in vitro, effectively preventing graft-versus-host disease (GVHD) in immunodeficient mouse models (143). Recently, the Sirpilla group demonstrated the therapeutic potential of CAR-MSCs in treating GVHD (144). Specifically, E-cadherin-targeted CAR-MSCs localized to colonic cells and improved symptoms and survival rates through the upregulation of immunosuppressive genes and cytokines.
Table 2 summarizes the main clinical progress of CAR-based cell therapies for the treatment of autoimmune diseases to date. CAR-based cell therapy has emerged as a revolutionary immunotherapy, achieving significant breakthroughs in the treatment of autoimmune diseases in recent years. These studies highlight the potential of CAR-based cell therapy to induce long-term remission and reduce disease activity in patients with severe autoimmune diseases. AS is an autoimmune disease characterized by immune system dysregulation. CAR-based cell therapy may offer new treatment opportunities for AS patients by targeting abnormal immune cells. However, the application of CAR-based cell therapy for AS is still in the research and exploration stage and has not yet reached a mature stage for clinical application.

Table 2. The summary of ongoing and planned clinical trials of CAR-based treatments for autoimmune diseases.
5.3 Potential of CAR-based therapy for AS treatment
In contrast to SLE, which is primarily driven by pathogenic B cells, AS is characterized by dysregulated T cell activation (145). In recent years, significant progress has been made in CD7- and CD5-targeted CAR-T cell therapy for T-cell malignancies (146, 147). However, the efficacy of these approaches in AS remains to be demonstrated. Considering the widespread distribution and critical role of T cell antigens in normal tissues, the design of CAR-T cell therapy for AS should emphasize precision to minimize potential off-target effects and preserve the integrity of the immune system. Pathogenic T cells, such as TRBV9+ T cells as reported, represent promising candidates for therapeutic targeting (20). Targeting these specific T cells may offer a more refined strategy for AS treatment, thereby minimizing the risk of off-target effects.
Utilizing CAR-T cells to target and eliminate pathogenic cells represents one potential therapeutic strategy for AS. Another approach involves harnessing immune regulatory cells to precisely modulate the immune microenvironment in AS. Given the limited accessibility of the disease site in AS, employing inflammation-suppressing cells such as Tregs and MSCs, with enhanced targeting capabilities, may also hold considerable promise for effectively treating AS. CAR-based therapies warrant further investigation in future studies. Leveraging CAR-based therapies to selectively eliminate the root causes of the disease while simultaneously modulating the excessive inflammatory microenvironment, without inducing significant systemic immune suppression, may offer a new generation of safe and effective therapies for curing AS.
6 Perspective on novel cell therapies for AS treatment
Despite the availability of diverse treatment modalities for AS, many therapeutic regimens are often accompanied by challenges such as adverse effects and the development of long-term drug resistance. These challenges require us to continuously explore and develop novel treatment approaches. The emergence and development of novel cell therapies, particularly MSC therapy and CAR-based therapy, have brought promising hope to the treatment of AS (Figure 5).

Figure 5. Schematic overview of current and future directions in cell therapy for AS. The left side illustrates the current schematic, mechanisms, and future directions of MSC therapy for AS, while the right side depicts the possible schematic, mechanisms, and future directions of CAR-based therapy for AS. Solid lines indicate ongoing research, and dashed lines suggest potential future directions.
6.1 Current challenges and next steps of MSCs for AS treatment
Extensive preclinical and clinical studies have demonstrated that MSCs exhibit high safety and efficacy in treating AS. These cells play a pivotal role in modulating overactivated immune cells, reducing chronic inflammation and promoting tissue repair through their anti-inflammatory and regenerative properties. However, before wide application of MSC treatment to AS, several challenges must be addressed.
6.1.1 Quality and cost control of MSCs
The origin of MSCs is a significant factor. For acquisition, umbilical cord-derived MSCs (UC-MSCs) provide a more convenient and non-invasive alternative to bone marrow-derived MSCs (BM-MSCs) and adipose tissue-derived MSCs (AD-MSCs) (148). Moreover, the heterogeneity of different MSC populations must be carefully considered for clinical applications. A systematic review and network meta-analysis revealed that, autologous BM-MSCs showed the most improvement in Range of Motion (ROM) and pain relief in knee osteoarthritis patients, UC-MSC were most effective for positive Whole-Organ Magnetic Resonance Imaging Score (WORMS), and AD-MSCs were most effective for Western Ontario McMaster Universities Osteoarthritis Index (WOMAC)-positive patients (149). However, which types of MSCs have the best therapeutic outcomes for AS remain uncertain.
Recently, the U.S. Food and Drug Administration (FDA) in the United States and the National Medical Products Administration (NMPA) in China approved two MSC drugs for treating GVHD (150). However, there is a significant price difference between the Ryoncil® (allogeneic BM-MSCs, Mesoblast) and Amimestrocel Injection (hUC-MSCs, Platinum Life). This discrepancy can primarily be attributed to variations in cell sources, research costs, manufacturing procedures, and market strategies. To address the cost and ensure consistent quality and efficacy, standardization of practices in culture, cryopreservation, and transportation of MSCs is essential in both preclinical and clinical settings (151). Moreover, the in vitro expansion of MSCs to achieve high cell yields is critical for advancing MSC therapy (151). This process involves cost challenges that must be addressed for feasible and scalable MSC treatments. Striking a balance between optimizing MSC proliferation and ensuring safety, efficacy and cost-effectiveness is essential for broader clinical application.
6.1.2 Optimization of the MSC administration procedure
The ideal treatment dosage, optimization of the administration route and determination of the optimal timing for MSC intervention in AS patients should be standardized and incorporated into a standardized operating procedure (SOP) to facilitate comparisons of MSC therapy efficacy.
In a rat model of osteoarthritis, MSC transplantation via both intra-articular injection and intravenous injection was explored, with results indicating that cells administered through intra-articular injection persisted in the knee joint for up to one week, highlighting the potential for sustained local therapeutic effects (152). Current clinical trials of MSC administration for AS predominantly utilize intravenous injection, which may be limited by insufficient cell homing and retention. Exploring alternative administration routes or evaluating the potential of repeated injections represents a critical direction for advancing future research. In addition, larger-scale and higher-quality studies are needed to comprehensively evaluate the feasibility and potential value of MSC therapy for AS.
6.1.3 Tracing MSC cell fate and effects in vivo
Although the safety of administering MSCs has been demonstrated in numerous clinical trials, the limited understanding of their dynamic biodistribution and fate within the body represents a significant challenge to the advancement of MSC therapies.
The majority of studies indicate that MSCs exhibit a relatively brief residence time in the body following intravenous administration, with most cells being sequestered in the lungs and remaining viable for 24–72 hours (153, 154). This rapid clearance is attributed to multiple factors, including apoptosis, autophagy, ferroptosis in MSCs, as well as phagocytosis by various immune cells (154–158). The fate of infused MSCs, including their interaction with the host immune system, is crucial for their therapeutic impact. MSCs are efficiently phagocytosed by innate immune cells, such as monocytes and macrophages, resulting in phenotypic and functional modifications in these cells, including the secretion of IDO and IL-10 (154, 155, 158). Innate immune cells may either remain at the initial site or migrate to other organs, thereby further regulating the adaptive immune response (154, 159). This intricate interplay of combined effects profoundly shapes the therapeutic potential of MSCs.
