 Desh Raj1,2,3Amit K. Verma1,2,3Risha Mathur1,2,3Jenna Woody1,2,3Mahesh Kathania1,2,3*
Desh Raj1,2,3Amit K. Verma1,2,3Risha Mathur1,2,3Jenna Woody1,2,3Mahesh Kathania1,2,3* K. Venuprasad1,2,3*
K. Venuprasad1,2,3*- 1Department of Internal Medicine, UT Southwestern Medical Center, Dallas, TX, United States
- 2Department of Immunology, UT Southwestern Medical Center, Dallas, TX, United States
- 3Harold C. Simmons Comprehensive Cancer Center, UT Southwestern Medical Center, Dallas, TX, United States
Interleukin-17A (IL-17A) is a pro-inflammatory cytokine that plays a pivotal role in immune responses, particularly in the pathogenesis of various autoimmune diseases and infections. Recent advances have highlighted the significance of post-translational modifications, particularly ubiquitination, in regulating IL-17A expression and IL-17A receptor signaling pathways. Here, we summarize the intricate relationship between IL-17A and ubiquitination, exploring how ubiquitin-mediated processes influence IL-17A production, receptor signaling, and downstream effector functions. We provide insights into the potential therapeutic implications of targeting IL-17A and its ubiquitination pathways in inflammatory diseases and autoimmune disorders. A clear understanding of this relationship could pave the way for novel strategies in immune modulation, potentially enhancing management and treatment efficacy in various human diseases.
Introduction
Interleukin 17A (IL-17A) is critical in the host immune response against bacterial and fungal infections, especially at the mucosal surface (1, 2). However, dysregulated IL-17A expression is strongly linked to several human diseases, such as multiple sclerosis (MS), psoriasis, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and asthma (2). Its biological effects are mediated by activating various signaling pathways that regulate the transcription of target genes involved in inflammation, tissue remodeling, and the recruitment of immune cells (2).
Ubiquitination, a post-translational modification whereby ubiquitin moieties are covalently attached to target proteins, regulates numerous cellular processes, including protein degradation, signal transduction, and cellular localization (3). It involves a multi-enzymatic biochemical reaction in which the Ub-activating (E1) enzyme activates Ub, which is then transferred to the Ub-conjugating (E2) enzyme. The Ub-ligating (E3) enzymes facilitate the formation of the isopeptide bond between the Ub (C-terminus) and specific substrate lysine residues (3). The human genome is estimated to encode 2 E1s, nearly 50 E2s, and over 600 E3s. Each E3 recognizes a set of substrates that share one or more ubiquitination signals, and an individual E3 cooperates with one or a few E2s. The E3 ligases are divided into two different groups based on their functional domains: the homology to the E6-associated protein carboxyl terminus (HECT) type E3s and the fascinating new gene (RING) type E3s (3). The ubiquitin molecules in a polyubiquitin chain are generally linked through the K48 or K63-linked polyubiquitin chains; however, other lysine residues in a ubiquitin molecule have been shown to participate in linkage (3). Interestingly, the different types of polyubiquitin chains have different effects on the substrate. The function of E3 ubiquitin ligases is reversed by the action of deubiquitinating enzymes (DUBs) (4). They specifically cleave the isopeptide bonds between ubiquitin and the Lys residue within the ubiquitinated substrate (4). The dynamic interplay between ubiquitination and the signaling pathways that lead to IL-17 expression is an area of growing interest, offering insights into the fine-tuning of immune responses. Ubiquitin ligases and deubiquitinases modulate the expression of IL-17A and the stability and activity of IL-17A signaling components, thereby influencing the intensity and duration of IL-17A-mediated cellular responses.
Here, we summarize the current knowledge regarding the relationship between IL-17A and ubiquitination, highlighting how this axis regulates immune responses and the implications for therapeutic interventions in inflammatory diseases.
IL-17 and inflammation
The IL-17 family of cytokines consists of six members (IL-17A to IL-17F) which binds to IL-17 receptors (IL-17RA to IL-17RE) and implement their physiological functions (5). Among the IL-17 family members, IL-17A is the most studied and highly significant cytokine. The human IL-17A is synthesized as a 155-amino-acid precursor which is then posttranscriptional modified by cleave of 23-amino-acid signal peptide at the N-terminus which is followed by dimerization via disulfide bonds to generate mature homodimer of 35 kDa (6). Among the members of the IL-17 family, IL-17F is most similar to IL-17A, with 55% sequence homology (7). IL-17F forms homodimers or heterodimers with IL-17A and binds to IL-17 receptors for signal transduction.
IL-17A is predominantly expressed by CD4+ T helper cells (Th17); however, natural killer T cells, CD8+ T cells, γδ T cells, innate lymphoid cells (ILCs), dendritic cells, macrophages, and other cells also produce this cytokine (8). The differentiation of Th17 cells depends on the presence of proinflammatory Interleukin-6 (IL-6), Transforming growth factor-beta (TGF-β), and Interleukin 1β (IL-1β) (8, 9). Th17 differentiation requires the activation of the transcription factors signal transducer and activator of transcription 3 (STAT3) and retinoic acid-related orphan receptor gamma t (RORγt) (10). After the initial differentiation, Th17 expresses the IL-23 receptor (IL-23R) and requires IL-23 for their proliferation and survival. Although T-cell receptor (TCR) activation is necessary for CD4+ and CD8+ T-cell IL-17A synthesis, innate immune cells primarily produce IL-17A in the presence of IL-6 and IL-23 (11).
