 Yu ZhangHaixia GuanXixuan FengMengyan LiuJinhuan ShaoMengchi LiuJialei HeYahui Jin*Jinglin Zhu*
Yu ZhangHaixia GuanXixuan FengMengyan LiuJinhuan ShaoMengchi LiuJialei HeYahui Jin*Jinglin Zhu* Chunli Zheng*
Chunli Zheng*- Yan’an Medical College, Yan’an University, Yan’an, Shaanxi, China
Colorectal cancer (CRC) is a prevalent malignancy of the digestive system, with metastatic CRC (mCRC) exhibiting persistently poor overall survival rates. Consequently, there is an urgent need to develop more effective and safer therapeutic strategies. In recent years, immunotherapy has emerged as a groundbreaking approach in CRC treatment. This review highlights the advancements in immune checkpoint Inhibitors (ICIs), cancer vaccines, oncolytic virotherapy, adoptive cell therapy(ACT), and matrix-depletion therapy. Additionally, we explore potential combinatorial immunotherapy strategies for CRC, emphasizing their clinical applications and addressing the challenges associated with CRC immunotherapy. By proposing strategies to overcome these limitations, this review aims to provide novel insights into the evolving landscape of CRC immunotherapy.
1 Introduction
CRC ranks as the third most common cancer globally and is the second leading cause of cancer-related mortality. Despite significant advances in diagnostic methods and treatment options, the prognosis for patients with mCRC continues to be poor, particularly for those diagnosed with late-stage disease. While early-stage CRC (stage I) exhibits a 5-year survival rate of 91%, it dramatically declines to approximately 14% for patients with metastatic involvement (1), emphasizing the urgent need for more effective therapeutic strategies. Traditional treatment modalities such as surgery, radiotherapy, and chemotherapy have demonstrated limited efficacy in advanced CRC, primarily due to the inherent tumor heterogeneity, the emergence of resistance mechanisms, and the lack of durable responses (2–5). More recently, immunotherapy has emerged as a promising fourth pillar in cancer treatment, complementing conventional therapies and offering substantial benefits in specific patient populations (6–9). Among these, ICIs have revolutionized the treatment landscape for solid tumors, including CRC (10). Additionally, personalized therapeutic strategies such as cancer vaccines and oncolytic virotherapy are under active investigation (11, 12). Yet, the application of these approaches is associated with a range of benefits and drawback(Table 1).
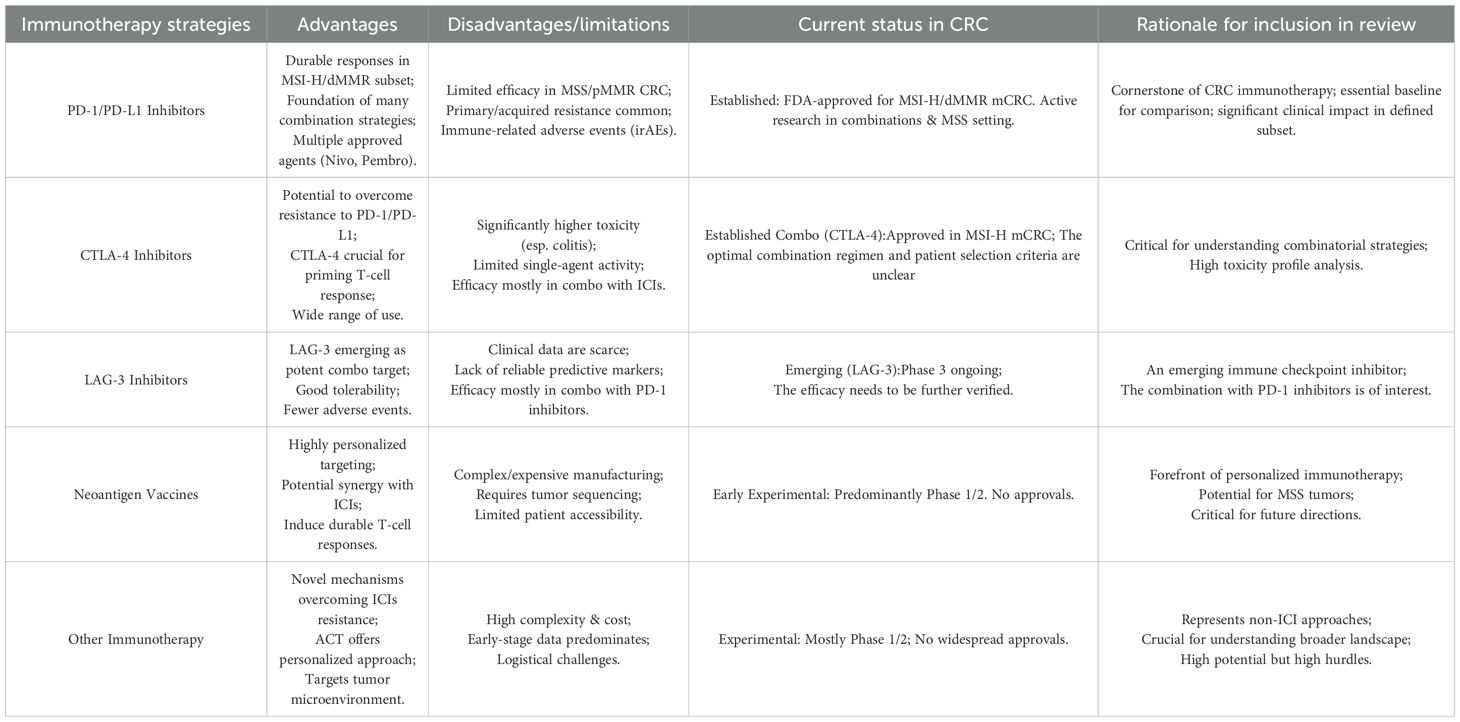
Table 1. Comparative analysis and rationale for included immunotherapy groups in colorectal cancer.
CRC has a unique tumor microenvironment (TME) compared to other cancer types, primarily due to microsatellite instability (MSI), which is often a phenotypic consequence of mismatch repair-deficient (dMMR) (13). In dMMR/MSI-H CRC, the TME is enriched with immune cells, particularly CD8+ T cells, driven by high tumor mutation burden (TMB) and neoantigen production (14, 15). These factors create an inflammatory TME, supporting the efficacy of ICIs like programmed death-1 (PD-1)/programmed death-ligand 1 (PD-L1) blockers. PD-L1 expression is also elevated in dMMR/MSI-H CRC, further enhancing ICIs effectiveness. However, immune suppressive elements, including Tregs, MDSCs, and factors like TGF-β and IL-10, can limit ICIs responses, causing resistance (13). Conversely, mismatch repair-proficient (pMMR)/MSS CRC shows sparse immune infiltration and low PD-L1 expression, leading to intrinsic ICIs resistance. Its TME may harbor more immunosuppressive factors, reducing immunotherapy efficacy (16). This contrast underscores the need for strategies to counteract the immunosuppressive TME, aiming to extend immunotherapy benefits to 85% of MSS CRC patients. Transforming “cold” into “hot” tumors mainly involves combining checkpoint inhibitors with treatments like radiotherapy, chemotherapy, and anti-angiogenic drugs. Other strategies include boosting tumor cell immunogenicity, enhancing antigen presentation, recruiting/activating immune cells, and reprogramming the TME. While early clinical trials show promise, challenges such as TME complexity and patient heterogeneity persist. Further research is crucial to optimize these approaches and enhance immunotherapy efficacy in a broader patient population.
In this review, we explore the molecular mechanisms underlying immune evasion in CRC, focusing on immune checkpoint inhibitors, cancer vaccines, oncolytic virotherapy, ACT, and matrix-depletion therapy (Figures 1, 2). We also discuss current challenges in CRC immunotherapy, including resistance mechanisms, limited clinical efficacy in MSS tumors, and the need for personalized treatment approaches. The review aims to provide a comprehensive overview of the current state of immunotherapy in CRC and highlight emerging strategies aimed at overcoming these barriers, ultimately improving patient outcomes.
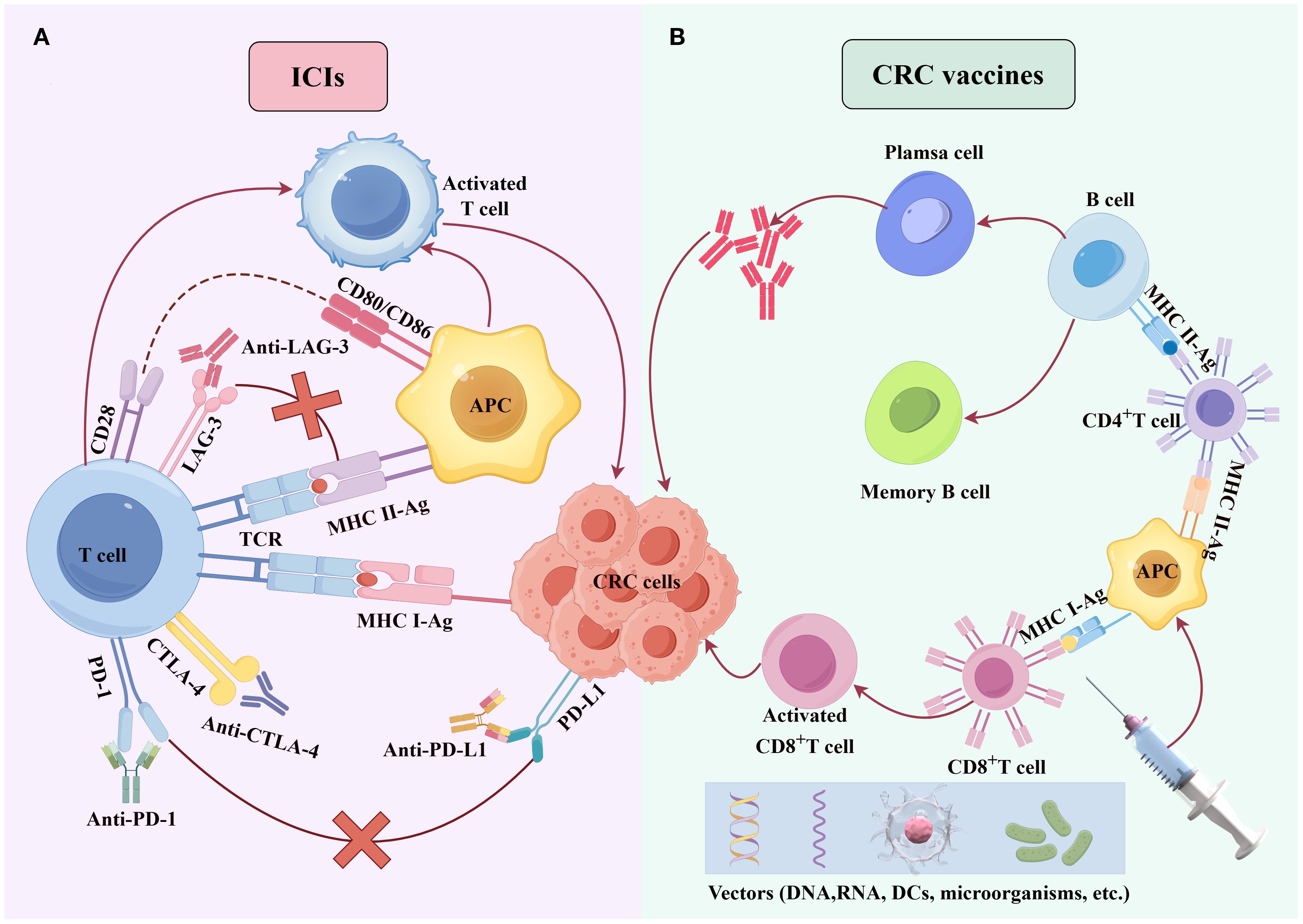
Figure 1. Molecular mechanisms of ICIs and cancer vaccines. (A) PD-1/PD-L1/LAG-3 inhibitors block the binding of PD-1/PD-L1/LAG-3 binding, counteracting CRC cell-mediated immune suppression of T cells and enhancing T-cell-mediated cytotoxicity against CRC cells. (B) Tumor antigens, delivered via diverse vectors (e.g., DNA, RNA, DCs, or microbes), are processed by dendritic cells, presented to T cells, and activate antigen-specific cytotoxic T cells to eliminate CRC cells.
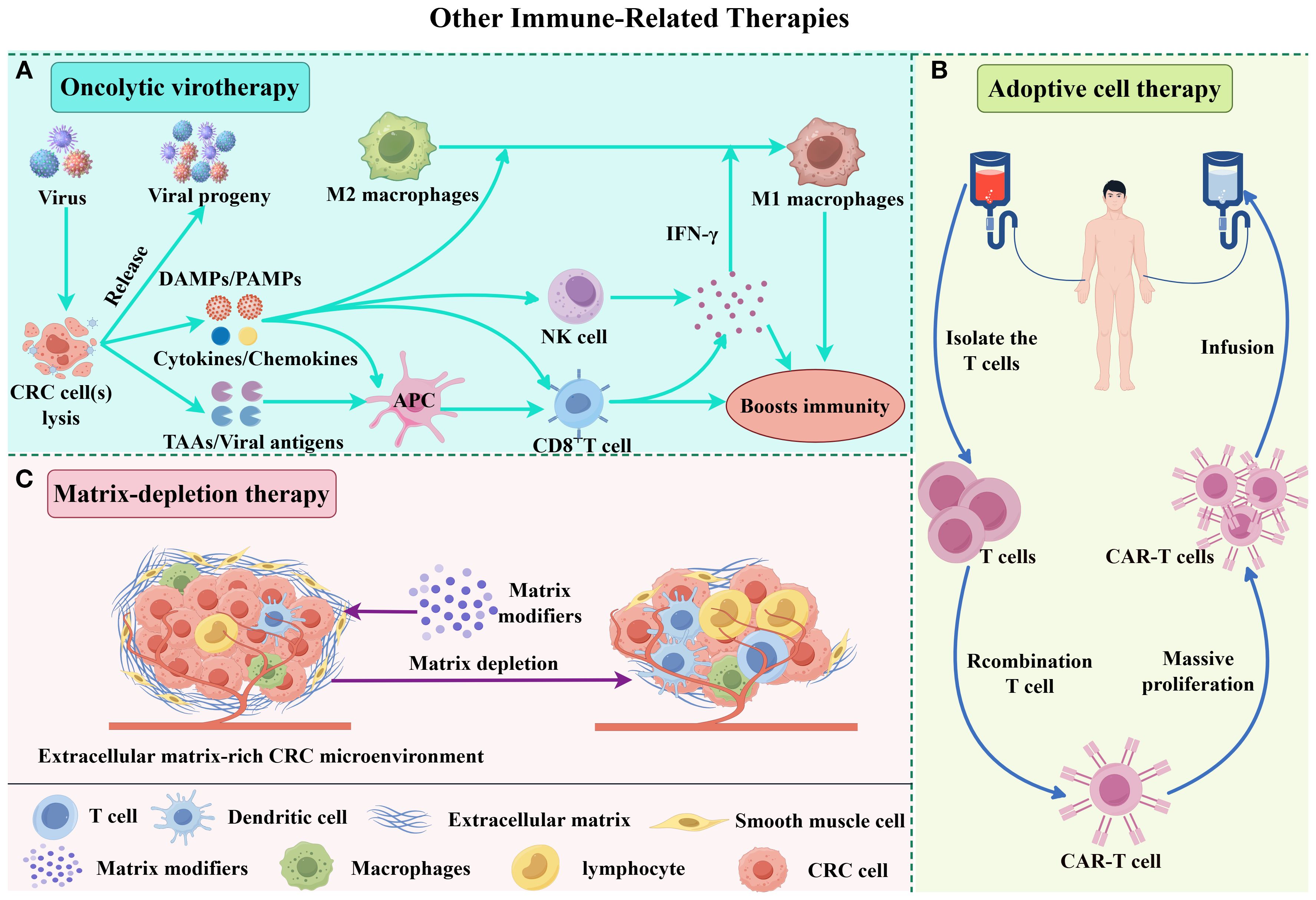
Figure 2. Molecular mechanisms of other immune-related combination therapies. (A) Upon infection by an oncolytic virus, CRC cells mount an antiviral response, secreting antiviral cytokines-particularly interferons (IFNs). These cytokines promote the maturation of antigen-presenting cells (APCs), such as dendritic cells (DCs), and stimulate CD8+ T cells and natural killer (NK) cells. As infected tumor cells lyse, they release viral progeny, damage-associated molecular patterns (DAMPs) like host cell proteins, pathogen-associated molecular patterns (PAMPs) from viral particles, and tumor-associated antigens (TAAs), including neoantigens. The viral progeny then go on to infect additional tumor cells. DAMPs and PAMPs activate the immune system via receptors like Toll-like receptors (TLRs). Meanwhile, APCs capture TAAs and neoantigens, triggering antigen- and virus-specific CD8+ T cell responses. This cascade creates an immune-stimulatory environment that drives tumor-supportive M2 macrophages to shift toward a pro-inflammatory M1 phenotype. (B) ACT involves isolating autologous T/NK cells, ex vivo expansion and/or genetic engineering to enhance tumor specificity, followed by reinfusion. These activated lymphocytes selectively recognize and eliminate CRC cells. (C) Matrix modulators deplete extracellular matrix components, disrupting the tumor microenvironment to enhance immune cell infiltration and drug penetration, thereby potentiating therapeutic efficacy.
