 Yuwei Hu1,2†Shuchang Dai1,2†Congchao Qiao1,2,3†Yifan Ye4Junyi Ren5Kai Wang5,6*Ling Li7,8*
Yuwei Hu1,2†Shuchang Dai1,2†Congchao Qiao1,2,3†Yifan Ye4Junyi Ren5Kai Wang5,6*Ling Li7,8* Zhong Liu1,2*
Zhong Liu1,2*- 1Clinical Transfusion Research Center, Institute of Blood Transfusion, Chinese Academy of Medical Sciences & Peking Union Medical College, Chengdu, China
- 2Key Laboratory of Transfusion Adverse Reactions, Chinese Academy of Medical Sciences, Chengdu, China
- 3Department of Transfusion Medicine, Dazhou Integrated Traditional Chinese Medicine and Western Medicine Hospital, Dazhou Second People’s Hospital, Dazhou, Sichuan, China
- 4State Key Laboratory of Experimental Hematology, National Clinical Research Center for Blood Diseases, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Tianjin, China
- 5School of Medicine, University of Electronic Science and Technology of China, Chengdu, Sichuan, China
- 6Department of Emergency, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu, China
- 7Department of Blood Transfusion, The Third People’s Hospital of Chengdu (Affiliated Hospital of Southwest Jiaotong University), College of Medicine, Southwest Jiaotong University, Chengdu, Sichuan, China
- 8School of Public Health, Anhui Medical University, Hefei, China
Platelets have long been acknowledged for their essential roles in hemostasis and thrombosis; however, recent insights highlight their broader involvement as key participants in host responses during infection. Beyond their classical functions, platelets exhibit diverse anti-infective capabilities, such as direct pathogen internalization, receptor-mediated pathogen recognition, the release of antimicrobial peptides, cytokines, and chemokines, and the generation of immunomodulatory extracellular vesicles. These intrinsic platelet attributes enable dynamic interactions with pathogens and immune cells, significantly contributing to pathogen capture, neutralization, and the orchestration of innate and adaptive immune responses. This review examines the multifaceted intrinsic roles of platelets and delineates the beneficial outcomes of their activation, providing an integrated perspective on platelet-driven immunity and defense mechanisms during infection.
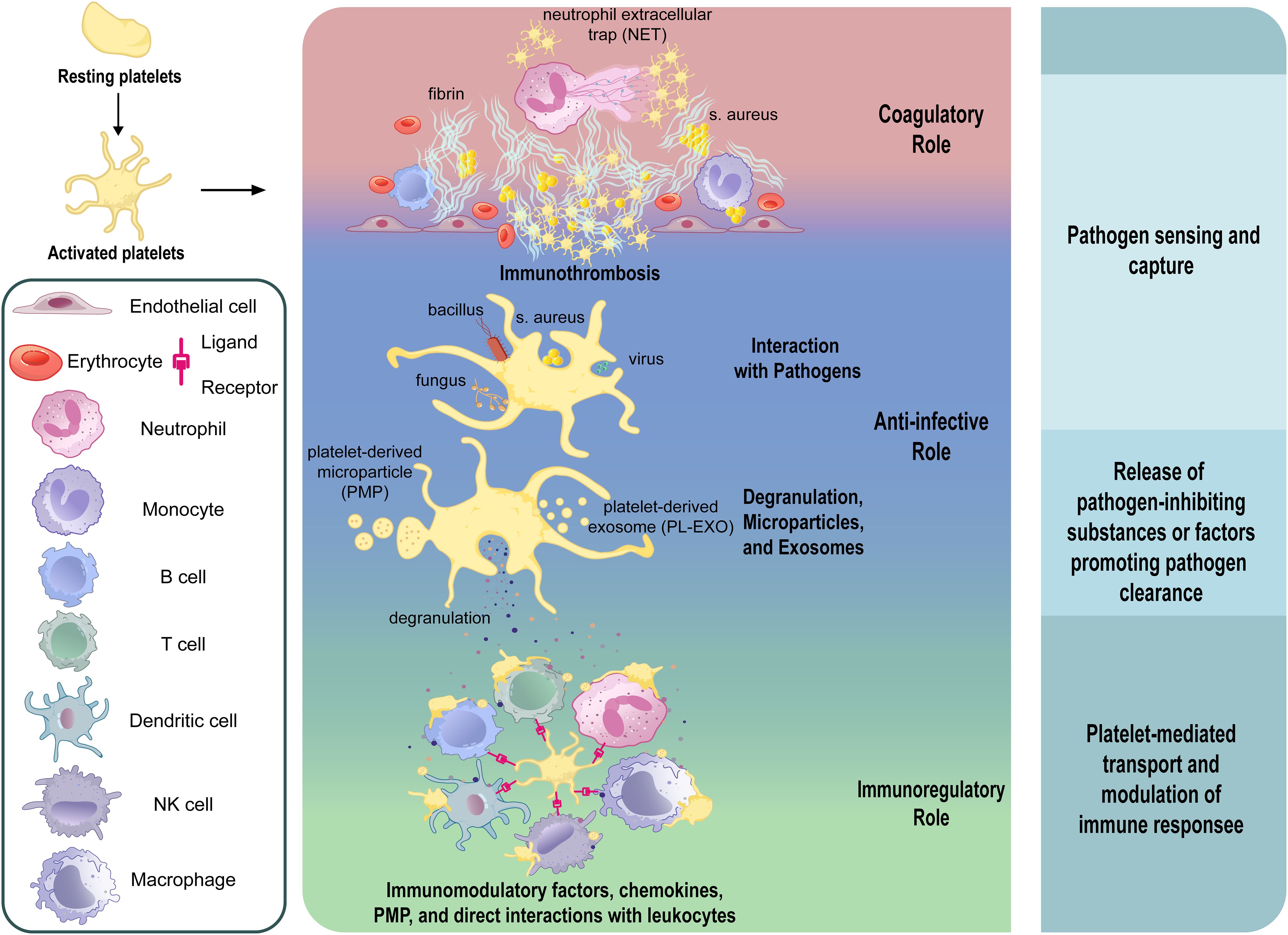
Graphical Abstract. Overview of platelet activation, its intrinsic roles and functional outcomes during infection. This schematic illustrates the transition of platelets from the resting to activated state and their multifaceted involvement in infection. Upon activation, platelets execute four major functions: (1) formation of immunothrombosis involving fibrin, neutrophil extracellular traps (NETs), and S. aureus to restrict pathogen spread; (2) direct interactions and internalization with various pathogens including bacteria, fungi, and viruses; (3) release of antimicrobial substances via degranulation and generation of platelet-derived microparticles (PMPs) and exosomes (PL-EXOs); and (4) modulation of immune responses through immunomodulatory factor release, chemokine secretion, and direct interaction with leukocytes. These activities are grouped into three functional roles—coagulatory, anti-infective, and immunoregulatory—ultimately contributing to pathogen sensing and capture, promotion of pathogen clearance, and modulation of immune responses.
Introduction
In recent years, accumulating evidence has highlighted that platelets actively participate in host immune responses—including pathogen recognition, inflammatory regulation, and tissue repair—alongside their classical roles in coagulation and thrombosis (1–7). They express an array of immune receptors, including pattern recognition receptors (PRRs) (8–11), adhesion molecules (12–15), and cytokine/chemokine receptors (16, 17), enabling platelets to directly sense and respond to pathogens and inflammatory stimuli (18–20). Upon activation by microbial components or inflammatory stimuli, platelets initiate immune defense by first capturing and internalizing pathogens through receptor-mediated recognition, targeting a broad range of microbes including bacteria, fungi, and viruses (4, 10, 21–27). They subsequently promote the formation of immunothrombosis, a coordinated intravascular response involving fibrin deposition, neutrophil extracellular traps (NETs), and platelet aggregation that serves to localize pathogens and prevent their systemic dissemination (28–30). Concurrently, activated platelets undergo degranulation, releasing antimicrobial peptides such as thrombocidins and defensins from α-granules, which exert direct microbicidal effects (31–34). In addition, platelets release platelet-derived microparticles (PMPs) and exosomes (PL-EXOs), which carry cytokines, chemokines, and immunomodulatory molecules, thereby amplifying the local immune response and facilitating crosstalk with other immune cells (35–37).
Clinically, platelet count and functionality have also emerged as significant prognostic markers in infectious diseases (38–41). Thrombocytopenia, commonly observed in severe infections and sepsis, is strongly associated with increased morbidity and mortality (39, 42–49). Additionally, disseminated intravascular coagulation (DIC), a severe complication characterized by systemic coagulation dysregulation commonly seen in advanced stages of sepsis, further underscores the critical interplay between platelets and immune-mediated pathological conditions (50, 51).
Physiologically, platelet counts range from 150 to 450 × 109/L. However, in clinical contexts such as hematologic malignancies or allogeneic hematopoietic progenitor cell transplant, a threshold of approximately 10 × 109/L is often sufficient for prophylactic platelet transfusion to prevent spontaneous bleeding (18, 52–55). This considerable functional reserve strongly suggests that platelets exert additional biological functions beyond hemostasis (18, 56–59).
Given that platelets exhibit intrinsic functional roles that extend beyond coagulation regulation to include coordinating immunological defense mechanisms during infection, understanding these diverse platelet roles and their functional outcomes in infectious diseases yields valuable insights that may inform the development of therapeutic interventions, such as platelet-mediated immunoregulatory, strategies to promote platelet regeneration, and transfusion guidance for managing thrombocytopenia in infectious conditions.
In this review, we summarize the multifaceted intrinsic roles of platelets and their functional outcomes and discuss the clinical implications of platelet depletion in infectious diseases.
Methods
To systematically summarize existing scientific evidence regarding the roles of platelets during infection, we conducted a targeted literature review using PubMed, Web of Science (ISI), and the Chinese Biomedical Literature Database (CBM), covering both English and Chinese publications up to 1 April 2025. The search strategy employed Boolean logic to combine three conceptual domains: (1) platelets and platelet transfusion-related products, (2) infection and sepsis-associated conditions, and (3) immunological mechanisms and related cellular processes. Search terms included both MeSH terms and free-text keywords and are provided in the Supplementary Material (Supplementary Table S1).
After initial retrieval, duplicate records were removed using EndNote and manual verification. We then screened titles and abstracts to identify studies of relevance, prioritizing original research and high-quality review articles that explored the intrinsic roles and functional outcomes of platelets during infection. Full-texts of eligible articles were obtained. This review synthesizes the current evidence, aiming to provide an integrated perspective on the intrinsic roles and functional outcomes of platelet activation during infection.
Roles
Coagulatory role of platelets in infection
In infection, particularly sepsis, the coagulation cascade is systemically activated through pathogen-driven and platelet-mediated mechanisms, contributing to immune thrombosis and DIC (7, 50, 60–62). Platelets activated by microbial pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS), release procoagulant factors and facilitate thrombin generation on their surfaces, significantly enhancing clot formation. Concurrently, bacterial-induced tissue damage and inflammation upregulate tissue factor (63), initiating the extrinsic pathway of coagulation (64, 65). Additionally, pathogens trigger activation of coagulation factor XII (FXII) through PAMPs, facilitating the intrinsic coagulation pathway, while activated platelets enhance NETosis, further amplifying immunothrombosis (28, 29, 66). Notably, recent evidence from SARS-CoV-2 infection demonstrates that fibrin forms complexes with the viral spike protein, generating proinflammatory clots that drive oxidative stress, immune dysregulation, and neuronal injury in both the lungs and brain (30). Despite compensatory fibrinolytic responses aimed at resolving excessive fibrin deposition, pathological impairment of fibrinolytic activity sustains persistent thrombotic states, resulting in DIC, characterized by systemic microthrombosis and bleeding, ultimately progressing toward multi-organ failure (67).
Anti-infective role of platelets in infection
Platelets exert multifaceted anti-infective capabilities through degranulation and interactions with pathogens, involving receptor-mediated binding, indirect interactions via plasma proteins, and rapid responses to microbial toxins and viral mediators.
Interactions with pathogens
Direct interactions
Platelets directly interact with pathogens through a broad repertoire of surface receptors and innate immune sensors, enabling rapid pathogen recognition, internalization, and immunoregulatory (Figure 1). For bacterial pathogens, classical integrins such as GPIIb-IIIa (68–70) and GPIbα (68, 71–73) mediate adhesion and internalization (Figure 1). IsdB protein of Staphylococcus aureus (S. aureus) (69, 74) and PadA of Streptococcus gordonii (S. gordonii) (70) directly bind GPIIb-IIIa independently of fibrinogen. GPIbα, part of a leucine-rich glycoprotein complex, binds bacterial proteins such as SrpA from Streptococcus sanguinis (72), GspB from S. gordonii (72), and SarP from S. aureus (75), facilitating pathogen capture and host defense. In addition, platelets recognize PAMPs through PRRs, including Toll-like Receptors(TLRs), C-type Lectin Receptors(CLRs), and NOD-like Receptors(NLRs) (76–79). TLR4 detects bacterial LPS, initiating platelet activation, cytokine release, and immune response modulation (80–86). Intracellular TLRs (TLR3 (87–89), TLR7 (10), TLR9 (90)) recognize viral nucleic acids, stimulating inflammatory responses (Figure 1). CLRs, notably CLEC2, mediate platelet activation critical in viral infections (91, 92), whereas NLRs, particularly NLRP3 and NOD2, enhance cytokine secretion and platelet functions upon pathogen detection (93, 94).
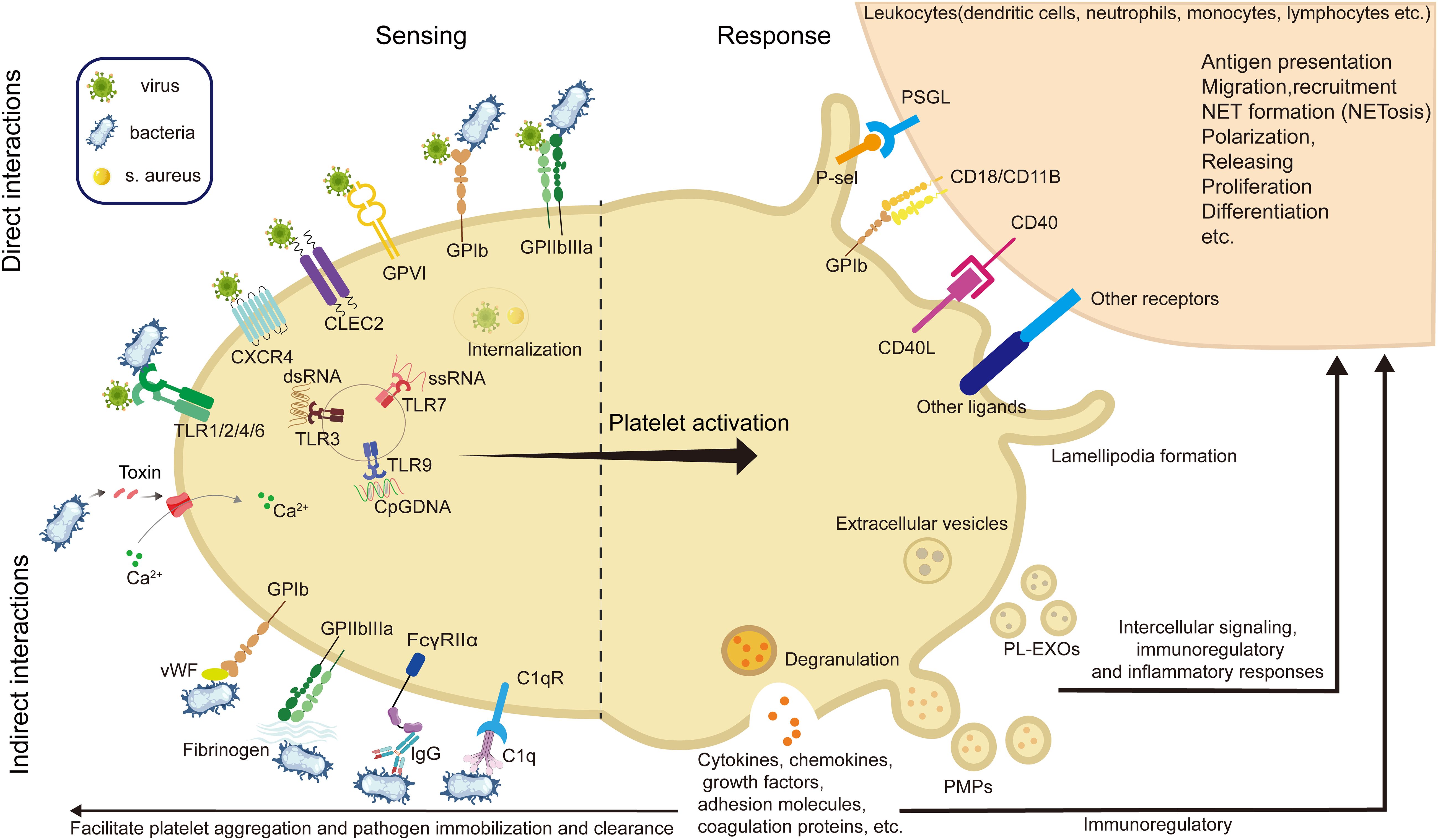
Figure 1. Diagram of platelet interactions with pathogens and downstream responses in infection. During the process of infection and inflammation, platelets sense pathogens such as bacteria and viruses through direct and indirect interactions, and respond by releasing granules and vesicles, and binding to immune cells, ultimately exerting anti-infective and immunoregulatory effects. (GPVI, glycoprotein VI; GPIb, glycoprotein Ib; GPIIb/IIIa, glycoprotein IIb/IIIa; CLEC-2, C-type lectin-like receptor 2; DC-SIGN, dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin; TLR, toll-like receptor; FcγRII, Fc gamma receptor II; CD40L, CD40 ligand; MHC-I, major histocompatibility complex class I; NETs, neutrophil extracellular traps; PMP, platelet-derived microparticles; PL-EXOs, platelet-derived exosomes).