IL-17A is a potent proinflammatory cytokine that induces neutrophil and monocyte recruitment to the site of inflammation by inducing the expression of chemokine (C-X-C motif) ligand 1 (CXCL1), CXCL2, and CCL20 (8). IL-17A also promotes neutrophil differentiation via the production of granulocyte colony-stimulating factors (G-CSF) and monocyte chemoattractant protein-1 (MCP-1) by non-hematopoietic target cells (11). Il17a deficiencies in mice result in defective neutrophils, leading to increased susceptibility to extracellular pathogens, including the bacteria Klebsiella pneumoniae, Candida albicans, and Toxoplasma gondii (12). In addition, IL-17A regulates the expression of molecules with antimicrobial activity, such as β-defensins, calgranulins, and mucins. Defensins act as natural antibiotics in the lungs, skin, and gut. Another IL-17A target gene is Chemokine (C-C motif) ligand 20 (CCL20), a chemokine that recruits dendritic cells (DCs) and T cells, thereby providing a positive feedback loop for IL-17A amplification by recruiting Th17 cells to inflamed sites (12). However, IL-17A is not always protective against infections. In schistosomiasis, IL-17A stimulates a pathogenic inflammatory response that can be alleviated with antibodies to IL-17A (13). Elevated IL-17A levels are also associated with severe periodontal disease. Importantly, elevated IL-17A is strongly linked to autoimmune pathology. Increased IL-17A levels were found in RA, SLE, and psoriasis patients (11). Consistent results suggest a pathogenic role for IL-17A in various mouse models of autoimmune disease. Similarly, dysregulated IL-17A-mediated inflammation is linked to graft vs host disease and some cancers (14, 15).
Intracellular events of IL-17A expression
The differentiation of Th17 cells requires coordinated activation of T cells in the presence of TGF-β, IL-6, IL-1β, and IL-23 (8). TCR stimulation-induced phosphatidylinositol 3-kinase (PI3K), Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), Nuclear Factor of Activated T cells (NFAT), and Mitogen-activated protein (MAP) kinase pathways are involved in IL-17A production (10). RAR-related orphan receptor gamma t (RORγt), a member of the nuclear receptor family of proteins, is a key transcriptional factor for IL-17A expression (16). It has been demonstrated that cholesterol derivatives, including desmosterol and oxysterols, serve as natural ligands and activate RORγt (17, 18). Whereas 3-oxoLC, a bile acid synthesized from cholesterol, acts as an inhibitory ligand of RORγt (19). Further, Raftlin1, a lipid raft protein, was shown to recruit specific phospholipids to RORγt and promote the transcriptional activity of RORγt and IL-17A expression (20). RORγt binds to RORE sequences within the CNS2 of the Il17a gene and mediates Il17a transcription by controlling the chromatin remodeling (10). In addition to RORγt, p300 and JmjC domain-containing protein (JMJD)3 also bind to CNS2 and mediate permissive histone acetylation and remove repressive histone marker H3K27me3 (21). CNS2 interacts with the Il17a promoter to induce Il17a transcription (10, 22). Runt-related transcription factor (RUNX)1 also binds to the CNS2 region of Il17a promoter (23). RUNX1 binds to RORgt to enhance expression of Il17a (10, 24).
STAT3 (another transcription factor), activated by IL-6, is involved in IL-17A expression by binding to the the Il17a promoter (25). Additionally, JunB was found to colocalize with interferon regulatory factor (IRF)4, which is involved in IL17A expression (26, 27). IRF4 binds to the regulatory elements of the Il17a promoter, which are co-bound by BATF, an AP-1 factor (10, 27). KLF4, a Kruppel-like factor, is involved in IL-17A expression by directedly binding to the Il17a promoter independently of RORgt (28). A metabolic sensor, Hypoxia-inducible factor (HIF)-1α, associates with RORgt, and binds to hypoxia response element located in the proximal region of the Rorc promoter (29). This suggests a complex network of transcriptional regulators is involved in generating Th17 cells.
Ubiquitination in IL-17A expression
Ubiquitin(Ub) conjugation was initially thought to be involved in proteasomal degradation of misfolded proteins (30). However, increasing evidence shows a broader implication in multiple subcellular processes, including the localization of proteins withing the cytoplasm, nuclear translocation, protein-protein interactions, cell membrane receptor turnover, and gene expression. Predictably, IL-17A expression is also regulated by ubiquitin conjugation (Figure 1).