2 ICIs
ICIs represent a paradigm shift in cancer immunotherapy, functioning by reversing immune suppression mediated by checkpoint molecules and reactivating T-cell-mediated antitumor responses (17, 18). The PD-1/PD-L1 axis and cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) have emerged as key targets in the treatment of various cancers, including CRC, demonstrating significant clinical efficacy (19, 20). More recently, lymphocyte activation gene 3 (LAG-3) has emerged as a next-generation immune checkpoint, offering promising potential for advancing cancer therapy (21).
PD-1, CTLA-4, and LAG-3 serve as negative regulators of T-cell activation and migration, playing pivotal roles in immune checkpoint blockade (10, 22). As such, inhibitors targeting these checkpoints have been pivotal in immune checkpoint blockade strategies, significantly enhancing immune-mediated tumor clearance and inducing durable responses across a variety of solid tumors and hematologic malignancies (23, 24). Notably, combination therapies involving LAG-3 inhibitors alongside PD-1/PD-L1 blockade have shown synergistic effects, further enhancing therapeutic outcomes in preclinical and clinical settings (25–28). Despite these promising developments, the clinical application of these inhibitors is not without challenges. Although PD−1/PD−L1 or CTLA−4 blockade significantly prolongs overall survival in patients with PD−L1–positive advanced non–small cell lung cancer (NSCLC), unresectable stage III–IV melanoma, and CRC—with the greatest benefit observed in dMMR/MSI−H CRC—these therapies can also induce immune−related adverse events (irAEs) in a subset of patients, potentially limiting their broader applicability (29–32). Furthermore, the development of resistance to these therapies remains a significant hurdle, with some patients exhibiting primary or acquired resistance (33, 34), underscoring the need for more personalized treatment strategies that consider individual tumor profiles and immune landscape variations. This section aims to provide a detailed overview of the mechanisms of action, clinical applications, and current limitations of PD-1/PD-L1, CTLA-4, and LAG-3 inhibitors, as well as the strategies being explored to overcome these barriers.
2.1 Anti-PD-1 and anti-PD-L1 inhibitors
PD−1 is an inhibitory receptor of the B7−CD28 family, expressed on activated T cells, B cells, and a subset of NK cells. Upon engagement by PD−L1, tyrosine residues within PD−1’s ITSM and ITIM motifs are phosphorylated, creating docking sites for SHP2. In its basal state, SHP2’s N−SH2 domain occludes its PTP catalytic pocket; ligand−induced phosphotyrosine binding displaces N−SH2, unleashing phosphatase activity (35). Activated SHP2 preferentially dephosphorylates the co−stimulatory receptor CD28—rather than TCR components such as CD3ζ or ZAP70—thereby attenuating downstream PI3K/Akt signaling, IL−2 production, and T−cell proliferation (36). In myeloid cells, PD−1–SHP2 signaling also impairs phosphorylation of transcription factors IRF8 and HOXA10, inhibiting monocyte differentiation and antigen presentation; intriguingly, SHP2 appears dispensable for PD−1–mediated T−cell exhaustion in chronic viral infection models, underscoring context−dependent pathway regulation (37, 38). Furthermore, numerous studies have established a correlation between PD-L1 upregulation and poor prognosis in CRC (39–41). PD-1/PD-L1 inhibitors counteract this immune suppression by blocking the PD-1/PD-L1 pathway, thereby enhancing T-cell-mediated antitumor immunity. To date, the U.S. Food and Drug Administration (FDA) has approved several PD-1 (Table 2) /PD-L1(Table 3) inhibitors, including pembrolizumab, nivolumab, cemiplimab, and avelumab, for the treatment of various malignancies (42).
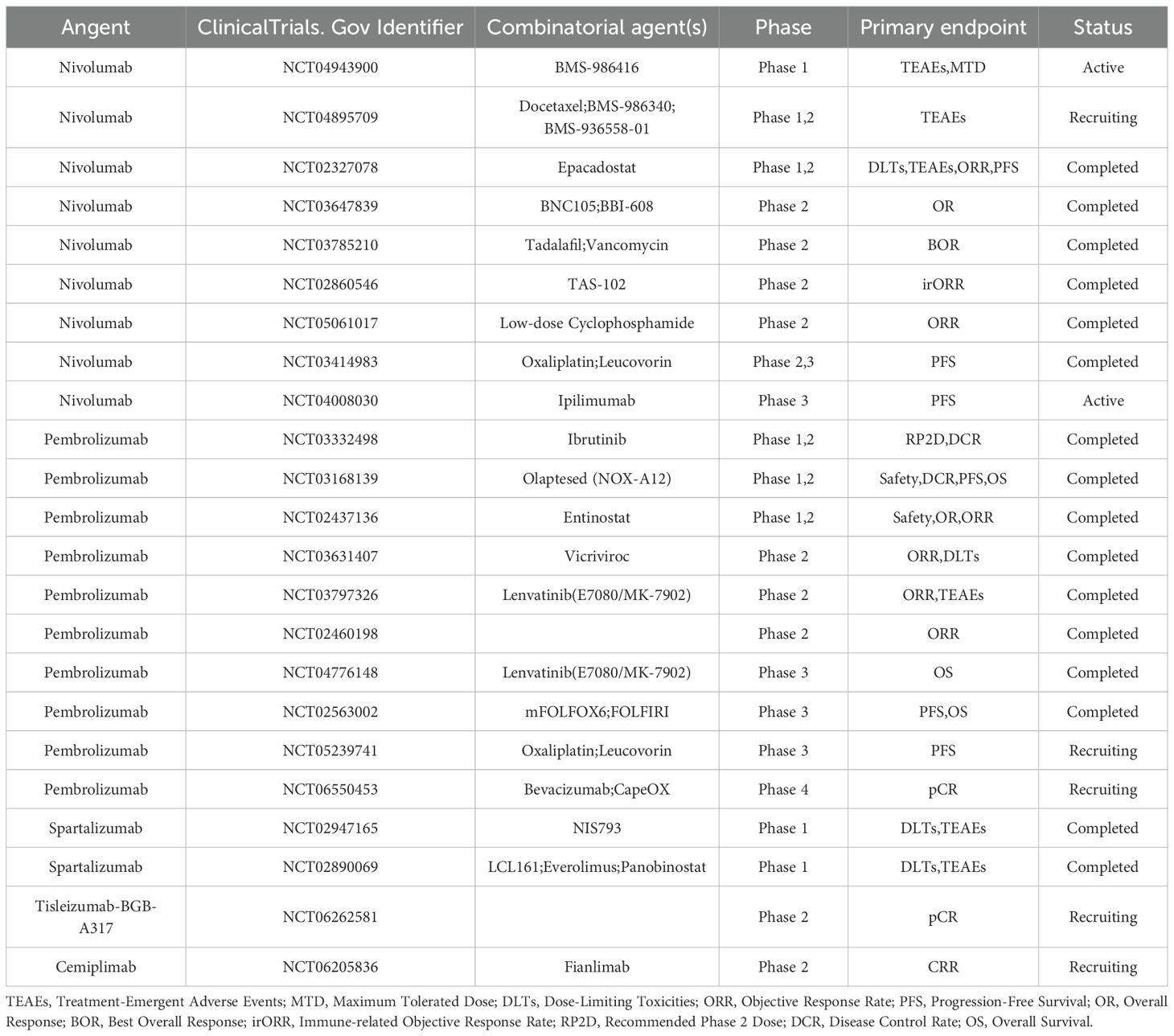
Table 2. Clinical trials based on PD-1 inhibitors in the treatment of colorectal cancer.
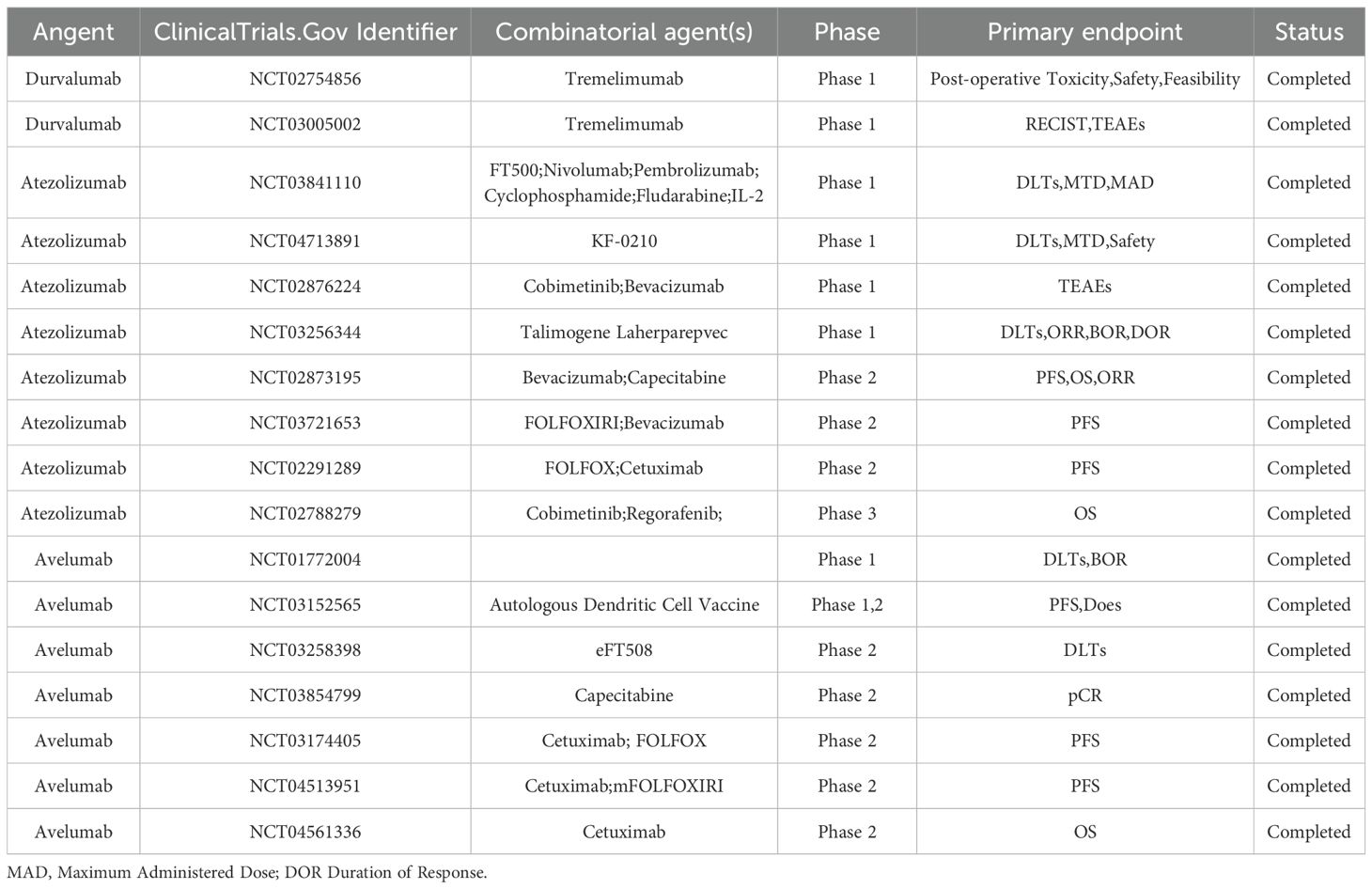
Table 3. Clinical trials based on PD-L1 inhibitors in the treatment of colorectal cancer.
The KEYNOTE-177 trial, a landmark international Phase III study, has drawn significant attention for its evaluation of pembrolizumab versus chemotherapy in patients with dMMR/MSI-H mCRC (32). The primary endpoints were OS and PFS. In the final analysis, the median PFS was 16.5 months for pembrolizumab monotherapy compared to 8.2 months for chemotherapy, although no statistically significant difference in OS was observed between the two groups (32). The favorable safety profile of pembrolizumab, as demonstrated in prior trials, further supports its clinical utility (43). These findings underscore the superiority of pembrolizumab as a first-line treatment for dMMR/MSI-H mCRC, offering prolonged PFS and reduced adverse events compared to conventional chemotherapy. Consequently, the FDA approved pembrolizumab for this indication, marking a significant milestone in CRC immunotherapy.
The Phase II CheckMate 142 trial evaluated the efficacy of nivolumab combined with relatlimab, a LAG-3 inhibitor, in patients with dMMR/MSI-H mCRC (44). After a median follow-up of 47.4 months, the ORR, DCR, median PFS, and 2-year PFS rate were 50%, 70%, 27.5 months, and 51%, respectively (44). Compared to pembrolizumab or nivolumab monotherapy, the combination of nivolumab and relatlimab yielded a higher ORR and a longer investigator-assessed PFS, highlighting the durable clinical benefits of this dual checkpoint blockade (43, 45). These results suggest that combining nivolumab with relatlimab represents a promising therapeutic strategy for dMMR/MSI-H mCRC.
Notably, two neoadjuvant trials illustrate the potential for combining immune checkpoint blockade with other modalities. In the randomized phase II PICC trial (NCT03926338), 34 patients with locally advanced dMMR/MSI-H CRC received neoadjuvant toripalimab either alone or alongside the COX-2 inhibitor celecoxib; the combination arm achieved an impressive pCR rate of 88% versus 65% with toripalimab alone, and no relapses were observed at a 12-month follow-up, with only 3% of patients experiencing grade ≥ 3 immune-related adverse events (46). In another ongoing phase II study (NCT04715633), 52 participants with dMMR/MSI-H CRC are being treated preoperatively with camrelizumab plus oral apatinib; interim analyses report an ORR of 42% and a favorable safety profile, with mature pCR and long−term outcomes eagerly awaited. Another ongoing phase II trial NCT06205836 is investigating the safety and efficacy of cemiplimab, both as a monotherapy and in combination with fianlimab, in patients aged 70 years and older with dMMR/MSI-H mCRC. The primary endpoint is the complete response rate (CRR), and the results are highly anticipated.
2.2 CTLA-4 inhibitors
Structurally, CTLA-4 has a high homology with CD28, but it has a greater affinity for B7 homologs than CD28: CTLA-4 has a monomeric affinity of about 0.2 μM for CD80 and a monomeric affinity of about 2 μM for CD86 (47). During transendocytosis, CD80 continues to bind to CTLA-4 and is endocytosed with CTLA-4 into advanced endosomes and lysosomes, inducing ubiquitination of lysine residues of CTLA-4, which ultimately leads to the degradation of CTLA-4, thereby reducing the amount of CTLA-4 on the cell surface, inhibiting the recycling of CTLA-4 and weakening its negative regulatory effect on the immune response (47). However, CD86 will rapidly dissociate and degrade after being captured by CTLA-4 and transendocytosis, while CTLA-4 will not be modified and can be recycled to the cell surface for further ligand capture, thus maintaining the negative regulatory function of CTLA-4 on the immune response to a certain extent (47). PD-L1 and CD80 can undergo cis-interaction within the cell, i.e., on the same cell surface, PD-L1 and CD80 can form heterodimers. This cis-interaction is able to inhibit the binding of PD-L1 to PD-1 and the interaction of CD80 with CTLA-4, thereby enhancing the immune response to some extent (48). In summary, CTLA-4 inhibitors may be used to treat CRC with a combination of drugs that modulate CD80 and CD86 expression, or in combination with PD-L1 inhibitors, may achieve better therapeutic outcomes (49). At the same time, according to the individual differences of patients (such as the expression levels of CD80, CD86 and PD-L1 on the surface of tumor cells), personalized combination treatment plans are formulated to improve the pertinence and effectiveness of treatment.
Leveraging the molecular choreography of CTLA-4, CD28, and PD-L1 offers a blueprint for precision therapy in CRC. Tumors with dominant CD80 signaling—readily stripped by CTLA-4 and routed to lysosomal oblivion—are primed for CTLA-4 blockade, whereas CD86-centric lesions, whose ligand dissociates and spares CTLA-4 recycling, may demand dual checkpoint inhibition to extinguish residual negative feedback. Concomitant quantification of PD-L1-CD80 cis-heterodimers on tumor cells stratifies patients further: where this inhibitory pair is abundant, adding a PD-L1 antagonist liberates both CD80 for CD28 co-stimulation and PD-L1 from PD-1 restraint, amplifying anti-tumor immunity. Thus, an integrative map of surface CD80, CD86, and PD-L1 densities—together with germline CTLA-4 variants that tune ligand avidity—can guide calibrated sequencing or combination of CTLA-4 and PD-L1 inhibitors, converting mechanistic insight into bespoke regimens that maximize efficacy while minimizing collateral toxicity. These clinical studies will provide key insights into the treatment of CRC (Table 4).