Furthermore, viral particles can also engage platelet integrins such as GPIIb and αVβ3, which facilitate adhesion and internalization, as observed with hantaviruses and adenoviruses (95–97). Other integrins, including α2β1, mediate binding to rotaviruses (98), while coxsackievirus B utilizes the Coxsackie and adenovirus receptor (CAR) for platelet interaction (99). Platelet GPVI, classically involved in collagen sensing, has been shown to recognize hepatitis C virus (HCV) (100). C-type lectin domain family 1-member B(CLEC-2) and Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin(DC-SIGN) also serve as viral interaction points, particularly in the context of HIV and dengue virus (DENV) (101) (Figure 1). These multifaceted receptor-mediated interactions highlight the direct role of platelets in viral recognition and immune activation, complementing their antibacterial functions and reinforcing their importance in antiviral defense.
The internalization of bacteria by platelets was initially observed with S. aureus, demonstrated to occur independently of the open canalicular system (OCS) in vacuoles expressing activation markers CD62P and GPIIb-IIIa (21–24). Bacterial internalization mechanisms differ among pathogens; S. aureus requires platelet activation (e.g. by ADP or thrombin) for internalization, whereas Porphyromonas gingivalis (P. gingivalis) can independently induce its internalization by platelets via aggregation mechanisms alone (23). Similarly, Platelets internalize various viruses, including HIV (10), influenza virus (eg.H1N1) (4), DENV (102), and HCV (103, 104), and these interactions lead to distinct downstream consequences (4, 10, 25–27). Viral internalization typically triggers platelet activation and degranulation, contributing to viral clearance; however, in certain contexts, this process may instead favor viral persistence. For instance, Koupenova et al. observed HIV localized within platelet vacuoles that fuse with α-granules rich in inflammatory peptides, potentially facilitating viral degradation (10). In contrast, HCV internalization by platelets appears to shield the virus from immune clearance, limiting uncoating without promoting elimination (27). Platelets harboring DENV have shown negative-stranded viral RNA, suggesting the possibility of limited replication, though no productive infection or viral transmission to other cells has been documented (27). This duality—clearance versus concealment—likely depends on the virus type and host context, representing a promising but still underexplored area of platelet immunobiology with potential therapeutic implications.
Indirect interactions
In addition to direct receptor–ligand binding, platelets participate in pathogen recognition and clearance through a variety of indirect mechanisms mediated by plasma proteins and soluble immune components. Key among these are immunoglobulins, von Willebrand factor (vWF), fibrinogen, and components of the complement system (105–111).
Some GPIIb-IIIa binds to microbial surface components indirectly (21–24, 68–73, 112), recognizing adhesive matrix molecules (MSCRAMMs) (113), including ClfA (114), ClfB (115) and Fnbp (A and B) proteins (116) of S. aureus, and Fbl of Streptococcus lugdunensis (S. lugdunensis) (117). And GPIbα and GPIIb-IIIa, the principal platelet adhesion receptors, contribute to indirect pathogen engagement through bridging molecules such as vWF and fibrinogen, respectively (106, 118). S. aureus protein A binds to vWF, subsequently interacting with platelet GPIbα, thus promoting bacterial adhesion and aggregation (109). Glycoprotein-fibrinogen interactions further contribute to pathogen capture, involving various bacterial surface proteins such as ClfA (116), ClfB (114), FnbpA/B (117) from S. aureus, and Fbl from S. lugdunensis (115), each interacting with distinct fibrinogen domains.
Complement receptor interactions, including C1q receptor gC1q-R expressed on platelets, bind complement-coated pathogens, enhancing pathogen clearance while also marking platelets as targets for complement-mediated lysis under pathological conditions, emphasizing the dual immune and hemostatic roles of platelets (119–124). For instance, Epstein-Barr virus (EBV) binds platelet complement receptor CR2 (110), while HIV engages multiple receptors on platelets, including CXCR4, CCR1, CCR3, and CCR4, contributing to platelet activation and modulation of the antiviral response (111).
Similarly, platelets express FcγRIIa, a low-affinity receptor for IgG, which enables them to recognize and internalize IgG-opsonized pathogens, including SARS-CoV-2 (125). This internalization process facilitates fusion with α-granules that contain antimicrobial peptides, thereby promoting intracellular degradation of the pathogens (105–108, 126). Moreover, FcγRIIa mediates platelet responses to virus–antibody immune complexes (124, 127). In DENV infection, such immune complexes trigger platelet activation and aggregation via FcγRIIa, leading to efficient clearance of virus-coated platelets in the spleen, a key organ for platelet turnover and immune surveillance (128). Similarly, HCV infection, immune complexes are cleared through platelet-mediated mechanisms in the liver, underscoring the importance of tissue-specific immune environments in antiviral defense (100).
Microbial toxins and viral soluble mediators
Platelets play a critical role in host defense by responding to a variety of pathogen-derived toxins and soluble factors.
Several bacterial toxins directly interact with platelet membranes or receptors, leading to platelet activation and degranulation. For instance, α-toxin from S. aureus (129) and protease-activated receptor (PAR) activators from P. gingivalis (130) directly target platelet receptors, initiating cellular activation similar to thrombin. Other toxins, including pneumolysin from Streptococcus pneumoniae (S. pneumoniae) and streptolysin from Streptococcus pyogenes (S. pyogenes) interact with platelet membranes, potentially inducing platelet activation and degranulation, thus mediating innate defense mechanisms (106, 127, 131–133).
In viral infections, platelets can be activated indirectly via host-virus interaction intermediates. HIV-derived Trans-Activator of Transcription (TAT) protein binds to platelet CCR3 and integrin β3, promoting activation (101, 134). Additionally, the non-structural protein 1 (NS1) of dengue virus directly binds to platelet TLR4, inducing platelet activation and contributing to dengue-associated thrombocytopenia and hemorrhage (135).
In viral infections, platelets can be activated indirectly via host-virus interaction intermediates. HIV-derived Trans-Activator of Transcription (TAT) protein binds to platelet CCR3 and integrin β3, promoting activation (101, 134). Additionally, the non-structural protein 1 (NS1) of dengue virus directly binds to platelet TLR4, inducing platelet activation and contributing to dengue-associated thrombocytopenia and hemorrhage (135).
Collectively, these findings underscore the sensitivity of platelets to microbial and viral products and their participation in both proinflammatory and antiviral host responses. More details are summarized in Table 1.
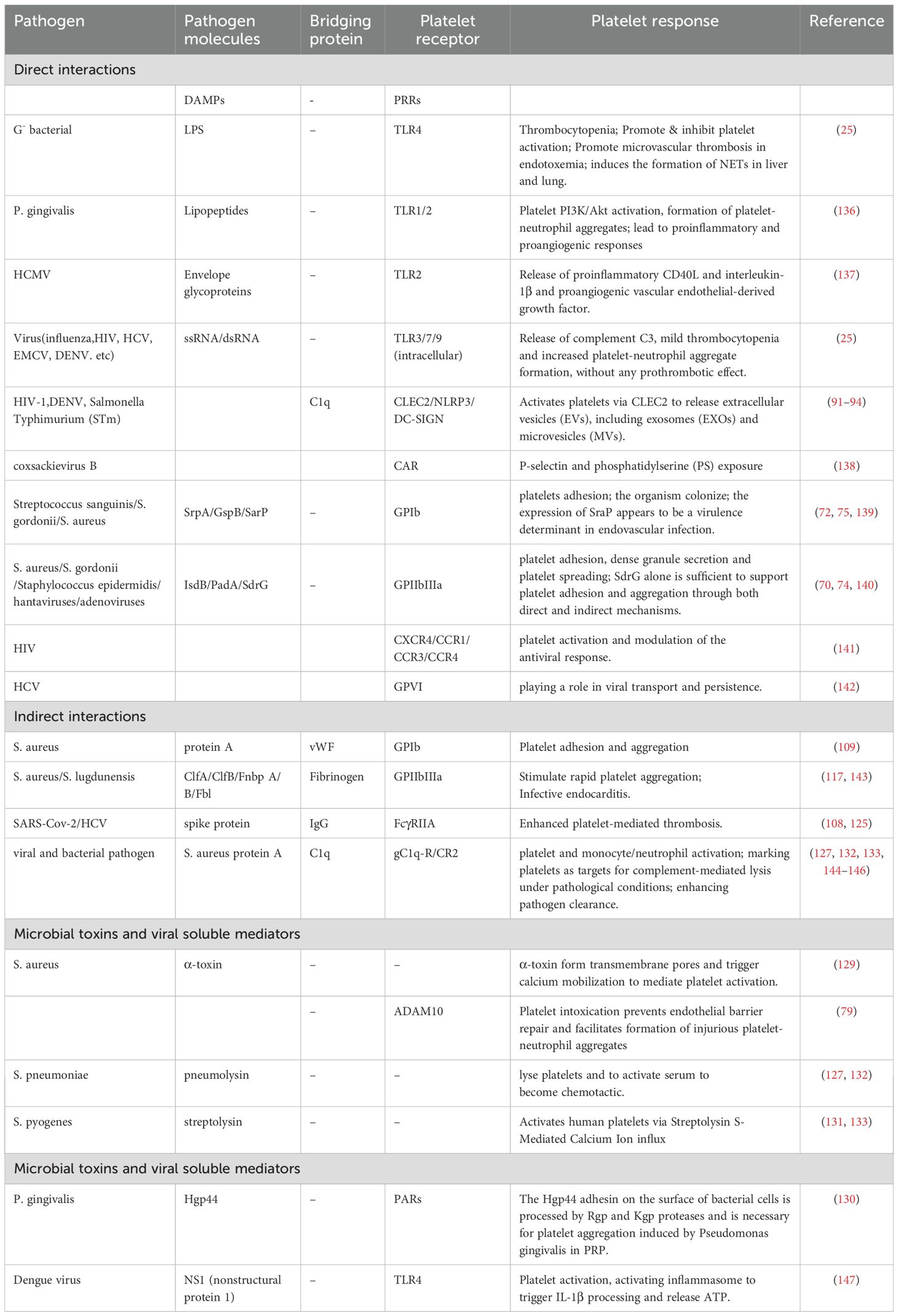
Table 1. Examples of the interaction between platelets and pathogens.
Degranulation
Upon activation, platelets release a spectrum of granule contents essential for their anti-infective functions. These granules include α-granules, dense granules, lysosomes, and specialized T-granules, each contributing distinct functions.
The α-granules, the most abundant platelet granules, contain various proteins, cytokines, chemokines, and growth factors, including adhesion molecules (GPVI, GPIIb/IIIa, GPIb-IX-V complex), coagulation proteins (e.g., vWF, fibrinogen, fibronectin), and cytokines/chemokines (e.g., platelet-derived growth factor [PDGF], transforming growth factor-β [TGF-β], CCL3, CCL5 (RANTES), and CXCL4/PF4 (31). These mediators facilitate not only platelet aggregation and pathogen immobilization but also immunoregulatory (see Section 3) (32, 33, 148). PF4 (CXCL4) binds to polyanionic structures on E. coli, undergoes conformational changes, and exposes neoepitopes that promote opsonization by anti-PF4/polyanion antibodies (128). PF4 also suppress HIV infection in some contexts (149, 150), while paradoxically enhancing HIV replication in others (151). CXCL7 is proteolytically cleaved into active fragments such as NAP-2 and connective tissue-activating peptide III (CTAP-III). Further C-terminal processing yields thrombocidins, a subclass of platelet-derived microbicidal peptides effective against S. aureus, B. subtilis, E. coli, and L. lactis (152). In addition, platelets also release Defensins, which exert direct antimicrobial effects upon release (153–155). For example, β-defensin 1 is stored in cytoplasmic compartments and released in response to S. aureus α-toxin (153), while α-defensin impairs the growth of E. coli (155). Platelets release antiviral peptides, such as PD1–PD4 and RW1–RW5, upon thrombin stimulation (156). Platelet-derived ROS and RNS (e.g., H2O2, NO3-, NO) also impair viral replication, including that of human cytomegalovirus (HCMV) (157).
Dense granules, containing smaller molecules such as ADP, serotonin, polyphosphates, histamine, and Ca²+, play a significant role in platelet activation and aggregation, thereby promoting rapid responses at infection sites (158). Platelet lysosomes, enriched with hydrolytic enzymes, are implicated in extracellular matrix degradation, receptor cleavage, and potentially autophagic clearance of pathogens (158). Additionally, platelets possess specialized T-granules containing TLR9, enabling recognition of bacterial DNA CpG sequences, thus actively participating in innate immune responses against pathogens (159).
The immunoregulatory role of platelets in infection
Platelet-derived immunoregulatory mediators
Soluble mediators and chemokines
Upon activation, platelets release a diverse array of immunomodulatory factors, including. vasoactive agents like serotonin (142) and platelet-activating lipids like TXA2 (160) and PAF (161), which modulate endothelial and immune cell activity. Growth factors such as PDGF (162) and TGF-β (141, 163–166) influence monocyte differentiation and lymphocyte regulation, while platelet-derived chemokines, including CXCL7, PF4, and CCL5, orchestrate neutrophil and monocyte recruitment, reinforcing host defense (167–170). Notably, PF4 also promotes the formation of neutrophil extracellular traps (NETs), thereby enhancing bacterial clearance (171), and supports antiviral immunity by recruiting leukocytes to the lungs during influenza infection (172) and modulating interferon responses during flavivirus infections such as dengue and Japanese encephalitis (173). Recognition of viral single-stranded RNA (e.g., from HIV or influenza) via TLR7 induces release of α-granule contents, CD40L, and P-selectin, also enhancing platelet-neutrophil aggregation and NET formation (174, 175). In parallel, activation of the NLRP3 inflammasome promotes cytokine secretion, while platelet-derived complement C3 amplifies neutrophil responses through further NET induction (4, 93, 119). Platelets also secrete antimicrobial peptides like thrombocidins and cytokines including IL-1β (146, 176), high mobility group box 1(HMGB1) (177, 178), and soluble CD40L ligand (sCD40L) (179) amplify immune signaling. As reservoirs of these immunomodulatory mediators, platelets not only regulate inflammation but also store and release a wide range of chemokines and cytokines that influence immune cell recruitment, wound healing, immune tolerance, and tumor metastasis (5, 111, 180, 181). Expressing multiple chemokine and cytokine receptors, platelets actively sense and respond to inflammatory signals, positioning them as key intermediaries linking innate and adaptive immunity.
Extracellular vesicles
Activated platelets also release extracellular vesicles classified into two main types: PMPs. and PL-EXOs (34), which are actively involved in immunoregulatory.
PMPs first reported in 1946 (182), represent the most abundant circulating microvesicle population, identifiable through enrichment via centrifugation and characterized by retained procoagulant activities (35, 36, 183). PMPs are heterogeneous vesicles released from the platelet plasma membrane, with diameters ranging from 100 nm to 1 µm (184). Under conditions of platelet activation or apoptosis, vesicles form through budding at specific sites of the cell membrane and eventually detach; some pseudopodia fragment, releasing debris into the bloodstream, thereby generating PMPs. This formation process involves calcium ion influx, phosphatidylserine exposure, and the Bax/caspase signaling pathway (185, 186). PMPs contain bioactive lipids (e.g., phosphatidylserine, tissue factor, arachidonic acid), surface proteins (e.g., CD41, CD31, P-selectin), and various microRNAs, supporting roles in coagulation, vascular repair, and immunoregulatory (187). Functionally, PMPs exert potent immunomodulatory effects. They activate neutrophils and endothelial cells via CD62P, enhancing neutrophil-endothelium adhesion (35), and facilitate monocyte recruitment through transfer of GPIbα (36). In viral infections, platelet activation by dengue virus (DENV) or SARS-CoV-2 triggers increased PMP release through CLEC-2 (91) or CD47 (188), respectively. These PMPs subsequently activate neutrophils and macrophages via TLR2 and TLR4 signaling, promoting NETosis and cytokine release, thereby amplifying inflammation (37). While PMP elevation is observed in many pathological states, the precise immunoregulatory mechanisms remain incompletely understood and merit further investigation.
PL-EXOs typically have diameters smaller than 100 nm, originating from early endosomes and multivesicular bodies (MVBs). They are released through fusion with the platelet cell surface and the OCS (184), a process dependent on ESCRT (138). PL-EXOs are enriched in tetraspanins (CD63, CD9, CD81) and endosomal sorting complex-related proteins (TSG101) (189). Additionally, exosomal cargo includes both mRNA and miRNA, exhibiting diverse types and abundant content (138). These vesicles mediate critical intercellular signaling, modulate immune and inflammatory responses, and facilitate tissue repair through miRNA delivery and receptor transfer (190, 191).
Moreover, PMPs and PL-EXOs can interact with distant tissues, including bone marrow, lymph nodes, and synovial fluids, highlighting their role in systemic communication during infections and other inflammatory conditions (138, 192).
Platelet-mediated immune cell interactions
Platelets profoundly modulate immune responses through dynamic interactions with. innate and adaptive immune cells, facilitated primarily by direct cell-cell contacts and the release of immunomodulatory mediators. Platelets significantly modulate immune responses through interactions with various leukocytes, including dendritic cells (144, 145, 193), neutrophils (35, 71, 194, 195), monocytes (58), lymphocytes (196), even mast cell (197).
Innate immunity
Among innate immune cells, platelets play a pivotal role in regulating neutrophil. function. by forming platelet–neutrophil aggregates via P-selectin/P-selectin glycoprotein ligand 1(PSGL-1) and glycoproteins such as GPIbα and GPIIb, which promote neutrophil adhesion and transmigration to sites of inflammation (138, 185, 193, 194). They also induce neutrophil extracellular trap (NET) formation, a critical mechanism for trapping and neutralizing pathogens. In addition, platelet-derived chemokines like PF4 and CCL5 (RANTES) enhance neutrophil recruitment and activity, particularly under conditions such as acute lung injury (35, 71, 194, 195, 198–202). Recent studies have unveiled key mechanisms linking platelet activation to NETosis and immunothrombosis during sepsis. For instance, STING signaling in platelets has been shown to amplify granule secretion and intravascular thrombosis (203), while gasdermin D(GSDMD)-mediated platelet pyroptosis (66), driven by S100A8/A9–TLR4 signaling, promotes the release of oxidized mitochondrial DNA that enhances NET formation. These mechanisms establish a pathogenic feedback loop between platelets and neutrophils, contributing to excessive inflammation and tissue injury (Figure 2).