The development of advanced imaging and tracking technologies is crucial for elucidating the fate of MSC. In preclinical studies, precise and effective detection methods, such as magnetic resonance imaging, fluorescence labeling, optical imaging, photoacoustic imaging, ultrasound imaging and quantitative gene detection, have been widely utilized to non-invasively track transplanted stem cells (160, 161). Despite these advancements, the clinical translation of these technologies faces significant challenges. Currently, there is a lack of robust and reliable methods for tracking MSCs and their production in clinical trials. To address this challenge, the integration of multiple imaging modalities may enhance precision and provide complementary information. The development of novel imaging techniques and the identification of specific markers for MSCs are equally critical. Future progress in integrated imaging platforms, coupled with in-depth mechanistic studies, will accelerate the clinical translation of MSC-based therapies in AS.
6.2 The potential of precision CAR-based cell therapies for AS
CAR-based cell therapies represent a highly specific and targeted treatment modality that aligns well with the complex pathophysiology of AS. However, several critical questions still require clarification.
6.2.1 Ideal targets for precision
The selection of CAR targets is of paramount importance in the development of CAR-based cell immunotherapy for AS. An ideal target antigen must exhibit high specificity and safety to minimize the risk of off-target effects leading to severe tissue damage. CAR-mediated target recognition is not limited to cell surface proteins but can also identify soluble protein ligands, post-translational modifications, and glycolipids. However, the complexity of autoimmune diseases requires careful consideration of antigen expression patterns and potential off-target effects. Unlike cancer, where CAR-T cells aim to eliminate malignant cells, CAR-based therapy for autoimmune diseases may have distinct or more complicated therapeutic mechanisms, i.e. immunomodulation. Therefore, how to selectively target pathogenic cells while sparing healthy tissues should be given more consideration. This necessitates a deep understanding of disease-specific antigen profiles and the development of CAR constructs with enhanced specificity. For instance, instead of targeting the overall T cells implicated in AS pathogenesis, targeting TRBV9+ T cells, a subset of T cells closed related to AS pathogenesis, provides a more precise strategy to meet the above ends.
Targeting pro-inflammatory cytokines is another strategy awaiting preclinical evaluation. A series of cytokines such as TNF-α, IL-6, and IL-17A are upregulated in AS and have a pathogenic role (162). Theoretically, using cytokine receptors as the extracellular domain of CARs could convert pro-inflammatory signals into CAR co-stimulatory signals. For instance, in tumor treatment, genetically modified CARs targeting TGF-β have been used to transmit TGF-β signals to the CD28 co-stimulatory domain, enhancing T-cell therapy (163).
6.2.2 Optimal cellular candidates
The choice of CAR cells is crucial for the success of AS treatment. Tregs, known for their immunomodulatory functions, can be activated in inflammatory environments and release inhibitory cytokines such as IL-10 and TGF-β. CAR-Tregs have the potential to achieve highly effective and durable immune modulation through direct or paracrine actions, which could positively impact the disease course and prognosis of AS. Macrophages, whose phenotypes can regulate immune responses, have shown promise in treating autoimmune diseases such as type 1 diabetes when using reparative M2 macrophages (164). CAR-modified M2 macrophages may become a novel immunotherapy option for AS. Additionally, MSCs, with their potent immunomodulatory properties, could offer a new treatment paradigm for AS after CAR modification, providing higher precision and specificity. Further exploration of the roles of these cells in AS, identification of specific phenotypic markers and optimization of their regulatory functions are essential for developing new CAR therapies.
6.2.3 Comprehensive preclinical validation
Before initiating multicenter clinical trials, extensive basic and preclinical research is necessary to evaluate the effects of CAR cell therapy for AS and optimize its safety and specificity. Key areas of focus include determining appropriate CAR designs and signaling mechanisms, assessing potential toxicity to normal tissues, and refining cell infusion techniques and treatment protocols. Additionally, a comprehensive evaluation of potential adverse events and long-term effects is crucial to ensure the controllability and sustainability of the treatment. By systematically conducting these preliminary studies, a solid scientific foundation can be laid for future multicenter clinical trials, thereby advancing the progress of CAR cell therapy in treating AS and making it a safer, more effective, and more sustainable treatment option.
7 Conclusion
The pathogenesis of AS is multifactorial, involving a complex interplay of genetic, immunological, and environmental factors. While the treatment landscape for AS has significantly evolved with the advent of advanced therapies, challenges remain in achieving long-term disease control and minimizing adverse effects. Traditional first-line treatments, such as NSAIDs and TNFis, remain the cornerstone of therapy but often fall short in addressing the heterogeneous nature of AS. The introduction of more biologic and targeted synthetic DMARDs, including IL-17A inhibitors and JAKis, has expanded therapeutic options.
Emerging cell therapies, such as MSCs and CAR-based cell therapy, offer novel approaches by targeting specific immune cells or providing regenerative benefits. These therapies hold promise in addressing the underlying pathophysiology of AS, potentially offering more durable and personalized treatment options. Nevertheless, their application in AS is still in its infancy, with ongoing clinical trials exploring their safety and efficacy.
Despite these advancements, several challenges persist. The high costs and accessibility issues associated with advanced therapies, particularly cell therapy, limit their widespread use. Furthermore, the long-term safety and efficacy of these novel approaches require further investigation through large-scale, randomized clinical trials. Future research should focus on optimizing treatment protocols, developing more precise targeting mechanisms, and exploring combination therapies to enhance efficacy and reduce side effects. Additionally, a deeper understanding of the pathogenesis of AS is crucial for the development of more effective and targeted treatments.
Author contributions
MK: Conceptualization, Data curation, Formal Analysis, Investigation, Writing – original draft, Visualization. WL: Conceptualization, Writing – original draft, Visualization. HL: Investigation, Writing – original draft, Visualization. XP: Investigation, Writing – original draft, Visualization. MW: Investigation, Writing – original draft, Visualization. NQ: Validation, Writing – review & editing. ZW: Investigation, Writing – original draft. YW: Funding acquisition, Project administration, Resources, Validation, Writing – review & editing. FZ: Funding acquisition, Project administration, Resources, Validation, Writing – review & editing.
Correction note
This article has been corrected with minor changes. These changes do not impact the scientific content of the article.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Revised: This work is supported by the Ili Kazakh Autonomous Prefecture Science and Technology Programs (No. YKX2023A11 and No. YKX2024A01) and the Enterprise Commissioned R&D Project (No. 21040400-J).
Acknowledgments
We acknowledge the use of ChatGPT version 4.0 for language editing and translation assistance.