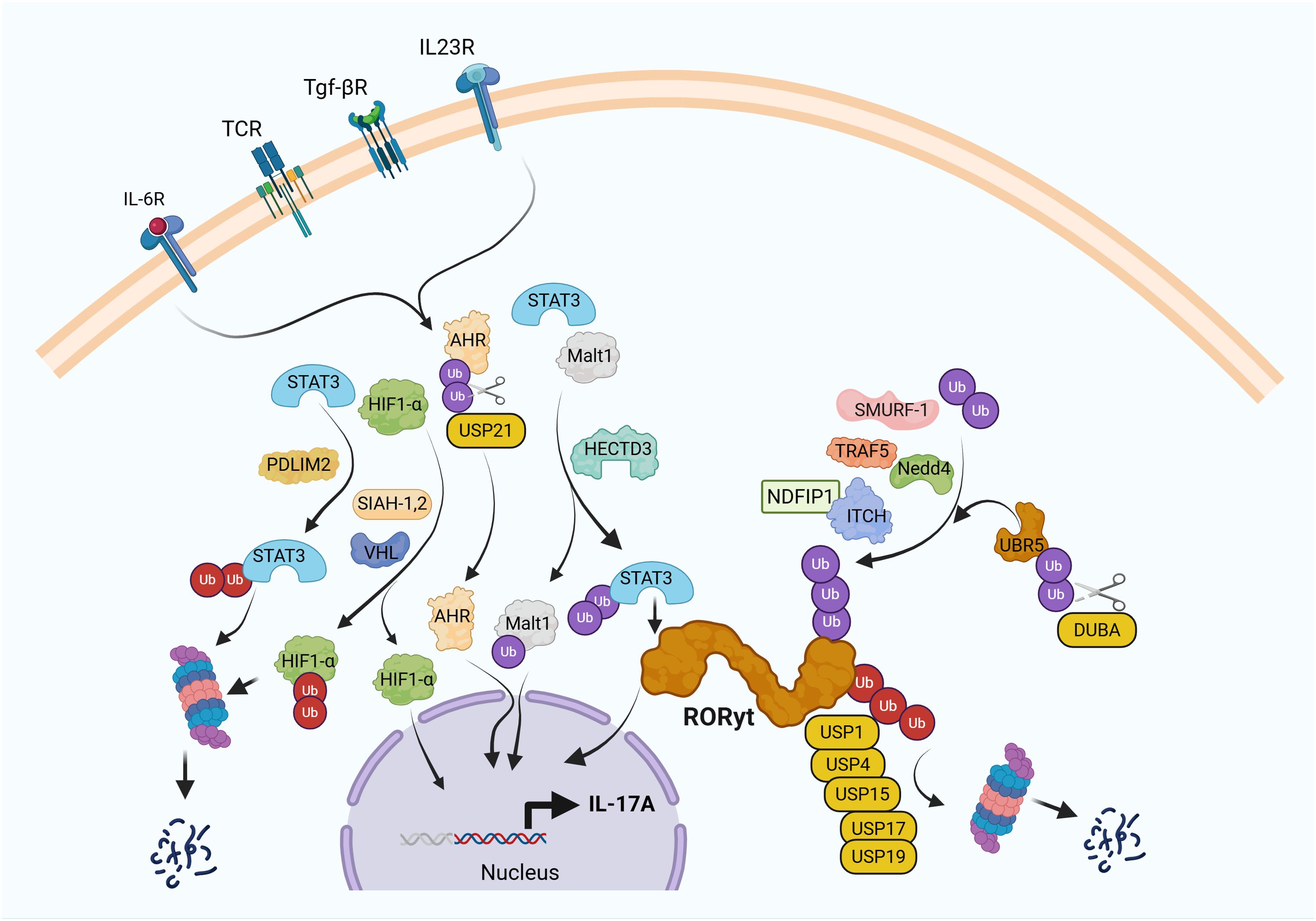
Figure 1. Ubiquitin pathway regulates IL-17A expression in Th17 cells. T cell receptor (TCR) ligation in the presence of IL-6, TGF-β, and IL-23 induces differentiation of naïve CD4+ T cells into Th17 cells through activation of key transcription factors, including RORγt, aryl hydrocarbon receptor (AHR), MALT1, and HIF-1α, which collectively drive IL-17A expression. Ubiquitin-mediated post-translational modifications tightly control this process. E3 ubiquitin ligases such as Itch and SMURF1 target RORγt for ubiquitination, thereby limiting chronic IL-17A production. In contrast, TRAF5 and NEDD4 promote IL-17A expression, supporting Th17 polarization. HECTD3 enhances Th17 pathogenicity by ubiquitinating STAT3 and MALT1, while SLIM/PDLIM2 facilitates proteasomal degradation of STAT3. SIAH1/2 stabilizes HIF-1α to promote Th17 differentiation, whereas the E3 ligase VHL paradoxically also contributes to Th17 development by ubiquitinating HIF-1α for degradation, reflecting the context-dependent roles of these factors. Deubiquitinases (DUBs) further fine-tune this regulatory network. DUBA interacts with UBR5 to modulate RORγt ubiquitination, acting as a negative regulator of IL-17A expression. Additional DUBs, including USP1, USP4, USP15, and USP17, contribute to the precise control of RORγt activity. USP21 stabilizes AHR, thereby indirectly enhancing Th17 differentiation. These findings underscore the complexity and specificity of ubiquitin-mediated regulation in IL-17A-driven immune responses, highlighting potential targets for therapeutic intervention in autoimmune diseases.
E3 Ligases and IL-17A
The PDZ-LIM domain protein PDLIM2, a nuclear ubiquitin E3 ligase, has been shown to inhibit Th17 cells by targeting STAT3 for polyubiquitination and proteasomal degradation (31, 32). Deficiency in PDLIM2 resulted in the accumulation of STAT3 in the nucleus, enhanced Th17 cell differentiation, and exacerbated IL-17A-mediated granuloma formation (32). whereas the E3 ubiquitin ligase HECTD3 promoted Th17 cells via non degradative K27-linked and K29-linked polyubiquitin chains on STAT3 and Malt1 (33). Hectd3-deficient mice exhibited reduced EAE severity and defective Th17 cell differentiation (33).
E3 ubiquitin protein ligase Itch regulates IL-17A production by ubiquitination of RORgt in Th17 cells (34). Itch recognizes the PPLY region on RORgt through its WW domain resulting in proteasomal degradation leading to inhibition of IL-17A expression. Itch deficiency resulted in spontaneous dermatitis and colitis, which was associated with elevated IL-17A expression (34). A defect in ITCH-mediated RORγt degradation was demonstrated in colorectal cancer (CRC) patients, where Colon rectal neoplasia differentially expressed(CRNDE-h ) protein was shown to associate with the conserved PPLY region within RORgt in Th17 cells infiltrated to tumors (35). CRNDE-h binding of to RORgt prevented ubiquitination of RORgt by blocking its binding to ITCH (35). Not surprisingly, a the percentage of Th17 cells among tumor-infiltrating lymphocytes (TILs) from CRC patients was positive correlated with CRNDE-h expression (35). This further supported the observed aggressive colon cancer growth in Itch−/− mice. This data further highlighted the involvement of ITCH in Th17-mediated tumor-promoting inflammation. Another report showed that Nedd4 targets RORγt for K27-linked ubiquitination, which promotes IL-17A, and Nedd4 deficiency resulted in attenuated IL-17A production and EAE (36). Further, HECTD3 family interacting protein 1 (Ndfip1), a co-activator of the E3 ubiquitin ligase Itch, attenuates the frequency and Th17 cells pathogenicity (37, 38). NDFIP1 binds to Itch and promotes its ligase activity in murine CD4+ T cells following TCR ligation via recruitment of ubiquitin-conjugating enzyme E2 (UBCH7) to Itch (39). Similar to Itch−/− Th17 cells, when adoptively transferred, Ndfip1 deficient Th17cells produced more IL-17A and induced severe colitis, indicating a pivotal role for the NDFIP1-ITCH pathway in the regulation of IL-17A-mediated inflammation (37, 38).