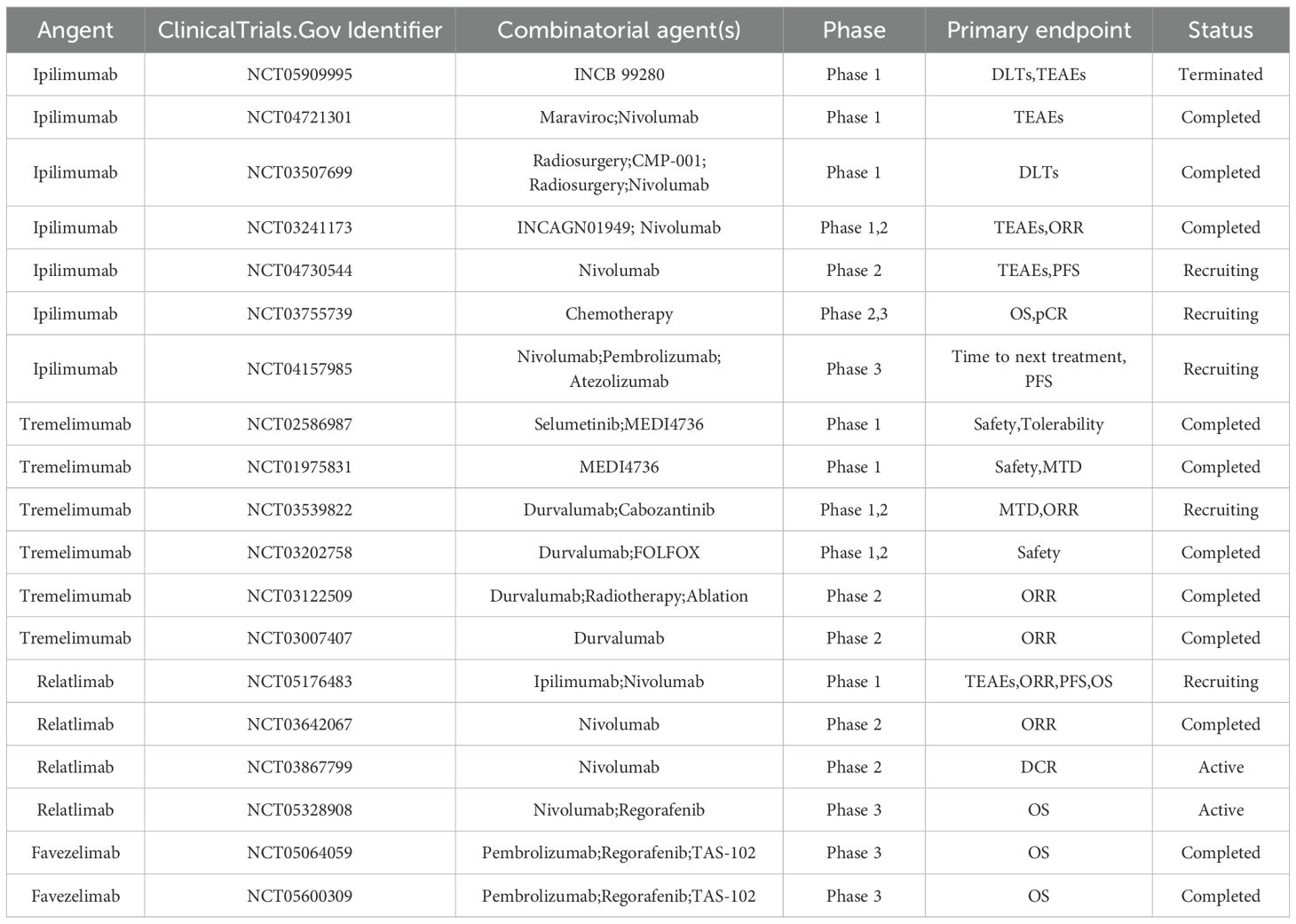
Table 4. Clinical trials based on CTLA-4 inhibitors in the treatment of colorectal cancer.
The TME harbors a significant population of regulatory T cells (Tregs), which express elevated levels of CTLA-4 and contribute to immunosuppression. In this context, CTLA-4 inhibitors, including ipilimumab and tremelimumab, have been developed and widely utilized in clinical practice. These agents exert their antitumor effects by alleviating T-cell suppression and depleting Tregs (50, 51). Building on this, Guo et al. (52) conducted a preclinical study on 2MW4691, a bispecific antibody designed to balance Treg depletion and T-cell activation. The experimental results suggest that 2MW4691 holds promise as a candidate for cancer therapy, warranting further evaluation of its tolerability in clinical trials.
In the CheckMate 142 trial, the combination of nivolumab with low-dose ipilimumab demonstrated a high response rate and reliable safety in 119 patients with dMMR/MSI-H CRC (53). After a median follow-up of 4 years, the ORR for the combination therapy was 65%, with a remarkable 48-month OS rate of 71%, underscoring the durable efficacy of this regimen (54). Another large-scale Phase III trial, CheckMate 8HW, enrolled 707 patients to evaluate the PFS of nivolumab plus ipilimumab versus nivolumab monotherapy in dMMR/MSI-H CRC. Across all treatment lines, the combination of nivolumab and ipilimumab outperformed nivolumab alone, offering a novel therapeutic option for patients with mCRC (55).
Tremelimumab, another well-characterized CTLA-4 inhibitor, differs from ipilimumab as an IgG2 monoclonal antibody. It has demonstrated promising therapeutic efficacy across various malignancies, including lung cancer (56), biliary tract cancer (57), and urothelial carcinoma (58). In a Phase Ib/II trial, the combination of tremelimumab and durvalumab with chemotherapy exhibited robust clinical activity in RAS-mutated mCRC (59). No safety concerns were observed in the Phase Ib portion, and the subsequent Phase II trial primarily assessed PFS, reporting a 3-month PFS rate of 90.7% and a secondary ORR of 64.5% (59).
2.3 LAG-3 inhibitors
LAG-3, a next-generation immune checkpoint, is highly expressed on exhausted T cells and has emerged as a promising therapeutic target. Initially identified for its ability to bind MHC class II molecules and inhibit T-cell activation (60). However, even in the absence of MHC II, LAG-3 binds to the TCR-CD3 complex and traces immune synapses, subsequently disrupting the binding of the tyrosine kinase p56lck (Lck) to CD4 and CD8 co-receptors, thereby limiting TCR signaling and downstream T cell activation (61). This effect is mediated by the EP motif, suggesting that TCR or CD3 is the primary cis-ligand for LAG-3 (61). LAG-3 regulates T-cell activation through steric modulation of antigen presentation: LAG-3 dimers compress the lateral spacing of MHC-II molecules on dendritic cells, occluding the CD4 co-receptor docking site and hindering TCR triggering (62). Epitope mapping of clinical-stage LAG-3 antagonists has revealed that blockade can be achieved without directly targeting the MHC-binding interface, suggesting alternative mechanisms to disrupt this inhibitory axis (62). These mechanistic insights not only deepen our understanding of checkpoint biology but also inform the design of next−generation immunotherapeutics. Furthermore, dual deficiency of PD-1 and LAG-3 in CD8+ T cells enhances tumor clearance, accompanied by increased interferon-γ (IFN-γ) release and upregulation of interferon-responsive genes (63).
The importance of alternative ligands such as galectin-3 (Gal-3), fibrinogen-like protein 1 (FGL1), LSECtin, and α-syn PFF needs to be further confirmed (64). Despite ongoing debates regarding LAG-3 signaling pathways and ligand interactions, the clinical synergy between LAG-3 inhibitors and other immune checkpoint blockers has been well-documented (65) (Table 5). Relatlimab (a LAG-3 monoclonal antibody) and nivolumab has received FDA approval for the treatment of unresectable or metastatic melanoma, marking a significant milestone in cancer immunotherapy (66).
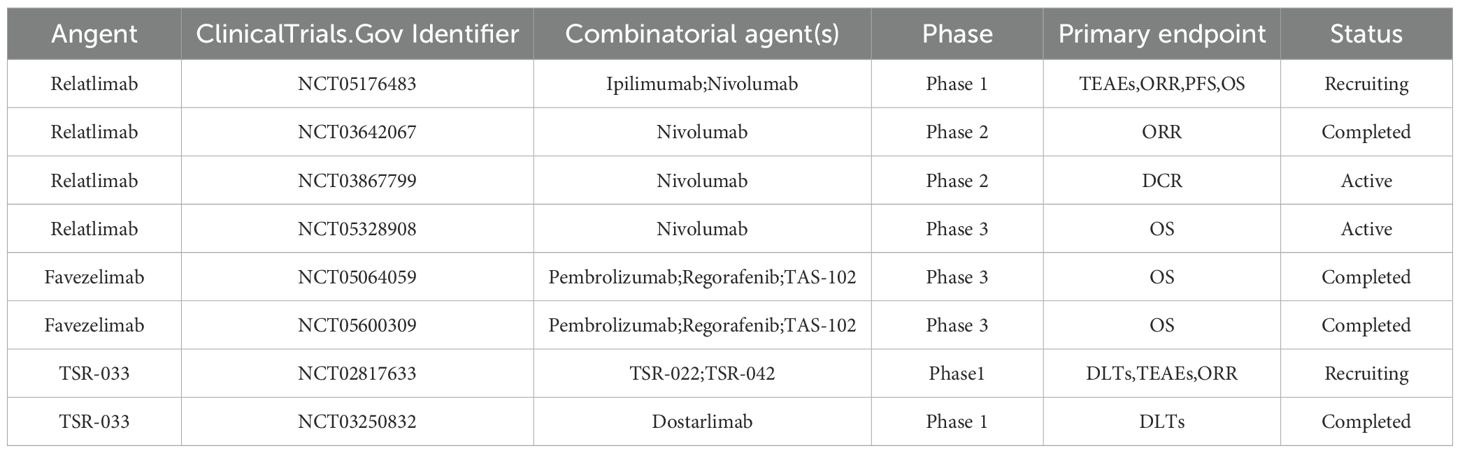
Table 5. Clinical trials based on LAG-3 inhibitors in the treatment of colorectal cancer.
In the context of CRC, the combination of nivolumab and relatlimab has demonstrated clinical benefits in patients with dMMR/MSI-H CRC, as evidenced by the CheckMate 142 trial (44). A Phase III trial evaluating favezelimab (a LAG-3 inhibitor) plus pembrolizumab in CRC reported no treatment-related deaths and notable antitumor activity in PD-L1-positive patients (67). Additionally, the global KEYFORM-007 trial is investigating the safety and efficacy of the co-formulated favezelimab/pembrolizumab (MK-4280A) in PD-L1-positive CRC patients, with an enrollment of 505 participants. This trial also compares the combination to standard-of-care treatments such as TAS-102 (trifluridine and tipiracil) and regorafenib, with results eagerly anticipated. Tebotelimab, a bispecific antibody targeting both PD-1 and LAG-3, has shown encouraging therapeutic responses in solid tumors and hematologic malignancies, with an ORR of 19% and a favorable safety profile (68). These findings highlight the potential of dual checkpoint blockade in overcoming immune resistance and enhancing antitumor immunity.
Currently, ICIs show more efficacy in dMMR/MSI-H CRC patients. However, the heterogeneity of CRC can restrict ICIs’ effectiveness. Moreover, ICIs’ use often leads to drug resistance and immune-related side effects. Thus, a comprehensive patient evaluation before treatment is necessary. Since ICIs have limited efficacy in pMMR/MSS CRC patients, they usually need to be used with other therapies.
3 Exploration of immunotherapy in pMMR/MSS CRC
Compared to dMMR/MSI-H CRC, pMMR/MSS CRC is characterized by a “cold” immune microenvironment, with limited immune cell infiltration and low TMB. This results in minimal benefits from monotherapy with immune checkpoint inhibitors, necessitating the development of combination strategies to overcome immune resistance (69). Below, we discuss several promising approaches to enhance immunotherapy efficacy in pMMR/MSS CRC.
3.1 ICIs plus chemotherapy
Chemotherapeutic agents, such as oxaliplatin and 5-fluorouracil, can induce immunogenic cell death, releasing tumor antigens and neoantigens that activate dendritic cells and enhance antigen presentation (70, 71). Additionally, chemotherapy modulates the immunosuppressive TME by reducing the population of Tregs and myeloid-derived suppressor cells (MDSCs) while increasing CD8+ T-cell infiltration, thereby converting “cold” tumors into “hot” tumors (72, 73). Furthermore, chemotherapy may upregulate PD-L1 expression on tumor cells, enhancing the targeting efficacy of ICIs (74, 75). This synergistic interplay between chemotherapy and ICIs creates a dual mechanism of “immune priming and immune maintenance,” offering a promising strategy for pMMR/MSS CRC.
The KEYNOTE-651 study evaluated the long-term safety and efficacy of pembrolizumab combined with mFOLFOX7/FOLFIRI in pMMR/MSS CRC patients, demonstrating favorable outcomes in Cohorts A, C, and E (76). In Cohorts B and D, the benefit of ORR appeared more pronounced in the KRAS wild-type subgroup, warranting further investigation (77). Another multicenter Phase II trial, GOIM 2802, showed that bevacizumab combined with the XELOX-2 regimen administered biweekly was effective and well-tolerated in mCRC patients, with an ORR comparable to that of the bevacizumab plus FOLFOX-4 group (78). The CAMILLA trial (NCT03539822), a Phase I/II clinical study, established the safety of cabozantinib plus durvalumab combined with chemotherapy in pMMR/MSS mCRC patients during its Phase I component (79). The Phase II portion, which included a CRC cohort, reported a DCR of 86.2%, highlighting the antitumor activity and manageable toxicity of this combination (80). These encouraging results have prompted the initiation of the Phase III STELLAR-303 trial, further exploring this therapeutic approach.
3.2 ICIs plus radiotherapy
Radiotherapy, like chemotherapy, can promote the release of tumor antigens and remodel the TME, enhancing systemic immunity through the abscopal effect (81, 82). Radiation-induced upregulation of cell surface molecules on tumor cells further augments the cytotoxic activity of NK cells and T cells (83). The release of type I interferons enhances T-cell priming and dendritic cell activation, contributing to a robust antitumor immune response (84, 85). These immunostimulatory effects make radiotherapy a valuable adjunct to ICIs in pMMR/MSS CRC.
A Phase II randomized trial reported impressive results, with short-course radiotherapy combined with the PD-1 inhibitor toripalimab and CAPOX achieving a complete response (CR) rate of 58.1% in patients with locally advanced rectal cancer (LARC) (86). Another study evaluated the efficacy and safety of tislelizumab (a PD-1 inhibitor) combined with chemoradiotherapy in LARC patients, demonstrating a CR rate of 40.0% (87). Although these results are promising, larger-scale trials are needed to validate these findings. Neoadjuvant chemoradiotherapy (NACRT) with or without sintilimab (a PD-1 inhibitor) achieved a CR rate of 44.8% in pMMR LARC patients, significantly higher than the 26.9% observed in the control group (88). Immunohistochemical analysis suggested that PD-L1-positive patients may derive greater benefit from this treatment, highlighting the importance of biomarker-driven patient selection.
A neoadjuvant therapy study involving 44 LARC patients treated with preoperative chemoradiotherapy (CRT) and nivolumab monotherapy reported 3-year relapse-free survival (RFS) outcomes. In MSS patients receiving CRT, those with high expression of PD-L1, PD-1, CTLA-4, and Ki-67, as well as an elevated CD8/eTreg ratio, exhibited a higher trend of 3-year RFS (89). These findings underscore the potential of combining radiotherapy with ICIs in pMMR/MSS CRC and suggest that neoadjuvant strategies may become a standard treatment option for this patient population.
3.3 ICIs plus anti-angiogenic agents
Anti-angiogenic agents, primarily tyrosine kinase inhibitors (TKIs) and antibodies targeting the vascular endothelial growth factor (VEGF)/VEGF receptor (VEGFR) pathway have demonstrated synergistic effects when combined with ICIs (90, 91). VEGF not only promotes angiogenesis but also exerts direct immunosuppressive effects by recruiting MDSCs and Tregs while inhibiting T-cell function (92, 93). Anti-angiogenic drugs, such as bevacizumab and ramucirumab, normalize tumor vasculature, improve blood perfusion, and alleviate hypoxia within the TME (94, 95). Vascular normalization enhances immune cell infiltration and reduces endothelial cell-induced T-cell apoptosis, creating a favorable environment for ICIs to exert their effects (96).