Conflict of interest
Authors MK, WL, HL, NQ, ZW, and FZ were employed by the company Asia Cell & Gene Therapeutics Co., Limited. Authors MK, WL, ZW, and FZ were employed by the company Horgos Stem Cell Therapy Co., Limited.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Correction note
This article has been corrected with minor changes. These changes do not impact the scientific content of the article.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. We acknowledge the use of ChatGPT version 4.0 for language editing and translation assistance. Specifically, ChatGPT version 4.0 was utilized to translate the original Chinese content into English. Additionally, the translated content was reviewed and confirmed by the authors to ensure its accuracy and consistency with the original meaning. The conception, literature review, analysis, and overall content of this manuscript remain the sole responsibility of the authors. The use of ChatGPT version 4.0 was limited to language refinement and translation support, and it did not influence the originality or intellectual content of the work.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bittar M and Mease P. Novel therapies in axial spondyloarthritis. Best Pract Res Clin Rheumatol. (2022) 36:101811. doi: 10.1016/j.berh.2022.101811
2. Bandrés Ciga S, Salvatierra J, López-Sidro M, García-Sánchez A, Durán R, Vives F, et al. An examination of the mechanisms involved in secondary clinical failure to adalimumab or etanercept in inflammatory arthropathies. JCR J Clin Rheumatol. (2015) 21:115–9. doi: 10.1097/rhu.0000000000000229
3. Dean LE, Jones GT, MacDonald AG, Downham C, Sturrock RD, and Macfarlane GJ. Global prevalence of ankylosing spondylitis. Rheumatology. (2013) 53:650–7. doi: 10.1093/rheumatology/ket387
4. Xiang Y-J and Dai S-M. Prevalence of rheumatic diseases and disability in China. Rheumatol Int. (2008) 29:481–90. doi: 10.1007/s00296-008-0809-z
5. Huang F, Zhu J, Wang YH, Zhang JL, Jin HT, Zhang W, et al. Recommendations for diagnosis and treatment of ankylosing spondylitis. Zhonghua Nei Ke Za Zhi. (2022) 61:893–900. doi: 10.3760/cma.j.cn112138-20211226-00913
6. van der Heijde D, Braun J, Deodhar A, Baraliakos X, Landewé R, Richards HB, et al. Modified stoke ankylosing spondylitis spinal score as an outcome measure to assess the impact of treatment on structural progression in ankylosing spondylitis. Rheumatology. (2019) 58:388–400. doi: 10.1093/rheumatology/key128
7. Khan MA. HLA-B*27 and ankylosing spondylitis: 50 years of insights and discoveries. Curr Rheumatol Rep. (2023) 25:327–40. doi: 10.1007/s11926-023-01118-5
8. Harrison SR and Marzo-Ortega H. Have therapeutics enhanced our knowledge of axial spondyloarthritis? Curr Rheumatol Rep. (2023) 25:56–67. doi: 10.1007/s11926-023-01097-7
9. Healey EL, Haywood KL, Jordan KP, Garratt A, and Packham JC. Impact of ankylosing spondylitis on work in patients across the UK. Scand J Rheumatol. (2011) 40:34–40. doi: 10.3109/03009742.2010.487838
10. Bakland G, Gran JT, and Nossent JC. Increased mortality in ankylosing spondylitis is related to disease activity. Ann Rheum Dis. (2011) 70:1921–5. doi: 10.1136/ard.2011.151191
11. Golder V and Schachna L. Ankylosing spondylitis: an update. Aust Fam Physician. (2013) 42:780–4. Available at: https://www.racgp.org.au/afp/2013/november/ankylosing-spondylitis/
12. Burgos-Varga R, Wei JC, Rahman MU, Akkoc N, Haq SA, Hammoudeh M, et al. The prevalence and clinical characteristics of nonradiographic axial spondyloarthritis among patients with inflammatory back pain in rheumatology practices: a multinational, multicenter study. Arthritis Res Ther. (2016) 18:132. doi: 10.1186/s13075-016-1027-9
13. Sarsenova M, Issabekova A, Abisheva S, Rutskaya-Moroshan K, Ogay V, and Saparov A. Mesenchymal stem cell-based therapy for rheumatoid arthritis. Int J Mol Sci. (2021) 22:11592. doi: 10.3390/ijms222111592
14. Hassan SH, Alshahrani MY, Saleh RO, Mohammed BA, Kumar A, Almalki SG, et al. A new vision of the efficacy of both CAR-NK and CAR-T cells in treating cancers and autoimmune diseases. Med Oncol. (2024) 41:127. doi: 10.1007/s12032-024-02362-0
15. Muzes G and Sipos F. CAR-based therapy for autoimmune diseases: A novel powerful option. Cells. (2023) 12:1534. doi: 10.3390/cells12111534
16. Mohammadi V, Maleki AJ, Nazari M, Siahmansouri A, Moradi A, Elahi R, et al. Chimeric antigen receptor (CAR)-based cell therapy for type 1 diabetes mellitus (T1DM); current progress and future approaches. Stem Cell Rev Rep. (2024) 20:585–600. doi: 10.1007/s12015-023-10668-1
17. van Onna M, Jurik AG, van der Heijde D, van Tubergen A, Heuft-Dorenbosch L, and Landewe R. HLA-B27 and gender independently determine the likelihood of a positive MRI of the sacroiliac joints in patients with early inflammatory back pain: a 2-year MRI follow-up study. Ann Rheumatic Dis. (2011) 70:1981–5. doi: 10.1136/annrheumdis-2011-200025
18. Chen B, Li J, He C, Li D, Tong W, Zou Y, et al. Role of HLA-B27 in the pathogenesis of ankylosing spondylitis (Review). Mol Med Rep. (2017) 15:1943–51. doi: 10.3892/mmr.2017.6248
19. Antoniou AN, Lenart I, and Guiliano DB. Pathogenicity of misfolded and dimeric HLA-B27 molecules. Int J Rheumatol. (2011) 2011:486856. doi: 10.1155/2011/486856
20. Yang X, Garner LI, Zvyagin IV, Paley MA, Komech EA, Jude KM, et al. Autoimmunity-associated T cell receptors recognize HLA-B*27-bound peptides. Nature. (2022) 612:771–7. doi: 10.1038/s41586-022-05501-7
21. Kollnberger S, Bird L, Sun MY, Retiere C, Braud VM, McMichael A, et al. Cell-surface expression and immune receptor recognition of HLA–B27 homodimers. Arthritis Rheumatism. (2002) 46:2972–82. doi: 10.1002/art.10605
22. Kollnberger S, Chan A, Sun MY, Ye Chen L, Wright C, di Gleria K, et al. Interaction of HLA-B27 homodimers with KIR3DL1 and KIR3DL2, unlike HLA-B27 heterotrimers, is independent of the sequence of bound peptide. Eur J Immunol. (2007) 37:1313–22. doi: 10.1002/eji.200635997
23. Payeli SK, Kollnberger S, Marroquin Belaunzaran O, Thiel M, McHugh K, Giles J, et al. Inhibiting HLA–B27 homodimer–driven immune cell inflammation in spondylarthritis. Arthritis Rheumatism. (2012) 64:3139–49. doi: 10.1002/art.34538
24. Colbert RA, Tran TM, and Layh-Schmitt G. HLA-B27 misfolding and ankylosing spondylitis. Mol Immunol. (2014) 57:44–51. doi: 10.1016/j.molimm.2013.07.013
25. Burr ML, Cano F, Svobodova S, Boyle LH, Boname JM, and Lehner PJ. HRD1 and UBE2J1 target misfolded MHC class I heavy chains for endoplasmic reticulum-associated degradation. Proc Natl Acad Sci U S A. (2011) 108:2034–9. doi: 10.1073/pnas.1016229108