What triggers the Itch-mediated ubiquitination of RORγt? It was shown that p21-activated kinase 2 (Pak2), a serine (S)/threonine (T) kinase, was shown to recognize a conserved KRLS motif within RORγt and phosphorylates the S-316 within this motif (40). Pak2-mediated phosphorylation enhanced RORγt ubiquitination. The genetic deletion of Pak2 in Th17 cells reduces RORγt phosphorylation and increases IL-17A expression. Similarly, the reconstitution of RORγt-S316A mutant in Rorc−/− Th17 cells enhanced IL-17A expression due to reduced ubiquitination (40). In silico analysis of the modeled structure of RORγt showed that S316 makes an H-bond (3.6 Å) with a side-chain amino group of asparagine (N) 253 of the neighboring α-helix and stabilizes the ligand-binding domain (LBD) of RORγt, which reduces the accessibility of the PPLY motif. When S316 was substituted with phospho-mimetic aspartic acid residues (D) 316, the H-bond interaction between S316 and N253 was abolished, suggesting that phosphorylation provides increased accessibility of the PPLY motif of RORγt to ITCH (40). This suggested that a crosstalk between phosphorylation and ubiquitination plays a critical role in regulating the stability of RORγt and IL-17A expression. While Itch promoted degradation of RORγt, tumor necrosis factor receptor-associated factor 5 (TRAF5) interacts with and ubiquitinates RORγt via Lys-63-linked polyubiquitination (41). TRAF5 stabilizes the RORγt protein level depending on its RING finger domain. Depletion of TRAF5 in Th17 cells destabilizes RORγt protein and downregulates Th17-related genes, including Il17a (41).
Screening of a cDNA library to identify specific modulators for Il17a promoter activity led to the identification of the E3 ubiquitin ligases SIAH1 and SIAH2, as positive regulators of Il17a promoter activity in a T-cell line and promoted Th17 development ex vivo (42). This enhancement was a consequence of increased HIF-1α protein. Without HIF-1α, both ubiquitin ligases had little effect on Th17 cell differentiation (42). These results suggest that SIAH1 and SIAH2 play a pivotal role in promoting Th17 cell differentiation via the modulation of the stability of HIF-1α protein. Also, deletion of von Hippel-Lindau (VHL), an E3 ubiquitin ligase that targets HIF-1α, promoted Th17 differentiation (43). Mice deficient in VHL in their T cells were resistant to EAE. In the absence of VHL, Th17 cells had decreased activation of STAT3 and SMAD2 (43) (Table 1).
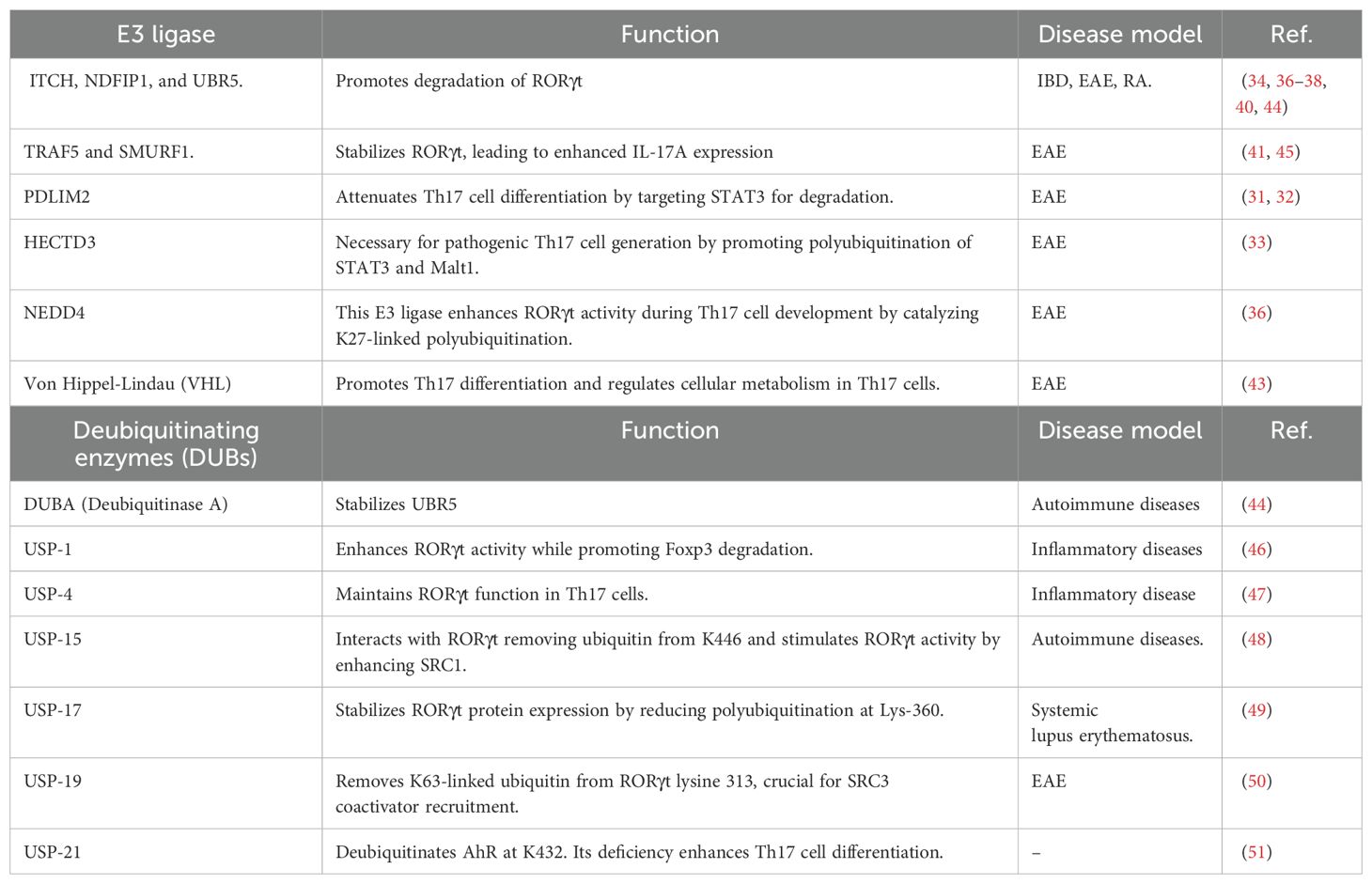
Table 1. List of E3 ligases and DUBs and their function in IL-17A expression.