BD0801, a humanized anti-VEGF monoclonal antibody, has shown superior antitumor effects compared to bevacizumab in preclinical models, likely due to its potent VEGF/VEGFR blockade and inhibitory effects on human umbilical vein endothelial cells (97). The REGOMUNE trial, a Phase II study, evaluated the safety and efficacy of regorafenib combined with avelumab in MSS CRC patients, demonstrating good tolerability and no unexpected adverse events (98). Biomarker analysis revealed that patients with higher CD8+ T-cell infiltration experienced improved median OS and PFS, further supporting the combination of anti-angiogenic agents with ICIs (98). Fruquintinib, a highly selective TKI targeting VEGFR1, 2, and 3, has shown promising results in mCRC patients who have undergone at least two prior lines of chemotherapy. An international Phase III double-blind trial reported a median OS of 9.3 months and a median PFS of 3.7 months in the fruquintinib group, significantly higher than the 6.6 months and 1.8 months observed in the placebo group (99). These findings suggest that fruquintinib represents a valuable therapeutic option for mCRC patients. Other TKIs, including anlotinib (100–102) and sorafenib (103), have also shown potential in CRC treatment. Additionally, the emergence of bispecific antibodies, such as ivonescimab (anti-PD-1/VEGF-A), has expanded the therapeutic arsenal. A Phase Ia clinical trial demonstrated the promising antitumor activity of ivonescimab, with further studies underway to evaluate its efficacy in combination with other therapies (104).
However, prolonged VEGF/VEGFR blockade may lead to excessive vascular pruning, resulting in reduced drug distribution and hypoxia. Therefore, the therapeutic efficacy of anti-angiogenic agents depends on achieving a balance between vascular normalization and immune activation (96). Exploring triple therapy combinations involving anti-angiogenic agents, ICIs, and other modalities may further modulate the TME and enhance treatment outcomes.
3.4 ICIs plus MEK inhibitors
The mitogen-activated protein kinase (MAPK) pathway regulates critical cellular processes, including proliferation, differentiation, and apoptosis. Dysregulation of MAPK signaling contributes to uncontrolled cell growth and tumor development, making MEK a clinically relevant target (105, 106). MEKi can reduce the release of immunosuppressive factors and decrease the recruitment of immunosuppressive cells. Preclinical data suggest that MEK inhibition upregulates PD-L1 and MHC-I expression, facilitating the subsequent blockade of the PD-1/PD-L1 pathway (107–109).
A Phase Ib trial evaluated the combination of atezolizumab and cobimetinib (a MEK inhibitor) in patients with advanced solid tumors (110). Among the 84 enrolled mCRC patients (62 pMMR/MSS and 2 dMMR/MSI−H), only seven achieved confirmed responses. Diarrhea was the most frequent adverse event. The regimen demonstrated suboptimal safety, with grade 3–4 treatment−related adverse events occurring in 44% of patients and approximately 70% requiring treatment discontinuation or dose reduction due to intolerability (110). Although initial synergistic activity was observed in mCRC, it was not substantiated in a subsequent Phase III trial. Consequently, despite cobimetinib’s potential to modulate the tumor microenvironment, the atezolizumab–cobimetinib combination proved insufficient to overcome immunotherapy resistance in MSS mCRC patients.
Similarly, the Phase III IMblaze370 trial compared atezolizumab + cobimetinib and atezolizumab monotherapy versus regorafenib in the third−line setting for mCRC2. This study enrolled 363 patients (with dMMR/MSI−H recruitment capped at ≤ 5%) and used OS as the primary endpoint (111). IMblaze370 failed to meet its primary endpoint, demonstrating no significant difference in OS, ORR, or PFS for either atezolizumab−containing arm compared with regorafenib (111). These results indicate that combining MEK inhibitors with immune checkpoint inhibitors offers no substantial clinical benefit in pMMR/MSS CRC and warrants further investigation.
3.5 Other prospective combinations
3.5.1 TGF-β: a double-edged sword in CRC
TGF-β is an pleiotropic cytokine whose signal is transduced from membrane to nucleus by SMAD proteins (112). Through the canonical TGF-β/SMAD4 axis, it governs virtually every facet of CRC-initiation, growth, apoptosis, differentiation and dissemination-yet its biological output is context-dependent (113). In early lesions, intact SMAD4 enforces cytostasis by inducing cell-cycle arrest and apoptosis, casting TGF-β as a bona-fide tumor suppressor (113, 114). Once genomic instability erodes this circuitry, malignant cells co-opt the pathway: autocrine TGF-β production fuels immune evasion, neovascularization and metastatic spread (113). Consequently, TGF-β overexpression marks a pivotal switch from tumor containment to progression (114, 115). Early clinical data now show that pharmacological blockade of TGF-β signaling dismantles this immunosuppressive scaffold, amplifies checkpoint-inhibitor efficacy and establishes a self-reinforcing loop of T-cell activation (112, 116). Dual targeting of TGF-β and immune checkpoints therefore constitutes a rational, mechanism-based strategy for microsatellite-stable CRC.
Bintrafusp alfa, a bifunctional fusion protein targeting PD-L1 and TGF-β, has shown promise in various cancers, including CRC (117), lung cancer (118), biliary tract cancer (119), and cervical cancer (120). Although a Phase III trial in advanced lung cancer was terminated early due to lack of superiority over pembrolizumab (121), preclinical studies suggest that bintrafusp alfa can reprogram the TME to overcome immune evasion and reduce radiation-induced fibrosis, highlighting its potential in combination therapies (122). A Phase I/Ib trial explored the use of NIS793, an anti-TGF-β monoclonal antibody, in combination with spartalizumab (a PD-1 inhibitor) in advanced solid tumors, demonstrating the potential for further development (123).
3.5.2 Gut microbiota
Gut microbiota and their metabolites play a crucial role in modulating host immunity, particularly through bile acid metabolism and short-chain fatty acid production. Dysbiosis of the gut microbiota has been linked to CRC development and progression (124, 125). Emerging evidence suggests a strong association between gut microbiota composition and immunotherapy efficacy, including effects on treatment response and toxicity (126–128). In chemo-refractory non-small cell lung cancer and RAS wild‐type (WT) mCRC patients treated with cetuximab and avelumab, the presence of butyrate-producing bacterial strains, such as Agathobacter M104/1 and Blautia SR1/5, was associated with improved treatment outcomes (129).
Furthermore, gut microbiota is emerging as both modulators and biomarkers of immunotherapy efficacy. In advanced non-small-cell lung cancer receiving chemo-immunotherapy, patients treated with nivolumab plus ipilimumab and platinum doublet (NIC) exhibited higher baseline abundances of Faecalibacterium and Butyricicoccus compared with those receiving pembrolizumab plus platinum doublet (PC); this microbial signature was associated with superior overall survival (130). This analytical approach was similarly employed in a recent trial investigating atezolizumab plus bevacizumab (Atz/Bev) for recurrent mesothelioma (131). Collectively, these findings underscore the potential of gut flora to serve as predictive indicators of response in colorectal and other cancers treated with immune-based regimens. Even more exciting is that fecal microbiota transplantation (FMT) has shown promise in enhancing immunotherapy efficacy and overcoming resistance to ICIs in melanoma patients (132, 133). These findings underscore the potential of modulating the gut microbiota to improve immunotherapy outcomes in CRC.
Combination therapies involving ICIs for pMMR/MSS CRC face significant challenges. The tumor microenvironment often resists immune activation, rendering ICIs less effective. Moreover, potential synergistic effects and irAEs require careful management. Additionally, many primary and secondary resistance mechanisms are present in the tumor microenvironment that prevent the efficacy of ICIs. For example, impairments in antigen presentation machinery and IFN-γ signaling pathways can lead to resistance to immune checkpoint blockade therapy. The integration of ICIs with other treatment approaches holds promise in enhancing the efficacy of immunotherapy in CRC patients, but further research is needed to confirm the synergistic effects of these combinations.
4 Cancer vaccines
Cancer vaccines hold promise for both pMMR/MSS CRC and dMMR/MSI CRC. Unlike dMMR/MSI CRC, pMMR/MSS CRC has a lower mutation load but still harbors specific mutations. Recent advances in vaccine development are reshaping therapeutic paradigms for CRC, with neoantigen-targeted vaccines emerging as particularly transformative agents. Conventional vaccines targeting Tumor-Associated Antigens (TAAs) face inherent limitations due to shared expression in normal tissues, raising risks of autoimmune sequelae (134). In contrast, neoantigen vaccines leverage tumor-specific somatic mutations to elicit precise antitumor immunity while sparing healthy tissues, thereby overcoming central tolerance mechanisms and enabling personalized therapeutic strategies (135, 136). The efficacy of such approaches correlates strongly with TMB, where hypermutated tumors exhibit enhanced immunogenic potential (137). While vaccines enrich the presentation of tumor-specific antigens, immune checkpoint inhibitors (ICIs) relieve immune system suppression. Their synergistic effects enhance anti - tumor immunity. Research is now moving toward combining cancer vaccines with ICIs rather than using vaccines alone. For instance, in the NCT04041310 trial conducted on dMMR/MSI CRC patients, the combination of cancer vaccines and ICIs was explored. Furthermore, advancements in delivery platforms spanning nucleic acid-based vectors (DNA/RNA), antigen-presenting cell (APC) systems, and engineered bacterial carriers have substantially expanded the therapeutic arsenal for CRC management (138). (Table 6).
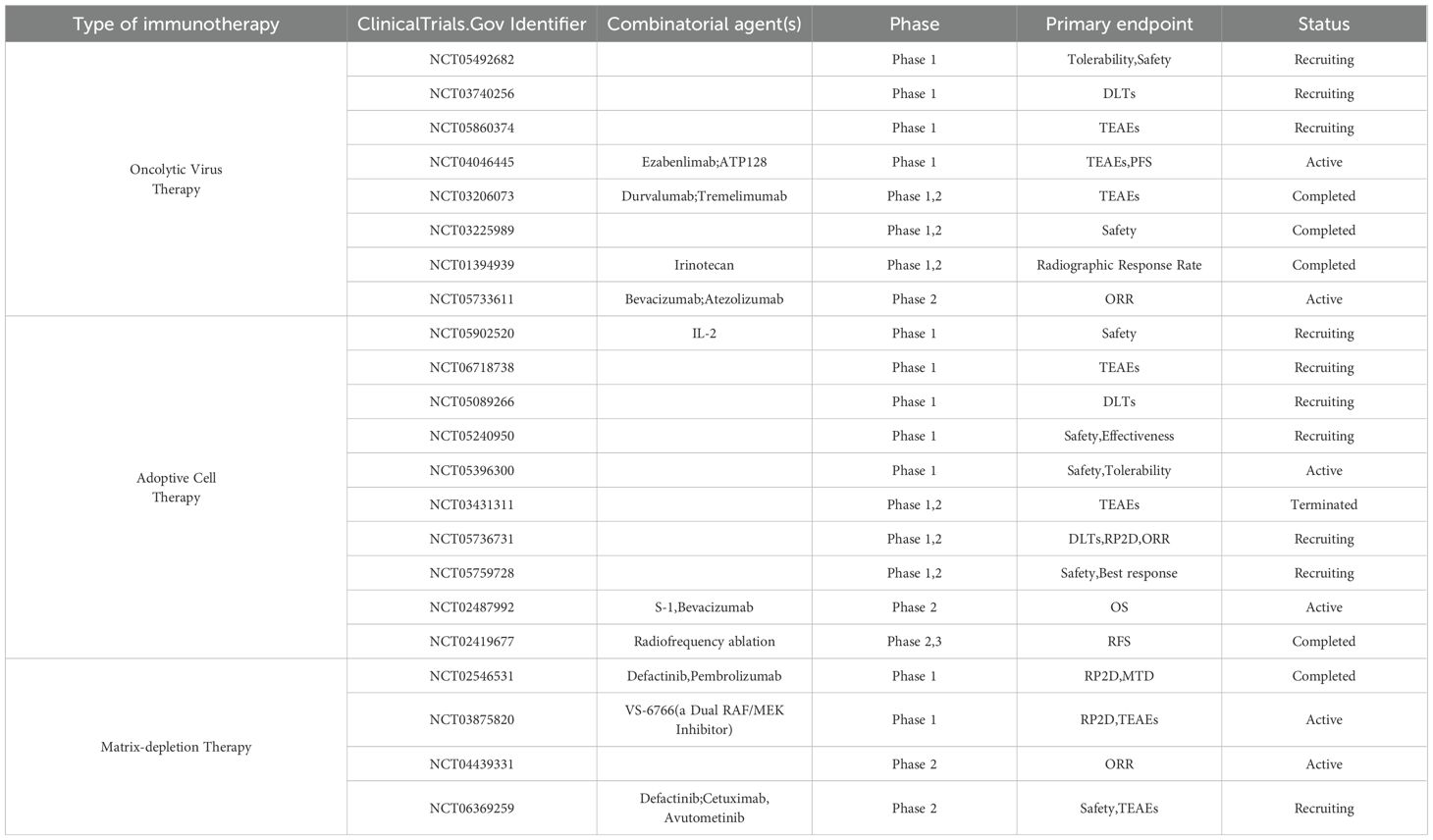
Table 6. Clinical trials based on other immunotherapy for the treatment of colorectal cancer.
Preclinical validation in CRC murine models demonstrated that engineered neoantigen vaccines induce robust antitumor activity, significantly suppressing metastatic progression and extending overall survival through tumor-specific T-cell priming (139). Translational studies in patients with recurrent or metastatic MSS CRC revealed clinically meaningful stratification: responders exhibiting neoantigen-specific immunity achieved superior progression-free survival (PFS:19 vs 11 months) compared to non-responders (140). Parallel breakthroughs in renal cell carcinoma, a malignancy sharing immunological features with MSS CRC, demonstrated universal neoantigen-specific T-cell activation across nine treated patients, with no disease recurrence or dose-limiting toxicities observed during follow-up (141). These findings have galvanized clinical exploration, with ongoing trials such as NCT05141721 evaluating neoantigen vaccines GRT-C901/GRT-R902 in combination with ICIs, with PFS as the primary endpoint in phase III evaluation.
Despite the high specificity and immunogenicity potential of novel antigen vaccines for pMMR/MSS CRC and dMMR/MSI CRC, significant barriers are still present (140, 142). The customization required for these vaccines demands intensive individualized analysis, which is not only cost-prohibitive but also time-intensive, thereby potentially deferring critical treatment initiation. Regarding clinical trials, while early-stage studies have yielded encouraging results, these outcomes have not been consistently replicated in large-scale Phase III trials. This discrepancy may stem from a variety of factors, including trial design limitations, suboptimal patient cohort selection, and the application of efficacy evaluation criteria that may not fully capture the complex biological and clinical impacts of new antigen vaccines.
5 Other immune-related therapies
In the realm of CRC immunotherapy, emerging breakthroughs such as oncolytic virotherapy, ACT, and matrix-depletion therapy have demonstrated significant therapeutic potential (Table 7). Although their mechanisms of action differ, these strategies collectively enhance immune function to combat CRC, each with distinct advantages and limitations. Oncolytic viruses (OVs) exhibit a remarkable ability to augment T-cell-mediated cytotoxicity, yet they face challenges such as inefficient delivery and limited tumor penetration (143, 144). ACT offers durable anti-tumor effects but is hindered by high costs and the risk of T-cell exhaustion (145, 146). Matrix-depletion therapy, on the other hand, disrupts the dense extracellular matrix (ECM) surrounding tumors, improving drug penetration and enhancing the efficacy of other treatments (147). The following sections will elaborate on these three immunotherapeutic approaches.
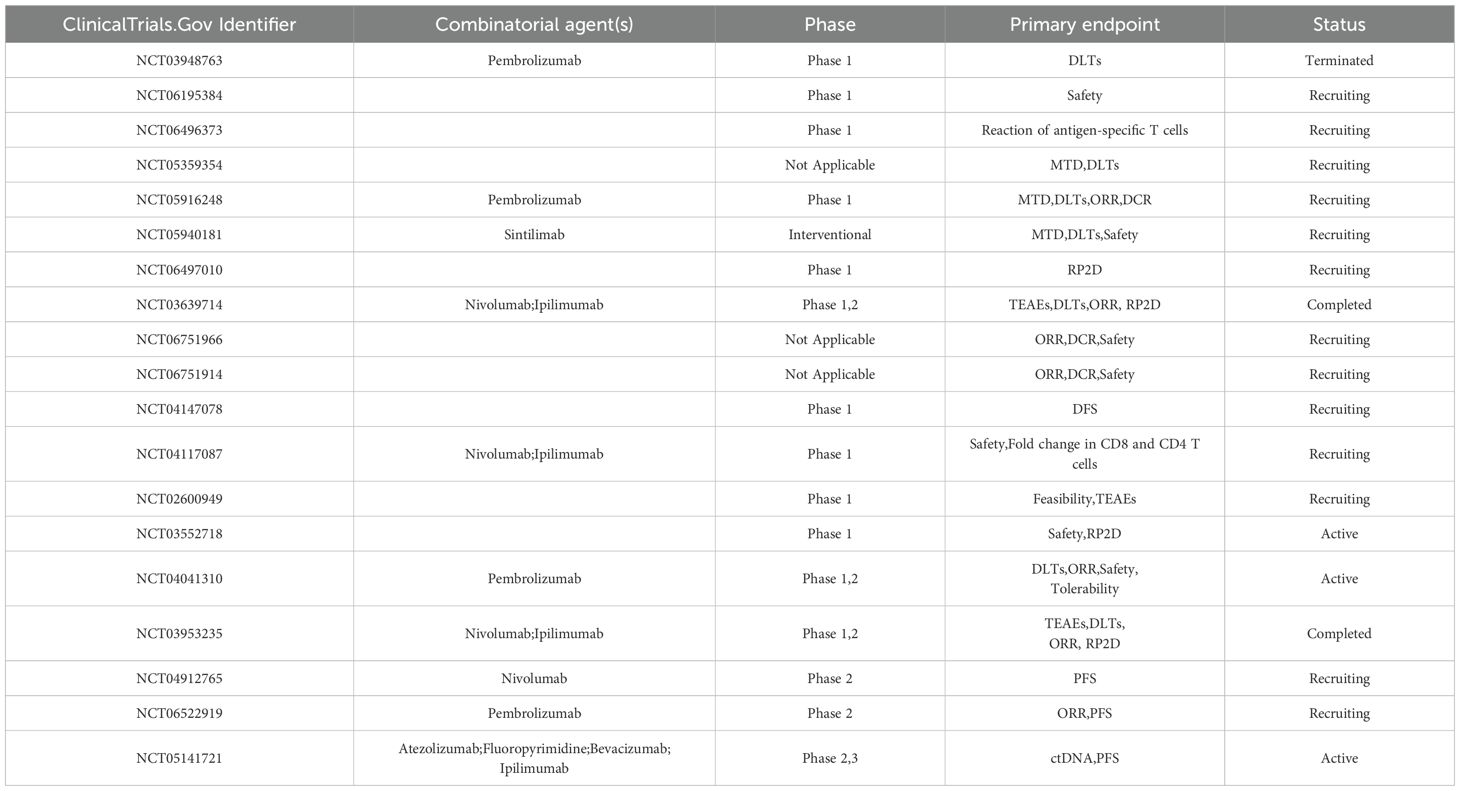
Table 7. Clinical trials based on neoantigen vaccines for the treatment of colorectal cancer.