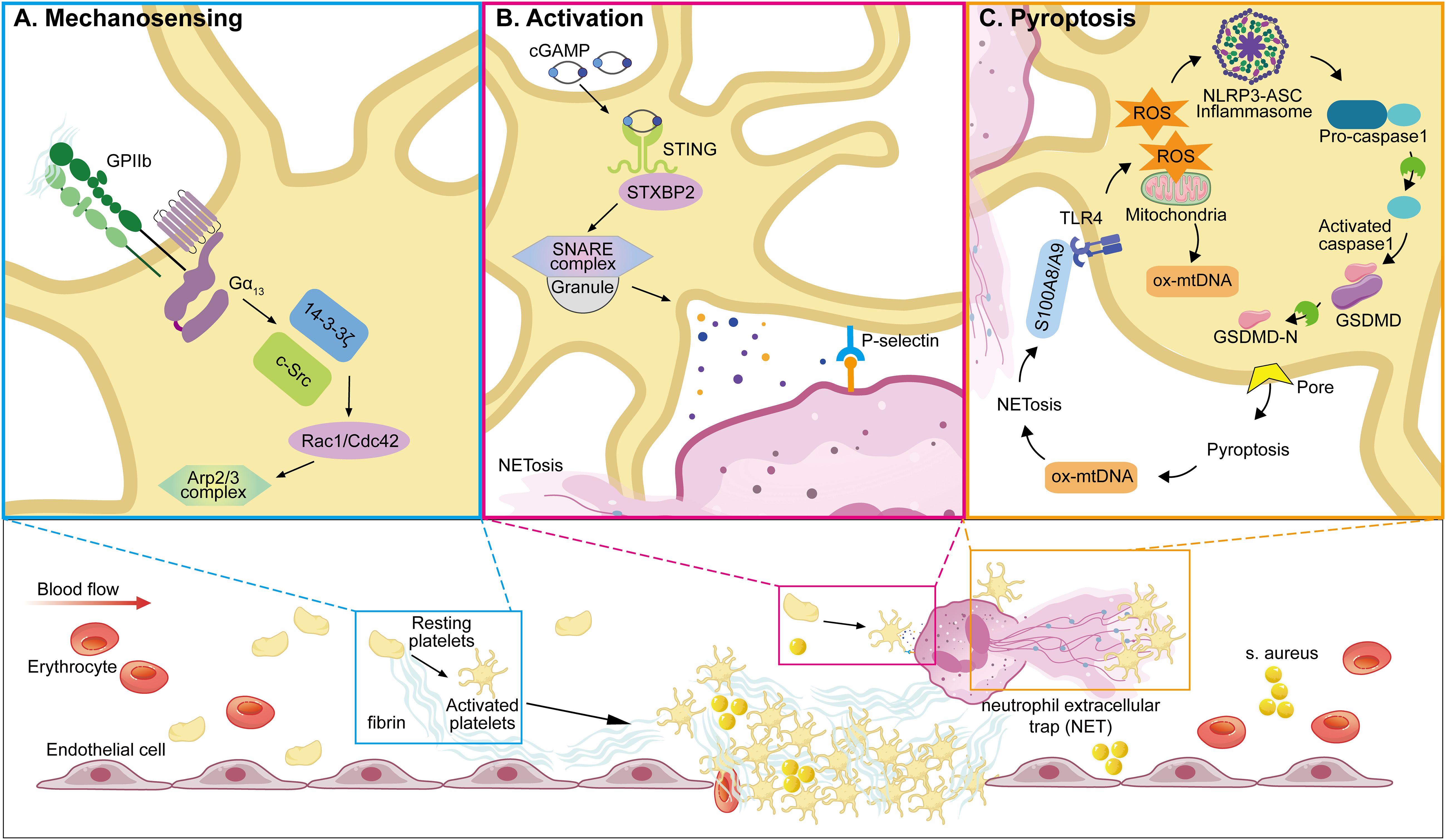
Figure 2. Representative molecular pathways involved in platelet resistance to infection. This illustration summarizes three recent reported, sequentially linked molecular pathways by which platelets participate in infection: (A) Mechanosensing-mediated migration (204). Upon vascular injury or inflammation, platelet integrin GPIIb senses fibrin(ogen)-rich matrix exposure and initiates mechanosensing through Gα13–c-Src–14-3-3ζ signaling. This promotes platelet polarization and lamellipodia formation, enabling directional migration toward sites of endothelial damage—a critical early step in immune hemostasis. (B) STING-dependent activation and NETosis (203). In sepsis, platelets are activated via STING (stimulator of interferon genes), which is triggered by cytosolic cGAMP or mitochondrial DNA. Activated STING interacts with the SNARE machinery through STXBP2 to promote granule secretion (e.g., P-selectin), facilitating platelet–neutrophil interactions and subsequent formation of NETs. (C) GSDMD-mediated pyroptosis (66). As inflammation escalates, platelet pyroptosis is induced through a S100A8/A9–TLR4–NLRP3 axis. Mitochondrial ROS trigger inflammasome assembly, leading to caspase-1 activation and cleavage of GSDMD. The resulting GSDMD-N fragments form membrane pores, causing pyroptotic cell death and the release of oxidized mitochondrial DNA (ox-mtDNA), which further enhances NET formation. NETs in turn release S100A8/A9, establishing a feedforward loop that amplifies platelet pyroptosis and inflammatory signaling. (GPIIb, glycoprotein IIb (integrin αIIb); Src, proto-oncogene tyrosine-protein kinase Src; 14-3-3ζ, 14-3–3 zeta protein; STING, stimulator of interferon genes; STXBP2, syntaxin-binding protein 2; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; cGAMP, cyclic GMP-AMP; NETs, neutrophil extracellular traps; TLR4, toll-like receptor 4; NLRP3, NOD-like receptor family pyrin domain-containing 3; GSDMD, gasdermin D; ox-mtDNA, oxidized mitochondrial DNA; ROS, reactive oxygen species; IL-1β, interleukin-1 beta; ASC, apoptosis-associated speck-like protein containing a CARD; PMNs, polymorphonuclear neutrophils).
For monocytes, activated platelets form platelet–monocyte aggregates (PMAs) primarily via P-selectin–PSGL-1 binding, with co-stimulatory interactions involving CD40L–Mac-1 and GPVI–CD147,driving monocyte polarization toward an inflammatory M1 phenotype, amplifying cytokine secretion and contributing to immune activation in conditions like sepsis (58, 205–207). Conversely, in a murine model of systemic inflammation, the interaction of CLEC-2 on platelets with podoplanin on tissue-resident macrophages establishes an anti-inflammatory axis that mitigates immune cell infiltration and preserves vascular integrity during sepsis (208).
Platelets also engage with dendritic cells (DCs) through axes such as CD40–CD40L, P-selectin–PSGL-1, and JAM-C–Mac-1, promoting DC maturation, antigen internalization, and subsequent presentation to T lymphocytes—thereby bridging innate and adaptive immunity (144, 145, 168, 193, 209–213). Additionally, platelet-derived mediators such as CXCL4 and soluble CD40L (sCD40L) reinforce DC functions by upregulating costimulatory molecules and pro-inflammatory cytokines (210, 211).
Although less extensively studied, mast cells are another innate immune population influenced by platelet interaction (197). These interactions may modulate early inflammatory signaling and histamine release, further integrating vascular responses into immune defense.
Adaptive immunity
In adaptive immunity, platelets bridge innate signals and adaptive responses by directly, store and express substantial amounts of functional major histocompatibility complex class I (MHC-I) molecules, facilitating antigen processing and presentation to CD8+ T cells upon activation, thus enhancing adaptive immune responses (214). In T cells, platelet-derived CXCL4 (PF4) and serotonin are key immunoregulatory factors (5, 8, 215–217), while surface molecules such as CD62P, GPIIb/IIIa, and CD40L facilitate direct interaction with activated T lymphocytes (218–220). These interactions influence T-cell activation, proliferation, and trafficking to secondary lymphoid organs, with context-dependent outcomes—platelets may enhance or suppress T-cell effector functions depending on the immune context (216, 221–225). Moreover, platelets interact with B-cells via the CD40-CD40L axis, significantly affecting B-cell proliferation, differentiation, class switching, and memory formation, thereby modulating antibody-mediated immunity (199, 217, 226, 227). Conversely, platelets also contribute to inflammation resolution. In a murine model of bacterial pneumonia, they promote Treg expansion and macrophage polarization via TGFβ and CD62P-sCD40L interactions (179). Platelets further enhance T helper (Th)1, Th17 differentiation of CD4+ T cells via cell–cell contacts and release of PF4, CCL5 (RANTES) and TGFβ (223).
Furthermore, in shaping humoral immunity, platelet interactions with natural killer (NK) cells—mediated by immune checkpoint molecules like GITRL and RANKL—can suppress NK cell cytotoxicity and IFN-γ production, modulating immune responses under both infectious and neoplastic conditions (211, 212).
Interestingly, lymphocytes can also influence platelet biology: through the use of platelet-derived prostaglandin H2 (PGH2), lymphocytes synthesize PGI2, a potent inhibitor of platelet activation. This bidirectional regulation highlights the complex interplay between platelets and lymphocytes in shaping adaptive immunity (228, 229).
Transfusion-related immunomodulation
Instead of immunoregulatory functions of endogenous platelets, transfused allogeneic platelets can also exert potent immunomodulatory effects, which may be either beneficial or detrimental depending on the clinical context. TRIM refers to immune alterations associated with blood transfusions, encompassing immunosuppressive and pro-inflammatory effects potentially contributing to adverse clinical outcomes such as cancer recurrence, postoperative infections, multiorgan failure, and increased mortality (230, 231).
A large single-center cohort study demonstrated that allogeneic platelet transfusion during cardiac surgery was significantly associated with an increased risk of bloodstream infections, but showed no significant correlation with hospital-acquired pneumonia or surgical site infections. This suggests that platelet transfusion may selectively increase the risk of specific infections through immunomodulatory mechanisms (232). Kah et al. reported a case of an acute lymphoblastic leukemia patient who developed disseminated Fusarium infection and endogenous fungal endophthalmitis following leukocyte-depleted platelet transfusion. This supports the hypothesis that platelet-derived immunomodulatory factors may compromise host immune defenses through TRIM mechanisms, thereby increasing susceptibility to opportunistic infections (233). Regarding immunosuppressive effects, Sadallah et al. revealed that extracellular vesicles from stored platelets can redirect monocyte differentiation towards immature dendritic cells (iDC), which subsequently mature into DC, while simultaneously downregulating inflammatory responses in human macrophages (234). Using an in vitro whole-blood transfusion model, Perros et al. demonstrated that platelets modulate immune responses by suppressing DC-associated pro-inflammatory cytokines through soluble mediators while enhancing the anti-inflammatory cytokine IL-10 (235).
Multiple factors underlie TRIM, including the transfusion of allogeneic monocytes, soluble leukocyte-derived mediators, and circulating soluble HLA peptides within allogeneic plasma (230). Platelets have emerged as significant contributors to TRIM, with transfusion-associated platelet-derived molecules such as sCD40L, soluble OX40 ligand (sOX40L), soluble MHC class I (sMHC-I), and soluble FAS ligand (sFASL) increasing during storage and potentiating proinflammatory and immunomodulatory effects following transfusion (5, 6, 230, 236–242).
Functional outcomes
Pathogen sensing and capture
Upon vascular injury in infection, platelet integrin GPIIb senses fibrin(ogen)-rich matrix exposure and initiates mechanosensing through Gα13–c-Src–14-3-3ζ signaling (204)(Figure 2). This promotes platelet polarization and lamellipodia formation, enabling directional migration toward sites of endothelial damage and forming immunothrombosis (204),which represents a critical component of the innate intravascular immune response, exerting multiple protective functions including pathogen containment, elimination, and immune coordination (243, 244). It achieves pathogen capture and confinement primarily through fibrin network formation within thrombi, preventing pathogen dissemination and tissue invasion (245). Additionally, the localized thrombotic environment supports innate immune cell recruitment and releases antimicrobial peptides at sites of pathogen entrapment, thereby enhancing pathogen clearance and immune defense mechanisms (3).
Platelets, which possess cellular structures facilitating virus attachment, internalization, and replication, further contribute to antiviral defense by sensing viral components through PRRs (3). Upon virus binding and internalization, platelets become activated, triggering granule secretion and promoting platelet-neutrophil interactions, which collectively enhance antiviral responses. For instance, in mouse models, platelets detect encephalomyocarditis virus via TLR7, resulting in significant platelet-neutrophil aggregate formation and rapid platelet consumption, leading to protective immunity (10). Similarly, during influenza infections, platelet-mediated virus internalization through TLR7 initiates the release of complement component C3, inducing neutrophil DNA release and aggregation, thus underscoring platelet-neutrophil crosstalk as a critical mechanism in orchestrating host immune and complement responses (4, 10).
Platelet-mediated transport and induction of immune responses
Platelets significantly contribute to adaptive immune responses by facilitating pathogen transport and antigen presentation. Some platelet-bound bacteria persist in circulation long enough to be transported to the spleen, where they are recognized by CD8α+ dendritic cells, subsequently eliciting cytotoxic T-cell responses (246, 247). Similarly, in DENV infection, immune complexes trigger platelet activation and aggregation via FcγRIIa, leading to the efficient clearance of virus-coated platelets in the spleen, a key organ for platelet turnover and immune surveillance (128). In the case of HCV infection, immune complexes are cleared through platelet-mediated mechanisms in the liver (100).
In addition to transport, platelets influence pathogen fate via surface receptors. Platelet-expressed GPIb, for example, modulates the handling of pathogens opsonized by complement factor C3b. While C3b-coated bacteria are typically cleared by macrophages in the spleen, engagement by platelet GPIb redirects these complexes toward splenic dendritic cells, thereby enhancing adaptive immune responses (248, 249).
Moreover, megakaryocytes—precursors of platelets—express MHC class I molecules and actively process and cross-present antigens on their surface, initiating CD8+ T-cell activation and proliferation; during thrombopoiesis, these antigen-loaded MHC class I complexes are transferred to proplatelets (250). Given that platelets and megakaryocytes harbor all components necessary for antigen processing and presentation, platelets can directly interact with T-cells and also facilitate B-cell maturation and antibody class switching. Collectively, these mechanisms underscore the intricate interactions among platelets, antigen-presenting cells, and lymphocytes, highlighting the crucial role of platelets in orchestrating pathogen-specific adaptive immunity.
Release of pathogen-inhibiting substances or factors promoting pathogen clearance
Upon activation, platelets release multiple substances that directly inhibit pathogen growth or facilitate pathogen clearance. Platelet-derived β-defensins, a group of cationic antimicrobial peptides, inhibit bacterial proliferation by disrupting membranes and induce NETosis (153); platelets aggregate around pathogens such as S. aureus, release β-defensins, and trigger NETosis to effectively trap and neutralize the bacteria (66, 153). Similarly, thrombocidins—truncated variants of neutrophil-activating peptide-2 (NAP-2) originally isolated from platelet granules—demonstrate potent bactericidal effects against diverse bacterial strains, including B. subtilis, L. lactis, and possess fungicidal activity against C. neoformans (152). Moreover, platelet-secreted cytokines such as IL-1β, released upon bacterial LPS stimulation or viral infection, augment bacterial phagocytosis (3, 93, 251) and further amplify macrophage-derived IL-1β production, reinforcing antimicrobial defenses (252). Additionally, platelet-expressed GPIb modulates the fate of pathogens opsonized by complement factor C3b; typically, macrophages clear C3b-coated bacteria in the spleen, yet if platelets engage bacteria through GPIb, the platelet-pathogen complexes are redirected towards splenic dendritic cells, enhancing adaptive immune responses (248, 249). Collectively, these platelet-driven antimicrobial pathways significantly contribute to pathogen containment and the orchestration of both innate and adaptive immunity.
Discussion
In this review, we have comprehensively outlined the intrinsic roles of platelets during infection and their corresponding functional outcomes. Platelets actively engage in immune defense by employing PRRs such as TLRs to detect pathogens (18, 19), initiating responses like immunothrombosis to limit pathogen spread (28, 29). Activated platelets undergo extensive degranulation, releasing antimicrobial peptides such as thrombocidins and β-defensins, as well as cytokines like IL-1β and various chemokines, thereby enhancing immune cell recruitment and effectively orchestrating innate and adaptive immune responses (31–34). These multifaceted mechanisms highlight platelets as central players not only in preserving vascular integrity but also in coordinating robust immune defenses during infectious states.
Platelets are essential intravascular sentinels capable of rapidly responding to pathogens or other abnormalities. Upon pathogen encounter, platelets initiate immunothrombosis, effectively restricting pathogen spread and promoting pathogen clearance through transport to immune-rich organs such as the liver and spleen (246, 247). Similarly, in malignant neoplasm pathophysiology, by shielding circulating tumor cells (CTCs) from shear stress, mediating immune evasion, and neoangiogenesis, platelets may facilitate metastasis (253), highlighting platelets’ broader biological functions beyond hemorrhage prevention and their intricate role in clinical scenarios like sepsis-associated thrombocytopenia (SAT).