DUBs and IL-17A
Ubiquitination is a highly dynamic and reversible process, and the removal of Ub chains bound to protein substrates is mediated by deubiquitinating enzymes (DUBs). DUBA is a deubiquitylating enzyme that negatively regulates IL-17A production in T cells (44). DUBA is associated with the UBR5 (a ubiquitin ligase), suppressing abundance of DUBA in naive T cells. Accumulated DUBA stabilized UBR5, which then ubiquitylated RORgt in response to TGF-β signaling in activated T cells (44). Th17 cells highly express the deubiquitinase ubiquitin-specific protease (USP)4, which is essential for maintaining RORγt and Th17 cell function. USP4 interacted and deubiquitinated K48-linked polyubiquitination of RORγt, thereby promoting RORγt function and Il17a transcription (47). Further, it was shown that USP17 stabilizes RORγt protein expression by reducing RORγt polyubiquitination at its Lys-360 residue (49). In contrast, knockdown of endogenous USP17 in Th17 cells resulted in decreased RORγt protein levels and downregulation of Th17-related genes. Furthermore, USP17 expression was upregulated in CD4+ T cells from systemic lupus erythematosus patients (49). USP19 was shown to suppress Th17 cells in vitro and Th17-mediated pathogenesis in vivo. Mechanistically, USP19 removed the K63-linked ubiquitin chain from RORγt lysine 313, which is essential for recruiting the coactivator SRC3 (50). In contrast, USP1 promoted Th17-cell differentiation by attenuating Treg-cell differentiation. USP1 in CD4+ T cells enhanced the activity of RORγt but promoted the proteasomal degradation of Foxp3 (46). USP15 interacts with RORγt and removes ubiquitin from K446, and stimulates RORγt activity by enhancing coactivator SRC1 recruitment. Knockdown of USP15 or expression of inactive USP15 impaired Th17 differentiation, suggesting a positive role for USP15-mediated deubiquitination of RORγt in Th17 differentiation (48). USP21 was shown to interact with and stabilize AhR by removing the K48-linked polyubiquitin chains from AhR (51). USP21 inhibits the transcriptional activity of AhR in a deubiquitinating-dependent manner. USP21 deubiquitinates at the K432 residue, and ubiquitination on this site is required for the transcriptional activity of AhR. Deficiency of USP21 enhanced the differentiation of Th17 cells in vitro and in vivo (51). The USP21-deficient T cells were more colitogenic upon adoptive transfer to Rag1−/− mice (51). Thus, IL-17A expression is tightly regulated by the ubiquitin pathway by targeting key signaling intermediates and transcription factors (Table 1).
Ubiquitination in IL-17A receptor signaling
Upon binding to the IL-17RA-IL-17RC receptor complex, IL-17A activates NF-κB to induce the expression of Il17a target genes (5). While IL-17RA is expressed ubiquitously, IL-17RC expression is restricted, which limits IL-17A signaling to epithelial and mesenchymal cells (5). The cytoplasmic tail of the IL-17R contains a conserved SEFIR domain (52). A SEFIR domain was also found in the adaptor protein Act1, which is implicated in the activation of NF-κB (52). Subsequent work showed that Act1 was recruited to IL-17RA in an IL-17-dependent manner. Act1 contains a tumor-necrosis factor receptor-associated factor (TRAF)-binding motif that recruits TRAF6 (53, 54). This results in K63-linked ubiquitination of TRAF6, activating the kinase TAK1 and NF-κB (53, 54). Unrestrained IL-17A signaling is prevented by K48-linked ubiquitination of Act1 by F-box E3 ubiquitin ligase β-TrCP (55). A20, a deubiquitinase, also fine-tunes IL-17A signaling (56). A20 is recruited via the CBAD to IL-17RA and removes the K63-linked ubiquitin chains on TRAF6, which tempers IL-17A signaling as a negative feedback mechanism (56). Similarly, the ubiquitin-specific protease USP25 was shown to deubiquitinate TRAF6 and prevent excessive IL-17A-induced signaling and IL-17A-dependent experimental autoimmune encephalomyelitis (EAE) (57). Thus, IL-17A-mediated inflammation is prevented by the ubiquitin pathway at multiple levels.
Conclusion
In conclusion, the regulation of IL-17-mediated inflammation by ubiquitination represents a critical layer of control in immune signaling, balancing host defense and the prevention of excessive inflammation. Ubiquitination modulates key components of IL-17A expression and signaling by targeting key signaling intermediates and transcription factors, such as RORγt and STAT3. This post-translational modification fine-tunes the intensity and duration of IL-17-driven responses, thereby shaping the overall immune milieu. Disruptions in this regulatory network are increasingly linked to the pathogenesis of autoimmune diseases, where unchecked IL-17 signaling contributes to chronic inflammation and tissue damage.
Targeting IL-17A and IL-17 receptors using antibodies (e.g., the IL-17 inhibitor secukinumab and the IL-17R inhibitor brodalumab) has achieved remarkable success in treating psoriasis (58). However, these agents have unexpectedly low efficacy in IL-17-related diseases such as rheumatoid arthritis (RA) and multiple sclerosis (MS). This was suggested to be due to autonomous activation of IL-17R signaling and resistance to IL-17-directed therapy (59). Moreover, there is a potential risk of systemic inactivation of IL-17A activity, which provides host defense and barrier function at mucosal surfaces (60–63). As a result, treatment with IL-17A inhibitors is linked to new-onset and exacerbations of inflammatory bowel disease and colitis (8).