5.1 Oncolytic virotherapy
OVs exert their antitumor effects through two distinct yet complementary mechanisms (1): direct cytopathic destruction of malignant cells via selective intracellular replication and subsequent cell lysis, and (2) indirect immunostimulatory effects mediated by the massive release of TAAs and neoantigens following viral-induced oncolysis, which initiates systemic antitumor immune responses (12). Importantly, the presentation of viral antigens additionally triggers robust antiviral immune responses that exhibit cross-reactivity with tumor cells, thereby establishing a bimodal attack on neoplastic tissues (148).
A groundbreaking clinical advance was achieved by Zhao Yongxiang’s team through the development of NDV-GT, a recombinant Newcastle disease virus engineered to express porcine α1,3-galactosyltransferase (α1,3GT). This modification induces tumor-specific hyperacute immune rejection via ectopic α-gal epitope expression (149). In a phase I/II trial involving 23 patients with advanced malignancies, NDV-GT elicited a DCR of 90.00% (18/20) with no severe adverse events. Notably, three CRC patients discontinued treatment due to COVID-19 pandemic-related logistical constraints, underscoring real-world challenges in clinical trial execution (149).
Genetic modification of adenoviral vectors enables precise modulation of the TME (150). Rongye Jing and colleagues developed rAd.mDCN.mCD40L, a novel oncolytic adenovirus co-expressing CD40 ligand and decorin, a stromal remodeling agent. In CT26 CRC models, this construct demonstrated potent suppression of hepatic metastasis (151). Another studies revealed that adenoviruses encoding glypican-3 core proteins synergized with NK cell adoptive transfer, enhancing NK-mediated tumor cytotoxicity and intratumoral lymphocyte infiltration in CRC xenografts (152). These findings validate OVs as potent adjuvants for cell-based immunotherapies through multidimensional immune activation (152–154).
While FOLFOXIRI chemotherapy remains a cornerstone in mCRC management, its limited efficacy has prompted exploration of OV-chemotherapy combinations. Girod et al. demonstrated enhanced tumoricidal activity in Colo320 CRC cells through co-administration of coxsackievirus B3 PD-H and chemotherapeutic agents, with combination therapy surpassing monotherapy efficacy via virus-mediated chemosensitization (155). To address targeting limitations and therapeutic resistance, a novel approach utilizing mesenchymal stem cell (MSC)-delivered coxsackievirus A21 achieved robust antitumor effects in CRC murine models, highlighting the potential of cellular vehicles for precise viral delivery (156).
Furthermore, the release of TAAs and cytokines following oncolysis is considered a potential mechanism for enhancing ICIs (157). Several clinical studies (158, 159) have evaluated the combination of oncolytic virotherapy with ICIs in CRC, demonstrating favorable clinical activity and manageable toxicity. In a first-in-human signal-seeking study, 34 patients with proficient-mismatch-repair/microsatellite-stable pMMR/MSS CRC received the oncolytic vaccinia virus pexastimogene devacirepvec (Pexa-Vec) in tandem with the PD-L1 blocker durvalumab, with or without the CTLA-4 antagonist tremelimumab (158). Although the combination was well tolerated, its antitumor impact was marginal: median progression-free survival edged from 2.1 to 2.3 months—incremental gains that fall squarely within the efficacy corridor of existing FDA-approved third-line therapies for mCRC (158). Separately, intratumoral delivery of the engineered herpes simplex virus-2 derivative OH2, combined with the PD-L1 inhibitor LP002, yielded an exceptional responder: a single responder experienced a 313-day remission and remains alive at 499 days (159). Such a solitary signal, however, cannot escape the statistical noise inherent to a four-patient cohort; confirmation awaits adequately powered, prospectively designed trials (159).
But, oncolytic virotherapy for CRC faces three main challenges. First, delivery difficulties. Most CRC tumors are internal, making direct injection tough. Second, immune response balance. Pre-existing immunity can block oncolytic virus efficacy. Repeated injections may trigger neutralizing antibodies. Third, lack of predictive biomarkers. Identifying patients most likely to benefit from oncolytic virotherapy is challenging due to the absence of reliable biomarkers. These issues limit oncolytic virotherapy’s effectiveness and broader application in CRC treatment (160).
5.2 ACT
ACT represents a highly innovative approach to cancer treatment, involving the extraction, modification, and expansion of immune cells ex vivo, followed by re-infusion into patients to enhance anti-tumor immunity. Among various ACT modalities, NK cell therapy, which activates innate immunity, and chimeric antigen receptor T (CAR-T) cell therapy, which offers precise tumor targeting, are the most prominent (161). In CRC, cytokine-induced killer (CIK) cell therapy and CAR-T cell therapy have shown promising results, while newer approaches, such as T-cell receptor-engineered T cells and CAR-macrophages, hold significant potential (161). As technology advances and more clinical trials are conducted, ACT is expected to further improve CRC treatment outcomes, offering hope for better patient survival.
CIK cells are a heterogeneous population of immune effector cells generated by stimulating and expanding peripheral blood mononuclear cells with cytokines ex vivo (162). This population includes conventional T cells, NK cells, and NKT-like cells, with NKT-like cells being the primary effectors that can recognize and kill tumors in an MHC-unrestricted manner (162). A meta-analysis of 70 clinical trials involving 6,743 CRC patients demonstrated that CIK cell therapy significantly improved clinical outcomes, particularly in terms of quality of life and survival (163). Most studies (66 trials) combined CIK therapy with chemotherapy (e.g., FOLFOX or XELOX regimens) to enhance chemotherapy efficacy and mitigate its side effects. Among these, 45 trials administered CIK cells concurrently with chemotherapy, with infusion timing varying across studies: early (3 trials), mid-chemotherapy (7 trials), late (13 trials), and unspecified (22 trials). Future studies should explore optimal infusion schedules to maximize therapeutic benefits. Additionally, personalized CIK cell therapies tailored to patients’ genetic mutations and immune profiles are under development.
CAR-T cell therapy involves genetically modifying patients’ T cells to express a chimeric antigen receptor (CAR) targeting specific tumor antigens. Upon re-infusion, CAR-T cells recognize and bind to tumor surface antigens, releasing cytokines that induce tumor cell apoptosis (164). In CRC, current trials focus on identifying suitable tumor antigens and improving CAR-T cell efficacy and safety. Common targets include carcinoembryonic antigen (CEA) and NKG2D ligands. Several trials (NCT02349724, NCT02416466, NCT02850536) have assessed the safety of CAR-T cells in CEA-positive patients. Notably, NCT02349724 showed that one patient survived for 23 months with elevated serum IFN-γ levels and no grade 3 or higher adverse events (165). Similarly, no severe adverse events were observed in the other trials, confirming the safety of CAR-T infusion (166, 167). Furthermore, NCT02850536 demonstrated improved CAR-T cell delivery to the liver, resulting in increased OS (167). Despite these promising advances, CAR-T therapy in CRC remains limited and requires further exploration to enhance its efficacy.
ACT for CRC faces several key challenges. The immunosuppressive TME of solid tumors poses a significant barrier to ACT efficacy by inhibiting T cell function (168). Additionally, identifying suitable targets that balance safety and efficacy is challenging. ACT is also associated with substantial toxicities, such as cytokine release syndrome, neurotoxicity, and target-related adverse effects (169). The generation of tumor-specific lymphocytes for each patient is technically demanding and economically costly, which limits the widespread application of ACT. Furthermore, T cell exhaustion, characterized by the loss of effector function and self-renewal capacity, further diminishes treatment effectiveness (169). These complexities collectively restrict the clinical application of ACT in CRC.
5.3 Matrix-depletion therapy
Matrix-depletion therapy is an emerging strategy that targets the ECM within the TME. This approach primarily addresses collagen metabolism dysregulation, aberrant activation of matrix metalloproteinases, and the dysregulated expression of factors such as TGF-β (170–172). In CRC, these imbalances are closely linked to TME reprogramming, making ECM components potential therapeutic targets (173, 174). Although matrix-depletion therapy is still in its early stages in CRC, it has shown promise in other stroma-rich malignancies, such as pancreatic cancer.
One key ECM component, hyaluronic acid, plays a critical role in tumor progression. A Phase II trial in metastatic pancreatic cancer patients tested the combination of pegvorhyaluronidase alfa (PEGPH20) with nab-paclitaxel, gemcitabine plus nab-paclitaxel, and gemcitabine alone (175). The results demonstrated that PEGPH20 significantly prolonged PFS and increased ORR, particularly in patients with high hyaluronic acid expression (175). However, a subsequent Phase III trial failed to show benefits in OS or PFS with PEGPH20, leading to its discontinuation in development (176).
Another promising target in matrix-depletion therapy is the Hedgehog signaling pathway, which is implicated in stromal formation. Inhibition of this pathway has been shown to reduce stromal density (177). A Phase II trial combining a Hedgehog pathway inhibitor with cemiplimab in metastatic basal cell carcinoma showed promising anti-tumor activity and safety (178). However, results in sarcoma and myeloma were less encouraging, necessitating further exploration in CRC (179, 180).
Focal adhesion kinase (FAK), a tyrosine kinase involved in integrin signaling, has emerged as another therapeutic target (181). A Phase II trial evaluating a FAK inhibitor in meningioma reported a 33% six-month PFS rate, highlighting its potential as a therapeutic strategy (182).
In summary, while matrix-depletion therapy shows promise, it faces several challenges, including limited specificity and tumor heterogeneity. Moreover, ECM degradation may facilitate circulating tumor cell dissemination, underscoring the need for further safety and efficacy evaluations in CRC.
6 Conclusion and future perspectives
There is no doubt that immunotherapy has achieved remarkable success in CRC treatment. ICIs have made significant strides, while cancer vaccines, oncolytic virotherapy, ACT, and matrix-depletion therapy hold immense potential. Each modality offers unique advantages but also presents challenges. For instance, prolonged ICI use may lead to resistance, and cancer vaccines face hurdles such as insufficient immunogenicity and tumor heterogeneity. OVs exhibit high oncolytic specificity but are limited by the availability of viral vectors and require further evaluation of their efficacy and safety. ACT, though promising, remains unpredictable in its outcomes. While certain matrix-modulating agents can improve the CRC immune microenvironment, the complex interactions among ECM components complicate therapeutic strategies.
In most studies, ICI monotherapy or ICI-ICI combination therapy had an ORR of less than 10% in patients with MSS/pMMR CRC. Thus, addressing the majority of patients with MSS/pMMR CRC is a key challenge (183). To enhance treatment efficacy, various ICI-based strategies, including combinations with radiotherapy, chemotherapy, anti-angiogenic agents, and MEK inhibitors, have been tested in this subgroup. Additionally, targeting TGF-β in combination with ICIs and leveraging the immunomodulatory role of gut microbiota are promising avenues. Breakthroughs in neoantigen vaccines and the emergence of next-generation immune checkpoints such as LAG-3, TIM-3, and TIGIT have further invigorated the field (141, 184, 185).
In conclusion, there are still some challenges that need to be overcome (1): Investigating the crosstalk among various immunosuppressive cells in the CRC immune microenvironment (2); Identifying precise predictive biomarkers (3); Optimizing combination therapy strategies in terms of efficacy, safety, and personalization. By deepening our understanding of the biology and immune microenvironment of CRC, and by integrating new technologies and treatments, we can advance CRC treatment and improve patient outcomes.
Author contributions
YZ: Writing – original draft, Writing – review & editing, Conceptualization. HG: Writing – review & editing, Writing – original draft. XF: Conceptualization, Writing – review & editing. MYL: Conceptualization, Writing – review & editing. JS: Conceptualization, Writing – review & editing. MCL: Writing – review & editing. JH: Writing – review & editing. YJ: Conceptualization, Writing – review & editing. JZ: Funding acquisition, Writing – review & editing. CZ: Supervision, Funding acquisition, Conceptualization, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Regional Project of the National Natural Science Foundation of China (grant number 32360156 and 32460186) and the Yan’an University Doctoral Research Project (Class A) (grant number YDBK-2022–73 and YDBK-2022-77).