Clinical evidences and implications
Thrombocytopenia is a common complication in patients with sepsis (254, 255). Clinical data demonstrates that the prevalence of SAT varies among different research in intensive care units (ICUs), ranging from 10% to 83.5% (44, 46, 48, 256–262). Studies have shown that thrombocytopenia in sepsis patients correlates with increased mortality rates and extended ICU stays (263, 264). Furthermore, evidence indicates that persistent thrombocytopenia has an association with poor clinical outcomes (263). Recent research underscores the prognostic significance of both static and dynamic platelet indices in sepsis. For instance, Chen et al. reported that a lower platelet count (PC) and higher mean platelet volume (MPV) were independently associated with increased risks of intraventricular hemorrhage and mortality in preterm infants (265), and that transfusions at higher PCs may paradoxically increase adverse outcomes (265–267). Wang et al. identified distinct platelet trajectory subphenotypes in adult septic patients, showing that stable or declining trajectories during the first four ICU days were independently associated with increased 28-day mortality compared to ascending patterns, with thrombocytopenia mediating up to 37% of this risk (268). Cheng et al. confirmed that SAT, particularly when severe or persistent, correlates with higher in-hospital mortality in patients with sepsis-induced coagulopathy, namely, DIC (42). Similarly, Ye et al. emphasized that dynamic monitoring of platelet counts, rather than single time-point measurements, enhances the predictive accuracy for hospital mortality in sepsis, underscoring the importance of incorporating longitudinal platelet indices into clinical risk stratification models (28).
Platelet transfusion remains the most effective and widely adopted intervention to raise circulating platelet levels and is considered a cornerstone of SAT management (269–271). However, the optimal prophylactic transfusion threshold remains controversial (268, 272–274), and current clinical guidelines are hindered by the lack of high-quality evidence to support a standardized approach (275–278). Furthermore, emerging evidence indicates that a lower transfusion threshold (platelet ≤ 20x109/L) may not confer significant clinical benefits in SAT patients (38, 271, 279, 280).
Regarding antiplatelet therapies, several clinical investigations have explored their potential role in infection-associated coagulopathy, particularly in the context of COVID-19. A meta-analysis involving 87,824 patients suggested that antiplatelet therapy might be associated with lower mortality in COVID-19 based on observational data (OR: 0.72, 95% CI: 0.61–0.85) (281). However, randomized controlled trials did not confirm a clinical benefit of adding antiplatelet therapy to standard care, regardless of baseline illness severity or concomitant anticoagulation (282–285).
Regarding anticoagulant therapies, recombinant human thrombomodulin (rhTM), an anticoagulant targeting excessive thrombin generation, has been widely used in Japan for sepsis-associated DIC (286). However, the SCARLET trial failed to demonstrate a mortality benefit of rhTM in a broader population with sepsis-associated coagulopathy, suggesting that the degree of coagulopathy in these patients may have been insufficient to benefit from this therapy (287). Complementary to pharmacologic strategies, Olas reviews the potential of natural phenolic compounds—such as resveratrol, curcumin, and quercetin—for their anti-platelet, antioxidant, and anticoagulant properties (288). Although these compounds show promise in modulating hemostasis and reducing oxidative stress in cardiovascular disease, their clinical relevance in COVID-19 remains speculative and largely unsubstantiated by in vivo evidence (288). About the immunoregulatory therapies for platelet, clopidogrel was recently used to successfully inhibit platelet inflammasome assembly and, thus, the release of pro-inflammatory IL-1β and IL-18 under septic conditions resulting in improved renal function (56, 176).
Hypothesis
Given the complex and dynamic roles of platelets in infection revealed in this review, we propose that platelet function during sepsis is not static, but evolves in tandem with disease progression. Early in sepsis, immune hyperactivation predominates, whereas in later stages, patients may transition to an immunosuppressed state (3–5, 289, 290), often complicated by the development of DIC (1, 2, 61, 291, 292). Therefore, the predominant role of platelets likely shifts over time—from immune surveillance and modulation in early disease to consumptive coagulopathy during advanced stages. This temporal heterogeneity underpins the need for stage-specific strategies.
Based on current evidence regarding the prognostic utility of platelet indices (38, 42, 263, 268)—including absolute counts and dynamic trajectories—we hypothesize that SAT may encompass two distinct phases. Initially, platelet consumption may reflect their active participation in anti-infective and immunoregulatory processes. In patients with disease progression, however, a secondary phase may emerge where platelets are rapidly consumed due to microthrombus formation in the context of overt or subclinical DIC.
This leads to a conceptual therapeutic implication: prophylactic platelet transfusion should be considered early to preserve platelet numbers and functionality, potentially preventing irreversible transition to DIC. Interventions aimed at preserving or restoring platelet function may be more effective in this “pre-DIC window”. In contrast, antiplatelet and anticoagulant agents—although beneficial in certain coagulopathic settings—may exacerbate functional platelet inhibition that is difficult to reverse in critically ill patients. Moreover, in the context of DIC progression, platelet dysfunction induced by pharmacologic agents which are lack of reversal agents (293), may not be mitigated by transfusion alone, possibly heightening the risk of spontaneous hemorrhage (293–296).
Limitations
Despite significant advances in understanding the intrinsic roles and functional outcomes during infection, several limitations remain that may impact the interpretation and generalizability of current findings.
First, a major constraint lies in the scarcity of human interventional data. Much of the mechanistic evidence regarding platelet immune functions originates from murine models or in vitro studies, which may not fully recapitulate the complexity of human immunopathology. Notably, species-specific differences in platelet receptor expression and signaling pathways can lead to divergent immune outcomes. For example, murine platelets express higher levels of TLR4—facilitating stronger responses to LPS—whereas human platelets exhibit reduced TLR4 expression and a less pronounced pro-inflammatory profile (297, 298). Moreover, discrepancies in intracellular signaling molecules (e.g., TLN1, CALM3, PRKCB in humans vs. RASGRP2, ITGB2, MYL9 in mice) may modulate platelet reactivity and immune crosstalk (299).
Second, variability in platelet preparation protocols, such as the use of different agonists in ex-vivo (e.g., thrombin, LPS), anticoagulants, and storage conditions, can influence platelet activation states and introduce inconsistencies across studies, whereas in vivo models like ARDS or S. pneumoniae infection demonstrate platelet-mediated tissue protection and resolution (300, 301). These technical variables may exaggerate or obscure specific immunological phenotypes.
Third, the inflammatory context and disease phase profoundly affect platelet function. In acute infection models such as CLP-induced or LPS-induced sepsis, platelets tend to exhibit pro-inflammatory behaviors, promoting NETosis, cytokine release, and immune cell recruitment (298, 302, 303). In contrast, chronic or resolving models—such as cancer-associated inflammation or post-viral recovery—often reveal platelet-mediated resolution and immune regulation, including promotion of Treg expansion or macrophage polarization (298, 302, 303). This functional dichotomy is not contradictory, but rather reflects the dynamic and context-dependent nature of platelet roles in immunity.
Finally, methodological heterogeneity across studies—including model choice, timing of sampling, and outcome assessment—further complicates data integration and cross-comparison.
Future investigations should aim for standardized protocols and longitudinal designs to better delineate the immunological spectrum of platelet function in infection.
Perspectives
Basic and translational research
Future research, including basic and translational research, needs to emphasize the primary roles and diverse functional outcomes of platelets in driving disease progression across various clinical conditions, particularly in bloodstream infections frequently observed in sepsis. And combining acute and chronic inflammation models, and employing a diverse array of experimental techniques—from in vivo imaging to single-cell omics—will be essential to dissect the multifaceted roles of platelets. Particular attention should be given to the temporal dynamics of platelet activity, as their contribution likely varies across different stages of inflammation, including initiation, propagation, and resolution. Cross-species comparative studies may also provide valuable insights into the evolutionary conservation and diversification of platelet-mediated immune regulation, thereby informing both basic mechanistic understanding and translational applicability in humans.
Diagnose
Emerging evidence suggests that platelets, beyond their classical hemostatic functions, possess diagnostic utility in infectious diseases through their immunological responsiveness. They express a range of PRRs, including (76–79), which enable the detection of microbial components and DAMP. Differential activation of these receptors—for example, TLR4 in sepsis or TLR7 in viral infections—may serve as a cellular signature of pathogen type. Then, upon activation, platelets release immunomodulatory mediators such as PF4, CD40L, P-selectin, and IL-1β (31, 146, 174–176, 179), which are detectable in plasma and correlate with disease severity, offering potential as accessible biomarkers. Furthermore, specific pathogens induce distinct platelet responses—for instance, dengue virus activates the NLRP3 inflammasome via TLR4 (93, 94), while viruses such as HIV and HCV interact through CLRs (91, 92). These mechanistic differences may help discriminate between bacterial and viral infections.
Together, these features position platelet-derived signatures as promising diagnostic indicators for pathogen profiling, immune status assessment, and infection severity stratification.
Therapy
Given the central involvement of platelets in inflammation, host defense, immune. modulation, and coagulation, it is imperative to refine evidence-based protocols for prophylactic platelet transfusion, as well as to evaluate the therapeutic use of platelet-activating agents and anti-platelet drugs. As underscored by the 2022 NHLBI and OASH Transfusion Medicine State of Science Symposium (304), a deeper understanding of how donor and recipient characteristics influence not only hemostatic but also non-hemostatic platelet functions—such as immune regulation, inflammatory response, and vascular repair—may yield critical insights for clinical practice. These evolving clinical and experimental observations also support the hypothesis that transfused platelets can modulate immune responses, urging further elucidation of the molecular mediators involved in transfusion-related immunomodulation (TRIM).
Targeting the immunoregulatory functions of platelets—beyond their traditional hemostatic roles—represents a promising frontier in therapeutic innovation. Interventions that modulate platelet–immune cell interactions, such as inhibition of inflammasome assembly or disruption of platelet–leukocyte aggregates, hold potential to deliver immune benefits without compromising hemostasis (305). Notably, clopidogrel has recently demonstrated efficacy in suppressing platelet inflammasome activation, thereby reducing the release of IL-1β and IL-18 and improving renal outcomes in sepsis models (56, 176). These findings exemplify the translational promise of platelet-directed immunotherapies and highlight the need for further mechanistic and clinical exploration.
Conclusion
In summary, platelets function far beyond hemostasis, actively bridging infection sensing, immune coordination, and pathogen elimination. These insights suggest novel avenues for immunomodulatory strategies in infection-related clinical scenarios. As our understanding of platelet immunobiology continues to evolve, targeting their immunoregulatory functions opens new avenues for diagnostics, prognostics, and therapeutics in infectious diseases. Future research should prioritize the temporal dynamics of platelet function and explore stage-specific interventions to optimize both immune support and vascular integrity in critically ill patients.
Author contributions
YH: Visualization, Project administration, Writing – original draft, Investigation, Conceptualization, Methodology. SD: Visualization, Writing – original draft. CQ: Writing – original draft, Funding acquisition. YFY: Investigation, Writing – original draft. JR: Investigation, Writing – original draft. KW: Writing – review & editing, Supervision, Funding acquisition. LL: Supervision, Writing – review & editing, Project administration, Funding acquisition. ZL: Conceptualization, Supervision, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the CAMS Innovation Fund for Medical Sciences (grant number 2021-I2M-1-060), the Dazhou Science and Technology Program Key Research and Development Project (project number 23ZDYF0024), the Sichuan Province Science and Technology Program (grant number 2023NSFSC1475), the Health Commission of Sichuan Province (grant number 2023-207), and the Scientific Research Project of the Third People’s Hospital of Chengdu (project number 2023PI10).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1616783/full#supplementary-material
Abbreviations
CLEC-2, C-type lectin-like receptor 2; CCR, CC chemokine receptor; CXCL, CXC chemokine ligand; CR,complement receptor; Chemokine (C-X-C motif) receptor 4(CXCR-4);GPVI, glycoprotein VI; GPIbα, glycoprotein Ib alpha; JAM-C, junctional adhesion molecule-C; MSCRAMM, microbial surface components recognizing adhesive matrix molecules; NET, neutrophil extracellular trap; NETosis, neutrophil extracellular trap formation; PL-EXO, platelet-derived exosome; PMP, platelet-derived microparticle; PMV, platelet microvesicle; SAT, sepsis-associated thrombocytopenia; TRIM, transfusion-related immunomodulation.
References
1. Foley JH and Conway EM. Cross talk pathways between coagulation and inflammation. Circ Res. (2016) 118:1392–408. doi: 10.1161/CIRCRESAHA.116.306853
2. Levi M, van der Poll T, and Büller HR. Bidirectional relation between inflammation and coagulation. Circulation. (2004) 109:2698–704. doi: 10.1161/01.CIR.0000131660.51520.9A
3. Hottz ED, Bozza FA, and Bozza PT. Platelets in immune response to virus and immunopathology of viral infections. Front Med. (2018) 5:121–. doi: 10.3389/fmed.2018.00121
4. Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun. (2019) 10:1780–. doi: 10.1038/s41467-019-09607-x
5. Maouia A, Rebetz J, Kapur R, and Semple JW. The immune nature of platelets revisited. Transfusion Med Rev. (2020) 34:209–20. doi: 10.1016/j.tmrv.2020.09.005
6. Aslam R, Speck ER, Kim M, Freedman J, and Semple JW. Transfusion-related immunomodulation by platelets is dependent on their expression of MHC Class I molecules and is independent of white cells. Transfusion. (2008) 48:1778–86. doi: 10.1111/j.1537-2995.2008.01791.x
7. Liesenborghs L, Meyers S, Vanassche T, and Verhamme P. Coagulation: At the heart of infective endocarditis. J Thromb Haemostasis. (2020) 18:995–1008. doi: 10.1111/jth.14736
8. Zakeri A and Russo M. Dual role of toll-like receptors in human and experimental asthma models. Front Immunol. (2018) 9:1027. doi: 10.3389/fimmu.2018.01027