26. DeLay ML, Turner MJ, Klenk EI, Smith JA, Sowders DP, and Colbert RA. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheumatol. (2009) 60:2633–43. doi: 10.1002/art.24763
27. Layh-Schmitt G, Yang EY, Kwon G, and Colbert RA. HLA-B27 alters the response to tumor necrosis factor alpha and promotes osteoclastogenesis in bone marrow monocytes from HLA-B27-transgenic rats. Arthritis Rheumatol. (2013) 65:2123–31. doi: 10.1002/art.38001
28. Chimenti MS, Perricone C, Novelli L, Caso F, Costa L, Bogdanos D, et al. Interaction between microbiome and host genetics in psoriatic arthritis. Autoimmun Rev. (2018) 17:276–83. doi: 10.1016/j.autrev.2018.01.002
29. Lim CSE, Sengupta R, and Gaffney K. The clinical utility of human leucocyte antigen B27 in axial spondyloarthritis. Rheumatology. (2018) 57:959–68. doi: 10.1093/rheumatology/kex345
30. Gill T, Brooks SR, Rosenbaum JT, Asquith M, and Colbert RA. Novel inter-omic analysis reveals relationships between diverse gut microbiota and host immune dysregulation in HLA–B27–induced experimental spondyloarthritis. Arthritis Rheumatol. (2019) 71:1849–57. doi: 10.1002/art.41018
31. Asquith M, Davin S, Stauffer P, Michell C, Janowitz C, Lin P, et al. Intestinal metabolites are profoundly altered in the context of HLA–B27 expression and functionally modulate disease in a rat model of spondyloarthritis. Arthritis Rheumatol. (2017) 69:1984–95. doi: 10.1002/art.40183
32. Fiorillo MT, Haroon N, Ciccia F, and Breban M. Editorial: ankylosing spondylitis and related immune-mediated disorders. Front Immunol. (2019) 10:1232. doi: 10.3389/fimmu.2019.01232
33. Dashti N, Mahmoudi M, Aslani S, and Jamshidi A. HLA-B*27 subtypes and their implications in the pathogenesis of ankylosing spondylitis. Gene. (2018) 670:15–21. doi: 10.1016/j.gene.2018.05.092
34. Martín-Esteban A, Gómez-Molina P, Sanz-Bravo A, and López de Castro JA. Combined effects of ankylosing spondylitis-associated ERAP1 polymorphisms outside the catalytic and peptide-binding sites on the processing of natural HLA-B27 ligands. J Biol Chem. (2014) 289:3978–90. doi: 10.1074/jbc.M113.529610
35. Evans DM, Spencer CCA, Pointon JJ, Su Z, Harvey D, Kochan G, et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet. (2011) 43:761–7. doi: 10.1038/ng.873
36. Gravallese EM and Schett G. Effects of the IL-23–IL-17 pathway on bone in spondyloarthritis. Nat Rev Rheumatol. (2018) 14:631–40. doi: 10.1038/s41584-018-0091-8
37. Garcia-Montoya L, Gul H, and Emery P. Recent advances in ankylosing spondylitis: understanding the disease and management. F1000Research. (2018) 7:F1000. doi: 10.12688/f1000research.14956.1
38. Linguang Z. Clinical development of tumor necrosis factor-alpha inhibitors for ankylosing spondylitis. J Chin Pharm Sci. (2018) 27:59–63. doi: 10.5246/jcps.2018.01.007
39. Kwon HY, Kim YJ, Kim T-H, and Ahn SJ. Comparison of incidence or recurrence of anterior uveitis in patients with ankylosing spondylitis treated with tumor necrosis factor inhibitors. J Clin Med. (2024) 13:912. doi: 10.3390/jcm13030912
40. Bindu S MS and Bandyopadhyay U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: a current perspective. Biochem Pharmacol. (2020) 180:114147. doi: 10.1016/j.bcp.2020.114147
41. Hu J, Mao Y, Zhang Y, Ye D, Wen C, and Xie Z. Moxibustion for the treatment of ankylosing spondylitis: a systematic review and meta-analysis. Ann Palliat Med. (2020) 9:709–20. doi: 10.21037/apm.2020.02.31
42. Baraliakos X, Kiltz U, Peters S, Appel H, Dybowski F, Igelmann M, et al. Efficiency of treatment with non-steroidal anti-inflammatory drugs according to current recommendations in patients with radiographic and non-radiographic axial spondyloarthritis. Rheumatology. (2017) 56:95–102. doi: 10.1093/rheumatology/kew367
43. Baraliakos X, Papagoras C, and Klavdianou K. JAK inhibitors for the treatment of axial spondyloarthritis. Mediterr J Rheumatol. (2023) 34:129–38. doi: 10.31138/mjr.34.2.129
44. Wang H, Zheng H, and Ma Y. Drug treatment of ankylosing spondylitis and related complications: an overlook review. Ann Palliat Med. (2020) 9:2279–85. doi: 10.21037/apm-20-277
45. Puig L. Methotrexate: new therapeutic approaches. Actas Dermosifiliogr. (2014) 105:583–9. doi: 10.1016/j.ad.2012.11.017
46. Altan L, Bingol U, Karakoc Y, Aydiner S, Yurtkuran M, and Yurtkuran M. Clinical investigation of methotrexate in the treatment of ankylosing spondylitis. Scand J Rheumatol. (2001) 30:255–9. doi: 10.1080/030097401753180318
47. Ebrahimiadib N, Berijani S, Ghahari M, and Pahlaviani FG. Ankylosing spondylitis. J Ophthalmic Vis Res. (2021) 16:462–9. doi: 10.18502/jovr.v16i3.9440
48. Ward MM, Deodhar A, Gensler LS, Dubreuil M, Yu D, Khan MA, et al. Update of the american college of rheumatology/Spondylitis association of america/Spondyloarthritis research and treatment network recommendations for the treatment of ankylosing spondylitis and nonradiographic axial spondyloarthritis. Arthritis Rheumatol. (2019) 71:1599–613. doi: 10.1002/art.41042
49. Sen R, Riofrio M, and Singh JA. A narrative review of the comparative safety of disease-modifying anti-rheumatic drugs used for the treatment of rheumatoid arthritis. Expert Opin Drug Saf. (2024) 23:687–714. doi: 10.1080/14740338.2024.2348575
50. Martínez-Ramos S, Rafael-Vidal C, Pego-Reigosa JM, and García S. Monocytes and macrophages in spondyloarthritis: functional roles and effects of current therapies. Cells. (2022) 11:515. doi: 10.3390/cells11030515
51. Egeberg A, Rosenø NAL, Thein D, Lørup EH, Nielsen M-L, Nymand L, et al. Drug survival of biologics and novel immunomodulators for rheumatoid arthritis, axial spondyloarthritis, psoriatic arthritis, and psoriasis - A nationwide cohort study from the DANBIO and DERMBIO registries. Semin Arthritis Rheumatism. (2022) 53:151979. doi: 10.1016/j.semarthrit.2022.151979
52. Garcia-Montoya L and Emery P. Disease modification in ankylosing spondylitis with TNF inhibitors: spotlight on early phase clinical trials. Expert Opin Investig Drugs. (2021) 30:1109–24. doi: 10.1080/13543784.2021.2010187
53. Zouris G, Evangelopoulos DS, Benetos IS, and Vlamis J. The use of TNF-alpha inhibitors in active ankylosing spondylitis treatment. Cureus. (2024) 16:e61500. doi: 10.7759/cureus.61500
54. Knight DM, Trinh H, Le J, Siegel S, Shealy D, McDonough M, et al. Construction and initial characterization of a mouse-human chimeric anti-TNF antibody. Mol Immunol. (1993) 30:1443–53. doi: 10.1016/0161-5890(93)90106-l