Acknowledgments
The figures were drawn by Figdraw.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fimmu.2025.1727580.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Wagle NS, Cercek A, Smith RA, and Jemal A. Colorectal cancer statistics, 2023. CA Cancer J Clin. (2023) 73:233–54. doi: 10.3322/caac.21772, PMID: 36856579
2. Shinji S, Yamada T, Matsuda A, Sonoda H, Ohta R, Iwai T, et al. Recent advances in the treatment of colorectal cancer: A review. J Nippon Med Sch. (2022) 89:246–54. doi: 10.1272/jnms.JNMS.2022_89-310, PMID: 35082204
3. Wang C, Yuan M, Gao Y, Hou R, Song D, and Feng Y. Changes in tumor immune microenvironment after radiotherapy resistance in colorectal cancer: A narrative review. Oncol Res Treat. (2023) 46:177–91. doi: 10.1159/000530161, PMID: 36948165
4. Kim S, Kang SI, Kim S, and Kim JH. Prognostic implications of chemotherapy-induced neutropenia in stage III colorectal cancer. J Surg Res. (2021) 267:391–6. doi: 10.1016/j.jss.2021.05.002, PMID: 34218138
5. Dhanyamraju PK, Schell TD, Amin S, and Robertson GP. Drug-tolerant persister cells in cancer therapy resistance. Cancer Res. (2022) 82:2503–14. doi: 10.1158/0008-5472.Can-21-3844, PMID: 35584245
6. Mendis S and Gill S. Cautious optimism-the current role of immunotherapy in gastrointestinal cancers. Curr Oncol. (2020) 27:S59–s68. doi: 10.3747/co.27.5095, PMID: 32368175
7. Zheng R, Liu X, Zhang Y, Liu Y, Wang Y, Guo S, et al. Frontiers and future of immunotherapy for pancreatic cancer: from molecular mechanisms to clinical application. Front Immunol. (2024) 15:1383978. doi: 10.3389/fimmu.2024.1383978, PMID: 38756774
8. Zhang Y and Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. (2020) 17:807–21. doi: 10.1038/s41423-020-0488-6, PMID: 32612154
9. Yu WD, Sun G, Li J, Xu J, and Wang X. Mechanisms and therapeutic potentials of cancer immunotherapy in combination with radiotherapy and/or chemotherapy. Cancer Lett. (2019) 452:66–70. doi: 10.1016/j.canlet.2019.02.048, PMID: 30902563
10. Naimi A, Mohammed RN, Raji A, Chupradit S, Yumashev AV, Suksatan W, et al. Tumor immunotherapies by immune checkpoint inhibitors (ICIs); the pros and cons. Cell Commun Signal. (2022) 20:44. doi: 10.1186/s12964-022-00854-y, PMID: 35392976
11. Sellars MC, Wu CJ, and Fritsch EF. Cancer vaccines: Building a bridge over troubled waters. Cell. (2022) 185:2770–88. doi: 10.1016/j.cell.2022.06.035, PMID: 35835100
12. Ma R, Li Z, Chiocca EA, Caligiuri MA, and Yu J. The emerging field of oncolytic virus-based cancer immunotherapy. Trends Cancer. (2023) 9:122–39. doi: 10.1016/j.trecan.2022.10.003, PMID: 36402738
13. Bai Z, Zhou Y, Ye Z, Xiong J, Lan H, and Wang F. Tumor-infiltrating lymphocytes in colorectal cancer: the fundamental indication and application on immunotherapy. Front Immunol. (2021) 12:808964. doi: 10.3389/fimmu.2021.808964, PMID: 35095898
14. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. (2017) 357:409–13. doi: 10.1126/science.aan6733, PMID: 28596308
15. De’ Angelis GL, Bottarelli L, Azzoni C, De’ Angelis N, Leandro G, Di Mario F, et al. Microsatellite instability in colorectal cancer. Acta Biomed. (2018) 89:97–101. doi: 10.23750/abm.v89i9-S.7960, PMID: 30561401
16. Vilar E and Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol. (2010) 7:153–62. doi: 10.1038/nrclinonc.2009.237, PMID: 20142816
17. Arrichiello G, Poliero L, Borrelli C, Paragliola F, Nacca V, Napolitano S, et al. Immunotherapy in colorectal cancer: is the long-awaited revolution finally happening? Cancer Treat Res Commun. (2021) 28:100442. doi: 10.1016/j.ctarc.2021.100442, PMID: 34391139
18. Dutta S, Ganguly A, Chatterjee K, Spada S, and Mukherjee S. Targets of immune escape mechanisms in cancer: basis for development and evolution of cancer immune checkpoint inhibitors. Biol (Basel). (2023) 12(2):218. doi: 10.3390/biology12020218, PMID: 36829496
19. Jin C, Zhu X, Huang X, Gong T, Wei Z, and You J. Efficacy and safety of PD-1/PD-L1 and CTLA-4 immune checkpoint inhibitors in colorectal cancer: a meta-analysis. J Comp Eff Res. (2022) 11:203–12. doi: 10.2217/cer-2021-0134, PMID: 35023361
20. Cai L, Chen A, and Tang D. A new strategy for immunotherapy of microsatellite-stable (MSS)-type advanced colorectal cancer: Multi-pathway combination therapy with PD-1/PD-L1 inhibitors. Immunology. (2024) 173:209–26. doi: 10.1111/imm.13785, PMID: 38517066
21. Huo JL, Wang YT, Fu WJ, Lu N, and Liu ZS. The promising immune checkpoint LAG-3 in cancer immunotherapy: from basic research to clinical application. Front Immunol. (2022) 13:956090. doi: 10.3389/fimmu.2022.956090, PMID: 35958563
22. Tang Q, Zhao S, Zhou N, He J, Zu L, Liu T, et al. PD−1/PD−L1 immune checkpoint inhibitors in neoadjuvant therapy for solid tumors (Review). Int J Oncol. (2023) 62(4):49. doi: 10.3892/ijo.2023.5497, PMID: 36866750
23. Spagnolo F, Boutros A, Cecchi F, Croce E, Tanda ET, and Queirolo P. Treatment beyond progression with anti-PD-1/PD-L1 based regimens in advanced solid tumors: a systematic review. BMC Cancer. (2021) 21:425. doi: 10.1186/s12885-021-08165-0, PMID: 33865350
24. Xing K, Zhou P, Li J, Liu M, and Zhang WE. Inhibitory effect of PD-1/PD-L1 and blockade immunotherapy in leukemia. Comb Chem High Throughput Screen. (2022) 25:1399–410. doi: 10.2174/1574893616666210707101516, PMID: 34238150
25. Du J, Chen H, You J, Hu W, Liu J, Lu Q, et alProximity between LAG-3 and the T cell receptor guides suppression of T cell activation and autoimmunity. Cell. (2025) 188(15):4025-42.e20. doi: 10.1016/j.cell.2025.06.004, PMID: 40592325
26. Jiang H, Ni H, Zhang P, Guo X, Wu M, Shen H, et al. PD-L1/LAG-3 bispecific antibody enhances tumor-specific immunity. Oncoimmunology. (2021) 10:1943180. doi: 10.1080/2162402x.2021.1943180, PMID: 34239776
27. Tobin JWD, Bednarska K, Campbell A, and Keane C. PD-1 and LAG-3 checkpoint blockade: potential avenues for therapy in B-cell lymphoma. Cells. (2021) 10(5):1152. doi: 10.3390/cells10051152, PMID: 34068762
28. Cillo AR, Cardello C, Shan F, Karapetyan L, Kunning S, Sander C, et al. Blockade of LAG-3 and PD-1 leads to co-expression of cytotoxic and exhaustion gene modules in CD8(+) T cells to promote antitumor immunity. Cell. (2024) 187:4373–88.e15. doi: 10.1016/j.cell.2024.06.036, PMID: 39121849
29. Kähler KC, Hassel JC, Heinzerling L, Loquai C, Thoms KM, Ugurel S, et al. Side effect management during immune checkpoint blockade using CTLA-4 and PD-1 antibodies for metastatic melanoma - an update. J Dtsch Dermatol Ges. (2020) 18:582–609. doi: 10.1111/ddg.14128, PMID: 32489011
30. Liu LL, Skribek M, Harmenberg U, and Gerling M. Systemic inflammatory syndromes as life-threatening side effects of immune checkpoint inhibitors: case report and systematic review of the literature. J Immunother Cancer. (2023) 11(3):e005841. doi: 10.1136/jitc-2022-005841, PMID: 36878533
31. Blum SM, Rouhani SJ, and Sullivan RJ. Effects of immune-related adverse events (irAEs) and their treatment on antitumor immune responses. Immunol Rev. (2023) 318:167–78. doi: 10.1111/imr.13262, PMID: 37578634
32. Diaz LA Jr., Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): final analysis of a randomised, open-label, phase 3 study. Lancet Oncol. (2022) 23:659–70. doi: 10.1016/s1470-2045(22)00197-8, PMID: 35427471
33. Kluger H, Barrett JC, Gainor JF, Hamid O, Hurwitz M, LaVallee T, et al. Society for Immunotherapy of Cancer (SITC) consensus definitions for resistance to combinations of immune checkpoint inhibitors. J Immunother Cancer. (2023) 11(3):e005921. doi: 10.1136/jitc-2022-005921, PMID: 36918224
34. Schoenfeld AJ and Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. (2020) 37:443–55. doi: 10.1016/j.ccell.2020.03.017, PMID: 32289269
35. Lin X, Kang K, Chen P, Zeng Z, Li G, Xiong W, et al. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol Cancer. (2024) 23:108. doi: 10.1186/s12943-024-02023-w, PMID: 38762484
36. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, and Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. (2012) 209:1201–17. doi: 10.1084/jem.20112741, PMID: 22641383
37. Christofides A, Katopodi XL, Cao C, Karagkouni D, Aliazis K, Yenyuwadee S, et al. SHP-2 and PD-1-SHP-2 signaling regulate myeloid cell differentiation and antitumor responses. Nat Immunol. (2023) 24:55–68. doi: 10.1038/s41590-022-01385-x, PMID: 36581713
38. Rota G, Niogret C, Dang AT, Barros CR, Fonta NP, Alfei F, et al. Shp-2 is dispensable for establishing T cell exhaustion and for PD-1 signaling In Vivo. Cell Rep. (2018) 23:39–49. doi: 10.1016/j.celrep.2018.03.026, PMID: 29617671
39. Jiang F, Yu W, Zeng F, Cheng G, Xu J, Yang S, et al. PD-1 high expression predicts lower local disease control in stage IV M0 nasopharyngeal carcinoma. BMC Cancer. (2019) 19:503. doi: 10.1186/s12885-019-5689-y, PMID: 31138162
40. Lee BH, Park Y, Kim JH, Kang KW, Lee SJ, Kim SJ, et al. PD-L1 expression in bone marrow plasma cells as a biomarker to predict multiple myeloma prognosis: developing a nomogram-based prognostic model. Sci Rep. (2020) 10:12641. doi: 10.1038/s41598-020-69616-5, PMID: 32724129
41. Hong Y, Chen Q, Wang Z, Zhang Y, Li B, Guo H, et al. Targeting Nuclear Receptor Coactivator SRC-1 Prevents Colorectal Cancer Immune Escape by Reducing Transcription and Protein Stability of PD-L1. Adv Sci (Weinh). (2024) 11(33):e2310037. doi: 10.1002/advs.202310037, PMID: 38953362
42. Twomey JD and Zhang B. Cancer immunotherapy update: FDA-approved checkpoint inhibitors and companion diagnostics. AAPS J. (2021) 23:39. doi: 10.1208/s12248-021-00574-0, PMID: 33677681
43. André T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N Engl J Med. (2020) 383:2207–18. doi: 10.1056/NEJMoa2017699, PMID: 33264544
44. Overman MJ, Gelsomino F, Aglietta M, Wong M, Limon Miron ML, Leonard G, et al. Nivolumab plus relatlimab in patients with previously treated microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: the phase II CheckMate 142 study. J Immunother Cancer. (2024) 12(5):e008689. doi: 10.1136/jitc-2023-008689, PMID: 38821718
45. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. (2017) 18:1182–91. doi: 10.1016/s1470-2045(17)30422-9, PMID: 28734759
46. Hu H, Kang L, Zhang J, Wu Z, Wang H, Huang M, et al. Neoadjuvant PD-1 blockade with toripalimab, with or without celecoxib, in mismatch repair-deficient or microsatellite instability-high, locally advanced, colorectal cancer (PICC): a single-centre, parallel-group, non-comparative, randomised, phase 2 trial. Lancet Gastroenterol Hepatol. (2022) 7:38–48. doi: 10.1016/s2468-1253(21)00348-4, PMID: 34688374
47. Kennedy A, Waters E, Rowshanravan B, Hinze C, Williams C, Janman D, et al. Differences in CD80 and CD86 transendocytosis reveal CD86 as a key target for CTLA-4 immune regulation. Nat Immunol. (2022) 23:1365–78. doi: 10.1038/s41590-022-01289-w, PMID: 35999394
48. Fehervari Z. Controlling PD-L1 in cis. Nat Immunol. (2019) 20:665. doi: 10.1038/s41590-019-0412-3, PMID: 31110310
49. Grebinoski S, Gocher-Demske AM, and Vignali DAA. Catch and release: freeing up PD-L1 ameliorates autoimmunity. Nat Immunol. (2022) 23:344–6. doi: 10.1038/s41590-022-01140-2, PMID: 35190718
50. Tay C, Tanaka A, and Sakaguchi S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell. (2023) 41:450–65. doi: 10.1016/j.ccell.2023.02.014, PMID: 36917950
51. Zong Y, Deng K, and Chong WP. Regulation of Treg cells by cytokine signaling and co-stimulatory molecules. Front Immunol. (2024) 15:1387975. doi: 10.3389/fimmu.2024.1387975, PMID: 38807592
52. Guo C, Dai X, Du Y, Xiong X, and Gui X. Preclinical development of a novel CCR8/CTLA-4 bispecific antibody for cancer treatment by disrupting CTLA-4 signaling on CD8 T cells and specifically depleting tumor-resident Tregs. Cancer Immunol Immunother. (2024) 73:210. doi: 10.1007/s00262-024-03794-3, PMID: 39123089
53. Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. (2018) 36:773–9. doi: 10.1200/jco.2017.76.9901, PMID: 29355075
54. André T, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Nivolumab plus low-dose ipilimumab in previously treated patients with microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: 4-year follow-up from CheckMate 142. Ann Oncol. (2022) 33:1052–60. doi: 10.1016/j.annonc.2022.06.008, PMID: 35764271
55. André T, Elez E, Lenz HJ, Jensen LH, Touchefeu Y, Van Cutsem E, et al. Nivolumab plus ipilimumab versus nivolumab in microsatellite instability-high metastatic colorectal cancer (CheckMate 8HW): a randomised, open-label, phase 3 trial. Lancet. (2025) 405:383–95. doi: 10.1016/s0140-6736(24)02848-4, PMID: 39874977
56. Goldman JW, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab, with or without tremelimumab, plus platinum-etoposide versus platinum-etoposide alone in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): updated results from a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. (2021) 22:51–65. doi: 10.1016/s1470-2045(20)30539-8, PMID: 33285097
57. Oh DY, Lee KH, Lee DW, Yoon J, Kim TY, Bang JH, et al. Gemcitabine and cisplatin plus durvalumab with or without tremelimumab in chemotherapy-naive patients with advanced biliary tract cancer: an open-label, single-centre, phase 2 study. Lancet Gastroenterol Hepatol. (2022) 7:522–32. doi: 10.1016/s2468-1253(22)00043-7, PMID: 35278356
58. Powles T, van der Heijden MS, Castellano D, Galsky MD, Loriot Y, Petrylak DP, et al. Durvalumab alone and durvalumab plus tremelimumab versus chemotherapy in previously untreated patients with unresectable, locally advanced or metastatic urothelial carcinoma (DANUBE): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. (2020) 21:1574–88. doi: 10.1016/s1470-2045(20)30541-6, PMID: 32971005
59. Thibaudin M, Fumet JD, Chibaudel B, Bennouna J, Borg C, Martin-Babau J, et al. First-line durvalumab and tremelimumab with chemotherapy in RAS-mutated metastatic colorectal cancer: a phase 1b/2 trial. Nat Med. (2023) 29:2087–98. doi: 10.1038/s41591-023-02497-z, PMID: 37563240
60. Huard B, Mastrangeli R, Prigent P, Bruniquel D, Donini S, El-Tayar N, et al. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc Natl Acad Sci U S A. (1997) 94:5744–9. doi: 10.1073/pnas.94.11.5744, PMID: 9159144
61. Guy C, Mitrea DM, Chou PC, Temirov J, Vignali KM, Liu X, et al. LAG3 associates with TCR-CD3 complexes and suppresses signaling by driving co-receptor-Lck dissociation. Nat Immunol. (2022) 23:757–67. doi: 10.1038/s41590-022-01176-4, PMID: 35437325
62. Ming Q, Antfolk D, Price DA, Manturova A, Medina E, Singh S, et al. Structural basis for mouse LAG3 interactions with the MHC class II molecule I-A(b). Nat Commun. (2024) 15:7513. doi: 10.1038/s41467-024-51930-5, PMID: 39209860
63. Andrews LP, Butler SC, Cui J, Cillo AR, Cardello C, Liu C, et al. LAG-3 and PD-1 synergize on CD8(+) T cells to drive T cell exhaustion and hinder autocrine IFN-γ-dependent anti-tumor immunity. Cell. (2024) 187:4355–72.e22. doi: 10.1016/j.cell.2024.07.016, PMID: 39121848
64. Aggarwal V, Workman CJ, and Vignali DAA. LAG-3 as the third checkpoint inhibitor. Nat Immunol. (2023) 24:1415–22. doi: 10.1038/s41590-023-01569-z, PMID: 37488429