9. Vogel S, Arora T, Wang X, Mendelsohn L, Nichols J, Allen D, et al. The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and Bruton tyrosine kinase. Blood Adv. (2018) 2:2672–80. doi: 10.1182/bloodadvances.2018021709
10. Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, et al. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood. (2014) 124:791–802. doi: 10.1182/blood-2013-11-536003
11. Shiraki R, Inoue N, Kawasaki S, Takei A, Kadotani M, Ohnishi Y, et al. Expression of Toll-like receptors on human platelets. Thromb Res. (2004) 113:379–85. doi: 10.1016/j.thromres.2004.03.023
12. Liu Y, Zhang Y, Ding Y, and Zhuang R. Platelet-mediated tumor metastasis mechanism and the role of cell adhesion molecules. Crit Rev Oncol Hematol. (2021) 167:103502. doi: 10.1016/j.critrevonc.2021.103502
13. Merten M, Dong JF, Lopez JA, and Thiagarajan P. Cholesterol sulfate: a new adhesive molecule for platelets. Circulation. (2001) 103:2032–4. doi: 10.1161/01.CIR.103.16.2032
14. Palabrica T, Lobb R, Furie BC, Aronovitz M, Benjamin C, Hsu YM, et al. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature. (1992) 359:848–51. doi: 10.1038/359848a0
15. Cheresh DA, Berliner SA, Vicente V, and Ruggeri ZM. Recognition of distinct adhesive sites on fibrinogen by related integrins on platelets and endothelial cells. Cell. (1989) 58:945–53. doi: 10.1016/0092-8674(89)90946-X
16. Kaur S, Singh A, Kaur J, Verma N, Pandey AK, Das S, et al. Upregulation of cytokine signalling in platelets increases risk of thrombophilia in severe COVID-19 patients. Blood Cells Mol Dis. (2022) 94:102653. doi: 10.1016/j.bcmd.2022.102653
17. Boehlen F and Clemetson KJ. Platelet chemokines and their receptors: what is their relevance to platelet storage and transfusion practice? Transfus Med. (2001) 11:403–17. doi: 10.1046/j.1365-3148.2001.00340.x
18. Rolla R, Puricelli C, Bertoni A, Boggio E, Gigliotti CL, Chiocchetti A, et al. Platelets: "multiple choice" effectors in the immune response and their implication in COVID-19 thromboinflammatory process. Int J Lab Hematol. (2021) 43:895–906. doi: 10.1111/ijlh.13516
19. Mandel J, Casari M, Stepanyan M, Martyanov A, and Deppermann C. Beyond hemostasis: platelet innate immune interactions and thromboinflammation. Int J Mol Sci. (2022) 23:3868. doi: 10.3390/ijms23073868
20. Cognasse F, Duchez AC, Audoux E, Ebermeyer T, Arthaud CA, Prier A, et al. Platelets as key factors in inflammation: focus on CD40L/CD40. Front Immunol. (2022) 13:825892. doi: 10.3389/fimmu.2022.825892
21. Clawson CC, White JG, and Good RA. Interaction of monocytes, platelets, and bacteria in vitro. Proceedings Annu meeting Electron Microscopy Soc America. (2020) 28:68–9. doi: 10.1017/S0424820100067339
22. Clawson CC, White JG, and Herzberg MC. Platelet interaction with bacteria. VI. contrasting the role of fibrinogen and fibronectin. Am J Hematol. (1980) 9:43–53. doi: 10.1002/ajh.2830090106
23. Li X, Iwai T, Nakamura H, Inoue Y, Chen Y, Umeda M, et al. An ultrastructural study of Porphyromonas gingivalis-induced platelet aggregation. Thromb Res. (2008) 122:810–9. doi: 10.1016/j.thromres.2008.03.011
24. Youssefian T, Drouin A, Masse JM, Guichard J, and Cramer EM. Host defense role of platelets: engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood. (2002) 99:4021–9. doi: 10.1182/blood-2001-12-0191
25. Carestia A, Godin LC, and Jenne CN. Step up to the platelet: Role of platelets in inflammation and infection. Thromb Res. (2023) 231:182–94. doi: 10.1016/j.thromres.2022.10.001
26. de Almeida AJ, Campos-de-Magalhaes M, Antonietti CL, Brandao-Mello CE, da Silva ML, de Oliveira RV, et al. Autoimmune thrombocytopenia related to chronic hepatitis C virus infection. Hematology. (2009) 14:49–58. doi: 10.1179/102453309X385106
27. Kar M, Singla M, Chandele A, Kabra SK, Lodha R, and Medigeshi GR. Dengue virus entry and replication does not lead to productive infection in platelets. Open Forum Infect Dis. (2017) 4:ofx051. doi: 10.1093/ofid/ofx051
28. Kopiński P, Kopytek M, Ząbczyk M, Undas A, and Natorska J. Neutrophil extracellular traps (NETs) in aortic stenosis: Comparison of methods for assessment of NETs formation. Postępy Higieny i Medycyny Doświadczalnej. (2023) 77:163–9. doi: 10.2478/ahem-2023-0018
29. Juang LJ, Mazinani N, Novakowski SK, Prowse ENP, Haulena M, Gailani D, et al. Coagulation factor XII contributes to hemostasis when activated by soil in wounds. Blood Adv. (2020) 4:1737–45. doi: 10.1182/bloodadvances.2019000425
30. Ryu JK, Yan Z, Montano M, Sozmen EG, Dixit K, Suryawanshi RK, et al. Fibrin drives thromboinflammation and neuropathology in COVID-19. Nature. (2024) 633:905–13. doi: 10.1038/s41586-024-07873-4
31. Sang Y, Roest M, de Laat B, de Groot PG, and Huskens D. Interplay between platelets and coagulation. Blood Rev. (2021) 46:100733. doi: 10.1016/j.blre.2020.100733
32. Gianazza E, Brioschi M, Baetta R, Mallia A, Banfi C, and Tremoli E. Platelets in healthy and disease states: from biomarkers discovery to drug targets identification by proteomics. Int J Mol Sci. (2020) 21:4541. doi: 10.3390/ijms21124541
33. Koupenova M, Kehrel BE, Corkrey HA, and Freedman JE. Thrombosis and platelets: an update. Eur Heart J. (2017) 38:785–91. doi: 10.1093/eurheartj/ehw550
34. Lazar S and Goldfinger LE. Platelets and extracellular vesicles and their cross talk with cancer. Blood. (2021) 137:3192–200. doi: 10.1182/blood.2019004119
35. Kuravi SJ, Harrison P, Rainger GE, and Nash GB. Ability of platelet-derived extracellular vesicles to promote neutrophil-endothelial cell interactions. Inflammation. (2019) 42:290–305. doi: 10.1007/s10753-018-0893-5
36. Chimen M, Evryviadou A, Box CL, Harrison MJ, Hazeldine J, Dib LH, et al. Appropriation of GPIbalpha from platelet-derived extracellular vesicles supports monocyte recruitment in systemic inflammation. Haematologica. (2020) 105:1248–61. doi: 10.3324/haematol.2018.215145
37. Zhang Y, Yue Y, Cheng Y, Jiao H, and Yan M. Antigen B from Echinococcus granulosus regulates macrophage phagocytosis by controlling TLR4 endocytosis in immune thrombocytopenia. Chem Biol Interact. (2025) 406:111350. doi: 10.1016/j.cbi.2024.111350
38. Ye Q, Wang X, Xu X, Chen J, Christiani DC, Chen F, et al. Serial platelet count as a dynamic prediction marker of hospital mortality among septic patients. Burns Trauma. (2024) 12:tkae016. doi: 10.1093/burnst/tkae016
39. Blumberg N. Faculty Opinions recommendation of Evaluation of the association of platelet count, mean platelet volume, and platelet transfusion with intraventricular hemorrhage and death among preterm infants. Faculty Opin – Post-Publication Peer Rev Biomed Literature: H1 Connect. (2022) 5(10):e2237588. doi: 10.3410/f
40. ElMaraghy AA, AbdelFattah EB, and Ahmed MS. Platelet count: Is it a possible marker for severity and outcome of community acquired pneumonia? Egyptian J Chest Dis Tuberculosis. (2016) 65:499–504. doi: 10.1016/j.ejcdt.2015.09.001
41. Verma S, Khana R, Khanna V, Kandula S, Rajgopal V, and Prasad V. Platelet-based biomarkers: A new frontier in inflammation monitoring for respiratory infections [version 1; peer review: awaiting peer review. F1000Research. (2025) 14:328. doi: 10.12688/f1000research.161124.1
42. Cheng JJ, Liufu R, Zhuang J, and Chen MY. Risk factors of sepsis-associated thrombocytopenia among patients with sepsis induced coagulopathy. Clin Appl Thromb Hemost. (2024) 30:10760296241283166. doi: 10.1177/10760296241283166
43. Martha JW, Wibowo A, and Pranata R. Prognostic value of elevated lactate dehydrogenase in patients with COVID-19: a systematic review and meta-analysis. Postgrad Med J. (2022) 98:422–7. doi: 10.1136/postgradmedj-2020-139542
44. Vandijck DM, Blot SI, De Waele JJ, Hoste EA, Vandewoude KH, and Decruyenaere JM. Thrombocytopenia and outcome in critically ill patients with bloodstream infection. Heart Lung. (2010) 39:21–6. doi: 10.1016/j.hrtlng.2009.07.005
45. Martin CM, Priestap F, Fisher H, Fowler RA, Heyland DK, Keenan SP, et al. A prospective, observational registry of patients with severe sepsis: the Canadian Sepsis Treatment and Response Registry. Crit Care Med. (2009) 37:81–8. doi: 10.1097/CCM.0b013e31819285f0
46. Sharma B, Sharma M, Majumder M, Steier W, Sangal A, and Kalawar M. Thrombocytopenia in septic shock patients—A prospective observational study of incidence, risk factors and correlation with clinical outcome. Anaesthesia Intensive Care. (2007) 35:874–80. doi: 10.1177/0310057X0703500604
47. Solves Alcaina P. Special series on platelet transfusion. Ann Blood. (2022) 7:35–. doi: 10.21037/aob-2021-04
48. Venkata C, Kashyap R, Farmer JC, and Afessa B. Thrombocytopenia in adult patients with sepsis: incidence, risk factors, and its association with clinical outcome. J Intensive Care. (2013) 1:9. doi: 10.1186/2052-0492-1-9
49. Sharma B, Sharma M, Majumder M, Steier W, Sangal A, and Kalawar M. Thrombocytopenia in septic shock patients–a prospective observational study of incidence, risk factors and correlation with clinical outcome. Anaesth Intensive Care. (2007) 35:874–80. doi: 10.1177/0310057X0703500604
50. Giustozzi M, Ehrlinder H, Bongiovanni D, Borovac JA, Guerreiro RA, Gąsecka A, et al. Coagulopathy and sepsis: Pathophysiology, clinical manifestations and treatment. Blood Rev. (2021) 50:100864. doi: 10.1016/j.blre.2021.100864
51. Iba T, Watanabe E, Umemura Y, Wada T, Hayashida K, Kushimoto S, et al. Sepsis-associated disseminated intravascular coagulation and its differential diagnoses. J Intensive Care. (2019) 7:32. doi: 10.1186/s40560-019-0387-z
52. Kaufman RM, Djulbegovic B, Gernsheimer T, Kleinman S, Tinmouth AT, Capocelli KE, et al. Platelet transfusion: a clinical practice guideline from the AABB. Ann Intern Med. (2015) 162:205–13. doi: 10.7326/M14-1589
53. Wandt H, Schaefer-Eckart K, Wendelin K, Pilz B, Wilhelm M, Thalheimer M, et al. Therapeutic platelet transfusion versus routine prophylactic transfusion in patients with haematological Malignancies: an open-label, multicentre, randomised study. Lancet. (2012) 380:1309–16. doi: 10.1016/S0140-6736(12)60689-8
54. Diedrich B, Remberger M, Shanwell A, Svahn BM, and Ringden O. A prospective randomized trial of a prophylactic platelet transfusion trigger of 10 x 10(9) per L versus 30 x 10(9) per L in allogeneic hematopoietic progenitor cell transplant recipients. Transfusion. (2005) 45:1064–72. doi: 10.1111/j.1537-2995.2005.04157.x
55. Schiffer CA, Anderson KC, Bennett CL, Bernstein S, Elting LS, Goldsmith M, et al. Platelet transfusion for patients with cancer: Clinical practice guidelines of the American society of clinical oncology. J Clin Oncol. (2001) 19(5):1519–38. doi: 10.1200/JCO.2001.19.5.1519
56. Ludwig N, Hilger A, Zarbock A, and Rossaint J. Platelets at the Crossroads of Pro-Inflammatory and Resolution Pathways during Inflammation. Cells. (2022) 11:1957. doi: 10.3390/cells11121957
57. Karakas D and Ni H. Unveiling platelets as immune regulatory cells. Am Heart Assoc;. (2024) 134:987–9. doi: 10.1161/CIRCRESAHA.124.324167
58. Li C, Ture SK, Nieves-Lopez B, Blick-Nitko SK, Maurya P, Livada AC, et al. Thrombocytopenia independently leads to changes in monocyte immune function. Circ Res. (2024) 134(8):970–86. doi: 10.1161/CIRCRESAHA.123.323662
59. Nicolai L, Pekayvaz K, and Massberg S. Platelets: Orchestrators of immunity in host defense and beyond. Immunity. (2024) 57:957–72. doi: 10.1016/j.immuni.2024.04.008
60. Swieringa F, Spronk HMH, Heemskerk JWM, and van der Meijden PEJ. Integrating platelet and coagulation activation in fibrin clot formation. Res Pract Thromb Haemost. (2018) 2:450–60. doi: 10.1002/rth2.12107
61. Boral BM, Williams DJ, and Boral LI. Disseminated intravascular coagulation. Am J Clin Pathol. (2016) 146:670–80. doi: 10.1093/ajcp/aqw195
62. Conway EM, Mackman N, Warren RQ, Wolberg AS, Mosnier LO, Campbell RA, et al. Understanding COVID-19-associated coagulopathy. Nat Rev Immunol. (2022) 22:639–49. doi: 10.1038/s41577-022-00762-9
63. Senini V, Amara U, Paul M, and Kim H. Porphyromonas gingivalis lipopolysaccharide activates platelet Cdc42 and promotes platelet spreading and thrombosis. J Periodontol. (2019) 90:1336–45. doi: 10.1002/JPER.18-0596
64. Hotchkiss RS and Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. (2003) 348:138–50. doi: 10.1056/NEJMra021333
65. Tapper H and Herwald H. Modulation of hemostatic mechanisms in bacterial infectious diseases. Blood. (2000) 96:2329–37. doi: 10.1182/blood.V96.7.2329
66. Su M, Chen C, Li S, Li M, Zeng Z, Zhang Y, et al. Gasdermin D-dependent platelet pyroptosis exacerbates NET formation and inflammation in severe sepsis. Nat Cardiovasc Res. (2022) 1:732–47. doi: 10.1038/s44161-022-00108-7
67. Iba T, Helms J, and Levy JH. Sepsis-induced coagulopathy (SIC) in the management of sepsis. Ann Intensive Care. (2024) 14:148. doi: 10.1186/s13613-024-01380-5
68. Brennan MP, Loughman A, Devocelle M, Arasu S, Chubb AJ, Foster TJ, et al. Elucidating the role of Staphylococcus epidermidis serine–aspartate repeat protein G in platelet activation. J Thromb Haemostasis. (2009) 7:1364–72. doi: 10.1111/j.1538-7836.2009.03495.x
69. Miajlovic H, Zapotoczna M, Geoghegan JA, Kerrigan SW, Speziale P, and Foster TJ. Direct interaction of iron-regulated surface determinant IsdB of Staphylococcus aureus with the GPIIb/IIIa receptor on platelets. Microbiology. (2010) 156:920–8. doi: 10.1099/mic.0.036673-0
70. Petersen HJ, Keane C, Jenkinson HF, Vickerman MM, Jesionowski A, Waterhouse JC, et al. Human platelets recognize a novel surface protein, PadA, on Streptococcus gordonii through a unique interaction involving fibrinogen receptor GPIIbIIIa. Infect Immun. (2010) 78:413–22. doi: 10.1128/IAI.00664-09
71. Carestia A, Kaufman T, and Schattner M. Platelets: new bricks in the building of neutrophil extracellular traps. Front Immunol. (2016) 7:271. doi: 10.3389/fimmu.2016.00271
72. Plummer C, Wu H, Kerrigan SW, Meade G, Cox D, and Ian Douglas CW. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br J Haematology. (2005) 129:101–9. doi: 10.1111/j.1365-2141.2005.05421.x
73. Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med. (2000) 192:193–204. doi: 10.1084/jem.192.2.193
74. Brennan MP, Loughman A, Devocelle M, Arasu S, Chubb AJ, Foster TJ, et al. Elucidating the role of Staphylococcus epidermidis serine-aspartate repeat protein G in platelet activation. J Thromb Haemost. (2009) 7:1364–72. doi: 10.1111/j.1538-7836.2009.03495.x
75. Siboo IR, Chambers HF, and Sullam PM. Role of SraP, a Serine-Rich Surface Protein of Staphylococcus aureus, in binding to human platelets. Infect Immun. (2005) 73:2273–80. doi: 10.1128/IAI.73.4.2273-2280.2005
76. Guo L and Rondina MT. The era of thromboinflammation: platelets are dynamic sensors and effector cells during infectious diseases. Front Immunol. (2019) 10:2204. doi: 10.3389/fimmu.2019.02204
77. Middleton EA, Weyrich AS, and Zimmerman GA. Platelets in pulmonary immune responses and inflammatory lung diseases. Physiol Rev. (2016) 96:1211–59. doi: 10.1152/physrev.00038.2015
78. O'Neill LA, Golenbock D, and Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol. (2013) 13:453–60. doi: 10.1038/nri3446
79. Powers ME, Becker RE, Sailer A, Turner JR, and Bubeck Wardenburg J. Synergistic Action of Staphylococcus aureus alpha-Toxin on Platelets and Myeloid Lineage Cells Contributes to Lethal Sepsis. Cell Host Microbe. (2015) 17:775–87. doi: 10.1016/j.chom.2015.05.011
80. Montrucchio G, Bosco O, Del Sorbo L, Fascio Pecetto P, Lupia E, Goffi A, et al. Mechanisms of the priming effect of low doses of lipopoly-saccharides on leukocyte-dependent platelet aggregation in whole blood. Thromb Haemost. (2003) 90:872–81. doi: 10.1160/TH03-02-0085
81. Rivadeneyra L, Carestia A, Etulain J, Pozner RG, Fondevila C, Negrotto S, et al. Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB. Thromb Res. (2014) 133:235–43. doi: 10.1016/j.thromres.2013.11.028
82. Carestia A, Rivadeneyra L, Romaniuk MA, Fondevila C, Negrotto S, and Schattner M. Functional responses and molecular mechanisms involved in histone-mediated platelet activation. Thromb Haemost. (2013) 110:1035–45. doi: 10.1160/TH13-02-0174
83. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. (2011) 118:1952–61. doi: 10.1182/blood-2011-03-343061