The advancements in the understanding of ubiquitin-mediated regulation could aid in developing strategies to inhibit selective aspects of IL-17A-mediated inflammation in a site-specific manner. Targeted modulation of ubiquitin-related enzymes within the IL-17A pathway holds tremendous promise for the treatment of autoimmune disorders. Small molecules that block or promote interactions between E3 ligases and their substrates could be developed to dampen pathological IL-17 activity without broadly compromising host defense. Moreover, the development of proteolysis-targeting chimeras (PROTACs) presents an exciting avenue for increasing the specificity of substrate degradation, thereby enabling the selective removal of pro-inflammatory signaling proteins. These strategies reflect a shift toward precision immunomodulation, where leveraging the specificity of the ubiquitin system may yield next-generation therapeutics capable of restoring immune balance in autoimmune conditions. Overall, targeting the ubiquitin machinery within the IL-17 axis holds promise for precision immunomodulation, offering opportunities to mitigate chronic inflammation while preserving protective immunity.
Author contributions
DR: Writing – original draft. AV: Writing – review & editing. RM: Writing – review & editing. JW: Writing – review & editing. MK: Writing – original draft. KV: Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by funds from the National Institutes of Health (R01-AI155786, R01-CA266072, and 1R01-CA282143) and a translational pilot project grant from the Harold C. Simmons Comprehensive Cancer Center, UT Southwestern Medical Center (Project #00013516) to K. Venuprasad.
Acknowledgments
We thank Dr. Ezra Burstein for the helpful discussions and Dr. Wayne Lancaster for the critical reading of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Schnell A, Littman DR, and Kuchroo VK. T(H)17 cell heterogeneity and its role in tissue inflammation. Nat Immunol. (2023) 24:19–29. doi: 10.1038/s41590-022-01387-9
2. Patel DD and Kuchroo VK. Th17 cell pathway in human immunity: lessons from genetics and therapeutic interventions. Immunity. (2015) 43:1040–51. doi: 10.1016/j.immuni.2015.12.003
3. Venuprasad K, Zeng M, Baughan SL, and Massoumi R. Multifaceted role of the ubiquitin ligase Itch in immune regulation. Immunol Cell Biol. (2015) 93:452–60. doi: 10.1038/icb.2014.118
4. Dewson G, Eichhorn PJA, and Komander D. Deubiquitinases in cancer. Nat Rev Cancer. (2023) 23:842–62. doi: 10.1038/s41568-023-00633-y
5. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. (2009) 9:556–67. doi: 10.1038/nri2586
6. Moseley TA, Haudenschild DR, Rose L, and Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. (2003) 14:155–74. doi: 10.1016/S1359-6101(03)00002-9
7. Tong Z, Yang XO, Yan H, Liu W, Niu X, Shi Y, et al. A protective role by interleukin-17F in colon tumorigenesis. PloS One. (2012) 7:e34959. doi: 10.1371/journal.pone.0034959
8. Kumar R, Theiss AL, and Venuprasad K. RORgammat protein modifications and IL-17-mediated inflammation. Trends Immunol. (2021) 42:1037–50. doi: 10.1016/j.it.2021.09.005
9. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
10. Capone A and Volpe E. Transcriptional regulators of T helper 17 cell differentiation in health and autoimmune diseases. Front Immunol. (2020) 11:348. doi: 10.3389/fimmu.2020.00348
11. Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol. (2023) 23:38–54. doi: 10.1038/s41577-022-00746-9
12. McGeachy MJ, Cua DJ, and Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. (2019) 50:892–906. doi: 10.1016/j.immuni.2019.03.021
13. Mbow M, Larkin BM, Meurs L, Wammes LJ, de Jong SE, Labuda LA, et al. T-helper 17 cells are associated with pathology in human schistosomiasis. J Infect Dis. (2013) 207:186–95. doi: 10.1093/infdis/jis654
14. Malard F, Gaugler B, Lamarthee B, and Mohty M. Translational opportunities for targeting the Th17 axis in acute graft-vs.-host disease. Mucosal Immunol. (2016) 9:299–308. doi: 10.1038/mi.2015.143
15. Zhang X, Li B, Lan T, Chiari C, Ye X, Wang K, et al. The role of interleukin-17 in inflammation-related cancers. Front Immunol. (2024) 15:1479505. doi: 10.3389/fimmu.2024.1479505
16. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
17. Soroosh P, Wu J, Xue X, Song J, Sutton SW, Sablad M, et al. Oxysterols are agonist ligands of RORgammat and drive Th17 cell differentiation. Proc Natl Acad Sci U.S.A. (2014) 111:12163–8. doi: 10.1073/pnas.1322807111
18. Santori FR, Huang P, van de Pavert SA, Douglass EF Jr., Leaver DJ, Haubrich BA, et al. Identification of natural RORgamma ligands that regulate the development of lymphoid cells. Cell Metab. (2015) 21:286–98. doi: 10.1016/j.cmet.2015.01.004
19. Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature. (2019) 576:143–8. doi: 10.1038/s41586-019-1785-z