55. van der Heijde D, Kivitz A, Schiff MH, Sieper J, Dijkmans BA, Braun J, et al. Efficacy and safety of adalimumab in patients with ankylosing spondylitis: results of a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. (2006) 54:2136–46. doi: 10.1002/art.21913
56. Gorman JD, Sack KE, and Davis JC Jr. Treatment of ankylosing spondylitis by inhibition of tumor necrosis factor alpha. N Engl J Med. (2002) 346:1349–56. doi: 10.1056/NEJMoa012664
57. Shealy DJ, Cai A, Staquet K, Baker A, Lacy ER, Johns L, et al. Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor alpha. MAbs. (2010) 2:428–39. doi: 10.4161/mabs.12304
58. Dhillon S. Certolizumab pegol: a review of its use in patients with axial spondyloarthritis or psoriatic arthritis. Drugs. (2014) 74:999–1016. doi: 10.1007/s40265-014-0239-z
59. Chimenti MS, Perricone C, D’Antonio A, Ferraioli M, Conigliaro P, Triggianese P, et al. Genetics, epigenetics, and gender impact in axial-spondyloarthritis susceptibility: an update on genetic polymorphisms and their sex related associations. Front Genet. (2021) 12:671976. doi: 10.3389/fgene.2021.671976
60. Benucci M, Damiani A, Li Gobbi F, Bandinelli F, Infantino M, Grossi V, et al. Correlation between HLA haplotypes and the development of antidrug antibodies in a cohort of patients with rheumatic diseases. Biol: Targets Ther. (2018) 12:37–41. doi: 10.2147/btt.S145941
61. Pascual-Salcedo D, Plasencia C, Ramiro S, Nuno L, Bonilla G, Nagore D, et al. Influence of immunogenicity on the efficacy of long-term treatment with infliximab in rheumatoid arthritis. Rheumatology. (2011) 50:1445–52. doi: 10.1093/rheumatology/ker124
62. Wolbink GJ, Aarden LA, and Dijkmans BAC. Dealing with immunogenicity of biologicals: assessment and clinical relevance. Curr Opin Rheumatol. (2009) 21:211–5. doi: 10.1097/BOR.0b013e328329ed8b
63. Alawadhi A, Alawneh K, and Alzahrani ZA. The effect of neutralizing antibodies on the sustainable efficacy of biologic therapies: what’s in it for African and Middle Eastern rheumatologists. Clin Rheumatol. (2012) 31:1281–7. doi: 10.1007/s10067-012-2040-2
64. Mann DL, McMurray JJV, Packer M, Swedberg K, Borer JS, Colucci WS, et al. Targeted anticytokine therapy in patients with chronic heart failure. Circulation. (2004) 109:1594–602. doi: 10.1161/01.Cir.0000124490.27666.B2
65. Chung ES, Packer M, Lo KH, Fasanmade AA, and Willerson JT. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-α, in patients with moderate-to-severe heart failure. Circulation. (2003) 107:3133–40. doi: 10.1161/01.Cir.0000077913.60364.D2
66. Min HK, Kim SH, Kim H-R, and Lee S-H. Therapeutic utility and adverse effects of biologic disease-modifying anti-rheumatic drugs in inflammatory arthritis. Int J Mol Sci. (2022) 23:13913. doi: 10.3390/ijms232213913
67. Minozzi S, Bonovas S, Lytras T, Pecoraro V, González-Lorenzo M, Bastiampillai AJ, et al. Risk of infections using anti-TNF agents in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: a systematic review and meta-analysis. Expert Opin Drug Safety. (2016) 15:11–34. doi: 10.1080/14740338.2016.1240783
68. Gossec L, Le Henanff A, Breban M, Vignon E, Claudepierre P, Devauchelle V, et al. Continuation of treatment with infliximab in ankylosing spondylitis: 2-yr open follow-up. Rheumatology. (2006) 45:859–62. doi: 10.1093/rheumatology/kel015
69. Russell MD, Stovin C, Alveyn E, Adeyemi O, Chan CKD, Patel V, et al. JAK inhibitors and the risk of Malignancy: a meta-analysis across disease indications. Ann Rheumatic Dis. (2023) 82:1059–67. doi: 10.1136/ard-2023-224049
70. Kim HW, Park JK, Yang J-A, Yoon YI, Lee EY, Song YW, et al. Comparison of tuberculosis incidence in ankylosing spondylitis and rheumatoid arthritis during tumor necrosis factor inhibitor treatment in an intermediate burden area. Clin Rheumatol. (2013) 33:1307–12. doi: 10.1007/s10067-013-2387-z
71. Bridgewood C, Watad A, Russell T, Palmer TM, Marzo-Ortega H, Khan A, et al. Identification of myeloid cells in the human enthesis as the main source of local IL-23 production. Ann Rheumatic Dis. (2019) 78:929–33. doi: 10.1136/annrheumdis-2018-214944
72. Mease PJ, Chohan S, Fructuoso FJG, Luggen ME, Rahman P, Raychaudhuri SP, et al. Efficacy and safety of tildrakizumab in patients with active psoriatic arthritis: results of a randomised, double-blind, placebo-controlled, multiple-dose, 52-week phase IIb study. Ann Rheumatic Dis. (2021) 80:1147–57. doi: 10.1136/annrheumdis-2020-219014
73. Mei Y, Pan F, Gao J, Ge R, Duan Z, Zeng Z, et al. Increased serum IL-17 and IL-23 in the patient with ankylosing spondylitis. Clin Rheumatol. (2010) 30:269–73. doi: 10.1007/s10067-010-1647-4
74. Glatt S, Baeten D, Baker T, Griffiths M, Ionescu L, Lawson ADG, et al. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann Rheumatic Dis. (2018) 77:523–32. doi: 10.1136/annrheumdis-2017-212127
75. van der Heijde D, Cheng-Chung Wei J, Dougados M, Mease P, Deodhar A, Maksymowych WP, et al. Ixekizumab, an interleukin-17A antagonist in the treatment of ankylosing spondylitis or radiographic axial spondyloarthritis in patients previously untreated with biological disease-modifying anti-rheumatic drugs (COAST-V): 16 week results of a phase 3 randomised, double-blind, active-controlled and placebo-controlled trial. Lancet. (2018) 392:2441–51. doi: 10.1016/s0140-6736(18)31946-9
76. van der Heijde D, Gensler LS, Deodhar A, Baraliakos X, Poddubnyy D, Kivitz A, et al. Dual neutralisation of interleukin-17A and interleukin-17F with bimekizumab in patients with active ankylosing spondylitis: results from a 48-week phase IIb, randomised, double-blind, placebo-controlled, dose-ranging study. Ann Rheumatic Dis. (2020) 79:595–604. doi: 10.1136/annrheumdis-2020-216980
77. Braun J, Brandt J, Listing J, Zink A, Alten R, Golder W, et al. Treatment of active ankylosing spondylitis with infliximab: a randomised controlled multicentre trial. Lancet. (2002) 359:1187–93. doi: 10.1016/s0140-6736(02)08215-6
78. Huang J-X, Zhang L-J, and Wei JC-C. Interleukin-17 inhibitors for the treatment of ankylosing spondylitis. Rheumatol Immunol Res. (2020) 1:25–9. doi: 10.2478/rir-2020-0004
79. Poizeau F, Nowak E, Kerbrat S, Le Nautout B, Droitcourt C, Drici M-D, et al. Association between early severe cardiovascular events and the initiation of treatment with the anti–interleukin 12/23p40 antibody ustekinumab. JAMA Dermatol. (2020) 156:1208–15. doi: 10.1001/jamadermatol.2020.2977