84. Sheu JR, Hung WC, Kan YC, Lee YM, and Yen MH. Mechanisms involved in the antiplatelet activity of Escherichia coli lipopolysaccharide in human platelets. Br J Haematol. (1998) 103:29–38. doi: 10.1046/j.1365-2141.1998.00938.x
85. Stahl AL, Svensson M, Morgelin M, Svanborg C, Tarr PI, Mooney JC, et al. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood. (2006) 108:167–76. doi: 10.1182/blood-2005-08-3219
86. Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, et al. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J Immunol. (2009) 182:7997–8004. doi: 10.4049/jimmunol.0802884
87. Anabel AS, Eduardo PC, Pedro Antonio HC, Carlos SM, Juana NM, Honorio TA, et al. Human platelets express Toll-like receptor 3 and respond to poly I:C. Hum Immunol. (2014) 75:1244–51. doi: 10.1016/j.humimm.2014.09.013
88. D'Atri LP, Etulain J, Rivadeneyra L, Lapponi MJ, Centurion M, Cheng K, et al. Expression and functionality of Toll-like receptor 3 in the megakaryocytic lineage. J Thromb Haemost. (2015) 13:839–50. doi: 10.1111/jth.12842
89. Pozner RG, Ure AE, Jaquenod de Giusti C, D'Atri LP, Italiano JE, Torres O, et al. Junin virus infection of human hematopoietic progenitors impairs in vitro proplatelet formation and platelet release via a bystander effect involving type I IFN signaling. PloS Pathog. (2010) 6:e1000847. doi: 10.1371/journal.ppat.1000847
90. Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, et al. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood. (2006) 107:637–41. doi: 10.1182/blood-2005-06-2202
91. Sung P-S, Huang T-F, and Hsieh S-L. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat Commun. (2019) 10:2402–. doi: 10.1038/s41467-019-10360-4
92. Perez-Toledo M, Beristain-Covarrubias N, Pillaye J, Persaud RR, Marcial-Juarez E, Jossi SE, et al. Discrete and conserved inflammatory signatures drive thrombosis in different organs after Salmonella infection. Nat Commun. (2025) 16:2356. doi: 10.1038/s41467-025-57466-6
93. Hottz ED, Lopes JF, Freitas C, Valls-de-Souza R, Oliveira MF, Bozza MT, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood. (2013) 122:3405–14. doi: 10.1182/blood-2013-05-504449
94. Brill A, Bourne J, Campos J, Hopkin S, Whitworth K, Palis J, et al. Megakaryocyte NLRP3 hyperactivation induces anemia and potentiates inflammatory response in mice. Res Square Platform LLC. (2023) 14:1226196. doi: 10.21203/rs.3.rs-2616664/v1
95. Gavrilovskaya IN, Gorbunova EE, and Mackow ER. Pathogenic hantaviruses direct the adherence of quiescent platelets to infected endothelial cells. J Virol. (2010) 84:4832–9. doi: 10.1128/JVI.02405-09
96. Kasirer-Friede A, Kahn ML, and Shattil SJ. Platelet integrins and immunoreceptors. Immunol Rev. (2007) 218:247–64. doi: 10.1111/j.1600-065X.2007.00532.x
97. Shimony N, Elkin G, Kolodkin-Gal D, Krasny L, Urieli-Shoval S, and Haviv YS. Analysis of adenoviral attachment to human platelets. Virol J. (2009) 6:25–. doi: 10.1186/1743-422X-6-25
98. Fleming FE, Graham KL, Takada Y, and Coulson BS. Determinants of the specificity of rotavirus interactions with the alpha2beta1 integrin. J Biol Chem. (2011) 286:6165–74. doi: 10.1074/jbc.M110.142992
99. Negrotto S, Jaquenod de Giusti C, Rivadeneyra L, Ure AE, Mena HA, Schattner M, et al. Platelets interact with Coxsackieviruses B and have a critical role in the pathogenesis of virus-induced myocarditis. J Thromb Haemostasis. (2015) 13:271–82. doi: 10.1111/jth.12782
100. Zahn A, Jennings N, Ouwehand WH, and Allain J-P. Hepatitis C virus interacts with human platelet glycoprotein VI. J Gen Virology. (2006) 87:2243–51. doi: 10.1099/vir.0.81826-0
101. Chaipan C, Soilleux EJ, Simpson P, Hofmann H, Gramberg T, Marzi A, et al. DC-SIGN and CLEC-2 mediate human immunodeficiency virus type 1 capture by platelets. J Virol. (2006) 80:8951–60. doi: 10.1128/JVI.00136-06
102. Simon AY, Sutherland MR, and Pryzdial ELG. Dengue virus binding and replication by platelets. Blood. (2015) 126:378–85. doi: 10.1182/blood-2014-09-598029
103. Ariede JR, Pardini MIDMC, Silva GF, and Grotto RMT. Platelets can be a biological compartment for the Hepatitis C Virus. Braz J Microbiol. (2015) 46:627–9. doi: 10.1590/S1517-838246220140553
104. Padovani JL, Corvino SM, Drexler JF, Silva GF, Pardini MIDMC, and Grotto RMT. In vitro detection of hepatitis C virus in platelets from uninfected individuals exposed to the virus. Rev da Sociedade Bras Medicina Tropical. (2013) 46:154–5. doi: 10.1590/0037-8682-1627-2013
105. Capel PJ, van de Winkel JG, van den Herik-Oudijk IE, and Verbeek JS. Heterogeneity of human IgG Fc receptors. Immunomethods. (1994) 4:25–34. doi: 10.1006/immu.1994.1004
106. Cox D, Kerrigan SW, and Watson SP. Platelets and the innate immune system: mechanisms of bacterial-induced platelet activation. J Thromb Haemostasis. (2011) 9:1097–107. doi: 10.1111/j.1538-7836.2011.04264.x
107. Reilly RF. The pathophysiology of immune-mediated heparin-induced thrombocytopenia. Semin Dial. (2003) 16:54–60. doi: 10.1046/j.1525-139X.2003.03013.x
108. Worth RG, Chien CD, Chien P, Reilly MP, McKenzie SE, and Schreiber AD. Platelet FcgammaRIIA binds and internalizes IgG-containing complexes. Exp Hematol. (2006) 34:1490–5. doi: 10.1016/j.exphem.2006.06.015
109. Vanassche T, Kauskot A, Verhaegen J, Peetermans WE, van Ryn J, Schneewind O, et al. Fibrin formation by staphylothrombin facilitates Staphylococcus aureus-induced platelet aggregation. Thromb Haemost. (2012) 107:1107–21. doi: 10.1160/TH11-12-0891
110. Ahmad A and Menezes J. Binding of the Epstein-Barr virus to human platelets causes the release of transforming growth factor-beta. J Immunol. (1997) 159:3984–8. doi: 10.4049/jimmunol.159.8.3984
111. Clemetson KJ, Clemetson JM, Proudfoot AEI, Power CA, Baggiolini M, and Wells TNC. Functional expression of CCR1, CCR3, CCR4, and CXCR4 chemokine receptors on human platelets. Blood. (2000) 96:4046–54. doi: 10.1182/blood.V96.13.4046
112. Colicchia M, Schrottmaier WC, Perrella G, Reyat JS, Begum J, Slater A, et al. S100A8/A9 drives the formation of procoagulant platelets through GPIbalpha. Blood. (2022) 140:2626–43. doi: 10.1182/blood.2021014966
113. Josefsson E, McCrea KW, Eidhin DN, O'Connell D, Cox J, Hook M, et al. Three new members of the serine-aspartate repeat protein multigene family of Staphylococcus aureus. Microbiol (Reading). (1998) 144:3387–95. doi: 10.1099/00221287-144-12-3387
114. McDevitt D, Francois P, Vaudaux P, and Foster TJ. Molecular characterization of the clumping factor (fibrinogen receptor) of Staphylococcus aureus. Mol Microbiol. (1994) 11:237–48. doi: 10.1111/j.1365-2958.1994.tb00304.x
115. Ní Eidhin D, Perkins S, Francois P, Vaudaux P, Höök M, and Foster TJ. Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol Microbiol. (1998) 30:245–57. doi: 10.1046/j.1365-2958.1998.01050.x
116. Flock JI, Fröman G, Jönsson K, Guss B, Signäs C, Nilsson B, et al. Cloning and expression of the gene for a fibronectin-binding protein from Staphylococcus aureus. EMBO J. (1987) 6:2351–7. doi: 10.1002/j.1460-2075.1987.tb02511.x
117. Mitchell J, Tristan A, and Foster TJ. Characterization of the fibrinogen-binding surface protein Fbl of Staphylococcus lugdunensis. Microbiology. (2004) 150:3831–41. doi: 10.1099/mic.0.27337-0
118. Kerrigan SW and Cox D. Platelet-bacterial interactions. Cell Mol Life Sci. (2010) 67:513–23. doi: 10.1007/s00018-009-0207-z
119. Hamad OA, Nilsson PH, Wouters D, Lambris JD, Ekdahl KN, and Nilsson B. Complement component C3 binds to activated normal platelets without preceding proteolytic activation and promotes binding to complement receptor 1. J Immunol. (2010) 184:2686–92. doi: 10.4049/jimmunol.0902810
120. Kim H and Conway EM. Platelets and complement cross-talk in early atherogenesis. Front Cardiovasc Med. (2019) 6:131. doi: 10.3389/fcvm.2019.00131
121. Ghebrehiwet B, Murphy T, and Peerschke E. Activation-dependent surface expression of gC1qR/p33 on human blood platelets. Thromb Haemostasis. (2017) 89:331–9. doi: 10.1055/s-0037-1613450
122. Peerschke EI, Yin W, and Ghebrehiwet B. Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol Immunol. (2010) 47:2170–5. doi: 10.1016/j.molimm.2010.05.009
123. Wang H, Ricklin D, and Lambris JD. Complement-activation fragment C4a mediates effector functions by binding as untethered agonist to protease-activated receptors 1 and 4. Proc Natl Acad Sci U S A. (2017) 114:10948–53. doi: 10.1073/pnas.1707364114
124. Myoung SS, Francis SJ, Chen J, Lee G, Rauova L, Poncz M, et al. Complement activation as a biomarker for platelet-activating antibodies in heparin-induced thrombocytopenia. J Thromb Haemost. (2025) 23:1066–76. doi: 10.1016/j.jtha.2024.12.015
125. Hoepel W, Chen HJ, Geyer CE, Allahverdiyeva S, Manz XD, de Taeye SW, et al. High titers and low fucosylation of early human anti-SARS-CoV-2 IgG promote inflammation by alveolar macrophages. Sci Transl Med. (2021) 13:eabf8654. doi: 10.1126/scitranslmed.abf8654
126. Antczak AJ, Vieth JA, Singh N, and Worth RG. Internalization of IgG-coated targets results in activation and secretion of soluble CD40 ligand and RANTES by human platelets. Clin Vaccine Immunol. (2011) 18:210–6. doi: 10.1128/CVI.00296-10
127. Johnson MK, Boese-Marrazzo D, and Pierce WA Jr. Effects of pneumolysin on human polymorphonuclear leukocytes and platelets. Infect Immun. (1981) 34:171–6. doi: 10.1128/iai.34.1.171-176.1981
128. Wang TT, Sewatanon J, Memoli MJ, Wrammert J, Bournazos S, Bhaumik SK, et al. IgG antibodies to dengue enhanced for FcγRIIIA binding determine disease severity. Sci (New York NY). (2017) 355:395–8. doi: 10.1126/science.aai8128
129. Arvand M, Bhakdi S, Dahlback B, and Preissner KT. Staphylococcus aureus alpha-toxin attack on human platelets promotes assembly of the prothrombinase complex. J Biol Chem. (1990) 265:14377–81. doi: 10.1016/S0021-9258(18)77312-2
130. Fitzpatrick RE, Wijeyewickrema LC, and Pike RN. The gingipains: scissors and glue of the periodontal pathogen, Porphyromonas gingivalis. Future Microbiol. (2009) 4:471–87. doi: 10.2217/fmb.09.18
131. Bryant AE, Bayer CR, Chen RY, Guth PH, Wallace RJ, and Stevens DL. Vascular dysfunction and ischemic destruction of tissue in Streptococcus pyogenes infection: the role of streptolysin O-induced platelet/neutrophil complexes. J Infect Dis. (2005) 192:1014–22. doi: 10.1086/jid.2005.192.issue-6
132. Fritsch KJ, Kruger L, Handtke S, Kohler TP, Ozhiganova A, Jahn K, et al. Pneumococcal neuraminidases increase platelet killing by pneumolysin. Thromb Haemost. (2025) 125:243–54. doi: 10.1055/a-2369-8680
133. Riegner A, Jahn K, Wesche J, Thiele T, and Siemens N. Streptococcus pyogenes Activates Human Platelets via Streptolysin S-Mediated Calcium Ion Influx. J Innate Immun. (2025) 17:198–210. doi: 10.1159/000544951
134. Wang J, Zhang W, Nardi MA, and Li Z. HIV-1 Tat-induced platelet activation and release of CD154 contribute to HIV-1-associated autoimmune thrombocytopenia. J Thromb Haemost. (2011) 9:562–73. doi: 10.1111/j.1538-7836.2010.04168.x
135. Chao C-H, Wu W-C, Lai Y-C, Tsai P-J, Perng G-C, Lin Y-S, et al. Dengue virus nonstructural protein 1 activates platelets via Toll-like receptor 4, leading to thrombocytopenia and hemorrhage. PloS Pathog. (2019) 15:e1007625–e. doi: 10.1371/journal.ppat.1007625
136. Assinger A, Laky M, Schabbauer G, Hirschl AM, Buchberger E, Binder BR, et al. Efficient phagocytosis of periodontopathogens by neutrophils requires plasma factors, platelets and TLR2. J Thromb Haemost. (2011) 9:799–809. doi: 10.1111/j.1538-7836.2011.04193.x
137. Assinger A, Kral JB, Yaiw KC, Schrottmaier WC, Kurzejamska E, Wang Y, et al. Human cytomegalovirus-platelet interaction triggers toll-like receptor 2-dependent proinflammatory and proangiogenic responses. Arterioscler Thromb Vasc Biol. (2014) 34:801–9. doi: 10.1161/ATVBAHA.114.303287
138. Spakova T, Janockova J, and Rosocha J. Characterization and therapeutic use of extracellular vesicles derived from platelets. Int J Mol Sci. (2021) 22:9701. doi: 10.3390/ijms22189701
139. Takamatsu D, Bensing BA, Cheng H, Jarvis GA, Siboo IR, López JA, et al. Binding of the Streptococcus gordonii surface glycoproteins GspB and Hsa to specific carbohydrate structures on platelet membrane glycoprotein Ibalpha. Mol Microbiol. (2005) 58:380–92. doi: 10.1111/j.1365-2958.2005.04830.x
140. Keane C, Petersen HJ, Tilley D, Haworth J, Cox D, Jenkinson HF, et al. Multiple sites on Streptococcus gordonii surface protein PadA bind to platelet GPIIbIIIa. Thromb Haemost. (2013) 110:1278–87. doi: 10.1160/TH13-07-0580
141. Sica A and Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
142. Kral JB, Schrottmaier WC, Salzmann M, and Assinger A. Platelet interaction with innate immune cells. Transfus Med Hemother. (2016) 43:78–88. doi: 10.1159/000444807
143. Kerrigan SW, Clarke N, Loughman A, Meade G, Foster TJ, and Cox D. Molecular basis for Staphylococcus aureus-mediated platelet aggregate formation under arterial shear in vitro. Arterioscler Thromb Vasc Biol. (2008) 28:335–40. doi: 10.1161/ATVBAHA.107.152058
144. Han P, Hanlon D, Arshad N, Lee JS, Tatsuno K, Robinson E, et al. Platelet P-selectin initiates cross-presentation and dendritic cell differentiation in blood monocytes. Sci Adv. (2020) 6:eaaz1580. doi: 10.1126/sciadv.aaz1580
145. Nakanishi T, Inaba M, Inagaki-Katashiba N, Tanaka A, Vien PT, Kibata K, et al. Platelet-derived RANK ligand enhances CCL17 secretion from dendritic cells mediated by thymic stromal lymphopoietin. Platelets. (2015) 26:425–31. doi: 10.3109/09537104.2014.920081
146. Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. (2010) 327:580–3. doi: 10.1126/science.1181928
147. Quirino-Teixeira AC, Rozini SV, Barbosa-Lima G, Coelho DR, Carneiro PH, Mohana-Borges R, et al. Inflammatory signaling in dengue-infected platelets requires translation and secretion of nonstructural protein 1. Blood Adv. (2020) 4:2018–31. doi: 10.1182/bloodadvances.2019001169
148. Moore CM, O'Reilly D, McCallion N, and Curley AE. Changes in inflammatory proteins following platelet transfusion in a neonatal population. Pediatr Res. (2023) 94:1973–7. doi: 10.1038/s41390-023-02731-x
149. Auerbach DJ, Lin Y, Miao H, Cimbro R, DiFiore MJ, Gianolini ME, et al. Identification of the platelet-derived chemokine CXCL4/PF-4 as a broad-spectrum HIV-1 inhibitor. Proc Natl Acad Sci. (2012) 109:9569–74. doi: 10.1073/pnas.1207314109
150. Solomon Tsegaye T, Gnirß K, Rahe-Meyer N, Kiene M, Krämer-Kühl A, Behrens G, et al. Platelet activation suppresses HIV-1 infection of T cells. Retrovirology. (2013) 10:48–. doi: 10.1186/1742-4690-10-48
151. Schwartzkopff F, Grimm TA, Lankford CSR, Fields K, Wang J, Brandt E, et al. Platelet factor 4 (CXCL4) facilitates human macrophage infection with HIV-1 and potentiates virus replication. Innate Immunity. (2009) 15:368–79. doi: 10.1177/1753425909106171
152. Krijgsveld J, Zaat SAJ, Meeldijk J, van Veelen PA, Fang G, Poolman B, et al. Thrombocidins, microbicidal proteins from human blood platelets, are C-terminal deletion products of CXC chemokines. J Biol Chem. (2000) 275:20374–81. doi: 10.1074/jbc.275.27.20374
153. Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, et al. Novel anti-bacterial activities of beta-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PloS Pathog. (2011) 7:e1002355. doi: 10.1371/journal.ppat.1002355
154. Yeaman MR. Platelets: at the nexus of antimicrobial defence. Nat Rev Microbiol. (2014) 12:426–37. doi: 10.1038/nrmicro3269
155. Valle-Jiménez X, Ramírez-Cosmes A, Aquino-Domínguez AS, Sánchez-Peña F, Bustos-Arriaga J, Romero-Tlalolini MDLÁ, et al. Human platelets and megakaryocytes express defensin alpha 1. Platelets. (2019) 31:344–54. doi: 10.1080/09537104.2019.1615612
156. Mohan KVK, Rao SS, and Atreya CD. Antiviral activity of selected antimicrobial peptides against vaccinia virus. Antiviral Res. (2010) 86:306–11. doi: 10.1016/j.antiviral.2010.03.012
157. Tilton C, Clippinger AJ, Maguire T, and Alwine JC. Human cytomegalovirus induces multiple means to combat reactive oxygen species. J Virol. (2011) 85:12585–93. doi: 10.1128/JVI.05572-11
158. Heijnen H and van der Sluijs P. Platelet secretory behaviour: as diverse as the granules … or not? J Thromb Haemostasis. (2015) 13:2141–51. doi: 10.1111/jth.13147