65. Chavanton A, Mialhe F, Abrey J, Baeza Garcia A, and Garrido C. LAG-3: recent developments in combinational therapies in cancer. Cancer Sci. (2024) 115:2494–505. doi: 10.1111/cas.16205, PMID: 38702996
66. Chocarro L, Bocanegra A, Blanco E, Fernández-Rubio L, Arasanz H, Echaide M, et al. Cutting-edge: preclinical and clinical development of the first approved lag-3 inhibitor. Cells. (2022) 11(15):2351. doi: 10.3390/cells11152351, PMID: 35954196
67. Garralda E, Sukari A, Lakhani NJ, Patnaik A, Lou Y, Im SA, et al. A first-in-human study of the anti-LAG-3 antibody favezelimab plus pembrolizumab in previously treated, advanced microsatellite stable colorectal cancer. ESMO Open. (2022) 7:100639. doi: 10.1016/j.esmoop.2022.100639, PMID: 36493599
68. Luke JJ, Patel MR, Blumenschein GR, Hamilton E, Chmielowski B, Ulahannan SV, et al. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: a phase 1 trial. Nat Med. (2023) 29:2814–24. doi: 10.1038/s41591-023-02593-0, PMID: 37857711
69. Khosravi GR, Mostafavi S, Bastan S, Ebrahimi N, Gharibvand RS, and Eskandari N. Immunologic tumor microenvironment modulators for turning cold tumors hot. Cancer Commun (Lond). (2024) 44:521–53. doi: 10.1002/cac2.12539, PMID: 38551889
70. Galluzzi L, Guilbaud E, Schmidt D, Kroemer G, and Marincola FM. Targeting immunogenic cell stress and death for cancer therapy. Nat Rev Drug Discov. (2024) 23:445–60. doi: 10.1038/s41573-024-00920-9, PMID: 38622310
71. Kepp O and Kroemer G. Immunogenic cell stress and death sensitize tumors to immunotherapy. Cells. (2023) 12(24):2843. doi: 10.3390/cells12242843, PMID: 38132163
72. Opzoomer JW, Sosnowska D, Anstee JE, Spicer JF, and Arnold JN. Cytotoxic chemotherapy as an immune stimulus: A molecular perspective on turning up the immunological heat on cancer. Front Immunol. (2019) 10:1654. doi: 10.3389/fimmu.2019.01654, PMID: 31379850
73. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. (2010) 70:3052–61. doi: 10.1158/0008-5472.Can-09-3690, PMID: 20388795
74. Dosset M, Vargas TR, Lagrange A, Boidot R, Végran F, Roussey A, et al. PD-1/PD-L1 pathway: an adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology. (2018) 7:e1433981. doi: 10.1080/2162402x.2018.1433981, PMID: 29872568
75. Luo H, Song G, Wang D, Li M, and Dai N. Combining PD-1 or PD-L1 inhibitors with chemotherapy is a good strategy for the treatment of extensive small cell lung cancer: A retrospective analysis of clinical studies. Front Immunol. (2022) 13:1059557. doi: 10.3389/fimmu.2022.1059557, PMID: 36544769
76. Chen EX, Kavan P, Tehfe M, Kortmansky JS, Sawyer MB, Chiorean EG, et al. Pembrolizumab plus binimetinib with or without chemotherapy for MSS/pMMR metastatic colorectal cancer: outcomes from KEYNOTE-651 cohorts A, C and E. Clin Colorectal Cancer. (2024) 23:183–93. doi: 10.1016/j.clcc.2024.03.002, PMID: 38653648
77. Kim R, Tehfe M, Kavan P, Chaves J, Kortmansky JS, Chen EX, et al. Pembrolizumab plus mFOLFOX7 or FOLFIRI for microsatellite stable/mismatch repair-proficient metastatic colorectal cancer: KEYNOTE-651 cohorts B and D. Clin Colorectal Cancer. (2024) 23:118–27.e6. doi: 10.1016/j.clcc.2024.03.001, PMID: 38762348
78. Maiello E, Di Maggio G, Cordio S, Cinieri S, Giuliani F, Pisconti S, et al. Bevacizumab in combination with either FOLFOX-4 or XELOX-2 in first-line treatment of patients with metastatic colorectal cancer: A multicenter randomized phase II trial of the gruppo oncologico dell’Italia meridionale (GOIM 2802). Clin Colorectal Cancer. (2020) 19:109–15. doi: 10.1016/j.clcc.2020.01.003, PMID: 32089455
79. Saeed A, Park R, Dai J, Al-Rajabi R, Kasi A, Baranda J, et al. Cabozantinib plus durvalumab in advanced gastroesophageal cancer and other gastrointestinal Malignancies: Phase Ib CAMILLA trial results. Cell Rep Med. (2023) 4:100916. doi: 10.1016/j.xcrm.2023.100916, PMID: 36702123
80. Saeed A, Park R, Pathak H, Al-Bzour AN, Dai J, Phadnis M, et al. Clinical and biomarker results from a phase II trial of combined cabozantinib and durvalumab in patients with chemotherapy-refractory colorectal cancer (CRC): CAMILLA CRC cohort. Nat Commun. (2024) 15:1533. doi: 10.1038/s41467-024-45960-2, PMID: 38378868
81. McLaughlin M, Patin EC, Pedersen M, Wilkins A, Dillon MT, Melcher AA, et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer. (2020) 20:203–17. doi: 10.1038/s41568-020-0246-1, PMID: 32161398
82. Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. (2004) 58:862–70. doi: 10.1016/j.ijrobp.2003.09.012, PMID: 14967443
83. Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. (2006) 203:1259–71. doi: 10.1084/jem.20052494, PMID: 16636135
84. Zhang F, Manna S, Pop LM, Chen ZJ, Fu YX, and Hannan R. Type I interferon response in radiation-induced anti-tumor immunity. Semin Radiat Oncol. (2020) 30:129–38. doi: 10.1016/j.semradonc.2019.12.009, PMID: 32381292
85. Lim JY, Gerber SA, Murphy SP, and Lord EM. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8(+) T cells. Cancer Immunol Immunother. (2014) 63:259–71. doi: 10.1007/s00262-013-1506-7, PMID: 24357146
86. Wang YQ, Shen LJ, Wan JF, Zhang H, Wang Y, Wu X, et al. Short-course radiotherapy combined with CAPOX and PD-1 inhibitor for the total neoadjuvant therapy of locally advanced rectal cancer: the preliminary single-center findings of a prospective, multicentre, randomized phase II trial (TORCH). Zhonghua Wei Chang Wai Ke Za Zhi. (2023) 26:448–58. doi: 10.3760/cma.j.cn441530-20230107-00010, PMID: 37217353
87. Yang Z, Gao J, Zheng J, Han J, Li A, Liu G, et al. Efficacy and safety of PD-1 blockade plus long-course chemoradiotherapy in locally advanced rectal cancer (NECTAR): a multi-center phase 2 study. Signal Transduct Target Ther. (2024) 9:56. doi: 10.1038/s41392-024-01762-y, PMID: 38462629
88. Xiao WW, Chen G, Gao YH, Lin JZ, Wu XJ, Luo HL, et al. Effect of neoadjuvant chemoradiotherapy with or without PD-1 antibody sintilimab in pMMR locally advanced rectal cancer: A randomized clinical trial. Cancer Cell. (2024) 42:1570–81.e4. doi: 10.1016/j.ccell.2024.07.004, PMID: 39094560
89. Tsukada Y, Bando H, Inamori K, Wakabayashi M, Togashi Y, Koyama S, et al. Three-year outcomes of preoperative chemoradiotherapy plus nivolumab in microsatellite stable and microsatellite instability-high locally advanced rectal cancer. Br J Cancer. (2024) 131:283–9. doi: 10.1038/s41416-024-02730-7, PMID: 38834744
90. Song Y, Fu Y, Xie Q, Zhu B, Wang J, and Zhang B. Anti-angiogenic agents in combination with immune checkpoint inhibitors: A promising strategy for cancer treatment. Front Immunol. (2020) 11:1956. doi: 10.3389/fimmu.2020.01956, PMID: 32983126
91. Huinen ZR, Huijbers EJM, van Beijnum JR, Nowak-Sliwinska P, and Griffioen AW. Anti-angiogenic agents - overcoming tumour endothelial cell anergy and improving immunotherapy outcomes. Nat Rev Clin Oncol. (2021) 18:527–40. doi: 10.1038/s41571-021-00496-y, PMID: 33833434
92. Tamura R, Tanaka T, Akasaki Y, Murayama Y, Yoshida K, and Sasaki H. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: perspectives for therapeutic implications. Med Oncol. (2019) 37:2. doi: 10.1007/s12032-019-1329-2, PMID: 31713115
93. Liu G, Chen T, Ding Z, Wang Y, Wei Y, and Wei X. Inhibition of FGF-FGFR and VEGF-VEGFR signalling in cancer treatment. Cell Prolif. (2021) 54:e13009. doi: 10.1111/cpr.13009, PMID: 33655556
94. Lorenc P, Sikorska A, Molenda S, Guzniczak N, Dams-Kozlowska H, and Florczak A. Physiological and tumor-associated angiogenesis: Key factors and therapy targeting VEGF/VEGFR pathway. BioMed Pharmacother. (2024) 180:117585. doi: 10.1016/j.biopha.2024.117585, PMID: 39442237
95. Patel SA, Nilsson MB, Le X, Cascone T, Jain RK, and Heymach JV. Molecular mechanisms and future implications of VEGF/VEGFR in cancer therapy. Clin Cancer Res. (2023) 29:30–9. doi: 10.1158/1078-0432.Ccr-22-1366, PMID: 35969170
96. Kuo HY, Khan KA, and Kerbel RS. Antiangiogenic-immune-checkpoint inhibitor combinations: lessons from phase III clinical trials. Nat Rev Clin Oncol. (2024) 21:468–82. doi: 10.1038/s41571-024-00886-y, PMID: 38600370
97. Xue L, Gao X, Zhang H, Tang J, Wang Q, Li F, et al. Antiangiogenic antibody BD0801 combined with immune checkpoint inhibitors achieves synergistic antitumor activity and affects the tumor microenvironment. BMC Cancer. (2021) 21:1134. doi: 10.1186/s12885-021-08859-5, PMID: 34686154
98. Cousin S, Cantarel C, Guegan JP, Gomez-Roca C, Metges JP, Adenis A, et al. Regorafenib-avelumab combination in patients with microsatellite stable colorectal cancer (REGOMUNE): A single-arm, open-label, phase II trial. Clin Cancer Res. (2021) 27:2139–47. doi: 10.1158/1078-0432.Ccr-20-3416, PMID: 33495314
99. Dasari A, Lonardi S, Garcia-Carbonero R, Elez E, Yoshino T, Sobrero A, et al. Fruquintinib versus placebo in patients with refractory metastatic colorectal cancer (FRESCO-2): an international, multicentre, randomised, double-blind, phase 3 study. Lancet. (2023) 402:41–53. doi: 10.1016/s0140-6736(23)00772-9, PMID: 37331369
100. Li H, Feng H, Zhang T, Wu J, Shen X, Xu S, et al. CircHAS2 activates CCNE2 to promote cell proliferation and sensitizes the response of colorectal cancer to anlotinib. Mol Cancer. (2024) 23:59. doi: 10.1186/s12943-024-01971-7, PMID: 38515149
101. Jia ZX, Zhang Z, Li Z, Li A, Xie YN, Wu HJ, et al. Anlotinib inhibits the progress of colorectal cancer cells by antagonizing VEGFR/JAK2/STAT3 axis. Eur Rev Med Pharmacol Sci. (2021) 25:2331–43. doi: 10.26355/eurrev_202103_25272, PMID: 33755971
102. Luo B, Zhang S, Tan D, Yu X, Lin J, and Wang M. Anlotinib benefits the αPDL1 immunotherapy by activating ROS/JNK/AP-1 pathway to upregulate PDL1 expression in colorectal cancer. Oxid Med Cell Longev. (2022) 2022:8965903. doi: 10.1155/2022/8965903, PMID: 36238642
103. Jeong KY, Park M, Sim JJ, and Kim HM. Combination antitumor effect of sorafenib via calcium-dependent deactivation of focal adhesion kinase targeting colorectal cancer cells. Molecules. (2020) 25(22):5299. doi: 10.3390/molecules25225299, PMID: 33202899
104. Frentzas S, Austria Mislang AR, Lemech C, Nagrial A, Underhill C, Wang W, et al. Phase 1a dose escalation study of ivonescimab (AK112/SMT112), an anti-PD-1/VEGF-A bispecific antibody, in patients with advanced solid tumors. J Immunother Cancer. (2024) 12(4):e008037. doi: 10.1136/jitc-2023-008037, PMID: 38642937
105. Frémin C and Meloche S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J Hematol Oncol. (2010) 3:8. doi: 10.1186/1756-8722-3-8, PMID: 20149254
106. Dennison L, Mohan AA, and Yarchoan M. Tumor and systemic immunomodulatory effects of MEK inhibition. Curr Oncol Rep. (2021) 23:23. doi: 10.1007/s11912-020-01008-4, PMID: 33547983
107. Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, et al. The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res. (2015) 21:1639–51. doi: 10.1158/1078-0432.Ccr-14-2339, PMID: 25589619
108. Rosen LS, LoRusso P, Ma WW, Goldman JW, Weise A, Colevas AD, et al. A first-in-human phase I study to evaluate the MEK1/2 inhibitor, cobimetinib, administered daily in patients with advanced solid tumors. Invest New Drugs. (2016) 34:604–13. doi: 10.1007/s10637-016-0374-3, PMID: 27424159
109. Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity. (2016) 44:609–21. doi: 10.1016/j.immuni.2016.01.024, PMID: 26944201
110. Hellmann MD, Kim TW, Lee CB, Goh BC, Miller WH Jr., Oh DY, et al. Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann Oncol. (2019) 30:1134–42. doi: 10.1093/annonc/mdz113, PMID: 30918950
111. Eng C, Kim TW, Bendell J, Argilés G, Tebbutt NC, Di Bartolomeo M, et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. (2019) 20:849–61. doi: 10.1016/s1470-2045(19)30027-0, PMID: 31003911
112. Liu S, Ren J, and Ten Dijke P. Targeting TGFβ signal transduction for cancer therapy. Signal Transduct Target Ther. (2021) 6:8. doi: 10.1038/s41392-020-00436-9, PMID: 33414388
113. Fasano M, Pirozzi M, Miceli CC, Cocule M, Caraglia M, Boccellino M, et al. TGF-β Modulated pathways in colorectal cancer: new potential therapeutic opportunities. Int J Mol Sci. (2024) 25(13):7400. doi: 10.3390/ijms25137400, PMID: 39000507
114. Su J, Morgani SM, David CJ, Wang Q, Er EE, Huang YH, et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature. (2020) 577:566–71. doi: 10.1038/s41586-019-1897-5, PMID: 31915377
115. Chakravarthy A, Khan L, Bensler NP, Bose P, and De Carvalho DD. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun. (2018) 9:4692. doi: 10.1038/s41467-018-06654-8, PMID: 30410077
116. Niu M, Yi M, Wu Y, Lyu L, He Q, Yang R, et al. Synergistic efficacy of simultaneous anti-TGF-β/VEGF bispecific antibody and PD-1 blockade in cancer therapy. J Hematol Oncol. (2023) 16:94. doi: 10.1186/s13045-023-01487-5, PMID: 37573354
117. Spira A, Wertheim MS, Kim EJ, Tan B, Lenz HJ, Nikolinakos P, et al. Bintrafusp alfa: A bifunctional fusion protein targeting PD-L1 and TGF-β, in patients with pretreated colorectal cancer: results from a phase I trial. Oncologist. (2023) 28:e124–e7. doi: 10.1093/oncolo/oyac254, PMID: 36576431
118. Vokes EE, Mornex F, Sezer A, Cheng Y, Fang J, Baz DV, et al. Bintrafusp alfa with CCRT followed by bintrafusp alfa versus placebo with CCRT followed by durvalumab in patients with unresectable stage III NSCLC: A phase 2 randomized study. J Thorac Oncol. (2024) 19:285–96. doi: 10.1016/j.jtho.2023.09.1452, PMID: 37797733
119. Yoo C, Javle MM, Verdaguer Mata H, de Braud F, Trojan J, Raoul JL, et al. Phase 2 trial of bintrafusp alfa as second-line therapy for patients with locally advanced/metastatic biliary tract cancers. Hepatology. (2023) 78:758–70. doi: 10.1097/hep.0000000000000365, PMID: 36999533
120. Birrer M, Li G, Yunokawa M, Lee JY, Kim BG, Oppermann CP, et al. Bintrafusp alfa for recurrent or metastatic cervical cancer after platinum failure: A nonrandomized controlled trial. JAMA Oncol. (2024) 10:1204–11. doi: 10.1001/jamaoncol.2024.2145, PMID: 39052242
121. Cho BC, Lee JS, Wu YL, Cicin I, Dols MC, Ahn MJ, et al. Bintrafusp alfa versus pembrolizumab in patients with treatment-naive, programmed death-ligand 1-high advanced NSCLC: A randomized, open-label, phase 3 trial. J Thorac Oncol. (2023) 18:1731–42. doi: 10.1016/j.jtho.2023.08.018, PMID: 37597750
122. Lan Y, Moustafa M, Knoll M, Xu C, Furkel J, Lazorchak A, et al. Simultaneous targeting of TGF-β/PD-L1 synergizes with radiotherapy by reprogramming the tumor microenvironment to overcome immune evasion. Cancer Cell. (2021) 39:1388–403.e10. doi: 10.1016/j.ccell.2021.08.008, PMID: 34506739
123. Bauer TM, Santoro A, Lin CC, Garrido-Laguna I, Joerger M, Greil R, et al. Phase I/Ib, open-label, multicenter, dose-escalation study of the anti-TGF-β monoclonal antibody, NIS793, in combination with spartalizumab in adult patients with advanced tumors. J Immunother Cancer. (2023) 11(11):e007353. doi: 10.1136/jitc-2023-007353, PMID: 38030303
124. Cai J, Sun L, and Gonzalez FJ. Gut microbiota-derived bile acids in intestinal immunity, inflammation, and tumorigenesis. Cell Host Microbe. (2022) 30:289–300. doi: 10.1016/j.chom.2022.02.004, PMID: 35271802