20. Singh AK, Kumar R, Yin J, Brooks Ii JF, Kathania M, Mukherjee S, et al. RORgammat-Raftlin1 complex regulates the pathogenicity of Th17 cells and colonic inflammation. Nat Commun. (2023) 14:4972. doi: 10.1038/s41467-023-40622-1
21. Liu X, Wang L, Zhao K, Thompson PR, Hwang Y, Marmorstein R, et al. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. (2008) 451:846–50. doi: 10.1038/nature06546
22. Wang X, Zhang Y, Yang XO, Nurieva RI, Chang SH, Ojeda SS, et al. Transcription of Il17 and Il17f is controlled by conserved noncoding sequence 2. Immunity. (2012) 36:23–31. doi: 10.1016/j.immuni.2011.10.019
23. Liu HP, Cao AT, Feng T, Li Q, Zhang W, Yao S, et al. TGF-beta converts Th1 cells into Th17 cells through stimulation of Runx1 expression. Eur J Immunol. (2015) 45:1010–8. doi: 10.1002/eji.201444726
24. Zhang F, Meng G, and Strober W. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol. (2008) 9:1297–306. doi: 10.1038/ni.1663
25. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. (2010) 32:605–15. doi: 10.1016/j.immuni.2010.05.003
26. Brustle A, Heink S, Huber M, Rosenplanter C, Stadelmann C, Yu P, et al. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol. (2007) 8:958–66. doi: 10.1038/ni1500
27. Hasan Z, Koizumi SI, Sasaki D, Yamada H, Arakaki N, Fujihara Y, et al. JunB is essential for IL-23-dependent pathogenicity of Th17 cells. Nat Commun. (2017) 8:15628. doi: 10.1038/ncomms15628
28. Lebson L, Gocke A, Rosenzweig J, Alder J, Civin C, Calabresi PA, et al. Cutting edge: The transcription factor Kruppel-like factor 4 regulates the differentiation of Th17 cells independently of RORgammat. J Immunol. (2010) 185:7161–4. doi: 10.4049/jimmunol.1002750
29. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. (2011) 146:772–84. doi: 10.1016/j.cell.2011.07.033
30. Venuprasad K, Yang C, Shao Y, Demydenko D, Harada Y, Jeon MS, et al. Immune regulation by ubiquitin conjugation. Adv Exp Med Biol. (2006) 584:207–17. doi: 10.1007/0-387-34132-3_15
31. Qu Z, Fu J, Ma H, Zhou J, Jin M, Mapara MY, et al. PDLIM2 restricts Th1 and Th17 differentiation and prevents autoimmune disease. Cell Biosci. (2012) 2:23. doi: 10.1186/2045-3701-2-23
32. Tanaka T, Yamamoto Y, Muromoto R, Ikeda O, Sekine Y, Grusby MJ, et al. PDLIM2 inhibits T helper 17 cell development and granulomatous inflammation through degradation of STAT3. Sci Signal. (2011) 4:ra85. doi: 10.1126/scisignal.2001637
33. Cho JJ, Xu Z, Parthasarathy U, Drashansky TT, Helm EY, Zuniga AN, et al. Hectd3 promotes pathogenic Th17 lineage through Stat3 activation and Malt1 signaling in neuroinflammation. Nat Commun. (2019) 10:701. doi: 10.1038/s41467-019-08605-3
34. Kathania M, Khare P, Zeng M, Cantarel B, Zhang H, Ueno H, et al. Itch inhibits IL-17-mediated colon inflammation and tumorigenesis by ROR-gammat ubiquitination. Nat Immunol. (2016) 17:997–1004. doi: 10.1038/ni.3488
35. Sun J, Jia H, Bao X, Wu Y, Zhu T, Li R, et al. Tumor exosome promotes Th17 cell differentiation by transmitting the lncRNA CRNDE-h in colorectal cancer. Cell Death Dis. (2021) 12:123. doi: 10.1038/s41419-020-03376-y
36. Zeng Q, Guo H, Tang N, Renavikar PS, Karandikar NJ, Lovett-Racke AE, et al. K27-linked RORgammat ubiquitination by Nedd4 potentiates Th17-mediated autoimmunity. J BioMed Sci. (2025) 32:26. doi: 10.1186/s12929-025-01120-2
37. Layman AAK, Sprout SL, Phillips D, and Oliver PM. Ndfip1 restricts Th17 cell potency by limiting lineage stability and proinflammatory cytokine production. Sci Rep. (2017) 7:39649. doi: 10.1038/srep39649
38. Ramon HE, Beal AM, Liu Y, Worthen GS, and Oliver PM. The E3 ubiquitin ligase adaptor Ndfip1 regulates Th17 differentiation by limiting the production of proinflammatory cytokines. J Immunol. (2012) 188:4023–31. doi: 10.4049/jimmunol.1102779
39. Oliver PM, Cao X, Worthen GS, Shi P, Briones N, MacLeod M, et al. Ndfip1 protein promotes the function of itch ubiquitin ligase to prevent T cell activation and T helper 2 cell-mediated inflammation. Immunity. (2006) 25:929–40. doi: 10.1016/j.immuni.2006.10.012
40. Kathania M, Kumar R, Lenou ET, Basrur V, Theiss AL, Chernoff J, et al. Pak2-mediated phosphorylation promotes RORgammat ubiquitination and inhibits colonic inflammation. Cell Rep. (2022) 40:111345. doi: 10.1016/j.celrep.2022.111345
41. Wang X, Yang J, Han L, Zhao K, Wu Q, Bao L, et al. TRAF5-mediated lys-63-linked polyubiquitination plays an essential role in positive regulation of RORgammat in promoting IL-17A expression. J Biol Chem. (2015) 290:29086–94. doi: 10.1074/jbc.M115.664573
42. Matsui-Hasumi A, Sato Y, Uto-Konomi A, Yamashita S, Uehori J, Yoshimura A, et al. E3 ubiquitin ligases SIAH1/2 regulate hypoxia-inducible factor-1 (HIF-1)-mediated Th17 cell differentiation. Int Immunol. (2017) 29:133–43. doi: 10.1093/intimm/dxx014
43. Chitrakar A, Budda SA, Henderson JG, Axtell RC, and Zenewicz LA. E3 ubiquitin ligase von hippel-lindau protein promotes th17 differentiation. J Immunol. (2020) 205:1009–23. doi: 10.4049/jimmunol.2000243