80. Veale DJ, McGonagle D, McInnes IB, Krueger JG, Ritchlin CT, Elewaut D, et al. The rationale for Janus kinase inhibitors for the treatment of spondyloarthritis. Rheumatology. (2019) 58:197–205. doi: 10.1093/rheumatology/key070
81. Traves PG, Murray B, Campigotto F, Galien R, Meng A, and Di Paolo JA. JAK selectivity and the implications for clinical inhibition of pharmacodynamic cytokine signalling by filgotinib, upadacitinib, tofacitinib and baricitinib. Ann Rheumatic Dis. (2021) 80:865–75. doi: 10.1136/annrheumdis-2020-219012
82. Akkoc N and Khan MA. JAK inhibitors for axial spondyloarthritis: what does the future hold? Curr Rheumatol Rep. (2021) 23:34. doi: 10.1007/s11926-021-01001-1
83. Ytterberg SR, Bhatt DL, Mikuls TR, Koch GG, Fleischmann R, Rivas JL, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. (2022) 386:316–26. doi: 10.1056/NEJMoa2109927
84. Shen N, Wu X, Guo Z, Yang S, Liu C, Guo Z, et al. Classification and treatment for cervical spine fracture with ankylosing spondylitis: A clinical nomogram prediction study. Pain Res Manage. (2022) 2022:7769775. doi: 10.1155/2022/7769775
85. Baig Mirza A, Fayez F, Rashed S, Georgiannakis A, Lam PY, Lysomirski A, et al. Surgical management and outcomes of ankylosing spondylitis fractures in adults: a systematic review and meta-analysis. Neurosurg Rev. (2025) 48:366. doi: 10.1007/s10143-025-03518-w
86. Gravaldi LP, Bonetti F, Lezzerini S, and De Maio F. Effectiveness of physiotherapy in patients with ankylosing spondylitis: A systematic review and meta-analysis. Healthc (Basel). (2022) 10:132. doi: 10.3390/healthcare10010132
87. Friedenstein AJ, Petrakova KV, Kurolesova AI, and Frolova GP. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. (1968) 6:230–47. doi: 10.1097/00007890-196803000-00009
88. Perrotta FM, Guerra G, De Socio A, Scriffignano S, and Lubrano E. Mesenchimal stem cells: a possible role in the pathogenesis and treatment of spondyloarthritis. Reumatismo. (2017) 69:1–8. doi: 10.4081/reumatismo.2017.976
89. Munir H and McGettrick HM. Mesenchymal stem cell therapy for autoimmune disease: risks and rewards. Stem Cells Dev. (2015) 24:2091–100. doi: 10.1089/scd.2015.0008
90. Selmani Z, Naji A, Zidi I, Favier B, Gaiffe E, Obert L, et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4+CD25highFOXP3+ regulatory T cells. Stem Cells. (2008) 26:212–22. doi: 10.1634/stemcells.2007-0554
91. Chinnadurai R, Copland IB, Patel SR, and Galipeau J. IDO-independent suppression of T cell effector function by IFN-gamma-licensed human mesenchymal stromal cells. J Immunol. (2014) 192:1491–501. doi: 10.4049/jimmunol.1301828
92. Yan L, Zheng D, and Xu RH. Critical role of tumor necrosis factor signaling in mesenchymal stem cell-based therapy for autoimmune and inflammatory diseases. Front Immunol. (2018) 9:1658. doi: 10.3389/fimmu.2018.01658
93. Yang Y, He X, Zhao R, Guo W, Zhu M, Xing W, et al. Serum IFN-gamma levels predict the therapeutic effect of mesenchymal stem cell transplantation in active rheumatoid arthritis. J Transl Med. (2018) 16:165. doi: 10.1186/s12967-018-1541-4
94. Liu H, Liu S, Song X, Jiang A, Zou Y, Deng Y, et al. Nanoparticle encapsulated CQ/TAM combination harmonizes with MSCs in arresting progression of severity in AP mice through iNOS (IDO) signaling. Mater Today Bio. (2022) 14:100226. doi: 10.1016/j.mtbio.2022.100226
95. Huang ZF, Zhu J, Lu SH, Zhang JL, Chen XD, Du LX, et al. Inhibitory effect of human umbilical cord-derived mesenchymal stem cells on interleukin-17 production in peripheral blood T cells from spondyloarthritis patients. Zhongguo Shi Yan Xue Ye Xue Za Zhi. (2013) 21:455–9. doi: 10.7534/j.issn.1009-2137.2013.02.041
96. Miao J and Zhu P. Functional defects of treg cells: new targets in rheumatic diseases, including ankylosing spondylitis. Curr Rheumatol Rep. (2018) 20:30. doi: 10.1007/s11926-018-0729-1
97. Guo H, Zheng M, Zhang K, Yang F, Zhang X, Han Q, et al. Functional defects in CD4(+) CD25(high) FoxP3(+) regulatory cells in ankylosing spondylitis. Sci Rep. (2016) 6:37559. doi: 10.1038/srep37559
98. English K, Ryan JM, Tobin L, Murphy MJ, Barry FP, and Mahon BP. Cell contact, prostaglandin E(2) and transforming growth factor beta 1 play non-redundant roles in human mesenchymal stem cell induction of CD4+CD25(High) forkhead box P3+ regulatory T cells. Clin Exp Immunol. (2009) 156:149–60. doi: 10.1111/j.1365-2249.2009.03874.x
99. Melief SM, Schrama E, Brugman MH, Tiemessen MM, Hoogduijn MJ, Fibbe WE, et al. Multipotent stromal cells induce human regulatory T cells through a novel pathway involving skewing of monocytes toward anti-inflammatory macrophages. Stem Cells. (2013) 31:1980–91. doi: 10.1002/stem.1432
100. Hong J, Hueckelhoven A, Wang L, Schmitt A, Wuchter P, Tabarkiewicz J, et al. Indoleamine 2,3-dioxygenase mediates inhibition of virus-specific CD8(+) T cell proliferation by human mesenchymal stromal cells. Cytotherapy. (2016) 18:621–9. doi: 10.1016/j.jcyt.2016.01.009
101. Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. (2002) 99:3838–43. doi: 10.1182/blood.v99.10.3838
102. Zhao J, Yuan W, Tao C, Sun P, Yang Z, and Xu W. M2 polarization of monocytes in ankylosing spondylitis and relationship with inflammation and structural damage. APMIS. (2017) 125:1070–5. doi: 10.1111/apm.12757
103. Shen D, Wang Z, Wang H, Zhu H, Jiang C, Xie F, et al. Evaluation of preclinical efficacy of human umbilical cord mesenchymal stem cells in ankylosing spondylitis. Front Immunol. (2023) 14:1153927. doi: 10.3389/fimmu.2023.1153927
104. Zvyagin IV, Mamedov IZ, Britanova OV, Staroverov DB, Nasonov EL, Bochkova AG, et al. Contribution of functional KIR3DL1 to ankylosing spondylitis. Cell Mol Immunol. (2010) 7:471–6. doi: 10.1038/cmi.2010.42
105. Sotiropoulou PA, Perez SA, Gritzapis AD, Baxevanis CN, and Papamichail M. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells. (2006) 24:74–85. doi: 10.1634/stemcells.2004-0359
106. Spaggiari GM, Capobianco A, Becchetti S, Mingari MC, and Moretta L. Mesenchymal stem cell-natural killer cell interactions: evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood. (2006) 107:1484–90. doi: 10.1182/blood-2005-07-2775
107. Dillon SM, Rogers LM, Howe R, Hostetler LA, Buhrman J, McCarter MD, et al. Human intestinal lamina propria CD1c+ dendritic cells display an activated phenotype at steady state and produce IL-23 in response to TLR7/8 stimulation. J Immunol. (2010) 184:6612–21. doi: 10.4049/jimmunol.1000041
108. Utriainen L, Firmin D, Wright P, Cerovic V, Breban M, McInnes I, et al. Expression of HLA-B27 causes loss of migratory dendritic cells in a rat model of spondylarthritis. Arthritis Rheumatol. (2012) 64:3199–209. doi: 10.1002/art.34561
109. Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-gammat+ CD3+CD4-CD8- entheseal resident T cells. Nat Med. (2012) 18:1069–76. doi: 10.1038/nm.2817
110. Zhang W, Ge W, Li C, You S, Liao L, Han Q, et al. Effects of mesenchymal stem cells on differentiation, maturation, and function of human monocyte-derived dendritic cells. Stem Cells Dev. (2004) 13:263–71. doi: 10.1089/154732804323099190
111. Jiang XX, Zhang Y, Liu B, Zhang SX, Wu Y, Yu XD, et al. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood. (2005) 105:4120–6. doi: 10.1182/blood-2004-02-0586
112. Spaggiari GM, Abdelrazik H, Becchetti F, and Moretta L. MSCs inhibit monocyte-derived DC maturation and function by selectively interfering with the generation of immature DCs: central role of MSC-derived prostaglandin E2. Blood. (2009) 113:6576–83. doi: 10.1182/blood-2009-02-203943
113. Wang K, Lu J, Song C, Qiao M, Li Y, Chang M, et al. Extracellular vesicles derived from ligament tissue transport interleukin-17A to mediate ligament-to-bone crosstalk in ankylosing spondylitis. Adv Sci (Weinh). (2024) 11:e2406876. doi: 10.1002/advs.202406876
114. Franz-Odendaal TA. Induction and patterning of intramembranous bone. Front Biosci (Landmark Ed). (2011) 16:2734–46. doi: 10.2741/3882
115. Ko FC and Sumner DR. How faithfully does intramembranous bone regeneration recapitulate embryonic skeletal development? Dev Dyn. (2021) 250:377–92. doi: 10.1002/dvdy.240
116. Kusuda M, Haroon N, and Nakamura A. Complexity of enthesitis and new bone formation in ankylosing spondylitis: current understanding of the immunopathology and therapeutic approaches. Mod Rheumatol. (2022) 32:484–92. doi: 10.1093/mr/roab057
117. Krajewska-Wlodarczyk M, Owczarczyk-Saczonek A, Placek W, Osowski A, Engelgardt P, and Wojtkiewicz J. Role of stem cells in pathophysiology and therapy of spondyloarthropathies-new therapeutic possibilities? Int J Mol Sci. (2017) 19:80. doi: 10.3390/ijms19010080
118. Yu T, Zhang J, Zhu W, Wang X, Bai Y, Feng B, et al. Chondrogenesis mediates progression of ankylosing spondylitis through heterotopic ossification. Bone Res. (2021) 9:19. doi: 10.1038/s41413-021-00140-6
119. Papagoras C, Chrysanthopoulou A, Mitsios A, Ntinopoulou M, Tsironidou V, Batsali AK, et al. IL-17A expressed on neutrophil extracellular traps promotes mesenchymal stem cell differentiation toward bone-forming cells in ankylosing spondylitis. Eur J Immunol. (2021) 51:930–42. doi: 10.1002/eji.202048878
120. Zhang X, Aubin JE, and Inman RD. Molecular and cellular biology of new bone formation: insights into the ankylosis of ankylosing spondylitis. Curr Opin Rheumatol. (2003) 15:387–93. doi: 10.1097/00002281-200307000-00004
121. Sieper J and van der Heijde D. Review: Nonradiographic axial spondyloarthritis: new definition of an old disease? Arthritis Rheumatol. (2013) 65:543–51. doi: 10.1002/art.37803
122. Gao WX, Sun YQ, Shi J, Li CL, Fang SB, Wang D, et al. Effects of mesenchymal stem cells from human induced pluripotent stem cells on differentiation, maturation, and function of dendritic cells. Stem Cell Res Ther. (2017) 8:48. doi: 10.1186/s13287-017-0499-0
123. Liu CH, Raj S, Chen CH, Hung KH, Chou CT, Chen IH, et al. HLA-B27-mediated activation of TNAP phosphatase promotes pathogenic syndesmophyte formation in ankylosing spondylitis. J Clin Invest. (2019) 129:5357–73. doi: 10.1172/JCI125212
124. Tsui HW, Inman RD, Reveille JD, and Tsui FW. Association of a TNAP haplotype with ankylosing spondylitis. Arthritis Rheumatol. (2007) 56:234–43. doi: 10.1002/art.22307
125. Daoussis D, Kanellou A, Panagiotopoulos E, and Papachristou D. DKK-1 is underexpressed in mesenchymal stem cells from patients with ankylosing spondylitis and further downregulated by IL-17. Int J Mol Sci. (2022) 23:6660. doi: 10.3390/ijms23126660
126. Xie Z, Wang P, Li Y, Deng W, Zhang X, Su H, et al. Imbalance between bone morphogenetic protein 2 and noggin induces abnormal osteogenic differentiation of mesenchymal stem cells in ankylosing spondylitis. Arthritis Rheumatol. (2016) 68:430–40. doi: 10.1002/art.39433
127. Tsukasaki M and Takayanagi H. Osteoimmunology: evolving concepts in bone-immune interactions in health and disease. Nat Rev Immunol. (2019) 19:626–42. doi: 10.1038/s41577-019-0178-8
128. Tang W, Zhao K, Li X, Zhou X, and Liao P. Bone marrow mesenchymal stem cell-derived exosomes promote the recovery of spinal cord injury and inhibit ferroptosis by inactivating IL-17 pathway. J Mol Neurosci. (2024) 74:33. doi: 10.1007/s12031-024-02209-3
129. Yang HK, Moon SJ, Shin JH, Kwok SK, Park KS, Park SH, et al. Regression of syndesmophyte after bone marrow transplantation for acute myeloid leukemia in a patient with ankylosing spondylitis: a case report. J Med Case Rep. (2012) 6:250. doi: 10.1186/1752-1947-6-250
130. Wang P, Li Y, Huang L, Yang J, Yang R, Deng W, et al. Effects and safety of allogenic mesenchymal stem cell intravenous infusion in active ankylosing spondylitis patients who failed NSAIDs: a 20-week clinical trial. Cell Transpl. (2014) 23:1293–303. doi: 10.3727/096368913X667727
131. Li A, Tao Y, Kong D, Zhang N, Wang Y, Wang Z, et al. Infusion of umbilical cord mesenchymal stem cells alleviates symptoms of ankylosing spondylitis. Exp Ther Med. (2017) 14:1538–46. doi: 10.3892/etm.2017.4687
132. Luo XL, Jiang Y, Li Q, Yu XJ, Ma T, Cao H, et al. hESC-derived epicardial cells promote repair of infarcted hearts in mouse and swine. Advanced Sci. (2023) 10:e2300470. doi: 10.1002/advs.202300470
133. Ramírez-Valle F, Maranville JC, Roy S, and Plenge RM. Sequential immunotherapy: towards cures for autoimmunity. Nat Rev Drug Discov. (2024) 23:501–24. doi: 10.1038/s41573-024-00959-8
134. Zhou J, Lei B, Shi F, Luo X, Wu K, Xu Y, et al. CAR T-cell therapy for systemic lupus erythematosus: current status and future perspectives. Front Immunol. (2024) 15:1476859. doi: 10.3389/fimmu.2024.1476859
135. Flemming A. CAR-T cells take aim at autoimmunity. Nat Rev Drug Discov. (2016) 15:603–3. doi: 10.1038/nrd.2016.180
136. Wang D, Shao Y, Zhang X, Lu G, and Liu B. IL-23 and PSMA-targeted duo-CAR T cells in Prostate Cancer Eradication in a preclinical model. J Transl Med. (2020) 18:23. doi: 10.1186/s12967-019-02206-w
137. Britanova OV, Lupyr KR, Staroverov DB, Shagina IA, Aleksandrov AA, Ustyugov YY, et al. Targeted depletion of TRBV9(+) T cells as immunotherapy in a patient with ankylosing spondylitis. Nat Med. (2023) 29:2731–6. doi: 10.1038/s41591-023-02613-z