125. Qu R, Zhang Y, Ma Y, Zhou X, Sun L, Jiang C, et al. Role of the gut microbiota and its metabolites in tumorigenesis or development of colorectal cancer. Adv Sci (Weinh). (2023) 10:e2205563. doi: 10.1002/advs.202205563, PMID: 37263983
126. Kroemer G and Zitvogel L. Cancer immunotherapy in 2017: The breakthrough of the microbiota. Nat Rev Immunol. (2018) 18:87–8. doi: 10.1038/nri.2018.4, PMID: 29379189
127. Alexander JL, Wilson ID, Teare J, Marchesi JR, Nicholson JK, and Kinross JM. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol. (2017) 14:356–65. doi: 10.1038/nrgastro.2017.20, PMID: 28270698
128. M. PC, Claudia W, Kaiser A SHC, Marcello C, Lydie C, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. (2007) 117:2197–204. doi: 10.1172/jci32205, PMID: 17657310
129. Martini G, Ciardiello D, Dallio M, Famiglietti V, Esposito L, Corte CMD, et al. Gut microbiota correlates with antitumor activity in patients with mCRC and NSCLC treated with cetuximab plus avelumab. Int J Cancer. (2022) 151:473–80. doi: 10.1002/ijc.34033, PMID: 35429341
130. Hakozaki T, Tanaka K, Shiraishi Y, Sekino Y, Mitome N, Okuma Y, et al. Gut microbiota in advanced NSCLC receiving chemoimmunotherapy: an ancillary biomarker study from the phase III trial JCOG2007 (NIPPON). J Thorac Oncol. (2025) 20:912–27. doi: 10.1016/j.jtho.2025.02.026, PMID: 40058642
131. Zhang M, Bzura A, Baitei EY, Zhou Z, Spicer JB, Poile C, et al. A gut microbiota rheostat forecasts responsiveness to PD-L1 and VEGF blockade in mesothelioma. Nat Commun. (2024) 15:7187. doi: 10.1038/s41467-024-49842-5, PMID: 39168966
132. Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. (2021) 371:595–602. doi: 10.1126/science.abf3363, PMID: 33542131
133. Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. (2021) 371:602–9. doi: 10.1126/science.abb5920, PMID: 33303685
134. Hollingsworth RE and Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines. (2019) 4:7. doi: 10.1038/s41541-019-0103-y, PMID: 30774998
135. Pounraj S, Chen S, Ma L, Mazzieri R, Dolcetti R, and Rehm BHA. Targeting tumor heterogeneity with neoantigen-based cancer vaccines. Cancer Res. (2024) 84:353–63. doi: 10.1158/0008-5472.Can-23-2042, PMID: 38055891
136. Hu Z, Leet DE, Allesøe RL, Oliveira G, Li S, Luoma AM, et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat Med. (2021) 27:515–25. doi: 10.1038/s41591-020-01206-4, PMID: 33479501
137. Blass E and Ott PA. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol. (2021) 18:215–29. doi: 10.1038/s41571-020-00460-2, PMID: 33473220
138. Ruzzi F, Riccardo F, Conti L, Tarone L, Semprini MS, Bolli E, et al. Cancer vaccines: Target antigens, vaccine platforms and preclinical models. Mol Aspects Med. (2025) 101:101324. doi: 10.1016/j.mam.2024.101324, PMID: 39631227
139. Redenti A, Im J, Redenti B, Li F, Rouanne M, Sheng Z, et al. Probiotic neoantigen delivery vectors for precision cancer immunotherapy. Nature. (2024) 635:453–61. doi: 10.1038/s41586-024-08033-4, PMID: 39415001
140. Yu YJ, Shan N, Li LY, Zhu YS, Lin LM, Mao CC, et al. Preliminary clinical study of personalized neoantigen vaccine therapy for microsatellite stability (MSS)-advanced colorectal cancer. Cancer Immunol Immunother. (2023) 72:2045–56. doi: 10.1007/s00262-023-03386-7, PMID: 36795124
141. Braun DA, Moranzoni G, Chea V, McGregor BA, Blass E, Tu CR, et al. A neoantigen vaccine generates antitumour immunity in renal cell carcinoma. Nature. (2025) 639:474–82. doi: 10.1038/s41586-024-08507-5, PMID: 39910301
142. Jia W, Zhang T, Huang H, Feng H, Wang S, Guo Z, et al. Colorectal cancer vaccines: The current scenario and future prospects. Front Immunol. (2022) 13:942235. doi: 10.3389/fimmu.2022.942235, PMID: 35990683
143. Webb MJ, Sangsuwannukul T, van Vloten J, Evgin L, Kendall B, Tonne J, et al. Expression of tumor antigens within an oncolytic virus enhances the anti-tumor T cell response. Nat Commun. (2024) 15:5442. doi: 10.1038/s41467-024-49286-x, PMID: 38937436
144. Guedan S and Alemany R. CAR-T cells and oncolytic viruses: joining forces to overcome the solid tumor challenge. Front Immunol. (2018) 9:2460. doi: 10.3389/fimmu.2018.02460, PMID: 30405639
145. Albarrán V, San Román M, Pozas J, Chamorro J, Rosero DI, Guerrero P, et al. Adoptive T cell therapy for solid tumors: current landscape and future challenges. Front Immunol. (2024) 15:1352805. doi: 10.3389/fimmu.2024.1352805, PMID: 38550594
146. Li W, Pan X, Chen L, Cui H, Mo S, Pan Y, et al. Cell metabolism-based optimization strategy of CAR-T cell function in cancer therapy. Front Immunol. (2023) 14:1186383. doi: 10.3389/fimmu.2023.1186383, PMID: 37342333
147. Ferrara B, Pignatelli C, Cossutta M, Citro A, Courty J, and Piemonti L. The extracellular matrix in pancreatic cancer: description of a complex network and promising therapeutic options. Cancers (Basel). (2021) 13(17):4442. doi: 10.3390/cancers13174442, PMID: 34503252
148. Lemos de Matos A, Franco LS, and McFadden G. Oncolytic viruses and the immune system: the dynamic duo. Mol Ther Methods Clin Dev. (2020) 17:349–58. doi: 10.1016/j.omtm.2020.01.001, PMID: 32071927
149. Zhong L, Gan L, Wang B, Wu T, Yao F, Gong W, et al. Hyperacute rejection-engineered oncolytic virus for interventional clinical trial in refractory cancer patients. Cell. (2025) 188:1119–36.e23. doi: 10.1016/j.cell.2024.12.010, PMID: 39826543
150. Sosnovtseva AO, Stepanova OV, Stepanenko AA, Voronova AD, Chadin AV, Valikhov MP, et al. Recombinant adenoviruses for delivery of therapeutics following spinal cord injury. Front Pharmacol. (2021) 12:777628. doi: 10.3389/fphar.2021.777628, PMID: 35082666
151. Rong Y, Ning Y, Zhu J, Feng P, Zhu W, Zhao X, et al. Oncolytic adenovirus encoding decorin and CD40 ligand inhibits tumor growth and liver metastasis via immune activation in murine colorectal tumor model. Mol Biomed. (2024) 5:39. doi: 10.1186/s43556-024-00202-1, PMID: 39306655
152. Li X, Zhang Y, Mao Z, Zhao H, Cao H, Wang J, et al. Decorin-armed oncolytic adenovirus promotes natural killers (NKs) activation and infiltration to enhance NK therapy in CRC model. Mol Biomed. (2024) 5:48. doi: 10.1186/s43556-024-00212-z, PMID: 39482550
153. McGrath K and Dotti G. Combining oncolytic viruses with chimeric antigen receptor T cell therapy. Hum Gene Ther. (2021) 32:150–7. doi: 10.1089/hum.2020.278, PMID: 33349123
154. Watanabe N, McKenna MK, Rosewell Shaw A, and Suzuki M. Clinical CAR-T cell and oncolytic virotherapy for cancer treatment. Mol Ther. (2021) 29:505–20. doi: 10.1016/j.ymthe.2020.10.023, PMID: 33130314
155. Girod M, Geisler A, Hinze L, Elsner L, Dieringer B, Beling A, et al. Combination of FOLFOXIRI drugs with oncolytic coxsackie B3 virus PD-H synergistically induces oncolysis in the refractory colorectal cancer cell line colo320. Int J Mol Sci. (2024) 25(11):5618. doi: 10.3390/ijms25115618, PMID: 38891807
156. Karbalaee R, Mehdizadeh S, Ghaleh HEG, Izadi M, Kondori BJ, Dorostkar R, et al. The effects of mesenchymal stem cells loaded with oncolytic coxsackievirus A21 on mouse models of colorectal cancer. Curr Cancer Drug Targets. (2024) 24:967–74. doi: 10.2174/0115680096273465231201115839, PMID: 38310465
157. Harrington K, Freeman DJ, Kelly B, Harper J, and Soria JC. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov. (2019) 18:689–706. doi: 10.1038/s41573-019-0029-0, PMID: 31292532
158. Monge C, Xie C, Myojin Y, Coffman K, Hrones DM, Wang S, et al. Phase I/II study of PexaVec in combination with immune checkpoint inhibition in refractory metastatic colorectal cancer. J Immunother Cancer. (2023) 11(2):e005640. doi: 10.1136/jitc-2022-005640, PMID: 36754451
159. Zhang H, Ren Y, Wang F, Tu X, Tong Z, Liu L, et al. The long-term effectiveness and mechanism of oncolytic virotherapy combined with anti-PD-L1 antibody in colorectal cancer patient. Cancer Gene Ther. (2024) 31:1412–26. doi: 10.1038/s41417-024-00807-2, PMID: 39068234
160. Chen L, Zuo M, Zhou Q, and Wang Y. Oncolytic virotherapy in cancer treatment: challenges and optimization prospects. Front Immunol. (2023) 14:1308890. doi: 10.3389/fimmu.2023.1308890, PMID: 38169820
161. Han J, Zhang B, Zheng S, Jiang Y, Zhang X, and Mao K. The progress and prospects of immune cell therapy for the treatment of cancer. Cell Transplant. (2024) 33:9636897241231892. doi: 10.1177/09636897241231892, PMID: 38433349
162. Moser LM, Heim C, KosChade SE, Wendel P, Bozkurt S, Harenkamp S, et al. CAR-CIK vs. CAR-T: benchmarking novel cytokine-induced killer cells as solid tumor immunotherapy in ErbB2+ rhabdomyosarcoma. Front Immunol. (2025) 16:1485817. doi: 10.3389/fimmu.2025.1485817, PMID: 39963129
163. Ying Li CM, Li R, Drew P, Price T, Smith E, Maddern GJ, et al. Clinical application of cytokine-induced killer (CIK) cell therapy in colorectal cancer: Current strategies and future challenges. Cancer Treat Rev. (2024) 122:102665. doi: 10.1016/j.ctrv.2023.102665, PMID: 38091655
164. Peng L, Sferruzza G, Yang L, Zhou L, and Chen S. CAR-T and CAR-NK as cellular cancer immunotherapy for solid tumors. Cell Mol Immunol. (2024) 21:1089–108. doi: 10.1038/s41423-024-01207-0, PMID: 39134804
165. Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ Liver metastases. Clin Cancer Res. (2015) 21:3149–59. doi: 10.1158/1078-0432.Ccr-14-1421, PMID: 25850950
166. Katz SC, Hardaway J, Prince E, Guha P, Cunetta M, Moody A, et al. HITM-SIR: phase Ib trial of intraarterial chimeric antigen receptor T-cell therapy and selective internal radiation therapy for CEA(+) liver metastases. Cancer Gene Ther. (2020) 27:341–55. doi: 10.1038/s41417-019-0104-z, PMID: 31155611
167. Katz SC, Moody AE, Guha P, Hardaway JC, Prince E, LaPorte J, et al. HITM-SURE: Hepatic immunotherapy for metastases phase Ib anti-CEA CAR-T study utilizing pressure enabled drug delivery. J Immunother Cancer. (2020) 8(2):e001097. doi: 10.1136/jitc-2020-001097, PMID: 32843493
168. Mohd Salim NH, Mussa A, Ahmed N, Ahmad S, Yean Yean C, Hassan R, et al. The immunosuppressive effect of TNFR2 expression in the colorectal cancer microenvironment. Biomedicines. (2023) 11(1):173. doi: 10.3390/biomedicines11010173, PMID: 36672682
169. Sun L, Su Y, Jiao A, Wang X, and Zhang B. T cells in health and disease. Signal Transduct Target Ther. (2023) 8:235. doi: 10.1038/s41392-023-01471-y, PMID: 37332039
170. Rømer AMA, Thorseth ML, and Madsen DH. Immune modulatory properties of collagen in cancer. Front Immunol. (2021) 12:791453. doi: 10.3389/fimmu.2021.791453, PMID: 34956223
171. Cabral-Pacheco GA, Garza-Veloz I, Castruita-De la Rosa C, Ramirez-Acuña JM, Perez-Romero BA, Guerrero-Rodriguez JF, et al. The roles of matrix metalloproteinases and their inhibitors in human diseases. Int J Mol Sci. (2020) 21(24):9739. doi: 10.3390/ijms21249739, PMID: 33419373
172. Gillesberg FS, Pehrsson M, Bay-Jensen AC, Frederiksen P, Karsdal M, Deleuran BW, et al. Regulation of fibronectin and collagens type I, III and VI by TNF-α, TGF-β, IL-13, and tofacitinib. Sci Rep. (2025) 15:1087. doi: 10.1038/s41598-024-84151-3, PMID: 39774197
173. Valkenburg KC, de Groot AE, and Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol. (2018) 15:366–81. doi: 10.1038/s41571-018-0007-1, PMID: 29651130
174. Feng Y, Ma W, Zang Y, Guo Y, Li Y, Zhang Y, et al. Spatially organized tumor-stroma boundary determines the efficacy of immunotherapy in colorectal cancer patients. Nat Commun. (2024) 15:10259. doi: 10.1038/s41467-024-54710-3, PMID: 39592630
175. Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, et al. HALO 202: randomized phase II study of PEGPH20 plus nab-paclitaxel/gemcitabine versus nab-paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J Clin Oncol. (2018) 36:359–66. doi: 10.1200/jco.2017.74.9564, PMID: 29232172
176. Van Cutsem E, Tempero MA, Sigal D, Oh DY, Fazio N, Macarulla T, et al. Randomized phase III trial of pegvorhyaluronidase alfa with nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J Clin Oncol. (2020) 38:3185–94. doi: 10.1200/jco.20.00590, PMID: 32706635
177. Karunasagara S, Taghizadeh A, Kim SH, Kim SJ, Kim YJ, Taghizadeh M, et al. Tissue mechanics and hedgehog signaling crosstalk as a key epithelial-stromal interplay in cancer development. Adv Sci (Weinh). (2024) 11:e2400063. doi: 10.1002/advs.202400063, PMID: 38976559
178. Lewis KD, Peris K, Sekulic A, Stratigos AJ, Dunn L, Eroglu Z, et al. Final analysis of phase II results with cemiplimab in metastatic basal cell carcinoma after hedgehog pathway inhibitors. Ann Oncol. (2024) 35:221–8. doi: 10.1016/j.annonc.2023.10.123, PMID: 38072158
179. Gounder MM, Rosenbaum E, Wu N, Dickson MA, Sheikh TN, D’Angelo SP, et al. A phase ib/II randomized study of RO4929097, a gamma-secretase or notch inhibitor with or without vismodegib, a hedgehog inhibitor, in advanced sarcoma. Clin Cancer Res. (2022) 28:1586–94. doi: 10.1158/1078-0432.Ccr-21-3874, PMID: 35110418
180. Tibes R, Kosiorek HE, Dueck AC, Palmer J, Sproat L, Bogenberger J, et al. Phase 1/1b study of azacitidine and hedgehog pathway inhibitor sonidegib in patients with myeloid neoplasms. Cancer. (2023) 129:2321–30. doi: 10.1002/cncr.34800, PMID: 37042080
181. Golubovskaya VM. Targeting FAK in human cancer: from finding to first clinical trials. Front Biosci (Landmark Ed). (2014) 19:687–706. doi: 10.2741/4236, PMID: 24389213
182. Brastianos PK, Twohy EL, Gerstner ER, Kaufmann TJ, Iafrate AJ, Lennerz J, et al. Alliance A071401: phase II trial of focal adhesion kinase inhibition in meningiomas with somatic NF2 mutations. J Clin Oncol. (2023) 41:618–28. doi: 10.1200/jco.21.02371, PMID: 36288512
183. Guven DC, Kavgaci G, Erul E, Syed MP, Magge T, Saeed A, et al. The efficacy of immune checkpoint inhibitors in microsatellite stable colorectal cancer: A systematic review. Oncologist. (2024) 29:e580–600. doi: 10.1093/oncolo/oyae013, PMID: 38309719
184. Takamatsu K, Tanaka N, Hakozaki K, Takahashi R, Teranishi Y, Murakami T, et al. Profiling the inhibitory receptors LAG-3, TIM-3, and TIGIT in renal cell carcinoma reveals Malignancy. Nat Commun. (2021) 12:5547. doi: 10.1038/s41467-021-25865-0, PMID: 34545095
Keywords: colorectal cancer immunotherapy, immune checkpoint inhibitors, cancer vaccines, adoptive cell therapy, matrix-depletion therapy
Citation: Zhang Y, Guan H, Feng X, Liu M, Shao J, Liu M, He J, Jin Y, Zhu J and Zheng C (2025) Emerging strategies in colorectal cancer immunotherapy: enhancing efficacy and survival. Front. Immunol. 16:1616414. doi: 10.3389/fimmu.2025.1616414
Received: 22 April 2025; Accepted: 17 September 2025;
Published: 02 October 2025; Corrected: 07 November 2025.
Edited by:
Jie Li, Peking Union Medical College Hospital (CAMS), ChinaReviewed by:
Thanh Huong Phung, Hanoi University of Pharmacy, VietnamValentina Lopardo, University of Salerno, Italy
Copyright © 2025 Zhang, Guan, Feng, Liu, Shao, Liu, He, Jin, Zhu and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunli Zheng, Y2h1bmxpemhlbmdAMTI2LmNvbQ==; Jinglin Zhu, emh1amluZ2xpbjI3QDE2My5jb20=; Yahui Jin, c3h4YWp5aEAxMjYuY29t