159. Ebermeyer T, Cognasse F, Berthelot P, Mismetti P, Garraud O, and Hamzeh-Cognasse H. Platelet innate immune receptors and TLRs: A double-edged sword. Int J Mol Sci. (2021) 22:7894. doi: 10.3390/ijms22157894
160. Pulcinelli FM, Riondino S, Celestini A, Pignatelli P, Trifiro E, Di Renzo L, et al. Persistent production of platelet thromboxane A2 in patients chronically treated with aspirin. J Thromb Haemost. (2005) 3:2784–9. doi: 10.1111/j.1538-7836.2005.01633.x
161. Yost CC, Weyrich AS, and Zimmerman GA. The platelet activating factor (PAF) signaling cascade in systemic inflammatory responses. Biochimie. (2010) 92:692–7. doi: 10.1016/j.biochi.2010.02.011
162. Krettek A, Ostergren-Lunden G, Fager G, Rosmond C, Bondjers G, and Lustig F. Expression of PDGF receptors and ligand-induced migration of partially differentiated human monocyte-derived macrophages. Influence IFN-gamma TGF-beta Atherosclerosis. (2001) 156:267–75. doi: 10.1016/s0021-9150(00)00644-4
163. Ehrhardt RO, Strober W, and Harriman GR. Effect of transforming growth factor (TGF)-beta 1 on IgA isotype expression. TGF-beta 1 induces a small increase in sIgA+ B cells regardless of the method of B cell activation. J Immunol. (1992) 148:3830–6. doi: 10.4049/jimmunol.148.12.3830
164. Santarlasci V, Maggi L, Capone M, Frosali F, Querci V, De Palma R, et al. TGF-beta indirectly favors the development of human Th17 cells by inhibiting Th1 cells. Eur J Immunol. (2009) 39:207–15. doi: 10.1002/eji.200838748
165. Takimoto T, Wakabayashi Y, Sekiya T, Inoue N, Morita R, Ichiyama K, et al. Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J Immunol. (2010) 185:842–55. doi: 10.4049/jimmunol.0904100
166. Warner GL, Nelson DO, and Scott DW. Synergy between TGF-beta and anti-IgM in growth inhibition of CD5+ B-cell lymphomas. Ann N Y Acad Sci. (1992) 651:274–6. doi: 10.1111/j.1749-6632.1992.tb24625.x
167. Chen Y, Zhong H, Zhao Y, Luo X, and Gao W. Role of platelet biomarkers in inflammatory response. biomark Res. (2020) 8:28. doi: 10.1186/s40364-020-00207-2
168. Scherlinger M, Richez C, Tsokos GC, Boilard E, and Blanco P. The role of platelets in immune-mediated inflammatory diseases. Nat Rev Immunol. (2023) 23:495–510. doi: 10.1038/s41577-023-00834-4
169. Shi G and Morrell CN. Platelets as initiators and mediators of inflammation at the vessel wall. Thromb Res. (2011) 127:387–90. doi: 10.1016/j.thromres.2010.10.019
170. Tokarz-Deptula B, Palma J, Baraniecki L, Stosik M, Kolacz R, and Deptula W. What function do platelets play in inflammation and bacterial and viral infections? Front Immunol. (2021) 12:770436. doi: 10.3389/fimmu.2021.770436
171. Palankar R, Kohler TP, Krauel K, Wesche J, Hammerschmidt S, and Greinacher A. Platelets kill bacteria by bridging innate and adaptive immunity via platelet factor 4 and FcγRIIA. J Thromb Haemostasis. (2018) 16:1187–97. doi: 10.1111/jth.13955
172. Guo L, Feng K, Wang YC, Mei JJ, Ning RT, Zheng HW, et al. Critical role of CXCL4 in the lung pathogenesis of influenza (H1N1) respiratory infection. Mucosal Immunol. (2017) 10:1529–41. doi: 10.1038/mi.2017.1
173. Ojha A, Bhasym A, Mukherjee S, Annarapu GK, Bhakuni T, Akbar I, et al. Platelet factor 4 promotes rapid replication and propagation of Dengue and Japanese encephalitis viruses. EBioMedicine. (2019) 39:332–47. doi: 10.1016/j.ebiom.2018.11.049
174. Diebold SS, Kaisho T, Hemmi H, Akira S, and Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. (2004) 303:1529–31. doi: 10.1126/science.1093616
175. Kim S-J, Davis R, and Jenne C. Platelets as modulators of inflammation. Semin Thromb Hemostasis. (2017) 44:091–101. doi: 10.1055/s-0037-1607432
176. Borges-Rodriguez M, Shields CA, Travis OK, Tramel RW, Baik CH, Giachelli CA, et al. Platelet inhibition prevents NLRP3 inflammasome activation and sepsis-induced kidney injury. Int J Mol Sci. (2021) 22:10330. doi: 10.3390/ijms221910330
177. Vogel S, Bodenstein R, Chen Q, Feil S, Feil R, Rheinlaender J, et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J Clin Invest. (2015) 125:4638–54. doi: 10.1172/JCI81660
178. Vogel S, Rath D, Borst O, Mack A, Loughran P, Lotze MT, et al. Platelet-derived high-mobility group box 1 promotes recruitment and suppresses apoptosis of monocytes. Biochem Biophys Res Commun. (2016) 478:143–8. doi: 10.1016/j.bbrc.2016.07.078
179. Rossaint J, Thomas K, Mersmann S, Skupski J, Margraf A, Tekath T, et al. Platelets orchestrate the resolution of pulmonary inflammation in mice by T reg cell repositioning and macrophage education. J Exp Med. (2021) 218:e20201353. doi: 10.1084/jem.20201353
180. Bakogiannis C, Sachse M, Stamatelopoulos K, and Stellos K. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine. (2019) 122:154157. doi: 10.1016/j.cyto.2017.09.013
181. Blair P and Flaumenhaft R. Platelet alpha-granules: basic biology and clinical correlates. Blood Rev. (2009) 23:177–89. doi: 10.1016/j.blre.2009.04.001
182. Chargaff E and West R. The biological significance of the thromboplastic protein of blood. J Biol Chem. (1946) 166:189–97. doi: 10.1016/S0021-9258(17)34997-9
183. Wolf P. The nature and significance of platelet products in human plasma. Br J Haematol. (1967) 13:269–88. doi: 10.1111/j.1365-2141.1967.tb08741.x
184. Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, and Sixma JJ. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood. (1999) 94:3791–9. doi: 10.1182/blood.V94.11.3791
185. Siljander PR. Platelet-derived microparticles - an updated perspective. Thromb Res. (2011) 127 Suppl 2:S30–3. doi: 10.1016/S0049-3848(10)70152-3
186. Schoenwaelder SM, Yuan Y, Josefsson EC, White MJ, Yao Y, Mason KD, et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood. (2009) 114:663–6. doi: 10.1182/blood-2009-01-200345
187. Guo J, Cui B, Zheng J, Yu C, Zheng X, Yi L, et al. Platelet-derived microparticles and their cargos: The past, present and future. Asian J Pharm Sci. (2024) 19:100907. doi: 10.1016/j.ajps.2024.100907
188. Maugeri N, De Lorenzo R, Clementi N, Antonia Diotti R, Criscuolo E, Godino C, et al. Unconventional CD147-dependent platelet activation elicited by SARS-CoV-2 in COVID-19. J Thromb Haemost. (2022) 20:434–48. doi: 10.1111/jth.15575
189. Lopez E, Srivastava AK, Burchfield J, Wang YW, Cardenas JC, Togarrati PP, et al. Platelet-derived- extracellular vesicles promote hemostasis and prevent the development of hemorrhagic shock. Sci Rep. (2019) 9:17676. doi: 10.1038/s41598-019-53724-y
190. Montemore C, Dobrydneva Y, and Firmani M. The impact of blood utilization guidelines on product usage. Am Soc Clin Lab Science. (2019) 32:49–55. doi: 10.29074/ascls.2019001636
191. Puhm F, Flamand L, and Boilard E. Platelet extracellular vesicles in COVID-19: Potential markers and makers. J Leukoc Biol. (2022) 111:63–74. doi: 10.1002/JLB.3MIR0221-100R
192. Qu M, Zou X, Fang F, Wang S, Xu L, Zeng Q, et al. Platelet-derived microparticles enhance megakaryocyte differentiation and platelet generation via miR-1915-3p. Nat Commun. (2020) 11:4964–. doi: 10.1038/s41467-020-18802-0
193. Nishat S, Wuescher LM, and Worth RG. Platelets Enhance Dendritic Cell Responses against Staphylococcus aureus through CD40-CD40L. Infect Immun. (2018) 86:e00186–18. doi: 10.1128/IAI.00186-18
194. Kaiser R, Escaig R, Erber J, and Nicolai L. Neutrophil-platelet interactions as novel treatment targets in cardiovascular disease. Front Cardiovasc Med. (2022) 8:824112–. doi: 10.3389/fcvm.2021.824112
195. Ueki H, Wang IH, Kiso M, Horie K, Iida S, Mine S, et al. Neutrophil adhesion to vessel walls impairs pulmonary circulation in COVID-19 pathology. Nat Commun. (2025) 16:455. doi: 10.1038/s41467-024-55272-0
196. Wang H, Liu C, Xie X, Niu M, Wang Y, Cheng X, et al. Multi-omics blood atlas reveals unique features of immune and platelet responses to SARS-CoV-2 Omicron breakthrough infection. Immunity. (2023) 56:1410–28 e8. doi: 10.1016/j.immuni.2023.05.007
197. De Giovanni M, Dang EV, Chen KY, An J, Madhani HD, and Cyster JG. Platelets and mast cells promote pathogenic eosinophil recruitment during invasive fungal infection via the 5-HIAA-GPR35 ligand-receptor system. Immunity. (2023) 56:1548–60 e5. doi: 10.1016/j.immuni.2023.05.006
198. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
199. Schrottmaier WC, Mussbacher M, Salzmann M, and Assinger A. Platelet-leukocyte interplay during vascular disease. Atherosclerosis. (2020) 307:109–20. doi: 10.1016/j.atherosclerosis.2020.04.018
200. Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. (2016) 128:2435–49. doi: 10.1182/blood-2016-04-710632
201. Panicker SR, Mehta-D'souza P, Zhang N, Klopocki AG, Shao B, and McEver RP. Circulating soluble P-selectin must dimerize to promote inflammation and coagulation in mice. Blood. (2017) 130:181–91. doi: 10.1182/blood-2017-02-770479
202. Vajen T, Koenen RR, Werner I, Staudt M, Projahn D, Curaj A, et al. Blocking CCL5-CXCL4 heteromerization preserves heart function after myocardial infarction by attenuating leukocyte recruitment and NETosis. Sci Rep. (2018) 8:10647. doi: 10.1038/s41598-018-29026-0
203. Yang M, Jiang H, Ding C, Zhang L, Ding N, Li G, et al. STING activation in platelets aggravates septic thrombosis by enhancing platelet activation and granule secretion. Immunity. (2023) 56:1013–26 e6. doi: 10.1016/j.immuni.2023.02.015
204. Kaiser R, Anjum A, Kammerer L, Loew Q, Akhalkatsi A, Rossaro D, et al. Mechanosensing via a GpIIb/Src/14-3-3ζ axis critically regulates platelet migration in vascular inflammation. Blood. (2023) 141:2973–92. doi: 10.1182/blood.2022019210
205. Carestia A, Mena HA, Olexen CM, Ortiz Wilczynski JM, Negrotto S, Errasti AE, et al. Platelets promote macrophage polarization toward pro-inflammatory phenotype and increase survival of septic mice. Cell Rep. (2019) 28:896–908 e5. doi: 10.1016/j.celrep.2019.06.062
206. Fu G, Deng M, Neal MD, Billiar TR, and Scott MJ. Platelet-monocyte aggregates: understanding mechanisms and functions in sepsis. Shock. (2021) 55:156–66. doi: 10.1097/SHK.0000000000001619
207. Loguinova M, Pinegina N, Kogan V, Vagida M, Arakelyan A, Shpektor A, et al. Monocytes of different subsets in complexes with platelets in patients with myocardial infarction. Thromb Haemost. (2018) 118:1969–81. doi: 10.1055/s-0038-1673342
208. Rayes J, Lax S, Wichaiyo S, Watson SK, Di Y, Lombard S, et al. The podoplanin-CLEC-2 axis inhibits inflammation in sepsis. Nat Commun. (2017) 8:2239–. doi: 10.1038/s41467-017-02402-6
209. Xu P, Jiang Y, Zuo H, Liu X, Xia T, Zhou R, et al. Vincristine-loaded platelets coated with anti-CD41 mAbs: a new macrophage targeting proposal for the treatment of immune thrombocytopenia. Biomater Sci. (2019) 7:4568–77. doi: 10.1039/C9BM01026B
210. Silva-Cardoso SC, Affandi AJ, Spel L, Cossu M, van Roon JAG, Boes M, et al. CXCL4 exposure potentiates TLR-driven polarization of human monocyte-derived dendritic cells and increases stimulation of T cells. J Immunol. (2017) 199:253–62. doi: 10.4049/jimmunol.1602020
211. Hamzeh-Cognasse H, Cognasse F, Palle S, Chavarin P, Olivier T, Delezay O, et al. Direct contact of platelets and their released products exert different effects on human dendritic cell maturation. BMC Immunol. (2008) 9:54. doi: 10.1186/1471-2172-9-54
212. Langer HF, Daub K, Braun G, Schonberger T, May AE, Schaller M, et al. Platelets recruit human dendritic cells via Mac-1/JAM-C interaction and modulate dendritic cell function in vitro. Arterioscler Thromb Vasc Biol. (2007) 27:1463–70. doi: 10.1161/ATVBAHA.107.141515
213. Lei W. Research progress of BAFF in primary immune thrombocytopenia. Adv Clin Med. (2023) 13:18522–6. doi: 10.12677/ACM.2023.13112603
214. Angenieux C, Dupuis A, Gachet C, de la Salle H, and Maitre B. Cell surface expression of HLA I molecules as a marker of young platelets. J Thromb Haemost. (2019) 17:1511–21. doi: 10.1111/jth.14537
215. Guo L, Shen S, Rowley JW, Tolley ND, Jia W, Manne BK, et al. Platelet MHC class I mediates CD8+ T-cell suppression during sepsis. Blood. (2021) 138:401–16. doi: 10.1182/blood.2020008958
216. Shi G, Field DJ, Ko KA, Ture S, Srivastava K, Levy S, et al. Platelet factor 4 limits Th17 differentiation and cardiac allograft rejection. J Clin Invest. (2014) 124:543–52. doi: 10.1172/JCI71858
217. Sprague DL, Elzey BD, Crist SA, Waldschmidt TJ, Jensen RJ, and Ratliff TL. Platelet-mediated modulation of adaptive immunity: unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood. (2008) 111:5028–36. doi: 10.1182/blood-2007-06-097410
218. León-Ponte M, Ahern GP, and O'Connell PJ. Serotonin provides an accessory signal to enhance T-cell activation by signaling through the 5-HT7 receptor. Blood. (2007) 109:3139–46. doi: 10.1182/blood-2006-10-052787
219. Li N, Ji Q, and Hjemdahl P. Platelet–lymphocyte conjugation differs between lymphocyte subpopulations. J Thromb Haemostasis. (2006) 4:874–81. doi: 10.1111/j.1538-7836.2006.01817.x
220. Liu CY, Battaglia M, Lee SH, Sun Q-H, Aster RH, and Visentin GP. Platelet factor 4 differentially modulates CD4+CD25+ (Regulatory) versus CD4+CD25– (Nonregulatory) T cells. J Immunol. (2005) 174:2680–6. doi: 10.4049/jimmunol.174.5.2680
221. Diacovo TG, Catalina MD, Siegelman MH, and von Andrian UH. Circulating activated platelets reconstitute lymphocyte homing and immunity in L-selectin-deficient mice. J Exp Med. (1998) 187:197–204. doi: 10.1084/jem.187.2.197
222. Fleischer J, Grage-Griebenow E, Kasper B, Heine H, Ernst M, Brandt E, et al. Platelet factor 4 inhibits proliferation and cytokine release of activated human T cells. J Immunol. (2002) 169:770–7. doi: 10.4049/jimmunol.169.2.770
223. Gerdes N, Zhu L, Ersoy M, Hermansson A, Hjemdahl P, Hu H, et al. Platelets regulate CD4+ T-cell differentiation via multiple chemokines in humans. Thromb Haemostasis. (2011) 106:353–62. doi: 10.1160/TH11-01-0020
224. Iannacone M, Sitia G, Narvaiza I, Ruggeri ZM, and Guidotti LG. Antiplatelet drug therapy moderates immune-mediated liver disease and inhibits viral clearance in mice infected with a replication-deficient adenovirus. Clin Vaccine Immunol. (2007) 14:1532–5. doi: 10.1128/CVI.00298-07
225. Lalor P and Nash GB. Adhesion of flowing leucocytes to immobilized platelets. Br J Haematology. (1995) 89:725–32. doi: 10.1111/j.1365-2141.1995.tb08408.x
226. Aloui C, Prigent A, Sut C, Tariket S, Hamzeh-Cognasse H, Pozzetto B, et al. The signaling role of CD40 ligand in platelet biology and in platelet component transfusion. Int J Mol Sci. (2014) 15:22342–64. doi: 10.3390/ijms151222342
227. Zamora C, Toniolo E, Diaz-Torne C, Canto E, Magallares B, Ortiz MA, et al. Association of platelet binding to lymphocytes with B cell abnormalities and clinical manifestations in systemic lupus erythematosus. Mediators Inflamm. (2019) 2019:2473164. doi: 10.1155/2019/2473164
228. Nicolaou A, Mauro C, Urquhart P, and Marelli-Berg F. Polyunsaturated Fatty Acid-derived lipid mediators and T cell function. Front Immunol. (2014) 5:75–. doi: 10.3389/fimmu.2014.00075
229. Wu KK, Papp AC, Manner CE, and Hall ER. Interaction between lymphocytes and platelets in the synthesis of prostacyclin. J Clin Invest. (1987) 79:1601–6. doi: 10.1172/JCI112995
230. Remy KE, Hall MW, Cholette J, Juffermans NP, Nicol K, Doctor A, et al. Mechanisms of red blood cell transfusion-related immunomodulation. Transfusion. (2018) 58:804–15. doi: 10.1111/trf.2018.58.issue-3
231. Opelz G, Mickey MR, and Terasaki PI. Blood transfusions and unresponsiveness to HL-A. Transplantation. (1973) 16:649–54. doi: 10.1097/00007890-197312000-00017
232. Mansour A, Massart N, Gouin-Thibault I, Seite T, Cognasse F, Anselmi A, et al. Impact of intraoperative allogeneic platelet transfusion on healthcare-associated infections in cardiac surgery: insights from a large single-center cohort study. J Cardiothorac Vasc Anesth. (2024) 38:1650–8. doi: 10.1053/j.jvca.2024.02.031
233. Kah TA, Yong KC, and Rahman RA. Disseminated fusariosis and endogenous fungal endophthalmitis in acute lymphoblastic leukemia following platelet transfusion possibly due to transfusion-related immunomodulation. BMC Ophthalmol. (2011) 11:30. doi: 10.1186/1471-2415-11-30
234. Sadallah S, Eken C, Martin PJ, and Schifferli JA. Microparticles (ectosomes) shed by stored human platelets downregulate macrophages and modify the development of dendritic cells. J Immunol. (2011) 186:6543–52. doi: 10.4049/jimmunol.1002788