44. Rutz S, Kayagaki N, Phung QT, Eidenschenk C, Noubade R, Wang X, et al. Deubiquitinase DUBA is a post-translational brake on interleukin-17 production in T cells. Nature. (2015) 518:417–21. doi: 10.1038/nature13979
45. Zhong W, Feng L, Tian W, Qu H, Xu H, Ning K, et al. SMURF1 inhibits the Th17 and Th17.1 polarization and improves the Treg/Th17 imbalance in systemic lupus erythematosus through the ubiquitination of RORgammat. Mol Immunol. (2023) 157:186–94. doi: 10.1016/j.molimm.2023.03.024
46. Zhu X, Wang P, Zhan X, Zhang Y, Sheng J, He S, et al. USP1-regulated reciprocal differentiation of Th17 cells and Treg cells by deubiquitinating and stabilizing TAZ. Cell Mol Immunol. (2023) 20:252–63. doi: 10.1038/s41423-022-00969-9
47. Yang J, Xu P, Han L, Guo Z, Wang X, Chen Z, et al. Cutting edge: Ubiquitin-specific protease 4 promotes Th17 cell function under inflammation by deubiquitinating and stabilizing RORgammat. J Immunol. (2015) 194:4094–7. doi: 10.4049/jimmunol.1401451
48. He Z, Wang F, Ma J, Sen S, Zhang J, Gwack Y, et al. Ubiquitination of RORgammat at lysine 446 limits th17 differentiation by controlling coactivator recruitment. J Immunol. (2016) 197:1148–58. doi: 10.4049/jimmunol.1600548
49. Han L, Yang J, Wang X, Wu Q, Yin S, Li Z, et al. The E3 deubiquitinase USP17 is a positive regulator of retinoic acid-related orphan nuclear receptor gammat (RORgammat) in Th17 cells. J Biol Chem. (2014) 289:25546–55. doi: 10.1074/jbc.M114.565291
50. Zhang J, Bouch RJ, Blekhman MG, and He Z. USP19 suppresses th17-driven pathogenesis in autoimmunity. J Immunol. (2021) 207:23–33. doi: 10.4049/jimmunol.2100205
51. Wang L, Cheng H, Wang X, Zhu F, Tian N, Xu Z, et al. Deubiquitination of aryl hydrocarbon receptor by USP21 negatively regulates T helper 17 cell differentiation. J Leukoc Biol. (2024) 117. doi: 10.1093/jleuko/qiae148
52. Zhang B, Liu C, Qian W, Han Y, Li X, and Deng J. Crystal structure of IL-17 receptor B SEFIR domain. J Immunol. (2013) 190:2320–6. doi: 10.4049/jimmunol.1202922
53. Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. (2007) 8:247–56. doi: 10.1038/ni1439
54. Liu C, Qian W, Qian Y, Giltiay NV, Lu Y, Swaidani S, et al. Act1, a U-box E3 ubiquitin ligase for IL-17 signaling. Sci Signal. (2009) 2:ra63. doi: 10.1126/scisignal.2000382
55. Shi P, Zhu S, Lin Y, Liu Y, Liu Y, Chen Z, et al. Persistent stimulation with interleukin-17 desensitizes cells through SCFbeta-TrCP-mediated degradation of Act1. Sci Signal. (2011) 4:ra73. doi: 10.1126/scisignal.2001653
56. Garg AV, Ahmed M, Vallejo AN, Ma A, and Gaffen SL. The deubiquitinase A20 mediates feedback inhibition of interleukin-17 receptor signaling. Sci Signal. (2013) 6:ra44. doi: 10.1126/scisignal.2003699
57. Zhong B, Liu X, Wang X, Chang SH, Liu X, Wang A, et al. Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat Immunol. (2012) 13:1110–7. doi: 10.1038/ni.2427
58. von Stebut E, Boehncke WH, Ghoreschi K, Gori T, Kaya Z, Thaci D, et al. IL-17A in psoriasis and beyond: cardiovascular and metabolic implications. Front Immunol. (2019) 10:3096. doi: 10.3389/fimmu.2019.03096
59. Luo Q, Liu Y, Shi K, Shen X, Yang Y, Liang X, et al. An autonomous activation of interleukin-17 receptor signaling sustains inflammation and promotes disease progression. Immunity. (2023) 56:2006–2020 e6. doi: 10.1016/j.immuni.2023.06.012
60. Deng Z, Wang S, Wu C, and Wang C. IL-17 inhibitor-associated inflammatory bowel disease: A study based on literature and database analysis. Front Pharmacol. (2023) 14:1124628. doi: 10.3389/fphar.2023.1124628
61. Grumme L, Dombret S, Knosel T, Skapenko A, and Schulze-Koops H. Colitis induced by IL-17A-inhibitors. Clin J Gastroenterol. (2024) 17:263–70. doi: 10.1007/s12328-023-01893-9
62. Ju J, Dai Y, Yang J, Liu C, Fan L, Feng L, et al. Crohn’s disease exacerbated by IL-17 inhibitors in patients with psoriasis: a case report. BMC Gastroenterol. (2020) 20:340. doi: 10.1186/s12876-020-01474-x
Keywords: IL-17A, ubiquitination, deubiquitination, RORgt, E3 ligases
Citation: Raj D, Verma AK, Mathur R, Woody J, Kathania M and Venuprasad K (2025) Fine-tuning immunity: ubiquitin-dependent regulation of interleukin-17A expression by Th17 cells. Front. Immunol. 16:1614409. doi: 10.3389/fimmu.2025.1614409
Received: 18 April 2025; Accepted: 25 June 2025;
Published: 01 August 2025.
Edited by:
Noah Isakov, Ben-Gurion University of the Negev, IsraelReviewed by:
Choong-Hyun Koh, Seoul National University, Republic of KoreaQiao Cheng, The First Affiliated Hospital of Soochow University, China
Copyright © 2025 Raj, Verma, Mathur, Woody, Kathania and Venuprasad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mahesh Kathania, bWFoZXNoLmthdGhhbmlhQHV0c291dGh3ZXN0ZXJuLmVkdQ==; K. Venuprasad, dmVudXByYXNhZC5wb29qYXJ5QFVUU291dGh3ZXN0ZXJuLmVkdQ==