138. Sun Y, Yuan Y, Zhang B, and Zhang X. CARs: a new approach for the treatment of autoimmune diseases. Sci China Life Sci. (2023) 66:711–28. doi: 10.1007/s11427-022-2212-5
139. Mougiakakos D, Kronke G, Volkl S, Kretschmann S, Aigner M, Kharboutli S, et al. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N Engl J Med. (2021) 385:567–9. doi: 10.1056/NEJMc2107725
140. Rejeski K, Subklewe M, and Locke FL. Recognizing, defining, and managing CAR-T hematologic toxicities. Hematol Am Soc Hematol Educ Program. (2023) 2023:198–208. doi: 10.1182/hematology.2023000472
141. Bahadorian D, Mollazadeh S, Mirazi H, Faraj TA, Kheder RK, and Esmaeili SA. Regulatory NK cells in autoimmune disease. Iran J Basic Med Sci. (2023) 26:609–16. doi: 10.22038/IJBMS.2023.68653.14969
142. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation. (2012) 9:112. doi: 10.1186/1742-2094-9-112
143. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. (2016) 126:1413–24. doi: 10.1172/jci82771
144. Sirpilla O, Sakemura RL, Hefazi M, Huynh TN, Can I, Girsch JH, et al. Mesenchymal stromal cells with chimaeric antigen receptors for enhanced immunosuppression. Nat BioMed Eng. (2024) 8:443–60. doi: 10.1038/s41551-024-01195-6
145. Bulliard Y, Freeborn R, Uyeda MJ, Humes D, Bjordahl R, de Vries D, et al. From promise to practice: CAR T and Treg cell therapies in autoimmunity and other immune-mediated diseases. Front Immunol. (2024) 15:1509956. doi: 10.3389/fimmu.2024.1509956
146. Lin H, Cheng J, Zhu L, Zeng Y, Dai Z, Zhang Y, et al. Anti-CD5 CAR-T cells with a tEGFR safety switch exhibit potent toxicity control. Blood Cancer J. (2024) 14:98. doi: 10.1038/s41408-024-01082-y
147. Hu Y, Zhang M, Yang T, Mo Z, Wei G, Jing R, et al. Sequential CD7 CAR T-cell therapy and allogeneic HSCT without GVHD prophylaxis. N Engl J Med. (2024) 390:1467–80. doi: 10.1056/NEJMoa2313812
148. Nagamura-Inoue T and He H. Umbilical cord-derived mesenchymal stem cells: Their advantages and potential clinical utility. World J Stem Cells. (2014) 6:195–202. doi: 10.4252/wjsc.v6.i2.195
149. Chen X, Zheng J, Yin L, Li Y, and Liu H. Transplantation of three mesenchymal stem cells for knee osteoarthritis, which cell and type are more beneficial? a systematic review and network meta-analysis. J Orthop Surg Res. (2024) 19:366. doi: 10.1186/s13018-024-04846-1
150. Remestemcel-L (Ryoncil) for graft-versus-host disease. Med Lett Drugs Ther. (2025) 67:e36–7. doi: 10.58347/tml.2025.1722i
151. Naji A, Eitoku M, Favier B, Deschaseaux F, Rouas-Freiss N, and Suganuma N. Biological functions of mesenchymal stem cells and clinical implications. Cell Mol Life Sci. (2019) 76:3323–48. doi: 10.1007/s00018-019-03125-1
152. Dias de Oliveira FB, Antonioli E, Dias OFM, de Souza JG, Agarwal S, Chudzinski-Tavassi AM, et al. Comparative effects of intra-articular versus intravenous mesenchymal stromal cells therapy in a rat model of osteoarthritis by destabilization of medial meniscus. Int J Mol Sci. (2023) 24:15543. doi: 10.3390/ijms242115543
153. Amadeo F, Hanson V, Liptrott NJ, Wilm B, Murray P, and Taylor A. Fate of intravenously administered umbilical cord mesenchymal stromal cells and interactions with the host’s immune system. BioMed Pharmacother. (2023) 159:114191. doi: 10.1016/j.biopha.2022.114191
154. de Witte SFH, Luk F, Sierra Parraga JM, Gargesha M, Merino A, Korevaar SS, et al. Immunomodulation by therapeutic mesenchymal stromal cells (MSC) is triggered through phagocytosis of MSC by monocytic cells. Stem Cells. (2018) 36:602–15. doi: 10.1002/stem.2779
155. Pang SHM, D’Rozario J, Mendonca S, Bhuvan T, Payne NL, Zheng D, et al. Mesenchymal stromal cell apoptosis is required for their therapeutic function. Nat Commun. (2021) 12:6495. doi: 10.1038/s41467-021-26834-3
156. Dang S, Xu H, Xu C, Cai W, Li Q, Cheng Y, et al. Autophagy regulates the therapeutic potential of mesenchymal stem cells in experimental autoimmune encephalomyelitis. Autophagy. (2014) 10:1301–15. doi: 10.4161/auto.28771
157. Song M, Lv K, Xu Z, Li J, Sun J, Shi J, et al. N6 methyladenosine eraser FTO suppresses Staphylococcus aureus-induced ferroptosis of bone marrow mesenchymal stem cells to ameliorate osteomyelitis through regulating the MDM2/TLR4/SLC7A11 signaling pathway. Cell Biol Int. (2024) 48:450–60. doi: 10.1002/cbin.12115
158. Galleu A, Riffo-Vasquez Y, Trento C, Lomas C, Dolcetti L, Cheung TS, et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci Transl Med. (2017) 9:eaam7828. doi: 10.1126/scitranslmed.aam7828
159. Preda MB, Neculachi CA, Fenyo IM, Vacaru AM, Publik MA, Simionescu M, et al. Short lifespan of syngeneic transplanted MSC is a consequence of in vivo apoptosis and immune cell recruitment in mice. Cell Death Dis. (2021) 12:566. doi: 10.1038/s41419-021-03839-w
160. Shan Y, Zhang M, Tao E, Wang J, Wei N, Lu Y, et al. Pharmacokinetic characteristics of mesenchymal stem cells in translational challenges. Signal Transduct Target Ther. (2024) 9:242. doi: 10.1038/s41392-024-01936-8
161. Chan AML, Sampasivam Y, and Lokanathan Y. Biodistribution of mesenchymal stem cells (MSCs) in animal models and implied role of exosomes following systemic delivery of MSCs: a systematic review. Am J Transl Res. (2022) 14:2147–61.
162. Zhang W, Feng J, Cinquina A, Wang Q, Xu H, Zhang Q, et al. Treatment of systemic lupus erythematosus using BCMA-CD19 compound CAR. Stem Cell Rev Rep. (2021) 17:2120–3. doi: 10.1007/s12015-021-10251-6
163. Kim SH and Lee SH. Updates on ankylosing spondylitis: pathogenesis and therapeutic agents. J Rheum Dis. (2023) 30:220–33. doi: 10.4078/jrd.2023.0041
Keywords: ankylosing spondylitis, mesenchymal stem cells, regenerative medicine, chimeric antigen receptor T-cell therapy, clinical progress, autoimmune inflammation
Citation: Ke M, Liu W, Lu H, Pan X, Wu M, Qi N, Wang Z, Wu Y and Zhang F (2025) Breaking boundaries in ankylosing spondylitis: how innovative cell therapies reshape immunity, drive cutting-edge advances, and face future challenges. Front. Immunol. 16:1613502. doi: 10.3389/fimmu.2025.1613502
Received: 17 April 2025; Accepted: 23 June 2025;
Published: 11 July 2025; Corrected: 20 August 2025.
Edited by:
Martin Johannes Hoogduijn, Erasmus University Medical Center Rotterdam, NetherlandsReviewed by:
Lifei Liu, The People’s Hospital of Liaoning Province, ChinaXinzhe Feng, Second Military Medical University, China
Copyright © 2025 Ke, Liu, Lu, Pan, Wu, Qi, Wang, Wu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuehong Wu, d3V5dWVob25nMjAwM0AxNjMuY29t; Feng Zhang, emhhbmdmZW5nMjI2N0BtZS5jb20=