235. Perros AJ, Christensen AM, Flower RL, and Dean MM. Soluble mediators in platelet concentrates modulate dendritic cell inflammatory responses in an experimental model of transfusion. J Interferon Cytokine Res. (2015) 35:821–30. doi: 10.1089/jir.2015.0029
236. Frazier SK, Higgins J, Bugajski A, Jones AR, and Brown MR. Adverse reactions to transfusion of blood products and best practices for prevention. Crit Care Nurs Clin North Am. (2017) 29:271–90. doi: 10.1016/j.cnc.2017.04.002
237. Geiger TL. Transfusion-associated immune modulation: a reason to TRIM platelet transfusions? Transfusion. (2008) 48:1772–3. doi: 10.1111/j.1537-2995.2008.01860.x
238. Vamvakas EC and Blajchman MA. Transfusion-related immunomodulation (TRIM): an update. Blood Rev. (2007) 21:327–48. doi: 10.1016/j.blre.2007.07.003
239. Shorr AF, Jackson WL, Kelly KM, Fu M, and Kollef MH. Transfusion practice and blood stream infections in critically ill patients. Chest. (2005) 127:1722–8. doi: 10.1378/chest.127.5.1722
240. Blajchman MA. Transfusion-associated immunomodulation and universal white cell reduction: are we putting the cart before the horse? Transfusion. (1999) 39:665–70. doi: 10.1046/j.1537-2995.1999.39070665.x
241. Kao KJ, Cook DJ, and Scornik JC. Quantitative analysis of platelet surface HLA by W6/32 anti-HLA monoclonal antibody. Blood. (1986) 68:627–32. doi: 10.1182/blood.V68.3.627.627
242. Lalezari P and Driscoll AM. Ability of thrombocytes to acquire HLA specificity from plasma. Blood. (1982) 59:167–70. doi: 10.1182/blood.V59.1.167.167
243. Engelmann B and Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. (2013) 13:34–45. doi: 10.1038/nri3345
244. Denorme F and Campbell RA. Procoagulant platelets: novel players in thromboinflammation. Am J Physiol Cell Physiol. (2022) 323:C951–C8. doi: 10.1152/ajpcell.00252.2022
245. Yeaman MR. Platelets in defense against bacterial pathogens. Cell Mol Life Sci. (2010) 67:525–44. doi: 10.1007/s00018-009-0210-4
246. Verschoor A, Neuenhahn M, Navarini AA, Graef P, Plaumann A, Seidlmeier A, et al. A platelet-mediated system for shuttling blood-borne bacteria to CD8α+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat Immunol. (2011) 12:1194–201. doi: 10.1038/ni.2140
247. Li J, Karakas D, Xue F, Chen Y, Zhu G, Yucel YH, et al. Desialylated platelet clearance in the liver is a novel mechanism of systemic immunosuppression. Res (Wash D C). (2023) 6:0236. doi: 10.34133/research.0236
248. Broadley SP, Plaumann A, Coletti R, Lehmann C, Wanisch A, Seidlmeier A, et al. Dual-track clearance of circulating bacteria balances rapid restoration of blood sterility with induction of adaptive immunity. Cell Host Microbe. (2016) 20:36–48. doi: 10.1016/j.chom.2016.05.023
249. Verschoor A, Neuenhahn M, Navarini AA, Graef P, Plaumann A, Seidlmeier A, et al. A platelet-mediated system for shuttling blood-borne bacteria to CD8alpha+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat Immunol. (2011) 12:1194–201. doi: 10.1038/ni.2140
250. Zufferey A, Speck ER, Machlus KR, Aslam R, Guo L, McVey MJ, et al. Mature murine megakaryocytes present antigen-MHC class I molecules to T cells and transfer them to platelets. Blood Adv. (2017) 1:1773–85. doi: 10.1182/bloodadvances.2017007021
251. Brown GT and McIntyre TM. Lipopolysaccharide signaling without a nucleus: kinase cascades stimulate platelet shedding of proinflammatory IL-1beta-rich microparticles. J Immunol. (2011) 186:5489–96. doi: 10.4049/jimmunol.1001623
252. Ali RA, Wuescher LM, Dona KR, and Worth RG. Platelets Mediate Host Defense against Staphylococcus aureus through Direct Bactericidal Activity and by Enhancing Macrophage Activities. J Immunol. (2017) 198:344–51. doi: 10.4049/jimmunol.1601178
253. Garcia-Leon MJ, Liboni C, Mittelheisser V, Bochler L, Follain G, Mouriaux C, et al. Platelets favor the outgrowth of established metastases. Nat Commun. (2024) 15:3297. doi: 10.1038/s41467-024-47516-w
254. Strauss R, Wehler M, Mehler K, Kreutzer D, Koebnick C, and Hahn EG. Thrombocytopenia in patients in the medical intensive care unit: Bleeding prevalence, transfusion requirements, and outcome*. Crit Care Med. (2002) 30:1765–71. doi: 10.1097/00003246-200208000-00015
255. Thiolliere F, Serre-Sapin AF, Reignier J, Benedit M, Constantin JM, Lebert C, et al. Epidemiology and outcome of thrombocytopenic patients in the intensive care unit: results of a prospective multicenter study. Intensive Care Med. (2013) 39:1460–8. doi: 10.1007/s00134-013-2963-3
256. Abe T, Kubo K, Izumoto S, Shimazu S, Goan A, Tanaka T, et al. Complement activation in human sepsis is related to sepsis-induced disseminated intravascular coagulation. Shock. (2020) 54:198–204. doi: 10.1097/SHK.0000000000001504
257. Arif SH, Ahmad I, Ali SM, and Khan HM. Thrombocytopenia and bacterial sepsis in neonates. Indian J Hematol Blood Transfus. (2012) 28:147–51. doi: 10.1007/s12288-011-0118-7
258. Bedet A, Razazi K, Boissier F, Surenaud M, Hue S, Giraudier S, et al. Mechanisms of thrombocytopenia during septic shock: A multiplex cluster analysis of endogenous sepsis mediators. Shock. (2018) 49:641–8. doi: 10.1097/SHK.0000000000001015
259. Claushuis TAM, van Vught LA, Scicluna BP, Wiewel MA, Klein Klouwenberg PMC, Hoogendijk AJ, et al. Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients. Blood. (2016) 127:3062–72. doi: 10.1182/blood-2015-11-680744
260. Li M-F, Li X-L, Fan K-L, Yu Y-Y, Gong J, Geng S-Y, et al. Platelet desialylation is a novel mechanism and a therapeutic target in thrombocytopenia during sepsis: an open-label, multicenter, randomized controlled trial. J Hematol Oncol. (2017) 10:104–. doi: 10.1186/s13045-017-0476-1
261. Martin CM, Priestap F, Fisher H, Fowler RA, Heyland DK, Keenan SP, et al. A prospective, observational registry of patients with severe sepsis: The Canadian Sepsis Treatment and Response Registry*. Crit Care Med. (2009) 37:81–8. doi: 10.1097/CCM.0b013e31819285f0
262. Ree IMC, Fustolo-Gunnink SF, Bekker V, Fijnvandraat KJ, Steggerda SJ, and Lopriore E. Thrombocytopenia in neonatal sepsis: Incidence, severity and risk factors. PloS One. (2017) 12:e0185581–e. doi: 10.1371/journal.pone.0185581
263. Dong F, Ma N, Chang S, Yang H, Gao M, Liu Q, et al. The role of recombinant human thrombopoietin in critically ill patients with sepsis-associated thrombocytopenia: a clinical study. Zhonghua wei zhong bing ji jiu yi xue. (2020) 32:1445–9. doi: 10.3760/cma.j.cn121430-20201130-00737
264. Menard CE, Kumar A, Houston DS, Turgeon AF, Rimmer E, Houston BL, et al. Evolution and impact of thrombocytopenia in septic shock: a retrospective cohort study. Crit Care Med. (2019) 47:558–65. doi: 10.1097/CCM.0000000000003644
265. Chen C, Wu S, Chen J, Wu J, Mei Y, Han T, et al. Evaluation of the association of platelet count, mean platelet volume, and platelet transfusion with intraventricular hemorrhage and death among preterm infants. JAMA network Open. (2022) 5:e2237588–e. doi: 10.1001/jamanetworkopen.2022.37588
266. Sola-Visner MC. Platelet transfusions in neonates — Less is more. New Engl J Med. (2019) 380:287–8. doi: 10.1056/NEJMe1813419
267. Curley A, Stanworth SJ, Willoughby K, Fustolo-Gunnink SF, Venkatesh V, Hudson C, et al. Randomized trial of platelet-transfusion thresholds in neonates. New Engl J Med. (2019) 380:242–51. doi: 10.1056/NEJMoa1807320
268. Wang K, Lu D, and Wang F. Subphenotypes of platelet count trajectories in sepsis from multi-center ICU data. Sci Rep. (2024) 14:20187–. doi: 10.1038/s41598-024-71186-9
269. Claushuis T, Van Vught L, Scicluna B, Wiewel M, Klein Klouwenberg P, Hoogendijk A, et al. Molecular Diagnosis and Risk Stratification of Sepsis Consortium. Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients. Blood. (2016) 127:3062–72. doi: 10.1182/blood-2015-11-680744
270. Zhang M-K, Xu T-Q, Zhang X-J, Rao Z-G, He X-X, Wu M-Q, et al. Thrombocytopenia in 737 adult intensive care unit patients: a real-world study of associated factors, drugs, platelet transfusion, and clinical outcome. SAGE Open Med. (2020) 8:2050312120958908. doi: 10.1177/2050312120958908
271. Nellis ME, Karam O, Mauer E, Cushing MM, Davis PJ, Steiner ME, et al. Platelet transfusion practices in critically ill children. Crit Care Med. (2018) 46:1309–17. doi: 10.1097/CCM.0000000000003192
272. He S, Fan C, Ma J, Tang C, and Chen Y. Platelet transfusion in patients with sepsis and thrombocytopenia: A propensity score-matched analysis using a large ICU database. Front Med. (2022) 9:830177–. doi: 10.3389/fmed.2022.830177
273. Maier CL, Stanworth SJ, Sola-Visner M, Kor D, Mast AE, Fasano R, et al. Prophylactic platelet transfusion: is there evidence of benefit, harm, or no effect? Transfusion Med Rev. (2023) 37:150751. doi: 10.1016/j.tmrv.2023.150751
274. Zhou W, Fan C, He S, Chen Y, and Xie C. Impact of platelet transfusion thresholds on outcomes of patients with sepsis: analysis of the MIMIC-IV database. Shock (Augusta Ga). (2022) 57:486–93. doi: 10.1097/SHK.0000000000001898
275. Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. (2017) 43:304–77. doi: 10.1007/s00134-017-4683-6
276. Estcourt LJ, Birchall J, Allard S, Bassey SJ, Hersey P, Kerr JP, et al. Guidelines for the use of platelet transfusions. Br J Haematology. (2016) 176:365–94. doi: 10.1111/bjh.2017.176.issue-3
277. Liu Z. Standard definitions for the use of whole blood and blood components. Beijing: People's Medical Publishing House (2019).
278. Stanworth SJ and Shah A. How I use platelet transfusions. Blood. (2022) 140:1925–36. doi: 10.1182/blood.2022016558
279. Wu S, Chen Q, Pan J, and Zhou A. Platelet transfusion and mortality in patients with sepsis-induced thrombocytopenia: A propensity score matching analysis. Vox Sanguinis. (2022) 117:1187–94. doi: 10.1111/vox.v117.10
280. Blumberg N. A randomized trial of neonatal platelet transfusion thresholds. New Engl J Med. (2019) 380:1584–. doi: 10.1056/NEJMc1902638
281. Zong X, Wang X, Liu Y, Li Z, Wang W, Wei D, et al. Antiplatelet therapy for patients with COVID-19: Systematic review and meta-analysis of observational studies and randomized controlled trials. Front Med (Lausanne). (2022) 9:965790. doi: 10.3389/fmed.2022.965790
282. Abdullah A, Froess J, Berger JS, Geraci MW, Gong MN, Kornblith L, et al. Effect of P2Y12 inhibitors, SGLT2 inhibitors and P-selectin inhibitors on one-year quality-of-life outcomes in critically ill patients hospitalized for COVID-19: A pre-specified secondary analysis of the ACTIV4a randomized clinical trial. Am J Respir Crit Care Med. (2025) 211:A2867–A. doi: 10.1164/ajrccm.2025.211.Abstracts.A2867
283. Aronow W. Faculty opinions recommendation of effect of antithrombotic therapy on clinical outcomes in outpatients with clinically stable symptomatic COVID-19: the ACTIV-4B randomized clinical trial. Faculty Opin – Post-Publication Peer Rev Biomed Literature: H1 Connect. (2021) 326(17):1703–12. doi: 10.3410/f
284. Athale J and Danner RL. In critically ill patients with COVID-19, antiplatelet therapy did not increase organ support–free days at 21 d. Ann Internal Med. (2022) 175:JC80. doi: 10.7326/J22-0046
285. Samama CM. Faculty Opinions recommendation of Aspirin in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Faculty Opin – Post-Publication Peer Rev Biomed Literature: H1 Connect. (2022). doi: 10.3410/f
286. Egi M, Ogura H, Yatabe T, Atagi K, Inoue S, Iba T, et al. The Japanese clinical practice guidelines for management of sepsis and septic shock 2020 (J-SSCG 2020). J Intensive Care. (2021) 9:53. doi: 10.1186/s40560-021-00555-7
287. van der Poll T. Recombinant human soluble thrombomodulin in patients with sepsis-associated coagulopathy. JAMA. (2019) 321:1978. doi: 10.1001/jama.2019.5792
288. Olas B. The antioxidant, anti-platelet and anti-coagulant properties of phenolic compounds, associated with modulation of hemostasis and cardiovascular disease, and their possible effect on COVID-19. Nutrients. (2022) 14(7):1390. doi: 10.3390/nu14071390
289. Cao M, Wang G, and Xie J. Immune dysregulation in sepsis: experiences, lessons and perspectives. Cell Death Discov. (2023) 9:465–. doi: 10.1038/s41420-023-01766-7
290. Pei F, Yao R-Q, Ren C, Bahrami S, Billiar TR, Chaudry IH, et al. Expert consensus on the monitoring and treatment of sepsis-induced immunosuppression. Mil Med Res. (2022) 9:74–. doi: 10.1186/s40779-022-00430-y
291. Gando S. Role of fibrinolysis in sepsis. Semin Thromb Hemostasis. (2013) 39:392–9. doi: 10.1055/s-0033-1334140
292. Hayakawa M, Sawamura A, Gando S, Jesmin S, Naito S, and Ieko M. A low TAFI activity and insufficient activation of fibrinolysis by both plasmin and neutrophil elastase promote organ dysfunction in disseminated intravascular coagulation associated with sepsis. Thromb Res. (2012) 130:906–13. doi: 10.1016/j.thromres.2012.01.015
293. De Luca L, Pugliese FR, Susi B, Navazio A, Corda M, Fabbri A, et al. ANMCO/SIMEU consensus document on the use of reversal agents for antithrombotic therapies in patients with ongoing bleeding or at high risk of haemorrhagic events. Eur Heart J Suppl. (2024) 26:ii211–ii20. doi: 10.1093/eurheartjsupp/suae033
294. Baharoglu MI, Al-Shahi Salman R, Cordonnier C, Koopman MM, Manson L, Susen S, et al. PATCH trial: explanatory analyses. Blood. (2020) 135:1406–9. doi: 10.1182/blood.2019003298
295. Lee RH, Kasthuri RS, and Bergmeier W. Platelet transfusion for patients with platelet dysfunction: effectiveness, mechanisms, and unanswered questions. Curr Opin hematology. (2020) 27:378–85. doi: 10.1097/MOH.0000000000000608
296. Prodan CI. Platelets after intracerebral haemorrhage: more is not better. Lancet. (2016) 387:2577–8. doi: 10.1016/S0140-6736(16)30478-0
297. Aslam R, Speck ER, Kim M, Crow AR, Bang KWA, Nestel FP, et al. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-α production in vivo. Blood. (2006) 107:637–41. doi: 10.1182/blood-2005-06-2202
298. Assinger A, Schrottmaier WC, Salzmann M, and Rayes J. Platelets in sepsis: an update on experimental models and clinical data. Front Immunol. (2019) 10:2019. doi: 10.3389/fimmu.2019.01687
299. Balkenhol J, Kaltdorf KV, Mammadova-Bach E, Braun A, Nieswandt B, Dittrich M, et al. Comparison of the central human and mouse platelet signaling cascade by systems biological analysis. BMC Genomics. (2020) 21:897. doi: 10.1186/s12864-020-07215-4
300. Abdulnour R-EE, Dalli J, Colby JK, Krishnamoorthy N, Timmons JY, Tan SH, et al. Maresin 1 biosynthesis during platelet-neutrophil interactions is organ-protective. Proc Natl Acad Sci United States America. (2014) 111:16526–31. doi: 10.1073/pnas.1407123111
301. Sugimoto MA, Sousa LP, Pinho V, Perretti M, and Teixeira MM. Resolution of inflammation: what controls its onset? Front Immunol. (2016) 7:160–. doi: 10.3389/fimmu.2016.00160
302. Sansbury BE and Spite M. Resolution of acute inflammation and the role of resolvins in immunity, thrombosis, and vascular biology. Circ Res. (2016) 119:113–30. doi: 10.1161/CIRCRESAHA.116.307308
303. Nathan C and Ding A. Nonresolving inflammation. Cell. (2010) 140:871–82. doi: 10.1016/j.cell.2010.02.029
304. Custer B, Bloch EM, Bryant BJ, D'Alessandro A, Delaney M, Goel R, et al. Proceedings of the 2022 NHLBI and OASH state of the science in transfusion medicine symposium. Transfusion. (2023) 63:1074–91. doi: 10.1111/trf.17296
Keywords: platelets, infection, immunoregulation, immunothrombosis, pathogen clearance, immune response, immunity, host defense
Citation: Hu Y, Dai S, Qiao C, Ye Y, Ren J, Wang K, Li L and Liu Z (2025) Platelets in infection: intrinsic roles and functional outcomes. Front. Immunol. 16:1616783. doi: 10.3389/fimmu.2025.1616783
Received: 23 April 2025; Accepted: 03 June 2025;
Published: 07 July 2025.
Edited by:
Guoxing Wang, Sanofi, United StatesReviewed by:
Huirui Wang, Second Affiliated Hospital of Henan University of Science and Technology, ChinaYun Zhao, BenQ Medical Center, China
Copyright © 2025 Hu, Dai, Qiao, Ye, Ren, Wang, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Wang, a3dhbmctc3VyZ2VvbkBmb3htYWlsLmNvbQ==; Ling Li, bGlsaW5nQHN3anR1LmVkdS5jbg==; Zhong Liu, bGl1ekBpYnQucHVtYy5lZHUuY24=
†These authors have contributed equally to this work